

Abstract
Post viral bacterial infections are a major healthcare challenge in coronavirus infections including COVID-19, however, the coronavirus-specific mechanisms of increased host susceptibility to secondary infections remain unknown. In humans, coronaviruses including SARS-CoV-2 infect lung immune cells including alveolar macrophages, a phenotype poorly replicated in mouse models of SARS-CoV-2. To overcome this, we used a mouse model of native murine β-coronavirus which infects both immune and structural cells to investigate coronavirus-enhanced susceptibility to bacterial infections. Our data show that coronavirus infection impairs the host ability to clear invading bacterial pathogen and potentiate lung tissue damage in mice. Mechanistically, coronavirus limits the bacterial killing ability of macrophages by impairing lysosomal acidification and fusion with engulfed bacteria. In addition, coronavirus-induced lysosomal dysfunction promotes pyroptotic cell death and the release of IL-1β. Inhibition of cathepsin B decreased cell death, IL-1β release, and promoted bacterial clearance in mice with post coronavirus bacterial infection.
Introduction:
The current COVID-19 pandemic is the third major disease outbreak caused by coronaviruses in the last 20 years, emphasizing the clinical relevance of coronaviruses (1–3). Mechanisms of coronavirus-mediated disease severity remain multifactorial and include old age, along with co-morbidities (1, 4). Post viral bacterial infections (bacterial superinfections) play a detrimental role in promoting the disease severity of COVID-19, and are a stronger predictor of death in COVID-19, compared to influenza infection (5). The presence of bacterial infections in COVID-19 is reported in approximately 14% of ICU patients in a meta-analysis (6), while a more recent study has shown its presence in approximately 50% of hospitalized patients (7), and as high as 100% in patients who succumb to COVID-19 (8). The presence of bacterial infections in such high numbers of patients despite extensive use of antibiotics indicates an impairment in host defense. Various mechanisms of host susceptibility to post viral bacterial infection have been proposed including impaired neutrophilic recruitment, impaired type 17 response, and decreased host tolerance (9–11), however, these studies are largely performed in animal models of influenza infections. Coronavirus-specific mechanisms that render the host susceptible to secondary bacterial infections are poorly understood.
β-coronaviruses including mouse hepatitis virus (MHV) and SARS-CoV-2, utilize a novel egress pathway that involves lysosomes to egress from infected cells (12). Given the central role of lysosomes in bacterial killing through phagocytic mechanisms (13, 14), we sought to determine how this unique aspect of the coronavirus life cycle affects the host’s ability to clear secondary bacterial infections. We used a mouse model of MHV lung infection, which shows acute lung pathology and systemic disease similar to the COVID-19 (15–18). Genetically modified mouse models of COVID-19, such as human ACE2 overexpressing mouse under K18 promoter (epithelium) (19), does not allow SARS-CoV-2 infection in macrophages, which is well described in COVID-19 patients (20, 21). The MHV model allowed us to investigate the post-coronaviral bacterial infection susceptibility in a natural host (mouse) and thus circumventing these limitations of mouse models of SARS-CoV-2 including infections to macrophages. Using this mouse model of post-coronaviral bacterial infection, we show that coronavirus impairs lysosomal acidification and fusion with bacteria in macrophages, leading to impaired bacterial killing. At the same time, coronavirus-mediated lysosomal swelling leads to lysosomal rupture and release of lysosomal enzymes, such as cathepsin B, rendering the immune cells susceptible to pyroptotic cell death and release of IL-1β during secondary bacterial infections.
Material and Methods:
Mice:
All experiments were performed as per approved protocols from Yale Animal Resource Center (2021–20044). Wild-type mice (C57/B6) were obtained from Jackson Laboratory and bred at Yale where they were maintained at a 12-hour dark/light cycle and ad libitum food and water supplies. Caspase1/11−/− mice were a kind gift from Dr. Jack Elias (Brown University).
Pathogens:
MHV was provided by Dr. Susan Compton (co-author) while PA-GFP was provided by Dr. Jonathan Audia (co-author). Influenza strain H3N2 was a kind gift of Dr. Adolfo García-Sastre. PA01 strain was provided by Dr. Barbara Kazmierczak at Yale University and Streptococcus strain was obtained from ATCC.
MHV Infection:
Mice were infected with 1 X 104 PFUs of MHV-A59 by intranasal route in 40 μl of PBS or received similar amounts of the vehicle as described previously (16).
Bacterial infection:
Mice were infected with either Pseudomonas aeruginosa (PA01 strain) or Streptococcus pneumoniae (TIGR4 strain) by intratracheal route as described previously (22, 23).
Alveolar macrophage isolation and infection:
Alveolar macrophages were obtained by lavaging naïve or MHV infected mice using antibiotic (Penicillin/Streptomycin) containing PBS to avoid any bacterial contamination. The cells were seeded in 96 well plates at 1 × 105 cells/well and allowed to adhere before either infecting with MHV (for naïve macrophages) and/or with bacteria.
Peritoneal macrophage isolation and infection:
Peritoneal macrophages were obtained from mice that were injected with 3% thioglycolate broth for 3 to 5 days. Macrophages were obtained by peritoneal lavage and then seeded into culture plates at desired concentrations.
In vitro post-viral bacterial infection:
Macrophages were cultured and infected with 0.01 MOI of MHV for 20 hours and followed by infection with either Pseudomonas or Streptococcus at MOI of 10 or 1 for various time points.
Lysotracker staining:
Lysotracker dye was diluted in phenol red free DMEM media and was incubated with the cells seeded either in a 12 -well plate for 15 mins before imaging or immediately before starting the kinetics of lysosomal acidification in a 96-well plate.
Immunofluorescence imaging:
Cells seeded into chamber slides were fixed using 2% normal buffered paraformaldehyde, permeabilized with 1% TritonX-100 and blocked with 1% BSA followed by incubation with anti-LAMP1 (10ug/ml) or anti-cathepsin B (1:800 dilution) antibodies overnight at 4-degree C. Cells were then incubated with fluorescence-labeled secondary antibody (1:1000 dilution) for 1 hour. The imaging was performed.
Reagents:
CA-074Me was purchased from ApexBio (Cat# A8239). Caspase-1 inhibitor Ac-YVAD-CMK was purchased from Cayman (Cat# 10014). Latex beads were purchased from Polysciences.com (Cat#17458–10). LysoTracker™ Red DND-99 was purchased from Thermo Scientific (Cat# L7528). Cathepsin B substrate Z-Arg-Arg-pNA was obtained from Sigma (Cat# SCP0108).
