

Abstract
Genome-wide association studies (GWAS) have identified more than 75 genetic variants associated with Alzheimer’s disease (ad). However, how these variants function and impact protein expression in brain regions remain elusive. Large-scale proteomic datasets of ad postmortem brain tissues have become available recently. In this study, we used these datasets to investigate brain region-specific molecular pathways underlying ad pathogenesis and explore their potential drug targets. We applied our new network-based tool, Edge-Weighted Dense Module Search of GWAS (EW_dmGWAS), to integrate ad GWAS statistics of 472 868 individuals with proteomic profiles from two brain regions from two large-scale ad cohorts [parahippocampal gyrus (PHG), sample size n = 190; dorsolateral prefrontal cortex (DLPFC), n = 192]. The resulting network modules were evaluated using a scale-free network index, followed by a cross-region consistency evaluation. Our EW_dmGWAS analyses prioritized 52 top module genes (TMGs) specific in PHG and 58 TMGs in DLPFC, of which four genes (CLU, PICALM, PRRC2A and NDUFS3) overlapped. Those four genes were significantly associated with ad (GWAS gene-level false discovery rate < 0.05). To explore the impact of these genetic components on TMGs, we further examined their differentially co-expressed genes at the proteomic level and compared them with investigational drug targets. We pinpointed three potential drug target genes, APP, SNCA and VCAM1, specifically in PHG. Gene set enrichment analyses of TMGs in PHG and DLPFC revealed region-specific biological processes, tissue-cell type signatures and enriched drug signatures, suggesting potential region-specific drug repurposing targets for ad.
Introduction
Alzheimer’s disease (ad) has surfaced as one of the most significant health perils of old age, with nearly half of the adults over 85 years old in the US affected with ad. Accordingly, ad is becoming one of the major health and economic hazards (1,2). It has been well established that many mechanisms are involved in ad etiology (3). However, beyond the accumulation of amyloid-β plaques and tangles, ad pathophysiology is still largely unknown. Furthermore, only one drug, aducanumab, focusing on amyloid-β plaque clearance, has been approved for ad treatment (4,5), although its beneficial effect on cognitive function remains controversial. More studies are needed to understand the complex mechanism underlying ad pathophysiology, as it involves genetic factors and multiple brain regions (6–8).
On the other hand, several large-scale ad genome-wide association studies (GWAS) with millions of case and control samples have identified more than 75 genetic susceptibility variants across different ancestries (9–12). However, it remains unclear where and how the identified GWAS variants manifest their conjunctive effects on ad pathophysiology at the transcriptomic and proteomic levels. Previous transcriptome analyses have highlighted upregulated neuroinflammation, downregulated neuronal functions and other alterations associated with ad (13–15). Critically, the transcriptome data might not fully represent the proteome-level expression in brain tissue because of the complex post-transcriptional regulations, such as translational, post-transcriptional regulation and protein degradation (16,17). An increasing amount of high-throughput proteomics datasets has been generated from brain samples of ad individuals and healthy controls using the latest tandem mass tag (TMT) method (18). Analyses using those proteomics datasets generated from different brain regions allow us to gain insight into ad mechanisms with a higher resolution. This is crucial because proteins serve as better drug targets than genes, and, at the same time, drugs that target proteins with genetic support have an increased odds of success in clinical trials (19,20). In this way, studies that search for altered proteins with a solid genetic basis are particularly important for understanding ad pathophysiologies and discovering novel and repurposing drug targets.
Previously, we successfully applied our network-based analysis algorithm, Edge-Weighted dense module search for Genome-Wide Association Studies (EW_dmGWAS), to integrate multi-omics data to reveal dense modules of genes associated with complex traits (21,22). For example, we applied EW_dmGWAS on multiple sclerosis GWAS and brain tissue transcriptome data and pinpointed validated target genes with potential druggability (23). Our EW_dmGWAS algorithm outperforms traditional analysis in identifying disease-associated subnetworks (21,23,24). The networks depicted by EW_dmGWAS are harmonized with both genetic variants data (GWAS) and transcriptomic profiles on the human protein–protein interactome (21,23,24). Most importantly, these subnetworks contain the genes with well-known signals from one layer of omics data and synergistic effects from the multi-omics data. However, transcriptomes cannot capture the post-transcriptional activity, and different brain regions might be involved in different pathophysiological mechanisms (25). Hence, there is a pressing need to uncover the novel mechanisms associated with ad through the integrative analysis of the proteomic profiles and genetic data at the brain region level.
To address this gap, we aimed to extend the EW_dmGWAS to integrate ad GWAS summary statistics and brain protein expression profiles from the two large ad cohorts to unveil the common and brain region-specific dense gene modules associated with ad (26,27). Specifically, two protein expression datasets were obtained from the parahippocampal gyrus (PHG) and the dorsolateral prefrontal cortex (DLPFC), and were analyzed as individual discovery datasets. Then, we reconstructed the gene networks from the top-ranked ad modules in each brain region, followed by network evaluations. Additionally, we highlighted the potential drug-targetable genes based on brain region-specific co-expression profiles and current investigational drugs. Moreover, we conducted functional enrichment analyses to prioritize key ad-associated biological functions, drug signatures and cell-type signatures in the brain.
Results
Gene network modules identified by EW_dmGWAS calculations
We identified brain region-specific gene modules by extending our EW_dmGWAS (version 3.1) algorithm to integrate ad GWAS by proxy summary statistics, brain protein expression data collected from two independent cohorts and human protein–protein interactions (PPIs). An overview of the study design is presented in Figure 1. The input for EW_dmGWAS contains two parts: node weights and edge weights. First, we calculated the node weights based solely on the ad GWAS summary statistics of 472 868 individuals of European descent (9). Specifically, we applied the Multi-marker Analysis of GenoMic Annotation (MAGMA) tool (28) to condense the ad GWAS summary statistics to 18 447 nearby genes (Supplementary Material, Table S1). Then, the gene node weight value was converted from the gene-level significance (see Materials and Methods). Second, the edge weights were calculated based on brain region-specific protein expression data in each cohort, including 9210 gene-level protein expression data of PHG from the Mount Sinai Brain Bank study (18,29) and 8251 gene-level protein expression data of DLPFC from the Religious Order Study and Memory and Aging Project (ROSMAP) (27,30). Data normalization, batch correction and missing data imputation were conducted within each dataset. Then, the edge weights were calculated based on the differentially co-expression profiles between ad cases and control samples in each dataset and were matched with PPIs from the BioGRID database (see Materials and Methods).
Figure 1.

Workflow of the study. Abbreviations: Alzheimer’s disease (AD), parahippocampal gyrus (PHG), dorsolateral prefrontal cortex (DLPFC), Mount Sinai Brain Bank (MSBB), Religious Order Study and Memory and Aging Project (ROSMAP), protein–protein interactions (PPIs), genome-wide association studies (GWAS), Edge-Weighted dense module Search of GWAS (EW_dmGWAS), Web-based Cell-type-Specific Enrichment Analysis of Genes (WebCSEA).
After data harmonization procedures within each dataset, a total of 8141 gene note-weights and 243 276 edge-weights were used for EW_dmGWAS analysis in the MSBB dataset (Supplementary Material, Tables S2 and S3), and 7715 node-weights and 236 618 edge-weights were utilized in the ROSMAP dataset (Supplementary Material, Tables S4 and S5). Furthermore, to balance the importance of genetic variants and human protein interactomes, we introduced a scaling factor (λMSBB = 2.05; λROSMAP = 2.23) calculated by the ratio of the variance of node weights and edge weights. Finally, we identified 1056 and 1002 dense gene modules from the gene-level protein expression dataset of PHG tissue and DLPFC tissues, respectively (Supplementary Material, Tables S6 and S7).
