

Abstract
Analysis of food is essential for safety, quality control, government regulations, and recommendations to answer basic research questions. Capillary electrophoresis-mass spectrometry (CE-MS) is a powerful hyphenated technique in food, beverages, and foodomics for analytes ranging from small organic ions and biochemical compounds to macromolecules. Advantages of CE-MS for food analysis include high efficiency, high resolution, low cost of reagent consumption, fast and green approach in various food research areas. This review offers a comprehensive evaluation of CE-MS application for food analysis published in the open literature in the last decade (July 2010-October 2020). The principles of various CE-MS modes, CE-inductively coupled plasma mass spectrometry, ionization interfaces, and sample preparation methods for multiple types of liquid and solid food analysis are compiled. The latest advances and potential trends are outlined in several food analysis areas where CE-MS could be beneficial.
Keywords: Food Analysis and foodomics, Evolution of CE-MS, Modes of CE-MS, CE-ICP-MS, Interfaces, Solid-phase extraction, Liquid-liquid extraction, QuEChERS Liquid and solid food analysis, Stereoselective food
1. Introduction
Food analysis involves the development, application, and study of analytical methodologies used for characterizing the properties of food. The examination ensures that food contains the correct type and amount of essential ingredients, and its label declarations are right or to determine the levels of nutrients present [1]. The information mentioned above is critical to evaluate the properties of food and control the quality of the produced food that is safe, desirable, and nutritious.
The analysis of food and food components is performed by scientists in all significant sectors of the food industry. Therefore, this community includes food manufacturers, food ingredient suppliers, governmental laboratories, analytical service laboratories, and academic research laboratories. There are many reasons for the analysis of food samples. The first is to ensure food safety. For example, a food may be considered unsafe if it contains toxic chemicals (e.g., pesticides, herbicides, and toxic metals), dangerous bacteria or other microorganisms (e.g., salmonella), or objectionable material (e.g., glass, wool, or insect). The second reason is quality control. The food manufacturer’s goals are to increase its market share continuously. Manufacturers routinely performed quality control to make a profit so that the food products are safe and nutritious to their customers. On the other hand, the food purchaser expects the food properties to be very similar (if not better) from its previous purchase. However, the raw ingredients and the process conditions of food may vary from batch to batch, resulting in changes in the food product's properties in an uncontrolled fashion. To control the food processing, the manufacturer must understand the final properties of food through research and development to consistently produce quality food products. The third reason for food analysis is government regulations and recommendations. There are several regulating agencies such as the United States department of agriculture (USDA), the food and drug Administration (FDA), the Environmental Protection Agency (EPA), and the National Marine Fisheries Service (NMFS). All of the agencies mentioned above are responsible for ensuring that the industrial sector provides safe and nutritional food, allowing consumers to make wise choices about their diet. To eliminate economic fraud and ensure food authenticity, these agencies strive to publish documents containing detailed information and recommendations about food testing and food safety. Finally, many scientists working in universities, government-funded national laboratories, and large food organizations performed food analysis to answer basic research questions. For example, much research is directed towards foodomics. Understanding the structure and interactions of food ingredients through omics technologies to improve public health and knowledge is essential. Thus, in both fundamental and applied food research, new and improved analytical techniques are needed to characterize foods' overall properties and ensure the role each food ingredient plays in determining food properties. The properties include food storage, heating, freezing, and mixing.
Analytical techniques typically used in food analysis includes spectroscopic methods [ultraviolet-visible, fluorescence, nuclear magnetic resonance spectroscopy (NMR), atomic and molecular mass spectrometry (MS)], and separation methods [gas chromatography (GC), high-performance liquid chromatography (HPLC), supercritical fluid chromatography (SFC) and capillary electrophoresis (CE)]. Besides, electrochemical and hyphenated methods based on combining separation to spectroscopy or spectrometry techniques are standard.
Although MS plays a crucial role in food analysis, food matrices' complexity does not allow MS to be a standalone technique. In contrast, HPLC, GC, SFC, and CE, combined with MS detector, provide sensitive, selective, and repeatable methods for food analysis applications [2]. While GC-MS is specifically applicable to the analysis of volatile organic compounds in food and food products, SFC-MS helps analyze thermally unstable and nonvolatile food components that are difficult to analyze by GC-MS. On the other hand, the analysis of polar compounds is complicated by SFC-MS. The use of HPLC-MS ensures several advantages include, quicker and less extensive extraction procedures.
Moreover, HPLC-MS ability to identify and measure a broader range of polar, semipolar, and non-polar food compounds with a wide range of molecular sizes allows this technique to be superior to GC-MS or SFC-MS. Also, LC-MS/MS is most commonly used for proteomic analysis of complex food samples where peptide masses may overlap even using a high-resolution mass spectrometer. The electromigration technique such as CE-MS in its various operational mode is particularly suited for the rapid separation of ionic, weakly ionic, and highly polar compounds with very high resolution. The main advantages include minimum sample and reagent consumption, low cost, and fast analysis. With the decrease in organic solvent consumption, it is environmentally friendly. Although CE-UV detectors' low sensitivity is the main drawback, the sensitivity is improved with CE-MS coupling. Moreover, the sensitivity of CE-MS can be further enhanced using preconcentration techniques and high-resolution mass spectrometry. However, CE-MS is not as stable and robust as LC-MS or GC-MS platforms.
1.1. Identification of studies in capillary electrophoresis-mass spectrometry
First, four databases (PubMed, Science Direct, Sci-Finder, and Web of Science) were searched with the following keywords “capillary electrophoresis mass spectrometry food analysis," "foodomics," "food analysis" "liquid food," "solid food," add meat from July 2010-Oct 2020. The flow chart for the review paper selection process and compounds involved in the food products is shown in Fig.S1. We included only studies involving electromigration techniques such as capillary zone electrophoresis (CZE), micellar electrokinetic chromatography (MEKC), capillary electrochromatography (CEC), capillary isoelectric focusing (CIEF), chiral capillary electrophoresis (CCE) coupled to electrospray ionization mass spectrometry (ESI)-MS, matrix-assisted laser desorption ionization (MALDI)-MS and inductively coupled plasma (ICP)-MS. The review excluded approaches using only NMR, UV-Vis or (MS), sensors, and ELISA. Besides, papers such as CE-UV detection and CE with conductivity or fluorescence detection are not reviewed. We removed duplicate findings using endnotes.
The two techniques, CE and MS, are fascinating areas for food analysis because, in an extensive range of applications, MS is employed than CE. The distribution of papers in CE-MS, MS, and CE represent 8.4% (87), 31.7% (328), and 59.9% (621), respectively (Fig.1). Interestingly, the bar plot comparison shows more papers published in the first seven years by CE than MS, whereas the reverse trend is seen in the last three years. Furthermore, a higher number of MS only papers than CE-MS is found for food analysis because MS is universal and started to be successfully employed earlier than CE-MS. Another reason could be that MS is widely applied in analytical chemistry (with or without separation). On the other hand, CE-MS is a relatively new technique introduced nearly 30 years ago [3]. This fact may account for the lack of well-defined food analysis studies related to CE-MS. In the future, as these coupled techniques become more widespread in food science, one may obtain a more well-defined comparison between CE and MS. As illustrated in Fig. S2, for a total of 87 publications found on CE-MS, 19 were associated with publication in Electrophoresis, 11 in Journal of Chromatography, 10 in Food Science, 6 in Analytica Chimica Acta, 5 in Talanta, and less than five in the various journals.
Fig. 1.

Bar plots showing trends of papers on CE, MS, and CE-MS in food analysis published in the last 10 years. Papers under CE only includes detectors such as UV, LIF, ECLD, indirect detection, and other miscellaneous detectors. Papers under CE-MS includes all CE modes coupled to ESI-MS, ESI-MS/MS, offline-MALDI-MS, and Chip-based CE-MS and DART-MS. Papers under MS only includes stand-alone ESI, APPI, APCI, and ICP-MS ionization methods with mass analyzers. On the other hand, GC-MS, LC-MS, CE-MS, PTR-MS, MALDI-TOF-MS, ambient mass spectrometry techniques such as direct analysis in real time (DART-MS) was excluded. All papers including review articles and publications before July 2010 are excluded.
This review evaluates electromigration techniques, sample preparation, and application developments regarding CE-MS for food analysis and foodomics. The articles published between July 2010 and October 2020 are reported, highlighting the critical milestones. First, the operation principles on using various electromigration techniques or CE modes coupled to MS include two standard interfaces (sheath and sheathless) and three different ion sources (ESI, MALDI, and ICP) presented. After providing an overview of the extraction methods, the various extraction methods for CE-MS are briefly discussed. The application and examples of the developed CE-MS methodologies in food analysis are correlated to food safety, food quality authenticity, bioactivity as well as food processing and storage conditions. To the best of our knowledge, no comprehensive review is conducted that integrates the papers published in the last ten years on CE-MS for food analysis in this journal.
2. Fundamentals of electromigration techniques coupled to mass spectrometry
This section will discuss the principles and mechanisms of CE-MS modes such as CZE-MS, MEKC-MS, CEC-MS, CIEF-MS, and CCE-MS used for food analysis. The basic operating principle of various electromigration techniques in CE involves applying an electric field to a submillimeter diameter capillary (such as an open tube) filled with buffer or any affinity reagent. Besides, the capillary can also be filled with micelles or a chiral reagent, packed with particles, or polymerized into a monolith. Therefore, depending on the solvent's nature or charge on the affinity reagent used to fill the capillary, an electroosmotic flow (EOF) is the bulk flow of either excess of positive or negative ions, is generated. The EOF can separate analytes based on the charge to mass ratio (z/m).
The papers published in the last decade on food analysis have utilized five capillary electromigration techniques coupled to MS. The modes include CZE-MS, MEKC-MS, CIEF-MS, CEC-MS, and CCE-MS. The benefits of on-line coupling of these techniques provide information-rich data, including highly efficient peaks in electropherogram and less noisy mass spectra. However, run times are typically longer in CE-MS than CE-UV as the commercial CE instruments required the use of capillaries with at least 50-55 cm length to reach the mass spectrometer. Besides electrospray ionization (ESI), there are reports for the on-line CE coupling to inductively coupled plasma mass spectrometry (ICP-MS) for food analysis [4].
The pie charts showing the frequency of applying various types of electromigration techniques, interfaces, ionization sources, and mass analyzers are summarized in Fig.2 (a)-(d), respectively. The pie chart in Fig.2a indicates that the CZE is the preferred separation mode (~90%) coupled to MS in the last ten years for food analysis, while MEKC-MS (7%), CCE-MS, CIEF-MS and CEC-MS (1% each) are less commonly used. Fig.2b shows that most food analysis publications have used the sheath-flow interface using ESI (~86%) and ICP (13%) as ionization sources for CE, respectively, but only 1% for on-line sheathless CE-ESI-MS. This low percentage could be because the sheathless interface is recently commercialized, whereas the sheath flow interface was commercialized twenty years ago. The CE-MS in various modes utilizes the ESI (total ~76%) as the most commonly used ionization source, followed by ICP (12 %) and MALDI (2%) (Fig.2c). Therefore, electromigration techniques have predominantly used ESI only. However, there is only one publication where DART is reported as an ionization source [5]. To this date, most CE-MS food analysis studies have been performed by the time of flight (TOF) mass analyzers (Fig.2d) because it is a relatively low-cost alternative to CE-HRMS, such as orbitrap MS. Besides, TOF-MS provides high mass resolution and high mass accuracy. The latter advantage is more important for unknown peak identification, particularly in food adulteration. The pie chart in Fig.2d indicates that the frequency of mass analyzers used decreases in the following order: TOF-MS (30%) < single quad-MS (14%) < ion-trap MS (11%) < ion-trap (MS/MS) (10%) < triple quad (9%) < QTOF (8%) whereas the remaining miscellaneous mass analyzers (1-2%) are less commonly used. The fundamental principles of the separation mechanism of CZE, MEKC, CEC, CIEF, and CCE, when coupled to MS, are discussed in the following sub-sections.
Fig. 2.


Pie charts showing distribution of various (a) separation modes, (b) interfaces, (c) ionization sources, (d) mass analyzers in CE-MS.
2.1. Capillary zone electrophoresis-mass spectrometry
The technique of CZE-MS is based on the addition of a volatile buffer solution to prevent interference of salts in the ionization process. Charged analytes are separated based on their electrophoretic mobility, which is dependent on the strength of the electric field and the rate of analyte migration. This rate is directly related to the buffer's pH, as positively charged analytes have effective mobility towards the anode while negatively charged analytes have effective mobility towards the cathode. As shown in Fig. 3, under neutral to basic pH conditions, the high mobility cations with a small mass (or highest charge to mass ratio, z/m) will elute first, followed by low mobility cations with small z/m as they migrate in the same direction as EOF. It is worth noting that the neutrals cannot separate from each other in CZE-UV, but one could still detect peaks in the CZE-MS electropherogram. For example, neutral compounds will remain unionized in CZE buffer solution, but if they have an ionizable functional group in the gas phase, they could ionize in the electrospray. Thus, peaks will be eluted and reach the detector simultaneously, but one can differentiate them based on different m/z in the extract ion electropherogram (EIE). On the other hand, low mobility anions with small z/m will elute fourth, while high mobility anions elute slowest and the last in the migration order. Thus, the migration order of cations, neutral, and anions in CZE can be summarized as follows: small cations< large cations<all neutrals< large anions< small anions (Fig.3).
Fig. 3.

Schematic of solute migration in capillary zone electrophoresis (CZE)-MS under positive polarity. Note that the organic cations migrate fastest because they travel to the cathodic end (MS end), which is the same direction as the EOF travel. Neutrals travel at the same velocity as the EOF velocity and are not separated from one another. Organic anions migrate slowest because they are attracted to the positive end of the anode but are eventually pulled by the EOF toward the MS detector. The separation capillary is inserted through a stainless-steel needle and the sheath liquid and drying gas flows through the two outer tubes of the nebulizer and mixes with the CE effluent at the separation capillary tip to assist electrospray ionization (ESI).
For CZE with ultraviolet detection, nonvolatile buffers such as borate, phosphate are typically used. However, to be compatible with the ESI process, buffers used in CZE-ESI-MS must be volatile. Ammonium formate (NH4COOH)/formic acid (HCOOH), ammonium acetate (NH4CH3COO)/acetic acid (CH3COOH), ammonium hydroxide/-NH4CH3COO are commonly used buffers, which cover the total acidic, neutral, and basic pH range, respectively. One of the main drawbacks of using volatile buffers for CZE-MS is the separation is often not optimal, limiting the selectivity for separation of charged analytes. One reason is the low concentration of volatile buffer (electrolyte) used in CZE-MS, causing peak broadening due to mismatch of conductivity difference between the background buffer ions and the analytes present in food. The buffer with high ionic strength is avoided as the resulting high current limits MS detection use. Therefore, either the ionic buffer strength is adjusted, or a lower voltage is applied. Although the use of very high concentration (e.g., 1 M formic acid) and organic solvents in CZE-MS are reported to improve the separation and MS detection, the combined use of lower buffer ionic strength and high voltage often results in fast separations.
In addition to the use of uncoated capillaries, physically coated or covalently bonded capillaries are also used in CZE-MS. This use is common for analyzing basic proteins and positively charge small molecules in food products [6]. However, the positively charged compounds have a strong electrostatic affinity for the negatively charge silanol groups on the fused silica capillary wall leading to peak tailing and loss of separation efficiency. The use of coated capillary not only stabilizes the EOF, but the capillary lifetime can be extended when the capillary tip is exposed to the conventional co-axial CE-MS interface. Some of the coated capillaries used in food analysis are polyvinyl alcohol [7], hydroxypropyl cellulose [8], (N,N,N’,N’-tetraethyldiethylenetriamine [9], N-(2-hydroxypropyl) methacrylamide [9], polybrene [10], and polyethylene glycol [11].
2.2. Micellar electrokinetic chromatography-mass spectrometry (MEKC-MS)
Separation by micellar electrokinetic chromatography (MEKC) is carried out using buffers or electrolytes containing surfactant (e.g., sodium dodecyl sulfate, SDS), forming micelles at a concentration higher than the critical micelle concentration (CMC). When dispersed in buffer or electrolyte solution, these micelles are filled inside the fused silica capillaries; two distinct phases are formed, an aqueous (buffer) and a micelle forming pseudostationary phase. However, in MEKC-MS, the pseudophase is established using a volatile surfactant such as ammonium perfluorooctanoate (APFO) [12][13] or a polymeric surfactant such as polysodium undecenoyl leucine sulfate [14]. Because both electrophoretic and chromatographic principles are involved in MEKC, this CE mode extends the application to include both charged and neutral molecules. Thus, MEKC-MS has excellent potential for sensitive analysis of a wide range of small and large molecular weight compounds found in food samples.
Separation in MEKC is based on the analyte distribution between the pseudo phase, formed by the dispersed micelles and the buffer solution. This distribution is shown in Fig. S3. where equilibrium is established between the free and complex form of the APFO micelles with the neutral analyte. Under the electric field application, the negatively charged volatile micelles such as APFO migrates towards the anode (injector side). The neutral analyte is pulled towards the cathode (MS detector side) with the EOF. Because MEKC-MS is typically carried out under a basic volatile buffer solution, the net migration velocity of APFO micelle or the micelle's complexed form is towards the MS end. Therefore, in MEKC-MS, hydrophilic neutral analyte such as thiourea do not interact with the APFO micelle and travel at the EOF rate to elute at a dead time (to). In contrast, moderately hydrophobic neutral analyte interacts with the micelle eluting at time ta. On the other hand, hydrophobic neutral analyte interacts very strongly with the APFO micelle to elute at the micellar migration time (tmc) or sometimes remains undetected in MEKC-MS (Fig. S3., bottom electropherogram).
Ion-dipole and ion-ion intermolecular forces (IMFs) are responsible for the interaction of positively and negatively charged analytes with the APFO micelle, whereas hydrophobic and hydrogen bonding IMFs are present between the neutral analyte and the micelle. The elution order in MEKC-MS depends on these partitioning interactions of the analyte in and out of the APFO micelle. In basic buffer solution and substantial EOF, the hydrophilic cation is more strongly attracted to the buffer and will elute immediately after neutral, whereas hydrophobic cation will bind strongly with the negatively charged APFO micelle due to combined electrostatic and hydrophobic IMFs to elute last. On the other hand, both hydrophilic and hydrophobic anions are repelled by the negative charge head group of APFO but will elute before hydrophobic cationic analyte. Thus, the elution in the basic solution would follow the order: hydrophilic cation < neutral < hydrophilic anion < hydrophobic anion <hydrophobic cation. Conversely, acidic buffer solution will reverse the elution order under zero EOF and negative polarity.
The on-line hyphenation of MEKC–MS is often hampered by incorporating nonvolatile surfactant such as sodium dodecyl sulfate (SDS). This surfactant results in fouling of the ion source, causing loss of sensitivity and interference with analytes in the low molecular mass region [14]. One possible approach to solving poor ionization efficiency limitation involves using a volatile or semi-volatile surfactant, such as APFO [12][13], surfactant discussed above. Other possible approach includes the use of polymeric surfactant [14], anodically migrating micelles [15], a partial filling technique [16] and recent use of direct analysis in real-time mass spectrometry (DART-MS) [5].
The type of buffer and buffer pH is selected in MEKC-MS to generate sufficiently high EOF, allowing transport of cationic, anionic, and neutral analytes to separate and reach the detector's (MS end). However, simultaneous detection of cationic and anionic analytes are difficult because most of the commercially available on-line CE-MS instrument does not have the option of simultaneously ionizing and detecting both positive and negative ions in the ESI-MS. The APFO is the most widely used volatile surfactant reported for food analysis [12][13][17]. D’Orazio et al. [12] employed APFO to spike and separate estrogenic compounds in fifteen milk and yogurt samples. Specifically, using full-filling MEKC-MS/-MS and ion trap, the separation and identification of all the estrogenic compounds of interest were achieved without any ion-source contamination. Also, APFO is anionic, resulting in the analyte's electrophoretic mobility opposite the EOF. Typically, a basic operating pH (8.0 to 11.0) with a positive polarity is used with APFO. At this pH, there is electrophoretic mobility towards the cathode. However, the strong EOF pushes the analytes towards the anode (detector), whereas the APFO micelles may not reach the MS detection as their migration is opposite to the EOF.
2.3. Capillary electrochromatography-mass spectrometry (CEC-MS)
The CEC is a hybrid technique, which stems from two separation techniques, namely HPLC and CZE. Thus, a capillary column in CEC contains a true stationary phase (like HPLC). Instead of being pushed by the high-pressure pump, the mobile phase is forced through the capillary by the electric field, generating EOF on the fused silica capillary surface. The CEC offers the advantage of a two to three-fold higher plate number (N), a sub microliter flow rate when coupled to ESI-MS, and less consumption and disposal of toxic organic solvents compared to the use of HPLC [18]. In addition, CEC provides greater peak capacity and higher selectivity compared to CZE. However, simultaneous separation of anionic, and neutral compounds is more challenging in CEC compared to MEKC.
Furthermore, the repeatability of separation in CEC (in particular CEC-MS) is still questionable. Another hindrance is the lack of commercial CEC columns and dedicated CEC instrumentation. Both remain scarce.
Separation in CEC-UV or CEC-MS can be performed either in open-tubular, particle packed, or monolithic columns. The stationary phase is physically coated or covalently bonded to the capillary column's inner wall in the open-tubular column. In the packed, CEC-UV configuration, the silica particles are connected with organic ligands and are packed under high pressure with two frits in CEC. On the other hand, in packed column CEC-MS, one frit is needed on the inlet end, whereas the outlet end exposed to the ESI-MS end is tapered to prevent particles' movement and control the EOF [19]. Although the use of internal taper design for the CEC-MS column is promising, such particle-based columns have advantages of robustness, improved spray stability, and lower noise. The technique requires a skilled operator to make internal tapered columns. The third type of column is a monolithic column, also known as the continuous bed with continuous skeleton and microscale throughpores. The monolithic materials' most appealing aspect is their ease of preparation through in-situ polymerization processes, avoiding frit formation and column packing.
The two types of monolithic columns include polymer-based and silica-based monoliths. Polymeric monolithic columns are made via in-situ polymerization by copolymerizing organic monomers and crosslinker in the presence of porogens (organic solvents), chemicals, or photoinitiator. Silica-based monoliths are prepared by bonding silica or ODS particle together by sintering or by a sol-gel process, including hydrolysis and polycondensations of tetraalkoxysilanes. The monolithic CEC-MS is slowly attracting more and more attention. This slow progress is because of technical difficulties such as poor wetting of the monolithic column, bubble formation at the MS end, unstable EOF and irreproducible peak area, and retention time.
When designing the monolithic column for CEC-MS, three critical factors, namely, separation selectivity of analytes, the magnitude and direction of EOF, and the compatibility with the mobile phases, must be considered. To obtain successful separation on the monolithic CEC column, one of the monomers must contain ionizable functionality. For example, in situ polymerization of butyl methacrylate (BMA) monomer with EDMA as crosslinker generate very little EOF (e.g., 1.0 x 10-9 cm2 V−1 sec−1) because no ionizable moieties exist on the monolithic bed, on this type of monolithic columns. However, as shown in Fig. S4, the direction of EOF is from cathode to anode when positively charged monomer such as vinyl-benzyl trimethyl ammonium (VBTA) is added to a monolithic mixture of vinyl/alkene. In contrast, cathodic EOF is obtained when a negatively charged monomer such as acrylamido-2-methylpropane sulfonic acid (AMPs) is added to the polymerized monolith. Thus, the use of VBTA and AMPS monolithic columns provide fast separation of anions and cations, respectively. The flow rate of EOF (i.e., porosity) of a monolithic column is a critical factor because it drives the mobile phase and analytes and supports electrospray formation. A suitable flow rate of EOF for CEC-ESI is in the range of 50–300 nL/min. In addition to separation selectivity and EOF, the mobile phase's role is crucial for the successful operation of CEC-ESI-MS. A suitable mobile phase for monolithic CEC-MS should meet high electrochromatography selectivity requirements for the stationary phase. Also, sufficient porosity dictates driving the mobile phase through the stationary phase's macropores and mesopores. Moreover, the mobile phase should support a stable ESI and provide low contamination for MS detection. Like any CE-MS modes, the mobile phase in CEC-MS must contain volatile buffer additives such as formic acid/ammonium formate, acetic acid/ammonium acetate, ammonia, or short-chain volatile organic amines. Finally, a lower conductivity mobile phase could prevent joule heating and electrical arcing upon electric field application. Inorganic salts, nonvolatile surfactants, and other additives should be avoided or eliminated.
Although the number of papers reporting CEC-ESI-MS mode for food analysis published in the last decade is scarce, this mode has been applied to the food analysis in at least one publication [20]. Overall, both atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI) are reliable ionization sources for the CEC-MS, which can be used for the potential application in food analysis.
2.4. Capillary Isoelectric focusing-mass spectrometry (CIEF-MS)
The technique of CIEF is a high-resolution technique for the separation of amphoteric molecules such as proteins and peptides containing ionizable acidic or basic groups. At a certain buffer pH, these amphoteric molecules become zwitterions. They are focused on the isoelectric point (pI) in coated capillaries filled with a mixture of ampholyte buffer, forming a pH gradient inside the capillary. Separations by CIEF is usually performed using dynamic or permanent coated capillaries containing a neutral and hydrophilic polymer to suppress EOF and prevent protein adsorption to the capillary wall [21]. A CIEF-MS system was reported to analyze milk whey protein of similar isoelectric points [22].
There is a significant effort to increase the power of CIEF by interfacing to MS to obtain structural information of the protein. However, there are several fundamental challenges associated with the coupling of CIEF with ESI-MS. First, the electrical continuity required for the electrophoretic separation has to be maintained. Second, the compatibility of nonvolatile anolyte or catholyte entering the MS detector constitutes another difficulty. Therefore, anolyte and catholyte solutions, which are classically composed of phosphoric acid and sodium hydroxide, respectively must be replaced by volatile acids or bases such as formic acid, acetic acid, and ammonia. Third, standard anticonvective gels diminished ionization efficiency resulting in unacceptable MS contamination. This issue has been recently resolved using MS-compatible glycerol [22], which reduces EOF and provides an adequate resolution of protein bands without the need for coated capillaries.
The CIEF was recently interfaced with MS using a flow-through microvial interface [23]. Two configurations were used in the CIEF-MS experiment. In the first configuration, a protocol involved a neutral coated capillary while the sample is dissolved in glycerol and sandwiched between anolyte and catholyte solution (also containing glycerol, Fig. S5A). The flow-through microvial connected to a syringe pump provided a smooth delivery of the sheath liquid (consisting of formic acid, methanol, and water). In the second configuration (Fig. S5B and S5C), the coated capillary was filled with the ampholytes–analyte mixture. The CE inlet vial contained 1% (v/v) acetic acid as anolyte, while the flow-through microvial filled by the alkaline 1.0 % (w/v) of ammonium hydroxide served as catholyte. The mobilization in the second configuration was accomplished using pressure on the inlet vial triggering the migration of formate ions in the direction of the positive electrode. This second approach allowed the protein bands to mobilize towards the MS providing a resolution of 0.02 pH units.
2.5. Chiral capillary electrophoresis-mass spectrometry (CCE-MS)
Chiral separation in the electromigration technique, based on the formation of two distinct diastereomeric complexes between the chiral selector (CS) and the two enantiomers. The fundamental basis of enantioseparation is the difference in free energy (δΔG) of the formation of two diastereomeric adducts, including both enthalpic and entropic parameters. Almost all electromigration techniques coupled to MS have been applied in chiral separations. However, MEKC-MS using polymeric surfactants [14] and electrokinetic chromatography (EKC)-MS using small molecular weight chiral selectors (vancomycin, crown ethers, and sulfated cyclodextrins) are two CE-MS techniques, which are used extensively [24]. Note that the use of polymeric surfactant in MEKC-MS is very convenient and provides good separation selectivity and sensitivity. Still, using the aforementioned low molecular weight chiral selectors in EKC-MS requires a partial filling of the capillary to avoid ion suppression.
A significant number of food components are chiral. Thus, the separation and analysis of chiral compounds in food is a topic of utmost importance. One excellent review is recently published in this journal, which provided an extensive overview of various separation techniques investigated for chiral analysis in food safety, food traceability, and bioactivity [25]. While HPLC and GC have been used for chiral analysis in food science, the use of electromigration techniques for food (e.g., analysis of amino acids in vinegar) by CCE-MS is reported in section 5.4 using chiral crown ether [26].
3. Interface Development and Ionization Sources
The interface for the coupling of CE to ESI-MS or ICP-MS instruments is located between the end of the CE column and the start of the ionization source (electrospray, inductively coupled plasma) of the mass spectrometer. A correct design interface must meet the following requirements: (a) provide good electrical contact at the outlet end of the capillary; (b) minimize laminar flow due to the suction effect of the nebulizer, (c) compensate for the flow rate incompatibility between the EOF generated inside the capillary and uptake flow of the nebulizer to prevent suction resulting in a laminar flow; (e) achieving high transport efficiency of the analyte transfer to the electrospray or plasma source. The coupling of CE to ESI-MS or ICP-MS continues to be in the developmental stage. The user must understand the operational parameters of new emerging interfaces to maximize operation sensitivity. In this section, the two commercially available sheath flow and sheathless interface for ESI, as well as one for ICP-MS, are described. Also, the use of offline-MALDI interface used in food analysis is discussed. It should be noted that there are many reported interfaces for each ionization source, but only the interfaces that have been used for application in food analysis are detailed below.
3.1. Sheath flow ESI-ionization interface
The electrospray ionization (ESI) is the most widely used ionization source for on-line CE-MS coupling in food analysis because of the ease of its ability to carry out ionization for many molecules ranging from small metabolites to intact proteins. As mentioned earlier, the sheath flow and sheathless are the two commercially available interfaced used for CE-ESI-MS with sheath flow interface (Fig.S6 A-B) being used in most publications food analysis.
The co-axial sheath liquid flow interface is the most common design used for pairing CE to MS detection. Smith et al. [27] were the first to propose the co-axial delivery of a solvent (aka. sheath liquid) to the end of the CE separation capillary as an outlet electrolyte reservoir. Following the aforementioned proposed design, a triple tube sprayer (aka. nebulizer) was commercialized by Hewlett-Packard (currently known as Agilent Technologies) in 1995. As shown in Fig. S6 A, the separation capillary "c" is surrounded by the sheath liquid, which in turn is surrounded by the nebulizer gas (nitrogen). Fig. S6 B shows the actual sprayer. The ribbed ring in the middle of the sprayer could be rotated to allow the axial direction's capillary adjustment. Because the EOF rate in CE typically varies from 1-100 nL per minute, a sheath liquid consisting of an alcohol-water mixture with volatile acids or bases is added to enhance the droplet formation at the capillary tip (labeled as c with an arrow in Fig. S6 B). The sheath liquid is added typically at a flow rate of 1-10 μL/min from port b of the actual sprayer using a pneumatic HPLC pump, promoting spray formation and completing the electrical circuit. Note that the sprayer needle protruding the sprayer body in Fig.S6 B is grounded, which means a CE and ESI current return path. Moreover, the voltage is applied to the inlet side of the ESI capillary. The application of this interface is compatible with APCI [28] as well as APPI [29], as reported in several publications [30][31]. The limit of detection (LOD) reported with sheath flow interface is at least 1 μM (without preconcentration), but this may change as the recent advances in jet stream ionization technology in MS have shown improved LOD.
