

SUMMARY
Therapeutic options for treatment of basal-like breast cancers remain limited. Here, we demonstrate that bromodomain and extra-terminal (BET) inhibition induces an adaptive response leading to MCL1 protein-driven evasion of apoptosis in breast cancer cells. Consequently, co-targeting MCL1 and BET is highly synergistic in breast cancer models. The mechanism of adaptive response to BET inhibition involves the upregulation of lipid synthesis enzymes including the rate-limiting stearoyl-coenzyme A (CoA) desaturase. Changes in lipid synthesis pathway are associated with increases in cell motility and membrane fluidity as well as re-localization and activation of HER2/EGFR. In turn, the HER2/EGFR signaling results in the accumulation of and vulnerability to the inhibition of MCL1. Drug response and genomics analyses reveal that MCL1 copy-number alterations are associated with effective BET and MCL1 co-targeting. The high frequency of MCL1 chromosomal amplifications (>30%) in basal-like breast cancers suggests that BET and MCL1 co-targeting may have therapeutic utility in this aggressive subtype of breast cancer.
Graphical Abstract
In brief
Yan et al. show that pharmacological co-targeting of MCL1 and BET is highly effective in breast cancer cells. The proposed combination therapy may be effective for treatment of patients with aggressive subtypes of breast cancers whose tumors carry genetic aberrations associated with cell-death evasion.
INTRODUCTION
Despite the success of targeted therapies in cancer treatment, response to monotherapies has been transient due to the almost inevitable emergence of resistance. Blocking the routes that lead to the emergence of resistance with combination therapy is the most promising strategy to date (Holohan et al., 2013; Kim et al., 2018). However, discovery of effective combination therapies is a daunting task due to the complexity of drug response landscapes. A key mechanism of resistance to targeting oncogenic processes is the adaptive activation of compensatory processes, e.g., feedback loops in the short term or oncogenic alterations in the long term (Taylor and Schlaepfer, 2018; Sun et al., 2017; Muranen et al., 2012; Duncan et al., 2012; O’Reilly et al., 2006). Systematic approaches are needed to identify adaptive responses, reveal drug-induced vulnerabilities, and thus enable discovery of effective rational combination therapies.
Basal-like breast cancers make up nearly 20% of breast cancer cases, yet treatment options are limited for this aggressive subtype (Cancer Genome Atlas Network, 2012). This necessitates the need for the development of effective targeted therapies. The only recurrent aberrations providing therapeutic opportunities in the basal-like subtype are in the homologous recombination (HR) pathway, including BRCA1/2 alterations (Farmer et al., 2005). Gene copy-number alteration (CNA) of MCL1, an anti-apoptotic mediator, represents an additional potential driver aberration that is recurrent in breast cancer with an enrichment in the basal subtype (Beroukhim et al., 2010). With the introduction of specific inhibitors, MCL1 has become an actionable therapeutic target. Broader impact of MCL1 inhibitors could be achieved by selectively inducing survival dependencies that make use of higher levels of MCL1 (Kotschy et al., 2016; Campbell et al., 2018).
The bromodomain and extra-terminal (BET) proteins, including BRD2, BRD3, and BRD4, represent promising targets based on pre-clinical and clinical data (Lovén et al., 2013; Rhyasen et al., 2018; Sun et al., 2018). BET proteins link epigenetic states to lineage-specific gene expression through binding to acetylated histones and regulating transcriptional elongation downstream of enhancer sites (Figure 1A). Preclinical efficacy of BET inhibitors (BETis) has led to clinical trials in diverse cancer types including triple-negative breast cancers (TNBCs) (Shu et al., 2016) (Figure 1B). Reduction in cellular robustness, cell-cycle progression, DNA-damage response (DDR), and replication stress have been proposed as some of the underlying mechanisms of response to BET inhibition (Sun et al., 2018; Doroshow et al., 2017; Zhang et al., 2018). BET molecules also regulate cellular plasticity as exemplified by the emergence of drug-tolerant persistent cells with BRD4 dependence and altered differentiation states after MEK and PI3K/mTOR inhibition in TNBC (Risom et al., 2018). The adaptive responses and therapeutic stress such as DDR associated with inhibition of BET activity provide an unexplored opportunity to discover drug-induced dependencies and synthetic lethal combination therapies.
Figure 1. Breast cancer cells have differential responses to BET inhibition.

(A) BETi binds to the acetyl histone binding cavity on BET proteins, prevents recruitment of BET to chromatin, and alters expression of lineage-specific genes.
(B) Chemical structures of the BETis used in the perturbation experiments.
(C) The relative responses to the BETis JQ1 and IBET151 across 27 breast cancer lines and the ovarian cancer line SKOV3 are quantified as AUC of the dose-response relationship (dose range 0–10 μM).
(D) Scatterplot of responses to IBET151 versus JQ1 across the cell lines in (C) demonstrates the similarity in phenotypic response to the two BETis.
(E) Landscape of potential driver oncogenic events in selected cell lines.
(F) Dose-dependent responses to JQ1 in cell lines cultured in 3D matrigel.
(G) Images of dose-dependent responses to JQ1 in 3D Matrigel-cultured cells.
(H) Proteomic response map of the cell lines to BET inhibition. Proteomic data was collected with RPPA using antibodies that quantify 217 total or phosphoprotein levels (two time points, 2D and 3D cultures, varying doses of the four BETis in B, mean of duplicates).
Here, we demonstrate through data-driven modeling and subsequent experimental validation that BET inhibition induces an adaptive survival mechanism involving lipid metabolism and kinase signaling converging on MCL1-dependent evasion of apoptosis. This combination may have therapeutic value as MCL1 CNAs, either as high-level amplifications or low-level gains, are frequent in breast cancer. Our integrated approach provides a generalizable protocol for identification of effective precision combination therapies based on analysis of multi-omics data.
RESULTS
Breast cancer cells have differential responses to BET inhibition
To address the efficacy spectrum, predictors of response, and mechanisms of resistance to BET inhibition in breast cancer, we profiled responses to BET targeting in 28 cell lines (Figures 1C and S1A-S1B) using JQ1 and IBET151, two structurally different and efficacious BETis. The high correlation between the responses to JQ1 and IBET151 (R = 0.91) suggests that both compounds have similar efficacies and on-target activity in vitro (Figure 1D). Consistent with previous studies, genomic correlates of response to BET inhibition do not indicate a significant enrichment for any recurrent/driver breast cancer genomic aberration within resistant or sensitive cell lines (Figures 1E and S1A) (Cancer Genome Atlas Network, 2012; Meric-Bernstam et al., 2014; Xu and Vakoc, 2017). The lack of genomic markers predictive of response to BETis in breast cancer necessitates deeper mechanistic analyses to discover biomarkers likely to identify patients who will benefit from BETis.