Modules network evaluation and module selection
We ranked all ad-associated dense modules based on 1000-permutation z scores and evaluated the network properties of genes within an increasing chosen number of top modules (see Materials and Methods). To assess the network feature, we calculated the coefficient of determination of the log–log relationship of frequency of edges over nodes, R2, to measure the degree of scale-free networks. As shown in Figure 2A and B, the scale-free network indexes are positively correlated with the number of modules selected at the beginning, and the index stabilized after more modules were merged. The median scale-free network index R2 was 0.84 for PHG top modules and 0.82 for DLPFC top modules, indicating approximately scale-free networks for both results.
Figure 2.

Characteristics of scale-free networks of resulting modules from brain region-specific EW_dmGWAS analysis. (A) Scatter plot of the scale-free index (R2) for networks constructed by top modules identified from EW_dmGWAS analysis of PHG. (B) Scatter plot of the scale-free index (R2) for networks constructed by top modules identified from EW_dmGWAS analysis of DLPFC. The red line in (A and B) represents the median R2 of all networks. (C) Density histogram shows the distribution of the number of edges of each gene within the gene network constructed by the TMGs in the PHG. (D) Density histogram shows the distribution of the number of edges of each gene within the gene network constructed by TMGs in the DLPFC.
We further assessed the conservativeness of genes in the top modules between the two datasets. We first calculated the Jaccard indexes to evaluate the membership of the top 100 modules (Supplementary Material, Fig. S1A). The mean Jaccard index was 0.006, indicating overall low consistency between gene modules of the two datasets. However, some gene modules between the two datasets had relatively high consistency. For example, the Jaccard index between the top-ranked module in the DLPFC dataset and the 22nd ranked module in the PHG dataset was 0.40. Considering the order of modules might not be the most important factor in our analysis, the membership between the accumulated small networks is the most crucial point. Therefore, we assessed the membership of the accumulating top gene modules. The range of Jaccard indexes was 0.034–0.063 when comparing the genes within the combined 25 dense modules in the two datasets (Supplementary Material, Fig. S1B).
For better visualization and interpretation, we empirically selected the genes in the top 25 modules from EW_dmGWAS results of each dataset as the top module genes (TMGs) for further evaluation. We identified 52 unique TMGs from the protein expression dataset in PHG tissue (MSBB) (Supplementary Material, Table S8). Figure 2C shows that more nodes within the network possess a lower number of edges. After log transformation, the edge density distribution shows an approximately negative linear relationship with R2 = 0.77. On the other hand, we identified 58 unique TMGs within the top 25 modules from the ROSMAP protein expression dataset of DLPFC tissue (Supplementary Material, Table S9). Figure 2D shows that a merged network of TMGs of DLPFC possessed an edge distribution similar to PHG. However, the density of nodes with fewer edges is lower, resulting in a lower R2 = 0.53 in the merged network of DLPFC.
Key overlapped genes identified between TMGs in PHG and DLPFC
We further compared the TMGs in PHG and DLPFC for identification of key overlapped risk genes. We identified five overlapped genes between two gene sets, including CLU [MAGMA Z score = 7.66, P-value = 9.44 × 10−15, false discovery rate (FDR) = 2.00 × 10−11], PICALM (MAGMA Z score = 6.78, P-value = 6.19 × 10−12, FDR = 3.42 × 10−9), PRRC2A (MAGMA Z score = 4.22, P-value = 1.21 × 10−5, FDR = 1.34 × 10−3), NDUFS3 (MAGMA Z score = 3.14, P-value = 8.38 × 10−4, FDR = 3.84 × 10−2) and DNM2 (MAGMA Z score = 2.18, P-value =1.45 × 10−2, FDR = 0.22). Four of the five overlapped genes had GWAS gene-level FDR less than 0.05. All four genes have been reported to be associated with ad (29–32), suggesting they might be ubiquitously associated with ad in different brain regions.
Differential co-expression network analysis revealed potential therapeutic targets of ad
Considering protein expression might better reflect the pathological progression of the disease, we analyzed the regulatory networks of the four key overlapped genes in each brain region at the protein expression level to gain novel insights into therapeutic targets for ad (33). Specifically, among the four overlapped genetic risk genes between TMGs of PHG and DLPFC datasets (CLU, PICALM, PRRC2A and NDUFS3), we identified the brain region-specific differentially co-expressed proteins (defined by edge weight > 1.96). We compared the proteins and their coding genes in each regulatory network with the known investigational ad drug targets (Fig. 3, Supplementary Material, Table S10).
Figure 3.

The brain region-specific differentially co-expressed genes of the key overlapped genes. (A) The workflow of the differentially co-expression analysis. The genes were ordered by GWAS gene-level significance. (B) The differentially co-expressed genes of the key overlapped genes in the PHG contain genetic targets of investigational drugs. (C) The differentially co-expressed genes of the key overlapped genes in the DLPFC. †: SLC6A4 was only identified in EW_dmGWAS in PHG. ‡: GJA1 was only identified by differentially co-expression analysis in PHG.
In the PHG region, we identified 83 differentially co-expressed genes with at least one of four key overlapped genes (Fig. 3B). Interestingly, four differently co-expressed genes, APP, SNCA, VCAM1 and GJA1, are therapeutic targets of existing investigational drugs (34–37). In the DLPFC region, we identified 38 differentially co-expressed genes with at least one of the four key overlapped genes (Fig. 3C). However, none of those genes were the therapeutic target of known drugs for ad, suggesting that the DLPFC region-specific genes and their co-expression patterns are not targeted by current treatment strategies.
Networks reconstructed with ReactomeFIVIz show enrichment of relevant biological pathways
The EW_dmGWAS algorithm was able to identify discrete gene modules, while some key biological pathways may be shared between modules. To further explore the comprehensive biological functions of identified brain region-specific gene modules associated with ad, we reconstructed the TMGs into a fully connected interaction network using ReactomeFIViz (38). Figure 4A shows the reconstructed network from the 52 TMGs identified in the PHG dataset and 20 linker genes inferred by ReactomeFIViz (Supplementary Material, Table S11). Seven linker genes overlapped with the genes in all 1056 modules identified by EW_dmGWAS in the PHG dataset. We performed an over-representation analysis of all genes within the Reactome network for biological explanations. As shown in Figure 4B, the most significantly enriched gene ontology (GO) biological processes (BP) term was ‘reactive oxygen species metabolic process’ (FDR = 1.99 × 10−10) with 15 overlapping genes. Interestingly, among the top 20 enriched terms, several immune-related terms were identified, including ‘neuroinflammatory response’ (FDR = 3.06 × 10−7), ‘leukocyte activation involved in inflammatory response’ (FDR = 6.49 × 10−7) and ‘immune response-regulating signaling pathway’ (FDR = 6.27 × 10−5).
Figure 4.

Brain region-specific interaction networks and functional enrichment analyses. The genetic interaction networks were constructed of the TMGs and linking genes (highlighted in red) from the PHG (A) or DLPFC (C) using the ReactomeFIVIz tool. The size of each node was positively associated with the number of connections within the network. The genes in diamond shapes represent the key overlapped genes between two interaction networks. (B and D) Top 20 enriched GO BP terms in all genes within genetic interaction networks of PHG (B) and DLPFC (D).
With 58 TMGs identified in DLPFC dataset, we reconstructed the fully connected network with additional 20 linker genes inferred by ReactomeFIViz (Fig. 4C, Supplementary Material, Table S12). Only four linker genes overlapped with the genes in the 1002 gene modules identified by EW_dmGWAS in the DLPFC dataset. Figure 4D lists the top 20 enriched GO BP terms for the network genes. The most significantly enriched term was ‘regulation of vesicle-mediated transport’ (FDR = 7.14 × 10−11). Notably, we found several GO BP terms related to protein folding and metabolism, such as ‘regulation of protein complex assembly’ (FDR = 1.45 × 10−8), ‘amyloid-β metabolic process’ (FDR = 2.78 × 10−8), ‘amyloid precursor protein metabolic process’ (FDR = 1.18 × 10−5), among others. The finding is consistent with previous studies that metabolism makers of amyloid precursor protein are related to neurodegeneration in the preclinical stage of ad (39).