While the co-axial sheath liquid interface with sheath liquid flow rates of 3 to 10 μL/min is still the commonly used interface for robust and high throughput analyses, it has three significant limitations. First, a substantial dilution of the CE effluent leads to low ionization efficiency resulting in lower sensitivity. The combined EOF and sheath liquid flow is higher in flow rates than nanoflow HPLC, and thus sensitivity is lower than nano ESI. Second, the nebulizer's suction often creates band broadening, especially when wider i.d. capillaries (75 and 100 μm) are used and decrease in electrophoretic resolution of the separated peaks. Third, suppose there is no common ion present in the BGE and the sheath liquid. In that case, it may result in the co- or counterions from the sheath liquid entering the separation capillary, causing depletion of separation buffer even during the electrophoretic run. Consequently, this results in changes in migration time and peak area. New development aims for the improvement of sensitivity using a sheathless CE-MS interface. However, only in the recent past sheathless interface has been reported in food analysis [32].
3.2. Sheathless ESI-ionization interface
Sheathless ESI-ionization interfaces allow for 10-100 folds enhanced sensitivity, low cost, and a relatively simple set-up because the separation buffer is the only liquid involved, and no dilution occurs. However, maintaining a proper and long term stable electrical contact is challenging. Moini showed a simple way to fabricate a sheathless interface by etching the capillary separation end with hydrofluoric acid making the fused silica capillary porous [33]. This porous section serves as the transport of small ions and electrons in and out of the capillary. As shown in the schematic in Fig. 4a, the capillary is inserted in a stainless steel ESI needle. A conductive liquid is delivered coaxially to the outside of the porous end, making the hydraulic electric contact between the needle and the separation capillary. This concept was implanted by Beckman Coulter in 2010 [34] but was commercialized later by Sciex Separations under the trade name of CESI-8000. In this set-up (Fig. 4b), the ESI voltage is applied to the sprayer or the MS inlet. The capillary is grounded via the conductive liquid placed in the outlet CE vial, whereas the current monitor maintains the ESI voltage.
Fig. 4.

The porous tip sheathless interface a schematic and b photograph of the prototype interface. Reproduced from [34] with permission from the American Chemical Society, copyright 2011.
One of the main limitations of porous tip design is the restriction in the availability of only 30 μm, i.d., capillary, which prevents the injection of a large sample amount. Another drawback is that only the separation capillaries with porous tips assembled with proper adapters manufactured by Sciex CESI 8000 can be used with a mass spectrometer. Furthermore, the porous tip capillaries' longevity is limited to only 200 injections. The replacement capillary can only be obtained by the manufacturer, which may increase the overall cost of analysis in food applications. Furthermore, researchers' flexibility to tune separation under different CE-MS conditions and modes is difficult as this interface can only operate under relatively low CE currents (below 10 μA). Therefore, one should carefully consider the composition and concentration of the BGE to optimize separation selectivity.
3.3. Inductively coupled plasma interface
Capillary electrophoresis interfaced with inductively coupled plasma (ICP) mass spectrometry is a powerful trace elemental analysis technique. Successful coupling of CE with ICP-MS requires maintaining a steady electrical contact at the end of the CE capillary, matching uL/min CE eluate capillary flow rate with the mL/min uptake rate of the ICP-MS, and efficient introduction of analytes from CE capillary to the plasma. To date, three types of interfaces reported in the literature are sheath-flow interface, sheathless interface, and hybrid generation interface [35]. Amongst the three types of interfaces, the sheath-flow interface is used more frequently by many researchers. Sheath liquid serves two purposes: completing the CE's electrical circuit and compensating for CE's low flow rate. Furthermore, the mixing of sheath liquid mixing could cause analyte dilution resulting in some loss of sensitivity. Some of the CE nebulizers developed for ICP-MS are concentric, cross flow, MCN-100, and ultrasonic nebulizers [36].
A typical CE-ICP-MS sheath-flow interface set up is shown in Fig. 5. This interface is the same design as the Agilent CE-ESI-MS sprayer (G1607A, Agilent Technologies, USA) used to couple CE with ICP-MS, serving as a nebulizer for the ICP-MS. The triple tube nebulizer is directly inserted into a homemade low volume chamber. The CE capillary is inserted through the sprayer's inner stainless-steel needle, and the outlet end of the CE capillary is protruded 0.1 mm beyond the sprayer tip. Sheath flow liquid is added through the sprayer's upper port (using a quaternary HPLC pump equipped with a 1:100 flow splitter), which then passes through the gap between the CE capillary and the inner stainless steel capillary and sprays together with the effluent emerging from the CE capillary. The carrier gas added through the sprayer's outermost tube for nebulization is mixed with make-up argon gas in a low-volume spray chamber and transported into the ICP torch operated at atmospheric pressure. The stainless-steel needle of the nebulizer was grounded to maintain a steady separation voltage. Within the spray chamber, larger droplets from aerosol are separated and removed. The standard spray chamber design can also cause additional dilution/loss of analytes affecting the amount of analyte transported to plasma. For these reasons, there is still much room for improvement in spray chamber designs and CE-ICP-MS interfaces. The ICP torch is typically a quartz torch in which an electromagnetic field[4][35][36] with a power of 600-1800 W is generated by high power radio frequency that ignites the argon gas (seeded with free electrons from discharge coil) producing high-temperature plasma (5000-10000 K). The analytes entering the plasma flame are ionized by collision with electrons and charged ions in the flame, then transferred to the mass analyzer.
Fig. 5.

Schematic diagram of the interface for coupling capillary electrophoresis (CE) with inductively coupled plasma–mass spectrometry (ICP-MS). Reproduced from [35] with permission from the American Chemical Society, copyright 2014.
3.4. Off-line CE-MALDI-MS interface
The coupling of CE with MALDI-MS has many advantages over CE-ESI-MS. First, it offers better tolerance to salts, enabling more selection of BGE and more comfortable adaptation of other CE separation modes. Second, the number of spectra acquired on the sample spot is not limited by the MS data acquisition rate or the time of the elution peak. Therefore, MS/MS identification from full scan MS can be easily performed for masses of interest. However, the most challenging problem for off-line CE-MALDI-MS coupling is maintaining an electrical connection of the CE effluent at the capillary terminal. An additional challenge is to minimize the decrease in sensitivity during the collection of CE effluent without disturbing the separation process and peak resolution.
The off-line hyphenation of CE with MALDI-MS enables additional sample treatment, including enrichment steps or enzymatic digestion. For food analysis, the Girault group introduced a silver-coated capillary outlet for iontophoretic sample deposition on the MALDI plate (Fig. S7) [8][37][38]. The CE effluent exiting the capillary is dipped on the MALDI target containing microliter droplets. The current breakdown typically encountered in the sheathless system is avoided using neutral coating with zero EOF. The capillary ends are lifted in and out of the MALDI droplet at 15 sec per step without compromising peak efficiency, as verified by the UV detector output.
4. Sample preparation in Food Analysis
Food products consist of a wide variety of components. For example, proteins, peptides, amino acids, fats, carbohydrates, vitamins, nucleotides, small metabolites, organic compounds, and minerals. Sample preparation is essential for analyzing samples containing complex food matrices (e.g., species identification in matrices composed of a blend of two or more species). Furthermore, food samples from different sources (solid and liquid) contain complex matrices that can interfere and compromise the sensitivity. The analysis of a specific compound in the food matrix requires homogenization, centrifugation, and interferences as the essential steps before extraction and preconcentration. These steps mentioned above should be environmentally safe, non-toxic, rapid, and automated, but high sensitivity, robustness, and cost-effectiveness are highly desirable in many cases. It isn't easy to meet all of the criteria mentioned above. However, in this section, some of the key extraction techniques reported for solid and liquid food analysis before injection in CE-MS is described. This includes, solid-phase extraction (SPE), solid-liquid extraction (SLE), liquid-liquid extraction (LLE), and QuEChERS. Other methods such as enzyme-assisted protein extraction (EPE) [39], enzyme-assisted water-phase extraction (EWME) [40], supercritical fluid extraction (SFE) [41], microwave-assisted extraction (ME) [42], ultrasound-assisted extraction (UE) [17][43], dispersive liquid-liquid microextraction (DLLME) [12][44], as well as other variants of conventional extraction techniques [8][17][41][45-49], have been reported to extract major components from food samples. Thus, only the principles behind the most commonly used sample preparation techniques for analysis for solid and liquid food samples are described in the following sub-sections. For more detailed information about the theory and applications, some excellent review articles on SPE [50], LLE [51], and QuEChERS [52] are recommended.
4.1. Solid-phase extraction (SPE)
Solid-phase extraction (SPE) is a preparative method used for selective extraction, purification, and preconcentration of analytes of interest in a wide variety of matrices from liquid to solid samples. The solid-phase extraction method is widely used in CE-MS to analyze different compounds in numerous food matrixes. The technique of SPE can either be performed off-line or on-line. Off-line SPE is carried out in cartridges/columns with a bed containing stationary phase (sorbents or cartridge) materials placed on a vacuum manifold (Fig.S8) [53]. The necessary procedure involves conditioning the cartridge with appropriate solvents, loading the sample, retaining the target analyte, washing/purifying, and finally eluting the retained analyte with a suitable strong solvent. Stationary phase materials with versatile chemistries (reversed phase, normal phase, mixed-mode, and ion-exchange) and various sorbents offer a wide range of selectivity. The SPE for food analysis improves detection, sample recovery, and quantitation accuracy and minimizes the source contamination. This improvement of SPE is because the food structure represents a complicated matrix and can be formed into different physical stages, such as concrete, viscous or liquid. Therefore, SPE steps particularly have a vital role in the determination of specific compounds in food.
4.1.1. On-line SPE
As mentioned above, SPE is customarily carried out in the off-line mode. However, several on-line attempts are reported for the CE-MS determination in food applications [54][55]. The schematic of an on-line SPE method using three different approaches is shown in Fig. S9 (a-c) [56]. In the first approach (Fig. S9a), a Teflon sleeve connects a 10 cm, latex-coated capillary (25 μm i.d., acting as a preconcentrator) to an open tubular separation capillary (75 μm i.d.,). In the second approach, small pack beds or sorbents are connected between the two pieces of open capillaries (Fig. S9b). While the use packed bed proved useful and provided higher retention and column capacity than the first approach, frits are needed to hold the packed bed in place, resulting in backpressure and irrepeatable EOF. The third on-line SPE approach (Fig. S9c) has used disks or membranes loaded with a solid sorbent. The impregnated membrane is placed in a Teflon sleeve center, which holds the two fused-silica capillaries. Care has to be taken to prevent membrane compression or any damage to the Teflon tubing wall as this may block the cartridge and decrease the liquid flow. The advantage of this fritless approach includes low pressure and higher flow rate (due to the use of smaller bed) and smaller elution volume. The potential of this on-line SPE procedure for the analysis of antibiotics [57] and alkaloids [55] is demonstrated in several CE-MS publications.
4.1.2. Off-line SPE
The application of off-line SPE to analyze versatile compounds in numerous food samples is discussed in section 5. The availability of various packings for SPE makes this technique one of the best selections for specific sample preparation needs. A brief description of different off-line SPE methods is given in the following sections, with some relevant applications in which they are used in combination with CE- MS.
The SPE columns for pesticide analysis in fresh fruits are necessary due to the diluted contaminants in the complex fruit matrix. Garcia et al. [58] have practiced SPE of pesticides by pressurized liquid extraction (PLE) using hot water at 60 °C, and 1500 psi, followed by a SPE clean-up using a mixed-mode (strong cation exchange-reversed phase) cartridges to determine seven pesticides spiked in Mediterranean summer fruits before CZE-MS analysis. At the least, 40% of watermelon, 25% of melon, 44.4% of peaches, and 33.3% of apricots presented higher minimum requirement levels.
Tong et al. [59] used C-18 SPE steps to determine one microcystin (MC-LR) peptide after removing the extra water from the crude algae sample. Based on the mass scan spectrum, MC-LR in crude algae in lake water samples was confirmed. According to Catala-Clariana et al. [60], milk-derived functional foods are precursors of many different bioactive peptides that may remain latent until being released by enzymatic proteolysis during gastrointestinal digestion or food processing. Milk emulsions were diluted with a citrate buffer containing dithiothreitol and urea followed by a comparison of different reversed-phase commercial C18 and C8 silica(50 mg of sorbent) and polymer-based (30 mg and 10 mg sorbent) Strata X and hydrophobic-lipophilic balance (HLB) SPE columns. The octadecylsilyl (C18) and StrataX cartridges could provide the best clean-up results for infant formula analysis, while HLB cartridges were specific for extraction of 13, 8, and 6 peptides of IF1, IF2, and IF3, respectively. Better extraction with C18 than with the C8 cartridges suggested that retention was mainly based on hydrophobic interactions. The polymeric sorbents, i.e., HLB and Strata-X, showed good HLB, but the average results obtained with HLB suggested that StrataX were more hydrophobic than HLB. Although the combined use of C18 and StrataX cartridges provided excellent coverage of the IF's low molecular mass peptides, the use of all three SPE columns was recommended for comprehensive coverage. In another communication [61], the same research group reported an identical SPE protocol to identify a total of three (788, 1007, and 868) low molecular mass peptides with C18 cartridges in IF1, IF2, and IF3, respectively, and another three (311, 1328, and 1959) peptides with StrataX cartridges by CZE-MS.
Two main classes of antibiotics, such as quinolones (QN) in milk [57] and aminoglycosides (AGS) in honey [62] was performed using on-line and off-line SPE procedures, respectively. Gonzalez et al. [57] evaluated molecularly imprinted polymers (MIP) as a sorbent for constructing an on-line SPE. One of the significant advantages of the MIP is that they allow the introduction of high volumes of milk sample. Using MIPs is very specific compared to the partitioning process carried out when other sorbents are used. The authors found that the retention of the QNs at high flow rates was not complete, and the use of a high injection pressure could lead to erroneous or irreproducible results. Nevertheless, the method was selective and allowed sensitive multi-residue determination of QNs antibiotics of veterinary use in milk. In the subsequent study [57], the MIPs was also used for the off-line SPE extraction of nine AGs from a complex matrix such as honey, providing a higher selectivity and a lower sample manipulation. The use of MIP was shown to be a powerful extraction tool in a simple sample clean-up step.
Two types of toxins, paralytic shellfish toxin (PST) [63] and neurotoxin [64], in seafood (muscles and lobster) are reported. In the first study [63], even when using a reversed-phase C-18 cartridge, the CZE with conductivity and MS method suffered from significant interferences from the shellfish matrix. On the other hand, MEKC with fluorescence detection was successfully used for PST screening of a mussel sample. The study demonstrated the superiority of MEKC with fluorescence detection for the screening of PSTs in shellfish samples. In the second study [64], using a mixed-mode cation exchange SPE column, lower conductivity of the extract allowed field amplified stacking in CE-MS. This stacking causes the final step of the clean-up after SPE involved redissolution in a low conductivity solvent such as methanol without deteriorating neurotoxin's peak shapes β-N-methylamino-L-alanine (BMAA) and its isomers.
Melamine (MEL), 2,4,6-triamino-1,3,5-triazine is an emerging contaminant, illegally added to dairy products such as milk, infant formula, and pet food to increase nitrogen content, artificially boosting the protein level [65]. Commercially synthesized MEL can contain structural by-products, such as ammeline (AMLN), ammelide (AMLD), and cyanuric acid (CYA). Two SPE columns were compared in spiked blank powdered milk and contaminated powdered milk. The mixed-mode cation exchange column allowed the extraction of MEL and AMLN, but not AMLD and CYA. A new SPE Strata column was also tested with a protocol developed by the manufacturer. Still, peak shapes obtained for MEL and AMLN were not satisfactory, while AMLD and CYA could not be detected at a concentration lower than ten ppm.
Endocrine-disrupting compounds (EDCs) are chemicals or mixtures of chemicals that interfere with how the hormones in the human work. Some EDCs such as chlorophenols, alkylphenols, bisphenol-A (BPA), 4-tertbutyl-phenol (t-BP), and 4-tertbutyl benzoic acid (t-BBA), could enter food at several stages of food production and via the plastic packaging material. The combined use of restricted access material (RAMs) and polymeric SPE column was reported for the extraction, preconcentration, and efficient removal of sugar from honey. Thus, allowing the preconcentration of a greater SPE column volume for analysis of several EDCs in honey [66].
The β-carboline alkaloids (HAlks), a family of compounds with antidepressant effects, are found at trace levels (<25 mg kg−1 algae) in the edible invasive algae Undaria pinnatifida, aka. wakame. Tascon and coworkers' studies for on-line SPE found that peak areas, hence SPE efficiency using the C18 sorbent, were shallow for harmaline, which was the most polar of the three alkaloids [55]. The authors concluded that on-line SPE-CE-MS using C-18 cartridges is a powerful method to detect and quantify trace levels of alkaloids in complex plant extracts, with potential application in quality control, food safety, and foodomics.
Human milk oligosaccharides (HMOs) and galactooligosaccharides (GOS), have a profound influence on the development of the neonatal gastrointestinal system. For example, these oligosaccharides affect how microbial colonization occurs in the intestine, which influences the intestinal absorption and metabolism of food components. Furthermore, the immune system act as receptor analogs for pathogenic bacteria, preventing constipation [67]. However, very little is known about oligosaccharides' fate in the breast- and formula-fed babies' colon. The oligosaccharides from feces were extracted after centrifugation, filtration, and inactivation of the fecal enzymes. The carbohydrates from fecal extracts were cleaned by SPE using a graphitized carbon column cartridge. The cartridge was first washed with 80/20 acetonitrile/water containing 0.1 % TFA to remove monomers and lactose by eluting 2% acetonitrile solution. In contrast, the remaining carbohydrate was eluted with 40 % ACN/60% water, 0.05% TFA, followed by drying the sample residue and redissolving in water for analysis by CE-MS [68].
The determination of organotin compounds at trace level in plastic food packaging materials is needed for food safety. Because of the complexity of organotin and sample treatment procedures, analysis is quite challenging. In a recent report [32], the extracts of four organotin compounds from food packaging samples were cleaned using ultrasonic extraction, and then preconcentration was performed using C18 SPE cartridges. Satisfactory recoveries (80.27–101.56%) for all four organotins were obtained at three different concentration levels with %RSDs in the range of 2.56–7.89 (n = 3). The authors found no significant matrix effect for the determination of the organotin residues in food packaging materials.
4.2. QuEChERS
QuEChERS, a type of SPE technique that stands for Quick, Easy, Cheap, Effective, Rugged, and Safe, is a specialized modern extraction technique for liquid samples or solid dissolved in water. Lehotay and Anastassiades first designed this method in 2003 to analyze pesticides in fruits and vegetables in 2003 [69]. Overall, QuEChERS is typically carried out in two main steps. First, the sample is added to a mixture of water and acetonitrile followed by extraction with salts (e.g., MgSO4, NaCl, sodium citrate, and NaCOOCH3) by vigorous shaking and centrifugation. The salts' addition results in phase separation between water and acetonitrile, and hydrophobic analytes are extracted in the organic layer. In the second step, the sample clean-up is accomplished with dispersive solid-phase extraction. In some cases, the organic layer is treated with sorbent materials (e.g., silica, primary-secondary amine, C-18, etc.) for cleaning up the residual matrix. Fig.S10 outlines the difference in recipes for QuEChERS in the Association of Analytical Chemistry (AOAC) and the European official methods (EOM) [70].
The QuEChERS method has many advantages over traditionally used extraction techniques. Some of the benefit of QuEChERS includes accurate analytical results and high extraction recoveries, saves time and labor, reduces the cost of waste disposal due to decrease in hazardous solvent consumption, requires a lower supply of laboratory glassware with a decreasing number of sample preparation steps. Rajczak et al. recently reviewed QuEChERS for food applications [52].
Although the QuEChERS method's popularity has skyrocketed in recent years for analysis of samples by LC-MS and GC-MS, there are few reports for its application in CE-MS analysis of food. For example, QuEChERS has been used as a sample preparation method for trace analysis in food samples by CE-MS [7][54][71][72]. Concerning salts' use to induce phase separation, MgSO4 and NaCl were tested in most applications where QuEChERS was used for CE-MS. However, ammonium formate as buffering salts (in place of sodium citrate) to improve polar compounds' recoveries has been reported only on one occasion [72]. The reported examples of QuEChERS are pesticides in sugarcane juice and tomato [54], pesticides in corn [7], benzimidazole in eggs [72], EDC in honey [73]. As mentioned earlier, all of the food samples have used AAOC to analyze isoflavones in soy biscuit [71], which have followed the EOM recipe.
4.3. Solid-liquid extraction (SLE)
Solid-liquid extraction (SLE) is a sample preparation method used to extract analytes of interest from solid samples. Typically, solid pieces are mixed in a suitable solvent using vortex, ultrasonication, or centrifugation to dissolve analytes into the solvent, which is further purified or filtered. A schematic model of SLE features a straightforward extraction process in which the solute (i.e., a compound of interest) is separated from a mixture of solids by dissolving it in a suitable solvent (Fig. S11). There are three constituents in SLE extraction. These constituents are composed of a solute, insoluble solids, and solvent. In the first step, a sample is dried and ground into a powder to increase its surface area, enhancing its extraction rate. In the next step after extraction, the soluble compounds are separated from the solids by filtration. Selection of a suitable solvent for use in extraction is mandatory. Various factors such as solvent polarity, the particle size, and solid-to-liquid ratio can affect the extraction efficiency. Also, extraction conditions such as time, temperature, and mechanic agitation need to be carefully considered.
In CE-MS of food analysis, SLE is the most widely used technique for extracting polar analytes in solid food materials using polar solvents. Typical SLE solvents used for food samples are water, methanol, ethanol, acetonitrile, acetone, dichloromethane, and dimethyl sulfoxide as pure solvents as well as the combination of two or more solvents. Food materials from various sources and types require processing/grinding into a fine powder before performing SLE. Application of SLE in CE-MS analysis of variety of food materials is demonstrated in the following publications: metabolites in edamame [74]; soybean [75]; avocado [76]; rat’s liver [77]; lettuce [11]; fish [78]; grounded coffee [79]; ham, [80]; and mice liver tissues [81]. Besides, SLE is also reported for fatty acids in cheese/freeze-dried coffee [82] and tea leaves [83]; Cr in yeast tablet [84]; Ziram and Zineb in cabbage [43], saxitoxin in fish [85]; peptides in soybean and nutraceutical tablets [86][87]; sudan dyes in chili products [88][89]; furosine in grains [90]; melamine in milk powder [20]; amino acids in royal jelly products[91]; caffeine in Chinese white tea leaves [5]; 5-OH-methyl-furfural in food products (cereals, barley, coffee) [92]; allergens in milk [38]; lysozyme in cheese [93]; nucleotides in baby foods [94]; alkaloids in algae [95]; thiamine and thiamine phosphate in meat [96].
4.4. Liquid-liquid extraction (LLE)
Liquid-liquid extraction is the second most widely used method for retrieving analytes from aqueous food samples by CE-MS. The LLE is based on the principle that a solute or an analyte is a partition in a particular proportion between two immiscible solvents. The solvents are typical, aqueous (water), and non-aqueous (organic). A mixture of aqueous/organic solvent is used for extracting the analyte of interest from interfering compounds. For CE-MS analysis, a small volume of the aqueous sample containing analytes is mixed with water-immiscible solvents in a centrifuge tube, mostly by vortex or sonication to allow analyte transfer from aqueous to the organic phase. Examples of typical organic solvents used in LLE of food samples are acetonitrile, methanol, ethanol, chloroform, etc. The LLE method's main advantages are the easy availability of solvents and the use of cost-effective apparatus. However, low sample recoveries, limited selectivity, and labor-intensive protocols have limited the use of LLE. Examples of the use of LLE in CE-MS of food samples are analysis of metabolites in seaweed [97]; rat plasma [98]; soybean [99]; milk [100]; mice plasma [101]; isoflavones in soy drink [102]; biogenic amines in beer/wine [103]; phenolic compounds in almond [104]; amino acids in oils [105]; betaines in oils [106]; EDCs in honey [73]. Three of the microscale variants of LLE used to extract liquid food samples are dispersive liquid-liquid microextraction (DLLME), vortex-assisted surfactant enhanced emulsification liquid-liquid microextraction (VSLLME), and freeze-out LLE, which are described briefly in the following sections.
4.3.1. DLLME
This type is a miniaturized version of the LLE technique that utilizes a ternary solvent system: aqueous sample, dispersion solvent (soluble in aqueous and extractor solvents), and extractor solvent (immiscible with water). This extraction technique is carried out by injecting a mixture of dispersion solvent (e.g., ACN) and extractor solvent (e.g., CHCl3) into a centrifuge tube containing liquid samples (in water) or solid samples (first extracted in aqueous solvent). The mixture is then shaken vigorously until droplets are formed and centrifuged. The sedimented droplets containing the analyte is dried, reconstituted in a suitable solvent, and finally injected into the CE-MS system. In DLLME, partitioning of analytes into the extraction phase happens immediately due to the droplets' very high collective surface area. As a result, high enrichment factors are obtained. Another benefit of DLLME is low solvent consumption relative to other liquid extraction methods. Dairy food products present a complex sample matrix, and the removal of interfering components from this matrix is challenging. Examples of DLLME applications are pointed out in CE-MS analysis of estrogenic compounds in milk/yogurt [12] and benzimidazole in meat [44].
4.3.2. VASLLME
This mini-extraction is a modified version of DLLME that utilizes surfactants such as SDS or Tween-20 solution as an emulsifier solvent. The procedure is performed precisely as DLLME. One example in food analysis is the extraction of pesticides in juice samples by MEKC-MS using 100 mM AFPOA as an emulsifier is reported [13]. A stepwise sample preparation procedure using vortex assisted surfactant enhanced liquid-liquid microextraction (VASLLME) is illustrated in Fig. S12. The process involves adding 5 g of juice in a 50-mL falcon tube and centrifuged for 10 min at 9509 rcf. Next, the upper aqueous layer was transferred into a 15-mL falcon tube with conical bottom. A mixture of 530 mL aliquot of 100 mM APFO, pH 9.0 (emulsifier, 530 μL), and 1300 mL of chloroform (extraction solvent) was quickly added into the falcon tube. The tube was closed and vortexed for 30 s, centrifuged for 10 min at 9509 rcf. The sedimented organic phase was removed using a syringe and collected in a glass vial. The chloroform was evaporated using a gentle nitrogen stream until dryness. The final residue was reconstituted with 250 mL of 75 mM APFO pH 9.0, vortex for 2 min. Finally, the solution is filtered and injected into the CE-MS. The procedure mentioned above resulted in a sample throughput of approximately 15 samples/h with a preconcentration factor of 20.
In general, the above-mentioned variants of LLE are both quick and straightforward, but they do have some drawbacks. The procedure needs sample pH adjustment, filtration, or centrifugation, depending on the sample pretreatment required, which increases the total sample preparation time.