We tested cell viability responses to BETis in three-dimensional (3D) spheroid cultures in five selected cell lines (Figure 1F). The cell lines (ordered from most resistant to sensitive), HCC1954 (basal-like, ERBB2 amplified), BT474 (HER2+, ER+), MDAMB468 (basal-like), SKBR3 (HER2+), and SKOV3 (serous ovarian, ERBB2 amplified), are selected to cover different levels in the BETi sensitivity spectrum. Despite being an ovarian cancer line, SKOV3 is included as a sensitive control to decode events exclusive to more resistant cells for the following reasons. First, SKOV3 originates from a patient with serous adenocarcinoma, an ovarian cancer subtype with similarities to basal-like breast cancers (Cancer Genome Atlas Network, 2012). Second, SKOV3 also carries major genomic similarities to breast cancers (e.g., TP53 null, ERBB2 amplified, PIK3CA mutated). The comparison of responses in 2D versus 3D media determines whether matrix attachment alters responses to BET inhibition (Figures 1E-1G). The impact of 3D culture on cell viability is most dramatic for the most sensitive cell line, SKOV3, in which the concentration of drug which exhibited 50% cell viability (IC50) increases from ~200 nM to 1.5 μM (Figure S1C). To identify processes that may potentially explain differential responses to BET inhibition, we measured changes in 217 key oncogenic signaling phosphoproteins using reverse-phase protein arrays (RPPAs) (Figure 1H; see Figures S1D and S1E for analysis of responses in 2D versus 3D cultures). The RPPA targets are chosen to monitor activity and levels of representative molecules from key pathways including PI3K, RAS-MAPK, Src/FAK, RTK signaling axes, DNA repair, cell cycle, apoptosis, immuno-oncology, and histone modifications (Akbani et al., 2014). The phosphoproteomic response to BET inhibition in diverse cell lines, time points, and conditions generates a comprehensive response map of the perturbations with BETis in breast cancer cells.
Computational network modeling identifies MCL1 upregulation as an adaptive response to BET inhibition
We analyzed the proteomic responses to BETis using our TargetScore network modeling method (Figure 2) (Wang et al., 2021). Our goal was to identify collective adaptive response mechanisms that can drive drug resistance and drug-induced vulnerabilities to combination therapies (Figures 2A and S2). Based on a reference network model of signaling (Figure S2A), we calculated the TargetScores for all proteins after treatment with the four BETi compounds (Figure 1B) in the five cell lines using the temporal phosphoproteomic data (Figure 1H).
Figure 2. Computational network modeling identifies MCL1 upregulation as an adaptive response to BET inhibition.

(A) The TargetScore algorithm (see STAR Methods) identifies network-level adaptive responses to targeted perturbations based on molecular drug response data. The method involves (1) molecular profiling of responses to perturbations in samples with varying sensitivity to the perturbation agent. (2) Construction of a reference network that captures potential relations between all measured proteomic entities. (3) Quantification of a sample-specific adaptation (target) score that links protein interactions to drug response on the reference network using proteomic data. (4) Identification of network modules that have a significantly high TargetScore (i.e., collectively participate in adaptive responses) in each sample. (5) Selection of actionable targets that participate in adaptive responses and experimental testing of drug combinations.
(B) Heatmaps capture the highest and lowest 10 TargetScores for 24 (left) and 48 (right) h post-treatment in 3D cultures. The ranking in the HCC1954 is used as reference, and cell lines are ordered from most resistant (HCC1954) to most sensitive (SKOV3).
(C) Analysis and statistical assessment of adaptive responses (positive TargetScore) and direct responses (negative TargetScore) in HCC1954 24 (left) and 48 h (right) post-treatment. TargetScores are based on the average values across the four BETis in Figure 1B. The Q values represent the FDR-adjusted p values against the TargetScore-null distribution.
(D) Network modules of adaptive responses and mechanisms of action 24 (left) and 48 h (right) post-treatment. Asterisk (*) denotes the significantly strong TargetScores according to the statistical validation (see STAR Methods).
(E) Association of MCL1 mean TargetScore across four BETis (top) and BETi sensitivity (bottom). The Spearman correlation between the predicted adaptive responses to BETi and resistance to BETi is 0.9 (p = 0.04).
(F) Copy-number and mRNA expression status of MCL1 in cell lines.
(G) The frequency of breast cancer samples with high- (red) and low- (pink) level copy-number increases in MCL1.
(H) Distribution of MCL1 mRNA levels with varying MCL1 copy-number statuses across breast cancer samples (TCGA).
Differential analysis of the TargetScores for JQ1 across cell lines identifies adaptive responses that are either exclusive to most resistant cell types or shared across all samples (Figures 2B-2D, S2B, and S2C). In the most resistant line, HCC1954, the anti-apoptotic protein MCL1 has the highest TargetScore value in response to JQ1 in both 24 and 48 h among the 217 proteins and phosphoproteins assessed. In HCC1954, 24 h after JQ1 treatment, TargetScore identifies a set of molecules downstream of RTK signaling such as mTOR, mTOR_pS2448, MIG6, MEK1/2_pS217/S221, P70S6K_pT389, and GSK3α/β_pS21_S9. The enrichment suggests involvement of RTK signaling in the adaptive response. At 48 h, more diverse adaptive responses are observed as proteins involved in apoptosis (MCL1, BCL2, BCL-XL), DNA repair (ATR, PMS2), lipid metabolism (SCD, FASN), AKT/mTOR signaling (mTOR_pS2448, INPP4B), and TP53, which is mutated in HCC1954, have high TargetScores. The TargetScore in BT474 carries important similarities to HCC1954 (Figure S2B). In BT474, MCL1 has a high TargetScore (fifth highest at 24 h and the highest at 48 h). At both 24- and 48-h time points, high TargetScores in BT474 are associated with total protein level changes in EGFR and its downstream targets (Figure S2C). The MCL1 TargetScore followed a near monotonously decreasing trend with increasing sensitivity to JQ1 (Figure 2E). The trend suggests MCL1 upregulation is correlated with resistance to BET inhibition. Although mTOR_pS2448 also had high TargetScores in HCC1954, there was no correlation between the scores and sensitivity to JQ1 across additional cell lines. In conclusion, TargetScore analysis nominates the upregulation of anti-apoptotic MCL1 molecule as a key adaptive response to BET inhibition exclusively in resistant lines.
To detect a statistically robust and consistent adaptive response signature, we analyzed the average TargetScores for each protein across four different BETis (Figures 2C and 2D). We eliminated high TargetScores that are solely driven by network connectivity biases without significant input from cell-type-specific response data using a bootstrapping-based statistical assessment method. The method generates a null model of TargetScores using randomized datasets (mixed protein labels) and the reference network model. On the null model, we identify the TargetScores with high values and low false discovery rate (FDR)-adjusted p values (Q < 0.1), an indication that the scores are driven by both the network and sample-specific data. MCL1 (at both 24 and 48 h), MIG6 (at 24 h), and mTORpS2448 (at 48 h) have the most significant (adjusted p value [padj] < 10−4) and highest TargetScores in HCC1954. At both time points, multiple markers of AKT pathway activation (P70S6K_pT389, PDK1_pS241, mTOR_PS2448), MAPK (MEK1/2_pS217/S221), and lipid metabolism (SCD and FASN) pathways have high TargetScores. The FDR-adjusted p values are <0.1 at 48 h, particularly for the lipid metabolism mediators SCD and FASN. In conclusion, statistical analysis results for JQ1, IBET151, IBET726, and IBET762 were consistent with JQ1-focused analyses, particularly in the nomination of MCL1 as a key driver of adaptive responses (Figure S3). An emerging hypothesis is that BET inhibition-induced MCL1 upregulation may be associated with and a consequence of altered MAPK and AKT/mTOR signaling, as well as the lipid metabolism.
