

Abstract
Background
Isoniazid (INH) metabolism depends on the N-acetyl transferase 2 (NAT2) enzyme, whose maturation process remains unknown in low birth weight (LBW) and preterm infants. We aimed to assess INH exposure and safety in infants receiving oral tuberculosis prevention.
Methods
This population pharmacokinetics (PK) analysis used INH and N-acetyl-isoniazid (ACL) concentrations in infants (BW ≤ 4 kg), including preterm, with follow-up for 6 months. PK parameters were described using nonlinear mixed effects modeling. Simulations were performed to assess INH exposure and optimal dosing regimens, using 2 targets: Cmax at 3–6 mg/L and area under the curve (AUC) ≥ 10.52 mg h/L.
Results
We included 57 infants (79% preterm, 84% LBW) in the PK analysis, with a median (range) gestational age of 34 (28.7–39.4) weeks. At the time of sampling, postnatal age was 2.3 (0.2–7.3) months and weight (WT) was 3.7 (0.9–9.3) kg. NAT2 genotype was available in 43 (75.4%) patients (10 slow, 26 intermediate, and 7 fast metabolizers). Ninety percent of NAT2 maturation was attained by 4.4 post-natal months. WT, postmenstrual age, and NAT2 genotype significantly influenced INH exposure, with a 5-fold difference in AUC between slow and fast metabolizers for the same dose. INH appeared safe across the broad range of exposure for 61 infants included in the safety analysis.
Conclusions
In LBW/preterm infants, INH dosing needs frequent adjustment to account for growth and maturation. Pharmacogenetics-based dosing regimens is the most powerful approach to deliver safe and equalized exposures for all infants, because NAT2 genotype highly impacts INH pharmacokinetic variability.
Keywords: neonate, pediatric, tuberculosis, N-acetyl-isoniazid, pharmacokinetics
Isoniazid dosing should be frequently adjusted to body weight in infants because growth and maturation are rapid. Given the 5-fold difference in isoniazid exposure between slow and fast NAT2 metabolizers, identification of metabolizer status can optimize dosing.
Tuberculosis (TB) chemoprophylaxis is recommended for infants born to mothers with TB. In the absence of prophylaxis, the risk to the young children of developing TB approaches 20% within 2 years of exposure, and mortality can reach up to 19% [1, 2]. Isoniazid (INH) is a first-line anti-TB drug and a cornerstone for TB treatment and prevention [3].
Although frequently used, the interindividual variability (IIV) is large due to a genetically determined metabolic pathway. INH is acetylated by the N-acetyl transferase 2 (NAT2) enzyme to N-acetyl-isoniazid (ACL). Individuals can be divided into slow, intermediate, and fast metabolizers using NAT2 genotyping [4] or its surrogate ACL/INH ratio [5, 6]. Adult studies showed that NAT2 genotype-guided dosing can improve INH exposure [7, 8] and reduce its toxicity [9]. Although there is no recommendation regarding pharmacogenetics-based therapy, NAT2 genotype could also be relevant in infant and children.
Only limited data described INH pharmacokinetics (PK) and NAT2 maturation in preterm (<37 weeks gestational age) and/or low-birth-weight (LBW, <2500 g) infants, despite such infants having physiological characteristics that influence pharmacokinetic (PK) parameters and distinct maturation function [10]. Developmental pharmacology highlights the evolution in PK through infancy and childhood, incorporating changes in body composition and enzyme maturation. For example, the ratio of liver weight to body weight (WT) is greater in infants, leading to a higher clearance for some drugs [11]. Because NAT2 metabolism is active in utero prior to delivery and matures during the first year of life [12], it is mandatory to understand the maturation of NAT2 activity in this vulnerable population to use INH effectively and safely. Pediatric INH PK studies have been mostly performed in young children and adolescents. Six PK studies demonstrated that NAT2 genotype influenced INH clearance, with four studies also showing maturation over time [13–18]. Bekker et al conducted the only PK study in preterm and/or LBW infants and demonstrated a 2.5-fold difference in INH exposure between slow and fast metabolizers for the same dose [19]. However, they could not build a population PK model incorporating a developmental pharmacogenetic approach and none of these studies reported ACL concentrations or ACL/INH ratio.
The present PK study was conducted in predominantly preterm infants receiving prophylactic INH. We aimed to describe the maturation of NAT2 activity, the major pathway for INH metabolism, enabling development of dosing regimen guidance in this vulnerable population.
METHODS
Study Population
The IMPAACT P1106 trial was a phase IV prospective, multi-arm trial conducted at two South African sites, the Family Centre for Research with Ubuntu (FAMCRU) in Tygerberg Hospital, Cape Town, and the Perinatal HIV Research Unit (PHRU) in Chris Hani Baragwanath Hospital, Johannesburg. Infants with birth weight ≤4000 grams (preterm or full-term) who were receiving INH as part of clinical care for TB prophylaxis were eligible for enrollment. Study infants were followed for 6 months from study entry. Patients with any severe congenital malformation or other medical condition incompatible with life or that would interfere with study participation or interpretation were excluded.
Study Design
Participants received approximately 10 mg/kg/day of INH, as oral suspension (50 mg/mL) formulated in each hospital pharmacy or as part of a 100-mg tablet crushed in water, as prescribed by their clinical care provider [20].