Cathepsin B inhibition:
For inhibition of cathepsin B, cells were treated with CA-074Me at 100 μM concentration 30 mins before PA or SP infection. For in vivo inhibition of cathepsin B, mice were injected with CA-074Me at 7-day post-MHV infection at 4 hours prior to and at the time of PA infection.
Western Blot and Enzyme-linked immunosorbent assay (ELISA):
Western blot and ELISA analysis were performed as described previously (22).
Phagocytosis assay:
Phagocytosis assay was performed using fluorescently labeled latex beads (1μm diameter) Fluoresbrite. Macrophages that were either infected with MHV or mock infection for 20 hours were incubated with these beads for an additional 1 hour at the MOI of 200. The numbers of beads per 100 cells were counted.
Bacterial Count:
Bacterial counts were performed as previously described by plating serially diluted BAL or lung homogenates (22, 23).
Broncho-alveolar lavage and tissue histology:
Mice were lavaged by injecting two aliquots of 750 ul of sterile PBS under anesthesia. Post lavaging, the left lung was either collected for bacterial counts in the lung tissue or was inflated with 0.5% low melting point agar and then fixed in 4% paraformaldehyde. The tissue was embedded, sectioned, and stained by hematoxylin and eosin by Yale Histology Core.
LDH Measurement and TUNEL staining:
LDH was measured in the phenol red free cell culture supernatants using the LDH detection kit and TUNEL staining was performed on lung tissue using In-situ Cell Death Detection kit (Roche) as per the instructions.
Propidium iodide staining:
20X dilution of propidium iodide and Hoechst stain were mixed and cells were imaged using fluorescence microscopy.
Electron Microscopy:
Cells were fixed in glutaraldehyde and sent to Yale Electron Microscopy Core.
Statistics:
All the experiments were performed at least twice to ensure reproducibility. Data were analyzed using Graph-pad prism software. The numbers of mice per group were determined based on our previous experience with coronavirus infections and bacterial infections where 5–6 mice per group were used. Data are either pooled from two or more experiments or presented as one of two or three experiments with similar outcomes. For comparing two groups, Student’s t-test was performed. For comparing multiple groups, one-way ANOVA was performed followed by Dunnett’s multiple comparisons. A p-value of <0.05 was considered statistically significant.
Results:
MHV infects and replicates within pulmonary immune cells:
As SARS-CoV-2 can infect and replicate within human lung immune cells, we sought to determine whether murine hepatitis virus (MHV) can infect lung immune cells. To confirm whether MHV infects the lung immune cells, we infected naïve alveolar macrophages with MHV (MOI of ~ 1) for 2 hours ex-vivo. The unbound MHV was washed off to remove the unbound virus and 300ul of fresh media was obtained. A 50 μl aliquot of this media was obtained at 0, 24, 48, and 72 hours to quantify MHV using a qPCR assay. Our data show that MHV levels significantly increased over time during the course of infection (Fig. 1A). To confirm the MHV infection of immune cells in vivo, we harvested lung immune cells by lavaging mock or MHV-infected mice at 3-day post-infection and performed immunostaining for MHV N-protein. Our data show the presence of MHV N protein in immune cells obtained from infected mice (Fig. 1A, Right panel), confirming the ability of MHV to infect the lung immune cells., further confirming the replication of MHV in lung immune cells.
Fig. 1. MHV infects immune cells and impairs bacterial clearance in Gram-positive and Gram-negative bacterial lung infections.
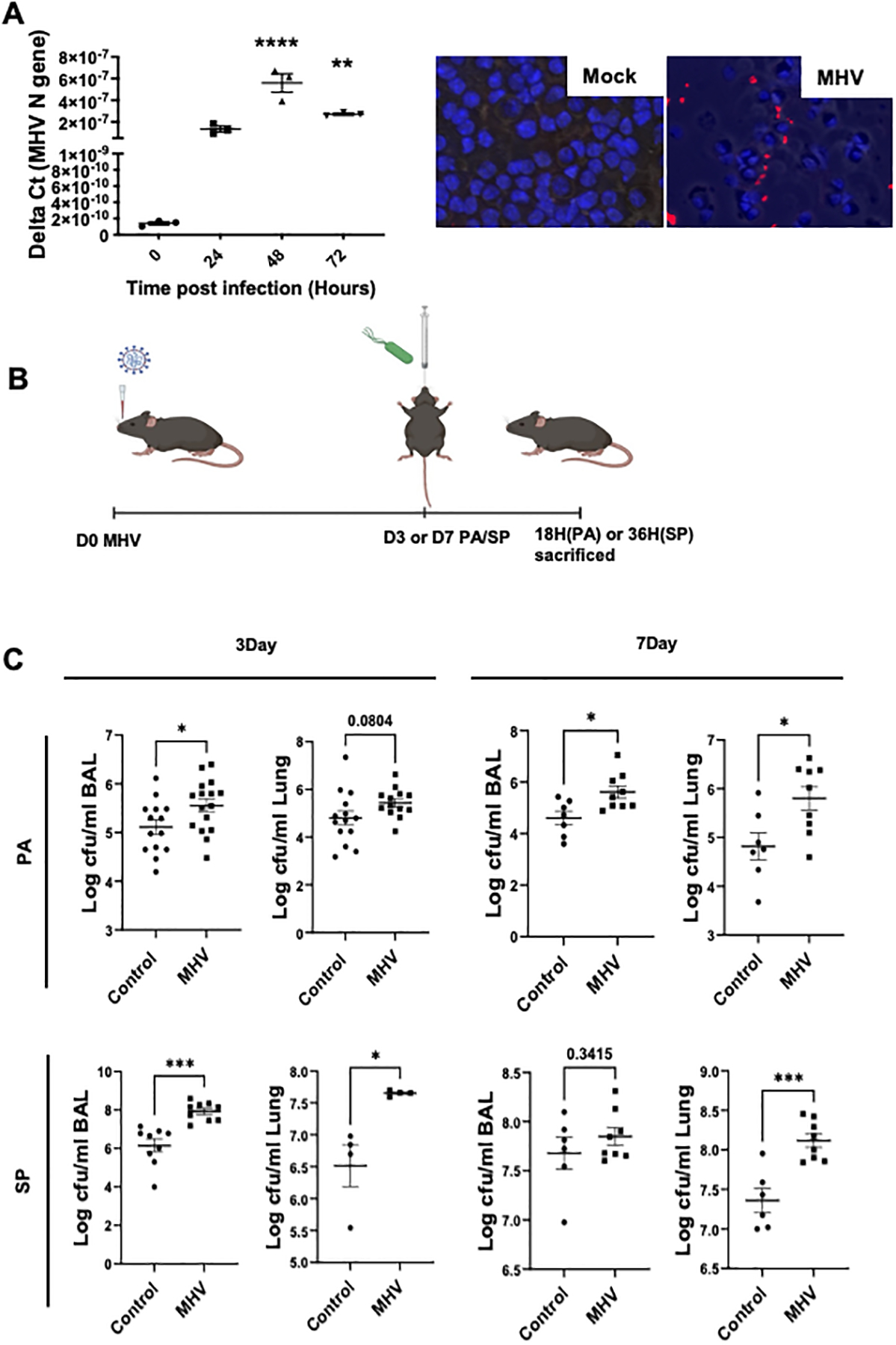