5. Applications of CE-MS in Safety, Quality, and Adulteration
Food evaluations involve assessment of food for the presence of harmful: (a) microbial agents (bacteria, virus, parasites, toxic microalgae) and (b) chemical compounds (pesticides, industrial and environmental contaminants, veterinary drugs, toxins, and allergens). Besides, adulteration and other fraudulent practice and packaging might also compromise food safety. The data obtained to meet these objectives as mentioned above by various modes of CE-MS for both new and established chemicals found in liquid and solid food samples are summarized in Table 1 and Table 2, respectively, and are discussed below:
Table 1:
List of the sample preparation methods used in CE-MS analysis of liquid food
| Sample Type |
Target Compounds |
Extraction Method |
Extraction Protocol | CE-MS Conditions | Ref |
|---|---|---|---|---|---|
| Rabbit serum | Whey proteins (β-lactalbumin, Lactoglobulin A, Lactoglobulin B, Bovine serum Albumin, Casein Lactoferrin, Ribonuclease A) | No pretreatment | N/A, except matrix effect was evaluated using 1 mL of 10 times-diluted serum added to 48 mL of glycerol–H2O containing 0.5 mL Pharmacia Biotech ampholine (pH 4–6) and 0.5 mL Beckman ampholyte (pH 3–10) | CIEF: bare fused-silica capillary (80 or 100 cm length, 50 μm i.d., 375 μm o.d.) BGE: 1% (v/v) ampholyte mixture (pH 3-10) (1% v/v) and 1% (v/v) ampholine mixture (pH 4-6) in 30: 70 (v/v) glycerol: H2O. Anolyte: 1 mM glutamic acid/50 mM formic acid in glycerol:H2O medium; Catholyte: 1 mM lysine/100 mM ammonia. Focusing: +30 kV for 16 min. Mobilization: cathodic (50 mbar pressure), constant voltage at +30 kV. SL:1% formic acid in 80:20 (v/v) MeOH/H2O; 6 μL/min; ESI-MS: Positive +4.5 kV; SC: DGT 350 °C, NGT 100 °C, NP: 55 kPa | 22 |
| Honey | Endocrine disruptors (4-t-BP, 4-t-BBA, BPA, PCP 2,4,5-TCP, 2,4-DCP) | RAM and SPE | RAM: 4 mL of honey injected in RAM (pump A). At 10 min, RAM set-up to elution and analyte collected in MeOH using pump B. SPE: Oasis HLB cartridge preconditioned with 5 mL AcOET, 5 mL ACN, 5 mL H2O. About ~3.5 mL of sample loaded and cartridge dried (15 min vacuum @ 15 mmHg). Elution with 0.5 mL ACN and 3 mL AcOET. Evaporate to dryness, reconstitute in 60/40 % MeOH/H2O |
CZE: Bare fused silica capillary (75 μm i.d), BGE: 15 mM NH4OAc, pH 11.0. Injection and voltage: 15 mbar,17s,+22kV. SL: 3% (v/v) NH4OH in MeOH, 0.78 mL/min; ESI-MS: Negative, −4.0 kV SC: ESI −4000 V; DGF: 2 L/min; DGT: 100 °C; NP: 2 psi |
66 |
| Chili tomato sauce | Sudan dyes (Sudan I Sudan II Sudan III Sudan IV) | LLE | 1.0 g sample mixed with 10 mL of (3:2:1, v/v/v) acetone/DCM/MeOH, vortexed (2 min), sonicated (5 min), and centrifuged (5 min @10000 rpm). Supernatant dried, residue suspended (1 mL of acetone containing IS), and diluted (50:50 (v/v) 60 mM NH4HCO3 / 50 mM SDS + 30% ACN) | MEKC: Uncoated fused silica capillary (80 cm length, 50 μm i.d). BGE: 40 mM NH4HCO3 + 25 mM SDS + 32.5% (v/v) ACN (pH 8.0). Injection and voltage: 0.5 psi, 6s; +25 kV. SL: 0.1% formic acid in 50: 50 IPA/H2O, 4 μL/min. ESI-MS: Positive, + 4.5 kV. SC: DGF: 4 L/min; DGT: 200 °C; NP: 4 psi | 89 |
| Vegetable oils | Non-protein AAs (Ornithine, b-alanine, GABA, Alloisoleucine, Citrulline, Pyroglutamic acid) | LLE | 40 g of oils extracted with 160 mL of MeOH/CHCl3 (2:1 v/v) and left at −20 °C overnight. Centrifuged (15 min @ 4000 x g, 4°C) and upper phase washed with 40 mL CHCl3 and 100 mL H2O. Aqueous phase dried (80°C) and derivatized with butanol before injection | CZE: Uncoated fused-silica capillary (50 μm id with 60 cm of length). BGE: 0.1 M formic acid (pH 2.0). Injection and voltage: 50 mbar, 50s; +25 kV. SL: 0.1% formic acid in 50:50 IPA/H2O, 3.3 μL/min; ESI-MS: Positive, +4.5 kV. SC: DGF: 3 L/min; DGT: 300 °C; NP: 2 psi | 105 |
| Vegetable oil | Betaines (Proline betaine, Glycine betaine, Carnitine, Rigonelline) | LLE | 40 g of oils extracted with 160 mL of 2:1 (v/v) MeOH/CHCl3 and left at −20 °C (overnight). Mixture centrifuged (15 min @ 4000 x g, 4°C) and upper phase washed with 40 mL CHCl3 and 100 mL H2O using centrifugation. Aqueous phase dried at 80°C and derivatized with ButOH before injection | CZE: Uncoated fused-silica capillary, 50 μm id, total length of 60 cm. BGE: 0.1 M formic acid (pH 2.0). Injection and voltage: 50 mbar 50 sec, +25 kV. SL: 0.1% FA in 50:50 IPA/H2O, 3.3 μL/min. ESI-MS: Positive, +4.5 kV; SC: ESI +4.5kV DGF: 3 L/min; DGT: 300 °C; NP: 2 psi | 106 |
| Honey | Endocrine disruptors | LLE and QuEChERS | LLE: 9.0 mL of n-hexane added to the spiked diluted honey and centrifuged (10 min @ 3900 RCF). Upper organic phase dried (N2 at 40 °C), reconstituted [300 μL of an aq. solution 14%(v/v) MeOH and 1%(v/v) of 28% (w/w) NH4OH solution], vortex and filtered (0.45-μm cellulose filter) collected directly in 100 mL CE vial. QuEChERS: 10mL spiked-diluted honey placed in centrifuge tubes containing a mixture of salts accompanying in the kit and centrifuged (10 min @ 3000 RCF). 1 mL of the upper aqueous-organic phases cleaned with PSA supplied with the kit by centrifugation (1 min @ 3000RCF) and dried. Reconstituted as explained in the LLE procedures | CZE: Fused-silica capillaries (100 or 50 μm id) with a total length of 87 cm to MS, 20 cm to the UV. BGE: 15 mM NH4OAc (pH 11.0 adjust with 28% w/w NH4OH); Injection and voltage: 50 mbar over either 16 s or 50 s for the 100-μm id and 50-μm id capillaries, respectively; ~13.2 and 12.4% of the total capillary length, +22kV with ramp of 7s. SL: 70:30 %(v/v) IPA/MeOH, 10 μL/min. ESI-MS Negative, −4 kV; SC: DGF: 7 L/min ; DGT: 350 °C ; NP: 4 psi | 73 |
| Japanese sake (wine) | Metabolites (organic acids, amino acids, peptides, and sugar) | Filtration | Sake samples centrifuged (15 min @ 9,100g), filtered (5 kDa cutoff membrane filter), and analyze immediately by CE-TOFMS | CZE of cationic metabolite: fused silica capillaries (100 cm × 50 μm I.D). BGE: 1 M formic acid. BGE: 5 mM NH4OAc (pH 8.5). Injection and voltage: 5 k Pa for 30s, +30 kV. SL: 1:1 MeOH/−H2O, 0.1 μM hexakis (2, 2-difluoro-ethoxy) phosphazene. 10 uL/min, ESI-MS: +4kV. CZE of anionic metabolite: COSMO(+) capillary (110 cm × 50 μm I.D),). BGE: 50 mM NH4OAc (pH 8.5). Injection and voltage: 50 mbar, 30s, −30 kV. SL: 5 mM NH4OAc, 0.1 μM hexakis (2,2-difluoroethoxy) phosphazene, in 1:1 MeOH/H2O,10 mL/min. ESI-MS: −3.5kV. SC:DGF: 10 L/min; DGT: 300 °C; NP: 69 kPa. | 45 |
| Soy drinks | Isoflavones (Daidzin, Genistin Daidzein, Genistein Formononetin, Biochanin A, Glycitein Apigenin) | LLE | Ethanol mixed with sample (2:1) centrifuged (30 min @ 5000 rpm), filtered (0.45 μm nylon syringe filter). A 100 μL of filtrate diluted with 1.0 mL H2O and injected in CE-MS | CZE: Uncoated 75 μm capillary with a total length 57 cm to MS, 50 cm to the UV. BGE: 15 mM NH4OAc (pH 11.0 adjust with NH4OH). Injection and voltage: 50 mbar 5s, +25 kV. SL: 0.5% HOAc in 1:1 (v/v) IPA/-H2O,10 μL/min. ESI-MS:Positive, +3.5 kV; SC: ESI 3500 V; DGF: 6 L/min; DGT: 350 °C; NP: 5 psi | 102 |
| Balsamic vinegar, liquid coffee, and soft beverages | 5-hydroxy-methylfurfural | LLE | 300 mg sample dissolved in 6 mL of 1% TCA solution, vortexed (2 min), ultrasonicated (10 min), vortexed (2 min), and centrifuged (10 min @ 9000 rpm, 4 °C). Extracts filtered (0.20 mm nylon filter) and mixed with 6 mL of 500 mg/L FMK solution to obtain a concentration of 5 mg/L in a final volume of 600 μL | CZE: Untreated fused-silica capillary (50 μm i.d. and effective length of 60 cm). BGE: 50 mM formic acid (pH 3.0 adjust with 0.5 M NH4OH). Injection and voltage: 50 mbar, 10s, +25 kV.SL: 0.1% FA in 50% MeOH, 3 μL/min. ESI-MS: Positive, +3.5 kV, SC: DGF: 2 L/min; DGT: 200 °C; NP: 10 psi | 92 |
| Bovine milk | Quinolones (Danofloxacin, sarafloxacin, difloxacin, enrofloxacin, ciprofloxacin flumequine marbofloxacin oxolinic acid) | In-line SPE | 25μL acetic acid added to 5 g of bovine milk and centrifuged (9000 rpm, 10 min) for protein precipitation. 1mL of supernatant mixed with 2 mL of 50 mM NH4OAc buffer pH 5.0 and defatted by centrifuging (5 min @ 9000 rpm) with 3mL of n-hexane. 0.5mL of aqueous phase mixed with 1mL of 50 mM NH4OAc (pH 5.0), filtered, and injected | CZE: Bare fused-silica capillary of 130 cm total length ×50 μm id (360 μm od). BGE: 50 mM NH4OAc (pH 9.1 adjust with 5 M NH4OH). Injection and voltage: 2bar, 15 min, +25 kV. SL: 50:49:1 (v/v/v) IPA/H2O/FA, 3 μL/min. ESI-MS: Negative, 4kV; SC: DGF: 6 L/min; DGT: 150 °C; NP: 4 psi | 57 |
| Banana, tomato, and peach juices | Carbamate pesticides | VSLLME | 5 g of juice centrifuged (10 min @ 9509 rcf) and the supernatant collected (15 mL falcon tube). Mixture of 100 mM APFO, pH 9.0 (emulsifier, 530 μL) and CHCl3 (extraction solvent, 1300 μL) injected into the sample tube, vortexed (30 s), and centrifuged (10 min @ 9509 rcf). Sedimented organic phase removed (with syringe) and CHCl3 evaporated under N2. Residues redissolved (250 mL of 75 mM APFO pH 9.0), filtered and injected into CE | MEKC: Bare fused-silica capillary(90 cm total length, 50 μm ID, 375 μm OD. BGE: 100 mM perfluorooctanoic acid (pH 9.0 adjust with 15 M NH4OH). Injection and voltage: 50 mbar, 30 sec, +23 kV. SL: 99.9:0.1 (v/v) IPA/FA, 1.66 μL/min; ESI-MS: Negative, −4.8 kV; SC: DGF: 8 L/min; DGT: 180 °C; NP: 0.082 MPa | 13 |
| Blueberry Juice | Organic acids (succinic acid, citric acid, salicylic acid malic acid, benzoic acid sorbic acid, ascorbic acid, tartaric acid) | Filtration | Juice sample diluted with BGE (1:10), centrifuged (10,000 r/min, 25,152 g for 10 min), and filtered (0.22 μm membrane) | CZE: Bare fused-silica capillary (50 μm i.d × 70 cm). BGE: 40 mM NH4OAc (pH 6.0 adjust with 1 M HOAc), +22 kV. SL: 7.5 mM HOAc in 70:30 (v/v) IPA/H2O, 6 μL/min. ESI-MS: Positive, +4 kV; SC: ESI 4 kV; DGF: 6 L/min; DGT: 350 °C; NP: 12 psi | 108 |
| Human milk | Free nucleotide monophosphate | CUF | 1 mL of milk sample mixed with 4 mL of H2O centrifuged (15 min @ 2800g). 5-mL supernatant collected (top fat layer avoided) into the CUF device (previously conditioned with 5 mL H2O) and ultrafiltered (30 min, 2800g). Filtrate injected into the CE–ESI-MS system | CZE: Fused-silica capillaries (50 and 100 μm id) with total lengths of 87.5 to MS and 20 cm to the UV detector. Injection and voltage: 50 mbar, 30 s, +30 kV. BGE: 30 mM NH4COOH (pH 9.6). SL: 50:50 (v/v) IPA/H2O,10 μL/min ESI-MS: Negative, −4 kV, SC:; DGF: 7 L/min; DGT: 350 °C; NP: 10 psi | 112 |
| Cow milk, goat milk | Estrogenic compounds (estriol, 17α-estradiol, 17β-estradiol, estrone, 17α–etynylestradiol, mycotoxin, 2-methoxyestradiol, α-zearalanol, β-zearalanol, α-zearalenol, and β-zearalenol) | DLLME | Milk sample (centrifuged, protein extracted, and defatted) diluted to a final volume of 7.5 mL (with H2O and NaCl added to final concentration of 30% w/v) and filtered (0.45 μm PTFE filter). Mixture of 500 μL ACN (as dispersion solvent) and 110 μL CHCl3 (as extraction solvent) mixed with aqueous extract (vortex 2 min). Resulting dispersion centrifuged (4000 rpm/2500×g, 5 min) observing later a droplet of CHCl3 in the bottom layer. Upper aqueous phase (~ 4 mL partially removed) (droplet transferred to a vial, dried (N2), reconstituted (75 μL BGE) and injected | MEKC: Fused-silica capillaries 60 cm (50 μm id × 363 μm od. BGE: 45 mM ammonium perflurooctanoic acid (pH 9.0) + 10% (v/v) MeOH. Injection and voltage: 0.5 psi, 25 s, +25 kV. SL: 96:4 (v/v) IPA/H2O, 1.7 μL/min ESI-MS: SC: ESI −4.6 kV V; DGF: 3 L/min; DGT: 200 °C; NP: 3 psi | 12 |
| Honey | Aminoglycosides Gentamicin, a mixture of GENT C1, GENT C1a and GENT C2, Neomycin, Apramycin, Paromomycin, Dihydrostreptomycin, Spectinomycin, Streptomycin | SPE | 10 g of honey spiked at different levels with working standards of AGs. Samples diluted (with 19.5 mL of 50 mM K3PO4 buffer pH 7.0) and final volume adjusted to 20.0 mL (with 50 mM K3PO4 buffer pH 7.0). Aliquot (3 mL) loaded onto Supelco MIP AG SPE column (pre-conditioned with 1 mL of MeOH and 1 mL of phosphate buffer pH 7.0 at a flow rate of 0.2 mL/min), washed (3 mL H2O and 1 mL 0.1% NH4OH, flow rate <0.5 mL/min), dried (vacuum 0.8 bar, 5 min), washed (1 mL 40:60 (v/v) ACN:H2O and 1 mL 50:50, (v/v) DCM:MeOH), and dried (vacuum, 10 s). Analytes eluted with 1 mL of 1% FA in MeCN:H2O (20:80, v/v), diluted (4x with H2O), filtered, and analyzed by CZE-MS/MS method. | CZE: a bare fused-silica capillary (90 cm total length, 50 μm i.d., 375 mm o.d). BGE: 200 mM formic acid, 7 mM NH4OH (pH 2.2). Injection and voltage: 25 mbar, 90 sec at 25 kV. SL: 50:49.9:0.1 (v/v/v) MeOH/H2O/-HOAc, 2 μL/min ESI-MS: −4.0 kV; SC: ESI −4000 V; DGF: 5 L/min; DGT: 150 °C; NP: 5 psi | 62 |
| Sugarcane and tomato juices | Halosulfuron-methyl herbicides | QuEChERS | 10.0 g sample placed in 50 mL centrifuge tube with 10.0 mL of ACN, 4.0 g of MgSO4 (anhydrous), and 1.0 g of NaCl (Agilent Bond Elut QuEChERS AOAC Extraction kit – PN 5982-5755). Tube shake (1 min) and centrifuged (5 min, RCF = 1521). 500 μL supernatant diluted (500 μL BGE) filtered (0.45 μm CE membrane, Agilent P/N 5190-5284) and analyzed | CZE: 58 cm long, 50 μ m i.d., 360 μm o.d. fused-silica capillary. Injection and voltage: 100 mbar, 12s, +25 kV.BGE: 20 mM NH4HCO3 (pH 8.5 adjust with NH4OH). SL: BGE diluted 5x with 1:1 (v/v) MeOH/H2O, 6 μL/min. ESI-MS: Positive, +5kV,SC: DGF: 3 L/min; DGT: 280 °C; NP: 7 psi | 54 |
| Beer and wine | Biogenic amines Spermine, Spermidine, Putrescine, Cadaverine, Histamine, 2-Phenylethylamine, Tryptamine, Tyramine, Urocanic acid, | Simple clean-up (LLE) | Phenolic compounds and other interferents removed from 10 mL sample with 0.5 g PVPP (vortex 3 min, centrifuge 5 min at RCF = 1521). Supernatant filtered (0.45 μm CE filter, Agilent P/N 5190-5284) and analyzed | CZE: 70cm long, 50 μm i.d., 360 μm o.d. polyvinyl alchol coated capillary (Agilent Technologies). BGE: 0.5 M HOAc (pH 2.5). Injection and voltage: 50 mbar, 10s, +25 kV. SL: BGE diluted 5x with 1:1 (v/v) MeOH/H2O, 6 μL/min. ESI-MS: Positive, +4.5 kV; SC: DGF: 6 L/min; DGT: 150 °C; NP: 12 psi | 103 |
| Fermented milk | Cationic and Anionic Metabolites, peptides | LLE | Fermented milk sample centrifuged (10 min @ 15,300 x g) and 100 μL supernatant mixed with 900 μL MeOH, 1000 μL CHCl3, and 400 μL H2O. Resulting mixture centrifuged (5 min @ 2,300 x g) and aqueous phase passed through ultrafiltration membrane (5-kDa cutoff filter for 2 h @ 9,100 x g, 4°C). Filtrate dried, redissolved (25 μL H2O) and analyzed | CZE: bare fused-silica capillary (50 μm, 80 cm). BGE: commercial cation solution (H3301-1001); anion solution (I3301-1023). Injection and voltage: 50 mbar, 10 s and 50 mbar , 25 s at 25 kV and 30 kV for cation and anion analysis. SL: 5 mM NH4OAc in 50% (v/v) MeOH/H2O, 1 μM reserpine, 10 μL/min. ESI-MS:+4 kV and 3.5 kV for cation and anion analysis, respectively. SC: ESI +4000 V and −3500 V; DGF: 10 L/min DGT: 300 °C, NP: 10 psi | 100 |
| Orange juice and wine | Anionic metabolites | Filtration | Samples filtered with 0.2 μm PES filter | CZE: cationic PTH capillary coating (50 μm i.d) composed of a poly-(N,N,N’,N’-tetraethyldiethylenetriamine, N-(2-hydroxypropyl) methacrylamide, TEDETAMA-co-HPMA (50:50) copolymer. BGE:1 M FA (pH 2.4); Injection and voltage: 34.5 mbar for 80s, + or −20 kV. SL: 50:50 IPA/H2O at 0.24 mL/h. ESI-MS: Positive and negative ion mode; SC: DGF: 4 L/min; DGT: 200 °C; NP: 0.4 bar | 9 |
| Japanese sake | Organic acids | Fractionation by ion exchange | pH of sake adjusted to 6.5 (1 N NaOH) and fractioned into basic AAs, neutral and acidic AAs, OAs, and sugars using columns IRC-76, IR-120BH, and IRA-96SB. OA fractions lyophilized and dissolved in H2O to 50-fold conc. Compared to original conc. OA fraction further diluted 50 x by ND96 Ringer’s solution (pH 7) | No CZE-MS conditions specified | 107 |
| Milk | Tetracyclines and quinolones | SPE | 1 g milk mixed with 4 mL of 0.2%FA in ACN and 1 mL Mcllvaine’s buffer; pH adjusted to 4 (1 M NaOH). Mixture vortexed (10s), centrifuged (6 min @ 7500 rpm), and supernatant passed at 1 mL/min through SPE cartridge (Oasis PRIME HLB preconditioned with 3 mL 0.2% FA in ACN and 1 mL Mcllvaine’s buffer). Extracts collected, dried (N2), reconstituted (10 mL of 1 M NH4OH), and filtered | CZE: a bare fused-silica capillary (90 cm total length, 50 μm i.d., 375 μm o.d.). BGE: 75 mM NH4OAc + 2.5 mM EDTA (pH 9.0). Injection and voltage, 50 mbar, 100s at +25 kV. SL: 5 μM purine and 1.25 μM HP-0921) in 70:29.9:0.1 (v/v/v) MeOH:H2O:formic acid, , 2 μL/min. ESI-MS: Positive +5.4 kV, SC: DGF: 3 L/min; DGT: 250 °C; NP: 5 psi | 109 |
| Wine | Biogenic amines | Filtration | Wine samples ultrasonicated (15 min), filtered (0.45 μm PTFE syringe filter), and injected into CE-MS. | CZE: fused silica capillary (internal diameter 75 μm, total length 100 cm); BGE: 100 mM formic acid. Injection and voltage: 0.7 psi, 6s at +30 kV. ESI-MS: +4.5 kV; SC: DGF: 4 L/min; DGT: 180 °C; NP: 0.4 bar | 81 |
| Wine | Lactic, succinic, malic, tartaric, shikimic, and citric acids | Filtration | Wine samples diluted with H2O (1:5 ratio), filtered (0.45 μm PVDF syringe filter), and injected into CE-MS | CZE: Fused -silica, 80 or 120 cm long (50 μm id), polybrene coated capillaries. BGE: 50 mM NH4OAc (pH 6.0). Injection and voltage: 50 mbar, 2s, +20 kV. SL: 1% (v/v) formic acid, 0.7 μL/min. ESI-MS: Positive, +4.5 kV; SC: DGF: 8 L/min; DGT: 325 °C; NP: 35 psi | 10 |
| Vinegars | Chiral amino acids | Filtration | 500 μL sample filtered (3 kD cutoff filter) by centrifugation (10 min @ 14,000 g) and diluted with H2O | CZE: (50 μm i.d.×358 μm o.d., 100cm in total length). BGE: 1 M formic acid + 30 mM (+)- and (−)-(18-crown-6)-2,3,11,12-tetracarboxylic acid. Injection and voltage: 50 mbar, 10s, +24 kV. SL: 1:1 (v/v) MeOH/ H2O,0.15 μM Hexakis (HP0921) and 0.075 μM purine; 10 μL/min. ESI-MS: Positive, +4 kV; SC: DGF:12 L/min; DGT: 200 °C | 26 |
| Chili sauce | Sudan dyes | Freeze-out cleanup | 2.0 g sample mixed with 5 mL ACN and sonicated (5 min, 35 kHz, 120 W). Mixture centrifuged (5 min @ 4025×g),supernatant mixed with 2.5 mL H2O and 1 g NaCl and placed at −20 °C for 3 h. 100 μL of the ACN layer diluted to 1:10 with 900 μL of 6:1:1:1 (v/v/v/v) H2O/ ACN/MeOH/THF mixture, leading to a final dilution of 1:50. Solution filtered (0.22 μm nylon syringe filter) and assayed by MEKC/MS/MS | MEKC: BGE: 60:20:10:10 (v/v/v/v), 25 mM ammonium perfluorooctanoic acid; /ACN/MeOH/THF (pH 9.0); Injection and voltage: 5 kPa, 50s, +25 kV. SL: 50:49.9:0.1 (v/v/v) IPA/ H2O/FA, 10 μL/min. ESI-MS: +3 kVSC: DGF: 6 L/min; DGT: 250 °C; NP: 206.8 kPa | 89 |
| Dietary supplement | Nanomaterials monitored isotope (m/z): 112Cd, 197Au, 195Pt, 105Pd. | No pretreatment | Suspensions injected directly to CE-ICP-MS | MEKC: Bare fused-silica capillaries (i.d. 50 μm; o.d. 360 μm; length 67 cm). BGE: 10 mM TRIS + 70 mM SDBS (pH 9.0). Injection and voltage: 30 mbar, 5s, +30 kV. Make up liquid: 1% nitric acid, 10 % MeOH in water. ICP-MS: RF power: 1500 W; plasma gas: 15 L/min, NGF: 0.9 L/min | 122 |
CZE: capillary zone electrophoresis; CIEF: capillary isoelectric focusing; MEKC: micellar electrokinetic chromatography; ESI-MS: electrospray ionization mass specorometery; ICP-MS: inductively coupled plasma mass spectrometery; BGE: background electrolyte; SL: sheath liquid; SC: spray chamber; DGT: drying gas temperature; DGF: drying gas flow; NGT: nebulizing gas temperature; NP: nebulizer pressure; AG: aminoglycoside; PVPP: poly(vinylpolypyrrolidone); PES: polyethersulfone; OA: organic acids; EDTA: ethylenediamine tetraacetate; PSA: primary secondary amine material; FMK: 2-Furylmethylketone; NGF: nebulizer gas flow.
Table 2.