Blood samples (0.2 mL) were collected, using a semi-intensive PK sampling strategy at birth, 4, 6, 10, 16 and 24 weeks of age. At each visit, 2–3 samples were collected (1.5 and 4 hours postdosing, and an optional 24-hour sample).
The NAT2 genotype was quantified and categorized into slow, intermediate, or fast metabolizers.
Patient characteristics were recorded, including sex, intrauterine growth, gestational age, postnatal age (PNA) defined as the time elapsed after birth, postmenstrual age (PMA) defined as gestational age at birth plus PNA, WT and height at birth and on the day of each visit, ethnicity, cotreatments (nevirapine, lopinavir-ritonavir, lamivudine, zidovudine and rifampicin), serum creatinine, hemoglobin, and alanine aminotransferase.
Assay
INH and ACL PK samples were analyzed at the Division of Clinical Pharmacology, University of Cape Town, using previously described methods [21].
DNA was extracted to analyze 7 NAT2 single-nucleotide polymorphisms (SNPs): rs1801279 (in position 191), rs1041983 (in position 282), rs1801280 (in position 341), rs1799929 (in position 481), rs1799930 (in position 590), rs1208 (in position 803), and rs1799931 (in position 857). The alleles were defined following the SNPedia classification [22].
Population Pharmacokinetics
Data were analyzed using nonlinear mixed effects modeling, using NONMEM (version 7.4). Data set manipulation and data visualization were performed using R software (version 3.5.3). The model was evaluated using Perl-speaks-NONMEM (PsN, version 4.9.0) and vpc package (version 1.2.1).
Both 1 and 2-compartment models with first-order absorption and elimination rates were tested. INH and ACL concentrations below the limit of quantification (LOQ at 0.1 mg/L for INH and 0.05 mg/L for ACL) were set at half the value of the LOQ. Inter-individual and inter-occasion variability (IIV and IOV) were modeled exponentially assuming a log-normal distribution. Proportional, additive, and combined error models were explored to model residual variability.
The tested covariates were WT, gestational age, PNA, PMA, height, intrauterine growth, sex, nevirapine cotreatment, serum creatinine, and NAT2 genotype. WT was tested using the typical weight-based allometric rule. For missing NAT2 genotypes, a mixture model was used to determine the most probable genotype [23].
A stepwise covariate model building procedure was used to evaluate covariates [24, 25], based on a forward inclusion followed by a backward deletion approach. A covariate was retained if its effect was biologically plausible, if it reduced the PK parameters IIV and if the effect was significant in both forward selection (P < .05) and backward elimination (P < .01).
Model performance was evaluated by graphical assessment of goodness-of-fit plots and objective function value (OFV). Simulation-based diagnostics, such as visual predictive checks (VPC) were used for model evaluation. Parameter precision was further evaluated performing 500 nonparametric bootstrap analyses, with 95% confidence interval (CI) of each parameter distribution [24].
Isoniazid Exposure and Dosing Evaluation
Monte Carlo simulations were performed to explore INH exposure for 2 targets: Cmax between 3 and 6 mg/L and AUC0-∞ ≥ 10.52 mg h/L, both corresponding to 90% of the maximum early bactericidal activity [26, 27]. An optimal dosing regimen was defined as a probability to attain the AUC0-∞ ≥90%.
For the first simulation, we evaluated mg/kg dosing, using WHO recommended doses at 10 (7–15) mg/kg/day [20]. For the second simulation, we evaluated a proposed weight-band dosing strategy, using the revised 2021 Department of Health South African TB drug dosing chart developed for dispersible pediatric fixed-dose combination (75/50 mg of rifampicin and INH). INH dosing recommendations were divided into three weight-bands (25 mg for 2–2.9 kg; 37.5 mg for 3–3.9 kg; and 50 mg for 4–7.9 kg) [28]. The demographics of simulated patients were representative of our initial cohort.
Molar acetyl-Isoniazid/ Isoniazid Ratio
Molar ACL/INH ratio was determined at 3-hours after dosing (H3), for comparison with NAT2 genotypes. We determined thresholds for slow and fast metabolizers when 100% of the infants were below or above the latter.
Safety
Adverse events (AEs) were classified as expected or unexpected. Expected AEs were pre-identified as events commonly associated with prematurity or LBW present on the day of study visit. Unexpected AEs were defined as unanticipated events not commonly associated with prematurity and graded according to the Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events [29]. AEs were evaluated from entry to week 24 and the primary safety endpoint included death and any Grade 3/4 AEs.
Ethics
The study was approved by the research ethics committees of Stellenbosch University (M13/08/037) and the University of the Witwatersrand (140303) and was registered at https://clinicaltrials.gov/ (NCT02383849). Written informed consent was obtained from the infant’s legal representatives after oral and written information.
RESULTS
Patients
Sixty-one infants received preventive INH from August 2015 to September 2019. Four infants were excluded from the PK analysis due to an absence of all samples. All were included in the safety analysis.
