

Abstract
Water solubility and structural stability are key merits for proteins defined by the primary sequence and 3D-conformation. Their manipulation represents important aspects of the protein design field that relies on the accurate placement of amino acids and molecular interactions, guided by underlying physiochemical principles. Emulated designer proteins with well-defined properties both fuel the knowledge-base for more precise computational design models and are used in various biomedical and nanotechnological applications. The continuous developments in protein science, increasing computing power, new algorithms, and characterization techniques provide sophisticated toolkits for solubility design beyond guess work. In this review, we summarize recent advances in the protein design field with respect to water solubility and structural stability. After introducing fundamental design rules, we discuss the transmembrane protein solubilization and de novo transmembrane protein design. Traditional strategies to enhance protein solubility and structural stability are introduced. The designs of stable protein complexes and high-order assemblies are covered. Computational methodologies behind these endeavors, including structure prediction programs, machine learning algorithms, and specialty software dedicated to the evaluation of protein solubility and aggregation, are discussed. The findings and opportunities for Cryo-EM are presented. This review provides an overview of significant progress and prospects in accurate protein design for solubility and stability.
1. Introduction
Proteins evolve through natural selection for their particular function and location in biological systems. For DNA replication, RNA transcription and ribosome protein translation, the proteins need to be water-soluble in aqueous cytosol. Likewise, secreted proteins also need to be in aqueous environments for diffusion or circulation.1,2 On the other hand, cells must have barriers to separate themselves from the environment and to concentrate cellular substances. Thus, dynamic membranes are necessary for all living systems to enclose the cytosols and establish concentration gradients for substances moving in and out of cells. In addition to the hydrophobic and nonselective lipid bilayer, membrane proteins are essential to various functions, such as photosynthesis systems, transporters, ion channels, ATP synthases, and membrane receptors, serving as communication systems embedded in the lipid bilayer that divide and regulate the internal and external cellular environments.1,2
In this regard, native proteins may be broadly categorized in two classes: (i) hydrophilic and (ii) hydrophobic, with exceptions such as fibrous proteins and transmembrane β-barrels.3 The hydrophilic class comprises water-soluble proteins that reside mostly in cytoplasm such as hemoglobin (Figure 1A), or circulate extracellularly such as lysozyme.4,5 The hydrophobic class comprises membrane proteins embedded in cellular and other membranes, which include G protein-coupled receptors (GPCRs) (Figure 1B), photosynthesis machines, etc.6,7 Although both hemoglobin and GPCRs are mostly composed of α-helices, the former is highly water-soluble and the latter are water-insoluble.8,9
Figure 1.
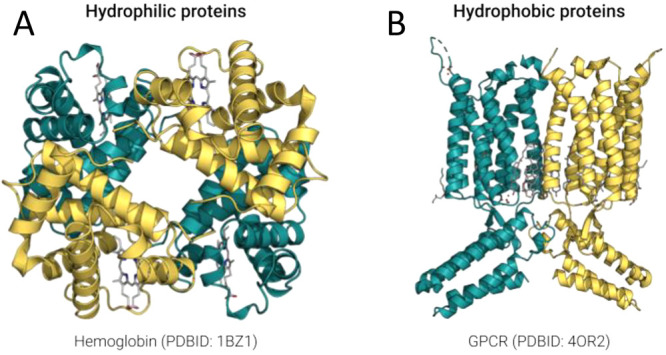
Molecular structures of representative hydrophilic and hydrophobic proteins with high α-helical content. (A) Molecular structure of hemoglobin (PDB ID: 1BZ1), which comprises ∼80% α-helices and is highly water-soluble in cytosol. (B) Molecular structure of G protein-coupled receptors (metabotropic glutamate receptor 1 bound to an allosteric modulator, PDB ID: 4OR2), which comprise ∼50–80% α-helices and are water insoluble. The side chain chemical properties of amino acids on these α-helices determine the proteins water solubility.
Solubility is a fundamental concept and crucial in protein science.10,11 As a trait of proteins determined by both their primary sequences and environmental conditions,12 the concept is important for structural and biophysical studies, and has been a major concern for protein chemists, structural biologists, pharmaceutical scientists, or any researcher who work with proteins in solution.13,14 Solubility has profound implications in biotechnological, biochemical and medical applications especially concerning the expression and purification of therapeutic proteins.15 High concentrations of protein formulations are often required for subcutaneous applications or based on the pathogenicity of target diseases.16
Central to solubility are the charge and polarity of amino acids, and their capability to interact with surrounding water molecules. All 20 natural amino acids can be found in both α-helices and β-sheets, albeit with varying propensities.1−3,17 They differ in the chemical structure of side chains and hence in hydrophilicity in aqueous environments. For instance, the side chains of leucine (L), valine (V), isoleucine (I), alanine (A), and phenylalanine (F) are nonpolar and cannot form any hydrogen bonds with water, rendering them hydrophobic. Aspartic acid (D), asparagine (N), glutamate (E), and glutamine (Q) can form four hydrogen bonds, while serine (S), threonine (T), and tyrosine (Y) can form three hydrogen bonds with water molecules, rendering them hydrophilic. Additionally, positively charged amino acids such as histidine (H), lysine (K), and arginine (R) not only can form two, three, and five hydrogen bonds with water molecules, respectively, but also will be protonated at acidic or neutral pH which can subsequently induce strong electrostatic interactions called ionic bonds, or salt bridges to stabilize the overall conformation of a protein (Figure 2).18
Figure 2.

Hydrogen bond formations of water molecules with amino acids’ side chains at neutral pH. Aspartic acid (D) and glutamic acid (E) (four water molecules), asparagine (N) and glutamine (Q) (four water molecules), serine (S), threonine (T), and tyrosine (Y) (three water molecules), arginine (R) (five water molecules), lysine (K) (three water molecules), histidine (H) (two water molecules), and tryptophan (W) (one water molecule).
Despite their categorization and differences in hydrophilicity, some pairs or groups of amino acids have strikingly similar side chain chemical structures and electron density maps, indicating similar steric interactions. This similarity occasionally triggers erroneous charging of tRNAs and mis-incorporation of amino acids into proteins. For example, the valine (V) tRNA synthetase (ValRS) mischarges threonine (T) and isoleucine (I) at a rate of one per 200–400.19−21 This observation was recently adopted in the solubilization of transmembrane proteins, as will be discussed in Section 3.4.
Similarly, all three types of α-helices, that is, hydrophobic, partially hydrophilic, and hydrophilic (Figure 3), share an identical molecular structural basis revealed through high-resolution X-ray crystallography and cryo-electron microscopy (Cryo-EM) studies, namely: (i) 1.5 Å per amino acid rise, (ii) 100° per amino acid rotation, (iii) 5.4 Å, 360°, and 3.6 amino acids per α-helical turn despite the difference in their solubility in water.22 Such similarities pose intriguing questions about the dependence of water solubility and structural stability of proteins on their molecular structures and interactions.
Figure 3.
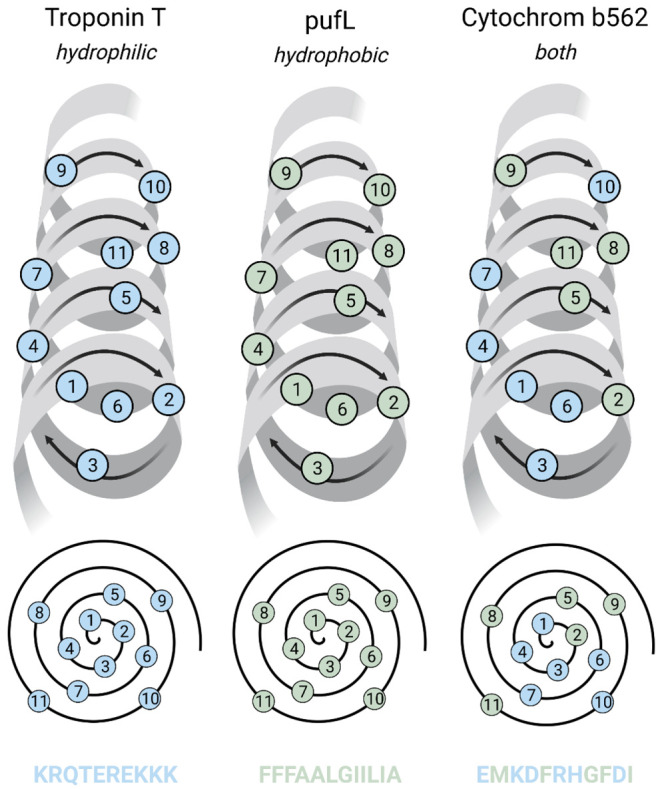
Three types of α-helices: hydrophilic (blue residues), hydrophobic (green residues), and partially hydrophilic. They all share the same structure: (a) 1.5 Å per amino acid rise, (b) 100° per amino acid rotation, (c) 5.4 Å, 360°, and 3.6 amino acids per α-helical turn, despite differences in their solubility in water.
Understanding and tuning protein water solubility and stability is crucial to elucidating their structure–function relation in fundamental biology, as well as to developing them as novel components in interdisciplinary applications. Numerous efforts have been devoted to designing protein sequences for enhanced solubility without disturbing the native structure and function. More recently, the increasing computing power and new algorithms enable protein scientists to turn the focus to more difficult targets that were not previously achievable, such as transmembrane proteins and novel structures like cross-α amyloids and β-barrels, or to incorporate functions into a de novo protein structure design.
This review is organized to present the most recent advances in the field of protein design with special emphasis placed on aspects of water solubility and structural stability. While we will also cover areas regarding protein functions and novel structures, this will not be an exhaustive review to summarize the entire field regarding biochemical, biophysical, structural or proteomic aspects of protein design. To obtain a broad understanding and a comprehensive view, interested readers should consult previous dedicated reviews or refer to the original literature for more detailed discussions.22−26
In this review, we will start with the definition of protein water solubility, followed by a brief introduction to the fundamentals of protein design. The main focus will be placed on the discussion of recent progress in the solubility redesign of naturally occurring transmembrane proteins, not only because of their critical roles in biological processes, but also because of the difficulties associated with solubilization and overexpression. De novo design of transmembrane proteins will then be covered to discuss transmembrane protein solubility from an inverse direction. Common approaches for solubility enhancements of other membrane-bound or nonmembrane-bound proteins will be reviewed. The design and tuning of protein functions will then be discussed. Designing complex soluble structures and higher-order protein assemblies through de novo approaches will also be covered. A supporting section on coiled-coil structures will be presented. Additionally, the commonly used algorithms and methodologies for protein design, structural predication and solubility evaluation will be discussed. Finally, findings and opportunities for Cryo-EM in native and designer proteins will be presented.
2. Solubility and Introduction to Protein Design
The solubility of a substance is chemically defined to be the maximum amount of mass that can be dissolved in a designated solvent, presumably water in an aqueous solution.27 Yet this definition is not directly applicable to proteins, especially when structures and functions are concerned. The absolute maximum solubility of a protein is defined by quantitatively measuring it, often by using condition extremes, such as pH, temperature variation or high concentration denaturants, to determine the thermodynamic equilibrium.28 However, high concentrations can also induce the protein to form soluble aggregates, or accommodate alternative conformations with similar energy states that diminish native functions without precipitation.29 The protein conformation associated with the desired functional performance does not necessarily align with that of the maximum solubility. Thus, in this review, we obscure the boundary between absolute solubility and protein stability for a better discussion of these biomolecules in a stabilized, functional form.
Proteins are long polypeptide chains folded into the functional forms with a hierarchical structure, namely primary, secondary, tertiary, and quaternary structures (Figure 4). In this regard, the sequence or the primary structure is the basic level for determining a protein’s solubility in the first place. Individual amino acids vary greatly in their hydrophilicity and thus contribute differently to the solubility of the chain.30 Polar or charged amino acids are more likely to interact with surrounding water molecules so as to increase a protein’s structural stability in aqueous environments in a cumulative manner. Secondary and tertiary structures form by the preferential association of different segments in the polypeptide chain to accommodate a 3D conformation that minimize the overall free energy. In the class of hydrophilic soluble proteins, burying hydrophobic amino acids into the core and avoiding their contact with environmental water molecules are the major driving forces for this process, called hydrophobic effect.31,32 While the conformation is subsequently stabilized by hydrogen bonds between surface hydrophilic residues and water molecules, exposed hydrophobic residues can create an energy penalty and negatively affect the structural stability. Maximizing polar interactions on the surface has been the guiding principle for soluble protein design over the past several decades.33 In this section, we will summarize the deterministic factors for this property, introduce traditional methods for protein solubilization and provide an overview of common design approaches adopted for solubility and stability enhancement.
Figure 4.

Hierarchy in protein structures. Clockwise: primary protein sequence with amino acid side chains, secondary structural motifs of α-helix (blue color) and β-sheet (green color), folded monomeric tertiary structure, and self-assembled trimeric quaternary structure with three subunits.
2.1. Parameters Affecting Solubility
2.1.1. Extrinsic Factors
The solubility and stability of proteins are affected by many extrinsic factors, such as pH, temperature, solvent, ionic strength, metal-ion cofactors, and the presence of surfactants.34,35 These environmental parameters synergistically interact with proteins’ intrinsic properties such as the size, hydrophobicity, electrostatics, charge distribution, and influence the apparent stability of the polypeptide chain in a given condition.36−38
Appropriate temperature and solvent are the prerequisite conditions for proteins to prevent aggregation and retain functions. Varying these parameters provides a straightforward way to determine proteins’ dependent solubility at physiological conditions and to control their folding/unfolding, inducing structure disruptions and functional inactivations.39,40 Native proteins often exhibit the highest solubility between 40 and 50 °C, whereas denaturation usually occurs when the temperature of the system is elevated for an extended amount of time.41 Proteins are denatured by the impact of temperature or solvent change on noncovalent interactions, and the disruption of their secondary and tertiary structures. The hydrophobic groups initially buried in the core including the sulfhydryl groups SH- are then forcibly exposed to water molecules after the protein structure is destroyed, which leads to aggregation, coagulation, and precipitation.42
On the other hand, the use of surfactants or organic solvents is extremely important both in the solubilization of the class of hydrophobic membrane bound proteins, and biocatalytic reactions with enzymes for chemical production in industrial applications.43,44
2.1.2. Hydrophobic Residues and Patches
Besides the contributions from extrinsic factors, the solubility and stability of a protein are principally defined by its primary sequence and hierarchical structure.45 Arrangements of nonpolar amino acids and exposed hydrophobic patches are determinative factors in this regard. These have previously been experimentally determined through a hydrophobic dye or tracer.46 They can also be predicted in silico using solvent-accessible surface area (SASA) and hydrogen bond estimation algorithms (DSSP), based on amino acid positions, and net charges.47 Highly hydrophobic proteins can be separated by purification with different salt concentrations and hydrophobic resins such as Phenyl-Sepharose.
Many prediction tools were developed based on the evaluation of hydrophobic residues. A technology named spatial aggregation propensity (SAP) was used to identify hot-spots on the protein surface for aggregation, based on the dynamic exposure of spatially adjacent hydrophobic amino acids.48 Jacak et al. developed a novel nonpairwise-decomposable scoring term named hpatch that penalizes surface hydrophobic patch formation and guides subsequent optimization of protein binding and solubility in unbound states, as shall be introduced with more details in a later section.49 Furthermore, a new knowledge-based interatomic potential of mean force was developed by Zhou et al. to assess impacts from individual buried residues.50 A strong correlation was found between buried accessible surface area (ASA) of residues and their contribution to protein stability, whereas regression slopes of all 20 amino acids (called the buriability) are positive (pro-burial). The burial of polar residues contributes less to stability than nonpolar ones, while buriability scale concerns both inter-residue and solvent interactions. The structural differences between the two groups of amino acids further contribute to their buriability gap, since the shapes of polar residues are evolutionarily optimized to enhance anisotropy of their interactions, whereas the opposite is true for nonpolar residues. The evolution of natural amino acids seems to have facilitated a tight packing of nonpolar residues and sterically repulse polar ones inside the dense core.
2.1.3. Isoelectric Point and pKa
The isoelectric point (pI) is the pH at which the net charge of a protein becomes zero, whereas surface charges of protein are modulated by environmental pH conditions.51,52 Proteins generally exhibit the lowest solubility at pI, which is determined cumulatively by their sequences.53 While the pI of a protein was traditionally determined by isoelectric focusing, several online servers also provide convenient calculation tools in silico.54−56
The pI for each protein greatly affects its solubility and functions in different environmental conditions. For instance, insulin has an acidic pI of 5.4 due to a majority of acidic functional groups in its composition, which poses difficulties for their applications at neutral pHs with the tendency to form microprecipitates in subcutaneous tissues. Herein, basal insulin analogues were created by the A21G mutation and addition of two R residues at the C-terminus to incorporate additional positive charges and improve its solubility at physiological conditions.57,58 Alternative mutations were also adopted to create a soluble insulin detemir,59 while ester tags were used by Nadendla and Friedman to block acidic group exposures in the native insulin with a photocleavable linker.60
Mathematically, pI is the average of pKa values for functional groups within an amino acid, and is calculated from the acid dissociation constant Ka, which is an equilibrium constant for dissociation.61 Similar to pH, chemists define pKa to be the negative logarithm of Ka to provide a more manageable number, where lower pKa values indicate stronger acids. In the context of proteins, their net charges varies with pH, and is determined cumulatively by amino acid contents and pKa of the ionizable groups, which is of great interest to scientists due to their relevance in catalysis, protein stability, and protein–protein interactions.62
There are many experimentally determined values of charges and pKa of individual amino acids (Table 1) and whole proteins.63 Yet it is not feasible to carry out such characterization every time the charge or pKa of a protein is needed. Many in silico methods have been developed for their prediction.
Table 1. Amino Acid pKa and pI Values188,189.
| pKa (25 °C) |
||||||
|---|---|---|---|---|---|---|
| amino acid | type | COOH | NH2 | side chain | pI (25 °C) | charge (pH 7) |
| Alanine | Neutral | 2.34 | 9.69 | 6.00 | 0 | |
| Arginine | Basic | 2.17 | 9.04 | 12.48 | 10.76 | +1 |
| Asparagine | Neutral | 2.02 | 8.80 | 5.41 | 0 | |
| Aspartic acid | Acidic | 1.88 | 9.60 | 3.65 | 2.77 | –1 |
| Cysteine | Neutral | 1.96 | 10.28 | 8.18 | 5.07 | 0 |
| Glutamic acid | Acidic | 2.19 | 9.67 | 4.25 | 3.22 | –1 |
| Glutamine | Neutral | 2.17 | 9.13 | 5.65 | 0 | |
| Glycine | Neutral | 2.34 | 9.60 | 5.97 | 0 | |
| Histidine | Basic | 1.82 | 9.17 | 6.00 | 7.95 | +1 |
| Isoleucine | Neutral | 2.36 | 9.60 | 6.02 | 0 | |
| Leucine | Neutral | 2.36 | 9.60 | 5.98 | 0 | |
| Lysine | Basic | 2.18 | 8.95 | 10.53 | 9.74 | +1 |
| Methionine | Neutral | 2.28 | 9.21 | 5.74 | 0 | |
| Phenylalanine | Neutral | 1.83 | 9.13 | 5.48 | 0 | |
| Proline | Neutral | 1.99 | 10.60 | 6.30 | 0 | |
| Serine | Neutral | 2.21 | 9.15 | 5.68 | 0 | |
| Threonine | Neutral | 2.09 | 9.10 | 5.60 | 0 | |
| Tryptophan | Neutral | 2.83 | 9.39 | 5.89 | 0 | |
| Tyrosine | Neutral | 2.20 | 9.11 | 10.07 | 5.66 | 0 |
| Valine | Neutral | 2.32 | 9.62 | 5.96 | 0 | |
One of the primary methods for this purpose is called PROPKA.64 It predicts the pKa values of ionizable groups in proteins and protein–ligand complexes. PROPKA relies on the protein’s physical desolvation and dielectric response description. Alternatively, MCCE (multiconformation continuum electrostatics) searches for various conformations and degrees of freedom in proteins through Monte Carlo calculations. It relies on different isosteric conformers, heavy atom rotamers and proton positions with different degrees of optimization, which are tested against a curated group of 305 experimental pKas in 33 proteins.65 One additional method is H++, which is an automated system that computes pK values of ionizable groups in macromolecules, particularly proteins, peptides, adds missing hydrogen atoms according to the specified pH of the environment.66 It automates the steps of correcting errors, adding missing atoms, filling valences with hydrogens, predicting amino acid pK values, assigning predefined partial charges and radii to all atoms, and generating force field parameters for molecular modeling and simulations.
Moreover, CpHMD (constant pH molecular dynamics) is a molecular dynamics (MD) technique that emerged over the past decade to consider the precise impact of pH during simulations.67 It simulates the titratable sites’ protonation states at a specified pH, which allows for detailed investigation into the mechanisms of pH-dependent conformational processes. PypKa is a flexible python module for Poisson–Boltzmann-based pKa calculations and titration of amino acid residues. The prediction accuracy of these methods was plotted against running speed and is shown in Figure 5.
Figure 5.
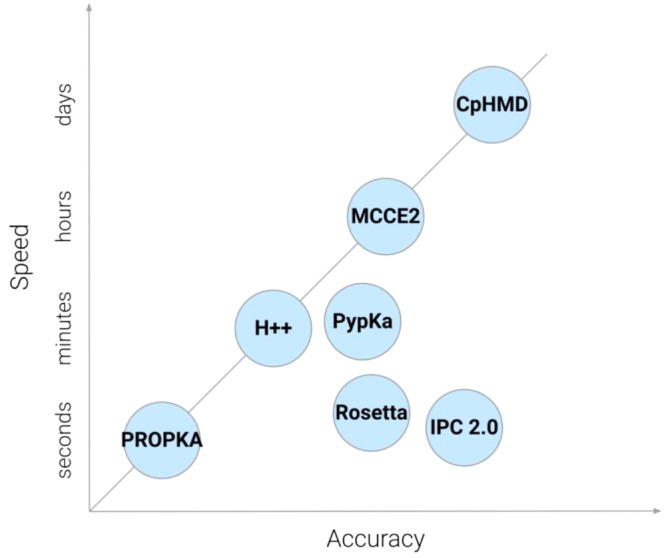
Plot of prediction accuracy versus speed on current computational methods for pKa calculations.
Recently, more methods of pKa and charge prediction were developed based on machine learning algorithms. DeepKa defines a protein’s pKa through training and validating with the pKa values derived from continuous constant pH molecular dynamics simulations of 279 soluble proteins.68 HDNNP (high-dimensional neural network potentials) predicts atomic partial charges. It is a neural network-based model trained on data from quantum mechanics calculations using the fragment molecular orbital method.69 IPC (isoelectric point calculator) calculates the isoelectric point of a protein using a mixture of deep learning and support vector regression models.56
2.1.4. Amino Acid Considerations
Besides the cumulative effects on protein solubility from properties such as pI and pKa, amino acids also individually contribute to the overall stability of proteins through alternative reaction mechanisms. Trevino and Pace conducted a systematic comparison of the relative contributions from each of the 20 amino acids to the protein solubility of ribonuclease Sa (RNase Sa) by point mutation at solvent-exposed site 76.70 The effect of the mutation at different pH or charge conditions was evaluated. A surprisingly wide variation was observed among different polar and charged amino acids whereas D, E, and S residues contributed more significantly to the stability of the protein especially at high net charges. It was suggested that their contribution to protein solubility depended more on amino acids’ hydration capability rather than hydrophobicity.
Warwicker et al. later computationally studied the sequence-based charged properties of K and R residues as well as their correlation with protein expression and solubility based on the available data set for E. coli proteins in a cell-free system.71,72 Although both amino acids contain positive charges in physiological conditions, their observed behaviors were quite different. A high propensity of R residues can promote both specific and nonspecific surface interactions, which was not favored for high concentration conditions and can be a hindrance in protein therapeutics.73 A lower ratio of R also presented at higher levels of mRNAs correlating with the high protein expression level. An effect named supercharging was also proposed and preferred more K residues to prevent the aggregation of partially folded proteins and increase solubility.74
Another residue playing a key role in protein stability is C, which contains thiol groups that can form disulfide bonds through oxidative folding.75 The disulfide bonds are considered important structural formation units76 and can stabilize protein folding when introduced at appropriate sites.77 Yet proteins with exposed C residues have a natural tendency to form intermolecular disulfide bonds that frequently result in their aggregation and precipitation.78 One example of this type are keratins, C-rich proteins mainly expressed in the epithelium and hair which constitute the largest subgroup of intermediate filaments.79−81 Recombinant keratins constantly misfold into inclusion bodies when expressed in E. coli with poor solubilities.80,82−84 Changes in buffer conditions are usually needed for determining their biophysical properties and for applications as hemostatic agents.85,86
Experimental data about the stability of proteins and their mutants are systematically summarized in the ProThermDB database, which is the latest version of Thermodynamic Database for Proteins and Mutants (ProTherm).87 Sarai and co-workers first established the ProTherm in 1999 to help scientists to understand the underlying mechanisms governing protein stability.88 The initial database contained more than 3300 data entries regarding parameters closely associated with the thermodynamic stability of proteins, which include the unfolding Gibbs free energy, enthalpy, heat capacity, transition temperature, activity, etc., for both native and mutant proteins. As of 2021, the number of entries had increased to ∼31,500 in its latest version. The ProThermDB has been used effectively to elucidate the relationship between physicochemical properties and protein stability, to predict the melting temperature and free energy changes upon mutation, and to understand the pathogenic mechanism of diseases causing mutations that result in changes of stability.87
2.1.5. Solubility Prediction
Accurate prediction of solubility in silico can greatly facilitate the optimization cycles for experimental protein solubilization. Scientists have devoted extensive efforts to the development of algorithms to correlate protein solubility with their primary sequences for precise predictions.89−91 For this task, SOLpro uses 23 feature groups which can be calculated from the primary sequence to design a two-stage support vector machine,92 whereas another method Protein-Sol combines 35 features in a linear model to predict protein solubility.93 There is also CamSol, which draws on structural data, solvent exposure and the intrinsic solubility profile, to create a protein’s intrinsic solubility profile and identify areas of low solubility with a self-assembly propensity.94 Alternatively, SoDoPE uses a “solubility-weighted index” based on the amino acid composition to predict protein solubility.95 Last but not least, DeepSol predicts solubility using a convolutional neural network based on several features of the sequence and structure.96
In addition to examining and predicting solubility, there are also several in silico methods dedicated to predicting protein aggregation. AbsoluRATE uses parameters such as environmental conditions, disorders and aggregation propensities for a machine learning model to carry out aggregation kinetic predictions.97 An alternative machine learning approach is VLAmY-Pred,98 and there is a Solubis method which also uses aggregation propensity for protein aggregation prediction.99
The technical details of these algorithms will be introduced in Section 9.
2.2. Approaches for Protein Solubilization
For molecular and structural studies and for biotechnology applications, large amounts of proteins are usually required. In heterogeneous protein expression systems, changing the expression conditions can be helpful in regulating the solubility of target proteins.12 Common methods include: (i) reducing expression speed by low concentration or using a weak promoter, (ii) lowering the expression temperature, (iii) optimizing growth media, (iv) coexpression with molecular chaperons, or (v) protein fusion.100
While altering the solution conditions is simple and straightforward, such an approach is not always appropriate, and often insufficient to increase protein solubility to the desired extent as the overall properties are still limited by the native sequence.45 It then becomes increasingly necessary to make modifications to proteins’ primary sequences and alter the properties intrinsically. These designer proteins should show enhanced inherent affinity to water molecules and form additional hydrogen bonds with aqueous solvents, through which their overall solubility and stability can be increased. The typical protein design methods include: (i) site-directed mutagenesis, (ii) directed evolution, and (iii) de novo protein designs. We will provide a discussion on the fundamentals of these methods in the following sections and focused on most recent advancements in proteins solubilization efforts in later sections.
2.3. Protein Folding and Design
The precise design of protein properties relies on a thorough understanding of the underlying design rules which determine how a polypeptide chain folds into its native structure. Protein folding and design are two sides of the same coin.101,102 The folding prediction aims to accurately determine a full 3D structure with a given amino acid sequence, whereas the design objective is to develop the sequence required to form a desired backbone structure. Since the two problems mostly follow the same underlying principles, understanding protein folding will predictably benefit the protein design field.
However, the folding problem is not easy to solve, since the number of possible conformational states in a given sequence is astronomical, as stated by the Levinthal’s paradox.103−105 For instance, a sequence composed of 100 amino acids have 3198 possible conformations when bonding and rotameric states are considered, which are hopeless for any modern computer to traverse. While a thermodynamic hypothesis proposed by Anfinsen suggests that proteins spontaneously fold into the minimal energy states accessible to the amino acid sequences,106,107 the vast conformational space poses a serious kinetic challenge to searching through all possible configurations which would necessitate an enormously long time, whereas native proteins fold on the order of seconds.108 Levinthal cannot reconcile the conflict between folding thermodynamics and kinetics, and thus postulated that special folding pathways are conserved in the evolutionary process, with its end at the native structure which is not necessarily at the global free energy minimum.109 Nevertheless, it is the common consensus that the native state of a protein must collectively outweigh adjacent non-native states and reside in a funnel-shaped energy minima across the landscapes, as shown in Figure 6.110−112
Figure 6.

Schematic of the folding dynamics in a funnel-like free energy landscape. Red dots represent unfolded states; yellow dots represent intermediates states visited with local energy minima; blue dot represents final native conformation residing in global minimum. Multiple parallel pathways (white arrows) might present in spontaneous folding. The number of representative unfolded conformations is related to the trajectory. The observed stretched exponential folding kinetics is the result of the superposition of the multiple pathways from the unfolded substates (Ui, i = 1 to N) to the folded state (F) via intermediate substates (U’i, i = 1 to N), indicating conformational heterogeneity. Reprinted with permission from ref (190). Copyright 2013 Elsevier.
Many theories have been proposed for protein folding to achieve this energy minimum, but only four of them are the most acknowledged. The majority of work related to nucleation or folding refers to these models (Figure 7), which include: (i) the framework (diffusion/collision) model - local elements of the secondary structure are formed first, then they diffuse together and collide; (ii) the hydrophobic collapse model - the protein folds around its hydrophobic residues, and rearranges from the limited conformations of this intermediate “molten globule”; (iii) the nucleation propagation model - local interactions form a small amount of secondary structures, which act as nuclei for the outward propagation of additional structures; (iv) the nucleation-condensation model - the presence of a metastable nucleus that cannot start folding until a sufficient number of stabilizing long-range interactions have accumulated; as soon as this happens, the native structure condenses quickly and the nucleus is not yet fully formed in the transition state.113
Figure 7.
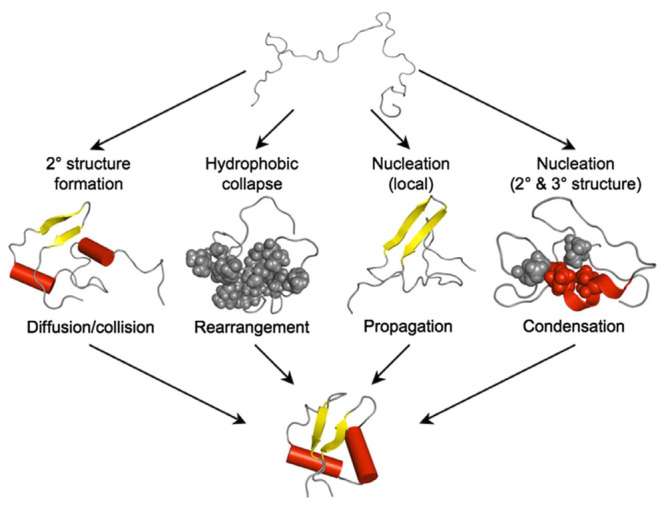
Description of the four “classical” folding mechanisms. Four most common models: (i) framework (diffusion/collision) model, (ii) hydrophobic collapse model, (iii) nucleation propagation model, (iv) nucleation–condensation model. Reprinted with permission from ref (113). Copyright 2010 Elsevier.
The hydrophobic collapse model has emerged to be the most acknowledged folding theory. It is proposed that the nascent polypeptide starts to form initial secondary structures after exiting the ribosome to create localized hydrophobic regions with thermodynamic pressure (hydrophobic effect) and avoid water interactions, which subsequently aggregates into a hydrophobic core with polar residues exposed to the aqueous solvent. The tertiary protein structure is obtained in which the surface facing the solvent consists of hydrophilic residues, and the inner surface of hydrophobic ones.114 The hydrophobic collapse in globular proteins occurs in nanoseconds and has two subprocesses: direct hydrophobic collapse; and formation of primary elements of the tertiary structure.115,116 Amphiphilic residues are required at the coagulated part of the proteins to contact both the polar solvent and nonpolar residues in the core, which also help to regulate intermolecular assemblies, as shall be discussed with more detail in later sections.117
Such an understanding of protein folding then translates into underlying rules to promote a stable structure followed by protein design processes: (i) burial of hydrophobic residues (like L, I, V, or F) in the protein’s core region, away from the aqueous solvent, (ii) minimization of the pocket the protein occupies in water, and (iii) maximization of van der Waals (vdW) forces between side chains of amino acids without steric clashes. Other considerations include: (i) a coherent interior core hydrogen bond network with all polar groups satisfied; (ii) compensated electrostatic charge interactions; and (iii) accommodation of favored torsional preferences. It is also worth noting that the stabilization of a native structure in the design can either be achieved through stabilizing (reducing the free energy of) the target folded conformation or destabilizing (inducing the energy penalty of) the alternative unfolded states.26,118
2.4. Mutagenesis-Based Protein Design
Site-directed mutagenesis is the most common engineering method for the rational modification of native proteins and to generate derivatives based on the prior biophysical characterization and crystal structure of target species, often through the insertion, deletion or point mutation of the DNA codon for amino acids at a specific site.119,120 It remains a tremendously powerful and relevant approach for probing protein properties in a highly precise manner. The technique is continuously developed and refined due to the increasing need for protein engineering in biomedical and pharmaceutical applications.121 With respect to protein solubility, the technology was utilized by biologists to identify nonfunctional hydrophobic residues exposed to the solvent where aggregation problems arose, and replace them by hydrophilic ones to increase surface polar interactions for the target protein. Pioneered in Alan Fersht’s lab in the 1980s, it has gradually become the most used technique to enhance protein solubility and stability.122−126
The successful regulation of protein solubility through site-directed mutagenesis relies heavily on the identification of critical residues and surface patches. Numerous computational programs have been developed for this task, including CamSol, DeepSol, and SoDoPE, which conduct preliminary surface charge simulations to identify residues in hydrophobic patches that negatively affect the protein solubility.95,96,127 The selected sites are then randomly mutated to alternative amino acids to determine the impact on the score for hydrophobicity. Technical details for these algorithms will be introduced in Section 9.
The site-directed mutation is especially useful for enhancing the solubility of antibodies since most of their sequences and the overall geometry are similar and well-defined.128,129 With an intact paratope for antigen binding, mutating or even redesigning for alternative sequences can efficiently increase the solubility of a target antibody and enhance its stability in vivo.
Xu et al. engineered the human catalytic antibody Se-scFv-B3 (selenium-containing single-chain Fv fragment of clone B3) with glutathione peroxidase (GPX) activity, but this resulted in lower catalytic performance. On the basis of docking analysis in the homologous model, the decreased catalytic efficiency was attributed to the poor hydrophilicity of presumptive GSH (GSH-S-DNPBu) binding sites where a critical residue of S was missing.130 Subsequent A44S and A180S mutations were introduced into the putative binding interfaces to be converted into a catalytic selenocysteine group. Mutated variants of scFv were expressed in soluble fraction of E. coli, whereas the A180S variant exhibited superior GPX activity compared to the original design.
Another example was reported by Pepinsky et al., who improved the solubility of anti-LINGO-1 Mab Li33 in the full antibody form due to the presence of extensive hydrophobic residues in the CDR (complementarity determining region).131 Site-directed mutagenesis was conducted on these residues which determined 3 out of 4 suitable sites (W94, W104 and I57) without affecting the protein functions. The mutations were then combined with isotype switching and glycosylation site insertion mutagenesis which resulted in a new Li33 Mab variant with significantly improved solubility (from 0.3 mg/mL to >50 mg/mL) and reduced aggregation.
With successes in similar practices, site-directed mutagenesis significantly facilitates the pharmaceutical industry’s development of treatments for protein aggregation-related diseases. Yet the technique is on a case-by-case basis, highly dependent on the physiological properties and structural contexts of individual proteins, which inherently limits the application of knowledge generated from one work to other targets.26 The approach can also be hindered by the absence of high-resolution crystal structures for certain groups of proteins such as transmembrane proteins.132 Thus, in this review, while selected reports based on site-directed mutagenesis will be briefly covered, we will not focus the discussion on this approach.
2.5. Directed Evolution
Directed evolution is an alternative synthetic method that mimics the natural evolution process. The procedure commits to iterative Darwinian optimization cycles of protein sequences with advised randomness of amino acid choices, designated screening and selection strategies.133−135 After identifying a starting protein, diversification of the gene sequence, expression and functional screening are subsequently implemented to achieve target protein properties. Directed evolution also benefits from the molecular insights gained from template proteins to identify optimal sparse sampling for beneficial mutations, since complete randomization is not possible. The approach has emerged as a key technology for generating highly stable, tailor-made enzymes as biocatalysts.133,134,136,137 It can also be used to develop stable supramolecular scaffolds with protein subunits,138 or as an investigation tool for structure–function relation of transmembrane proteins in complement with computational modeling.139
The most representative successes of directed evolution are from the enzyme engineering field in their design and optimization for better reproducibility and catalytic efficiency.34 Many of the efforts of this kind have had enormous impact in protein engineering with significant potential in clinical practices. Common examples include but are not limited to protease engineering with tuned substrate specificity,140 molecular probe development for noninvasive bioimaging,141 antibody screening to target growth factors in cancers,142 and high-efficiency enzyme engineering with reduced immunogenicity in cancer therapies.143
As a methodology to improve protein solubility and stability, directed evolution is especially advantageous over site-directed mutagenesis without the necessity for prior knowledge of its structure or function when screening soluble variants of a target protein.144 However, a series of in vivo and in vitro quality control techniques are necessary to ensure the folding and function of selected variants. A typical example is the solubilization of tobacco etch virus (TEV) reported by van den Berg et al.145 Mutants were generated by directed evolution to enhance its expression in E. coli hampered by the low solubility. A library created with error-prone PCR (polymerase chain reaction) was combined with the Gateway system for easy gene transfer into adapted vectors. One mutant was identified with a 5-fold increase in the yield of purified and active TEV protease compared to the parental gene.
High-efficiency directed evolution methodologies are constantly being developed to meet the growing demands of research and industrial applications. A highly efficient screening system named SE-PACE (soluble expression phage-assisted continuous evolution) was recently developed by David Liu’s group to improve soluble expression of rapidly evolving proteins.146 The system integrates an AND logic gate that enables two concurrent selections for protein evolution with or without the selection for protein functions. SE-PACE was tested in the evolution of antibody single-chain variable fragments (scFv) that misfolded and aggregated in E. coli. The system produced soluble variants with improved cytosolic expression between 2- and 6-fold, largely retained binding activity and enhanced thermodynamic stability.
However, while directed evolution has been a widely adopted technique in enhancing the stability and promoting efficiency especially for biocatalysts, we will not be discussing this approach extensively in this review due to page limitations. For a more detailed discussion of different methodologies in directed evolution as well as their applications in enzyme and catalyst design, please refer to previous reviews.133,134,137,147
2.6. De Novo Protein Design
“Where nature finishes producing its own species, man begins, using natural things and in harmony with this very nature, to create an infinity of species.”
- Leonardo da Vinci
We have mentioned in Section 2.3 Levinthal’s paradox and the astronomical number of possibilities for even a short polypeptide chain to accommodate.103,104 The whole proteome in all living organisms generated through natural evolution, albeit highly diverse and efficiently supporting life-forms as we know it, still only occupies very sparsely the total sequence space available, which forms protein clusters with similar structure, function and conserved sequences.26 Protein design techniques such as site-directed mutagenesis and directed evolution sample incrementally outside the clusters occupied by native proteins but are still bound by their original templates. On the other hand, the ever-expanding knowledge-base in biophysics, biochemistry, and the folding and structures of existing architectures in native proteins has provided fruitful resources such as the Protein Data Bank (PDB),148 which lays a solid ground for scientists to proceed beyond those evolutional methods and build functional polypeptide chains from scratch.149,150
This approach was named de novo protein design, indicating the generation of new protein variants from physicochemical principles and molecular interactions without sequences directly related to those from native species. The subject emerged only about four decades ago but has progressed into an enormous field in protein design.25 It has undergone several distinct periods, from early models with simple combinations of amino acids, to parametrically generated computational designs guided by biophysics, and more recently, fragment-based structure designs.151,152 Following the underlying design principles, many algorithms and computer programs have been developed to obtain novel protein species with high design accuracy.153−156 The programs are also responsible for initial in silico verifications of these designer proteins through structure prediction and multiple rounds of optimization to achieve a final design with minimized energy functions. The major features of the de novo design process are illustrated in Figure 8 including backbone sampling, sequence optimization, functional site design, and scoring to select low energy sequences.157
Figure 8.

Major aspects of the de novo protein design. The design of a functional de novo protein, for example, a binder (middle, magenta) to a target protein (middle, gray), requires sampling of the backbone structure space to find a backbone compatible with the function, sequence optimization to stabilize the backbone, and designing the functional site interactions. A scoring function is necessary to select designs with desired properties, typically by identifying low-energy sequence-structure combinations. Reprinted with permission from ref (157). Copyright 2021 Elsevier.
The major achievements of the field to date still lie within the design of new structures, from early idealized models of helical bundles158 or simple α/β folds,112,159 to recent complex assemblies or difficult targets like helical barrels,160,161 fibril amyloids,162−164 multidimensional assemblies,165−170 all-β structures,171,172 or transmembrane proteins.173−175 Success in the accurate design of these structures helps to elucidate many aspects of protein folding and interactions.176 More recently, building on structural models, scientists are pivoting their focus from designing novel proteins incorporated with biological functions to opening up possibilities based on non-natural sequences to solve new challenges in a variety of biomedical,177−179 biotechnological,170,180−183 and catalytical applications.184−186
Many of the de novo designed structures are highly relevant in elucidating fundamental rules of protein interactions that contribute significantly to the solubility and stability. Although these works will be extensively discussed in greater detail in later sections, it is impossible to carry out a thorough review of efforts in the whole de novo protein design subject in limited pages. There are numerous reviews of this very active field with more technical details.25,26,102,157,176,187 Interested readers should consult those articles for a more thorough view of the field.
3. Soluble Variant Design of Transmembrane Proteins
Structural and functional studies of transmembrane proteins are at the frontier of molecular biology. Approximately 30% of the open reading frames in the genomes of higher eukaryotes code for proteins that span or are associated with cell membranes.191−193 They represent indispensable sets of enzymatic, signaling and molecular transporting functions in the human body, which hold enormous significance in diseases and make up over 60% of therapeutic targets for drugs.139,194−196 Yet large-scale synthesis, characterization, and utilization of these functional molecules lag far behind soluble proteins as they are difficult to overexpress and tend to aggregate in aqueous conditions due to molecules’ hydrophobicity.196−198 The traditional methods to overcome these issues include detergent screening or applying lipid/micelles such as nanodiscs to stabilize their natural conformations.199 However, due to their structural and functional diversity, each type of transmembrane protein requires individual effort, so finding a suitable cosolvent for a specific membrane protein can be time-consuming, expensive, and arduous.200 Approaches utilizing detergent-like amphiphiles are also expensive for any follow-up application beyond research purposes.
In contrast to optimizing the environment to suit the proteins, developments in structural biology and steadily increasing computing power now allow scientists to accurately make changes to the proteins to suit the environment.201 Redesigning transmembrane proteins for higher solubility without disrupting the structure or diminishing the function can provide valuable insights into the fundamental principles of how they perform in vivo and be used in vitro. While either α-helical bundles or β-barrels can be the main structural component, the former class bears greater functional diversity and thus attracts significantly more interest from scientists.25 In this section, we review past attempts and successes in the transmembrane protein solubilization as well as the prospects for this field.
3.1. Multiple Attempts on Phospholamban
In the early 2000s, phospholamban became the first transmembrane protein that caught general attention for solubilization studies from multiple groups, primarily due to (i) its biological importance and necessity for structural study at the time; (ii) its simplicity in the primary sequence and pentameric structure; and (iii) the large data set available for critical amino acids through mutagenesis.43,202,203 Phospholamban is a 52-amino acid integral membrane protein on the cardiac sarcoplasmic reticulum membrane. It is a critical regulator for cardiac muscle contraction and relaxation by forming a complex with Ca2+-ATPase and modulating Ca2+ cycling between the cytoplasm and sarcoplasmic reticulum.204,205 The protein contains one helical membrane-spanning segment and exists in equilibrium between monomeric and pentameric states in the natural environment.206 It was proposed to have a left-handed, heptad repeat conformation, with a leucine/isoleucine zipper motif at the interface.207
The efforts to solubilize phospholamban were built on the hypothesis by Eisenberg and Rees who suggested that transmembrane and soluble proteins share similar structure features and physiological properties in the core while only solvent-exposed residues differ.208,209 Similarities were found in (1) the protein surface area, (2) the packing density of buried atoms, and (3) the relative hydrophobicity of buried residues, by structural comparison. These findings suggest that, from a stability point of view, changing the lipid-exposed residues of a transmembrane protein into polar or charged residues might not significantly destabilize the protein. Thus, theoretically scientists should be able to tune the solubility of a target protein by tweaking its surface residues to enhance the hydrophilicity and prevent it from entering the membrane but not significantly alter the conformation.
As the first attempt, Frank et al. removed the outer hydrophobic patches on phospholamban by substituting amino acids with those from the soluble cartilage oligomeric matrix protein (COMPcc), which shares a homologous pentameric parallel coiled-coil oligomerization.43 The redesigned protein could be expressed in the soluble fraction of bacteria and exhibited helical structures under circular dichroism (CD). However, although the design was capable of forming a higher order oligomerization state when fused with maltose-binding protein (Mal-bp), it did not show the strict pentameric assembly exhibited by the native phospholamban. In contrast, Li and co-workers took a more aggressive approach and swapped phospholamban’s lipid-facing residues (L, I, and V) with charged K and E.202 As there were two reported helical bundle models offset by one position in the heptad repeat, two soluble phospholamban variants were designed based on both models (Figure 9A).207,210,211 The design effort helped to identify the correct model as only one of them produced a stable pentamer. A total of 10 out of 52 amino acids were converted. The redesigned protein, SIM, exhibited high solubility in aqueous solution and had helical structures similar to the native protein. It attained a predominantly pentameric state with minor content of dimers. The pentamer can be disrupted when sensitive residues, such as C41 and I40 were mutated, which agreed with mutagenesis studies on the native protein. However, NMR (nuclear magnetic resonance) data suggested that the designed SIM protein showed dynamic molten-globule-like properties, which posed uncertainty in its conformational rigidity. The phenomenon was attributed to the introduction of charged residues that can disrupt local interactions between helices and side chains, affecting the packing of core residues.
Figure 9.

Solubilization attempts on phospholamban. (A) Sequence of canine wild-type phospholamban compared to several redesigned soluble peptides from Engle (PLB-COMP-1, PLB-COMP-2), Engelman (SIM-FULL, ADA-FULL), and DeGrado lab (WSPLB). The differences between WSPLB and phospholamban are shown in red, the differences between SIMM-FULL and WSPLB are shown in blue, and the differences between PLB-COMP-1 (and 2) and SIMM-FULL are shown in green. Reprinted with permission from ref (203). Copyright 2003 John Wiley and Sons. (B) Antiparallel tetrameric helical bundle (left), representing the topology of WSPLB tetrameric peptides, with e–e and g–g contacts labeled, and a parallel pentameric WSPLB (right) with side chain identities. Reprinted with permission from ref (214). Copyright 2005 John Wiley and Sons. (C) X-ray crystal structure of WSPLB (21–52, PDB ID: 1YOD) solved to 1.8 Å (left). A model of full-length pentameric WSPLB shows interactions between Q22(g) and N27(e) in the polar switch (right). Reprinted with permission from ref (215). Copyright 2005 Elsevier.
Slovic et al. further developed the redesign mechanism for phospholamban by a fully automated computational approach.203 In their model, a perturbation index was introduced to evaluate the appropriate mutation sites to avoid critical residues.212 Exposed hydrophobic amino acids (b, c, f position in the heptad, as shown in Figure 9B) were replaced by polar ones guided by a design algorithm based on the pairwise scoring potential. Without focusing on the rotameric states of individual sites, a Monte Carlo simulation was carried out on substitutions against fixed residues to minimize the energy function and promote intra- and interhelical interactions. The designed water-soluble phospholamban (WSPLB) exhibited a helical secondary structure and monomer-pentamer equilibrium similar to the native protein. A destabilizing effect from phosphorylation at S16 and T17 sites was observed on both phospholamban and WSPLB, suggesting packing similarity between the two. In addition, the group was the first to point out that L(a)–I(d) repeats commonly posited for the pentameric oligomerization in phospholamban preferentially formed an antiparallel tetrameric coiled-coil similar to that observed in GCN4.213
By comparing full-length and multiple truncations of WSPLB, Slovic and co-workers suggested that cytoplasmic residues (1–20) served as a “polar switch” and promoted the pentamer formation.214 The argument was supported by the protein’s X-ray crystal structure.215 WSPLB (21–52, PDB ID: 1YOD) attained an antiparallel coiled-coil structure with three heptads offset (Figure 9C), which was attributed to a close-packed hydrophobic core and the nature of the residues on the e and g positions, as smaller side chains preferred to interact with main chain carbonyl groups rather than with those from a neighboring helix. The model of full length pentameric WSPLB contained extensive interhelical hydrogen-bonding that helped to stabilize the core. Molecular dynamic (MD) models of phospholamban and WSPLB revealed that the N-terminal cytoplasmic region and the transmembrane region from both proteins acquired helical structures with a flexible linker in between.216 The two helices were found to be perpendicular to each other and tended to accommodate an open conformation (T state) that allowed the binding site to come into contact with Ca2+ ATPase, whereas the two phosphorylation sites were constantly exposed to the solvent. The simulation cross-referenced with crystal structures determined for native phospholamban and provided insights for the structure–function relation of the protein.217−220
3.2. Ion Channels and Other Pore-Forming Proteins
In parallel with efforts to solubilize phospholamban, another class of membrane proteins that attracted wide attention due to their important biological transportation functions is pore-forming proteins, primarily ion channels. Despite the species diversity, many channel proteins of the same type share similar structural segments and oligomerization states.221 Tailoring the solubility of selected channel-forming monomers and their assembly can elucidate structure, functional selectivity and practical implications for broader variants.
3.2.1. Single Mutation on Aerolysin
The first report of solubility design on channel forming proteins discussed single point mutations in a bacterial toxin named aerolysin. The protein accommodates a heptameric oligomerization state and forms a pore in the lipid membrane. Tsitrin et al. made several single point mutations (Y221G, Y221F, Y298G, or F410G) that had a pronounced effect on its solubility.222 The mutations, occurring in domain 4 of the protein, disrupted the formation of “aromatic belts” at the upper boundary of the pore, which are typically believed to anchor and stabilize transmembrane structures in the bilayer.223 The mutations altered the protein’s function by partially or completely blocking its hemolytic activity but the mutated protein still bound to target cells and exhibited similar secondary structures to their native counterpart. One variant, Y221G, formed a regular complex of two funnel-shaped heptamers joined by their larger bases and was stable in the aqueous environment without any detergent, which provided a higher resolution structure for the previously obscured transmembrane region.224 While the disruption of the aromatic cap on membrane structures yields valuable insights in the study of aerolysin and other transmembrane protein targets, such an approach based on single point mutations is hardly generalizable since the specific protein is also secreted as a water-soluble precursor and is amphipathic in nature.225
3.2.2. KcsA Potassium Channel
A more generalizable example on pore-forming proteins was later reported by Slovic et al., by redesigning a soluble variant of KcsA, namely, WSK-3.226 KcsA is one type of potassium ion channel that shares the common tetrameric structure and conduction pathway in the center of the symmetric axis, with sequence and structure similarities to both prokaryotic and eukaryotic proteins.221 Its monomer consists of an N-terminal transmembrane helix, a short helix selection filter and a C-terminal transmembrane helix. A conserved sequence motif TXGYG accounts for ion selectivity. Since there were high-resolution X-ray crystal structures reported on KcsA (PDB ID: 1K4C and 1K4D), the redesign of the protein provided a good structural model for potassium ion channels in general.227,228 Valiyaveetil et al. had reported the semisynthesis of the first 125 amino acids of KcsA with the C-terminus truncated earlier.229 The full-length crystal structure in its close conformation was later reported by Kossiakoff’s group (PDB ID: 3EFF and 3EFD).230
Slovic and co-workers divided the sequence of KcsA to exposed and buried groups. A statistical entropy-based algorithm was then used to predict the compatibility of amino acids in specific sites, so as to minimize the environmental energy within a given backbone.231 Pore-lining residues, extracellular (EC) loops, intracellular (IC) regions, toxin binding sites and buried residues were retained for the proper drive of folding and function. A total of 35 exposed residues were analyzed using the algorithm with consideration of amino acids’ propensities in specific secondary structures as well as their respective polarities.232 An in silico verification was conducted by comparing the final sequence with 47 aligned potassium channel proteins in living organisms. Their first iteration WSK-1 contained 29 mutations with respect to the wild type KcsA, which exhibited binding toward the toxin but formed aggregations. Two additional mutations L81R and L116R (WSK-3) were then introduced to further design exposed hydrophobic patches without disrupting the protein folding, as shown in Figure 10A. WSK-3 showed a comparable secondary structure to KcsA, existed primarily in a tetrameric state, and could specifically bind the AgTx2 toxin with an affinity comparable to the native protein. A small molecule channel blocker TEA also effectively inhibited AgTx2 binding, suggesting a correct conformation in the channel region.
Figure 10.

Solubilization attempts on various ion channels. (A) Depiction of KcsA and WSK-3. From left to right: KcsA with exposed residues to be mutated in the design, KcsA structure with side chains of mutated residues removed, and WSK-3. Reprinted with permission from ref (226). Copyright 2004 National Academy of Sciences. (B) NMR structure of WSK-3 (green) superimposed with KcsA crystal structure (PDB ID: 1K4C). Mutations made to facilitate solubility and AgTx2 binding are highlighted in orange and black, respectively. Reprinted with permission from ref (234). Copyright 2008 National Academy of Sciences. (C) Ribbon diagram of MscL residues chosen for replacement to cysteine for polyethylene glycol-polyamide oligomer modifications. Reprinted with permission from ref (237). Copyright 2004 Elsevier.
In a follow-up work, Bronson et al. carried out MD simulation of WSK-3, resulting in comparable or smaller final RMSD (root-mean-square deviation) values with respect to native KcsA simulations with different initial potassium configurations.233 However, they found a reoriented side chain in the central cavity of WSK-3 to allow water to permeate through the cavity wall, which was not present in KcsA. The NMR structure of this protein was later determined by Ma et al., proving a similar structure of WSK-3 with well-defined outer, inner, pore helices and the selectivity filter.234 Its main deviation from KcsA located in the loops between outer and pore helices, is shown in Figure 10B. Ma also confirmed the tetrameric state of WSK-3 protein under acidic conditions, in agreement with previous binding assays and simulations. However, collective domain motions with loose motif interactions were observed for WSK-3, suggesting a more fluidic and intrinsically less stable structure as compared to the native protein.
3.2.3. Alternative Approaches
Following the KcsA work, Roosild and Choe reported the redesign of another K+ channel protein, KchAfu104, in the absence of any reported crystal structure.235 They made predictions on the putative lipid-facing and channel-forming residues for the target protein by a systematic conservation pattern analysis based on the sequence alignment of homologous proteins. No homologous structural model was generated. It was reasoned that lipid-facing residues would show less conservation compared to interface and core amino acids responsible for folding and function. After several iterations, they substituted 10 out of 104 residues with additional deletions of 19 amino acids and Mal-bp fusion on the N-terminus. The design resulted in a construct that can be expressed in the soluble fraction of bacterial culture without apparent aggregations. The modified protein also exhibited a tetrameric oligomerization which was the presumed state for many potassium channels.236
Aside from the computational redesign efforts on primary sequences of proteins, Becker and co-workers took an alternative approach to solubilizing a channel protein by covalently modifying it with amphiphiles at a precise stoichiometry.237 The crystal structure was used to guide the selection and mutation of four surface residues to cysteines to anchor polyethylene glycol-polyamide oligomers on each site, on the large conductance mechanosensitive ion channel (MscL) from Mycobacterium tuberculosis.238 Changes were made on the lipid-facing second transmembrane helix, as shown in Figure 10C. The modified subunits formed native-like secondary structures and oligomerization states without any detergent. A pentameric assembly was also observed under an electron microscope. Yet the composites still exhibited a tendency to aggregate and no functional assays nor analyses on potentially introduced disulfide bonds due to cysteine mutations were conducted.
More recently, Andrews et al. modified a proton conduction channel protein, that is, motility protein B (MotB), to enhance its expression and stability for structural studies.239 MotB is a single-span transmembrane protein that commonly forms a dimer and, together with four surrounding MotAs, functions as a stator complex of the proton driven motor in bacterial flagellum.240,241 To prevent the degradation associated with truncation methods and preserve the native conformation of the “plug” and linker regions, Andrews and co-workers used leucine zippers to replace the transmembrane helices, leaving the rest of the sequence untouched. This was named chimMotB. It was reasoned that the parallel coiled-coil state of the leucine zipper can resemble the open state of the native protein.242ChimMotB was found to form a monodisperse dimer, and exhibited an α-helical structure with content close to the predicted value, a melting temperature similar to the native variant, and stability against proteolysis shown in the truncated proteins without the transmembrane regions. The X-ray crystallographic structure of chimMotB was later reported by the same group, presenting a pseudoatomic model of active full-length MotB that was not available before.243
3.3. Multipass Transmembrane Receptors
3.3.1. Bacteriorhodopsin
As simple transmembrane protein structures were successfully solubilized, scientists moved to more complicated targets such as multipass transmembrane receptors like G protein-coupled receptors (GPCRs). Bacteriorhodopsin, a light-driven proton pump that converts light energy to chemical energy in the purple membrane of Halobacterium halobium, was the first to attract wide attention.244 There were multiple reasons for the interest: (i) it is the first well-characterized seven transmembrane protein with resolved crystal structure, which made their surface residues identifiable;245,246 (ii) the structure also serves as a prototype for the entire family of seven-transmembrane GPCRs, so was assumed for the mechanism developed for redesign; (iii) it can accommodate many mutations;247 and (iv) it has a simple color-changing assay that can be used to evaluate folding and function.248 However, despite all these advantages and efforts, success in bacteriorhodopsin solubilization was very limited.
Dating back to 1993, Sirokman and Fasman led the attempts to increase the solubility of bacteriorhodopsin by chemically conjugating it with methoxypolyethylene glycol, with a putative anchor point on the side of the transmembrane region.249 Yet the conjugation only resulted in a partially denatured protein which did not show the desired secondary structure until 50% trifluoroethanol (TFE) was added. The modified protein did show proton pumping capability comparable to its native counterpart. Gibas and Subramaniam then took a sequence modification approach to distribute polar and charged residues onto bacteriorhodopsin’s surface to resemble that of a soluble helical protein.250 A cumulative fractional polar surface area was used to evaluate the protein’s hydrophilicity. They edited out W and replaced G with A and S in the transmembrane sequences, while retaining function or folding related, highly preserved or buried, and loop residues. Sixty-eight residue candidates were identified by comparing polar counterparts found in homologous sequences, other similar fragments, or arbitrarily if no resemblance can be found. They designed several variants with 24 to 49 modifications out of the 234-amino acid sequence. C and H were avoided in the substitution to prevent potential forming of disulfide bonds or inducing undesired reactivity. Gibas extensively simulated the conformational characteristics of redesigned sequences to provide computational indications of the approach, although no experimental verifications were performed.
A similar but more thorough attempt was then revisited by Mitra et al.251 A solvent accessibility criterion named FRACS was used on the reported bacteriorhodopsin structure to determine the surface exposure of individual amino acids.252 Replacements were made following a series of rules relating to the amino acids’ polarity, charge, helix propensity, molecular contact and position. S and T were preserved so as not to disrupt possible hydrogen bonds, but aromatic pairs were replaced by E and R when possible. Negatively and positively charged amino acids were placed near the N- and C- terminus of the helix, respectively, to stabilize helical macrodipoles by opposite charges. While they were not able to obtain a significant soluble expression by redesigning bacteriorhodopsin as a trimeric oligomer with 14.9% surface residue change based on the native model as shown in Figure 11A (PDB ID: 2BRD), their monomeric design with 13.5–24.3% mutagenesis resulted in expression with yields in ∼100 mg/L range which remained soluble in buffer containing 2M-8 M urea. However, all purified proteins misfolded into primarily β-sheet structures and precipitated when the organic solvent or surfactants were completely removed. Attempts to crystallize the proteins were not successful. The water-soluble bacteriorhodopsin variants not only failed to retain their function but also did not show a purple color or bind the retinal to induce red-shifts in the visible spectrum.
Figure 11.