List of the sample preparation methods used in the CE-MS analysis of solid food
| Sample Type | Target Compounds |
Extraction Method |
Extraction Protocol | CE-MS Conditions | Ref |
|---|---|---|---|---|---|
| Almond Skin | Phenolic compounds | LLE | 10 g dry almond skin mixed with 150 mL hexane, stirred (40 min), and gravity filtered (Whatman #4). Solids refluxed with 100 mL of 70% MeOH (60 °C, 40 min), filtered, and dried (vacuum rotary evaporator 40 °C). Residue dissolved (2 mL of 50:50 (v/v) MeOH/H2O) and analyzed. | CZE: Uncoated capillaries 95 cm long and 50 μm inner diameter (360 μm outer diameter). BGE: 200 mM (NH4)3BO3 (pH 10). Injection and voltage: 50 mbar, 15s, +30 kV. SL: 0.1% (v/v) TEA in 60:40 IPA/H2O; 0.2 mL/min. ESI-MS: Negative, −4.5 kV. SC: DGF: 4 L/min, DGT: 190 °C, NP: 0.4 bar | 104 |
| Algae | Microcystins | SPE | Algae sample centrifuged to remove extra water and mixed with 50 mL of HOAc: H2O (5:95, v/v), ultrasonicated (10 min) and centrifuged (10 min @ 4000 RPM). Supernatant loaded on SPE cartridge (C-18 phase), pre-conditioned with 10 mL MeOH and 15 mL H2O. Sample washed with 15 mL MeOH: H2O (20: 80), eluted with 10 mL (100% MeOH), evaporated to dryness and reconstituted in 1 mL MeOH. | CZE: A 90 cm length of 50 μmi.d., 360 μm o.d. uncoated fused-silica capillary. BGE: 40 mM NH4OAc (pH 9.86). Injection: 20 kV, 20s. Separation voltage: +20 kV. SL: 7.5 mM HOAc in 50:50 IPA/H2O; 3 μL/min. ESI-MS: +5 kV, SC: DGF: 6 L/min, DGT: 150 °C, NP: 10 psi. | 59 |
| Edamame | Charged metabolites | SLE | Edamame beans homogenized without solvent (60s @2000 rpm), mixed with 2.5 mL MeOH (containing 200 μM IS), 2.5 mL CHCl3 and 1.0 mL H2O and centrifuged (20 min @ 3000 rpm). 250 μL aq. solution filtered (3 min @ 10 000 rpm through 5 kDa cutoff filter). 100 μL filtrate centrifugally concentrated and dissolved in 50 μL H2O. | Cationic analyte. BGE: 1 M formic acid, SL: MeOH/H2O (50% v/v) and 0.1 M hexakis; 10 μL/min. Anionic analyte BGE: 50 mM NH4OAc (pH 8.5) SL: 5 mM NH4OAc in 50% (v/v) MeOH/H2O and 0.1 μM Hexakis; SC: ESI 3500 V, DGF: 10 μL/min, DGT: 300 °C, NP: 10 psi | 72 |
| Melon, watermelon, apricot, and peach | Pesticides Simazine, Haloxyfop, Acifluorfen, Picloram Ioxynil, Dinoseb, Flutolanil |
PLE, SPE | PLE: preheat (2 min), heating cell (5 min), solvent in contact with the sample (5 min, static time), pressure 1500 psi, temperature 60 °C, N2 purging (60 s), solvent volume flushing respect the cell size (60%) and one times cycled. H2O used as extraction solvent. H2O extracts pumped (2 mL/min) through Strata TM cartridge (pre-conditioned with 5 mL MeOH, 5 mL H2O). Retained pesticides eluted with 4 mL MeOH (1 mL/min) followed by 0.5 mL air. Eluate dried (10 min with N2 @ 40 °C), redissolved in1 mL BGE and injected | CZE: Uncoated fused-silica capillary with 50 cm total length, 75 μm ID, and 375 μm OD. BGE: 35 mM NH4HCOO/formic acid (90:10 v/v), pH 9.7. Injection and voltage: 0.5 psi, 5s, 23 kV. SL: 35 mM NH4HCOO/-FA (90:10 v/v), pH 9.7; 5 μL/min ESI-MS: Positive, + 4kV; SC: ESI , DGF: 7 L/min, DGT: 200 °C, NP: 13 psi | 58 |
| Soybean | Peptides | SLE | 150 mg grounded beans + 1.5 mL extraction buffers (ACN/H2O (80:20 v/v), vortexed 10 min, sonicated 3 min, and centrifuged 5 min @ 9000 g). 50 μL supernatant boiled (10 min), and 50 μL bovine pancreas trypsin solution (50mM NH4HCO3, pH 8.0) + (1:50, 1:25 and 1:5 w/w) enzyme:substrate incubated overnight at 37 °C, shaking @ 600 rpm). Reaction stopped by heating (90 °C) for 5 min, suspension centrifuged (5 min @ 14 000 g), supernatant fraction collected and stored at 20°C prior to analysis. | CZE: Uncoated fused-silica capillary (50 μm id, 90 cm total length). BGE: 0.5 M formic acid. Injsction and voltage: 34.5 mbar, 20 s, +25 kV. SL: 50:50 (v/v) IPA/H2O; 3 μL/min. ESI-MS: positive ion mode, SC: DGF: 4 L/min, DGT: 200 °C, NP: 0.4 bar | 86 |
| Infant milk formula | Bioactive peptides | SPE | 4.7 g IF dissolved in 30mL H2O,0.5 mL of the resultant emulsions diluted with 2.5 mL of reduction buffer (73 mg of trisodium citrate dihydrate, 38mg of DTT in 37.5 mL of 8 M urea; pH 8 adjusted with NaOH),incubated (1 h at RT). Incubation samples centrifuged (30 min @ 4600 rpm), fat layer removed, clear solution filtered (0.22 μm nylon filters) before SPE. Sep-Pack® C18 cartridge conditioned (2 mL MeOH, and 2 mL H2O), 2mL of sample loaded, retained compounds eluted with 200 μL solution (80:20 (v/v) MeOH:H2O and 0.1% (v/v) of FA) at 1 mL/min. Eluate dried (air at RT) and reconstituted with 200 μL H2O before CE–MS. | CZE: Uncoated 57 cm× 50 μm I.D. fused silica capillary. BGE: 1 M HOAc. Injection and voltage, 33.5 mbar, 15s, +15 kV. SL: 0.05% (v/v) FA in 60:40 (v/v) IPA/H2O; 3.3 μL/min. ESI-MS: Positive +4 kV; SC: DGF: 4 L/min, DGT: 200 °C, NP: 7 psi. | 60 |
| Feces | Oligosaccharides | SPE | Fecal slurries (50 mg/mL) kept overnight at 4 °C, centrifuged (15 min, 3500 x g) and filtered (0.22 μm membrane). Fecal enzymes inactivated by heat (5 min, 100 °C). Carbohydrates extracts purified by SPE on graphitized carbon column cartridges (150 mg bed weight, 4 mL tube size; Cartridges washed with 0.1% (v/v) TFA in 80/20 (v/v) ACN/H2O followed by H2O. Extracts loaded onto cartridge; monomers/lactose removed using aq. 2% (v/v) ACN, and carbohydrates eluted with 0.05% (v/v) TFA in 40/ 60 (v/v) ACN/H2O. Dried and rehydrated with H2O. | CZE: Bare fused-silica capillary (50 μm ID x 85 cm L) BGE: 0.3% (v/v) formic acid (pH 2.4). Injection and voltage: 10 psi, 2s, −20 kV. SL: 50:50 IPA/H2O; 2 μL/min. ESI-MS: Negative, −1.9 kV SC: ESI −1.9 kV, DGF: μL/min, MS capillary T: 190 °C. | 68 |
| Milk powder | Melamine and its analogs (Melamine, Ammeline, Ammelide, Cyanuric Acid) | SPE | Delipidated milk powder reconstituted (instructions on package). Protocol (A): 1 mL sample + 100 μL of 0.2 M HCl + 33 mL ACN (vortex, centrifuge 10 min @ 6000 rpm). Supernatant loaded onto Strata cartridge (preconditioned 3 mL MeOH and 3 mL H2O (1 mL/min); washed (1 mL ACN/H2O (50:50, v/v), 500 μL MeOH/ H2O (50:50, v/v)); dried (2 min, 10 psi); eluted (500 μL MeOH and 1 mL MeOH-NH4OH (95:5, v/v)); dried (N2); reconstituted in 1 mL ACN/20 mM NH4OAc (95:5, v/v); injected into CE/MS. Protocol (B): 10 mL (0.12 M) HCl + 5 g sample (vortexed 45 s, centri-fuged 5 min @ 4000 rpm); supernatant loaded onto MCX cartridge (preconditioned 5 mL MeOH, 5 mL H2O (1 mL/min)); washed (5 mL (0.1 M HCl), 2 mL MeOH); eluted (5 mL ACN-NH4OH (95:5, v/v); injected into CE/MS | CZE: Bare fused-silica capillary (total length of 80 cm and an internal diameter of 50 μm). BGE: 25 mM NH4OAc (pH 5.2). Injection and voltage: 50 mbar, 25 s, +30 kV. SL: 25 mM HOAc in 50:50:2 (v/v/v) IPA/H2O/NH4OH; 3 μL/min. ESI-MS: Positive, +4.3 kV and negative, −3.4 kV; SC: DGF: 4 mL/min, DGT: 250 °C, NP: 4 psi. | 65 |
| Rice | Selenium monitored isotope (m/z): 78Se, 80Se | EAE | 0.5 g rice (crushed)+ 40 mg protease + 5 mL H2O (incubated 16 h @ 37 °C) centrifuged (10 min @ 5000 rpm); supernatant filtered (0.22 μm); diluted with H2O and analyzed by CE-ICP-MS. | CZE: Bare fused silica capillary i.d. 75 μm; o.d. 365 μm; 80 cm long. BGE: 20 mM NaH2PO4 + 10 mM Na2B4O7 + 0.2 mM CTAB (pH 8.6). Injection and voltage:10 s,−16 kV. ICP-MS: velocity: 12 μL/min (pump 1), 200 μL/min (pump 2); RF power: 1300 W; plasma gas: 15 L/min (outer), 0.9 L/min (intermediate); carrier gas: 0.7 L/min; makeup gas: 0.3 L/min; DRC reaction gas: H2 (3 mL/min); nebulizer: MCN (50-200 μL/min) | 39 |
| Milk powder | melamine | SLE | 0.5 g sample + 1 mL NH4OH (0.1 M) + 3.5 mL ACN (vortexed 20s, sonicated 30 min, and centrifuged 20 min @ 6000 rpm) and clear liquid dried (oven @ 70 °C), mixed with 1 mL NH4OH (0.1 M), and analyzed by CEC-MS | CEC: (100 μm i.d., 60 cm L). a polymerization solution (divinyl benzene and alkene (1-octene, 1-dodecene or 1-octadecene), porogenic solvents water, cyclohexanol and N,N-dimethylacetamide charged monomer (vinyl-benzyltrimethylammonium and initiator (AIBN) was used toprepare the monolithic columns. BGE: 80:20 (v/v) 5 mM NH4OAc/ACN (pH 8). Injection and voltage: −5 kV (3s), 0 kV. SL: 2.5 mM NH4OAc in 80:20 IPA/H2O; 220 μL/h. ESI-MS: Positive, +4 kV , SC: DGF: 4 L/min, DGT: 180 °C, NP: 5.8 psi | 20 |
| Tea leaves | Metabolites | SLE | 100 mg powdered leaf + 1 mL MeOH (containing 50 μM IS) + 1 mL CHCl3 + 400 μL H2O (vortexed 20s, sonicated 30 min, centrifuged 20 min @ 6000 rpm); aqueous layer recovered; ultrafiltered (5 kDa-cutoff filter by centrifugation 120 min @ 9,100 g, 4 °C); dried and dissolved in 50 μL H2O (containing 100 μM IS). | CZE: Bare fused-silica capillary (50 μm X 80 cm). BGE: 1 M formic acid (pH 1.8) (cationic analytes); 50 mM NH4OAc (pH 8.5) (anionic analytes). SL: 5 mM NH4OAc in 50% (v/v) MeOH/H2O; 10 μL/min. ESI-MS: Positive (+4 kV) and Negative (−3.5 kV) ion modes. SC: DGF: 10 L/min, DGT: 300 °C. | 83 |
| Fermented soybean | Metabolites | SLE | 5 mg sample + 5 mg/mL MeOH centrifuged (10 min @ 5000 rpm, 25 °C), extracts filtered (0.22 μm), and analyzed by CE-MS | CZE: Bare fused-silica capillary (50 μm i.d. × 100 cm total length). BGE: 1 M formic acid (cation analysis); 20 mM NH4HCOO, pH 10 (anion analysis). Injection and voltage: I5 mbar,5s, +30 kV, SL: 0.5 M reserpine in 1:1 (v/v) MeOH/H2O; 10 μL/min ESI-MS: + 4 kV (cation) −4kV (anion), SC: DGF: 10 psig, DGT: 300 °C. | 75 |
| Flour, pasta, bread | Furosine | Acid hydrolysis | 100 mg samples hydrolyzed with 5 mL (8 N) HCl (23 h @ 110 °C), filtered (0.45 μm PTFE); dried (N2), resuspended (2.5 mL BGE), and re-filtered (0.20 μm PTFE). Aliquot diluted with 14 μL (25 mg/L) quinine to obtain conc. 0.5 mg/L in 700 μL final volume. | CZE: Bare fused-silica capillary 50 μm id and effective length of 90 cm and 60 cm. (BGE: 50 mM formic acid (pH 2.7 adjust with 0.5 M NH4OH) Injection and voltage: 50mbar, 5s, +25 kV. SL: 0.1% FA in 50:50 (v/v) MeOH/H2O; 3 μL/min ESI-MS: Positive, +3.5 kV, SC: DGF: 5 L/min, DGT: 200 °C, NP: 10 psi. | 90 |
| Dietary polyphenols Treated with HT29 colon cancer cells | Metabolites | Cell-disruption | Cells (equal volume) washed (PBS solution) and centrifuged. Pellets suspended with homogenization buffer and protease inhibitor cocktail. Cells disrupted (Polytron homogenizer), centrifuged (14 min @14 000 × g, 4 °C), pellet (nuclear and mitochondrial fractions) discarded, supernatants centrifuged (1 h @ 100,000 × g, 4 °C), 400 μL of cytosolic fraction ultra-filtrated (40 min @14,000 × g, 20 °C). | CZE: Uncoated fused-silica capillary (50 μm id, 363 μm od and 80 cm total length). BGE: 1 M formic. Injection: 34.5 mbar, 80s, +25 kV. SL: 50:50 (v/v) IPA/H2O; 0.2 mL/min ESI-MS: Positive ion mode, −4.0 kV. SC: DGF: 4 L/min, DGT: 250 °C, NP: 0.4 bar. | 41 |
| K562 leukemia cells | Metabolites | Cell-disruption | Cells (equal volume) washed (PBS solution) and centrifuged. Pellets suspended with homogenization buffer and protease inhibitor cocktail. Cells disrupted (Polytron homogenizer), centrifuged (14 min @14 000 × g, 4 °C), supernatants centrifuged (1 h @ 100,000 × g, 4 °C) (cytoplasmic fraction). 400 μL of cytoplasmic fraction ultra-filtrated (40 min @14,000 × g, 20 °C). Fractions with molecular weight <3 kDa aliquoted and stored at −80m°C until CE-MS analysis. | CZE: Uncoated fused-silica capillary (50 μm id, 363 μm od and 80 cm total length). BGE: 1 M formic acid. Injection and voltage: 0.5 psi, 60s, +25 kV. SL: 50:50 (v/v) IPA/H2O; 0.2 mL/min ESI-MS: Positive, +4.0 kV , SC: DGF: 4 L/min, DGT: 250 °C, NP: 0.4 bar. | 46 |
| Milk powder | β-lactoglobulin and α-lactalbumin | Protein purification | 20 mg/mL milk powder solution centrifuged (5 min @15 000 × g). 2 mL of defatted milk pH adjusted to 4.5 with 1M HCl and centrifuged (5 min @15 000 × g), precipitated casein removed. supernatant diluted 50 x with sample BGE before analysis. | CZE: Uncoated fused silica capillaries (50 μm id, 375 μm od, 50 cm total length. BGE: 10% HOAc (pH 1.5); Sample BGE: 10 mM PBS + 0.1% Tween 20. Injection and voltage: 39 mbar, 600s, +24 kV. MALDI : positive, matrix: sample collected on MALDI plate, buffer droplets dried, 2 μL of matrix (2 mg/mL SA in 70% ACN, 0.1% TFA) spotted above each sample droplet, dried at RT. | 8 |
| Fish | Hg(II), MeHg, and EtHg (monitored isotope (m/z): 199Hg, 201Hg, 202Hg); | MSLE | 0.5 g dried fish + 20.0 mL of 1 M HCl solution in 100 mL Teflon beaker/screw covered placed in microwave digester (5 min @ 70 °C, 400 W). Sample cooled (RT), extracts separated by filtering (0.22 μm membrane filter), and dried (pressured N2 blowing concentrator). Residue diluted to the appropriate volume with BGE and used for the CE–ICP–MS analysis with continuous sample-introduction mode. | CZE: i.d. 75 μm; o.d. 365 μm; 85cm long. BGE: 50 mM H3BO3 + 12.5 mM Na2B4O7 (pH 9.2). Inject: 10s, +18 kV; sep voltage: +18 kV. ICP-MS: velocity: 11 μL/min (pump 1), 6 μL/min (pump 2); RF power: 1300 W; plasma gas: 15 L/min (outer), 0.9 L/min (intermediate); carrier gas: 0.75 L/min; makeup gas: 0.3 L/min; DRC reaction gas: H2 (3 mL/min); nebulizer: ACN (20-35 μL/min) | 42 |
| Chili powder | Sudan dyes | SLE | 1.0 g chilli powder diluted (10 mL 3:2:1, (v/v/v), acetone/DCM/MeOH), vortexed (2 min), sonicated (5 min), and centrifuged (5 min @ 10,000 rpm). supernatant dried, suspended (5 mL acetone) and diluted (50:50 v/v) with BGE (60mM NH4HCO3, 50mM SDS, and 30% ACN). | MEKC: Bare fused silica capillary (a total length of 80 cm and 50 μm id, 375 μm od). BGE: 40mM NH4HCO3, 32.5% ACN partially filled 25mM SDS (20 psi, 13.2s). Injection and voltage: 0.5 psi, 6s, +25 kV. SL: o.1% FA in 50:50 (v/v) IPA/H2O; 4 μL/min. ESI-MS: Positive, +4.5 kV, SC: DGF: 4 L/min, DGT: 200 °C, NP: 4 psi. | 88 |
| Infant milk formula | Peptides | SPE | 4.7 g of IF + 30 mL H2O (warm) water (45 °C). Resultant emulsion (0.5 mL) diluted with 2.5 mL reduction buffer (73 mg of trisodium citrate + 38 mg DTT in 37.5 mL (8 M) urea; pH 8 adjusted with dil. NaOH; make up to 50 mL with H2O); incubated (1 h at RT); centrifuged (30 min @ 2500 × g); fat layer removed; clear solutions filtered (0.22 μm nylon filter. 2 mL solution loaded onto SPE cartridge (preconditioned with 2 mL MeOH, 2 mL H2O); compounds eluted with 200 μL of 80:20 (v/v) MeOH/-H2O and 0.1% (v/v) FA at 1 mL/min. Eluate dried with air (RT) and reconstituted with 200 μL H2O before CE-MS analysis. | CZE: A 57 cm × 50 μm id × 365 μm od fused silica capillary. BGE: 1 M HOAc (pH 2.3). Injection and voltage: 33.5 mbar, 15 s, +15 kV. SL: 0.05% HF in 60:40 (v/v) IPA/H2O; 3.3 μL/min ESI-MS: Positive, + 4 kV, SC: DGF: 4 L/min, DGT: 200 °C, NP: 7 psi. | 61 |
| Egg | Benzimidazoles | QuEChERS | 3 g egg yolk + 10 g H2O + 10 mL ACN (mix 1 min in centrifuge tube). 4.0 g MgSO4, 1.0 g of NaCl and 1.0 mL NH4HCOO solution (2.5 mM, pH 7.5) added to tube (shake 1 min) and centrifuged (5 min @ 1500 × g). DSPE cleanup: 1 mL supernatant + 0.025 g PSA + 0.150 g MgSO4, centrifuged (2 min @ 1500 x g), dried (N2) and dry residue vortex-shaken in 1 mL of an ACN/formic acid (99:1, vol.) solution. | CZE: A 75 μm I.D. with a total length of 57 cm and 50cm to the UV detector. BGE: 6 M formic acid in 70:30 H2O/IPA. Injection and voltage: 50 mbar, 30 s, +22 kV. SL: 50:50 IPA/H2O; 10 μL/min ESI-MS: Positive, +4 kV , SC: DGF: 7 L/min, DGT: 350 °C, NP: 4 psi. | 72 |
| Soy biscuit | Peptides | QuEChERS | 2.5 g of grounded biscuit extracted (10.0 mL hexane), organic layer discarded and solid portion mixed with 10 mL 50:50 (v/v) ACN:H2O (shaken 5 min, RT) followed by addition of mixture (4 g MgSO4 + 1 g NaCl + 1 g Na3Cit.2H2O + 0.5 g Na2HCit. 1.5H2O), tube shaken vigorously (1 min to prevent MgSO4 conglomerates), and centrifuged (5 min @ 5000 rpm). 1.0 mL ACN fraction subjected to DSPE using a mixture of 150 mg MgSO4, 150 mg of silica sorbent, 25 mg of PSA and 25 mg of C18 sorbent, shaking vigorously (1 min), and centrifuging (5 min @ 5000 rpm). Extracts filtered (0.22 μm PVDF syringe filter) and 100 μL filtered extract diluted (H2O) to a volume of 1.0 mL before analysis. | CZE: Uncoated fused silica capillary 87.5-cm total-length, 21 cm to the UV detector. BGE: 15 mM NH4OAc (pH 11), Injection and voltage: 50 mbar, 50 s, +25 kV. SL: 90:10 (v/v) IPA/H2O; 1 μL/min. ESI-MS: Positive, 3.5 kV, DGF: 6 L/min, DGT: 350 °C, NP: 4 psi. | 71 |
| Royal jelly products | AAs | SLE | Tablets were grounded, capsules opened to release the contents. 1.0 g aliquot mixed with 9 mL of 75% (v/v) EtOH, ultrasonicated (5 min, RT) and centrifuged (5 min @3000 rpm/1200 x g). 0.9 mL supernatant mixed with 0.1 mL of 1 M HCl, and 0.1 mL portion of the IS (1 mg/mL Cys) added prior to CE-MS/MS analysis. | CZE: Uncoated fused-silica capillary (50 μm, I.D., 100 cm in length). BGE: 1 M formic acid (pH 1.8). Injection and voltage: 50 mbar, 5s, +30 kV. SL: 50:50 (v/v) MeOH/H2O; 8 μL/min. ESI-MS: +4 kV. SC: DGF: 10 L/min, DGT: 300 °C, NP: 10 psi. | 91 |
| Avocado fruits | 10 Metabolites | SLE | 4 g freeze-dried (and homogenized) sample mixed with 40 mL MeOH and shaken (vortex 30). Supernatants centrifuged (10 min @ 3000 rpm), resulting supernatants dried and redissolved in 1 mL of 50/50 (v/v) MeOH/H2O. | CZE: Bare fused-silica capillaries with 50 μm id and a total length of 85 cm. BGE: 40 mM NH4OAc (pH 9.5). Injection and voltage: 0.5 psi, 5s, +30 kV. SL: 60:40 (v/v) IPA/H2O; 0.24 mL/h ESI-MS: Negative, −3.5 kV , DGF: 5 L/min, DGT: 250 °C, NP: 5 psi. | 76 |
| Milk powder | Allergens (IgE) | SLE | Milk powder dissolved in H2O at 20 mg/ mL and 2 mL of solution mixed with 50 mM NH4OAc buffer (pH 4.5), centrifuged (2 min @ 12,000g), and supernatant with whey proteins removed from the casein precipitate. Casein precipitate washed (NH4OAc buffer) and dissolved in 2 mL of 25 mM NH4HCO3 buffer (pH 8.5). UHT treated milk defatted by centrifugation (5 min @12 000g). Whey and casein fraction solutions diluted 20x with the sample buffer prior to IACE analysis. | CZE: Capillary (50 μm i.d., 41.5 cm effective length, 50 cm total length) coated with 5% hydroxypropylcellulose (HPC) solution, BGE: 10% HOAc (pH 2); sample buffer: 10 mM PBS + Tween 20 (pH 7.4). Injection and voltage: 40 mbar, 300s, +24 kV. MALDI matrix: 2 mg/ml SA in 70% ACN, 29.9%H2O, 0.1% TFA) | 38 |
| Tea leaves | Caffeine | SLE | 2 g of Chinese white tea leaves + 10 mL MeOH/H2O (95/5, v/v) ultrasonicated (20 min), filtered and injected for online CZE-DART-MS analysis. | CZE: 80 cm fused silica capillary with 75 μm i.d. and 360 μm o.d.. BGE: 15 mM sodium borate, 15 mM SDS, 18% acetonitrile. Injection and voltage: 50 mbar, 10s,+25 kV. SL: 25:25:50 (v/v/v) MeOH/acetone/H2O; 10 μL/min. DART-MS: −3.5 kV, SC: DGF: 0.12 L/h, DGT: 300 °C, NP: 2 psi. | 5 |
| Infant formula | Five Riboucleotides (AMP, GMP, CMP, IMP, UMP) | SLE, CUF | 0.5 g of infant milk mixed with H2O up to 15 g, stored at 5°C (15 min) followed by centrifugation (15 min @ 2800 × g). 10 mL supernatant collected (avoid collecting top fat layer) and passed through the CUF device (30min, 2800×g), pre-conditioned with 5 mL H2O (15 min, 2800 × g). | CZE: Uncoated fused-silica capillaries (50 and 100 μm id) with total lengths of 87.5 (MS) and 20 cm to the UV. BGE: 30 mM NH4HCOO/NH4OH (pH 9.6). Injection and voltage: 50 mbar, 30s, +25 kV. SL: 50:50 (v/v) IPA/H2O; 10 μL/min. ESI-MS: −4.0 k V, DGF: 7 L/min, DGT: 350 °C, NP: 10 psi. | 94 |
| Mussel | Paralytic shellfish toxins | SPE | 3 mL 1% HOAc solution + 5.0 g homogenized raw meat vortexed (1 min), boiled (100°C, 5 min); revortexed, cooled (5°C, 5 min), and centrifuged (10 min @ 4500 rpm). Supernatant stored at −20°C. Prior to analysis, 0.5 mL extract cleaned using a C18 cartridge (Waters). Effluent containing toxins collected and column rinsed with 2 mL H2O to remove residual PSTs from the column. A 0.5 μL 1 M NaOH added to extract. | CZE: Bare fused silica capillaries (57 cm-long 50 μm id). BGE: 35 mM morpholine (pH 5 adjust with formic acid). Separation and voltage: 5 kV for 10s, +20 kV. SL: 0.1% formic acid in 80:20 (v/v) IPA/H2O; 4 μL/min. ESI-MS: Positive, 4.2 kV, SC: DGF: 2 L/min, DGT: 250 °C, NP: 10 psi. | 63 |
| Cheese | Lysozyme | SLE | 2 g cheese sample and 70 mL of 1 M HOAc (in H2O) added into a 100 mL volumetric flask and placed in thermal bath (40 °C). Sample homogenized (1 h @ 25,000 rpm), H2O added to the flask to 100 mL mark. Extracted samples filtered (0.45 μm) prior to analysis. | CZE: Polyacrylamide coated fused silica capillary, BGE: 100 mM formic acid. Injection and voltage: 100 mbar, 5s, +20 kV. SL: 50:49.5:0.5 (v/v/v) MeOH/H2O/FA; 4 μL/min. ESI-MS: Positive, +4 kV SC: DGF: 10 L/min, DGT: 200 °C, NP: 10 psi. | 93 |
| Cereals, barley | 5-hydroxymethylfurfural | SLE | 300 mg sample dissolved in 6 mL of 1% TCA solution, vortexed (2 min), ultrasonicated (10 min), vortexed (2 min), and centrifuged (10 min @ 9000 rpm, 4 °C). Extracts filtered (0.20 μm nylon filter) and mixed with 6 mL of 500 mg/L FMK solution to obtain a concentration of 5 mg/L to a final volume of 600 μL. | CZE: Untreated fused-silica capillaries of 50 μm i.d. total length 60 cm. BGE:50 mM formic acid (pH adjust with 0.5 M NH4OH). Injection and voltage: 50 mbar (10s), +25 kV. SL: 0.1% formic acid in 50% MeOH, 3 μL/min. ESI-MS: Positive, +3.5 kV. SC: DGF: 2 L/min; DGT: 200 °C; NP: 10 psi | 92 |
| Soybean | Cationic and anionic metabolites | LLE | 70 mg dry sample + 20 volumes of MeOH containing 8 μM reference compounds (methionine sulfone for cation and camphor 10-sulfonic acid for anion analyses) mixed (Retsch mixer mill MM310 @ 27 Hz for 1 min). Extracts centrifuged (3 min @15,000 × g, 4 °C), 500 μL supernatant mixed (500 μL of CHCl3 + 200 μL H2O), upper layer removed and evaporated (30 min @ 45°C) by centrifugal concentrator to obtain two layers. High-molecular-weight compounds (oligosugars) removed by centrifugal filtration (5-kDa cutoff filter @ 9,100 g and 4°C for 120 min) of upper layer and filtrate dried (120 min) by centrifugal concentrator | CZE: Bare fused silica capillary (50 μm i.d. × 100 cm total length). BGE: 1 M formic acid (cation analysis); 20 mM NH4HCOO, pH 10 (anion analysis). Separation and voltage: 50 mbar, 50 s, +30 kV. SL: 0.5 μM reserpine in 50:50 (v/v) MeOH/H2O; 10 μL/min. ESI-MS: +4 kV for cation and −4 kV for anions, SC: DGF: 10 psi, DGT: 300 °C | 99 |
| Algae | Harmala alkaloids | SLE | 0.1 g sieved powder extracted (500 μL MeOH and 500 μL of 3.5 M HCl), mixture incubated (6 h at 80 °C) in a shaker. Sample vortexed (30 s), centrifuged (15 min @10,000g, 25 °C), supernatant ultrafiltered (3 kDa cut-off,15 min @16,000g, 25 °C),washed with 50 μL H2O (centrifuged,10 min @12,000g). H2O added to a final volume of 250 μL | CZE: A 60 cm total length, 75 μm (i.d), and 360 μm o.d bare silica capillary. BGE: 25 mM NH4OAc + 10 (v/v) MeOH (pH 7.8). Injection and voltage: 35 mbar, 3s, +20 kV. SL: 0.05% formic acid in 60:40 (v/v) IPA/H2O; 3.3 μL/min. ESI-MS: Positive, +4 kV and −4 kV (cation and anion); SC: DGF: 5 mL/min, DGT: 250 °C | 95 |
| Yoghurt | Estrogenic compounds | DLLME | Yoghurt sample (centrifuged, protein extracted, and defatted) diluted to a final volume of 7.5 mL (with H2O and NaCl added to final concentration of 30% w/v) and filtered (0.45 μm PTFE filter). Mixture of 500 μL ACN (as dispersion solvent) and 110 μL CHCl3 (as extraction solvent) mixed with aqueous extract (vortex 2 min). Resulting dispersion centrifuged (4000 rpm/2500×g, 5 min) observing later a droplet of CHCl3 in the bottom layer. Upper aqueous phase partially removed (about 4 mL), droplet transferred to a vial, dried (N2), reconstituted (75 μL BGE), and injected. | MEKC: Fused-silica capillaries of 60 cm (50 μm id × 363 μmid). BGE: 45 mM ammonium perfluorooctanoic acid (pH 9.0) + 10% (v/v) MeOH. Injection and voltage: 0.5 psi (25 s), +25 kV. SL: 96:4 (v/v) IPA/H2O, 1.7 μL/min. ESI-MS: Negative, −4.6 kV; SC: DGF: 3 L/min; DGT: 200 °C; NP: 3 psi | 12 |
| Yeast tablet | Cr(III), Cr(VI), Cr picolinate | SLE | 0.1 g sample + 4 mL (1:3, v/v) MeOH/H2O, ultrasonicated (20 min, 200 W). whole solution cooled (RT), extract separated by filtering (0.22 μm membrane filter), residue diluted (H2O) and used for CE-ICP-MS analysis. | CZE: BGE: 25 mM Na2HPO4 + 6.26 mM Na2B4O7 + 0.6 mM CTAB (pH 7.8) ICP-MS: velocity: 20 μL/min (pump 1), 280 μL/min (pump 2); RF power: 1400 W; plasma gas: 15 L/min (outer), 0.9 L/min (intermediate); carrier gas: 0.8 L/min; makeup gas: 0.2 L/min; DRC reaction gas: H2 (2 mL/min); monitored isotope (m/z): Cr52; nebulizer: MCN (100-400 μL/min) | 84 |
| Rice | Arsenic species monitored isotope (m/z): 75 (As+); 77 (ArCl+) | EWPMD | Rice and rice cereal ground with an 8000 M mixer/mill, sieved (0.1 mm mesh), oven-dried (2 h @ 90 °C). 0.2 g of rice powder + 0.1 g of 0.5% (w/w) α- amylase mixed (15 mL glass tube) and diluted to final weight (2 g with H2O). Tube capped and placed in a microwave digestion system (1 h @ 80 °C and 2 h @ 90 °C with stirring). Centrifuge to remove remaining solids and 30 μL of o-ASA (50 ng/g) added to the 270 μL rice extract. | CZE: A 60 cm long coated fused silica capillary, 100 μm i.d., 360 μm o.d. BGE: 8 mM Na2CO3 (pH 11). Injection and voltage: 15 mbar (8s), + 20 kV. ICP-MS: velocity: 20 μL/min (pump 1), 280 μL/min (pump 2); RF power: 1500 W; plasma gas: 15 L/min; carrier gas: 0.9 L/min; makeup gas: 0.45 L/min; DRC reaction gas: H2 (2 mL/min); nebulizer: Mira Mist CE. | 40 |
| Cheese | Lysozyme | SLE | 2 g of grounded cheese mixed with 20 mL NaCl 1M (10 min at 30 Hz). pH of mixer raised to 6.0 with 1M NaOH, and temp. maintained (40°C for 30 min). pH decreased 4.3 with 1M HCl and mixture filtered ( filter paper and 0.22 μm filter). | CZE: Fused-silica capillaries of 50 μm id with poly-(TEDETAMA-co-HPMA) 50:50 copolymer-coating. BGE: 35 mM NH4OAc (pH 4.8 adjust with HOAc). Injection and voltage: 0.5 psi (5s), −20 kV.ESI-MS: SC: DGF: 4 L/min, DGT: 200 °C, NP: 0.4 bar. | 93 |
| Baby foods | Nucleotides | SLE | 0.50 g + H2O (4.5 g), shaken manually, centrifuged (15 min @ 2800g, RT). 4.0-mL supernatant and passed through CUF device (30 min @ 2800g) previously conditioned with 5.0 mL H2O (15 min @ 2800g). 0.5 mL of H2O added to the CUF device and centrifuged again (30 min @ 2800g),filtrate directly analyzed by CE-MS. | CZE: Bare fused silica capillary, 50 μm I.D., BGE: 76 mM hexafluoro-2-propanol (pH 10 adjust with conc. NH4OH). Injection and voltage: 50 mbar (40s), +30 kV. SL: 1.6% (v/v) hexafluoro-2-propanol in H2O; 10 μL/min. ESI-MS: 3.5 kV, SC: DGF: 7 L/min, DGT: 350 °C, NP: 7 psi. | 94 |
| Onion fed rat’s liver | Metabolites | SLE | 50 mg liver tissue homogenized, cold MeOH/H2O (1:1, w/v) at 1:10 (w/v). 100 μL homogenate + 100 μL of 0.2 M formic acid (FA) centrifuged (10 min @16,000 x g, 4 °C) and added to a centrifree ultracentrifugation (30 kDa protein cutoff filter) for deproteinization by centrifu-gation (70 min @ 2000 x g, 4 °C). Filtrate dried (Speed-Vac Concentrator), resuspended in 100 μL of 0.1 M FA, 0.2 mM methionine sulfone (IS) before analysis. | CZE: Uncoated fused silica capillary (total length, 96 cm; i.d., 50 μm). BGE: 0.8 M formic acid in 190% MeOH. Injection and voltage: 50 mbar (50 s), +30 kV. SL: 10 mM formic acid in 1:1 (v/v) MeOH/−H2O; 6 μL/min. ESI-MS: Positive, +3.5 kV, SC:.5 kV, DGF: 10 L/min, DGT: 200 °C, NP: 10 psi. | 77 |
| Algae | Harmala alkaloids | Online-SPE | 0.1 g (±0.0001 g) of dried algae + 500 mL MeOH + 500 ml (3.5 M) HCl incubated (6 h @ 80 °C, Thermo-Shaker TS-100), vortexed (30 s) and centrifuged (15 min @ 10,000 g, 25 °C). Solid discarded and supernatant (approx. 950 ml) evaporated to dryness, reconstituted (H2O to a final volume of 250 mL) and filtered (0.22 μm nitrocellulose membrane) before ultrafiltration (3 KDa cut-off, 15 min @16,000 g, 25 °C). 50 ml H2O added to filtrate and centrifuged (10 min @12,000 g). Filtrate reconstituted with H2O to final volume of 100 ml and analyzed by SPE-CE-MS. | CZE: 60 cm total length, 75 μm id, and 360 μm od bare fused silica capillary. BGE: 10 mM NH4OAc, 10% MeOH (pH 7.8). Injection and voltage: 35 mbar (3 s), +20 kV. SL: 0.05% (v/v) FA in 60;40 (v/v) IPA/H2O; 3.3 μL/min. ESI-MS: +4 kV; SC: DGF: 4 L/min, DGT: 200 °C, NP: 5 psi. | 55 |
| Lobster | Neurotoxin (β-N-methylamino-L-alanine) | SPE | 50-mg freeze-dried samples hydrolyzed with 1 mL 6 M HCl in flame-sealed glass ampoule (16 h @ 110 °C). Hydrolysates dried (N2 @ 55 °C), spiked with BMAA-d3 and reconstituted in 2.5 mL (20 mM) HCl. A portion of this crude extract (25 mg tissue equivalent) loaded onto an Oasis-MCX cartridge (pre-conditioned with 3 mL MeOH and 3 mL (20 mM) HCl), washed (3 mL H2O and 4 mL MeOH) and BMAA eluted with 7 mL (5% w/v) NH4OH. Eluent dried (N2), reconstituted (0.5 mL of 2 mM HCl) and filtered (0.22 μm Ultrafree-MC filter) | CZE: The exit of the CE capillary was stripped of polyimide coating, heated, pulled and cut. The tapered section (ca. 4.5 μm long), 100 μm OD and 30 μm ID. BGE: 5 M formic acid in 9:1 (v/v) H2O/can. Injection and voltage: 50 mbar (60 s), + 15 kV to +30 kV. SL: 50;50:0.1 (v/v/v), MeOH/−H2O/formic acid; 1 μL/min. ESI-MS: 4.2 kV. | 64 |
