

Abstract
The field of single-atom catalysis (SAC) has expanded greatly in recent years. While there has been much success developing new synthesis methods, a fundamental disconnect exists between most experiments and the theoretical computations used to model them. The real catalysts are based on powder supports, which inevitably contain a multitude of different facets, different surface sites, defects, hydroxyl groups, and other contaminants due to the environment. This makes it extremely difficult to determine the structure of the active SAC site using current techniques. To be tractable, computations aimed at modeling SAC utilize periodic boundary conditions and low-index facets of an idealized support. Thus, the reaction barriers and mechanisms determined computationally represent, at best, a plausibility argument, and there is a strong chance that some critical aspect is omitted. One way to better understand what is plausible is by experimental modeling, i.e., comparing the results of computations to experiments based on precisely defined single-crystalline supports prepared in an ultrahigh-vacuum (UHV) environment. In this review, we report the status of the surface-science literature as it pertains to SAC. We focus on experimental work on supports where the site of the metal atom are unambiguously determined from experiment, in particular, the surfaces of rutile and anatase TiO2, the iron oxides Fe2O3 and Fe3O4, as well as CeO2 and MgO. Much of this work is based on scanning probe microscopy in conjunction with spectroscopy, and we highlight the remarkably few studies in which metal atoms are stable on low-index surfaces of typical supports. In the Perspective section, we discuss the possibility for expanding such studies into other relevant supports.
1. Introduction
The field of “single-atom” catalysis has expanded rapidly over recent years with highly efficient and active catalysts demonstrated for a wide variety of chemical,1−6 photochemical,7,8 and electrochemical9 reactions. While the concept seems well established by the sheer number of studies and has been extensively reviewed,4,10−24 in many cases it is not really clear if and how the single atom really catalyzes the reaction.11 Recent advances in transmission electron microscopy have made it possible to routinely demonstrate the existence of isolated heavy atoms on an as-synthesized catalyst,25 and the location of the heavy atom can be determined relative to the lattice of the support. One must remember, however, that the TEM image is a 2D projection of a 3D object, so it is not possible to know whether any atom is located at the surface without multiple projections. Moreover, the support lattice shows columns of atoms from the bulk of the material, not the surface atoms to which the metal atom is bound. For the oft-used metal oxide supports, the oxygen sublattice is typically not resolved at all, and it is to these atoms that the metal atom is proposed to bind. Even if they were visible, metal oxide surfaces are often not simple truncations of the bulk structure (i.e., they reconstruct to a minimum energy configuration) and typically contain a variety of different defects. Such sites are thought to bind metal adatoms strongly (knowledge derived from surface-science-type studies) and thus likely play a significant role in SAC. Further guidance on the type of site comes from complementary spectroscopies such as XANES, IRAS, and XPS, but these area-averaging techniques do not necessarily give information on the active site (which may be a minority species) and are somewhat indirect, as will be discussed in this review.
A second major issue in SAC research is that even if the state of the as-synthesized catalyst can be determined, proving that the system remains atomically dispersed during catalytic reactions remains challenging.11,26,27 It is possible that the system evolves in the reactive environment to form small nanoparticles and that these are really the active site. “Postmortem” imaging of samples is not routinely performed, but even in cases where it is, doubts linger as to whether the system could have redispersed once outside the reaction environment. As a consequence, SAC remains controversial and there remains significant scope for fundamental insights.
Ultimately, atomic-scale details regarding the active sites and reaction mechanisms are proposed primarily on the basis of density functional theory (DFT) calculations. Periodic slab calculations based on a low-index facet of the support material are used, which may or may not appear on the powder catalyst. A suitable adsorption site for the metal adatom is then commonly selected based on a strong binding energy relative to other possible sites in DFT with some guidance from experiment. For example, if CO-IRAS measurements suggest the metal is cationic and XANES suggests coordination to oxygen then cation-like sites on or within an idealized surface may be tested. With a site selected, the reaction pathway is studied and a mechanism is proposed. Given the assumptions made about the nature of the support surface and the educated guess at an adsorption site, these calculations represent a plausibility argument, which shows that the reaction could proceed in this way. It is not proof that it does so. Similar caveats exist for the results of theoretical screening studies, which attempt to determine the best metal atom for a particular reaction, because the result depends strongly on the site and mechanism assumed, as seen recently for CO oxidation on FeOx-supported SACs.28−30
Clearly then, the complexity of the catalyst makes it difficult to assess whether the site and reaction pathways proposed on the basis of theory are realistic. In this review, we will cover the pertinent literature from the surface-science community that can help to understand how SAC works. In the surface-science approach, a single-crystalline sample exhibiting a low-index surface orientation is prepared under UHV conditions (typically by Ar+ sputtering and high-temperature annealing) until it is free of contamination such as OH groups and carbon. In this state, the system resembles an experimental analogue of that simulated by periodic slab calculations. Crucially, the atomic-scale structure of the surface has been determined with sub-Angstrom precision for several common support materials such as TiO2, CeO2, MgO, Fe3O4, and Fe2O3. While such “model” surfaces necessarily lack some of the complexity of applied SAC systems, they can serve as a solid basis for experimental studies of SAC mechanisms and as a benchmark for the theoretical approach used in high surface area catalytic studies. Typically, the metal atoms are evaporated directly onto the surface, meaning there are no ligands, and no calcination or activation of the system is performed prior to study.
In what follows, we will discuss what work exists in the surface-science literature that is relevant to SAC, ordered by different support materials. Five main sections give an overview of TiO2, the iron oxides FeOx, CeO2, MgO, and Cu2O. At the end of each section, we summarize the state of the surface-science research for SACs on a given oxide, including key takeaways. The section on TiO2 is further split into rutile and anatase TiO2, while the section on iron oxides in turn addresses three different FeOx facets α-Fe2O3(0001), α-Fe2O3(11̅02), and Fe3O4(001). When discussing adatoms on the oxides that have been most extensively studied by surface-science methods (TiO2, Fe3O4, and CeO2), we loosely group elements by position in the periodic table or by similar observed behavior of adatoms. Finally, in the Perspective, we summarize the state of research on these different model systems, discuss it in the context of other work, and give an overview of promising directions for further research.
2. Titania (TiO2)
2.1. Rutile TiO2(110)
Rutile TiO2(110) is one of the most intensively studied systems in surface science.31−33 This is partly because single-crystal samples of high quality are inexpensive and widely available and partly because UHV preparation can be easily achieved by in situ cycles of inert gas sputtering followed by annealing. This treatment results in a slight reduction of the sample, which is sufficient to allow experiments based on electron transfer (STM, XPS, etc.). The structure of the as-prepared surface is precisely known from quantitative structural techniques (SXRD/LEED-I(V)),34,35 and the results agree with DFT calculations36 and fit well with the results of scanning probe studies.37 Interestingly, the contrast observed in STM is reversed from the topography, the low-lying Ti5c atoms are imaged bright, and the protruding “bridging” O2c rows are imaged dark (see Figure 1a and 1b).31 This is related to the electronic structure and the fact that the Ti atoms have electronic states close to the Fermi level (EF). Specifically, the samples are n-type due to the sample reduction, and the Ti states are located at the conduction band minimum. The newest generation of scanning probe instruments allows simultaneous imaging by STM and noncontact AFM, allowing one to image both the atomic and the electronic structures of the surface. Due to the reduction of the sample, surface oxygen vacancies (VO) are common defects. They are imaged as bright protrusions in the dark O2c rows in STM and as missing atoms in the O2c rows in ncAFM (Figure 1a and 1c, respectively). The reduced surface is often denoted r-TiO2(110). One of the most important conclusions from the work on TiO2(110) has been the importance of VO as active sites for both chemical reactions and the nucleation of metal nanoparticles.31−33 For example, water molecules react with the oxygen vacancies creating two “surface hydroxyl” groups, i.e., a pair of H atoms adsorbed at the O2c rows. This reaction is highly efficient, and the residual water in a UHV environment is sufficient to fill all of the VO sites over several hours. When saturation exposure to water creates a partially hydroxylated surface, it is often referred to as h-TiO2(110). Similarly, exposing the r-TiO2(110) surface to very small amounts of molecular oxygen results in the repair of the vacancy and the adsorption of an O atom atop the Ti5c rows. The resulting surface is then termed o-TiO2(110). It is important to note that realistic catalytic environments will thus not have surface VO present in an appreciable number.
Figure 1.

STM and nc-AFM imaging of the rutile TiO2 (110) surface. Same area of the sample is shown with (a) empty states STM, (b) filled states STM, and (c) AFM. All images were measured at a sample temperature T = 78 K. (d) Structural model of the surface. STM and AFM images were measured in constant-height mode. Adapted from ref (37). Copyright 2017 American Physical Society under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
2.1.1. Cu, Ag, and Au on TiO2(110)
It seems well established that Au1 prefers to bind at VO sites on r-TiO2(110).38,39 Thornton and co-workers39 imaged the VO sites directly by STM (see Figure 2) and then observed Au atoms to occupy exactly these positions after deposition in UHV. Moreover, they performed voltage pulses with the STM tip which caused the Au atom to hop out of the vacancy, which could be clearly observed after the Au had left.
Figure 2.

STM images of the as-prepared TiO2(110) surface with oxygen vacancies (black circles in a) and following deposition of Au metal at room temperature. Au atoms clearly occupy the original VO sites. Reprinted with permission from ref (39). Copyright 2017 American Chemical Society under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
Recently, a very nice experiment/theory paper revisited the Au1/TiO2(110) system with a particular focus on the photocatalytic properties.40 The authors confirmed that Au1 adatoms preferentially occupy VO sites, and once these were filled, they observed that the Ti5c sites became occupied at 80 K. Interestingly, the authors mentioned that the adsorption configuration obtained in DFT is affected by the size of the computational supercell as well as by the inclusion of a VO in the model. With a VO concentration corresponding to the experimental conditions, a vertical Ti–Au bond is obtained rather than the tilted geometry obtained previously on stoichiometric slabs.41 A particularly interesting aspect of this study was the measurement of localized metal-induced gap states below EF by scanning tunneling spectroscopy, which the authors showed provides a dedicated channel for the transfer of a photoexcited hole from the TiO2 substrate to the adsorbed Au atoms. The hole transfer could be accomplished by UV light exposure or by the STM tip and was found to weaken the Ti–Au bond at the 5-fold-coordinated Ti site, allowing the Au atoms to diffuse across the surface at 80 K. Atoms adsorbed at the VO sites were unaffected by UV exposure.
Other interesting insights into adatom behavior can be inferred from surface-science studies of nanoparticle nucleation. A comparison of Au42 and Ag43 nanoparticle growth on the r-, h-, and o- TiO2(110) surfaces was performed by Wendt and co-workers, who found that the presence of hydroxyl groups accelerates sintering for both metals. Interestingly, a surface oxygen adatom on the o-TiO2(110) surface was suggested to provide an even stronger binding defect site than a VO. DFT calculations show that the metal atoms bind between the oxygen adatom and a surface “bridging” oxygen atom in a 2-fold coordination. Note that Ag has a 2-fold coordination in the Ag2O bulk oxide. For Ag, a bond strength of 1.35 eV was obtained with a diffusion barrier of approximately 0.95 eV.
STM investigations of Ag on r-TiO2(110) find no evidence for stable Ag single atoms. Indeed, Ag clusters seem to be formed already at 100 K44 even if the surface is bombarded by Ar to induce additional surface defects. Cu immediately forms clusters on TiO2 upon deposition at room temperature,45 but oxygen exposure leads to the formation of 2D islands.
Very little experimental data exists for single Cu atoms on rutile TiO2(110). DFT-based calculations predict that Cu binds in a bridging position on oxygen terminal rows on the stoichiometric surface with a binding energy in the range from −230 to −265 kJ mol–1.46,47 This is greater than the adsorption energy at oxygen vacancy sites, suggesting a difference in behavior from Au. In general,41 it seems that noble metal atoms can bind as cations on unsaturated oxygen atom sites or as anions on unsaturated Ti atom sites. Giordano et al.47 commented that the stronger binding of Cu and Ag over Au stems from the lower ionization potential of these elements, which more readily give away the outermost s electron and become cationic. Au, on the other hand, is extremely electronegative and readily accepts electrons from surface VO sites leading to a stronger bond in this location. This trend can also be viewed through the absolute Lewis acid hardness.48 Since Au (Lewis hardness 3.5) is harder than Cu (3.25), it is expected to bind more strongly than Cu on reduced rutile TiO2, where it acts as a Lewis acid in its interaction with a VO. The opposite is then true on stoichiometric TiO2, where Au acts as a Lewis base in its interaction with bridging oxygen atoms. The latter concept is useful in understanding how doping the TiO2(110) surface affects noble metal atom binding. The presence of Cl defects makes the surface more basic and thus electron transfer more difficult, which reduces the Au bond strength accordingly48 and explains why the presence of Cl leads to enhanced sintering of Au clusters on TiO2(110).49 Interestingly, recent DFT calculations have shown that iodine doping might be able to enhance the binding of Ag, Cu, and Pd adatoms on TiO2(110). While no additional electron transfer was found that could explain a significant increase in the adsorption energies for the metal at the O2c site in comparison to other halogen-doped surfaces, the authors note significant hybridization between the metal, O2c, and I states. This suggests that I forms covalent bonds to the metals through the TiO2 surface.48 It will be fascinating if this stabilization could be verified by experiment, as to date the doping of the metal oxide is a little studied strategy to affect the stabilization of metal adatoms.
2.1.2. Ni, Pd, and Pt on TiO2(110)
A room-temperature STM study of Ni adsorption on r-TiO2(110) reported 3D Ni clusters on the terrace sites even at extremely low coverage.50 Interestingly, annealing these Ni clusters in O2 resulted in their breakup and the formation of 2D oxidized Ni islands.45 An EXAFS study published the same year51 by the same group suggested that Ni atoms preferentially occupy step edges in a Ti substitutional site. Several DFT studies calculated the favored position for hypothetical Ni adatoms on r-TiO2(110),52,53 with the most modern calculations favoring adsorption directly above an in-plane oxygen atom. It seems likely that this could only be stabilized at low temperature.
Pd adsorption was studied by STM by Goodman and co-workers, who reported that the smallest stable species was a Pd dimer54 and found a distinct preference for Pd clusters to occupy the step edges. There have been several theoretical studies55 which find that Pd atoms would, in principle, be most stable in a VO site. On a stoichiometric surface, the Pd adatom adsorbs in a similar way to Ni,56 described above, in a hollow site between bridging and in-plane O atoms. The difference to the site directly above the in-plane O atom is negligible, and the diffusion barrier for diffusion along the direction of the bridging oxygen rows is less than 0.05 eV. This well explains why dimers and larger clusters rapidly form in the Pd/TiO2(110) system.
Onishi and co-workers were one of the first groups to intentionally image Pt atoms adsorbed on r-TiO2(110).57 They identified three adsorption sites using noncontact AFM: atop the 5-fold Ti atoms, atop the bridging O rows, and in bridging VO sites. Only the atoms in the VO sites were immobile during room-temperature imaging. Subsequent studies by Perez et al.58,59 using ncAFM and DFT calculations suggested that the mobile species observed on the bridging oxygen rows by Onishi were most likely OH groups, which form through reaction of residual water with VO.
Wang and co-workers studied the adsorption of 0.01 ML Pt atoms on r-TiO2(110) using STM and compared their data to theoretical calculations (see Figure 3).60 They found Pt atoms to adsorb solely at the VO sites at 80 K with no evidence for occupation of the Ti5c sites or the bridging oxygen rows. Accompanying theoretical calculations suggested the Pt atoms trapped at a VO protrude higher than the neighboring bridging oxygen atoms and bond to two 6-fold-coordinated Ti atoms forming a symmetric Ti–Pt–Ti configuration along the [001] direction (Figure 3d). The excess electrons from the vacancy accumulate at the Pt atom, leading to a Bader charge of 0.9 e–. Note that this differs from the general idea that the metal adatoms prefer substitutional cation sites on oxide materials and are positively charged. Exposure to CO led to Pt–CO species situated at the VO sites, but the CO leans toward one of the neighboring Ti rows (Figure 3e and 3f). While the β-Pt–CO configuration was found to be the more stable configuration by almost 0.5 eV, the α-Pt–CO seemingly fits better to what is observed in STM. The authors also measured STM after O2 exposure and found evidence for molecular adsorption at the Pt sites at 78 K. Unfortunately, no study of the thermal stability of the adatoms was conducted as part of this work, so it was not clear if Pt situated at VO sites would be stable at elevated temperatures. Extremely recently, the present authors’ group performed a study using STM and confirmed that Pt atoms are indeed stable at VO sites at room temperature.61
Figure 3.

STM images at 80 K showing 0.01 ML Pt adsorbed on the r-TiO2(110) surface before (a) and after (b) exposure to CO. Red and white crosses in a show the position of VO on the surface prior to Pt deposition. (b) Pt-related protrusions become larger (red) due to the adsorption of CO and move away from the vacancy site. This suggests the CO tilts toward the neighboring Ti row. Yellow arrows in b highlight CO adsorbed on the Ti rows. DFT models of the clean surface (c), Pt (orange) adsorbed in the VO site (d), and two variants of the Pt–CO species that were calculated (e, f). CO carbon and oxygen atoms are drawn in gray and black, respectively. Reprinted with permission from ref (60). Copyright 2017 AIP Publishing.
Room-temperature ncAFM experiments of Pt adsorption on a h-TiO2(110) surface (i.e., all VO sites removed by reaction with water) were performed by Pérez and co-workers.59 Following the deposition of Pt, large but uniform protrusions were observed atop the Ti5c rows, which the authors suggest are due to Pt atoms mobile around a single Ti5c site. Theoretical calculations suggest that the Pt atom can interact with a number of different nearby surface oxygen sites. The features assigned as Pt atoms appear as the brightest protrusions on the surface irrespective of the AFM imaging mode (i.e., the nature of the tip termination), partly because they interact strongly with the AFM tip, a feature which allows one to easily distinguish them from the surface OH groups in ncAFM studies. In a follow up paper,58 the same group also acquired KPFM images of the system in which the species assigned as Pt atoms appear significantly darker than the surrounding TiO2 support. This suggests charge transfer from the Pt atom into the surface, consistent with their simulations. It is nevertheless surprising that Pt adatoms would be stable on the Ti rows given that Pt atoms are seemingly able to diffuse at 80 K and find the available VO sites on the r-TiO2(110) surface. It is of course possible that the presence of the hydroxyl species on the surface affects the mobility of the Pt species, and it would be interesting to know the diffusion barrier along the Ti rows in this situation from DFT.
This conclusion of stable Pt on the h-TiO2(110) surface seems at odds with the results of Wendt and co-workers,62 who compared Pt nanoparticle nucleation on the r-TiO2(110), o-TiO2(110), and h-TiO2(110) surfaces at 90–110 K, 300 K, and after annealing at 800 K. The location of the Pt atoms was not the central focus of the study, but nonetheless, the observation of the nanoparticles and accompanying theory sheds light on the differing behavior on these three surfaces. At low temperature, Pt nanoparticles were already observed at a coverage of 0.025 ML in all cases with no apparent preference for step edge or terrace sites. Similar results were found at room temperature for the reduced and oxidized surfaces, but the surface with hydroxyl groups exhibited significantly larger nanoparticles more frequently found at step edges. DFT calculations predicted low diffusion barriers of 0.33 and 0.39 eV on the stoichiometric and reduced surface, respectively, suggesting that the as-deposited metal would diffuse even at 150 K. Pt1 adatoms were considered to be trapped at a point VO (−4.28 eV vs a gas-phase Pt atom), while an oxygen atom can also function as a trapping site. Here, the Pt atom is bound between the oxygen adatom and a surface bridging oxygen in a 2-fold configuration. Interestingly, the authors predicted that the major difference with the h-TiO2(110) surface is the immobility of the surface defect, as the Pt atom can diffuse as a Pt1H entity with a barrier less than 0.5 eV. The VO and O adatom, in contrast, do not diffuse at these temperatures. After annealing at 800 K, no difference is observed because trapping is not effective at this temperature.
One further possibility for a Pt adsorption site at the TiO2(110) surface was proposed by Chang et al. in 2014 on the basis of high-angle annular dark-field (HAADF) STEM data.63 The authors suggested that Pt atoms can occupy 5 different sites at room temperature, with most residing in oxygen vacancy sites located within the Ti–O basal plane (rather than the bridging oxygen vacancy row). The authors rationalized this surprising finding using DFT calculations, which showed that while the formation energy of the bridging oxygen vacancy is lower than the in-plane vacancy by 0.15 eV, the total energy gained by placing Pt in an in-plane vacancy is greater. Later, they performed DFT+U calculations that suggested that in-plane oxygen vacancies could coexist with bridging oxygen vacancies in a dilute defect regime. Nevertheless, it is important to note that this defect and/or metal occupation has not been observed in scanning probe studies in UHV, and it is possible that the preparation of the surface by in-air annealing or the electron beam utilized in the experiments might have had an effect. It is known, for example, that annealing a TiO2(110) sample in O2 results in an irregular termination due to the oxidation of Ti interstitials at the surface.64
As mentioned above, the Thornton group39 published a paper in which it was clearly demonstrated using STM that Au atoms occupy bridging oxygen vacancies, not in-plane vacancies. The seemingly incontrovertible proof of the Au position was addressed in a second HAADF-STEM study,65 which agreed that Au indeed occupies a regular bridging VO site. The authors finally concluded that strong hybridization between Pt-5d6s/Obr-2p orbitals and Pt-5d/Ti5c-3d orbitals is responsible for a 1 eV gain in adsorption energy for occupation of a bridging oxygen vacancy over an in-plane vacancy, despite charge transfer clearly being higher in the latter case. It would certainly be very nice to see a similar study to that performed by Thornton and co-workers for Pt atoms to ascertain once and for all if in-plane VO occupation is possible.
2.1.3. Co, Rh, and Ir on TiO2(110)
No studies of Ir or Rh adsorption on single-crystal TiO2(110) in a low-coverage regime could be found. However, a recent study of Rh atoms on rutile TiO2 nanoparticles is accompanied by a thorough theoretical analysis with predictions made for what might be observed.66Figure 4 shows that Rh atoms prefer to substitute a Ti cation in a 6-fold surface site under oxidizing conditions (beneath the bridging oxygen row, black line in Figure 4). This is consistent with the experiment insofar as no CO can be adsorbed to conduct an IRAS experiment after the sample was annealed in an oxygen atmosphere. As mentioned above, annealing a TiO2(110) sample in oxygen leads to the oxidation of Ti interstitials at the surface and the growth of new material, i.e., growth of new TiO2 at step edges or as new terraces.64 At O2 chemical potentials between −1.7 and −2.5 eV, a VO is predicted to form above the Rh cation, which reduces its coordination to 5-fold (green line in Figure 4). Interestingly, in extremely reducing conditions, the Rh atom is found to prefer a “supported geometry”, which is a site above an in-plane oxygen atom in which the Rh is coordinated to both Ti and O atoms from the surrounding surface (blue line in Figure 4). Note that viewed from above (as in a STEM experiment), this site could look very much like adsorption in an in-plane VO. The authors go on to discuss the influence of H2 and CO atmospheres and show that adatom geometries can compete with the stable substitutional ones in realistic catalytic conditions. Finally, the authors show that the undercoordinated Rh configuration can coordinate two CO molecules, while the substitutional geometry cannot. IRAS experiments conducted in a CO-rich atmosphere clearly show the signature of the Rh(CO)2 dicarbonyl, consistent with the theoretical predictions. It is important to recognize, however, that the experiments were performed on nanoparticles with a 30 nm diameter, while the calculations utilized terrace sites on a TiO2(110) slab using periodic boundary conditions. The direct comparison is thus not ideal but does nevertheless serve to validate the theory that the Rh adatoms change their adsorption site depending on the environment.
Figure 4.