Drug repurposing analysis with CMap signatures
Besides biological pathways, to examine the enriched drug signatures for TMGs in each brain region, we applied the co-expressed gene-set enrichment analysis method (Cogena) implemented in the Bioconductor R package (see Materials and Methods). As shown in Table 1, for the 52 TMGs identified in PHG tissue, the top enriched drugs from the downregulated 100 connectivity map (CMap) gene set included midecamycin (−log2 FDR = 10.5) and Trolox C (−log2 FDR = 10.5). The top enriched drugs from the upregulated 100 CMap gene set of PHG tissue included abamectin (−log2 FDR = 9.7), tanespimycin (−log2 FDR = 9.7) and chlorphenamine (−log2 FDR = 9.7). On the other hand, for the 58 TMGs identified in DLPFC tissue (Table 2), the top enriched drugs from the downregulated 100 CMap gene set included proglumide (−log2 FDR = 15.2), methazolamide (−log2 FDR = 12.3) and molecular phenazopyridine (−log2 FDR = 12.3). Furthermore, the drug signature enrichment analysis of upregulated 100 CMap gene set in DLPFC tissue identified ‘0198306-0000’ small molecules (−log2 FDR = 10.9), dinoprostone (−log2 FDR = 9.5) and disulfiram (−log2 FDR = 9.5), among others.
Table 1.
Drug repositioning for the top module genes from the parahippocampal gyrus dataset
| Drug name | Cell line | Regulatory direction | −log2 FDR | Genes enriched |
|---|---|---|---|---|
| Midecamycin | MCF7 | Upregulated | 10.5 | DNM2, NF1, PICALM |
| Prasterone | HL60 | Upregulated | 10.5 | DNM2, PICALM, PTK2B |
| Trolox C | MCF7 | Upregulated | 10.5 | DNM2, NF1, PICALM |
| Mafenide | MCF7 | Upregulated | 10.5 | CHP1, NF1, PICALM |
| Triflupromazine | MCF7 | Upregulated | 10.5 | CHP1, PICALM, PRRC2A |
| Conessine | HL60 | Upregulated | 10.5 | ATP1B1, DNM2, NF1 |
| Meteneprost | MCF7 | Upregulated | 10.5 | CHP1, NF1, PICALM |
| Abamectin | PC3 | Downregulated | 9.7 | AIF1, EGFR, PCOLCE, UBXN1 |
| Tanespimycin | PC3 | Downregulated | 9.4 | CLU, EGFR, HSPA5 |
| Chlorphenamine | PC3 | Downregulated | 9.4 | AIF1, EGFR, ITGB1 |
| Thalidomide | MCF7 | Downregulated | 9.4 | CD74, CPS1, EGFR |
| Pridinol | HL60 | Downregulated | 9.4 | AIF1, DDAH2, PDPR |
| Metyrapone | HL60 | Downregulated | 9.4 | EGFR, HLA-DRB1, UBXN1 |
| Lysergol | MCF7 | Downregulated | 9.4 | AIF1, EGFR, GSTP1 |
| Primidone | MCF7 | Downregulated | 9.4 | EGFR, PXN, UBXN1 |
| Androsterone | MCF7 | Downregulated | 9.4 | EGFR, PXN, UBXN1 |
| (−)-Isoprenaline | HL60 | Downregulated | 9.4 | HLA-DRA, HLA-DRB1, SLC6A12 |
| Sitosterol | MCF7 | Downregulated | 9.4 | CD74, PDPR, PXN |
| Dexpanthenol | MCF7 | Downregulated | 9.4 | AIF1, EGFR, GSTP1 |
| Suramin sodium | PC3 | Downregulated | 9.4 | EGFR, ITGB1, NUP160 |
| Levcycloserine | PC3 | Downregulated | 9.4 | ITGB1, PXN, REL |
| Etamivan | PC3 | Downregulated | 9.4 | EGFR, HLA-DRB1, PCOLCE |
| Gibberellic acid | MCF7 | Downregulated | 9.4 | ITGB1, PXN, REL |
FDR: false discovery rate.
Table 2.
Drug repositioning for the top module genes from the dorsolateral prefrontal cortex dataset
| Drug name | Cell line | Regulatory direction | −log2 FDR | Genes enriched |
|---|---|---|---|---|
| Proglumide | PC3 | Upregulated | 15.2 | BCAM, FNDC3A, FUS, PICALM, RABEP1 |
| Methazolamide | MCF7 | Upregulated | 12.3 | DNM2, PICALM, TLN2, ZYX |
| Phenazopyridine | PC3 | Upregulated | 12.3 | BCAM, BIN1, RABEP1, RPS9 |
| Sulindac | MCF7 | Upregulated | 9.9 | BCAM, FKBPL, RPS9 |
| Nordihydroguaiaretic acid | MCF7 | Upregulated | 9.9 | BCAM, RPS9, ZYX |
| Troglitazone | MCF7 | Upregulated | 9.9 | PICALM, RABEP1, ZYX |
| Trimethoprim | MCF7 | Upregulated | 9.9 | CNN2, PICALM, TLN2 |
| Benperidol | MCF7 | Upregulated | 9.9 | PICALM, RPS9, TLN2 |
| Terconazole | MCF7 | Upregulated | 9.9 | DNM2, PICALM, TLN2 |
| Ciprofibrate | MCF7 | Upregulated | 9.9 | BRAF, RABEP1, ZYX |
| 0198306-0000 | PC3 | Downregulated | 10.9 | APP, DMWD, SQSTM1, VASP |
| Dinoprostone | MCF7 | Downregulated | 9.5 | BMPR2, CSNK1A1, VASP |
| Disulfiram | PC3 | Downregulated | 9.5 | APP, CLU, SQSTM1 |
| Aceclofenac | PC3 | Downregulated | 9.5 | APP, PRRC2A, VASP |
| Zomepirac | HL60 | Downregulated | 9.5 | APOE, DMWD, MAPT |
| Monastrol | MCF7 | Downregulated | 9.5 | DMWD, SQSTM1, VASP |
| Carteolol | HL60 | Downregulated | 9.5 | BMPR2, DMWD, SQSTM1 |
FDR: false discovery rate.
Tissue-cell type specificity of TMGs in brain
Our TMGs were derived from the bulk proteomic data from two brain regions. To characterize the cellular context of these top module genes, we applied our newly developed tool, Web-based Cell-type-Specific Enrichment Analysis of Genes (40, 41), with TMGs of each brain region, respectively. We found that TMGs of both PHG and DLPFC were enriched in excitatory neuron 3e sub-cell types in the brain [PHG TMGs raw P-value < 0.01 (combined P-value = 0.01), DLPFC TMGs raw P-value = 0.01(combined P-value = 0.21)] on the single-cell panel from disease-relevant tissue (frontal cortex) (42), suggesting that our top modules from both brain regions captured the underlying genetic risks and were enriched in the excitatory neuron 3e sub-cell type (Supplementary Material, Fig. S2). Previous studies have observed the imbalance of excitatory and inhibitory neurons in experimental models and ad patients (43,44). It has been hypothesized that alternation of excitatory and inhibitory neuron activities may disrupt the cortical regions’ cognitive-related functions, contributing to the cognitive function decline in ad (43,44). Interestingly, enriched excitatory neuron genes were also a potential target for drugs identified in the Cogena drug repurposing analysis. In PHG, tanespimycin (−log2 FDR = 9.4), a potent heat shock protein 90 (HSP90) inhibitor, was identified in the Cogena analysis of TMGs (Table 1). The enriched genes contained CLU and HSPA5, which were also the enriched gene signature of excitatory neurons. A recent model study demonstrated the protective effect of tanespimycin against seizures and cognition decline in both mice and cynomolgus monkey models (45). In DLPFC, disulfiram (−log2 FDR = 9.5) was identified as a potential repurposing drug through Cogena analysis (Table 2). The three enriched genes of disulfiram, APP, CLU and SQSTM1, were also enriched signature genes of excitatory neurons. It has been suggested that disulfiram may contribute to increased ADAM10 expression, which may ameliorate the amyloid-β accumulation and cognition deficits in the ad mouse model (46).