| Head lettuce | Metabolites (50 target compounds (anions: organic acids, phosphorylated compounds, cations: amino acids) | SLE | 50 mg of frozen leaves grounded and homogenized in liquid N2. Samples mixed with 0.2 mL MeOH (100%) for enzyme inactivation. 0.2 mL H2O containing IS (50 μM (PIPES) and 50 μM methionine sulfone) added to homogenates, centrifuged (4 min @ 22,000g, 4 °C), supernatant ultrafiltered (3 kDa cut-off filter) with centrifugation (30 min @ 14,000g, 4 °C) and analyzed by CE-MS | CZE: For anions polyethylene glycol-coated capillary (DB-WAX, Agilent Technologies, Palo Alto, CA., USA, 100 cm , 50 μm i.d.) with 20 mM ammonium acetate (pH 8.5) as a running buffer. For cations, uncoated fused silica capillary (90 cm, 50 μm i.d.),1 M formic acid (FA) pH 1.9) as the running buffer. Injection and voltage: ±25 kV voltage was carried out for anions in negative mode or cations in positive mode. ESI-MS: + 3.5 kV for cations, −3.5 kV for anions. SL: 5 mM NH4OAc in 50% (v/v) MeOH(for anions); 0.1% FA in 50% (v/v) MeOH (for cations); 8 μL/min. SC: DGF: 8 L/min, DGT: 320 °C | 11 |
| Mussel | Saxitoxin monitored isotope (m/z): 151Eu; 153Eu; | SLE | 0.5 g vacuum freeze dried (−46°C) dried mussel powder mixed with 1.0 mL DMSO under ultrasonic oscillation (30 min @ 30°C). DMSO solution (with extracts) separated by centrifugation. DMSO extracting solution concentrated to 500 μL (N2-blowing concentrator) and used for CE-ICP-MS analysis | CZE: 60 cm length × 75 μm i.d. × 375 μm o.d. bare fused-silica capillary. BGE: 20 mM NaH2PO4 + 5 mM Na2B4O7 + 0.2 mM CTAB (pH 6 adjusted with 0.1 M HCl). Injection and voltage: ICP-MS: velocity: 20 μL/min (pump 1), 200 μL/min (pump 2); RF power: 1300 W; outer plasma gas: 15 L/min; auxiliary plasma gas: 0.9 L/min carrier gas: 0.8 L/min; makeup gas: 0.2 L/min; acq. mode: time-resolved mode; nebulizer: 60 psid, MCN (100-400 μL/min); DGF: 10 L/min. | 85 |
| Cabbage leaves | Zinc dimethyldithiocarbamate, zinc ethylenebisdithiocarbamate monitored isotope (m/z): Zn66; | SLE | 1.0 g sample + 5 mL (0.1 M) NaOH sonicated (20 min, 200 W), cooled to RT, and filtered (0.22 μm membrane filter) and diluted to desired volume with H2O (according to the zinc content in the sample), and final solution was used for CE–ICP-MS analysis. | CZE: 50 cm length × 75 μm id × 375 μm od fused silica capillary. BGE: 50 mM H4BO3 + 12.5 mM Na2B4O7 (pH 9). ICP-MS: velocity: 12 μL/min (pump 1), 240 μL/min (pump 2); RF power:1400W; outer plasma gas: 15 L/min; auxiliary plasma: 0.9 L/min carrier gas: 0.8 L/min; makeup gas: 0.2 L/min; 60 psi, MCN (100-400 μL/min), DGF: 10 L/min. | 43 |
| Cheese, coffee | Saturated fatty acids | SLE | 0.5 g cheese pulverized (liquid N2) + 1 mL H2O + 3 mL ACN sonicated (30 min @ 35 °C) and centrifuged (10 min @ 14,000 rpm). Supernatant filtered (3 kDa cutoff) by centrifugation (20 min @ 14,000 rpm). 0.5 g coffee + 5 mL H2O + 15 mL of ACN (treated as above). 100 mL of filtered aliquot used for CE-MS analysis | CZE: Fused-silica capillary (50 μm i.d. x 358 μm o.d., 85 cm in length). BGE: 30 mM NH4HCOO in 40% ACN (pH 10 adjust with 25% NH4OH). Injection and voltage: 50 mbar (20 s), +30 kV. SL: 250 μM ionic liquids as ion pairing reagent in 1:1 (v/v) MeOH/-H2O; 10 μL/min. ESI-MS: +3.5 kV; SC: DGF: 11 mL/min, DGT: 230 °C | 82 |
| Seaweed (O. japonicus A. Berger) | Metabolites (Anions and Cations) | LLE | Fermented samples (4 days) kept on ice (5±10 min) and centrifuged (20 min @ 3,000 rpm at 4 °C). Pellets retained for further processing. Supernatants mixed with equal volume EtOAc and shaken for 2 h (RT). EtOAc separated from supernatants using sep. funnel, and dried (rotary evaporator). Dried compounds dissolved (1.6 mL MeOH), stored on ice. Retained pellets washed with 10 mL H2O (vortex 30s), centrifuged (5 min @ 3000 rpm) and supernatant discarded. MEOH extract solution (1.6 mL) added to pellets , sonicated (30s) along with 1.1 mL IS solution. Mixture left at RT (30s), centrifuged (20 min @ 3000 rpm, 4°C). Supernatant transferred to ultra-centrifugal filter (5-kDa-cutoff), centrifuged (2±5 h @ 9,000 rpm, 4°C), dried under vacuum (3 h @ 1,500 rpm) | CZE: Cationic and anionic metabolites study were carried out with a fused silica capillary of 50 μm × 80 cm. BGE : cation buffer solution (p/n: H3301-1001), anion buffer solution (p/n: I3302-1023); Injection and voltage, 50 mbar for 10 s and 25 s using 27 kV and 30 kV, respectively. ESI-MS: Positive, 4.0 kV and Neaative, −3.5 kV for cations and anions. SL: HMT (p/n: H3301-1020) | 97 |
| Pork meat | Benzimidazole | DLLME | samples crushed and homogenized. Portions of 1 g (crushed/homogenized) sample + 1mL of H2O agitated (vortex) and 2mL ACN added (mix 30s). 0.5 g MgSO4 and 0.1 g NaCl added to mixture (agitate 5 min), centrifuged (10 min @ 9000rpm) and 1700 μL of upper phase mixed with 950 μL CHCl3 (extractant) and injected in 5mL of H2O for DLLME. Ternary system vigorously shaken by hand (60s) and stable cloudy solution formed centrifuged (2 min @ 5000rpm) for phase separation. CHCl3 layer collected and dried with N2. Residue reconstituted with 250 μL of 30:70 (v/v) ACN/H2O and filtered before injection into CE-MS | CZE: Uncoated fused-silica capillary (100 cm total length, 50 μm i.d., 375 μm o.d.). BGE: 500 mM formic acid (pH 2.2). Injection and voltage: 50 mbar (25 s), 25 kV. SL: 50:49.5:0.5 (v/v/v) EtOH/H2O/formic acid ; 0.1 mL/h. ESI-MS: −4.5 kV; SC: DGF: 8 L/min, DGT: 250 °C, NP: 6 psi | 44 |
| Fish | Metabolites | SLE | Fish feed pellet (20 mg) supplemented with or without probiotic crushed (mortar), mixed with 600 μL MeOH containing IS (50 μM) and homogenized (1,500 rpm, 120 sec × 2 times). CHCl3 (600 μL), H2O (240 μL) added to homogenate, mixed thoroughly, and centri-fuged (2,300 x g for 5min, 4 °C). H2O layer (200 μL × 2) filtrated (5-kDa cut-off filter). Filtrate centrifugally concentrated and re-suspended in 50 μL of H2O | CZE: A fused silica capillary with inner diameter and total length of 50 μm and 80 cm. BGE: electrophoresis buffer (Solution ID H3301-1001 for cation mode and H3302-1021 for anion mode; HMT Inc., Tsuruoka, Japan). Injection: 50 mbar (10 s) in cation mode and 50 mbar (25 s) in anion mode. ESI -MS operated in positive ion mode (4.5 kV) and negative ion mode (3.5 kV) for cat-ionic and anionic metabolites, respectively | 78 |
| Citrus tumida peel fed mice liver | Metabolites | SLE | 50 mg frozen liver sample immersed in 1800 μL 50% ACN in H2O containing IS (H3304-1002, HMT). Tissue homogenized (BMS-M10N21 homogenizer) and centrifuged (2300 g, 5 min,4 °C). 800 μL of the upper layer filtered by centrifugation (9100 g, 120 min, 4 °C) using (HMT 5-kDa cut-off filter). Filtrate resuspended in 50 μL H2O for CE-MS analysis | CZE: BGE: H3301-1001 for cation SL: 5 mM NH4OAc in 50% (v/v) MeOH/H2O; 10 μL/min. ESI-MS: 4.0 kV; SC: DGF: 10 L/min, DGT: 300 °C | 101 |
| Nutraceutical Tablets | Antihypertensive peptides | SLE | Nutraceutical tablets grounded (mortar and pestle) to fine powder. 0.15 g powder mixed with 1mL of warm H2O (at 45 °C), mixture shaken (1 h at 45 °C, 700 rpm) and centrifuged (7000 x g, 10 min, 25 °C). Supernatant filtered (a 0.22 μm nylon filter) before analysis by CE-MS | CZE: A 72cm total length x 75 μm id x 360 μm od bare fusedsilica capillary. BGE: 1 M HOAc (pH 2.3). Injection and voltage: 34 mbar (15s), +10 kV. SL: 0.05% fir nut in 60:40 (v/v) IPA/H2O; 3.3 μL/min. ESI-MS: +4.0 kV; SC: DGF: 4 L/min, DGT: 200 °C, NP: 7 psi. | 87 |
| Pork meat | Thiamine, thiamine phosphate | SLE | Frozen muscle tissue immediately plunged into MeOH containing 50 μM IS (H33041002) at 0 °C and homogenized 3 times (1500 rpm for 120 s). H2O and CHCl3 added at a ratio of 2:5 to the samples, mixed, and centrifuged (4 min at 2300×g, 4 °C). Upper aqueous layer centrifugally filtered (5-kDa cutoff filter) to remove proteins. Filtrate lyophilized and suspended in H2O before CE-TOFMS analysis | CZE: A fused silica capillary (50 μm i.d. ×80 cm total length), with commercial Cation and Anion electrophoresis buffers (Solution ID: H3301-1001 and H3302-1021 used. SL: cationic metabolites: MeOH/H2O (50% v/v) and 0.1 μM Hexakis, for anionic metabolites: 5 mM NH4OAc in MeOH/H2O 50% (v/v) containing 0.1 μM Hexakis; 10 μL/min SC: ESI-MS : Positive , 4.0 kV and Negative, 3.5 kV, DGF: 10 psi, DGT: 300 °C | 96 |
| Sea foods | Neurotoxin (β-N-methylamino-L-alanine) | Hydrolysis | Lysis buffer (1% sodium deoxycholate in 50 mM NH4HCO3, pH 7.5–8.0) added to 1 g seafood and homogenized. 1 mL of homogenate centrifuged (5 min @ 21,000×g), supernatant probe sonicated (20% amplitude, 20 s) twice and centrifuged (5 min @ 21,000×g). protein concentration measured (Nano-Drop 2000c, absorbance at 280 nm). Appropriate volume of sample containing 250 μg of protein transferred to a 10-kDa MWCO-filter unit (pre-washed with 200 μL H2O), centrifuged (14,000×g,15 min) and washed with 400 μL H2O (14,000×g and 20 °C) until <100 μL remained in filter unit. Eluate collected for free fraction analysis. Retained volume in filter unit washed with 100 μL H2O, filter unit inverted and spun (14,000×g and 20 °C) to collect protein fraction. Appropriate volume containing 250 μg sample added to clean vials for total sample analysis. Separated fractions stored at −20 °C until hydrolysis. Total and free protein fraction samples hydrolyzed with 40 μL of 6 N HCl and 5 μL of 4.7mM SIL-BMAA reconstituted in 50 μL H2O. Hydrolyzed samples incubated overnight (95 °C), lyophilized, and reconstituted in 50 μL of diluent (110 mM NH4OAc + 2% FA in 50% MeOH). Final samples diluted 1:100 prior to analysis | BGE: 2% FA in 50% MeOH in H2O | 47 |
| Coffee | Monosaccharides | Acid hydrolysis | 1.0 g dried coffee + 10 mL of 1 M H2SO4 heated (90 °C, 150 min) and manually stirred every 30 min. Solution cooled (tapwater) until RT, filtered, and the final volume make up to 10 mL H2O. 1 mL of 1 M Ba(OH)2 suspension added to 1 mL of sample solution for neutralization and sulfate precipitation (as BaSO4). Samples centrifuged (1 min @ 6000 rpm), filtered (0.2 μm PVDF/PPmembrane), diluted 10x before injecting | CZE: 90 cm long, 20 μm i.d. and 360 μm o.d. fused-silica capillary. BGE: 500 mM TEA (pH 12.3). Injection and voltage: 100 mbar (30s), +25 kV. SL: BGE diluted 50-folds with 1:1 (v/v) MeOH/H2O; 6 μL/min. ESI-MS: Positive, +4.5 kV; SC: ESI 4.5 kV, DGF: 6 L/min, DGT: 200 °C, NP: 10 psi | 48 |
| Fish | Hg and MeHg monitored isotope: 200Hg and 202Hg | MAE | Fish muscle sample extracted by MAE method reported in previous paper [Ref 21]. Extract filtered (0.22 μm membrane filter), evaporated to dryness (vacuum freeze drying, −46°C) and residue quantitatively dissolved into appropriate volume of BGE before analysis with CE–ICP-MS | CZE: a 70 cm length × 75 μm id × 365 μm od fused-silica capillary. BGE: 0.72 mM Tris + 0.72 mM H3BO3 + 16 mM EDTA + 0.3 mM CTAB (pH 8.0). Injection: −18 kV (10s), separation voltage: +20 kV. ICP-MS: velocity: 14 rpm (pump 1), 6 rpm (pump 2); RF power: 1500 W; outer plasma gas: 15 L/min; auxiliary plasma gas: 0.9 L/min carrier gas: 0.72 L/min; makeup gas: 0.27 L/min | 117 |
| Mussel | Paralytic shellfish toxins, tetrodotoxins, and domic acid | DE | Wet tissue homogenate + equal mass of 0.1 M HCl homogenized (10,000 rpm), boiled (water bath, 5 min), cooled to RT, centrifuged (6700 g, 10 min). Final volumes determined gravimetrically. A 100-μL sub-sample of supernatant vortexed with 300 μL of ACN insoluble protein, removed by spin filtration (0.22-μm polyvinylidene difluoride filter), at 2500 g for 5 min. 250 μL filtrate spiked with 16 μL (25 μM BMAA-d3) | CZE: Bare fused-silica capillary tubing with 100 cm total length (50-μm i.d., 363-μm o.d) BGE: 5 M formic acid in 10% (v/v) ACN/H2O. Separation and voltage: 50 mbar (30 s), +30 kV. SL: 0.1% FA in 1:1 (v/v) MeOH/H2O; 4 μL/min. ESI-MS: + 4 kV; SC: ESI 4000 V, CGP: 40 psi, DGT: 250 °C, NP: 4 psi. | 110 |
| Green/roasted coffee | Metabolites | SLE | 5 mg of grounded coffee mixed with 1.5mL 25% (v/v) MeOH in H2O and SLE carried out in Thermomixer Compact (700rpm, 15 min @ 25 °C). After centrifugation (3500 rpm, 10 min, 25 °C) the supernatant fraction directly analyzed by CE–MS | CZE: Uncoated fused-silica capillaries of 50 μm ID with a total length of 100 cm. BGE: 1 M formic acid (pH 1.8). Injection and voltage: +30 k 50 mbar (80 s), +30 kV. SL: 1 M HOAc in 50:50 (v/v) MeOH/H2O; 8 μL/min. ESI-MS: Positive, +3 kV., SC: DGF: 5 L/min, DGT: 180 °C, NP: 10 psi; SG (jet stream): 3 L/min, 150 °C | 79 |
| Corn | Pesticides | QuEChERS | 15-g homogenized sample (fortified with QC spiking solution (100 μL) if needed and vortexed for 1 min) + 15.0 mL ACN containing 1% (v/v) of HOAc added to each tube as well an Agilent-buffered QuEChERS extraction salt packet contenting 6 g MgSO4 and 1.5 g NaOAc. Sample tubes capped tightly, hand-shaken vigorously (1 min), and centrifuged (5000 rpm, 5 min). 1 mL supernatant diluted with BGE 1:1 (v/v), filtered (0.2 μm PVDF and PP membrane) and analyzed. | CZE: A 58-cm long, 50 μm i.d., 360 μm o.d. fused-silica capillary (Agilent, Redmond, OR, USA) with the inner wall coated with poly(vinyl alcohol) (PVA). BGE: 0.1 M formic acid (pH 2.4). Separation and voltage: 100 mbar (12 s), +28 kV. SL: BGE diluted 5x with 1:1 (v/v) MeOH/H2O; 5 μL/min ESI-MS: Positive, +4 kV; SC: DGF: 6 L/min, DGT: 250 °C, NP: 7 psi. | 7 |
| Barley grain and malt | Hordeins | SLE | 15 g barley grain/malt grounded in a coffee grinder and stored at RT. 250 mg ground sample mixed with 1 mL of 50:50 (v/v) IPA:H2O, incubated (30 min, constant shaking) and centrifuged (15,000×g, 5 min, RT). Supernatant filtered (0.20 μm nylon filter) before the analysis. | CZE: A 72 cm LT × 75 μm id × 365 μm od fused silica capillary coated with hydroxypropyl cellulose. BGE: 1 M HOAc (pH 2.3). Injection and voltage: 50 mbar (20 s), +25 kV. SL: 0.05% (v/v) FA in 60:40 (v/v) IPA/H2O; 3.3 μL/min. ESI-MS: +4 kV, SC: DGF: 4 L/min, DGT: 300 °C, NP: 7 psi. | 49 |
| Dry-cured ham | Metabolites (amino acids, organic acids and nucleotides) | SLE | Ham sample extracted by plunging the samples into 500 μL MeOH containing (20 μM each methionine sulfone, D-camphor-10-sulfonic acid, and 2-(N-morpholino) ethanesulfonic acid as IS. Samples homogenized (1,500 rpm, 5 min) using cell disruption device. Homogenized sample mixed with 500 μL CHCl3 and 200 μL H2O, centrifuged (4,600g, 15 min, 4 °C) and upper aqueous layer (300 μL) centrifugally filtered (5-kDa cutoff filter) at 9,100g for 4 h at 20 °C. Filtrate (300 μL) lyophilized and dissolved in 50 μL of H2O containing reference compounds (200 μM 3-aminopyrrolidine and trimesate) before analysis. | CZE: BGE: 1 M formic acid, 50 mM NH4OAc (pH 8.5) SL: 1:1 (v/v) MeOH/H2O + 0.1 μM Hexakis (cation analysis; 5 mM NH4OAc in 1:1 (v/v) MeOH/H2O + 0.1 μM Hexakis (anion analysis); 10 μL/min. ESI-MS: Positive, +4 kV; SC: DGF: 7 L/min, DGT: 300 °C. | 80 |
| Chili powder | Sudan dyes Sudan I, Sudan II, Sudan III and Sudan IV | Freeze-out cleanup | 2.0 g sample + 5 mL ACN sonicated 5 min (35 kHz, 120 W), centrifuged (4025×g, 5 min) and supernatant transferred to 15 mL centrifuge tube (solid residue reextracted). Combined supernatants mixed with 2.5 mL H2O and 1 g NaCl and placed at −20 °C for 3 h. 100 μL of the ACN layer diluted to 1:10 with 900 μL of 6:1:1:1 (v/v/v/v) H2O/ ACN/MeOH/THF mixture, resulting in final dilution of 1:50. Diluted solution filtered (0.22 μm nylon syringe filter) and analyzed by MEKC/MS/MS. | MEKC: BGE: 60:20:10:10 (v/v/v/v), 25 mM ammonium perfluorooctanoic acid; /ACN/MeOH/THF (pH 9.0); Injection and voltage: 5 kPa, 50s, +25 kV. SL: 50:49.9:0.1 (v/v/v) IPA/H2O/FA, 10 μL/min. ESI-MS: +3 kV; SC: DGF: 6 L/min; DGT: 250 °C; NP: 206.8 kPa | 89 |
| Plastic food packaging materials | Organotin | SPE | Food packaging sample rinsed (H2O), dried, and cut into small pieces (approx. 0.4 × 0.4 cm). 0.5 g sample extracted with 5 mL DCM (ultrasonic,30 min @ 25 °C), evaporated to dryness (rotary evaporator @ 45 °C) and residue reconstituted (3 mL of 10 : 90 (v/v) ACN/H2O). 30 mL extracts (pH 2 adjusted with 1 M HCl) passed (1 mL/min) through C18 SPE extraction column (pre-conditioned with 5 mL MeOH 5 mL H2O), rinsed (10 mL H2O), air dried, and eluted with 3 mL of 5% HOAc in ACN. Eluted analyte evaporated to dryness (N2) and reconstituted (200 μL of 5% HOAc in ACN) for CE-ESI-MS analysis. | CZE: A fused-silica capillary (30 μm i.d. and 150 μm o.d., with a length of 90 cm, smoothened with 2000 mesh sand paper and etched with HF solution to form a porous section of approximately 5 cm using a sheathless interface (made in house). BGE: 10% HOAc in 20:80 (v/v) MeOH/ACN. Sheathless ESI-MS: Positive, +1.6 kV; SC: DP: 60 eV, CGP:10 psi, interface heating: 50 °C | 32 |
Hexakis: hexakis (2,2-difluoroethoxy)phosphazene; PLE: pressurized liquid extraction; EAE: enzyme-assisted extraction; CTAB: cetyltrimethylammonium bromide; SA: sinapinic acid; TFA: trifluoroacetic acid; TEA: trimethylamine; IS: internal standard; MSLE: microwave-assisted solid-liquid extraction; AA: amino acid; FA: formic acid; DSPE: dispersive solid phase extraction; PSA: primary-secondary amine; UHT: Ultrahigh temperature; PST: paralytic shellfish toxins; MAE: microwave-assisted extraction; DE: dispersive extraction; CGP: curtain gas pressure: PIPES: 1,4-piperazine di-ethane sulfonic acid. AMP: adenosine 5-monophosphate; CMP, cytidine 5-monophosphate; CUF, centrifugal ultrafiltration; GMP: guanosine 5_-monophosphate; IMP: inosine 5-monophosphate; PNP, programmed nebulizer pressure; UHQ, ultra high quality; UMP, uridine 5-monophosphate
5.1. Analysis of liquid food
The bar plot distribution of type of liquid and solid food used in CE-MS in years 2010-2020 is shown in Fig. S13 with an increasing trend till 2017, but a somewhat surprising decline in the past several years. Furthermore, more papers were published on liquid food over 2010-2015 compared to the last five years (2016-2020). The liquid food such as beer, wine, milk, honey, oils, and sauces represent nearly 25% (22) out of the total 87 papers dealing with CE-MS analysis of food samples published in the last decade (Fig. S13, inset pie chart).
5.1.1. Beverages and drinks
Biogenic amines and organic acids
Analysis of biogenic amines (BAs) and organic acids in alcoholic drinks such as beer and wines are of significant interest because the high concentration of BAs is an indicator for food spoilage and food poisoning caused by microorganisms. For example, when food is poisoned, the concentration of BAs and enzymes are high. Therefore, BAs can be used as a marker for hygienic conditions of the raw material and during the manufacturing of alcoholic drinks because their high concentration in the drink is connected to harmful bacteria (e.g., Lactobacilli, Escherichia, Salmonella). The aforementioned types of bacteria are precursors of BA, which means that they are responsible for forming BAs, causing food poisoning.
A recent study reported BAs analysis using CZE-ESI with a triple quadrupole mass spectrometer [103]. While nine biogenic amines (putrescine, spermine, spermidine, cadaverine, histamine, tryptamine, tyramine, phenylethylamine, and urocanic acid) were not simultaneously separated in the TIC mode, and perhaps this is not required as not all nine amines are present in each beer and wine sample. The BAs were still quantitatively analyzed in beer and wine using polyvinyl alcohol (PVA) coated capillary and ESI-MS detection in MRM mode. The separation was achieved in 10 min, and quantitation was carried out using the standard addition method (recovery 87-113% and LOD 1-2 μg/mL, 16 nL injections). In the proposed CE-MS method, the use of PVA coated capillary combined with triple quadrupole mass analyzer provides improved separation efficiency, precision, and sensitivity for BAs analysis. Compared to other reported LC and GC-MS methods, CE-MS was reported as a rapid method with a low amount of sample requirements, producing less waste.
In another study [81], twelve BAs in eight different grape wines and four fruit were analyzed in 15 min on bare fused silica capillary using CZE-ESI-MS. A lower LOD of 15 ng/mL was achieved, and recovery was between 65-117%. The BAs analysis in different wines revealed an exciting correlation between the type of BAs versus acidity, wine age, and kind of fruits used in wine manufacturing. In addition to the advantages mentioned above, CE-MS's greenness was evaluated and found to be consistent with the safety standards set by National Environmental Methods Index.
The organic acids (OAs) content is essential for wine stability. Their concentration should be monitored during the entire verification process starting from grapes juices to the end process of alcohol fermentation and stabilization. The OAs are analyzed in beverages such as beer, wine, and blueberry juices by CZE interfaced to quadrupole time of flight (QTOF)-MS [10]. Six OAs (lactic, succinic, malic, tartaric, shikimic, and citric acids) were partially separated using ammonium acetate buffer on a polybrene coated capillary reversed polarity and reverse EOF in 4 min. The quantitation of OA (e.g., citric acid) in red wine is crucial to control because it impacts both the import and export quality of this type of wine. Also, malolactic fermentation is related to converting malic acid into lactic acid, and the quantitation of these two acids provided the fermentation stage. OAs such as lactic, succinic, malic, tartaric, shikimic, and citric acids were analyzed in 17 Macedonian red wines by CE-QTOF-MS. The limit of quantitation of OAs range between 3.4-107 mM and recovery in the range of 95.8-102.7%. The CE-MS method provides shorter analysis than the previously reported HPLC and CE-UV methods with much rapid quality control of wine making processes and the detection of wine alterations. In the next reported study [107], about 64 organic acids, some of which are known to activate GABAA receptors, were identified and quantified in 6 Japanese sake (a traditional Japanese alcoholic beverage) by CE-time of flight (TOF)-MS.
Eight OAs (succinic, citric, salicylic, malic, benzoic, sorbic, ascorbic, and tartaric acids were identified by CZE and quantitated by ion-trap (IT)-MS in blueberry juices[108]. While malic acid, succinic acid, and tartaric acid are the main ingredients in blueberry, the other six organic acids are used as additives to promote digestion and appetite. The legislation is in place in various countries for the maximum allowable concentration of these OAs as additives in blueberry juice. A mixture of eight OAs standards was baseline separated in 8 min with five OAs (succinic, citric, malic, ascorbic, and tartaric acid) identified by retention time and mass spectra (data not shown) in blueberry juice (Fig. 6 A-B). Quantitation was achieved with a limit of detection (LOD) and recoveries of 0.8-4.5 μg/mL and 86.8-99.8%, respectively. For the first time, hexamethrine bromide was utilized as pseudostationary phase (PSPs) by CE-MS. Compared to other CE-MS modes, the published article claimed to achieve rapid separation, required no specialized column preparation procedure, and without the use of any surfactants, which could interfere in MS detection.
Fig. 6.

Total ion electropherograms for CE-ESIMS analysis of a standard solution of the eight organic acid (a) and a 1# blueberry juice sample (b). Experimental conditions: capillary 50 μm × 70 cm; separation voltage:22 kV; buffer solution: 40.0 mmol/L ammonium acetate (pH 6.0); sheath liquid, 70:30,(v/v) isopropanol/water containing 7.5 mmol/L acetic acid with a flow rate of 6.0 μL/min; flow of drying gas: 10.0 L/min; temperature of drying gas: 350 °C; nebulizing gas pressure:12 psi; Analyte identification: succinic acid (1), citric acid (2), salicylic acid (3), malic acid (4), benzoic acid (5), sorbic acid (6), ascorbic acid (7), tartaric acid (8). Reproduced from [108] with permission from Springer Link, copyright 2014.
Peptides and proteins in milk
As a part of quality control and food safety program, analysis of milk proteins and peptides is of significant interest because many proteins are known allergens in milk. When these proteins are consumed as food, they are hydrolyzed in the biological system, releasing peptides that could produce favorable or unfavorable biological activities. Milk products such as cow milk, infant milk formula, soy milk, and skimmed milk samples were analyzed for qualitative identification and quantitative measurements of bioactive peptides and proteins by CE-MS. In three hypoallergic infant milk formulas, 24 to 38 peptides were identified with considerably lower analysis time by CE-TOF-MS [61] compared to HPLC-MS. Most of the peptides were found to be ACE inhibitors, antihypertensive, antithrombotic, hypercholesterolemic, immunomodulation, cytotoxicity, antioxidant, antimicrobial, antigenic, and opioid. In another study, CZE-TOF-MS and CZEIT-MS [60] were compared for analysis of infant formulae. Although both extracted ion electropherograms (EIE) and isotopic patterns were very similar, the mass spectra obtained by CZE-TOF-MS showed improved mass accuracy and resolution. The electropherograms showed about nine bioactive peptides were identified in hypoallergenic infant milk, and five were confirmed to be ACE inhibitors and antimicrobials by CZE-ITP-MS. The identification was confirmed by CZE-TOF-MS, which provided less than 1.5 ppm of mass accuracy.
Two major whey proteins, α-lactalbumin, β-lactoglobulin are proteins that can result in allergy even at concentration requiring quality control [8]. The magnetic beads were coated inside the hydroxypropyl cellulose capillary to capture the two proteins. Next, the two proteins were eluted in less than 18 min using immunoaffinity capillary electrophoresis (IACE), which combines immunoaffinity extraction with CE separation. Off-line MALD-TOF-MS analyzed the collected CE fractions. These two proteins mentioned above were identified and quantified in skimmed milk, and soy-milk samples with LOD of ≤ 2.1 nM were achieved for both whey proteins. In another study performed by the same research group, lactoferrin, bovine serum albumin, α-lactalbumin, β-lactoglobulin components were analyzed in milk extracts in component resolved diagnostic format of cow's milk allergy [38]. The separation was performed by IACE protocol with UV detection followed by off-line MALDI-TOF-MS. The combined use of IACE when coupled with MALDI-TOF-MS improves the sensitivity to as low as 2 nM. The CE fraction spotting on the MALDI plate opens new avenues for allergens structural characterization in proteomics.
Antibiotics and steroids in milk and honey
Antibiotics (quinolones, tetracyclines, and aminoglycosides) and steroids are used in dairy, farming, and beekeeping for safety against microbial infections, livestock farms, and increasing milk productions in cows. When exposed to animals and farms, these chemicals impact products that originate from these sources, thereby affecting human health. Sensitive detection and quantification of such chemicals are essential for safety and quality control in liquid food products. Residues of eight quinolones (oxolinic acid, flumequine, ciprofloxacin, danofloxacin, enrofloxacin, marbofloxacin, sarafloxacin, and difloxacin) and seven tetracyclines (methacycline, doxycycline, tetracycline, 4-epitetra-cycline, minocycline, demeclocycline, and chlortetracycline) were analyzed in cow milk by CZE-QTOF-MS/MS after SPE clean-up [109]. All fifteen antibiotics were quantified in whole cow milk, goat milk, and semi-skimmed milk developed method. The LOQ in the range 1.6-9.7 μg/kg and recoveries of 72.6-105.8% was achieved. For this study, a combination of Oasis HLB with CZE-QTOF enables trace analysis of antibiotics in milk samples. One of CE's main drawbacks with triple quadrupole MS/MS is the low-resolution power, which increases the number of false positives. These drawbacks can be overcome by using a high-resolution mass spectrometer, which improves selectivity and provides a higher mass resolution under full scan conditions. Thus, the use of QTOF-MS combines the advantages of the TOF accurate mass and resolution and high sensitivity of the MS/MS technique. Another critical advantage of QTOF-MS is the availability of full MS/MS spectra after a single injection for peak identification and confirmation, allowing the data to be analyzed retrospectively for new substances of interest in food samples.