Redesign of multipass transmembrane receptors. (A) Natural trimer model basis for the redesign of soluble bacteriorhodopsin (PDB ID: 2BRD, created by Mol*294). (B) Top: structure superimposition of murine MUR (PDB ID: 4DKL, yellow color) and wsMUR-TM receptor (blue color). Bottom: mutated positions in wsMUR are depicted as blue spheres, the majority of which (50 out of 55) are located at the exterior of the structure. Reprinted with permission from ref (262). Copyright 2013 Perez-Aguilar et al. (C) Multiple views of particle envelope reconstruction calculated ab initio on ΔspMal-bp-EmrE-ApoAI* from SAXS data. ΔspMal-bp crystal structure (PDB ID: 1NL5); ApoAI lipid-free crystal structure (PDB ID: 2A01); and electron microscopy-derived structure of dimeric EmrE (PDB ID: 2I68). The MBP label in the graph refers to maltose-binding protein (Mal-bp) defined in the manuscript. Reprinted with permission from ref (267). Copyright 2015 Mizrachi et al.
One last attempt for bacteriorhodopsin solubilization was made by Gohon et al., who used an amphipol A8–35 to substitute detergents for native bacteriorhodopsin.253 The amphipol-associated complex could carry out a complete photocycle and also helped to isolate monomers of bacteriorhodopsin that gradually associates into ordered fibrils.
3.3.2. Nicotinic Acetylcholine Receptor
Despite the lack of notable success in the design of water-soluble variants of bacteriorhodopsin, a similar rationale in the solubilization of KcsA was later adopted by Cui et al. to redesign nicotinic acetylcholine receptor (nAChR, PDB ID: 2BG9).254 This receptor has four transmembrane domains that form a pentameric assembly to serve as neurotransmitter-gated ion channels and mediate fast synaptic transmissions.255 On the basis of a low-resolution structural template, Cui and co-workers predicted the solvent exposure of nAChR amino acids using the same algorithm as in the KcsA effort.256 Only hydrophobic residues not located at the interfaces between helices in the transmembrane region were targeted. Two considerations were then taken for the mutation: (i) the probability of an amino acid at a specific site, and (ii) the amino acid diversity of the overall sequence. Additional modifications were conducted on the extracellular domain in the native protein by replacing it with a poly glycine linker to connect TM3 and TM4. A final 23 amino acids were substituted corresponding to a 17% change from the total sequence. The designed WSA (water-soluble acetylcholine receptor channel) was expressed in inclusion bodies of an E. coli system and refolded at high pH. However, 2% lyso-lipid LPPG was necessary to maintain WSA’s monodispersity at lower pH for characterization. The NMR structure of WSA resembled that of previously reported GLIC (Gloeobacter violaceus pentameric ligand-gated ion channel) transmembrane domains rather than the design template and contained less helical content than the theoretical calculated value. This was attributed to the distortion caused by the lack of loop regions. Dual NMR peaks were observed for several residues, indicating the coexistence of two conformational states that were slowly interchanging over time, which was uncharacteristic in the native nAChR. In general, a more flexible structure was observed in the loop region of the WSA that could facilitate transmembrane helix movements. Functionally, the protein resembled GLIC for its capability to bind selected anesthetics, indicating a similar anesthetic binding site with proper conformation in the structure.
3.3.3. Mu Opioid Receptor
One of the most important families of integral membrane proteins is GPCRs, which represents the largest family of proteins in the human genome. GPCRs are involved in the regulation of an incredible range of physiological functions.257 They are targets for ∼35% of the total approved drugs available in market.258 Yet GPCRs remain among the most challenging groups of membrane proteins to study.
Saven’s group and Liu’s group, working in collaboration at the University of Pennsylvania, were the first to partially solubilize seven-transmembrane GPCRs through sequence modification. The receptor studied was mu opioid receptor (MUR), which regulates pain response in the human body.259 While the molecular mechanism for protein function was not fully elucidated at the time, high-resolution structures were available to guide the redesign effort.260,261 Over a period of seven years, Saven’s group reported multiple iterations on their receptor design, including the transmembrane region only design (wsMUR-TM),262 the murine MUR based design (wsMUR-TM_v2),263 and the full sequence design (wsMUR-FL).264 Across the different designs, they followed the same comparative homology strategy, whereas sequences of similar GPCRs with known structures were aligned to model and identify potential exterior hydrophobic residues of MUR. An entropy-based approach similar to the one in the KcsA work was adopted to evaluate probabilities of amino acids at specific sites and their rotameric states. All amino acids besides P and C were evaluated at the variable positions to determine mutations compatible with the rest of the sequence in steric, electrostatic, and hydrogen bond interactions, while fulfilling the environmental energy requirement for solvation.
Figure 11B shows their initial water-soluble design wsMUR-TM, in which only the TM region and interconnected loops were considered with both N- and C-termini truncated. An iterated optimization was then conducted based on the murine MUR structure, resulting in wsMUR-TM_v2. Seven residues in the helical core were reverted back to native states to eliminate possible disruptions on the ligand recognition and internal packing in the first model.261 The third design, wsMUR-FL, contained the full-length protein with both N- and C- termini. The percentages of mutation for the three designs were ∼18% (53/288), ∼16% (46/288), and ∼12% (49/400), respectively. All designs were expressed and purified through the E. coli system. Yet an initial addition of 0.1% SDS and final 0.01% SDS were needed to prevent the aggregation. In comparison to the ∼40% α-helix in the native protein, helical contents of the three soluble proteins were ∼48%, ∼57%, and ∼37.6%, respectively. All wsMUR variants can specifically bind antagonist naltrexone with affinity in the tens of nanomolar range, which was comparable to native receptors. Although intended for structural optimization, wsMUR-TM_v2 rather exhibited the lowest thermostability, was the most prone to aggregation, required a higher concentration of SDS to stabilize and had the lowest affinity toward ligands, suggesting that the initial design was superior compared to structural based reversions which unfavorably decreased the solubility of the redesigned protein. These computational approaches alone might not be sufficient to completely solubilize GPCRs as detergent SDS was still needed for the structural stabilization. Besides the structural and biophysics studies, Saven and Liu were the first groups to explore these new variants of membrane receptors in various practical applications. The solubilization of GPCRs sheds light on how such previously unattainable biological species can be utilized in biomedical applications, such as in high-specificity biosensors.265,266
3.3.4. SIMPLEx
An alternative approach for transmembrane protein solubilization was reported by the DeLisa group at Cornell University in 2015.267,268 Compared to the commonly practiced fusion tag strategy which was also used to enhance membrane protein expression, DeLisa and co-workers took a step forward by introducing a protein that can function as a pseudodetergent to cover hydrophobic patches when connected to the membrane target.269 The amphipathic fusion partner, namely apolipoprotein A-I lacking the 43 residues of N-terminal domain (ApoAI*), has a highly fluidic structure that can accommodate various sizes of proteins and significant geometry changes.270 The strategy was named SIMPLEx (solubilization of integral membrane proteins with high levels of expression), which constructs chimeras of membrane proteins with ΔspMal-bp (Mal-bp lacking native export signal peptide) fusion at the N-terminus and ApoAI* at the C-terminus. The N-terminus fusion was needed to prevent undesired membrane integration during synthesis.
The wide applicability of the methodology was demonstrated with targets from various host systems that were not structurally relevant, including ethidium multidrug resistant protein E (EmrE), human cytochrome b5 (cyt b5) and a panel of 10 additional membrane proteins. The full chimeric proteins including all three segments can be readily expressed in the soluble fraction of E. coli. The absence of either fusion often lead to aggregation during expression, but cleavage of the N-terminus Mal-bp after synthesis did not precipitate the complex. The folding of the chimeras was verified via a quality control mechanism inherent in E. coli.271 Further characterization revealed that solubilized EmrE (PDB ID: 2I68) retained ligand affinity toward several natural substrates, while chimeric cyt b5 still performed octameric oligomerization. The flexibility of ApoAI* appeared to be sufficient to accommodate considerable protein conformational change during both of these processes. It was proposed that the ApoAI* actually formed an amphipathic shield to prevent membrane proteins from directly contacting water molecules but their plasticity still allowed oligomerization and ligand binding, as shown in Figure 11C.
In their follow-up work, the group further demonstrated the application of SIMPLEx in a membrane-bound enzyme DsbB to convert it to a water-soluble biocatalyst, that promoted disulfide-bond formation in various substrate proteins.272 Both the catalytic activity and substrate specificity of the protein were preserved in several E. coli strains, although an oxidizing cytoplasm was required for proper folding. The work opened up potential applications by transplanting enzymatic activities previously only associated with membrane enzymes to different cellular environments in vivo and under nonbiological conditions in vitro. However, despite the wide applicability of the SIMPLEx system, there have not been many reports of follow-up work that further scale up the utilization of transmembrane proteins using this approach.
3.4. QTY Code
While substantial achievements have been made in solubilizing transmembrane proteins via the modification of surface hydrophobic amino acids and the minimization of energy function through complicated computational means, success relies heavily on the availability of the crystal structure for the target or at least accurate homologous prediction models. Each target also requires extensive efforts that are usually not directly adaptable by other candidates. The stringent requirement for a skillset in computation and prior knowledge of the proteins of interest to build a template limited the widespread use of such methodology, and the utilization thereafter of soluble variants of transmembrane proteins in practical applications.
Following the question “Can you convert a hydrophobic α-helix to a hydrophilic one?” by the late Alexander Rich, Shuguang Zhang conceived of a simple mechanism for direct solubility regulation of α-helices, without the necessity of arduous computational optimization.273 The methodology, which he named the QTY code, is based on the side chain structure and electron density map similarities in certain pairs of 20 amino acids, regardless of their polarity and charge.274 In nature, such similarity has resulted in occasional erroneous charging of tRNAs during protein synthesis.20 Zhang and co-workers recognized this “nature’s confusion” concept and identified corresponding polar and nonpolar amino acid pairs that can be interchanged. Specifically, the hydrophobic L, I, V, and F were correlated with the hydrophilic residues of Q, T, and Y, denoted as L ⇔ Q, I/V ⇔ T, and F ⇔ Y, and shown in Figure 12A. Q, T, and Y were selected over charged residues to prevent potential surface charge alteration. Q was chosen over N due to its higher propensity in α-helices as N existed primarily at turns. It was reasoned that the exchange of structurally similar nonpolar residues to polar ones can promote hydrogen bond formation without disturbing the native steric packing or the overall conformation, so as to solubilize the transmembrane protein target, which can be used for drug discovery, deorphanization studies and disease therapies.
Figure 12.

QTY code for transmembrane protein redesign. (A) Crystallographic electron density maps for amino acids involved in the pairwise substitution of QTY code. Hydrophobic amino acid LIVF are replaced by hydrophilic QTY to stabilize GPCRs in aqueous environment. Reprinted with permission from ref (277). Copyright 2019 National Academy of Sciences. (B) Computer simulations of CCR5QTY (cyan) and CXCR4QTY (blue) in explicit water environment (24.85 °C at pH 7.4 and 0.9% NaCl) are superimposed with the crystal structures of CCR5 (PDB ID: 4MBS, magenta) and CXCR4 (PDB ID: 3ODU, green). Two side views and one top view are shown. Reprinted with permission from ref (274). Copyright 2018 National Academy of Sciences. (C) De novo predicted structure superimpositions for CCR5QTY (cyan) with CCR5 (green) by AlphaFold2. (D) De novo predicted structure superimpositions for CCR2QTY (cyan) with CCR2 (green) by AlphaFold2.
The demonstration of this methodology was first reported in 2018 on four chemokine receptors, namely CCR5, CXCR4, CXCR7, and CCR10, all belonging to the GPCR family.274 QTY substitutions were applied to all identified L, I, V, and F sites within the receptors’ transmembrane regions but not any of the terminus, EC or IC regions, which were in contrast to the common approach of altering only the hydrophobic amino acids in the outer perimeter. Despite the substantial sequence change (20–30%), QTY designed receptors were readily expressed in both SF9 insect cells and E. coli systems and purified without any detergent or lipid additives. Yet either or both of arginine and dithiothreitol were needed in the refolding or assay buffer due to the large number of C residues in these receptors. These water-soluble QTY variants retained native-like secondary structures, melting temperature profiles, and affinities toward respective ligands. Slightly reduced ligand binding activities were observed in a 50% human serum environment due to the complex nature of biofluids, which also suggested the physiological relevance of solubilized receptors. While no crystal structure has yet been reported for QTY proteins to date, MD simulation in the physiological condition resulted in stable structural models closely resembling native proteins with RMSD of ∼2 Å, as shown in Figure 12B. Predicted structures of QTY variant receptors using AlphaFold2 also showed remarkably close superimpositions with their native counterparts (Figure 12C,D).275
QTY substitutions in the interior of GPCR helical bundles are likely to diminish the “hydrophobic effect” that drives the folding of globular soluble proteins. MD simulations reveal the extensive generation of inter- or intra- helical hydrogen bonds, which might provide QTY proteins with an atypical polar core and help to stabilize the conformation.276 Such hypotheses still need to be experimentally verified. The technique was further developed by Qing et al., where additional QTY modifications were introduced at IC segments of receptors for enhanced solubility.277 On the basis of the common topology of seven-transmembrane segments, Qing and co-workers explored the functional tuning of soluble GPCRs by swapping the EC domains of two receptors to tailor its ligand affinity as chimera proteins. The details for this work will be discussed in Section 6.4. QTY-designed chemokine receptors exhibited high thermostability in the presence of arginine, which further opened up possibilities for biotechnological applications.
In a follow-up study, Hao et al. expanded the use of QTY code to several types of cytokine receptors including CXCR2, CCR9, IL10Rα, IL4Rα, IFNγR1, and IFNλR1.278 They proposed that soluble receptors fused with IgG-Fc fragments can function as molecular scavengers against excessive cytokine release in various pathogenic conditions. On the other hand, taking advantage of the interchangeability between receptors in native and QTY forms, Qing et al. discovered through the functions of short QTY proteins that native non-full length GPCRs with significant sequence deletions might provide another level of bioregulatory functions by serving as negative regulators for full length proteins.279 The work serves as a demonstration of how the study on solubilized proteins can benefit the understanding of native transmembrane receptors from a fundamental science perspective.
In a recent paper, Tegler et al. described an early attempt in the development of the QTY code, where only 29 lipid-facing residues in CXCR4 were substituted, named as CXCR4QTY29.280 The design provided a middle ground for QTY-based redesign where the receptor became more hydrophilic than the native protein, but still required detergent to stabilize in aqueous solution. It retained partial membrane-based functions. Expressed and isolated through a cell-free system, CXCR4QTY29 exhibited an α-helical structure and ligand affinity comparable to a native protein. When transfected into HEK293 cells, CXCR4QTY29 successfully inserted into the cell membrane and carried out cell signaling at higher concentrations of the ligand. This report bridged the gap between native receptors and fully substituted QTY receptors, indicating that the QTY methodology can be gradually applied to achieve a compromise of solubility and function on cell membranes.
3.5. Summary and Prospect
Figure 13 summarizes a timeline for major milestones in the quest for transmembrane protein solubilization. While more attempts have been undertaken than reported achievements in this field, not all efforts, especially those not entirely successful, have been well-documented apart from the bacteriorhodopsin studies discussed above.251 Combined with structural and functional diversity of transmembrane proteins, the lack of past experiences has hindered the pace of advances. Transmembrane protein solubilization is still among the most difficult challenges to date in the protein design field.
Figure 13.

Timeline for major achievements in the field of transmembrane protein solubilization.
Transmembrane proteins have domains naturally embedded in the double lipids, with extensive nonpolar contact between surface residues and the bilayer. The outward-facing surface is primarily hydrophobic, lacking hydrogen bond interactions, and thus folding differently from globular soluble proteins. Despite the decades of efforts on various transmembrane proteins, most work has followed the underlying rules based on the argument by Eisenberg and Rees in 1989, that soluble and transmembrane proteins share similar cores but have different surface residues.252 The statement led to a consensus that researchers need only to identify lipid/solvent facing residues and replace them with hydrophilic ones to solubilize transmembrane proteins. Innovations have mainly been focused on developing efficient methods for identifying and substituting those residues, including the sequence alignment, crystallization, or homologous modeling based on proteins with similar structures. The lack of crystal structures resulted in much greater reliance on computational modeling and solvent-exposed residue predictions, where several entropy-based algorithms were developed and optimized.203,226,254,262 While quite effective on a given target, extensive computational work was often required and many of the solubilized transmembrane protein exhibited more fluidic structures compared to their native counterparts, indicating decreased conformational stability.202,234,254 It is possible that the hydrophobic effect and steric interactions sufficient to maintain structure rigidity in a lipid environment might not be enough when stronger polar and ionic interactions are introduced to the proteins’ outer perimeter in aqueous solutions. Alternatively, an effective fusion protein pathway was also developed in which an amphipathic fusion partner served as a universal biological detergent for transmembrane proteins against interfacing with water molecules, though not many follow-up studies were conducted.267
The QTY code proposed by Zhang and co-workers follows a pattern rather than relying on computational simulations to conduct amino acid substitutions in the transmembrane region.274,277 The application of this simple method successfully solubilized over 10 chemokine receptors and cytokine receptors with native-like ligand affinity. How this code will affect proteins with functional transmembrane domains still requires further study. There are currently no crystal structures reported to date for any of the redesigned proteins.
Nonetheless, there are several areas of scientific and technological development that can further benefit the progress in this field. First, the continuously increasing computational power in the past few decades has already enabled the redesign from short transmembrane peptides like phospholamban to multipass proteins like GPCRs. It is reasonable to pursue studies of more sophisticated targets and complexes involving multiprotein interactions. Besides hardware, novel algorithms are constantly being developed either for sequence-based structure or solubility prediction,127,281−283 and general protein design mechanisms,284−286 some of which already included membrane protein packs.287,288 Recent years have witnessed the integration of machine learning (ML) algorithms in biological science, and molecular biology is no exception. The DeepMind team of Google reported their consecutive successes in ML based protein structure prediction algorithm called AlphaFold2,289,290 which can achieve experiment-like accuracies.291 While AlphaFold2 has not been utilized for purposes other than structure predictions, several other groups have already taken advantage of the neural networks to develop models for protein design.292,293 Moreover, the recent development of the QTY code, which supplements the early presumptions in 1989 on which most of transmembrane protein solubilization efforts are based, can benefit the entire field. Combined with rapid advances in computation, the QTY code can serve as an underlying principle to facilitate the design effort and enable high-throughput transmembrane protein redesign not applicable before. Finally, while early transmembrane protein solubilization was mainly aimed at the elucidation of protein structure and functional mechanisms, efforts are already begun to use solubilized proteins for subsequent development of various biomedical and technological applications.265,266,278 Cost-efficient and high-throughput production of soluble equivalents of transmembrane proteins can enable practical applications that were not previously possible. The potential for industrialization can form a positive feedback loop which further stimulates the interest and efforts in this field.
4. Transmembrane Protein Design
Numerous efforts have been made toward designing synthetic polypeptides that can be inserted into a membrane, mostly through a de novo approach.295,296 Simplified models were developed to resemble the large variety of transmembrane proteins in nature, and to minimize complexity in order to illustrate the fundamental connections.297 Understanding folding and insertion mechanisms for these models helped to elucidate the relation of sequence, structure, and function in native transmembrane proteins. Similar to solubilization efforts discussed in the previous section, α-helical bundle-based transmembrane structures have attracted wider interest due to their high biological relevance in living organisms and technical feasibility.
In this section, we will briefly introduce the lipid environment for membrane proteins and models for their folding and insertion. The focus will then be placed on common membrane protein design motifs and recent work on designing structural and functional transmembrane proteins. Detailed discussions on the fundamentals of membrane protein folding and stability can be found in previous reviews.209,298
4.1. Folding and Stability of Membrane Proteins
4.1.1. Lipid Membrane Environment
Native transmembrane proteins fold and function in the fluid membrane lipids. The hierarchical structure of the membrane provides several environments with distinct properties that correlate to and interact with different domains of a transmembrane protein.209 As shown in Figure 14A, membranes are composed of two layers opposing each other that can be divided into three main regions with well-defined borders: the hydrophobic center (∼25–35 Å); the hydrophilic head groups (each ∼10–15 Å); and the outer aqueous region.
Figure 14.

(A) MD simulation of a hydrated lipid bilayer, including a hydrocarbon layer ∼30 Å and two headgroup layers ∼15 Å each. Reprinted with permission from ref (209). Copyright 2000 Annual Reviews. (B) Schematic for membrane protein folding. For α-helical proteins, helix insertion and packing can be separated; stable transmembrane helices can present without tertiary structure. For β-barrel proteins, folding and insertion are likely to be coupled. Mismatch between the hydrophobic width of the protein (green region) and the bilayer induces distortions in either the protein or the bilayer. Reprinted with permission from ref (311). Copyright 2004 National Academy of Sciences. (C) Factors affecting the affinity of transmembrane helices with putative “dimerization motifs” such as GX3G. A generic transmembrane dimer model is used for illustration, based on a chimera of BNIP3 transmembrane dimer and QSOX (quiescin/sulfhydryl oxidase) soluble domain. Reprinted with permission from ref (339). Copyright 2015 American Chemical Society. (D) Structure for GASright motif of a right-handed helical dimer and a crossing angle of approximately −40°. GX3G sequence enables backbone contact at the crossing point (red), and formation of interhelical H bonds between Cα-H donors and carbonyl oxygen acceptors. Reprinted with permission from ref (347). Copyright 2017 American Chemical Society.
The thickness of the lipid bilayer varies by the length of the hydrocarbon chains attached to a given headgroup. Interactions between both head groups and chains define the area occupied by each lipid molecule.299,300 The hydrophobic core region exhibits a partially ordered structure which is mostly impermeable to polar molecules and ions. It exhibits a low viscosity, liquid-like state that lacks long-range order, which provides solvation for transmembrane proteins and largely define their properties. The hydrophilic head groups have a close packing which isolate the alkyl chains from contacting water by creating a permeability barrier. They vary in chemical distribution, exhibit horizontally distinguished composition, thickness and fluidity, and create lateral forces across the lipid bilayer.301 The dense packing of the head groups also results in the depletion of free water-molecules in this area which makes aromatic amino acids the preferred associations, creating “aromatic rings” as mentioned in the previous section. All factors combine to preferentially accommodate particular types of transmembrane proteins across the lipid layer while affecting their folding, stability, and function.302 The presence of transmembrane proteins in turn decreases the fluidity of the lipid while their hydrophobic domains perturb the local conformations and compositions.303,304
4.1.2. Two-Stage Model and Beyond
The folding of a membrane protein is different from that of water-soluble proteins as the common “hydrophobic effect” is negligible in lipids.305 The vdW interactions and solvophobic exclusion play important roles during the process.306,307 After being synthesized by the ribosome, transmembrane proteins fold concurrently during the coupling with a transmembrane chaperone named translocon, the exit from the translocon, and the entrance into the lipid membrane. However, recent reviews argued that these additional machineries and the membrane environment are not required for proteins to attain their native conformations.308,309 A traditional view for the process is a two-stage model proposed by Popot and Engelman.310 It is an oversimplification of the actual process but summarizes key steps for the conformational establishment of transmembrane proteins and serves as guidance for their designs. In this model, individual helices were proposed to form, reach thermodynamic equilibrium and stably interact with the lipid environment before contacting with other helices and self-assembling into the native state, as shown in the schematic of Figure 14B.311 The conclusion was drawn from observing the folding of bacteriorhodopsin, where fragments of helices were formed before they attained the final multipass functional conformation.312 The model also indicates that the folding and insertion of helices into the membrane are irreversible since unfolding or leaving the membrane would demand to overcome a large energy penalty. Considering the well-defined hierarchical structure of the lipid bilayer, this model generally restricts interactions between different helical segments to lateral associations.298 The model was further refined by Engelman to add a possible third stage in which the formation of higher order structures helps partitioning additional peptide regions or prosthetic groups.313 The third stage is believed to involve the accommodation of polar backbones, creating internal space through helix interaction, and creating binding surfaces.
4.1.3. Formation of a Stable Conformation
There are several factors that contribute to a stable α-helix spanning the lipid bilayer. Most transmembrane domains contain hydrophobic stretches typically of 15–20 amino acids, consistent with the approximate thickness of the core region in the bilayer (∼1.5 Å per rise). Shorter or longer helices can also exist in the transmembrane region for a single pass, albeit creating a “hydrophobic mismatch”. The exposure of hydrophobic residues to water is highly unfavorable and creates an energy penalty. While the bilayer can distort to accommodate the mismatch, helical domains longer than 26 residues will also kink, tilt or simply extend into the interfacial region. The presence of a single proline can induce the formation of hairpin bends in the helix, inducing multipass geometries.314,315 In some cases, increased hydrophobic mismatch can induce interhelical contacts to lower the free energy associated with protein–lipid interaction, or even change the thickness of the bilayer in the Angstrom scale.303
Composition-wise, transmembrane helices contain over half hydrophobic residues such as L, I, V, F, M, and A. Gromiha analyzed 295 transmembrane helical segments in 70 membrane proteins and found that residues including A, C, F, G, I, L, M, S, T, V, W, and Y are all prevalent in the helices.316 Strong hydrophobic residues like L are heavily favored but K appears to be sufficient for membrane insertion meeting the minimum requirement in hydrophobicity.317,318 For simple helical models comprising only L and A, a minimum of five L residues were needed for a 21 amino acid sequence. Peptides with alternating L and A residues resulted in stable transmembrane segments.319,320 Polar and especially charged residues are relatively rare but can still be found in the membrane domain.321,322 Charged residues usually flank the helix to interact with aqueous and interfacial regions of the bilayer, which help to modulate the structural stability and orientation. Aromatic residues have a higher propensity for the lipid interface, suggesting their role in the protein anchoring.323,324 A “positive inside” rule renders the intracellular regions of transmembrane proteins often enriched in positively charged residues.325 A higher proportion of W and Y is found in the headgroup region to assist helix positioning and dynamics, where K and R are also observed due to the “snorkeling” effect.223,326 With all 20 amino acids, P exhibits a more subtle effect, as it opposes helix formation in soluble proteins but plays a supporting role in membrane environments.327,328 It is also found to assist the helix insertion into membranes in certain scenarios.329 More differences arise when the folding mechanism of multipass transmembrane proteins is taken into account. A higher proportion of β-branched residues with low helix propensities exists in the interior of helical bundles compared to soluble proteins, which seems to drive the secondary structure formation by forming hydrogen bonds in low dielectric environments, such as in bacteriorhodopsin.252
In terms of energetics, transmembrane proteins reside in a free energy minimum that can stabilize their native conformations, similar to soluble proteins. The energy minimum is defined by the native sequence of the protein, the lipid environment and aqueous interfaces.330 Although extensive molecular machineries are required to direct transmembrane proteins to accommodate their native structures in living cells, once folded, the transmembrane domains are highly stable and resistant to full denaturation by means commonly applicable to water-soluble proteins.331 Elaborate means were developed by cells to handle the nonnative transition states when proteins are unfolded or partially folded during their synthesis. Transmembrane chaperone translocons prevent these nascent polypeptides from aggregation and translocate them to the lipid bilayer where the free energy minimum is finally reached.309 The integration process is mediated by allowing the partially folded polypeptide chain to pass through the pore region, equilibrate and reside in either the membrane lipid or aqueous region depending on their “biological hydrophobicity scale”.332
4.2. Design of Transmembrane Proteins
Knowledge acquired from the folding and stabilization of natural membrane proteins has long served as the guidance and restraints that have been incorporated into computational programs for synthetic transmembrane helical peptide design.287,288,333,334 These helical peptides in turn serve as simplified models to help elucidate folding and intra- or intermolecular interaction mechanisms for common helical bundles found in the nature.
4.2.1. Common Design Motifs
Despite the structural and functional diversity of α-helical transmembrane proteins, many of them share similar multimeric interaction units that fall within six classes of topologies and conformations, with helical trimers being the most common basic unit as determined by Feng and Barth.335 These classes cover a large fraction of protein topology and are enriched in recurrent sequence motifs that can be deconstructed from the evolutionarily conserved interhelical contacts. They are constantly observed to promote helix–helix interactions, and are consequently adopted in the design processes. As side chains along the helices help to define the specificity of helical interactions, simplifying matters by abstracting design factors is straightforward to predict structural interactions.
The GX3G motif, first identified by Russ and Engelman through library analysis of ∼107 possible sequences, plays an important role in the dimerization of glycophorin A.336,337 It represents the most abundant pairwise interaction of residues in transmembrane helices compared to random spacing.338 The motif exists in over 50% of single-pass transmembrane proteins to promote interhelical interactions, while its presence in multipass transmembrane proteins can contribute to the folding process.339 GX3G is more generally represented by small-X3-small where the small amino acid occurring at i and i+4 positions is typically G, A, or S.340 The motif maximizes interfacial vdW forces or hydrogen bonds by bringing the backbones of two helices into close proximity.337,341 However, the presence of the motif does not necessarily imply interhelical interaction and the affinity between GX3G-containing helices depends strongly on the sequence context, as shown in Figure 14C.342−344
One of the most representative forms of GX3G that can promote dimerization is the GASright motif. It is named from the parallel, right-handed helices at a crossing angle of ∼40°, whereas GX3G can also accommodate left-handed, antiparallel forms or at lipid binding sites.345 The strength of the helical association by GASright is modulated by amino acids adjacent to the motif and the geometry, rendering a great variation in the structural stability.346 While there are several determinant factors for this interaction, the major contributor is believed to be the interhelical hydrogen bond network between multiple Cα-H donors and C=O acceptors when two helices come close, as shown in Figure 14D.347 In fact, the unique geometrical features of GASright make it the optimal form for concurrent Cα-H hydrogen bond formation among all possible GX3G configurations, which has been utilized by scientists in de novo transmembrane peptide designs.348
The leucine zipper motif is an antiparallel coiled-coil structure first identified in yeast transcriptional activator GCN4.349 The motif contains a periodic repeat of L every seven residues, with nonpolar residues preferentially residing in a and d positions of the heptad packed in the core of the coiled-coil.350,351 The leucine zipper can drive self-assembly of transmembrane segments without the requirement of polar residues for helical interactions.352 The self-association follows a “knobs-into-holes” (KIH) type of interaction regime found in native proteins and can accommodate mutations to other hydrophobic residues.352 An iteration of the leucine zipper, namely (GX6)n, was extensively used for membrane protein designs. Similar to the GX3G motif, (GX6)n contains a small amino acid defined to be G, A, or S, and serves the function of close packing.345,353 The motif resembles the alanine-coil found in the water-soluble proteins with a crossing angle between −10° to −20°.354 A computationally designed coiled-coil based on (GX6)n, namely MS1, showed an interhelical interaction strength in the order of G > A > V > I for the small residue in the a position, which is opposite to the hydrophobicity of respective amino acids.351 Among the four variants of peptides, MS1-G predominately formed antiparallel dimers, MS1-A formed a mixture of parallel and antiparallel dimers at the monomer–dimer equilibrium, while MS1-V and MS1-I were mostly monomeric with scarce parallel dimers. Computation and conformational optimization revealed that smaller residues in the a position can increase the vdW interaction by efficient close packing as well as facilitate interhelical electrostatic interactions.
4.2.2. Design of Transmembrane Structures
Early designs of transmembrane proteins were focused on the rational arrangement of precise hydrophobic patterning using simple amino acid combinations. Lear et al. constructed a 21-residue peptide with only L and S to fabricate a leucine zipper based structure that can span the membrane.355 The design was chemically synthesized onto a solid support to perform as a selective ion channel. Another success from the von Heijine group primarily used L and A to fabricate one, two or four transmembrane segments with three LAALLAL repeats minus one L at the C-end of the helix. The whole sequence can be inserted efficiently into the inner membrane of E. coli during expression.356 The group also utilized positively charged K residues to control the protein orientation and topology in lipid bilayers through the precise placement of amino acids in the loop region. More recently, an antiparallel four-helix bundle with sequence LLLSGLGLLLLSLLGLLLLS in the helix region was reported by Lalaurie et al. to represent an abstracted version of the commonly found sequences in small multidrug resistance group proteins.357 The single 133-residue chain can be expressed in E. coli onto the cytoplasmic membrane with the intended secondary structure and topology, or stably extracted into maltoside detergent micelles. Yet the lack of tertiary packing design folded the protein in a dynamic molten-globular state.
An alternative approach is to use water-soluble proteins as the template, in contrast to the solubilization efforts discussed in Section 3. The methodology was adopted especially for the purpose of interpreting association mechanisms for coiled-coils. For the leucine zipper motif in GCN4, a buried N residue was responsible for the dimerization of helices.358 Transmembrane analogues of the protein were then designed to investigate the role of the amino acid in membranes by changing the surface residues to nonpolar ones, by both the DeGrado and the Engelman groups.359,360 Strong hydrogen bond association was observed in both models and mediated by the same mechanism, but trimers were the predominant species in DeGrado’s design. The interaction provided a strong thermodynamic driving force for stabilized structures by avoiding the exposure of polar residues to the lipid, whereas the mutation from N to V negated the oligomerization in designed peptides. Subsequently, studies on helix self-assembly by polar side chains at the interface were pursued by both groups.361,362 Polar residues such as N, D, Q, and E, promoted hydrogen bonding and stable oligomer formations, while residues with fewer polar atoms showed a weaker association. The energetics of the interaction was found to be stronger when the residues were located in the middle of the transmembrane helix rather than the headgroup region.363 In the follow-up work, DeGrado’s lab further explored the potential of hydrogen bond networks in the system by introducing V13S or V13T mutations at one position before the crucial N residue.364 Both S and T simultaneously behaved as an electron acceptor to the N in the neighboring helix and donor to the residue at i-3 in its own helix. An intricate local hydrogen bond network was formed to stabilize the trimerization of the peptides, as shown in Figure 15A.
Figure 15.

(A) Model of the trimeric structure of MS1 and MS1-V13T. Side chains from residues 13 and 14 are shown. Hydrogen bonds are represented as dashed lines. The polar network is shown in detail on the right. Interhelical hydrogen bonds are shown as black dashes, and the intrahelical hydrogen bond is shown in red. Reprinted with permission from ref (364). Copyright 2006 American Chemical Society. (B) Design models with IC and EC regions of four transmembrane proteins (top). From left to right, TMHC2 (transmembrane hairpin C2), TMHC2_E (elongated, PDB ID: 6B87), TMHC2_L (long span), and TMHC2_S (short span). Horizontal lines demarcate the membrane regions. Ribbon diagrams are at left, electrostatic surfaces are at right. Confocal microscopy images for HEK293T cells transfected with fluorescent tag fused TMHC2 proteins are at bottom. Line scans show fluorescence change across the membrane. Reprinted with permission from ref (173). Copyright 2018 The American Association for the Advancement of Science. (C) Sequence alignment of phospholamban and PL5. Phospholamban’s polar region (orange) and nonpolar region (yellow) highlighted; transmembrane helix underlined. The heptad repeat is labeled abcdefg; LxxIxxx motif, green. Red arrows indicate polar-to-nonpolar mutations. Superimposition shows a close match between the PL5 crystal structure (PDB ID: 6MQU, orange) and the MD-refined design model (Cyan). (D) Side chain steric packing at PL5′s helical interface. The repeated symmetrical interaction of helices provides the primary stabilization for PL5. Geometric complementary residues interact across the interface, roughly in layers characterized by two categories: a/g and e/g layers. Cα–Cβ and Cβ–Cϒ bond vectors point at opposite directions within the two layers. C, D: Reprinted with permission from ref (372). Copyright 2018 The American Association for the Advancement of Science.
The precise design of de novo multipass transmembrane proteins with well-defined and resolved high-resolution structures was not realized until recently, as reported by Lu et al.173 On the basis of the theory that soluble and transmembrane proteins share similar cores and on the stabilizing effect of internal hydrogen bonds on transmembrane helix association as discussed above, the group repurposed a previously designed soluble four-helix bundle generated with parametric equations with buried hydrogen bond networks.365 Surface residues were restricted to hydrophobic amino acids in the designs, whereas aromatic ring caps and the “positive inside” rule were utilized to define protein topology and orientation. Using the Rosetta algorithm, Lu and co-workers were able to design multiple two-helix and four-helix transmembrane proteins containing 76 to 215 amino acids that can accommodate multimeric oligomerization states. Most variants besides the 15-residue short version expressed well and localized onto membranes in E. coli and HEK293 cells, as shown in Figure 15B. The designed proteins are highly stable, and retain most of the helical structure when heated to 95 °C. With the extended intracellular region to assist the folding and stability, the crystal structure of TMHC2_E (PDB ID: 6B87) was resolved and aligned well with the design model (RMSD: 0.60–0.84 Å).366 Topologies with higher complexity were also attempted, including a trimeric six-helix assembly and a C4 tetramer by two transmembrane proteins with extensive cytoplasmic bowl-shaped domains, the latter of which showed 3.3 to 3.6 Å deviation between the crystal structure and the design model. The work demonstrated a design pathway that is exactly the opposite to native transmembrane protein solubilization, through designing soluble proteins with built-in hydrogen bond networks and converting surface residues to match the lipid requirement.
However, despite consecutive successes in designing transmembrane proteins with buried hydrogen bonds, small molecule motifs or extracellular domain templates, such interactions are not general features in native proteins and the primary force for transmembrane structure stabilization is insufficiently understood.173,367 The question remains whether the solvent/lipid exclusion and steric packing with vdW interactions are sufficient to drive the interhelical association and protein folding, as more efficient side chain packing was commonly observed in transmembrane proteins.368,369 Mutations of interior residues in the transmembrane region may disrupt vdW packing by creating voids or steric clashes, and subsequently destabilize the overall structure.370,371 Yet the design of transmembrane proteins folded solely by steric packing had not been successful until the recent work by Mravic et al., who built a phospholamban-based model stabilized by pairwise nonpolar interaction alone.372 Within its characteristic LxxIxxx heptad repeats, the core-facing a and d positions and interfacial e and g positions were considered during the design to render PL5 peptide variants. The N-terminus of phospholamban was redesigned by converting N residues to less polar ones, more sterically favored in an ideal structure, while the C-terminus was kept unchanged except for several synthesis facilitating residues. PL5 formed stable pentamers that retained their secondary structure at boiling temperatures in SDS and accommodated crystallographic structures in agreement with the MD model (PDB ID: 6MQU, RMSD 0.6 Å, Figure 15C). The bundle exhibited a tight interhelical packing but formed a pore in the central core. Changing the residue in the e and g positions did not disrupt the oligomerization but changed the pore radius. A KIH mechanism was clearly shown in the PL5 structure. As presented in Figure 15D, the a/g and d/e interfaces showed reverse positioning of the side chains that can maximize the packing density. Intrahelical hydrogen bonds also helped the packing when S and C were present. However, the author found that such packing required a remarkably stringent complementarity in the geometry. A single mutation from L to I at the g position resulted in a steric clash between adjacent helices and completely abolished the assembly, which was usually tolerated in water-soluble coiled-coils. At the e position, β-branched amino acids I, V or T and also C can promote the pentameric association but substitutions of A or L resulted in monomeric peptides, indicating that the assembly is highly specific. The group then conducted a database search and found structurally similar interhelical geometries recurring in the transmembrane proteome, demonstrating a widespread stabilizing mechanism through tight steric packing in native proteins.
In a recent effort, Gromiha and co-workers established an explicit database called MPTherm for the thermodynamics of membrane proteins and their mutants.373 MPTherm provides data for around 7000 mutants in more than 300 membrane proteins based on literature and ProThermDB database. Protein sequences, structural information, and topologies are associated with their thermodynamic parameters including melting temperature, free energy, enthalpy etc. while cross-linking with PDB and Uniprot entries. The database provides valuable resources on the understanding of relations between the structure, stability and function of membrane proteins.
4.2.3. Design of Functional Transmembrane Proteins
On the basis of the insight gained from transmembrane protein studies, in parallel with designs for defined structural characteristics, significant accomplishments have also been made to specific-function designs including the proton, ion, and electron transport, cofactor binding, and transmembrane helix targeting. In the 1970s, Goodall and Urry built peptides with AAG repeats that can be inserted into synthetic membranes as conductive ion channels.374 The methodology was extended by Kennedy et al. in 1977, using LSLG repeats to construct channels of varying lengths.375 A major milestone of these efforts was made by Lear and co-workers who designed a self-assembling 21-residue peptide with three full repeating heptads of LSSLLSL or LSLLLSL.355,376 The leucine zipper style design featured L as lipid-facing residues and S in the interior, with peptides associated into parallel helical bundles with left-handed crossing angles and tightly packed interfaces, as shown in Figure 16A. The inner core allowed conduction of either ions or protons. The single-channel conduction of the peptide resembles that from an acetylcholine receptor. The LSSLLSL repeats formed a hexametric channel, while the hydroxyl side chain of the S interacted with water to create a pore large enough to accommodate a solvated ion in the bundle. A substitution was made from S to L in the later repeats, which rendered the oligomeric state decreasing to tightly packed trimers or tetramers, resulting in a much smaller channel size with a more stringent selection rule that only conducted protons through the water hopping mechanism.
Figure 16.

(A) Computational model of (LSLLLSL)3 ion channel. Side chains of S are shown in ball-and-stick, L residues are shown with green sticks. Reprinted with permission from ref (25). Copyright 2020 Cambridge University Press. (B) Computational design and MD simulations of Rocker. (Top) Schematic of the conformational exchange between two opposite asymmetric states without being trapped in a symmetric state with both sites simultaneously occupied. (Bottom) From left to right: Metal binding site with ExxH motifs and a single E from each dimer; and the repacking algorithm with A at the tight interface; and F at the loose interface. Reprinted with permission from ref (377). Copyright 2014 The American Association for the Advancement of Science. (C) From left to right: superposition of the backbones of the crystal structure (blue) and the design model (gray) of WSHC6 (RMSD: 0.89 Å, PDB ID: 6TJ1 and 6TMS) with red sphere representing a water molecule; the cross-section of WSHC6 channel with a chain of water molecules occupying the central pore; superposition of the octameric assembly of the crystal structure (blue) and the design model (gray) of WSHC8 (RMSD: 2.51 Å, PDB ID: 6O35). Reprinted with permission from ref (174). Copyright 2020 Springer Nature. (D) From left to right: X-ray crystal structure (1.9 Å) of CC-Type2-(TaId)5 (PDB ID: 6YAZ) with internal T side chains (cyan) shown as sticks and hydroxyl groups (red) highlighted; hydrogen-bonding water (red spheres) network in the lumen of CC-Type2-(TaId)5; crystal structure of the octameric assembly for CCTM-VbIc with N to C terminus colored from blue to red. Reprinted with permission from ref (390). Copyright 2021 Springer Nature. (E) Top and side views of computational models of PRIME. The carbon atoms of the porphyrin cofactor are shown in purple. Reprinted with permission from ref (25). Copyright 2020 Cambridge University Press.
As one important aspect of transmembrane protein functions, interest has arisen in the design of ion/proton transporters. Following DeGrado’s first work on the ion channel design, his lab reported the design of an antiporting Zn2+/proton transporter named Rocker.377 The peptide contains 25 amino acids that form antiparallel heterotetramers with a 10° to 20° angle between helices and two nonequivalently packed interfaces, as shown in Figure 16B. It was built on a previously designed water-soluble DF protein with four E-two H di-Zn2+ binding sites, which were largely protonated in metal-free states to neutralize buried negative charges in the protein interior.378 Zn2+ binding displaced the protons to achieve desired thermodynamic coupling. The complex was designed to adopt two energetically degenerate asymmetric configurations with a negative cooperativity to expose sites on either the periplasmic or the cytoplasmic side of the membrane. A fully symmetric state was destabilized to prevent the simultaneous ion occupation at both sites which inhibited the transport. The mechanism is similar to that of the native transmembrane protein EmrE, which also rocks between asymmetrical homodimeric states.379 Transition metal ions Co2+ but not Ca2+ can also be transported down their concentration gradients with linked reverse proton flux. The transport efficiency of Rocker was not comparable to native proteins but provided a notable milestone for de novo design of ion transporters without traditional optimizations such as the directed evolution. The non-Zn2+ form structure of the designed protein was solved by X-ray crystallography and NMR, which suggested two closely interacting helix dimers, agreeing with the design model.
Representing a more stringent thermodynamic challenge from the channel design, the focus was turned to fabricating transmembrane pores with controllable geometries, which has enormous potential in biomedical applications, such as single-molecule sensing or sequencing.380−384 Such nanopores usually require five or more α-helices in the final assembly, as oligomerization with fewer subunits resulted in tighter packing and preluded their use as large molecule transporters.385 Several groups first achieved the goal by modifying naturally occurring peptide templates, such as cWza and pPorA. On the basis of their early success in building water-soluble α-helical barrels,386,387 the Woolfson and Bayley group designed a transmembrane nanopore assembly through the consensus sequence derivation from the E. coli outer membrane protein Wza.161 The design was based on the C-terminal D4 domain of the protein which associated into parallel homo-octamers with weak KIH interactions in its native form. The 35-residue cWza was derived from 94 sequences from the database and chemically synthesized, which accommodated ∼40% α-helical structure in micelles. The lipid insertion, pore formation and state change were represented by single-channel current recordings under a given voltage. Several conformational states with different inner pore diameters were suggested. CWza-K375C mutation was found to lengthen the open H state for conduction. The channel can also be blocked by cationic cyclodextrins in a concentration dependent manner. A follow-up work was reported by Mahendran’s group, who adopted the same methodology and designed a 40-amino acid derivative peptide pPorA as an ion-selective transmembrane pore.388 Similar characterizations were conducted to verify the insertion and assembly of designed peptide into lipid bilayer. The group also demonstrated ion selectivity tuning between K+ and Cl–, and blockage by cyclodextrins. The pPorA helical barrel was later developed for selective single-molecule sensing of cationic and anionic peptides.389 However, no crystal structure was reported from either group to support their claims on pore conformations.
More recently, several groups reported the design of α-helical barrels pores through de novo approaches, all of which utilized water-soluble design models and conducted transmembrane transitions. Xu et al. developed integrated nanopore structures that formed double concentric rings from subunits with two or four helices.174 The concentric design was reasoned to stabilize the structure by shielding the inner conductive polar network from undesired interaction with lipid-facing nonpolar residues. Starting with parametrically generated water-soluble backbones, Xu and co-workers conducted hydrogen bond network searches and combinatorial optimization to determine the side chain sequences, and used SAXS to evaluate the highest score candidates. Transmembrane transition was done by surface residue replacement and adding one E and two K residues at central channel openings to increase polarity. The ion conductor TMHC6 had core residues from a previously reported single ring structure, which can conduct Na+ in both ways and be blocked by gadolinium from the extracellular side.160 The pore also exhibited significantly higher conductance toward K+ ions. Their octameric transmembrane pore TMHC8 design, however, was less stable and additional linkage was added to stabilize the assembly. The resulting TMH4C4 formed a 16-helix association with strict stoichiometry. The pore had a central channel size of 10 Å in diameter and a transmembrane span of 31 Å, which was able to transport a large fluorophore named biotinylated Alexa Fluor 488. Both designs from this report had close agreement between the crystal structure and the design model (Figure 16C).
The de novo design of an α-helical barrel pore without buttress was later realized by Scott et al.390 They utilized the same water-soluble template to start with as had been adopted in Xu’s effort. In contrast to Xu’s method, screening was conducted to optimize the amino acid selection at all positions in the heptad. The β-branched amino acid T at the a position combined with S at the d position were found to be optimal for the KIH type interior packing to maintain an open barrel while providing a hydrogen bond network to accommodate free moving water molecules from lined hydroxyl groups (Figure 16D). Small hydrophobic A residues were kept at the e and g interfacial positions to support the high-order bundle formation. The hydrophobic V/I residue at the exterior b/c positions performed best among all combinations for monodisperse, stable membrane associated peptide channels. The designed peptide, namely CCTM-VbIc (or CC-Type2-(TaId)5, PDB ID: 6YAZ, in water-soluble form), predominantly showed the α-helical structure, and could be inserted into a planar lipid membrane. It exhibited cation-selectivity with a permeability ratio of ∼ 5:1 between K+ and Cl–, and was also capable of conducting Na+ and Cs+. Computational simulations suggested that it has a hexametric assembly based on the structure of the water-soluble channel that is 7 Å wide and 20 Å long. Yet a tetrameric antiparallel helical dimer bundle was observed from X-ray crystallography, as shown in Figure 16D. The disparity between the crystal structure and design model was attributed to potential multiple states accessible to the sequence with close free energy. The author assumed that tetramers were not likely to be the conducting state as the cavity inside was too narrow and the energy barrier was too high to perform ion conduction. This assumption was supported by both MD and electrostatic simulations, where water molecules were constrained in dimeric, isolated states in the tetrameric conformation. Some of the natural membrane-spanning pore-forming peptides also accommodate a similar antiparallel structure, which might not necessarily be their active states.391,392 Yet a high-resolution structure of the hexametric assembly would be needed to confirm the multistate theory.
Another element of major interest in transmembrane protein function is the cofactor binding and electron transfer, which is a critical part of bioenergetic processes. Electrons hop between redox-active cofactors during the transfer. One approach is the design of water- or membrane-soluble maquettes to bind heme for light harvesting, which will be covered in Section 6.2. Korendovych et al. designed a 24-residue transmembrane protein PRIME (PoRphyrins In MEmbrane) based on a water-soluble peptide backbone to selectively bind non-natural porphyrin-based cofactors in bis-His geometry that was close enough to perform multicenter electron transfer across the lipids.393 PRIME exists predominantly as monomers but forms a tetrameric helical bundle upon cofactor binding with antiparallel D2 symmetry and interhelical “Ala-coil” motif, as shown in Figure 16E.394 D2 symmetry is ideal for transfer and transport proteins as it places an axis of symmetry parallel to the membrane and resembles the antiparallel homodimer found in natural transporters.395 The peptide contains two H-binding sites that can specifically bind two FeIIIDPP cofactors in the expected stoichiometry. A second-shell hydrogen bond network was designed to stabilize the reaction center with a main chain carbonyl and a T18 from the adjacent helix. Several characterizations revealed consistency between the design model and the peptide. The work presented a pathway through which the functional design of soluble proteins can be tweaked to make transmembrane variants that enable the nonbiological redox active cofactor integration and electron conduction.
Utilizing the GASright motif, Yin et al. developed an algorithm named CHAMP (computed helical antimembrane protein) for the design of sequence-specific transmembrane-helix targeting peptides.367 Their first designs can modulate the activity of closely related human integrins αIIbβ3 and αvβ3 by competitive lateral association with active transmembrane domains in α subunits. They compared sequences of the target transmembrane domains with existing structural motifs to identify suitable backbone geometries from a library of structural defined helix pairs in energy minima with backbone interactions. Highly stringent shape-complementary vdW interactions were considered to differentiate fine topographical differences in target helices with similar motifs but minor conformational differences.396 Sequence-specific interactions were observed between the designed peptide and targets that lead to the formation of dimers in micelles. The peptides also activated their respective targets in mammalian cells and induced platelet aggregation, without any detectable cross-reactivity. Further use of the peptides was demonstrated with isolated full-length integrin αIIbβ3.397 Utilizing the same methodology but with the integration of the Rosetta molecular modeling suite, Mravic et al. extended the design on a α5β1 integrins targeting peptide in endothelial cell activations.398 However, while both of the new CHAMP (PDB ID: 2W2E and 1KPL) peptides can activate α5β1 integrins in vivo and in vitro, they preferentially bind either to the β1 or both α5 and β1 transmembrane domains, instead of the α5 domain originally targeted. The inferior sequence specificity was attributed to the L-rich AX3G motif in α5, whereas the bulky F residue inhibited close interhelical packing. Instead, the GX3G motif with additional small residues and β-branched amino acids on the flank in β1 domain was more preferred by peptide binding. Besides DeGrado’s effort, a report by Heim et al. also showed that a selected sequence in a series of 26-residue single-transmembrane proteins (LIL) containing only L and I following the initial M residue can activate platelet-derived growth factor β receptor to transform cells and change subsequent cell biology.399
4.3. Implication for Design of Water Solubility
From the discussion above, it is evident that the various approaches to transmembrane and water-soluble proteins designs exhibit great similarities with some interchangeable methodologies (loosely defined). With the same underlying rule as stated by Eisenberg and Rees that both protein variants share similar cores but with different surface residues, the natural or synthetic variants of either type of proteins have served as the template for structural and functional designs of the other, with opposite modifications.252 While the computational solubilization of native transmembrane proteins calls for the replacement of lipid-exposed residues by polar or charged amino acids, the design of the transmembrane structure often adopts cores from water-soluble proteins. Approaches from both directions have seen great success for the past several decades as discussed in the past two sections.173,174,203,226,239,262,372,390
However, despite the resemblance between these two fields, the distinct properties and folding mechanisms for water-soluble and transmembrane proteins still dictate significant considerations, especially when fine-tuning of structure and function is needed. The main driving force of water-soluble protein folding is the hydrophobic effect, which is negligible in transmembrane environments. With a similar hydrophobic core, the folding of transmembrane proteins has proved to require a more stringent complementarity in sequence geometry for optimal vdW interactions,372 especially when the natural machineries are not accessible.308 On the other hand, upon folding, transmembrane proteins exhibit greater stability compared to water-soluble variants with the well-defined hierarchical structure of the lipid bilayer. A lower fluidity in hydrocarbon chains and consequently a larger energy barrier for conformational disruption in the bilayer pose additional requirements for insertion during the design process but are likely to be more tolerant for subsequent applications. Such differences often result in the solubilized transmembrane proteins acquiring a more fluidic “molten globe”-like structure due to the destabilized interior interactions and conformations.202,234,254 The hydrogen bond network is one of the most commonly adopted features for transmembrane core stabilization. Yet with the realization of higher requirement on steric packing in the lipids, similar questions might arise: whether the design of the hydrogen bond network can always be transplanted from one variant to another, or will more stringent interactions be needed to compensate for the more dynamic environment in aqueous solutions? Such concerns might be more relevant to the energy landscape of folding or when multiple states with close energy profiles exist. With constant progress and success in the de novo design of both water-soluble and transmembrane proteins, scientists are moving their targets from simplified peptide models to long-chain proteins with more complicated higher-order structures. More delicate considerations will be needed to handle the issues that were previously negligible or nonexistent in simple models.
Thanks to recent advances in computing hardware and algorithms, both computer-assisted redesign and de novo design of water-soluble and transmembrane proteins have significantly progressed in the past decades. Novel machine learning algorithms have achieved reasonably high accuracy for protein structure prediction,290,400,401 which, combined with the development of single particle imaging by Cryo-EM,402 have profound implications for the field of structural biology. Utilizing their AlphaFold2 neural network algorithm, DeepMind has published a database containing more than 350,000 predicted structures representing 98.5% of known proteins in the human genome as well as in 20 model organisms.403 Although the prediction confidence is only ∼58% overall, the availability of the vast sequence space with previously totally unknown structures still provides referable data sets from which putative protein topologies can be estimated and utilized in the design work. Predicted surface motifs and exposed residues can potentially serve as the starting point for native protein redesign or functional domain extractions. With new structural information generated by various means, it is possible for protein scientists to take a top-down approach for protein design and functional manipulation to produce novel species that can benefit the fundamental understanding of the field and enable new applications.
5. Design of Proteins with Enhanced Solubility
Besides the limited but ever-progressing successes in the solubilization and design of transmembrane proteins, altering solubility through engineering approaches still has profound implications and enormous potential in other species within the protein sequence space. In this section, we will introduce common strategies for the solubilization of membrane-bound protein (MBP) in addition to transmembrane proteins, expression and solubility enhancements for integral soluble proteins, and characteristic ways to increase the stability and safety of monoclonal antibodies as required for therapeutic agents.
5.1. Membrane Bound Proteins
MBPs refer to proteins that are associated with and travel within the biological membranes, which can be released by disrupting the lipid bilayer. MBPs are characterized either as integral membrane proteins (IMP) or peripheral membrane proteins, referring to species permanently or temporarily anchored to the lipid bilayer.404 The transmembrane proteins discussed in previous sections are one type of IMP that spans the full width of membrane lipids. Other types of MBPs embed part of their structural segments on the surface of the lipids to participate in cell processes, many of which are of great pharmaceutical relevance.405,406 Herein, we will focus on discussing the nontransmembrane MBPs in this section and the use of “MBP” will be exclusively referring to such protein types.
Similar to the transmembrane proteins, the in vitro expression of MBPs often results in misfolding and protein aggregations.407,408 This phenomenon poses serious obstacles for the large-scale heterologous expression and structural study of MBPs. Detergents, organic solvents or lipids need to be utilized for satisfactory protein stabilization.409 Moreover, MBPs can exhibit both water-soluble and membrane-bound forms, the latter of which serves as a drug target related to various pathogeneses.405,410 Some membrane-bound enzymes can be directly used for disease treatments. Many efforts were undertaken to solubilize MBPs via protein design approaches without altering their functions, to enable the overexpression and facilitate subsequent studies or applications.
5.1.1. Encoding Gene Modification
One design approach to this issue is the direct modification on the encoding gene of MBPs to enable in-host water-soluble expression. For instance, penicillin-binding proteins (PBP) play a key role in the metabolism of murein. Yet their crystal structures were difficult to resolve by X-ray analysis due to the need for detergent additives to stabilize the isolated proteins. Ferreria et al. constructed a water-soluble variant of PBP5 with truncated C-terminus by directly modifying its genetic sequence.407 The excised ducA gene in PBP5 was shortened at the carboxyl-coding end, and cloned into a runaway-replication-control plasmid pWKL-61. The redesigned sequence contained stop codons in all three possible reading frames and replaced the original translation stop codon. The constructed plasmid pWKL-61 allowed gene amplification and overproduction of water-soluble proteins at an elevated temperature of 35 °C during culturing. Compared to the recombinant full-length PBP5 which was highly unstable, the water-soluble PBP5 can be overproduced in the absence of any detergent. Their enzymatic activity and interaction with [14C] benzylpenicillin were also retained.
5.1.2. Redesign of Exposed Residues
The solubilization of MBPs can also be achieved by redesigning the hydrophobic residues embedded in the lipid membrane. Many pathogenic bacteria utilize quorum-sensing systems to regulate the expression of their virulence genes and promote biofilm formations. Thus, the use of enzymes to disrupt their signaling is a promising way to abolish the biological activity of quorum-sensing molecules. One enzyme of this type, the water-insoluble mammalian paraoxonases, is membrane or high-density-lipoprotein (HDL)-associated, and can hydrolyze lactones with various modifications or carbon chain lengths. Sarkar et al. took three different approaches to redesign and increase the solubility of human PON1 (Figure 17), which included (i) removal of the hydrophobic N-terminal leader sequence (ΔN-huPON1), (ii) mutations of hydrophobic amino acids in the presumptive HDL binding site to polar residues (ΔHDL-huPON1), and (iii) mutations of surface residues to be more polar during the directed evolution of G2E6 PON1 (g2e6p-huPON1).411 All three engineered variants were more soluble than the native huPON1. The fluorescence for ΔN-huPON1 was about twice that of huPON1 and g2e6p-huPON1, whereas ΔHDL-huPON1 were about four- and six-times more fluorescent, respectively.
Figure 17.

PON1 solubilizing mutations. Left: the surface of PDB ID: G2E6, with hydrophobic amino acids (VGMCILYFW) shown in red. Residues 1–15 are not resolved in the X-ray crystal structure, but the ΔN-huPON1 variant removed residues 4–17. Mid: positions modified in the ΔHDL-huPON1 variant are shown in spheres. These residues compose much of the hydrophobic surface patch near the N-terminus. The Ca2+ ions are shown as pink spheres, and a phosphate bound in the presumed active site is shown in orange sticks. Right: the 59 positions that differ between huPON1 and G2E6 are shown. The positions that were modified in g2e6p-huPON1 are spheres, and the other 43 positions are sticks. Position 166, which was modified because of its proximity of 192, is noted. (PDB ID: 1V04). Reprinted with permission from ref (411). Copyright 2012 American Society for Biochemistry and Molecular Biology.
In a follow-up work, water-soluble huPON2 variants were designed based on the crystal structure of rabbit-human hybrid PON1.412 The N-terminal H1 region of huPON2 was removed, and the H2 region was replaced with a degenerate tripeptide linker carrying a proline turn flanked by one flexible residue at both sides G/S–P-G/S. A flexible and hydrophilic degenerate dipeptide linker D/N/S/G-D/N/S/G was used to replace the H3 region of huPON2. Two water-soluble clones of D2 and E3 showed significant quorum quenching bioactivities and effectively impeded biofilm formation.
5.2. Nonmembrane Bound Proteins
Besides transmembrane proteins and MBPs, water-soluble proteins can also suffer from poor solubility especially when large-scale synthesis and applications with high concentrations are concerned. Overexpression in both prokaryotic and eukaryotic cells can lead to misfolding and improper intramolecular interactions of proteins which often result in their inadequate expression or inclusion body formation.413 Many efficient approaches, such as affinity tags, protein fusions, and rational designs, have been adopted to enhance the protein solubility and promote proper folding.414 Herein, we will review strategies commonly applied to increase the solubility of nonmembrane bound proteins and facilitate their expression, which can serve as a general toolbox for protein solubility design.
5.2.1. Solubility Enhancing Fusion Tags
The affinity tag is a polypeptide fusion partner widely used for recombinant protein purification that has minimal effects on the tertiary structure and biological activity of proteins, and easy to remove.415 Some affinity tags can also simultaneously enhance the solubility and expression of selected proteins, individually or in combination with 6 × His-tag (Figure 18A), as summarized in Table 2.414
Figure 18.