Fifty-seven patients had 624 measured plasma INH and ACL concentrations in the PK analysis (Table 1). Forty-five (79%) infants were preterm and 48 (84%) were LBW. Median (range) INH doses were 10.1 (8.1–38.3) mg/kg/day at first visit and 10.7 (2.9–18.1) mg/kg/day at last visit. Also, 8 (14%) patients received the same INH dose throughout the study, without any adaptation based on weight gain. NAT2 genotype was determined for 43 (75.4%) patients, with 10 slow, 26 intermediate, and 7 fast metabolizers. Supplementary Figure 1 depicts the INH and ACL concentrations versus time, according to NAT2 genotype.
Table 1.
Patients’ Characteristics in the Pharmacokinetics Analysis
| Parameter | Cohort, n = 57 n (%) or Median (Range) |
|---|---|
| Demographic data | |
| Sex ratio M/F | 24/33 |
| Age at birth (GA weeks + daysa) | 34 (28.7–39.4) |
| 28 w – 29 w + 6 d | 4 (7) |
| 30 w – 33 w + 6 d | 23 (40.3) |
| 34 w – 36 w + 6 d | 18 (31.6) |
| ≥37 GA | 12 (21.1) |
| Postnatal age at first visitb (months) | 0.6 (0.2–2.9) |
| Body weight at first visitb (kg) | 2.1 (0.9–4.7) |
| Height at first visitb (cm) | 45.1 (34–55.5) |
| Intrauterine growthc | |
| 10th–90th percentile | 38 (66.7) |
| <10th percentile | 8 (14) |
| <3rd percentile | 11 (19.3) |
| Postnatal age throughout the study (months) | 2.3 (0.2–7.3) |
| Postmenstrual age throughout the study (months) | 10.1 (7.2–15.8) |
| Body weight throughout the study (kg) | 3.7 (0.9–9.3) |
| Height throughout the study (cm) | 50.1 (34–73) |
| Clinical data | |
| Ethnicity (Black/colored/other) | 39/17/1 |
| Infants born to HIV-infected mother | 34 (59.6) |
| Treated with lopinavir | 1 (1.8) |
| Treated with nevirapine | 32 (56.1) |
| Treated with lamivudine | 2 (3.5) |
| Treated with zidovudine | 25 (43.9) |
| Treated with rifampicin | 4 (7) |
| Biological data for all visits | |
| Serum creatinine (mg/dL) | 0.42 (0.2–0.85) |
| Hemoglobin (g/dL) | 9.6 (7.6–12.6) |
| ALT (IU/L) | 10 (3–30) |
| Pharmacological data | |
| Samples per patient | 12 (2–14) |
| No. of occasions per patient | 6 (1–7) |
| Daily dosing regimen (mg/kg) | 10.3 (2.9–38.3) |
| Treated with oral suspension | 44 (77.2) |
| Treated with tablet | 28 (49.1) |
| Pharmacogenomics data (NAT2 genotype) | |
| Slow metabolizer | 10 (17.5) |
| Intermediate metabolizer | 26 (45.6) |
| Fast metabolizer | 7 (12.3) |
| Unknown classification | 4 (7) |
| Missing data | 10 (17.5) |
Abbreviations: ALT, alanine aminotransferase; GA, gestational age; HIV, human immunodeficiency virus; NAT2, N-acetyl transferase 2.
In sum, 28 weeks equal to 6.5 months, 30 weeks equal to 7 months, 34 weeks equal to 8 months, and 37 weeks equal to 8.6 months (term).
For 39 patients, the first visit was at birth.
Using the Fenton chart.
Population Pharmacokinetic Modeling
Data was best fitted by a one-compartment model for INH and ACL, with first-order absorption and elimination. Absorption constant (KA) was fixed to 3, according to literature revision and sensitivity analysis [18]. IIV was estimated for INH and ACL clearance (CLp/F and CLm/(Fxfm), respectively) with a significant correlation between them. Residual variability was described by a combined (proportional plus additive) error model for INH, and a proportional error model for ACL.
Allometric scaling based on WT was significant, with exponents fixed at 0.75 for clearances and 1 for V (P < .001). NAT2 genotype had a significant impact on CLp/F, using 3 genotypes (P < .01). A mixture model was used for the 14 (24.6%) infants with unknown genotype, resulting in assignment of 9 as slow metabolizers, 5 as intermediate metabolizers, and none as fast metabolizers. The probability for being a slow metabolizer was 58%. CLp/F was estimated to be 1.1 L/h in a 3.7 kg slow metabolizer and increased up to 2.7 L/h (145%) and 5.5 L/h (400%) for the intermediate and fast metabolizers, respectively (P < .001). IOV was implemented on CLm/(Fxfm) using 3 different occasions. Finally, integration of the effect of PMA on CLp/F was significant (P < .01), using the same maturation function for all NAT2 metabolizers [30]. Ninety percent of NAT2 maturation was attained by 4.4 postnatal months (Supplementary Figure 2). The final PK parameters estimates are summarized in Table 2. The VPC for INH and ACL, according to the NAT2 genotype, are shown in Figure 1.
Table 2.