Discussion
In this work, we utilized our network-based method to capture the convergent signal from ad genetic data and multi-brain region proteomics. As a result, we prioritized TMGs in the PHG and DLPFC. We pinpointed four overlapped genes (CLU, PICALM, PRRC2A and NDUFS3) with GWAS gene-level significance (FDR < 0.05) between TMGs. Moreover, the regulatory network of the four overlapped genes was analyzed, highlighting therapeutic target genes of existing investigational drugs in PHG. Furthermore, we found heterogeneous enriched biological pathways and drug signatures and similar neuron signatures in TMGs of two brain regions.
The current study applied the EW_dmGWAS algorithm to harness the evidence from one of the largest ad genome-wide meta-analyses to date to search dense gene modules associated with ad (9). However, the strength of genome-wide associations varied between different ad GWAS as the characteristics of samples included were diverse. We, therefore, estimated the variation of using a separated large-scale ad GWAS on the resulting gene modules (11). The newly calculated node weights using ad GWAS by Wightman et al. (11) were moderately correlated with those calculated from ad GWAS by Schwartzentruber et al. (r = 0.646, Supplementary Material, Fig. S3). As a result, the identified dense gene modules varied between independent EW_dmGWAS analyses using two ad GWAS. We subsequently examined the consistency of the gene in the accumulating top modules in two settings. We found higher gene consistencies within the same protein expression dataset (Jaccard scores, 0.147–0.213) compared to the gene consistencies across the protein expression datasets (Jaccard scores, 0.048–0.099) (Supplementary Material, Table S13). Thus, we believe the variation between protein expression profiles from two brain regions was greater than the variation between the two ad GAWS. Moreover, we believe the EW_dmGWAS algorithm was able to detect the variations caused by inputted GWAS signals.
Our brain region-specific network analyses pinpointed four overlapped promising candidate ad genetic risk genes (CLU, PICALM, PRRC2A and NDUFS3) between TMGs identified from PHG and DLPFC datasets independently. Among these overlapped genes, CLU (encoding clusterin) gene had the highest GWAS gene-level significance (P-value =9.44 × 10−15, FDR = 2.00 × 10−11). CLU has been identified as the third most significant risk gene associated with late-onset ad (47) and might explain approximately 9% of ad attributable risk (48). Although it is still largely unclear how CLU contributes to ad risk, some have suggested that its respective glycoprotein, clusterin, is associated with amyloid-β aggregation (29), toxicity (49) and clearance (50). Our analyses revealed enrichment of biological pathways related to the amyloid-β process and clearance. Clusterin is involved in regulating cell survival and cell death pathways (51). Accordingly, we found in our analyses several pathways related to cell death and mitochondria enriched, such as ‘neuron death,’ ‘protein localization to mitochondria,’ ‘regulation of mitochondria organization’ and ‘cell–cell signaling by WNT.’ Specifically, ‘signaling by WNT’ is a well-known biological pathway implicated in ad, being found downregulated in ad, leading to an increase in GSK-3β and increased tau phosphorylation and synapse loss. In addition, one study reported colocalization of clusterin and amyloid-β plaques surrounded by p-tau deposits in the temporal cortex of ad individuals (52). Furthermore, clusterin has been reported to be involved in oxidative stress and is increased by proteolytic stress. Interestingly, the top enriched biological pathways in our analysis of PHG proteomic data were ‘reactive oxygen species metabolic process’ and ‘response to oxidative stress,’ further supporting the importance of clusterin for oxidative stress in ad.
Another key overlapped gene between TMGs in two brain regions, PICALM, which encodes Phosphatidylinositol Binding Clathrin Assembly Protein, was identified (gene-level P-value =6.19 × 10−12, FDR = 3.42 × 10−9) by EW_dmGWAS in both proteomic datasets. It has been reported that PICALM methylation in blood might contribute to the cognitive function decline in ad (53). PICALM modulates the production, transportation and clearance of β-amyloid peptides (30). One study has indicated that increasing Phosphatidylinositol Binding Clathrin Assembly Protein levels may rescue the APOE4-induced endocytic defect (54). A recent publication reported that the PRRC2A (encoding proline-rich coiled-coil 2A) gene plays an important role in regulating oligodendrocyte specification and myelination by functioning as a newly identified N6-methyladenosine (m6A) reader (55). Further, PRRCA2 was found to be downregulated in microglia and astrocytes of aging mice brains (31). Finally, ubiquinone oxidoreductase core subunit S3, encoded by the NDUFS3 gene, and other members of ubiquinone oxidoreductase, such as NDUFA2 and NDUFA3, were downregulated in individuals with late-onset ad (32).
Differential co-expression network analysis highlighted four therapeutic target genes of existing investigational drugs in PHG, including APP, VCAM1, GJA1 and SNCA. Most AD drugs under development or testing devote significant efforts to interfering with the production and accumulation of amyloid-β peptide, the product of APP (34). VCAM1, a member of the No. 14 of the top 25 gene module from EW_dmGWAS, is one of the drug targets of Carvedilol (35), which can attenuate brain Aβ content and cognitive deterioration in animal models (56). GJA1 is also the target gene of Carvedilol (36), but our EW_dmGWAS analysis did not capture it. Carvedilol has been assessed for its effectiveness and saf-ety in one phase IV clinical trial. Although the clinical trial failed to show a significant difference between the Hopkins Verbal Learning Test (HVLT) score changes between the treatment group and placebo group, the results showed a slight reduction of amyloid-β42 level in cerebrospinal fluid in the treatment group versus an increase of amyloid-β42 in the placebo group (https://clinicaltrials.gov/, NCT01354444). Besides, SNCA is the therapeutic target of resveratrol and was identified within the top 100 (No. 71) modules of EW_dmGWAS analysis (37). Multiple studies have shown that resveratrol presents neuroprotective effects in animal models. However, such protective effects were not observed in clinical trials (57). In addition, a recent review study has indicated that resveratrol derivatives might serve as a potential ad treatment (57). Apart from co-expression candidates of overlapped risk genes, we identified the SLC6A4 gene, which is included within the top 150 modules in our EW_dmGWAS analysis of the PHG dataset. SLC6A4 is the potential therapeutic target of multiple drugs developed or repurposed for psychiatric disorders among ad patients, including Aripiprazole (58,59), AVP-786 (60), AXS-05 (61) and Escitalopram (62,63).
The results of the current study should be interpreted with caution. First, our study might suffer from the heterogeneous data of cohort-specific proteomics expression datasets. To remove the potential variation caused by confounding factors, we adopted strict criteria for sample labeling based on each study’s clinical diagnosis and pathological diagnostic information, following the National Institute on Aging and Alzheimer’s Association (NIA-AA) diagnostic recommendations (3). Batch effect correction and normalization procedures were conducted within proteomics data in each cohort. Moreover, the same data imputation method was applied to proteomics expression datasets. Therefore, the remaining variation across different cohorts may mainly represent brain region-specific proteomics expression variation. Second, we cannot assess the regulatory relationship between the PHG region and the DLPFC region with the current proteomic data. The disconnection between these two brain regions might be a pathological pathway for cognitive dysfunction in ad (64). Then, we applied a scale-free network index to the top modules identified from the EW_dmGWAS analysis. However, the current network constructed by TMGs could not meet the empirical scale-free network index of 0.8. We believe a more robust network could be built with a more holistic human PPI database. Lastly, the current study results are derived from European population GWAS and proteome data. Thus, our findings might not be extrapolated to non-European populations.