(A) Schematic representation of the pathway from protein expression to purification using solubility tags. The “MBP” tag in the graph refers to maltose-binding protein (Mal-bp) defined in the manuscript. Reprinted with permission from ref (414). Copyright 2006 Elsevier. (B) Molecular chaperones provide assistance to folding by lowering free energy barriers and preventing aberrant intermolecular interactions (red), which can lead to various forms of aggregates. Reprinted with permission from ref (467). Copyright 2016 The American Association for the Advancement of Science.
Table 2. Commonly Used Solubility Enhancing Fusion Tags.
| tags | protein | source organisms | MW (kDa) | ref |
|---|---|---|---|---|
| Mal-bp | Maltose-binding protein | Escherichia coli | 43 | (416, 417) |
| GST | Glutathione S-transferase | Schistosoma japonicum | 26 | (418) |
| TRX | Thioredoxin | Escherichia coli | 12 | (418) |
| SlyD | FKBP-type peptidyl–prolyl cis–trans isomerase SlyD | Escherichia coli | 196 | (419) |
| DsbA | Disulfide-bond A oxidoreductase | Escherichia coli | 21 | (423) |
| NusA | N-Utilization substance | Escherichia coli | 55 | (91) |
| T7PK | Phage T7 protein kinase | Bacteriophage T7 | 17 | (414) |
| Skp | Seventeen kilodalton protein | Escherichia coli | 17 | (428) |
| HaloTag7 | Modified haloalkane dehalogenase | Escherichia coli | 34 | (420) |
| CBDengD | CBD of noncellulosomal cellulase EngD | Clostridium cellulovorans | 20 | (426) |
| SEP(C9R) | GFP-based superecliptic pHluorin | Aequorea victoria | 1.6 | (424, 425) |
| MOCR | monomeric Ocr | Escherichia coli | 16 | (455) |
The maltose-binding protein (Mal-bp) from E. coli is a widely useful tag of this kind, which can simultaneously improve the translational expression, provide an affinity purification strategy, and enhance the solubility of partner proteins.416 The ligand-binding cleft in Mal-bp is considered a likely site for peptide binding, and four solubilization mechanisms have been proposed.417 One model is that fusion proteins form soluble, micelle-like, multiprotein complexes with hydrophobic regions of Mal-bp; while in another possible model, Mal-bp is believed to restrict the motion and reduce the number of possible conformations in a slow-folding passenger protein. A third possible model suggests that Mal-bp may act as a molecular chaperone, physically interacting with and sequestering its fusion partners from self-association during the folding process. It is also proposed that Mal-bp can inhibit protein aggregation in their native states by intramolecular association and sequestration. Fox et al. used the Mal-bp tag to fuse the green fluorescent protein (GFP), p16, and E6. There was little or no effect on the solubility of the fusion proteins by single point mutations on W62E, A63E, Y155E, W230E, and W340E within the maltose binding cleft, but a significant decrease was observed when three amino acids were mutated near one end of the cleft (W232E, Y242E, and I317E).417 In a separate work, two other water-soluble fusion tags, namely GST and TRX, were used to inhibit the aggregation of six different proteins (TIMP, E6, p16, CATΔ9, GFP, and TEV) and compared to the Mal-bp tag. Mal-bp tag showed better solubility enhancement effects comparing to GST and TRX tags.418
Another solubility enhancing fusion tag is SlyD, which is a stress-responsive protein originally used for purifications in the immobilized metal affinity chromatography. Han et al. explored the use of SlyD as an N-terminus fusion partner and cis-acting folding enhancer for the expression of several recombinant proteins that commonly formed inclusion bodies during synthesis.419 Their test objects included: (i) minipro-insulin, (ii) human epidermal growth factor, (iii) human prepro-ghrelin, (iv) human interleukin-2, (v) human activation induced cytidine deaminase, (vi) deletion mutant of human glutamate decarboxylase, (vii) human ferritin light chain, (viii) human G-CSF, (ix) human cold autoinflammatory syndrome 1 protein NACHT domain, and (x) Pseudomonas putida cutinase. The cytoplasmic solubilities of these proteins were all observed to increase when fused with SlyD. It is believed that SlyD can shield the interactive surfaces of heterologous proteins from nonspecific interactions that lead to the aggregation.
Alternatively, HaloTag7 tag was engineered through molecular evolution for maximal soluble expressions of fusion proteins in E. coli, which increased the stability and negative charges relative to the parental dehalogenase, resulting in the improved expression of properly folded proteins with high solubility. Twenty-three human-origin proteins were fused with HaloTag7, Mal-bp, GST, or 6 × His Tag, and expressed in E. coli. Around 74% of the HaloTag7 fused proteins were produced in soluble form, while 52%, 39%, and 22% of soluble fraction expressions were observed when the proteins were fused to Mal-bp, GST, and 6 × His-Tag, respectively.420
Davis et al. compared the soluble expression of human interleukin-3 (hIL-3) with four fusion tags, including NusA (most soluble), GrpE, BFR, and TRX (least soluble).91 Expression experiments suggested that NusA/hIL-3 fusion protein was completely in soluble fraction, while GrpE/hIL-3 and BFR/hIL-3 displayed partial solubility, and Thioredoxin/hIL-3 was mostly in the insoluble fraction at 37 °C.
The activity of many functional proteins depends on not only its solubility, but also the correct disulfide bond formation. Lipase B (PalB) from Pseudozyma antarctica, a cold active microbial lipase with transesterification activity, was primarily expressed in fungi and yeasts. Xu et al. reported the expression of functional PalB in the cytoplasm of E. coli by fusing it with different tags including GST, Mal-bp, NusA, TRX, T7PK, Skp, and DsbA.421 Among all tested variants, only the DsbA tag significantly improved both the solubility and activity of PalB, while Mal-bp and T7PK enhanced solubility but not activity. The result was attributed to DsbA’s capability to catalyze direct bridging of disulfide bonds in translocated proteins. The chaperone activity of DsbA assisting the folding of several periplasmic proteins was also noted in other reports.422,423 Alternatively, the SEP (solubility enhancement peptide) tag fusion was reported to help in producing large quantities of proteins containing multiple disulfide bonds.424,425
Additionally, the cellulose-binding domain (CBD) of noncellulosomal cellulase EngD from Clostridium cellulovorans was used as an affinity tag for the soluble expression of catalytic domains of EngB and EngD.426,427 The Skp tag can also help partner protein folding, even though it was not expressed in the periplasm.428 In general, the solubility enhancement by fusion tags is an efficient approach, although the screening of tags is needed as specific proteins react differently to different tags.
5.2.2. Molecular Chaperone Fusion
Another approach to enhance the soluble protein expression is through the molecular chaperone fusion. Molecular chaperones are proteins that interact with and aid the folding of another protein without being part of its final structure, which also prevent undesired intermolecular interactions leading to aggregations (Figure 18B).429 Detailed models and mechanism of molecular chaperone on protein folding can be found in previous reviews.429,430
E. coli employs two major chaperone systems: the DnaK (Hsp70) system and the GroEL (Hsp60)/GroES system.431 DnaK, a major E. coli Hsp70 family member, consists of a 44kD N-terminal ATPase domain and a 27kD C-terminal peptide-binding domain, whereas GroEL and its cofactor GroES represent the paradigmatic Group I chaperonin system.430 Kyratsous et al. used the DnaK and GroEL chaperonins to facilitate the large-scale production of soluble recombinant mouse prion protein (PrP).431 The target sequence was fused to the C-terminus of either DnaK or GroEL, and a poly-His tag was added for the affinity purification. A thrombin cleavage site was placed at the fusion junction to separate the target protein with the chaperone. Both Dnak-PrP and GroEL-PrP fused sequences yielded large amounts of soluble proteins, although the target protein was also found in inclusion bodies. Interestingly, the DnaK fusion variants were more soluble than the GroEL fusion variants.
Furthermore, the artificial chaperone fusion can be designed using water-soluble proteins. Zou et al. constructed a two-module chaperone system RS-mTEV, which includes a mutant protease domain of tobacco etch virus (mTEV) with the canonical recognition sequence but no proteolytic activity, and a solubility-enhancing fusion partner, namely the C-terminus of E. coli lysyl-tRNA synthetase (RS).432 The RS-mTEV protein showed higher solubility than mTEV alone, and was tested against GFP fused hepatitis B virus X protein (HBx). Both the yield and solubility of L-EGFP-HBx were improved when coexpressed with RS-mTEV, whereas a recognition sequence (ENLYFQG) of mTEV was fused to the N-terminus of EGFP-HBx, denoted as L. Moreover, the antiaggregation and solubility enhancement capabilities of RS-mTEV were also observed in the in vitro refolding experiments. The refolding yield of L-EGFP-HBx increased with an RS-mTEV concentration dependence in buffers containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl and 5 mM MgCl2. In a different study, Stull et al. reported an ATP-independent chaperone Spy from E. coli that can rapidly associate with immunity protein 7 and eliminate its aggregation tendency.433
5.2.3. Soluble Protein Partner Fusion
The solubility of fusion proteins can be predicted from their primary sequences with good confidence levels. Thus, it is a common practice to link a soluble fragment to an insoluble protein to increase its solubility. The soluble fusion partner can additionally pose steric hindrances against particle collisions to prevent aggregations.
Ribosomes are extremely soluble ribonucleoprotein complexes, which can be an ideal partner for such applications. Sørensen et al. tested the efficacy of ribosomal protein L23 (rpL23) as a solubilizing fusion partner for (i) GFP, (ii) streptavidin (SA), (iii) murine interleukin-6 (mIL-6), (iv) yeast elongation factor 1A, and (v) a ScFV antibody, in an rpL23 deficient E. coli strain.434 Proteins were fused to rpL23 at the N-terminus and a 6 × His-tag at the C-terminus. Active GFP, SA, and mIL-6 were cleaved off the engineered ribosome, whereas high yields of recombinant proteins were obtained using this coupling method. Intrinsically disordered proteins (IDP) are also rich in polar amino acids, which can enhance the solubility of fusion proteins. In addition, the bulky IDP segments can occupy a large physical space and entropically exclude fusion partners against close contacts due to the Brownian motion. Segments with this property were named “entropic bristles” (EBs).435 Santner et al. designed four EB fusion polypeptides with different lengths (from 60 to 250 amino acids), and the results indicated that the length of the peptide is more important for solubility enhancement than its composition. EB60A and EB60B (6.8 kDa) with the same length but different compositions showed similar solubility enhancing capabilities, which were less than the longer EB144 (15 kDa) and EB250 (26.1 kDa) fusions. The use of IDPs as a solubility enhancer was then proved with several insoluble partner proteins.436
5.2.4. Charge-Mediated Solubility Enhancement
Besides steric repulsion and hydrogen bonds, the solubility of recombinant proteins can also be tailored by introducing charges to the fusion complex, often through polyanionic tags. For instance, a highly negatively charged RNA-mediated chaperone was developed to be fused to the N-terminus of aggregation-prone target proteins, which subsequently bound to the folded N-terminal RNA-binding domain to improve the intermolecular electrostatic repulsion and solubility.437 Similarly, a super negatively charged GFP (negGFP) was fused to the sterile alpha motif domains to enhance their soluble expressions.438 Many molecular chaperones with negative charges are beneficial for the passenger protein’s solubility. Wayne et al. found that the antiaggregation efficacy of Hsp90 significantly decreased when its charge-rich regions (CX and CL regions) were removed in the fusion with citrate synthase.439
5.2.5. Structure-Based Mutation
Cysteine is the only natural amino acid that contains a reactive sulfhydryl group capable of forming intra- and intermolecular disulfide bonds, which are closely related to the protein folding and aggregation. Therefore, a rational and effective strategy to improve protein solubility is through systematic scanning and mutagenesis against C residues.440 LovD is a 46 kDa acyltransferase found in the lovastatin biosynthetic pathway. More than 50% of total LovD exists in insoluble pellets, which hinders the whole-cell biocatalytic activity. Xie et al. identified two surface-exposed C residues in LovD that are responsible for the undesired disulfide bond formation and oligomerization which lead to aggregation.440 The group found that residue replacements on C40 and C60 sites can significantly improve the protein’s solubility and the whole-cell biocatalytic activity. Further mutagenesis revealed C40A and C60N mutations to be most beneficial, resulting in 27% and 26% increase in whole-cell activities, respectively. Other works also suggest that the global or site-specific replacement of C residues with amino acids of comparable size or polarity is an efficient approach to noteworthily enhance the solubility of recombinant proteins.441−443
Sequence substitution for hydrophobic residues was also adopted to enhance the solubility of proteins. One such effort was conducted on a series of consensus ankyrin repeat proteins (1ANK, 2ANK, 3ANK and 4ANK), which were designed by Peng and co-workers to serve as a generalized scaffold for engineering protein–protein interactions.444 The repeats exhibited inadequate solubility at physiological pH, which hindered their subsequent applications. The group then replaced solvent-exposed L by R residues in target 4ANK sequences to increase the surface charge and electrostatic interactions.445 The resulting 4ANK-TALR sequence (terminal all L to R) was soluble and stable over a large pH range, demonstrating the feasibility of this approach, which also helped to elucidate the impact of introduced surface charges on protein solubility and salt dependence.
5.3. Monoclonal Antibodies
Antibodies are glycoproteins tailor-made to seek out, attach to, and eliminate specific antigens as a source of pathogenesis. It is considered one of the most rapidly growing class of pharmaceuticals with the continuously increasing market for biogenic drugs in modern precision medicine.446−448 Albeit generally in soluble forms, antibodies still suffer from aggregation issues in vivo that are of great concern to antibody-based therapies. It can lead to the decrease in the bioactivity and elicit undesired immunological responses, which is often the main reason behind high development costs.449 To fulfill the stability and safety requirements before the drugs can receive FDA (Food and Drug Administration) approval and reach the market, many engineering approaches were adopted to overcome antibodies’ solubility issues.450
There are several typical structure-based approaches for this effort, including (i) modifying the pI, (ii) decreasing the surface hydrophobicity, and (iii) introducing an N-linked carbohydrate moiety within a complementarity-determining region sequence, as employed by Wu and co-workers.451 In practice, the solubility of anti-IL-13 monoclonal antibody CNTO607 had a 2-fold improvement by modifying the pI compared to the native protein, and several mutants with decreased overall surface hydrophobicity also exhibited moderately improved solubilities. In contrast, the introduction of the consensus N-glycosylation site into H-CDR2 shielded the aggregation “hot spot” in H-CDR3, and significantly improved the solubility of CNTO607. The affinities toward IL-13 from modified CNTO607 variants were similar to the natural antibody.
In parallel, Chennamsetty et al. developed a prediction algorithm on aggregation-prone regions of antibodies using a SAP technology.452 The atomistic simulation of a full antibody molecule in an explicit solvent was performed to evaluate dynamic fluctuations and determine the extent of hydrophobic patch exposure on the antibody surface. The aggregation can be subsequently inhibited by mutating particular hydrophobic residues exposed on the surface.
More recently, Liu et al. adopted a solubilization partner strategy to enhance the solubility of a novel heavy and light chain fusion protein, namely single-chain variable fragment (ScFv), against fibroblast growth factor receptor 3. ScFv was attached with Sumo (Small ubiquitin-related modifier) by polymerase chain reaction (PCR) and cloned into the pET-20b vector to achieve the soluble expression.453 In another work, RNA was developed as a molecular chaperone (chaperna: chaperone + RNA), where the soluble expression of receptor-binding domain on Middle East respiratory syndrome-coronavirus was achieved by fusing with the RNA-interaction domain and bacterioferritin.454
5.4. Summary and Prospect
In summary, the solubilization methodologies of MBPs resemble those for transmembrane proteins discussed in previous sections, but exhibit a higher freedom in flexibility due to their less stringent interactions with the lipid bilayer and also the occasional existence of water-soluble forms in their conformations. Besides the commonly used surface residue redesign for lipid exposed amino acids, more direct approaches such as truncations or the swapping of membrane associated segments have also been adopted to realize their solubilization and overexpression, which can be of great assistance in the crystal structure study and functional investigations.409
The task is relatively “easier” where water-soluble proteins are concerned, even though many of them still suffer from solubility issues. Compared to the strict requirements for structural information or the accurate identification of lipid-exposed hydrophobic patches in transmembrane proteins and MBPs, the enhancement of solubility can be more conveniently achieved through tag/chaperone/protein fusion strategies for soluble targets. An alternative aggregation prevention strategy through charge repulsions can also be implemented to achieve this goal. Similar strategies have been adopted in combination with sequence modifications in the solubilization of transmembrane proteins but were rarely sufficient by their own.43,222,267 The analysis of hydrophobic residues or aggregation-prone regions can still benefit this process but is not as critical as membrane-bound variants. When more stringent requirements for protein stability and safety need to be met, such as in monoclonal antibodies, additional measures will need to be taken into consideration.
Although we have covered many traditional approaches to enhance protein solubility, novel methodologies keep emerging with the ever-increasing amount of structural information across the proteome for living organisms and protein structure–function relations, which subsequently contribute to the knowledge-base of solubility limiting factors. It is anticipated that continuously increasing computing power and algorithms will provide us with the essentials to carry out advanced individual protein analysis and enable them to meet stringent criteria in more kinds of applications.
In Table 3, we summarize the in vitro techniques covered in the past few sections and in silico methods to be introduced in the latter section, to provide readers a listing of all relevant resources for protein solubility enhancement and evaluation that are included in this review.
Table 3. In Vitro and In Silico Methods for Protein Solubility Enhancement and Evaluation.
| category | methodology/tool name |
|---|---|
| Expression condition optimization | Reducing speed, lowering temperature, optimizing growth media.100 |
| Protein design based approaches | Site-directed mutagenesis, directed evolution, rational redesign on lipid exposed residues,208,226,254,262 rational design based on segment swapping,239,412 QTY Code,274 encoding gene modification.407 |
| Protein fusion based approaches | Protein partner fusion: SIMPLEx,267 ribosomal protein,434 IDP;435 Solubility enhancing fusion tags: Mal-bp,416 GST,418 TRX,418 SlyD,419 DsbA,421 NusA,91 T7PK,421 Skp,428 HaloTag7,420 CBDengD,426 SEP(C9R),424,425 MOCR;455 Molecular chaperone fusion: DnaK,431 GroEL,430 RS-mTEV,432 Spy.433 |
| Charges-mediated enhancement | negGFP.438 |
| Surface hydrophobic residue/patch identification | Experimental: hydrophobic dye or tracer;46In silico prediction: SAP,48 hPatch.49 |
| Protein solubility prediction | GraphSol,456 ProGan,457 DeepSol,96 PaRSnIP,458 Protein-Sol,93 Cam-Sol,94 PON-Sol,459 ESPRESSO,460 PROSO II,461 CCSOL,462,463 SCM,464 Samak-Wang,465 SOLpro,92 PRSP,466 SoDoPE.95 |
6. Design Function of Soluble Proteins
“What I cannot create, I do not understand.”
-Richard Feynman
Proteins are the fundamental minuscule molecular machines that undertake the majority of functions in living organisms. Significant efforts have been devoted to the elucidation of their physiological properties, structures, and biological functions. However, while native proteins have been highly efficient and indispensable in every aspect of biological processes, they neither occupy the full sequence space nor are they always the optimal biomolecules to undertake a target function. The existence of orphan receptors,468 and certain groups of people who are deficient in certain proteins,469 indicate that nature’s solutions might not always be the optimal ones. With the growing knowledge in structure–function relations for the proteome, scientists have started designing novel protein species with dedicated functions, either with a natural template, or from a de novo approach.
One important prerequisite for engineering new functional proteins is their stability in a predefined environment. It is an underlying consideration that newly designed species need to behave sufficiently well before they can be functionally characterized. The insolubility of recombinant proteins produced in heterologous expression systems often results in nonfunctional biomolecules. While their solubility can be improved by optimizing the expression conditions, such as the temperature, pH, or salt condition in the solution, consideration of the primary sequence during the design phase can also contribute to this goal. In this section, we will review recent progress in functional protein design, and efforts to enhance their stability in solution. The design of water-soluble metal binding proteins and maquettes in catalytic and light-harvesting applications will be discussed in detail. Redesigning native proteins for higher stability while defining their function will also be covered.
6.1. Protein Design with Specific Binding Domain
Starting from the 1970s, advances in de novo protein design have brought considerable successes in making idealized polypeptide structures that did not exist in nature, especially with the exponential increases in computing power and algorithm developments.25,470 The growing knowledge about protein thermodynamics and available crystal structures for native proteins over the past six decades enable protein architects to design de novo species with desired structures, and tailored functions. Early works were primarily based on the physical principles and molecular mechanic force fields on proteins generated by parametric functions. The design process has then been facilitated by integrated packages for protein structure prediction and design such as Rosetta, which consolidated the essential design steps into a single framework in more recent studies.471,472
6.1.1. De Novo Design of Metal Binding Proteins
One important aspect of protein function is to bind metal ion cofactors and form active complexes in various biological processes. These protein variants, namely metalloproteins, account for almost half of the proteome in nature and play important roles in living organisms for structural stabilization, chemical signaling and catalysis.473,474 The de novo protein design provides a feasible pathway to test our knowledge about their structures and functions by reconstructing them in non-native species.475 Such practice is more than simply duplicating native metalloproteins. The design approach can help to elucidate from the top-down important structural features that might have been neglected in the study of native proteins, or produce novel species with improved stability and efficacy beyond the native counterparts.476
The design of metalloproteins has proven to be more challenging than structural proteins due to the variety in number and geometry for metal ions. Metal ion binding sites also serve as nucleation sites to guide the folding of small proteins, such as zinc fingers, where the stable form of the protein complex depends on metal–ligand interactions. The backbone structure and overall folding are restrained by the ligation geometry, energetically accessible side chain conformations, and second-shell hydrogen bonds.25 Pecoraro’s lab designed three-stranded α-helical bundles to study the metal binding sites in the trigonal geometry (Figure 19A).477 The three-coordinate site was engineered into a trimeric model of the peptide TRI, a peptide based on a three-stranded coiled-coil CoilSer. The TRI family has a general sequence of Ac-G(LKALEEK)4G-CONH2 with four heptad repeats containing hydrophobic residues at a and d positions, charged residues at e and g positions to promote hydrogen bonding and salt bridge formations, and helix-inducing or polar residues at the remaining positions. In an earlier effort, Lombardi et al. designed a minimal diiron protein (DF) with C2 symmetry containing four E-two H sites, which can bind two transition metal ions in a four-helix bundle (Figure 19B).475 The structure of DF was confirmed by X-ray crystallography and NMR following the initial report, while robust and saturable catalytic activities were demonstrated (Figure 19C).184 The binding site design was later adopted in other synthetic proteins such as in transmembrane Zn2+ antiporter named Rocker, as discussed in the previous section.377
Figure 19.

Desired geometry of the metal ion-binding site dictates the overall 3D structure in de novo protein design. (A) Trigonal three-Cys site dictates the backbone of a three-helix bundle in the TRI series of peptides. (B) More complex C2 symmetrical site is formed from four E-two H residues, which binds two transition metal ions in a four-helix bundle in the DF series of proteins. A, B: Reprinted with permission from ref (25). Copyright 2020 Cambridge University Press. (C) Milestones in the development of DF proteins. Reprinted with permission from ref (184). Copyright 2019 American Chemical Society.
In the effort from Pecoraro and co-workers, a L16C substitution at the a position was adopted to generate the thiolate site in both the parallel and antiparallel (up–up–down) topologies. The group designed a mercury-binding two- and three-helical bundles to elucidate the interplay between metal and protein conformations, which proved to be advantageous over small molecule models.478 The peptides can be designed to accommodate well-defined and soluble secondary and tertiary structures in aqueous environments, which were used to generate distorted coordination conditions often difficult to achieve in small molecule complexes as they were formed by the spontaneous assembly. A subsequent work reported the de novo design of defined chemistry of Cd(II), Hg(II), As(III), and Pb(II) in thiolate-rich environments, with particular emphasis on controlling the ion coordination numbers.479 When the three-cystine metal-binding site was placed at the C-terminal end of the existing three-helix bundle, a predefined coordination geometry and stable construct was obtained, which can encapsulate heavy metals Hg(II), Cd(II), and Pb(II) with a high affinity and binding constants >107 M–1 determined through titration data.480 The spectroscopic properties of the metal–peptide complexes were similar to those of parallel three-stranded coiled-coils, indicating an agreement between the binding complex and the design model.
Tanaka et al. later conducted the de novo design of peptides which can bind two transitional metal ions simultaneously.481 Two peptides with 30 amino acids, IZ-3adH and IZ-3aH were designed based on the backbone of IZ, [YGG(IEKKIEA)4] with defgabc heptad positions, which formed three-stranded coiled-coils. IZ-3adH had two H residues and can bind Ni(II), Zn(II), or Cu(II), whereas IZ-3aH had one H residue and can bind Cu(II), Zn(II), but not Ni(II). A follow-up iteration IZ(5)-2a3adH had 38 residues including three H and was able to bind Ni(II) and Cu(II) simultaneously in the hydrophobic core.
More recently, the functional activities beyond the catalysis of trimeric coiled-coils have been investigated. Berwick et al. designed a de novo peptide MB1 as luminescent probes (Tb) and magnetic resonance imaging contrast agents in the presence of a trivalent lanthanide ion.482 The coiled-coil was designed based on the sequence Ac-G(IaAbAcIdEeQfKg)xG-NH2 with heptad repeats. The Ln(III) ion was coordinated through three D side chains and three carbonyl oxygen atoms from N residues at the layer above. The W14 indole residue adjacent to the binding site served as a sensitizer for Tb(III) luminescence. In a separate work, the DNA-binding domain from the heat shock protein and the metal ion induced three-stranded coiled-coil were combined to design a new protein with the heat shock element DNA binding function controlled by metal ions.483
6.1.2. Helical Bundle to Bind Complex Cofactors
Beyond the binding of single type metal ions, functional helical bundles were also designed to associate with complex cofactors as catalysis often requires the orchestration of a series of side chains, substrates and cofactors.484 The design of a water-soluble, 62-residue, di-α-helical peptide was recently reported by Polizzi et al. to mimic the key structural features of cytochrome bc1 which accommodates two bis-histidyl heme groups. The peptide assembled into a four-helix dimer with 2-fold symmetry and four parallel hemes.484 Spectral and electrochemical properties comparable to native hemes, including heme–heme redox interaction, were observed from the designed helical bundles. These simplified metalloproteins allowed direct elucidation of deterministic factors for electrochemical properties of heme in native proteins and subsequent rational tuning of the redox potential for cofactors, whereas the positive charges of two ferric hemes electrostatically assist the reduction process.485 The knowledge generated on binding site mechanisms also provides insights in the subsequent studies of actual functional processes ranging from light capture to catalysis.486,487
Polizzi et al. then designed a porphyrin-binding sequence 1 (PS1), which can bind electron-deficient non-native porphyrins at elevated temperatures.488 The design of PS1 used a parametrically generated backbone SCRPZ-2 and allowed flexible-backbone design with sequence optimization for all interior and substrate-binding site residues. The first design variant succeeded without any experimental screening process. PS1 exhibited a high binding affinity toward the insoluble cofactor (CF3)4PZn, and the complex formed within seconds of the mixture, which showed excellent stability under 1% w/v octylglucopyranoside detergent for temperatures as high as 100 °C. The high-resolution structure of holo-PS1 was in sub-Å agreement with the design model. The conformational specificity of PS1 revealed the importance of unifying core packing and binding-site definition simultaneously as a central principle of protein design for ligand binding.
Burton et al. also provided a strategy for the predictable and robust construction of de novo biocatalysts.185 They incorporated a catalytic motif that can promote hydrolytic activities to an engineered protein framework. A de novo designed heptameric α-helical barrel was used as the structural template whereas the C–H–E triads were installed onto each individual helix to form a catalytic center in the assembly. The side chains from C or S residue can act as the nucleophile in the imidazole group whereas a proximal H deprotonates the thiol or hydroxyl moiety for hydrolysis.489 The core-facing a and d positions in heptad repeats with previously hydrophobic L and I residues were targeted for mutation, with 21 possible contiguous d–a–d or a–d–a sites at which C–H–E or E–H–C triads can be installed. The group tested variants with single, double and triple mutations at the target sites. Only the design with full triad incorporation showed a significant increase in catalytic activity compared to the established baseline. X-ray crystal structure analysis on variants with single C mutation revealed free-thiol from the residue with Sγ–Sγ distances of 4.2–4.7 Å, which was too distant to form structure-disrupting disulfide bonds (∼2 Å). Although the catalytic efficiency of the designed helical barrel was still ten times weaker than natural catalysts, the report provided a design framework where the biocatalytic activity can be installed and further optimized in subsequent studies.
Another effort was reported from Baker’s lab, where Tinberg et al. designed specific small-molecule-binding sites to the steroid digoxigenin (DIG) as protein binders with high affinities through the Rosetta algorithm.490 Disembodied binding sites were created by placing amino acids with optimal orientation and geometrical complementarity in a set of protein scaffolds, followed by subsequent side chain optimizations for additional buttressing to define molecular interactions.491 An ideal DIG-binding site contains W or H for hydrogen bond interaction with the polar groups of DIG and steric vdW interactions between Y, F or W and the steroid ring, which is embedded in the designed scaffold. Seventeen variants were selected for experimental verification based on the computed binding affinity, shape complementarity, and the extent of binding site preorganization in the unbound state. A design named DIG10 exhibited the highest binding affinity as determined by the yeast surface display, flow cytometry and isothermal titration calorimetry measurements. The variant also had the most favorable computed protein–ligand interaction energy and preorganized binding sites. Tinberg and co-workers further optimized the design by selected hydrophobic amino acid replacements which resulted in DIG10.1 with a 75-fold increase in binding affinity. Subsequent designs DIG10.2 and DIG10.3 benefited from generating a library of site-directed mutagenesis on interface residues or positions as determined by deep sequencing, the latter of which exhibited picomolar affinity toward the target DIG protein.
6.1.3. Functional Protein Design Based on Natural Scaffolds
Besides the work in de novo metalloprotein design, early successes were reported on functional redesign efforts based on native proteins. Der et al. designed MID1, a zinc-binding dimeric two-helix protein with a connecting loop, which hydrolyzed p-nitrophenol ester or phosphor-ester with good efficiency.492 Rosetta Match was used to screen the design of two-residue zinc binding sites out of 600 known monomeric protein scaffolds. Hits on the same scaffold were combinatorially enumerated and grafted to create a C2-symmetric dimer with two metal sites at the interface. The second chain was rotated around the zinc axis to maintain symmetry while searching for rigid-body alignments with proper coordination geometry but not clashes, followed by the Monte Carlo simulated annealing. The design models were filtered by two primary metrics: computed binding energy excluding zinc contribution, and binding energy per unit of interface area. A final group of eight designs was tested experimentally which resulted in MID1 that functioned well.
Another more recent notable success in enzyme design based on native protein modifications was reported by Song and Tezcan.493 A monomeric redox protein cytochrome bc562 was selected as the starting template, where zinc binding sites were introduced to promote controlled self-assembly into tetrameric structures. The resulting assembly displayed the β-lactamase activity, the primary mechanism of antibiotic resistance, and enabled E. coli cells to survive ampicillin treatment.
The aforementioned peptide designs generally exhibited well-defined structures and a high stability with metal cofactors in aqueous solution. Polar residues were placed at solvent-exposed positions to provide water solubility, while hydrophobic pockets were constructed into the assembly cores.475 Successful engineering of stable and active metalloproteins can enable the integration of various functions into more complicated protein structures.478
6.2. Water-Soluble Light-Harvesting Maquettes Design
Sunlight is the most abundant sustainable energy source available to humanity.494 Natural organisms developed efficient mechanisms for converting light to biological energy. Antenna-protein complexes are used to transfer photons to membrane-bound photosynthetic reaction center proteins that conduct charge separation, drive cellular ion/metabolite transport across the membrane, and initiate ATP synthesis.495−497 The successful modeling and reproduction of antenna-protein complexes can enable novel means of harvesting sustainable energy through biosynthesis.498,499 Great efforts have been made to design artificial light harvesting membrane proteins that can bind a variety of cofactors such as heme, and synthetic tetrapyrroles.496,500 However, similar to transmembrane proteins as discussed in previous sections, water solubility is also an issue for these proteins. Detergents were frequently used for solubilization of both proteins and cofactors.496,501 In this section, we will describe recent advances in water-soluble light-harvesting proteins designed for biophotosynthesis. The ability to precisely control their surface hydrophilicity depending on the target location without disturbing functions serves a perfect example of the integration of both functional and water solubility manipulation in the protein design process. Table 4 summarizes the most common designs for light-harvesting maquettes as discussed below.
Table 4. Primary Amino Acid Sequences and Features of Light-Harvesting Maquettes.
| type | name | sequence | features | ref |
|---|---|---|---|---|
| Hydrophilic maquettes | H10H24 | CGGGELWKLHEELLKKFEELLKLHEERLKKL | Disulfide linked loop between helices | (484) |
| BB | CGGGEIWKLHEEFLKKFEELLKLHEERLKKL | Disulfide linked loop between helices | (504, 505) | |
| BBC16 | C16- | Disulfide linked loop between helices | (504, 506) | |
| CGGGEIWKLHEEFLKKFEELLKLHEERLKKL | ||||
| HP1 | CGGGEIWKQHEEALKKFEEALKQFEELKKL | Disulfide linked loop between helices | (507, 508) | |
| HP7 | GEIWKQHEDALQKFEEALNQFEDLKQL- | Disulfide links two loops | (509, 510) | |
| GGSG CGSGG- | ||||
| EIWKQHEDALQKFEEALNQFEDLKQL | ||||
| HP8 | CGGGEIWKLHEEFLKKFEELLKLHEERLKKL | Disulfide linked loop between helices | (511) | |
| PS1 | EFEKLRQTGDELVQAFQRLREIFDKGDDDSLE | Porphyrin binding | (488) | |
| QVLEEIEELIQKHRQLFDNRQEAADTEAAKQG | ||||
| DQWVQLFQRFREAIDKGDKDSLEQLLEELEQA | ||||
| LQKIRELAEKKN | ||||
| Amphiphilic maquettes | AP0 | EIWKLHEEFLKKFEELLKLHEERLKKLLLLALLQLLLALLQLGGC- | Cofactor binding | (513) |
| AP1 | SSDPLVVAASIIGILHFILWILDRGGNGEIFKQHEEALKKFE | Cofactor binding | (512, 514) | |
| AP2 | IIMAIAMVHLLFFFEIWKEFEEALKKFEEALKEFEELKKL | Cofactor binding | (512, 515) | |
| AP3 | CGGGIIMAIAMVHLLFLFEIWKQFEEALKKFE | Disulfide linked loop between helices and cofactor binding | (512, 515) | |
| Proteins 1 | CGGGEIWKQHEEALKKFFAFHFILPFIIMAIAWVHLLFLFGEGL | Disulfide linked loop between helices | (516) | |
| Proteins 2 | GEIWKQHEDALQKFFALLLLLALLLLLALLLHLLAFE GGSGGGSGG | Single sequence peptide | (516) | |
| KFLLLLALLALLLLALLLHLLAFWEALNQFEDLAKQ GGSGGGSGG | ||||
| EIWKQHEDALQKFFALLLLLALLLLLALLLHLLAFK GGSGGGSGG | ||||
| EFLLLLALLALLLLALLLHLLAFWEALNQFEDLAKQ | ||||
| PRIME | AIYGILAHSLASILALLTGFLTIW | Porphyrin binding | (393, 518) | |
| C-type cytochrome maquettes (CTMs) | C2 | GMTPEQIWKQHEDALQKFEEALNQFEDLKQLG | Single sequence peptide | (520, 521) |
| GSGSGSGGECIACHEDALQKFEEALNQFEDLK | ||||
| QLGGSGGSGGSGGEIWKQHEDALQKFEEALNQ | ||||
| FEDLKQLGGSGSGSGGEIWKQHEDALQKFEEA | ||||
| LNQFEDLKQL | ||||
| C45 | GMTPEQIWKQFEDALQKFEEALNQFEDLKQLG | Single sequence peptide | (486) | |
| GSGSGSGGEIWKQFEDALQKFEEALNQFEDLK | ||||
| QLGGSGGSGGSGGECIACHEDALQKFEEALNQ | ||||
| FEDLKQLGGSGSGSGGEIWKQFEDALQKFEEA | ||||
| LNQFEDLKQL |
6.2.1. Hydrophilic Maquettes Design
Maquettes are common templates for synthetic proteins that can be given desired functional features from native proteins, or peptide-based synthetic analogues.485 The typical structure of a maquette is a four-helix bundle, with each helix 30–40 amino acids in length, and with central histidine residues for ligation.184 The bundle contains nonpolar residues in the core region and polar residues for helical interactions on the surface.501,502 Three nonpolar residues at a, d, and e positions drive the four-helix bundle formation.503
The first report of light-harvesting maquettes was designed in Dutton’s lab in 1994. A water-soluble dihelical peptide named [H10H24]2 was synthesized to accommodate two bis-histidyl heme groups.484 The initial design comprised a 27-residue helix and a flexible tether (CGGG), which formed interhelical disulfide bonds and resulted in a four-helical bundle in aqueous solution. H residues were placed at positions 10 and 24 of each helix to provide axial ligands for heme. The [H10H24]2 maquettes exhibited predominantly α-helical structure and micromolar to submicromolar KD values for heme binding.
Subsequently, a more amphiphilic peptide derivative named BB was designed based on [H10H24]2, with L6I and L13F mutations.504,505 The four-helix bundle coordinated hemes by bishistidyl ligation to form the heme protein maquette (heme2-α-SS-α)2, with a dissociation constant ∼10–12 M in the aqueous solution. Furthermore, one palmitoyl (C16) chain was added to the N-terminus of BB to enhance its amphiphilicity and stability at the interface, which resulted in a soluble BBC16 peptide. The reorientation of the ZnPPIX-BBC16 complex in the Langmuir monolayer changed under different surface pressures.504,506
The de novo hydrophilic (HP) four-helix bundle named HP1 was designed based on the X-ray crystal structure of the apomaquette L31M, which is an apomaquette derived from the structurally heterogeneous tetraheme-binding H10H24 prototype (Figure 20A).507,508 The second heme-binding site was removed by replacing H24 with F and delete R27, to simplify the holomaquette structure and avoid steric clashes between bound hemes. L9 and L23 were replaced by Q to attenuate the surface hydrophobicity, and F13 and L20 were converted to A to increase the conformational specificity. The redesigned HP1 maquette can dissolve in aqueous solution, and form an antiparallel symmetric diheme four-helix bundle via disulfide bridges. Yet the protein was susceptible to water access when heme was ligated. An iteration, HP7 was then designed by connecting the loops via a disulfide link (“candelabra” motif) and changing the sequence of surface amino acids.509 HP7 became progressively structured upon heme binding and adopted unique structures with hemes other than protoporphyrin IX. The time of hydrogen/deuterium exchange in the backbone amides of HP7 increased from minutes to hours, indicating a significant blockage of water access to the hydrophobic domain compared to HP1.510
Figure 20.

(A) Schematic representations of HP1 and HP7 where the hydrophobic interior is purple, the alternating positive and negative charges on the protein surface are shown as blue and pink, and glutamate residues are red. HP1 is oriented in an antitopology with a poorly defined intermonomer interface that allows water access into the core. The candelabra motif of HP7 prohibits water access to the core through the use of hemes and a disulfide bond to fix the intermonomer interface. Reprinted with permission from ref (510). Copyright 2008 Portland Press. (B) Schematic of the AP family design and assembly. Positive residues are colored blue, negative is red, polar uncharged are yellow, and nonpolar residues are purple. The incorporation of heme is a brown box. The HP1 sourced HP domain was linked to an LP domain by a flexible linker (AP1, top) or directly (AP3, bottom). AP maquettes can readily assemble with detergents to form micelles or with lipids to form membranes. Reprinted with permission from ref (512). Copyright 2005 American Chemical Society.
Another iteration in redesign, namely HP8, transformed a non-native peptide into native-like proteins derived from considerations in biological evolution coupled with 1H NMR as a selection criterion.511 The variant with L6I and L13F mutations at d position exhibited improved stability and increased the chemical shift dispersion.508,511
6.2.2. Amphiphilic Maquettes Design
To build a platform with functions across an interface, amphiphilic (AP) maquettes were designed by combining the hydrophilic and originally lipophilic transmembrane domains with a nonpolar core or a soft interface between polar and nonpolar media.512 The redesign of external residues in a particular module could also change its solubility in membrane.359 Solubilization of water-insoluble amphiphilic peptides and the assembled maquettes need common detergents near or above critical micellar concentration.
AP0 and AP1 were AP maquettes that incorporated the heme binding site derived from HP1 maquette with two different sequences for the LP domain (Figure 20B). The hydrophobic segment of AP0 was based on the heptad repeat in the de novo designed LS2 proton channel,513 while AP1 was based on the transmembrane domain of the M2 proton channel of the influenza virus.512,514 AP0 and AP1 both possessed bis-histidyl metalloporphyrin binding sites in the hydrophilic domain of the four-helix bundle.515 In addition, the heme bH binding site sequence (188–200) from bovine mitochondrial cytochrome bc1 was used as the LP domain of AP2 and AP3 maquettes, with hydrophilic segments from HP1 having the loop region deleted or additional H to F mutations, respectively.512 Both AP2 and AP3 possessed bis-histidyl metalloporphyrin binding sites within the hydrophobic domain that could be dissolved in an organic solvent such as methanol, but exhibited poor solubility and required detergents in water.515 Chlorophylls, bacteriochlorophylls, and their metal-substituted analogues did not bind to HP1, AP0, and AP1 maquettes. Yet LP domains of AP2 and AP3 significantly enhanced the BChl solubility and affinity in the presence of detergents.
Goparaju et al. carried out the de novo design on two adaptable and amphiphilic four-helix transmembrane protein frames (Protein 1 and Protein 2) to recreate and modulate core transmembrane bioenergetic electron-transfer functions.516 Protein 1 was a symmetric design with four separate and identical helices, while protein 2 was a single-chain four-helix transmembrane protein. The transmembrane domain of protein 2 is rich in L and A due to their high α-helical propensity, while the L to A ratio matched the profile in cytochrome b helices.
The roles of individual amino acids in the structure–function relation was investigated by Farid et al. by designing elementary single-chain maquettes for diverse oxidoreductase functions.517 Maquettes with several lengths yield monomers with no sign of multimers or aggregates below 150 μM. The cofactor binding affinities and redox midpoint potentials varied with minimal change, sometimes as small as one amino acid.
Furthermore, an amphiphilic protein PRIME was designed based on the backbone of a water-soluble multiporphyrin binding peptide, which binds two Fe(II/III) diphenylporphyrins in a bis-His geometry from a D2-symmetrical bundle.393,518 Amphiphilic PRIME is insoluble in water and could assemble with Fe(II/III) diphenylporphyrin cofactor in the presence of detergent. The details of this work are covered in Section 4.2.3.
6.2.3. C-type Cytochrome Maquettes Design
C-type cytochromes are a ubiquitous class of electron transfer proteins and oxidoreductase enzymes that contain a variant of iron protoporphyrin IX (heme C).519 A synthetic c-type cytochrome maquette named C2 can covalently graft heme onto the protein backbone in vivo after the post-translational modification in E. coli.520 The N-cap sequence was added to the N-terminus of the protein to increase its thermal stability, and the consensus CIACH was selected as the c-type incorporation motif. The C2 maquettes were soluble in water, and produced a nascent electron transfer chain for defined vectoral electron transfer. In addition, the C2 scaffold showed flexibility and structural plasticity to create a suite of in vivo-assembled mono- and diheme above the maquettes, which was beneficial for subsequent de novo design on oxygen-activating oxidoreductases.521
A subsequent iteration was designed for water-soluble c-type cytochrome maquette with heme covalently appended to helix-4 (C4).486 H to F mutations were conducted (C46) at the second noncovalent tetrapyrrole-binding site to restrict conformational flexibility and improve core rigidity, which also prevented binding of a second tetrapyrrole and increased the protein’s Tm. The distal H on helix-2 was also replaced by F to obtain the mono histidine-ligated c-type cytochrome maquette (C45). C45 exhibited kinetics that surpassed natural peroxidases, and was resilient against both elevated temperatures and organic solvents.
6.2.4. Water-Soluble Cofactors Design
In addition to the design of water-soluble maquettes, the modification on the insoluble tetrapyrrole cofactors is also needed to achieve the solubilization of light-harvesting protein complex.485,488,522 Therefore, the binding of Zn tetrapyrroles was explored with different patterns of polar and nonpolar substitutions on the four-helix maquettes.501 The maquette frames with two H residues at interior positions 7 and 112 were constructed following the principles of protein folding. The solubility of the maquettes was tuned by adjustments at the meso positions. Thirteen Zn(II) tetrapyrroles were designed and tested, some of which exhibited higher solubility and enhanced binding toward maquettes in the aqueous solution. An artificial photosystem was then designed based on the amphiphilic Zn(II) tetrapyrroles binding with do novo maquettes, which were immobilized onto a TiO2 electrode in CHES buffer.523 Improved photo voltage was observed with a lower porphyrin requirement and more efficient photocurrent generation from a smaller dye loading.
6.2.5. Light-Harvesting Complex Design
In addition to cofactors, the reconstitution of core light harvesting complexes also need detergents, which were constructed based on the histidine interactions between light harvesting proteins and cofactors.524,525 Removal of hydrophobic domains and incorporation of Chl and BChl into de novo maquettes are effective approaches to design the water-soluble light-harvesting complexes.496,526 Zeng et al. constructed hybrid fusion proteins from water-soluble fragments of ApcE(1–240/Δ77–153) and HP7.527 The hydrophobic loop of ApcE(1–240) between residues 80 and 150 was removed while a modified HP7 sequence was fused to the N- or the C-terminus of ApcEΔ, or inserted between residues 76 and 78. The resulting fusion proteins were soluble in water, and complexes exhibited significant intramolecular fluorescence resonance energy transfers with yields ranging from 21–50%. In another effort, a de novo protein PS1 was designed based on the D2-symmetrical parametrized backbone of SCRPZ-2 to bind a water-insoluble cofactor (CF3)4PZn.488 The complex formed within seconds of adding (CF3)4PZn from the organic solution to aqueous PS1 at temperatures up to 100 °C.
6.3. Redesigning Native Proteins
With continuous successes in designing do novo proteins with dedicated reaction centers, scientists turned their interests to functions that involved more complicated domain structures. One of the recent efforts was reported by the Baker group on redesigning the backbone of interleukin-2 (IL-2) for higher stability and more specific functions.528 IL-2 is one type of human cytokine that regulates white blood cell activities and is considered a key central immune cytokine for cancer treatments.529 However, the cytokine can interact with multiple receptor subunits (IL-2Rαβγc) and has nonideal structural features that compromise its stability, both of which hinder its therapeutic uses, primarily due to interactions with IL-2Rα. Silva et al. designed an IL-2 mimic with only essential binding sites toward the target receptor without otherwise any resemblance to the native protein in topology or the amino acid sequence. As shown in Figure 21, the de novo IL-2 mimic idealized the recapitulated binding interfaces with a parametric construction of disembodied helices and knowledge-based loop closure. In native proteins, the H3 and H4 helices interact with the β- and γ-subunits of the IL-2 receptor, respectively, while the H1 helix interacts with both β- and γ-subunits. The H2 helix holds the above three helices in place and has an irregular structure that interacts with the α-subunit, which was targeted for redesign.
Figure 21.

(A) Structure of human IL-2 (HsIL-2 in the graph) in complex with its receptor IL-2Rαβγc (surface representation) (PDB ID: 2B5I). (B) Designed mimics have four helices; three (blue, yellow, and red) mimic IL-2 interactions with IL-2Rβγc, whereas the fourth (green) holds the first three in place. Top, first iteration: each of the core elements of IL-2 (helices H1–H4) were independently idealized by the assembly of four residue clustered protein fragments. Bottom, second iteration: the core elements were built using parametric equations that recapitulate the shape of each disembodied helix, allowing changes in the length of each helix by up to ±8 amino acids. (C) Pairs of helices were reconnected using ideal loop fragments. (D) Combinations of helix hairpins in C to generate fully connected protein backbones. (E) Rosetta flexible backbone sequence design. (F) Binding and activity of selected designs (solid symbols). The green arrow originates at the parent of the optimized design Neo-2/15. Reprinted with permission from ref (528). Copyright 2019 Springer Nature.
Two iterations of the design were made. In the first iteration, each of the helices was independently idealized by assembling four residue clustered protein fragments, while the fragment-derived loops were used to generate fully connected backbones. A Rosetta combinatorial flexible backbone sequence design was performed for each backbone, and a more regular structure for H2 (H2′) than in IL-2 was obtained. The second iteration improved on the stability of the models by repeating the computational design protocol on the backbone with the highest affinity in the first-round design and coupling the loop building process with a parametric variation of the helix lengths (up to ±8 amino acids). The Neo-2/15, a 100-residue protein, was obtained by the second iteration, with highest affinity for both human and mouse IL-2Rβγc. It can be expressed in the prokaryotic system while its complex structure with mouse IL-2Rβγc aligned well to the human IL-2 receptor.530 The in vivo therapeutic efficacy of Neo-2/15 was tested in B16F10 (melanoma) and CT26 (colon cancer) mouse models. Dose-dependent delays of tumor growth in both cancer models were observed in single agent treatments. The rationale in the redesign of IL-2 functional mimics can potentially be widely applicable for developing next-generation therapeutics for enhanced stability, robustness and specific interaction surfaces.
6.4. QTY Design of Chimeric or Truncated Chemokine Receptors
We introduced in Section 3.4 the recently reported simple and powerful tool named QTY code for the water-soluble GPCR design.274 It is a useful methodology for functional investigations of transmembrane receptors under detergent-free condition, as native receptors require detergents which can potentially distort their structures. When combined with methods such as chimeric design or truncation, previously unattainable fine-tuning of receptor functions can be achieved.
Qing et al. designed chimeric proteins by switching the N-terminus and three EC loops between different chemokine receptors to help elucidating the native ligand interaction mechanism.277 Specifically, CCR5QTY and CXCR4QTY were redesigned to construct chimera A (replacing EC loops of CCR5QTY with GS linkers of the same length) and chimera B (replacing N-terminus and EC loops of CCR5QTY with the N-terminus and EC loops of CXCR4), as shown in Figure 22A. The chimeras were expressed with high yields in the E. coli system, exhibited α-helical secondary structures, and showed similar Tm profiles to CCR5QTY, indicating similar folding in the backbone although the EC components were different.
Figure 22.

(A) Schematic of the chimera receptor designs. CCR5QTY’s seven-transmembrane regions (yellow) and IC loops were chosen as the backbone of designs. In Chimera A (bottom left) the three EC loops of CCR5QTY were replaced by GS linkers (red) with the same lengths (upper, yellow loops), but CCR5QTY’s N-terminus (yellow line) was unchanged. In Chimera B (bottom right), the N-terminus and three EC loops of CCR5QTY were replaced by those of CXCR4QTY (green). Reprinted with permission from ref (277). Copyright 2019 National Academy of Sciences. (B) Schematic illustration of possible ligand interactions for truncated CXCR4 and CCR5 receptors. The ligand-binding motifs in the N-terminus and three EC loops are simplified and represented with cartoon blocks. Reprinted with permission from ref (279). Copyright 2020 Qing et al.
Functionally, chimera B exhibited reduced (3-fold) affinity to CXCL12, which is the natural ligand of CXCR4, and significantly decreased (20-fold) affinity to CCL5, which is the natural ligand of CCR5. Chimera A showed an 8-fold decrease in CCL5 affinity and no affinity toward CXCL12. The results agreed with previous computational models on ligand docking and helped to illustrate the relative contributions from N-terminus, EC loops and transmembrane regions for interactions in native receptors.531,532 The findings also prove the feasibility of constructing hybrid proteins, which do not exist in nature, that can integrate and fine-tune functions from multiple receptor templates with a rigid backbone structure.
A more recent work reported the modification of chemokine receptors with significant sequence deletions.279 On the basis of the yeast-2-hybrid screening and the convenient transition between native and QTY variant transmembrane receptors, Qing et al. found that the affinities toward respective ligands from CXCR4QTY and CCR5QTY were still retained with up to 58% loss of amino acid sequences. It was proposed that despite the loss of many domains and the inevitable change of receptors’ conformation, the remaining sequences still contained segments capable of capturing their native ligands, albeit with reduced affinities (Figure 22B). Reconverted non-QTY variants of truncated receptors can be produced in HEK293T cell and inserted onto the membrane, where they negatively regulated the function of full-length receptors, or showed a reduced Ca2+ signaling at an elevated ligand concentration for one of the truncations. Considering the producibility of QTY receptors in E. coli, the truncation approach can potentially enable scale-up production of minimal GPCRs for therapeutic or bioelectronic applications.
6.5. Summary and Prospect
In summary, the protein design with targeted functions has profound implications and enormous potential in diverse areas of biology, chemistry, materials and medicine. There are several approaches to realize these goals. Whereas the common “bottom-up” strategy builds on natural templates by optimizing local structures and global topologies with given functional motifs, a de novo “top-down” approach is also widely adopted, via selecting or building a simplified stable scaffold, and installing reaction centers or grafting known functional motifs in it.533 Both strategies can endow the designed protein with target folding and desired functions. The solubility is an underlying important aspect, with considerations on both the stability of designed proteins and the active complexes in environments required to perform functions, such as for enzymes, where minor changes in the conformation can disrupt the whole catalytic process. During our review, it was noted that multiple rounds of structural optimizations or screenings were usually conducted to identify final designs which function biophysically well enough in solution before moving on to activity characterizations. The stability enhancement itself is sometimes another design goal. Many design variants also exhibited high thermostability against elevated temperatures. This may be due to the relatively simple structures and well-defined interactions within the molecules or assemblies, although such properties are rarely observed in native proteins. The methodologies and rationale adopted in designing novel species contribute to our understanding of native proteins while providing biomolecules with extraordinary properties not available in nature that can be used in numerous applications.
7. Novel Structures, Assembly, and Interaction Designs
One important result of our growing knowledge in protein interactions is the ability to design higher-order assemblies beyond individual structures with well-coordinated placement of amino acids. More delicate interactions need to be considered with stringent interaction patterns to ensure the successful design of complexes that might otherwise fail due to residue mismatch at interfaces and subsequent energy penalty for deviating from the ideal conformation. The accurate design of specific geometries for engineered proteins with arbitrary size, shape and function remains an exciting and challenging prospect for protein scientists.534
In this regard, we select and review characteristic higher-order protein assemblies of recent reports on naturally occurring but previously difficult to reproduce, or newly discovered structures in protein structural biology. Besides the general focus of this review, that is, α-helix based designs, we will also briefly cover β-strand or β-sheet based structures in this section, but only to the extent where sequence determined interactions and designs of solubilities are concerned. More systematic reviews of this class of proteins can be found in previous publications and will not be the focus here.535−537 Following that, a dedicated section will be presented on the de novo design of delicately tailored multidimensional assemblies, primarily through the Rosetta algorithm, which can serve as the structural basis for future design of functional complexes. Finally, the role and design of hydrogen bond networks and polar cores in either water-soluble or transmembrane proteins will be discussed.
7.1. Novel Structures Based on α-Helices
7.1.1. Water-Soluble α-Helical Barrels
Coiled-coils are among the most well-characterized structures in the protein design field, which can be conveniently generated by Crick’s parametric equations and share relatively straightforward sequence repeats.538 Multimeric coiled-coils from dimers to tetramers have served as ideal templates on which a variety of structural and functional proteins have been engineered, as discussed in previous sections. On the other hand, the higher order oligomerization states of coiled-coils have long been recognized for their potential channel-forming capability and convenience to install functions in the central pore, but it was not until recently that the accurate design of these α-helical barrels could be readily achieved.539
The first report of this type was from Liu et al. in 2006, who designed a seven-helix pseudo coiled-coil based on the GCN4 leucine zipper (PDB ID: 2R2V).540 The group replaced all eight e and g position residues in the heptads by A, while keeping other positions intact, and was able to obtain a water-soluble helical heptamer named GCN4-pAA (PDB ID: 2HY6). The interfacial A residues promoted close contacts between adjacent helices to associate into a left-handed superhelix. However, the crystal structure study revealed an offset by one amino acid between adjacent helix pairs which resulted in a deviation of one full heptad between the first and seventh helix in a closed heptamer. The barrel consisted of a 7 Å central channel lined with core hydrophobic side chains while N17 formed a buried hydrogen bond network that also helped to stabilize the assembly.
The Woolfson group then led efforts to design blunt end soluble α-helical barrels. Zaccai et al. first reported the realization of a hexametric peptide assembly by CC-Hex, derived from a de novo designed 32 amino-acid parallel tetrameric coiled-coil CC-Tet (PDB ID: 3R4A).541 The design was based on extending hydrophobic interactions to previous charge-complementary interfaces, namely K at the e position and E at the g position, as shown in Figure 23A. Since the e and g sites were progressively being buried with the increasing oligomeric state, swaps between A at the b position and K at the e position were adopted in the CC-Hex design to establish new peripheral KIH interactions. The substitutions resulted in two complete offset heptads, namely LxxxAxx and xxxIxxE, in the abcdefg sequence, which were believed to cooperatively stabilize the oligomerization. The hexamer contained a 6 Å central channel that allowed the passage of water molecules despite the hydrophobic inner linings of methyl groups from L and I. The design also tolerated the incorporation of charged D or H in lieu of L24. The stoichiometric mixture of L24D (PDB ID: 3R46) and L24H (PDB ID: 3R47) variants resulted in a symmetric heterohexamer with alternating helices, where H and D residues tilted toward each other to form internal hydrogen bond contacts. However, it was later indicated that CC-Hex’s oligomeric state was prone to change with polar mutations.542 They tested alternative polar residues at the L24 site, which resulted in multiple accessible coiled-coil conformations with similar energy states, including parallel and antiparallel tetramers, open barrel and collapsed hexamers, and intermediate equilibrium states controlled by the solvent pH. The structural plasticity of CC-Hex rendered charged residues especially disfavored in the parallel barrel state, which switched the assembly to tetramers for a tighter packing.
Figure 23.

(A) Helical wheel representation for parallel four-helix coiled-coil. The uppermost wheel carries the sequence used as the basis for the de novo coiled-coil CC-Tet. Residues are listed using single-letter codes. Reprinted with permission from ref (541). Copyright 2011 Macmillan Publishers Ltd. (B) Left: Helical-wheel diagram for a type II pentamer, illustrating heterotypic interfaces between cdga and deab. The approximate primary contacts in α-helical barrels are c–b′, d–e′, g–a′, and a–d′. Key geometric parameters are shown: coiled-coil radius (r), oligomeric state (N = M + 4), and helical offset (ω1). Right: Section through a coiled-coil pentamer crystal structure (PDB ID: 1MZ9). (C) X-ray crystal structures (top and middle) and conserved packing of L (a, red) and I (d, green) residues with steric variation of e and g residues for three de novo α-helical barrels (bottom). From left to right, CC-Pent (PDB ID: 4PN8), CC-Hex2 (PDB ID: 4PN9), and CC-Hept (PDB ID: 4PNA). B, C: Reprinted with permission from ref (160). Copyright 2014 The American Association for the Advancement of Science. (D) Optimal template geometry designed to target the SWCNT surface (the array of adjacent benzenoid rings in black illustrates the helical pattern of the SWCNT). Reprinted with permission from ref (181). Copyright 2011 The American Association for the Advancement of Science.
Subsequently, Thomson et al. improved the design by developing a parametric computational framework to deliver water-soluble α-helical barrels with tunable oligomerizations, including pentamers, new hexamers, and a heptamer.160 In the new approach, the oligomeric state was defined by the angular offset between interfaces of hydrophobic seams, as shown in Figure 23B. Sequences were selected based on a scoring system to encode these hydrophobic seams. Destabilizing polar residues were removed at the a and d positions, whereas one hydrophobic residue was positioned at the e or g position. With multiple rounds of screening, 12 pentameric sequences, seven hexametric sequences, two heptameric sequences and one octameric sequence were experimentally tested, among which four pentamers, six hexamers and one heptamer accommodated target assemblies (Figure 23C). The crystal structures for selected variants (CC-Pent, PDB ID: 4PN8; CC-Hex2, PDB ID: 4PN9; and CC-Hept, PDB ID: 4PNA) were resolved and aligned well with design models with low RMSDs. These α-helical barrels and their interior linings were more robustly adaptable to sequence mutations and were widely adopted by multiple groups for subsequent rational design of functions either in soluble form or embedded in the membrane lipids, to carry out enzymic activities,185,543 and serve as small molecule receptors544 or conducting nanopores.174,390 The hydrophilicity of the barrels was tuned primarily by changing the exposed f position residues to either hydrophobic or hydrophilic amino acids in the membrane and soluble designs, respectively.
An alternative effort was contributed by Grigoryan et al., who provided a general framework for parametrizing the backbone of helical bundles with arbitrary orientations using modified Crick equations, as well as guiding the design of de novo coiled-coils.545 The method was used to delineate sequence spaces to highly restricted designable conformation clusters, representing a 160-fold reduction in geometrically feasible structures, whereas most of the natural coiled-coils were found close to the idealized Crick backbone within 1 Å RMSD. The rationale was then utilized by the same group to design helical bundles to form a tubular structure that can wrap around and solubilize single-wall carbon nanotubes (SWCNT), which featured both surface recognition and favorable interhelical packings.181 One of the designs, HexCoil-Gly, accommodated a tentative antiparallel hexametric state with longitudinal continuum, as shown in Figure 23D. However, the peptide only assembled in the presence of SWCNTs and its crystal structure was not resolved. Similar parametric models were then adopted and extended by Huang et al. to design hyper-thermostable helical bundles with various geometries by an automated process.546 Helix backbones were generated with the Crick’s equation, optimized in sequence energy function and connected by loop building. Several three-, four-, and five-helix bundles were obtained and closely matched their design models. The capability to accurately design high order α-helical bundles with a central channel had a considerable impact in biotechnology, particularly in sensing and nanopore applications.
7.1.2. Supramolecular Fibrils with α-Helical Subunits
Aside from the lateral bundle association, α-helices can also be assembled into high aspect-ratio fibrous structures.547 One characteristic native protein exhibiting this behavior is keratin, an abundant protein source from living organisms and widely used as wound-healing agents.83,84 They contain highly conserved structural features including N-terminal, C-terminal loops and a central rod domain including four α-helices connected by three linkers.548 Keratins associate into continuous filaments by interactions between “sticky ends” at the helical regions, primarily through disulfide bonds with additional contributions from hydrogen bonds and vdW interactions.549,550 Similar end-to-end connections were adopted by Pandya et al. to build fibrils through staggered interactions from flanking ion pairs in helical coiled-coils.551 A computational illustration for this incremental assembly is shown in Figure 24A. While the fibers spanned several hundred μm in length, their diameter appeared to be thicker compared to helical dimers, indicating simultaneous unspecific lateral associations. The rationale was also utilized in Wagner and Fairman’s design but with hydrophobic interfaces.552 Moreover, the misalignment of the seven-helix bundle design based on GCN4 as discussed in Section 7.1.1 provided an ideal model to engineer noncovalent interactions into a lock-washer structure for 1D nanotubes with a hollow core, as shown in Figure 24B.164 A similar effort was conducted on blunt-end CC-Hex with introduced electrostatic interactions at each terminus of the barrel, namely, positive charges at N-terminus and negative charges at C-terminus, to align the core channel of bundles and obtain protein nanotubes with a high aspect-ratio.163 Nambiar et al., on the other hand, fabricated a rectangular shape, natural crystal-like structure with trimeric α-helical bundle single units by not only designing interactions at the end of the helix but also installing aromatic moieties in the middle region to induce π-stacking and lateral associations (Figure 24C).553 Such types of interactions, which include hydrophobic, metal–ligand, and electrostatic, can also assist the crystallization of target proteins when they are not strong enough to induce the formation of elongated fibers.554
Figure 24.

(A) Computer modeling of the designed self-assembling fiber SAF-p1 (colored yellow-to-red from the N- to the C-terminus) and SAF-p2 (colored blue-to-cyan from the N- to the C-terminus) to form two strands of a staggered, parallel, coiled-coil fiber (left). Negatively charged E (red) and positively charged K (blue) residues form complementary charge interactions (right). Reprinted with permission from ref (551). Copyright 2000 American Chemical Society. (B) Lock-washer structure derived from the GCN4-based seven-helix bundle (PDB ID: 2HY6) in helical nanotubes. Blue and red surfaces represent N- and C-terminus heptads at the interfaces. Reprinted with permission from ref (164). Copyright 2013 American Chemical Society. (C) TEM image with inset depicting the coiled-coil trimer packing within the microstructure, and cross-sectional view of hexagonal close-packing. Reprinted with permission from ref (553). Copyright 2018 American Chemical Society. (D–G) Design of soluble cross-α amyloid fibrils. Reprinted with permission from ref (162). Copyright 2018 Zhang et al. (D) Structure of α-amyloid assembly αAmS (PDB ID: 6C4Z). The N- and C-termini are colored in blue and red, respectively. (E) Three types of helix–helix interfaces with small-residue packing exist in the αAmmem (PDB ID: 6C4X). A17 (magenta), A13 (green), and S11 (orange) are involved in the interhelical packing with larger hydrophobic residues (white sticks) occurring at positions filling the space as helices diverge from the point of closest approach near small residues. (F) Illustration of interhelical d–d′, g–g′, and e–e′ interfaces for the amyloid-like structure with a parallel dimer as the subunit. The small residues and the corresponding interfaces in the helix wheels are boxed. (G) Designed sequences for water-soluble amyloid-like structures compared to αAmmem and sequence changes between the crystal structures of αAmmem (top) versus αAmS (bottom) in the ball-and-stick representation. Hydrophobic residues on the surface of αAmmem are colored green, while the designed residues at the same locations of αAmS are colored cyan and pink for positively and negatively charged, respectively. The mutation on A11 at e–e′ interface is varied to examine its size effect, as shown in red in αAmG (PDB ID: 6C4Y), αAmA, αAmS, αAmL (PDB ID: 6C51), and αAmF. The synergistic effects of varying three small residues to L are tested by αAm3L. A nonaggregating water-soluble αTet (PDB ID: 6C52) is also designed. (H) Six representative designs of repeat protein assembly with design models (top) and computed energy landscapes (bottom). All six landscapes are strongly funneled into the designed energy minimum. Reprinted with permission from ref (559). Copyright 2011 Huang et al.
While fibrils made of parallel α-helices seems to be a reasonable deduction following the widespread cross-β structures, its existence in nature was not discovered until recently, although several human-origin proteins such as ankyrin repeat domains show reminiscent interactions.555 An α-helical peptide from Staphylococcus aureus, namely PSMα3 (PDB ID: 5I55), was found to accommodate this cross-α amyloid-like spiral structure through tight mating, whereas the helix dimers aligned perpendicular to the fibril axis.556 The architecture served as a physical barrier of rigid microbial biofilms that contributed to the resilience and resistance of the organism.
A soluble variant of the 25 amino-acid long Rocker peptide discussed in Section 4.2.3 was then designed by Zhang et al. to reproduce this structure in vitro and obtain characterizable cross-α fibril assemblies.162 The transmembrane analogue αAmmem (PDB ID: 6C4X) was an antiparallel helix dimer that can form a counterclockwise twisted cross-α fibril with two tightly associated layers (Figure 24D). High geometric complementarity with KIH interactions presented in the side chain packing to ensure the longitudinal extension. Small A17, S11 and S13 residues helped to mediate close interhelical contacts at d–d′, e–e′, and g–g′ interfaces, respectively, with a left-handed crossing angle between 15 and 20° to account for the spiraling shape (Figure 24E,F). This unique tristabilization mechanism resulted in straight rather than curved helices.
The water-soluble peptides were designed by substituting solvent exposed nonpolar residues to polar or charged E, K, and R, as underlined in Figure 24G. Core residues in interfaces and nonstructure defining residues from αAmmem were kept intact. The soluble peptides showed a crystal structure comparable to the transmembrane template. Additional mutations on A11 were then conducted to determine the role of e–e′ interaction on the longitudinal assembly. Small G, A, and S residues at A11 position along with A11L mutation all resulted in observable fibril formation, while the substitution to larger F residue or L placements on all three interfaces resulted in amorphous aggregations. Small residues also had minimal impact on the assemblies’ rotational angle, varying from 18 dimers/turn to 20 dimers/turn. However, the A11L substitution altered the single unit of fibrils to antiparallel four-helix coiled-coils that was longer and wider but more loosely packed and can dissociate into isolated forms with further solubilization. In this case, soluble peptide designs enabled scientists to tune and investigate key interactions for newly discovered protein structures.
Although Zhang’s work represented the first report on strictly defined, longitudinally extended, cross-α fibril structures, designs with similar lateral association of α-helices have been reported prior by Brunette et al. using tandem repeat proteins as components.557,558 A fully automated program was used to design protein repeats with helix–loop–helix−loop motif that can pack into superstructures through interhelical associations with defined curvature and rise.559 The starting backbone conformations were generated through the Monte Carlo fragment assembly with symmetry preservation, followed by the all atom sequence optimization to find low energy sequences with good core packing. A pseudo energy term was added to penalize undesired geometries. Sampled conformations mapped out an energy landscape showing the design trajectories. In the second-round design, exposed hydrophobic residues in N- and C-terminal repeats were switched to polar residues for better solubility. Figure 24H shows several representative designs and their corresponding energy landscapes with a funnel minimum. Out of 83 experimentally characterized designs, the authors obtained crystal structures for 15 assemblies that exhibited a broad range of curvatures and agreed closely with corresponding design models. A parallel effort using the same methodology with protein repeats resulted in concentric or buttressed α-solenoids with rotational symmetries and a central pore as presented in helical bundles. The work differed from the open architecture work by adding a geometric constraint to juxtapose N- and C- termini of the assembly.167 More recently, Shen et al. extended this methodology to produce micron size filament arrays with different diameters that were comprised by de novo helical bundles and can be controlled by capping monomer units. Six out of 124 designs were structurally characterized and agreed with the design models.180
7.2. Design of Water-Soluble β-Strands Based Structures
While α-helix based structures are the main targets for the protein interaction study, functional inhibitions, and uses in biomedical applications, β-strand and β-sheet motifs are also widespread in native proteins that contribute structurally and functionally.560 The GFP primarily composed of β-barrels is one example of this kind that has become an inseparable part of modern cell biology.561 However, the engineering of β-strand based structures have not obtained a similar level of successes compared to α-helical targets. This is especially the case for the design of all-β structures such as β-barrels, β-sandwiches, or cross-β sheets. The natural propensity of β-strands or sheets for undesirable intermolecular associations often leads to the collapse of soluble structures and aggregations without a delicately controlled register.562
7.2.1. β-Sandwich and β-Barrels
In the mid-1980s, Richardson and Erickson collaborated on the designs of “betabellin”s, meant to mimic the structure of a β-sandwich protein.563 The class of proteins was among the first de novo designed species with a target structure based on the simple binary code of hydrophilicity in amino acids that defined alternating polar and nonpolar interactions. However, the initial peptides constantly suffered from poor solubility and diversion from the design models due to the sticky edges prone to undesired interactions, which was proved later to engage in the amyloid-like fibrils formation.564 An iteration of the design increased solubility and reduced aggregations of the “betabellin” but also rendered highly fluidic structures, which prevented the crystallization and structure determination.565
The design of β-proteins with a well-defined tertiary structure was not achieved until recently by Dou and co-workers who were able to obtain a water-soluble β-barrel with incorporated active sites in its central cavity for built-in fluorescent molecule binding.171,566 The work started with idealized parametric models to generate an eight-stranded hyperboloid backbone with a shear number (total shift between the first and last strands) of ten. Local twists were introduced to adjust angles between strands and maximize interstrand hydrogen bonds, followed by a combinatorial sequence optimization to minimize the free energy. However, none of the initial sequences attained the target all-β structure and were insoluble with extensive broken hydrogen bonds on the closing of barrels. The conflicts were attributed to backbone strains mediated by steric clashes along the strips of side chains and unfavorable amino acid chirality, which was eliminated by introducing “glycine kinks” via central V to G mutations.567 A subsequent 2D-to-3D approach was adopted to map out torsional and distance constraints, with “glycine kinks” placement to control shape, curvature and interior volume of the backbone. Additional incorporation of β-bulges on the bottom and β-turns on the top completely eliminated steric strains and stabilized the interactions.568 Four out of 500 designs with low energy and a coherent hydrogen bond network were experimental tested, one of which presented as a stable monomer (BB1) with characteristic β-sheet CD spectra and crystal structure close to the design model (RMSD: 1.4 Å). On the basis of BB1, the group further incorporated a binding pocket in the center of the cavity for DFHBI, which was a derivative of the intrinsic chromophore of GFP. Energy minimization for sequences around the binding sites and the total complex was conducted to build a pocket buttressed by the underlying hydrophobic core. The designed barrel was smaller and structurally simpler than GFP, and exhibited fluorescence activation both in vitro and in vivo, although with intensities more than one magnitude lower compared to GFP. The report represented a design pathway where scientists began with idealized draft backbones but accommodated significant symmetry-breaking to build a continuous hydrogen bond network without strains so as to grant water solubility and incorporate active sites to the protein.
From another aspect of the topic, a more recent work reported the de novo design of transmembrane β-barrel from the same group.175 The membrane soluble β-barrel had sequences closely resembling those from the aforementioned water-soluble variants but with predominantly lipid-exposed residues and additional membrane anchoring residues. Figure 25A shows the careful placement of aromatic residues at trans (translocating side, extracellular, long loops) and cis (nontranslocating side, intracellular, short loops), and glycine kinks of the design. In the initial design, all residues were polar in the interior and nonpolar in the exterior of the barrel, resulting in a binary pattern similar to β-sheets but opposite to the water-soluble barrel designs. The rationale aligned well with a recent analysis on native proteins suggesting that membrane barrels are taller, fatter and inside-out soluble barrels.569 However, experimental success was not achieved until additional destabilization with β-hairpins on the trans side and reduced β-sheets propensity were incorporated to slow down undesired nucleation and allowed proper folding of the structure. Compared to the soluble variants, transmembrane designs had a smaller population of glycine kinks and preglycine hydrogen bonds which were disfavored due to the lipid environments and lack of water molecules to stabilize exposed carbonyls. Twenty new designs were tested experimentally. The hydrogen bond networks and crystal structures were in agreement with the design models for selected variants (TMB2.3 and TMB2.17). An earlier work from Nanda’s group reported a similar design effort but with a template protein OmpA, where a significant portion (<60%) of lipid-facing residues were automatedly redesigned judging by a statistical potential, without disturbing protein folding and its ability to be inserted into lipid membranes.570
Figure 25.