Estimates of the Final Population Pharmacokinetics Parameters of Isoniazid and Acetyl Isoniazid, Standardized for a Body Weight of 3.7 kg
| Parameter | Estimate (RSE, %) | Bootstrap (95% CI) |
|---|---|---|
| KA (/h) | 3a | … |
| CLp/F slow (L/h) | 1.1 (12) | .8–2.6 |
| CLp/F intermediate (L/h) | 2.7 (11.3) | 2–6.3 |
| CLp/F fast (L/h) | 5.5 (12.5) | 3.7–13.9 |
| Vp/F (L) | 6.3 (4.9) | 5.8–6.9 |
| CLm/(Fxfm) (L/h) | 0.8 (10.8) | .6–1 |
| Vm/(Fxfm) (L) | 7.9 (12.1) | 6.5–10.6 |
| HILL | 4 (14.6) | 1.4–10 |
| PMA50 (months) | 7.7 (6.9) | 6.9–16.7 |
| Mixture model probabilityb (%) | 58 (27.6) | 22–89 |
| Proportional residual error INH(%) | 45 (5.5) | .4–.5 |
| Additive residual error INH (mg/L) | 0.2 (20.6) | .04–.3 |
| Proportional residual error ACL(%) | 35 (6.5) | .3–.4 |
| IIV CLp/F (CV, %) | 40.4 (22) | 29.1–51.5 |
| IIV CLm/(Fxfm) (CV, %) | 67.1 (19.2) | 40.2–97.9 |
| Correlation IIV CLp/F-CLm/(Fxfm) (%) | 60.3 (0.1) | 40.2–97.9 |
| BOV (CV, %) | 51.3 (20.2) | 27.8–89.8 |
The typical parameters refer to an infant weighing 3.7 kg, where ,, and.
Abbreviations: ACL, acetyl isoniazid; BOV, between-occasion variability; CI, confidence interval; CLm/(Fxfm), ACL clearance; CLp/F, INH clearance; IIV, inter-individual variability; CV, coefficient of variation calculated with where is the estimated variance; fm, fraction metabolized from INH to ACL, fixed to 1; HILL, hill coefficient; INH, isoniazid; PMA50, age for which CLp/F has attained half of its adult value in postmenstrual age; RSE, relative standard error; Vm/(Fxfm), ACL volume of distribution; Vp/F, INH volume of distribution.
Fixed parameter.
Probability to be assigned to the slow metabolizer group with the mixture model.
Figure 1.

Visual predictive check for the parent (INH) and the metabolite (ACL), according to the metabolizer status (slow, intermediate, and fast), obtained from 500 simulated data sets. Solid red lines represent the 50th percentile of the observations, dotted blue lines represent the 2.5th and 97.5th percentiles of the observations; red areas represent the 95% prediction intervals for the 50th percentile, blue areas represent the 95% prediction intervals for the 2.5th and 97.5th percentiles. Abbreviations: Abbreviations: ACL, N-acetyl-isoniazid; INH, isoniazid.
Evolution of Clearance
The cohort was followed until 6 months of age. The distribution of CLp/F and its evolution during follow-up is depicted in Figure 2. We defined ΔCLp/F as the difference of CLp/F between the last and first visit. Median ΔCLp/F was near zero for slow and intermediate metabolizers and was 0.13 (0.06–0.2) L/h/kg for fast metabolizers (P < .001), corresponding to 15% increase. The typical CLp/F for an individual weighing 3.7 kg depending on their NAT2 genotype and PMA is depicted in Supplementary Figure 2.
Figure 2.

Distribution of the INH clearance, according to the NAT2 genotype (in red, blue, and green for slow, intermediate, and fast metabolizers, respectively), at first and last visit of follow-up. Each line represents 1 individual. For 39 patients, the first visit was at birth. For 48 patients, the last visit of follow-up was at 6 months of age. Abbreviations: INH, isoniazid; NAT2, N-acetyl transferase 2.
Isoniazid Exposure
Based on the individual PK parameters, median (range) for INH Cmax was 4.8 (1.2–19.9) mg/L and for AUC0-∞ 21.1 (3.9–237.5) mg h/L. Fifteen (26.3%) patients, all of whom received <10 mg/kg/day, experienced a Cmax <3 mg/L. Sixteen (28%) patients, all intermediate or fast metabolizers, experienced an AUC0-∞ <10.52 mg h/L.