In a second study, bovine milk samples were analyzed by CE-MS with the use of an in-line molecularly imprinted polymer (MIP)-SPE column for the determination of eight quinolones antibiotics residues [57]. As expected, the MIP column's use resulted in improved sensitivity with LOD in the range of 3.8-4.7 μg/kg and recoveries in the range of 70-102%. The utility of MIPs for in-line concentration, preconcentration, and sample clean-up was demonstrated for the first time to analyze antibiotic residues in milk.
The aminoglycoside antibiotics such as gentamycin (GENT C1, GENT C1a, and GENT C2), neomycin, apramycin, paromomycin, dihydrostreptomycin, spectinomycin, and streptomycin were identified and quantified simultaneously in a commercial honey sample using MIP-SPE for extraction and sample clean up followed by CZE with ion trap in MRM mode [62]. Spiked samples' recoveries in the honey range between 88.2-99.8% and LODs range from 0.4-28.5 μg/kg. In addition to the MIP-SPE, field amplified sample stacking (FASS) as a preconcentration strategy improves sensitivity and selectivity.
Steroidal compounds (endoestrogens: estriol, 17α-estradiol, 17β-estradiol, and estrone; exoestrogen: 17 α-etynylestradiol; mycotoxin: zearalenone; primary metabolites: 2-methoxyestradiol, α-zearalanol, β-zeralanol, α-zearalenol, and β-zearalenol) were analyzed in whole and skimmed cow milk as well as semi-skimmed goat milk by MEKC-MS [12]. The total ion chromatogram (TIC), base peak chromatogram (BPC), and extracted ion chromatograms (EIC) is overlaid and compared in Fig. S14. Due to the low sensitivity of MEKC-MS, the TIC mode and BPC, as expected, are relatively noisy, but all spiked steroids in whole cow milk were identified in EIC mode. The two peaks marked with an asterisk in the EIC at m/z 303 was also found to be in the standard mixture and was assigned to the loss of water molecule from α-Zel and β-Zel steroidal compounds in the electrospray. Using a volatile and MS compatible APFO surfactant combined with DLLME, clean-up the LOQs within 3-32 μg/L (whole cow milk), 1-21 μg/L (skimmed cow milk), and 4-61 μg/L (semi-skimmed goat milk) were achieved with the developed MEKC-MS/MS method. This research reports the first publication combining DLLME with MEKC-MS. The benefits of MEKC-MS involve a low consumption of organic solvents and samples and a relatively low cost of analysis.
Endogenous compounds
Analysis of endogenous compounds in food samples can provide essential insights into food quality and indications of storage conditions or thermal treatments. Furosine and 5-hydroxymethylfurfural are used as markers for assessing the quality and freshness of liquid foods. CE-MS/MS carried out a qualitative and quantitative analysis of furosine in milk samples with high sensitivity and precision without requiring sample pretreatment [90]. The validated method provided LOD and LOQ of 0.07 and 0.25 mg/L with recovery between 77-97%. Another vital food quality marker, 5-hydroxymethylfurfural (HMF) was quantitatively determined in balsamic vinegar, coffee, and beverages by CE-MS/MS in ESI positive mode [92]. The best peak shape and maximum intensity were achieved using 50 mM ammonium formate as BGE at pH 3. The LOD and LOQ of 0.03 and 0.1 mg/mL were respectively achieved. The results of the developed method were compared with that of reversed phase HPLC with UV detection. The agreement between the two data sets suggested good reliability of the proposed method. Isoflavones such as diadzin, genistein, daidzein, genistein, formononetin, biochanin, and glycitein were analyzed in soy drink by CE-MS [102]. The optimized separation method allowed separation of all seven isoflavones in less than 16 min with LODs between 0.65 and 4.8 μg/mL. During CE-MS runs, application of the programmed nebulizing gas pressure provided improved resolution and prevented current drops.
5.1.2. Oils and sauces
Olive oil is of high nutritional value, provides several health benefits, and is safer than other cheaper edible oils. However, olive oil is often adulterated with sunflower, corn, and soybean oils. Several amino acids and betaine compounds are useful markers of such adulterations in vegetable oils. Six non-protein amino acids such as ornithine, β-alanine, GABA, alloisoleucine, citrulline, and pyroglutamic acid were detected and quantified in vegetable oils by CZE with ion-trap MS/MS detection [105]. All six amino acids were separated in less than 15 min with high resolutions (>5), and LODs, as well as LOQs, were achieved as low as 0.04 and 0.06 ng/g, respectively. This optimized and validated method allowed a highly sensitive quantitation of six amino acids in sunflower, corn, and soybean oils using LLE and butanol derivatization. In another report, betaines such as glycine betaine, trigonelline, proline betaine, and carnitines were quantified in the mixture of olive oil and seed oil by optimized CZE-MS/MS (ion-trap) [106]. Using butanol derivatization, baseline separation of four betaines was achieved in 10 min. The recoveries of 80-99% and LODs and LOQs of 0.1 ppb were achieved. Non-protein amino acids and betaines were identified by unique fingerprint fragmentation obtained from MS/MS.
Sudan dyes (I-IV) are food colorants used in chili products illegally and are known to be carcinogens. Fukuji et al. optimized a partial filling MEKC-MS method for the separation of sudan dyes. The four dyes were resolved in 8 min, in which 25 % of the capillary was partially filled with 25 mM sodium dodecyl sulfate (SDS), 40 mM ammonium bicarbonate, and 32.5% ACN (v/v); and 75% of the capillary was filled with a CZE buffer [40 mM ammonium bicarbonate, 32.5% ACN (v/v)] [89]. The commercial tomato sauce sample did not found to contain any measurable amount of sudan dyes. Still, the method provided good recovery (85-99%) with LOD in the range of 0.57-0.71 μg/mL, illustrating the method's potential for quality control of the chili sauce sample. In a related MEKC-MS/MS study, Moreno-Gonzalez et al. used an MS-friendly, volatile surfactant, APFO, in BGE to separate 4 Sudan dyes in 4 chili sauces and six chili pastes by MEKC-MS/MS [17]. This fluorocarbon-based surfactant allows researchers to overcome the significant difficulty of MS contamination, which is related to nonvolatile surfactants' use without compromising the efficiency and selectivity. The overlayed EIC for the MEKC-MS of four sudan dyes spiked in chili powder showed fair resolution of four dyes within 17 min (Fig. S15). The developed method achieved the LOD of 22 μg/kg and recovery of 84.4-99.6%.
Contaminants such as chlorophenols (2,4-dichlorophenol, 2,4,5-trichlorophenol, and pentachlorophenol), bisphenol-A, 4-tert-butyl-phenol, and 4-tert-butyl benzoic acid are harmful EDCs. Two CE-MS methods were reported for the simultaneous quantitation of these contaminants in honey samples. The first method utilized restricted access materials and polymeric sorbent based SPE clean-up before injection on the CE-MS instrument [66]. The simultaneous analysis of six contaminants was achieved in 11 min. The LODs in the range of 5-31 ng/g and recoveries in the range of 94-114% were obtained in the optimized method. In the second method, LLE sample clean-up combined with programmed nebulizing gas pressure (PNP) provided high sensitivity and high resolution of EDCs in honey. The PNP used provided high resolution and stable CE current, but using capillaries wider than the 50–75 μm conventional ones [73]. Simultaneous analysis of 6 EDCs was achieved in 16 min. With the LLE clean-up method, a LOD of 1-5 ng/g was reported.
5.2. Analysis of solid food
Solid food such as meat, seafood, fish, edible plants, dairy, supplements, package food, pesticides, residues, and food packaging materials represent nearly 65% (55) out of the total 87 papers dealing with CE-MS analysis published in the last decade (Fig. S13, inset pie chart). Analysis of various organic and biochemical compounds in the aforementioned food sample is highlighted in the following sections.
5.2.1. Meat, Seafood and Fish
Thiamine accumulation is a reliable indicator of meat aging. Thiamine and thiamine phosphate levels in pork muscles were studied as a measure of postmortem pork aging. The muscle samples were analyzed at time intervals of 4, 24, and 168 h postmortem by CE-TOF-MS [96]. The studies revealed that thiamine triphosphate level is approximately 1.8 times higher in longissimus lumborum (LL) than vastus intermedius (VI) pork muscles at 0 h postmortem but declined within 24 hrs, with no detectable level at 168 hr. However, thiamine phosphate level in both types of pork muscles increases after 24 hr. Benzimidazole (BZ) is an anthelmintic drug used in agriculture for the treatment of parasites and fungi. These drugs, when exposed to humans from animal foods, are known to be harmful to health. DLLME-CZE-MS/MS using fragmentation experiments allow the unequivocal identification of 12 BZ in poultry and porcine muscles [44]. The simultaneous analysis was achieved in 30 min with LOD < 3 g/kg, LOQs < 16 g/kg, and recoveries > 70%. The combination of both DLLME with CZE-MS/MS leads to a hyphenated method that is low cost, rapid, and has a low environmental impact, which is some of the main criteria needed for any analytical approach for drug residues in food samples.
Polar marine toxins (paralytic shellfish toxins (PSTs), tetrodotoxins, and domic acid) and β-N-methylamino-L-alanine (BMAA) neurotoxins produced by algae, cyanobacteria, dinoflagellates, etc. can be exposed to several kinds of seafood (e.g., fish, mussel, scallop, oysters, lobsters). These contaminated kinds of seafood pose serious health risks to humans when consumed. At least five CE-MS-based methods were reported for the screening of these toxins in a variety of seafood. In the first report, a speedy chip-based CE-MS/MS method was developed to separate and detect BMAA in scallop samples [47]. The BMAA and its two structural isomers, N-2-aminoethyl glycine (AEG) and 2,4-diaminobutyric acid (2,4-DAB), were baseline separated in less than 1 min (Fig. 7) using 2% formic acid in 50% methanol as BGE. The LLOD of 4.8 ng/mL and LLOQ of 48 ng/mL were achieved. In a second report, CZE-MS/MS method was developed for quantitative analysis of BMAA in cycad, mussel, and lobster samples [64]. The four isomers of BMAA and interfering matrix components were separated in 38 min using a formic acid buffer containing 10% acetonitrile. The SPE with a strong cation exchange cartridge provided efficient sample clean-up, and a LOD of 0.8 ng/mL was reported for BMAA. In a third report, similar to the second one, the same methodology was utilized to separate and quantitate polar marine toxins (paralytic shellfish toxins, tetrodotoxins, and domoic acid) in mussel tissue samples [110]. The analysis was carried out in 45 min, and the LODs in mussel tissue samples were 0.0052, 0.160, and 0.0018-0.120 mg/kg for tetrodotoxins, domoic acid, and paralytic shellfish toxins, respectively. Another two reports in the literature are on paralytic shellfish toxins such as saxitoxin (STX), its analogs decarbamoylsaxitoxin (dcSTX), neosaxitoxin (NEO), decabamoyl-neosaxitoxin (dcNEO), gonyautoxin 1 to 5 (GTX1-GTX5), decarbamoylgonyautoxin 2 to 3 (dcGTX2-dcGTX3), and C toxins [59][61]. All of the toxins above were analyzed in mussel samples by CZE-MS and compared with other separation and detection methods, including UV, contactless conductivity (C4D), and MEKC with fluorescence detection (FLD) [61]. While CZE-MS provided LOD in the range of 517-5670 ng/mL, C4D and MEKC-FLD provided better LODs in the range of 140-715 ng/mL and 60.9-104 ng/mL, respectively. Thus, the developed CZE-UV and CZE-MS methods failed to provide the regulatory limits required LODs.
Fig. 7.
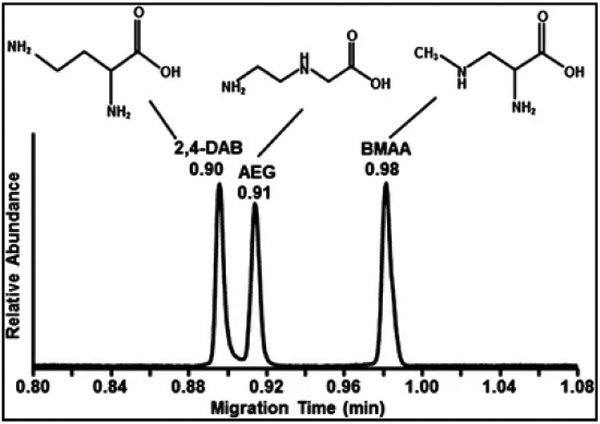
Baseline separation of BMAA from its structural isomers AEG and 2, 4-DAB in less than 1 min of analysis time. Identity of BMAA was confirmed by comigration with SIL-BMAA standard, and identity of AEG and 2, 4-DAB were determined by migration times indexed to that of BMAA (0.93 and 0.92, respectively). Reproduced from [47] with permission from Springer Link, copyright 2018.
Microcystins (MC) are well-known cyanotoxins found in algae, amongst which toxic heptapeptides (MC-LR, MC-RR, and MC-YR) are known to intoxicate animals and humans. CZE-MS using both scan and single ion monitoring method was developed by Tong et al. (2010) for sensitive analysis of MC isomers in crude algae samples [59]. The MCs have a common structure with seven amino acids, of which two are variable (MC-Leu-Arg and MC-Tyr-Arg). Two MCs (MC-LR and MC-YR) were separated in 9 min using optimized separation conditions. Analytes were extracted by SPE using a C-18 cartridge from algae samples. The LODs of 0.05 and 0.08 μg/mL for MC-LR and MC-YR were obtained, and recovery was in the range of 95.8-108%. Harmala alkaloids (HA) are alkaloids with antidepressant activity and are found in several plants and algae extracts. Two methods were reported for trace level detection and quantification of 6 harmala alkaloids (harmane, norharmane, harmalol hydrochloride dihydrate, harmine, harmaline, and harmol). The first method utilized CZE with ion trap MS. The SLE with 50:50 methanol and 3.5 M hydrochloric acid was used to extract edible algae wakame samples from different commercial brands [95]. Six HAs were separated in less than 8 min, and the LODs found were in the range of 1.5-10 ng/mL. Harmalol, harmine, and harmaline were quantified in algae extracts and found at a concentration of 7-24 μg/kg of dry algae. The second method based on CZE with TOF-MS utilized an on-line SPE C-18 cartridge resulting in preconcentration with improved detection at parts per trillion level of 6 HAs in edible algae [55]. The SPE preconcentration provided LODs between 2-77 pg/kg for standards, 1000 folds lower than CE-MS without SPE. Three HAs out of six were detected, whereas polar harmol and harmalol suffered profound matrix effect and could not be detected. Harmane was detected at a very low concentration (70 ng/kg) in dry algae.
5.2.2. Solid Tea and Coffee Beans
The on-line coupling of CE with ambient direct analysis in real-time mass spectrometry (DART-MS) was developed and used for the case study of endogenous caffeine in Chinese white tea leaves [5]. One of CE-DART-MS benefits is that it can tolerate higher concentrations of detergents and nonvolatile salts, which was first illustrated by CZE and MEKC separation of a mixture of 4-aminoantipyrine, zolmitriptan, and quinine. The CZE-DART-MS was successfully applied to caffeine analysis, which was detected using 15 mM sodium borate as BGE and further identified by MS/MS. Roasted ground coffee is often adulterated by cheap quality materials such as soybean and corn. Coffee beans contain higher amounts of polysaccharides that are responsible for the aroma and color of the coffee. The monosaccharides resulting from the hydrolysis of polysaccharides are used to indicate adulteration with soybean and corn. The CZE-MS/MS method was used for the first time for simultaneous detection and quantitation of nine monosaccharides in coffee beans [48]. The optimized method using 500 mM TEA at pH 12.3 allowed fast analysis (< 12 min) of nine monosaccharides, and the LOQ was 0.01 mM.
5.2.3. Dairy
Various modification in CE-MS techniques has been used to analyze several solid dairy products including milk powders, cheese, eggs, and infant formulas. Lysozyme is used as preservatives and antibacterial in several food products. Monitoring lysozyme concentration is essential as it can cause allergy in humans. CZE-QqQ MS method was utilized for quantitation of lysozyme in cheese samples [93]. Lysozyme was extracted using acetic acid, and the analysis was performed using polyacrylamide coated capillary with 100 mM formic acid as BGE. The LOD and LOQ of the developed method were 3.6 and 10.9 mg/kg. The usefulness of poly-(N, N, N', N’-tetraethydiethylenetriamine-co-N-(2-hydroxypropyl) methacrylamide) 50:50 coated capillary for CE-MS was demonstrated in qualitative analysis of lysozyme in cheese samples.
Dietary saturated fatty acids are known to increase cholesterol levels in the blood. Quantitative determination of saturated fatty acids can be useful for assessing food quality. A wide range of fatty acids (C2-C16) was analyzed in cheese samples by CE-QTOF-MS using paired ESI [82]. The simultaneous separation of eleven fatty acids in the cheese sample was achieved in 23 min. The CE-MS method allows monoanions of fatty molecules to form ion-pair associated complexes by adding multiple charged ionic liquids in the sheath liquid after separation. Thus, the positively charged complexes of fatty acids are detected using positive ESI mode with high sensitivity and stability. A variety of Ionic liquids were added to the sheath liquid to enhance detection sensitivity, but N, N’-dibutyl 1, 1’-pentylenedipyrrolidium ionic liquid provided the best ESI-MS sensitivity due to more favorable ion-pairing with negatively charged fatty acids. The optimized method provided LODs and LOQs in the range of 0.13-2.88 μg/mL and 0.44-9.62 μg/mL, respectively, with the recovery of spiked cheese sample in the range of 78-108.2%.
The BZs are drugs used in poultry animals to prevent certain infections that often appear in meat and eggs at trace levels. A CZE-MS method was proposed for simultaneous qualitative and quantitative determination of ten BZs in egg samples [72]. QuEChERS based clean-up and preconcentration before CE-MS analysis allowed sensitive analysis of BZs. The LOD between 3-51 μg/L was achieved, and the recoveries were in the range of 74-112%.
Ribonucleotides that are found in human breast milk are being added to infant formulas (IF). CE-MS based method was developed to quantify five ribonucleotide 5’-monophosphates in powdered infant formula [111][112]. Simple centrifugal ultrafiltration treatment of infant formula samples before analysis provided LODs in the range of 0.8-1.8 μg/g and recoveries between 90-106%.
Melamine is a nitrogen-rich compound that is found to be added illegally in dairy products. High nitrogen content is exploited to show the high protein content in foods. Because of the severe health risks caused by melamine, several methods were developed to analyze melamine and related product derivatives. Kohler et al. developed the CE-MS method using a single quadrupole mass analyzer and ESI voltage switching for the simultaneous analysis of melamine, ammeline, ammelide, and cyanuric acid powdered milk samples [65]. Using optimized BGE 25 mM ammonium acetate at pH 5.2 allowed separation of both cationic (melamine and ammeline) and anionic (ammelide and cyanuric acid) analytes in a single run by polarity switching. For example, the use of 2% ammonia in the sheath liquid permitted simultaneous protonation of MEL and AMLN and deprotonation of AMPLD and CYN in the electrospray. Therefore, all compounds were separated and detected together by CE and MS, respectively, with LOD as low as 200 ppb.
The CEC-TOF-MS was developed by Huang et al., who first tested a series of monolithic columns by a simple in situ polymerization of divinylbenzene (DVB), vinylbenzyl trimethylammonium chloride (VBTA), and different alkene monomers (1-octene, 1-dodecene, or 1-octadecene) for simultaneous separation and quantitation of melamine and three related compounds [20]. Fig. S16(a-b) compares the CEC-UV versus CEC-MS electropherograms using the optimal poly-(DVB-1-dodecene-VBTA) monolithic column. Note that shorter run times by ~1.5 min was observed by CEC-MS compared to CEC-UV without any decrease in resolution using the same BGE containing 80: 20 (v/v) 5 mM ammonium acetate/acetonitrile at pH 8. Furthermore, compared to the CEC-UV, the sensitivity by CEC-MS was 31-372 folds better. Comparable LOD of the CEC-MS method with LC-MS instrument was obtained. Finally, the milk powder samples were treated with simple SLE and analyzed using an optimized method. The LOD was achieved in the range of 2.2-19.4 μg/L with recoveries between 68-85%.
5.2.4. Grains and spices
Hordeins are the main storage proteins found in barley grains and malt. A CZE-TOF-MS method has been developed for simultaneous separation and characterization of four hordeins: ϒ, B, C, and D-hordeins in barley grain and malt [49]. Barley grain and malt were extracted with 50% (v/v) isopropanol and analyzed by CE-MS on hydroxypropyl cellulose coated column using 1 M acetic acid (pH 2.3) as BGE. Furosine is found in several grains and related food products because of the reaction between lysine and reducing carbohydrates. Analysis of furosine levels in foods can provide information on thermal treatment. In another study, a CE-IT-MS/MS method was developed for the qualitative and quantitative determination of furosine in several types of flour (wheat, chestnut, lupin, einkorn, and chickpeas), pasta, and bread [90]. The CE-MS, when compared to HPLC- showed advantages in terms of cost-effectiveness and faster run times. Other benefits of CE-MS over HPLC-MS include the economic impact of the former hyphenation technique as a low amount of solvent is required, and low-cost capillaries are used instead of dedicated columns with shelf-life limited under strongly acidic mobile phase. Extraction-free sample treatment with simple hydrolysis and filtration allowed CZE-MS analysis with LOD and LOQ of 0.07 and 0.25 mg/L, respectively, with recovery between 77-97%.
Food coloring dyes are used in a wide range of food products. Dyes such as sudan I-IV are relatively cheap and readily available and are often used to adulterate many foods such as spices even though they are known carcinogens. A highly sensitive and selective MEKC-MS/MS method has been developed to determine these dyes in chili powders, sauces, and pastes [17]. The optimized process allows the determination of four sudan dyes in chili products with LOQ of 22 μg/kg and recoveries of spiked samples between 84.4-99.6%.
Plant polyphenolic compounds are essential antioxidants found in many fruits, herbs, vegetables, spices such as almond, etc. The LLE was used for isolating fractions containing phenolic compounds from the crude extracts and nine phenolic compounds in 35 min by CZE-TOF-MS. The sensitivity, mass accuracy, and true isotopic pattern of TOF-MS allowed the identification and quantitation of a broad series of known phenolic compounds present in almond-skin extracts using CZE as separation component [104]. Three of these compounds were phenolic acids (p-hydroxybenzoic, protocatechuic, and vanillic acids), and six were flavonoids (isorhamnetin-3-rutinoside, kaempferol-3-rutinoside, naringenin-7-O-glucoside, quercetin-3-O-glucoside or galactoside, and isorhamnetin-3-glucoside or galactoside).
5.2.5. Supplements and packaged food
CE-MS has analyzed food supplements such as baby foods to determine the contents, quality, and freshness. Nucleotides are essential building blocks of DNA and RNA, which play a vital role in energy metabolism. Nucleotide mono, di-, and triphosphates, as well as unmodified nucleosides, were separated and quantified simultaneously in baby foods (containing fish, yogurt, meat, fruit, dairy desserts) by CE-MS using hexafluoro-2-propanol (HFIP) as a new additive to both background electrolyte (BGE) and sheath liquid [94]. The authors noted that the use of HFIP provided high efficiency and high sensitivity, which was not lost when switching to the analysis of complex food matrix. The instrumental LODs were in the range of 14-53 ng/mL for nucleosides and 7-124 ng/mL for nucleotides.
The chemical 5-hydroxymethylfurfural (HMF) is produced in food products by the Maillard reaction or sugar decomposition. The concentration of HMF is often used as an indicator of food quality, storage conditions, and food processing. A CE-IT-MS/MS method was utilized for the quantification of HMF in baby cereals [92]. Selected MS/MS ion monitoring (SMIM) was selected as acquisition mode, and continuous MS/MS scans were performed for a precursor ion. Comparable results were obtained on quantitative data with HPLC.
The free amino acid content in royal jelly requires derivatization, resulting in a time-consuming HPLC method of limited utility. Because several free amino acids are useful markers to determine food quality in royal jelly products and pharmaceutical formulation, a label-free CE-MS method will determine these amino acids without any significant matrix effect. A simple CE-MS/MS without any preconcentration and sample preparation were reported for all 16 amino acids without derivatization [91]. The LODs were between 0.61-10.5 μg/g, and recoveries ranged from 88.3-108.6% for the optimized method. Furthermore, the proposed method was applied to various types of royal jelly products. The content of free amino acid in royal jelly was compared to that in honey samples to assess the differences between the two products.
Isoflavones are phytoestrogens that are protective against many diseases and are mostly found in soy products. A CE-MS method was developed for the simultaneous analysis of both glycosylated forms (glycitin, daidzin, and genistin) and the aglycones (formononetin, biochanin A, glycitein, daidzein, and genistein) in soy biscuit sample [71]. QuEChERS based extraction was utilized for fast and efficient sample clean-up. The LOD and LOQs were achieved in the range of 0.21-2.0 μg/L and 0.69-6.6 μg/L, respectively, with recoveries between 91-109%. The use of CE-MS offers simplicity and ease of use, especially considering the complexity of food matrices in soy biscuit samples.
CE-TOF-MS analyzed commercial antihypertensive nutraceutical tablets, and 17 peptides were identified with advanced chemometric software [87]. The results suggest that CE-MS has excellent potential for analyzing bioactive peptides in nutraceuticals and mild derived food products in quality control and authentication of the antihypertensive nutraceutical.
5.2.6. Pesticides residues and food packaging materials
Pesticides have been used in fruits, vegetables, and most crops to ensure pests' safety and yield. Due to the growing quality of produce, several new pesticides are being used. As a result, various pesticide residues are exposed to humans through fruits, vegetables, grains, etc. Several CE-MS based methods have been developed for trace analysis of both charged and neutral pesticides food products.
Seven pesticides such as flutolanil, simazine, haloxyfop, acifluorfen, dinoseb, picloram, and ioxynil were analyzed in melon, watermelon, apricot, peach samples by CE-MS [58]. Samples were treated with pressurized liquid extraction followed by SPE using polymeric StrataX cartridge before analysis. The developed method provided LOD and LOQ of 0.01 and 0.05 mg/kg, respectively, and recoveries between 58-88%. There are many published extraction methods for pesticide analysis in various matrices, but the pressurized liquid extraction (PLE) method using only hot water as extractant solvent combined with CE-MS provides an ecological, selective, fast, and straightforward approach. This CE-MS method can be conveniently applied as a routine laboratory procedure to identify and quantify pesticide residues in fruits. It detects in a single run several of these pesticides in different fruit samples at concentrations below their maximum residue limit established by the European regulations.
A total of nine pesticides, including thiabendazole, aminocarb, imazalil, atrazine, metazachlor, metoxuron, carbofuron, metosulam, and imazapyr, were analyzed and separated in 6 min in corn samples by CZE-MS/MS combined with QuEChERS extraction [7]. Some of the pesticides mentioned above are very hydrophilic and are poorly retained by reversed-phase HPLC, making the accurate quantitation difficult by HPLC-UV or HPLC-MS. On the other hand, CZE-MS offers one such alternative because it uses this ionic nature to mobilize and separate these ionic/hydrophilic pesticides. For analysis of ionic pesticides, CE-MS has several advantages: high peak resolution, short separation time, and low reagent consumption. Nine polar pesticides were separated and analyzed using PVA coated capillary and 0.1 M formic acid at pH 2.4 as BGE, which provided stable and repeatable EOF. The optimized method provided LODs and LOQs of 0.03-0.28 μg/kg and 0.10-0.93 μg/kg, respectively, and recoveries were in the range of 70-110%. The results suggest that the CE/MS attends the international legislation for all the analyzed polar pesticides in corn.
Neutral carbamate pesticides, which CZE cannot separate, were separated and quantitated in fruit and vegetable juices by MEKC coupled to ion-trap (IT)-MS/MS employing VASLLME based sample extraction [13]. Simultaneous determination of 25 pesticides was carried out using 100 mM volatile surfactant (AFPO, pH 9) in BGE. The LODs and LOQs of 0.8-1.4 μg/kg and 2.3-4.7 μg/kg, respectively, with recoveries between 81-104%, were obtained by the developed method. The CE-MS/MS constitutes a simple, rapid, and reliable assay to determine CRB pesticides in juices. The assay has low operational time and costs, requires a very low consumption of organic solvents and other reagents, and, more importantly, fulfills the current green chemistry demands.
A CZE-MS/MS-based method was developed to determine halosulfuron-methyl herbicide residue in sugar cane juice and tomato samples [54]. Due to the instability and low volatility of the sulfonylureas, GC or GC-MS is not appropriate. However, CE and CE-MS are easily adaptable to quantify this herbicide in complex food samples due to the ionic nature. Besides, as mentioned in many articles, CE-MS is considered environmentally friendly compared to LC-MS. The QuEChERS based extraction was used before electrophoretic separation using ammonium carbonate as BGE. The obtained LODs for both food samples were two ppb, and the recoveries were between 96-104%. The developed CE-MS method generates low waste and could be considered environmental-friendly.
Besides pesticides, plasticizers such as organotin used in plastic food packaging are known to contaminate food products. The first application of the sheathless CZE-TOF-MS method combined with ultrasonic extraction has been used to analyze organotin, such as dibutyltin, tributyl tin, diphenyl tin, and triphenyl tin in five food packaging materials and five edible plant oil samples [32]. Representative EIC of three food packaging materials and three edible oil samples is shown in Fig. S17. The results suggest that packaging boxes and edible oils are prone to organotin contamination. For example, DPhT and TBT were detected in low ng (0.1- 0.5 ng/g) levels in plastic cake packaging and milk packaging boxes.
On the other hand, DBT was detected in corn oil and peanut oil, whereas TBT was found in soybean oil but all at the higher concentration range of 0.7-0.9 ng/g compared to the packaging material. CE-ESI-MS using a sheathless interface significantly improve the detection limits because it does not require any sheath liquid to establish an electrical connection, which would dilute the samples. The analytical method's LODs and LOQs were 0.002-0.05 ng/mL and 0.04-0.614 ng/mL for four organotin compounds. Therefore, it is capable of achieving highly-sensitive detection of trace-amount organotins. The recoveries of organotins in spiked food packaging materials and edible oils were in the range of 80.27-101.56% and 80.70-108.52%, respectively. The developed sheathless CE-ESI-MS method combined with ultrasonic extraction and SPE allowed ultrasensitive determination of organotins in food packaging materials and edible oils.
5.3. Bioanalysis and Foodomics
Bioanalysis is a sub-discipline of analytical chemistry that involves quantitative measurements of drugs/metabolites, proteins, macromolecules, etc., in a biological system. Omics refers to the studies such as genomics, proteomics, metabolomics, etc. Foodomics is a discipline that involves the analysis of food and nutrition domains using omics tools. Foodomics aims to provide valuable information regarding food safety, food quality, and effects of food components that can be utilized to improve consumer health and knowledge. Advanced analytical techniques with high specificity, selectivity, and sensitivity are needed for bioanalysis and omics studies because of the increased complexity and variations of matrices as well as the chemical nature and concentrations of target analytes. CE-based techniques, especially with MS detection, can be useful for bioanalysis and foodomics applications, as discussed below.