MCL1 mRNA and protein levels inversely correlate with BETi sensitivity
The analysis of MCL1 copy-number, transcriptomic, and proteomic status provides orthogonal evidence on the roles of MCL1 in drug-resistant cells. MCL1 is copy-number amplified and mRNA overexpressed in the resistant lines HCC1954 and BT474 (Figure 2F) (Barretina et al., 2012). MCL1 is copy-number gained and has intermediate mRNA expression in the MDAMB468 line, which has a slightly lower JQ1 resistance. More sensitive cell lines, SKBR3 and SKOV3, have low mRNA expression and/or diploid copy-number states for MCL1. The MCL1 aberration patterns in cell lines also reflect the distribution of MCL1 status in patient cohorts. High-level copy-number amplifications and low-level gains in the MCL1 locus are observed in 15% and 57% of breast cancer cases in the TCGA cohort (n = 1,084 patients), respectively (Figures 2G and 2H). MCL1 aberrations are particularly enriched in the aggressive basal subtype, with 32% of cases being copy-number amplified. We profiled MCL1 levels in BETi-treated and drug-naive cells with varying JQ1 sensitivity (Figure 3A). As expected from copy-number and transcriptomic states, HCC1954 and BT474 cells (MCL1 amplified) express substantial levels of MCL1 protein in their drug-naive states, whereas SKOV3 cells (MCL1 wild type) have lower MCL1 protein expression. In BT474 and HCC1954, which already have relatively high MCL1 levels, treatment with JQ1 leads to a time-dependent increase of MCL1 levels (Figure 3A). In contrast, MCL1 protein levels do not increase in BETi-sensitive SKOV3 cells in response to short-term BETi treatment. Motivated by these evidence lines, we next experimentally tested the effect of co-targeting BET and MCL1 in breast cancer cells. We focused on lines that are most resistant to BET inhibition, where predicted effects are most likely to be manifest.
Figure 3. BET and MCL1 inhibitors are synergistic in breast cancer cells.

(A) MCL1 protein expression in drug-naive BETi-resistant (HCC1954, BT474) and BETi-sensitive (SKOV3) cells in 3D spherical cultures (left). MCL1 protein level changes in response to BET inhibition (2 μM of JQ1, 0 to 48 h).
(B) Cell viability response (mean of triplicates) to inhibitors of MCL1 (S63845) and BET (JQ1) in HCC1954, BT474, and SKOV3 cells in 3D spherical cultures supplemented with 2% Matrigel.
(C) Interactions between BETi and MCL1i are quantified using the Bliss independence method.
(D) Overexpression of MCL1 protein in SKOV3 cells confers resistance to BETi. The western blots comparing the MCL1 expression in cells transformed with empty versus MCL1-expressing pcDNA3.1 plasmid (left). The cell viability responses to BETi in wild-type versus MCL1-overexpressing SKOV3 cells (right).
(E) MCL1 knockdown (KD) with siRNA in HCC1954 cells leads to increased BETi sensitivity. The western blot confirms KD of MCL1 (left). The cell viability responses to JQ1 in HCC1954 with WT-MCL1 versus MCL1-KD (right).
(F) BRD4 KD with siRNA in HCC1954 sensitizes cells to MCL1 i. The western blots confirm KD of BRD4 and subsequent increase in MCL1 levels (left). The BRD4 KD and S63845 cell viability responses (right).
(G) The shifts in cell-cycle stage distribution in HCC1954, BT474, and SKOV3 in response to JQ1 (2 μM) and S63845 (0.3 μM) combination 48 h post-treatment are quantified using flow cytometry.
(H) Western blot analysis of cleaved PARP levels monitors apoptotic response to JQ1 and S63845 2 and 12 h post-treatment.
In (D)–(F), the error bars represent ±SEM over 4 replicates.
BET and MCL1 inhibitors are synergistic in breast cancer cells
We treated BETi-resistant HCC1954 and BT474 and BETi-sensitive SKOV3 cells with combinations of BETi (JQ1) and a highly selective MCL1 inhibitor (MCL1i; S63845) (Kotschy et al., 2016) (Figure 3). We measured cell viability in response to the combination of MCL1i and BETi (Figure 3B). Consistent with our previous experiments, increasing doses of BETi in HCC1954 and BT474 do not affect cell viability. We observe a substantial response to MCL1 inhibition (0.9 μM) in HCC1954, while BT474 is relatively more resistant. Both cell lines are highly responsive to combination treatment as evidenced by more than 80% reduction in cell viability when JQ1 and S83645 are introduced at 10 and 0.9 μM, respectively. In contrast, SKOV3 is not responsive to the S83645, and the combination does not alter the already high activity of JQ1. (Figure 3C). Based on the analysis of Bliss independence scores, the combination is highly synergistic (Bliss score < 0.5) at diverse doses in both BT474 and HCC1954. In SKOV3, the two compounds are antagonistic in multiple-dose configurations with Bliss scores as high as 1.45. The knockdown and overexpression of MCL1 as well as knockdown of BRD4 support the observations from pharmacological inhibition of BRD4 and MCL1. Overexpression of MCL1 confers significant resistance to the BETi-sensitive SKOV3 cells (Figure 3D), supporting our previous observation that MCL1 levels can predict BETi sensitivity, while in the already resistant and MCL1-amplified HCC1954 cells, the BETi response is not impacted by MCL1 overexpression (Figure S4A). Similarly, small interfering RNA (siRNA)-based knockdown of MCL1 sensitizes cells to BET inhibition in the HCC1954 and MDAMB486 cells carrying pre-existing MCL1 amplification and BETi resistance (Figures 3E and S4B). In the BETi-sensitive SKOV3 cells, MCL1 knockdown has no impact, as expected, since the cells are already low in MCL1 levels (Figure S4B). The knockdown of BRD4 recapitulates the adaptive response (MCL1 level increase) and cellular impact (increased sensitivity to MCL1 targeting) of pharmacological inhibition of BET in HCC1954 but not in SKOV3 (Figures 3F and S4C). The results of siRNA and pharmacological perturbation experiments validates the TargetScore-based predictions that co-targeting MCL1 and BET is effective in BETi-resistant cells.
To identify mechanisms underlying synergy, we measured the effects of BET and MCL1 targeting on cell-cycle progression based on DNA content measurements with flow cytometry (Figure 3G). Consistent with the TargetScore calculations, which suggested a reduction of cell-cycle pathway activities, JQ1 induces an S-phase cell-cycle arrest in all samples. In both HCC1954 and BT474, the S-phase population is substantially enriched upon JQ1 treatment. Next, we monitored apoptotic responses through PARP-cleavage detection and flow cytometry (Figures 3H and S4D). Importantly, 12 h of JQ1 treatment induces PARP cleavage in SKOV3 but not in drug-resistant HCC1954 and BT474. The observed cell-cycle arrest across all cell lines but lack of apoptosis in drug-resistant cell populations explains the static cell viability profiles in response to monotherapy. Single-agent treatment with MCL1i induces partial PARP cleavage in HCC1954 but not in SKOV3 and BT474. The combination of JQ1 (BETi) and S8364 (MCL1i) induces PARP cleavage in all tested cell lines, particularly in HCC1954 and BT474. The flow cytometry-based apoptosis profiling in HCC1954, BT474, and SKOV3 also confirms the observations from the PARP-cleavage assay that in SKOV3, BET inhibition is sufficient to induce apoptosis, while in the HCC1954 and BT474, the combination is necessary (Figure S4D). The apoptotic shift and reduced cell count in response to MCL1 and BET co-targeting provides strong evidence that resistance to BETi is driven by the increased anti-apoptotic MCL1 activity in breast cancer (Figures 3E and 3F). This finding also further validates the predictions from the TargetScore algorithm.
MCL1 copy-number status is a predictor of response to co-targeting MCL1 and BET
Next, we investigated predictors of response to MCL1 inhibition in combination with BET inhibition. We focused on MCL1 copy-number status motivated by the computational predictions and the subsequent experimental validation. We tested the effect of MCL1 and BET co-targeting on cell viability in 10 cell lines with varying genomic backgrounds (Figures 4A-4D). Cell lines with either low-level gain or high-level amplification, as well as higher mRNA expression of the MCL1 gene, are more responsive to MCL1 inhibition, with effective eradication of tumor cells at high doses and a significantly lower area under the dose-response curve (AUC) compared with MCL1 diploid cells (Figures 4B-4D; Table S1). We did not observe any significant enrichment of MCL1i and/or MCL1i + BETi sensitivity in tumors with other recurrent oncogenic alterations (Figure S5A).