Atomistic thermodynamics for substitutional (@) and supported (/) Rh geometries at the TiO2(110) surface. (a) Relative stability as a function of oxygen chemical potential Δμ(O). Optimal structures for substitutional (b) and supported (c) Rh SAs on the considered TiO2 surfaces. Lowest energy structures are also shown as insets in a. Color code: O, red; Ti, blue; Rh, green. Adapted with permission from ref (66). Copyright 2019 Springer Nature under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
Very recently, the present authors’ group studied Rh adsorption on r-TiO2(110) using room-temperature STM in UHV conditions. Isolated Rh atoms were observed after deposition at 100 K with no preference for the VO sites. Annealing to 150 K was already sufficient to sinter the adatoms into small clusters, which grew larger upon annealing to 300 K. Consequently, it seems that Rh diffusion is facile on TiO2(110), and this will prevent the stabilization of Rh1 species at temperatures relevant for SAC.61
Chen and co-workers67 compared the nucleation and growth of Co particles to other metals (Au, Ni, and Pt). In similar conditions, cluster sizes increase in the order of Co < Pt < Ni < Au, suggesting Co has a stronger interaction with the TiO2(110) surface.
2.1.4. Fe on TiO2(110)
STM images of Fe at the TiO2(110) surface suggest that Fe can be stabilized at the VO sites.68 Small clusters are also imaged after room-temperature Fe deposition, and while significant sintering occurred after heating to 473 K, the features assigned to single Fe atoms survived.
2.2. Anatase TiO2
Most, but not all, surface-science studies of anatase TiO2 focus on the most stable (101) surface. After preparation in UHV, the surface exhibits a sawtooth-like surface termination that exposes a mixture of fully coordinated and undercoordinated Ti and O atoms69 (see Figure 5). The surface is well characterized in terms of its structure70,71 and molecular adsorption72−74 and is thus in principle suitable as a model system for SAC purposes. One of the key differences between anatase TiO2(101) and rutile TiO2(110) is that surface oxygen vacancies are not stable on the former surface. Even if such species are created artificially (by low-energy electron bombardment), they diffuse into the bulk already at low temperature and a stoichiometric surface is recovered.74,75 Thus, such sites would not be expected to be available to stabilize metal atoms under ambient conditions.
Figure 5.

Bulk-terminated surfaces of (a) anatase (101) and (b) anatase (001). Adapted from ref (76). Copyright 2018 MPDI under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
2.2.1. Au and Pt on Anatase TiO2
Experimental surface-science studies of metal adsorption on anatase TiO2(101) are limited and mostly predate a particular interest in obtaining isolated single atoms. Diebold, Selloni, and co-workers studied Pt and Au evaporated onto anatase TiO2(101) using STM and found that both systems sinter already at low coverage, resulting in nanoparticles.77 Au was found to interact weakly with the surface (adsorption energy just 0.25 eV) with adatoms computed to be most stable directly above a Ti5c atom. The weak interaction with Au leads to large nanoparticles in experiment, and a strong preference for the step edge was observed in room-temperature STM images. When the support was irradiated with electrons to create VO sites prior to Au deposition, smaller clusters were observed on the terraces, suggesting nanoparticle nucleation occurred at the VO sites. This is consistent with the much larger adsorption energy of 3.16 eV computed for Au1 at a VO site.
In the case of Pt, clusters nucleated both on the terrace and at the step edges (see Figure 6), and the observation of atom-sized protrusions was interpreted as Pt single atoms (Figure 6, inset i). However, the site of these species atop surface O2c atoms disagreed with the DFT+U-predicted site, and the protrusions appear similar to those observed subsequently for adsorbed water. Slightly larger protrusions were tentatively assigned to dimers and trimers. Theoretical calculations suggest that the optimal location for a Pt atom is in between two O2c sites, close to two Ti5c and two Ti6c surface atoms. The average Pt–O2c and Pt–Ti distances were ∼2.04 and 2.78 Å, respectively. The Pt1 adsorption energy was found to be fairly strong at 2.2 eV, but no diffusion barrier was computed. Pt deposition on a reduced anatase TiO2(101) surface with O2c vacancies results in smaller Pt clusters, and the accompanying DFT calculations suggest Pt would have a much stronger adsorption energy (4.7 eV) at the VO site.
Figure 6.
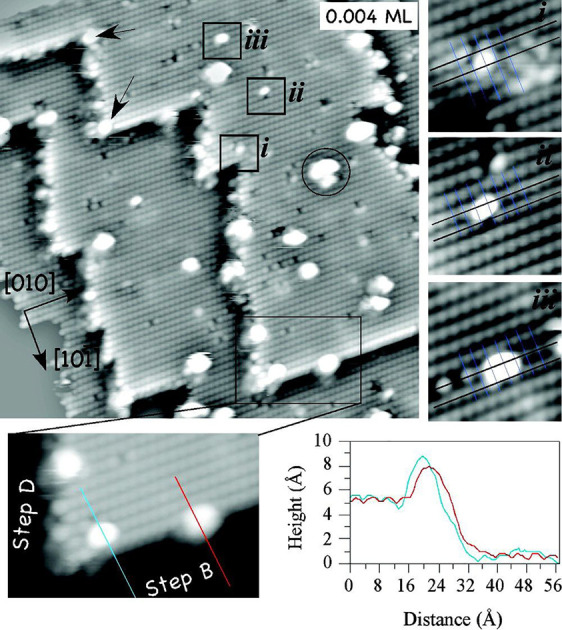
STM images of the anatase(101) surface after deposition of 0.004 ML Pt. Inset and line profiles at the bottom show that Pt clusters at step edges are mostly located at the upper terraces, with kink sites being preferred nucleation sites (see black arrows). Clusters in the insets (i–iii) were tentatively assigned as a (i) Pt monomer, (ii) Pt dimer, and (iii) Pt trimer. Reprinted with permission from ref (77). Copyright 2008 American Chemical Society.
More recently, an extremely thorough computational analysis of the Pt1/anatase TiO2(101) system was performed by Pacchioni’s group to accompany experiments performed on a powder catalyst. Interestingly, the Pt1/TiO2 catalyst was prepared with an extremely low loading such that most particles of the support would contain just one atom, thus avoiding the possibility for agglomeration into clusters.78−81 The system was characterized by IRAS as a function of temperature, allowing both the CO-stretch signature and the CO binding energy of the Pt species to be simultaneously determined. These parameters can be extracted from DFT-based models of the system and were used to test the validity of the results. Occupation of different sites clearly makes a large difference in the properties of the metal atom and through this the CO binding properties. However, none of the simple candidate positions for Pt on an anatase TiO2(101) surface (adatom bound to surface oxygen, substitutional cation site, occupying an oxygen vacancy, etc.) reproduced the CO-stretch frequencies and binding energies observed experimentally. Ultimately, the authors proposed that additional coordination to surface OH groups could produce good agreement. Such species are omnipresent on metal oxide surfaces in realistic conditions but are almost never considered in SAC calculations. It is important to note however that this does not constitute proof that this configuration is actually present on the samples. Nevertheless, this work clearly shows that the use of simplistic models is not sufficient to capture the complexity of real SAC systems.
2.2.2. Rh on Anatase TiO2(101)
There is a dearth of experimental surface-science studies of Rh, Ir, or Ni atoms on anatase TiO2(101). However, Christopher and Pacchioni performed an important study that shows how the typical pretreatments performed in SAC affect the properties of a Rh/TiO2 powder system.80 Following a standard solution-based synthesis procedure, the catalyst was heated in O2 at 350 °C, ostensibly to remove the ligands. The resulting material did not adsorb CO at room temperature at all, suggesting that the Rh does not reside at the surface of the TiO2 nanoparticles. The authors assumed that Rh atoms substitute cations in the bulk TiO2 lattice, which seems reasonable (note, the present authors’ group has shown this phenomenon directly for Fe3O4 and α-Fe2O3).82,83 In any case, reducing the sample by heating in either H2 or CO atmosphere modifies the catalyst such that CO adsorption becomes possible at Rh sites, and the authors clearly see the IRAS signature of the Rh dicarbonyl. This suggests the presence of isolated Rh atoms. While the room-temperature IRAS signature was the same whether CO or H2 was used as the reductant, the thermal evolution of the systems was clearly different. Following CO pretreatment, both CO molecules desorb simultaneously from the dicarbonyl at 240 °C, whereas following H2 reduction, some fraction of the species forms a distinct intermediate. It makes sense that the surface reduced in H2 might exhibit hydroxyl groups, and DFT calculations were indeed able to show that Rh(OH)(CO)2 could produce the properties observed in experiment. The key takeaways from this study, however, are that the pretreatment makes a significant difference in the resulting properties of the catalyst and that room-temperature IRAS alone is not sufficient to distinguish the different species on the surface.
2.3. Conclusions
Overall, TiO2 remains an interesting model system for SAC because of the rich interaction of adatoms with its various defects. The surface-science experiments performed on rutile TiO2(110) to date reveal that VO’s are the most stable sites to stabilize electronegative metal atoms. VO sites react strongly with water, however, and thus will not be present at the surface during wet synthesis of real systems. Moreover, the calcination treatment typically employed in the synthesis of a real SAC system is oxidizing and thus will not lead to the generation of VO sites. It is likely, however, that VO sites will be created during activation of the catalyst (which generally involves heating in a reducing atmosphere) and that metal atoms previously stabilized in other sites, or in clusters, could migrate to VO sites and remain stable there in a reactive environment. As such, it is interesting to consider what catalytic properties the resulting negatively charged metal adatoms might have, and there are several computational investigations where Au1/r-TiO2(110), for example, is predicted to be a good catalyst system.84−87 As yet, there have not been experimental investigations to confirm these predictions. More fundamentally, it would be interesting to study how a UHV-prepared TiO2(110) surface is modified by realistic calcination and reduction treatments and if isolated Au or Pt adatoms reside in VO sites afterward. Other metals of interest, for example, Rh, which do not preferentially occupy VO sites, seem to diffuse and sinter too readily to be promising SAC systems unless they could be stabilized by coordination to additional ligands.
The studies to date on anatase (101) provide little evidence for stable metal adatoms. In contrast to rutile (110), VO’s are not stable on the surface, but it is possible that they could be formed during reduction and rapidly occupied by diffusing metal adatoms. It is important to note that the experiments performed by the Christopher group that suggest the Pt/anatase system to be active for CO oxidation utilized a very low loading that ensured each anatase particle supported only one metal adatom. As such, the sintering observed at room temperature in surface-science experiments was avoided. It would of course be possible to investigate the most stable site at cryogenic temperatures, but combining structural studies of reactivity will inevitably be difficult. In the present authors’ opinion, it seems that the TiO2 surfaces primarily studied so far are not ideal model systems for SAC research. It is the stable nature of these surfaces that makes them somewhat unreactive, however, and it would be interesting to learn if the different structures and potential coordinations presented by other less stable facets could enhance adatom stabilization.
3. Iron Oxides (FeOx)
Much of the pioneering work on single-atom catalysis from the group of Zhang and co-workers utilizes iron oxide as the support.29,88,89 In their 2011 Nature Chemistry paper,90 for example, it was shown that Pt atoms bound to iron oxide particles are cationic and that these species catalyze CO oxidation and preferential oxidation of CO (PROX) as efficiently as Pt nanoparticles. The iron oxide support employed by Zhang and co-workers is nominally α-Fe2O3, but the FeOx notation acknowledges that the surface is likely reduced following activation (i.e., heating in a reducing atmosphere). Iron has three stable oxides (FeO, Fe3O4, Fe2O3), but intermediate stoichiometries are possible, and it is relatively easy to change between them, particularly at the surface.91 Depending on the conditions, it is possible that a hematite (α-Fe2O3) surface can even change phase locally to form Fe3O4, even if the bulk remains Fe2O3. Nevertheless, for simplicity, the theoretical calculations accompanying studies of FeOx-based catalysts utilize an idealized α-Fe2O3(0001) structure. However, surface-science studies indicate a highly complex surface phase diagram for the α-Fe2O3(0001) facet, and the idealized (1 × 1) iron or oxygen terminations assumed in most DFT studies may be too simplistic. In what follows, we briefly discuss what is known about metal adsorption on α-Fe2O3(0001) before discussing recent work on a different low-energy facet: α-Fe2O3(1102). Following this, we describe what has been learned from what we consider to be the best characterized model system for SAC: Fe3O4(001).
3.1. α-Fe2O3(0001)
The (0001) facet of hematite has been studied extensively using the surface-science approach, but significant disagreement remains about its possible terminations and especially their respective stability regions.91 Most studies report iron- or oxygen-terminated bulk truncation models,92,93 but experimental evidence for a ferryl termination has also been reported.94,95 More critically, UHV preparation also often results in a complex superstructure with ∼4 nm periodicity, generally referred to as the “biphase” termination.96−99 The nature of this biphase structure remains poorly understood at an atomic scale and is effectively inaccessible to theory due to the large number of atoms per unit cell. DFT modeling of FeOx-based SAC systems generally assumes one of the bulk-truncated (1 × 1) terminations. Adatoms are placed in 3-fold hollow sites of the oxygen-terminated surface,90 which is equivalent to adatoms substituting iron in the iron-terminated surface. Catalytic activity has been theoretically screened for a range of different metals in this configuration.28,100 However, the experimental evidence for the adatom site comes mainly from TEM, which shows the single atoms in cation-like positions with respect to the hematite bulk.90 This makes the assignment of an Fe substitution site plausible, but other explanations are also still possible.
In the surface-science literature, there is no experimental evidence for single atoms being stabilized on the α-Fe2O3(0001) surface. Few deposition experiments have been attempted, likely due to the overall complexity of the surface. In a recent study, Lewandowski et al. deposited Fe and Au on the biphase termination and showed that both metals initially accumulate in only one of the three distinct regions found in the superstructure.98 Small gold clusters prepared in this manner have subsequently been shown to be active for CO oxidation.101 However, even if some single atoms were present in these experiments, they are clearly not stabilized at high loadings, and the biphase likely is not a good representation of nanoparticle catalyst surfaces.
Overall, there remains a significant experimental gap between theoretical models assuming a simple (1 × 1) bulk truncation and actual catalysts, where the surface termination is unknown. However, both the variety of reported terminations and the difficulty in theoretical treatment of the “biphase” make α-Fe2O3(0001) an extremely challenging and ultimately unattractive model system for surface science.
3.2. α-Fe2O3(11̅02)
The α-Fe2O3(1102) surface is nonpolar and exists in a (1 × 1) bulk truncation after UHV preparation, although a reduced (2 × 1) termination is formed in reducing conditions.102 This well-defined structure is in principle ideal for SAC studies, and the first such investigation was published recently. Upon deposition at room temperature, Rh was shown to form clusters (Figure 7).83 However, when the sample is heated, the clusters disappear and the Rh atoms become incorporated in the lattice of the support. On the basis of a combination of STM, low-energy ion scattering (LEIS), and DFT, it was concluded that the redispersed Rh atoms are located in the immediate subsurface layer.83 Of course, this means that the Rh atoms will be inaccessible for reactants, and thus, this system will likely not be active as a single-atom catalyst. However, it illustrates that great care must be taken in identifying single atoms as active sites, as the subsurface Rh atoms may easily be identified as single atoms in cation-like sites at the surface by TEM.
Figure 7.

STM images of 0.025 ML Rh on α-Fe2O3(1102). (a) 0.025 ML Rh as deposited on the clean α-Fe2O3(1102)-(1 × 1) surface at room temperature (Usample = +3 V, Itunnel = 0.3 nA) and (b) after annealing at 500 °C for 15 min in UHV (Usample = −2.8 V, Itunnel = 0.1 nA). (c) 0.025 ML Rh as deposited on the clean α-Fe2O3(1102)-(2 × 1) surface (Usample = −3 V, Itunnel = 0.1 nA) and (d) after annealing at 300 °C for 10 min in UHV (Usample = −2.8 V, Itunnel = 0.1 nA). Reproduced with permission from ref (83). Copyright 2021 Wiley VCH under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
While Rh on α-Fe2O3(1102) forms clusters at room temperature in UHV, it can be stabilized by coadsorption with water, which is stable on this surface up to 345 K.103 This results in Rh(OH)2 species (Figure 8), which are mobile at room temperature but do not agglomerate to clusters.104 This is interesting because water and hydroxyl groups are omnipresent on metal oxides in atmospheric conditions, so their presence should be generally taken into account in the modeling of oxide-supported SAC systems.
Figure 8.

Rh on α-Fe2O3(1102) stabilized by coadsorbed water. (a) ncAFM image acquired at liquid He temperature of 0.05 ML Rh on α-Fe2O3(1102), deposited at room temperature in a partial pressure of 2 × 10–8 mbar H2O, then heated to 80 °C to desorb all water not coordinated to Rh. (b) Schematic model (top view) for the features indicated by green arrows in a. Rh adatom (gray) is stabilized by two OH groups (Owater in blue, hydrogen in white). Zigzag rows of surface oxygen are marked in blue in both panels. Orange arrow highlights a third protrusion that is sometimes present, which was attributed to an additional water molecule atop a surface Fe and hydrogen bonded to one of the OH groups. Reproduced with permission from ref (104). Copyright 2022 American Chemical Society.
3.3. Fe3O4(001)
Fe3O4(001) is an ideal model system to study isolated adatoms under UHV conditions. Following preparation by Ar+ sputter/anneal cycles in UHV, the surface exhibits a (√2 × √2)R45° [also known as c(2 × 2)] periodicity. STM images reveal undulating rows of Fe atoms running in the [110] directions, consistent with a termination at the plane containing both octahedrally coordinated Fe and oxygen (see Figure 9). The surface layer is bulk-like in terms of stoichiometry but is distorted due to an ordered array of cation vacancies and interstitials in the immediate subsurface. The proposed structure was confirmed by a combination of quantitative LEED105 and DFT+U calculations as well as SXRD106 measurements. Crucially for our purposes here, the reconstruction has been shown to stabilize ordered arrays of metal adatoms of almost any variety91 and is thus an ideal model system to study fundamental processes in SAC. In what follows, we summarize the main results from almost 10 years of work on this surface. However, the focus will be on publications post 2015, as an extensive summary of prior work already exists as part of a review of iron oxide surfaces.91
Figure 9.

(A) Schematic model of a bulk-terminated Fe3O4(001) surface. (B) Schematic model of the surface structure obtained following preparation in ultrahigh vacuum. Interstitial Fe atom with tetrahedral coordination (Feint) replaces two Fe with octahedral coordination (Feoct) from the third layer. (C) Top view of the subsurface cation vacancy (SCV) model showing with a yellow × the site in which metal adatoms adsorb on the Fe3O4(001) surface. (D) DFT+U calculation showing Au adatoms adsorbed on Fe3O4(001) at the maximum coverage (defined as 1 monolayer). (E) Surface energy versus O2 chemical potential for the two terminations shown in A and B, as calculated by DFT. Subsurface cation vacancy structure is more stable in all conditions reachable experimentally. Reproduced with permission from ref (105). Copyright 2014 American Association for the Advancement of Science.
3.3.1. Cu, Ag, and Au on Fe3O4(001)
The stabilization of metal adatoms on Fe3O4(001) was first reported for Au in 2012.107 It was shown that Au adatoms adsorb midway between the rows of Fe atoms imaged in STM. This location is consistent with 2-fold coordination to surface oxygen atoms (see Figures 9 and 10) in a site which is essentially where the next tetrahedrally coordinated Fe atom would be if the spinel structure were continued outward. The Au atoms were stationary in STM movies at room temperature and remained stable against thermal sintering to temperatures as high as 700 K. On the basis of a Monte Carlo simulation, it was suggested that the coverage threshold coincided with the probability for two adatoms to be deposited directly into the same unit cell. The thermal sintering at 700 K (also observed for other metals) seems to be linked to an order–disorder transition that occurs in the surface reconstruction at this temperature. For coverages in excess of 2.1 × 1013 cm–2, Au clusters were observed to coexist with adatoms after deposition at room temperature.108,109 STM images of the surface after heating the mixed system were suggestive of a “rolling snowball mechanism” of cluster growth, whereby the clusters diffuse at elevated temperature and pick up otherwise stable adatoms they encounter.
Figure 10.