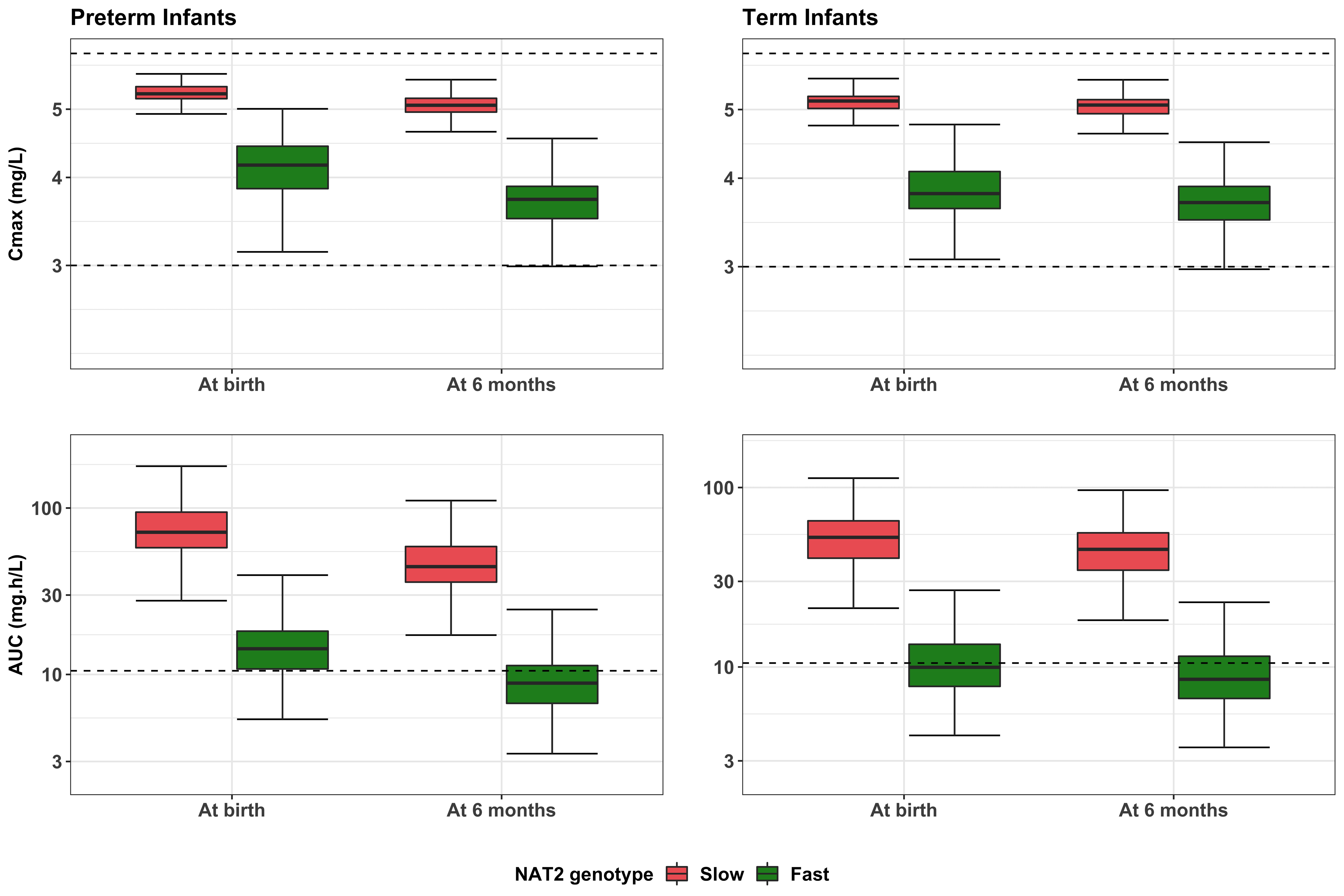
Cmax and AUC0-∞ depended on the NAT2 genotype (Figure 3). Slow metabolizers had the highest Cmax (5.9 [2–19.9] mg/L) and AUC0-∞ (48.8 [17–237] mg h/L). Fast metabolizers exceeded the Cmax target (3.7 [2.6–6] mg/L) but had low AUC0-∞ (8.3 [4–21.2] mg h/L) even with 15 mg/kg/day.
Figure 3.

Distribution of the Cmax and AUC0-∞ of INH, with respect to the dosing regimen and the NAT2 genotype (slow, n = 19, intermediate, n = 31 and fast, n = 7). Each dot represents an occasion, at first and last visit. Each line represents one individual. For 39 patients, the first visit was at birth. For 48 patients, the last visit of follow-up was at 6 months of age. Dotted black lines represent the target (Cmax between 3 and 6 mg/L and AUC0-∞ above 10.52 mg h/L). Abbreviations: AUC, area under the curve; INH, isoniazid.
Simulations
We performed 500 Monte Carlo simulations on a built dataset of 54 simulated patients (Figure 4). Standard dosing regimens of 10 mg/kg ensured PK target attainment for over 92% of the slow and intermediate metabolizers. For fast metabolizers, 20 mg/kg was necessary to attain the AUC target for 90% of the infants. The proposed weight-bands dosing gave a wider range of Cmax and AUC0-∞ in the upper weight-band, with fast metabolizers always at higher risk for underexposure. The most impactful parameter on INH exposure was NAT2 genotype.
Figure 4.

Simulations (n = 500 on a built data set of 54 simulated patients) showing the distribution of Cmax and AUC0-∞ of INH, with respect to the dosing regimen and the NAT2 genotype. Dotted black lines represent the target (Cmax between 3 and 6 mg/L and AUC0-∞ above 10.52 mg h/L). The exact dose explored the median WHO recommended dosing regimen at 10 mg/kg. Flat dose explored the revised 2020 Western Cape government tuberculosis drug dosing chart, using 3 weight-bands and dose, 2–2.9 kg infants treated with 25 mg, 3–3.9 kg infants treated with 37.5 mg, 4–7.9 kg infants treated with 50 mg. We explored the model-informed dose that allowed to attain the AUC target for all infants. Abbreviations: AUC, area under the curve; INH, isoniazid; NAT2, N-acetyl transferase 2.