(A) Top: sequence features defining de novo transmembrane β-barrel fold and shape. Hydrogen bonds (dashed lines) connect β-strands in the designs. Side chains are shown as gray spheres and G residues as yellow dots. Aromatic girdle motifs are shown in red, Y residues of the mortise-tenon motifs in blue, and P residues as black pentagons. Glycine kinks bend the β-sheets into four corners (arrows). Bottom: hydrogen bond geometries between pairs of residues involving a glycine kink, comparing crystal structures of water-soluble (PDB ID: 6CZH) and transmembrane β-barrels (PDB ID: 1BXW). Glycine residues are in yellow. Water molecules are shown as red dots. Distributions of the C–O–H–N and O–H–N angles are shown to describe the corresponding hydrogen bond geometry. Reprinted with permission from ref (175). Copyright 2021 The American Association for the Advancement of Science. (B) Structures of the amyloid fibril MAX1 showing: strands align perpendicular to the main fibril axis (top, PDB ID: 2N1E), and residue arrangements (bottom) with K (blue sticks) on the wet interface and V (green ball and sticks) on the dry interface. Reprinted with permission from ref (25). Copyright 2020 Cambridge University Press. (C) Schematic of architecture for PSAMs (PDB ID: 3EC5, 2OY7, 2OY8, 2OYB) with the SLB colored blue and the N- and C-terminal domains colored gray. The side chains of the two cross-strand ladders used as hosts are shown as stick models. The table summarizes the sequences of the ladders in PSAMs. (D) X-ray crystal structures of cross-β PSAMs. The overall structure of the YY/LF PSAM dimer is shown in cartoon representations, in two orthogonal views. The two molecules are related by a pseudo 2-fold symmetry (dashed line). C, D: Reprinted with permission from ref (172). Copyright 2010 National Academy of Sciences.
7.2.2. Water-Soluble Cross-β Assembly
Compared to the recently discovered cross-α assembly introduced in the previous section, the cross-β motif represents a more thoroughly investigated class of higher-order architectures, in which β-strands align perpendicular to their associated axis and pair along the nonpolar interface.571 This is primarily known as the underlying structure for amyloid fibrils occurring in different kinds of neurodegenerative diseases, prion strain variations,572 and protein-misfolding diseases.573
There have been many studies on the short peptide design capable to accommodate the cross-β geometry, starting from the early work in which simple LKLKLKL heptapeptides assembled into a well-defined β-sheet in solution.574 Rich and Zhang later recognized the potential of self-assembling β-peptide scaffolds with enormous use cases in applications including wound healing and drug delivery.273 Many cross-β designs haven been subsequently reported over the past decades with a variety of functions including catalysts,575,576 stimuli-responsive hydrogels,577,578 graphene-binders,579 and phosphopeptides.580
However, there has been only one single report to date on the successful design of cross-β motifs in a fully water-soluble form. Biancalana et al. reproduced the minimal components of the motif with rows of hydrophobic residues running across each β-strand layers, resembling ladders due to its characteristic periodic pattern.172 The ladders have a dry interface with tight steric interactions and a wet interface that is hydrated and loosely packed similar to those in amyloid fibril MAX1, as shown in Figure 25B.581,582 The design was named PSAM (peptide self-assembly mimic, PDB ID: 3EC5, 2OY7, 2OY8, 2OYB), which contained N- and C-terminal globular domains of OspA and a single-layer β-sheet (SLB) in the middle, which, in this case, was derived from the sequence of Alzheimer’s amyloid-β (Aβ) core region, LVFFA. Several sequence combinations were iterated from hydrophobic ladders and tested, as shown in Figure 25C. An alternating L and F residue design was adopted to induce an antiparallel assembly.583 The designed YY/LF and FL/LF PSAMs were expressed in the soluble fraction of E. coli and formed a dimer, consistent with the cross-β architecture. Further crystallographic data confirmed their structures as designed (Figure 25D). Also, YY/LF and FL/LF variants exhibited similar conformational states and the same head-to-tail dimerization (backbone RMSD: 2.4 Å) suggesting the energetic preference in this orientation. The soluble design of PSAM provides an opportunity to dissect interactions in cross-β structures with fewer constraints imposed by amyloid formation, as well as insights on structural tolerance to accommodate amino acid replacements without significant conformational changes.
7.3. Multidimensional Assembly Design
As previously noted, the growing computing power and recently developed algorithms have enabled the design of many new 2D and 3D assembly structures that were not previously attainable. One great representation of this is the series of single- or multicomponent protein structures with DNA origami-like precision designed through the Rosetta algorithm. The assemblies relied on the accurate design of interfaces between protein subunits.26 While each specific structure had their own niche concerns needing to be resolved to achieve the target association, a general workflow was developed to be applicable to a wide range of targets. Using the early design of dual tetrahedral architecture as an example, the procedures implemented by the Rosetta software suite can be summarized as follows (Figure 26A):153,169,584
-
(1)
Identification of the stoichiometry and symmetry elements for the building blocks and the unit cells; screening from the database for qualified backbone candidates.
-
(2)
In silico docking analysis of building blocks as rigid bodies with limited degrees of freedom along the symmetry axis to isolate suitable candidates and docking configurations for further design. Large contact areas and minimal conformation changes are favored.
-
(3)
Side chain optimization on the interface residues of both subunits to obtain low energy sequences with native protein-like features to generate new building blocks with preferential assembly in the computed interfaces.
-
(4)
Experimental verification and further optimization (if necessary) to determine the final designs.
Figure 26.

(A) Overview of the multicomponent computational assembly design. (i) Architecture comprises two trimeric building blocks (green and blue) with tetrahedral point group symmetry. Each building block has two rigid-body degrees of freedom, one translational and one rotational. The systematic docking exploration identifies large interfaces with high densities of contacting residues formed by well-anchored regions. (ii) Sequences are designed at the new interface to stabilize the modeled configuration and drive coassembly of the two components. Reprinted with permission from ref (169). Copyright 2014 Macmillan Publishers Ltd. (B) Crystal structure representation of de novo designed self-assembling structures. Reprinted with permission from ref (26). Copyright 2016 Macmillan Publishers Ltd. (C) Averaged TEM negative staining image superimposed with the design model of binary 2D crystalline arrays with (bottom) and without (top) GFP fusion. Reprinted with permission from ref (170). Copyright 2021 Ben-Sasson et al.
By tweaking the symmetric requirements and target assemblies of the building blocks, a series of de novo structures were successfully designed and verified (Figure 26B), which included polyhedrons,585 multicomponent nanocages,169 60-subunit,165 and two component 120-subunit icosahedron,166 as well as 2D crystals with protein building blocks (Figure 26C).168,170,586 Subsequent developments of the algorithm have also enabled the design of antibody-incorporated nanocages587 and minimal flexibility protein assemblies with rigid linkers through multipoint anchoring.588 Many of these designed structures encompassed a hollow interior with nanometer size vacancies to serve as potential vehicle of other biomolecules, or multivalent anchoring sites for functional motifs, which subsequently enabled the fabrication of novel protein-based nanomaterials tailored for specific biotechnological applications.
7.4. Hydrogen Bond Network and Polar Core
Besides the growing complexity of accurately designed protein assemblies, one other fundamental aspect of protein design we would like to revisit in this section is the design of polar interactions and the hydrogen bond network, with a focus on its integration in the core of proteins or assemblies, and whether it can be a driving force for protein folding and stabilization.
Hydrogen bonds are one of the most prominent interactions present in protein biology, accounting for a significant portion of intermolecular associations, that is, stabilizing ∼40% of natural dimers. They are the core of supramolecular structure constructions.589 One example of its utilization in molecular biology is DNA origami, which enables the folding and sculpturing of DNA assemblies into precisely controlled arbitrary 3D nanostructures.590,591 The strict pairing of DNA nucleotides forms self-contained central hydrogen bonds through all buried polar atoms in the near-ideal geometry leading to high specificity heterodimers of double helices. While it is tempting to adopt the programming analogy and transplant these ideal assemblies to proteins, more difficulties arise in reality due to the variety of polar amino acids (Figure 2), their numerous rotameric states, and highly variable backbone geometries that add up to an astronomical number of network possibilities. Loosely defined interfaces that do not fully satisfy bonding capabilities for every participating residue may also exist, adding an extra layer of complexity. Exposed polar residues are usually compensated by multiple amino acids in a local network to meet the electrostatic and shape complementarity, or otherwise pose a considerable energy penalty against folding and structural stability.592−594
The design of these hydrogen bonds was first explored to promote protein–protein interactions, which was less demanding due to the presence of water molecules and solvent exposures.595 Joachimiak et al. reported the design of DNase-immunity protein and new binding partners with affinity 300-fold higher to unspecific bindings, by identifying the critical interface residues and sampling alternate rigid body orientations to obtain iterative structure-based mutations.596 However, a later work by Stranges and Kuhlman suggested the limitations of this approach by comparing five successful cases against 158 failures, both sets of which were designed by Rosetta.597 Fewer polar residues seem to be a key feature at the interfaces of the successful designs as compared with many of the failed designs, or in native proteins. Attempts to integrate extensive sets of interfacial hydrogen bond interactions often lead to no detectable binding, indicating that the algorithm had not been able to account for all the unsatisfied residues which induced mismatch energetic penalties to prevent the ligand binding. A later report suggested that the pairwise correlation between hydrogen bond donors and acceptors depending on the bonding strength can improve the interface efficiency by minimizing the solvent interaction and promoting a strong binding between the protein and ligands.598
Recently Boyken et al. reported building self-coherent hydrogen bond networks in coiled-coil assemblies by a Rosetta based algorithm, HBNet.365 The algorithm systematically searched all hydrogen bonds and steric repulsions for residues at possible rotameric states, which were represented by nodes and edges in a simplified diagram (Figure 27A). The graph was traversed to identify connected low energy networks and reject unsatisfied polar residues, which were then redesigned until all side chain hydrogen bonds in the backbone structure were enumerated and satisfied. The methodology was tested using the concentric ring geometry to isolate the designed network from solvents and avoid the complication of designing side chain interactions with both residues and solvents.599 As outlined by Figure 27B, the coiled-coils were parametrically built, with hydrogen bond networks searched and satisfied by HBNet, and subjected to the rotamer optimization for remaining positions to meet the oligomerization and symmetry states. The approach had a high success rate in α-helical bundle design (66/114) with eight different topologies, C2, C3, or C4 symmetry and various supercoiling. The selected peptides tested by SAXS and crystallography showed close agreements to their design models in both the structure and hydrogen bond network. The algorithm was later further developed to design large, asymmetric structures and complexes.600
Figure 27.

(A) Left: all side chain conformations (rotamers) of polar amino acid types considered for design at each residue position (oxygen colored red, nitrogen blue); Middle: many combinations of hydrogen-bonding rotamers are possible, and the challenge is to traverse this space and extract. Right: networks of connected hydrogen bonds. (B) Design strategy of internal hydrogen bond network in helical bundles. From top to bottom: the parametric generation of C3 symmetric two-ring coiled-coil backbones; the HBNet application to parametric backbones to identify the best hydrogen bond networks; and remaining residue design in the context of the assembled symmetric oligomer. A, B: Reprinted with permission from ref (365). Copyright 2016 The American Association for the Advancement of Science. (C) Close up view of the core region of Top7 before (left) and after (middle) hydrogen bond network incorporation, and the model of disulfide-bonded Top7_PC with core hydrogen bond network (right). Reprinted with permission from ref (276). Copyright 2016 The Protein Society. (D) Additional internal hydrogen bonds in simulated CXCR4QTY. Notation: ‘s’ denotes a side chain bond and ‘b’ denotes a backbone bond. Thus, T152s-T148b denotes that the side chain of T at location 152 forms a hydrogen bond with the backbone of T at position 148. From left to right, top to bottom: Q260s-S260s-Y256b, T215b-Q216s-Q246s, Y249s-Q253, Q167s-H203b, T169s- Q165b, T204s-Q208s, Q78s-Q69s-Q69b, T112s-Q108b, Q290s-T287b. Reprinted with permission from ref (277). Copyright 2019 National Academy of Sciences.
Since hydrophobic cores are commonly adopted in the water-soluble proteins that drive the folding, polar interactions in the core region can be destabilizing due to the competing hydrogen bond interactions from water molecules. Traditional protein designs featured hydrophobic residues such as L and I in the buried interior, whereas the integration of polar residues in the all-hydrophobic core was usually for functional incorporations. Yet these modifications were energetically unfavorable and needed to be accommodated by the hyper-stability of ideal de novo models,160,174,185 or otherwise, collapsed the structure.542 The study of native proteins did not always result in the same conclusion, as buried hydrogen bonds can either hinder the kinetics of protein folding,601 or stabilize the structure against denaturation.602 The question remains whether these polar interactions and the internal hydrogen bond network can drive the folding of a single polypeptide chain and stabilize its final conformation.
The question was probed by Baker’s lab through engineering an internally polar version of the de novo designed α/β protein Top7.159,276 Their initial target was to make an “inside-out” protein with polar and nonpolar residues in the core and surface, respectively. Yet low energy candidates obtained through all-residue redesign from the fixed backbone failed to express in E. coli. A subsequent iteration combining the computed five-residue polar interior with surface residues of the original Top7 predictably ended up with a destabilized core. Further integration of a 12 to 45 disulfide bond into the structure was needed to produce a soluble protein, namely Top7_PC, which was expressed in the stable form. The several iterations of the design are shown in Figure 27C. Top7_PC exhibited similar folding kinetics to Top7, had a similar crystal structure to the design model but was less stable compared to Top7 with the same disulfide bond mutations. Additionally, two of the five interior polar residues failed to form the designed hydrogen bonds and were more exposed to the solvent. The work proved that the hydrogen bond network can be designed de novo in the core of a soluble protein, but needs to compete with solvent interactions and requires additional strong linkages like disulfide bonds to drive the folding and stabilize the structure.
On the other hand, compared with the inhospitable environments for water-soluble proteins, transmembrane proteins seem to be a natural and sounding habitant for a polar core.353,603 Expectedly, a widespread presence of internal hydrogen bond networks was predicted within the interior of the proteins, rarely exposed to lipids, and bordered internal water filled cavities involved in the small compound binding and transport.604 Native transmembrane helices generally have at least one interhelical hydrogen bond in their structures.353 Multipass transmembrane proteins like GPCRs and active transporters contain higher densities of buried polar residues compared to other variants that participate in water mediated signal transductions.605 These internal hydrogen bond networks are highly conserved in contrast to interfacial ligand interactions likely due to the necessity to meet electrostatic complementarity and functional requirements.
However, despite the consensus that nonpolar solvents can strengthen naked hydrogen bonds relative to polar solvents, a prior study suggested that hydrogen-bonded side chain interactions only contribute modestly in the stability of native membrane proteins.606 Statistically a similar propensity of polar residues and ratio of unsatisfied partners were observed in membrane protein cores as compared to water-soluble proteins.604 This less-than-expected condition was attributed to (i) the much stronger backbone hydrogen bond in nonpolar solvents which served as alternative competitors similar to the function of water molecules in water-soluble proteins, and (ii) a high dielectric polarizable environments that weaken side chain interactions in transmembrane proteins.607 Similar to the internal hydrogen bond network installations in water-soluble proteins and assemblies, design efforts with less complexity were also made on the integration of interfacial polar interactions for de novo designed transmembrane α-helical oligomers, which was discussed in details in Section 4.2.2. Stabilizing effects were observed from hydrogen bond networks in these designs, whereas both the side chain and backbone of amino acids were involved in the interactions.358,361−364
An interesting phenomenon was observed by Qing et al. on QTY code solubilized GPCR proteins, which showed superior heat tolerance against elevated temperatures without losing function in arginine-containing solvents.277 The high thermostability was attributed to the formation of an internal hydrogen bond network due to polar residue substitutions based on their molecular dynamic simulations, as shown in Figure 27D. However, the crystal structures of QTY proteins need to be determined to support the hypothesis. In contrast, a more recent publication showed that the soluble BH3 interacting-domain death agonist protein with similar QTY substitutions at the core region resulted in mutations with comparable secondary structures but was more prone to chemical denaturation only when surface hydrogen bond interactions were negated by a high concentration guanidine hydrochloride.608
7.5. Molecular Dynamics Simulations
7.5.1. Molecular Dynamics Models
MD simulations predict the evolution in time of the system under study at femtosecond and angstrom resolution.609 It generates atomic trajectories describing the system’s behavior. The accuracy of the simulation directly depends on a description of how the molecules will interact with, namely a force field, which is a functional form and parameter set used to calculate the potential energy of a system of atoms or coarse-grained particles. The general pipeline for these types of simulations is illustrated in Figure 28, which can be done with a number of software packages. The most widespread ones are CHARMM,610 Amber,611 NAMD,612 GROMACS,613 ACEMD,614 and Desmond.615
Figure 28.
Molecular dynamics simulation pipeline.
Despite classical MD is appropriate for soluble protein studies, some special MD methods were dedicatedly designed for the simulation of this class of proteins.616 CpHMD emerged over the past decade to consider precisely the pH effects during simulations.67 It allows determining the titratable sites’ protonation states during the MD simulation at a specified pH, which enables the mechanism investigation on pH-dependent conformational processes. For example, this method provided insights into the physical basis of protein solubility via RNase Sa and insulin simulations.617 Another MD method is subvolume-KB molecular dynamics, which determines solution structures based on the Kirkwood-Buf theory for closed systems and cluster analysis. The method identifies molecular associations based on hydrophobic interactions, hydrate formation, hydrogen bonds, and electrostatic forces affecting solution nonideality in different types of aqueous systems. However, it has not yet been applied to proteins.618 Solvent-shells featurization is based on MD calculations with Markov state models. It includes the degree of freedom for solvents in the analysis, and was successfully applied to two-domain protein BphC to identify functional water molecules.619 In addition, enhanced sampling MD simulations such as steered MD and replica-exchange umbrella sampling are widely used in membrane proteins or protein aggregates. Such methods allow the simulation of the membrane permutation process of cyclic peptides across the lipid bilayer.620
Alternatively, coarse-grained MD models provide a less structured protein representation making two or more atoms a single interaction unit. This group of methods is highly applicable for large systems such as protein aggregates and assemblies since it significantly speeds up the calculation time due to fewer degrees of freedom, simpler, softer potentials, and larger time steps. CG MARTINI models are successful in describing many physical properties of bilayers. Also, they shed light on the GPCR-arrestin complex formation621 and TNF receptor family members’ transmembrane organization.622
7.5.2. MD Simulation of Protein Assemblies
Despite the continuous increase in computational power, the MD simulation of protein assemblies remains challenging due to the system’s complexity and size. Nevertheless, there are some MD techniques appropriate for this purpose. One widely used technique is the enhanced MD sampling (EMDS), which contains four main types: Replica-Exchange MD simulation (REMD), accelerated MD (aMD), metadynamics or adaptive biasing force (MetaD), and Markov state (MC) models (Figure 29).623 REMD concurrently runs many replicas of the system in parallel with various temperatures or Hamiltonian energies, that are exchanged or swapped replicas at fixed intervals based on the Metropolis criterion. It generates a generalized ensemble and enhances the convergence of MD simulation. aMD facilitates the sampling process by running simulations on modified potential energy functions to expedite the molecules overcoming local minimums. MetaD continuously changes potential energy function in particular and energy profile in general on a regular basis during the simulation. The sampling of unexplored conformational space and avoidance of repeated states are encouraged by adding potential energy to visited states. MC generates a kinetic model from a long unbiased MD trajectory by building microstates from the initial trajectory and calculating a transition matrix from the states with a lag time.624
Figure 29.

Overview of enhanced sampling methods: (A) replica-exchange MD simulation, (B) accelerated MD, (C) metadynamics or adaptive biasing force, and (D) Markov state model. Reprinted with permission from ref (624). Copyright 2020 Multidisciplinary Digital Publishing Institute.
EMDS was successfully applied to simulate the reversible association of five structurally and functionally diverse protein systems. Pan et al. used a tempered binding modification of EMDS which allows scaling all interactions uniformly and simulating unbiased for any particular protein assembly. It provided repeated association and dissociation steps of the complex near the native interface.625 Alternatively, classical MD simulations were used in studies of HIV-1 and Bin/Amphiphysin/Rvs protein assemblies, which suggested its applicability for protein assembly studies.626
7.6. Summary and Prospect
Molecular assemblies are one of the most important driving forces to sustain the living organisms, undertaking essential roles in numerous biological processes related to structure, genetics, communication and transportation both within the cells and at an organism level.627 As the fundamental molecular machines, proteins and peptide assemblies are extensively used in nature to build functional complexes with intriguing applications. Herein, extensive efforts were devoted into understanding and reproducing naturally occurring assemblies from a molecular level. Although accurate design with nature-like complexity and specificity remains a challenging task, much progress has been made in de novo design of the idealized multidimensional models with cyclic, helical, and lattice symmetries.628 The engineering efforts in turn contributed to the understanding of underlying biophysical principles and predictive models, transforming to methodological advances in a fruitful area of protein design.
In this section, we reviewed the most recent progress on the design of complex structures and assemblies building on prior knowledge from simpler models. Finely tailored arrangements of residues and functional groups account for precise interfacial interactions to determine the geometry of assemblies spanning a range of time, length, and complexity scales. One characteristic approach was to expand the prior coiled-coil models by either integrating more subunits into a bundle, or introducing multiple rigid contact points and adjusting the helix spatial orientation for preferential structure extensions. Among all binding mechanisms, hydrophobic effects and close helical contacts through small G, A, and S residues are still the most prominent to stabilize multiple α-helical barrels and cross-α fibrils.162,541 Electrostatic interactions were utilized to create “adhesive ends” for associating large complex structures with precise geometrical alignments.163 Alternative mechanisms like disulfide bond and π-stacking were also introduced to assist the higher-order structure formation.276,553
However, interestingly, while being one of the most indispensable interactions in protein biology, the design of hydrogen bond networks has been a challenging task for both water-soluble and transmembrane proteins. The difficulties lie in the necessity to satisfy every participating residue in a highly complementary manner, both electrostatically, morphologically or even concerning their bonding strength. Unsatisfied residues induce unfavorable polar interactions that negatively affect the protein and complex stability.597 Because of its critical roles in protein biology, there have been many design attempts to integrate hydrogen bond networks in either protein–ligand binding pairs or in the core of a protein, with different levels of complexity.276,358,361−365,596,600 Yet in many cases hydrogen bonds were built into an ideal design model or natural templates to introduce functional features and did not always play a stabilizing role.185,276 Even with recent successes in installing more complicated coherent networks into coiled-coils, much work is still needed to enable the accurate design of polar interactions with nature-like precision with respect to their structural and functional impacts.
With the continuous development of computational algorithms and more accurate calculation of energy functions, breaking the symmetry and designing structures beyond idealized models is becoming increasingly possible.171,175 This is especially important for all-β proteins since (i) the ideal parametric backbone models can be difficult for natural amino acids to strictly follow without conflicts; and (ii) the sticky nature of β-strands can render undesired aggregations and amyloids. The introduction of asymmetrical motifs like kinks/turns induced by glycine or proline can be crucial to release strains and steric clashes in a more specific manner for structure stabilizations. On the other hand, many of the designed α-helical structures have exhibited superb thermostabilities and denaturant resistance.26,168,169,173,546,585,600 While such characteristics can be beneficial for their use as nanomaterials, it is rarely the case in native proteins. Breaking the symmetry to integrate specific binding motifs or tailored functions can help to elucidate how native proteins work and expand their use-case in a variety of applications.
We mentioned in the Introduction section that the boundary between protein solubility and stability was obscured in this review, which is especially the case for this section. In complex structures, the consistency in oligomerization and structure stability are more important merits to evaluate a design. For supramolecular structures such as cross-α or cross-β amyloid fibrils, the concept of solubility for whole structures is barely relevant while the long-range order and interaction mechanisms are important considerations. Yet it is also evident that the soluble design of their building blocks can be of great assistance to achieve uniformity in higher-order structures and elucidate critical residues to tune the interactions.162,172 The soluble design also potentially opens up possibilities for these molecular assemblies to be used in a variety of biomedical and nanotechnological applications including hydrogels and bioscaffolds.
In prospect, one primary target of protein complexes and assemblies design is for interaction mechanism studies, which are especially important for disease-related residue misplacements and native protein malfunctions.629,630 With design tools becoming more intuitive and predictions of structures becoming more accurate, designs based on ideal models can be further fine-tuned to resemble native proteins in pathogenic biostructures and enable the development of precision therapeutics. With the highly accurate structural prediction for individual proteins in light of recent algorithms such as AlphaFold2, it is increasingly possible to predict the association and long-range order of supramolecular structures based on the characteristics of their subunits and subsequently contribute to the design process for more robust functional assemblies.631,632
From another aspect, the capability to engineer well-defined protein complexes and assemblies with superior robustness compared to nature’s offering enables their potential use as new polymeric materials that can modulate biological functions both in vitro and in vivo. More sophisticated subunits further allow the delicate tuning of assemblies’ structure and function with a higher degree of freedom in design. For instance, stable protein scaffolds with functional incorporation can be used as novel therapeutics or delivery vehicles.166,587 Low level association with nanomaterials can either be utilized to enhance the performance or solubility of those nonbiological molecules,181 or even create a highly integrated bioelectronic interfaces with molecular level communications.633,634 While extremely challenging, the functional nature of protein building blocks provides their assemblies with vast possibilities beyond the potential of short peptides and DNA origami.
8. Coiled-Coils: Design and Prediction
Arguably, α-helical coiled-coils are one of the most understood structures in protein science, primarily due to their (i) abundance in nature; (ii) high degree of symmetry; and (iii) easiness for structural characterization.539,635 They are formed with two or more α-helices winding around each other into a supercoil. Coiled-coil structures comprise 3–5% of all protein-encoding DNA, whereas almost 10% proteins in eukaryotic proteome have coiled-coil domains.636 They undertake a variety of biological tasks, ranging from supporting structural rigidity and transducing conformational changes, to transporting biomolecules, binding DNAs, as well as driving protein folding and interactions.637 Because of the simplicity in describing and defining the structures, they are widely adopted in mechanism studies to elucidate and predict interhelical side chain interactions that also fuels the knowledge-base for the design of other proteins.638,639 The design of coiled-coil based peptides and proteins render fruitful results both in the soluble and transmembrane forms.
Since many of the designed structures covered in previous sections are based on coiled-coils, we think it is beneficial to add an extra supporting section to introduce fundamentals of the structure so that readers can consult and better understand the molecular basis of those design efforts. However, the design of coiled-coils comprises such an enormous field that we have no intention of reviewing it thoroughly since it does not align with the primary target of the current article. Interested readers should refer to other dedicated papers and books for more detailed information.539,635,637,640−644 We will focus on introducing the fundamentals and crucial structural considerations of coiled-coils related to their sequence, limiting our discussion on the molecular and design basis of structures introduced in previous sections and several new opportunities, as well as covering prediction mechanisms and computational design tools.
8.1. Structure of Coiled-Coils
8.1.1. Canonical Model
The name of “coiled-coil” was first used by Francis Crick, to describe a ubiquitous quaternary fold where two or more α-helices wrap around each other and interlock their side chains with varied stoichiometries and periodicities.645 This structure was parallelly observed and reported by Linus Pauling.646 Crick parametrized the description for this type of structure, using a simple set of equations to compute the coordinates of helical backbones.151 The model accounts for 97% of existing coiled-coils in nature and is considered the canonical model through which a large number of structural and functional diversity can be achieved from a small set of quantifiable principles. These parametric equations obviate the need to sample all conformational spaces and are widely adopted for protein structure prediction and design through calculation.
In this canonical view, coiled-coils are generated through a repeating sequence unit of seven amino acids, namely heptads and denoted abcdefg, as mentioned earlier. Heptad residues at the interfaces form KIH types of interactions and generate stabilized supercoils with defined oligomeric states corresponding to their primary sequences (Figure 30A).647 A classic representation of heptads has a regular pattern of HPPHPPP where H represents hydrophobic amino acids and P represents hydrophilic ones. The helices are amphipathic with a hydrophobic stripe along their long axis. “Knob” residues on one helix project into shape-complementary “holes” formed by four-residue diamonds on an adjacent helix.
Figure 30.

(A) Structure of α-helical coiled-coils. Left: helical-wheel showing heptad projection and representation of geometric parameters. Right: representation of KIH interactions. Reprinted with permission from ref (710). Copyright 2018 John Wiley and Sons. (B) Transitions in periodicity and supercoiling caused by the insertion or deletion of residues. Top: insertion of a stammer causes overwinding of the supercoil; insertion of a stutter relaxes the supercoil and change its handedness; further insertion of a stutter tightens it in the other direction. Bottom: insertions of two or 2 × 3 residues strain the helix beyond the breaking point, leading to the formation of β-layers; α/β fold formed with repeated insertion. Reprinted with permission from ref (637). Copyright 2017 Elsevier. (C) From left to right: side and end views of the hydrophobic interior of dimeric coiled-coil GCN4 (PDB ID: 2ZTA), trimeric coiled-coil GCN4 derivative (PDB ID: 1GCM), and tetrameric GCN4 derivative (PDB ID: 1GCL) with corresponding helical wheels. Reprinted with permission from ref (25). Copyright 2020 Cambridge University Press.
Structural specificity in coiled-coils is dependent on interactions between core-facing and interhelical residues. In a site-specific manner, positions a and d generally comprise nonpolar residues facing the interior of the supercoil with packing complementarity, which form the hydrophobic core that drives the oligomerization. Aliphatic hydrophobic residues are preferred to aromatic ones due to the bulk and steric constraints of the latter.640 The flanking positions, e and g, usually comprise polar or charged residues that induce interhelical electrostatic interactions. These play a significant role in folding kinetics and the partner selection, which may be used as a negative design principle.648,649 The former interactions are important in establishing a tight KIH packing while the latter determine the formation of salt bridges between two helices, although their influences on the overall structural integrity have remain controversial.650 Other positions appear to be more permissive compared to these sites, although they mostly favor polar and helix-forming amino acids in soluble proteins, both in nature and by protein designers. They promote the hydrogen bond formation and electrostatic interactions with solvents and external species to stabilize the oligomerization and overall conformation.643 These interactions cumulatively render coiled-coils one of the most stable protein folds with high resistance to both chemicals and temperatures, especially for short structures with only four or five heptads.546,651 Both the composition and the length define the conformational specificity and stability of a supercoil to several kcal/mol.
The discrepancy between the perfect periodicity (3.5 residues/turn) and amino acid placements in an undistorted α-helix (3.6 residues/turn) requires structural accommodations to achieve repeatable packing. Thus, a supercoil is formed to adjust the relative periodicity of each helix toward the central axis to 3.5 and allow residues to occupy the same position in each repeat. Such twisting is left-handed with an angle of about 13° per turn counterclockwise.637 Alternative conformations also present in nature but are much rarer compared to structures following the parametrized model, since they depart from the ideal KIH geometry.652 Yet such noncanonical coiled-coils with repeats longer than seven residues are increasingly discovered and reported.653 Common repeat types include stammers (three additional residues) and stutters (four additional residues), which cause an angle change of 17° and −17°, respectively. Stammers tighten the supercoil (30°) while stutters relax and change it into a right-handed one (−4°). Such irregularity can happen individually or combined in heptads to yield a variety of noncanonical structures. In general, all periodic coiled-coils without disruption of helices can be described as three- and four-residue combinations starting with a core residue.654 Short β-strands can also be integrated into the geometry and result in a new variant of α/β coiled-coils (Figure 30B).
Both parallel and antiparallel conformations can be adopted by coiled-coils, while parallel forms are more commonly observed in nature, probably due to certain interhelical interactions such as hydrogen bonds. The interface between a–a and d–d positions in the parallel conformation becomes a–d and d–a in antiparallel conformations, although packing geometries between the side chains of the core residues still appear to be parallel and perpendicular. The side chain interactions in the flanking e and g positions are significantly different, turning from gn–en+1 to g–g and e–e, providing an additional rule of consideration for coiled-coil designs.640 Antiparallel coiled-coils in the PDB are also found to contain more charged residues in the core than parallel ones.655
8.1.2. Oligomerization
The oligomeric state is an important characteristic of coiled-coils. About 93% (937/1012, with <50% sequence identity) of the current coiled-coils are dimers, trimers or tetramers (Figure 30C) according to the CC+ database.656,657
The difference in oligomerization may be determined by sequence variations in heptad repeats, where the types of side chains at the a and d positions have a significant impact. For instance, a combination of β-branched residues (I, V) at a and γ-branched residues (L) at d was found to favor dimers, whereas occupation at inverse positions favors tetramer and a mixed occupation results in trimers, based on GCN4 study.213,637 Larger side chains in aromatic residues can induce higher oligomeric states, while smaller residues contribute to the formation of more closely spaced antiparallel supercoils.354,658 Such preferences reflect the engagement between knobs and holes in a Cα–Cβ bond vector through shape complementary packing, as I and V prefer parallel packing while L prefers perpendicular packing.637,640 Positions e and g help to define the oligomeric state by the types of residues and their interactions. In dimers and trimers, charged residues usually occupy these positions and provide electrostatic interactions to shield the hydrophobic core from the solvent. The inclusion of hydrophobic residues at either or both of the positions and in KIH interactions expands the core area of coiled-coils to form tetramers or attain higher oligomerization.659 Broadening of the interface also provides a wider shield for the hydrophobic core, which can introduce a loci in the center of the bundle, allowing the installation of molecular channels or catalytic centers, as demonstrated in the design of α-helical barrels described in previous sections.160,539,542,546
Higher order coiled-coils involve additional seams beyond a–d interaction in the bundle. They are characterized into three types, depending on the relative placements of these hydrophobic seams. These are Type I (seams with a sharing position); Type II (adjacent seams) and Type III (separated seams), as shown in Figure 31.637 Their heptad repeats deviate from the canonical representation of HPPHPPP, to HPPHPPH or HPPHHPP; HPPHHPH; and HPHHPHP or HHPHPHP; respectively. The strategic placement of β-branched residues in the lumen is suggested to be essential for structural stabilization and to maintain the symmetry, since the inner pore challenges the close hydrophobic packing in the folding process.647
Figure 31.

Coiled-coil interactions in higher oligomers. Type N represents the standard model. Type I helices interact through two hydrophobic seams that share a central position; Type II helices through two adjacent seams; and Type III helices through seams separated by one position (which projects into the pore of the barrel). Note that this figure does not show all helices of the oligomers. Reprinted with permission from ref (637). Copyright 2017 Elsevier.
Yet the hydrophobic effect is not specific and cannot independently define the exact number and orientation of helices especially in high order oligomerizations. For instance, Type II sequences can lead to the formation of coiled-coils with oligomeric states from pentamers to heptamers. Different conformations may be separated by only small energy differences, resulting in multiple accessible isoenergetic states defined by external factors that allow switching with a trigger element.540,660,661 The transition between parallel and antiparallel states is also commonly observed especially when charged residues are present at helical interfaces, where higher order oligomers are disfavored and turn into lower order conformations with antiparallel dimers as components.542 Lizatović et al. obtained such transitions in a pentameric coiled-coil design through the protonation of E residue using pH as a control.662 Another example covered in this review is the carbon nanotube solubilizing peptides, which accommodate different conformations with and without the presence of SWCNT.181
8.1.3. Other Structural Considerations
Although coiled-coils are generally considered to be stable due to numerous core interactions, some special residues still play a significant role in their topology. For instance, C, G, and P are less frequently found in the structure unless specific interactions are needed.663 G and P are considered α-helical breakers and are generally not favored in helices. C has free thiols that can form disulfide bonds and induce undesired interactions unless appropriately designed. The polar residue N, traditionally presumed to be buried and to form stable self-compensating hydrogen bonds in the core when occupying position a, has recently been revealed to be highly dynamic in its conformation that broadens the backbone albeit remaining specific. Placing N at the d position disfavors the dimeric state and is better accommodated as a trimer.664
The structural stability of coiled-coils increases with the length of the chain in a nonlinear manner, with increasing hydrophobic burial, hydrogen bonding and polar interactions.665,666 For soluble coiled-coils, three or four heptad repeats with 20–30 amino acids are sufficient for a stable folding,667,668 while a two-heptad trimer design was also reported.669 However, longer coiled-coils are often not extensively enriched with nonpolar residues in the core, but rather with clusters of small polar or charged residues. Such residues’ destabilizing effects can be compensated by the stability afforded from the chain length, while inducing local flexibility and unfolding to allow integration of specific biological functions.644,670
8.2. Coiled-Coils Designs
Coiled-coils are one of the primary structural models in the protein design field due to their simple folding and well-established design rules.671,672 It is approached empirically from a minimalist aspect by repetitive peptide design with defined symmetry and limited interactions, which skips the challenge of sampling vast conformational spaces. The field also benefited significantly from recent increases in computing power which enable the construction of complex structures integrated with various geometries and functional components. More thorough coverage for this fruitful field can be found in previous reviews.25,635,639,640,643,644
One of the areas with extensive interests is the design of high order coiled-coil oligomers as α-helical barrels, as has already been largely introduced in previous sections. A tailored broadening of the hydrophobic seams results in a hollow core at the center which allows the installation of molecular channels or functional components both in soluble and transmembrane forms. The efforts were led by the Woolfson group through building a basis set of de novo coiled-coil peptides with controllable geometries.671 With a few modifications on the initial models following rules established through the study of leucine-zipper peptides (GCN4) as described above,213,673 they were able to obtain helical coiled-coils adopting well-defined dimeric (CC-Di), trimeric (CC-Tri) and tetrameric states (CC-Tet). The sequences contain four heptads and are capped with a G residue at each end to provide sufficient structural stability. While the initial tetramers and trimers agreed well with the designs, a disparity was observed for the proposed dimeric peptide where a trimer was instead observed. The mismatch suggested that the KIH interaction between I at the a position and L at the d position alone is not sufficient for oligomeric specificity, which was then resolved by changing an I to N in the middle a site, based on database analysis. The successful design of the so-named CC-Di peptide was explained by a negative design rule: rather than forming well-defined interhelical interactions, placing an N residue at the core position forcibly buries its amide containing side chain in the hydrophobic region and destabilizes the oligomerization, where the penalty is least severe for dimers. Building on CC-Tet, numerous high-order oligomers such as CC-Pent, CC-Hex, and CC-Hept, were subsequently designed by expanding the hydrophobic interfaces to flanking charge-complementary heptad positions.160,541,546 CC-Hex designs were then used to form high aspect-ratio nanotubes by introducing electrostatic interactions at the peptide termini.163 The methodology or the models from the basis set of de novo coiled-coils were also adopted to build α-helical barrel conducting pore without buttress both in transmembrane174,390 and soluble forms,390 or a biocatalyst integrated with hydrolytic activities.185 These structures have already been covered in previous sections and will not be discussed again in detail here.
Another area of interest for coiled-coil based designs is their utilization in novel nanobiomaterials.641 Numerous efforts based on their tunable self-assembly were reported in various biomedical applications.661,674−679 There is also interest in their integration with inorganic materials, where the selected sensing or control maybe achieved through conformational changes.680−682 For instance, based on the isoleucine zipper GCN-pII, Dublin et al. constructed a staggered trimeric coiled-coil TZ1H, with an Ag+ conformational switch, that can be integrated into long aspect-ratio helical fibers and guide the assembly of metal nanowires encased.683 Shlizerman et al. modulated the gold work function on devices through surface functionalization of coiled-coil dimers that can switch between parallel and antiparallel states. At similar surface protein coverage, chemical composition and height of the complex structures, different conformations of coiled-coils induced distinguishable charge transport behavior, electrical responses and hysteresis in the I/V curve, which were attributed to net molecular dipoles of different magnitudes and direction.684 Moreover, by integrating a Zn2+ binding site, Aupic et al. was able to construct a homodimeric coiled-coil capable of performing a drastic conformational change reversibly between the assembled state and random conformation in response to environmental clues such as pH or ions.685
Protein origami and assembly with DNA origami-like precision and diversity remain challenging due to the complexity in amino acid interactions. Careful designs of coiled-coil peptides were proposed as a proxy to mimic the straightforward pairwise complementarity of nucleic acid building blocks.643 The concept was validated by Gradišar et al., who designed a self-assembling tetrahedron based on orthogonal dimeric coiled-coils.686 Although constructed from a single polypeptide chain, the sequence contained 12 concatenated segments separated by flexible linkers. The path of chain folding was guided by defined helices traversing tetrahedron edges to form dimers with corresponding partners, whereas vertexes were induced by the reconstitution of split fluorescent proteins. The design methodology was then further extended to polyhedrons formed with 16 and 18 comprising units, folding into a four-sided pyramid and a triangular prism, respectively.687 The self-assembly of the tetrahedral structure was also demonstrated in bacteria, mammalian cells and mice without apparent immunogenetic responses. The discrete long-range interactions in the folding pathway were later elucidated by Aupič et al. through MD simulations and Förster resonance energy transfer (FRET) measurements. It showed a stepwise mechanism with dimeric coiled-coil intermediates that can be employed in diverse final folds, allowing a modular design approach and relaxing the overall stringency of requirements.688 In another recent effort, well-defined tetrahedral, octahedral, and icosahedral cages were constructed through a symmetry-directed approach by fusing a trimeric protein (TriEst) to a pentameric de novo designed coiled-coil through optimized linkers. The coiled-coils provided modularity in this approach and guided the assembly of the cages, which showed extreme stability against thermal and chemical denaturation, especially compared to the individual components.689
8.3. Computational Tools for Prediction and Designs
As one of the first protein motifs well-described structurally and theoretically, numerous computational algorithms and web-based bioinformatic tools were developed for the identification of coiled-coil domains,538,690−692 prediction of oligomeric states,693−695 and analysis of their crystal structures,696 from proteins’ primary sequences and experimental data.635
8.3.1. Coiled-Coil Prediction
Lupas et al. first reported a widely used program called COILS, which is a statistically controlled predictor based on the amino acid profile of the protein and a scoring matrix, that was translated to the probability of motif presence.538,690 It delineates and predicts discontinuity in coiled-coil domains by comparing unknown sequences with confirmed structures, and was used to identify more than 200 proteins with possible motifs. Additional inputs were introduced with the upgraded version, PCOILS, which substitutes sequence comparison for profile comparison in COILS, and significantly outperforms the former version.697 Other methods with similar analyzing concepts in the same era include COILER,663 PairCoil698 and MultiCoil.699
Walshaw and Woolfson then developed SOCKET, a program that identifies characteristic KIH side chain packing in 3D structures to differentiate coiled-coils from regular helical interactions.700 SOCKET can determine the structural boundaries and orientations, oligomeric states, and heptad registers. The program was used to find coiled-coil domains in the PDB. Outputs were collated into a relational database named CC+, providing resources and underlying principles of coiled-coils.657 The 2.0 version of the program was recently released as a Web server, which increases the number of helices that can be classified, and the proteinogenic or non-natural amino acids that can be analyzed at KIH interfaces.701 Additional user-friendly features such as a visualization module was also incorporated to enhance its accessibility to a wider community.
MultiCoil grew out of the program PairCoil, which was initially dedicated to predicting dimeric structures using a scoring function on “pairwise probabilities” calculated from the frequency of occurrence for each amino acid pair in a protein database.692,698 MultiCoil added a database of trimeric coiled-coils for accurate oligomeric classification.699 MultiCoil2 further integrated Hidden Markov Models for sequence-dependent location and oligomerization predictions.694 An optimized scoring function through multinomial logistic regression is used to produce Markov Random Field Potentials with pairwise correlations localized in sequences. It significantly outperformed the original program both at the coiled-coil detection and oligomeric state prediction, thanks to an expanded database containing 2105 sequences and 124,088 residues, compared to 1013 sequences and 6319 residues in the previous one. A relatively earlier method that also utilizes Hidden Markov Models is CCHMM_PROF.702 It extrapolates the evolutionary information in multiple sequence alignments to detect potential coiled-coil regions. The reported accuracy of CCHMM_PROF on a data set containing 104 sequences and 10,724 residues was higher than MultiCoil but slightly lower compared to MultiCoil2, although a different data set was used for the evaluation of the latter.
Additional programs that conduct oligomerization predictions include SCORER and SCORER 2.0, the latter of which takes advantage of the coiled-coil database established with SOCKET to achieve much-improved accuracy on sequences with unknown oligomeric states.663,693 RFCoil distinguishes dimers from trimers through amino acid indices and the random forest classification algorithm,703 whereas LOGICOIL is also capable of predicting tetramers and the topology of dimers (parallel vs antiparallel).695
In more specialized applications, a CCFold algorithm was developed by Guzenko et al. to create models of intermediate filaments through the prediction of dimeric coiled-coil fragments.704 The method extends the analysis beyond common heptad repeats in the canonical model to other hydrophobic repeat patterns and periodicities by approaching the problem from a protein-folding aspect. The input sequences are dissected for overlapping analysis against coiled-coil fragments to determine the probability of the presence of a certain structural feature. Waggawagga provides a web-based comparative visualization tool of coiled-coil predictions based on many of the algorithms introduced above.705 Comparisons of the effectiveness for these computational algorithms on defined data sets were also performed by multiple groups.706
Machine learning based algorithms are increasingly used for the prediction and analysis of coiled-coils. DeepCoil represents such an effort based on either protein sequences or sequence profiles.707 A data set was generated by SOCKET on biological assemblies obtained from the PDB to provide both training and test sets for the model, which were carefully separated with less than 30% shared identities. DeepCoil was later used for the genome-wide identification of coiled-coil domains and found 35 regions not predicted by other methods. The same group developed a fully automated software, namely SamCC-Turbo, to survey the PDB and build a database containing ∼50,000 fragmented coiled-coil regions. The data set was proposed to provide a reference for fragment-based modeling tools like CCFold, and neural network training for DeepCoil.
8.3.2. Coiled-Coil Design
In addition to prediction and analysis tools, several computational tools have been devised to facilitate the coiled-coil design and increase their accessibility to nonspecialist users through user-friendly interfaces. One pioneering effort was from DeGrado’s lab, who devised a web interface for coiled-coil parameter fitting and backbone generation named CCCP (coiled-coil Crick parametrization). The method was covered in Section 7.1.1.545
Another widely acknowledged program is CCBuilder, an interactive web-based application for generating coiled-coil models.154 The program was based on MakeCCSC, a backbone modeling tool following Crick’s original parametrization.708 CCBuilder creates a fully atomistic model from a given sequence and structural parameters. The generated backbone is then passed to Rosetta for side chain packing. The quality of the model, including the KIH packing and interaction energies under all-atom force fields, is subsequently evaluated. A supplemental tool namely CCScanner was also developed to automate parameter fitting and optimization for a given sequence. Both methods were built into an ISAMBARD software package for integrated applications.709 The program was later upgraded to optimize the original architecture and interface for improved usability and scalability.710 CCBuilder 2.0 employs a generalized parametric description for coiled-coils with enhanced flexibility and modeling accuracy, including a modern implementation of SOCKET for geometric analysis. It provides the capability of building assembly models including oligomeric states beyond eight and collagens. The highly integrated package enables the direct feeding of structural data from native proteins to design models or predictions based on primary sequences.
General-purpose software suites such as Rosetta have also been employed for the design of coiled-coil based structures, the detailed discussion of which can be found in previous sections and will not be repeated here.180,546,661
The prediction and design tools introduced in this section are summarized in Table 5.
Table 5. Computational Tools for Coiled-Coil Prediction and Design.
| category | name | feature | ref |
|---|---|---|---|
| Coiled-coil prediction | COILS | Statistically controlled predictor based on the primary sequence. | (538, 690) |
| SOCKET | Identification of KIH packing to differentiate coiled-coils structures. | (700,701) | |
| PairCoil | Dimer prediction based on pairwise probabilities. | (692, 698) | |
| MultiCoil | Coiled-coil domain and dimeric/trimeric state prediction. | (694, 699) | |
| CCHMM_PROF | Hidden Markov Model based detection with evolutionary information. | (702) | |
| SCORER | Oligomerization predictions based on databased established by SOCKET. | (663, 693) | |
| RFCoil | Dimer/Trimer prediction through the random forest classification. | (703) | |
| LOGICOIL | Tetramers and dimers topology prediction. | (695) | |
| CCFold | Noncanonical model and repeats prediction. | (704) | |
| Waggawagga | Web-based comparative visualization tool. | (705) | |
| DeepCoil | Machine learning based algorithm to detects both canonical and noncanonical coiled-coil domains. | (707) | |
| Coiled-coil design | CCCP | Web interface for coiled-coil parameter fitting and backbone generation. | (545) |
| CCBuilder | Web-based application for generating coiled-coil models, built into an ISAMBARD software package with other tools including CCScanner, SOCKET, etc. | (154, 710) | |
| Rosetta | General-purpose software that can be used to design coiled-coils. | (180, 546, 661) |
8.4. Summary
Coiled-coils represent one of the most well-understood structures in protein science. Their abundance in nature and functional diversity provides us a rich database through which many of the fundamental principles of protein interactions and performances are elucidated. In the protein design field, coiled-coils serve as the molecular basis for many of the difficult or complex structures discussed in previous sections. Their simplistic parametrization and achievable superb structural stability provide well-defined models that enable further engineering without extensive concerns on disrupting the original conformations. The design of coiled-coil based peptides and proteins represents an extremely fruitful field both in soluble and transmembrane forms. While solubility is less of a discussion in this section, related topics were selectively covered so as to provide supporting information through which models and designs introduced in previous sections can be better explained. Nevertheless, with the help of greater computing power and novel algorithmic methodologies, the protein design field is likely to continuously benefit from structural and interaction considerations extrapolated through coiled-coil studies, as well as the integration of short coiled-coil fragments themselves. Novel designs with high order oligomerization, or as components for protein origami or bioelectronic interfaces, can be envisioned with high tunability and stability to enable new opportunities and applications in multiple fields.
9. Computational Algorithms and Application of Machine Learning Models
We have repeatedly stated in the previous sections that the development of protein design and accurate manipulation of protein solubility and stability have benefited significantly from the increasing computing power and novel algorithms, since it is a computation intensive task. This is a 2-fold statement. On the one hand, the continuous advances in electrical engineering and nanotechnologies are enabling hardware evolutions that increase the calculation power by orders of magnitude, as predicted by Moore’s Law.711 On the other hand, the continuous increase in the knowledge-base for structural information and biophysiochemical principles of proteins forms a positive feedback loop to computational biology allowing the development of novel algorithms to use calculation powers more efficiently, both to gain insights in native proteins and to design new species with predefined properties and functions. The most recent addition to the toolbox is the artificial intelligence/machine learning based approach, which not only advances our ability to predict structures and evaluate biophysical properties of proteins, but also offers a range of new options for the design of protein sequences and structures, from either natural templates or de novo.
In this section, we will discuss most recent developments in related computational programs. Multipurpose programs that can be adopted to a variety of tasks in structure prediction and design like Rosetta will be covered, with discussions dedicated to the design of water-soluble proteins.153,285−287,559,712 The recent achievement in deep learning-based algorithms, AlphaFold2, will be introduced with extensive discussion of its potential and limitations in protein design.290,401 Specialty algorithms dedicated to the prediction and design of protein solubility and stability will also be covered.95,96,127 Since we are limited by the length of this article, many other useful programs for both structural prediction and protein design will not be included in this review. Interested readers should consult previous papers for their methodology and applications.96,153−156
9.1. Rosetta Software Suite
Originally developed for protein folding analysis and structure prediction in 1997, Rosetta has emerged undisputedly as one of the most recognized and extensively used software suites for macromolecular modeling and protein designs today, as shown in Figure 32.584 Over the past 20 years, the application of the Rosetta package has expanded to diverse tasks which include the protein–protein, protein–ligand or biomineral surface docking, loop modeling, experimental data assisted (such as NMR) modeling and protein designs. It is widely applicable to a variety of systems with different target molecules and complexity levels, ranging from noncanonical chemistries, peptides, membrane proteins, and protein assemblies to DNA and RNA. The RosettaCommons, a hub and virtual community of the software, expanded from a single academic institution to laboratories at over 70 institutions wordwide.713 Herein, we will provide a brief discussion on the software suite, its basic principles and major applications, and relevance to the topic of this review on the design of water-soluble proteins. For a thorough discussion on the Rosetta suite and its applications, readers should consult other dedicated reviews.584,714−716
Figure 32.
Characteristic tasks (blue) and major systems that can be computationally modeled (red) in the Rosetta software suite.
9.1.1. Basic Principles and Developments
Rosetta is a unified software package that is fundamentally based on the hypothesis by Christian Anfinsen that proteins spontaneously fold into minimal energy states accessible to the amino acid sequences.26,106 Similar to other structure prediction and design algorithms, there are two main tasks associated with this process, namely, (i) sampling all relevant conformational spaces in the case of prediction, or sequence spaces in the case of design, and (ii) ranking accurately the energies of the resulting structural models.714 A Monte Carlo annealing algorithm is adopted in this case to determine native protein conformations.717,718 The algorithm randomly inserts fragments from known protein structures into the models. The energy function is defined as the Bayesian probability of structure/sequence and exploited as a custom scoring function.719 For de novo structure prediction, Rosetta begins with an extended polypeptide chain and starts the folding by inserting backbone fragments with low resolution energy functions and extensive sampling. The number of resulting models is reduced based on the main chain RMSD before performing atomic-detail refinements to deliver the final predictions.719 For the protein design task, a target protein backbone is generated first, followed by the design and optimization of sequences with the lowest energy states.176
The software was first applied on short polypeptide chains with sequence similarity to a known nine-amino acid-long structural fragment to simulate its residue–residue interactions.720 With the limited amount of available structural knowledge for parametrization, the algorithm was able to make reasonable predictions of α-helical fragments and β-sheets, though to a lesser extent. The software was then extensively modified and improved to incorporate new sets of scoring functions that are more sensitive for hydrogen bond and electrostatic interaction computations,285,721 small- and biomolecule ligands docking,712,722 and a framework for membrane protein modeling (RosettaMP).723 The most significant progress was made with the program in the area of biomolecular designs.724,725
To date, the RosettaCommons provides flexible molecular modeling libraries to accomplish specific modeling tasks that are routinely used in the everyday “wet” or “dry” lab tasks such as the assessment of the effect for single point mutations in terms of the ddG (delta–delta-G) energies for both soluble and membrane proteins, and the design of fixed or variable-length loops for structural or substrate-binding applications, up to the whole protein or even protein heterodimer designs (Table 6). Most of the aforementioned tools can be run in a cloud using the Robetta server or as stand-alone applications.726 The different protocols can also be used individually or linked together to accomplish complex tasks.
Table 6. Featured Rosetta Software Suite Design Tools.
| • SEWING: | Build new protein structures from large elements (e.g., helix–loop–helix motifs) of native proteins. |
| • Loopmodel: | Design of fixed/variable size loops. |
| • Enzyme Design: | Design a protein around a small molecule, with catalytic constraints. |
| • Pepspec: | Evaluate and design peptide–protein interactions. |
| • Match: | Place a small molecule into a protein pocket so it satisfies given geometric constraints. |
Though versatile and flexible, the canonical Rosetta software suite has some inherent limitations. The first issue is related to scoring functions - the key tools for evaluating configurations and interactions of individual residues. Even the most sophisticated scoring functions rely heavily on the statistics obtained from the PDB.148 Though its size is estimated to approach 200,000 by 2023, less than half of this quantity represents truly unique sequences, with extreme cases such as human thrombin contributing over 450 structures to date. The other obstacle is the enormous number of unrestrained degrees of freedom due to the presence of rotatable bonds in proteins. Even with the most advanced force fields and newest computational techniques, specialized accelerating hardware such as Anton supercomputer or general-purpose graphical processing units had to be used to achieve sampling convergence. The computational complexity is especially pronounced for the de novo design of loops or whole proteins.727
9.1.2. Water-Soluble Protein Design with Rosetta
As has been discussed earlier, the buried hydrophobic amino acids are a primary driving force for protein folding.114 Increasing the difference in the buried hydrophobic surface between folded and unfolded states will stabilize proteins, whereas exposed hydrophobic residues impose energy penalties. However, in many cases, the Rosetta scoring function fails to prevent the formation of large hydrophobic patches on protein surfaces in its designs, although the average hydrophobic area (∼28%) is similar to naturally occurring proteins.49 The tendency reflects the favorable energetics of placing amino acids with similar properties together, but poses extra risks of inducing undesired hydrophobic effects that can result in aggregations.728
To solve this problem, Jacak et al. developed a new Rosetta scoring function in the algorithm using an hpatch score that explicitly disfavored large patches rather than the total hydrophobic surface area, so as to reduce the average size of surface hydrophobic patches on designed proteins.49 There are two implementations of the concept, namely, hpatch-fast, which assigns scores to surface residues based on the amount of exposed hydrophobic surface area (hSASA) within 10 Å of their vicinity, and hpatch-SASA which uses the exact SASA for each atom in the protein and scores explicit patches that span many residues. The hpatch-fast score starts with precomputed values for the calculation of average areas from each residue and often results in large hydrophobic patches since the contribution of individual residues depends extensively on their context and can extend beyond the 10 Å threshold. In contrast, the application of hpatch-SASA renders an obvious change on the surfaces of designed proteins, with a marked decrease in the size and number of hydrophobic patches as well as the proportion of hydrophobic residues, which resembles those from a water-soluble protein. Furthermore, the weight of the hpatch score can be adjusted to favor either the design of solubility or the maximization of free energy for protein folding, depending on the emphasis of the design.
The Rosetta algorithms have an impressive track record in the design of water-soluble proteins and complexes with high stability.288 These efforts date back to the design of Top7, a water-soluble de novo α/β protein with 93 amino acids, with its X-ray crystal structure in close agreement to the design model.159 The work conducted by Kuhlman et al. marked the first report of such efforts, outlining a general procedure to identify low free energy designs with multiple rounds of iterations cycling between the sequence and backbone optimizations with in silico structural predictions. The designed Top7 is highly soluble (at 25 to 60 mg mL–1) and thermally stable, similar to many of the aforementioned structures introduced in previous sections.
The software suite was later used for the design of water-soluble enzymes. Jiang et al. developed a general method to construct active sites for multistep catalytic reactions in a de novo enzyme with retro-aldolase activity.22 The key steps for the design included defining catalytic mechanisms and identifying scaffolds that can accommodate designed transition states. RosettaMatch was then used to reduce active-site possibilities in a given scaffold with poor catalytic geometries or significant steric clashes. Successful designs were selected based on predicted transition state binding energies, the catalytic geometry, and the consistency of side chain conformations. The designs are mostly soluble and exhibit atomically accurate X-ray crystal structures. However, whereas designs with explicit water molecules to mediate proton shuffling showed higher catalytic efficiencies compared to other models, none of the designs had a similar performance comparable to native proteins. Yet a similar approach to the design of catalysis for Kemp elimination resulted in enzymes comparable to the most active natural catalysts, that is, 5-nitro-benzisoxazole.729,730 Besides, many other water-soluble structures have been designed based on the Rosetta algorithms,169,283,730 whereas the scoring function and sampling methods were tailored for each specific target.288
9.2. Machine Learning Based Algorithms
An elegant way to overcome the excess of unrestrained degrees of freedom, specifically in the case of protein structure prediction, was again borrowed from evolution. Residues coupled through structural or functional links tend to coevolve together.731 This relation ought to be universal for both pro- and eukaryotes,732 and allows one to abstract extract spatial pairing information from evolution related sequences,733 even to actual pairwise distances between residues which later can be used as additional constraints during the molecular modeling.734 This type of inference became possible due to two main reasons: the advances of next generation sequencing that resulted in the accumulation of huge sequence databases of evolution related sequences,735 and advances in the artificial intelligence software. Altogether, this led to the breakthrough in the precision of protein structure prediction in 2018 during the CASP13 challenge.
Yet it was not until 2020 and CASP14 when the real revolution happened with the introduction of the end-to-end models and self-attention mechanisms.736 The main drawback of the previous implementation was due to crucial pivot points during structure prediction. After the generation of pairwise constraints or even probable distributions of torsion angles,737 all the information was passed to classic physics-based modeling methods such as the L-BFGS optimization algorithm to perform actual folding and local refinements.738 Thus, there was not a direct link between input data and the final result which is crucial for efficient neural network training.739 The gap was closed with the introduction of SE(3)-Transformers, specifically, neural network equivariant under continuous 3D roto-translations. Equivariance is important to ensure the stable and predictable performance during geometrical operations on atoms’ coordinates which are required for protein structure generation.740 SE(3)-Transformers also natively support a self-attention mechanism, a computational approach based on the introduction of additional feedback loops between multiple data representations (for example, coevolution data extracted from multiple sequences alignments, predicted pairwise distances, and data from structural templates and crude models) to distill only the meaningful signals and to enhance the signal-to-noise ratio.400
Combined, the most recent advances in the area of highly accurate protein structure prediction resulted in two main algorithms: AlphaFold2 by DeepMind which dominated the CASP14 challenge, and RoseTTAFold by Baker and colleagues which was released later, but achieve comparable performance in terms of the prediction quality.290 Both algorithms were released as source code and web-services. Moreover, DeepMind in collaboration with the European Bioinformatics Institute released a database of predictions for the reference proteomes of 21 model species including mammals, bacteria, including pathogens, and plants.403
9.2.1. AlphaFold
AlphaFold was based on convolutional neural networks (CNNs).289,741 A protein-specific potential was trained and subjected to minimization by gradient descent to make accurate predictions on backbone torsion angles and pairwise distances between residues. Distance predictions provided richer training signals for the model with more specific structural information compared to contact predictions. In CASP13, the first generation AlphaFold created high-accuracy structures (with template modeling scores of 0.7 or higher, which measures the degree of match of a predicted backbone to the native structure)742 for 24 out of 43 free modeling domains, whereas the next best method, which used sampling and contact information, achieved such accuracy for only 14 out of 43 domains.289
As mentioned above, the latest version of AlphaFold published in 2021, namely AlphaFold2, represents a revolutionary milestone with structure prediction at the near experimental accuracy.290 In CASP14, AlphaFold2 vastly outperformed competing methods with a median backbone accuracy of 0.96 Å RMSD95 (Cα root–mean–square deviation at 95% residue coverage), compared to 2.8 Å RMSD95 from the closest competitor, as shown in Figure 33A.743 The all-atom accuracy was 1.5 Å RMSD95 for AlphFold2 with improved side chain conformation prediction over template-based methods, compared to 3.5 Å RMSD95 from the closest competitor. The method was also scalable to simulate longer polypeptide chains with pack between different domains within a protein.
Figure 33.