(A) Mouse alveolar macrophages were obtained by lavaging naïve mice and cells were seeded at 1 × 105 cells/well in 96 well plate. Cells were then infected with MHV (MOI of 10) for 2 hours and washed twice to remove the residual viruses and replenished with 300 μl of fresh media. A 50 μl aliquot was obtained at 0, 24, 48, and 72 hours and RNA was purified to run qPCR using primers for the MHV N gene. Data are from one of two independent experiments with similar outcomes and represented as Delta Ct values (A, left). To confirm that MHV infects lung immune cells in vivo, we stained the BAL cells obtained from either mock or MHV-infected mice on day 3. Representative images are shown (A, right) from two independent experiments. Red- MHV N protein, blue-DAPI. (B) Mouse model of post coronaviral bacterial infection. Mice infected with MHV (intranasal) for 3 or 7 days were superinfected with Pseudomonas aeruginosa (PA, 2.5 × 106 CFUs) or Streptococcus pneumoniae (SP, 1 × 104 CFUs) through the intratracheal route and then euthanized at 18 hours (PA) or 36 hours (SP) post-bacterial infection. (C) Bacterial load in the BAL and lung tissue (left lobe) was measured. Data are pooled from two or three independent experiments. *, P < 0.05, **, P < 0.01, ***, P < 0.005, ****, P < 0.0001 using either t-test or one-way ANOVA followed by Dunnett’s multiple comparisons test, as appropriate.
MHV infection impairs bacterial clearance in mice:
To determine the impact of coronavirus infection on host immunity against secondary bacterial infections, mice were infected with a sublethal dose of MHVA59 (104 PFUs/mouse by intranasal route) or vehicle and then infected with Pseudomonas aeruginosa (PA, 2.5 × 106 CFUs/mouse) or Streptococcus Pneumoniae (SP, 1 × 104 CFUs/mouse), at either 3- or 7-days post-MHV infection (Fig. 2A). Our data reveal that MHV-infected mice had significantly elevated bacterial load in broncho-alveolar lavage (BAL) and lung tissue compared to mice with mock viral infection (Fig. 2B). The SP infected mouse BAL on day 7 post-MHV had larger variation due to extensive lung consolidation that did not allow us to properly lavage, especially in the superinfection group. This impaired ability of mice to clear bacterial pathogen after coronavirus was not associated with impaired inflammatory cell recruitment. At most of the time points, we observed an increase in the inflammatory cell recruitment in the BAL (Supplemental Fig. 1A & B). These immune cells were largely composed of neutrophils and no significant differences were observed in macrophage numbers (Supplemental Fig. C &D). Additionally, we found that levels of neutrophil chemoattractants including KC and MIP-2 were not decreased during bacterial infection post-coronaviral infection (Supplemental Fig.1E & F). These data suggest that coronavirus impairs the host ability to clear a secondary bacterial infection independent of the inflammatory response.
Fig. 2. MHV exacerbates lung injury during secondary bacterial infections.
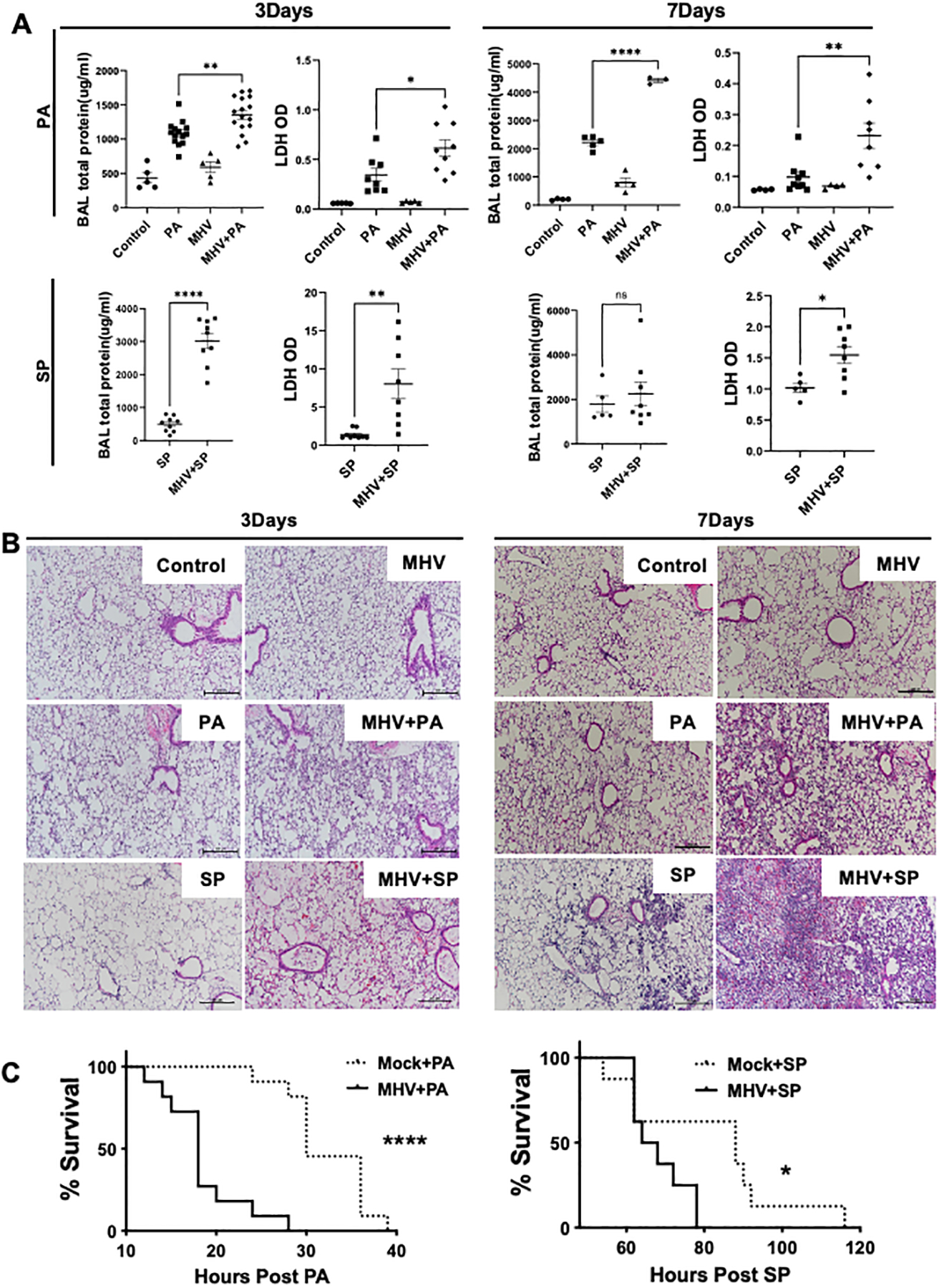
(A) Total protein content and LDH levels in the BAL samples of mice infected with PA or SP at day 3 or 7 post-MHV infection and their respective histological analyses (B) indicated increased tissue injury in the superinfection group. Mouse survival was measured to compare the effect of MHV infection on host survival post-PA or SP infections (C). Data are pooled from two independent experiments for each survival curve. N = 11 each group for PA and 8–9 for SP. *, P <0.05, **, P < 0.01, ****, and P < 0.001 using one-way ANOVA followed by Dunnett’s multiple comparisons test. Survival curves were compared using Log-rank (Mantel-Cox) test.