(A) Representative STM images (Usample = +1–1.5 V, Itunnel = 0.1–0.3 nA) showing metal adatoms adsorbed between the Fe rows of the Fe3O4(001) support. Text in each panel indicates the DFT+U-derived adsorption energies, Bader charges, and heights of the Me1 adatom (z) above the surface Fe atoms in the 2-fold adsorption geometry as well as the CO-induced vertical displacement (Δz). (B–D) DFT+U-derived minimum energy structure for the 2-fold-coordinated Cu1/Fe3O4(001) adatom before (B) and after (C) adsorption of CO as well as the Pd1CO carbonyl (D), which is lifted from the surface. (E) IrCO replaces a 5-fold-coordinated surface Fe atom during the TPD ramp, meaning that CO desorption ultimately occurs from the depicted 5-fold Ir1 geometry. Adapted from ref (112). Copyright 2021 American Association for the Advancement of Science.
Experimental observations for Ag were somewhat similar to Au, although the coverage threshold for cluster formation was significantly higher (ca. 7 × 1013 cm–2 or 50% of the available sites occupied).110 Using DFT+U calculations, it was shown that the Ag dimer is unstable compared to two Ag adatoms on Fe3O4(001). This was consistent with the observation of Ag adatom mobility at room temperature in STM movies, suggesting that cluster nucleation required a particular minimum size.
Cu adatoms can be stabilized to even higher coverages than Ag and Au,111 and there is evidence that a second adsorption site can be occupied once the standard configuration (2-fold coordinated to lattice oxygen) becomes saturated. The protrusion appears to be located in the “wide” phase of the surface reconstruction, which puts the adatom directly above the Feint atom in Figure 9c. It is not clear if the Feint remains in place, and it seems more likely that it moves to occupy one of the neighboring Feoct vacancies in the layer below.
DFT+U-based calculations suggest that all three noble metal atoms take a 1+ oxidation state when adsorbed on Fe3O4(001). This makes sense because all three metals are 2-fold coordinated to oxygen in their native oxides, where they take a 1+ oxidation state. The assignment is supported by XPS binding energies, which compare well between Cu and Ag adatoms and literature values for the metal oxides.111 Quantitative normal incidence X-ray standing wave experiments further support the theoretical model of the adsorption geometry with excellent agreement for the position of the Ag and Cu adatoms with respect to the surface. However, this agreement is contingent on the theoretical lattice parameter being constrained to the experimental value, as expanding the lattice widens the separation of the surface oxygen atoms to which the adatoms bind, causing them to sink lower into the surface to obtain the same binding length. This issue was not encountered with the hybrid functional calculations, primarily because the theoretical lattice parameter comes out very close to the experimental value.111
3.3.2. 3d Transition Metals (Ti, Mn, Co, and Ni) on Fe3O4(001)
All of the 3d transition metals excluding Cu exhibit a similar behavior.113,114 Upon deposition, the adatoms occupy the same 2-fold coordination as shown for Au in Figure 9d, but this is unstable against incorporation in the Fe3O4 lattice. This is straightforward to understand because all of these metals form stable solid solution ferrite compounds with the spinel structure (MeFe2O4, where Me = metal). The temperature at which incorporation occurs increases from left to right in the periodic table, and a significant proportion of Ti is already incorporated upon deposition at room temperature. This is possible because Fe vacancies exist in the subsurface, and these can be occupied by either the adatom itself or an Fe atom displaced from the first layer. If the sample is heated above 700 K then all of the foreign metal diffuses to the bulk of the sample and is undetectable by XPS. Thus, in contrast to the noble metals discussed above, which sinter to clusters, the 3d transition metals are thermodynamically driven to disperse within the oxide.
3.3.3. Rh and Ir on Fe3O4(001)
Rhodium and iridium adsorb in the standard 2-fold coordination on Fe3O4(001). These metals are not 2-fold coordinated in their stable bulk oxides however and thus prefer to incorporate within the Fe3O4 lattice where octahedral coordination to oxygen can be achieved (see Figure 10E). In both cases, a temperature of at least 500 K is required to initiate the transition. Despite this similarity, Rh ultimately disperses within the support bulk after prolonged heating, whereas Ir leaves the lattice and forms large metallic clusters (see Figure 11). This reflects the higher cohesive energy of Ir metal versus Rh and the greater oxophilicity of Rh (as judged by the heat of formation of the most stable oxide). For both metals, 6-fold coordination in the subsurface layer is energetically favored over 5-fold coordination in the surface layer. This has implications for SAC because an atom with 6-fold coordination is inaccessible and will not interact strongly with reactants.
Figure 11.

(A) Ir1 atoms evaporated directly onto the Fe3O4(001) surface at 300 K are imaged as bright protrusions between the Fe rows of the support (red circle in STM image). Double protrusions are metastable Ir2 dimers (orange arrow). (B) DFT-derived minimum-energy structure of the 2-fold-coordinated Ir adatom on Fe3O4(001). (Inset in A) STM simulation based on this structure. (C) After annealing at 623 K, Ir atoms appear as bright protrusions within the Fe row in STM images (green circle). (D) DFT-derived minimum energy structure of the 5-fold-coordinated Ir atom incorporated within the Fe3O4(001) surface. (Inset in C) Corresponding STM simulation. (E) At 723 K, some of the bright protrusions within the row are replaced by extended bright protrusions in STM (yellow circle). Some small irregular clusters are also observed. (F) DFT-derived minimum energy structure of the 6-fold-coordinated Ir adatom incorporated in the subsurface layer of Fe3O4(001). (Inset in E) STM simulation based on this structure. (G) Annealing at 973 K leads to formation of metallic Ir clusters with an apparent height of ∼3 nm. Reproduced with permission from ref (115). Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
CO adsorption has been studied on both Rh and Ir adatoms on Fe3O4(001) using surface-science techniques.82,112,115 It was found that CO binds strongly but that adsorption does not lead to destabilization and sintering (as is the case for Pd and Pt). Indeed, the adsorption of a single CO molecule causes the adatom to relax toward the surface, which is linked to the formation of a weak bond to a subsurface oxygen atom. With this, the metal atom takes a pseudosquare-planar environment, akin to the structure found in Ir(I) and Rh(I) complexes. The adsorption of a second CO molecule leads to the formation of a dicarbonyl, and together with the bonds to the support, the metal adatom achieves a square-planar configuration. It is interesting to note that the CO desorption observed in TPD experiments ultimately occurs from a metal atom with 5-fold coordination, because the switch to octahedral coordination in the surface layer occurs with the CO still attached.
For Rh, it was observed that exposure to O2 even at very low pressures leads to destabilization and sintering. The resulting RhOx clusters are active for CO oxidation and are difficult to distinguish from Rh adatoms in XPS. This suggests one must be careful assigning Rh1 species on the basis of cationic signature in spectroscopy.
3.3.4. Pt and Pd on Fe3O4(001)
Pt116 and Pd117 both form 2-fold-coordinated adatoms on Fe3O4(001) upon room-temperature deposition. The adatoms are stable to high coverage and temperature (700 K), but both are destabilized by CO, which is omnipresent even in the UHV environment. PdCO and PtCO species are mobile at room temperature, which leads to agglomeration. In the case of Pd,117 STM movies suggest that PdCO can be immobilized if they encounter a surface hydroxyl group before a second PdCO species, which suggests that Pd1 might be more stable in a realistic environment where the support is hydroxylated. The alternative possibility, which happens often in the movies, is the nucleation of a Pd cluster and further sintering into Pd nanoparticles. In the case of Pt,116 the most likely event is the formation of a Pt2(CO)2 species. These are stable and immobile in UHV at room temperature but break apart if the sample is heated to approximately 500 K. The final state is a mixture of clusters and adatoms, suggesting that further diffusion occurs at this temperature. In addition to destabilization by CO, the same effect has also been reported for Pd adatoms in the presence of methanol.118
Very recently,119 it was shown that the decomposition of the (PtCO)2 dimer is linked to the production of CO2. On the basis of quantitative, isotopically labeled TPD measurements and DFT, it was concluded that Pt dimers react with the surface through extraction of oxygen from the lattice. Interestingly, the DFT calculations (see Figure 12) show that both the (PtCO)2 and the Fe3O4(001) supports must adopt a metastable configuration for the reaction to proceed at the temperature observed, because this ultimately reduces the energy required to stabilize the Pt2CO intermediate. This then breaks into a Pt atom and a PtCO, which can diffuse at elevated temperature, explaining why a mixture of adatoms and larger clusters remains in STM images after the reaction.
Figure 12.

Reaction scheme for (PtCO)2 species on Fe3O4(001). Reaction occurs via a metastable configuration of the (PtCO)2 and Fe3O4(001) support, which allows extraction of lattice oxygen at minimum energetic cost. In the schematics, the Feoct and Fetet of the Fe3O4(001) support are dark blue and cyan, respectively. O atoms are red, Pt are white, and C and O in CO are black and red, respectively. Reproduced from ref (119). Copyright 2022 American Association for the Advancement of Science under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
3.3.5. CO Adsorption Trends on Fe3O4(001)-Based SACs
Recently, a systematic investigation of CO adsorption on Fe3O4(001)-based Au, Ag, Cu, Ni, Pt, Rh, and Ir SACs was published.112 The CO desorption temperature observed in TPD was converted to desorption energy assuming an ideal lattice gas and compared directly to the results of DFT calculations. The results reveal similar trends to those observed for close-packed metal surfaces (see Figure 13) with some key differences. The noble metals bind weakest and Rh and Ir strongest. In most cases, the single atoms bind CO stronger than the close-packed metal surface, with Ni being the exception where a weaker interaction is observed. The differences were interpreted using the density of states extracted from DFT+U calculations, and it was concluded that the proximity of the d states to EF was a primary factor, as it is for metal surfaces. Adatoms in the 2-fold coordination geometry are all close to a 1+ oxidation state, so the electronic structure differs from a metal surface. In most cases, this results in a shift in the d-band center of mass to higher energies. However, this electronic effect is modulated by a couple of factors that play only a minor role for metal surfaces. For example, CO adsorption causes significant relaxations, which lower the adsorption energy from that expected on the basis of the d-band position alone. Also, there is the possibility, discussed above, that adsorption weakens the adatom–support interaction so much that a mobile metal carbonyl is created, which leads to sintering.
Figure 13.
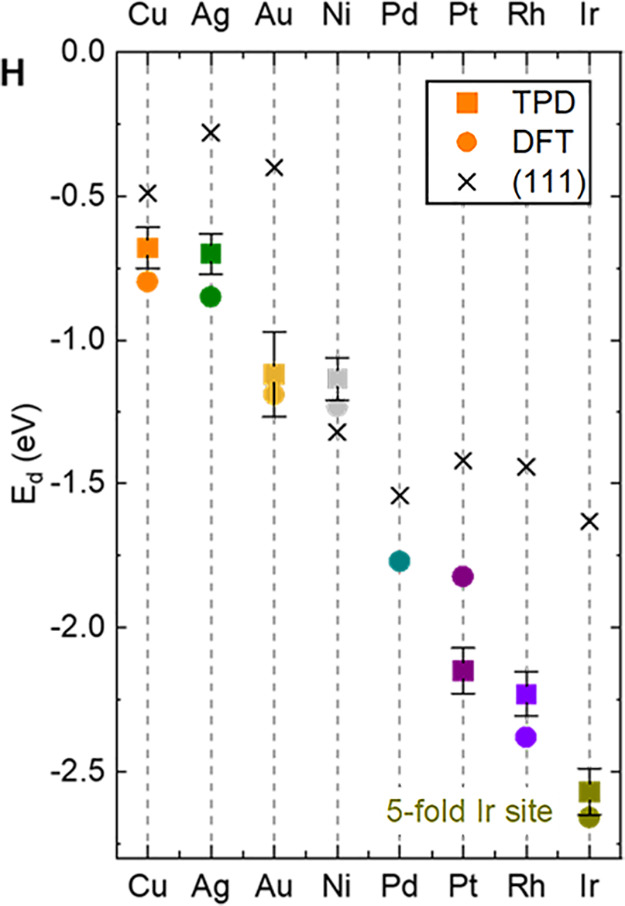
Plot of experimental and calculated CO adsorption/desorption energies alongside experimental values for respective metal (111) surfaces. Error bars for the experimental data assume a temperature uncertainty of ±10 K (±20 K for Au). Figure adapted with permission from ref (112). Copyright 2021 American Association for the Advancement of Science.
Overall, it was shown that the observed behavior can be understood in some cases by comparison to metal oxide surfaces if similar oxygen coordination exists. In this scenario, CO competes with the oxide to bind the metal adatom, and strong CO binding destabilizes the atom on the surface. In the case of Rh and Ir, however, the preference for square-planar and octahedral environments when CO adsorbs leads to a strengthening of the interaction with the support, and the 2-fold coordination of the adatom is easily modified by adsorption. Ultimately, the results clearly demonstrate that the adsorption properties of SAC systems are more closely related to coordination complexes than metal nanoparticles, which should influence the metals selected for a specific reaction.
3.3.6. H2 Activation Trends on Fe3O4(001)-Based SACs
Dohnalek and co-workers studied H2 activation on the Pd1/Fe3O4(001) system using STM and DFT120 and found that a very high density of surface hydroxyls is created when H2 is exposed to a surface with a low density of adatoms. This suggests that H2 dissociation occurs followed by spillover onto the oxide support to form surface OH groups. While OH diffusion is slow on the surface, it can be assisted by water, and this mechanism led to the migration of OH groups away from the Pd atoms. The experimental results for Pd matched well to the barriers predicted by DFT-based calculations, which gives confidence in the accuracy of the theory for this system. Similar calculations were then performed for a variety of metals (see Figure 14), and it was predicted that Pt will behave similarly to Pd, while heterolytic dissociation can also occur on Rh and Ir, leading to a possible equilibrium with hydride–hydroxyl pairs. For Ru, the hydride–hydroxyl pair becomes strongly preferred.
Figure 14.
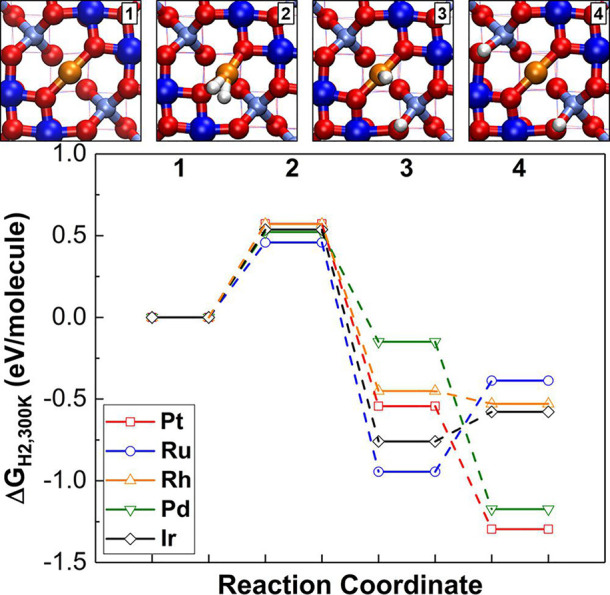
DFT-predicted energy pathway of H2 dissociation mechanism on a single metal (Pd, Pt, Rh, Ir, and Ru) atom on Fe3O4(001), corrected by gas-phase entropy at 300 K. Color code: oxygen, red; Fe, blue; 2-fold-coordinated metal adatom, orange; H, white. Reprinted with permission from ref (120). Copyright 2019 American Chemical Society.
3.4. Conclusions
Of the iron oxide surfaces, α-Fe2O3(0001) is most widely assumed in high surface area studies, but the adatom geometry generally utilized in DFT studies has not been reported by even a single experimental work. This is mainly because α-Fe2O3(0001) exhibits a variety of complex superstructures when prepared in UHV and is thus extremely challenging to prepare and poorly suited as a model system. However, it is questionable how representative such UHV-specific terminations of hematite would be in realistic conditions. Experiments have shown hydroxylation of α-Fe2O3(0001) surfaces in the presence of even low background pressures (<10–4 Torr) of water,121 and theory also predicts the high stability of hydroxylated surfaces.122 Therefore, a more realistic approach to construct a model system for this termination may be to intentionally hydroxylate it, though this may be challenging in UHV.
At the other extreme, Fe3O4(001) has proved to be an excellent model system in that it stabilizes high loadings of single atoms at room temperature. This has facilitated fundamental studies of a range of SAC properties with good agreement between DFT calculations and experimental work, including adsorption trends for simple molecules on a range of different elements. Unfortunately, the structural modifications induced by water mean that it will be difficult to correlate local coordination to chemical reactivity at elevated pressures.
Finally, we have recently begun investigating the α-Fe2O3(1102) surface as a SAC model system and found it to be highly promising. This facet is much easier to prepare than the (0001), and single Rh adatoms were stabilized at room temperature by coadsorbed water.104 Since the support in most powder catalyst works is hematite, rather than magnetite, we find this to currently be the most promising iron oxide model system for bridging high surface area studies and theory.
4. Ceria (CeO2)
Cerium oxide is a clear example of a catalyst support that strongly participates in reactions, in terms of not only electronic effects modifying the catalytic properties of supported metals but also acting as an oxygen reservoir. Due to the capability of Ce to reversibly transform between the two stable oxidation states of Ce4+ and Ce3+, ceria can easily exchange oxygen with the environment. This has multiple implications for single-atom catalysis: An abundance of different types of defects related to oxygen vacancies in the surface, in the subsurface, or at steps supplies potential sites to stabilize adatoms at low coverage. However, the low barriers for creating and repairing these defects also implies that they might change significantly under reaction conditions, potentially destabilizing the adatoms. Facile oxygen vacancy creation can also be relevant if oxygen for the reaction is supplied directly from the surface, i.e., in a Mars–van Krevelen mechanism.
The typical defects on CeO2(111) have been studied extensively. Surface and subsurface oxygen vacancies can easily be introduced by annealing in reducing conditions and give rise to Ce3+ ions at the surface.123 For many metals, the most relevant adsorption sites appear to be at step edges,124 which have also been studied in detail.125,126
Adsorption studies on a wide variety of metals on stoichiometric or reduced CeO2(111) can be found in the literature, though mostly at high coverages. Generally, these can be divided into metals that become fully oxidized and form mixed oxides with ceria at room temperature, such as Al, Ga, and Sn,127−129 and metals that quickly form metallic clusters. With few exceptions, clusters tend to preferentially decorate step edges on stoichiometric ceria130−132 or nucleate at surface defect sites on oxygen-deficient ceria films.133,134 It is worth noting that whether charge transfer occurs and whether the admetals are reduced or oxidized appears to be highly dependent on both the metal and the oxidation state of the ceria film.135−139
4.1. Pt, Pd, and Ni on CeO2
Stabilization of single Pt adatoms on CeO2 was studied extensively by the groups of Matolín and Libuda. A detailed review of the Pt/CeO2 system already exists,140 but we will summarize the findings here to put them in the context of SAC. While the ideal CeO2(111) surface does not provide sites to trap single Pt atoms, Matolín and Libuda used SRPES and DFT to identify a “nanopocket” stabilizing Pt2+ in a square-planar coordination on {100} nanofacets with an exceptionally high adsorption energy of −678 kJ/mol.141 TEM shows that such nanofacets indeed exist on CeO2 particles prepared by magnetron sputtering. In the surface-science studies, the nanofacet sites were prepared by codepositing Ce and Pt in an oxygen atmosphere on a CeO2(111) film under conditions suitable for forming CeO2 nanoparticles. While these nanoparticles on the CeO2(111) surface can still be imaged by STM, the roughness of the surface does not allow direct confirmation of the Pt adsorption site by scanning probe techniques. However, the Pt2+ particles are shown to resist reduction, sintering, and bulk diffusion up to the highest temperatures that could be achieved on that surface (ca. 750 K), in agreement with the high stability predicted by DFT. Interestingly, the calculated adsorption energy of Pt in the nanopocket exceeds the cohesive energy of bulk Pt (−564 kJ/mol), and DFT predicts that abstracting Pt from metallic clusters is possible (Figure 15).141 Indeed, at least partial redispersion from metallic particles to Pt2+ was later shown in experiment.140
Figure 15.

Structure and energetics of the anchored Pt2+ species on ceria nanoparticles determined by theory. Pt2+ is strongly bound at the {100} nanofacets of the ceria nanoparticle. Color coding of atoms: red, O; beige, Ce4+; brown, Ce3+; blue, Pt; white, H. Reproduced with permission from ref (141). Copyright 2014 John Wiley and Sons.
The high stability of Pt2+ in a surface site seems to recommend this system as a single-atom catalyst. However, the Pt atoms appear to be rather inactive: No CO is adsorbed above 110 K,142 and no dissociation of molecular hydrogen is observed.143 This is not surprising as the square-planar “surface pocket” already provides an ideal environment for the Pt2+ ion in d8 configuration, which disfavors further bonds. However, the catalysts can be activated by reducing Ce4+ to Ce3+, which destabilizes the single-atom sites (Figure 16). Annealing in hydrogen (when some metallic Pt is present to initialize hydrogen dissociation)143 or methanol144 or depositing Sn as a reducing agent145 thus accumulates the Pt2+ single atoms to subnanometer clusters, which serve as a “working state” for reaction. The authors speculate that a Pt–CeOx catalyst with ideal Pt loading will be able to reversibly cycle between atomically dispersed Pt2+ species and the active subnanometer particles.141 In this scenario, the single-atom sites are not involved in SAC in a literal sense but are still highly relevant for the long-term stability of the catalyst, as the regular redispersion prevents accumulation to larger clusters in the long run.
Figure 16.