(A) AlphaFold2’s performance on the CASP14 set (N = 87 protein domains) relative to the top-15 entries (out of 146), group numbers correspond to the numbers assigned to entrants by CASP; error bars represent the 95% confidence interval of the median, estimated with 10,000 bootstrap samples. (B) Prediction of CASP14 target T1049 (blue, PDB ID: 6Y4F) compared to the experimental structure (green). (C) Example of a well predicted zinc binding site (PDB ID: 6YJ1). (D) CASP target T1044 (PDB ID: 6VR4), a 2,180-residue single chain, predicted with correct domain packing. (E) Model architecture of AlphaFold2. Arrows show the information flow among the various components. Array shapes are shown in brackets with s: number of sequences, r: number of residues and c: number of channels. Reprinted with permission from ref (290) under Creative Commons licenses.
The additional evolutionary, physical, and geometric constraints incorporated into the neural network architecture accounted for the accuracy improvement, which consisted two main modules, Evoformer and a structure prediction module. The methodology started from multiple sequence alignments (MSAs), and replaced 2D convolution with an attention mechanism better representing amino acid interactions. The 3D coordinates for residues were generated by a two-track network where information at sequence level and distance map level was iteratively transformed back and forth for optimization. An SE(3)-equivariant transformer was then used to refine the atomic coordinates. Accurate end-to-end structure prediction was achieved in which all parameters were optimized by the reverse propagation from final coordinates back to the input sequence. The schematic workflow of the algorithm and example predictions are shown in Figure 33.
With demonstrated utility to the experimentalist community, the use of AlphaFold2 was expanded to the human proteome. In a concurrent report, the structures of almost the entire human proteome (98.5% of human proteins) were predicted using AlphaFold2, with an upper length limit of 2700 residues, whereas only 17% of the total residues in human sequences have been covered by experimentally determined structures within last few decades.403,744 While previous large-scale prediction efforts mainly focused on domains, AlphaFold2 processed the full-length of proteins to mitigate the risk of missing unannotated structural regions and to include interdomain packing predictions. A per-residue confidence metric named pLDDT (predicted local distance difference test) was introduced to evaluate the quality of predictions on a scale from 0 to 100, whereas pLDDT > 90 was the high accuracy cutoff. The resulting data set covers 58% of residues with a decent confidence level (pLDDT > 70). The predictions were released to the community via a public database (https://alphafold.ebi.ac.uk/), along with the source code on GitHub and web service of AlphaFold2 (https://github.com/deepmind/alphafold/tree/main/alphafold).
9.2.2. Structure Prediction with RoseTTAFold
In parallel, BaeK et al. published the RoseTTAFold package, which was inspired by AlphaFold and performed nearly as well as AlphaFold2.400 As a “three-track” neural network, RoseTTAFold simultaneously considers protein sequence patterns, amino acid interactions, and proteins’ possible 3D structures with atomic coordinates (Figure 34). Similar to AlphaFold2, the architecture allows the network to transform and integrate information between sequences, distances, and coordinates. Improved performance of the three-track model over two-track model was demonstrated with identical training sets. The inferior performance of RoseTTAFold compared to AlphaFold2 in the CASP14 was attributed to a deficiency in the deep learning algorithm and computing power limitations. The source code for RoseTTAFold is also freely available on web servers, which allows scientists to build on their effort and explore areas of intense interests both for the structure prediction and protein design.745
Figure 34.

Network architecture and performance of RoseTTAFold. (A) RoseTTAFold architecture with 1D, 2D, and 3D attention tracks. Multiple connections between tracks allow the network to simultaneously learn relationships within and between sequences, distances, and coordinates. (B) Average template modeling score of prediction methods on CASP14 targets. Zhang-server and BAKER-ROSETTASERVER were the top 2 server groups, while AlphaFold2 and BAKER were the top 2 human groups in CASP14; BAKER-ROSETTASERVER and BAKER predictions were based on trRosetta. Predictions with the two-track model and RoseTTAFold (both end-to-end and pyRosetta version) were completely automated. (C) Blind benchmark results on CAMEO medium and hard targets; model accuracies are template modeling score values from the CAMEO Web site. Reprinted with permission from ref (400). Copyright 2021 The American Association for the Advancement of Science.
9.2.3. Prospect of Using AlphaFold2 for Protein Design
It is tempting to consider AlphaFold2 a once and for all solution for protein folding, but there are inherent issues associated with the algorithm. First, not every part of a structure is predicted with the same reliability in AlphaFold2. The pLDDT is a good metric to assess this but does not provide information beyond references, although low and very low (less than 70 and 50, respectively) scores might indicate the presence of intrinsically disordered regions. Another metric is the predicted alignment error (PAE), which refers to AlphaFold’s expected position error at residue x if the predicted and true structures were aligned on residue y. Low PAE scores means that a spatial configuration of residues, especially if they belong to remote parts of the protein, can be believed.
Second, due to the nature of the evolutionary inference, AlphaFold2 relies on high quality MSAs. Thus, de novo designs, which have several to no related native sequences and shallow alignments can present a challenge for the implementation. With the self-attention component of the neural network, high-quality predictions might still be possible with a customized procedure with an increased number of internal prediction cycles. The other consequence of the MSA usage is the inability to capture the effect of single point mutations. The linkage between residues in the alignment will outweigh one or multiple single point mutations. However, other predictions tools such as FoldX can be used on AlphaFold2 structure templates.746
One other major deficiency of AlphaFold2 is the total unawareness of environmental conditions such as the pH, ion strength, ligand presence, membrane or solvent. This is an important consideration when the optimization of protein constructs, such that they would have no steric conflicts and additional groups, is concerned, especially when engineering disulfide-rich proteins. For example, AlphaFold2 was not able to capture the effect of 6 × His tag on the network of disulfide bridges. Experimental evidence showed, when placed at the N-terminus, 6 × His would disturb the native pattern of disulfide bridges, while AlphaFold2 predicted almost identical structures (Figure 35A).747 The same also goes for the pH, as the raw AlphaFold2 output contains only heavy atoms positions, with hydrogens added afterward with a common structural tool under the neutral pH assumption. However, those factors seem to be partially and implicitly captured since structures of transmembrane proteins such as GPCRs seem to be in agreement with the experimental structures obtained in conditions emulating membrane environments, as shown in Figure 35B.
Figure 35.

(A) Structures of rat corticotropin-releasing factor receptor type 2a predicted by AlphaFold2. Structures with 6 × His tag on the N-terminus, without one, and on the C-terminus are presented from left to right. AlphaFold2 failed to predicted the disturbance of the first disulfide bridge. (B) AlphaFold2 output structure on human CXCR2 chemokine receptor colored according to pLDDT scores with PAE plot on the bottom. (C) Conformational ensemble generated by AlphaFold2 compared to X-ray structures of the “open” (dark gray) and “closed” (white) AmiN kinase states. The AlphaFold2 structure resembles the “open” state.
The other practical limitations are very implementation dependent. When an object of interest is a flexible protein with multiple well separated conformations, such as GPCRs, enzymes, and especially kinases, even extended ensembles at the current stage do not guarantee sufficient sampling of possible conformational states (Figure 35C). Since MSAs of evolution related sequences stores enough information to evenly estimate the dynamics of structural domains, the ability to compare multiple predictions with different setup settings is essential.733
9.3. Algorithms to Predict Protein Solubility and Aggregation
Beyond structural predictions on a full-sequence level, there are more intriguing aspects of protein design that specifically concern their properties including the overexpression with native conformations.152,748 Besides the careful selection of host, optimization of expression condition, genetic codon usages and the denaturants utility, tweaking the amino acid sequences is a common method to enhance the solubility and expression of target proteins.749 While ultimately the effectiveness of modifications is determined by experimental verifications, in silico solubility predictions can greatly facilitate this process, reducing both the cost and labor in this trial and error procedure. Many computer algorithms were developed in the past decades to accurately and efficiently predict protein solubility/aggregation to enable subsequent optimization and synthesis.750
9.3.1. Solubility Prediction Based on Primary Sequence
Since the primary sequence fundamentally defines proteins’ behavior in solvents, the solubility prediction from amino acid composition seems to be a reasonable route and was extensively developed.750,751 Several sequence-based features including the extent of charged and turn-forming residues, the level of hydrophobic stretches, and the length of the sequence have shown a strong correlation with the prediction results.96,458
Rawi et al. developed PRotein SolubIlity Predictor (PaRSnIP) to predict protein solubility using a gradient boosting machine.458 Two types of features were used as inputs in PaRSnIP to differentiate the soluble and insoluble sequences. First, characteristic sequences that can directly define frequencies of mono-, di- or tripeptides, the absolute charge, or frequencies of turn-forming residues, were included. Second, the structural information such as secondary structure and relative solvent accessibility were predicted using the SCRATCH suite.752 An overall prediction accuracy of ∼74% was achieved by PaRSnIP. Alternative algorithms such as CCSOL,462,463 Samak-Wang,465 PROSO,12 SOLpro,92 and Niu-Li753 were also used in this task with the support vector machine (SVM) as the core discriminative model.
More recently, Khurana and co-workers developed a deep learning -based solubility predictor called DeepSol without any feature engineering, which was the hallmark for prior efforts.96 DeepSol was directly applied to raw protein sequences, whereas the CNN framework exploited k-mer structures in the input sequence, and simplified the solubility prediction workflow by avoiding the procedure of extensive feature engineering. DeepSol and PaRSnIP used the same training set, and 3.5% higher accuracy was obtained with the DeepSol algorithm.
The primary sequence-based computational tools used for protein solubility predictions are summarized in Table 7.
Table 7. Computer Algorithms to Predict Protein Solubility Based on Protein Sequence.
| year | name | database | accuracy | Matthew’s correlation coefficient | ref |
|---|---|---|---|---|---|
| 2021 | GraphSol | 2737 | 0.78 | (456) | |
| 2019 | ProGan | 3148 | (457) | ||
| 2018 | DeepSol | 129643 | 0.77 | 0.55 | (96) |
| 2018 | PaRSnIP | 129643 | 0.74 | 0.48 | (458) |
| 2017 | Protein-Sol | 2359 | 0.90a | (93) | |
| 2016 | PON-Sol | 1080 | 0.49 | (459) | |
| 2014 | Niu-Li | 9500 | 0.88 | 0.76 | (753) |
| 2013 | ESPRESSO | 4922 | 0.68 | 0.42 | (460) |
| 2012 | PROSO II | 82299 | 0.75 | 0.39 | (461) |
| 2012 | CCSOL | 3043 | 0.74 | (462, 463) | |
| 2012 | SCM | 957/16902/82299 | 0.72 | (464) | |
| 2009 | Samak-Wang | 3173 | 0.8 | (465) | |
| 2009 | SOLpro | 17408 | 0.74 | 0.49 | (92) |
| 2007 | PROSO | 14200 | 0.72 | 0.44 | (12) |
| 1991 | PRSP | 81 | 0.88 | (466) |
Accuracy at 58% solubility prediction threshold.
9.3.2. Solubility Prediction Based on Protein Structure
In addition to the information encoded in proteins’ primary structures, the 3D conformation of proteins largely determines their interactions from different domains with the surrounding environment. Undoubtedly, the precision of in silico solubility prediction is likely to be improved with considerations of protein surface features.
The flexibility of protein structures plays an important role in the ligand binding, conformal variations, functions and stability. Bhandari et al. demonstrated that this structural flexibility can also be used as a feature in protein solubility predictions.95 They developed the SoDoPE (Soluble Domain for Protein Expression) algorithm with a predictor of “Solubility-Weighted Index”, which is a proxy for global structural flexibility. The program is also capable of conducting prediction on proteins with solubility-enhancing tags. An alternative methodology named SOLart utilizes solubility-dependent distance potentials to reveal the relation between residue–residue interactions and protein solubility. Hou et al. defined the solubility-dependent potentials in terms of the torsion angle values and solvent accessibility, and then integrated it with other features into a random forest model to predict the protein solubility and aggregations.754
MD simulation was also used to predict the recombinant expression of four-helix bundles and reported by Schaller and co-workers.755 The statistical models with a SVM classifier were established by determining the stability-related parameters using a thermal unfolding MD simulation.
While site-directed mutations are extensively used for the solubility enhancement in recombinant proteins, the accurate selection and rational design of mutation sites remain a challenge. Sormanni and co-workers developed a CamSol algorithm to solve the issue, which requires structural knowledge from the target protein to conduct accurate predictions.127 An initial solubility score based on the hydrophobicity, electrostatic charges and interplay within the protein is defined, while appropriate mutations with the maximal solubility and fundamental properties are identified by screening amino acid substitutions or insertions.
9.3.3. Aggregation Prediction
Aggregations are one of the most detrimental processes that negatively affect protein solubility and stability, which often happen when proteins fail to fold into their native structures. The tendency to aggregate can be alleviated by identifying and redesigning aggregation-prone regions on protein surfaces. One important area of research is the undesired aggregation into β-amyloids which is a ubiquitous process in biology observed in both physiological and pathological settings, as has been discussed in previous sections.756 Computational tools for aggregation prediction of proteins are summarized in Table 8.98,757
Table 8. Computational Tools for Aggregation Prediction of Proteins.
| year | name | characteristics | applications | ref |
|---|---|---|---|---|
| 2022 | AGGRESCAN3D 2.0 | Extends A3D to previously inaccessible proteins and incorporates new modules. | Prediction and optimization of protein solubility | (763) |
| 2015 | AGGRESCAN3D | Protein 3D structure as input, energetically minimized using the FoldX force field. | β-aggregate-prone region prediction | (762) |
| 2007 | AGGRESCAN | Aggregation prediction based on an aggregation-propensity scale for natural amino acids. | Aggregation-prone segment prediction | (761) |
| 2022 | TAPASS | A pipeline for annotation of protein amyloidogenicity in the context of other structural states. | Amyloidogenicity prediction | (779) |
| 2021 | SolupHred | The first dedicated software for prediction of pH-dependent protein aggregation. | pH-dependent protein aggregation prediction | (780) |
| 2021 | TISIGNER.com | Including TIsigner, SoDoPE and Razor for production improvement. | Protein expression and solubility optimization | (781) |
| 2021 | iAMY-SCM | A scoring card method-based predictor. | Amyloid protein prediction | (776) |
| 2021 | ANuPP | Taking into account atomic-level features of hexapeptides. | Aggregation nucleating region prediction | (775) |
| 2021 | AbsoluRATE | SVM based regression model to predict absolute rates of aggregation using experimental conditions and sequence-based properties. | Protein aggregation rate prediction | (97) |
| 2020 | WALTZ-DB 2.0 | The largest open-access repository for amyloid fibril formation determinants. | Aggregation-prone sequence prediction | (765) |
| 2010 | WALTZ | Combining the position-specific amyloid sequence with structural information. | Amyloid-forming sequence prediction | (764) |
| 2020 | CORDAX | Use crystal contact information to generate fibril cores from isolated PDB structures. | Aggregation-prone region prediction | (777) |
| 2020 | PATH | Comparative modeling, query sequence threaded into seven templates representing different structural classes. | Amyloidogenicity prediction | (778) |
| 2020 | AgMata | Unsupervised tool to predict β-aggregation in proteins. | β-aggregate-prone region prediction | (756) |
| 2018 | RFAmy | Feature extraction algorithms and classification algorithms improvement. | Amyloid protein prediction | (767) |
| 2015 | APPNN | Based on recursive feature selection and feed-forward neural networks. | Amyloid formation prediction | (769) |
| 2014 | PASTA 2.0 | Energy function rederived on a larger data set of globular protein domains. | β-sheet structure aggregation prediction | (772) |
| 2007 | PASTA | An energy function from the hydrogen bonding statistics on β-strands. | β-sheet structure aggregation prediction | (771) |
| 2014 | FISH Amyloid | Based on site specific co-occurrence of amino acids. | Amyloidogenic segment prediction | (766) |
| 2011 | AmyloidMutants | Discrimination of topologically dissimilar amyloid conformations. | β-sheet structure aggregation prediction | (773) |
| 2010 | FoldAmyloid | Packing density and the probability of hydrogen bond formation. | Amyloidogenic region prediction in protein chains | (760) |
| 2009 | NetCSSP | Latest version of CSSP algorithm and a Flash chart-based graphic interface. | Chameleon sequence and amyloid fibril formation prediction | (774) |
| 2006 | 3D profile | Base on the crystal structure of the peptide NNQQNY. | Fibril-forming segment prediction | (799) |
| 2005 | Zyggregator | Protein aggregation prediction based on α-helix and β-sheet propensities, hydrophobicity, net charge of polypeptide, hydrophobic/hydrophilic patterns, and presence of Gatekeeper residues. | Aggregate-prone region prediction | (758, 759) |
| 2005 | PAGE | Aromaticity, β-propensity, charge, polar-nonpolar surfaces, and solubility are the factors employed for APR identification. | Protein aggregation rate and β-aggregate-prone region prediction | (768) |
| 2004 | TANGO | Protein aggregation prediction based on physicochemical principles of β-sheet formation in term of concentration, pH, ionic strength and TFE content. | β-aggregate-prone region prediction | (770) |
Many algorithms were developed for the prediction of amyloidogenic domains from the protein sequences. Tartaglia et al. proposed a Zyggregator method to predict the aggregation propensity of peptides and proteins.758,759 This algorithm can identify regions in the sequence of an unstructured peptide or unfolded proteins critical for aggregation based on the contributions from α-helix and β-sheet propensities, hydrophobicity, net charge of polypeptide, hydrophobic/hydrophilic patterns, and presence of gatekeeper residues.757
Additional parameters generated from protein structures were introduced in the FoldAmyloid algorithm.760 It was demonstrated that high packing density and propensity of backbone hydrogen bonds had an observable impact on amyloid fibril formation. The expected packing density and hydrogen bonding probability for each residue in the spatial structure were obtained first, and the average values for 20 amino acids were then calculated and used as expected values in the input sequence. FoldAmyloid classified correctly 75% of amyloidogenic peptides and 74% of nonamyloidogenic ones.
AGGRESCAN is based on an aggregation-propensity scale for natural amino acids derived from in vivo experiments.761 This algorithm evaluates the protein solubility and aggregation from the primary sequence and provides strategies for amyloidogenesis treatments. In addition, most in silico aggregation predictors failed to perform accurate predictions for the aggregation-prone region burial within native globular proteins, as they used the linear sequence as the input and assumed all aggregation-prone regions to be on protein surfaces. To circumvent this limitation, Zambrano et al. developed the AGGRESCAN3D (A3D) server using a structure-based approach, which enabled spatially adjacent aggregation-prone amino acids to be specifically detected regardless of their position.762 The identified aggregation-prone residues can be mutated to design the soluble variants or to test the impact of pathogenic mutations by adding the A3D server. More recently, Ventura and co-workers published AGGRESCAN3D 2.0, which extended the application of A3D to previously inaccessible proteins and incorporated new modules to assist protein redesign, including stability calculation and solubility optimization using an experimentally validated computational pipeline.763
Many additional algorithms were developed for protein aggregation and amyloid formation predictions based on the sequence pattern. Maurer-Stroh and co-workers developed the WALTZ algorithm by combining the position-specific amyloid sequence information with structural information to identify amyloid-forming sequences.764 The updated database, WALTZ-DB 2.0, contains structural information on experimentally validated amyloid-forming peptides, which includes an in-house developed data set of 229 hexapeptides, manual curation of 98 amyloid-forming peptides, and novel structural information for peptide entries.765 It is the largest publicly available repository for experimentally determined amyloid-forming peptide sequences. The FISH Amyloid algorithm for the detection of amyloidogenic segments was proposed to identify the positions of residues with correlated occurrence over a sliding window of a specified length.766 Niu et al. also established a novel amyloid predictor named RFAmy using random forest algorithm based on compositional and physicochemical features from primary sequences.767 Tartaglia et al. developed PAGE to predict the aggregation rates and β-aggregate prone regions, based on combining known physicochemical properties of amino acids, along with computational simulations of β-aggregating peptides.768 Phoenix and co-workers developed a highly accurate and effective method named APPNN to predict the amyloid propensity from the polypeptide sequence, based on a small subset of highly relevant physicochemical and biochemical amino acid properties.769 The predictor showed sensitivity, specificity, positive predictive value, negative predictive value, and overall accuracy of 87.4%, 78.9%, 90.9%, 72.3%, and 84.9%, respectively.
Alternatively, the aggregation predictions were performed based on the secondary structure propensities. A predictor named TANGO was developed by Fernandez-Escamilla et al. to predict protein aggregations based on physicochemical principles of β-sheet formation in terms of concentration, pH, ionic strength, and TFE content.770 Four conformational states and different energy terms including hydrophobicity and solvation energetics, electrostatic interactions, and hydrogen bonds were incorporated into the algorithm, which correctly predicted 87% of the peptides corresponding to sequence fragments from 21 different proteins.
The PASTA server was developed by Trovato et al. to predict the most aggregation-prone domains based on the hypothesis that the mechanisms and underlying physics governing β-sheet formation in native proteins also hold for β-sheet formation in amyloid aggregates.757,771 This approach associates energies to corresponding β-strand intermolecular hydrogen bonds between amyloidogenic sequence stretches, and further assumes that distinct protein molecules involved in fibril formation will adopt the minimum energy β-pairings to better stabilize the cross-β core. Furthermore, the PASTA 2.0 server, which was rederived on a larger data set of globular protein domains, was developed to evaluate the stability of putative cross-β pairings between different sequence stretches.772
AmyloidMutants was developed by O’Donnell et al. to carry out de novo prediction of wild-type and mutant amyloid structures that can discriminate between the dissimilar amyloid conformations with the same sequence locations, which energetically quantifies the effect of sequence mutations on fibril conformation and stability.773 Kim et al. proposed a unique and sensitive contact-dependent secondary structure propensity (CSSP) algorithm to detect local conformational changes in protein sequences, whereas NetCSSP provided an efficient approach to calculate the CSSP values for amyloid fibril formation prediction.774
More recently, a web server called ANuPP (aggregation nucleation prediction in peptides and proteins) was developed by Prabakaran et al. to predict both amyloidogenic hexapeptides and aggregation-prone regions in proteins using atomic-level features.775 ANuPP was trained using a data set of both amyloidogenic and nonamyloidogenic hexapeptides, which was divided into two parts: 90% for training and cross-validation and 10% as a blind test evaluation set. It showed an accuracy of 83% with an AUC of 0.883 in the blind test data set of 142 hexapeptides and an average segment overlap score of 48.7% for identifying aggregation-prone regions in 37 proteins. It is the first sequence-based algorithm that uses atom-based features and considers diversity of aggregation mechanisms. Alternatively, Orlando et al. proposed the AgMata algorithm to calculate the aggregation propensity by predicting regions primed to form strong β-sheet-like interactions.756 Unlike previous approaches, AgMata is completely unsupervised without any trained aggregation set and can define the interaction energy between residues beyond amino acid types. A scoring card method-based predictor, namely iAMY-SCM, was also proposed by Charoenkwan et al. to predict and analyze the amyloid protein based on a simple weighted-sum function in conjunction with the propensity score of dipeptides.776
Louros et al. developed the CORDAX algorithm to explore the amyloid sequence using the high-resolution structural information in amyloid cores currently available in the PDB.777 It not only detects aggregation-prone regions in proteins, but also predicts the structural topology, orientation and overall architecture of the resulting putative fibril core. Wojciechowski and Kotulska developed a novel structure-based method for predicting amyloidogenicity called PATH based on threading potentially amyloidogenic sequences on zipper-like amyloid structures, corresponding to all representative and experimentally confirming structural classes of short amyloids.778 An affinity of sequences to each structural class was evaluated with regard to the total energy, and a model with the minimal statistical potential was assumed to be the most stable and accurate.
Rawat et al. developed an AbsoluRATE algorithm to predict the aggregation kinetics of native proteins, which used an SVM based regression model to calculate aggregation rates using parameters of environmental conditions, disorders, and aggregation propensities.97 Moreover, TAPASS developed by Falgarone et al. carries out amyloidogenicity prediction in the context of other known or predicted structural states, which was used in the proteome-wide analysis to discover new amyloid-forming proteins.779 Ventura and co-worker developed a web-based interface called SolupHred that implements the aforementioned theoretical framework to compute aggregation propensities of intrinsically disordered proteins as a function of pH.780 This algorithm calculates the sequence lipophilicity and net charge at each pH through an empirical equation. Moreover, Bhandari et al. developed a web service named TISIGNER.com, combining TIsigner (Translation Initiation coding region designer), SoDoPE (Soluble Domain for Protein Expression), and Razor for recombinant protein production improvements, which specializes in the synonymous optimization of recombinant protein expression, as well as solubility and signal peptide analysis.780,781
9.4. MD Simulations on Protein Solubility and Aggregation
MD simulations was first developed in the late 70s and became an indispensable part of computational biology in proteins.782 The simulation objects progressed from hundreds of atoms to biological systems, including soluble proteins in explicit solvents, membrane proteins, or large macromolecular complexes.783,784 The improvement of calculation capacity is attributed to the development of both high performance computing and MD algorithms including the fine-tuning of energy calculations and parallelization.785 MD simulations are useful tools to elucidate protein structures and kinetics, as well as interactions with substrates and solvents at the atomic-level. MD simulations also include important sequence-dependent features or physicochemical properties during the computation of protein behaviors, which provided them more biological relevance comparing to deep learning based algorithms such as AlphaFold2.755
As important aspects of protein properties, solubility and aggregation of proteins were also analyzed by MD based algorithms. Taiji and co-workers investigated the role of individual amino acids to the solubility of proteins and peptides through MD simulations.786 Twenty-seven tetra-peptides were regularly positioned in 106 Å3 cubic boxes containing ∼3 × 104 water molecules and simulated for 100 ns using Amber and a standard force field without introducing any artificial hydrophobic interactions. The results correlated well with experimental data on solubility of amino acids and hydrophobicity scales.787,788
The solvation mechanism is crucial for solubility and aggregation prediction of proteins since solvent dynamics strongly influence the fibrillation of an amyloidogenic system. The most effective and simple approach is the explicit representation of solvent molecules, which can recover most of the solvation effects such as hydrophobic interactions induced from the entropic origin.785 Small- and wide-angle X-ray scattering (SAXS/WAXS) methods are commonly used as a structural probe to provide information about biomolecules in a given solution.789 Knight et al. developed a web server named WAXSiS (WAXS in solvent) to simulate SAXS/WAXS curves based on the explicit solvent molecular dynamics, the model of which accounts for both the hydration layer of biomolecules and the excluded solvent.790 Thermal fluctuations of water, side chains, and counterions are also considered during the computation. A MD based approach was adopted by Kumar et al. to reveal the inhibitive effects of bioactive molecules on I113T induced aggregation in superoxide dismutase type 1 and their capability to revert structural changes in terms of hydrogen bond pattern, flexibility, and conformational stability, which suggested a potential treatment for amyotrophic lateral sclerosis.791
Although explicit solvent models provide the most detailed and complete description of hydration phenomena, they demand high computational power due to the large number of atoms involved and the necessity to average over many solvent configurations for meaningful thermodynamic data.792 In contrast, implicit solvent models offer inexpensive alternatives for simulations in terms of protein folding, native fold recognition, small molecule hydration free energy prediction, and binding affinity prediction, which are widely used despite with less accuracy.793,794 Pronk et al. developed a high-throughput and parallel open source molecular simulation toolkit called GROMACS 4.5, which integrated several implicit solvent models, and new free energy algorithms.613 The algorithms can handle various biomolecules, including proteins, nucleic acids and lipids, and provide cost-effective, high-throughput computations. Implicit solvent simulations can reach a performance in excess of ms/day for small proteins. Gallicchio and Levy also developed an analytic implicit model AGBNP based on the pairwise descreening generalized Born (GB) approximation.792 The estimator decomposes the nonpolar free energy into a cavity component, proportional to surface area, and an attractive dispersion energy term, assuming that the solvent density outside the solute is homogeneous. Onufriev and co-workers then compared the speed of different conformational transitions using GB implicit model and particle mesh Ewald (PME) explicit model based MD simulations.795 The results indicated that speedups in conformational sampling for GB relative to PME are dependent on the actual system and cases, where one-fold to ∼100-fold speedup were achieved at same simulation temperatures. Two main factors are expected to contribute to this process, including effective solvent viscosity reduction and energy landscape alterations.
In addition, MD models were used to elucidate the solvent contribution at protein interfaces. Samsonov et al. developed a database of SCOWLP to investigate the contribution from solvent to interface definition for all protein complexes in the PDB.796 The dynamic and energetic properties of water-mediated protein interactions were characterized by comparing different interfacial interactions at residue and solvent levels, whereas the mobility, free energy, and conservation of interactions were investigated, proving the necessity of integrating solvent parameters in the development of energetic functions to describe protein–protein interactions. Bhunia and co-workers developed a tool of solvent relaxation NMR for the real-time monitoring of water dynamics in the protein aggregation landscape, and all-atom classical MD simulation was used to validate this method.797 MD simulation was applied to reveal a partially resembling pattern of water dynamics for fragment AV20 (A21-V40) of amyloid system Aβ40.
Furthermore, since the overexpressed recombinant proteins frequently misfold into inclusion bodies and deviate from the desired soluble and active forms, MD simulations were also used to evaluate the tendency of such processes. Schaller and co-workers established a statistical model using a thermal unfolding MD simulation to determine stability-related parameters and predict the recombinant expression of de novo four-helix bundles.755 The model successfully linked the in silico data and in vitro expression results. Besides, Zhang and Lazim adopted the MD simulation to predict the protein stability in specific buffer conditions. The stability of four apomyoglobin mutants were investigated in explicit 2 M urea solution at pH 4.2. Variations in the RMSD, native contacts and solvent accessible surface area were calculated to evaluate the effect of mutations on proteins’ overall conformation, whereas experimental results agreed with the in silico prediction.
In the pharmaceutical industry, aggregations are the leading cause of decreased antibody activity which activate undesired immunological responses. A SAP tool was developed by Chennamsetty et al. to identify the aggregation-prone regions of antibodies through atomistic simulation in an explicit solvent.452 The calculation was performed to characterize the extent of hydrophobic patches exposed on the antibody surface, which gave a unique SAP value to every atom and averaged to a final value.
9.5. Summary and Prospect
With the recent revolution in the field of highly accurate protein structure prediction, the full potential of machine learning based computational biology is yet to be revealed. Despite the aforementioned limitations in current approaches, the combination of these novel algorithms together with well-established molecular modeling techniques can alleviate the prior shortcomings and achieve previously unattainable accuracy. With additional biophysiochemical considerations, AlphaFold2 may open up new prospects for predictions of disordered regions, protein solubility, availability of proteolysis sites, localization signals, or oligomeric states. Combining these molecular modeling with experimental data from NMR, X-ray crystallography and Cryo-EM can significantly benefit the development of integrative structural biology.631
Beyond the prediction tasks themselves, high accuracy in simulated structures can provide valuable insights into the biological functions and mechanisms of proteins before their molecular structures are experimentally determined. This is especially the case for small molecule and protein binder designs, as well as the computational discovery of new ligands for targets of interest. Similarly important is the design for protein solubility. As mentioned in previous sections, it is crucial in this task to precisely identify the exterior and interior of protein surfaces, through which corresponding amino acid substitutions can be conducted to tune the solubility without disrupting core interactions and leading to the structure collapse.
On the other hand, besides the general-purpose prediction algorithms, many efforts have been made to develop specialty methodologies to evaluate specific aspects of protein properties, such as the programs we have introduced above on solubility and aggregation predictions. MD models are indispensable in this aspect, which provide accurate simulations on diverse range of protein structures, properties and interactions with high physiological relevance, especially when environmental factors are considered. Deep learning algorithms were recently extensively investigated, with helps to establish correlations between amino acid sequences and protein solubility/stability in a database of over 350,000 proteins from humans and 20 model organisms, including E. coli, yeast, and fruit flies.403,798 These specific in silico approaches can significantly facilitate empirical experimental optimizations, which are time-consuming and labor-intensive.
10. Cryo-Electron Microscopy
The introduction of high-resolution direct electron detectors for Cryo-EM, developed by Richard Henderson and colleagues at MRC-Laboratory of Molecular Biology, represents a breakthrough in structural biology.800,801 The efficiency of electron detectors combined with the imaging methods and computer programs for image processing have significantly accelerated high-resolution molecular structural determination.802,803
Structure information is not only critical in elucidating the molecular mechanism of naturally occurring proteins but also essential to guide the design of novel species. While traditional methods for structure determination include X-ray crystallography and NMR, Cryo-EM is an increasingly popular new player in the field.804 The technique allows near atomic resolution analysis of large and complex biological systems and biomolecules that are difficult for X-ray or NMR. Changes in the conformation or composition from the same sample can also be obtained to unravel dynamic states of macromolecules in native processes, providing insight into their conformational and energy landscapes.805,806 Prior to 2014, only 16 of Cryo-EM structures were deposited in the PDB, while 1753 structures have been collected at 2020 (Figure 36A). The proportion of structures with resolution higher than 4.0 and 3.5 Å increased from 36% and 12% in 2015 to 75% and 50% in 2020, respectively (Figure 36B).807 Because of its unique advantages, the technique is widely adopted in a variety of applications, especially for protein structure determination and drug discovery.
Figure 36.

(A) Absolute number and (B) percentage of newly deposited Cryo-EM structures in the PDB above given resolutions. Reprinted with permission from ref (807). Copyright 2021 Elsevier.
In this section, we will provide a brief discussion on this still under-development technique, in regard to the properties of proteins. We start with introduction on its background and technical workflow related to the stability of biomolecules under inspection. Instrumental limitations in resolution and biomolecule sizes along with efforts for improvements will then be reviewed. The unique advantages of Cryo-EM over traditional methods will also be presented. Circumventing back to the focus of our review, the application of Cryo-EM in the structural determination of membrane proteins will be covered. Future implications and potential impacts on the structural biology and protein design field will also be discussed.
10.1. Introduction of Cryo-EM
10.1.1. General Background
Cryo-EM is based on the transmission electron microscope (TEM) first developed in 1931 by Max Knoll and Ernst Ruska, and the rapid cryo-cooling technique introduced by Jacques Dubochet and Alasdair McDowall in 1981 when imaging macromolecular complexes.808 It involves flash freezing sample solutions and bombarding them with jet of electrons to produce microscopic images of individual molecules. Cryo-EM overcomes the main obstacles to the structure determination of biomolecules using TEM, namely (i) the impossibility of imaging biological samples in high vacuum and (ii) the damage induced by high energy electron beams on biomolecules. By freezing samples at specific time intervals, it can also visualize different states of molecules along a dynamic conformational transition. Jacques Dubochet, Joachim Frank and Richard Henderson were awarded the 2017 Nobel Prize in chemistry “for developing Cryo-EM for the high-resolution structure determination of biomolecules in solution”.809
In protein structure determination by single particle analysis (SPA) using Cryo-EM, the general workflow includes: sample optimization, EM grid treatment, image collection, and image processing.810 Among them, sample preparation poses the principal challenge since it determines the final state of biomolecules in the image acquisition.811 The step involves both sample optimization and grid preparation, the latter of which is broken down into sample carriers and treatments, deposition and vitrification (Figure 37A).
Figure 37.

(A) General procedures in the sample preparation for Cryo-EM SPA. Reprinted with permission from ref (811). Copyright 2021 Springer Nature. (B) Histogram of Cryo-EM structures deposited in the PDB between 2010 and 2019. The bar shows the number of structures deposited in each year, divided based on the reported resolution: lower than 4 Å (dark blue), 4 to 3.5 Å (pink), 3.5 to 3 Å (green), 3 to 2.5 Å (yellow), and above 2.5 Å (red). Reprinted with permission from ref (821). Copyright 2020 Elsevier.
10.1.2. Protein Stability Considerations
The quality of data is closely related to both the protein stability and the vitrification procedure. While the protein stability determines its orientation and dispersion states, proper vitrification can prevent the generation of ice crystals which disrupt both the conformation and spatial distribution of biomolecules. Denaturation or aggregation of proteins in the bulk solution can nullify efforts to get high-quality single-particle images. The salt concentration is an essential consideration in this regard since it can shield charges. Aggregation is also induced during the preparation of Cryo-EM grid due to the heterogeneous environment, which is especially the case for membrane proteins with their large hydrophobic surface patches. The denaturation and dissociation of large multisubunit protein complexes were commonly observed at the air–water interface.812 Membrane proteins often need further stabilization, by detergent, amphipols or nanodiscs to mimic the lipid bilayer environment.
Water evaporation is another factor that diminishes the sample integrity during grid preparation. It changes the temperature, pH, and salt and particle concentrations, which can subsequently induce conformational changes of proteins.813 The level of evaporation is dependent on the environmental humidity, temperature, exposure time and grid area. Under laboratory conditions at 20 °C and a relative humidity of 40%, the evaporation rate is in the order of 40–50 nm/s.814 While it can be reduced by increasing the humidity or working at a lower temperature, alternative approaches were also used to facilitate the handling process for better sample control and to minimize water evaporation.
For instance, Peters and co-workers developed a device named VitroJet to automate grid handling and minimize human interventions, which integrated the glow-discharge module, pin-printing for sample application and jet vitrification.815 The system can reduce the sample waste by subnanoliter surface deposition, and mitigate the evaporation by dewpoint control feedback loops. The VitroJet was used to prepare samples of apoferritin, GroEL, worm hemoglobin and beta-galactosidase proteins for high-resolution SPA. In another effort, Arnold et al. reported an integrated liquid handling setup named cryoWriter, where 3–20 nL sample solution was transferred by a capillary and subsequently delivered at the grid surface.816 The temperature of the stage and the grid was kept above the dewpoint in the room throughout the process to minimize water evaporation. The method also allowed rapid introduction of additives, such as detergent, at small volumes (<5 nL) by a diffusion driven process.817 The cryoWriter was used to prepare high-quality Cryo-EM grids of different biological samples, including soluble and membrane proteins, filamentous assemblies, viruses and cell lysates.
The shear force generated at the air–water interface is another detrimental factor contributing to protein denaturation and often considered impossible to avoid. It can cause flow-induced reorientation of filamentous macromolecular assemblies such as tobacco mosaic virus, microtubules, and actin filaments.818,819 Glaeser summarized common approaches used to protect particles against damages induced by such interactions at the interface, which include stabilizing the structure in solution, blocking the air–water interface using surfactants, minimizing interaction by ultrafast thinning and quenching, and immobilizing the particles on structure-friendly affinity films.818
10.2. Recent Developments of Cryo-EM Single Particle Analysis
10.2.1. Improvement on the Resolution
While both electron beams and X-rays can damage biological samples, a coherently packed large crystal is more tolerant to radiation compared to single particles. Since cumulative diffraction from more biomolecules render a higher signal-to-noise (SN) ratio and hence higher resolution, Cryo-EM was at first used only as a complementary technique to X-ray crystallography for the structure determination of large protein assemblies and dynamic complexes that are difficult or impossible to crystallize. Yet recent technological developments have enabled Cryo-EM to achieve resolutions comparable to the best of macromolecular X-ray structures, reaching as high as 1.2 Å.820 The number of publicly available high-resolution Cryo-EM structures has significantly increased in the past decades (Figure 37B).821 Nearly half of the ∼1200 Cryo-EM structures deposited in the PDB are better than 3.5 Å resolution in 2019.
Laverty and co-workers reported the effects of three technological developments, including a new cold field emission gun (CFEG), a new energy filter and the latest generation of Falcon direct electron camera, on data acquisition for a ∼200 kDa membrane protein, namely homopentameric β3 γ-aminobutyric acid type-A receptor (GABAAR).822 It belongs to a large family of pentameric ligand-gated chloride channels and are targeted by a wide range of psychoactive drugs.823 Yet the structure of GABAAR was previously limited to 2.5–3 Å resolution, which hampered their therapeutic developments.824 By alleviating the electron beam-induced sample damage, the combined setup as described above increased the SN ratio of Cryo-EM images for GABAAR particles and achieved 2 Å resolution in the resolved structure. Similarly, the mouse apoferritin was imaged on the CFEG microscope, with the Falcon-4 camera and 10 eV slit width in the energy filter, where a structure reconstruction with 1.22 Å resolution was obtained from 3370 videos.820
10.2.2. Improvement on the Size Limit
Another technological limit imposed by Cryo-EM SPA is its lower size limitation on the sample being inspected. By applying instrumental developments such as new energy filters and Volta phase plate technologies, such limits are continuously challenged.809 Yet most of the protein structures determined through Cryo-EM are still above 100 kDa and with a lower limit currently at ∼50 kDa, which is larger than the average size of monomeric cellular proteins.825 Recently, Chiu and co-workers expanded the applicability of Cryo-EM to RNAs with a minimum MW of ∼40 kDa, which is lower than the smallest protein structure (Streptavidin, 52 kDa) determined.826,827 While it is generally difficult to obtain high-resolution structures of pure RNAs due to their high flexibility, the structures of SAM-IV riboswitches (119-nt, ∼40 kDa), a metabolite-binding downstream regulator, were determined in complex with apo and ligand at 3.7 and 4.1 Å resolution, respectively. The study demonstrates the use of Cryo-EM to determine the structural basis for ligand recognition by noncoding RNAs, in addition to illustrating its application beyond the current size limit of proteins.
An approach to overcoming this size limitation is to form a rigid complex between the small protein and a monoclonal antigen-binding fragment (Fab), which increases the effective mass and facilitates the image alignment. Small proteins can also be designed to self-assemble into large and symmetric structures resembling geometric solids, through multiple copies of a single subunit.828,829 For instance, Liu et al. designed a modular, symmetrical scaffold to connect a small 17 kDa protein (DARPin), and the resulting construct is amenable to be visualized using Cryo-EM SPA with local resolutions ranging from 3.5 to 5 Å (Figure 38).830 The approach was based on a modular 24-unit protein cage design comprising two types of subunits, each of which formed four trimers.169 DARPin was fused to the α-helical terminus of the cage protein to create a semirigid and geometrically predictable helical connection.831 The 3D Cryo-EM structure reconstruction was performed on the fused complex, and a total of 3665 movies were recorded, resulting in 229,953 particles for analysis. An atomic resolution of ∼2.5 Å at core and an overall resolution of ∼3.1 Å were obtained based on a subset of 34,650 particles. Furthermore, the loop sequences in DARPin can be designed to introduce additional target proteins to the scaffold in their native forms, which creates a distinct strategy to obtain high-resolution structures of small proteins.
Figure 38.

(A) Schematic diagram for a scaffolding system built on a designed symmetric protein cage. (B) Cryo-EM structure of DARP14 symmetric cage core with subunits colored as in A (PDB ID: 6C9K). (C) Representative motion-corrected micrographs of DARP14. (Inset) Fourier transformation showing visible thon rings to ∼3 Å. (D) Reference-free 2D class averages highlighting good alignment of the cage and clear density for fused 17 kDa DARPins. (E) Overview of a ∼3.1 Å reconstruction of the cage core. Left: representations of unfiltered local resolution viewing down the 3-fold and 2-fold symmetry axis highlighting extensive areas at an atomic resolution of ∼2.5 Å. Top right: Fourier shell correlation curves of unmasked and masked reconstructions. Bottom right: refined models fit into density for subunit A (yellow) and subunit B (blue) of the cage core. Reprinted with permission from ref (830). Copyright 2018 National Academy of Sciences.
Similarly, Yao et al. implemented a DARPin-Aldolase platform in which a small protein can be attached to a large and symmetric base via a selectable adapter.832 DARPin was rigidly fused to the first α-helix of tetrameric rabbit muscle aldolase through a helical linker to form the platform base. The Cryo-EM study of GFP complex using the platform rendered an overall 3 Å resolution, with 5–8 Å resolution in the GFP region. Another approach to generate large enough complexes was developed by Carlo Petosa and co-workers, by fusing a monomeric target to a homo-oligomeric protein for Cryo-EM SPA.833 The complex was constructed by fusing native maltose-binding protein to glutamine synthetase, and subsequently connecting with the target monomer via a short linker.
10.2.3. Other Advantages
Besides the apparent advantage in determining the structure for assemblies and difficult-to-crystallize proteins, the nature of data acquisition by Cryo-EM also allows more flexible sample manipulations. For instance, a DNA origami approach was adopted to design a molecular support with a defined interaction for transcription factor p53 in one Cryo-EM study.834 A hollow pillar with a honeycombed motif of 82 parallel double-stranded DNA helices and a height of 26 nm was constructed, where tailored positioning of the p53-binding DNA sequence in the central helix spanning the hollow support allowed specific binding and partial orientation control of the protein. The 3D support also shielded the target protein against shear forces on the air–water interface through the sample preparation process, and spontaneously formed monolayers in water to allow easy particle picking.
Another technological advantage of Cryo-EM over traditional X-ray crystallography is its rapid response capability to new and emerging targets since it does not require time for the crystal growth condition optimization. The recent outbreak of COVID-19 presented a perfect example. The structure of the spike protein and the antibody or ACE2 binding to the spike protein of SARS-CoV-2 were resolved within several months.835−837 Later, the structure of Remdesivir binding to RNA polymerase (RdRp) of coronaviruses was also rapidly determined using Cryo-EM SPA.838 Cryo-EM has become the primary technique for fast and high-resolution structure determination of biomolecules and complexes.
10.3. Cryo-EM Applications for Membrane Proteins
One of the forefront challenges in biology that has benefited most from Cryo-EM SPA is the structural determination of transmembrane proteins.807 As mentioned in previous sections, these groups of proteins are indispensable in biological processes, many of which, including GPCRs, ion channels, and transporters, are potential drug targets and shed profound implications in therapeutics. Despite wide interests on their structures and functional mechanisms, transmembrane proteins are hard to study due to the hydrophobic nature and tendency to aggregate in solution, nevertheless to say, the difficulty in forming ordered crystals. Since protein molecules are individually prepared as vitrified suspensions, Cryo-EM supersedes X-ray crystallography in the aspect of sample preparation and quickly become the favorite technique for the structural study of membrane proteins, especially combined with recent developments in nanodiscs and nanobodies.839 Before 2014, all deposited Cryo-EM PDB entries of membrane proteins were from electron crystallography rather than SPA, whereas less than 5% of total membrane protein structures were determined by Cryo-EM.840 The number increased rapidly and was higher than that from X-ray crystallography in 2019. Seventy-seven out of the ninety-nine GPCR structures deposited in the PDB last year (Jan–Jul 2021) were determined by Cryo-EM (Figure 39A,B).841
Figure 39.