Increased lung damage in mice with superinfection:
Next, we sought to determine whether the increased bacterial burden in MHV-infected mice leads to an elevated lung injury. Total protein content in the BAL, a marker of capillary permeability, and lactate dehydrogenase (LDH) release, a marker of cell death, was measured in the BAL samples of mice that were infected with a bacterial pathogen in the presence or absence of MHV pre-infection. Our data show that mice infected with either PA or SP post-viral infection had significantly elevated protein and lactate dehydrogenase (LDH) in their BAL compared to only bacteria or virus-infected mice (Fig. 2A). These data were corroborated by histological analysis of lung tissue (Fig. 2B). To further confirm the cell death data as indicated by elevated LHD levels, we performed the TUNEL staining of the lung tissue, which showed a dramatic increase in the TUNEL-positive cells in the post-coronaviral bacterial infection group compared to either viral or bacterial infections (Supplemental Fig. 1G & H). To assess the consequences of elevated injury in mice, we measured the host survival post bacterial infection in the presence or absence of a prior MHV infection. Our data show that MHV infection significantly impaired the host survival in PA infection (Fig. 2C). To determine whether this increased lung injury is due to the enhanced bacterial burden, we investigated the inflammatory response and tissue injury in a post-coronaviral lipopolysaccharide (LPS) model to eliminate any effects of differential pathogen burden. Our data show that MHV infection exacerbated LPS-induced inflammation and tissue injury as evident by an increased inflammatory response, increased BAL total protein content, and LDH levels (Supplemental Fig. 1I–K). These data show that coronavirus exacerbates tissue injury during secondary bacterial infections independent of impaired bacterial clearance.
Coronavirus impairs the bacterial killing ability of macrophages by causing lysosomal dysfunction:
To decipher the mechanism of coronavirus-impaired pathogen clearance, we investigated the bacterial killing ability of macrophages, a key immune cell type that is both resident to the lung tissue and recruited during infection. Peritoneal macrophages infected with MHV for 24 hours followed by a PA or SP infection show that MHV infection significantly impaired the bacterial killing ability of these cells (Fig. 3A). Similar findings were observed in a macrophage cell line RAW264.7 cells (Supplemental Fig. 2A). To further investigate the mechanism of coronavirus-impaired bacterial killing in macrophages, we measured the phagocytic ability and found no decrease in the overall phagocytic ability of macrophages post MHV infection (Supplemental Fig. 2B). These data suggest an impairment in the bacterial killing ability of macrophages independent of phagocytic mechanisms. Next, we evaluated macrophage lysosomal acidification and fusion with bacterial pathogens. Our data show that mock-infected cells upregulated lysosomal acidification in response to PA or SP as detected by the lysotracker which gives red fluorescence in the acidic compartment (Fig. 3B & C). MHV caused lysosomal deacidification and lowered bacteria-induced lysosomal acidification. We also observed impaired fusion of acidified lysosomes with a bacterial pathogen using confocal microscopy and green fluorescence-expressing PA (Fig. 3D). As lysotracker can only visualize acidified lysosomes, we performed immunostaining using LAMP1 antibody to visualize the total lysosomal compartment in the cells to show that MHV infection led to significantly increased visibility of LAMP1-coated enlarged vesicles that did not contain lysosomal enzyme cathepsin B (Fig. 3E), potentially indicating lysosomal swelling and rupture; releasing the lysosomal enzymes.
Fig. 3. MHV-induced lysosomal dysfunction impairs the bacterial killing ability of macrophages.