Development of Pt 4f (a) and Pd 3d (b) spectra obtained from Pt–CeO2 and Pd–CeO2 films, respectively, following annealing in UHV (top), under H2 (middle), and O2 (bottom) atmosphere. Reproduced with permission from ref (140). Copyright 2017 Royal Society of Chemistry.
A structural motif similar to the “nanopockets” was identified by Matolín’s group at monolayer-high step edges on CeO2(111).124 By adjusting the step density and the density of surface oxygen vacancies (Figure 17), they showed with STM and PES that a high step density allows accommodation of large amounts (0.05 ML) of Pt2+ without any cluster formation. In contrast, surface oxygen vacancies only serve as anchoring points for metallic clusters but do not prevent their formation. While the Pt atoms are again not visible in STM, the authors identify likely adsorption sites at steps by DFT. These sites again feature square-planar PtO4 moieties and very high adsorption energies (5.0–6.7 eV). There is both theoretical146 and experimental147 evidence that stable surface peroxo units form after exposure of CeO2(111) to molecular oxygen and that accommodating excess O atoms at steps allows one to increase the amount of Pt2+ species established after the deposition of Pt, thus maximizing the “single-atom” capacity of the surface. Very recently, cycling between single Pt atoms and Pt clusters has also been demonstrated experimentally for Pt anchored at step sites.148 It should be noted that since steps are present on any CeO2(111) film, the step-supported type of Pt2+ species identified by Matolín would almost certainly also be present on the CeO2(111) films supporting CeO2 nanoparticles discussed above. Even if no step-like sites exist on the nanoparticles directly, they should still be abundant on the underlying film. This implies that the results on Pt2+ in “nanopockets”, showing their high stability but also inactivity,141−143 are transferable to Pt supported at monolayer steps.
Figure 17.

Nucleation of Pt and stabilization of Pt2+ on ceria surfaces containing a controlled amount of surface defects. (a–c) CeO2(111) surface with a low density of surface oxygen vacancies and ML-high steps. (d–f) CeO1.7 surface with increased density of surface oxygen vacancies. (g–i) CeO2(111) surface with increased density of ML-high steps. (a, d, g) STM images of clean surfaces before deposition of Pt. (b, e, h) STM images after deposition of 0.06 ML Pt and annealing at 700 K in UHV. All STM images 45 × 45 nm2, tunneling current 25–75 pA, sample bias voltage 2.5–3.5 V. Scale bar, 20 nm (a). (c, f, i) PES spectra of the Pt deposit after annealing. All PES spectra were acquired with photon energy hν = 180 eV (black points). Fits indicate metallic (Pt0, blue line) and ionic (Pt2+, red line) contributions to Pt 4f signal. EB is the photoelectron binding energy. Reproduced with permission from ref (124). Copyright 2016 Springer Nature under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
Even more generally, many nanoparticle studies of Pt/CeO2 report Pt to be situated on facets other than [111], which have not been as thoroughly investigated by surface-science methods. However, a recent study of Pt on a realistic ceria support, combining DFT with X-ray absorption spectroscopy (XAS), IRAS, and XPS,149 suggests that the results of Libuda and Matolín are also generalizable to other facets. While this study finds higher stability for Pt on the [110] and [100] facets, the stable site is always a 4-fold-coordinated pocket, and single-site Pt is initially inactive toward CO, C3H6, and CH4 oxidation. Catalytic activity again only sets in once Pt is significantly reduced and sinters to small clusters.
Generalizing these results from Pt, a DFT investigation of adsorption energies at the {100} nanofacet site found that for 11 different metals the nanofacet site was always preferred to adsorption on a metal nanoparticle.150 The largest differences in adsorption energies were found for group X metals (Ni, Pd, and Pt) and for Fe, Co, and Os, indicating that these metals should be stabilized against sintering by the “nanopocket”. Investigations on Pd and Ni indeed show that these metals are likewise stabilized in a 2+ state.151 However, unlike Pt, both Pd and Ni segregate to their native oxides under some conditions, and both can be stabilized in ceria bulk sites, leading to bulk diffusion above 600 K. Furthermore, annealing in hydrogen did not lead to a change of oxidation state for Ni, suggesting that the “active state” of metallic particles is inaccessible, likely because the Ni2+ species is too stable.
While the experimental results discussed above suggest that single Pt atoms on ceria are only activated by cluster formation, a more recent theoretical study has instead proposed a reaction pathway in which single Pt atoms on CeO2(100) can be both stable and active after a reductive activation step in sufficiently high pressures of H2 or CO, which puts them in a transient 2-fold-coordinated state.152 Crucially, the reaction pathway involves phonon-assisted switching of the platinum charge state during reaction through electron injection to (and recovery from) the support. While these transient configurations and charge states may be difficult to characterize experimentally, the authors argue that dynamic charge transfer needs to be taken into account in modeling, rather than assuming a fixed Pt charge state.152 Such a pathway could in principle be reconciled with the studies showing clustering if active single Pt atoms exist in a preparation window in which ceria is sufficiently reduced but at low enough temperature to prevent agglomeration.
Very recently, Wan and co-workers explored the effect of pretreating CeO2(111) films with oxygen plasma. They concluded that the plasma treatment causes nanostructuring of the surface as well as formation of peroxo species in the surface, as shown schematically in Figure 18. Even relatively high loadings of 0.2 ML Pt are apparently stabilized as single atoms on surfaces prepared in this way. Crucially, while at least some of the Pt atoms are in inactive “nanopocket” sites, other adsorption geometries are also present. The Pt single atoms are thermally stable and active for CO oxidation, as the authors demonstrate both for the thin film model system and for powder catalysts.
Figure 18.
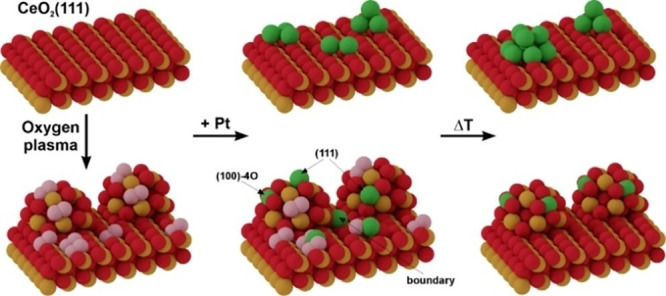
Schematic representation of the interaction of Pt atoms with pristine and oxygen plasma-treated CeO2(111) films. Upon deposition on a stoichiometric CeO2(111) surface, Pt forms small clusters which aggregate into larger Pt nanoparticles at elevated temperatures. Plasma pretreatment of the CeO2 surface produces peroxo species and induces surface restructuring, resulting in small ceria nanoparticles, which act as anchoring sites either directly upon Pt adsorption or through surface migration of peroxo-stabilized Pt single atoms. Color code: Ce, gold; O, red; Pt, green; peroxo O22–, pink. Reproduced with permission from ref (153). Copyright 2022 Wiley-VCH GmbH under CC-BY license (https://creativecommons.org/licenses/by/4.0/).
4.2. Au, Ag, and Cu on CeO2
While gold supported on ceria has been shown to be a promising single-atom catalyst,154 its coordination to the ceria support in the active state is contentious. On the stoichiometric CeO2(111) surface, theory predicts adsorption on oxygen bridge sites to be slightly preferred over top sites.155 However, it has been pointed out that the preferred site and the oxidation state of the gold atom on ceria strongly depend on the chosen theoretical method.156 Experimentally, when gold is deposited on stoichiometric CeO2(111) at 10 K, it is found in both oxygen top and oxygen bridge sites.157 Since gold accumulates at step edges at room temperature,130 this is likely due to limited mobility at 10 K. On the terrace sites, accumulating gold forms upright dimers and compact 3D clusters. From this and the absence of characteristic fingerprints of charged species in STM, the authors infer a close-to-neutral charge state of the aggregates.157
On oxygen-deficient CeO2(111), surface and subsurface oxygen vacancies are present, introducing Ce3+ sites at the surface.123 When gold was dosed on this surface at 10 K, some Au got trapped in surface oxygen vacancy sites, from which it could not be removed with the STM tip anymore.158 However, a later report139 points out that these defect sites are only rarely occupied. Rather, gold mostly binds to bridge sites when deposited at 15 K and then clusters at step edges upon annealing to 400 K, while undecorated surface oxygen vacancies remain visible (Figure 19). The authors conclude that while the oxygen vacancy is thermodynamically favored, diffusion into the vacancy site is kinetically hindered up to above 395 K. According to their DFT calculations, the large diffusion barrier (∼1.0 eV) is a consequence of Au having to change its charge state from +1 on the pristine surface, to 0 near the vacancy, to −1 in the vacancy in order to diffuse there.139
Figure 19.
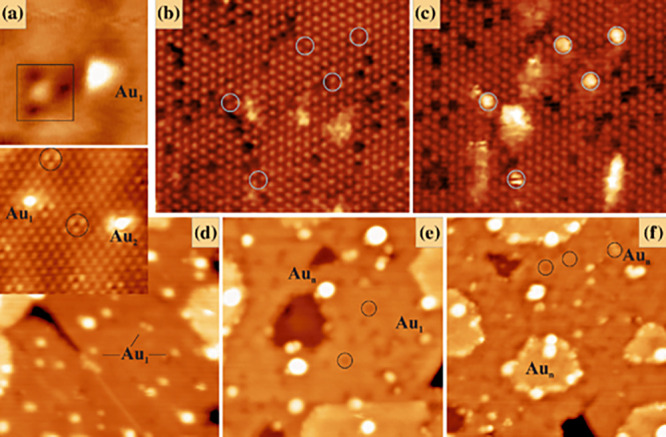
(a) High-resolution STM image of a regular and a defect-bound Au atom (box) deposited on CeO2–x/Ru(0001) at 15 K (+2.5 V, 3.8 × 3.0 nm2). (b, c) Identical surface region on a √3-reconstructed CeO2–x /Pt(111) film before and after exposure of 0.05 ML Au at 15 K (−3.0 V, 10.5 × 8.0 nm2). Adatoms and their binding sites on the pristine surface are marked by circles. (d) CeO2–x/Ru(0001) after gold deposition at 15 K and after annealing to (e) 200 and (f) 400 K (2.5 V, 30 × 30 nm2). (Inset in d) Close-up of the main image; some VOS defects are encircled. Reprinted with permission from ref (139). Copyright 2016 by the American Physical Society.
Interestingly, gold was found to interact not only with the surface oxygen vacancies directly but also with the Ce3+ ions introduced by subsurface oxygen vacancies.158 Dosed onto the defective surface at 10 K, Au atoms frequently appeared in pairs at a mean distance of 7.6 Å (two surface lattice constants), which DFT identifies as the expected spacing of the Ce3+ ions. The appearance of the paired features and the DFT results further suggest that charge transfer occurs at these sites, creating Au– species.
Ag and Cu single atoms on CeO2(111) have been explored much less than Au, but they appear to follow similar trends. Both accumulate mainly at step edges, with Ag interacting more strongly with the reduced surface,132,159,160 as is the case for Au. The experimental and theoretical evidence suggests that for both Ag132 and Cu137 single atoms and small clusters are oxidized on the pristine CeO2(111) surface but adopt a negative charge state when they are localized at surface oxygen vacancies. Interestingly, calorimetric studies show that while Ag interacts more strongly with reduced films than with the stoichiometric surface,133 the opposite is true for Cu,161 indicating different interaction with surface oxygen vacancies or with Ce3+ sites.
In the model studies discussed so far, the potential host sites considered for the catalyst metal are usually oxygen vacancies or adsites. In contrast, in nanoparticle studies, the active sites are often assigned as cerium substitution sites based on TEM.154,162 Such configurations have been considered for Cu and Ag in two recent works,163,164 although the applied doping levels (ca. 10 atom %) were much higher than those in SAC. Experimentally, both films were found to be more reducible by annealing in UHV than pure CeO2. This is in agreement with DFT, which predicts spontaneous formation of surface oxygen vacancies near the modifier cations.163 However, the Ag-modified films showed a lower concentration of Ce3+ cations than pristine ceria even in the presence of more oxygen vacancies.164 This is explained by reduction of Ag2+ to Ag+ being more favorable than reduction of Ce4+. This again highlights the difficulty in assigning sites based only on TEM in combination with DFT.
Short-lived single Au atoms have also been proposed as the active species in CO oxidation over ceria-supported Au nanoparticles based on molecular dynamics simulations.165 In the proposed mechanism, adsorbed CO induces gold atoms to break away from a nanoparticle as Au+–CO and diffuse on the surface, occupying on-top positions on surface oxygen atoms. Such a mechanism would probably not be recognized as a “single-atom” catalyst in most experiments, and indeed, it is debatable whether it should be considered as such as the dynamic Au+–CO complex can likely only be created in the presence of nanoparticles. Nonetheless, like the dependence of single-atom stability on the ceria oxidation state, this again highlights the strong influence of the environment on ceria-supported metals.
4.3. Rh on CeO2
The case of rhodium is interesting as a PES study from 2016 has shown more clearly than for other metals that it has the capacity to either oxidize or reduce a CeO2(111) film, depending on the film’s initial stoichiometry (Figure 20).138 While there are no atomic-scale images, the SRPES data in combination with DFT strongly suggest that Rh can either be in a cationic state on the stoichiometric surface or become anionic when occupying an oxygen vacancy. It is interesting to note, however, that in an STM study of larger amounts of Rh deposited on CeO2(111) at room temperature, clusters preferentially decorated steps with no difference between stoichiometric and reduced films.131 This may suggest that in contrast to what has been described for gold,134 Rh ions at oxygen vacancy sites do not act as nucleation centers for cluster formation.
Figure 20.

Evolution of the degree of CeOx reduction for various cerium oxide stoichiometries (1.98 > x > 1.67) during the consequent depositions of rhodium estimated from (a) XPS and (b) SRPES measurements. Reprinted with permission from ref (138). Copyright 2016 American Chemical Society.
4.4. Conclusions
On one hand, ceria is an excellent SAC model system in that a variety of elements can be stabilized as single atoms at steps or nanofacets when the surface is sufficiently oxidized. On the other hand, these stable sites appear to be universally inactive, and activity is obtained only when the single atoms are destabilized and sinter to small clusters.140 Recent work showing Ag and Cu in Ce substitutional sites seems promising,163,164 but it remains to be seen whether these dopant atoms remain at the surface and are accessible to adsorbates. Single atoms adsorbed on CeO2(111) have been studied at low temperatures, and in particular, the interrelation of charge state and adatom diffusion, which prevents Au adatoms from reaching the energetically favorable VO sites,139 is an interesting fundamental result. However, these adatoms generally sinter at room temperature and so are probably not the active site observed in high surface area studies. To our knowledge, there has not been any experimental work investigating stabilization or destabilization of adatoms by coadsorbates.
5. Magnesium Oxide (MgO)
MgO has received extensive attention over the years both as a catalyst support and as a thin insulating oxide. In fact, MgO was the basis for arguably one of the earliest works demonstrating single-atom catalysis on a model system, namely, acetylene cyclotrimerization on Pd1 at 300 K.166 Several types of defects which may play a role in stabilizing single adatoms and small clusters have been identified. Oxygen vacancies are found mainly at steps and can appear in the form of F0, F+ or F2+ color centers, where F+ and F0 correspond to one and two electrons trapped in the vacancy site, respectively. It has been demonstrated that existing vacancy sites can be charged by electron irradiation or simply by scanning with an STM tip,167−169 and new defect sites are created by higher electron doses (Figure 21).168
Figure 21.
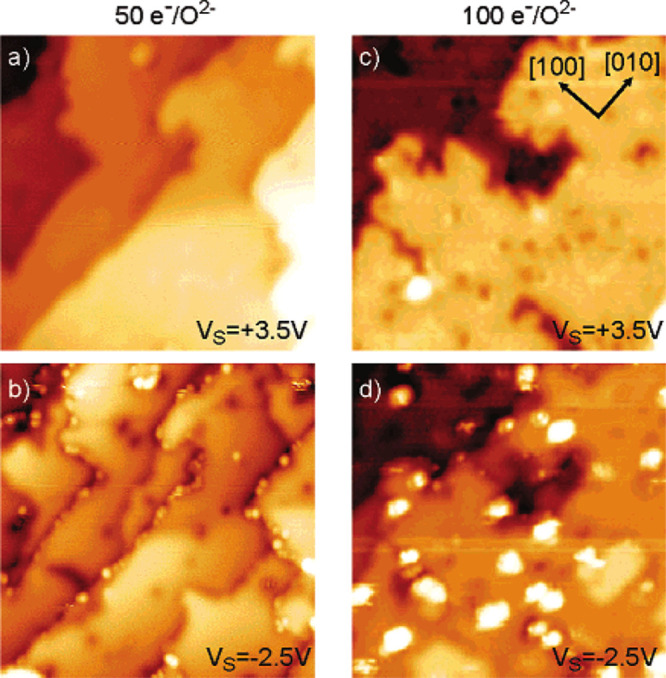
STM images (30 × 30 nm) of a 4 ML MgO(001) film grown on Ag(001) after bombardment with low (a, b) and high (c, d) electron doses. (a and c) Obtained at a sample bias of VS = +3.5 V, and (b and d) obtained at VS = −2.5 V. Reprinted with permission from ref (168). Copyright 2006 American Chemical Society.
A second family of defects are the so-called (H+)(e–) color centers, where deep electron traps are created in the vicinity of adsorbed protons. These defects also occur mainly at edges and kinks and can be created either by first adsorbing hydrogen, splitting it heterolytically, and then oxidizing the hydride by UV irradiation or by first creating an O– center through UV irradiation and then splitting H2 there to obtain H+ and a hydrogen radical.170−172 It is worth noting that while the existence of these (H+)(e–) centers has been demonstrated clearly by a combination of theory and electron paramagnetic resonance (EPR) spectroscopy, they have to the best of our knowledge not been identified by scanning probe techniques.
Finally, a defect predicted by theory to be relevant for trapping adatoms is the neutral divacancy in which an entire MgO unit is removed at the surface.173,174 Experimentally, evidence for this defect is scarce, though this may in part be explained by the fact that it may not be visible with most spectroscopic techniques (including EPR). Noncontact AFM images of cleaved MgO do indeed show pits that may correspond to divacancies, but unambiguous evidence is missing.175,176
5.1. Au on MgO
Probably the most extensively studied admetal on MgO is gold. An early experimental work on size-selected clusters demonstrated that charge (∼0.5 e) is transferred into adsorbed gold clusters and single adatoms on defect-rich films.177 This was tentatively attributed to charge transfer from F+ centers. While single atoms were essentially inert, Au8 was shown to be active for CO oxidation on defect-rich films, where charge transfer occurs, but not on defect-poor films, where it does not.177 In this, gold behaves differently from, e.g., Pd8, which was shown to be active no matter the defect concentration of the film.178 The negative charge of Au in this work is contrasted by more recent findings by the Freund group (Figure 22),179 who showed initial formation of positively charged gold, which they—also tentatively—attributed to charge transfer from gold into existing deep electron traps on the surface, such as grain boundaries of the MgO film. While these results seem contradictory at first glance, it seems entirely possible that the exact interaction of adatoms with a given MgO film depends on the type and density of defects as well as on the initial charge state of these defects.
Figure 22.