Figure 4. Molecular determinants of responses to MCL1 and BET inhibition.

(A) Dose-response curves of BETi (JQ1) and MCL1i (S63845) in breast and ovarian cancer cell lines (error bars represent ±SEM over triplicates).
(B) The normalized AUC (AUC/AUC100% viability) for S63845 and combination of JQ1 and S63845. Cell lines are classified based on the MCL1 copy-number status. The p values represent the significance of the response differences in cells with MCL1 copy-number WT/HETLOSS versus gain/amplification based on an unpaired t test. Error bars represent the ±SD over triplicates.
(C) Amax (response at maximum dose) for S63845(1 μM) and combination of JQ1 (10 μM) and S63845 (1 μM), classified according to MCL1 copy-number status. Error bars represent the ±SD over triplicates.
(D) The relation between combination therapy Amax, MCL1 mRNA levels, and copy-number status (source: CCLE). The surface represents the linear regression between Amax (dependent) and MCL1 copy-number and mRNA (independent) variables across cell lines.
(E) The synergy between JQ1 and S63845 is quantified with Bliss independence score for increasing doses of drugs for cell lines with varying MCL1 copy-number statuses.
(F) Cell viability responses to JQ1 (1 μM) and S63845 (0.1 μM) in 3D cultures of select cell lines. Images are collected 72 h after drug treatment. The p values are calculated with a t test over the triplicates between the combination and most effective single-agent arms. Error bars represent the ±SD over triplicates.
(G) Tumor volume curves in the MCL1-high MDAMB468 xenografts treated with BETi and MCL1i (each agent at 20 mg/kg, intraperitoneal injections 3 times/week). N = 9/arm for control, 8/arm for treatment cohort. The statistical differences were assessed with Mann-Whitney U test, and error bars represent the ±SEM of the responses in replicates.
(H) Representative images of IHC analysis for Ki67 and cleaved caspase 3 in xenografts 10 days after onset of drug treatments. The percentage of positive cells is quantified based on counts from 3 regions of interest (ROIs) of equal sizes in each condition. Error bars represent standard deviations over ROIs. Error bars represent the ±SD over triplicates.
(I) Representative images of IHC analysis for MCL1 response to BET and MCL1 inhibition 10 days after onset of in vivo drug treatments. The bar chart (right) quantifies the MCL1 positivity based on counts from 3 equal sized ROIs per condition. Error bars represent the ±SD over triplicates.
In (H) and (I), the statistical significance between groups of interest are demonstrated with a t test over the replicates, each representing an ROI on the corresponding IHC slide.
Through analysis of Bliss synergy scores, we detected three clusters with distinct BETi and MCL1i synergy (Figure 4E). The first cluster (HCC1937, SKOV3, and HCC1419) consists of MCL1i-resistant lines and is characterized by additive drug interactions between BETi and MCL1i (0.5 < Bliss score < 1.0). In the second cluster (SKBR3 and MCF7), there is an antagonistic interaction (Bliss score > 1.0), which can be attributed to the high efficacy of BETi as a single agent. In the third cluster (HCC70, BT20, MDAMB468, HCC1954, and BT474), there is a strong synergy (Bliss synergy < 0.5). The cells in the third cluster are resistant to BETi and also carry MCL1 copy-number increases. Finally, we confirmed that synergistic interactions between JQ1 and S63845 were also preserved in 3D spheroid cultures (Figure 4F). In summary, a robust and strong synergy exists between targeting MCL1 and BET in the BETi-resistant and MCL1-amplified context. We conclude that MCL1 copy-number status is a likely predictor of response to the drug combination.
BET and MCL1 co-targeting is highly effective in vivo
We investigated the efficacy of the combination in the MDAMB468 xenograft model in nude athymic NCr mice. The MDAMB468 model represents the basal breast cancer subtype with an amplification in EGFR, loss of PTEN, TP53 R273H mutation, and a chromosomal gain in the MCL1 gene (Figures 4G and S5B). Although each single agent generates a partial response, the drug combination induces a significantly stronger effect with a marked decrease in tumor volume compared with each single agent after 3 weeks of treatment. The drug-induced changes in cell proliferation and apoptotic markers in vivo are consistent with the in vitro results (Figure 4H). The proliferation marker Ki67 levels are lowest in tumors of mice treated with the drug combination, while we observe intermediate Ki67 levels in response to treatments with each single agent. Similarly, the level of the apoptosis marker cleaved caspase 3 is highest in combination-treated samples. As predicted by the computational models and in vitro experiments, MCL1, but not BCL2, protein levels are increased in response to BET targeting (Figures 4I and S5C). Although, the MCL1 level increase is highest in response to the combination, the change is not significant compared with the change due to the BETi treatment alone. BRD4, the main target of the BETi, is ubiquitously expressed in control and drug-treated states (Figures S5D and S5E). No animal weight loss is observed in any of the treatment arms, suggesting that the agents had no observable toxicity as single agents or in combination (Figure S5F). In summary, BETi increases MCL1 protein levels and induces a therapeutic vulnerability to MCL1 inhibition in vivo.
BETi-induced MCL1 upregulation is mediated by EGFR/HER2 signaling activity
To elucidate signaling mechanisms underlying the adaptive MCL1 protein increase, we searched for phosphorylation cascades that could link BET inhibition to MCL1 protein accumulation and subsequent apoptosis evasion. We quantified differential phosphorylations of each protein and focused on the most deviant proteins based on RPPA data from resistant and sensitive lines (Figure 5A). Statistical analysis suggests enrichment of HER2/EGFR downstream signaling in BETi-resistant cells as phosphorylation of HER2, EGFR, SHC, SRC, AKT, GSK3α/β, MEK1/2, and 4EBP1 increases in response to BETi in HCC1954 but not in SKOV3. In addition to RTK signaling, we observed increased phosphorylation of myosin II and NDRG1 in HCC1954. The differential analysis is consistent with the TargetScore analyses, which identify a module of BETi adaptive response centered around MCL1 and enriched with signaling molecules downstream of EGFR/HER2 (e.g., MAPK, AKT, SHC, mTOR, PKC) (Figure 2D). Based on the results, we hypothesize that BETi-induced MCL1 increase and subsequent apoptosis evasion can occur through activation of HER2/EGFR and downstream signaling.
Figure 5. Molecular mechanisms of response to BET and MCL1 co-targeting.

(A) Differential analysis of phosphoproteomic responses to BETis in resistant (HCC1954, BT474) and sensitive (SKOV3) cells. The heatmap represents the average log fold difference (log[Xperturbed/Xunperturbed]) across the four BETis (Figure 1B). “R to S deviation” is the fold changes between responses in resistant (HCC1954 and BT474) and sensitive (SKOV3) cells. The proteins with significantly different expressions between HCC1954 (most resistant) and SKOV3 (sensitive line) are listed (padj < 0.05).
(B) MCL1 and signaling activity changes in response to cocktails of pathway and BET inhibitors are monitored to demonstrate the role of signaling pathways linking BET and MCL1 activity.
(C) Differential analysis of transcriptomic responses (mean of duplicates) to BETi (1 μM, 24 h). The first three columns demonstrate fold changes of mRNA species that change in opposite directions in resistant versus sensitive lines. The red/white columns on right represent the involvement of each gene in different molecular processes according to a GO-term enrichment analysis.
(D) Transcriptomic responses (mean of duplicates) in key genes of the lipid metabolism pathway as identified by the differential response analysis from the RNA sequencing (RNA-seq) data (left) and confirmed based on qPCR analyses for the key rate-limiting enzyme SCD. RNA-seq analysis: error bars represent the ±SEM. pPCR analysis: error bars represent the ±SD.