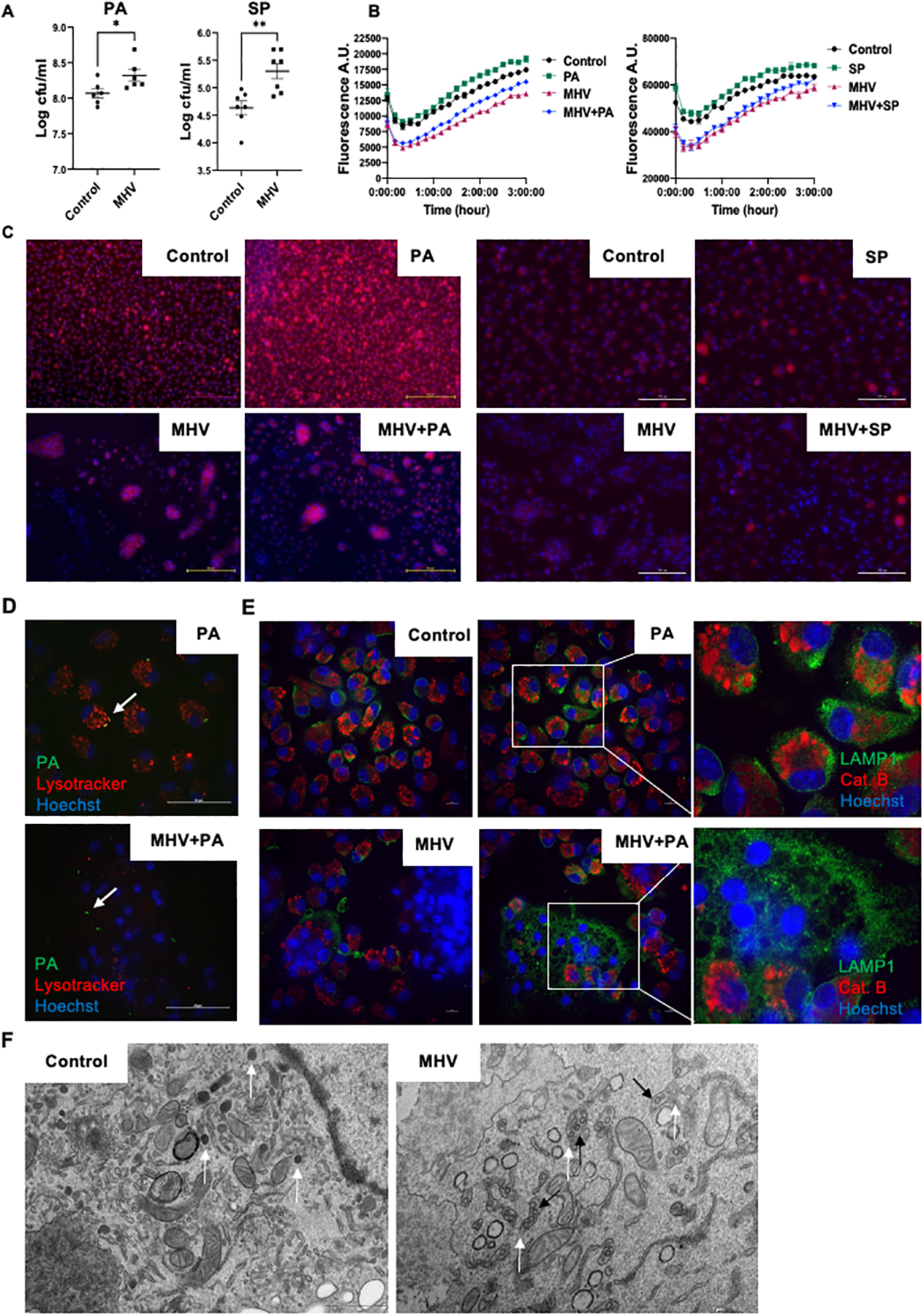
(A) Peritoneal macrophages were infected with MHV for 24 hours and then superinfected with a bacterial pathogen for 6 hours. Surviving bacteria were enumerated. (B) Lysosomal acidification of different groups of cells was measured using lysotracker for 3 hours post PA or SP infection. Cells were infected with MHV or vehicle for 20 hours before PA or SP infection according to different groups. (C) Representative images of acidified lysosomes detected by lysotracker after treatment of peritoneal macrophages with PA/SP, MHV, MHV+PA/SP or vehicle; red = lysotracker and blue = nucleus (Hoechst) at 2 hours. (D) Confocal microscopic analysis of phagocytosis and acidified lysosomal fusion in the presence or absence of MHV in the peritoneal macrophages. PA-GFP were (MOI of 20) used to infect the cells and lysosomes were stained using lysotracker (red). Blue = Hoechst. Pictures were taken at 2 hours post bacterial infection. Arrow points to fused acidified lysosomes and bacteria in the PA group while only bacteria in the MHV+PA group. Scale bar = 50 μm. (E) Confocal microscopic analysis of lysosomal enzyme release from peritoneal macrophages treated with PA, MHV, MHV+PA, or vehicle at 2 hours post bacterial infection. LAMP1 was stained in green as a lysosomal marker; cathepsin B was stained in red while blue indicates nucleus. (F) Transmission electron microscopic analysis of lysosomal structure in presence or absence of MHV infection at 20 hours post-MHV infection. Lysosomes are identified by their distinct morphology and dark color due to lysosomal enzymes. White arrows indicate normal lysosomal structures in the control cells and swollen lysosomes like vesicles in MHV infected cells. Black arrows indicate coronavirus in these vesicles. A.U. = arbitrary unit, Scale bar = 10 μm. *, P <0.05, **, P < 0.01 using t-test.
To confirm if lysosomes were indeed ruptured by MHV infection, we performed transmission electron microscopy to visualize lysosomal ultrastructure. Our data show that lysosomes appeared as dark, small vesicles in uninfected macrophages where the dark color is due to dense lysosomal enzymes. In contrast, MHV-infected cells had large lysosome-like vesicles that show signs of membrane rupture, and their faint color indicated the escape of lysosomal enzymes. Of significance, we found the presence of viral particles in these lysosome-like vesicles (Fig. 3F), which is in agreement with recent findings demonstrating that coronaviruses utilize lysosomes to egress from infected cells (12). The normal lysosomal structure was absent in the infected cells. Together, these data indicate that coronavirus impairs lysosomal function at multiple levels to cause lysosomal swelling, rupture, and release of lysosomal enzymes.