(A) Percentage of GPCR structures in the PDB determined by Cryo-EM per year. (B) Cumulative numbers of GPCR structures determined by X-ray crystallography and Cryo-EM, which include multiple structures of the same receptor bound to different ligands, intracellular binding partners, or non-native species. The data for 2021 includes only the first seven months of the year. A, B: Reprinted with permission from ref (841) under Creative Commons licenses. (C) Schematic of the activation of a class B GPCR by extracellular peptide agonist via a “two-domain” binding mechanism. (D) Cryo-EM density map of the GLP-1R:Gs complex, colored by subunits (transmembrane domains in light green, NTD in dark green, GLP1 peptide in orange, Gαs Ras-like in gold, Gβ in light blue, Gγ in dark blue, and Nb35 in gray). GLP-1R (PDB ID: 5EE7). (E) Structure of the activated GLP-1R:Gs complex in the same view and color scheme as shown in D. C–E: Reprinted with permission from ref (842). Copyright 2017 Springer Nature.
Although Cryo-EM has proven to be a suitable approach for determining the structure of membrane proteins, technical limitations such as the protein size limit still posed difficulties in its applications. It is challenging to determine 35–45 kDa GPCRs in the inactive state using the technique.841 Hence, a fusion strategy between GPCRs and binding partners was commonly adopted to prepare protein complexes with molecular weight high enough for Cryo-EM SPA. Zhang and co-workers determined the structure of glucagon-like peptide 1 receptor (GLP-1R) through a complex of the active-state GLP-1R and heterotrimeric G protein Gs (150 kDa) (Figure 39C–E).842 The GLP-1R:Gs complex was formed before the membrane extraction and purification to stabilize GLP-1R, using maltose neopentyl glycol as the detergent. A 4.1 Å global resolution and 3.9 Å nominal resolution were subsequently achieved on the 3D reconstruction of the complex structure. More recently, Cao et al. determined the agonist-stabilized structures of MRGPRX2 coupled to Gi1 and Gq in ternary complexes with the endogenous peptide cortistatin-14 or a synthetic agonist probe.843 MRGPRX2 is one type of mas-related GPCR that can effectively couple to α-subunits of both Gq and Gi in nearly all G protein families. Using (R)-ZINC-3573 and cortistatin-14 as agonists, the complex structures of MRGPRX2 with Gq and Gi1 were determined with global resolutions of 2.9 Å, 2.6 Å, 2.45 Å, and 2.54 Å, respectively. In another effort, a GPCR-G protein-β-arrestin (βarr) megacomplex was constructed to determine the structure of β2 adrenergic receptor (β2AR). The β2V2R-βarr complex was coexpressed in SF9 cells and stabilized by Fab30 with the detergent lauryl maltose neopentyl glycol. Two nanobodies, that is, nanobody 32 (Nb32) and nanobody 35 (Nb35), and the heterotrimeric Gs proteins were added to form the final megacomplex. However, the resolution of GPCR-G protein-βarr complex did not exceed 7 Å due to the flexibility of each component. Furthermore, Shaye et al. conducted a mechanism study by determining structures of human full-length GB1 (86 kDa) - GB2 (88 kDa) heterodimers that belong to the Metabotropic γ-aminobutyric acid receptors (GABAB).844 The unique activation mechanism of GABAB was elucidated through resolving four structures representing different activation states, namely an inactive apo form at 4 Å resolution, two intermediate agonist-bound forms at 3.6 Å resolution and an active form.845,846
On the other hand, although Cryo-EM SPA has abolished the necessity for crystallization, the protein stability can still be an issue especially for membrane proteins like GPCRs. Although detergents or detergent-like amphipols can be added to enhance their stability, such a substantial amount of unstructured mass around the transmembrane domain decreases the SN ratio and hamper the overall resolution in structures.840 Alternative approaches to enhance protein stability include binding a high-affinity ligand, engineering the receptor through amino acid mutations or binding an antibody,847−849 to obtain biochemically well-behaving proteins for analysis. Stable biochemical properties and a homogeneous conformation among vitrified particles can significantly facilitate data analysis and lead to high-quality structures.840 Recent developments in nanobodies and nanodiscs have significantly contributed in this aspect.850,851 They both provided platforms to stabilize the conformation of membrane proteins through interacting with different regions of the target. Zhang et al. determined the complex structure of neurotensin and neurotensin receptor 1 with Gαi1β1γ1 embedded in lipid nanodiscs at resolutions of 4.1 and 4.2 Å, respectively,852 whereas nanobodies also helped to determine structures of multiple GPCRs in activated, inactive, or transient signal transduction states.849
10.4. Summary and Prospect
Overall, Cryo-EM has become a favorite tool in structural biology, especially with membrane proteins and large protein assemblies. While the technique falls short in the resolution and limitation on target MW which prevent directly imaging average size cellular proteins, it outperforms X-ray crystallography in the response time and capability of monitoring dynamic states of biomolecules through flash freezing, and possesses distinct advantages on new and emerging targets. Such characteristics also drew extensive interests beyond the scope of biological applications and expanded its use in other fields.853
As a rapidly growing methodology with continuous instrumental developments contributing to overcome its technical drawbacks, Cryo-EM also benefited significantly through combining with computational methods. The algorithmic integration of CNNs into density map refinements has proven effective for the automated structure determination of assembled protein fragments and likely to enhance the analysis accuracy for unknown targets.854 It was also suggested that the AlphaFold2 algorithm, as introduced in the last section, can be used to build atomic models in Cryo-EM using ChimeraX. Such combinations can render a 2-fold advantage when protein designs are concerned. On one hand, computational designs can help to tune the solubility and stability of proteins and complexes to suffice the sample requirements for quality data acquisition by Cryo-EM SPA, especially when difficult targets such as membrane proteins are studied. On the other hand, Cryo-EM offers a fast-turnout approach for the rapid verification of synthesized proteins as compared to their design models. It is likely that the combination of the two can provide an effective approach to produce novel biomolecular species with tailored functions while fueling the knowledge of molecular biology in general.
Both inside cells and in solution, protein structures are likely very dynamic especially when they perform their biological functions including catalysis, signal transduction, ligand binding, interactions with other molecules, and work processing such as in the DNA replication, RNA transcription and protein synthesis. X-ray crystallography can capture a single and perhaps the most stable state of the protein, like a single frame in a movie. However, it is unable to capture the rapidly changing structures like in a film moving at 24 frames per second. On the other hand, Cryo-EM may record many states of a single molecule, like many frames in a movie. We eagerly look forward to future Cryo-EM experiments that can reveal the whole mechanistic procedures and multistates of a single protein in active biological processes.
11. Conclusion and Outlook
The last 70 years have witnessed the rapid advancement of human knowledge in understanding in ever-increasing detail how biology works at the molecular level. There are many remarkable findings and technological advancement which include: (i) discovery of the structure of the DNA double helix, as Francis called it “discovery of the secret of life”, thus understanding how heredity is passed on to generations, (ii) deciphering the universal genetic code, (iii) molecular biology of recombinant DNA, namely, gene cloning and expression in heterologous systems, (iv) precise mutagenesis to alter a single base pair in a gene at will, (v) rapid affinity protein purifications, (vi) rapid DNA sequencing and gene synthesis at a significantly reduced cost that completely transformed the protein design field so that researchers are able to simultaneously change 100 amino acids in a single protein in one run, (vii) high-resolution determination of protein structures by synchrotron X-ray crystallography, NMR, and Cryo-EM, (viii) synthetic biology to design genes, gene circuits, and genomes as well as de novo protein designs, (ix) significant increase in the computing power and databases, and (x) the advent of artificial intelligence and machine learning that ushered the highly accurate protein structure predictions freely available by AlphaFold2 and RoseTTAFold. All these technologies contributed enormously to the current protein design field, enabling ever-more precise understanding and manipulation of protein structures and functions.
In this review, we focused on the water solubility and structural stability of protein design (see summary of research milestones in Table 9), which are the prerequisites for native functions of these biomolecules. The review covered two major aspects of this subject, namely, the manipulation of solubility and stability in native proteins, and the design of de novo structures with target structures and functions.
Table 9. Timeline of Important Research on Water Solubility Design of Proteins.
| 1981 | Determination of affinities of amino acid side chains for solvent water (Wolfenden et al.).30 |
| 1988 | De novo design of a globular α-helical protein capable of adopting a stable, folded structure in aqueous solution (DeGrado’s lab).855 |
| 1989 | (i) Laying the ground for transmembrane protein design through the solvent-exposed hydrophobic residue exchange, and suggesting that transmembrane and soluble proteins share similar core structures with different solvent-exposed residues (Eisenberg and Rees).208 |
| (ii) Use of a short hydrophilic sequence to solubilize penicillin-binding protein 5 with the C-terminus truncation for crystallization (Ferrerra et al.).407 | |
| 1993 | De novo design of soluble four-helix bundle proteins by the binary patterning of polar and nonpolar amino acids (Hecht’s lab).856 |
| 1994 | De novo design of water-soluble multiheme proteins (DeGrado’s lab and Dutton’s lab).484 |
| 1997 | (i) Computer assisted fully automatic sequence design and validation of soluble FSD-1 protein (Mayo’s lab).857 |
| (ii) All theoretical design of soluble bacteriorhodopsin (Gibas and Subramaniam).250 | |
| 2000 | Design of water-soluble variants for phospholamban thru mutating lipid facing amino acids (Engel’s lab, Engelman’s lab, and DeGrado’s lab).43,359,360 |
| 2002 | (i) Rational design of water-soluble bacteriorhodopsin with 14.9% sequence change and limited solubility, still required detergents and lost purple color (not functional) (Engelman’s lab).251 |
| (ii) Conversion of transmembrane toxin aerolysin to a soluble complex through single point mutations (Van der Goot’s lab).222 | |
| 2003 | Enhancing solubility at the physiological pH and folding condition of ankyrin repeat proteins by substituting surface exposed leucine with arginine (Peng’s lab).444 |
| 2004 | (i) Design of water-soluble analogues for potassium channel KcsA, 160 aa protein (33/160 aa changes equal to 20.6%) that conducted ion transport (DeGrado’s lab).226 |
| (ii) MscL channel protein solubilization through the stoichiometric covalent modification with amphiphiles (Becker and Kochendoerfer).237 | |
| 2005 | Determination of the tetrameric structure for the water-soluble truncated phospholamban and elucidation of sequence determinants that defined coiled-coil symmetry (DeGrado’s lab).215 |
| 2006 | Redesign of a potassium channel with unknown structure by conservation pattern analysis (Roosild and Choe).235 |
| 2008 | Design and NMR structural study of solubilized KcsA analogue (33/81 aa changes = 40.7%) (Xu’s lab).234 |
| 2010 | Design and X-ray crystal structure analysis of a water-soluble form of cross-β architecture (Koide’s lab).172 |
| 2012 | Design and NMR structure determination of water-soluble nicotinic acetylcholine receptor (23/140 aa changes = ∼17%) (Xu’s lab).254 |
| 2013 | (i) Design of truncated water-soluble human mu opioid receptor (53/288 aa changes = 18%), detergent still needed for purification (Saven’s lab and Liu’s lab).262 |
| (ii) Design and functional study of MotB by transmembrane segment swapping with leucine zipper (Andrews and Roujeinikova).239 | |
| 2014 | (i) De novo design and X-ray characterization of water-soluble α-helical barrels with central pore diameter related to the oligomeric state (Woolfson’s lab).160 |
| (ii) Optimization on the transmembrane region design of water-soluble human mu opioid receptor (46/288 aa changes = 16%), with deteriorated protein performance (Saven’s lab and Liu’s lab).265,266 | |
| 2015 | Development of a protein (ApoAI*) fusion strategy (SIMPLEx) to solubilize 10 types of structurally irrelevant transmembrane proteins (DeLisa’s lab).267 |
| 2016 | (i) Engineering soluble human paraoxonase 2 without disturbing its folding for quorum quenching (Ge’s lab).412 |
| (ii) Design of internal hydrogen bond networks in water-soluble concentric coiled-coils (Baker’s lab).365 | |
| 2017 | (i) De novo design of water-soluble variants of light-harvesting protein maquettes (Moser’s lab).495 |
| (ii) Solubilization of membrane bound enzyme DsbB with the SIMPLEx strategy for in vivo and in vitro applications (DeLisa lab).272 | |
| 2018 | (i) Design and crystal structure determination of water-soluble coiled-coils (6/25 aa = 24%) that forms super helical cross-α amyloids (DeGrado’s lab).162 |
| (ii) Use of the QTY code to design five chemokine receptors (20–29% aa change) with native-like ligand specificity and Tm (Zhang’s Lab).274 | |
| (iii) De novo design of the water-soluble β-barrel structure with built-in affinity toward fluorescently activate DFHBI (Baker’s lab).171 | |
| 2019 | (i) Design of water-soluble GPCR based chimera receptors with tunable functions and high thermostability (Zhang’s Lab).277 |
| 2020 | (i) Design of full length water-soluble human mu opioid receptor with enhanced stability (Saven’s lab and Liu’s lab).264 |
| (ii) Design of truncated QTY chemokine receptors for the non-full length native receptor study (Zhang’s Lab).279 | |
| (iii) QTY code based partial solubilization of CXCR4 that retains cell function (Zhang’s Lab).280 |
The main focus of the first discussion was placed on transmembrane proteins, which represent indispensable sets of biofunctions in living organisms but are notoriously understudied due to the difficulty of overexpression and aggregation issues. In our review, the series of solubilization efforts were introduced including early transmembrane peptide assemblies, ion channels, to multipass membrane proteins and recent works on GPCRs. The reports marked continuous developments on the energy and scoring functions in computational algorithms, although mostly based on an early assumption proposed by Eisenberg and Rees in 1989, that soluble proteins and transmembrane proteins share similar core structures but different surface residues. Efforts from another direction, the de novo design of membrane inserting proteins, mark the inverse application of the same guiding rules that cross-reference with solubilization efforts. Under the common consensus of building a hydrophobic core with nonpolar residues buried in the interior of the designs, water-soluble and transmembrane models pose different requirements for the degree of complementarity in the close steric packing and internal hydrogen bonds, representing the differentiation in polar and nonpolar interactions at the interior of proteins. Most efforts built on or investigated such distinctions within the same framework. It was not until recently that an alternative methodology, the QTY code, was proposed and demonstrated. In contrast to the Eisenberg and Rees assumption, the QTY code is built on the hypothesis that the pairwise substitution between polar and nonpolar residues with similar side chain structures can accurately control the water solubility of a given protein, regardless of their locations in the structure. A correlation involving seven-amino acids was built to resemble those in the DNA code (and hence the name). Additionally, traditional solubilization techniques with fusion partners were also introduced in the review, which are widely adopted in other-than-transmembrane proteins. The successful design of transmembrane proteins by either method not only contributes significantly to the knowledge-base of this important class of proteins from a fundamental science perspective but also provides new pathways for further investigations and subsequent applications that were previously not attainable.
It should also be noted that beyond being individually functional, proteins are also capable of forming complexes, or supramolecular structures that account for higher-order symmetries or long-range ordering and functions. The accurate design of these structures relies on the precise installation of residue–residue interactions, whether steric, ionic, or hydrogen bonding. High complementarity and well-defined multipoint interactions are usually needed to ensure the correct formation of target structures with rigidity against distortions. The accurate design of the hydrogen bond network, especially in the interior of water-soluble proteins, remains technically challenging with limited success, as those interactions are otherwise disrupted and overpowered by solvent interactions if not well compensated and fully satisfied. With assistance in the design of soluble building blocks, many previously difficult or newly discovered structures, such as cross-α and cross-β amyloid fibrils, were successfully built and highlighted in this review. These examples demonstrate the proportional correlation between successful complex designs and our understanding of the structure–function relations in the field, which are likely to motivate subsequent interest and efforts in the study of fundamental protein biology.
Currently, the subjects of either discussion consist primarily of fundamental research, although opportunities to turn findings in those areas to biomedical and nanotechnological applications are beginning to be recognized. There is still remarkable potential to extend the frontier of the field beyond solubility and structural stability mediations. Many successful design models were reported with native or de novo integration of functional domains in proteins. We reviewed characteristic functional productions out of these design efforts with water solubility considerations, which ranged from integral soluble proteins with poor expression or stability, such as monoclonal antibodies, to light-harvesting maquettes with tunable regions of hydrophilicity, solubilized chemokine receptors with high thermostability and controllable ligand affinity, or de novo helical bundles with internal functional centers. As the design process becomes more intuitive with highly accurate structure predictions, the construction of new functional proteins becomes increasingly feasible. The simultaneous design of water solubility, stability and function in proteins opened up pathways for the utilization of these novel variants of functional biomolecules both in vivo and in vitro. We suggested that the potential applications for these new designs might include but are not limited to (i) novel therapeutics which utilize functional equivalents of native receptors, rather than targeting them, (ii) integrated vehicles for drug delivery and personalized medicine, (iii) biomimetic sensing platforms with high specificity and diagnostic precision, (iv) biocompatible human-electronic interfaces with molecular level integrations, and (v) functional supramolecular nanomaterials with hyperstability.
Over the last few decades, many computational tools and algorithms were developed by researchers to carry out protein folding and design tasks. We introduced two of the major players in this league, the Rosetta software suite and the recently released AlphaFold2, both of which marked the integration of deep learning algorithms into protein folding predictions. AlphaFold2 leads in the accuracy department but has yet to incorporate more versatile functions into their program. Either software is freely distributable and available as a web service to the wider scientific community, as well as to nonspecialty citizen scientists. Such availability is likely to significantly expand the overall protein biology field for application-oriented studies and opens up directions that might be neglected by protein biologists. The combination between widely accessible computer simulations and rational solubilization methodologies like the SIMPLEx and QTY code can further accelerate the design for water-soluble proteins and functions through the accurate identification of key interaction residues and protein fusion or swift rational substitutions. We also introduced programs such as CamSol and DeepSol that are designed for protein solubility and stability prediction and foresee their contributions in this aspect.
The water solubility and structural stability of proteins concern the most fundamental level of structures and functions for these minuscule molecular machines that are foundational to all life forms. The understanding of underlying biophysiochemical rules marks our capability to fine-tune their structures, functions and higher-order assemblies that can extend these biomolecules to biomedical and nanotechnological applications. We hope to provide a general overview of the field that can motivate other researchers to explore new methodologies for the water-soluble protein design and develop them into new variants of functional biological entities and biomaterials.
Acknowledgments
This work was supported by Avalon GloboCare Corp. We gratefully acknowledge Dorrie Langsley for careful and dedicated editing and making many constructive suggestions to significantly improve this review. We also thank Yinping Pan for help in the PDB entry identifications, as well as Haojie Zhang, Peiying Deng, Yao Hou, Ziwei Wang, Wenqing Zhang, Wenjie Zhong, Tangmin Lai, and Xiaoyun Xu for their help in checking the references.
Glossary
Abbreviations
- aa
amino acid
- AgTx2
agitoxin 2
- aMD
accelerated MD
- ApoAI*
apolipoprotein A-I lacking 43-residue of N-terminal domain
- ATP
adenosine triphosphate
- Aβ
amyloid-β
- Ca2+-ATPase
calcium ATPase
- CBD
cellulose-binding domain
- CD
circular dichroism
- CDR
complementarity determining region
- CFEG
cold field emission gun
- CHAMP
computed helical antimembrane protein
- COMPcc
cartilage oligomeric matrix protein
- CNN
convolutional neural network
- Cryo-EM
cryo-electron microscopy
- cyt b5
cytochrome b5
- ΔspMal-bp
maltose-binding protein lacking native export signal peptide
- DIG
digoxigenin
- DF
diiron protein
- DFHBI
mimic of green fluorescent protein fluorophore for imaging RNA in living cells
- DNA
deoxyribonucleic acid
- EB
entropic bristle
- EC
extracellular
- E. coli
Escherichia coli
- EGFP
enhanced green fluorescent protein
- EMDS
enhanced MD sampling
- EmrE
ethidium multidrug resistance protein E
- FDA
Food and Drug Administration
- GABAAR
homopentameric β3 γ-aminobutyric acid type-A receptor
- GLIC
gloeobacter violaceus pentameric ligand-gated ion channel
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- GrpE
Gro-P like protein E
- GST
glutathione S-transferases
- HDL
high density lipoprotein
- HEK293T
human embryonic kidney 293 cells
- Hbx
hepatitis B virus X protein
- hIL-3
human interleukin-3
- HP
hydrophilic
- IC
intracellular
- IDP
intrinsically disordered protein
- IL-2
interleukin-2
- IMP
integral membrane protein
- KIH
knobs-into-holes
- LPPG
lipid phosphate phosphatase gamma
- MBP
membrane-bound protein
- Mal-bp
maltose-binding protein
- MD
molecular dynamics
- MetaD
metadynamics or adaptive biasing force
- mIL-6
murine interleukin-6
- MotB
motility protein B
- MS1
computational designed coiled-coil based on (GX6)n
- MscL
large conductance mechanosensitive ion channel
- MUR
mu opioid receptor
- nAChR
nicotinic acetylcholine receptor
- negGFP
supernegatively charged GFP
- NMR
nuclear magnetic resonance
- PalB
lipase B from Pseudozyma Antarctica
- PBP
penicillin-binding proteins
- PCR
polymerase chain reaction
- PDB
Protein Data Bank
- pH
potential of hydrogen
- pI
isoelectric point
- PPO
polyamide oligomers
- PrP
prion protein
- PS1
porphyrin-binding sequence 1
- PSAM
peptide self-assembly mimic
- QTY
glutamine, threonine, tyrosine
- REMD
replica-exchange MD simulation
- RMSD
root-mean-square deviation
- RNA
ribonucleic acid
- PRIME
porphyrins in membrane
- RS
E. coli lysyl-tRNA synthetase
- SA
streptavidin
- SAF
self-assembling fiber
- SAM
sterile alpha motif
- SAP
spatial aggregation propensity
- SASA
solvent-accessible surface area
- SAXS
small-angle X-ray scattering
- ScFV
single-chain fragment variable
- SEP
solubility enhancement peptide
- SF9
clonal isolate of Spodoptera frugiperda
- SIMPLEx
solubilization of IMPs with high levels of expression
- SLB
single-layer β-sheet
- SN
signal-to-noise
- SPA
single particle analysis
- SVM
support vector machine
- SWCNT
single-wall carbon nanotube
- TEV
Tobacco Etch Virus
- TFE
trifluoroethanol
- TIMP
tissue inhibitors of metalloproteinase
- TM
transmembrane
- tRNA
transfer ribonucleic acid
- TRX
thioredoxin
- ValRS
valine tRNA synthetase
- vdW
van der Waals
- WSA
water-soluble acetylcholine receptor channel
- WSK-3
soluble variant of KcsA
- WSPLB
water-soluble phospholamban
Biographies
Rui Qing is an Associate Professor and PI of Qing Lab in State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology at Shanghai Jiao Tong University. He earned his Ph.D. degree in Materials Science and Engineering and worked on advanced energy storage and conversion technology at University of Florida, received postdoctoral training in the Media Lab, and was a research scientist at Koch Institute for Integrative Cancer Research at MIT. Dr. Qing has a multidisciplinary scientific background in both materials science and molecular biology. He codeveloped with Dr. Shuguang Zhang the QTY code for membrane protein solubilization, and expanded its applications in therapeutics and biomimetic sensing platforms. The research focus of Qing Lab is to advance the design methodology of membrane proteins and the molecular level integration in bioelectronics so as to address the societal challenges in human health and sustainability.
Shilei Hao received his Ph.D. in Biomedical Engineering from Chongqing University, China in 2014, joined the College of Bioengineering at Chongqing University as a lecturer, and is currently an Associate Professor. He worked at the Laboratory of Molecular Architecture, Media Lab, MIT as a Visiting Scientist from 2019–2020. His research focuses on protein materials and the drug design for biomedical applications, specifically on keratin-based functional materials and the drug design for the tissue engineering and therapeutics. Meanwhile, revealing the functional mechanisms of keratin materials through a recombinant protein approach is also his research interest.
Eva Smorodina is an undergraduate student with bioengineering and bioinformatics majors. She worked in the Virtual Structural Biology Group at FBB MSU under supervision of Andrey Golovin. There she studied molecular modeling and computational structural biology of proteins and nucleic acids. Since 2020, Eva has been investigating the effect of ions on the aggregation of amyloid fibrils at the Strodel lab at Forschungszentrum Jülich IBI-7. Since 2021, she has joined Greiff lab in Oslo, where she is working on antibody developability and several other projects related to computational structural immunology. Also, she is studying water-soluble GPCRs using various bioinformatic methods at the Laboratory of Molecular Architecture at MIT. Later Eva became a member of Sergey Ovchinnikov lab at Harvard, where she is currently investigating AlphaFold2 applications in the contest of protein de novo design. She has participated in many international scientific conferences, symposia, and schools.
David Jin, MD, Ph.D. is currently the Chief Executive Officer, President, and cofounder of Avalon GloboCare Corp., a clinical-stage biotechnology company dedicated to develop and deliver innovative and transformative cellular therapeutics in the fields of immuno-oncology and regenerative medicine. Dr. Jin is a strong advocate and catalyst of high-impact international biomedical partnership, collaboration, and academia–industry alliance. He has been principal investigator in more than 15 IND-enabling R&D programs and clinical trials as well as author/coauthor of over 80 peer-reviewed scientific articles, abstracts, reviews, and book chapters. His discovery of “Hemangiocytes”, a unique population of bone marrow-derived stem/progenitor cells endowed with dual hematopoietic and angiogenic phenotypes, has brought a new horizon in regenerative medicine. Dr. Jin received his combined MD–Ph.D. degree from SUNY Downstate College of Medicine in Brooklyn, NY. He received his clinical training and subsequent faculty tenure at Weill Cornell Medicine and New York-Presbyterian Hospital in the areas of internal medicine, hematology, and clinical oncology. At Weill Cornell, Dr. Jin also served as a senior translational clinician-scientist at the Howard Hughes Medical Institute and the Ansary Stem Cell Institute. Dr. Jin was honored as Top Chief Medical Officer by ExecRank in 2012 as well as recognized as Leading Physicians of the World in 2015 and 2018.
Arthur Zalevsky is a junior research fellow at the Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry. He received his Ph.D. degree in Mathematical Biology and Bioinformatics from the Lomonosov Moscow State University in Russia in 2019. His research interests include the design of biomolecules using machine learning and multiscale modeling.
Shuguang Zhang is at MIT Media Lab, Massachusetts Institute of Technology. His current research focuses on designs of biological molecules, particularly proteins and peptides that are short fragment of proteins. He received his B.S. from Sichuan University, China and Ph.D. in Biochemistry & Molecular Biology from University of California at Santa Barbara. He was an American Cancer Society Postdoctoral Fellow and a Whitaker Foundation Investigator at MIT. His work of designer self-assembling peptide scaffold won 2004 R&D100 award. He won a 2006 Guggenheim Fellowship and spent academic sabbatical in University of Cambridge, Cambridge, UK. He won 2006 Wilhelm Exner Medal of Austria. He was elected to Austrian Academy of Sciences in 2010. He was elected to American Institute of Medical and Biological Engineering in 2011, elected to US National Academy of Inventors in 2013, and elected to the European Academy of Science and Arts in 2021. He won the 2020 Emil Thomas Kaiser Award from the Protein Society. He is an honorary member of the Erwin Schrodinger Society and gave for the 20th Erwin Schrödinger Colloquium at the Austrian Academy of Sciences in 2021. He published >180 scientific papers that have been cited over 35,800 times with a h-index of 90. He is also a cofounder and board member of the Molecular Frontiers Foundation. Molecular Frontiers Foundation organizes the annual Molecular Frontiers Symposia in Sweden and around the world. The Foundation encourages young people to ask big and good scientific questions about nature. The selected winners will be awarded a Molecular Frontiers Inquiry Prize.
The authors declare the following competing financial interest(s): Shilei Hao, Eva Smorodina and Arthur Zalevsky declare non-competing financial interest. David Jin declares competing financial interest. Dr. David Jin is the President and CEO of Avalon GloboCare that specializes in cellular therapeutics in the fields of immuno-oncology and regenerative medicine. Avalon GloboCare is a sponsor of S. Zhangs research at MIT. Following MIT rules and regulations, David Jin and Avalon GloboCare do not interfere the publications of all articles. MIT filed patents based on an application for QTY solubilized membrane proteins to be used as decoy receptors in therapeutics. David Jin, Rui Qing, and Shuguang Zhang are inventors. MIT licensed its patents to Avalon GloboCare. Rui Qing declares competing financial interest from MIT licensing to Avalon GloboCare even though Rui Qing has no shares in Avalon GloboCare. MIT also licensed early QTY code patents to OH2Laboratories. Shuguang Zhang declares competing financial interest. Shuguang Zhang is a scientific advisor of OH2Laboratories and has shares in the company. Shuguang Zhang has no shares in Avalon GloboCare, nor advises the company.
References
- Stryer L.Gene rearrangements, recombination, transferase and cloning. Biochemistry, 2nd ed.; WH Freeman and Company:San Francisco, CA, 1981; pp 751–770. [Google Scholar]
- Lodish H.; Berk A.; Zipursky S.; Matsudaira P.; Baltimore D.; Darnell J.. The genetic basis of cancer. Mol. Cell. Biol., 5th ed.; WH Freeman and Company:New York, 2003; pp 943–949. [Google Scholar]
- Fersht A.Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding; Macmillan, 1999. [Google Scholar]
- Bell J. A.; Wilson K. P.; Zhang X. J.; Faber H. R.; Nicholson H.; Matthews B. W. Comparison of the crystal-structure of bacteriophage T4-lysozyme at low, medium, and high ionic strengths. Proteins 1991, 10, 10–21. 10.1002/prot.340100103. [DOI] [PubMed] [Google Scholar]
- Muirhead H.; Perutz M. Structure of hæemoglobin: A three-dimensional fourier synthesis of reduced human haemoglobin at 5.5 Å resolution. Nature 1963, 199, 633–638. 10.1038/199633a0. [DOI] [PubMed] [Google Scholar]
- Vinothkumar K. R.; Henderson R. Structures of membrane proteins. Q. Rev. Biophys. 2010, 43, 65–158. 10.1017/S0033583510000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A.; Frolow F.; Nelson N. Crystal structure of plant photosystem I. Nature 2003, 426, 630–635. 10.1038/nature02200. [DOI] [PubMed] [Google Scholar]
- Lin S. H.; Guidotti G. Purification of membrane proteins. Meth. Enzymol. 2009, 463, 619–629. 10.1016/S0076-6879(09)63035-4. [DOI] [PubMed] [Google Scholar]
- Weed R. I.; Berg G.; Reed C. F. Is hemoglobin an essential structural component of human erythrocyte membranes. J. Clin. Invest. 1963, 42, 581–588. 10.1172/JCI104747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R. M.; Shende V. R.; Motl N.; Pace C. N.; Scholtz J. M. Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys. J. 2012, 102, 1907–1915. 10.1016/j.bpj.2012.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S.; Ventura S. Computational re-design of protein structures to improve solubility. Expert Opin. Drug. Dis. 2019, 14, 1077–1088. 10.1080/17460441.2019.1637413. [DOI] [PubMed] [Google Scholar]
- Smialowski P.; Martin-Galiano A. J.; Mikolajka A.; Girschick T.; Holak T. A.; Frishman D. Protein solubility: sequence based prediction and experimental verification. Bioinformatics 2007, 23, 2536–2542. 10.1093/bioinformatics/btl623. [DOI] [PubMed] [Google Scholar]
- Bagby S.; Tong K. I.; Ikura M. Optimization of protein solubility and stability for protein nuclear magnetic resonance. Meth. Enzymol. 2001, 339, 20–41. 10.1016/S0076-6879(01)39307-2. [DOI] [PubMed] [Google Scholar]
- Caldwell G. W.; Ritchie D. M.; Masucci J. A.; Hageman W.; Yan Z. The new pre-preclinical paradigm: compound optimization in early and late phase drug discovery. Curr. Top. Med. Chem. 2001, 1, 353–366. 10.2174/1568026013394949. [DOI] [PubMed] [Google Scholar]
- Garidel P. Protein solubility from a biochemical, physicochemical and colloidal perspective. Am. Pharm. Rev. 2013, 2, 26–28. [Google Scholar]
- Garidel P.; Bassarab S. Impact of formulation design on stability and quality. Quality for biologics: Critical quality attributes, process and change control, product variation, characterisation, impurities and regulatory concerns 2009, 94–113. [Google Scholar]
- Chou P. Y.; Fasman G. D. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978, 47, 251–276. 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- Liljas A.; Liljas L.; Lindblom G.; Nissen P.; Kjeldgaard M.; Ash M.-R.. Textbook of structural biology; World Scientific, 2016. [Google Scholar]
- Fersht A. R.; Kaethner M. M. Enzyme hyperspecificity. Rejection of threonine by the valyl-tRNA synthetase by misacylation and hydrolytic editing. Biochemistry 1976, 15, 3342–3346. 10.1021/bi00660a026. [DOI] [PubMed] [Google Scholar]
- Lin L.; Hale S. P.; Schimmel P. Aminoacylation error correction. Nature 1996, 384, 33–34. 10.1038/384033b0. [DOI] [PubMed] [Google Scholar]
- Lin L.; Schimmel P. Mutational analysis suggests the same design for editing activities of two tRNA synthetases. Biochemistry 1996, 35, 5596–5601. 10.1021/bi960011y. [DOI] [PubMed] [Google Scholar]
- Branden C. I.; Tooze J.. Introduction to protein structure; Garland Science, 2012. [Google Scholar]
- Creighton T. E.Proteins: Structures and molecular properties; Macmillan, 1993. [Google Scholar]
- Twyman R.Principles of proteomics; Taylor & Francis, 2004. [Google Scholar]
- Korendovych I. V.; DeGrado W. F. De novo protein design, a retrospective. Q. Rev. Biophys. 2020, 53, e3. 10.1017/S0033583519000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P. S.; Boyken S. E.; Baker D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- McNaught A. D.; Wilkinson A.. Compendium of chemical terminology; Blackwell Science: Oxford, 1997. [Google Scholar]
- Privalov P. L. In Advances in protein chemistry; Elsevier, 1979; Vol. 33. [DOI] [PubMed] [Google Scholar]
- Ryan K. N.; Foegeding E. A. Formation of soluble whey protein aggregates and their stability in beverages. Food Hydrocoll. 2015, 43, 265–274. 10.1016/j.foodhyd.2014.05.025. [DOI] [Google Scholar]
- Wolfenden R.; Andersson L.; Cullis P. M.; Southgate C. C. B. Affinities of amino-acid side-chains for solvent water. Biochemistry 1981, 20, 849–855. 10.1021/bi00507a030. [DOI] [PubMed] [Google Scholar]
- Shaytan A. K.; Shaitan K. V.; Khokhlov A. R. Solvent accessible surface area of amino acid residues in globular proteins: correlation of apparent transfer free energies with experimental hydrophobicity scales. Biomacromolecules 2009, 10, 1224–1237. 10.1021/bm8015169. [DOI] [PubMed] [Google Scholar]
- Rose G. D.; Geselowitz A. R.; Lesser G. J.; Lee R. H.; Zehfus M. H. Hydrophobicity of amino-acid residues in globular-proteins. Science 1985, 229, 834–838. 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- Baumann G.; Froommel C.; Sander C. Polarity as a criterion in protein design. Protein Eng. 1989, 2, 329–334. 10.1093/protein/2.5.329. [DOI] [PubMed] [Google Scholar]
- Eijsink V. G. H.; Gaseidnes S.; Borchert T. V.; van den Burg B. Directed evolution of enzyme stability. Biomol. Eng. 2005, 22, 21–30. 10.1016/j.bioeng.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Song J. X. Insight into ″insoluble proteins″ with pure water. Febs Lett. 2009, 583, 953–959. 10.1016/j.febslet.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Timasheff S. N. Theory of protein solubility. Meth. Enzymol. 1985, 114, 49–77. 10.1016/0076-6879(85)14005-X. [DOI] [PubMed] [Google Scholar]
- Tayyab S.; Qamar S.; Islam M. Protein solubility - an old issue gaining momentum. Med. Sci. Res. 1993, 21, 805–809. [Google Scholar]
- Wu S. J.; Gilliland G. L.; Feng Y. Solubility and early assessment of stability for protein therapeutics. Biophysical Methods for Biotherapeutics: Discovery and Development Applications 2014, 65–91. 10.1002/9781118354698.ch3. [DOI] [Google Scholar]
- Daniel R. M.; Danson M. J. Temperature and the catalytic activity of enzymes: A fresh understanding. Febs Lett. 2013, 587, 2738–2743. 10.1016/j.febslet.2013.06.027. [DOI] [PubMed] [Google Scholar]
- D’Amico S.; Marx J. C.; Gerday C.; Feller G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003, 278, 7891–7896. 10.1074/jbc.M212508200. [DOI] [PubMed] [Google Scholar]
- Damodaran S.Protein: denaturation. Handbook of Food Science, Technology, and Engineering-4 Volume Set; CRC Press, 2005. [Google Scholar]
- Pelegrine D.; Gasparetto C. Whey proteins solubility as function of temperature and pH. LWT 2005, 38, 77–80. 10.1016/j.lwt.2004.03.013. [DOI] [Google Scholar]
- Frank S.; Kammerer R. A.; Hellstern S.; Pegoraro S.; Stetefeld J.; Lustig A.; Moroder L.; Engel J. Toward a high-resolution structure of phospholamban: Design of soluble transmembrane domain mutants. Biochemistry 2000, 39, 6825–6831. 10.1021/bi0000972. [DOI] [PubMed] [Google Scholar]
- Kirk O.; Borchert T. V.; Fuglsang C. C. Industrial enzyme applications. Curr. Opin. Biotechnol. 2002, 13, 345–351. 10.1016/S0958-1669(02)00328-2. [DOI] [PubMed] [Google Scholar]
- Kramer R. M.Towards a molecular understanding of protein solubility; Texas A&M University, 2011. [Google Scholar]
- Schein C. H. Solubility as a function of protein structure and solvent components. Biotechnology 1990, 8, 308–317. 10.1038/nbt0490-308. [DOI] [PubMed] [Google Scholar]
- Ali S. A.; Hassan M. I.; Islam A.; Ahmad F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. 10.2174/1389203715666140327114232. [DOI] [PubMed] [Google Scholar]
- Voynov V.; Chennamsetty N.; Kayser V.; Helk B.; Trout B. L. Predictive tools for stabilization of therapeutic proteins. MAbs 2009, 1, 580–582. 10.4161/mabs.1.6.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacak R.; Leaver-Fay A.; Kuhlman B. Computational protein design with explicit consideration of surface hydrophobic patches. Proteins 2012, 80, 825–838. 10.1002/prot.23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Zhou Y. Quantifying the effect of burial of amino acid residues on protein stability. Proteins 2004, 54, 315–322. 10.1002/prot.10584. [DOI] [PubMed] [Google Scholar]
- Pihlasalo S.; Auranen L.; Hanninen P.; Harma H. Method for estimation of protein isoelectric point. Anal. Chem. 2012, 84, 8253–8258. 10.1021/ac301569b. [DOI] [PubMed] [Google Scholar]
- Pergande M. R.; Cologna S. M. Isoelectric point separations of peptides and proteins. Proteomes 2017, 5, 4. 10.3390/proteomes5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y. J.; Khoo K. S.; Chung M. C. M.; Li S. F. Y. Determination of isoelectric points of acidic and basic-proteins by capillary electrophoresis. J. Chromatogr. A 1994, 680, 431–435. 10.1016/0021-9673(94)85140-9. [DOI] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P.; Longden I.; Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Kozlowski L. P. IPC 2.0: prediction of isoelectric point and pKa dissociation constants. Nucleic Acids Res. 2021, 49, W285–W292. 10.1093/nar/gkab295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon K.; King A. B. Glargine and detemir: Safety and efficacy profiles of the long-acting basal insulin analogs. Drug Healthc. Patient Saf. 2010, 2, 213–223. 10.2147/DHPS.S7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn C. J.; Plosker G. L.; Keating G. M.; McKeage K.; Scott L. Insulin glargine. Drugs 2003, 63, 1743–1778. 10.2165/00003495-200363160-00007. [DOI] [PubMed] [Google Scholar]
- Havelund S.; Plum A.; Ribel U.; Jonassen I.; Vølund A.; Markussen J.; Kurtzhals P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm. Res. 2004, 21, 1498–1504. 10.1023/B:PHAM.0000036926.54824.37. [DOI] [PubMed] [Google Scholar]
- Nadendla K.; Friedman S. H. Light control of protein solubility through isoelectric point modulation. J. Am. Chem. Soc. 2017, 139, 17861–17869. 10.1021/jacs.7b08465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu S. C.; David V.. Properties of analytes and matrices determining HPLC selection. Selection of the HPLC Method in Chemical Analysis; Moldoveanu S. C., David V., Eds; ScienceDirect, 2017; pp 189–230. [Google Scholar]
- Thurlkill R. L.; Grimsley G. R.; Scholtz J. M.; Pace C. N. pK values of the ionizable groups of proteins. Protein Sci. 2006, 15, 1214–1218. 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahari S.; Sun L.; Alexov E. PKAD: a database of experimentally measured pKa values of ionizable groups in proteins. Database (Oxford) 2019, 2019, baz024. 10.1093/database/baz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski M.; Olsson M. H.; Søndergaard C. R.; Jensen J. H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11, 1–6. 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Mao J.; Gunner M. R. MCCE2: improving protein pKa calculations with extensive side chain rotamer sampling. J. Comput. Chem. 2009, 30, 2231–2247. 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Morrow B. H.; Shi C.; Shen J. K. Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. 10.1080/08927022.2014.907492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z.; Luo F.; Wang Y.; Li E.; Huang Y. Protein pKa prediction with machine learning. ACS Omega 2021, 6, 34823–34831. 10.1021/acsomega.1c05440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K.; Masuda T.; Watanabe C.; Miyagawa N.; Mizouchi H.; Nagase S.; Kamisaka K.; Oshima K.; Ono S.; Ueda H.; et al. High-precision atomic charge prediction for protein systems using fragment molecular orbital calculation and machine learning. J. Chem. Inf. Model. 2020, 60, 3361–3368. 10.1021/acs.jcim.0c00273. [DOI] [PubMed] [Google Scholar]
- Trevino S. R.; Scholtz J. M.; Pace C. N. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007, 366, 449–460. 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwicker J.; Charonis S.; Curtis R. A. Lysine and arginine content of proteins: computational analysis suggests a new tool for solubility design. Mol. Pharmaceutics 2014, 11, 294–303. 10.1021/mp4004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T.; Ying B. W.; Saito K.; Jin W.; Takada S.; Ueda T.; Taguchi H. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4201–4206. 10.1073/pnas.0811922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov D. S. Therapeutic Proteins. Therapeutic Proteins 2012, 899, 1–26. 10.1007/978-1-61779-921-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der B. S.; Kluwe C.; Miklos A. E.; Jacak R.; Lyskov S.; Gray J. J.; Georgiou G.; Ellington A. D.; Kuhlman B. Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS One 2013, 8, e64363. 10.1371/journal.pone.0064363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige M. J.Oxidative folding of proteins: Basic principles, cellular regulation and engineering; Royal Society of Chemistry, 2018. [Google Scholar]
- Wiedemann C.; Kumar A.; Lang A. R.; Ohlenschlager O.. Cysteines and disulfide bonds as structure-forming units: insights from different domains of life and the potential for characterization by NMR. Front. Chem. 2020, 8. 10.3389/fchem.2020.00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Wang Y.; Luo X. Z.; Li J.; Reed S. A.; Xiao H.; Young T. S.; Schultz P. G. Enhancing protein stability with extended disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 5910–5915. 10.1073/pnas.1605363113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli P.; Navarro S.; Grana-Montes R.; Bano-Polo M.; Fernandez M. R.; Papaleo E.; Ventura S. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biol. 2018, 14, 566–575. 10.1016/j.redox.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Li W.; Wang Y.; Yin H.; Ding Y.; Ji J.; Wang B.; Hao S. Fabrication of an expandable keratin sponge for improved hemostasis in a penetrating trauma. Colloids Surf., B 2019, 182, 110367. 10.1016/j.colsurfb.2019.110367. [DOI] [PubMed] [Google Scholar]
- Gao F.; Li W.; Deng J.; Kan J.; Guo T.; Wang B.; Hao S. Recombinant Human Hair Keratin Nanoparticles Accelerate Dermal Wound Healing. ACS Appl. Mate.r Inter. 2019, 11, 18681–18690. 10.1021/acsami.9b01725. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Chen X.; Zhai D.; Gao F.; Guo T.; Li W.; Hao S.; Ji J.; Wang B. Development of keratin nanoparticles for controlled gastric mucoadhesion and drug release. J. Nanobiotechnol. 2018, 16, 24. 10.1186/s12951-018-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Li W.; Wang J.; Luo T.; Lou D.; Wang B.; Hao S. Recombinant human hair keratin proteins for halting bleeding. Artif. Cells Nanomed. Biotechnol. 2018, 46, 456–461. 10.1080/21691401.2018.1459633. [DOI] [PubMed] [Google Scholar]
- Kan J. L.; Li W. F.; Qing R.; Gao F. Y.; Wang Y. M.; Zhu L. C.; Wang B. C.; Hao S. L. Study of mechanisms of recombinant keratin solubilization with enhanced wound healing capability. Chem. Mater. 2020, 32, 3122–3133. 10.1021/acs.chemmater.0c00124. [DOI] [Google Scholar]
- Gao F. Y.; Li W. F.; Kan J. L.; Ding Y.; Wang Y. M.; Deng J.; Qing R.; Wang B. C.; Hao S. L. Insight into the regulatory function of human hair keratins in wound healing using Proteomics. Adv. Biosyst. 2020, 4, 1900235. 10.1002/adbi.201900235. [DOI] [PubMed] [Google Scholar]
- Zimmerman S. B.; Trach S. O. Effects of macromolecular crowding on the association of E. coli ribosomal particles. Nucleic Acids Res. 1988, 16, 6309–6326. 10.1093/nar/16.14.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerle C. T.; Frieden C. Effect of pH on the mechanism of actin polymerization. Biochemistry 1988, 27, 7766–7772. 10.1021/bi00420a027. [DOI] [PubMed] [Google Scholar]
- Nikam R.; Kulandaisamy A.; Harini K.; Sharma D.; Gromiha M. M. ProThermDB: thermodynamic database for proteins and mutants revisited after 15 years. Nucleic Acids Res. 2021, 49, D420–D424. 10.1093/nar/gkaa1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha M. M.; An J.; Kono H.; Oobatake M.; Uedaira H.; Sarai A. ProTherm: thermodynamic database for proteins and mutants. Nucleic Acids Res. 1999, 27, 286–288. 10.1093/nar/27.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi N.; Hashim S. Z. M.; Norouzi A.; Samian M. R. A review of machine learning methods to predict the solubility of overexpressed recombinant proteins in Escherichia coli. BMC Bioinform. 2014, 15, 134. 10.1186/1471-2105-15-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. L.; Harrison R. G. Predicting the solubility of recombinant proteins in Escherichia coli. Biotechnology 1991, 9, 443–448. 10.1038/nbt0591-443. [DOI] [PubMed] [Google Scholar]
- Davis G. D.; Elisee C.; Newham D. M.; Harrison R. G. New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 1999, 65, 382–388. . [DOI] [PubMed] [Google Scholar]
- Magnan C. N.; Randall A.; Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- Hebditch M.; Carballo-Amador M. A.; Charonis S.; Curtis R.; Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormanni P.; Amery L.; Ekizoglou S.; Vendruscolo M.; Popovic B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci. Rep. 2017, 7, 1–9. 10.1038/s41598-017-07800-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari B. K.; Gardner P. P.; Lim C. S. Solubility-Weighted Index: fast and accurate prediction of protein solubility. Bioinformatics 2020, 36, 4691–4698. 10.1093/bioinformatics/btaa578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S.; Rawi R.; Kunji K.; Chuang G. Y.; Bensmail H.; Mall R. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics 2018, 34, 2605–2613. 10.1093/bioinformatics/bty166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat P.; Prabakaran R.; Kumar S.; Gromiha M. M. AbsoluRATE: An in-silico method to predict the aggregation kinetics of native proteins. Biochim. Biophys. Acta. Proteins Proteom. 2021, 1869, 140682. 10.1016/j.bbapap.2021.140682. [DOI] [PubMed] [Google Scholar]
- Prabakaran R.; Rawat P.; Thangakani A. M.; Kumar S.; Gromiha M. M. Protein aggregation: in silico algorithms and applications. Biophys. Rev. 2021, 13, 71–89. 10.1007/s12551-021-00778-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Durme J.; De Baets G.; Van Der Kant R.; Ramakers M.; Ganesan A.; Wilkinson H.; Gallardo R.; Rousseau F.; Schymkowitz J. Solubis: a webserver to reduce protein aggregation through mutation. Protein Eng. Des. Sel. 2016, 29, 285–289. 10.1093/protein/gzw019. [DOI] [PubMed] [Google Scholar]
- Trevino S. R.; Scholtz J. M.; Pace C. N. Measuring and increasing protein solubility. J. Pharm. Sci. 2008, 97, 4155–4166. 10.1002/jps.21327. [DOI] [PubMed] [Google Scholar]
- DeGrado W. F. Introduction: Protein design. Chem. Rev. 2001, 101, 3025–3026. 10.1021/cr000663z. [DOI] [PubMed] [Google Scholar]
- Khoury G. A.; Smadbeck J.; Kieslich C. A.; Floudas C. A. Protein folding and de novo protein design for biotechnological applications. Trends Biotechnol. 2014, 32, 99–109. 10.1016/j.tibtech.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwanzig R.; Szabo A.; Bagchi B. Levinthal’s paradox. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 20–22. 10.1073/pnas.89.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal C.Proceedings of a meeting held at Allerton House; Debrunner P., Tsibris J. C. M., Munck E., Eds.; University of Illinois Press: Urbana, IL, 1969. [Google Scholar]
- Levinthal C. Are there pathways for protein folding?. J. chim. phys. 1968, 65, 44–45. 10.1051/jcp/1968650044. [DOI] [Google Scholar]
- Epstein C. J.; Goldberger R. F.; Anfinsen C. B. Cold Spring Harbor symposia on quantitative biology 1963, 28, 439–449. 10.1101/SQB.1963.028.01.060. [DOI] [Google Scholar]
- Anfinsen C. B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Gelman H.; Gruebele M. Fast protein folding kinetics. Q. Rev. Biophys. 2014, 47, 95–142. 10.1017/S003358351400002X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankov D. N.; Finkelstein A. V. Solution of Levinthal’s Paradox and a Physical Theory of Protein Folding Times. Biomolecules 2020, 10, 250. 10.3390/biom10020250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold P. E.; Montal M.; Onuchic J. N. Protein folding funnels - a kinetic approach to the sequence structure relationship. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 8721–8725. 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K. A.; Chan H. S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997, 4, 10–19. 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Koga N.; Tatsumi-Koga R.; Liu G. H.; Xiao R.; Acton T. B.; Montelione G. T.; Baker D. Principles for designing ideal protein structures. Nature 2012, 491, 222–227. 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickson A. A.; Clarke J. What lessons can be learned from studying the folding of homologous proteins?. Methods 2010, 52, 38–50. 10.1016/j.ymeth.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet D.; Voet J. G.; Pratt C. W.. Fundamentals of biochemistry: Life at the molecular level; Wiley, 2013. [Google Scholar]
- Zhou R. H.; Huang X. H.; Margulis C. J.; Berne B. J. Hydrophobic collapse in multidomain protein folding. Science 2004, 305, 1605–1609. 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- Lapidus L. J.; Yao S. H.; McGarrity K. S.; Hertzog D. E.; Tubman E.; Bakajin O. Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer. Biophys. J. 2007, 93, 218–224. 10.1529/biophysj.106.103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharadadevi A.; Sivakamasundari C.; Nagaraj R. Amphipathic alpha-helices in proteins: Results from analysis of protein structures. Proteins: Struct. Funct. Genet. 2005, 59, 791–801. 10.1002/prot.20459. [DOI] [PubMed] [Google Scholar]
- Kazlauskas R. Engineering more stable proteins. Chem. Soc. Rev. 2018, 47, 9026–9045. 10.1039/C8CS00014J. [DOI] [PubMed] [Google Scholar]
- Dubey K. K.; Pramanik A.; Yadav J.; et al. Enzyme Engineering. Advances in Enzyme Technology 2019, 325–347. 10.1016/B978-0-444-64114-4.00012-1. [DOI] [Google Scholar]
- Hsieh P. C.; Vaisvila R. Protein engineering: Single or multiple site-directed mutagenesis. Methods Mol. Biol. 2013, 978, 173–186. 10.1007/978-1-62703-293-3_13. [DOI] [PubMed] [Google Scholar]
- Walker K.Site-directed mutagenesis. In Encyclopedia of Cell Biology; 2016, 122–127.
- Carter P. J.; Winter G.; Wilkinson A. J.; Fersht A. R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 1984, 38, 835–840. 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- Winter G.; Fersht A. R.; Wilkinson A. J.; Zoller M.; Smith M. Redesigning enzyme structure by site-directed mutagenesis: tyrosyl tRNA synthetase and ATP binding. Nature 1982, 299, 756–758. 10.1038/299756a0. [DOI] [PubMed] [Google Scholar]
- Mohammadian H.; Mahnam K.; Sadeghi H. M.; Ganjalikhany M. R.; Akbari V. Rational design of a new mutant of tobacco etch virus protease in order to increase the in vitro solubility. Res. Pharm. Sci. 2020, 15, 164. 10.4103/1735-5362.283816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Lv Y.; Yu H.; Cao H.; Wang L.; Wen B.; Gu T.; Wang F.; Sun L.; Xin F. Site-directed mutagenesis of coenzyme-independent carotenoid oxygenase CSO2 to enhance the enzymatic synthesis of vanillin. Appl. Microbiol. Biotechnol. 2020, 104, 3897–3907. 10.1007/s00253-020-10433-1. [DOI] [PubMed] [Google Scholar]
- Mosavi L. K.; Peng Z. Y. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 2003, 16, 739–745. 10.1093/protein/gzg098. [DOI] [PubMed] [Google Scholar]
- Sormanni P.; Aprile F. A.; Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015, 427, 478–490. 10.1016/j.jmb.2014.09.026. [DOI] [PubMed] [Google Scholar]
- Pindrus M.; Shire S. J.; Kelley R. F.; Demeule B.; Wong R.; Xu Y. R.; Yadav S. Solubility challenges in high concentration monoclonal antibody formulations: relationship with amino acid sequence and intermolecular interactions. Mol. Pharmaceutics 2015, 12, 3896–3907. 10.1021/acs.molpharmaceut.5b00336. [DOI] [PubMed] [Google Scholar]
- Kang T. H.; Seong B. L.. Solubility, stability, and avidity of recombinant antibody fragments expressed in microorganisms. Front. Microbiol. 2020, 11. 10.3389/fmicb.2020.01927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Song J.; Yan F.; Chu H.; Luo J.; Zhao Y.; Cheng X.; Luo G.; Zheng Q.; Wei J. Improving GPX activity of selenium-containing human single-chain Fv antibody by site-directed mutation based on the structural analysis. J. Mol. Recognit. 2009, 22, 293–300. 10.1002/jmr.946. [DOI] [PubMed] [Google Scholar]
- Pepinsky R. B.; Silvian L.; Berkowitz S. A.; Farrington G.; Lugovskoy A.; Walus L.; Eldredge J.; Capili A.; Mi S.; Graff C.; et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci. 2010, 19, 954–966. 10.1002/pro.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter E. P.; Beis K.; Cameron A. D.; Iwata S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. 2008, 18, 581–586. 10.1016/j.sbi.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Maranas C. D. From directed evolution to computational enzyme engineering-A review. AIChE J. 2020, 66, e16847. 10.1002/aic.16847. [DOI] [Google Scholar]
- Zeymer C.; Hilvert D. Directed evolution of protein catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]
- Roodveldt C.; Aharoni A.; Tawfik D. S. Directed evolution of proteins for heterologous expression and stability. Curr. Opin. Struct. 2005, 15, 50–56. 10.1016/j.sbi.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Petrounia I. P.; Arnold F. H. Designed evolution of enzymatic properties. Curr. Opin. Biotechnol. 2000, 11, 325–330. 10.1016/S0958-1669(00)00107-5. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Sharma R. Directed evolution: An approach to engineer enzymes. Crit. Rev. Biotechnol. 2006, 26, 165–199. 10.1080/07388550600851423. [DOI] [PubMed] [Google Scholar]
- Worsdorfer B.; Woycechowsky K. J.; Hilvert D. Directed evolution of a protein container. Science 2011, 331, 589–592. 10.1126/science.1199081. [DOI] [PubMed] [Google Scholar]
- Lluis M. W.; Godfroy J. I.; Yin H. Protein engineering methods applied to membrane protein targets. Protein Eng. Des. Sel. 2013, 26, 91–100. 10.1093/protein/gzs079. [DOI] [PubMed] [Google Scholar]
- Pogson M.; Georgiou G.; Iverson B. L. Engineering next generation proteases. Curr. Opin. Biotechnol. 2009, 20, 390–397. 10.1016/j.copbio.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M. G.; Westmeyer G. G.; Romero P. A.; Szablowski J. O.; Kuster B.; Shah A.; Otey C. R.; Langer R.; Arnold F. H.; Jasanoff A. Directed evolution of a magnetic resonance imaging contrast agent for noninvasive imaging of dopamine. Nat. Biotechnol. 2010, 28, 264–U120. 10.1038/nbt.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B. J.; Neil J. R.; White F. M.; Wittrup K. D. Epidermal growth factor receptor downregulation by small heterodimeric binding proteins. Protein Eng. Des. Sel. 2012, 25, 47–57. 10.1093/protein/gzr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor J. R.; Panayiotou V.; Agnello G.; Georgiou G.; Stone E. M. Engineering reduced-immunogenicity enzymes for amino acid depletion therapy in cancer. Meth. Enzymol. 2012, 502, 291–319. 10.1016/B978-0-12-416039-2.00015-X. [DOI] [PubMed] [Google Scholar]
- Waldo G. S. Genetic screens and directed evolution for protein solubility. Curr. Opin. Chem. Biol. 2003, 7, 33–38. 10.1016/S1367-5931(02)00017-0. [DOI] [PubMed] [Google Scholar]
- Van Den Berg S.; Löfdahl P.-Å.; Härd T.; Berglund H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006, 121, 291–298. 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Wang T. N.; Badran A. H.; Huang T. P.; Liu D. R. Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol. 2018, 14, 972–980. 10.1038/s41589-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R.; Schwaneberg U.; Roccatano D. Computer-aided protein directed evolution: a review of web servers, databases and other computational tools for protein engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209008 10.5936/csbj.201209008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohne M.; Bornscheuer U. T. Protein engineering from ″scratch″ is maturing. Angew. Chem. 2014, 53, 1200–1202. 10.1002/anie.201309591. [DOI] [PubMed] [Google Scholar]
- Lutz S. Beyond directed evolution-semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. 10.1016/j.copbio.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. H. The Fourier transform of a coiled-coil. Acta Crystallogr. 1953, 6, 685–689. 10.1107/S0365110X53001952. [DOI] [Google Scholar]
- Kuhlman B.; Bradley P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. 10.1038/s41580-019-0163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A.; Tyka M.; Lewis S. M.; Lange O. F.; Thompson J.; Jacak R.; Kaufman K.; Renfrew P. D.; Smith C. A.; Sheffler W.; et al. Rosetta3: An object-oriented software suite for the simulation and design of macromolecules. Meth. Enzymol. 2011, 487, 545–574. 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C. W.; Bruning M.; Ibarra A. A.; Bartlett G. J.; Thomson A. R.; Sessions R. B.; Brady R. L.; Woolfson D. N. CCBuilder: an interactive web-based tool for building, designing and assessing coiled-coil protein assemblies. Bioinformatics 2014, 30, 3029–3035. 10.1093/bioinformatics/btu502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negron C.; Keating A. E. Multistate protein design using CLEVER and CLASSY. Meth. Enzymol. 2013, 523, 171–190. 10.1016/B978-0-12-394292-0.00008-4. [DOI] [PubMed] [Google Scholar]
- Smadbeck J.; Peterson M. B.; Khoury G. A.; Taylor M. S.; Floudas C. A. Protein WISDOM: A workbench for in silico de novo design of bioMolecules. J. Vis. Exp. 2013, 25, 50476. 10.3791/50476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Kortemme T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558 10.1016/j.jbc.2021.100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill R. B.; Raleigh D. P.; Lombardi A.; Degrado W. F. De novo design of helical bundles as models for understanding protein folding and function. Acc. Chem. Res. 2000, 33, 745–754. 10.1021/ar970004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B.; Dantas G.; Ireton G. C.; Varani G.; Stoddard B. L.; Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- Thomson A. R.; Wood C. W.; Burton A. J.; Bartlett G. J.; Sessions R. B.; Brady R. L.; Woolfson D. N. Computational design of water-soluble alpha-helical barrels. Science 2014, 346, 485–488. 10.1126/science.1257452. [DOI] [PubMed] [Google Scholar]
- Mahendran K. R.; Niitsu A.; Kong L. B.; Thomson A. R.; Sessions R. B.; Woolfson D. N.; Bayley H. A monodisperse transmembrane alpha-helical peptide barrel. Nat. Chem. 2017, 9, 411–419. 10.1038/nchem.2647. [DOI] [PubMed] [Google Scholar]
- Zhang S. Q.; Huang H.; Yang J. J.; Kratochvil H. T.; Lolicato M.; Liu Y. X.; Shu X. K.; Liu L. J.; DeGrado W. F. Designed peptides that assemble into cross-alpha amyloid-like structures. Nat. Chem. Biol. 2018, 14, 870–875. 10.1038/s41589-018-0105-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess N. C.; Sharp T. H.; Thomas F.; Wood C. W.; Thomson A. R.; Zaccai N. R.; Brady R. L.; Serpell L. C.; Woolfson D. N. Modular design of self-assembling peptide-based nanotubes. J. Am. Chem. Soc. 2015, 137, 10554–10562. 10.1021/jacs.5b03973. [DOI] [PubMed] [Google Scholar]
- Xu C. F.; Liu R.; Mehta A. K.; Guerrero-Ferreira R. C.; Wright E. R.; Dunin-Horkawicz S.; Morris K.; Serpell L. C.; Zuo X. B.; Wall J. S.; et al. Rational design of helical nanotubes from self-assembly of coiled-coil lock washers. J. Am. Chem. Soc. 2013, 135, 15565–15578. 10.1021/ja4074529. [DOI] [PubMed] [Google Scholar]
- Hsia Y.; Bale J. B.; Gonen S.; Shi D.; Sheffler W.; Fong K. K.; Nattermann U.; Xu C. F.; Huang P. S.; Ravichandran R.; et al. Design of a hyperstable 60-subunit protein icosahedron. Nature 2016, 535, 136–139. 10.1038/nature18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale J. B.; Gonen S.; Liu Y. X.; Sheffler W.; Ellis D.; Thomas C.; Cascio D.; Yeates T. O.; Gonen T.; King N. P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. 10.1126/science.aaf8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle L.; Hallinan J.; Bolduc J.; Parmeggiani F.; Baker D.; Stoddard B. L.; Bradley P. Rational design of alpha-helical tandem repeat proteins with closed architectures. Nature 2015, 528, 585–588. 10.1038/nature16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen S.; DiMaio F.; Gonen T.; Baker D. Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces. Science 2015, 348, 1365–1368. 10.1126/science.aaa9897. [DOI] [PubMed] [Google Scholar]
- King N. P.; Bale J. B.; Sheffler W.; McNamara D. E.; Gonen S.; Gonen T.; Yeates T. O.; Baker D. Accurate design of co-assembling multi-component protein nanomaterials. Nature 2014, 510, 103–108. 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson A. J.; Watson J. L.; Sheffler W.; Johnson M. C.; Bittleston A.; Somasundaram L.; Decarreau J.; Jiao F.; Chen J. J.; Mela I.; et al. Design of biologically active binary protein 2D materials. Nature 2021, 589, 468–473. 10.1038/s41586-020-03120-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J. Y.; Vorobieva A. A.; Sheffler W.; Doyle L. A.; Park H.; Bick M. J.; Mao B. C.; Foight G. W.; Lee M. Y.; Gagnon L. A.; et al. De novo design of a fluorescence-activating beta-barrel. Nature 2018, 561, 485–491. 10.1038/s41586-018-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancalana M.; Makabe K.; Koide S. Minimalist design of water-soluble cross-beta architecture. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3469–3474. 10.1073/pnas.0912654107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P. L.; Min D. Y.; DiMaio F.; Wei K. Y.; Vahey M. D.; Boyken S. E.; Chen Z. B.; Fallas J. A.; Ueda G.; Sheffler W.; et al. Accurate computational design of multipass transmembrane proteins. Science 2018, 359, 1042–1046. 10.1126/science.aaq1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C. F.; Lu P. L.; El-Din T. M. G.; Pei X. Y.; Johnson M. C.; Uyeda A.; Bick M. J.; Xu Q.; Jiang D. H.; Bai H.; et al. Computational design of transmembrane pores. Nature 2020, 585, 129–134. 10.1038/s41586-020-2646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobieva A. A.; White P.; Liang B. Y.; Horne J. E.; Bera A. K.; Chow C. M.; Gerben S.; Marx S.; Kang A.; Stiving A. Q.; et al. De novo design of transmembrane beta barrels. Science 2021, 371, 801. 10.1126/science.abc8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. What has de novo protein design taught us about protein folding and biophysics?. Protein Sci. 2019, 28, 678–683. 10.1002/pro.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva D. A.; Yu S.; Ulge U. Y.; Spangler J. B.; Jude K. M.; Labao-Almeida C.; Ali L. R.; Quijano-Rubio A.; Ruterbusch M.; Leung I.; et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 2019, 565, 186–191. 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier A.; Silva D. A.; Rocklin G. J.; Hicks D. R.; Vergara R.; Murapa P.; Bernard S. M.; Zhang L.; Lam K. H.; Yao G. R.; et al. Massively parallel de novo protein design for targeted therapeutics. Nature 2017, 550, 74–79. 10.1038/nature23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L. X.; Goreshnik I.; Coventry B.; Case J. B.; Miller L.; Kozodoy L.; Chen R. E.; Carter L.; Walls A. C.; Park Y. J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H.; Fallas J. A.; Lynch E.; Sheffler W.; Parry B.; Jannetty N.; Decarreau J.; Wagenbach M.; Vicente J. J.; Chen J. J.; et al. De novo design of self-assembling helical protein filaments. Science 2018, 362, 705–709. 10.1126/science.aau3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan G.; Kim Y. H.; Acharya R.; Axelrod K.; Jain R. M.; Willis L.; Drndic M.; Kikkawa J. M.; DeGrado W. F. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science 2011, 332, 1071–1076. 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. B.; Kibler R. D.; Hunt A.; Busch F.; Pearl J.; Jia M. X.; VanAernum Z. L.; Wicky B. I. M.; Dods G.; Liao H.; et al. De novo design of protein logic gates. Science 2020, 368, 78–84. 10.1126/science.aay2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan R. A.; Boyken S. E.; Ng A. H.; Samson J. A.; Dods G.; Westbrook A. M.; Nguyen T. H.; Lajoie M. J.; Chen Z. B.; Berger S.; et al. De novo design of bioactive protein switches. Nature 2019, 572, 205–210. 10.1038/s41586-019-1432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A.; Pirro F.; Maglio O.; Chino M.; DeGrado W. F. De novo design of four-helix bundle metalloproteins: One scaffold, diverse reactivities. Acc. Chem. Res. 2019, 52, 1148–1159. 10.1021/acs.accounts.8b00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A. J.; Thomson A. R.; Dawson W. M.; Brady R. L.; Woolfson D. N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. 10.1038/nchem.2555. [DOI] [PubMed] [Google Scholar]
- Maglio O.; Nastri F.; Pavone V.; Lombardi A.; DeGrado W. F. Preorganization of molecular binding sites in designed diiron proteins. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3772–3777. 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floudas C. A.; Fung H. K.; McAllister S. R.; Monnigmann M.; Rajgaria R. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci. 2006, 61, 966–988. 10.1016/j.ces.2005.04.009. [DOI] [Google Scholar]
- Haynes W. M.; Lide D. R.; Bruno T. J.. CRC handbook of chemistry and physics; CRC Press, 2016. [Google Scholar]
- Idrees M.; Mohammad A. R.; Karodia N.; Rahman A. Multimodal Role of Amino Acids in Microbial Control and Drug Development. Antibiotics (Basel) 2020, 9, 330. 10.3390/antibiotics9060330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. W.; Lee S. J.; Jo J.; Kim J. G.; Ki H.; Kim C. W.; Cho K. H.; Choi J.; Lee J. H.; Wulff M.; et al. Protein folding from heterogeneous unfolded state revealed by time-resolved X-ray solution scattering. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 14996–15005. 10.1073/pnas.1913442117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T. J.; Arkin I. T. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins: Struct. Funct. Genet. 2000, 39, 417–420. . [DOI] [PubMed] [Google Scholar]