(E) The differential analysis focusing only on total protein changes performed as in (A). The heatmaps represent the proteomic responses averaged across four BETis.
(F) The analysis of SCD, MCL1, and p-EGFR/p-HER2 levels in response to inhibitors of BET (JQ1) and SCD (A939572), as well as BRD4 KDs with siRNA.
(G) Representative images of IHC analysis of in vivo SCD expression in the mouse xenograft model (MDA-MB468) treated with BET and MCL1 inhibitors. Error bars represent the ±SD.
(H) ChIP-seq analysis of BRD4 binding on SCD gene regulatory sites in breast cancer cell lines treated with BETis (JQ1) (see Figure S5 for FASN and SREBF1).
(I) Correlation analysis of proteomic and transcriptomic levels involved in the associations linking BET to EGFR/HER2 within tumors of patients with ERBB2-amplified, basal breast, and ovarian cancer. All correlations are in the range of −0.5 to 0.5. As a reference, the average correlation between mRNA and protein levels of a given gene is estimated within a range of 0.3–0.6 across the whole genome (Liu et al., 2016; Gry et al., 2009) (data from TCGA breast and ovarian cancer projects).
To test this hypothesis, we profiled MCL1 levels and signaling under systematic perturbations on the signaling pathways (Figure 5B and S6A-S6C). We treated HCC1954, BT474, and SKOV3 cells with inhibitors of BET (JQ1), EGFR/HER2 (lapatinib), AKT (MK-2206), P38/MAPK14 (doramapimod), PKC (Dequalinium Chloride), and MEK1/2 (trametinib). In BT474 and HCC1954, both MCL1 and GSK3α/β_pS9/S21 levels increase in response to BETi, confirming the role of MCL1 in the adaptive response, and substantially decrease in response to lapatinib, suggesting a causal link from HER2/EGFR to MCL1 (Figures 5B and S7A-S7D). Inhibitors of downstream effector molecules (AKT, MAPK14, MEK1/2, PKC) have either partial or no impact on MCL1 levels, particularly in HCC1954, suggesting that no individual mechanism downstream of HER2/EGFR can single handedly regulate MCL1. Next, to test whether the inhibition of individual signaling routes downstream of EGFR/HER2 can reverse adaptive responses to BET inhibition, we co-targeted BET proteins and signaling pathways. In BT474 and HCC1954, BETi-driven increases in MCL1 and GSK3α/β_pS9/S21 are reversed by lapatinib, consistent with the role of HER2/EGFR activation in the mechanism. In contrast, MCL1 and GSK3α/β_pS9/S21 levels modestly increase (HCC1954) or do not change (BT474) in response to the combinations of BETi with all other agents (i.e., inhibitors of AKT, MEK1/2, PKC, and MAPK14/P38), suggesting that multiple signaling axes are required for the BETi adaptive response or, alternatively, inhibition of one mediator results in activation of the other pathways. Indeed, adaptive responses are blocked only with a cocktail of three signaling inhibitors with BETi (i.e., BETi with PKCi + P38i + AKTi or PKCi + P38i + MEK1/2i). The molecular responses to combinatorial perturbations suggest that EGFR/HER2 activation is necessary and sufficient for increased MCL1 in HCC1954 and BT474. However, none of the tested signaling inhibitors other than lapatinib can block the adaptive response to BET inhibition, indicating highly redundant signaling cascades downstream of EGFR/HER2 that converge on MCL1.
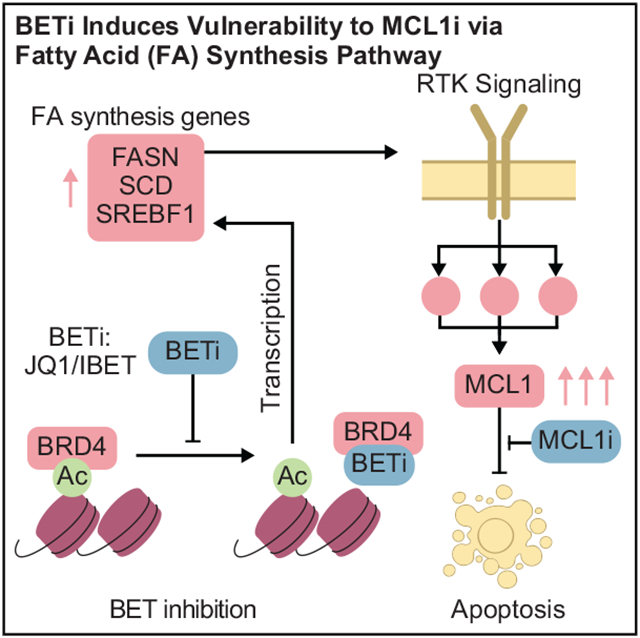
A BETi-induced transcriptional program alters fatty acid metabolism to activate EGFR/HER2 signaling
To explore the mechanisms of EGFR/HER2 activation by BET inhibition, we analyzed the mRNA and total-protein changes in response to BET inhibition. Transcriptomic responses were profiled with JQ1 treatment in HCC1954, BT474, and SKOV3 cells and mRNA sequencing (Figure 5C). The mRNA analysis identifies 127 differentially responding transcripts in HCC1954/BT474 versus SKOV3. A Gene Ontology (GO)-term-based gene enrichment analysis of differentially expressed genes identifies 85 statistically significant pathways with enrichment of biosynthetic pathways, particularly lipid metabolism, cell adhesion, and motility in resistant lines (Table S2). The SCD, which catalyzes the rate-limiting step in monounsaturated fatty acid (MUFA) synthesis and regulates lipid membrane fluidity in cells (Röhrig and Schulze, 2016; Stubbs and Smith, 1984; Roongta et al., 2011), is in 43 out of 85 significant pathways in the GO-term analysis. Other key lipid metabolism enzymes, including SREBF1—a master transcription factor that regulates expression of key fatty acid enzymes (e.g., SCD, FASN)—ELOVL7, SC5D, ACSF2, and ACAT1 are differentially overexpressed in response to BETi in resistant cells (Figures 5C and 5D). A qPCR experiment targeting the SCD transcript in HCC1954, BT474, and SKOV3 also confirms that BET inhibition increases expression of SCD gene in HCC1954, and to a lesser extent in BT474, but not in SKOV3. Moreover, differential analysis of total protein expression demonstrates the involvement of cell adhesion and fatty acid metabolism in BETi response, with E-cadherin and SCD as the highest-ranked differentially expressed proteins in resistant cells (Figure 5E). BETi also increases MyosinII_pS1943 and decreases AMPKa_pT172, supporting that BET inhibition alters motility and lipid metabolism.
Motivated by the roles of SCD as a rate-limiting step in MUFA synthesis and a regulator of membrane fluidity, we focused on SCD involvement in modulating EGFR/HER2 activity and MCL1 protein levels in response to BET inhibition (Figure 5F). First, we have validated that BET targeting leads to increased SCD protein levels in BETi-resistant HCC1954 cells but not in -sensitive SKOV3 cells. Treatment of HCC1954 cells with the SCD inhibitor (SCDi; A939572) decreases both p-HER2/EGFR and MCL1 levels, suggesting a role of SCD upstream of the HER2/EGFR-MCL1 signaling axis. To identify whether the BETi-induced MCL1 accumulation depends on SCD, we treated HCC1954 with JQ1 and A939572. Both HER2 phosphorylation and MCL1 decreases upon BET and SCD co-targeting, suggesting that SCD inhibition can block adaptive responses to BET inhibition. Consistent with the pharmacological inhibition, siRNA-based knockdown of BRD4 gene also leads to an increase in the SCD protein in the HCC1954, but not SKOV3, cells (Figure 5F). In MDAMB468 xenograft models, consistent with in vitro analyses, BET inhibition, but not MCL1 inhibition, leads to increased SCD protein levels (Figure 5G). Thus, BETi-induced expression of lipid metabolism genes, especially SCD, is likely an intermediate event between BET inhibition and HER2/EGFR to the MCL1 signaling axis.