(a) IR spectra of CO adsorbed on 0.02 ML Au/13 ML MgO(001)/Ag(001) as a function of temperature. Spectra were collected at the indicated temperature. Lower panel presents the results as an image plot with red being intense and blue representing no absorption. (b) IR spectra of CO adsorbed on 0.02 ML Au/13 ML MgO(001)/Ag(001) as a function of annealing temperature. Spectra were collected after recooling to 90 K and dosing with CO. (c) Model of the Au/MgO(001) surface representing the nature of Au species formed at various annealing temperatures as deduced from the IR spectra shown in part b. Reprinted with permission from ref (179). Copyright 2011 American Chemical Society.
Concerning nucleation sites for small clusters, theory predicts strong trapping of Au adatoms in oxygen vacancies and divacancies.180 This seems to be confirmed by a low-temperature STM study in which a thin MgO film was first irradiated by electrons to introduce color centers and Au was then deposited at 5–8 K.181 At these lowest temperatures, Au atoms initially adsorb as single atoms and dimers at terrace sites (Figure 23). After annealing to 30 K, the EPR signal corresponding to color centers is quenched, suggesting that small gold clusters have formed there.
Figure 23.
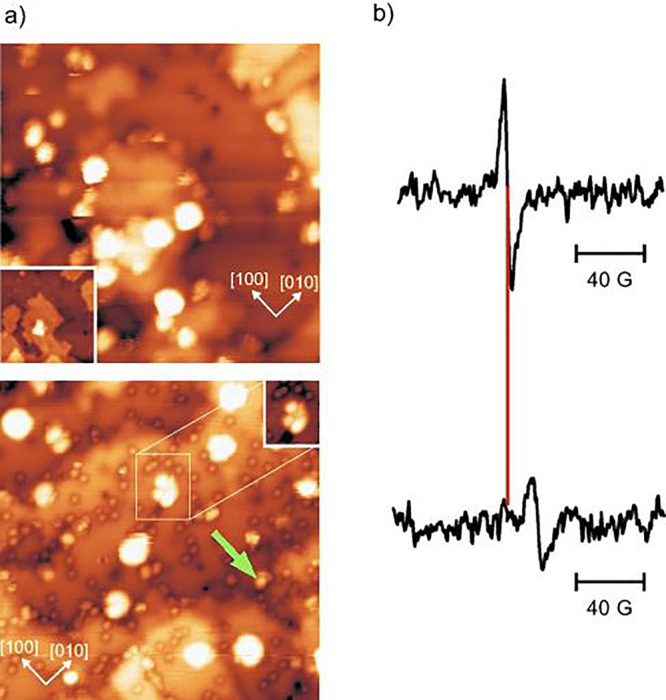
(a) STM images (30 × 30 nm2). (Top) 3–4 ML MgO(001)/Ag(001) taken after electron bombardment, Vs = −3.0 V, It = 8 pA; (inset) same area Vs = 3.5 V, It = 9 pA; (bottom) same preparation after deposition of 0.035 ML Au at 5–8 K; dimer is indicated by a green arrow; (inset) nucleation on a color center, with adjusted contrast; Vs = 1.3 V, It = 10 pA; 30 × 30 nm2. (b) EPR spectra around g = 2, (top) MgO(001) film on Mo(001) after low-dose electron bombardment; (bottom) same preparation after deposition of 0.015 ML Au at 30 K. Red line indicates the position of the color-center signal in both spectra. Reproduced with permission from ref (181). Copyright 2006 John Wiley and Sons.
Apart from the defects present, the properties of gold adsorption on MgO thin films also depend strongly on the thickness of the film. Theory predicts partial charge transfer from the metal substrate to gold for ultrathin MgO films on Ag or Mo and similar binding energies of Au on O and Mg terrace sites.182,183 Again, this is in good agreement with low-temperature STM results, which find Au exclusively adsorbing on O ions on 8 ML MgO films but about equal occupancies of O and Mg sites on 3 ML films (Figure 24).184 The underlying metal may contribute even more strongly if cations diffuse into the film and act as dopants. For example, MgO films grown on Mo(100) have been shown to contain Mo(V) centers, which appear to be situated at the surface.185,186 It seems likely that this would also influence the properties of adatoms on the MgO film, as Mo dopants in CaO(001) films, for example, have been shown to donate charge to adsorbed Au clusters, changing their shape.187
Figure 24.
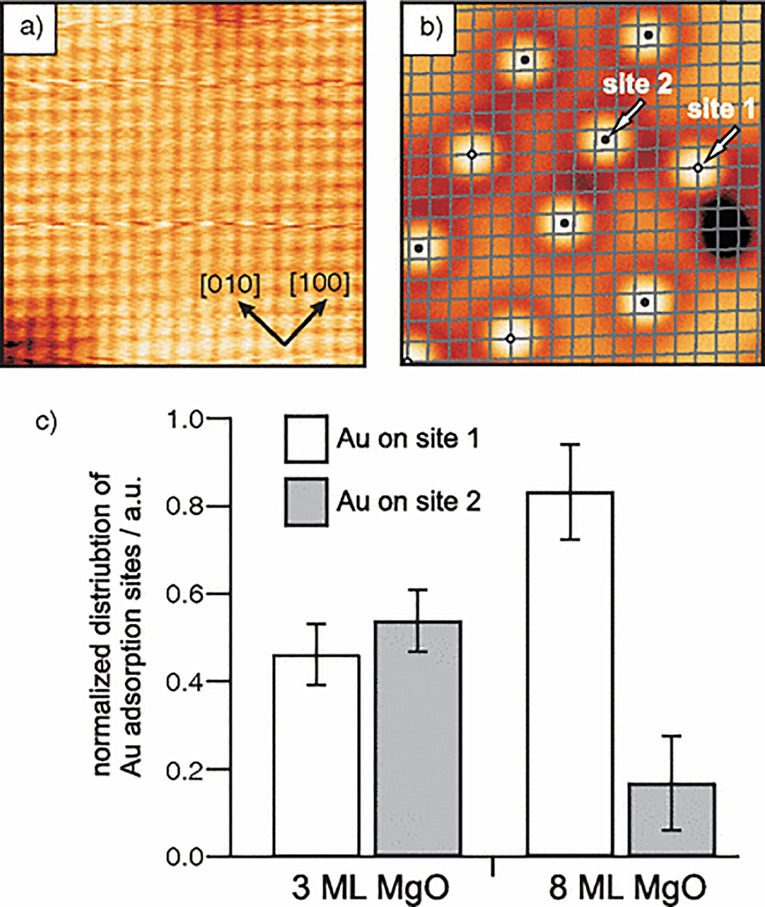
(a) Atomically resolved STM image (5 nm × 5 nm) on 3 ML MgO/Ag(001), VS = −0.02 V, IT = 7 nA; only one ionic sublattice is resolved. (b) STM image (5 nm × 5 nm) of Au atoms deposited at 5–10 K on 3 ML MgO/Ag(001), VS = −0.5 V, IT = 10 pA; ionic sublattice extracted from a is superimposed revealing the different adsorption sites. (c) Distribution of adsorption sites for Au on 3 and 8 ML thin MgO films. Reproduced with permission from ref (184). Copyright 2007 American Physical Society.
5.2. Pd on MgO
Apart from Au, the admetal that has been explored in most detail (at low coverages) is Pd. Interestingly, an AFM study on Pd adsorption on MgO over a wide temperature range concluded that nucleation kinetics are governed by point defects, most of which are found on MgO terraces.188 This is an interesting contrast to what one would expect for the case of Au: If Au mostly nucleates at color centers181 and color centers are found mostly at steps,168 this would suggest clusters should predominantly decorate step edges. Therefore, it seems that Pd either shows quite different nucleation behavior from Au or the role of other defects (e.g., uncharged divacancies at terraces) in cluster nucleation is generally underestimated. Divacancies and F+ centers have been identified as the most plausible nucleation sites by a theoretical study,174 which also predicts steps to be poor trapping sites, but it should be noted that F centers at steps were not taken into account in this comparison.
As mentioned above, catalytic activity for single Pd atoms on MgO was demonstrated already 20 years ago by TPD and IRAS.166 Size-selected clusters were deposited on MgO at 90 K and then saturated with C2H2. Upon heating, single Pd atoms catalyzed reaction to benzene at 300 K (Figure 25). This was attributed to charge transfer from defects into the Pd adatoms. Unfortunately, to our knowledge, the exact binding site of these Pd atoms has never been determined.
Figure 25.