- Lander E. S.; Consortium I. H. G. S.; Linton L. M.; Birren B.; Nusbaum C.; Zody M. C.; Baldwin J.; Devon K.; Dewar K.; Doyle M.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Wallin E.; von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J. T.; Chen Y. B.; Pu F.; Sun P. P.; He F.; Zhang L.; Li Y. W.; Ma Z. Q.; Wang H. Understanding membrane protein drug targets in computational perspective. Curr. Drug Targets 2019, 20, 551–564. 10.2174/1389450120666181204164721. [DOI] [PubMed] [Google Scholar]
- Rajendran L.; Knolker H. J.; Simons K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discovery 2010, 9, 29–42. 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- Rawlings A. E. Membrane proteins: always an insoluble problem?. Biochem. Soc. Trans. 2016, 44, 790–795. 10.1042/BST20160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S.; Bader M. L.; Drew D.; de Gier J. W. Rationalizing membrane protein overexpression. Trends Biotechnol. 2006, 24, 364–371. 10.1016/j.tibtech.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Loll P. J. Membrane protein structural biology: the high throughput challenge. J. Struct. Biol. 2003, 142, 144–153. 10.1016/S1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- Lv X. C.; Liu J. L.; Shi Q. Y.; Tan Q. W.; Wu D.; Skinner J. J.; Walker A. L.; Zhao L. X.; Gu X. X.; Chen N.; et al. In vitro expression and analysis of the 826 human G protein-coupled receptors. Protein Cell 2016, 7, 325–337. 10.1007/s13238-016-0263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate C. G. Practical considerations of membrane protein instability during purification and crystallisation. Heterologous Expression of Membrane Proteins: Methods and Protocols 2010, 601, 187–203. 10.1007/978-1-60761-344-2_12. [DOI] [PubMed] [Google Scholar]
- Rawlings A. E. Membrane protein engineering to the rescue. Biochem. Soc. Trans. 2018, 46, 1541–1549. 10.1042/BST20180140. [DOI] [PubMed] [Google Scholar]
- Li H. M.; Cocco M. J.; Steitz T. A.; Engelman D. M. Conversion of phospholamban into a soluble pentameric helical bundle. Biochemistry 2001, 40, 6636–6645. 10.1021/bi0026573. [DOI] [PubMed] [Google Scholar]
- Slovic A. M.; Summa C. M.; Lear J. D.; DeGrado W. F. Computational design of a water-soluble analog of phospholamban. Protein Sci. 2003, 12, 337–348. 10.1110/ps.0226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman H. K. B.; Jones L. R. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- Cornea R. L.; Autry J. M.; Chen Z. H.; Jones L. R. Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the Ca2+ pump of cardiac sarcoplasmic reticulum. J. Biol. Chem. 2000, 275, 41487–41494. 10.1074/jbc.M008195200. [DOI] [PubMed] [Google Scholar]
- Li M.; Reddy L. G.; Bennett R.; Silva N. D.; Jones L. R.; Thomas D. D. A fluorescence energy transfer method for analyzing protein oligomeric structure: Application to phospholamban. Biophys. J. 1999, 76, 2587–2599. 10.1016/S0006-3495(99)77411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman H. K. B.; Kobayashi Y. M.; Autry J. M.; Jones L. R. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 1996, 271, 5941–5946. 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Deantonio L.; Eisenberg D. Hydrophobic organization of membrane-proteins. Science 1989, 245, 510–513. 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 2000, 69, 881–922. 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- Arkin I. T.; Adams P. D.; Mackenzie K. R.; Lemmon M. A.; Brunger A. T.; Engelman D. M. Structural organization of the pentameric transmembrane alpha-helices of phospholamban, a cardiac ion-channel. EMBO J. 1994, 13, 4757–4764. 10.1002/j.1460-2075.1994.tb06801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Arkin I. T.; Engelman D. M.; Brunger A. T. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 1995, 2, 154–162. 10.1038/nsb0295-154. [DOI] [PubMed] [Google Scholar]
- Dieckmann G. R.; DeGrado W. F. Modeling transmembrane helical oligomers. Curr. Opin. Struct. 1997, 7, 486–494. 10.1016/S0959-440X(97)80111-X. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Zhang T.; Kim P. S.; Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 1993, 262, 1401–1407. 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Slovic A. M.; Lear J. D.; DeGrado W. F. De novo design of a pentameric coiled-coil: decoding the motif for tetramer versus pentamer formation in water-soluble phospholamban. J. Pept. Res. 2005, 65, 312–321. 10.1111/j.1399-3011.2005.00244.x. [DOI] [PubMed] [Google Scholar]
- Slovic A. M.; Stayrook S. E.; North B.; DeGrado W. F. X-ray structure of a water-soluble analog of the membrane protein phospholamban: Sequence determinants defining the topology of tetrameric and pentameric coiled coils. J. Mol. Biol. 2005, 348, 777–787. 10.1016/j.jmb.2005.02.040. [DOI] [PubMed] [Google Scholar]
- Houndonougbo Y.; Kuczera K.; Jas G. S. Structure and dynamics of phospholamban in solution and in membrane bilayer: computer simulations. Biochemistry 2005, 44, 1780–1792. 10.1021/bi0488404. [DOI] [PubMed] [Google Scholar]
- Oxenoid K.; Chou J. J. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10870–10875. 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardi R.; Shi L.; Traaseth N. J.; Walsh N.; Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9101–9106. 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth N. J.; Shi L.; Verardi R.; Mullen D. G.; Barany G.; Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 10165–10170. 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeazzetto S.; Saponaro A.; Young H. S.; Moncelli M. R.; Thiel G. Structure-function relation of phospholamban: modulation of channel activity as a potential regulator of SERCA activity. PLoS One 2013, 8, e52744. 10.1371/journal.pone.0052744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R.; Cohen S. L.; Kuo A. L.; Lee A.; Chait B. T. Structural conservation in prokaryotic and eukaryotic potassium channels. Science 1998, 280, 106–109. 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- Tsitrin Y.; Morton C. J.; El Bez C.; Paumard P.; Velluz M. C.; Adrian M.; Dubochet J.; Parker M. W.; Lanzavecchia S.; van der Goot F. G. Conversion of a transmembrane to a watersoluble protein complex by a single point mutation. Nat. Struct. Biol. 2002, 9, 729–733. 10.1038/nsb839. [DOI] [PubMed] [Google Scholar]
- Killian J. A.; von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000, 25, 429–434. 10.1016/S0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- Wilmsen H. U.; Leonard K. R.; Tichelaar W.; Buckley J. T.; Pattus F. The aerolysin membrane channel is formed by heptamerization of the monomer. EMBO J. 1992, 11, 2457–2463. 10.1002/j.1460-2075.1992.tb05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L.; Fivaz M.; van der Goot F. G. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 2000, 8, 168–172. 10.1016/S0966-842X(00)01722-4. [DOI] [PubMed] [Google Scholar]
- Slovic A. M.; Kono H.; Lear J. D.; Saven J. G.; DeGrado W. F. Computational design of water-soluble analogues of the potassium channel KcsA. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 1828–1833. 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral J. M.; Pfuetzner R. A.; Kuo A. L.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Zhou Y. F.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 angstrom resolution. Nature 2001, 414, 43–48. 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil F. I.; MacKinnon R.; Muir T. W. Semisynthesis and folding of the potassium channel KcsA. J. Am. Chem. Soc. 2002, 124, 9113–9120. 10.1021/ja0266722. [DOI] [PubMed] [Google Scholar]
- Uysal S.; Vasquez V.; Tereshko V.; Esaki K.; Fellouse F. A.; Sidhu S. S.; Koide S.; Perozo E.; Kossiakoff A. Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6644–6649. 10.1073/pnas.0810663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J. M.; Saven J. G. Statistical theory of combinatorial libraries of folding proteins: Energetic discrimination of a target structure. J. Mol. Biol. 2000, 296, 281–294. 10.1006/jmbi.1999.3426. [DOI] [PubMed] [Google Scholar]
- Munoz V.; Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994, 1, 399–409. 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- Bronson J.; Lee O. S.; Saven J. G. Molecular dynamics simulation of WSK-3, a computationally designed, water-soluble variant of the integral membrane protein KcsA. Biophys. J. 2006, 90, 1156–1163. 10.1529/biophysj.105.068965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D. J.; Tillman T. S.; Tang P.; Meirovitch E.; Eckenhoff R.; Carnini A.; Xu Y. NMR studies of a channel protein without membranes: Structure and dynamics of water-solubilized KcsA. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16537–16542. 10.1073/pnas.0805501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosild T. P.; Choe S. Redesigning an integral membrane K+ channel into a soluble protein. Protein Eng. Des. Sel. 2005, 18, 79–84. 10.1093/protein/gzi010. [DOI] [PubMed] [Google Scholar]
- Jan L. Y.; Jan Y. N. Cloned potassium channels from eukaryotes and prokaryotes. Annu. Rev. Neurosci. 1997, 20, 91–123. 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- Becker C. F. W.; Strop P.; Bass R. B.; Hansen K. C.; Locher K. P.; Ren G.; Yeager M.; Rees D. C.; Kochendoerfer G. G. Conversion of a mechanosensitive channel protein from a membrane-embedded to a water-soluble form by covalent modification with amphiphiles. J. Mol. Biol. 2004, 343, 747–758. 10.1016/j.jmb.2004.08.062. [DOI] [PubMed] [Google Scholar]
- Chang G.; Spencer R. H.; Lee A. T.; Barclay M. T.; Rees D. C. Structure of the MscL homolog from Mycobacterium tuberculosis: A gated mechanosensitive ion channel. Science 1998, 282, 2220–2226. 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- Andrews D. A.; Xie M.; Hughes V.; Wilce M. C.; Roujeinikova A. Design, purification and characterization of a soluble variant of the integral membrane protein MotB for structural studies. J. R. Soc. Interface 2013, 10, 20120717. 10.1098/rsif.2012.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann K. M.; Lowenthal A. C. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect. Immun. 2002, 70, 1984–1990. 10.1128/IAI.70.4.1984-1990.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T.; Imada K.; Namba K. Molecular motors of the bacterial flagella. Curr. Opin. Struct. 2008, 18, 693–701. 10.1016/j.sbi.2008.09.006. [DOI] [PubMed] [Google Scholar]
- O'Shea E. K.; Klemm J. D.; Kim P. S.; Alber T. X-Ray structure of the gcn4 leucine zipper, a 2-stranded, parallel coiled coil. Science 1991, 254, 539–544. 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- Andrews D. A.; Nesmelov Y. E.; Wilce M. C.; Roujeinikova A. Structural analysis of variant of Helicobacter pylori MotB in its activated form, engineered as chimera of MotB and leucine zipper. Sci. Rep. 2017, 7, 13435. 10.1038/s41598-017-13421-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorana H. G. 2 light-transducing membrane-proteins - bacteriorhodopsin and the mammalian rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 1166–1171. 10.1073/pnas.90.4.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N.; Ceska T. A.; Downing K. H.; Baldwin J. M.; Henderson R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J. Mol. Biol. 1996, 259, 393–421. 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- PebayPeyroula E.; Rummel G.; Rosenbusch J. P.; Landau E. M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 1997, 277, 1676–1681. 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- Khorana H. G. Bacteriorhodopsin, a membrane-protein that uses light to translocate protons. J. Biol. Chem. 1988, 263, 7439–7442. 10.1016/S0021-9258(18)68514-X. [DOI] [PubMed] [Google Scholar]
- Mollaaghababa R.; Steinhoff H. J.; Hubbell W. L.; Khorana H. G. Time-resolved site-directed spin-labeling studies of bacteriorhodopsin: Loop-specific conformational changes in M. Biochemistry 2000, 39, 1120–1127. 10.1021/bi991963h. [DOI] [PubMed] [Google Scholar]
- Sirokman G.; Fasman G. D. Refolding and proton-pumping activity of a polyethylene-glycol bacteriorhodopsin water-soluble conjugate. Protein Sci. 1993, 2, 1161–1170. 10.1002/pro.5560020711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibas C.; Subramaniam S. Knowledge-based design of a soluble bacteriorhodopsin. Protein Eng. 1997, 10, 1175–1190. 10.1093/protein/10.10.1175. [DOI] [PubMed] [Google Scholar]
- Mitra K.; Steitz T. A.; Engelman D. M. Rational design of ’water-soluble’ bacteriorhodopsin variants. Protein Eng. 2002, 15, 485–492. 10.1093/protein/15.6.485. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Wesson M.; Yamashita M. Interpretation of protein folding and binding with atomic solvation parameters. Chem. Scr. 1989, 29a, 217–221. [Google Scholar]
- Gohon Y.; Dahmane T.; Ruigrok R. W. H.; Schuck P.; Charvolin D.; Rappaport F.; Timmins P.; Engelman D. M.; Tribet C.; Popot J. L.; et al. Bacteriorhodopsin/amphipol complexes: Structural and functional properties. Biophys. J. 2008, 94, 3523–3537. 10.1529/biophysj.107.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T. X.; Mowrey D.; Bondarenko V.; Tillman T.; Ma D. J.; Landrum E.; Perez-Aguilar J. M.; He J.; Wang W.; Saven J. G.; et al. NMR structure and dynamics of a designed water-soluble transmembrane domain of nicotinic acetylcholine receptor. Biochim. Biophys. Acta Biomembr. 2012, 1818, 617–626. 10.1016/j.bbamem.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger J. E.; Corringer P. J. 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 2011, 60, 116–125. 10.1016/j.neuropharm.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Miyazawa A.; Fujiyoshi Y.; Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003, 423, 949–955. 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Liapakis G.; Cordomi A.; Pardo L. The G-protein coupled receptor family: actors with many faces. Curr. Pharm. Des. 2012, 18, 175–185. 10.2174/138161212799040529. [DOI] [PubMed] [Google Scholar]
- Sriram K.; Insel P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs?. Mol. Pharmacol. 2018, 93, 251–258. 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak G. W.; Pan Y. X. Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Abola E.; Stevens R. C. Recent progress in the structure determination of GPCRs, a membrane protein family with high potential as pharmaceutical targets. Membrane Protein Structure Determination: Methods and Protocols 2010, 654, 141–168. 10.1007/978-1-60761-762-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–U170. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Aguilar J. M.; Xi J.; Matsunaga F.; Cui X.; Selling B.; Saven J. G.; Liu R. Y. A computationally designed water-soluble variant of a G-protein-coupled receptor: the human mu opioid receptor. PLoS One 2013, 8, e66009. 10.1371/journal.pone.0066009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. L.; Perez-Aguilar J. M.; Matsunaga F.; Lerner M.; Xi J.; Selling B.; Johnson A. T. C.; Saven J. G.; Liu R. Y. Characterization of a computationally designed water-soluble human μ-opioid receptor variant using available structural information. Anesthesiology 2014, 121, 866–875. 10.1097/ALN.0000000000000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J.; Xiao J.; Perez-Aguilar J. M.; Ping J. L.; Johnson A. T. C.; Saven J. G.; Liu R. Y. Characterization of an engineered water-soluble variant of the full-length human mu opioid receptor. J. Biomol. Struct. Dyn. 2020, 38, 4364–4370. 10.1080/07391102.2019.1677502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner M. B.; Matsunaga F.; Han G. H.; Hong S. J.; Xi J.; Crook A.; Perez-Aguilar J. M.; Park Y. W.; Saven J. G.; Liu R. Y.; et al. Scalable production of highly sensitive nanosensors based on graphene functionalized with a designed G protein-coupled receptor. Nano Lett. 2014, 14, 2709–2714. 10.1021/nl5006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Johnson A. C.; Saven J. G. Water soluble G-protein coupled receptor enabled biosensors. Transl. perioper. pain med. 2019, 6, 98. 10.31480/2330-4871/095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrachi D.; Chen Y. J.; Liu J. Y.; Peng H. M.; Ke A. L.; Pollack L.; Turner R. J.; Auchus R. J.; DeLisa M. P. Making water-soluble integral membrane proteins in vivo using an amphipathic protein fusion strategy. Nat. Commun. 2015, 6, 98–103. 10.1038/ncomms7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrachi D. Creation of water-soluble integral membrane proteins using an engineered amphipathic protein “shield. Biophys. J. 2015, 108, 38a–38a. 10.1016/j.bpj.2014.11.237. [DOI] [Google Scholar]
- Roosild T. P.; Greenwald J.; Vega M.; Castronovo S.; Riek R.; Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science 2005, 307, 1317–1321. 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- Gursky O.; Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: Implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 2991–2995. 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa M. P.; Tullman D.; Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6115–6120. 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrachi D.; Robinson M. P.; Ren G. P.; Ke N.; Berkmen M.; DeLisa M. P. A water-soluble DsbB variant that catalyzes disulfide-bond formation in vivo. Nat. Chem. Biol. 2017, 13, 1022–1028. 10.1038/nchembio.2409. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang S. G. Self-assembling peptides: From a discovery in a yeast protein to diverse uses and beyond. Protein Sci. 2020, 29, 2281–2303. 10.1002/pro.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. G.; Tao F.; Qing R.; Tang H. Z.; Skuhersky M.; Corin K.; Tegler L.; Wassie A.; Wassie B.; Kwon Y.; et al. QTY code enables design of detergent-free chemokine receptors that retain ligand-binding activities. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E8652–E8659. 10.1073/pnas.1811031115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuhersky M. A.; Tao F.; Qing R.; Smorodina E.; Jin D.; Zhang S. Comparing native crystal structures and AlphaFold2 predicted water-soluble G protein-coupled receptor QTY variants. Life (Basel) 2021, 11, 1285. 10.3390/life11121285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanta B.; Chan K. K.; Barth P.; King T.; Sosnick T. R.; Hinshaw J. R.; Liu G. H.; Everett J. K.; Xiao R.; Montelione G. T.; et al. Introduction of a polar core into the de novo designed protein Top7. Protein Sci. 2016, 25, 1299–1307. 10.1002/pro.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing R.; Han Q. Y.; Skuhersky M.; Chung H.; Badr M.; Schubert T.; Zhang S. G. QTY code designed thermostable and water-soluble chimeric chemokine receptors with tunable ligand affinity. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 25668–25676. 10.1073/pnas.1909026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S.; Jin D.; Zhang S.; Qing R. QTY code-designed water-soluble Fc-fusion cytokine receptors bind to their respective ligands. QRB Discovery 2020, 1, e4. 10.1017/qrd.2020.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing R.; Tao F.; Chatterjee P.; Yang G.; Han Q.; Chung H.; Ni J.; Suter B. P.; Kubicek J.; Maertens B.; et al. Non-full-length water-soluble CXCR4QTY and CCR5QTY chemokine receptors: Implication for overlooked truncated but functional membrane receptors. Iscience 2020, 23, 101670. 10.1016/j.isci.2020.101670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegler L.; Corin K.; Pick H.; Brookes J.; Skuhersky M.; Vogel H.; Zhang S. G. The G protein coupled receptor CXCR4 designed by the QTY code becomes more hydrophilic and retains cell signaling activity. Sci. Rep. 2020, 10, 21371. 10.1038/s41598-020-77659-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkun M. C.; Koelman J. M. V. A.; Er S. Pushing the limits of solubility prediction via quality-oriented data selection. Iscience 2021, 24, 101961. 10.1016/j.isci.2020.101961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Garcia M.; Bano-Polo M.; Varejao N.; Jarnroz M.; Kuriata A.; Diaz-Caballero M.; Lascorz J.; More B.; Navarro S.; Reverter D.; et al. Combining structural aggregation propensity and stability predictions to redesign protein solubility. Mol. Pharmaceutics 2018, 15, 3846–3859. 10.1021/acs.molpharmaceut.8b00341. [DOI] [PubMed] [Google Scholar]
- Lai J. K.; Ambia J.; Wang Y. M.; Barth P. Enhancing structure prediction and design of soluble and membrane proteins with explicit solvent-protein interactions. Structure 2017, 25, 1758–1770. 10.1016/j.str.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. F.; Panaitiu A. E.; Grigoryan G. A general-purpose protein design framework based on mining sequence-structure relationships in known protein structures. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 1059–1068. 10.1073/pnas.1908723117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford R. F.; Leaver-Fay A.; Jeliazkov J. R.; O’Meara M. J.; DiMaio F. P.; Park H.; Shapovalov M. V.; Renfrew P. D.; Mulligan V. K.; Kappel K.; et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gront D.; Kulp D. W.; Vernon R. M.; Strauss C. E. M.; Baker D. Generalized fragment picking in Rosetta: design, protocols and applications. PLoS One 2011, 6, e23294. 10.1371/journal.pone.0023294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman J. K.; Mueller B. K.; Gray J. J. Expanding the toolkit for membrane protein modeling in Rosetta. Bioinformatics 2017, 33, 754–756. 10.1093/bioinformatics/btw716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A. M.; Meiler J. Computational design of membrane proteins using RosettaMembrane. Protein Sci. 2018, 27, 341–355. 10.1002/pro.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W.; Bridgland A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service R. F. The game has changed.’AI triumphs at protein folding. Science 2020, 370, 1144–1145. 10.1126/science.370.6521.1144. [DOI] [PubMed] [Google Scholar]
- Han X.; Ning W.; Ma X.; Wang X.; Zhou K. Improving protein solubility and activity by introducing small peptide tags designed with machine learning models. Metab. Eng. Commun. 2020, 11, e00138 10.1016/j.mec.2020.e00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C. H.; Qin Z.; Martin-Martinez F. J.; Buehler M. J. A self-consistent sonification method to translate amino acid sequences into musical compositions and application in protein design using artificial intelligence. ACS Nano 2019, 13, 7471–7482. 10.1021/acsnano.9b02180. [DOI] [PubMed] [Google Scholar]
- Sehnal D.; Bittrich S.; Deshpande M.; Svobodova R.; Berka K.; Bazgier V.; Velankar S.; Burley S. K.; Koca J.; Rose A. S. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. 10.1093/nar/gkab314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian P. Computational protein design, from single domain soluble proteins to membrane proteins. Chem. Soc. Rev. 2010, 39, 2071–2082. 10.1039/b810924a. [DOI] [PubMed] [Google Scholar]
- Barth P.; Senes A. Toward high-resolution computational design of the structure and function of helical membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 475–480. 10.1038/nsmb.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnow P. Designing minimalist membrane proteins. Biochem. Soc. Trans. 2019, 47, 1233–1245. 10.1042/BST20190170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie K. R. Folding and stability of α-helical integral membrane proteins. Chem. Rev. 2006, 106, 1931–1977. 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- Lewis B. A.; Engelman D. M. Lipid bilayer thickness varies linearly with acyl chain-length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 1983, 166, 211–217. 10.1016/S0022-2836(83)80007-2. [DOI] [PubMed] [Google Scholar]
- Marsh D. Lateral pressure in membranes. Biochim. Biophys. Acta 1996, 1286, 183–223. 10.1016/S0304-4157(96)00009-3. [DOI] [PubMed] [Google Scholar]
- Harder T.; Scheiffele P.; Verkade P.; Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 1998, 141, 929–942. 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q.; Ma Y. Q. Organization of membrane-associated proteins in lipid bilayers. Eur. Phys. J. E 2008, 25, 129–138. 10.1140/epje/i2007-10272-6. [DOI] [PubMed] [Google Scholar]
- Mouritsen O. G.; Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys. J. 1984, 46, 141–153. 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corin K.; Bowie J. U. How bilayer properties influence membrane protein folding. Protein Sci. 2020, 29, 2348–2362. 10.1002/pro.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford C. The hydrophobic effect and the organization of living matter. Science 1978, 200, 1012–1018. 10.1126/science.653353. [DOI] [PubMed] [Google Scholar]
- Hong H. Toward understanding driving forces in membrane protein folding. Arch. Biochem. Biophys. 2014, 564, 297–313. 10.1016/j.abb.2014.07.031. [DOI] [PubMed] [Google Scholar]
- Langosch D.; Heringa J. Interaction of transmembrane helices by a knobs-into-holes packing characteristic of soluble coiled coils. Proteins: Struct. Funct. Genet. 1998, 31, 150–159. . [DOI] [PubMed] [Google Scholar]
- Popot J.-L.; Engelman D. M. Membranes do not tell proteins how to fold. Biochemistry 2016, 55, 5–18. 10.1021/acs.biochem.5b01134. [DOI] [PubMed] [Google Scholar]
- White S. H.; von Heijne G. How translocons select transmembrane helices. Annu. Rev. Biophys. 2008, 37, 23–42. 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- Popot J.-L.; Engelman D. M. Membrane protein folding and oligomerization: the two-stage model. Biochemistry 1990, 29, 4031–4037. 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- Bowie J. U. Membrane proteins: A new method enters the fold. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 3995–3996. 10.1073/pnas.0400671101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovjak H.; Muller D. J.; Humphris A. D. L. Molecular force modulation spectroscopy revealing the dynamic response of single bacteriorhodopsins. Biophys. J. 2005, 88, 1423–1431. 10.1529/biophysj.104.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman D. M.; Chen Y.; Chin C. N.; Curran A. R.; Dixon A. M.; Dupuy A. D.; Lee A. S.; Lehnert U.; Matthews E. E.; Reshetnyak Y. K.; et al. Membrane protein folding: beyond the two stage model. Febs Lett. 2003, 555, 122–125. 10.1016/S0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- Nilsson I.; von Heijne G. Breaking the camel’s back: Proline-induced turns in a model transmembrane helix. J. Mol. Biol. 1998, 284, 1185–1189. 10.1006/jmbi.1998.2219. [DOI] [PubMed] [Google Scholar]
- Monne M.; von Heijne G. Effects of ’hydrophobic mismatch’ on the location of transmembrane helices in the ER membrane. Febs Lett. 2001, 496, 96–100. 10.1016/S0014-5793(01)02415-2. [DOI] [PubMed] [Google Scholar]
- Gromiha M. M. A simple method for predicting transmembrane α helices with better accuracy. Protein Eng. 1999, 12, 557–561. 10.1093/protein/12.7.557. [DOI] [PubMed] [Google Scholar]
- Zhang Y. P.; Lewis R. N. A. H.; Hodges R. S.; Mcelhaney R. N. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane-protein with phosphatidylcholine bilayers - differential scanning calorimetric and ftir spectroscopic studies. Biochemistry 1992, 31, 11579–11588. 10.1021/bi00161a042. [DOI] [PubMed] [Google Scholar]
- Lewis R. N. A. H.; Zhang Y. P.; Hodges R. S.; Subczynski W. K.; Kusumi A.; Flach C. R.; Mendelsohn R.; McElhaney R. N. A polyalanine-based peptide cannot form a stable transmembrane alpha-helix in fully hydrated phospholipid bilayers. Biochemistry 2001, 40, 12103–12111. 10.1021/bi010555m. [DOI] [PubMed] [Google Scholar]
- Chen H. F.; Kendall D. A. Artificial transmembrane segments - requirements for stop transfer and polypeptide orientation. J. Biol. Chem. 1995, 270, 14115–14122. 10.1074/jbc.270.23.14115. [DOI] [PubMed] [Google Scholar]
- Weiss T. M.; van der Wel P. C. A.; Killian J. A.; Koeppe R. E.; Huang H. W. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 2003, 84, 379–385. 10.1016/S0006-3495(03)74858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Delgado C.; Marti-Renom M. A.; Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur. Biophys. J. 2013, 42, 199–207. 10.1007/s00249-012-0813-9. [DOI] [PubMed] [Google Scholar]
- Saidijam M.; Azizpour S.; Patching S. G. Comprehensive analysis of the numbers, lengths and amino acid compositions of transmembrane helices in prokaryotic, eukaryotic and viral integral membrane proteins of high-resolution structure. J. Biomol. Struct. Dyn. 2018, 36, 443–464. 10.1080/07391102.2017.1285725. [DOI] [PubMed] [Google Scholar]
- Lew S.; Caputo G. A.; London E. The effect of interactions involving ionizable residues flanking membrane-inserted hydrophobic helices upon helix–helix interaction. Biochemistry 2003, 42, 10833–10842. 10.1021/bi034929i. [DOI] [PubMed] [Google Scholar]
- Von Heijne G. Membrane-proteins - from sequence to structure. Annu. Rev. Biophys. 1994, 23, 167–192. 10.1146/annurev.bb.23.060194.001123. [DOI] [PubMed] [Google Scholar]
- Vonheijne G. The distribution of positively charged residues in bacterial inner membrane-proteins correlates with the trans-membrane topology. EMBO J. 1986, 5, 3021–3027. 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojemalm K.; Higuchi T.; Lara P.; Lindahl E.; Suga H.; von Heijne G. Energetics of side-chain snorkeling in transmembrane helices probed by nonproteinogenic amino acids. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 10559–10564. 10.1073/pnas.1606776113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil K. T.; Degrado W. F. A Thermodynamic scale for the helix-forming tendencies of the commonly occurring amino-acids. Science 1990, 250, 646–651. 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- Li S.-C.; Goto N. K.; Williams K. A.; Deber C. M. Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6676–6681. 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley W. C.; Corboy M. J.; Cutler T. D.; Thibodeau P. H.; Oldan J.; Lee M. G.; Rizo J.; Hunt J. F.; Thomas P. J. A protein sequence that can encode native structure by disfavoring alternate conformations. Nat. Struct. Biol. 2002, 9, 381–388. 10.1038/nsb784. [DOI] [PubMed] [Google Scholar]
- Fleming K. G. Energetics of membrane protein folding. Annu. Rev. Biophys. 2014, 43, 233–255. 10.1146/annurev-biophys-051013-022926. [DOI] [PubMed] [Google Scholar]
- Haltia T.; Freire E. Forces and factors that contribute to the structural stability of membrane proteins. Biochim. Biophys. Acta 1995, 1228, 1–27. 10.1016/0005-2728(94)00161-W. [DOI] [PubMed] [Google Scholar]
- van den Berg B.; et al. X-ray structure of a protein conducting channel. Cell Struct. Funct. 2005, 30, 9–9. 10.1038/nature02218. [DOI] [Google Scholar]
- Caputo G. A.; London E. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic alpha-helices. Biochemistry 2003, 42, 3275–3285. 10.1021/bi026697d. [DOI] [PubMed] [Google Scholar]
- Barth P.; Schonbrun J.; Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 15682–15687. 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Barth P. A topological and conformational stability alphabet for multipass membrane proteins. Nat. Chem. Biol. 2016, 12, 167–173. 10.1038/nchembio.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. A transmembrane helix dimer: Structure and implications. Science 1997, 276, 131–133. 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- Senes A.; Gerstein M.; Engelman D. M. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 2000, 296, 921–936. 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- Teese M. G.; Langosch D. Role of GxxxG motifs in transmembrane domain interactions. Biochemistry 2015, 54, 5125–5135. 10.1021/acs.biochem.5b00495. [DOI] [PubMed] [Google Scholar]
- Dawson J. P.; Weinger J. S.; Engelman D. M. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 2002, 316, 799–805. 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- Smith S. O.; Song D.; Shekar S.; Groesbeek M.; Ziliox M.; Aimoto S. Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry 2001, 40, 6553–6558. 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- Duong M. T.; Jaszewski T. M.; Fleming K. G.; MacKenzie K. R. Changes in apparent free energy of helix-helix dimerization in a biological membrane due to point mutations. J. Mol. Biol. 2007, 371, 422–434. 10.1016/j.jmb.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langosch D.; Arkin I. T. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009, 18, 1343–1358. 10.1002/pro.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobus F. J.; Fleming K. G. The GxxxG-containing transmembrane domain of the CCK4 oncogene does not encode preferential self-interactions. Biochemistry 2005, 44, 1464–1470. 10.1021/bi048076l. [DOI] [PubMed] [Google Scholar]
- Walters R. F. S.; DeGrado W. F. Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13658–13663. 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E.; Wimley W. C.; Hristova K. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim. Biophys. Acta - Biomembr. 2012, 1818, 183–193. 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S. M.; Mueller B. K.; Lange E. J.; Senes A. Combination of Cα–H hydrogen bonds and van der Waals packing modulates the stability of GxxxG-mediated dimers in membranes. J. Am. Chem. Soc. 2017, 139, 15774–15783. 10.1021/jacs.7b07505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane proteins up for grabs. Nat. Biotechnol. 2007, 25, 646–647. 10.1038/nbt0607-646. [DOI] [PubMed] [Google Scholar]
- Alber T.; Oshea E. K.; Klemm J. D.; Kim P. S. X-Ray crystal-structure of the Gcn4 leucine zipper, an autonomously folding protein subdomain. FASEB J. 1991, 5, A782–A782. [Google Scholar]
- O'Shea E. K.; Rutkowski R.; Kim P. S. Evidence that the leucine zipper is a coiled coil. Science 1989, 243, 538–542. 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Kulp D. W.; Lear J. D.; DeGrado W. F. Experimental and computational evaluation of forces directing the association of transmembrane helices. J. Am. Chem. Soc. 2009, 131, 11341–11343. 10.1021/ja904625b. [DOI] [PubMed] [Google Scholar]
- Gurezka R.; Laage R.; Brosig B.; Langosch D. A heptad motif of leucine residues found in membrane proteins can drive self-assembly of artificial transmembrane segments. J. Biol. Chem. 1999, 274, 9265–9270. 10.1074/jbc.274.14.9265. [DOI] [PubMed] [Google Scholar]
- Adamian L.; Liang J. Interhelical hydrogen bonds and spatial motifs in membrane proteins: Polar clamps and serine zippers. Proteins: Struct. Funct. Genet. 2002, 47, 209–218. 10.1002/prot.10071. [DOI] [PubMed] [Google Scholar]
- Gernert K. M.; Surles M. C.; Labean T. H.; Richardson J. S.; Richardson D. C. The Alacoil: A very tight, antiparallel coiled-coil of helices. Protein Sci. 1995, 4, 2252–2260. 10.1002/pro.5560041102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear J. D.; Wasserman Z. R.; Degrado W. F. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. 10.1126/science.2453923. [DOI] [PubMed] [Google Scholar]
- Whitley P.; Nilsson I. M.; Von Heijne G. De-novo design of integral membrane-proteins. Nat. Struct. Biol. 1994, 1, 858–862. 10.1038/nsb1294-858. [DOI] [PubMed] [Google Scholar]
- Lalaurie C. J.; Dufour V.; Meletiou A.; Ratcliffe S.; Harland A.; Wilson O.; Vamasiri C.; Shoemark D. K.; Williams C.; Arthur C. J. The de novo design of a biocompatible and functional integral membrane protein using minimal sequence complexity. Sci. Rep. 2018, 8, 14564. 10.1038/s41598-018-31964-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury P. B.; Zhang T.; Kim P. S.; Alber T. A Switch between 2-stranded, 3-stranded and 4-stranded coiled coils in gcn4 leucine-zipper mutants. Science 1993, 262, 1401–1407. 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Choma C.; Gratkowski H.; Lear J. D.; DeGrado W. F. Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 2000, 7, 161–166. 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- Xiao Zhou F.; Cocco M. J.; Russ W. P.; Brunger A. T.; Engelman D. M. Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat. Struct. Biol. 2000, 7, 154–160. 10.1038/72430. [DOI] [PubMed] [Google Scholar]
- Gratkowski H.; Lear J. D.; DeGrado W. F. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 880–885. 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. X.; Merianos H. J.; Brunger A. T.; Engelman D. M. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 2250–2255. 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear J. D.; Gratkowski H.; Adamian L.; Liang J.; DeGrado W. F. Position-dependence of stabilizing polar interactions of asparagine in transmembrane helical bundles. Biochemistry 2003, 42, 6400–6407. 10.1021/bi020573j. [DOI] [PubMed] [Google Scholar]
- Tatko C. D.; Nanda V.; Lear J. D.; DeGrado W. F. Polar networks control oligomeric assembly in membranes. J. Am. Chem. Soc. 2006, 128, 4170–4171. 10.1021/ja055561a. [DOI] [PubMed] [Google Scholar]
- Boyken S. E.; Chen Z. B.; Groves B.; Langan R. A.; Oberdorfer G.; Ford A.; Gilmore J. M.; Xu C. F.; DiMaio F.; Pereira J. H.; et al. De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science 2016, 352, 680–687. 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristian L.; Nanda V.; Lear J. D.; DeGrado W. F. Synergistic interactions between aqueous and membrane domains of a designed protein determine its fold and stability. J. Mol. Biol. 2005, 348, 1225–1233. 10.1016/j.jmb.2005.03.053. [DOI] [PubMed] [Google Scholar]
- Yin H.; Slusky J. S.; Berger B. W.; Walters R. S.; Vilaire G.; Litvinov R. I.; Lear J. D.; Caputo G. A.; Bennett J. S.; DeGrado W. F. Computational design of peptides that target transmembrane helices. Science 2007, 315, 1817–1822. 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- Oberai A.; Joh N. H.; Pettit F. K.; Bowie J. U. Structural imperatives impose diverse evolutionary constraints on helical membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17747–17750. 10.1073/pnas.0906390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Q.; Kulp D. W.; Schramm C. A.; Mravic M.; Samish I.; DeGrado W. F. The membrane-and soluble-protein helix-helix interactome: similar geometry via different interactions. Structure 2015, 23, 527–541. 10.1016/j.str.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doura A. K.; Kobus F. J.; Dubrovsky L.; Hibbard E.; Fleming K. G. Sequence context modulates the stability of a GxxxG-mediated transmembrane helix-helix dimer. J. Mol. Biol. 2004, 341, 991–998. 10.1016/j.jmb.2004.06.042. [DOI] [PubMed] [Google Scholar]
- Guo R. Q.; Gaffney K.; Yang Z. Y.; Kim M.; Sungsuwan S.; Huang X. F.; Hubbell W. L.; Hong H. Steric trapping reveals a cooperativity network in the intramembrane protease GlpG. Nat. Chem. Biol. 2016, 12, 353–360. 10.1038/nchembio.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mravic M.; Thomaston J. L.; Tucker M.; Solomon P. E.; Liu L. J.; DeGrado W. F. Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 2019, 363, 1418–1423. 10.1126/science.aav7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulandaisamy A.; Sakthivel R.; Gromiha M. M. MPTherm: database for membrane protein thermodynamics for understanding folding and stability. Brief. Bioinformatics 2021, 22, 2119–2125. 10.1093/bib/bbaa064. [DOI] [PubMed] [Google Scholar]
- Goodall M. C.; Urry D. W. A synthetic transmembrane channel. Biochim. Biophys. Acta - Biomembr. 1973, 291, 317–320. 10.1016/0005-2736(73)90425-2. [DOI] [PubMed] [Google Scholar]
- Kennedy S. J.; Roeske R. W.; Freeman A. R.; Watanabe A. M.; Besche H. R. Synthetic peptides form ion channels in artificial lipid bilayer membranes. Science 1977, 196, 1341–1342. 10.1126/science.867034. [DOI] [PubMed] [Google Scholar]
- Degrado W. F.; Wasserman Z. R.; Lear J. D. Protein design, a minimalist approach. Science 1989, 243, 622–628. 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- Joh N. H.; Wang T.; Bhate M. P.; Acharya R.; Wu Y. B.; Grabe M.; Hong M.; Grigoryan G.; DeGrado W. F. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 2014, 346, 1520–1524. 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.; Kaplan S.; Lear J. D.; DeGrado W. F. Proton and metal ion-dependent assembly of a model diiron protein. Protein Sci. 2001, 10, 958–969. 10.1110/ps.52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison E. A.; DeKoster G. T.; Dutta S.; Vafabakhsh R.; Clarkson M. W.; Bahl A.; Kern D.; Ha T.; Henzler-Wildman K. A. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 2012, 481, 45–50. 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majd S.; Yusko E. C.; Billeh Y. N.; Macrae M. X.; Yang J.; Mayer M. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 2010, 21, 439–476. 10.1016/j.copbio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley H. Nanopore sequencing: from imagination to reality. Clin. Chem. 2015, 61, 25–31. 10.1373/clinchem.2014.223016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L. Q.; Braha O.; Conlan S.; Cheley S.; Bayley H. Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 1999, 398, 686–690. 10.1038/19491. [DOI] [PubMed] [Google Scholar]
- Bayley H.; Cremer P. S. Stochastic sensors inspired by biology. Nature 2001, 413, 226–230. 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- Gilbert R. J. C.; Bayley H.; Anderluh G. Membrane pores: from structure and assembly, to medicine and technology. Philos. Trans. R. Soc. London, B, Biol. Sci. 2017, 372, 20160208 10.1098/rstb.2016.0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niitsu A.; Heal J. W.; Fauland K.; Thomson A. R.; Woolfson D. N. Membrane-spanning alpha-helical barrels as tractable protein-design targets. Philos. Trans. R. Soc. London, B, Biol. Sci. 2017, 372, 20160213 10.1098/rstb.2016.0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Moeyaert B.; Cheley S.; Bayley H.; Maglia G. An engineered ClyA nanopore detects folded target proteins by selective external association and pore entry. Nano Lett. 2012, 12, 4895–4900. 10.1021/nl3024438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K.; Caaveiro J. M. M.; Morante K.; Gonzalez-Manas J. M.; Tsumoto K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 2015, 6, 6337. 10.1038/ncomms7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R. S.; Satheesan R.; Puthumadathil N.; Kumar K. S.; Jayasree P.; Mahendran K. R. Autonomously assembled synthetic transmembrane peptide pore. J. Am. Chem. Soc. 2019, 141, 2949–2959. 10.1021/jacs.8b09973. [DOI] [PubMed] [Google Scholar]
- Krishnan R. S.; Puthumadathil N.; Shaji A. H.; Kumar K. S.; Mohan G.; Mahendran K. R. Designed alpha-helical barrels for charge-selective peptide translocation. Chem. Sci. 2021, 12, 639–649. 10.1039/D0SC04856A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A. J.; Niitsu A.; Kratochvil H. T.; Lang E. J.; Sengel J. T.; Dawson W. M.; Mahendran K. R.; Mravic M.; Thomson A. R.; Brady R. L. Constructing ion channels from water-soluble α-helical barrels. Nat. Chem. 2021, 1–8, 643. 10.1038/s41557-021-00688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.; Weichbrodt C.; Salnikov E. S.; Dynowski M.; Forsberg B. O.; Bechinger B.; Steinem C.; de Groot B. L.; Zachariae U.; Zeth K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 4586–4591. 10.1073/pnas.1214739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S. P. The biophysics of peptide models of ion channels. Prog. Biophys. Mol. Biol. 1991, 55, 139–235. 10.1016/0079-6107(91)90004-C. [DOI] [PubMed] [Google Scholar]
- Korendovych I. V.; Senes A.; Kim Y. H.; Lear J. D.; Fry H. C.; Therien M. J.; Blasie J. K.; Walker F. A.; DeGrado W. F. De novo design and molecular assembly of a transmembrane diporphyrin-binding protein complex. J. Am. Chem. Soc. 2010, 132, 15516–15518. 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran F. V.; Wu S. P.; Wang W.; Nanda V.; Saven J. G.; Therien M. J.; DeGrado W. F. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc. 2005, 127, 1346–1347. 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- Forrest L. R. Structural symmetry in membrane proteins. Annu. Rev. Biophys. 2015, 44, 311–337. 10.1146/annurev-biophys-051013-023008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samish I.; MacDermaid C. M.; Perez-Aguilar J. M.; Saven J. G. Theoretical and computational protein design. Annu. Rev. Phys. Chem. 2011, 62, 129–149. 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- Caputo G. A.; Litvinov R. I.; Li W.; Bennett J. S.; DeGrado W. F.; Yin H. Computationally designed peptide inhibitors of protein-protein interactions in membranes. Biochemistry 2008, 47, 8600–8606. 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mravic M.; Hu H. L.; Lu Z. W.; Bennett J. S.; Sanders C. R.; Orr A. W.; DeGrado W. F. De novo designed transmembrane peptides activating the alpha(5)beta(1) integrin. Protein Eng. Des. Sel. 2018, 31, 181–190. 10.1093/protein/gzy014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim E. N.; Marston J. L.; Federman R. S.; Edwards A. P. B.; Karabadzhak A. G.; Petti L. M.; Engelman D. M.; DiMaio D. Biologically active LIL proteins built with minimal chemical diversity. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E4717–E4725. 10.1073/pnas.1514230112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C. L.; Zidek A.; Nelson A. W. R.; Bridgland A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Bai X. C.; McMullan G.; Scheres S. H. W. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Tunyasuvunakool K.; Adler J.; Wu Z.; Green T.; Zielinski M.; Žídek A.; Bridgland A.; Cowie A.; Meyer C.; Laydon A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Ghirlanda G. Design of membrane proteins: toward functional systems. Curr. Opin. Chem. Biol. 2009, 13, 643–651. 10.1016/j.cbpa.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Scarlata S. The effect of hydrostatic pressure on membrane-bound proteins. Braz. J. Med. Biol. 2005, 38, 1203–1208. 10.1590/S0100-879X2005000800007. [DOI] [PubMed] [Google Scholar]
- FERREIRA L. C.; Schwarz U.; Keck W.; Charlier P.; Dideberg O.; GHUYSEN J. M. Properties and crystallization of a genetically engineered, water-soluble derivative of penicillin-binding protein 5 of Escherichia coli K12. Eur. J. Biochem. 1988, 171, 11–16. 10.1111/j.1432-1033.1988.tb13751.x. [DOI] [PubMed] [Google Scholar]
- Pedro A.; Bonifácio M. J.; Queiroz J.; Maia C.; Passarinha L. A novel prokaryotic expression system for biosynthesis of recombinant human membrane-bound catechol-O-methyltransferase. J. Biotechnol. 2011, 156, 141–146. 10.1016/j.jbiotec.2011.08.022. [DOI] [PubMed] [Google Scholar]
- Fusco G.; De Simone A.; Gopinath T.; Vostrikov V.; Vendruscolo M.; Dobson C. M.; Veglia G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 1–8. 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magarkar A.; Parkkila P.; Viitala T.; Lajunen T.; Mobarak E.; Licari G.; Cramariuc O.; Vauthey E.; Róg T.; Bunker A. Membrane bound COMT isoform is an interfacial enzyme: general mechanism and new drug design paradigm. Chem. Commun. 2018, 54, 3440–3443. 10.1039/C8CC00221E. [DOI] [PubMed] [Google Scholar]
- Sarkar M.; Harsch C. K.; Matic G. T.; Hoffman K.; Norris J. R.; Otto T. C.; Lenz D. E.; Cerasoli D. M.; Magliery T. J. Solubilization and humanization of paraoxonase-1. J. Lipids 2012, 2012, 610937. 10.1155/2012/610937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. C.; Wang C.; Mulchandani A.; Ge X. Engineering soluble human paraoxonase 2 for quorum quenching. ACS Chem. Biol. 2016, 11, 3122–3131. 10.1021/acschembio.6b00527. [DOI] [PubMed] [Google Scholar]
- Mamipour M.; Yousefi M.; Hasanzadeh M. An overview on molecular chaperones enhancing solubility of expressed recombinant proteins with correct folding. Int. J. Biol. Macromol. 2017, 102, 367–375. 10.1016/j.ijbiomac.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D.; Chatterjee D. K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 2006, 17, 353–358. 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- Walker I. H.; Hsieh P.-c.; Riggs P. D. Mutations in maltose-binding protein that alter affinity and solubility properties. Appl. Microbiol. Biotechnol. 2010, 88, 187–197. 10.1007/s00253-010-2696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J. D.; Kapust R. B.; Waugh D. S. Single amino acid substitutions on the surface of Escherichia coli maltose-binding protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 2001, 10, 622–630. 10.1110/ps.45201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust R. B.; Waugh D. S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999, 8, 1668–1674. 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K. Y.; Song J. A.; Ahn K. Y.; Park J. S.; Seo H. S.; Lee J. Solubilization of aggregation-prone heterologous proteins by covalent fusion of stress-responsive Escherichia coli protein. SlyD. Protein Eng. 2007, 20, 543–549. 10.1093/protein/gzm055. [DOI] [PubMed] [Google Scholar]
- Ohana R. F.; Encell L. P.; Zhao K.; Simpson D.; Slater M. R.; Urh M.; Wood K. V. HaloTag7: a genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr. Purif. 2009, 68, 110–120. 10.1016/j.pep.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Yasin A.; Tang R.; Scharer J. M.; Moo-Young M.; Chou C. P. Heterologous expression of lipase in Escherichia coli is limited by folding and disulfide bond formation. Appl. Microbiol. Biotechnol. 2008, 81, 79–87. 10.1007/s00253-008-1644-6. [DOI] [PubMed] [Google Scholar]
- Messens J.; Collet J.-F.; Van Belle K.; Brosens E.; Loris R.; Wyns L. The oxidase DsbA folds a protein with a nonconsecutive disulfide. J. Biol. Chem. 2007, 282, 31302–31307. 10.1074/jbc.M705236200. [DOI] [PubMed] [Google Scholar]
- Martin J. L.; Bardwell J. C.; Kuriyan J. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 1993, 365, 464–468. 10.1038/365464a0. [DOI] [PubMed] [Google Scholar]
- Rathnayaka T.; Tawa M.; Nakamura T.; Sohya S.; Kuwajima K.; Yohda M.; Kuroda Y. Solubilization and folding of a fully active recombinant Gaussia luciferase with native disulfide bonds by using a SEP-Tag. Biochim. Biophys. Acta 2011, 1814, 1775–1778. 10.1016/j.bbapap.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Nautiyal K.; Kuroda Y. A SEP tag enhances the expression, solubility and yield of recombinant TEV protease without altering its activity. N. Biotechnol. 2018, 42, 77–84. 10.1016/j.nbt.2018.02.006. [DOI] [PubMed] [Google Scholar]
- Murashima K.; Kosugi A.; Doi R. H. Solubilization of cellulosomal cellulases by fusion with cellulose-binding domain of noncellulosomal cellulase engd from Clostridium cellulovorans. Proteins: Struct. Funct. Genet. 2003, 50, 620–628. 10.1002/prot.10298. [DOI] [PubMed] [Google Scholar]
- Foong F. C. F.; Doi R. H. Characterization and comparison of clostridium-cellulovorans endoglucanases-xylanases Engb and Engd hyperexpressed in Escherichia-coli. J. Bacteriol. 1992, 174, 1403–1409. 10.1128/jb.174.4.1403-1409.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D. K.; Esposito D. Enhanced soluble protein expression using two new fusion tags. Protein Expr. Purif. 2006, 46, 122–129. 10.1016/j.pep.2005.07.028. [DOI] [PubMed] [Google Scholar]
- Kim Y. E.; Hipp M. S.; Bracher A.; Hayer-Hartl M.; Ulrich Hartl F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Hartl F. U.; Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 2002, 295, 1852–1858. 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Kyratsous C. A.; Silverstein S. J.; DeLong C. R.; Panagiotidis C. A. Chaperone-fusion expression plasmid vectors for improved solubility of recombinant proteins in Escherichia coli. Gene 2009, 440, 9–15. 10.1016/j.gene.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon S. B.; Ryu K.; Son A.; Jeong H.; Lim K. H.; Kim K. H.; Seong B. L.; Choi S. I. Conversion of a soluble protein into a potent chaperone in vivo. Sci. Rep. 2019, 9, 2735. 10.1038/s41598-019-39158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull F.; Koldewey P.; Humes J. R.; Radford S. E.; Bardwell J. C. Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat. Struct. Mol. Biol. 2016, 23, 53–58. 10.1038/nsmb.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen H. P.; Kristensen J. E.; Sperling-Petersen H. U.; Mortensen K. K. Soluble expression of aggregating proteins by covalent coupling to the ribosome. Biochem. Biophys. Res. Commun. 2004, 319, 715–719. 10.1016/j.bbrc.2004.05.081. [DOI] [PubMed] [Google Scholar]
- Hoh J. H. Functional protein domains from the thermally driven motion of polypeptide chains: a proposal. Proteins: Struct. Funct. Genet. 1998, 32, 223–228. . [DOI] [PubMed] [Google Scholar]
- Santner A. A.; Croy C. H.; Vasanwala F. H.; Uversky V. N.; Van Y. Y. J.; Dunker A. K. Sweeping away protein aggregation with entropic bristles: intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. 10.1021/bi300653m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. I.; Han K. S.; Kim C. W.; Ryu K. S.; Kim B. H.; Kim K. H.; Kim S. I.; Kang T. H.; Shin H. C.; Lim K. H.; et al. Protein solubility and folding enhancement by interaction with RNA. PLoS One 2008, 3, e2677 10.1371/journal.pone.0002677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight M. J.; Leettola C.; Gingery M.; Li H.; Bowie J. U. A human sterile alpha motif domain polymerizome. Protein Sci. 2011, 20, 1697–1706. 10.1002/pro.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne N.; Bolon D. N. Charge-rich regions modulate the anti-aggregation activity of Hsp90. J. Mol. Biol. 2010, 401, 931–939. 10.1016/j.jmb.2010.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Pashkov I.; Gao X.; Guerrero J. L.; Yeates T. O.; Tang Y. Rational improvement of simvastatin synthase solubility in Escherichia coli leads to higher whole-cell biocatalytic activity. Biotechnol. Bioeng. 2009, 102, 20–28. 10.1002/bit.22028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramopoulou V.; Mamalaki A.; Tzartos S. J. Soluble, oligomeric, and ligand-binding extracellular domain of the human alpha7 acetylcholine receptor expressed in yeast: replacement of the hydrophobic cysteine loop by the hydrophilic loop of the ACh-binding protein enhances protein solubility. J. Biol. Chem. 2004, 279, 38287–38293. 10.1074/jbc.M402533200. [DOI] [PubMed] [Google Scholar]
- Cozzolino M.; Amori I.; Pesaresi M. G.; Ferri A.; Nencini M.; Carrì M. T. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu, Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 866–874. 10.1074/jbc.M705657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ems-McClung S. C.; Benmoussa M.; Hainline B. E. Mutational analysis of the maize gamma zein C-terminal cysteine residues. Plant Sci. 2002, 162, 131–141. 10.1016/S0168-9452(01)00549-0. [DOI] [Google Scholar]
- Mosavi L. K.; Minor D. L.; Peng Z. Y. Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 16029–16034. 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosavi L. K.; Peng Z. y. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 2003, 16, 739–745. 10.1093/protein/gzg098. [DOI] [PubMed] [Google Scholar]
- Aggarwal S. What’s fueling the biotech engine?. Nat. Biotechnol. 2007, 25, 1097–1104. 10.1038/nbt1007-1097. [DOI] [PubMed] [Google Scholar]
- Elgundi Z.; Reslan M.; Cruz E.; Sifniotis V.; Kayser V. The state-of-play and future of antibody therapeutics. Adv. Drug Delivery Rev. 2017, 122, 2–19. 10.1016/j.addr.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Kaplon H.; Muralidharan M.; Schneider Z.; Reichert J. M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormanni P.; Aprile F. A.; Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem. Soc. Rev. 2018, 47, 9137–9157. 10.1039/C8CS00523K. [DOI] [PubMed] [Google Scholar]
- Waldmann H. Human monoclonal antibodies: the benefits of humanization. Methods Mol. Biol. 2019, 1904, 1–10. 10.1007/978-1-4939-8958-4_1. [DOI] [PubMed] [Google Scholar]
- Wu S. J.; Luo J.; O’Neil K. T.; Kang J.; Lacy E. R.; Canziani G.; Baker A.; Huang M.; Tang Q. M.; Raju T. S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. 10.1093/protein/gzq037. [DOI] [PubMed] [Google Scholar]
- Chennamsetty N.; Voynov V.; Kayser V.; Helk B.; Trout B. L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 11937–11942. 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Zhang J.; Fan H.; Yin R.; Zheng Z.; Xu Q.; Liu Q.; He H.; Peng X.; Wang X.; et al. Expression and purification of soluble single-chain Fv against human fibroblast growth factor receptor 3 fused with Sumo tag in Escherichia coli. Electron. J. Biotechnol. 2015, 18, 302–306. 10.1016/j.ejbt.2015.05.006. [DOI] [Google Scholar]
- Kim Y.-S.; Son A.; Kim J.; Kwon S. B.; Kim M. H.; Kim P.; Kim J.; Byun Y. H.; Sung J.; Lee J.; et al. Chaperna-mediated assembly of ferritin-based Middle East respiratory syndrome-coronavirus nanoparticles. Front. Immunol. 2018, 9, 1093. 10.3389/fimmu.2018.01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelProposto J.; Majmudar C. Y.; Smith J. L.; Brown W. C. Mocr: a novel fusion tag for enhancing solubility that is compatible with structural biology applications. Protein Expr. Purif. 2009, 63, 40–49. 10.1016/j.pep.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zheng S.; Zhao H.; Yang Y. Structure-aware protein solubility prediction from sequence through graph convolutional network and predicted contact map. J. Cheminformatics 2021, 13, 1–10. 10.1186/s13321-021-00488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Zhang L.; Zhou K.; Wang X. ProGAN: Protein solubility generative adversarial nets for data augmentation in DNN framework. Comput. Chem. Eng. 2019, 131, 106533. 10.1016/j.compchemeng.2019.106533. [DOI] [Google Scholar]
- Rawi R.; Mall R.; Kunji K.; Shen C. H.; Kwong P. D.; Chuang G. Y. PaRSnIP: sequence-based protein solubility prediction using gradient boosting machine. Bioinformatics 2018, 34, 1092–1098. 10.1093/bioinformatics/btx662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Niroula A.; Shen B.; Vihinen M. PON-Sol: prediction of effects of amino acid substitutions on protein solubility. Bioinformatics 2016, 32, 2032–2034. 10.1093/bioinformatics/btw066. [DOI] [PubMed] [Google Scholar]
- Hirose S.; Noguchi T. ESPRESSO: a system for estimating protein expression and solubility in protein expression systems. Proteomics 2013, 13, 1444–1456. 10.1002/pmic.201200175. [DOI] [PubMed] [Google Scholar]
- Smialowski P.; Doose G.; Torkler P.; Kaufmann S.; Frishman D. PROSO II--a new method for protein solubility prediction. FEBS J. 2012, 279, 2192–2200. 10.1111/j.1742-4658.2012.08603.x. [DOI] [PubMed] [Google Scholar]
- Agostini F.; Cirillo D.; Livi C. M.; Ponti R. D.; Tartaglia G. G. ccSOL omics: a webserver for large-scale prediction of endogenous and heterologous solubility in E. coli. Bioinformatics 2014, 30, 2975–2977. 10.1093/bioinformatics/btu420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini F.; Vendruscolo M.; Tartaglia G. G. Sequence-based prediction of protein solubility. J. Mol. Biol. 2012, 421, 237–241. 10.1016/j.jmb.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Huang H. L.; Charoenkwan P.; Kao T. F.; Lee H. C.; Chang F. L.; Huang W. L.; Ho S. J.; Shu L. S.; Chen W. L.; Ho S. Y. Prediction and analysis of protein solubility using a novel scoring card method with dipeptide composition. BMC Bioinform. 2012, 13, 1–14. 10.1186/1471-2105-13-S17-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samak T.; Gunter D.; Wang Z.. 2012 IEEE 8th International Conference on E-Science; IEEE, 2012; pp 1–8.
- Wilkinson D. L.; Harrison R. G. Predicting the solubility of recombinant proteins in Escherichia coli. Nat. Biotechnol. 1991, 9, 443–448. 10.1038/nbt0591-443. [DOI] [PubMed] [Google Scholar]
- Balchin D.; Hayer-Hartl M.; Hartl F. U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Tang X. L.; Wang Y.; Li D. L.; Luo J.; Liu M. Y. Orphan G protein-coupled receptors (GPCRs): biological functions and potential drug targets. Acta Pharmacol. Sin. 2012, 33, 363–371. 10.1038/aps.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. K.; McDermott D. H.; Lisco A.; Foster G. A.; Krysztof D.; Follmann D.; Stramer S. L.; Murphy P. M. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 2010, 201, 178–185. 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. P.; DeGrado W. F. Design of a 4-helix bundle protein: synthesis of peptides which self-associate into a helical protein. J. Am. Chem. Soc. 1987, 109, 6751–6758. 10.1021/ja00256a032. [DOI] [Google Scholar]
- Maguire J. B.; Haddox H. K.; Strickland D.; Halabiya S. F.; Coventry B.; Griffin J. R.; Pulavarti S. V. K.; Cummins M.; Thieker D. F.; Klavins E.; et al. Perturbing the energy landscape for improved packing during computational protein design. Proteins: Struct. Funct. Genet. 2021, 89, 436–449. 10.1002/prot.26030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga N.; Koga R.; Liu G.; Castellanos J.; Montelione G. T.; Baker D. Role of backbone strain in de novo design of complex α/β protein structures. Nat. Commun. 2021, 12, 1–12. 10.1038/s41467-021-24050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Cangelosi V. M.; Zastrow M. L.; Tegoni M.; Plegaria J. S.; Tebo A. G.; Mocny C. S.; Ruckthong L.; Qayyum H.; Pecoraro V. L. Protein design: toward functional metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Yeung N.; Sieracki N.; Marshall N. M. Design of functional metalloproteins. Nature 2009, 460, 855–862. 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A.; Summa C. M.; Geremia S.; Randaccio L.; Pavone V.; DeGrado W. F. Retrostructural analysis of metalloproteins: application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6298–6305. 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker P. D. Designing redox metalloproteins from bottom-up and top-down perspectives. Curr. Opin. Struct. 2003, 13, 490–499. 10.1016/S0959-440X(03)00108-8. [DOI] [PubMed] [Google Scholar]
- Mocny C. S.; Pecoraro V. L. De novo protein design as a methodology for synthetic bioinorganic chemistry. Acc. Chem. Res. 2015, 48, 2388–2396. 10.1021/acs.accounts.5b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann G. R.; McRorie D. K.; Tierney D. L.; Utschig L. M.; Singer C. P.; O’Halloran T. V.; Penner-Hahn J. E.; DeGrado W. F.; Pecoraro V. L. De novo design of mercury-binding two-and three-helical bundles. J. Am. Chem. Soc. 1997, 119, 6195–6196. 10.1021/ja964351i. [DOI] [Google Scholar]
- Dieckmann G. R.; McRorie D. K.; Lear J. D.; Sharp K. A.; DeGrado W. F.; Pecoraro V. L. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J. Mol. Biol. 1998, 280, 897–912. 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Yudenfreund Kravitz J.; Thulstrup P. W.; Hemmingsen L.; DeGrado W. F.; Pecoraro V. L. Design of a Three-Helix Bundle Capable of Binding Heavy Metals in a Triscysteine Environment. Angew. Chem. 2011, 50, 2049–2053. 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T.; Mizuno T.; Fukui S.; Hiroaki H.; Oku J.-i.; Kanaori K.; Tajima K.; Shirakawa M. Two-metal ion, Ni (II) and Cu (II), binding α-helical coiled coil peptide. J. Am. Chem. Soc. 2004, 126, 14023–14028. 10.1021/ja047945r. [DOI] [PubMed] [Google Scholar]
- Berwick M. R.; Lewis D. J.; Jones A. W.; Parslow R. A.; Dafforn T. R.; Cooper H. J.; Wilkie J.; Pikramenou Z.; Britton M. M.; Peacock A. F. De novo design of Ln (III) coiled coils for imaging applications. J. Am. Chem. Soc. 2014, 136, 1166–1169. 10.1021/ja408741h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S.; Ishino S.; Ishino Y.; Tanaka T. Control of enzyme reaction by a designed metal-ion-dependent α-helical coiled-coil protein. J. Biol. Inorg. Chem. 2012, 17, 791–799. 10.1007/s00775-012-0896-x. [DOI] [PubMed] [Google Scholar]
- Robertson D. E.; Farid R. S.; Moser C. C.; Urbauer J. L.; Mulholland S. E.; Pidikiti R.; Lear J. D.; Wand A. J.; DeGrado W. F.; Dutton P. L. Design and synthesis of multi-haem proteins. Nature 1994, 368, 425–432. 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- Reedy C. J.; Gibney B. R. Heme protein assemblies. Chem. Rev. 2004, 104, 617–650. 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- Watkins D. W.; Jenkins J. M.; Grayson K. J.; Wood N.; Steventon J. W.; Le Vay K. K.; Goodwin M. I.; Mullen A. S.; Bailey H. J.; Crump M. P.; et al. Construction and in vivo assembly of a catalytically proficient and hyperthermostable de novo enzyme. Nat. Commun. 2017, 8, 1–9. 10.1038/s41467-017-00541-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W. J.; Yu J.; Song W. J. Proteins as diverse, efficient, and evolvable scaffolds for artificial metalloenzymes. Chem. Commun. 2020, 56, 9586–9599. 10.1039/D0CC03137B. [DOI] [PubMed] [Google Scholar]
- Polizzi N. F.; Wu Y.; Lemmin T.; Maxwell A. M.; Zhang S. Q.; Rawson J.; Beratan D. N.; Therien M. J.; DeGrado W. F. De novo design of a hyperstable non-natural protein–ligand complex with sub-Å accuracy. Nat. Chem. 2017, 9, 1157–1164. 10.1038/nchem.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. J.; Müller R.; Toscano M. D.; Kast P.; Hellinga H. W.; Hilvert D.; Houk K. Structural reorganization and preorganization in enzyme active sites: comparisons of experimental and theoretically ideal active site geometries in the multistep serine esterase reaction cycle. J. Am. Chem. Soc. 2008, 130, 15361–15373. 10.1021/ja803213p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinberg C. E.; Khare S. D.; Dou J.; Doyle L.; Nelson J. W.; Schena A.; Jankowski W.; Kalodimos C. G.; Johnsson K.; Stoddard B. L.; et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature 2013, 501, 212–216. 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghellini A.; Jiang L.; Wollacott A. M.; Cheng G.; Meiler J.; Althoff E. A.; Röthlisberger D.; Baker D. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006, 15, 2785–2794. 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der B. S.; Machius M.; Miley M. J.; Mills J. L.; Szyperski T.; Kuhlman B. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc. 2012, 134, 375–385. 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W. J.; Tezcan F. A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 2014, 346, 1525–1528. 10.1126/science.1259680. [DOI] [PubMed] [Google Scholar]
- Blankenship R. E.; Tiede D. M.; Barber J.; Brudvig G. W.; Fleming G.; Ghirardi M.; Gunner M.; Junge W.; Kramer D. M.; Melis A.; et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 2011, 332, 805–809. 10.1126/science.1200165. [DOI] [PubMed] [Google Scholar]
- Mancini J. A.; Kodali G.; Jiang J.; Reddy K. R.; Lindsey J. S.; Bryant D. A.; Dutton P. L.; Moser C. C. Multi-step excitation energy transfer engineered in genetic fusions of natural and synthetic light-harvesting proteins. J. R. Soc. Interface 2017, 14, 20160896 10.1098/rsif.2016.0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson K. J.; Anderson J. R. Designed for life: biocompatible de novo designed proteins and components. J. R. Soc. Interface 2018, 15, 20180472 10.1098/rsif.2018.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce R.; Van Amerongen H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 2014, 10, 492–501. 10.1038/nchembio.1555. [DOI] [PubMed] [Google Scholar]
- Otsuki J. Supramolecular approach towards light-harvesting materials based on porphyrins and chlorophylls. J. Mater. Chem. A 2018, 6, 6710–6753. 10.1039/C7TA11274B. [DOI] [Google Scholar]
- Gaut N. J.; Adamala K. P. Toward artificial photosynthesis. Science 2020, 368, 587–588. 10.1126/science.abc1226. [DOI] [PubMed] [Google Scholar]
- Koebke K. J.; Kühl T.; Lojou E.; Demeler B.; Schoepp-Cothenet B.; Iranzo O.; Pecoraro V. L.; Ivancich A. The pH-Induced Selectivity Between Cysteine or Histidine Coordinated Heme in an Artificial α-Helical Metalloprotein. Angew. Chem. 2021, 133, 4020–4024. 10.1002/ange.202012673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali G.; Mancini J. A.; Solomon L. A.; Episova T. V.; Roach N.; Hobbs C. J.; Wagner P.; Mass O. A.; Aravindu K.; Barnsley J. E.; et al. Design and engineering of water-soluble light-harvesting protein maquettes. Chem. Sci. 2017, 8, 316–324. 10.1039/C6SC02417C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda V.; Koder R. L. Designing artificial enzymes by intuition and computation. Nat. Chem. 2010, 2, 15–24. 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser C. C.; Ennist N. M.; Mancini J. A.; Dutton P. L.. Mechanisms of Primary Energy Transduction in Biology; The Royal Society of Chemistry, 2018. [Google Scholar]
- Chen X.; Moser C. C.; Pilloud D. L.; Dutton P. L. Molecular orientation of Langmuir–Blodgett films of designed heme protein and lipoprotein maquettes. J. Phys. Chem. B 1998, 102, 6425–6432. 10.1021/jp981855p. [DOI] [Google Scholar]
- Strzalka J.; Chen X.; Moser C. C.; Dutton P. L.; Ocko B. M.; Blasie J. K. X-ray scattering studies of maquette peptide monolayers. 1. Reflectivity and grazing incidence diffraction at the air/water interface. Langmuir 2000, 16, 10404–10418. 10.1021/la000264z. [DOI] [Google Scholar]
- Tronin A.; Strzalka J.; Chen X.; Dutton P.; Ocko B.; Blasie J. Orientational distributions of the di-α-helical synthetic peptide ZnPPIX-BBC16 in Langmuir monolayers by x-ray reflectivity and polarized epifluorescence. Langmuir 2001, 17, 3061–3066. 10.1021/la001489c. [DOI] [Google Scholar]
- Huang S. S.; Koder R. L.; Lewis M.; Wand A. J.; Dutton P. L. The HP-1 maquette: from an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5536–5541. 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koder R. L.; Dutton P. L. Intelligent design: the de novo engineering of proteins with specified functions. Dalton Trans. 2006, 3045–3051. 10.1039/b514972j. [DOI] [PubMed] [Google Scholar]
- Koder R. L.; Valentine K. G.; Cerda J.; Noy D.; Smith K. M.; Wand A. J.; Dutton P. L. Nativelike structure in designed four α-helix bundles driven by buried polar interactions. J. Am. Chem. Soc. 2006, 128, 14450–14451. 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- Anderson J. R.; Koder R. L.; Moser C. C.; Dutton P. L. Controlling complexity and water penetration in functional de novo protein design. Biochem. Soc. Trans. 2008, 36, 1106–1111. 10.1042/BST0361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney B. R.; Rabanal F.; Skalicky J. J.; Wand A. J.; Dutton P. L. Iterative protein redesign. J. Am. Chem. Soc. 1999, 121, 4952–4960. 10.1021/ja9833117. [DOI] [Google Scholar]
- Discher B. M.; Noy D.; Strzalka J.; Ye S.; Moser C. C.; Lear J. D.; Blasie J. K.; Dutton P. L. Design of amphiphilic protein maquettes: controlling assembly, membrane insertion, and cofactor interactions. Biochemistry 2005, 44, 12329–12343. 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear J.; Wasserman Z.; DeGrado W. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. 10.1126/science.2453923. [DOI] [PubMed] [Google Scholar]
- Nishimura K.; Kim S.; Zhang L.; Cross T. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry 2002, 41, 13170–13177. 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- Ye S.; Strzalka J. W.; Discher B. M.; Noy D.; Zheng S.; Dutton P. L.; Blasie J. K. Amphiphilic 4-helix bundles designed for biomolecular materials applications. Langmuir 2004, 20, 5897–5904. 10.1021/la0363884. [DOI] [PubMed] [Google Scholar]
- Goparaju G.; Fry B. A.; Chobot S. E.; Wiedman G.; Moser C. C.; Dutton P. L.; Discher B. M. First principles design of a core bioenergetic transmembrane electron-transfer protein. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 503–512. 10.1016/j.bbabio.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid T. A.; Kodali G.; Solomon L. A.; Lichtenstein B. R.; Sheehan M. M.; Fry B. A.; Bialas C.; Ennist N. M.; Siedlecki J. A.; Zhao Z.; et al. Elementary tetrahelical protein design for diverse oxidoreductase functions. Nat. Chem. Biol. 2013, 9, 826–833. 10.1038/nchembio.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister K. A.; Zou H.; Cochran F. V.; Bender G. M.; Senes A.; Fry H. C.; Nanda V.; Keenan P. A.; Lear J. D.; Saven J. G.; et al. Using α-helical coiled-coils to design nanostructured metalloporphyrin arrays. J. Am. Chem. Soc. 2008, 130, 11921–11927. 10.1021/ja800697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D. W.; Armstrong C. T.; Anderson J. R. De novo protein components for oxidoreductase assembly and biological integration. Curr. Opin. Chem. Biol. 2014, 19, 90–98. 10.1016/j.cbpa.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Anderson J. R.; Armstrong C. T.; Kodali G.; Lichtenstein B. R.; Watkins D. W.; Mancini J. A.; Boyle A. L.; Farid T. A.; Crump M. P.; Moser C. C.; et al. Constructing a man-made c-type cytochrome maquette in vivo: electron transfer, oxygen transport and conversion to a photoactive light harvesting maquette. Chem. Sci. 2014, 5, 507–514. 10.1039/C3SC52019F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D. W.; Armstrong C. T.; Beesley J. L.; Marsh J. E.; Jenkins J. M.; Sessions R. B.; Mann S.; Anderson J. R. A suite of de novo c-type cytochromes for functional oxidoreductase engineering. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 493–502. 10.1016/j.bbabio.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Chen M.; Eggink L. L.; Hoober J. K.; Larkum A. W. Influence of structure on binding of chlorophylls to peptide ligands. J. Am. Chem. Soc. 2005, 127, 2052–2053. 10.1021/ja043462b. [DOI] [PubMed] [Google Scholar]