Lysosomal damage promotes inflammasome activation and pyroptotic cell death to impair host immunity and exacerbate tissue damage:
Next, we sought to determine the mechanisms of enhanced tissue injury in post-coronaviral bacterial infections. First, we determined the mode of cell death in the lung tissue that was exacerbated in post-coronaviral bacterial infection. We observed activation of various types of cell death pathways in the lungs of mice that were infected either by MHV or bacterial pathogen including markers of apoptosis (Caspase-3), pyroptosis (Caspase-1), and necroptosis (RIP-3 and MLKL). However, only cleaved caspase-1 levels show a further increase in superinfection compared to single infections (Fig. 4A), indicating exacerbation of the inflammasome/pyroptotic pathway in mediating cell death. We confirmed exacerbated inflammasome activation during post-coronaviral bacterial infections by measuring IL-1β levels in the BAL of mice infected with either PA or SP post-MHV infection at both day 3 and day 7-time points (Fig. 4B). We also show that MHV exacerbated cell death caused by both PA and SP infections in macrophages in vitro (Fig. 4C). Similar observations were made using LPS stimulation, where increased cell death and IL-1β release was observed after MHV infection, indicating the independence of these effects from bacterial burden (Supplemental Fig. 3 A–C). Unlike MHV, influenza infection did not exacerbate cell death during secondary bacterial infection (Supplemental Fig. 3D & E). To determine whether pyroptosis is a key mediator of cell death during post-coronaviral bacterial infection, we utilized caspase-1 deficient peritoneal macrophages. We observed that Caspase1−/− macrophages were significantly protected from coronavirus-exacerbated cell death during bacterial infections by PI staining and LDH release (Fig. 4C & D) in both PA and SP infections. The role of caspase-1 in mediating post-coronaviral bacterial infection-induced cell death was confirmed using a pharmacological inhibitor of caspase-1 (Supplemental Fig. 4A & B). Further, we investigated whether increased inflammasome signaling is triggered by lysosomal damage and the release of lysosomal enzymes. We specifically focused on cathepsin B; a prominent lysosomal enzyme that has been shown to play a role in the inflammasome activation (24). Our data show that inhibition of lysosomal enzyme cathepsin B prevented cell death caused by a post-coronaviral bacterial infection in the peritoneal macrophages as indicated by decreased PI staining and reduced levels of LDH in cell supernatants (Fig. 4C & D). As expected, CA-074Me treatment significantly attenuated MHV-induced cathepsin B activity during PA infection in macrophage supernatants (Supplemental Fig. 4C). Further, we confirmed that decreased cell death by cathepsin B inhibitor was due to a decrease in the inflammasome pathway activation, as indicated by decreased IL-1β release (Fig. 4E). We also confirmed whether a similar phenotype was observed in the pulmonary immune cells. To determine this, we obtained lung immune cells from the BAL of either PBS or MHV-infected mice on day 3 post-infection. These cells were treated with either DMSO (vehicle) or CA-074Me for 1 hour before infecting them with PA (MOI of 10). Our data show that MHV exacerbated cell death which was ameliorated by CA-074Me treatment (Fig.4F).
Fig. 4. Pyroptosis is a key pathological cell death mechanism potentiated by MHV in presence of bacterial superinfection.
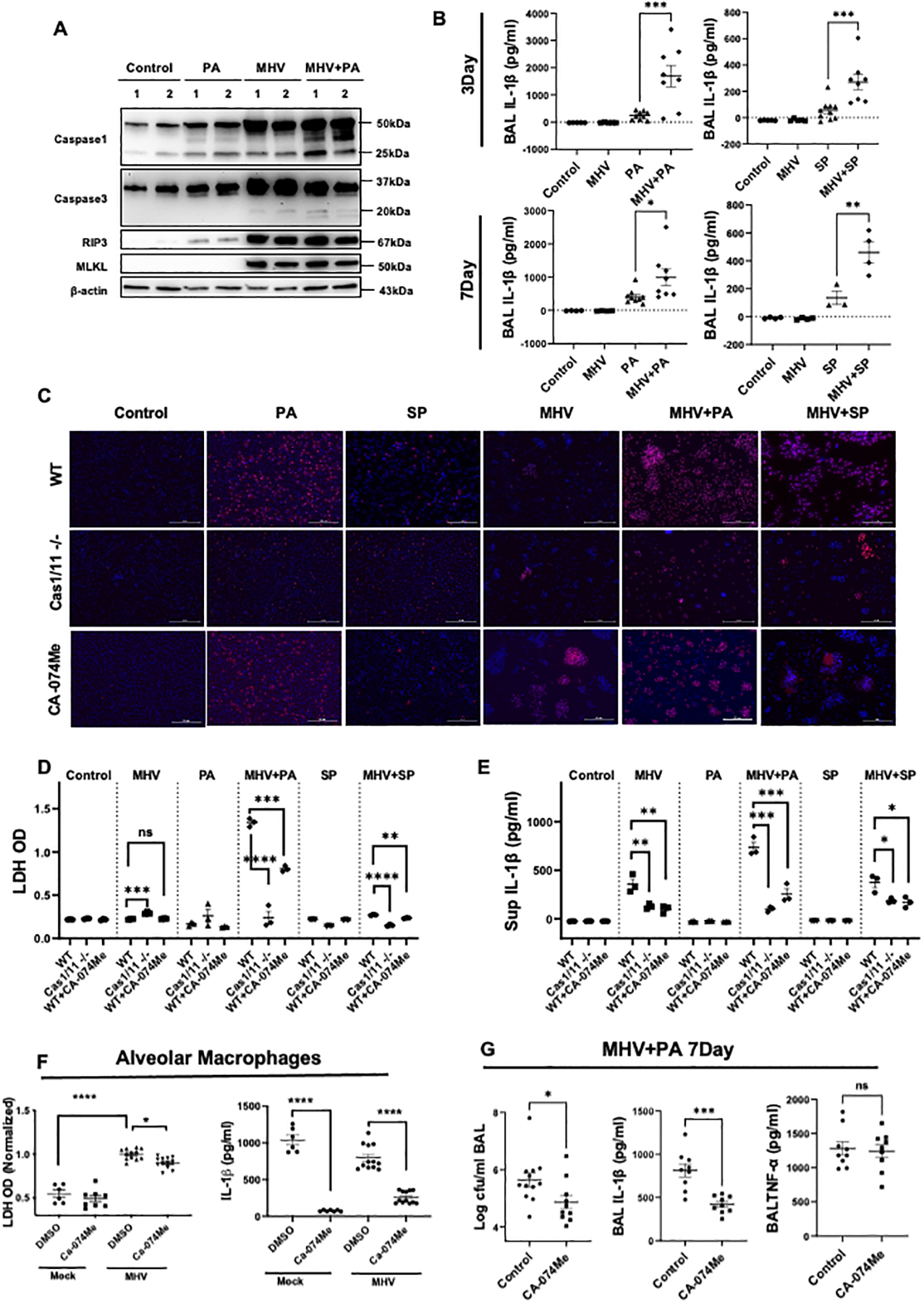
(A) Western Blot analysis to detect markers of cell death pathways in the lung tissue, including pyroptosis (Caspase-1), apoptosis (caspase-3), and necroptosis (RIP3 and MLKL) activation in lung lysates during PA infection post-MHV infection. (B) Levels of IL-1β in BAL samples from mice with bacterial infection with or without MHV. Data are pooled from two experiments. (C) Representative pictures of PI staining showing cell death in wild-type peritoneal macrophages treated with or without CA-074Me and caspase1/11−/− peritoneal macrophages in response to MHV+PA. MHV-infected cells appear to be larger given the formation of syncytia. Experiments were repeated 3 times and data from a representative experiment is shown. Scale bars are 100 μm. (D & E) The quantification of LDH and IL1-β in cell supernatants from (C). (F) LDH and IL-1β levels were also measured in the alveolar macrophages obtained from either mock or MHV infected mice that were treated with either vehicle or CA-074Me 30 minutes prior to PA infection (MOI of 10). (G) Bacterial load, IL-1β, and TNF-α in BAL from superinfected mice with the treatment of CA-074Me or vehicle were measured. Data are pooled from two independent experiments. PI = red and blue = nuclei. *, P <0.05, **, P < 0.01 ***, P <0.001 using either t-test or one-way ANOVA followed by Dunnett’s multiple comparisons test, as appropriate.