Catalytic C6H6 formation for different Pd cluster sizes obtained from temperature-programmed reaction experiments. Bottom spectrum shows that for clean MgO(100) films no benzene is formed. Dots, data; full line, data smoothing with adjacent averaging (25 points). Cluster coverage is 0.28% of a monolayer for all cluster sizes, where one monolayer corresponds to 2.25 × 1015 atoms/cm2. Reprinted with permission from ref (166). Copyright 2000 American Chemical Society.
5.3. Conclusions
MgO is clearly a challenging system to investigate with surface-science methods, most of all with STM. This is documented by the large body of work studying only the defects of the bare surface and the disagreements that remain despite this effort. An additional challenge in studying SAC on MgO is that in addition to the atomic makeup of the surface, the availability of adsorption sites may also depend on the initial charge state of defects and the final charge state of adatoms may vary accordingly. Furthermore, the experimental limit on film thickness means that some interaction with the underlying metal substrate is hard to rule out entirely. Overall, these challenges make it extremely difficult to compare the thin films to powder catalysts. In our view, MgO is certainly the most challenging of the model systems discussed here, and its interaction with adatoms remain the most poorly understood.
6. Copper Oxides (Cu2O)
The Sykes group at Tufts University has been a trailblazer in the study of SAC systems using surface-science methods. Ever since their seminal paper on “single-atom alloy” systems,189 they have shown that mechanisms understood using atomically precise surface-science methods can be directly applied to design active powder-based catalysts.190 Direct collaboration with the Stephanopoulos group, in particular, has shown that this type of synergy can be highly successful. In addition to their work on single-atom alloys, Sykes and co-workers developed a model oxide support based on the monolayer “29” oxide.191−194 This surface is formed by controlled oxidation of a Cu(111) single crystal in 5 × 10–6 mbar O2 at 650 K and has a Cu2O stoichiometry. The structure is ring-like, with a unit cell size 29 times that of the (1 × 1) (hence the name), and is close to that of Cu2O(111). It is worth noting that the model is quite complex, and while the agreement between STM and simulated STM is excellent, confirmation by a quantitative structural technique such as LEED-I(V) or SXRD would provide the ideal basis for the comparison to theory.
Pt adsorbs on the surface as isolated atoms at low temperature, and both XPS and CO-IRAS data suggest that the atoms are close to neutral. Upon heating, isotopically labeled TPD shows that approximately 1/3 of the CO is oxidized to CO2 in a Mars–van Krevelen-type process between 300 and 350 K. The remainder of the CO molecules desorb, and irrespective of the reaction pathway, the Pt atoms end up under the Cu2O film. This renders them inaccessible for further reactions. Studies of water adsorption on the same system demonstrated dissociative adsorption at the adatom site, and evidence for scrambling between the oxygen atoms from the water and oxide suggests a dynamic rearrangement during TPD acquisition.193
The group of Weissenrieder recently demonstrated a different approach to prepare a stable CuOx-based SAC model system.195 Evaporating Fe metal directly onto a Cu2O(100) single-crystal surface196 at room temperature leads to Fe clusters, so they instead deposited FeCp2 molecules, which prevents aggregation of the metal. On the basis of STM and XPS results, the molecule adsorbs dissociatively into FeCp and Cp fragments. It was then observed that heating the sample to 473 K in a partial pressure of 1 × 10–6 mbar O2 led to the removal of the ligands and an Fe 2p signal characteristic of Fe3+ cations. STM images clearly show that the surface is covered in isolated protrusions with a uniform height and position within the surface unit cell, and these remain stable up to 573 K. Comparing the data with STM simulations based on DFT calculations, it was concluded that the Fe is coordinated to 2 surface oxygen atoms from the surface with an additional O ligand provided by the reaction with a gas-phase O2 molecule. The O atom is also bound to two surface Cu sites. It should be noted however that the surface structure has not been confirmed by a quantitative structural technique. The authors performed CO oxidation at 473 K but found that the Fe soon diffuses into the support and becomes inactive. The biggest single takeaway from this paper is that the oxidation of a molecular precursor can lead to stable SAC sites on a surface where metal evaporation does not. It will be very interesting to see if the same method can be applied with other metals and more commonly utilized oxide supports.
7. Perspective
This summary of existing data from the surface-science community shows quite clearly that metal atoms deposited on low-index metal oxide surfaces under UHV conditions are typically unstable against agglomeration into clusters. This occurs because the cohesive energy of the metal is higher than the adsorption energy of an adatom on the metal oxide surface. In such cases, isolated atoms can be found at strongly binding defect sites, stabilized by kinetic limitations, or stabilized by an interaction with other surface species (e.g., surface hydroxyl or peroxo groups). There are specific cases in which the metal–host system form a bulk solid solution, as is the case with Fe3O4, where most metals form a stable MxFe3–xO4 ferrite. However, if the foreign metal is more oxophilic than the host metal, there will be a tendency to move into the subsurface and ultimately to the bulk to reach a higher coordination to oxygen than can be achieved in the surface layer. Of course, metal atoms in the bulk or even the immediate subsurface are unavailable for reactants, which implies incorporation is as likely a path to deactivation as thermal sintering. In this context, it is important to remember that TEM images do not provide information on the depth of the metal atom being imaged and that proving a particular atom really resides directly at the surface is extremely difficult.
The lack of virtually any evidence for the occupation of stable bulk-continuation cationic sites is sobering because such sites are often assumed for theoretical modeling of the reaction mechanism. One of the major recommendations to emerge from this analysis is that the barrier for diffusion should be calculated in addition to the adsorption energy before such a geometry is claimed. Then, there is the question of environment: The adsorption of reactants can destabilize otherwise stable metal adatoms,116−118 and there is growing evidence that interactions with water/OH groups can lead to stabilization.78,104 It may therefore be necessary to revisit some of the assumptions made in theoretical modeling, testing both more complex adsorption environments and possible coadsorbates before attempting to predict reaction barriers. Efforts to find true global minima for both surface termination and adatom coordination may be aided by recent advances in applying machine learning methods for surface science.
In powder studies, the SAC systems are typically synthesized using some kind of metal-containing salt and are further prepared by calcination and/or reduction. In this regard, the work of Wang et al. on Cu2O195 is particularly interesting because they show quite clearly that the stable Fe1 species they create from ferrocene is an adspecies with additional coordination to oxygen supplied during calcination. Similar processes to stabilize metal atoms have been shown by others,197−200 and one imagines that such supported geometries with non-native ligands could be commonplace in SAC. In the current authors’ opinion, it would be prescient to conduct similar studies on common oxide supports to determine to what extent the calcination/reduction step is responsible for adatom stability on surfaces and to determine to which extent such geometries should be considered in SAC modeling in the future.
While not much has been done regarding the mechanism of most reactions using the surface-science approach, there is some information regarding CO oxidation. The experience from surface science so far is that the energy required to extract lattice oxygen from low-index metal oxide surfaces is high and too high to account for reactions observed at room temperature and below. On Fe3O4(001), for example, ca. 550 K is required to remove lattice oxygen from the terrace, so any MvK process likely involves sites with lower coordinated oxygen such as steps. It is important to note that the MvK mechanism is critically dependent on the oxygen vacancy formation energy, and this depends very much on the surface termination assumed. If one calculates an MvK pathway on an unrealistically oxygen-rich surface termination, it will not be difficult to remove lattice oxygen and the pathway will appear to be energetically favorable. Clearly, theoretical calculations should primarily consider surface terminations that have been shown to exist experimentally. Nonetheless, it may also be wise to more routinely consider SAC environments other than surface continuation sites, which may yield a more oxidized local environment.
It generally seems reasonable to assume a MvK pathway for SAC systems because it is difficult to imagine O2 dissociating at a single-atom site, but there is some evidence that associative mechanisms featuring CO–OO intermediates30 or Eley–Rideal-type processes can occur.201,202 There is also evidence that oxide-supported single atoms can dissociate H2,120 but there is no report yet of the resulting hydroxyls being used for hydrogenation of any species. It would certainly be advisable for surface-science studies to move on from the focus on CO oxidation, particularly as the focus of applied SAC research moves into electrocatalysis.
We have focused here on oxides that can either be studied as single crystals or be prepared as thin films with a bulk-like structure. Notable exceptions to this are SiO2 and Al2O3. Like MgO, both are insulating and would provide an interesting contrast to the other systems discussed here in terms of their interaction with adatoms. However, in both cases, the thin films that have been prepared do not appear to be representative of the respective bulk oxides when it comes to adatom adsorption. For SiO2, quasi-2D monolayer and bilayer films have been grown on a variety of metal substrates.203,204 Pd adatoms deposited on bilayer SiO2/Ru(0001) at room temperature were shown to diffuse through nanopores in the film to the metal support even in the crystalline phase in the absence of defects. Au adatoms were slightly more stable but similarly diffused through the film at defects.205 On monolayer SiO2/Mo(112), Pd, Ag, and Au adatoms have been stabilized by first embedding Pd adatoms in some of the SiO2 nanopores.206 However, since the adatoms form covalent bonds to the embedded Pd, which in turn is essentially a part of the metal substrate, this is clearly not a good model for single adatoms supported on bulk SiO2. A similar behavior was observed for Al2O3 thin films outgrown from Ni3Al(111), where single atoms were shown to diffuse to the metal support through pores in the oxide film but pores already filled by Pd atoms act as cluster nucleation sites.207 In both cases, it thus seems that the available thin films are not yet suitable as model systems for single atoms supported on oxides and that studying SAC on these supports would first require developing more bulk-like samples.
One important issue with metal oxide materials that has emerged in the past decade is the presence of polarons, i.e., localized charge associated with lattice distortions.208 For example, the formation of an oxygen vacancy in a metal oxide leaves two electrons, which will often localize on cation sites, leading to a local distortion of the lattice. Such polarons have been directly imaged in scanning probe experiments37 and are known to interact with adsorbates such as CO, substantially changing the adsorption energy.209 As such, they could also interact with and affect the stability of metal adatoms. Correctly modeling polarons in theoretical calculations is challenging, and almost all of the theoretical work described in this review does not take them into account. This issue will also likely be important for realistic surfaces because repair of a vacancy by water can result in two-electron polarons due to the formation of two surface OH groups. Very recently,61 the group of Franchini, who are experts in calculating the effect of polarons in metal oxides, have turned their attention to SAC and demonstrated an approach to account for their presence. Specifically, they studied Au, Pt, and Rh on TiO2(110) and found that each metal interacts differently with polarons. While polarons are transferred to Au and Pt when they adsorb at VO sites, Rh adsorbs atop the surface Ti row and polarons are situated in the first subsurface Ti layer.
In recent years, there has been a concerted effort to develop the methods of surface science such that they can be applied to UHV-prepared samples moved into ambient pressure and electrochemical regimes. As such, the scene is set for model studies, if suitable model systems can be created. Much of the work with single-atom electrocatalysis is based on carbon or N-doped carbon supports, but as yet there is no surface-science study to investigate the coordination effects on the stability of such systems or the adsorption properties. Certainly, the ability exists to prepare graphene in situ and to precisely fabricate N-defects in graphene layers, so depositing metal adatoms and investigating with scanning probe techniques seems like a relatively small but potentially rewarding step. Similarly, single-atom photocatalysis is emerging as an exciting field, and it would be critical to determine what the role of the atom actually is. In principle, it can provide new active sites, increase the concentration of existing active sites, provide trap sites that influence recombination rates, and change the electronic structure locally or globally. For this we need to develop model systems based on model photocatalysts. The lack of stability of metal adatoms on titania seems problematic, and we suggest that Fe2O3(1102)102−104 (band gap ≈ 2 eV) can be a fertile model system for fundamental photocatalysis work. Other systems where the surface structures are understood, such as the perovskite SrTiO3, could be ideal if single-metal adatoms can be stabilized there.
Acknowledgments
G.S.P. and F.K. acknowledge funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement No. 864628) and by the Austrian Science Fund (FWF, Y847-N20, START Prize).
Glossary
List of Acronyms
- AFM
atomic force microscopy
- DFT
density functional theory
- EF
Fermi level
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- HAADF
high-angle annular dark-field [STEM]
- IRAS
infrared reflection absorption spectroscopy
- KPFM
Kelvin probe force microscopy
- LEED
low-energy electron diffraction
- LEED-I(V)
quantitative low-energy electron diffraction (I intensity, V acceleration voltage)
- ML
monolayer
- MvK
Mars–van Krevelen [mechanism]
- ncAFM
noncontact atomic force microscopy
- PES
photoelectron spectroscopy
- PROX
preferential oxidation of CO
- UHV
ultrahigh vacuum
- SAC
single-atom catalysis
- SCV
subsurface cation vacancy
- SRPES
synchrotron radiation photoelectron spectroscopy
- STEM
scanning transmission electron microscopy
- STM
scanning tunneling microscopy
- SXRD
surface X-ray diffraction
- [S]TEM
[scanning] transmission electron microscopy
- TPD
temperature-programmed desorption
- VO
oxygen vacancy
- XANES
X-ray absorption near-edge structure
- XPS
X-ray photoelectron spectroscopy
Biographies
Florian Kraushofer received his Ph.D. degree in Physics from the Technical University of Vienna in 2021 under the supervision of Gareth Parkinson, where he worked on iron oxide surfaces as model systems for single-atom catalysis. He is now a postdoctoral researcher at the Technical University of Munich.
Gareth Parkinson received his Ph.D. degree in Physics at the University of Warwick, UK. He worked as a postdoctoral researcher at PNNL and Tulane University in the United States before moving to Technische Universitat Wien in Vienna as an assistant professor in 2010. Since 2021 he has been Full Professor of surface reactivity in the Institute of Applied Physics, Technische Universitat Wien.
Special Issue
This paper is an additional review for Chem. Rev. 2020, volume 120, issue (21), , “Heterogeneous Single-Atom Catalysis”.
Author Contributions
CRediT: Florian Kraushofer writing-original draft, writing-review & editing; Gareth S. Parkinson supervision, writing-original draft, writing-review & editing.
Open Access is funded by the Austrian Science Fund (FWF).
The authors declare no competing financial interest.
References
- Yang X.-F.; Wang A.; Qiao B.; Li J.; Liu J.; Zhang T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740. 10.1021/ar300361m. [DOI] [PubMed] [Google Scholar]
- Liang S.; Hao C.; Shi Y. The Power of Single-Atom Catalysis. ChemCatChem. 2015, 7, 2559. 10.1002/cctc.201500363. [DOI] [Google Scholar]
- Liu J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34. 10.1021/acscatal.6b01534. [DOI] [Google Scholar]
- Cui X.; Li W.; Ryabchuk P.; Junge K.; Beller M. Bridging Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-Metal-Site Catalysts. Nat. Catal. 2018, 1, 385. 10.1038/s41929-018-0090-9. [DOI] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Zhang H.; Liu G.; Shi L.; Ye J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. 10.1002/aenm.201701343. [DOI] [Google Scholar]
- Wang B.; Cai H.; Shen S. Single Metal Atom Photocatalysis. Small Methods 2019, 3, 1800447. 10.1002/smtd.201800447. [DOI] [Google Scholar]
- Gao C.; Low J.; Long R.; Kong T.; Zhu J.; Xiong Y. Heterogeneous Single-Atom Photocatalysts: Fundamentals and Applications. Chem. Rev. 2020, 120, 12175. 10.1021/acs.chemrev.9b00840. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Fu S.; Shi Q.; Du D.; Lin Y. Single-Atom Electrocatalysts. Angew. Chem., Int. Ed. 2017, 56, 13944. 10.1002/anie.201703864. [DOI] [PubMed] [Google Scholar]
- Liu J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34. 10.1021/acscatal.6b01534. [DOI] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S.; Vorobyeva E.; Pérez-Ramírez J. The Multifaceted Reactivity of Single-Atom Heterogeneous Catalysts. Angew. Chem., Int. Ed. 2018, 57, 15316. 10.1002/anie.201806936. [DOI] [PubMed] [Google Scholar]
- Samantaray M. K.; Pump E.; Bendjeriou-Sedjerari A.; D’Elia V.; Pelletier J. D. A.; Guidotti M.; Psaro R.; Basset J.-M. Surface Organometallic Chemistry in Heterogeneous Catalysis. Chem. Soc. Rev. 2018, 47, 8403. 10.1039/C8CS00356D. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang X.; Zhang J.; Huang Y.; Liu B. In Situ/Operando Techniques for Characterization of Single-Atom Catalysts. ACS Catal. 2019, 9, 2521. 10.1021/acscatal.8b04937. [DOI] [Google Scholar]
- Wu J.; Xiong L.; Zhao B.; Liu M.; Huang L. Densely Populated Single Atom Catalysts. Small Methods 2020, 4, 1900540. 10.1002/smtd.201900540. [DOI] [Google Scholar]
- Feng S.; Lin X.; Song X.; Liu Y.; Jiang Z.; Hemberger P.; Bodi A.; Ding Y. The Role of H2 on the Stability of the Single-Metal-Site Ir1/AC Catalyst for Heterogeneous Methanol Carbonylation. J. Catal. 2020, 381, 193. 10.1016/j.jcat.2019.10.032. [DOI] [Google Scholar]
- Ishida T.; Murayama T.; Taketoshi A.; Haruta M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464. 10.1021/acs.chemrev.9b00551. [DOI] [PubMed] [Google Scholar]
- Zang W.; Kou Z.; Pennycook S. J.; Wang J. Heterogeneous Single Atom Electrocatalysis, Where “Singles” Are “Married. Adv. Energy Mater. 2020, 10, 1903181. 10.1002/aenm.201903181. [DOI] [Google Scholar]
- Hannagan R. T.; Giannakakis G.; Flytzani-Stephanopoulos M.; Sykes E. C. H. Single-Atom Alloy Catalysis. Chem. Rev. 2020, 120, 12044. 10.1021/acs.chemrev.0c00078. [DOI] [PubMed] [Google Scholar]
- Gusmão R.; Veselý M.; Sofer Z. Recent Developments on the Single Atom Supported at 2D Materials Beyond Graphene as Catalysts. ACS Catal. 2020, 10, 9634. 10.1021/acscatal.0c02388. [DOI] [Google Scholar]
- Li L.; Chang X.; Lin X.; Zhao Z.-J.; Gong J. Theoretical Insights into Single-Atom Catalysts. Chem. Soc. Rev. 2020, 49, 8156. 10.1039/D0CS00795A. [DOI] [PubMed] [Google Scholar]
- Tosoni S.; Pacchioni G. Bonding Properties of Isolated Metal Atoms on Two-Dimensional Oxides. J. Phys. Chem. C 2020, 124, 20960. 10.1021/acs.jpcc.0c05958. [DOI] [Google Scholar]
- Kaiser S. K.; Chen Z.; Faust Akl D.; Mitchell S.; Pérez-Ramírez J. Single-Atom Catalysts across the Periodic Table. Chem. Rev. 2020, 120, 11703. 10.1021/acs.chemrev.0c00576. [DOI] [PubMed] [Google Scholar]
- Thang H. V.; Maleki F.; Tosoni S.; Pacchioni G.. Vibrational Properties of CO Adsorbed on Au Single Atom Catalysts on TiO2(101), ZrO2(101), CeO2(111), and LaFeO3(001) Surfaces: A DFT Study. Top. Catal. 2021, 10.1007/s11244-021-01514-0. [DOI] [Google Scholar]
- Liu J. Aberration-Corrected Scanning Transmission Electron Microscopy in Single-Atom Catalysis: Probing the Catalytically Active Centers. Chin. J. Catal. 2017, 38, 1460. 10.1016/S1872-2067(17)62900-0. [DOI] [Google Scholar]
- Tieu P.; Yan X.; Xu M.; Christopher P.; Pan X. Directly Probing the Local Coordination, Charge State, and Stability of Single Atom Catalysts by Advanced Electron Microscopy: A Review. Small 2021, 17, 2006482. 10.1002/smll.202006482. [DOI] [PubMed] [Google Scholar]
- Duan S.; Wang R.; Liu J. Stability Investigation of a High Number Density Pt1/Fe2O3 Single-Atom Catalyst under Different Gas Environments by HAADF-STEM. Nanotechnology 2018, 29, 204002. 10.1088/1361-6528/aab1d2. [DOI] [PubMed] [Google Scholar]
- Liang J.; Yu Q.; Yang X.; Zhang T.; Li J. A Systematic Theoretical Study on FeOx-Supported Single-Atom Catalysts: M1/FeOx for CO Oxidation. Nano Res. 2018, 11, 1599. 10.1007/s12274-017-1775-0. [DOI] [Google Scholar]
- Liang J.; Yang X.; Xu C.; Zhang T.; Li J. Catalytic Ativities of Single-Atom Catalysts for CO Oxidation: Pt1/FeOx Vs. Fe1/FeOx. Chin. J. Catal. 2017, 38, 1566. 10.1016/S1872-2067(17)62879-1. [DOI] [Google Scholar]
- Li F.; Li Y.; Zeng X. C.; Chen Z. Exploration of High-Performance Single-Atom Catalysts on Support M1/FeOx for CO Oxidation Via Computational Study. ACS Catal. 2015, 5, 544. 10.1021/cs501790v. [DOI] [Google Scholar]