Two specific situations (LBW and preterm infant born at 28 weeks and 0.58 kg and full-term infant born at 3.5 kg) were also simulated 500 times according to NAT2 genotype, using the median WHO recommended dose of 10 mg/kg/day (Supplementary Figure 3). Again, NAT2 genotype was more impactful on INH exposure than WT or age.
Molar acetyl-Isoniazid/ Isoniazid Ratio
Molar ACL/INH ratio at 3-hours after dosing (H3) is presented in Supplementary Figure 4. All infants with a ratio <0.4 were slow metabolizers, and all >4.1 were fast metabolizers.
Safety
Safety data were available for 61 infants, of which 53 (87%) were LBW and 47 (77%) were preterm infants. INH was administered for a median (range) of 167 (22–224) days.
No infant on INH developed TB disease. Across the broad range of INH exposure and NAT2 genotype, INH appeared to be safe and well tolerated. One infant had possibly related Grade 1 gynecomastia, which resolved spontaneously after 3 weeks of discontinuation.
Twenty-five (41%) infants met the composite safety endpoint of death and/or Grade 3/4 AEs, and none of these events were assessed to be related to INH. Three infants died, 2 from sudden infant death syndrome, and 1 from severe pneumonia. In the 13 infants with 19 Grade 3/4 expected AEs, 11 were neonatal sepsis. In the 13 infants with 37 Grade 3/4 unexpected AEs, 16 events were infection (8 pneumonia and 8 gastroenteritis).
DISCUSSION
To our knowledge, this is the first time that the population PK characteristics of INH and its metabolite ACL are reported for preterm and term infants. WT using allometric scaling, NAT2 genotype and PMA significantly affected the PK parameters of INH and ACL. As simulations showed that NAT2 genotype was the most impactful parameter on INH exposure, pharmacogenetics-based dosing should be implemented to optimize INH exposure. We emphasized the existence of 3 subgroups for the NAT2 genotype: slow, intermediate, and fast metabolizers, with an impact on CLp/F. Only 2 previously reported population PK models differentiated the intermediate metabolizers [13, 18]. This group is often combined with fast metabolizers since these categories overlap. However, this group is of interest to illustrate the large variability in the metabolizer status, causing a wide diversity in INH clearance (CL). In pediatric literature, INH CL ranges from 0.1 to 0.38 L/h/kg and from 0.34 to 0.9 L/h/kg for slow and fast metabolizers, respectively [13–18]. CLp/F estimates were 0.3, 0.73, and 1.48 L/h/kg for slow, intermediate, and fast metabolizers, respectively. We assumed that all INH was metabolized into ACL, with CLm/(Fxfm) estimated at 0.22 L/h/kg. These values are lightly above those previously reported, which is consistent with a greater liver mass in younger children [31].
We also highlighted the maturation effect on CL, with an increase of CL between the first and last visit, and therefore a decrease in Cmax and AUC0-∞. Unfortunately, we could not show different maturation functions specific to NAT2 genotype. Previous studies exploring NAT2 activity at different ages indicated that most infants are slow metabolizers, with a transition to fast metabolism during childhood and definitive metabolic status at the age of 4 years [32]. We question this assumption because we showed intermediate and fast metabolizers at very young age, even in preterm infants. As the maturation process is still not well defined from birth through childhood, additional analyses are needed with data from a wider age range of infants and children.
According to our model, INH dosing regimens should be adapted with 3 parameters: NAT2 genotype, WT, and age. The most impactful covariate is the NAT2 genotype. In our study, fast metabolizers were at risk for subtherapeutic exposure, as previously demonstrated in adults [33]. For this reason, 2 adults studies and a systematic review described adjusting INH dosing based on NAT2 genotype [7, 27, 34]. This strategy could also benefit infants, because AUC among fast metabolizers was around 5-fold lower than in slow metabolizers, above that usually reported and poses a risk for subtherapeutic exposure [35]. In contrast, the slow and intermediate metabolizers could be at risk for toxicity at high INH dose, even if not described in our cohort. Even though NAT2 genotyping can be conducted worldwide [36], it is not universally available and can be substituted through using the molar ACL/INH ratio. Previous pediatric studies had validated molar ratio thresholds at 3 hours post dose: <0.48 and >0.77 for slow and intermediate/fast metabolizers, respectively [5], and 0.48 differentiating slow and fast metabolizers [6]. We propose new thresholds for which all infants would be affected: molar ratio < 0.4 for slow metabolizers and >4.1 for fast metabolizers. Regarding WT, some patients received a fixed amount of INH throughout the study without any adaptation. Because infants gain almost 1 kg per month, the absence of dose adjustment for growth might lead to a subtherapeutic exposure [37]. Finally, one might remain cautious about the use of fixed-dose combination, because INH might need independent adaptation.
Our study has several limitations. First, we could not include human immunodeficiency virus (HIV) status in our model, because no infant was living with HIV, and we did not find any drug-drug interaction (especially with antiretrovirals) in this study. Second, we could not study the absorption phase because no PK sample was collected during this period, and no data on food intake were available. We could not evaluate the consequences of breastfeeding due to the passage of INH in human milk. Also, we could not assess the impact of drug formulation on the absorption constant and thus on INH exposure. Finally, because no adverse events or unfavorable treatment outcome (ie, TB disease) occurred, we could not evaluate the safety of an optimized dosing regimen nor the validity of the PK target.