Foods are complex matrices, and food composition can be modified through changes in environmental conditions, food alteration, transport, storage, or fermentation, impacting human health. In this regard, CE–time-of-flight mass spectrometry (CE-TOF MS) have been utilized to study the effects of functional foods on a biological system and the effects of temperature, storage time, and ripening on components of food samples. For example, CE-TOF-MS was used for metabolic fingerprinting of the liver of hypercholesterolemic Wistar rats [77] and plasma of Wister Rats [98] after onion supplementation. About 33 metabolites, including amino acids, organic acids, purines, and pyridine compounds, were detected in liver samples, and 16 metabolites were identified in rat plasma samples. Another study of metabolites in mice fed with Japanese citrus fruit by CE-TOF-MS revealed 191 metabolites in plasma and 250 metabolites in liver samples [101].
The utility of capillary isoelectric focusing (CIEF) with ESI-MS detection was demonstrated for milk protein allergies by performing qualitative and quantitative analysis of milk whey protein in rabbit serum samples [22]. The MS detection provided high sensitivity (LOD 10-70 nM), which is adequate for detecting milk allergens in the multiplatform study for comprehensive metabolomic analysis. CE-TOF-MS studied the antiproliferative effects of dietary polyphenol on human colon cancer cells [41] and K562 leukemia cells[46]. Interesting CE-LIF-MS application was demonstrated for broad range screening of oligosaccharides in feces of infant formula-fed and breast milk-fed babies [68].
Besides foodomics applications, CE-MS is also very useful for determining food quality, freshness, and safety of solid and liquid foods using metabolites and sugar profiles. CE-TOF-MS has mainly been used in metabolomics in various food samples due to the high-resolution power and sensitive detection. The studies include metabolite profiling in fermented Orostachys japonicus A. Berger [97], probiotic fed fish [78], green and roasted coffee [79], fermented bean paste [75], tea leaves (during manufacture) [83], genetically modified soybean [99], lettuce head [11], edamame (in different storage time and temp) [74], avocado fruit (during the developmental and ripening process) [76], and GABA creamy fermented milk [100]. Besides, CE-MS sugar profiles of wine samples before and after pasteurization have been identified [45]. A powerful and fast CE-TOF-MS method was developed for proteomics of complex soybean protein mixtures[86]. The developed method allowed simultaneous analysis of 150 peptides in soybean protein fraction in 30 min. An exciting application of cationic coated capillary (poly(N, N, N', N’-tetraethyldiethylenetriamine, N-(2-hydroxypropyl) methacrylamide, TEDETAMA-co-HPMA (50:50) copolymer) was demonstrated for profiling of anionic metabolites in red wine and orange juice samples [9]. The optimized method allowed fast separation of 16 standard anionic metabolites in 12 min with high efficiency (N = 92,000 plates/meter). Using the developed CE-MS method, 87 and 142 metabolites were successfully identified in orange juice and wine sample respectively.
5.4. Analysis of stereoselective food
Many food products may contain chiral molecules (enantiomers) or racemates due to food processing or changes in the enantiomeric ratio caused by fermentation processes. For example, raspberries' natural flavor is composed exclusively of R-enantiomer, whereas the synthetic flavoring of raspberries contains a mixture of both R- and S- enantiomer of ionone. Similarly, the L-isomer of vitamin C is active and beneficial to human health, whereas the D-form is excreted and has no known benefits. D-amino acids (AAs) are mostly formed in food products because fermentation plays a vital role in taste and aroma. Several D-AAs such as alanine, leucine, and phenylalanine produces a sweeter taste, whereas L isomers have bitter tastes [113][114][115][116]. The analysis of such chiral AAs may provide information about food quality, including any adulteration. Therefore, the determination of enantiomeric ratios in food and beverages is a research area of significant interest.
To the best of our knowledge, the only food application based on CCE-MS reported in the last decade is the analysis of L-AAs in the presence of trace levels of D-AAs in vinegar. According to the fermentation process, AAs' enantiomeric ratio determines the taste quality studies of vinegar [26]. One challenge that has hindered the analysis of stereoselective food is that most chiral selectors are nonvolatile substances that are not compatible with CE-MS. The contamination of the ionization source of ESI-MS by nonvolatile chiral selectors results in a decreased detection sensitivity. Partial filling CCE-MS in which a plug of chiral selector such as 18-crown-6)-2,3,11,12-tetracarboxylic acid (18C6H4) operates by filling part of the capillary with separation buffer can still differentiate between D- and L-isomer of AAs, resulting in two peaks of the same m/z. Chiral separation of 17 amino acids with baseline resolution of eleven AAs in EICs was achieved using optimized BGE containing 30 mM 18C6H4 (Fig.8). Clearly, a partial filling method provided higher efficiency without any significant loss of ESI-MS sensitivity. The optimized CE-MS method provided LOD of 0.07-1.03 μg/mL and recoveries between 75-119.9% in various vinegar types.
Fig. 8.

Extracted ion electropherograms of 20 DL-AAs (20 μg/ml) obtained in a positive ESI mode using CCE-MS. BGE: 30 mM (+)-18C6H4 in water at 24 kV of a normal polarity by the partial filling technique. The separation buffer was injected at 50 mbar for 17 min to fill 70% of the capillary. The ions extracted for the AAs were [M+H]+. Reproduced from [26] with permission from Elsevier, copyright 2019.
6. Applications of CE-ICP-MS
Compared to the chromatographic method, CE coupled to inductively coupled plasma mass spectrometer (ICP-MS) provides much higher efficiency, selectivity, and mass resolution for a wide range of elemental speciation analysis. Small volume requirements result in less reagent and sample consumption and have a low operating cost.
In recent years, CE-ICP-MS has become one of the valuable techniques for speciation analysis, and the number of reports on its use in food safety has increased. In particular, CE-ICP-MS has been used for speciation analysis of heavy metals such as mercury, arsenic, chromium, selenium, zinc, and nanoparticles (gold, platinum, and palladium) in a variety of foods. Besides elemental analysis, CE-ICP-MS is also useful for sensitive analysis of organic molecules using metallic labeling.
Two methods were developed for trace analysis of mercury species in dried fish samples. The first method was developed for speciation analysis of mercury (Hg2+) and methyl mercury (CH3Hg+) in fish muscles using DNA sequence binding [116]. Because Hg2+ and CH3Hg+ are neutral molecules in solution, separation by CE is challenging. However, when these species are complexed with DNA, the different binding force between Hg2+ and CH3Hg+ makes the formed DNA-Hg2+ and DNA-CH3Hg+conjugates possess different negative charges. Therefore, the two mercury species were separated with reversed polarity CE-MS using cetyltrimethylammonium bromide as an EOF reversal agent. DNA-Hg2+ and DNA-CH3Hg+ were baseline separated in 11 min using optimized BGE containing 0.72 mmol/L Tris, 0.72 mmol/L H3BO3, 16 mmol/L EDTA, and 0.3 mmol/L cetyltrimethylammonium bromide (pH = 8.0) was selected as the optimal running buffer. The LOD of 0.12 and 0.10 ng/mL for DNA-Hg2+ and DNA-CH3Hg+, respectively, with recovery between 95-101%. The second method was developed for the ultrasensitive analysis of mercury, methyl mercury, and ethyl mercury in fish samples in 25 min [42]. The microwave-assisted extraction allowed detection of mercury species as low as 0.021-0.032 ng/mL with a recovery of 94-103%. However, in fish samples, only methyl mercury was detected.
Many arsenic species are known to accumulate in seafood and crops affecting human health. The arsenic species such as trivalent methylated arsenic species are highly toxic to humans, while species such as As (III) and As (V) are known as class I carcinogens. A highly sensitive CE-ICP-MS method was reported for analysis of arsenic species in seaweeds [118], scomberomorus niphonius (Japanese seer fish, a type of mackerel) [119], as well as rice and cereal samples [40]. Six arsenic species (arsenocholine (AsC), arsenobetaine (AsB), As(III),dimethylarsinic (DMA), monomethylarsonic (MMA), and As (V) were separated within 20 min [119]. The LOD of the optimized method was 0.08-0.12 μg/L, and recoveries were 90-103%. In another report, five arsenic species [DMA, MMA, As (III), As V, and o-arsanilic acid (o-ASA)] were separated within 10 min (Fig.9) with LOD less than 0.3 ng/g [40]. Increased migration time was observed in the column when injected with rice extracts (bottom two electropherograms, Fig. 9) compared to the standard arsenic mixture (top electropherogram, Fig. 9) due to adsorption of sugar on the capillary column. The first and second rice extract was digested with nitric acid and enzyme assisted microwave digestion, respectively. Moreover, unknown arsenic was also seen as unidentified peaks in both rice extracts (rice and rice cereal).
Fig. 9.
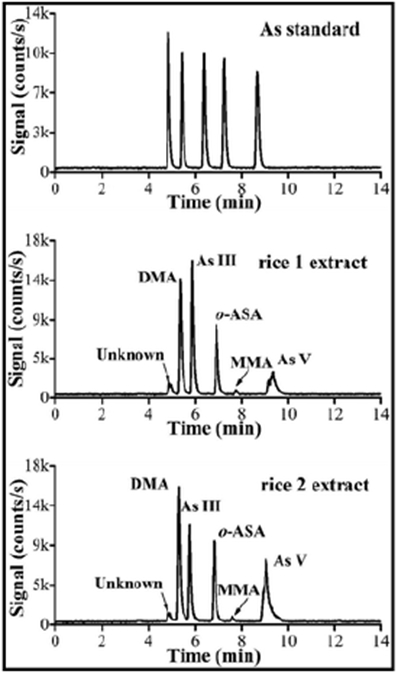
Electropherogram of arsenic standards and rice extracts by CE-ICP-MS with post extraction addition of o-AsA. Reproduced from [40] with permission from the American Chemical Society, copyright, 2015
Selenium is an essential element found at trace levels in several foods requiring nutritional and toxicological evaluation of different selenium compounds. Four selenium species Se (VI), Se2 (IV), SeCys (selenocysteine), and SeMet (selenomethionine), were separated and quantified in rice samples by CE-ICP-MS [39]. Electrophoretic separation was achieved within 18 min with baseline resolution. Highly efficient enzyme-assisted extraction allowed trace-level analysis with LOD 0.1-0.9 ng/mL with 90-103% recovery.
Chromium (Cr) is found in two oxidation states trivalent [Cr (III)] and hexavalent [Cr(VI)]. The later oxidation state is more toxic because it is a potent oxidizing agent with higher membrane transport [120]. The trivalent [i.e., Cr (III)] influence glucose metabolism and help insulin bind to its receptors [121]. Therefore, trivalent chromium has been used as a nutritional supplement in the form of Cr (III)-picolinate (CrPIC) for diabetic nephropathy. Developing a sensitive CE-ICP-MS method for the quantity of different Cr species is crucial in nutritional supplement safety and human health. CE-ICP-MS separated three chromium species (Cr (VI), Cr (III), and CrPic) in 8 min, and the concentrations were determined in yeast tablets with LOD in the range of 0.10-0.20 ng/mL, and recovery between 93-103% was obtained. The results demonstrated that CrPic is the main speciation form of chromium in the nutritional supplement, with a concentration of 1514.6 μg Cr/g.[84].
Two zinc fungicides, zinc dimethyl dithiocarbamate (Ziram) and zinc ethylene bisdithiocarbamate (Zineb), are used to protect fruits and vegetable crops from diseases. Ziram and Zineb were analyzed in cabbage leaves samples using electrokinetic chromatography (EKC)-ICP-MS [43]. Both analytes were separated in 12 min using 20 mM α-cyclodextrin in BGE of pH 9.0. The LOD of 1.9 and 3.0 ng/mL for Ziram and Zineb, respectively, was obtained with recovery between 95-107%.
Highly sensitive CE-ICP-MS was developed for trace analysis of saxitoxins in mussel samples using metal chelate labeling [85]. A combination of diethylenetriamine-N, N, N', N", N"-pentaacetic acid (DTAP) as a chelating agent and europium as a labeling element allowed the developed CE-ICP-MS method to provide LOD of 0.38 fmol. The application of CE-ICP-MS for separation and characterization of nanomaterials in the dietary supplement was demonstrated by Qu et al. [122]. Separation of gold nanoparticles with 5, 15, 20, and 30 nm size was achieved using 70 mM sodium dodecyl benzenesulfonate (SDBS) in BGE (10 mM TRIS, pH 9), and the optimized method was successfully used for the analysis of gold, platinum, and palladium nanoparticles in dietary supplements.
7. Conclusions
Hyphenated technologies such as HPLC-MS and GC-MS are still considered standard methodologies for analyzing food samples. The evolution of CE-MS in the past decade for food analysis suggests that this later hyphenation is slowly but gradually maturing. This improvement is mainly due to the development of new interface technology, volatile separation reagents, and column chemistry, which has allowed high resolution and fast analysis of food substances with a wide range of polarities. Applications of CE-MS and CE-ICP-MS hyphenated food safety techniques, food quality, food authenticity, toxicity, and food bioactivity (collectively known as foodomics) are discussed in this review. The data obtained suggest that the various platforms of these techniques (CZE, CEC, MEKC, CIEF, and CCE) will provide a massive amount of information. These platforms will have a significant impact on the future development of foodomics. The sensitivity and robustness are still the choking points of CE-MS, but this should improve with the recent development of robust and sensitive nanospray, sheathless interfaces, and jet stream technology with improved column chemistries CE-MS.
Future outlooks for CE-MS can be related to the new advances in stable and low cost open tubular CZE columns. While 90% of papers published for food analysis involve CZE-MS, there are neutral compounds in food products requiring MEKC-MS or CEC-MS. The applications of MEKC-MS for food analysis are reported only in 10% of the publication in the last decade, but this will change as new volatile surfactants with unique chromatographic selectivity becomes commercially available. For example, new MS-compatible (achiral/chiral surfactants and reagents) for MEKC-MS of neutral and stereoselective food components are envisaged. Similarly, CEC-MS will find its niche for the separation and sensitive detection of neutral compounds with the development of high porosity monolithic phases.
The technique of CIEF-MS is reported several years ago in only one publication but provided high sensitivity of proteins in milk samples. However, the recent development of CIEF-MS analysis facilitated by the flow-through microvial interface may benefit researchers as these new CE-MS provide separation of protein in food components which small differences in small pI value.
The use of CZE-TOF-MS was to intensively investigate for food analysis and foodomics in the past ten years. However, in the future, this hyphenation technology may be replaced by CZE-QTOF-MS, which combines the advantages of the high mass resolution, high mass accuracy, along with superior sensitivity and quantitation for the new substance of interest in food samples. The high-resolution mass analyzers will be worth for laboratory analysis of detecting contaminants in food. Moreover, CE-MS authentication and food samples screening will only become routine if sensitivity, dynamic range, and overall quantitative characteristics improve, and the instrument cost is reduced.
As documented through various applications in this review, CE-MS has firmly established itself as a "green method" for food analysis. However, the use of microchip CE-MS and monolithic CEC-MS can further minimize the use of solvents and reagents (mobile phase), making these two technologies not only low cost and ultrafast methods but also environmentally friendly. Unlike HPLC-MS, the number of dedicated commercial instruments for CE-MS is minimal, significantly restricting the development and application of this promising hyphenated technology. Therefore, it is essential to develop reliable, low-cost CE-MS instruments, ideally with thermostat cooling capacity, solvent gradient capability, efficient and stable capillary columns, and autosampler. Besides, online SPE will be engaging in the future that will minimize sample handling and maximize sensitivity.
Finally, a quest to understand at the molecular level in foodomics and strict monitoring of food contaminants at trace levels place stringent demands on hyphenation technology development. In this regard, the development of open tubular and monolithic versions of CEC-QTOF-MS and MEKC-QTOF-MS will play a significant role in meeting the challenges by offering high separation selectivity, efficiency, and peak capacity as well as high mass resolution and accuracy along with high sensitivity. Nonetheless, we fully expect that the use of new, innovative, and more dedicated CE-MS instruments, more variety of CEC columns and MS compatible surfactants, and reagents for killer and challenging applications will provide the much-needed popularity of CE-MS in food analysis.
Supplementary Material
Highlights.
Capillary electrophoresis mass spectrometry (CE-MS) is reviewed for food analysis
Principles of various modes of CE-MS and interfaces are discussed
Sample preparation methods for food analysis
Analysis of solid and liquid food
Application of CE-ESI-MS, CE-ICP-MS and offline CE-MALDI MS of solid and liquid food
7. Acknowledgments
The authors gratefully acknowledge the financial support of this project by the support from National Institute of Health (1 R21 MH107985-01).
Abbreviations
- 2,4-DAB
2,4-diaminobutyric acid
- I8-C6H4
(18-crown-6)-2,3,11,12-tetracarboxylic acid
- AAs
amino acids
- ACE
angiotensin converting enzyme
- ACN
acetonitrile
- AEG
N-2-aminoethylglycine
- AFPO
ammonium perfluorooctanoate
- AGs
aminoglycosides
- APCI
atmospheric pressure chemical ionization
- APPI
atmospheric pressure photoionization
- AMPS
acrylamido-2-methylpropane sulfonic acid
- AMLN
ammeline
- AMPLD
Ammelide
- AAOC
Association of Analytical Chemistry
- AsC
arsenocholine
- AsB
arsenobetaine
- BAs
biogenic amines
- BGE
background electrolyte
- BMA
butyl methacrylate
- BMAA
β-N-methylamino-L-alanine
- BZ
benzimidazole
- CD
cyclodextrin
- C4D
contactless conductivity detection
- CCE
chiral capillary electrophoresis
- CE
capillary electrophoresis
- CE-MS
capillary electrophoresis-mass spectrometry
- CEC
capillary electrochromatography
- CIEF
capillary isoelectric focusing
- CZE
capillary zone electrophoresis
- CrPIC
chromium picolinate
- CS
chiral selector
- CYN
cyanuric acid
- DART
direct analysis in real time
- DBT
dibutyl tin
- DPT
diphenyl tin
- dcSTX
decarbamoylsaxitoxin
- dcNEO
decarbamoylneosaxitoxin
- dcGTX
decarbamoylgonyautoxin
- DLLME
dispersive liquid-liquid microextraction
- DMA
dimethylarsenic
- DTAP
diethylenetriamine-N, N, N’, N”, N”-pentaacetic acid
- DVB
divinylbenzene
- EDCs
endocrine-disrupting compounds
- EDTA
ethylenediaminetetraacetate
- EDMA
ethylene glycol dimethacrylate
- EIE
extract ion electropherogram
- EKC
electrokinetic chromatography
- ELISA
enzyme-linked immunosorbent assay
- EOF
electroosmotic flow
- EOM
European Official Method
- EPE
enzyme-assisted protein extraction
- ESI
electrospray ionization
- WME
enzyme-assisted water-phase extraction
- FA
formic acid
- FLD
fluorescence detection
- GABA
gamma aminobutyric acid
- GC
gas chromatography
- GNT
gentamycin
- GOS
galactooligosaccharides
- GTX
gonyautoxin
- HAlKs
harmala alkaloids
- HFIP
hexafluoro-2-propanol
- HMOs
human milk oligosaccharides
- HMF
5-hydroxymethylfurfural
- HOAc
acetic acid
- HPMA
N-(2-hydroxypropyl) methacrylamide
- HPLC
high-performance liquid chromatography
- IACE
immunoaffinity capillary electrophoresis
- ICP
inductively coupled plasma
- IT
ion trap
- LOD
limit of detection
- LOQ
limit of quantitation
- LLE
liquid-liquid extraction
- MS
mass spectrometry
- ME
microwave-assisted extraction
- MEKC
micellar electrokinetic chromatography
- MALDI
matrix-assisted laser desorption ionization
- MMA
monomethylarsonic
- MEL
melamine
- MCs
microcystins
- MIP
molecularly imprinted polymer
- NEO
neosaxitoxin
- OAs
organic acids
- o-ASA
o-arsanilic acid
- PCR
polymerase chain reaction
- PLE
pressurized liquid extraction
- PNP
programmed nebulizing gas pressure
- PSTs
paralytic shellfish toxins
- PVA
polyvinyl alcohol
- QN
quinolones
- QTOF
quadrupole time of flight
- QuEChERS
quick, easy, cheap, effective, rugged, sensitive
- SDBS
sodium dodecyl benzenesulfonate
- SDS
sodium dodecyl sulfate
- SeCys
selenocysteine
- SeMet
selenomethionine
- SFE
supercritical fluid extraction
- SFC
supercritical fluid chromatography
- SLE
solid-liquid extraction
- SPE
solid-phase extraction
- STX
saxitoxin
- TBT
tributyl tin
- TPT
triphenyl tin
- TEDETAMA
poly(N, N, N’, N’-tetraethyldiethylenetriamine)
- TFA
trifluoroacetic acid
- TIC
total ion chromatogram
- TOF
time of flight
- UE
ultrasound-assisted extraction
- VASLLME
vortex-assisted surfactant enhanced liquid-liquid microextraction
- VBTA
vinylbenzyltrimethyl ammonium
- Ziram
zinc dimethyl dithiocarbamate
- Zineb
zinc ethylenebisdithiocarbamate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors of this review article has no conflict of interest
8. References
- [1].Rocco A, Aturki Z, Fanali S, Chiral separations in food analysis, TrAC - Trends Anal. Chem 52 (2013) 206–225. 10.1016/j.trac.2013.05.022. [DOI] [Google Scholar]
- [2].Nielsen SS, Food Analysis, Fourth, Springer US, Boston, MA, 2010. 10.1007/978-1-4419-1478-1. [DOI] [Google Scholar]
- [3].Olivares JA, Nguyen NT, Yonker CR, Smith RD, On-line mass spectrometric detection for capillary zone electrophoresis, Anal. Chem 59 (1987) 1230–1232. 10.1021/ac00135a034. [DOI] [Google Scholar]
- [4].Cubadda F, Chapter 19. Inductively coupled plasma mass spectrometry, in: Food Toxicants Anal., 2007: pp. 697–751. 10.1016/B978-044452843-8/50020-1. [DOI] [Google Scholar]
- [5].Chang C, Xu G, Bai Y, Zhang C, Li X, Li M, Liu Y, Liu H, Online coupling of capillary electrophoresis with direct analysis in real time mass spectrometry, Anal. Chem 85 (2013) 170–176. 10.1021/ac303450v. [DOI] [PubMed] [Google Scholar]
- [6].Qu H, Mudalige TK, Linder SW, Capillary Electrophoresis/Inductively-Coupled Plasma-Mass Spectrometry: Development and Optimization of a High Resolution Analytical Tool for the Size-Based Characterization of Nanomaterials in Dietary Supplements, Anal. Chem 86 (2014) 11620–11627. 10.1021/ac5025655. [DOI] [PubMed] [Google Scholar]
- [7].Daniel D, do Lago CL, Determination of Multiclass Pesticides Residues in Corn by QuEChERS and Capillary Electrophoresis Tandem Mass Spectrometry, Food Anal. Methods 12 (2019) 1684–1692. 10.1007/s12161-019-01501-y. [DOI] [Google Scholar]
- [8].Gasilova N, Gassner AL, Girault HH, Analysis of major milk whey proteins by immunoaffinity capillary electrophoresis coupled with MALDI-MS, Electrophoresis. 33 (2012) 2390–2398. 10.1002/elps.201200079. [DOI] [PubMed] [Google Scholar]
- [9].Acunha T, Simó C, Ibáñez C, Gallardo A, Cifuentes A, Anionic metabolite profiling by capillary electrophoresis-mass spectrometry using a noncovalent polymeric coating. Orange juice and wine as case studies, J. Chromatogr. A 1428 (2016) 326–335. 10.1016/j.chroma.2015.08.001. [DOI] [PubMed] [Google Scholar]
- [10].Ivanova-Petropulos V, Naceva Z, Sándor V, Makszin L, Deutsch-Nagy L, Berkics B, Stafilov T, Kilár F, Fast determination of lactic, succinic, malic, tartaric, shikimic, and citric acids in red Vranec wines by CZE-ESI-QTOF-MS, Electrophoresis. 39 (2018) 1597–1605. 10.1002/elps.201700492. [DOI] [PubMed] [Google Scholar]
- [11].Miyagi A, Uchimiya H, Kawai-Yamada M, Synergistic effects of light quality, carbon dioxide and nutrients on metabolite compositions of head lettuce under artificial growth conditions mimicking a plant factory, Food Chem. 218 (2017) 561–568. 10.1016/j.foodchem.2016.09.102. [DOI] [PubMed] [Google Scholar]
- [12].D’Orazio G, Asensio-Ramos M, Hernández-Borges J, Rodíguez-Delgado MÁ, Fanali S, Evaluation of the combination of a dispersive liquid-liquid microextraction method with micellar electrokinetic chromatography coupled to mass spectrometry for the determination of estrogenic compounds in milk and yogurt, Electrophoresis. 36 (2015) 615–625. 10.1002/elps.201400452. [DOI] [PubMed] [Google Scholar]
- [13].Moreno-González D, Huertas-Pérez JF, García-Campaña AM, Gámiz-Gracia L, Vortex-assisted surfactant-enhanced emulsification liquid-liquid microextraction for the determination of carbamates in juices by micellar electrokinetic chromatography tandem mass spectrometry, Talanta. 139 (2015) 174–180. 10.1016/j.talanta.2015.02.057. [DOI] [PubMed] [Google Scholar]
- [14].Rizvi SAA, Zheng J, Apkarian RP, Dublin SN, Shamsi SA, Polymeric Sulfated Amino Acid Surfactants: A Class of Versatile Chiral Selectors for Micellar Electrokinetic Chromatography (MEKC) and MEKC-MS, Anal. Chem 79 (2007) 879–898. 10.1021/ac061228t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang L, Harrata AK, Lee CS, On-Line Micellar Electrokinetic Chromatography–Electrospray Ionization Mass Spectrometry Using Anodically Migrating Micelles, Anal. Chem 69 (1997) 1820–1826. 10.1021/ac961202+. [DOI] [PubMed] [Google Scholar]
- [16].Molina M, Wiedmer SK, Jussila M, Silva M, Riekkola M-L, Use of a partial filling technique and reverse migrating micelles in the study of N-methylcarbamate pesticides by micellar electrokinetic chromatography–electrospray ionization mass spectrometry, J. Chromatogr. A 927 (2001) 191–202. 10.1016/S0021-9673(01)01053-6. [DOI] [PubMed] [Google Scholar]
- [17].Moreno-González D, Jáč P, Švec F, Nováková L, Determination of Sudan dyes in chili products by micellar electrokinetic chromatography-MS/MS using a volatile surfactant, Food Chem. 310 (2020) 125963. 10.1016/j.foodchem.2019.125963. [DOI] [PubMed] [Google Scholar]
- [18].Norton D, Zheng J, Danielson ND, Shamsi SA, Capillary Electrochromatography–Mass Spectrometry of Zwitterionic Surfactants, Anal. Chem 77 (2005) 6874–6886. 10.1021/ac050838a. [DOI] [PubMed] [Google Scholar]
- [19].Bragg W, Norton D, Shamsi SA, Optimized separation of β-blockers with multiple chiral centers using capillary electrochromatography–mass spectrometry, J. Chromatogr. B 875 (2008) 304–316. 10.1016/j.jchromb.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang HY, Lin CL, Jiang SH, Singco B, Cheng YJ, Capillary electrochromatography–mass spectrometry determination of melamine and related triazine by-products using poly(divinyl benzene-alkene-vinylbenzyl trimethylammonium chloride) monolithic stationary phases, Anal. Chim. Acta 719 (2012) 96–103. 10.1016/j.aca.2011.12.073. [DOI] [PubMed] [Google Scholar]
- [21].Doherty EAS, Meagher RJ, Albarghouthi MN, Barron AE, Microchannel wall coatings for protein separations by capillary and chip electrophoresis, Electrophoresis. 24 (2003) 34–54. 10.1002/elps.200390029. [DOI] [PubMed] [Google Scholar]
- [22].Lecoeur M, Gareil P, Varenne A, Separation and quantitation of milk whey proteins of close isoelectric points by on-line capillary isoelectric focusing-Electrospray ionization mass spectrometry in glycerol-water media, J. Chromatogr. A 1217 (2010) 7293–7301. 10.1016/j.chroma.2010.09.043. [DOI] [PubMed] [Google Scholar]
- [23].Wang L, Bo T, Zhang Z, Wang G, Tong W, Da Yong Chen D, High Resolution Capillary Isoelectric Focusing Mass Spectrometry Analysis of Peptides, Proteins, And Monoclonal Antibodies with a Flow-through Microvial Interface., Anal. Chem 90 (2018) 9495–9503. 10.1021/acs.analchem.8b02175. [DOI] [PubMed] [Google Scholar]
- [24].Tanaka Y, Otsuka K, Terabe S, Separation of enantiomers by capillary electrophoresis–mass spectrometry employing a partial filling technique with a chiral crown ether, J. Chromatogr. A 875 (2000) 323–330. 10.1016/S0021-9673(99)01334-5. [DOI] [PubMed] [Google Scholar]
- [25].Alvarez-Rivera G, Bueno M, Ballesteros-Vivas D, Cifuentes A, Chiral analysis in food science, TrAC Trends Anal. Chem 123 (2020) 115761. 10.1016/j.trac.2019.115761. [DOI] [Google Scholar]