- Diebold U. The Surface Science of Titanium Dioxide. Surf. Sci. Rep. 2003, 48, 53. 10.1016/S0167-5729(02)00100-0. [DOI] [Google Scholar]
- Dohnálek Z.; Lyubinetsky I.; Rousseau R. Thermally-Driven Processes on Rutile TiO2(110)-(1 × 1): A Direct View at the Atomic Scale. Prog. Surf. Sci. 2010, 85, 161. 10.1016/j.progsurf.2010.03.001. [DOI] [Google Scholar]
- Lun Pang C.; Lindsay R.; Thornton G. Chemical Reactions on Rutile TiO2(110). Chem. Soc. Rev. 2008, 37, 2328. 10.1039/b719085a. [DOI] [PubMed] [Google Scholar]
- Lindsay R.; Wander A.; Ernst A.; Montanari B.; Thornton G.; Harrison N. M. Revisiting the Surface Structure of TiO2(110): A Quantitative Low-Energy Electron Diffraction Study. Phys. Rev. Lett. 2005, 94, 246102. 10.1103/PhysRevLett.94.246102. [DOI] [Google Scholar]
- Cabailh G.; Torrelles X.; Lindsay R.; Bikondoa O.; Joumard I.; Zegenhagen J.; Thornton G. Geometric Structure of TiO2(110)(1 × 1): Achieving Experimental Consensus. Phys. Rev. B 2007, 75, 241403. 10.1103/PhysRevB.75.241403. [DOI] [Google Scholar]
- Busayaporn W.; Torrelles X.; Wander A.; Tomić S.; Ernst A.; Montanari B.; Harrison N. M.; Bikondoa O.; Joumard I.; Zegenhagen J.; et al. Geometric Structure of TiO2(110)(1 × 1): Confirming Experimental Conclusions. Phys. Rev. B 2010, 81, 153404. 10.1103/PhysRevB.81.153404. [DOI] [Google Scholar]
- Reticcioli M.; Setvin M.; Hao X.; Flauger P.; Kresse G.; Schmid M.; Diebold U.; Franchini C. Polaron-Driven Surface Reconstructions. Phys. Rev. X 2017, 7, 031053. 10.1103/PhysRevX.7.031053. [DOI] [Google Scholar]
- Wahlström E.; Lopez N.; Schaub R.; Thostrup P.; Rønnau A.; Africh C.; Lægsgaard E.; Nørskov J. K.; Besenbacher F. Bonding of Gold Nanoclusters to Oxygen Vacancies on Rutile TiO2(110). Phys. Rev. Lett. 2003, 90, 026101. 10.1103/PhysRevLett.90.026101. [DOI] [PubMed] [Google Scholar]
- Mellor A.; Humphrey D.; Yim C. M.; Pang C. L.; Idriss H.; Thornton G. Direct Visualization of Au Atoms Bound to TiO2(110) O-Vacancies. J. Phys. Chem. C 2017, 121, 24721. 10.1021/acs.jpcc.7b09608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S.; Li B.; Cui X.; Tan S.; Wang B. Photoresponses of Supported Au Single Atoms on TiO2(110) through the Metal-Induced Gap States. J. Phys. Chem. Lett. 2019, 10, 4683. 10.1021/acs.jpclett.9b01527. [DOI] [PubMed] [Google Scholar]
- Iachella M.; Le Bahers T.; Loffreda D. Diffusion Kinetics of Gold and Copper Atoms on Pristine and Reduced Rutile TiO2 (110) Surfaces. J. Phys. Chem. C 2018, 122, 3824. 10.1021/acs.jpcc.7b08183. [DOI] [Google Scholar]
- Matthey D.; Wang J. G.; Wendt S.; Matthiesen J.; Schaub R.; Lægsgaard E.; Hammer B.; Besenbacher F. Enhanced Bonding of Gold Nanoparticles on Oxidized TiO2(110). Science 2007, 315, 1692. 10.1126/science.1135752. [DOI] [PubMed] [Google Scholar]
- Hansen J. Ø.; Lira E.; Galliker P.; Wang J.-G.; Sprunger P. T.; Li Z.; Lægsgaard E.; Wendt S.; Hammer B.; Besenbacher F. Enhanced Bonding of Silver Nanoparticles on Oxidized TiO2(110). J. Phys. Chem. C 2010, 114, 16964. 10.1021/jp101714r. [DOI] [Google Scholar]
- Luo K.; St. Clair T. P.; Lai X.; Goodman D. W. Silver Growth on TiO2(110) (1 × 1) and (1 × 2). J. Phys. Chem. B 2000, 104, 3050. 10.1021/jp993062o. [DOI] [Google Scholar]
- Zhou J.; Kang Y. C.; Ma S.; Chen D. A. Adsorbate-Induced Dissociation of Metal Clusters: TiO2(110)-Supported Cu and Ni Clusters Exposed to Oxygen Gas. Surf. Sci. 2004, 562, 113. 10.1016/j.susc.2004.05.094. [DOI] [Google Scholar]
- Pillay D.; Wang Y.; Hwang G. S. Nucleation and Growth of 1B Metal Clusters on Rutile TiO2(110): Atomic Level Understanding from First Principles Studies. Catal. Today 2005, 105, 78. 10.1016/j.cattod.2005.04.016. [DOI] [Google Scholar]
- Giordano L.; Pacchioni G.; Bredow T.; Sanz J. F. Cu, Ag, and Au Atoms Adsorbed on TiO2(110): Cluster and Periodic Calculations. Surf. Sci. 2001, 471, 21. 10.1016/S0039-6028(00)00879-7. [DOI] [Google Scholar]
- Tada K.; Koga H.; Hayashi A.; Kondo Y.; Kawakami T.; Yamanaka S.; Okumura M. Effects of Halogens on Interactions between a Reduced TiO2 (110) Surface and Noble Metal Atoms: A DFT Study. Appl. Surf. Sci. 2017, 411, 149. 10.1016/j.apsusc.2017.03.113. [DOI] [Google Scholar]
- Takei T.; Akita T.; Nakamura I.; Fujitani T.; Okumura M.; Okazaki K.; Huang J.; Ishida T.; Haruta M. In Advanes in Catalysis; Gates B. C., Jentoft F. C., Eds.; Academic Press, 2012; Vol. 55. [Google Scholar]
- Fujikawa K.; Suzuki S.; Koike Y.; Chun W.-J.; Asakura K. Self-Regulated Ni Cluster Formation on the TiO2(110) Terrace Studied Using Scanning Tunneling Microscopy. Surf. Sci. 2006, 600, 117. 10.1016/j.susc.2006.03.019. [DOI] [Google Scholar]
- Koike Y.; Ijima K.; Chun W. J.; Ashima H.; Yamamoto T.; Fujikawa K.; Suzuki S.; Iwasawa Y.; Nomura M.; Asakura K. Structure of Low Coverage Ni Atoms on the TiO2(110) Surface - Polarization Dependent Total-Reflection Fluorescence Exafs Study. Chem. Phys. Lett. 2006, 421, 27. 10.1016/j.cplett.2006.01.045. [DOI] [Google Scholar]
- Castillo-Robles J. M.; Orgaz E. Structural and Optical Properties of Ni Atoms and Ni55 Cluster Adsorbed on a Rutile TiO2(110) Surface. Theor. Chem. Acc. 2018, 137, 31. 10.1007/s00214-018-2211-6. [DOI] [Google Scholar]
- Cao P. L.; Ellis D. E.; Dravid V. P. First-Principles Study of Initial Stage of Ni Thin-Film Growth on a TiO2 (110) Surface. J. Mater. Res. 1999, 14, 3684. 10.1557/JMR.1999.0497. [DOI] [Google Scholar]
- Xu C.; Lai X.; Zajac G.; Goodman D. Scanning Tunneling Microscopy Studies of the Surface: Structure and the Nucleation Growth of Pd. Phys. Rev. B 1997, 56, 13464. 10.1103/PhysRevB.56.13464. [DOI] [Google Scholar]
- Sanz J. F.; Márquez A. Adsorption of Pd Atoms and Dimers on the TiO2 (110) Surface: A First Principles Study. J. Phys. Chem. C 2007, 111, 3949. 10.1021/jp0639952. [DOI] [Google Scholar]
- Ong S. V.; Khanna S. N. Theoretical Studies of the Stability and Oxidation of Pdn (n = 1–7) Clusters on Rutile TiO2(110): Adsorption on the Stoichiometric Surface. J. Phys. Chem. C 2012, 116, 3105. 10.1021/jp212504x. [DOI] [Google Scholar]
- Sasahara A.; Pang C. L.; Onishi H. Probe Microscope Observation of Platinum Atoms Deposited on the TiO2(110)-(1 × 1) Surface. J. Phys. Chem. B 2006, 110, 13453. 10.1021/jp062000c. [DOI] [PubMed] [Google Scholar]
- Yurtsever A.; Fernández-Torre D.; Onoda J.; Abe M.; Morita S.; Sugimoto Y.; Pérez R. The Local Electronic Properties of Individual Pt Atoms Adsorbed on TiO2(110) Studied by Kelvin Probe Force Microscopy and First-Principles Simulations. Nanoscale 2017, 9, 5812. 10.1039/C6NR07550A. [DOI] [PubMed] [Google Scholar]
- Fernández-Torre D.; Yurtsever A.; Onoda J.; Abe M.; Morita S.; Sugimoto Y.; Pérez R. Pt Atoms Adsorbed on TiO2(110)-(1 × 1) Studied with Noncontact Atomic Force Microscopy and First-Principles Simulations. Phys. Rev. B 2015, 91, 075401. 10.1103/PhysRevB.91.075401. [DOI] [PubMed] [Google Scholar]
- Dong S.-h.; Wang A.-l.; Zhao J.; Tan S.-j.; Wang B. Interaction of CO and O2 with Supported Pt Single-Atoms on TiO2(110). Chin. J. Chem. Phys. 2020, 33, 349. 10.1063/1674-0068/cjcp1911198. [DOI] [Google Scholar]
- Sombut P.; Puntscher L.; Atzmueller M.; Jakub Z.; Reticcioli M.; Meier M.; Parkinson G. S.; Franchini C.. Role of Polarons in Single-Atom Catalysts: Case Study of Me1 [Au1, Pt1, and Rh1] on TiO2(110). Top. Catal. 2022, 10.1007/s11244-022-01651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieboldt F.; Vilhelmsen L. B.; Koust S.; Lauritsen J. V.; Helveg S.; Lammich L.; Besenbacher F.; Hammer B.; Wendt S. Nucleation and Growth of Pt Nanoparticles on Reduced and Oxidized Rutile TiO2(110). J. Chem. Phys. 2014, 141, 214702. 10.1063/1.4902249. [DOI] [PubMed] [Google Scholar]
- Chang T.-Y.; Tanaka Y.; Ishikawa R.; Toyoura K.; Matsunaga K.; Ikuhara Y.; Shibata N. Direct Imaging of Pt Single Atoms Adsorbed on TiO2 (110) Surfaces. Nano Lett. 2014, 14, 134. 10.1021/nl403520c. [DOI] [PubMed] [Google Scholar]
- Li M.; Hebenstreit W.; Diebold U.; Henderson M. A.; Jennison D. R. Oxygen-Induced Restructuring of Rutile TiO2(110): Formation Mechanism, Atomic Models, and Influence on Surface Chemistry. Faraday Discuss. 1999, 114, 245. 10.1039/a903598b. [DOI] [Google Scholar]
- Matsunaga K.; Chang T.-Y.; Ishikawa R.; Dong Q.; Toyoura K.; Nakamura A.; Ikuhara Y.; Shibata N. Adsorption Sites of Single Noble Metal Atoms on the Rutile TiO2(110) Surface Influenced by Different Surface Oxygen Vacancies. J. Phys.: Condens. Matter 2016, 28, 175002. 10.1088/0953-8984/28/17/175002. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Asokan C.; Xu M.; Graham G. W.; Pan X.; Christopher P.; Li J.; Sautet P. Rh Single Atoms on TiO2 Dynamically Respond to Reaction Conditions by Adapting Their Site. Nat. Commun. 2019, 10, 4488. 10.1038/s41467-019-12461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galhenage R. P.; Yan H.; Tenney S. A.; Park N.; Henkelman G.; Albrecht P.; Mullins D. R.; Chen D. A. Understanding the Nucleation and Growth of Metals on TiO2: Co Compared to Au, Ni, and Pt. J. Phys. Chem. C 2013, 117, 7191. 10.1021/jp401283k. [DOI] [Google Scholar]
- Madej E.; Spiridis N.; Socha R. P.; Wolanin B.; Korecki J. The Nucleation, Growth and Thermal Stability of Iron Clusters on a TiO2(110) Surface. Appl. Surf. Sci. 2017, 416, 144. 10.1016/j.apsusc.2017.04.114. [DOI] [Google Scholar]
- Setvín M.; Daniel B.; Mansfeldova V.; Kavan L.; Scheiber P.; Fidler M.; Schmid M.; Diebold U. Surface Preparation of TiO2 Anatase (101): Pitfalls and How to Avoid Them. Surf. Sci. 2014, 626, 61. 10.1016/j.susc.2014.04.001. [DOI] [Google Scholar]
- Hengerer R.; Bolliger B.; Erbudak M.; Grätzel M. Structure and Stability of the Anatase TiO2 (101) and (001) Surfaces. Surf. Sci. 2000, 460, 162. 10.1016/S0039-6028(00)00527-6. [DOI] [Google Scholar]
- Treacy J. P. W.; Hussain H.; Torrelles X.; Grinter D. C.; Cabailh G.; Bikondoa O.; Nicklin C.; Selcuk S.; Selloni A.; Lindsay R.; et al. Geometric Structure of Anatase TiO2(101). Phys. Rev. B 2017, 95, 075416. 10.1103/PhysRevB.95.075416. [DOI] [Google Scholar]
- Setvin M.; Hulva J.; Wang H.; Simschitz T.; Schmid M.; Parkinson G. S.; Di Valentin C.; Selloni A.; Diebold U. Formaldehyde Adsorption on the Anatase TiO2(101) Surface: Experimental and Theoretical Investigation. J. Phys. Chem. C 2017, 121, 8914. 10.1021/acs.jpcc.7b01434. [DOI] [Google Scholar]
- Setvin M.; Buchholz M.; Hou W.; Zhang C.; Stöger B.; Hulva J.; Simschitz T.; Shi X.; Pavelec J.; Parkinson G. S.; et al. A Multitechnique Study of CO Adsorption on the TiO2 Anatase (101) Surface. J. Phys. Chem. C 2015, 119, 21044. 10.1021/acs.jpcc.5b07999. [DOI] [Google Scholar]
- Setvín M.; Aschauer U.; Scheiber P.; Li Y.-F.; Hou W.; Schmid M.; Selloni A.; Diebold U. Reaction of O2 with Subsurface Oxygen Vacancies on TiO2 Anatase (101). Science 2013, 341, 988. 10.1126/science.1239879. [DOI] [PubMed] [Google Scholar]
- Scheiber P.; Fidler M.; Dulub O.; Schmid M.; Diebold U.; Hou W.; Aschauer U.; Selloni A. (Sub)Surface Mobility of Oxygen Vacancies at the TiO2 Anatase (101) Surface. Phys. Rev. Lett. 2012, 109, 136103. 10.1103/PhysRevLett.109.136103. [DOI] [PubMed] [Google Scholar]
- Kashiwaya S.; Morasch J.; Streibel V.; Toupance T.; Jaegermann W.; Klein A. The Work Function of TiO2. Surfaces 2018, 1, 73. 10.3390/surfaces1010007. [DOI] [Google Scholar]
- Gong X.-Q.; Selloni A.; Dulub O.; Jacobson P.; Diebold U. Small Au and Pt Clusters at the Anatase TiO2(101) Surface: Behavior at Terraces, Steps, and Surface Oxygen Vacancies. J. Am. Chem. Soc. 2008, 130, 370. 10.1021/ja0773148. [DOI] [PubMed] [Google Scholar]
- Thang H. V.; Pacchioni G.; DeRita L.; Christopher P. Nature of Stable Single Atom Pt Catalysts Dispersed on Anatase TiO2. J. Catal. 2018, 367, 104. 10.1016/j.jcat.2018.08.025. [DOI] [Google Scholar]
- DeRita L.; Resasco J.; Dai S.; Boubnov A.; Thang H. V.; Hoffman A. S.; Ro I.; Graham G. W.; Bare S. R.; Pacchioni G.; et al. Structural Evolution of Atomically Dispersed Pt Catalysts Dictates Reactivity. Nat. Mater. 2019, 18, 746. 10.1038/s41563-019-0349-9. [DOI] [PubMed] [Google Scholar]
- Asokan C.; Thang H. V.; Pacchioni G.; Christopher P. Reductant Composition Influences the Coordination of Atomically Dispersed Rh on Anatase TiO2. Catal. Sci. Technol. 2020, 10, 1597. 10.1039/D0CY00146E. [DOI] [Google Scholar]
- DeRita L.; Dai S.; Lopez-Zepeda K.; Pham N.; Graham G. W.; Pan X.; Christopher P. Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150. 10.1021/jacs.7b07093. [DOI] [PubMed] [Google Scholar]
- Jakub Z.; Hulva J.; Ryan P. T. P.; Duncan D. A.; Payne D. J.; Bliem R.; Ulreich M.; Hofegger P.; Kraushofer F.; Meier M.; et al. Adsorbate-Induced Structural Evolution Changes the Mechanism of CO Oxidation on a Rh/Fe3O4(001) Model Catalyst. Nanoscale 2020, 12, 5866. 10.1039/C9NR10087C. [DOI] [PubMed] [Google Scholar]
- Kraushofer F.; Resch N.; Eder M.; Rafsanjani-Abbasi A.; Tobisch S.; Jakub Z.; Franceschi G.; Riva M.; Meier M.; Schmid M.; et al. Surface Reduction State Determines Stabilization and Incorporation of Rh on α-Fe2O3(1102). Adv. Mater. Interfaces 2021, 8, 2001908. 10.1002/admi.202001908. [DOI] [Google Scholar]
- Chrétien S.; Metiu H. Density Functional Study of the CO Oxidation on a Doped Rutile TiO2(110): Effect of Ionic Au in Catalysis. Catal. Lett. 2006, 107, 143. 10.1007/s10562-005-0014-6. [DOI] [Google Scholar]
- Lv C.-Q.; Liu J.-H.; Guo Y.; Li X.-M.; Wang G.-C. DFT+U Investigation on the Adsorption and Initial Decomposition of Methylamine by a Pt Single-Atom Catalyst Supported on Rutile (110) TiO2. Appl. Surf. Sci. 2016, 389, 411. 10.1016/j.apsusc.2016.07.111. [DOI] [Google Scholar]
- Li L.; Li W.; Zhu C.; Mao L.-F. A DFT+U Study About Agglomeration of Au Atoms on Reduced Surface of Rutile TiO2 (110). Mater. Chem. Phys. 2021, 271, 124944. 10.1016/j.matchemphys.2021.124944. [DOI] [Google Scholar]
- Wang X.; Zhang L.; Bu Y.; Sun W. Interplay between Invasive Single Atom Pt and Native Oxygen Vacancy in Rutile TiO2(110) Surface: A Theoretical Study. Nano Res. 2022, 15, 669. 10.1007/s12274-021-3542-5. [DOI] [Google Scholar]
- Liang J.-X.; Lin J.; Liu J.; Wang X.; Zhang T.; Li J. Dual Metal Active Sites in an Ir1/FeOx Single-Atom Catalyst: A Redox Mechanism for the Water-Gas Shift Reaction. Angew. Chem., Int. Ed. 2020, 59, 12868. 10.1002/anie.201914867. [DOI] [PubMed] [Google Scholar]
- Lin J.; Qiao B.; Li N.; Li L.; Sun X.; Liu J.; Wang X.; Zhang T. Little Do More: A Highly Effective Pt1/FeOx Single-Atom Catalyst for the Reduction of NO by H2. Chem. Commun. 2015, 51, 7911. 10.1039/C5CC00714C. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Wang A.; Yang X.; Allard L. F.; Jiang Z.; Cui Y.; Liu J.; Li J.; Zhang T. Single-Atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 2011, 3, 634. 10.1038/nchem.1095. [DOI] [PubMed] [Google Scholar]
- Parkinson G. S. Iron Oxide Surfaces. Surf. Sci. Rep. 2016, 71, 272. 10.1016/j.surfrep.2016.02.001. [DOI] [Google Scholar]
- Wang X. G.; Weiss W.; Shaikhutdinov S. K.; Ritter M.; Petersen M.; Wagner F.; Schlögl R.; Scheffler M. The Hematite (α-Fe2O3) (0001) Surface: Evidence for Domains of Distinct Chemistry. Phys. Rev. Lett. 1998, 81, 1038. 10.1103/PhysRevLett.81.1038. [DOI] [Google Scholar]
- Shaikhutdinov S. K.; Weiss W. Oxygen Pressure Dependence of the α-Fe2O3(0001) Surface Structure. Surf. Sci. 1999, 432, L627. 10.1016/S0039-6028(99)00643-3. [DOI] [Google Scholar]
- Barbier A.; Stierle A.; Kasper N.; Guittet M.-J.; Jupille J. Surface Termination of Hematite at Environmental Oxygen Pressures: Experimental Surface Phase Diagram. Phys. Rev. B 2007, 75, 233406. 10.1103/PhysRevB.75.233406. [DOI] [Google Scholar]
- Lemire C.; Bertarione S.; Zecchina A.; Scarano D.; Chaka A.; Shaikhutdinov S.; Freund H.-J. Ferryl (Fe=O) Termination of the Hematite α-Fe2O3(0001) Surface. Phys. Rev. Lett. 2005, 94, 166101. 10.1103/PhysRevLett.94.166101. [DOI] [PubMed] [Google Scholar]
- Lad R. J.; Henrich V. E. Structure of α-Fe2O3 Single Crystal Surfaces Following Ar+ Ion Bombardment and Annealing in O2. Surf. Sci. 1988, 193, 81. 10.1016/0039-6028(88)90324-X. [DOI] [Google Scholar]
- Tang Y.; Qin H.; Wu K.; Guo Q.; Guo J. The Reduction and Oxidation of Fe2O3(0001) Surface Investigated by Scanning Tunneling Microscopy. Surf. Sci. 2013, 609, 67. 10.1016/j.susc.2012.11.005. [DOI] [Google Scholar]
- Lewandowski M.; Groot I. M.; Qin Z.-H.; Ossowski T.; Pabisiak T.; Kiejna A.; Pavlovska A.; Shaikhutdinov S.; Freund H.-J.; Bauer E. Nanoscale Patterns on Polar Oxide Surfaces. Chem. Mater. 2016, 28, 7433. 10.1021/acs.chemmater.6b03040. [DOI] [Google Scholar]
- Redondo J.; Lazar P.; Procházka P.; Průša S.; Mallada B.; Cahlík A.; Lachnitt J.; Berger J.; Šmíd B.; Kormoš L.; et al. Identification of Two-Dimensional Feo2 Termination of Bulk Hematite α-Fe2O3(0001) Surface. J. Phys. Chem. C 2019, 123, 14312. 10.1021/acs.jpcc.9b00244. [DOI] [Google Scholar]
- Li F.; Li Y.; Zeng X. C.; Chen Z. Exploration of High-Performance Single-Atom Catalysts on Support M1/FeOx for CO Oxidation Via Computational Study. ACS Catal. 2015, 5, 544. 10.1021/cs501790v. [DOI] [Google Scholar]
- Qiu H.; Kuhlenbeck H.; Bauer E.; Freund H.-J. Gold-Decorated Biphase α-Fe2O3(0001): Activation by CO-Induced Surface Reduction. J. Phys. Chem. C 2019, 123, 8221. 10.1021/acs.jpcc.8b08521. [DOI] [Google Scholar]
- Kraushofer F.; Jakub Z.; Bichler M.; Hulva J.; Drmota P.; Weinold M.; Schmid M.; Setvin M.; Diebold U.; Blaha P.; et al. Atomic-Scale Structure of the Hematite α-Fe2O3(1102) ″R-Cut″ Surface. J. Phys. Chem. C 2018, 122, 1657. 10.1021/acs.jpcc.7b10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakub Z.; Kraushofer F.; Bichler M.; Balajka J.; Hulva J.; Pavelec J.; Sokolovic I.; Müllner M.; Setvin M.; Schmid M.; et al. Partially Dissociated Water Dimers at the Water-Hematite Interface. ACS Energy Lett. 2019, 4, 390. 10.1021/acsenergylett.8b02324. [DOI] [Google Scholar]
- Kraushofer F.; Haager L.; Eder M.; Rafsanjani-Abbasi A.; Jakub Z.; Franceschi G.; Riva M.; Meier M.; Schmid M.; Diebold U.; et al. Single Rh Adatoms Stabilized on α-Fe2O3(1102) by Coadsorbed Water. ACS Energy Lett. 2022, 7, 375. 10.1021/acsenergylett.1c02405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliem R.; McDermott E.; Ferstl P.; Setvin M.; Gamba O.; Pavelec J.; Schneider M. A.; Schmid M.; Diebold U.; Blaha P.; et al. Subsurface Cation Vacancy Stabilization of the Magnetite (001) Surface. Science 2014, 346, 1215. 10.1126/science.1260556. [DOI] [PubMed] [Google Scholar]
- Arndt B.; Bliem R.; Gamba O.; van der Hoeven J. E. S.; Noei H.; Diebold U.; Parkinson G. S.; Stierle A. Atomic Structure and Stability of Magnetite Fe3O4(001): An X-Ray View. Surf. Sci. 2016, 653, 76. 10.1016/j.susc.2016.06.002. [DOI] [Google Scholar]
- Novotny Z.; Argentero G.; Wang Z.; Schmid M.; Diebold U.; Parkinson G. S. Ordered Array of Single Adatoms with Remarkable Thermal Stability: Au/Fe3O4(001). Phys. Rev. Lett. 2012, 108, 216103. 10.1103/PhysRevLett.108.216103. [DOI] [PubMed] [Google Scholar]
- Bartelt N. C.; Nie S.; Starodub E.; Bernal-Villamil I.; Gallego S.; Vergara L.; McCarty K. F.; de la Figuera J. Order-Disorder Phase Transition on the (100) Surface of Magnetite. Phys. Rev. B 2013, 88, 235436. 10.1103/PhysRevB.88.235436. [DOI] [Google Scholar]
- Arndt B.; Lechner B. A. J.; Bourgund A.; Grånäs E.; Creutzburg M.; Krausert K.; Hulva J.; Parkinson G. S.; Schmid M.; Vonk V.; et al. Order-Disorder Phase Transition of the Subsurface Cation Vacancy Reconstruction on Fe3O4(001). Phys. Chem. Chem. Phys. 2020, 22, 8336. 10.1039/D0CP00690D. [DOI] [PubMed] [Google Scholar]
- Bliem R.; Kosak R.; Perneczky L.; Novotny Z.; Gamba O.; Fobes D.; Mao Z.; Schmid M.; Blaha P.; Diebold U.; et al. Cluster Nucleation and Growth from a Highly Supersaturated Adatom Phase: Silver on Magnetite. ACS Nano 2014, 8, 7531. 10.1021/nn502895s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier M.; Jakub Z.; Balajka J.; Hulva J.; Bliem R.; Thakur P. K.; Lee T. L.; Franchini C.; Schmid M.; Diebold U.; et al. Probing the Geometry of Copper and Silver Adatoms on Magnetite: Quantitative Experiment Versus Theory. Nanoscale 2018, 10, 2226. 10.1039/C7NR07319D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulva J.; Meier M.; Bliem R.; Jakub Z.; Kraushofer F.; Schmid M.; Diebold U.; Franchini C.; Parkinson G. S. Unraveling CO Adsorption on Model Single-Atom Catalysts. Science 2021, 371, 375. 10.1126/science.abe5757. [DOI] [PubMed] [Google Scholar]
- Bliem R.; Pavelec J.; Gamba O.; McDermott E.; Wang Z.; Gerhold S.; Wagner M.; Osiecki J.; Schulte K.; Schmid M.; et al. Adsorption and Incorporation of Transition Metals at the Magnetite Fe3O4(001) Surface. Phys. Rev. B 2015, 92, 075440. 10.1103/PhysRevB.92.075440. [DOI] [Google Scholar]
- Gargallo-Caballero R.; Martín-García L.; Quesada A.; Granados-Miralles C.; Foerster M.; Aballe L.; Bliem R.; Parkinson G. S.; Blaha P.; Marco J. F.; et al. Co on Fe3O4(001): Towards Precise Control of Surface Properties. J. Chem. Phys. 2016, 144, 094704. 10.1063/1.4942662. [DOI] [PubMed] [Google Scholar]
- Jakub Z.; Hulva J.; Meier M.; Bliem R.; Kraushofer F.; Setvin M.; Schmid M.; Diebold U.; Franchini C.; Parkinson G. S. Local Structure and Coordination Define Adsorption in a Model Ir1/Fe3O4 Single-Atom Catalyst. Angew. Chem., Int. Ed. 2019, 58, 13961. 10.1002/anie.201907536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliem R.; van der Hoeven J. E.; Hulva J.; Pavelec J.; Gamba O.; de Jongh P. E.; Schmid M.; Blaha P.; Diebold U.; Parkinson G. S. Dual Role of CO in the Stability of Subnano Pt Clusters at the Fe3O4(001) Surface. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 8921. 10.1073/pnas.1605649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson G. S.; Novotny Z.; Argentero G.; Schmid M.; Pavelec J.; Kosak R.; Blaha P.; Diebold U. Carbon Monoxide-Induced Adatom Sintering in a Pd-Fe3O4 Model Catalyst. Nat. Mater. 2013, 12, 724. 10.1038/nmat3667. [DOI] [PubMed] [Google Scholar]
- Marcinkowski M. D.; Yuk S. F.; Doudin N.; Smith R. S.; Nguyen M.-T.; Kay B. D.; Glezakou V.-A.; Rousseau R.; Dohnálek Z. Low-Temperature Oxidation of Methanol to Formaldehyde on a Model Single-Atom Catalyst: Pd Atoms on Fe3O4(001). ACS Catal. 2019, 9, 10977. 10.1021/acscatal.9b03891. [DOI] [Google Scholar]
- Meier M.; Hulva J.; Jakub Z.; Kraushofer F.; Bobić M.; Bliem R.; Setvin M.; Schmid M.; Diebold U.; Franchini C.; et al. CO Oxidation by Pt2/Fe3O4: Metastable Dimer and Support Configurations Facilitate Lattice Oxygen Extraction. Sci. Adv. 2022, 8, eabn4580 10.1126/sciadv.abn4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudin N.; Yuk S. F.; Marcinkowski M. D.; Nguyen M.-T.; Liu J.-C.; Wang Y.; Novotny Z.; Kay B. D.; Li J.; Glezakou V.-A.; et al. Understanding Heterolytic H2 Cleavage and Water-Assisted Hydrogen Spillover on Fe3O4(001)-Supported Single Palladium Atoms. ACS Catal. 2019, 9, 7876. 10.1021/acscatal.9b01425. [DOI] [Google Scholar]