In summary, we conducted integrative analysis on genetic variants and protein expression profiles of PHG and DLPFC from ad and control samples to identify brain region-specific dense gene modules associated with ad. We identified brain region-specific TMGs in PHG and DPLFC datasets and highlighted four key overlapped genes. We found that their differentially co-expressed proteins in PHG contained potential genetic targets of known investigational drugs of ad. Lastly, we identified that specific excitatory neuron was highly enriched by TMGs of both brain regions. These findings provided new insights into ad’s complex pathology and the development of drugs targeting those molecular alterations.
Materials and Methods
Analysis datasets and data preprocessing
We obtained two independent brain region-specific protein expression datasets from the Synapse portal (65). We obtained protein expression data of the PHG region of 127 ad and 63 control subjects from the MSBB study (18,66); the protein expression dataset of the DLPFC region was obtained from the ROSMAP study (27,67), which included 108 ad and 84 control subjects (3). The diagnostic labels of both datasets were determined following the National Institute on Aging and Alzheimer’s Association (NIA-AA) diagnostic recommendations (3).
Protein expression profiles were measured by the TMT mass spectrometry-based quantification approach (26,27). Batch effect correction, normalization and imputation procedures for the MSBB protein expression dataset were preprocessed and reported in the original publication (66). For the ROSMAP protein expression dataset, we adapted the batch effect corrected and normalized data from the original publication (27). In addition, we applied a linear mixed-effects model, the same data imputation strategy used in the MSBB dataset, to impute the missing data in the ROSMAP protein expression dataset (68,69). One outlier was excluded from the ROSMAP dataset based on principal component analysis (Supplementary Material, Fig. S4). Finally, the protein expression profiles were mapped to corresponding gene symbols based on their respective UniProt IDs. Overall, the module search analysis used 9210 protein expression data from the MSBB dataset and 8251 protein expression data from the ROSMAP dataset.
GWAS summary statistics and node-weight calculation
We obtained the GWAS summary statistics from a comprehensive genome-wide meta-analysis study by Schwartzentruber et al. (9). This study applied a GWAS by proxy approach by including 472 868 (75 024 individuals diagnosed with ad or individuals with one or both parents diagnosed with ad and 397 844 controls) individuals of European descent. The GWAS has been approved as a powerful method for discovering genetic variants of complex traits, especially for late-onset diseases, in large cohort biobanks (70). Moreover, another large-scale ad GWAS using proxy cases suggested that the magnitude of the associations did not change substantially when only clinically diagnosed ad cases were included (12). Considering that the strengths of genome-wide association varied across studies, we obtained another large-scale genome-wide meta-analysis by Wightman et al. (11) for sensitivity analysis. We applied a state-of-the-art tool, MAGMA, using an SNP window of up 50 kb and down 35 kb for each gene to calculate the sum of squared SNP Z-statistics as χ2 statistic. A gene P-value was estimated using a known approximation of the sampling distribution for χ2 statistic, SNP covariance and degree of freedom (Supplementary Material, Table S1) (28). Node weight (v) for each gene was then calculated by 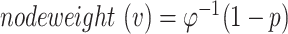 , where
, where 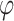 is the two-sided standard normal distribution function and p represents the gene-level P-value (21).
is the two-sided standard normal distribution function and p represents the gene-level P-value (21).
ad edge-weight calculation
The preprocessed protein expression profiles of PHG and DLPFC regions were used for edge-weight calculation. Edge weights for each gene pair were calculated based on the change of gene-level protein co-expression between ad cases and control samples. Briefly, we calculated the Pearson’s correlation coefficient values for each gene pair by using the gene-level protein expression dataset among cases and controls. Next, we performed Fisher transformation and Fisher’s test of the difference between cases and controls for edge-weight calculation in each dataset (21). Finally, all protein pairs were mapped to an experimentally validated PPI database and the BioGRID database (version 4.4.203) (71). We preprocessed the PPIs by removing non-human and redundant data, resulting in 19 094 genes and 539 890 unique human PPIs. After harmonizing each protein expression dataset with the non-redundant PPIs, the calculated edge weights could reflect the differential co-expression profiles between ad cases and control samples in brain regions. Following our previous work (23), we defined differential co-expression by edge weight transferring below the nominal P-value (edge weight > 1.96). The detailed methods are described in the original publication (21).
Network-based dense gene module identification
We applied an updated network-assisted analysis algorithm, the EW_dmGWAS tool, to integrate GWAS and proteomic signals of two brain regions separately (21). A greedy algorithm was implemented to search dense gene modules based on module score S calculated as:
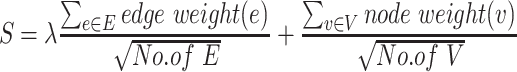 |
where node weights (e) and edge weights (v) were calculated in the previous step; E in the formula represents the set of edges; and V represents the set of nodes. λ in the formula is a scaling factor for GWAS and protein expression values. In this study, we used the variance ratio between edge weights and node weights as the scaling factor for each brain region. Finally, the resulting modules were ranked by EW_dmGWAS based on 1000 permutations of signal-enrichment from both genomic and gene expression profiling of ad samples and controls.
Top modules evaluation and selection
To explore the critical functions of modules identified from EW_dmGWAS, we selected top modules based on their significance ranking and evaluated them using a scale-free network index. First, all modules were ranked based on the 1000-permutation z-scores calculated in EW_dmGWAS. Second, we selected the top modules and assessed their scale-free network properties. Specifically, we curated all genes within the selected modules and reconstructed the merged network based on the BioGRID PPI. Scale-free network indexes were calculated and plotted to assess the network properties with the increasing size of the network. We assume the distribution of nodes and frequency of edges within a reconstructed biological network follow the power law, 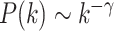 , where k stands for the number of node edges (72). After applying the log–log transformation, a linear fit,
, where k stands for the number of node edges (72). After applying the log–log transformation, a linear fit, 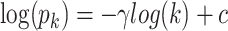 , can be found, and the coefficient of determination of the regression (R2) can be used to estimate the scale-free network property. Considering the network size and interpretability, we prioritized the top 25 modules for each brain region-specific protein expression dataset for visualization, key biological function exploration and downstream analyses.
, can be found, and the coefficient of determination of the regression (R2) can be used to estimate the scale-free network property. Considering the network size and interpretability, we prioritized the top 25 modules for each brain region-specific protein expression dataset for visualization, key biological function exploration and downstream analyses.
Reconstruction of the regulatory network
We performed downstream Reactome pathway analysis to explore the functional interactions within the identified genes of the top dense modules and linker genes. The EW_dmGWAS algorithm tends to prioritize discrete gene modules associated with ad, while the comprehensive regulatory networks might not be fully captured. Thus, we implemented a Cytoscape application, ReactomeFIViz, to reconstruct the regulatory network with genes of interest (38). The input of ReactomeFIViz was the gene sets from top-ranked modules that we obtained from the previous network dense module search. We used the latest Reactome FI Network version 2020 for network construction. In addition, the build-in linker genes deduction function was implemented to complement the network.
Functional enrichment analysis
To explore the brain region-specific functional enrichment of genes within selected top modules from EW_dmGWAS, we performed over-representation analyses using the R package WebGestaltR. We used GO, no redundant BP annotation and all human protein-coding genes as the reference (73). The final results were filtered based on Benjamini–Hochberg (BH) adjusted P-value < 0.05 (74).