Next, we demonstrated a direct epigenetic link from BET inhibition to the expression of the target fatty acid synthesis genes (SCD, FASN, and SREBF1). We analyzed the anti-BRD4 chromatin immunoprecipitation sequencing (ChIP-seq) data in breast cancer cell lines (SUM159, HCC1395, T47D) treated with BETi (Sun et al., 2018) (Figures 5H and S5D). We mapped the promoter/enhancer regions using GeneHancer (Fishilevich et al., 2017) and then validated that they were active in breast cancer cells using paired anti-H3K27ac ChIP-seq data. The BRD4 protein binds to the enhancer/promoter sites of target genes, and BET inhibition displaces BRD4 from the target regulatory sites by more than 75% in each cell line. We observe similar promoter/enhancer sites (and thus potential BRD4 binding sites) on the HCC1954 cell line based on the analysis of H3K27ac data (GSM2258830), suggesting that the above observations can be extrapolated to HCC1954.
Next, we explored whether the mechanism may be valid in patient cohorts. We quantified the associations between levels of BRD4, key members of the fatty acid pathway, and p-RTK in tumors of patients with basal-like and ERBB2-amplified breast cancers as well patients with serous ovarian cancer using TCGA data (Figure 5I). In all cancer types, BRD4 protein levels negatively correlate with SREBF1, SCD, and FASN mRNA levels (−0.43 < R < −0.05). This is consistent with the increased expression of fatty acid synthesis genes in response to BET inhibition. Consistent with the key role of SREBF1 on the transcription of lipid metabolism genes (Kim and Spiegelman, 1996), mRNA levels of SREBF1 and the two key enzymes FASN and SCD are strongly correlated (0.28 < R < 0.59). We analyzed RTK activation using available p-RTK measurements (i.e., p-HER2, p-EGFR, and p-MET). The relation between SCD and RTK phosphorylation is context dependent: in tumors with ERBB2 gene amplification, SCD is highly correlated with p-HER2 (R = 0.42) but not with p-EGFR or p-MET. In basal cancers, in which HER2 is usually not a driver, the correlation between SCD and p-HER2 is reversed (R = −0.22), while a strong correlation between SCD and p-MET is observed (R = 0.47). In ovarian cancers, no p-RTK species demonstrates a strong association (0.06 < R < 0.2). We do not observe similar associations between BRD4 and fatty acid metabolism in luminal A and B breast tumors. Thus, TCGA analysis suggests that the mechanistic link between BRD4, fatty acid metabolism, and RTK activation is likely relevant across ERBB2-amplified and basal-like breast cancers.
Finally, we profiled BETi-induced cellular phenotypes associated with transcriptomic and proteomic responses. In HCC1954, but not in SKOV3, BETi treatment induces morphology changes involving a transformation from a less structured and spread form to a more ordered and contracted state (Figure 6A). Wound scratch assays in monolayer cultures show that BET inhibition increases cell motility in BETi-resistant cells (HCC1954) but not in -sensitive cells (SKOV3) (Figure 6B). Consistent with BETi-induced SCD upregulation, HCC1954 membrane fluidity increases upon JQ1 treatment (48 h, 1 μM JQ1), as quantified by the ratio of monomer to excimer fluorescence of lipophilic pyrene probes, which undergo excimer formation with increasing membrane fluidity (Figure 6C). SKOV3 membrane fluidity decreases in response to BETi, consistent with the observed cell motility decrease. In drug-resistant HCC1954 cells, EGFR/HER2 protein molecules are co-localized and polarized on the cell membrane after BETi treatment, leading to an increased local concentration of EGFR/HER2 and likely downstream signaling (Figure 6D). No change in HER2 localization is observed in BETi-sensitive cells (SKOV3) upon BETi treatment. Thus, BET inhibition induces membrane re-modeling including cell morphology, motility, fluidity, and RTK localization changes associated and consistent with the observed molecular changes in drug-resistant cells.
Figure 6. Phenotypic responses to BET inhibition in resistant cells.

(A) Representative images (20×, bright field) of cellular morphology changes in response to BETi (48 h, 1 μM JQ1).
(B) The BETi induces increased motility of HCC1954 (top) and no substantial difference in SKOV3 (bottom) as monitored across time points with a wound scratching assay.
(C) Drug-induced changes in membrane fluidity (mean of triplicates, error bars represent ±SEM) in HCC1954 and SKOV3 cells based on fluorescent lipophilic pyrene probes that are enriched in excimer state (emission = 470 nm) with increasing membrane fluidity and enriched in monomers (emission = 400 nm) with decreasing membrane fluidity.
(D) The representative images of HER2/EGFR localization changes in response to BET inhibition (48 h, 1 μM). Images are collected using fluorescent confocal microscopy (20×). The fluorescence signals from EGFR/HER2 (green) in BETi-treated HCC1954 cells suggest increased localization of the receptors. The EGFR/HER2 signal was unchanged in response to BETi in SKOV3.
(E) The proposed mechanism of BET inhibition-induced vulnerability to BET and MCL1 co-targeting in the context of MCL1 chromosomal amplifications or gains based on the integrated multi-omics, network, and perturbation analyses. The marked genes and proteins are identified and validated in molecular perturbation response analyses.
DISCUSSION
We demonstrate that BET inhibition induces a cascade of adaptive responses that leads to MCL1-driven evasion of apoptosis and resulting vulnerability to co-targeting BET and MCL1, particularly in cells with amplified MCL1. MCL1 amplification is a well-characterized prognostic factor in breast cancer, although relatively little is known of the significance of low-level gains (Campbell et al., 2018; Berger et al., 2018). We identified the MCL1 copy-number gain and amplification as a predictor for response to co-targeting MCL1 and BET. From a translational perspective, this is particularly important as both high-level amplifications and low-level gains of the MCL1 gene are highly enriched in the aggressive basal subtype at frequencies of 32% and 57%, respectively. In the current paradigm, precision therapies usually rely on targeting high-level gene amplifications (e.g., ERBB2 amplifications) or mutations. The demonstration that a low-level gain is correlated with response to a combination therapy may profoundly improve the fraction of potentially benefiting patients in the clinic.