In summary, we showed that WT, PMA, and NAT2 genotype influenced INH and ACL PK parameters during the first weeks of life. Three NAT2 phenotypes were highlighted in this population and impacted INH exposure. Pharmacogenetics-based dose should be implemented to optimize INH exposure, using DNA or ACL/INH ratio, with 4 to 20 mg/kg/day of INH for slow and fast metabolizers. Where metabolizer status cannot be determined 10 mg/kg/day should be recommended, with therapeutic drug monitoring. To optimize INH dosing regimen for all children, we recommend an individual participant-level data meta-analysis using all the published PK studies in children.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. The authors thank the infants and the parents who accepted to participate in this study, making this work possible. They thank the pediatrics group within the Savic lab for their discussions related to this work.
Financial support. Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with cofunding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under award mumbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC), and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contributor Information
Agathe Béranger, Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California, USA.
Adrie Bekker, Family Center for Research with Ubuntu, Department of Paediatrics and Child Health, Stellenbosch University and Tygerberg Hospital, Cape Town, South Africa.
Belén P Solans, Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California, USA.
Mark F Cotton, Family Center for Research with Ubuntu, Department of Paediatrics and Child Health, Stellenbosch University and Tygerberg Hospital, Cape Town, South Africa.
Mark Mirochnick, Division of Neonatology, Department of Pediatrics, Boston University School of Medicine, Boston, Massachusetts, USA.
Avy Violari, Perinatal HIV Research Unit, University of the Witwatersrand, Johannesburg, South Africa.
Jiajia Wang, Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Heath, Boston, Massachusetts, USA.
Mae Cababasay, Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Heath, Boston, Massachusetts, USA.
Lubbe Wiesner, Division of Clinical Pharmacology, Department of Medicine, University of Cape Town, Cape Town, South Africa.
Renee Browning, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, Maryland, USA.
Jack Moye, Division of Extramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, USA.
Edmund V Capparelli, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California, USA; Pediatrics Department, Rady Children’s Hospital San Diego, University of California San Diego, La Jolla, California, USA.
Radojka M Savic, Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California, USA; UCSF Center for Tuberculosis, University of California San Francisco, San Francisco, California, USA.
References
- 1. Martinez L, Cords O, Horsburgh CR, et al. . The risk of tuberculosis in children after close exposure: a systematic review and individual-participant meta-analysis. The Lancet 2020; 395:973–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathad JS, Gupta A.. Tuberculosis in pregnant and postpartum women: epidemiology, management, and research gaps. Clin Infect Dis 2012; 55:1532–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. WHO. Global tuberculosis report. 2020. Available at: https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf. Accessed 2 February 2021.
- 4. Isoniazid. Drug Bank online. Available at: https://go.drugbank.com/drugs/DB00951. Accessed 17 March 2021.
- 5. Keller GA, Fabian L, Gomez M, Gonzalez CD, Diez RA, Girolamo GD.. Age-distribution and genotype-phenotype correlation for N-acetyltransferase in Argentine children under isoniazid treatment. Int J Clin Pharmacol Ther 2014; 52:292–302. [DOI] [PubMed] [Google Scholar]
- 6. Rey E, Gendrel D, Treluyer JM, et al. . Isoniazid pharmacokinetics in children according to acetylator phenotype. Fundam Clin Pharmacol 2001; 15:355–59. [DOI] [PubMed] [Google Scholar]
- 7. Jing W, Zong Z, Tang B, et al. . Population pharmacokinetic analysis of isoniazid among pulmonary tuberculosis patients from China. Antimicrob Agents Chemother 2020; 64:e01736–19,/aac/64/3/AAC.01736-19.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gausi K, Ignatius EH, Sun X, et al. . A semi-mechanistic model of the bactericidal activity of high-dose isoniazid against multi-drug-resistant tuberculosis: results from a randomized clinical trial. Am J Respir Crit Care Med 2021; 204:1327–35. doi: 10.1164/rccm.202103-0534OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pharmacogenetics-based tuberculosis therapy research group, Azuma J, Ohno M, et al. . NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur J Clin Pharmacol 2013; 69:1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu H, Rosenbaum S.. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther JPPT Off J PPAG 2014; 19:262–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van den Anker J, Reed MD, Allegaert K, Kearns GL.. Developmental changes in pharmacokinetics and pharmacodynamics. J Clin Pharmacol 2018; 58:S10–S25. [DOI] [PubMed] [Google Scholar]
- 12. McCarver DG, Hines RN.. The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther 2002; 300:361–66. [DOI] [PubMed] [Google Scholar]