- Hobbs C. J.; Roach N.; Wagner P.; van der Salm H.; Barnsley J. E.; Gordon K. C.; Kodali G.; Moser C. C.; Dutton P. L.; Wagner K.; et al. Emulating photosynthetic processes with light harvesting synthetic protein (maquette) assemblies on titanium dioxide. Mater. Adv. 2020, 1, 1877–1885. 10.1039/D0MA00427H. [DOI] [Google Scholar]
- Son M.; Pinnola A.; Gordon S. C.; Bassi R.; Schlau-Cohen G. S. Observation of dissipative chlorophyll-to-carotenoid energy transfer in light-harvesting complex II in membrane nanodiscs. Nat. Commun. 2020, 11, 1–8. 10.1038/s41467-020-15074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.; Watanabe A.; Sato R.; Okajima K.; Minagawa J. pH-responsive binding properties of light-harvesting complexes in a photosystem II supercomplex investigated by thermodynamic dissociation kinetics analysis. J. Phys. Chem. Lett. 2019, 10, 3615–3620. 10.1021/acs.jpclett.9b01208. [DOI] [PubMed] [Google Scholar]
- Cohen-Ofri I.; van Gastel M.; Grzyb J.; Brandis A.; Pinkas I.; Lubitz W.; Noy D. Zinc-bacteriochlorophyllide dimers in de novo designed four-helix bundle proteins. A model system for natural light energy harvesting and dissipation. J. Am. Chem. Soc. 2011, 133, 9526–9535. 10.1021/ja202054m. [DOI] [PubMed] [Google Scholar]
- Zeng X.-L.; Tang K.; Zhou N.; Zhou M.; Hou H. J.; Scheer H.; Zhao K.-H.; Noy D. Bimodal intramolecular excitation energy transfer in a multichromophore photosynthetic model system: hybrid fusion proteins comprising natural phycobilin-and artificial chlorophyll-binding domains. J. Am. Chem. Soc. 2013, 135, 13479–13487. 10.1021/ja405617c. [DOI] [PubMed] [Google Scholar]
- Silva D.-A.; Yu S.; Ulge U. Y.; Spangler J. B.; Jude K. M.; Labão-Almeida C.; Ali L. R.; Quijano-Rubio A.; Ruterbusch M.; Leung I.; et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 2019, 565, 186–191. 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth M. J.; Cretney E.; Kershaw M. H.; Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol. Rev. 2004, 202, 275–293. 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- Wang X. Q.; Rickert M.; Garcia K. C. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gamma(c) receptors. Science 2005, 310, 1159–1163. 10.1126/science.1117893. [DOI] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Elucidating a Key Component of Cancer Metastasis: CXCL12 (SDF-1 alpha) Binding to CXCR4. J. Chem. Inf. Model. 2014, 54, 1174–1188. 10.1021/ci500069y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; Floudas C. A. Elucidating a Key Anti-HIV-1 and Cancer-Associated Axis: The Structure of CCL5 (Rantes) in Complex with CCR5. Sci. Rep. 2015, 4, 5447. 10.1038/srep05447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Sesterhenn F.; Bonet J.; van Aalen E. A.; Scheller L.; Abriata L. A.; Cramer J. T.; Wen X.; Rosset S.; Georgeon S.; et al. Bottom-up de novo design of functional proteins with complex structural features. Nat. Chem. Biol. 2021, 17, 492–500. 10.1038/s41589-020-00699-x. [DOI] [PubMed] [Google Scholar]
- Ljubetic A.; Gradisar H.; Jerala R. Advances in design of protein folds and assemblies. Curr. Opin. Chem. Biol. 2017, 40, 65–71. 10.1016/j.cbpa.2017.06.020. [DOI] [PubMed] [Google Scholar]
- Nowick J. S. Exploring beta-sheet structure and interactions with chemical model systems. Acc. Chem. Res. 2008, 41, 1319–1330. 10.1021/ar800064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman C. J.; Nilsson B. L. Review self-assembly of amphipathic beta-sheet peptides: Insights and applications. Biopolymers 2012, 98, 169–184. 10.1002/bip.22058. [DOI] [PubMed] [Google Scholar]
- Nesloney C. L.; Kelly J. W. Progress towards understanding beta-sheet structure. Bioorg. Med. Chem. 1996, 4, 739–766. 10.1016/0968-0896(96)00051-X. [DOI] [PubMed] [Google Scholar]
- Lupas A. Coiled coils: New structures and new functions. Trends Biochem. Sci. 1996, 21, 375–382. 10.1016/S0968-0004(96)10052-9. [DOI] [PubMed] [Google Scholar]
- Woolfson D. N.; Bartlett G. J.; Bruning M.; Thomson A. R. New currency for old rope: from coiled-coil assemblies to alpha-helical barrels. Curr. Opin. Struct. 2012, 22, 432–441. 10.1016/j.sbi.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zheng Q.; Deng Y. Q.; Cheng C. S.; Kallenbach N. R.; Lu M. A seven-helix coiled coil. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15457–15462. 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai N. R.; Chi B.; Thomson A. R.; Boyle A. L.; Bartlett G. J.; Bruning M.; Linden N.; Sessions R. B.; Booth P. J.; Brady R. L.; et al. A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol. 2011, 7, 935–941. 10.1038/nchembio.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhys G. G.; Wood C. W.; Beesley J. L.; Zaccai N. R.; Burton A. J.; Brady R. L.; Thomson A. R.; Woolfson D. N. Navigating the structural landscape of de novo α-helical bundles. J. Am. Chem. Soc. 2019, 141, 8787–8797. 10.1021/jacs.8b13354. [DOI] [PubMed] [Google Scholar]
- Burton A. J.; Thomas F.; Agnew C.; Hudson K. L.; Halford S. E.; Brady R. L.; Woolfson D. N. Accessibility, reactivity, and selectivity of side chains within a channel of de novo peptide assembly. J. Am. Chem. Soc. 2013, 135, 12524–12527. 10.1021/ja4053027. [DOI] [PubMed] [Google Scholar]
- Thomas F.; Dawson W. M.; Lang E. J.; Burton A. J.; Bartlett G. J.; Rhys G. G.; Mulholland A. J.; Woolfson D. N. De novo-designed α-helical barrels as receptors for small molecules. ACS Synth. Biol. 2018, 7, 1808–1816. 10.1021/acssynbio.8b00225. [DOI] [PubMed] [Google Scholar]
- Grigoryan G.; DeGrado W. F. Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol. 2011, 405, 1079–1100. 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P. S.; Oberdorfer G.; Xu C. F.; Pei X. Y.; Nannenga B. L.; Rogers J. M.; DiMaio F.; Gonen T.; Luisi B.; Baker D. High thermodynamic stability of parametrically designed helical bundles. Science 2014, 346, 481–485. 10.1126/science.1257481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman R.; Akerfeldt K. S. Peptides as novel smart materials. Curr. Opin. Struct. 2005, 15, 453–463. 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Jacob J. T.; Coulombe P. A.; Kwan R.; Omary M. B. Types I and II keratin intermediate filaments. Cold Spring Harb. Perspect. Biol. 2018, 10, a018275 10.1101/cshperspect.a018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser J.; Grabmayr H.; Harasim M.; Herrmann H.; Bausch A. R. Assembly kinetics determine the structure of keratin networks. Soft Matter 2012, 8, 8873–8879. 10.1039/c2sm26032h. [DOI] [Google Scholar]
- Lee C. H.; Kim M. S.; Chung B. M.; Leahy D. J.; Coulombe P. A. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 2012, 19, 707–715. 10.1038/nsmb.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya M. J.; Spooner G. M.; Sunde M.; Thorpe J. R.; Rodger A.; Woolfson D. N. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry 2000, 39, 8728–8734. 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- Wagner D. E.; Phillips C. L.; Ali W. M.; Nybakken G. E.; Crawford E. D.; Schwab A. D.; Smith W. F.; Fairman R. Toward the development of peptide nanofilaments and nanoropes as smart materials. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12656–12661. 10.1073/pnas.0505871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar M.; Wang L. S.; Rotello V.; Chmielewski J. Reversible hierarchical assembly of trimeric coiled-coil peptides into banded nano-and microstructures. J. Am. Chem. Soc. 2018, 140, 13028–13033. 10.1021/jacs.8b08163. [DOI] [PubMed] [Google Scholar]
- Lanci C. J.; MacDermaid C. M.; Kang S. G.; Acharya R.; North B.; Yang X.; Qiu X. J.; DeGrado W. F.; Saven J. G. Computational design of a protein crystal. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7304–7309. 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M. D.; Harrison S. C. Structure of an I kappa B alpha/NF-kappa B complex. Cell 1998, 95, 749–758. 10.1016/S0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- Tayeb-Fligelman E.; Tabachnikov O.; Moshe A.; Goldshmidt-Tran O.; Sawaya M. R.; Coquelle N.; Colletier J. P.; Landau M. The cytotoxic Staphylococcus aureus PSM alpha a3 reveals a cross-alpha amyloid-like fibril. Science 2017, 355, 831–833. 10.1126/science.aaf4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunette T. J.; Parmeggiani F.; Huang P. S.; Bhabha G.; Ekiert D. C.; Tsutakawa S. E.; Hura G. L.; Tainer J. A.; Baker D. Exploring the repeat protein universe through computational protein design. Nature 2015, 528, 580–584. 10.1038/nature16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmeggiani F.; Huang P. S. Designing repeat proteins: a modular approach to protein design. Curr. Opin. Struct. 2017, 45, 116–123. 10.1016/j.sbi.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Huang P. S.; Ban Y. E. A.; Richter F.; Andre I.; Vernon R.; Schief W. R.; Baker D. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One 2011, 6, e24109 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins A. M.; Arora P. S. Anatomy of β-strands at protein–protein interfaces. ACS Chem. Biol. 2014, 9, 1747–1754. 10.1021/cb500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J. K. M.; Jackson S. E. The discovery and development of the green fluorescent protein, GFP. Chem. Soc. Rev. 2009, 38, 2821–2822. 10.1039/b917331p. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.; Richardson D. C. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2754–2759. 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J. S.; Richardson D. C. The de novo design of protein structures. Trends Biochem. Sci. 1989, 14, 304–309. 10.1016/0968-0004(89)90070-4. [DOI] [PubMed] [Google Scholar]
- Lim A.; Saderholm M. J.; Makhov A. M.; Kroll M.; Yan Y. B.; Perera L.; Griffith J. D.; Erickson B. W. Engineering of betabellin-15D: A 64 residue beta sheet protein that forms long narrow multimeric fibrils. Protein Sci. 1998, 7, 1545–1554. 10.1002/pro.5560070708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn T. P.; Tweedy N. B.; Williams R. W.; Richardson J. S.; Richardson D. C. Betadoublet - de-novo design, synthesis, and characterization of a beta-sandwich protein. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 8747–8751. 10.1073/pnas.91.19.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde J. M.; Bernlohr D. A.; Banaszak L. J. The up-and-down beta-barrel proteins. FASEB J. 1994, 8, 1240–1247. 10.1096/fasebj.8.15.8001736. [DOI] [PubMed] [Google Scholar]
- Murzin A. G.; Lesk A. M.; Chothia C. Principles determining the structure of beta-sheet barrels in proteins 0.1. a theoretical-analysis. J. Mol. Biol. 1994, 236, 1369–1381. 10.1016/0022-2836(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.; Getzoff E. D.; Richardson D. C. The beta bulge: a common small unit of nonrepetitive protein structure. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 2574–2578. 10.1073/pnas.75.6.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar R.; Feehan R.; Slusky J. S. Membrane barrels are taller, fatter, inside-out soluble barrels. J. Phys. Chem. B 2021, 125, 3622–3628. 10.1021/acs.jpcb.1c00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. A.; Whitehead T. A.; Nanda V. Computational redesign of the lipid-facing surface of the outer membrane protein OmpA. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 9632–9637. 10.1073/pnas.1501836112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D. S.; Sawaya M. R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- Sunde M.; Serpell L. C.; Bartlam M.; Fraser P. E.; Pepys M. B.; Blake C. C. F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Degrado W. F.; Lear J. D. Induction of peptide conformation at apolar water interfaces 0.1. a study with model peptides of defined hydrophobic periodicity. J. Am. Chem. Soc. 1985, 107, 7684–7689. 10.1021/ja00311a076. [DOI] [Google Scholar]
- Friedmann M. P.; Torbeev V.; Zelenay V.; Sobol A.; Greenwald J.; Riek R. Towards prebiotic catalytic amyloids using high throughput screening. PLoS One 2015, 10, e0143948 10.1371/journal.pone.0143948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Garawi Z. S.; McIntosh B. A.; Neill-Hall D.; Hatimy A. A.; Sweet S. M.; Bagley M. C.; Serpell L. C. The amyloid architecture provides a scaffold for enzyme-like catalysts. Nanoscale 2017, 9, 10773–10783. 10.1039/C7NR02675G. [DOI] [PubMed] [Google Scholar]
- Shigemitsu H.; Hamachi I. Design strategies of stimuli-responsive supramolecular hydrogels relying on structural analyses and cell-mimicking approaches. Acc. Chem. Res. 2017, 50, 740–750. 10.1021/acs.accounts.7b00070. [DOI] [PubMed] [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- Mustata G. M.; Kim Y. H.; Zhang J.; DeGrado W. F.; Grigoryan G.; Wanunu M. Graphene symmetry amplified by designed peptide self-assembly. Biophys. J. 2016, 110, 2507–2516. 10.1016/j.bpj.2016.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellach M.; Mondal S.; Harlos K.; Mance D.; Baldus M.; Gazit E.; Shimon L. J. W. A two-tailed phosphopeptide crystallizes to form a lamellar structure. Angew. Chem. 2017, 56, 3252–3255. 10.1002/anie.201609877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.; Sawaya M. R.; Balbirnie M.; Madsen A. O.; Riekel C.; Grothe R.; Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; McFarlane H. T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Gordon D. J.; Balbach J. J.; Tycko R.; Meredith S. C. Increasing the amphiphilicity of an amyloidogenic peptide changes the beta-sheet structure in the fibrils from antiparallel to parallel. Biophys. J. 2004, 86, 428–434. 10.1016/S0006-3495(04)74119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman J. K.; Weitzner B. D.; Lewis S. M.; Adolf-Bryfogle J.; Alam N.; Alford R. F.; Aprahamian M.; Baker D.; Barlow K. A.; Barth P.; et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 2020, 17, 665–680. 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N. P.; Sheffler W.; Sawaya M. R.; Vollmar B. S.; Sumida J. P.; Andre I.; Gonen T.; Yeates T. O.; Baker D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 2012, 336, 1171–1174. 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. B.; Johnson M. C.; Chen J. J.; Bick M. J.; Boyken S. E.; Lin B. H.; De Yoreo J. J.; Kollman J. M.; Baker D.; DiMaio F. Self-assembling 2D arrays with de novo protein building blocks. J. Am. Chem. Soc. 2019, 141, 8891–8895. 10.1021/jacs.9b01978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divine R.; Dang H.; Ueda G.; Fallas J. A.; Vulovic I.; Sheffler W.; Saini S.; Zhao Y. T.; Raj I. X.; Morawski P. A.; et al. Designed proteins assemble antibodies into modular nanocages. Science 2021, 372, 47. 10.1126/science.abd9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulovic I.; Yao Q.; Park Y. J.; Courbet A.; Norris A.; Busch F.; Sahasrabuddhe A.; Merten H.; Sahtoe D. D.; Ueda G.; et al. Generation of ordered protein assemblies using rigid three-body fusion. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2015037118 10.1073/pnas.2015037118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas L. J.; Wu C. H.; Das R.; Wu J. I. C. Hydrogen bond design principles. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1477 10.1002/wcms.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. F.; Meyer T. A.; Pan V.; Dutta P. K.; Ke Y. G. The beauty and utility of DNA origami. Chem. 2017, 2, 359–382. 10.1016/j.chempr.2017.02.009. [DOI] [Google Scholar]
- Chen Y. J.; Groves B.; Muscat R. A.; Seelig G. DNA nanotechnology from the test tube to the cell. Nat. Nanotechnol. 2015, 10, 748–760. 10.1038/nnano.2015.195. [DOI] [PubMed] [Google Scholar]
- Xu D.; Tsai C. J.; Nussinov R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- Sheinerman F. B.; Honig B. On the role of electrostatic interactions in the design of protein-protein interfaces. J. Mol. Biol. 2002, 318, 161–177. 10.1016/S0022-2836(02)00030-X. [DOI] [PubMed] [Google Scholar]
- Kuroda D.; Gray J. J. Shape complementarity and hydrogen bond preferences in protein-protein interfaces: implications for antibody modeling and protein-protein docking. Bioinformatics 2016, 32, 2451–2456. 10.1093/bioinformatics/btw197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T.; Baker D. Computational design of protein-protein interactions. Curr. Opin. Chem. Biol. 2004, 8, 91–97. 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Joachimiak L. A.; Kortemme T.; Stoddard B. L.; Baker D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J. Mol. Biol. 2006, 361, 195–208. 10.1016/j.jmb.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Stranges P. B.; Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013, 22, 74–82. 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. L.; Oezguen N.; Urvil P.; Ferguson C.; Dann S. M.; Savidge T. C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240 10.1126/sciadv.1501240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey D. L.; Malashkevich V. N.; Kim P. S. Buried polar residues in coiled-coil interfaces. Biochemistry 2001, 40, 6352–6360. 10.1021/bi002829w. [DOI] [PubMed] [Google Scholar]
- Maguire J. B.; Boyken S. E.; Baker D.; Kuhlman B. Rapid sampling of hydrogen bond networks for computational protein design. J. Chem. Theory Comput. 2018, 14, 2751–2760. 10.1021/acs.jctc.8b00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldburger C. D.; Jonsson T.; Sauer R. T. Barriers to protein folding: Formation of buried polar interactions is a slow step in acquisition of structure. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 2629–2634. 10.1073/pnas.93.7.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J. K.; Oas T. G. Contribution of a buried hydrogen bond to lambda repressor folding kinetics. Biochemistry 1999, 38, 6761–6768. 10.1021/bi990088x. [DOI] [PubMed] [Google Scholar]
- White S. H. How hydrogen bonds shape membrane protein structure. Adv. Protein Chem. 2005, 72, 157–172. 10.1016/S0065-3233(05)72006-4. [DOI] [PubMed] [Google Scholar]
- Illergard K.; Kauko A.; Elofsson A. Why are polar residues within the membrane core evolutionary conserved?. Proteins: Struct. Funct. Genet. 2011, 79, 79–91. 10.1002/prot.22859. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Ma A. K.; Fonseca R.; Latorraca N. R.; Kelly B.; Betz R. M.; Asawa C.; Kobilka B. K.; Dror R. O. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 3288–3293. 10.1073/pnas.1809251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh N. H.; Min A.; Faham S.; Whitelegge J. P.; Yang D.; Woods V. L.; Bowie J. U. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature 2008, 453, 1266–1273. 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J. U. Membrane protein folding: how important are hydrogen bonds?. Curr. Opin. Struct. 2011, 21, 42–49. 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C. L.; Kuo Y. H.; Lee S. W.; Chiang Y. W. Protein stability depends critically on the surface hydrogen-bonding network: A case study of Bid protein. J. Phys. Chem. B 2021, 125, 8373–8382. 10.1021/acs.jpcb.1c03245. [DOI] [PubMed] [Google Scholar]
- Frenkel D.; Smit B.; Tobochnik J.; McKay S. R.; Christian W. Understanding molecular simulation. Comput. Phys. 1997, 11, 351–354. 10.1063/1.4822570. [DOI] [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. T.; Humphrey W.; Gursoy A.; Dalke A.; Kalé L. V.; Skeel R. D.; Schulten K. NAMD: a parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. 1996, 10, 251–268. 10.1177/109434209601000401. [DOI] [Google Scholar]
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M. J.; Giupponi G.; Fabritiis G. D. ACEMD: Accelerating bio-molecular dynamics in the microsecond time-scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); IEEE, 2006; p 43.
- Lemke O.; Gotze J. P. On the stability of the water-soluble chlorophyll-binding protein (WSCP) studied by molecular dynamics simulations. J. Phys. Chem. B 2019, 123, 10594–10604. 10.1021/acs.jpcb.9b07915. [DOI] [PubMed] [Google Scholar]
- Tjong H.; Zhou H. X. Prediction of protein solubility from calculation of transfer free energy. Biophys. J. 2008, 95, 2601–2609. 10.1529/biophysj.107.127746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lbadaoui-Darvas M.; Takahama S. Water activity from equilibrium molecular dynamics simulations and Kirkwood-Buff theory. J. Phys. Chem. B 2019, 123, 10757–10768. 10.1021/acs.jpcb.9b06735. [DOI] [PubMed] [Google Scholar]
- Harrigan M. P.; Shukla D.; Pande V. S. Conserve water: A method for the analysis of solvent in molecular dynamics. J. Chem. Theory Comput. 2015, 11, 1094–1101. 10.1021/ct5010017. [DOI] [PubMed] [Google Scholar]
- Sugita M.; Sugiyama S.; Fujie T.; Yoshikawa Y.; Yanagisawa K.; Ohue M.; Akiyama Y. Large-scale membrane permeability prediction of cyclic peptides crossing a lipid bilayer based on enhanced sampling molecular dynamics simulations. J. Chem. Inf. Model. 2021, 61, 3681–3695. 10.1021/acs.jcim.1c00380. [DOI] [PubMed] [Google Scholar]
- Pluhackova K.; Wilhelm F. M.; Müller D. J. Lipids and phosphorylation conjointly modulate complex formation of β2-adrenergic receptor and β-arrestin2. Front. Cell Dev. Biol. 2021, 9, 807913. 10.3389/fcell.2021.807913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica M. P.; Smulski C. R. Coarse grained molecular dynamic simulations for the study of TNF receptor family members’ transmembrane organization. Front. Cell Dev. Biol. 2021, 8, 577278. 10.3389/fcell.2020.577278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. I.; Shao Q.; Zhang J.; Yang L.; Gao Y. Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902 10.1063/1.5109531. [DOI] [PubMed] [Google Scholar]
- Lazim R.; Suh D.; Choi S. Advances in molecular dynamics simulations and enhanced sampling methods for the study of protein systems. Int. J. Mol. Sci. 2020, 21, 6339. 10.3390/ijms21176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan A. C.; Jacobson D.; Yatsenko K.; Sritharan D.; Weinreich T. M.; Shaw D. E. Atomic-level characterization of protein-protein association. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 4244–4249. 10.1073/pnas.1815431116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi M.Self-assembling Biomaterials; Elsevier, 2018. [Google Scholar]
- Wilson C. J.; Bommarius A. S.; Champion J. A.; Chernoff Y. O.; Lynn D. G.; Paravastu A. K.; Liang C.; Hsieh M. C.; Heemstra J. M. Biomolecular assemblies: moving from observation to predictive design. Chem. Rev. 2018, 118, 11519–11574. 10.1021/acs.chemrev.8b00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norn C. H.; Andre I. Computational design of protein self-assembly. Curr. Opin. Struct. 2016, 39, 39–45. 10.1016/j.sbi.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Partridge A. W.; Therien A. G.; Deber C. M. Missense mutations in transmembrane domains of proteins: Phenotypic propensity of polar residues for human disease. Proteins: Struct. Funct. Genet. 2004, 54, 648–656. 10.1002/prot.10611. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrati G.; Landau M.; Ben-Tal N.; Lupas A.; Kosloff M.; Kosinski J. Integrative structural biology in the era of accurate structure prediction. J. Mol. Biol. 2021, 433, 167127. 10.1016/j.jmb.2021.167127. [DOI] [PubMed] [Google Scholar]
- Kinch L. N.; Pei J. M.; Kryshtafovych A.; Schaeffer R. D.; Grishin N. V. Topology evaluation of models for difficult targets in the 14th round of the critical assessment of protein structure prediction. Proteins: Struct. Funct. Genet. 2021, 89, 1673–1686. 10.1002/prot.26172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson A.; Song Z. Y.; Nilsson D.; Meyerson B. A.; Simon D. T.; Linderoth B.; Berggren M. Therapy using implanted organic bioelectronics. Sci. Adv. 2015, 1 (4), e1500039 10.1126/sciadv.1500039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Qing R.; Breitwieser A.; Sleytr U.. S-layer protein 2D lattice coupled detergent-free GPCR bioelectronic interfaces, devices, and methods for the use thereof. U.S. Patent US11293923B2, 2022.
- Woolfson D. N. Coiled-coil design: updated and upgraded.. Fibrous Proteins: Structures and Mechanisms 2017, 82, 35–61. 10.1007/978-3-319-49674-0_2. [DOI] [PubMed] [Google Scholar]
- Rose A.; Schraegle S. J.; Stahlberg E. A.; Meier I. Coiled-coil protein composition of 22 proteomes-differences and common themes in subcellular infrastructure and traffic control. BMC Evol. Biol. 2005, 5, 66. 10.1186/1471-2148-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A. N.; Bassler J. Coiled coils–a model system for the 21st century. Trends Biochem. Sci. 2017, 42, 130–140. 10.1016/j.tibs.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Mier P.; Alanis-Lobato G.; Andrade-Navarro M. A. Protein-protein interactions can be predicted using coiled coil co-evolution patterns. J. Theor. Biol. 2017, 412, 198–203. 10.1016/j.jtbi.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Kohn W. D.; Hodges R. S. De novo design of α-helical coiled coils and bundles: models for the development of protein-design principles. Trends Biotechnol. 1998, 16, 379–389. 10.1016/S0167-7799(98)01212-8. [DOI] [Google Scholar]
- Woolfson D. N. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 2005, 70, 79–112. 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- Woolfson D. N. Building fibrous biomaterials from alpha-helical and collagen-like coiled-coil peptides. Biopolymers 2010, 94, 118–127. 10.1002/bip.21345. [DOI] [PubMed] [Google Scholar]
- Grigoryan G.; Keating A. E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008, 18, 477–483. 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenta F.; Aupic J.; Strmsek Z.; Jerala R. Coiled coil protein origami: from modular design principles towards biotechnological applications. Chem. Soc. Rev. 2018, 47, 3530–3542. 10.1039/C7CS00822H. [DOI] [PubMed] [Google Scholar]
- Mason J. M.; Müller K. M.; Arndt K. M.. Protein Engineering Protocols; Springer, 2007; Vol. 352, pp 35–70. [Google Scholar]
- Crick F. The packing of α-helices: simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. 10.1107/S0365110X53001964. [DOI] [Google Scholar]
- Pauling L.; Corey R. B. Compound helical configurations of polypeptide chains: structure of proteins of the α-keratin type. Nature 1953, 171, 59–61. 10.1038/171059a0. [DOI] [PubMed] [Google Scholar]
- Rhys G. G.; Wood C. W.; Lang E. J. M.; Mulholland A. J.; Brady R. L.; Thomson A. R.; Woolfson D. N. Maintaining and breaking symmetry in homomeric coiled-coil assemblies. Nat. Commun. 2018, 9, 4132. 10.1038/s41467-018-06391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz M. O.; Jelesarov I.; Matousek W. M.; Honnappa S.; Jahnke W.; Missimer J. H.; Frank S.; Alexandrescu A. T.; Kammerer R. A. Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7062–7067. 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root B. C.; Pellegrino L. D.; Crawford E. D.; Kokona B.; Fairman R. Design of a heterotetrameric coiled coil. Protein Sci. 2009, 18, 329–336. 10.1002/pro.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matousek W. M.; Ciani B.; Fitch C. A.; Garcia-Moreno B.; Kammerer R. A.; Alexandrescu A. T. Electrostatic contributions to the stability of the GCN4 leucine zipper structure. J. Mol. Biol. 2007, 374, 206–219. 10.1016/j.jmb.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.; Nitsch M.; Kuhlmorgen B.; Golbik R.; Lupas A.; Kellermann J.; Engelhardt H.; Pfander J. P.; Muller S.; Goldie K.; et al. Tetrabrachion: a filamentous archaebacterial surface protein assembly of unusual structure and extreme stability. J. Mol. Biol. 1995, 245, 385–401. 10.1006/jmbi.1994.0032. [DOI] [PubMed] [Google Scholar]
- Gruber M.; Lupas A. N. Historical review: another 50th anniversary–new periodicities in coiled coils. Trends Biochem. Sci. 2003, 28, 679–685. 10.1016/j.tibs.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Gruber M. The structure of alpha-helical coiled coils. Adv. Protein Chem. 2005, 70, 37–78. 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- Hicks M. R.; Walshaw J.; Woolfson D. N. Investigating the tolerance of coiled-coil peptides to nonheptad sequence inserts. J. Struct. Biol. 2002, 137, 73–81. 10.1006/jsbi.2002.4462. [DOI] [PubMed] [Google Scholar]
- Straussman R.; Ben-Ya’acov A.; Woolfson D. N.; Ravid S. Kinking the coiled coil–negatively charged residues at the coiled-coil interface. J. Mol. Biol. 2007, 366, 1232–1242. 10.1016/j.jmb.2006.11.083. [DOI] [PubMed] [Google Scholar]
- Boyle A. L.Peptide applications in biomedicine, Biotechnology and Bioengineering; Elsevier, 2018. [Google Scholar]
- Testa O. D.; Moutevelis E.; Woolfson D. N. CC+: a relational database of coiled-coil structures. Nucleic Acids Res. 2009, 37, D315–322. 10.1093/nar/gkn675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yong W.; Deng Y.; Kallenbach N. R.; Lu M. Atomic structure of a tryptophan-zipper pentamer. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 16156–16161. 10.1073/pnas.0405319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshaw J.; Woolfson D. N. Extended knobs-into-holes packing in classical and complex coiled-coil assemblies. J. Struct. Biol. 2003, 144, 349–361. 10.1016/j.jsb.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Beck K.; Gambee J. E.; Kamawal A.; Bächinger H. P. A single amino acid can switch the oligomerization state of the α-helical coiled-coil domain of cartilage matrix protein. EMBO J. 1997, 16, 3767–3777. 10.1093/emboj/16.13.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman E. H.; Xu C.; DiMaio F.; Magnotti E.; Modlin C.; Yu X.; Wright E.; Baker D.; Conticello V. P. Structural plasticity of helical nanotubes based on coiled-coil assemblies. Structure 2015, 23, 280–289. 10.1016/j.str.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizatovic R.; Aurelius O.; Stenstrom O.; Drakenberg T.; Akke M.; Logan D. T.; Andre I. A de novo designed coiled-coil peptide with a reversible pH-induced oligomerization switch. Structure 2016, 24, 946–955. 10.1016/j.str.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Woolfson D. N.; Alber T. Predicting oligomerization states of coiled coils. Protein Sci. 1995, 4, 1596–1607. 10.1002/pro.5560040818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas F.; Niitsu A.; Oregioni A.; Bartlett G. J.; Woolfson D. N. Conformational dynamics of asparagine at coiled-coil interfaces. Biochemistry 2017, 56, 6544–6554. 10.1021/acs.biochem.7b00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain D. L.; Woods H. L.; Oakley M. G. Design and characterization of a heterodimeric coiled coil that forms exclusively with an antiparallel relative helix orientation. J. Am. Chem. Soc. 2001, 123, 3151–3152. 10.1021/ja004099l. [DOI] [PubMed] [Google Scholar]
- Litowski J. R.; Hodges R. S. Designing heterodimeric two-stranded alpha-helical coiled-coils: the effect of chain length on protein folding, stability and specificity. J. Pept. Res. 2001, 58, 477–492. 10.1034/j.1399-3011.2001.10972.x. [DOI] [PubMed] [Google Scholar]
- Su J. Y.; Hodges R. S.; Kay C. M. Effect of chain length on the formation and stability of synthetic alpha-helical coiled coils. Biochemistry 1994, 33, 15501–15510. 10.1021/bi00255a032. [DOI] [PubMed] [Google Scholar]
- Talbot J. A.; Hodges R. S. Tropomyosin: a model protein for studying coiled-coil and. alpha.-helix stabilization. Acc. Chem. Res. 1982, 15, 224–230. 10.1021/ar00079a006. [DOI] [Google Scholar]
- Burkhard P.; Meier M.; Lustig A. Design of a minimal protein oligomerization domain by a structural approach. Protein Sci. 2000, 9, 2294–2301. 10.1110/ps.9.12.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok S. C.; Hodges R. S. Stabilizing and destabilizing clusters in the hydrophobic core of long two-stranded alpha-helical coiled-coils. J. Biol. Chem. 2004, 279, 21576–21588. 10.1074/jbc.M401074200. [DOI] [PubMed] [Google Scholar]
- Fletcher J. M.; Boyle A. L.; Bruning M.; Bartlett G. J.; Vincent T. L.; Zaccai N. R.; Armstrong C. T.; Bromley E. H.; Booth P. J.; Brady R. L.; et al. A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth. Biol. 2012, 1, 240–250. 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Plecs J. J.; Tidor B.; Alber T.; Kim P. S. High-resolution protein design with backbone freedom. Science 1998, 282, 1462–1467. 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Kim P. S.; Alber T. Crystal structure of an isoleucine-zipper trimer. Nature 1994, 371, 80–83. 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Collier J. H. alpha-Helical coiled-coil peptide materials for biomedical applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1424. 10.1002/wnan.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelain F.; Luo Z.; Zhang S. Self-assembling peptide EAK16 and RADA16 nanofiber scaffold hydrogel. Chem. Rev. 2020, 120, 13434–13460. 10.1021/acs.chemrev.0c00690. [DOI] [PubMed] [Google Scholar]
- Utterstrom J.; Naeimipour S.; Selegard R.; Aili D. Coiled coil-based therapeutics and drug delivery systems. Adv. Drug Delivery Rev. 2021, 170, 26–43. 10.1016/j.addr.2020.12.012. [DOI] [PubMed] [Google Scholar]
- Bilgicer B.; Kumar K. De novo design of defined helical bundles in membrane environments. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15324–15329. 10.1073/pnas.0403314101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potekhin S. a.; Melnik T.; Popov V.; Lanina N.; Vazina A. a.; Rigler P.; Verdini A.; Corradin G.; Kajava A. De novo design of fibrils made of short α-helical coiled coil peptides. Chem. Biol. 2001, 8, 1025–1032. 10.1016/S1074-5521(01)00073-4. [DOI] [PubMed] [Google Scholar]
- Tunn I.; Harrington M. J.; Blank K. G. Bioinspired histidine–Zn2+ coordination for tuning the mechanical properties of self-healing coiled coil cross-linked hydrogels. Biomimetics 2019, 4, 25. 10.3390/biomimetics4010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reches M.; Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Kasotakis E.; Mossou E.; Adler-Abramovich L.; Mitchell E. P.; Forsyth V. T.; Gazit E.; Mitraki A. Design of metal-binding sites onto self-assembled peptide fibrils. Biopolymers 2009, 92, 164–172. 10.1002/bip.21163. [DOI] [PubMed] [Google Scholar]
- Scheibel T.; Parthasarathy R.; Sawicki G.; Lin X. M.; Jaeger H.; Lindquist S. L. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 4527–4532. 10.1073/pnas.0431081100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dublin S. N.; Conticello V. P. Design of a selective metal ion switch for self-assembly of peptide-based fibrils. J. Am. Chem. Soc. 2008, 130, 49–51. 10.1021/ja0775016. [DOI] [PubMed] [Google Scholar]
- Shlizerman C.; Atanassov A.; Berkovich I.; Ashkenasy G.; Ashkenasy N. De novo designed coiled-coil proteins with variable conformations as components of molecular electronic devices. J. Am. Chem. Soc. 2010, 132, 5070–5076. 10.1021/ja907902h. [DOI] [PubMed] [Google Scholar]
- Aupic J.; Lapenta F.; Jerala R. SwitCCh: metal-site design for controlling the assembly of a coiled-coil homodimer. Chembiochem 2018, 19, 2453–2457. 10.1002/cbic.201800578. [DOI] [PubMed] [Google Scholar]
- Gradisar H.; Bozic S.; Doles T.; Vengust D.; Hafner-Bratkovic I.; Mertelj A.; Webb B.; Sali A.; Klavzar S.; Jerala R. Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments. Nat. Chem. Biol. 2013, 9, 362–366. 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubetič A.; Lapenta F.; Gradišar H.; Drobnak I.; Aupič J.; Strmšek Ž.; Lainšček D.; Hafner-Bratkovič I.; Majerle A.; Krivec N.; et al. Design of coiled-coil protein-origami cages that self-assemble in vitro and in vivo. Nat. Biotechnol. 2017, 35, 1094–1101. 10.1038/nbt.3994. [DOI] [PubMed] [Google Scholar]
- Aupic J.; Strmsek Z.; Lapenta F.; Pahovnik D.; Pisanski T.; Drobnak I.; Ljubetic A.; Jerala R. Designed folding pathway of modular coiled-coil-based proteins. Nat. Commun. 2021, 12, 940. 10.1038/s41467-021-21185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristie-David A. S.; Chen J.; Nowak D. B.; Bondy A. L.; Sun K.; Park S. I.; Banaszak Holl M. M.; Su M.; Marsh E. N. G. Coiled-coil-mediated assembly of an icosahedral protein cage with extremely high thermal and chemical stability. J. Am. Chem. Soc. 2019, 141, 9207–9216. 10.1021/jacs.8b13604. [DOI] [PubMed] [Google Scholar]
- Lupas A.; Van Dyke M.; Stock J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Delorenzi M.; Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 2002, 18, 617–625. 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- McDonnell A. V.; Jiang T.; Keating A. E.; Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 2006, 22, 356–358. 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- Armstrong C. T.; Vincent T. L.; Green P. J.; Woolfson D. N. SCORER 2.0: an algorithm for distinguishing parallel dimeric and trimeric coiled-coil sequences. Bioinformatics 2011, 27, 1908–1914. 10.1093/bioinformatics/btr299. [DOI] [PubMed] [Google Scholar]
- Trigg J.; Gutwin K.; Keating A. E.; Berger B. Multicoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PloS one 2011, 6, e23519 10.1371/journal.pone.0023519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T. L.; Green P. J.; Woolfson D. N. LOGICOIL-multi-state prediction of coiled-coil oligomeric state. Bioinformatics 2013, 29, 69–76. 10.1093/bioinformatics/bts648. [DOI] [PubMed] [Google Scholar]
- Rose P. W.; Prlic A.; Bi C.; Bluhm W. F.; Christie C. H.; Dutta S.; Green R. K.; Goodsell D. S.; Westbrook J. D.; Woo J.; et al. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–356. 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber M.; Soding J.; Lupas A. N. Comparative analysis of coiled-coil prediction methods. J. Struct. Biol. 2006, 155, 140–145. 10.1016/j.jsb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Berger B.; Wilson D. B.; Wolf E.; Tonchev T.; Milla M.; Kim P. S. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 8259–8263. 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E.; Kim P. S.; Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997, 6, 1179–1189. 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshaw J.; Woolfson D. N. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 2001, 307, 1427–1450. 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Woolfson D. N. Socket2: a program for locating, visualizing and analyzing coiled-coil interfaces in protein structures. Bioinformatics 2021, 37, 4575–4577. 10.1093/bioinformatics/btab631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoli L.; Fariselli P.; Krogh A.; Casadio R. CCHMM_PROF: a HMM-based coiled-coil predictor with evolutionary information. Bioinformatics 2009, 25, 2757–2763. 10.1093/bioinformatics/btp539. [DOI] [PubMed] [Google Scholar]
- Li C.; Wang X. F.; Chen Z.; Zhang Z.; Song J. Computational characterization of parallel dimeric and trimeric coiled-coils using effective amino acid indices. Mol. Biosyst. 2015, 11, 354–360. 10.1039/C4MB00569D. [DOI] [PubMed] [Google Scholar]
- Guzenko D.; Strelkov S. V. CCFold: rapid and accurate prediction of coiled-coil structures and application to modelling intermediate filaments. Bioinformatics 2018, 34, 215–222. 10.1093/bioinformatics/btx551. [DOI] [PubMed] [Google Scholar]
- Simm D.; Hatje K.; Kollmar M. Waggawagga: comparative visualization of coiled-coil predictions and detection of stable single alpha-helices (SAH domains). Bioinformatics 2015, 31, 767–769. 10.1093/bioinformatics/btu700. [DOI] [PubMed] [Google Scholar]
- Li C.; Ching Han Chang C.; Nagel J.; Porebski B. T.; Hayashida M.; Akutsu T.; Song J.; Buckle A. M. Critical evaluation of in silico methods for prediction of coiled-coil domains in proteins. Brief Bioinform. 2016, 17, 270–282. 10.1093/bib/bbv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwiczak J.; Winski A.; Szczepaniak K.; Alva V.; Dunin-Horkawicz S. DeepCoil-a fast and accurate prediction of coiled-coil domains in protein sequences. Bioinformatics 2019, 35, 2790–2795. 10.1093/bioinformatics/bty1062. [DOI] [PubMed] [Google Scholar]
- Offer G.; Sessions R. Computer modelling of the α-helical coiled coil: packing of side-chains in the inner core. J. Mol. Biol. 1995, 249, 967–987. 10.1006/jmbi.1995.0352. [DOI] [PubMed] [Google Scholar]
- Wood C. W.; Heal J. W.; Thomson A. R.; Bartlett G. J.; Ibarra A. A.; Brady R. L.; Sessions R. B.; Woolfson D. N. ISAMBARD: an open-source computational environment for biomolecular analysis, modelling and design. Bioinformatics 2017, 33, 3043–3050. 10.1093/bioinformatics/btx352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C. W.; Woolfson D. N. CCBuilder 2.0: Powerful and accessible coiled-coil modeling. Protein Sci. 2018, 27, 103–111. 10.1002/pro.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller R. R. Moore’s law: past, present and future. IEEE spectrum 1997, 34, 52–59. 10.1109/6.591665. [DOI] [Google Scholar]
- Meiler J.; Baker D. ROSETTALIGAND: Protein-small molecule docking with full side-chain flexibility. Proteins: Struct. Funct. Genet. 2006, 65, 538–548. 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- Koehler Leman J.; Weitzner B. D.; Renfrew P. D.; Lewis S. M.; Moretti R.; Watkins A. M.; Mulligan V. K.; Lyskov S.; Adolf-Bryfogle J.; Labonte J. W.; et al. Better together: Elements of successful scientific software development in a distributed collaborative community. PLoS Comput. Biol. 2020, 16, e1007507 10.1371/journal.pcbi.1007507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K. W.; Lemmon G. H.; DeLuca S. L.; Sheehan J. H.; Meiler J. Practically useful: what the ROSETTA protein modeling suite can do for you. Biochemistry 2010, 49, 2987–2998. 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos E.; Silva D. A. Essentials of de novo protein design: Methods and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1374. 10.1002/wcms.1374. [DOI] [Google Scholar]
- Coluzza I. Computational protein design: a review. J. Phys.: Condens. Matter 2017, 29, 143001. [DOI] [PubMed] [Google Scholar]
- Alam N.; Schueler-Furman O.. Modeling Peptide-Protein Interactions; Springer, 2017. [Google Scholar]
- Rohl C. A.; Strauss C. E.; Misura K. M.; Baker D. Protein structure prediction using Rosetta. Meth. Enzymol. 2004, 383, 66–93. 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Khor B. Y.; Tye G. J.; Lim T. S.; Choong Y. S. General overview on structure prediction of twilight-zone proteins. Theor. Biol. Medical Model. 2015, 12, 15. 10.1186/s12976-015-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. T.; Kooperberg C.; Huang E.; Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997, 268, 209–225. 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- O’Meara M. J.; Leaver-Fay A.; Tyka M. D.; Stein A.; Houlihan K.; DiMaio F.; Bradley P.; Kortemme T.; Baker D.; Snoeyink J.; et al. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh B.; London N.; Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins: Struct. Funct. Genet. 2010, 78, 2029–2040. 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- Alford R. F.; Leman J. K.; Weitzner B. D.; Duran A. M.; Tilley D. C.; Elazar A.; Gray J. J. An integrated framework advancing membrane protein modeling and design. PLoS Comput. Biol. 2015, 11, e1004398 10.1371/journal.pcbi.1004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan V. K.; Workman S.; Sun T.; Rettie S.; Li X.; Worrall L. J.; Craven T. W.; King D. T.; Hosseinzadeh P.; Watkins A. M.; et al. Computationally designed peptide macrocycle inhibitors of New Delhi metallo-β-lactamase 1. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2012800118 10.1073/pnas.2012800118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan C. M.; Rocklin G. J.; Bick M. J.; Ford A.; Majri-Morrison S.; Kroll A. V.; Miller C. J.; Carter L.; Goreshnik I.; Kang A.; et al. Computational design of a synthetic PD-1 agonist. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2102164118 10.1073/pnas.2102164118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. E.; Chivian D.; Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasha D.; Lee W.; Thomsen A.; Geerts Y.; Marques A.; Steyaert M.; Sansen W.; Edwards C.; Redman-White W.; Bracey M.. Special issue on the 1998 European solid-state circuits conference, 1998.
- Pokala N.; Handel T. M. Energy functions for protein design: Adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J. Mol. Biol. 2005, 347, 203–227. 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Kemp D.; Casey M. L. Physical organic chemistry of benzisoxazoles. II. Linearity of the Broensted free energy relation for the base-catalyzed decomposition of benzisoxazoles. J. Am. Chem. Soc. 1973, 95, 6670–6680. 10.1021/ja00801a024. [DOI] [Google Scholar]
- Strauch E.-M.; Fleishman S. J.; Baker D. Computational design of a pH-sensitive IgG binding protein. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 675–680. 10.1073/pnas.1313605111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 1999, 286, 295–299. 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Morcos F.; Pagnani A.; Lunt B.; Bertolino A.; Marks D. S.; Sander C.; Zecchina R.; Onuchic J. N.; Hwa T.; Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, E1293–E1301. 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata D.; Ponzoni L.; Micheletti C.; Carnevale V. Patterns of coevolving amino acids unveil structural and dynamical domains. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E10612–E10621. 10.1073/pnas.1712021114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. B. Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 16856–16865. 10.1073/pnas.1821309116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisetty H.; Ovchinnikov S.; Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18734–18734. 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine E.; Eismann S.; Elofsson A.; Grudinin S. Protein sequence-to-structure learning: Is this the end(-to-end revolution)?. Proteins: Struct. Funct. Genet. 2021, 89, 1770–1786. 10.1002/prot.26235. [DOI] [PubMed] [Google Scholar]
- Yang J. Y.; Anishchenko I.; Park H.; Peng Z. L.; Ovchinnikov S.; Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 1496–1503. 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. C.; Nocedal J. On the limited memory Bfgs method for large-scale optimization. Math. Program. 1989, 45, 503–528. 10.1007/BF01589116. [DOI] [Google Scholar]
- AlQuraishi M. End-to-end differentiable learning of protein structure. Cell Syst. 2019, 8, 292–301. 10.1016/j.cels.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs F. B.; Worrall D. E.; Fischer V.; Welling M.. Se (3)-transformers: 3D roto-translation equivariant attention networks. arXiv:2006.10503 2020. 10.48550/arXiv.2006.10503 [DOI] [Google Scholar]
- Paul D.; Sanap G.; Shenoy S.; Kalyane D.; Kalia K.; Tekade R. K. Artificial intelligence in drug discovery and development. Drug Discovery Today 2021, 26, 80–93. 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- Simpkin A. J.; Rodríguez F. S.; Mesdaghi S.; Kryshtafovych A.; Rigden D. J. Evaluation of model refinement in CASP14. Proteins: Struct. Funct. Genet. 2021, 89, 1852. 10.1002/prot.26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E.DeepMind’s AI for protein structure is coming to the masses. Nature 2021. 10.1038/d41586-021-01968-y [DOI] [PubMed] [Google Scholar]
- Schymkowitz J.; Borg J.; Stricher F.; Nys R.; Rousseau F.; Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005, 33, W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose J.; Wendt N.; Kubald S.; Krause E.; Fechner K.; Beyermann M.; Bienert M.; Rudolph R.; Rothemund S. Hexa-histidin tag position influences disulfide structure but not binding behavior of in vitro folded N-terminal domain of rat corticotropin-releasing factor receptor type 2a. Protein Sci. 2004, 13, 2470–2475. 10.1110/ps.04835904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazes R. J.; Grisewood M. J.; Maranas C. D. Recent advances in computational protein design. Curr. Opin. Struc. Biol. 2011, 21, 467–472. 10.1016/j.sbi.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Wingfield P. T. Overview of the purification of recombinant proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1–6.1.35. 10.1002/0471140864.ps0601s80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor K.; Broom A.; Meiering E. M. Exploring the relationships between protein sequence, structure and solubility. Curr. Opin. Struct. Biol. 2017, 42, 136–146. 10.1016/j.sbi.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Idicula-Thomas S.; Balaji P. V. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. 2005, 14, 582–592. 10.1110/ps.041009005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Baldi P. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. 10.1093/bioinformatics/btu352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiaohui N.; Feng S.; Xuehai H.; Jingbo X.; Nana L. Predicting the protein solubility by integrating chaos games representation and entropy in information theory. Expert Syst. Appl. 2014, 41, 1672–1679. 10.1016/j.eswa.2013.08.064. [DOI] [Google Scholar]
- Hou Q.; Kwasigroch J. M.; Rooman M.; Pucci F. SOLart: a structure-based method to predict protein solubility and aggregation. Bioinformatics 2019, 36, 1445–1452. 10.1093/bioinformatics/btz773. [DOI] [PubMed] [Google Scholar]
- Schaller A.; Connors N. K.; Oelmeier S. A.; Hubbuch J.; Middelberg A. P. Predicting recombinant protein expression experiments using molecular dynamics simulation. Chem. Eng. Sci. 2015, 121, 340–350. 10.1016/j.ces.2014.09.044. [DOI] [Google Scholar]
- Orlando G.; Silva A.; Macedo-Ribeiro S.; Raimondi D.; Vranken W. Accurate prediction of protein beta-aggregation with generalized statistical potentials. Bioinformatics 2020, 36, 2076–2081. 10.1093/bioinformatics/btz912. [DOI] [PubMed] [Google Scholar]
- Meric G.; Robinson A. S.; Roberts C. J. Driving forces for nonnative protein aggregation and approaches to predict aggregation-prone regions. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 139–159. 10.1146/annurev-chembioeng-060816-101404. [DOI] [PubMed] [Google Scholar]
- Tartaglia G. G.; Pawar A. P.; Campioni S.; Dobson C. M.; Chiti F.; Vendruscolo M. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 2008, 380, 425–436. 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Pawar A. P.; Dubay K. F.; Zurdo J.; Chiti F.; Vendruscolo M.; Dobson C. M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Garbuzynskiy S. O.; Lobanov M. Y.; Galzitskaya O. V. FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. 10.1093/bioinformatics/btp691. [DOI] [PubMed] [Google Scholar]
- Conchillo-Solé O.; de Groot N. S.; Avilés F. X.; Vendrell J.; Daura X.; Ventura S. AGGRESCAN: a server for the prediction and evaluation of″ hot spots″ of aggregation in polypeptides. BMC Bioinform. 2007, 8, 1–17. 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano R.; Jamroz M.; Szczasiuk A.; Pujols J.; Kmiecik S.; Ventura S. AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. 10.1093/nar/gkv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujols J.; Iglesias V.; Santos J.; Kuriata A.; Kmiecik S.; Ventura S.. Insoluble Proteins; Springer, 2022. [Google Scholar]
- Maurer-Stroh S.; Debulpaep M.; Kuemmerer N.; De La Paz M. L.; Martins I. C.; Reumers J.; Morris K. L.; Copland A.; Serpell L.; Serrano L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- Louros N.; Konstantoulea K.; De Vleeschouwer M.; Ramakers M.; Schymkowitz J.; Rousseau F. WALTZ-DB 2.0: an updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res. 2020, 48, D389–D393. 10.1093/nar/gkz758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior P.; Kotulska M. FISH Amyloid–a new method for finding amyloidogenic segments in proteins based on site specific co-occurence of aminoacids. BMC Bioinform. 2014, 15, 1–8. 10.1186/1471-2105-15-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M.; Li Y.; Wang C.; Han K. RFAmyloid: a web server for predicting amyloid proteins. Int. J. Mol. Sci. 2018, 19, 2071. 10.3390/ijms19072071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G.; Cavalli A.; Pellarin R.; Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 2005, 14, 2723–2734. 10.1110/ps.051471205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Família C.; Dennison S. R.; Quintas A.; Phoenix D. A. Prediction of peptide and protein propensity for amyloid formation. PLoS one 2015, 10, e0134679 10.1371/journal.pone.0134679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla A.-M.; Rousseau F.; Schymkowitz J.; Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Trovato A.; Seno F.; Tosatto S. C. The PASTA server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- Walsh I.; Seno F.; Tosatto S. C.; Trovato A. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. 10.1093/nar/gku399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell C. W.; Waldispühl J.; Lis M.; Halfmann R.; Devadas S.; Lindquist S.; Berger B. A method for probing the mutational landscape of amyloid structure. Bioinformatics 2011, 27, i34–i42. 10.1093/bioinformatics/btr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Choi J.; Lee S. J.; Welsh W. J.; Yoon S. NetCSSP: web application for predicting chameleon sequences and amyloid fibril formation. Nucleic Acids Res. 2009, 37, W469–473. 10.1093/nar/gkp351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran R.; Rawat P.; Kumar S.; Gromiha M. M. ANuPP: a versatile tool to predict aggregation nucleating regions in peptides and proteins. J. Mol. Biol. 2021, 433, 166707. 10.1016/j.jmb.2020.11.006. [DOI] [PubMed] [Google Scholar]
- Charoenkwan P.; Kanthawong S.; Nantasenamat C.; Hasan M. M.; Shoombuatong W. iAMY-SCM: Improved prediction and analysis of amyloid proteins using a scoring card method with propensity scores of dipeptides. Genomics 2021, 113, 689–698. 10.1016/j.ygeno.2020.09.065. [DOI] [PubMed] [Google Scholar]
- Louros N.; Orlando G.; De Vleeschouwer M.; Rousseau F.; Schymkowitz J. Structure-based machine-guided mapping of amyloid sequence space reveals uncharted sequence clusters with higher solubilities. Nat. Commun. 2020, 11, 1–13. 10.1038/s41467-020-17207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski J. W.; Kotulska M. Path-prediction of amyloidogenicity by threading and machine learning. Sci. Rep. 2020, 10, 1–9. 10.1038/s41598-020-64270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgarone T.; Villain É.; Guettaf A.; Leclercq J.; Kajava A. V. TAPASS: Tool for Annotation of Protein Amyloidogenicity in the context of other Structural States. J. Struct. Biol. 2022, 214, 107840. 10.1016/j.jsb.2022.107840. [DOI] [PubMed] [Google Scholar]
- Pintado C.; Santos J.; Iglesias V.; Ventura S. SolupHred: a server to predict the pH-dependent aggregation of intrinsically disordered proteins. Bioinformatics 2021, 37, 1602–1603. 10.1093/bioinformatics/btaa909. [DOI] [PubMed] [Google Scholar]
- Bhandari B. K.; Lim C. S.; Gardner P. P. TISIGNER. com: web services for improving recombinant protein production. Nucleic Acids Res. 2021, 49, W654–W661. 10.1093/nar/gkab175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M. Dynamics of folded proteins. Nature 1977, 267, 585–590. 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Ahammad F.; Alam R.; Mahmud R.; Akhter S.; Talukder E. K.; Tonmoy A. M.; Fahim S.; Al-Ghamdi K.; Samad A.; Qadri I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 22, bbab098. 10.1093/bib/bbab098. [DOI] [PubMed] [Google Scholar]
- Nagy T.; Tóth Á.; Telbisz Á.; Sarkadi B.; Tordai H.; Tordai A.; Hegedus T. The transport pathway in the ABCG2 protein and its regulation revealed by molecular dynamics simulations. Cell. Mol. Life Sci. 2021, 78, 2329–2339. 10.1007/s00018-020-03651-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A.; Goñi J. R.; Orozco M.; Gelpí J. L. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y.; Suenaga A.; Sato Y.; Kosuda S.; Taiji M. All-atom molecular dynamics analysis of multi-peptide systems reproduces peptide solubility in line with experimental observations. Sci. Rep. 2016, 6, 1–11. 10.1038/srep19479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. M.; Khan M. A.; Kuroda Y. Analysis of amino acid contributions to protein solubility using short peptide tags fused to a simplified BPTI variant. Biochim. Biophys. Acta. Proteins Proteom. 2012, 1824, 1144–1150. 10.1016/j.bbapap.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Khan M. A.; Islam M. M.; Kuroda Y. Analysis of protein aggregation kinetics using short amino acid peptide tags. Biochim. Biophys. Acta. Proteins Proteom. 2013, 1834, 2107–2115. 10.1016/j.bbapap.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Putnam C. D.; Hammel M.; Hura G. L.; Tainer J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Knight C. J.; Hub J. S. WAXSiS: a web server for the calculation of SAXS/WAXS curves based on explicit-solvent molecular dynamics. Nucleic Acids Res. 2015, 43, W225–W230. 10.1093/nar/gkv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Bhardwaj V. K.; Singh R.; Purohit R. Explicit-solvent molecular dynamics simulations revealed conformational regain and aggregation inhibition of I113T SOD1 by Himalayan bioactive molecules. J. Mol. Liq. 2021, 339, 116798. 10.1016/j.molliq.2021.116798. [DOI] [Google Scholar]
- Gallicchio E.; Levy R. M. AGBNP: An analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J. Comput. Chem. 2004, 25, 479–499. 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- Dinner A. R.; Lazaridis T.; Karplus M. Understanding β-hairpin formation. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (16), 9068–9073. 10.1073/pnas.96.16.9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow C. D.; Nguyen H.; Pande V. S.; Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 2002, 420, 102–106. 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R.; Drozdetski A.; Walker R. C.; Onufriev A. V. Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015, 108, 1153–1164. 10.1016/j.bpj.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonov S.; Teyra J.; Pisabarro M. T. A molecular dynamics approach to study the importance of solvent in protein interactions. Proteins: Struct. Funct. Genet. 2008, 73, 515–525. 10.1002/prot.22076. [DOI] [PubMed] [Google Scholar]
- Chakraborty I.; Kar R. K.; Sarkar D.; Kumar S.; Maiti N. C.; Mandal A. K.; Bhunia A. Solvent Relaxation NMR: A Tool for Real-Time Monitoring Water Dynamics in Protein Aggregation Landscape. ACS Chem. Neurosci. 2021, 12, 2903–2916. 10.1021/acschemneuro.1c00262. [DOI] [PubMed] [Google Scholar]
- Service R. F. Huge protein structure database could transform biology. Science 2021, 373, 478. 10.1126/science.373.6554.478. [DOI] [PubMed] [Google Scholar]
- Thompson M. J.; Sievers S. A.; Karanicolas J.; Ivanova M. I.; Baker D.; Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4074–4078. 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R.; Chen S.; Chen J. Z.; Grigorieff N.; Passmore L. A.; Ciccarelli L.; Rubinstein J. L.; Crowther R. A.; Stewart P. L.; Rosenthal P. B. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J. Mol. Biol. 2011, 413, 1028–1046. 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; McMullan G.; Faruqi A. R.; Murshudov G. N.; Short J. M.; Scheres S. H.; Henderson R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 2013, 135, 24–35. 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar K. R.; Henderson R. Single particle electron cryomicroscopy: trends, issues and future perspective. Q. Rev. Biophys. 2016, 49, e13 10.1017/S0033583516000068. [DOI] [PubMed] [Google Scholar]
- Subramaniam S.; Kühlbrandt W.; Henderson R. CryoEM at IUCrJ: a new era. IUCrJ. 2016, 3, 3–7. 10.1107/S2052252515023738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin G.; Potter C. S.; Carragher B. Cryo-EM for small molecules discovery, design, understanding, and application. Cell Chem. Biol. 2018, 25, 1318–1325. 10.1016/j.chembiol.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh S. H.; Hryc C. F.; Jeong H. H.; Fei X.; Jakana J.; Lorimer G. H.; Chiu W. Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 8259–8264. 10.1073/pnas.1704725114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E.; Scheres S. H. Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol. Cell 2015, 58, 677–689. 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M. J.; Meyerowitz J. G.; Skiniotis G. Drug discovery in the era of cryo-electron microscopy. Trends Biochem. Sci. 2022, 47, 124–135. 10.1016/j.tibs.2021.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J.; McDowall A. Vitrification of pure water for electron microscopy. J. Microsc. 1981, 124, 3–4. 10.1111/j.1365-2818.1981.tb02483.x. [DOI] [Google Scholar]
- Renaud J.-P.; Chari A.; Ciferri C.; Liu W. T.; Rémigy H. W.; Stark H.; Wiesmann C. Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discovery 2018, 17, 471–492. 10.1038/nrd.2018.77. [DOI] [PubMed] [Google Scholar]
- Doerr A. Single-particle cryo-electron microscopy. Nat. Methods 2016, 13, 23–23. 10.1038/nmeth.3700. [DOI] [PubMed] [Google Scholar]
- Weissenberger G.; Henderikx R. J.; Peters P. J. Understanding the invisible hands of sample preparation for cryo-EM. Nat. Methods 2021, 18, 463–471. 10.1038/s41592-021-01130-6. [DOI] [PubMed] [Google Scholar]
- Kampjut D.; Steiner J.; Sazanov L. A. Cryo-EM grid optimization for membrane proteins. Iscience 2021, 24, 102139. 10.1016/j.isci.2021.102139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore L. A.; Russo C. J. Specimen preparation for high-resolution cryo-EM. Meth. Enzymol. 2016, 579, 51–86. 10.1016/bs.mie.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederik P. M.; Hubert D. Cryoelectron microscopy of liposomes. Meth. Enzymol. 2005, 391, 431–448. 10.1016/S0076-6879(05)91024-0. [DOI] [PubMed] [Google Scholar]
- Ravelli R. B.; Nijpels F. J.; Henderikx R. J.; Weissenberger G.; Thewessem S.; Gijsbers A.; Beulen B. W.; López-Iglesias C.; Peters P. J. Cryo-EM structures from sub-nl volumes using pin-printing and jet vitrification. Nat. Commun. 2020, 11, 1–9. 10.1038/s41467-020-16392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S. A.; Albiez S.; Bieri A.; Syntychaki A.; Adaixo R.; McLeod R. A.; Goldie K. N.; Stahlberg H.; Braun T. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. J. Struct. Biol. 2017, 197, 220–226. 10.1016/j.jsb.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Arnold S. A.; Albiez S.; Opara N.; Chami M.; Schmidli C.; Bieri A.; Padeste C.; Stahlberg H.; Braun T. Total sample conditioning and preparation of nanoliter volumes for electron microscopy. ACS Nano 2016, 10, 4981–4988. 10.1021/acsnano.6b01328. [DOI] [PubMed] [Google Scholar]
- Glaeser R. M. Proteins, interfaces, and cryo-EM grids. Curr. Opin. Colloid Interface Sci. 2018, 34, 1–8. 10.1016/j.cocis.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Lin Z.; Zakin J. L.; Talmon Y.; Davis H. T.; Scriven L. E. Cryo-TEM Imaging the Flow-Induced Transition from Vesicles to Threadlike Micelles. J. Phys. Chem. B 2000, 104, 5263–5271. 10.1021/jp0002998. [DOI] [Google Scholar]
- Nakane T.; Kotecha A.; Sente A.; McMullan G.; Masiulis S.; Brown P. M.; Grigoras I. T.; Malinauskaite L.; Malinauskas T.; Miehling J.; et al. Single-particle cryo-EM at atomic resolution. Nature 2020, 587, 152–156. 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Drie J. H.; Tong L. Cryo-EM as a powerful tool for drug discovery. Bioorg. Med. Chem. lett. 2020, 30, 127524. 10.1016/j.bmcl.2020.127524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty D.; Desai R.; Uchański T.; Masiulis S.; Stec W. J.; Malinauskas T.; Zivanov J.; Pardon E.; Steyaert J.; Miller K. W.; et al. Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 2019, 565, 516–520. 10.1038/s41586-018-0833-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R. W. Extrasynaptic GABAA receptors in the nucleus accumbens are necessary for alcohol drinking. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 4699–4700. 10.1073/pnas.1102818108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiulis S.; Desai R.; Uchański T.; Serna Martin I.; Laverty D.; Karia D.; Malinauskas T.; Zivanov J.; Pardon E.; Kotecha A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. 10.1038/s41586-018-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Huynh D. T.; Yeates T. O. A 3.8 A resolution cryo-EM structure of a small protein bound to an imaging scaffold. Nat. Commun. 2019, 10, 1864. 10.1038/s41467-019-09836-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Li S.; Kappel K.; Pintilie G.; Su Z.; Mou T.-C.; Schmid M. F.; Das R.; Chiu W. Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNA at 3.7 Å resolution. Nat. Commun. 2019, 10, 1–6. 10.1038/s41467-019-13494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Wang J.; Zhang X.; Yang Z.; Zhang J.-C.; Zhao L.; Peng H.-L.; Lei J.; Wang H.-W. Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Nat. Commun. 2019, 10, 1–11. 10.1038/s41467-019-10368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.-T.; Cascio D.; Yeates T. O. Structure of a 16-nm cage designed by using protein oligomers. Science 2012, 336, 1129–1129. 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- Sciore A.; Su M.; Koldewey P.; Eschweiler J. D.; Diffley K. A.; Linhares B. M.; Ruotolo B. T.; Bardwell J. C.; Skiniotis G.; Marsh E. N. G. Flexible, symmetry-directed approach to assembling protein cages. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 8681–8686. 10.1073/pnas.1606013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Gonen S.; Gonen T.; Yeates T. O. Near-atomic cryo-EM imaging of a small protein displayed on a designed scaffolding system. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 3362–3367. 10.1073/pnas.1718825115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz H. K.; Stumpp M. T.; Forrer P.; Amstutz P.; Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. 10.1016/S0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- Yao Q.; Weaver S. J.; Mock J.-Y.; Jensen G. J. Fusion of DARPin to aldolase enables visualization of small protein by cryo-EM. Structure 2019, 27, 1148–1155. 10.1016/j.str.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F.; Estrozi L. F.; Hans F.; Malet H.; Noirclerc-Savoye M.; Schoehn G.; Petosa C. Fusion to a homo-oligomeric scaffold allows cryo-EM analysis of a small protein. Sci. Rep. 2016, 6, 1–11. 10.1038/srep30909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T. G.; Bharat T. A.; Joerger A. C.; Bai X.-c.; Praetorius F.; Fersht A. R.; Dietz H.; Scheres S. H. Design of a molecular support for cryo-EM structure determination. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E7456–E7463. 10.1073/pnas.1612720113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X.; Yan R.; Zhang J.; Zhang G.; Zhang Y.; Hao M.; Zhang Z.; Fan P.; Dong Y.; Yang Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Zhang Y.; Li Y.; Xia L.; Guo Y.; Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokic G.; Hillen H. S.; Tegunov D.; Dienemann C.; Seitz F.; Schmitzova J.; Farnung L.; Siewert A.; Höbartner C.; Cramer P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 1–7. 10.1038/s41467-020-20542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januliene D.; Moeller A. Single-particle Cryo-EM of membrane proteins. Methods Mol. Biol. 2021, 2302, 153–178. 10.1007/978-1-0716-1394-8_9. [DOI] [PubMed] [Google Scholar]
- Cheng Y. Membrane protein structural biology in the era of single particle cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 58–63. 10.1016/j.sbi.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nafría J.; Tate C. G. Structure determination of GPCRs: cryo-EM compared with X-ray crystallography. Biochem. Soc. Trans. 2021, 49, 2345–2355. 10.1042/BST20210431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Sun B.; Feng D.; Hu H.; Chu M.; Qu Q.; Tarrasch J. T.; Li S.; Kobilka T. S.; Kobilka B. K.; et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546, 248–253. 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C.; Kang H. J.; Singh I.; Chen H.; Zhang C.; Ye W.; Hayes B. W.; Liu J.; Gumpper R. H.; Bender B. J.; et al. Structure, function and pharmacology of human itch GPCRs. Nature 2021, 600, 170–175. 10.1038/s41586-021-04126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaye H.; Ishchenko A.; Lam J. H.; Han G. W.; Xue L.; Rondard P.; Pin J.-P.; Katritch V.; Gati C.; Cherezov V. Structural basis of the activation of a metabotropic GABA receptor. Nature 2020, 584, 298–303. 10.1038/s41586-020-2408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. H.; Thomsen A. R.; Cahill T. J.; Huang R.; Huang L.-Y.; Marcink T.; Clarke O. B.; Heissel S.; Masoudi A.; Ben-Hail D.; et al. Structure of an endosomal signaling GPCR–G protein−β-arrestin megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. 10.1038/s41594-019-0330-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. H.; Lefkowitz R. J. Signaling at the endosome: cryo-EM structure of a GPCR–G protein–beta-arrestin megacomplex. FEBS J. 2021, 288, 2562–2569. 10.1111/febs.15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Stevens R. C.; Xu F. The importance of ligands for G protein-coupled receptor stability. Trends Biochem. Sci. 2015, 40, 79–87. 10.1016/j.tibs.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Magnani F.; Serrano-Vega M. J.; Shibata Y.; Abdul-Hussein S.; Lebon G.; Miller-Gallacher J.; Singhal A.; Strege A.; Thomas J. A.; Tate C. G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–1571. 10.1038/nprot.2016.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kobilka B. K.; Steyaert J. Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. 10.1146/annurev-pharmtox-010716-104710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Rapoport T. A. Cryo-EM structure determination of small proteins by nanobody-binding scaffolds (Legobodies). Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2115001118 10.1073/pnas.2115001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov R. G.; Gatsogiannis C.; Raunser S. Lipid Nanodiscs as a Tool for High-Resolution Structure Determination of Membrane Proteins by Single-Particle Cryo-EM. Meth. Enzymol. 2017, 594, 1–30. 10.1016/bs.mie.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Gui M.; Wang Z.-F.; Gorgulla C.; Yu J. J.; Wu H.; Sun Z.-y. J.; Klenk C.; Merklinger L.; Morstein L.; et al. Cryo-EM structure of an activated GPCR–G protein complex in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 258–267. 10.1038/s41594-020-00554-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng S.; Li Y.; Wang X. Cryo-EM for battery materials and interfaces: Workflow, achievements, and perspectives. iScience 2021, 24, 103402. 10.1016/j.isci.2021.103402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhang B.; Freddolino P. L.; Zhang Y. CR-I-TASSER: assemble protein structures from cryo-EM density maps using deep convolutional neural networks. Nat. Methods 2022, 19, 195–204. 10.1038/s41592-021-01389-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan L.; DeGrado W. F. Characterization of a helical protein designed from first principles. Science 1988, 241, 976–978. 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- Kamtekar S.; Schiffer J. M.; Xiong H.; Babik J. M.; Hecht M. H. Protein design by binary patterning of polar and nonpolar amino acids. Science 1993, 262, 1680–1685. 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- Dahiyat B. I.; Mayo S. L. De novo protein design: fully automated sequence selection. Science 1997, 278, 82–87. 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]