BET inhibition is linked to MCL1 accumulation through a multistep signaling cascade and in the context of MCL1 amplifications (Figure 6E). Consistent with previous reports (Hölscher et al., 2018; Korkut et al., 2015; Sun et al., 2018), TargetScore analysis and our cell-cycle measurements suggest that BETi induces an arrest in the S/G2 state. In parallel, the proteomic and transcriptomic perturbation response analyses as well as a ChIP-seq analysis suggest that BET inhibition removes BRD4 molecules from the promoter sites and leads to increased expression of the lipid genes including SCD (Figures 5C-5F). The increase in SCD expression in response to BETi suggests a potential repressor role for BET, which is less common than the transcriptional activator roles. However, BET protein action as a repressor has been demonstrated in diverse contexts including AML cells, where BRD4 binds to the promoter sites of TP53 and results in transcription repression (Latif et al., 2021; Song et al., 2020; Wu et al., 2006). SCD has been suggested as a modulator of cell motility and survival in breast cancer as well as cell membrane fluidity and EGFR activation in lung cancer (Angelucci et al., 2018; Nashed et al., 2012). Cellular analysis of SCD perturbations suggests that SCD protein activates HER2/EGFR signaling through membrane remodeling and increased cell membrane fluidity accompanied by increased cell motility. Interestingly, in resistant cells, increased cell motility accompanies apoptosis evasion, whereas in sensitive cells, a cytostatic response is followed by apoptosis. A compelling speculation is that when under stress, resistant cells not only escape apoptosis but also display a tendency to depart from the cellular locations that mediate stress, potentially reflecting processes inherited through phylogeny. The resulting spatial changes and polarization of EGFR/HER2 on the cell membrane are accompanied with increased EGFR/HER2 phosphorylation and downstream pathway activation consistent with previous reports (Puri et al., 2005; Turk et al., 2012). Downstream of EGFR/HER2, a combined and redundant effect from multiple upstream pathways (AKT/PI3K, RAF/MEK, MAPK14, PKC) is likely necessary as demonstrated by pathway inactivation and reversal of adaptive responses with cocktails of up to four drugs. Our integrated analyses suggest concerted roles for MCL1 copy-number-driven gene/protein expression and BETi-induced signaling plasticity changes. Together, they maintain high levels of MCL1 to evade apoptosis and confer resistance to BET inhibition. The complementarity and interplay between hard-coded genetic structure (e.g., MCL1 amplification) and drug-induced plasticity (e.g., activation of lipid and RTK pathways) may provide a solution to the long-standing conundrum on the relative dominance of genomics versus signaling activity in driving resistance to targeted agents (Yaffe, 2019). The genomic analyses suggest that the observed mechanisms involving BET, fatty acid synthesis, and RTK signaling may be represented in large cohorts of patients with breast cancer. Therefore, the observed drug responses are likely to be extrapolatable to clinical settings.
Advancing pre-clinical discoveries to clinical strategies requires a thorough understanding of biomarkers that can identify patients most likely to benefit, mechanisms of drug action, and evidence for generalizability. Despite the advances in sequencing technologies, durable responses to genetically matched targeted therapies in eligible patient cohorts are limited. Yet, robust molecular and cellular profiling methods coupled to bioinformatics analyses can tailor decision and improve patient benefit. Combined with clinical studies involving analysis of on-treatment biopsies, we argue that this computational/experimental study is a prototype for future translational applications, which will induce durable responses based on combination therapy decisions that are dynamically informed by multi-omics profiles of tumors evolving under cancer therapy.
Limitations of the study
For effective clinical translation of our findings, more comprehensive pre-clinical testing in patient-derived xenograft (PDX) models with diverse genomic backgrounds is needed. Another important but not adequately addressed aspect is drug toxicity, which is critical for successful translation. Despite the multiple lines of evidence and comprehensive validations, there remain few mechanistic points not covered in this study. First, it is not entirely possible, like in most biological research, to rule out the roles of other signaling molecules. Although we focused on p-HER/EGFR as the key RTK, other RTKs and downstream pathways may also contribute in other contexts as indicated by the involvement of p-MET in tumors of patients with no ERBB2 amplification (Figure 5I). There is a high degree of plasticity and redundancy between downstream pathways that link RTK activity to MCL1. The differential and complementary activities of such pathways may be highly dependent on the cellular contexts and feedback loops. GSK3-associated proteins such as FBXW7 or GSK3-independent mechanisms may also contribute to modulation of MCL1 protein levels (Inuzuka et al., 2011; Nifoussi et al., 2012). Although our studies, as well as a rich literature, (Wang et al., 2013; Maurer et al., 2006) suggest MCL1 protein stabilization is critical, it is possible that diverse MCL1 alterations (e.g., mRNA expression and stability changes, acetylation, alternative splicing) may also contribute to maintenance of an anti-apoptotic activity in response to BETi. Such mechanisms are not mutually exclusive and may cooperate to maintain MCL1 levels. Another intriguing question is why the BETi-induced upregulation of lipid metabolism genes and downstream signaling are driving the resistance in MCL1-amplified (or gained) cells but not in the MCL1 wild-type (WT), BETi-sensitive cells. A hypothesis that needs to be tested is that the pre-existing MCL1 protein may provide an early survival advantage, which enables the secondary transcriptional response leading to cellular plasticity and sustained resistance. We anticipate that future studies on both BRD4 and MCL1 biology will provide additional insights into mechanistic details and guide the strategies for more effective clinical translation of epigenetic and apoptotic agents for cancer treatment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for computational tools, resources and reagents should be directed to and will be fulfilled by the lead contact, Anil Korkut (akorkut@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq data have been deposited at GEO (GSE209934) and are publicly available as of the date of publication. The accession code is available in the key resources table. Proteomics data has been deposited to MDACC RPPA repository. The link to proteomics data is available in the key resources table. Microscopy and western blot data reported in this paper will be shared by the lead contact upon request.
The TargetScore code is shared as open source. The link is provided in the key resources table.
Any additional information, software and scripts required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MCL1 | Cell Signaling Technology | Cat#4572,RRID:AB_2281980 |
| MCL1 | Cell Signaling Technology | Cat# 39224,RRID:AB_2799149 |
| BCL2 | Cell Signaling Technology | Cat# 4223, RRID:AB_1903909 |
| BCL2 | Cell Signaling Technology | Cat# 15071,RRID:AB_2744528 |
| BRD4 | Cell Signaling Technology | Cat# 13440,RRID:AB_2687578 |
| Cleaved PARP | Cell Signaling Technology | Cat# 9541, RRID:AB_331426 |
| p-AKT(T308) | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| p-AKT (S473) | Cell Signaling Technology | Cat# 9271, RRID:AB_329825 |
| p-PKC | Cell Signaling Technology | Cat# 9375, RRID:AB_2284224 |
| P-MEK1/2 | Cell Signaling Technology | Cat# 9121, RRID:AB_331648 |
| p-GSK3 | Cell Signaling Technology | Cat# 9331, RRID:AB_329830 |
| p-HER2(T1248)/p-EGFR (Tyr1173) | Cell Signaling Technology | Cat# 2244, RRID:AB_331705 |
| p-P38 | Cell Signaling Technology | Cat# 4511, RRID:AB_2139682 |
| SCD | Cell Signaling Technology | Cat# 2794, RRID:AB_2183099 |