- 13. Zvada SP, Denti P, Donald PR, et al. . Population pharmacokinetics of rifampicin, pyrazinamide and isoniazid in children with tuberculosis: in silico evaluation of currently recommended doses. J Antimicrob Chemother 2014; 69:1339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guiastrennec B, Ramachandran G, Karlsson MO, et al. . Suboptimal antituberculosis drug concentrations and outcomes in small and HIV-coinfected children in India: recommendations for dose modifications. Clin Pharmacol Ther 2018; 104:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aruldhas BW, Hoglund RM, Ranjalkar J, et al. . Optimization of dosing regimens of isoniazid and rifampicin in children with tuberculosis in India. Br J Clin Pharmacol 2019; 85:644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horita Y, Alsultan A, Kwara A, et al. . Evaluation of the adequacy of WHO revised dosages of the first-line antituberculosis drugs in children with tuberculosis using population pharmacokinetic modeling and simulations. Antimicrob Agents Chemother 2018; 62:e00008–18,/aac/62/9/e00008-18.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Panjasawatwong N, Wattanakul T, Hoglund RM, et al. . Population pharmacokinetic properties of antituberculosis drugs in Vietnamese children with tuberculous meningitis. Antimicrob Agents Chemother 2020; 65:e00487–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu R, Kiser JJ, Seifart HI, et al. . The pharmacogenetics of NAT2 enzyme maturation in perinatally HIV exposed infants receiving isoniazid. J Clin Pharmacol 2012; 52:511–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bekker A, Schaaf HS, Seifart HI, et al. . Pharmacokinetics of isoniazid in low-birth-weight and premature infants. Antimicrob Agents Chemother 2014; 58:2229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. World Health Organization. Guidance for national tuberculosis programmes on the management of tuberculosis in children. Second edition. 2014. Available at: https://apps.who.int/iris/bitstream/handle/10665/112360/9789241548748_eng.pdf;jsessionid=E03BC29810CD0B4D0529DCB922CCF071?sequence=1. [PubMed] [Google Scholar]
- 21. Chirehwa MT, McIlleron H, Wiesner L, et al. . Effect of efavirenz-based antiretroviral therapy and high-dose rifampicin on the pharmacokinetics of isoniazid and acetyl-isoniazid. J Antimicrob Chemother 2019; 74:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. NAT2 genotype - SNPedia . Available at: https://www.snpedia.com/index.php/NAT2. Accessed 9 February 2021.
- 23. Carlsson KC, Savic RM, Hooker AC, Karlsson MO.. Modeling subpopulations with the $MIXTURE subroutine in NONMEM: finding the individual probability of belonging to a subpopulation for the use in model analysis and improved decision making. AAPS J 2009; 11:148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindbom L, Pihlgren P, Jonsson N.. PsN-Toolkit—A collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 2005; 79:241–57. [DOI] [PubMed] [Google Scholar]
- 25. Jonsson EN, Karlsson MO.. Automated covariate model building within NONMEM. Pharm Res 1998; 15:1463–68. [DOI] [PubMed] [Google Scholar]
- 26. Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis: drugs. 2002; 62:2169–2183. [DOI] [PubMed] [Google Scholar]
- 27. Donald PR, Parkin DP, Seifart HI, et al. . The influence of dose and N-acetyltransferase-2 (NAT2) genotype and phenotype on the pharmacokinetics and pharmacodynamics of isoniazid. Eur J Clin Pharmacol 2007; 63:633–39. [DOI] [PubMed] [Google Scholar]
- 28. Department of Health, South Africa. 2021. Dosing Guidelines for use of paediatric fixed-dose formulations for management of drug-sensitive tuberculosis. Circular 2021/06/24/EDP/01. South Africa. Available at: https://www.knowledgehub.org.za/system/files/elibdownloads/2021-06/Circular%20Paediatric%20TB%20FDCs-dosing%20tables_June2021_Final.pdf. Accessed 4 February 2022. [Google Scholar]
- 29. U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 1.0. Updated August 2009. Available at: https://rsc.niaid.nih.gov/sites/default/files/table-for-grading-severity-of-adult-pediatric-adverse-events.pdf. Accessed December 2004.
- 30. Anderson BJ, Holford NHG.. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 2008; 48:303–32. [DOI] [PubMed] [Google Scholar]
- 31. Schaaf HS. Isoniazid pharmacokinetics in children treated for respiratory tuberculosis. Arch Dis Child 2005; 90:614–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blake MJ, Castro L, Leeder JS, Kearns GL.. Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med 2005; 10:123–38. [DOI] [PubMed] [Google Scholar]
- 33. Wilkins JJ, Langdon G, McIlleron H, Pillai G, Smith PJ, Simonsson USH.. Variability in the population pharmacokinetics of isoniazid in South African tuberculosis patients: population pharmacokinetics of isoniazid. Br J Clin Pharmacol 2011; 72:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong B-L, D’Cunha R, Li P, et al. . A Systematic review and meta-analysis of isoniazid pharmacokinetics in healthy volunteers and patients with tuberculosis. Clin Ther 2020; 42:e220–e41. [DOI] [PubMed] [Google Scholar]
- 35. McIlleron H, Chirehwa MT.. Current research toward optimizing dosing of first-line antituberculosis treatment. Expert Rev Anti Infect Ther 2019; 17:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sabbagh A, Darlu P, Crouau-Roy B, Poloni ES.. Arylamine N-acetyltransferase 2 (NAT2) genetic diversity and traditional subsistence: a worldwide population survey. PLoS One 2011; 6:e18507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fenton TR, Kim JH.. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr 2013; 13:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.