- [26].Lee S, Kim SJ, Bang E, Na YC, Chiral separation of intact amino acids by capillary electrophoresis–mass spectrometry employing a partial filling technique with a crown ether carboxylic acid, J. Chromatogr. A 1586 (2019) 128–138. 10.1016/j.chroma.2018.12.001. [DOI] [PubMed] [Google Scholar]
- [27].Smith RD, Barinaga CJ, Udseth HR, Improved Electrospray Ionization Interface for Capillary Zone Electrophoresis-Mass Spectrometry, Anal. Chem 60 (1988) 1948–1952. 10.1021/ac00169a022. [DOI] [Google Scholar]
- [28].Hommerson P, Khan AM, de Jong GJ, Somsen GW, Capillary electrophoresis-atmospheric pressure chemical ionization-mass spectrometry using an orthogonal interface: Set-up and system parameters, J. Am. Soc. Mass Spectrom 20 (2009) 1311–1318. 10.1021/jasms.8b03512. [DOI] [PubMed] [Google Scholar]
- [29].Gu C, Shamsi SA, CEC-atmospheric pressure ionization MS of pesticides using a surfactant-bound monolithic column, Electrophoresis. 31 (2010) 1162–1174. 10.1002/elps.200900739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhao SS, Chen DDY, Applications of capillary electrophoresis in characterizing recombinant protein therapeutics., Electrophoresis. 35 (2014) 96–108. 10.1002/elps.201300372. [DOI] [PubMed] [Google Scholar]
- [31].Maxwell EJ, Zhong X, Zhang H, van Zeijl N, Chen DDY, Decoupling CE and ESI for a more robust interface with MS, Electrophoresis. 31 (2010) 1130–1137. 10.1002/elps.200900517. [DOI] [PubMed] [Google Scholar]
- [32].He H, Tian M, Hu L, Yang L, Ultrasensitive determination of organotin compounds in plastic food packaging and edible oils by sheathless capillary electrophoresis-electrospray ionization-mass spectrometry, Analyst. 145 (2020) 2286–2296. 10.1039/c9an02331c. [DOI] [PubMed] [Google Scholar]
- [33].Moini M, Simplifying CE–MS Operation. 2. Interfacing Low-Flow Separation Techniques to Mass Spectrometry Using a Porous Tip, Anal. Chem 79 (2007) 4241–4246. 10.1021/ac0704560. [DOI] [PubMed] [Google Scholar]
- [34].Faserl K, Sarg B, Kremser L, Lindner H, Optimization and Evaluation of a Sheathless Capillary Electrophoresis–Electrospray Ionization Mass Spectrometry Platform for Peptide Analysis: Comparison to Liquid Chromatography–Electrospray Ionization Mass Spectrometry, Anal. Chem 83 (2011) 7297–7305. 10.1021/ac2010372. [DOI] [PubMed] [Google Scholar]
- [35].Liu L, Yun Z, He B, Jiang G, Efficient Interface for Online Coupling of Capillary Electrophoresis with Inductively Coupled Plasma–Mass Spectrometry and Its Application in Simultaneous Speciation Analysis of Arsenic and Selenium, Anal. Chem 86 (2014) 8167–8175. 10.1021/ac501347d. [DOI] [PubMed] [Google Scholar]
- [36].Kannamkumarath SS, Wrobel K, Wrobel K, B’Hymer C, Caruso JA, Capillary electrophoresis-inductively coupled plasma-mass spectrometry: an attractive complementary technique for elemental speciation analysis., J. Chromatogr. A 975 (2002) 245–266. 10.1016/s0021-9673(02)01218-9. [DOI] [PubMed] [Google Scholar]
- [37].Busnel J-M, Josserand J, Lion N, Girault HH, Iontophoretic Fraction Collection for Coupling Capillary Zone Electrophoresis with Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry, Anal. Chem 81 (2009) 3867–3872. 10.1021/ac900128q. [DOI] [PubMed] [Google Scholar]
- [38].Gasilova N, Girault HH, Component-resolved diagnostic of Cows milk allergy by immunoaffinity capillary electrophoresis-matrix assisted laser desorption/ionization mass spectrometry, Anal. Chem 86 (2014) 6337–6345. 10.1021/ac500525n. [DOI] [PubMed] [Google Scholar]
- [39].Zhao Y, Zheng J, Yang M, Yang G, Wu Y, Fu F, Speciation analysis of selenium in rice samples by using capillary electrophoresis-inductively coupled plasma mass spectrometry, Talanta. 84 (2011) 983–988. 10.1016/j.talanta.2011.03.004. [DOI] [PubMed] [Google Scholar]
- [40].Qu H, Mudalige TK, Linder SW, Arsenic speciation in rice by capillary electrophoresis/inductively coupled plasma mass spectrometry: Enzyme-assisted water-phase microwave digestion, J. Agric. Food Chem 63 (2015) 3153–3160. 10.1021/acs.jafc.5b00456. [DOI] [PubMed] [Google Scholar]
- [41].Ibáñez C, Simó C, García-Cañas V, Gómez-Martínez Á, Ferragut JA, Cifuentes A, CE/LC-MS multiplatform for broad metabolomic analysis of dietary polyphenols effect on colon cancer cells proliferation, Electrophoresis. 33 (2012) 2328–2336. 10.1002/elps.201200143. [DOI] [PubMed] [Google Scholar]
- [42].Zhao Y, Zheng J, Fang L, Lin Q, Wu Y, Xue Z, Fu F, Speciation analysis of mercury in natural water and fish samples by using capillary electrophoresis-inductively coupled plasma mass spectrometry, Talanta. 89 (2012) 280–285. 10.1016/j.talanta.2011.12.029. [DOI] [PubMed] [Google Scholar]
- [43].Chen J, Fu F, Wu S, Wang J, Wang Z, Simultaneous detection of zinc dimethyldithiocarbamate and zinc ethylenebisdithiocarbamate in cabbage leaves by capillary electrophoresis with inductively coupled plasma mass spectrometry, J. Sep. Sci 40 (2017) 3898–3904. 10.1002/jssc.201700455. [DOI] [PubMed] [Google Scholar]
- [44].Tejada-Casado C, Moreno-González D, Lara FJ, García-Campaña AM, del Olmo-Iruela M, Determination of benzimidazoles in meat samples by capillary zone electrophoresis tandem mass spectrometry following dispersive liquid–liquid microextraction, J. Chromatogr. A 1490 (2017) 212–219. 10.1016/j.chroma.2017.02.023. [DOI] [PubMed] [Google Scholar]
- [45].Sugimoto M, Kaneko M, Onuma H, Sakaguchi Y, Mori M, Abe S, Soga T, Tomita M, Changes in the charged metabolite and sugar profiles of pasteurized and unpasteurized Japanese sake with storage, J. Agric. Food Chem 60 (2012) 2586–2593. 10.1021/jf2048993. [DOI] [PubMed] [Google Scholar]
- [46].Valdés A, Simó C, Ibáñez C, Rocamora-Reverte L, Ferragut JA, García-Cañas V, Cifuentes A, Effect of dietary polyphenols on K562 leukemia cells: A Foodomics approach, Electrophoresis. 33 (2012) 2314–2327. 10.1002/elps.201200133. [DOI] [PubMed] [Google Scholar]
- [47].Beri J, Kirkwood KI, Muddiman DC, Bereman MS, A novel integrated strategy for the detection and quantification of the neurotoxin β-N-methylamino-l-alanine in environmental samples, Anal. Bioanal. Chem 410 (2018) 2597–2605. 10.1007/s00216-018-0930-0. [DOI] [PubMed] [Google Scholar]
- [48].Daniel D, Lopes FS, dos Santos VB, do Lago CL, Detection of coffee adulteration with soybean and corn by capillary electrophoresis-tandem mass spectrometry, Food Chem. 243 (2018) 305–310. 10.1016/j.foodchem.2017.09.140. [DOI] [PubMed] [Google Scholar]
- [49].Pont L, Compte I, Sanz-Nebot V, Barbosa J, Benavente F, Analysis of Hordeins in Barley Grain and Malt by Capillary Electrophoresis-Mass Spectrometry, Food Anal. Methods 13 (2020) 325–336. 10.1007/s12161-019-01648-8. [DOI] [Google Scholar]
- [50].Zwir-Ferenc A, Biziuk M, Solid phase extraction technique - Trends, opportunities and applications, Polish J. Environ. Stud 15 (2006) 677–690. [Google Scholar]
- [51].Pedersen-Bjergaard S, Rasmussen KE, Grønhaug Halvorsen T, Liquid–liquid extraction procedures for sample enrichment in capillary zone electrophoresis, J. Chromatogr. A 902 (2000) 91–105. 10.1016/S0021-9673(00)00738-X. [DOI] [PubMed] [Google Scholar]
- [52].Rejczak T, Tuzimski T, A review of recent developments and trends in the QuEChERS sample preparation approach, Open Chem. 13 (2015) 980–1010. 10.1515/chem-2015-0109. [DOI] [Google Scholar]
- [53].Moldoveanu S, David V, Chapter 7 - Solid-Phase Extraction, in: Moldoveanu S, V.B.T.-M.S.P. for David C (Eds.), Elsevier, Amsterdam, 2015: pp. 191–286. 10.1016/B978-0-444-54319-6.00007-4. [DOI] [Google Scholar]
- [54].Daniel D, Dos Santos VB, Vidal DTR, Do Lago CL, Determination of halosulfuron-methyl herbicide in sugarcane juice and tomato by capillary electrophoresis-tandem mass spectrometry, Food Chem. 175 (2015) 82–84. 10.1016/j.foodchem.2014.11.137. [DOI] [PubMed] [Google Scholar]
- [55].Tascon M, Gagliardi LG, Benavente F, Parts-per-trillion detection of harmala alkaloids in Undaria pinnatifida algae by on-line solid phase extraction capillary electrophoresis mass spectrometry, Anal. Chim. Acta 954 (2017) 60–67. 10.1016/j.aca.2016.12.012. [DOI] [PubMed] [Google Scholar]
- [56].Puig P, Borrull F, Calull M, Aguilar C, Sorbent preconcentration procedures coupled to capillary electrophoresis for environmental and biological applications, Anal. Chim. Acta 616 (2008) 1–18. 10.1016/j.aca.2008.03.062. [DOI] [PubMed] [Google Scholar]
- [57].Moreno-González D, Lara FJ, Gámiz-Gracia L, García-Campaña AM, Molecularly imprinted polymer as in-line concentrator in capillary electrophoresis coupled with mass spectrometry for the determination of quinolones in bovine milk samples, J. Chromatogr. A 1360 (2014) 1–8. 10.1016/j.chroma.2014.07.049. [DOI] [PubMed] [Google Scholar]
- [58].Juan-García A, Font G, Juan C, Picó Y, Pressurised liquid extraction and capillary electrophoresis–mass spectrometry for the analysis of pesticide residues in fruits from Valencian markets, Spain, Food Chem. 120 (2010) 1242–1249. 10.1016/j.foodchem.2009.11.071. [DOI] [Google Scholar]
- [59].Tong P, Zhang L, He Y, Tang S, Cheng J, Chen G, Analysis of microcystins by capillary zone electrophoresis coupling with electrospray ionization mass spectrometry, Talanta. 82 (2010) 1101–1106. 10.1016/j.talanta.2010.05.045. [DOI] [PubMed] [Google Scholar]
- [60].Català-Clariana S, Benavente F, Giménez E, Barbosa J, Sanz-Nebot V, Identification of bioactive peptides in hypoallergenic infant milk formulas by capillary electrophoresis-mass spectrometry, Anal. Chim. Acta 683 (2010) 119–125. 10.1016/j.aca.2010.10.002. [DOI] [PubMed] [Google Scholar]
- [61].Català-Clariana S, Benavente F, Giménez E, Barbosa J, Sanz-Nebot V, Identification of bioactive peptides in hypoallergenic infant milk formulas by CE-TOF-MS assisted by semiempirical model of electromigration behavior, Electrophoresis. 34 (2013) 1886–1894. 10.1002/elps.201200547. [DOI] [PubMed] [Google Scholar]
- [62].Moreno-González D, Lara FJ, Jurgovská N, Gámiz-Gracia L, García-Campaña AM, Determination of aminoglycosides in honey by capillary electrophoresis tandem mass spectrometry and extraction with molecularly imprinted polymers, Anal. Chim. Acta 891 (2015) 321–328. 10.1016/j.aca.2015.08.003. [DOI] [PubMed] [Google Scholar]
- [63].Keyon ASA, Guijt RM, Gaspar A, Kazarian AA, Nesterenko PN, Bolch CJ, Breadmore MC, Capillary electrophoresis for the analysis of paralytic shellfish poisoning toxins in shellfish: Comparison of detection methods, Electrophoresis. 35 (2014) 1496–1503. 10.1002/elps.201300353. [DOI] [PubMed] [Google Scholar]
- [64].Kerrin ES, White RL, Quilliam MA, Quantitative determination of the neurotoxin β-N-methylamino-l-alanine (BMAA) by capillary electrophoresis–tandem mass spectrometry, Anal. Bioanal. Chem 409 (2017) 1481–1491. 10.1007/s00216-016-0091-y. [DOI] [PubMed] [Google Scholar]
- [65].Kohler I, Cognard E, Marchi I, Ortelli D, Edder P, Veuthey JL, Rudaz S, Schappler J, Single-run separation of closely related cationic and anionic compounds by CE-ESI-MS: Application to the simultaneous analysis of melamine and its analogs in milk, Chimia (Aarau). 65 (2011) 389–395. 10.2533/chimia.2011.389. [DOI] [PubMed] [Google Scholar]
- [66].Rodríguez-Gonzalo E, Domínguez-Álvarez J, García-Gómez D, García-Jiménez MG, Carabias-Martínez R, Determination of endocrine disruptors in honey by CZE-MS using restricted access materials for matrix cleanup, Electrophoresis. 31 (2010) 2279–2288. 10.1002/elps.200900715. [DOI] [PubMed] [Google Scholar]
- [67].Cummings JH, Macfarlane GT, Role of intestinal bacteria in nutrient metabolism, Clin. Nutr 16 (1997) 3–11. 10.1016/S0261-5614(97)80252-X. [DOI] [PubMed] [Google Scholar]
- [68].Albrecht S, Schols HA, Van Zoeren D, Van Lingen RA, Groot Jebbink LJM, Van Den Heuvel EGHM, Voragen AGJ, Gruppen H, Oligosaccharides in feces of breast- and formula-fed babies, Carbohydr. Res 346 (2011) 2173–2181. 10.1016/j.carres.2011.06.034. [DOI] [PubMed] [Google Scholar]
- [69].Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ, Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce., J. AOAC Int 86 (2003) 412–431. [PubMed] [Google Scholar]
- [70].Ronald SJL Majors E, Anastassiades Michelangelo, QuEChERS, a Sample Preparation Technique that Is “Catching On”: An Up-to-Date Interview with the Inventors, LCGC North Am. (2010) 504–516. https://www.chromatographyonline.com/view/quechers-sample-preparation-technique-catching-date-interview-inventors. [Google Scholar]
- [71].Bustamante-Rangel M, Delgado-Zamarreño MM, Pérez-Martín L, Carabias-Martínez R, QuEChERS method for the extraction of isoflavones from soy-based foods before determination by capillary electrophoresis-electrospray ionization-mass spectrometry, Microchem. J 108 (2013) 203–209. 10.1016/j.microc.2012.10.023. [DOI] [Google Scholar]
- [72].Domínguez-álvarez J, Mateos-Vivas M, García-Gómez D, Rodríguez-Gonzalo E, Carabias-Martínez R, Capillary electrophoresis coupled to mass spectrometry for the determination of anthelmintic benzimidazoles in eggs using a QuEChERS with preconcentration as sample treatment, J. Chromatogr. A 1278 (2013) 166–174. 10.1016/j.chroma.2012.12.064. [DOI] [PubMed] [Google Scholar]
- [73].Domínguez-Álvarez J, Rodríguez-Gonzalo E, Hernández-Méndez J, Carabias-Martínez R, Programed nebulizing-gas pressure mode for quantitative capillary electrophoresis-mass spectrometry analysis of endocrine disruptors in honey, Electrophoresis. 33 (2012) 2374–2381. 10.1002/elps.201100577. [DOI] [PubMed] [Google Scholar]
- [74].Sugimoto M, Goto H, Otomo K, Ito M, Onuma H, Suzuki A, Sugawara M, Abe S, Tomita M, Soga T, Metabolomic profiles and sensory attributes of edamame under various storage duration and temperature conditions, J. Agric. Food Chem 58 (2010) 8418–8425. 10.1021/jf101471d. [DOI] [PubMed] [Google Scholar]
- [75].Kim J, Choi JN, John KMM, Kusano M, Oikawa A, Saito K, Lee CH, GC-TOF-MS- and CE-TOF-MS-based metabolic profiling of cheonggukjang (fast-fermented bean paste) during fermentation and its correlation with metabolic pathways, J. Agric. Food Chem 60 (2012) 9746–9753. 10.1021/jf302833y. [DOI] [PubMed] [Google Scholar]
- [76].Contreras-Gutiérrez PK, Hurtado-Fernández E, Gómez-Romero M, Hormaza JI, Carrasco-Pancorbo A, Fernández-Gutiérrez A, Determination of changes in the metabolic profile of avocado fruits (Persea americana) by two CE-MS approaches (targeted and non-targeted), Electrophoresis. 34 (2013) 2928–2942. 10.1002/elps.201200676. [DOI] [PubMed] [Google Scholar]
- [77].González-Peña D, Dudzik D, García A, de Ancos B, Barbas C, Sánchez-Moreno C, Metabolomic fingerprinting in the comprehensive study of liver changes associated with onion supplementation in hypercholesterolemic Wistar rats, Int. J. Mol. Sci 18 (2017) 1–16. 10.3390/ijms18020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nguyen TL, Chun WK, Kim A, Kim N, Roh HJ, Lee Y, Yi M, Kim S, Il Park C, Kim DH, Dietary probiotic effect of Lactococcus lactis WFLU12 on low-molecular-weight metabolites and growth of olive flounder (Paralichythys olivaceus), Front. Microbiol 9 (2018) 1–17. 10.3389/fmicb.2018.02059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pérez-Míguez R, Sánchez-López E, Plaza M, Marina ML, Castro-Puyana M, Capillary electrophoresis-mass spectrometry metabolic fingerprinting of green and roasted coffee, J. Chromatogr. A 1605 (2019). 10.1016/j.chroma.2019.07.007. [DOI] [PubMed] [Google Scholar]
- [80].Sugimoto M, Sugawara T, Obiya S, Enomoto A, Kaneko M, Ota S, Soga T, Tomita M, Sensory properties and metabolomic profiles of dry-cured ham during the ripening process, Food Res. Int 129 (2020) 108850. 10.1016/j.foodres.2019.108850. [DOI] [PubMed] [Google Scholar]
- [81].Woźniakiewicz, Woźniakiewicz A, Nowak PM, Kłodzińska E, Namieśnik J, Płotka-Wasylka J, CE-MS and GC-MS as “Green” and Complementary Methods for the Analysis of Biogenic Amines in Wine, Food Anal. Methods 11 (2018) 2614–2627. 10.1007/s12161-018-1219-9. [DOI] [Google Scholar]
- [82].Lee JH, Kim SJ, Lee S, Rhee JK, Lee SY, Na YC, Saturated fatty acid determination method using paired ion electrospray ionization mass spectrometry coupled with capillary electrophoresis, Anal. Chim. Acta 984 (2017) 223–231. 10.1016/j.aca.2017.06.052. [DOI] [PubMed] [Google Scholar]
- [83].Yang Z, Kobayashi E, Katsuno T, Asanuma T, Fujimori T, Ishikawa T, Tomomura M, Mochizuki K, Watase T, Nakamura Y, Watanabe N, Characterisation of volatile and non-volatile metabolites in etiolated leaves of tea (Camellia sinensis) plants in the dark, Food Chem. 135 (2012) 2268–2276. 10.1016/j.foodchem.2012.07.066. [DOI] [PubMed] [Google Scholar]
- [84].Chen Y, Chen J, Xi Z, Yang G, Wu Z, Li J, Fu F, Simultaneous analysis of Cr(III), Cr(VI), and chromium picolinate in foods using capillary electrophoresis-inductively coupled plasma mass spectrometry, Electrophoresis. 36 (2015) 1208–1215. 10.1002/elps.201500015. [DOI] [PubMed] [Google Scholar]
- [85].He Y, Mo F, Chen D, Xu LJ, Wu Y, Fu FF, Capillary electrophoresis inductively coupled plasma mass spectrometry combined with metal tag for ultrasensitively determining trace saxitoxin in seafood, Electrophoresis. 38 (2017) 469–476. 10.1002/elps.201600411. [DOI] [PubMed] [Google Scholar]
- [86].Simó C, Domínguez-Vega E, Marina ML, García MC, Dinelli G, Cifuentes A, CE-TOF MS analysis of complex protein hydrolyzates from genetically modified soybeans - A tool for foodomics, Electrophoresis. 31 (2010) 1175–1183. 10.1002/elps.200900448. [DOI] [PubMed] [Google Scholar]
- [87].Benavente F, Pero-Gascon R, Pont L, Jaumot J, Barbosa J, Sanz-Nebot V, Identification of antihypertensive peptides in nutraceuticals by capillary electrophoresis-mass spectrometry, J. Chromatogr. A 1579 (2018) 129–137. 10.1016/j.chroma.2018.10.018. [DOI] [PubMed] [Google Scholar]
- [88].Fukuji TS, Castro-Puyana M, Tavares MFM, Cifuentes A, Sensitive and fast determination of Sudan dyes in chilli powder by partial-filling micellar electrokinetic chromatography-tandem mass spectrometry, Electrophoresis. 33 (2012) 705–712. 10.1002/elps.201100272. [DOI] [PubMed] [Google Scholar]
- [89].Fukuji TS, Castro-Puyana M, Tavares MFM, Cifuentes A, Fast determination of Sudan dyes in chilli tomato sauces using partial filling micellar electrokinetic chromatography, J. Agric. Food Chem 59 (2011) 11903–11909. 10.1021/jf203201b. [DOI] [PubMed] [Google Scholar]
- [90].Bignardi C, Cavazza A, Corradini C, Determination of furosine in food products by capillary zone electrophoresis-tandem mass spectrometry, Electrophoresis. 33 (2012) 2382–2389. 10.1002/elps.201100582. [DOI] [PubMed] [Google Scholar]
- [91].Akamatsu S, Mitsuhashi T, Development of a simple analytical method using capillary electrophoresis-tandem mass spectrometry for product identification and simultaneous determination of free amino acids in dietary supplements containing royal jelly, J. Food Compos. Anal 30 (2013) 47–51. 10.1016/j.jfca.2013.01.006. [DOI] [Google Scholar]
- [92].Bignardi, Cavazza A, Corradini C, Selected product ion monitoring for quantification of 5-hydroxymethylfurfural in food products by capillary zone electrophoresis-tandem ion trap mass spectrometry, Food Control. 46 (2014) 41–48. 10.1016/j.foodcont.2014.04.049. [DOI] [Google Scholar]
- [93].Kondeková M, Maier V, Ginterová P, Marák J, Ševčík J, Analysis of lysozyme in cheese samples by on-line combination of capillary zone electrophoresis and mass spectrometry, Food Chem. 153 (2014) 398–404. 10.1016/j.foodchem.2013.12.078. [DOI] [PubMed] [Google Scholar]
- [94].Mateos-Vivas M, Domínguez-Álvarez J, Rodríguez-Gonzalo E, Carabias-Martínez R, Capillary electrophoresis coupled to mass spectrometry employing hexafluoro-2-propanol for the determination of nucleosides and nucleotide mono-, di- and tri-phosphates in baby foods, Food Chem. 233 (2017) 38–44. 10.1016/j.foodchem.2017.04.060. [DOI] [PubMed] [Google Scholar]
- [95].Tascon M, Benavente F, Sanz-Nebot VM, Gagliardi LG, Fast determination of harmala alkaloids in edible algae by capillary electrophoresis mass spectrometry, Anal. Bioanal. Chem 407 (2015) 3637–3645. 10.1007/s00216-015-8579-4. [DOI] [PubMed] [Google Scholar]
- [96].Muroya S, Oe M, Ojima K, Thiamine accumulation and thiamine triphosphate decline occur in parallel with ATP exhaustion during postmortem aging of pork muscles, Meat Sci. 137 (2018) 228–234. 10.1016/j.meatsci.2017.11.035. [DOI] [PubMed] [Google Scholar]
- [97].Das G, Patra JK, Lee SY, Kim C, Park JG, Baek KH, Analysis of metabolomic profile of fermented Orostachys japonicus A. Berger by capillary electrophoresis time of flight mass spectrometry, PLoS One. 12 (2017) 1–13. 10.1371/journal.pone.0181280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].González-Peña D, Dudzik D, Colina-Coca C, de Ancos B, García A, Barbas C, Sánchez-Moreno C, Evaluation of onion as a functional ingredient in the prevention of metabolic impairments associated to diet-induced hypercholesterolaemia using a multiplatform approach based on LC-MS, CE-MS and GC-MS, J. Funct. Foods 19 (2015) 363–375. 10.1016/j.jff.2015.09.033. [DOI] [Google Scholar]
- [99].Kusano M, Baxter I, Fukushima A, Oikawa A, Okazaki Y, Nakabayashi R, Bouvrette DJ, Achard F, Jakubowski AR, Ballam JM, Phillips JR, Culler AH, Saito K, Harrigan GG, Assessing metabolomic and chemical diversity of a soybean lineage representing 35 years of breeding, Metabolomics. 11 (2015) 261–270. 10.1007/s11306-014-0702-6. [DOI] [Google Scholar]
- [100].Hagi T, Kobayashi M, Nomura M, Metabolome analysis of milk fermented by γ-aminobutyric acid-producing Lactococcus lactis, J. Dairy Sci 99 (2016) 994–1001. 10.3168/jds.2015-9945. [DOI] [PubMed] [Google Scholar]
- [101].Toyoda A, Sato M, Muto M, Goto T, Miyaguchi Y, Inoue E, Metabolomic analyses of plasma and liver of mice fed with immature Citrus tumida peel, Biosci. Biotechnol. Biochem 84 (2020) 1098–1104. 10.1080/09168451.2020.1719821. [DOI] [PubMed] [Google Scholar]
- [102].Bustamante-Rangel M, Delgado-Zamarreño M, Carabias-Martínez R, Domínguez-Álvarez J, Analysis of isoflavones in soy drink by capillary zone electrophoresis coupled with electrospray ionization mass spectrometry, Anal. Chim. Acta 709 (2012) 113–119. 10.1016/j.aca.2011.10.015. [DOI] [PubMed] [Google Scholar]
- [103].Daniel D, dos Santos VB, Vidal DTR, do Lago CL, Determination of biogenic amines in beer and wine by capillary electrophoresis–tandem mass spectrometry, J. Chromatogr. A 1416 (2015) 121–128. 10.1016/j.chroma.2015.08.065. [DOI] [PubMed] [Google Scholar]
- [104].Arráez-Román D, Fu S, Sawalha SMS, Segura-Carretero A, Fernández-Gutiérrez A, HPLC/CE-ESI-TOF-MS methods for the characterization of polyphenols in almond-skin extracts, Electrophoresis. 31 (2010) 2289–2296. 10.1002/elps.200900679. [DOI] [PubMed] [Google Scholar]
- [105].Sánchez-Hernández L, Marina ML, Crego AL, A capillary electrophoresis-tandem mass spectrometry methodology for the determination of non-protein amino acids in vegetable oils as novel markers for the detection of adulterations in olive oils, J. Chromatogr. A 1218 (2011) 4944–4951. 10.1016/j.chroma.2011.01.045. [DOI] [PubMed] [Google Scholar]
- [106].Sánchez-Hernández L, Castro-Puyana M, Luisa Marina M, Crego AL, Determination of betaines in vegetable oils by capillary electrophoresis tandem mass spectrometry - application to the detection of olive oil adulteration with seed oils, Electrophoresis. 32 (2011) 1394–1401. 10.1002/elps.201100005. [DOI] [PubMed] [Google Scholar]
- [107].Izu H, Shigemori K, Eguchi M, Kawane S, Fujii S, Kitamura Y, Aoshima H, Yamada Y, Direct activation of GABAA receptors by substances in the organic acid fraction of Japanese sake, Food Chem. 214 (2017) 354–359. 10.1016/j.foodchem.2016.07.048. [DOI] [PubMed] [Google Scholar]
- [108].Li B, Yongku L, Wang X, Wang F, Wang X, Wang Y, Meng X, Simultaneous separation and determination of organic acids in blueberry juices by capillary electrophoresis- electrospray ionization mass spectrometry, J. Food Sci. Technol 52 (2015) 5228–5235. 10.1007/s13197-014-1611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Moreno-González D, Hamed AM, Gilbert-López B, Gámiz-Gracia L, García-Campaña AM, Evaluation of a multiresidue capillary electrophoresis-quadrupole-time-of-flight mass spectrometry method for the determination of antibiotics in milk samples, J. Chromatogr. A 1510 (2017) 100–107. 10.1016/j.chroma.2017.06.055. [DOI] [PubMed] [Google Scholar]
- [110].Beach DG, Kerrin ES, Thomas K, Quilliam MA, McCarron P, Capillary electrophoresis–tandem mass spectrometry for multiclass analysis of polar marine toxins, Anal. Bioanal. Chem 410 (2018) 5405–5420. 10.1007/s00216-018-1089-4. [DOI] [PubMed] [Google Scholar]
- [111].Rodríguez-Gonzalo E, Domínguez-Álvarez J, Mateos-Vivas M, García-Gómez D, Carabias-Martínez R, A validated method for the determination of nucleotides in infant formulas by capillary electrophoresis coupled to mass spectrometry, Electrophoresis. 35 (2014) 1677–1684. 10.1002/elps.201300550. [DOI] [PubMed] [Google Scholar]
- [112].Mateos-Vivas M, Rodríguez-Gonzalo E, Domínguez-Álvarez J, García-Gómez D, Ramírez-Bernabé R, Carabias-Martínez R, Analysis of free nucleotide monophosphates in human milk and effect of pasteurisation or high-pressure processing on their contents by capillary electrophoresis coupled to mass spectrometry, Food Chem. 174 (2015) 348–355. 10.1016/j.foodchem.2014.11.051. [DOI] [PubMed] [Google Scholar]
- [113].Schiffman SS, Sennewald K, Gagnon J, Comparison of taste qualities and thresholds of D- and L-amino acids, Physiol. Behav 27 (1981) 51–59. 10.1016/0031-9384(81)90298-5. [DOI] [PubMed] [Google Scholar]
- [114].Kawai M, Sekine-Hayakawa Y, Okiyama A, Ninomiya Y, Gustatory sensation of (L)- and (D)-amino acids in humans., Amino Acids. 43 (2012) 2349–2358. 10.1007/s00726-012-1315-x. [DOI] [PubMed] [Google Scholar]
- [115].Okada K, Gogami Y, Oikawa T, Principal component analysis of the relationship between the d-amino acid concentrations and the taste of the sake, Amino Acids. 44 (2013) 489–498. 10.1007/s00726-012-1359-y. [DOI] [PubMed] [Google Scholar]
- [116].Sardella R, Lisanti A, Marinozzi M, Ianni F, Natalini B, Blanch GP, Ruiz del Castillo ML, Combined monodimensional chromatographic approaches to monitor the presence of d-amino acids in cheese, Food Control. 34 (2013) 478–487. 10.1016/j.foodcont.2013.05.026. [DOI] [Google Scholar]
- [117].Chen Y, Chen L, Wang X, Xi Z, Wu Y, Fu F, DNA binding in combination with capillary electrophoresis and inductively coupled plasma mass spectrometry for the rapid speciation analysis of mercury, Sep. Sci. Plus 1 (2018) 52–58. 10.1002/sscp.201700015. [DOI] [Google Scholar]
- [118].Zhao Y, Zheng J, Yang M, Fu F, [Speciation analysis of arsenic in seaweeds by capillary electrophoresis-inductively coupled plasma mass spectrometry], Se Pu = Chinese J. Chromatogr 29 (2011) 111–114. 10.3724/sp.j.1123.2011.00111. [DOI] [PubMed] [Google Scholar]
- [119].Chen FR, Zheng L, Wang ZG, Sun J, Han LH, Wang XR, Determination of arsenic speciation in scomberomorus niphonius by capillary electrophoresis-inductively coupled plasma mass spectrometry, Guang Pu Xue Yu Guang Pu Fen Xi/Spectroscopy Spectr. Anal 34 (2014) 1675–1678. 10.3964/j.issn.1000-0593(2014)06-1675-04. [DOI] [PubMed] [Google Scholar]
- [120].Andersson MA, V Petersson Grawé K, Karlsson OM, Abramsson-Zetterberg LAG, Hellman BE, Evaluation of the potential genotoxicity of chromium picolinate in mammalian cells in vivo and in vitro, Food Chem. Toxicol 45 (2007) 1097–1106. 10.1016/j.fct.2006.11.008. [DOI] [PubMed] [Google Scholar]
- [121].Barceloux DG, Chromium., J. Toxicol. Clin. Toxicol 37 (1999) 173–194. 10.1081/clt-100102418. [DOI] [PubMed] [Google Scholar]
- [122].Qu H, Mudalige TK, Linder SW, Capillary electrophoresis/inductively-coupled plasma-mass spectrometry: Development and optimization of a high resolution analytical tool for the size-based characterization of nanomaterials in dietary supplements, Anal. Chem 86 (2014) 11620–11627. 10.1021/ac5025655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.