Drug signature enrichment analysis
We performed drug signature enrichment analyses using the Cogena R package (75). Cogena is a framework that calculates the co-expression of inputted genes to determine gene expression signatures and creates clusters associated with the disease mechanism. Cogena uses hypergeometric tests to perform gene set enrichment analysis of biological pathways or curated drug signature gene sets. We separately constructed gene-level protein expression matrices of all genes in the top 25 modules for the MSBB and ROSMAP datasets. The protein expression matrices were inputted into Cogena for brain region-specific analysis. The default parameters were used to perform drug signature enrichment analyses: 10 clusters, 2 cores, hierarchical and pam methods for clustering methods and correlation for distance metric. We used two curated gene sets from the Cogena R package to perform these analyses: the CMap gene set for the top 100 downregulated genes per drug and the CMap gene sets for the top 100 upregulated genes per drug. Lastly, the −log2 FDR was reported for the enriched drug signature from the Cogena hypergeometric tests.
Web-based Cell-type-Specific Enrichment Analysis of Genes (WebCSEA)
We further assessed the cell-type specificity of top module genes of PHG and DLPFC proteomic expression through our newly developed application, (40,41). In WebCSEA, we adapted our previous decoding of the tissue specificity (deTS) algorithm on single-cell RNA-seq data of human tissues (76). In this work, we evaluated the top 25 module genes of PHG and DLPFC on the single-cell reference panel from disease-relevant tissue (frontal cortex) (42). We adapted both results from raw P-value mode and combined P-value mode, which were calculated to avoid potential bias from the gene set length and tissue-cell type difference by leveraging the permutation result from ~ 20 000 gene sets with moderate TC specificity derived from genetic studies. The detailed method can be found in the tutorial of the original web service.
Abbreviations
- ad
Alzheimer’s disease
- BP– biological process
- CMap—connectivity map
- Cogena—co-expressed gene-set enrichment analysis
- DLPFC—dorsolateral prefrontal cortex
- EW_dmGWAS—Edge-Weighted dense module Search of GWAS
- FDR—false discovery rate
- GO—gene ontology
- GWAS—genome-wide association studies
- MAGMA—Multi-marker Analysis of GenoMic Annotation
- PHG—parahippocampal gyrus
- PPI—protein–protein interaction
- ROSMAP—Religious Order Study and Memory and Aging Project
- TMG—top module genes
- TMT—tandem mass tag
- WebCSEA—Web-based Cell-type-Specific Enrichment Analysis of Genes
Author Contributions
Z.Z., Y.D., P.J. and A.L. contributed to the conception and design of the study; A.L., A.A.M. and Y.D. collected the data and performed the analysis; A.L., A.A.M., Y.D., B.S.F. and Z.Z. interpreted the results; A.L., A.A.M., Y.D., B.S.F., N.E., P.J. and Z.Z. wrote the manuscript.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This research is based on datasets available in online repositories. In addition, the proteomic expression profiles of PHG and DLPFC for supporting the findings of this study are available from the Synapse portal (https://adknowledgeportal.synapse.org/).
Conflict of Interest statement: The authors declare no conflict of interest.
Contributor Information
Andi Liu, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, Houston, TX 77030, USA; Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Astrid M Manuel, Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Yulin Dai, Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Brisa S Fernandes, Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Nitesh Enduru, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, Houston, TX 77030, USA; Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Peilin Jia, Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA.
Zhongming Zhao, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, Houston, TX 77030, USA; Center for Precision Health, School of Biomedical Informatics, Houston, TX 77030, USA; Human Genetics Center, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Funding
National Institutes of Health (NIH; grant R01LM012806). Z.Z. was partially supported by NIH grant (R01DE030122). We thank the technical support from the Cancer Genomics Core funded by the Cancer Prevention and Research Institute of Texas (CPRIT RP180734 and RP210045). A.L. was supported by a training fellowship from the Gulf Coast Consortia on Training in Precision Environmental Health Sciences (TPEHS) Training Grant (T32ES027801). A.M.M. was supported by a training fellowship from the Gulf Coast Consortia on the NIH National Library of Medicine (NLM) Training Program in Biomedical Informatics & Data Science (T15LM007093). The funder had no role in the study design, data collection, analysis, decision to publish or preparation of the manuscript.
References
- 1. Winblad, B., Amouyel, P., Andrieu, S., Ballard, C., Brayne, C., Brodaty, H., Cedazo-Minguez, A., Dubois, B., Edvardsson, D., Feldman, H.et al. (2016) Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurol., 15, 455–532. [DOI] [PubMed] [Google Scholar]
- 2. Hurd, M.D., Martorell, P., Delavande, A., Mullen, K.J. and Langa, K.M. (2013) Monetary costs of dementia in the United States. N. Engl. J. Med., 368, 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jack, C.R., Jr., Bennett, D.A., Blennow, K., Carrillo, M.C., Dunn, B., Haeberlein, S.B., Holtzman, D.M., Jagust, W., Jessen, F., Karlawish, J.et al. (2018) NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement., 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P.H., Williams, L., Maier, M., Dunstan, R., Salloway, S., Chen, T., Ling, Y.et al. (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature, 537, 50–56. [DOI] [PubMed] [Google Scholar]
- 5. Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P.H., Williams, L., Maier, M., Dunstan, R., Salloway, S., Chen, T., Ling, Y.et al. (2017) Addendum: the antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature, 546, 564. [DOI] [PubMed] [Google Scholar]
- 6. Badhwar, A., McFall, G.P., Sapkota, S., Black, S.E., Chertkow, H., Duchesne, S., Masellis, M., Li, L., Dixon, R.A. and Bellec, P. (2020) A multiomics approach to heterogeneity in Alzheimer's disease: focused review and roadmap. Brain, 143, 1315–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sengoku, R. (2020) Aging and Alzheimer's disease pathology. Neuropathology, 40, 22–29. [DOI] [PubMed] [Google Scholar]
- 8. Rodriguez-Arellano, J.J., Parpura, V., Zorec, R. and Verkhratsky, A. (2016) Astrocytes in physiological aging and Alzheimer's disease. Neuroscience, 323, 170–182. [DOI] [PubMed] [Google Scholar]
- 9. Schwartzentruber, J., Cooper, S., Liu, J.Z., Barrio-Hernandez, I., Bello, E., Kumasaka, N., Young, A.M.H., Franklin, R.J.M., Johnson, T., Estrada, K.et al. (2021) Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer's disease risk genes. Nat. Genet., 53, 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kunkle, B.W., Grenier-Boley, B., Sims, R., Bis, J.C., Damotte, V., Naj, A.C., Boland, A., Vronskaya, M., van derLee, S.J., Amlie-Wolf, A.et al. (2019) Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet., 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wightman, D.P., Jansen, I.E., Savage, J.E., Shadrin, A.A., Bahrami, S., Holland, D., Rongve, A., Borte, S., Winsvold, B.S., Drange, O.K.et al. (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat. Genet., 53, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bellenguez, C., Küçükali, F., Jansen, I.E., Kleineidam, L., Moreno-Grau, S., Amin, N., Naj, A.C., Campos-Martin, R., Grenier-Boley, B., Andrade, V.et al. (2022) New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat. Genet., 54, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heneka, M.T., Carson, M.J., El Khoury, J., Landreth, G.E., Brosseron, F., Feinstein, D.L., Jacobs, A.H., Wyss-Coray, T., Vitorica, J., Ransohoff, R.M.et al. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol., 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bossers, K., Wirz, K.T., Meerhoff, G.F., Essing, A.H., vanDongen, J.W., Houba, P., Kruse, C.G., Verhaagen, J. and Swaab, D.F. (2010) Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer's disease. Brain, 133, 3699–3723. [DOI] [PubMed] [Google Scholar]