Finally, we determined whether inhibition of lysosomal enzyme cathepsin B can provide an advantage in an in vivo model of post-coronaviral bacterial infection, by either promoting bacterial clearance or limiting pyroptotic cell death. Our data show that inhibition of cathepsin B using in vivo bioavailable drug CA-074Me improved bacterial clearance by the host in the post-MHV PA infection model. Mice treated with CA-074Me had significantly decreased BAL bacterial burden along with a twofold decrease in the IL-1β levels in the BAL (Fig. 4G), but not TNFα levels (Fig. 4G), indicating a specific impact on the inflammasome pathway. Taken together, these data indicate that lysosomal dysfunction impairs the bacterial killing ability of the host and promotes tissue damage through pyroptotic cell death.
Discussion:
Despite the extensive presence of secondary bacterial infections in severe COVID-19, mechanistic studies investigating how coronaviruses contribute to impaired host immunity during secondary bacterial infections remain limited. In this study, using a murine β-coronavirus infection, which has native infectivity to mice, we shed light on coronavirus-mediated lysosomal dysfunction as a key contributor to both impaired host defense and exacerbated tissue injury during secondary bacterial infection.
Our study shows that coronavirus-induced lysosomal dysfunction is multifactorial and includes deacidification, swelling, and rupture, leading to the release of lysosomal components. This dysfunction does not only impair the ability to clear invading pathogens but renders the cell susceptible to pyroptotic cell death. Pyroptosis is a known cell death pathway that is associated with exacerbated inflammation and inflammatory tissue injury in multiple disease models (25, 26). In the model of post-coronaviral bacterial infection, we show that pyroptosis and excessive IL-1β release was associated with cell death and extensive tissue injury in in vitro and in vivo models of post-coronaviral bacterial infection. Interestingly, targeting lysosomal enzyme cathepsin B was sufficient to block IL-1β release and cell death in macrophages.
Lysosomes are well-known for their highly concentrated acidic enzymes, which upon release have the potential to kill the cell (27–29). These enzymes are instrumental in digesting cellular debris, damaged organelles, as well as pathogenic bacteria that are ingested by phagocytic mechanisms. MHV, through its unique life cycle, which is shared with other β-coronaviruses such as SARS-CoV-2 (12), manipulates the lysosomal compartments to render them ineffective in their bacterial killing ability, while causing cell death. The mechanism by which lysosomal enzymes lead to inflammasome activation remains unclear, however, a previous study has shown that cathepsin B activates inflammasome through the NLRP3 adapter molecule (24). Inhibition of cathepsin B was sufficient to decrease IL-1β release in vivo and in vitro indicating a direct role of cathepsin B in promoting pyroptotic cell death during post-coronaviral bacterial infection.
In conclusion, we show that murine β-coronavirus causes lysosomal dysfunction and the release of lysosomal enzymes, which impair the host’s ability to clear secondary bacterial pathogens and render the host susceptible to exacerbated tissue injury during bacterial superinfection. Minimizing the deleterious effect of lysosomal damage by therapeutic targeting of lysosomal enzymes by CA-074Me can limit post-coronaviral susceptibility to secondary infection.
Limitations:
In this study we used a murine pathogen MHV, which does not infect humans, to investigate the host susceptibility to secondary bacterial infections, limiting its direct translation to the human coronavirus infections including COVID-19. However, MHV causes both lung and systemic disease, as manifested in COVID-19, and can cause lethality in a dose-dependent manner (16, 18, 30, 31). MHV model also recapitulated the essential role of type I interferons during early infection (16), which has been demonstrated in COVID-19 (21, 32), and in a hamster model, which are naturally susceptible to SARS-CoV-2 infections (33). In contrast, a mouse model of ACE2 overexpression by adeno-associated virus did not show beneficial roles of type I interferons, indicating limitations of studying pathogens outside their natural host (34). A humanized mouse model containing human immune cells will be a more relevant model of SARS-CoV-2 to study immune function in secondary bacterial infections (35). In addition, our study did not investigate whether systemic infection of MHV, especially that of the liver (16), has any effect on the pulmonary host defense against secondary bacterial infections. Further, our study was performed on only one mouse strain (C57/Bl6) and the applicability of these findings to other mouse strains remains unknown. However, we observed impaired ability to kill bacteria post-coronaviral infection in RAW264.7 cell line generated from BALB/c mice, indicating similar mechanisms may exist in other mouse strains. Despite these limitations, our study shows a key mechanism by which a model β-coronavirus affects the host’s ability to fight off bacterial infections.
Supplementary Material
Key Points:
Coronavirus impairs host immunity against secondary bacterial infections.
Coronavirus-induced lysosomal dysfunction impairs bacterial killing.
Targeting lysosomal enzyme cathepsin B limits pyroptotic cell death.
Acknowledgments:
We want to thank Dr. Adolfo García-Sastre for providing the influenza H3N2 strain.
Funding:
Lokesh Sharma is supported by a Parker B. Francis Foundation award and a Catalyst Award from the American Lung Association. Charles Dela Cruz is supported by NIH grant HL126094, VA BX004661, Department of Defense.