- Liu P.; Kendelewicz T.; Brown G. E.; Nelson E. J.; Chambers S. A. Reaction of Water Vapor with α-Al2O3(0001) and α-Fe2O3(0001) Surfaces: Synchrotron X-Ray Photoemission Studies and Thermodynamic Calculations. Surf. Sci. 1998, 417, 53. 10.1016/S0039-6028(98)00661-X. [DOI] [Google Scholar]
- Guo H.; Barnard A. S. Environmentally Dependent Stability of Low-Index Hematite Surfaces. J. Colloid Interface Sci. 2012, 386, 315. 10.1016/j.jcis.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Esch F.; Fabris S.; Zhou L.; Montini T.; Africh C.; Fornasiero P.; Comelli G.; Rosei R. Electron Localization Determines Defect Formation on Ceria Substrates. Science 2005, 309, 752. 10.1126/science.1111568. [DOI] [PubMed] [Google Scholar]
- Dvořák F.; Farnesi Camellone M.; Tovt A.; Tran N.-D.; Negreiros F. R.; Vorokhta M.; Skála T.; Matolínová I.; Mysliveček J.; Matolín V.; et al. Creating Single-Atom Pt-Ceria Catalysts by Surface Step Decoration. Nat. Commun. 2016, 7, 10801. 10.1038/ncomms10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbrügge S.; Cranney M.; Reichling M. Morphology of Step Structures on CeO2(111). Appl. Phys. Lett. 2008, 93, 073112. 10.1063/1.2969790. [DOI] [Google Scholar]
- Nilius N.; Kozlov S. M.; Jerratsch J.-F.; Baron M.; Shao X.; Viñes F.; Shaikhutdinov S.; Neyman K. M.; Freund H.-J. Formation of One-Dimensional Electronic States Along the Step Edges of CeO2(111). ACS Nano 2012, 6, 1126. 10.1021/nn2036472. [DOI] [PubMed] [Google Scholar]
- Skála T.; Šutara F.; Prince K. C.; Matolín V. Cerium Oxide Stoichiometry Alteration Via Sn Deposition: Influence of Temperature. J. Electron Spectrosc. Relat. Phenom. 2009, 169, 20. 10.1016/j.elspec.2008.10.003. [DOI] [Google Scholar]
- Skála T.; Šutara F.; Cabala M.; Škoda M.; Prince K. C.; Matolín V. A Photoemission Study of the Interaction of Ga with CeO2(111) Thin Films. Appl. Surf. Sci. 2008, 254, 6860. 10.1016/j.apsusc.2008.04.102. [DOI] [Google Scholar]
- Skála T.; Tsud N.; Prince K. C.; Matolín V. Formation of Alumina-Ceria Mixed Oxide in Model Systems. Appl. Surf. Sci. 2011, 257, 3682. 10.1016/j.apsusc.2010.11.107. [DOI] [Google Scholar]
- Lu J. L.; Gao H. J.; Shaikhutdinov S.; Freund H. J. Morphology and Defect Structure of the CeO2(111) Films Grown on Ru(0001) as Studied by Scanning Tunneling Microscopy. Surf. Sci. 2006, 600, 5004. 10.1016/j.susc.2006.08.023. [DOI] [Google Scholar]
- Zhou J.; Baddorf A. P.; Mullins D. R.; Overbury S. H. Growth and Characterization of Rh and Pd Nanoparticles on Oxidized and Reduced CeOx(111) Thin Films by Scanning Tunneling Microscopy. J. Phys. Chem. C 2008, 112, 9336. 10.1021/jp711198c. [DOI] [Google Scholar]
- Luches P.; Pagliuca F.; Valeri S.; Illas F.; Preda G.; Pacchioni G. Nature of Ag Islands and Nanoparticles on the CeO2(111) Surface. J. Phys. Chem. C 2012, 116, 1122. 10.1021/jp210241c. [DOI] [Google Scholar]
- Farmer J. A.; Baricuatro J. H.; Campbell C. T. Ag Adsorption on Reduced CeO2(111) Thin Films. J. Phys. Chem. C 2010, 114, 17166. 10.1021/jp104593y. [DOI] [Google Scholar]
- Baron M.; Bondarchuk O.; Stacchiola D.; Shaikhutdinov S.; Freund H. J. Interaction of Gold with Cerium Oxide Supports: CeO2(111) Thin Films Vs CeOx Nanoparticles. J. Phys. Chem. C 2009, 113, 6042. 10.1021/jp9001753. [DOI] [Google Scholar]
- Wilson E. L.; Chen Q.; Brown W. A.; Thornton G. CO Adsorption on the Model Catalyst Pd/CeO2-x(111)/Rh(111). J. Phys. Chem. C 2007, 111, 14215. 10.1021/jp0737449. [DOI] [Google Scholar]
- Zhou Y.; Zhou J. Interactions of Ni Nanoparticles with Reducible CeO2(111) Thin Films. J. Phys. Chem. C 2012, 116, 9544. 10.1021/jp300259y. [DOI] [Google Scholar]
- Szabová L.; Skála T.; Matolínová I.; Fabris S.; Farnesi Camellone M.; Matolín V. Copper-Ceria Interaction: A Combined Photoemission and DFT Study. Appl. Surf. Sci. 2013, 267, 12. 10.1016/j.apsusc.2012.04.098. [DOI] [Google Scholar]
- Ševčíková K.; Szabová L.; Kettner M.; Homola P.; Tsud N.; Fabris S.; Matolín V.; Nehasil V. Experimental and Theoretical Study on the Electronic Interaction between Rh Adatoms and CeOx Substrate in Dependence on a Degree of Cerium Oxide Reduction. J. Phys. Chem. C 2016, 120, 5468. 10.1021/acs.jpcc.5b11431. [DOI] [Google Scholar]
- Lustemberg P. G.; Pan Y.; Shaw B.-J.; Grinter D.; Pang C.; Thornton G.; Pérez R.; Ganduglia-Pirovano M. V.; Nilius N. Diffusion Barriers Block Defect Occupation on Reduced CeO2(111). Phys. Rev. Lett. 2016, 116, 236101. 10.1103/PhysRevLett.116.236101. [DOI] [PubMed] [Google Scholar]
- Lykhach Y.; Bruix A.; Fabris S.; Potin V.; Matolínová I.; Matolín V.; Libuda J.; Neyman K. M. Oxide-Based Nanomaterials for Fuel Cell Catalysis: The Interplay between Supported Single Pt Atoms and Particles. Catal. Sci. Technol. 2017, 7, 4315. 10.1039/C7CY00710H. [DOI] [Google Scholar]
- Bruix A.; Lykhach Y.; Matolínová I.; Neitzel A.; Skála T.; Tsud N.; Vorokhta M.; Stetsovych V.; Ševčíková K.; Mysliveček J.; et al. Maximum Noble-Metal Efficiency in Catalytic Materials: Atomically Dispersed Surface Platinum. Angew. Chem., Int. Ed. 2014, 53, 10525. 10.1002/anie.201402342. [DOI] [PubMed] [Google Scholar]
- Neitzel A.; Lykhach Y.; Skála T.; Tsud N.; Vorokhta M.; Mazur D.; Prince K. C.; Matolín V.; Libuda J. Surface Sites on Pt-CeO2 Mixed Oxide Catalysts Probed by CO Adsorption: A Synchrotron Radiation Photoelectron Spectroscopy Study. Phys. Chem. Chem. Phys. 2014, 16, 24747. 10.1039/C4CP03346A. [DOI] [PubMed] [Google Scholar]
- Lykhach Y.; Figueroba A.; Camellone M. F.; Neitzel A.; Skála T.; Negreiros F. R.; Vorokhta M.; Tsud N.; Prince K. C.; Fabris S.; et al. Reactivity of Atomically Dispersed Pt2+ Species Towards H2: Model Pt-CeO2 Fuel Cell Catalyst. Phys. Chem. Chem. Phys. 2016, 18, 7672. 10.1039/C6CP00627B. [DOI] [PubMed] [Google Scholar]
- Neitzel A.; Johánek V.; Lykhach Y.; Skála T.; Tsud N.; Vorokhta M.; Matolín V.; Libuda J. Reduction of Pt2+ Species in Model Pt-CeO2 Fuel Cell Catalysts Upon Reaction with Methanol. Appl. Surf. Sci. 2016, 387, 674. 10.1016/j.apsusc.2016.06.156. [DOI] [Google Scholar]
- Lykhach Y.; Figueroba A.; Skála T.; Duchoň T.; Tsud N.; Aulická M.; Neitzel A.; Veltruská K.; Prince K. C.; Matolín V.; et al. Redox-Mediated Conversion of Atomically Dispersed Platinum to Sub-Nanometer Particles. J. Mater. Chem. A 2017, 5, 9250. 10.1039/C7TA02204B. [DOI] [Google Scholar]
- Huang M.; Fabris S. Role of Surface Peroxo and Superoxo Species in the Low-Temperature Oxygen Buffering of Ceria: Density Functional Theory Calculations. Phys. Rev. B 2007, 75, 081404. 10.1103/PhysRevB.75.081404. [DOI] [Google Scholar]
- Tovt A.; Bagolini L.; Dvořák F.; Tran N.-D.; Vorokhta M.; Beranová K.; Johánek V.; Farnesi Camellone M.; Skála T.; Matolínová I.; et al. Ultimate Dispersion of Metallic and Ionic Platinum on Ceria. J. Mater. Chem. A 2019, 7, 13019. 10.1039/C9TA00823C. [DOI] [Google Scholar]
- Farnesi Camellone M.; Dvořák F.; Vorokhta M.; Tovt A.; Khalakhan I.; Johánek V.; Skála T.; Matolínová I.; Fabris S.; Mysliveček J. Adatom and Nanoparticle Dynamics on Single-Atom Catalyst Substrates. ACS Catal. 2022, 12, 4859. 10.1021/acscatal.2c00291. [DOI] [Google Scholar]
- Maurer F.; Jelic J.; Wang J.; Gänzler A.; Dolcet P.; Wöll C.; Wang Y.; Studt F.; Casapu M.; Grunwaldt J.-D. Tracking the Formation, Fate and Consequence for Catalytic Activity of Pt Single Sites on CeO2. Nat. Catal. 2020, 3, 824. 10.1038/s41929-020-00508-7. [DOI] [Google Scholar]
- Figueroba A.; Kovács G.; Bruix A.; Neyman K. M. Towards Stable Single-Atom Catalysts: Strong Binding of Atomically Dispersed Transition Metals on the Surface of Nanostructured Ceria. Catal. Sci. Technol. 2016, 6, 6806. 10.1039/C6CY00294C. [DOI] [Google Scholar]
- Neitzel A.; Figueroba A.; Lykhach Y.; Skála T.; Vorokhta M.; Tsud N.; Mehl S.; Ševčíková K.; Prince K. C.; Neyman K. M.; et al. Atomically Dispersed Pd, Ni, and Pt Species in Ceria-Based Catalysts: Principal Differences in Stability and Reactivity. J. Phys. Chem. C 2016, 120, 9852. 10.1021/acs.jpcc.6b02264. [DOI] [Google Scholar]
- Daelman N.; Capdevila-Cortada M.; López N. Dynamic Charge and Oxidation State of Pt/CeO2 Single-Atom Catalysts. Nat. Mater. 2019, 18, 1215. 10.1038/s41563-019-0444-y. [DOI] [PubMed] [Google Scholar]
- Wan W.; Geiger J.; Berdunov N.; Lopez Luna M.; Chee S. W.; Daelman N.; Lopez N.; Shaikhutdinov S.; Roldan Cuenya B. Highly Stable and Reactive Platinum Single Atoms on Oxygen Plasma-Functionalized CeO2 Surfaces: Nanostructuring and Peroxo Effects. Angew. Chem., Int. Ed. 2022, 61, e202112640 10.1002/anie.202112640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao B.; Liu J.; Wang Y.-G.; Lin Q.; Liu X.; Wang A.; Li J.; Zhang T.; Liu J. Highly Efficient Catalysis of Preferential Oxidation of CO in H2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249. 10.1021/acscatal.5b01114. [DOI] [Google Scholar]
- Zhang C.; Michaelides A.; King D. A.; Jenkins S. J. Structure of Gold Atoms on Stoichiometric and Defective Ceria Surfaces. J. Chem. Phys. 2008, 129, 194708. 10.1063/1.3009629. [DOI] [PubMed] [Google Scholar]
- Branda M. M.; Castellani N. J.; Grau-Crespo R.; de Leeuw N. H.; Hernandez N. C.; Sanz J. F.; Neyman K. M.; Illas F. On the Difficulties of Present Theoretical Models to Predict the Oxidation State of Atomic Au Adsorbed on Regular Sites of CeO2(111). J. Chem. Phys. 2009, 131, 094702. 10.1063/1.3216102. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Cui Y.; Stiehler C.; Nilius N.; Freund H.-J. Gold Adsorption on CeO2 Thin Films Grown on Ru(0001). J. Phys. Chem. C 2013, 117, 21879. 10.1021/jp407605m. [DOI] [Google Scholar]
- Pan Y.; Nilius N.; Freund H.-J.; Paier J.; Penschke C.; Sauer J. Titration of Ce3+ Ions in the CeO2(111) Surface by Au Adatoms. Phys. Rev. Lett. 2013, 111, 206101. 10.1103/PhysRevLett.111.206101. [DOI] [PubMed] [Google Scholar]
- Kong D.; Wang G.; Pan Y.; Hu S.; Hou J.; Pan H.; Campbell C. T.; Zhu J. Growth, Structure, and Stability of Ag on CeO2(111): Synchrotron Radiation Photoemission Studies. J. Phys. Chem. C 2011, 115, 6715. 10.1021/jp112392y. [DOI] [Google Scholar]
- Hu S.; Wang Y.; Wang W.; Han Y.; Fan Q.; Feng X.; Xu Q.; Zhu J. Ag Nanoparticles on Reducible CeO2(111) Thin Films: Effect of Thickness and Stoichiometry of Ceria. J. Phys. Chem. C 2015, 119, 3579. 10.1021/jp511691p. [DOI] [Google Scholar]
- James T. E.; Hemmingson S. L.; Ito T.; Campbell C. T. Energetics of Cu Adsorption and Adhesion onto Reduced CeO2(111) Surfaces by Calorimetry. J. Phys. Chem. C 2015, 119, 17209. 10.1021/acs.jpcc.5b04621. [DOI] [Google Scholar]
- Nie L.; Mei D.; Xiong H.; Peng B.; Ren Z.; Hernandez X. I. P.; DeLaRiva A.; Wang M.; Engelhard M. H.; Kovarik L.; et al. Activation of Surface Lattice Oxygen in Single-Atom Pt/CeO2 for Low-Temperature CO Oxidation. Science 2017, 358, 1419. 10.1126/science.aao2109. [DOI] [PubMed] [Google Scholar]
- Gasperi G.; Brugnoli L.; Pedone A.; Menziani M. C.; Valeri S.; Luches P. Reducibility of Ag- and Cu-Modified Ultrathin Epitaxial Cerium Oxide Films. J. Phys. Chem. C 2019, 123, 13702. 10.1021/acs.jpcc.9b02378. [DOI] [Google Scholar]
- Benedetti S.; Righi G.; Luches P.; D’Addato S.; Magri R.; Selloni A. Surface Reactivity of Ag-Modified Ceria to Hydrogen: A Combined Experimental and Theoretical Investigation. ACS Appl. Mater. Interfaces 2020, 12, 27682. 10.1021/acsami.0c03968. [DOI] [PubMed] [Google Scholar]
- Wang Y.-G.; Mei D.; Glezakou V.-A.; Li J.; Rousseau R. Dynamic Formation of Single-Atom Catalytic Active Sites on Ceria-Supported Gold Nanoparticles. Nat. Commun. 2015, 6, 6511. 10.1038/ncomms7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbet S.; Sanchez A.; Heiz U.; Schneider W. D.; Ferrari A. M.; Pacchioni G.; Rösch N. Acetylene Cyclotrimerization on Supported Size-Selected Pdn Clusters (1 ≤ n ≤ 30): One Atom Is Enough!. J. Am. Chem. Soc. 2000, 122, 3453. 10.1021/ja9922476. [DOI] [Google Scholar]
- Sterrer M.; Heyde M.; Novicki M.; Nilius N.; Risse T.; Rust H.-P.; Pacchioni G.; Freund H.-J. Identification of Color Centers on MgO(001) Thin Films with Scanning Tunneling Microscopy. J. Phys. Chem. B 2006, 110, 46. 10.1021/jp056306f. [DOI] [PubMed] [Google Scholar]
- Sterrer M.; Fischbach E.; Heyde M.; Nilius N.; Rust H.-P.; Risse T.; Freund H.-J. Electron Paramagnetic Resonance and Scanning Tunneling Microscopy Investigations on the Formation of F+ and F0 Color Centers on the Surface of Thin MgO(001) Films. J. Phys. Chem. B 2006, 110, 8665. 10.1021/jp060546t. [DOI] [PubMed] [Google Scholar]
- König T.; Simon G. H.; Martinez U.; Giordano L.; Pacchioni G.; Heyde M.; Freund H.-J. Direct Measurement of the Attractive Interaction Forces on F0 Color Centers on MgO(001) by Dynamic Force Microscopy. ACS Nano 2010, 4, 2510. 10.1021/nn100443n. [DOI] [PubMed] [Google Scholar]
- Chiesa M.; Paganini M. C.; Giamello E.; Di Valentin C.; Pacchioni G. First Evidence of a Single-Ion Electron Trap at the Surface of an Ionic Oxide. Angew. Chem. 2003, 115, 1801. 10.1002/ange.200250811. [DOI] [PubMed] [Google Scholar]
- Chiesa M.; Paganini M. C.; Spoto G.; Giamello E.; Di Valentin C.; Del Vitto A.; Pacchioni G. Single Electron Traps at the Surface of Polycrystalline MgO: Assignment of the Main Trapping Sites. J. Phys. Chem. B 2005, 109, 7314. 10.1021/jp044783c. [DOI] [PubMed] [Google Scholar]
- Sterrer M.; Berger T.; Diwald O.; Knözinger E.; Sushko P. V.; Shluger A. L. Chemistry at Corners and Edges: Generation and Adsorption of H Atoms on the Surface of MgO Nanocubes. J. Chem. Phys. 2005, 123, 064714. 10.1063/1.1997108. [DOI] [PubMed] [Google Scholar]
- Ojamäe L.; Pisani C. Theoretical Characterization of Divacancies at the Surface and in Bulk MgO. J. Chem. Phys. 1998, 109, 10984. 10.1063/1.477793. [DOI] [Google Scholar]
- Giordano L.; Di Valentin C.; Goniakowski J.; Pacchioni G. Nucleation of Pd Dimers at Defect Sites of the MgO (100) Surface. Phys. Rev. Lett. 2004, 92, 096105. 10.1103/PhysRevLett.92.096105. [DOI] [PubMed] [Google Scholar]
- Ashworth T. V.; Pang C. L.; Wincott P. L.; Vaughan D. J.; Thornton G. Imaging in Situ Cleaved MgO(100) with Non-Contact Atomic Force Microscopy. Appl. Surf. Sci. 2003, 210, 2. 10.1016/S0169-4332(02)01469-1. [DOI] [Google Scholar]
- Barth C.; Henry C. R. Atomic Resolution Imaging of the (001) Surface of UHV Cleaved MgO by Dynamic Scanning Force Microscopy. Phys. Rev. Lett. 2003, 91, 196102. 10.1103/PhysRevLett.91.196102. [DOI] [PubMed] [Google Scholar]
- Sanchez A.; Abbet S.; Heiz U.; Schneider W. D.; Häkkinen H.; Barnett R. N.; Landman U. When Gold Is Not Noble: Nanoscale Gold Catalysts. J. Phys. Chem. A 1999, 103, 9573. 10.1021/jp9935992. [DOI] [Google Scholar]
- Heiz U.; Sanchez A.; Abbet S.; Schneider W. D. Tuning the Oxidation of Carbon Monoxide Using Nanoassembled Model Catalysts. Chem. Phys. 2000, 262, 189. 10.1016/S0301-0104(00)00268-8. [DOI] [Google Scholar]
- Brown M. A.; Ringleb F.; Fujimori Y.; Sterrer M.; Freund H.-J.; Preda G.; Pacchioni G. Initial Formation of Positively Charged Gold on MgO(001) Thin Films: Identification by Experiment and Structural Assignment by Theory. J. Phys. Chem. C 2011, 115, 10114. 10.1021/jp201254s. [DOI] [Google Scholar]
- Del Vitto A.; Pacchioni G.; Delbecq F.; Sautet P. Au Atoms and Dimers on the MgO (100) Surface: A DFT Study of Nucleation at Defects. J. Phys. Chem. B 2005, 109, 8040. 10.1021/jp044143+. [DOI] [PubMed] [Google Scholar]
- Sterrer M.; Yulikov M.; Fischbach E.; Heyde M.; Rust H.-P.; Pacchioni G.; Risse T.; Freund H.-J. Interaction of Gold Clusters with Color Centers on MgO(001) Films. Angew. Chem., Int. Ed. 2006, 45, 2630. 10.1002/anie.200504443. [DOI] [PubMed] [Google Scholar]
- Giordano L.; Baistrocchi M.; Pacchioni G. Bonding of Pd, Ag, and Au Atoms on MgO(100) Surfaces and MgO/Mo(100) Ultra-Thin Films: A Comparative DFT Study. Phys. Rev. B 2005, 72, 115403. 10.1103/PhysRevB.72.115403. [DOI] [Google Scholar]
- Giordano L.; Pacchioni G. Charge Transfers at Metal/Oxide Interfaces: A DFT Study of Formation of Kδ+ and Auδ- Species on MgO/Ag(100) Ultra-Thin Films from Deposition of Neutral Atoms. Phys. Chem. Chem. Phys. 2006, 8, 3335. 10.1039/B604288K. [DOI] [PubMed] [Google Scholar]
- Sterrer M.; Risse T.; Heyde M.; Rust H.-P.; Freund H.-J. Crossover from Three-Dimensional to Two-Dimensional Geometries of Au Nanostructures on Thin MgO(001) Films: A Confirmation of Theoretical Predictions. Phys. Rev. Lett. 2007, 98, 206103. 10.1103/PhysRevLett.98.206103. [DOI] [PubMed] [Google Scholar]
- Gonchar A.; Lian J.; Risse T.; Freund H. J.; Di Valentin C.; Pacchioni G. Characterization of O–-Centers on Single Crystalline MgO(001)-Films. Top. Catal. 2015, 58, 811. 10.1007/s11244-015-0421-x. [DOI] [Google Scholar]
- Gonchar A.; Risse T. Characterisation of Paramagnetic Mo Impurities on MgO(100) Single-Crystalline Films Grown on Mo(100). Mol. Phys. 2013, 111, 2708. 10.1080/00268976.2013.811305. [DOI] [Google Scholar]
- Shao X.; Prada S.; Giordano L.; Pacchioni G.; Nilius N.; Freund H.-J. Tailoring the Shape of Metal Ad-Particles by Doping the Oxide Support. Angew. Chem., Int. Ed. 2011, 50, 11525. 10.1002/anie.201105355. [DOI] [PubMed] [Google Scholar]
- Haas G.; Menck A.; Brune H.; Barth J.; Venables J.; Kern K. Nucleation and Growth of Supported Clusters at Defect Sites: Pd/MgO(001). Phys. Rev. B 2000, 61, 11105. 10.1103/PhysRevB.61.11105. [DOI] [Google Scholar]
- Kyriakou G.; Boucher M. B.; Jewell A. D.; Lewis E. A.; Lawton T. J.; Baber A. E.; Tierney H. L.; Flytzani-Stephanopoulos M.; Sykes E. C. H. Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations. Science 2012, 335, 1209. 10.1126/science.1215864. [DOI] [PubMed] [Google Scholar]
- Hannagan R. T.; Giannakakis G.; Réocreux R.; Schumann J.; Finzel J.; Wang Y.; Michaelides A.; Deshlahra P.; Christopher P.; Flytzani-Stephanopoulos M.; et al. First-Principles Design of a Single-Atom Alloy Propane Dehydrogenation Catalyst. Science 2021, 372, 1444. 10.1126/science.abg8389. [DOI] [Google Scholar]
- Jensen F.; Besenbacher F.; Lægsgaard E.; Stensgaard I. Oxidation of Cu(111): Two New Oxygen Induced Reconstructions. Surf. Sci. 1991, 259, L774. 10.1016/0039-6028(91)90550-C. [DOI] [Google Scholar]
- Jensen F.; Besenbacher F.; Stensgaard I. Two New Oxygen Induced Reconstructions on Cu(111). Surf. Sci. 1992, 269–270, 400. 10.1016/0039-6028(92)91282-G. [DOI] [Google Scholar]
- Therrien A. J.; Groden K.; Hensley A. J. R.; Schilling A. C.; Hannagan R. T.; Marcinkowski M. D.; Pronschinske A.; Lucci F. R.; Sykes E. C. H.; McEwen J.-S. Water Activation by Single Pt Atoms Supported on a Cu2O Thin Film. J. Catal. 2018, 364, 166. 10.1016/j.jcat.2018.04.024. [DOI] [Google Scholar]
- Therrien A. J.; Hensley A. J. R.; Marcinkowski M. D.; Zhang R.; Lucci F. R.; Coughlin B.; Schilling A. C.; McEwen J.-S.; Sykes E. C. H. An Atomic-Scale View of Single-Site Pt Catalysis for Low-Temperature CO Oxidation. Nat. Catal. 2018, 1, 192. 10.1038/s41929-018-0028-2. [DOI] [Google Scholar]
- Wang C.; Tissot H.; Stenlid J. H.; Kaya S.; Weissenrieder J. High-Density Isolated Fe1O3 Sites on a Single-Crystal Cu2O(100) Surface. J. Phys. Chem. Lett. 2019, 10, 7318. 10.1021/acs.jpclett.9b02979. [DOI] [PubMed] [Google Scholar]
- Tissot H.; Wang C.; Stenlid J. H.; Brinck T.; Weissenrieder J. The Surface Structure of Cu2O(100): Nature of Defects. J. Phys. Chem. C 2019, 123, 7696. 10.1021/acs.jpcc.8b05156. [DOI] [Google Scholar]
- Chun W.-J.; Koike Y.; Ijima K.; Fujikawa K.; Ashima H.; Nomura M.; Iwasawa Y.; Asakura K. Preparation of Atomically Dispersed Cu Species on a TiO2 (110) Surface Premodified with an Organic Compound. Chem. Phys. Lett. 2007, 433, 345. 10.1016/j.cplett.2006.11.080. [DOI] [Google Scholar]
- Asakura K.; Takakusagi S.; Ariga H.; Chun W.-J.; Suzuki S.; Koike Y.; Uehara H.; Miyazaki K.; Iwasawa Y. Preparation and Structure of a Single Au Atom on the TiO2(110) Surface: Control of the Au-Metal Oxide Surface Interaction. Faraday Discuss. 2013, 162, 165. 10.1039/c2fd20131c. [DOI] [PubMed] [Google Scholar]
- Takakusagi S.; Kunimoto A.; Sirisit N.; Uehara H.; Ohba T.; Uemuara Y.; Wada T.; Ariga H.; Chun W.-J.; Iwasawa Y.; et al. A New Indicator for Single Metal Dispersion on a TiO2(110) Surface Premodified with a Mercapto Compound. J. Phys. Chem. C 2016, 120, 15785. 10.1021/acs.jpcc.5b11630. [DOI] [Google Scholar]
- Takakusagi S.; Iwasawa Y.; Asakura K. Premodified Surface Method to Obtain Ultra-Highly Dispersed Metals and Their 3D Structure Control on an Oxide Single-Crystal Surface. Chem. Rec. 2019, 19, 1244. 10.1002/tcr.201800088. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lu Y.; Liu L.; Yu L.; Yang C.; Delferro M.; Hoffman A. S.; Bare S. R.; Karim A. M.; Xin H. Catalytic CO Oxidation on MgAl2O4-Supported Iridium Single Atoms: Ligand Configuration and Site Geometry. J. Phys. Chem. C 2021, 125, 11380. 10.1021/acs.jpcc.1c02287. [DOI] [Google Scholar]
- Lu Y.; Wang J.; Yu L.; Kovarik L.; Zhang X.; Hoffman A. S.; Gallo A.; Bare S. R.; Sokaras D.; Kroll T.; et al. Identification of the Active Complex for CO Oxidation over Single-Atom Ir-on-MgAl2O4 Catalysts. Nat. Catal. 2019, 2, 149. 10.1038/s41929-018-0192-4. [DOI] [Google Scholar]
- Lewandowski A. L.; Tosoni S.; Gura L.; Yang Z.; Fuhrich A.; Prieto M. J.; Schmidt T.; Usvyat D.; Schneider W.-D.; Heyde M.; et al. Growth and Atomic-Scale Characterization of Ultrathin Silica and Germania Films: The Crucial Role of the Metal Support. Chem. Eur. J. 2021, 27, 1870. 10.1002/chem.202001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büchner C.; Heyde M. Two-Dimensional Silica Opens New Perspectives. Prog. Surf. Sci. 2017, 92, 341. 10.1016/j.progsurf.2017.09.001. [DOI] [Google Scholar]
- Büchner C.; Lichtenstein L.; Stuckenholz S.; Heyde M.; Ringleb F.; Sterrer M.; Kaden W. E.; Giordano L.; Pacchioni G.; Freund H.-J. Adsorption of Au and Pd on Ruthenium-Supported Bilayer Silica. J. Phys. Chem. C 2014, 118, 20959. 10.1021/jp5055342. [DOI] [Google Scholar]
- Ulrich S.; Nilius N.; Freund H.-J.; Martinez U.; Giordano L.; Pacchioni G. Modifying the Adsorption Characteristic of Inert Silica Films by Inserting Anchoring Sites. Phys. Rev. Lett. 2009, 102, 016102. 10.1103/PhysRevLett.102.016102. [DOI] [PubMed] [Google Scholar]
- Schmid M.; Kresse G.; Buchsbaum A.; Napetschnig E.; Gritschneder S.; Reichling M.; Varga P. Nanotemplate with Holes: Ultrathin Alumina on Ni3Al (111). Phys. Rev. Lett. 2007, 99, 196104. 10.1103/PhysRevLett.99.196104. [DOI] [PubMed] [Google Scholar]
- Franchini C.; Reticcioli M.; Setvin M.; Diebold U. Polarons in Materials. Nat. Rev. Mater. 2021, 6, 560. 10.1038/s41578-021-00289-w. [DOI] [Google Scholar]
- Zitolo A.; Goellner V.; Armel V.; Sougrati M.-T.; Mineva T.; Stievano L.; Fonda E.; Jaouen F. Identification of Catalytic Sites for Oxygen Reduction in Iron- and Nitrogen-Doped Graphene materials. Nat. Mater. 2015, 14, 937. 10.1038/nmat4367. [DOI] [PubMed] [Google Scholar]