- 15. Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J.Z., Menon, M., He, L., Abdurrob, F., Jiang, X.et al. (2019) Single-cell transcriptomic analysis of Alzheimer's disease. Nature, 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vogel, C. and Marcotte, E.M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet, 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao, J., Cheng, F. and Zhao, Z. (2017) Tissue-specific signaling networks rewired by major somatic mutations in human cancer revealed by proteome-wide discovery. Cancer Res., 77, 2810–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bai, B., Wang, X., Li, Y., Chen, P.C., Yu, K., Dey, K.K., Yarbro, J.M., Han, X., Lutz, B.M., Rao, S.et al. (2020) Deep multilayer brain proteomics identifies molecular networks in Alzheimer's disease progression. Neuron, 106, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao, J., Cheng, F., Wang, Y., Arteaga, C.L. and Zhao, Z. (2016) Systematic prioritization of druggable mutations in approximately 5000 genomes across 16 cancer types using a structural genomics-based approach. Mol. Cell. Proteomics, 15, 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santos, R., Ursu, O., Gaulton, A., Bento, A.P., Donadi, R.S., Bologa, C.G., Karlsson, A., Al-Lazikani, B., Hersey, A., Oprea, T.I.et al. (2017) A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov., 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang, Q., Yu, H., Zhao, Z. and Jia, P. (2015) EW_dmGWAS: edge-weighted dense module search for genome-wide association studies and gene expression profiles. Bioinformatics, 31, 2591–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jia, P., Zheng, S., Long, J., Zheng, W. and Zhao, Z. (2011) dmGWAS: dense module searching for genome-wide association studies in protein–protein interaction networks. Bioinformatics, 27, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manuel, A.M., Dai, Y., Freeman, L.A., Jia, P. and Zhao, Z. (2021) An integrative study of genetic variants with brain tissue expression identifies viral etiology and potential drug targets of multiple sclerosis. Mol. Cell. Neurosci., 115, 103656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan, F., Dai, Y., Iwata, J., Zhao, Z. and Jia, P. (2020) An integrative, genomic, transcriptomic and network-assisted study to identify genes associated with human cleft lip with or without cleft palate. BMC Med. Genet., 13, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manzoni, C., Kia, D.A., Vandrovcova, J., Hardy, J., Wood, N.W., Lewis, P.A. and Ferrari, R. (2018) Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief. Bioinform., 19, 286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bai, B., Wang, X., Li, Y., Chen, P.C., Yu, K., Dey, K.K., Yarbro, J.M., Han, X., Lutz, B.M., Rao, S.et al. (2020) Deep multilayer brain proteomics identifies molecular networks in Alzheimer's disease progression. Neuron, 105, 975, e977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson, E.C.B., Dammer, E.B., Duong, D.M., Ping, L., Zhou, M., Yin, L., Higginbotham, L.A., Guajardo, A., White, B., Troncoso, J.C.et al. (2020) Large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med., 26, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Leeuw, C.A., Mooij, J.M., Heskes, T. and Posthuma, D. (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol., 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Foster, E.M., Dangla-Valls, A., Lovestone, S., Ribe, E.M. and Buckley, N.J. (2019) Clusterin in Alzheimer's disease: mechanisms, genetics, and lessons from other pathologies. Front. Neurosci., 13, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu, W., Tan, L. and Yu, J.T. (2015) The role of PICALM in Alzheimer's disease. Mol. Neurobiol., 52, 399–413. [DOI] [PubMed] [Google Scholar]
- 31. Pan, J., Ma, N., Yu, B., Zhang, W. and Wan, J. (2020) Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflammation, 17, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adav, S.S., Park, J.E. and Sze, S.K. (2019) Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer's disease. Mol Brain, 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tasaki, S., Xu, J., Avey, D.R., Johnson, L., Petyuk, V.A., Dawe, R.J., Bennett, D.A., Wang, Y. and Gaiteri, C. (2022) Inferring protein expression changes from mRNA in Alzheimer's dementia using deep neural networks. Nat. Commun., 13, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao, J., Liu, X., Xia, W., Zhang, Y. and Wang, C. (2020) Targeting amyloidogenic processing of APP in Alzheimer's disease. Front. Mol. Neurosci., 13, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen, J.W., Lin, F.Y., Chen, Y.H., Wu, T.C., Chen, Y.L. and Lin, S.J. (2004) Carvedilol inhibits tumor necrosis factor-alpha-induced endothelial transcription factor activation, adhesion molecule expression, and adhesiveness to human mononuclear cells. Arterioscler. Thromb. Vasc. Biol., 24, 2075–2081. [DOI] [PubMed] [Google Scholar]
- 36. Yeh, H.I., Lee, P.Y., Su, C.H., Tian, T.Y., Ko, Y.S. and Tsai, C.H. (2006) Reduced expression of endothelial connexins 43 and 37 in hypertensive rats is rectified after 7-day carvedilol treatment. Am. J. Hypertens., 19, 129–135. [DOI] [PubMed] [Google Scholar]
- 37. Xia, D., Sui, R. and Zhang, Z. (2019) Administration of resveratrol improved Parkinson's disease-like phenotype by suppressing apoptosis of neurons via modulating the MALAT1/miR-129/SNCA signaling pathway. J. Cell. Biochem., 120, 4942–4951. [DOI] [PubMed] [Google Scholar]
- 38. Wu, G., Dawson, E., Duong, A., Haw, R. and Stein, L. (2014) ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. F1000Res, 3, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alcolea, D., Martinez-Lage, P., Sanchez-Juan, P., Olazaran, J., Antunez, C., Izagirre, A., Ecay-Torres, M., Estanga, A., Clerigue, M., Guisasola, M.C.et al. (2015) Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology, 85, 626–633. [DOI] [PubMed] [Google Scholar]
- 40.Dai, Y., Hu, R., Liu, A., Cho, K.S., Manuel, A.M., Li, X., Dong, X., Jia, P. and Zhao, Z. (2022) WebCSEA: web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Research, in press, gkac392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pei, G., Yan, F., Simon, L.M., Dai, Y., Jia, P. and Zhao, Z. (2022) deCS: A tool for systematic cell type annotations of single-cell RNA sequencing data among human tissues. Genomics, Proteomics & Bioinformatics, in press. [DOI] [PubMed] [Google Scholar]
- 42. Lake, B.B., Chen, S., Sos, B.C., Fan, J., Kaeser, G.E., Yung, Y.C., Duong, T.E., Gao, D., Chun, J., Kharchenko, P.V.et al. (2018) Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol., 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lauterborn, J.C., Scaduto, P., Cox, C.D., Schulmann, A., Lynch, G., Gall, C.M., Keene, C.D. and Limon, A. (2021) Increased excitatory to inhibitory synaptic ratio in parietal cortex samples from individuals with Alzheimer's disease. Nat. Commun., 12, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vico Varela, E., Etter, G. and Williams, S. (2019) Excitatory-inhibitory imbalance in Alzheimer's disease and therapeutic significance. Neurobiol. Dis., 127, 605–615. [DOI] [PubMed] [Google Scholar]