References:
- 1.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, and Xiong Y. 2020. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. Jama 323: 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang D, Lin M, Wei L, Xie L, Zhu G, Cruz CSD, and Sharma L. 2020. Epidemiologic and clinical characteristics of novel coronavirus infections involving 13 patients outside Wuhan, China. Jama 323: 1092–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guan W. j., Ni Z.-y., Hu Y, Liang W.-h., Ou C.-q., He J.-x., Liu L, Shan H, Lei C.-l., and Hui DS. 2020. Clinical characteristics of coronavirus disease 2019 in China. New England journal of medicine 382: 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan W. j., Liang W.-h., Zhao Y, Liang H.-r., Chen Z.-s., Li Y.-m., Liu X.-q., Chen R.-c., Tang C.-l., and Wang T. 2020. Comorbidity and its impact on 1590 patients with Covid-19 in China: A Nationwide Analysis. European Respiratory Journal 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shafran N, Shafran I, Ben-Zvi H, Sofer S, Sheena L, Krause I, Shlomai A, Goldberg E, and Sklan EH. 2021. Secondary bacterial infection in COVID-19 patients is a stronger predictor for death compared to influenza patients. Scientific reports 11: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lansbury L, Lim B, Baskaran V, and Lim WS. 2020. Co-infections in people with COVID-19: a systematic review and meta-analysis. Journal of Infection 81: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cataño-Correa JC, Cardona-Arias JA, Porras Mancilla JP, and García MT. 2021. Bacterial superinfection in adults with COVID-19 hospitalized in two clinics in Medellín-Colombia, 2020. Plos one 16: e0254671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharifipour E, Shams S, Esmkhani M, Khodadadi J, Fotouhi-Ardakani R, Koohpaei A, Doosti Z, and Golzari SE. 2020. Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC infectious diseases 20: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, and Deng JC. 2009. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. The Journal of clinical investigation 119: 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, Di YP, Clay ME, Enelow RI, and Dubin PJ. 2015. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. American Journal of Physiology-Lung Cellular and Molecular Physiology 309: L158–L167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, and Medzhitov R. 2013. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 340: 1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, Takvorian PM, Bleck C, Hsu VW, and Fehr AR. 2020. β-Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183: 1520–1535. e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorne K, Oliver R, and Barrett A. 1976. Lysis and killing of bacteria by lysosomal proteinases. Infection and Immunity 14: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeya H, and Spitznagel J. 1966. Cationic proteins of polymorphonuclear leukocyte lysosomes II. Composition, properties, and mechanism of antibacterial action. Journal of bacteriology 91: 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W, Kandula S, Huynh M, Greene SK, Van Wye G, Li W, Chan HT, McGibbon E, Yeung A, and Olson D. 2021. Estimating the infection-fatality risk of SARS-CoV-2 in New York City during the spring 2020 pandemic wave: a model-based analysis. The Lancet Infectious Diseases 21: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma L, Peng X, Qing H, Hilliard BK, Kim J, Swaminathan A, Tian J, Israni-Winger K, Zhang C, and Habet V. 2021. Distinct Roles of Type I and Type III Interferons During a Native Murine β Coronavirus Lung Infection. Journal of Virology: JVI. 01241–01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qing H, Sharma L, Hilliard BK, Peng X, Swaminathan A, Tian J, Israni-Winger K, Zhang C, Leao D, and Ryu S. 2020. Type I Interferon Limits Viral Dissemination-Driven Clinical Heterogeneity in a Native Murine Betacoronavirus Model of COVID-19. bioRxiv. [Google Scholar]
- 18.Andrade A. C. d. S. P., Campolina-Silva GH, Queiroz-Junior CM, de Oliveira LC, Lacerda L. d. S. B., Gaggino JCP, de Souza FRO, de Meira Chaves I, Passos IB, and Teixeira DC. 2021. A Biosafety Level 2 Mouse Model for Studying Betacoronavirus-Induced Acute Lung Damage and Systemic Manifestations. Journal of Virology 95: e01276–01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, Fox JM, Chen RE, Earnest JT, and Keeler SP. 2020. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nature immunology 21: 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, Abbott DA, Donnelly HK, Donayre A, and Goldberg IA. 2021. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 590: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida M, Worlock KB, Huang N, Lindeboom RGH, Butler CR, Kumasaka N, Conde CD, Mamanova L, Bolt L, Richardson L, Polanski K, Madissoon E, Barnes JL, Allen-Hyttinen J, Kilich E, Jones BC, de Wilton A, Wilbrey-Clark A, Sungnak W, Pett JP, Weller J, Prigmore E, Yung H, Mehta P, Saleh A, Saigal A, Chu V, Cohen JM, Cane C, Iordanidou A, Shibuya S, Reuschl AK, Herczeg IT, Argento AC, Wunderink RG, Smith SB, Poor TA, Gao CA, Dematte JE, Reynolds G, Haniffa M, Bowyer GS, Coates M, Clatworthy MR, Calero-Nieto FJ, Göttgens B, O’Callaghan C, Sebire NJ, Jolly C, de Coppi P, Smith CM, Misharin AV, Janes SM, Teichmann SA, Nikolić MZ, and Meyer KB. 2021. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma L, Amick AK, Vasudevan S, Lee SW, Marion CR, Liu W, Brady V, Losier A, Bermejo SD, and Britto CJ. 2018. Regulation and role of chitotriosidase during lung infection with Klebsiella pneumoniae. The Journal of Immunology 201: 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruz CSD, Liu W, He CH, Jacoby A, Gornitzky A, Ma B, Flavell R, Lee CG, and Elias JA. 2012. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell host & microbe 12: 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevriaux A, Pilot T, Derangère V, Simonin H, Martine P, Chalmin F, Ghiringhelli F, and Rébé C. 2020. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Frontiers in cell and developmental biology 8: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergsbaken T, Fink SL, and Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nature Reviews Microbiology 7: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yap JK, Moriyama M, and Iwasaki A. 2020. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. The Journal of Immunology 205: 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Gómez‐Sintes R, and Boya P. 2018. Lysosomal membrane permeabilization and cell death. Traffic 19: 918–931. [DOI] [PubMed] [Google Scholar]
- 28.Guicciardi ME, Leist M, and Gores GJ. 2004. Lysosomes in cell death. Oncogene 23: 2881–2890. [DOI] [PubMed] [Google Scholar]
- 29.Brojatsch J, Lima H Jr, Palliser D, Jacobson LS, Muehlbauer SM, Furtado R, Goldman DL, Lisanti MP, and Chandran K. 2015. Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lysosome-destabilizing agents. Cell cycle 14: 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss SR 2020. Forty years with coronaviruses. The Journal of experimental medicine 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, and Weiss SR. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. Journal of virology 80: 5768–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, and Chenevier-Gobeaux C. 2020. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bessière P, Wasniewsk M, Picard-Meyer E, Servat A, Figueroa T, Foret-Lucas C, Coggon A, Lesellier S, Boué F, and Cebron N. 2021. Intranasal type I interferon treatment is beneficial only when administered before clinical signs onset in the SARS-CoV-2 hamster model. Biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, Alfajaro MM, Wei J, Dong H, Homer RJ, Ring A, Wilen CB, and Iwasaki A. 2020. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sefik E, Israelow B, Mirza H, Zhao J, Qu R, Kaffe E, Song E, Halene S, Meffre E, and Kluger Y. 2021. A humanized mouse model of chronic COVID-19. Nature biotechnology: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.