- 45. Sha, L., Chen, T., Deng, Y., Du, T., Ma, K., Zhu, W., Shen, Y. and Xu, Q. (2020) Hsp90 inhibitor HSP990 in very low dose upregulates EAAT2 and exerts potent antiepileptic activity. Theranostics, 10, 8415–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reinhardt, S., Stoye, N., Luderer, M., Kiefer, F., Schmitt, U., Lieb, K. and Endres, K. (2018) Identification of disulfiram as a secretase-modulating compound with beneficial effects on Alzheimer's disease hallmarks. Sci. Rep., 8, 1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan, L., Wang, H.F., Tan, M.S., Tan, C.C., Zhu, X.C., Miao, D., Yu, W.J., Jiang, T., Tan, L., Yu, J.T.et al. (2016) Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer's disease cohorts. Sci. Rep., 6, 26027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bertram, L., McQueen, M.B., Mullin, K., Blacker, D. and Tanzi, R.E. (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet., 39, 17–23. [DOI] [PubMed] [Google Scholar]
- 49. Narayan, P., Orte, A., Clarke, R.W., Bolognesi, B., Hook, S., Ganzinger, K.A., Meehan, S., Wilson, M.R., Dobson, C.M. and Klenerman, D. (2011) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat. Struct. Mol. Biol., 19, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tarasoff-Conway, J.M., Carare, R.O., Osorio, R.S., Glodzik, L., Butler, T., Fieremans, E., Axel, L., Rusinek, H., Nicholson, C., Zlokovic, B.V.et al. (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol., 11, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moldoveanu, T., Follis, A.V., Kriwacki, R.W. and Green, D.R. (2014) Many players in BCL-2 family affairs. Trends Biochem. Sci., 39, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martin-Rehrmann, M.D., Hoe, H.S., Capuani, E.M. and Rebeck, G.W. (2005) Association of apolipoprotein J-positive beta-amyloid plaques with dystrophic neurites in Alzheimer's disease brain. Neurotox. Res., 7, 231–242. [DOI] [PubMed] [Google Scholar]
- 53. Mercorio, R., Pergoli, L., Galimberti, D., Favero, C., Carugno, M., Dalla Valle, E., Barretta, F., Cortini, F., Scarpini, E., Valentina, V.B.et al. (2018) PICALM gene methylation in blood of Alzheimer's disease patients is associated with cognitive decline. J. Alzheimers Dis., 65, 283–292. [DOI] [PubMed] [Google Scholar]
- 54. Narayan, P., Sienski, G., Bonner, J.M., Lin, Y.T., Seo, J., Baru, V., Haque, A., Milo, B., Akay, L.A., Graziosi, A.et al. (2020) PICALM rescues endocytic defects caused by the Alzheimer's disease risk factor APOE4. Cell Rep., 33, 108224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu, R., Li, A., Sun, B., Sun, J.G., Zhang, J., Zhang, T., Chen, Y., Xiao, Y., Gao, Y., Zhang, Q.et al. (2019) A novel m(6)a reader Prrc2a controls oligodendroglial specification and myelination. Cell Res., 29, 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang, J., Ono, K., Dickstein, D.L., Arrieta-Cruz, I., Zhao, W., Qian, X., Lamparello, A., Subnani, R., Ferruzzi, M., Pavlides, C.et al. (2011) Carvedilol as a potential novel agent for the treatment of Alzheimer's disease. Neurobiol. Aging, 32(2321), e2321–e2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arbo, B.D., Andre-Miral, C., Nasre-Nasser, R.G., Schimith, L.E., Santos, M.G., Costa-Silva, D., Muccillo-Baisch, A.L. and Hort, M.A. (2020) Resveratrol derivatives as potential treatments for Alzheimer's and Parkinson's disease. Front. Aging Neurosci., 12, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. De Deyn, P.P., Drenth, A.F., Kremer, B.P., Oude Voshaar, R.C. and Van Dam, D. (2013) Aripiprazole in the treatment of Alzheimer's disease. Expert. Opin. Pharmacother., 14, 459–474. [DOI] [PubMed] [Google Scholar]
- 59. Zhu, L., Wu, G., Heng, W. and Zang, X. (2021) A comparative study of olanzapine, aripiprazole and risperidone in the treatment of psychiatric and behavioral symptoms of Alzheimer's disease. Pak. J. Pharm. Sci., 34, 2053–2057. [PubMed] [Google Scholar]
- 60. Khoury, R., Marx, C., Mirgati, S., Velury, D., Chakkamparambil, B. and Grossberg, G.T. (2021) AVP-786 as a promising treatment option for Alzheimer's disease including agitation. Expert. Opin. Pharmacother., 22, 783–795. [DOI] [PubMed] [Google Scholar]
- 61. O’Gorman, C., Jones, A., Cummings, J.L. and Tabuteau, H. (2020) Efficacy and safety of AXS-05, a novel, oral, NMDA-receptor antagonist with multimodal activity, in agitation associated with Alzheimer’s disease: results from ADVANCE-1, a phase 2/3, double-blind, active and placebo-controlled trial. Alzheimers Dement., 16, 9. [Google Scholar]
- 62. An, H., Choi, B., Park, K.W., Kim, D.H., Yang, D.W., Hong, C.H., Kim, S.Y. and Han, S.H. (2017) The effect of escitalopram on mood and cognition in depressive Alzheimer's disease subjects. J. Alzheimers Dis., 55, 727–735. [DOI] [PubMed] [Google Scholar]
- 63. Porsteinsson, A.P., Drye, L.T., Pollock, B.G., Devanand, D.P., Frangakis, C., Ismail, Z., Marano, C., Meinert, C.L., Mintzer, J.E., Munro, C.A.et al. (2014) Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA, 311, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sampath, D., Sathyanesan, M. and Newton, S.S. (2017) Cognitive dysfunction in major depression and Alzheimer's disease is associated with hippocampal-prefrontal cortex dysconnectivity. Neuropsychiatr. Dis. Treat., 13, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Greenwood, A.K., Montgomery, K.S., Kauer, N., Woo, K.H., Leanza, Z.J., Poehlman, W.L., Gockley, J., Sieberts, S.K., Bradic, L., Logsdon, B.A.et al. (2020) The AD knowledge portal: a repository for multi-omic data on Alzheimer's disease and aging. Curr Protoc Hum Genet, 108, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang, M., Beckmann, N.D., Roussos, P., Wang, E., Zhou, X., Wang, Q., Ming, C., Neff, R., Ma, W., Fullard, J.F.et al. (2018) The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data, 5, 180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. De Jager, P.L., Ma, Y., McCabe, C., Xu, J., Vardarajan, B.N., Felsky, D., Klein, H.U., White, C.C., Peters, M.A., Lodgson, B.et al. (2018) A multi-omic atlas of the human frontal cortex for aging and Alzheimer's disease research. Sci Data, 5, 180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang, X., Smits, A.H., vanTilburg, G.B., Ovaa, H., Huber, W. and Vermeulen, M. (2018) Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc., 13, 530–550. [DOI] [PubMed] [Google Scholar]
- 69. Chen, L.S., Wang, J., Wang, X. and Wang, P. (2017) A mixed-effects model for incomplete data from labeling-based quantitative proteomics experiments. Ann. Appl. Stat., 11, 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu, J.Z., Erlich, Y. and Pickrell, J.K. (2017) Case-control association mapping by proxy using family history of disease. Nat. Genet., 49, 325–331. [DOI] [PubMed] [Google Scholar]
- 71. Stark, C., Breitkreutz, B.J., Reguly, T., Boucher, L., Breitkreutz, A. and Tyers, M. (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res., 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Khanin, R. and Wit, E. (2006) How scale-free are biological networks. J. Comput. Biol., 13, 810–818. [DOI] [PubMed] [Google Scholar]
- 73. Harris, M.A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C.et al. (2004) The gene ontology (GO) database and informatics resource. Nucleic Acids Res., 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., 57, 289–300. [Google Scholar]
- 75. Jia, Z., Liu, Y., Guan, N., Bo, X., Luo, Z. and Barnes, M.R. (2016) Cogena, a novel tool for co-expressed gene-set enrichment analysis, applied to drug repositioning and drug mode of action discovery. BMC Genomics, 17, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pei, G., Dai, Y., Zhao, Z. and Jia, P. (2019) deTS: tissue-specific enrichment analysis to decode tissue specificity. Bioinformatics, 35, 3842–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.