| BRD4 (IHC) | EMD Millipore. | Catalog No. ABE1391 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| β-actin | Sigma-Aldrich | Cat# A5316, RRID:AB_476743 |
| hERBB2/Her2 AF488 | R and D Systems | Cat# FAB9589G, RRID:AB_2800468 |
| Ki67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| V5-Tag (D3H8Q) | Cell signaling | Cat #13202, RRID:AB_2687461 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#:11668027 |
| PrestoBlue Cell Viability Assay kit | Life Technologies | Cat#:A13261 |
| Cellbrite orange | Biotium | Cat#:30022 |
| Prolong gold antifade mountant with DAPI | ThermoFisher Scientific | Cat#:P36935 |
| APO-BRDU™ Kit | BD Pharmingen | Cat#:556405 |
| Membrane Fluidity Kit | abcam | Cat#:Ab189819 |
| RNeasy Plus Mini Kit | Qiagen | Cat#:74134 |
| DharmaFECT Duo Transfection Reagent (Dharmacon) | Horizon Discovery | Cat#:T-2010-03 |
| RNeasy Mini kit | QIAGEN | Cat#: 74104 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat#:1708890 |
| Universal SYBR Green Fast qPCR Mix | ABclonal, | Cat#: #RK21203 |
| R.T.U. Vectastain Universal Elite ABC kit Anti-mouse IgG/Rabbit IgG | Vector Laboratories | Cat#: K-7100 |
| DAB (diaminobenzidine) Peroxidase (HRP) Substrate Kit | Vector Laboratories | Cat#: SK-4100 |
| Mayer’s hematoxylin | Electron Microscopy Sciences. | Cat#: 26173-03 |
| JQ1 | Selleckchem | Cat#: S7110 |
| SCD-inhibitor | MedChemExpress | Cat#: A939572 |
| MCL1 inhibitor-S63845 | Chemietek | Cat#: CT-S63845 |
| EGFR inhibitor-Lapatinib | Selleckchem | Cat#: S2111 |
| AKT inhibitor- MK-2206 2HCl | Selleckchem | Cat#: S1078 |
| PKC inhibitor- Go 6983 | Selleckchem | Cat#: S2911 |
| P38 MAPK inhibitor- Doramapimod (BIRB 796) | Selleckchem | Cat#: S1574 |
| MEK inhibitor- Trametinib (GSK1120212) | Selleckchem | Cat#: S2673 |
| Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) | Avanti Lipids | Cat#: 850345 |
| pegylated distearoyl-phosphatidyl ethanolamine (DSPE-PEG-2000) | Avanti Lipids | Cat#: 880128 |
| Deposited data | ||
| Gene expression changes occurring in response to treatment with JQ1 in breast cancer cell lines | This study | GEO: GSE209934 |
| Response and resistance to BET bromodomain inhibitors in triple negative breast cancer [ChIP-Seq] | Shu et al. (2016) | GEO: GSE63581 |
| Protein expression changes occurring in response to treatment with JQ1 in breast cancer cell lines | This study | https://tcpaportal.org/mclp/#/download |
| Gene expression and mRNA profiles in CCLE | CCLE | https://portals.broadinstitute.org/ccle/data/browseData?conversationPropagation=begin |
| mRNA and protein expression data in patients with breast cancer | TCGA |
https://www.cbioportal.org/study/summary?id=brca_tcga
https://www.cbioportal.org/study/summary?id=ov_tcga |
| Experimental models: Cell lines | ||
| BT474 | MD Anderson Cancer Center Cell Line Repository | N/A |
| BT20 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1954 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1937 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1419 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC70 | MD Anderson Cancer Center Cell Line Repository | N/A |
| MDAMB468 | MD Anderson Cancer Center Cell Line Repository | N/A |
| MCF7 | MD Anderson Cancer Center Cell Line Repository | N/A |
| SKBR3 | MD Anderson Cancer Center Cell Line Repository | N/A |
| SKOV3 | MD Anderson Cancer Center Cell Line Repository | N/A |
| Experimental models: Organisms/strains | ||
| Nude athymic NCr female mice | Department of Experimental Radiation Oncology, MD Anderson Cancer Center | N/A |
| Oligonucleotides | ||
| SCD1 (NM_005063): Forward primer: CCTGGTTTCACTTGGAGCTGTG | Sigma | N/A |
| SCD1 (NM_005063):Reverse primer: TGTGGTGAAGTTGATGTGCCAGC | Sigma | N/A |
| GAPDH (NM_002046): Forward primer: GTCTCCTCTGACTTCAACAGCG | Sigma | N/A |
| GAPDH (NM_002046): Reverse primer: ACCACCCTGTTGCTGTAGCCAA | Sigma | N/A |
| Human MCL1 siRNA | Sigma Aldrich | SASI HS01-00162656, SASI HS01-00162657 |
| Human BRD4 siRNA | Sigma Aldrich | SASI_HS01_00037409, SASI_Hs01_00037410 |
| Non-targeting siRNA Universal Negative Control | Sigma Aldrich | SIC001 |
| Recombinant DNA | ||
| pcDNA3.1 | Invitrogene | V79020 |
| Software and algorithms | ||
| Prism | GraphPad Software | N/A |
| Microsoft Excel | Microsoft | N/A |
| ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| TargetScore | Wang et al. (2021). https://doi.org/10.1101/2021.04.14.439861 | https://hub.docker.com/repository/docker/cannin/targetscore |
| rtracklayer 1.52.0 | Lawrence et al. (2009). | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html |
| Gviz 1.28.3 | Hahne and Ivanek (2016) | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| FACSDIV8.1 software | MDACC flow cytometry core facility | BD Biosciences (https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software) |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HCC1954, HCC1937, HCC1419, HCC70, BT20, BT474, SKOV3, SKBR3, MDAMB468 and MCF7 lines were obtained from MDACC Cell Line Repository and Gordon Mills and were thawed two weeks before experiments. All human cell lines were authenticated by fingerprinting using short tandem repeat testing and were verified to be free of mycoplasma contamination. There were less than 10 passages between thawing of the cells and the experiments described in this study. MCF-7 cells were maintained in DMEM supplemented with 10% FBS plus antibiotic/antimycotic solution (100U/mL streptomycin and 100U/mL penicillin) (Invitrogen). BT20 were maintained in EMEM supplemented with 10% FBS. Other cell lines were maintained in RPMI1640 supplemented with 10% FBS. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Breast cancer xenograft models
Nude athymic NCr female mice (5 weeks old) were obtained from the Department of Experimental Radiation Oncology, MDACC. All studies were conducted according to the experimental protocol approved by the MDACC IACUC. At the termination of the xenograft experiment, animals were euthanized according to guidelines by the Institutional Animal Care and Use Committee, adopted from the AVMA Guidelines for the Euthanasia of Animals: 2013 Edition. MDAMB468 cells (2×106 in 20% Matrigel) were orthotopically injected into the mammary fat pad of each mouse. After approximately 2 weeks, when tumor sizes reach 3-5 mm, mice were randomized in the treatment groups, including empty liposomes, liposomal JQ1, S83445 or combination groups. Treatments were administered at 20 mg/kg dose by i.p. injection (9 mice/per group, 10 in control arm) 3 times/week. Liposomes were prepared based on our previously published method (Hamurcu et al., 2016). Two animals from each arm were sacrificed at day 10 for histological analysis.
METHOD DETAILS
Drugs and antibodies
The inhibitors were purchased from Sellekchem (JQ1, IBETs, Lapatinib, MK-2206, Dequalinium Chloride, Trametinib, Doramapimod), ChemiEtek (S63845) and MedChemExpress (JQ1 and A939572). Specific antibodies against MCL1, BCL2, BCL-XL, BRD4, cleaved PARP, p-AKT(T308), p-AKT (S473), p-PKC, p-P38, p-MEK1/2, p-GSK3, p-HER2 (T1248)/p-EGFR (Tyr1173), V5-Tag (D3H8Q), EGFR, GSK3 and AKT were purchased from Cell Signaling Technology, MA. Antibody against β-actin is from Sigma-Aldrich. Anti-hERBB2/Her2 AF488 antibody was purchased from R&D systems Biotechne.
RPPA (reverse phase protein array)
The cells were washed 3 times with cold PBS and then suspended in RIPA buffer supplemented with proteinase inhibitor and phosphatase inhibitor (Pierce, Rockford, IL, USA). The cell suspension was vortexed for 15 s, placed on an end-over-end rotator for 30 min at 4°C and centrifuged at 14,000 x g for 15 min at 4°C. The lysates were prepared to provide 1–1.5 mg/mL of total protein lysate. RPPA analysis samples were prepared by adding SDS Sample Buffer, β-mercaptoethanol and RPPA Working Solution to obtain a final concentration of 0.5mg/mL. Samples were heated for 8 min at 100°C, centrifuged 2 min at 14,000 x g and stored at −80°C. The RPPA was performed at the MD Anderson Cancer Center Functional Proteomics core facility.
Immunoblotting
HCC1954, BT474 and SKOV3 cells were lysed in RIPA buffer (50mM Tris-HCl pH7.4, 150mM NaCl, 1mM EDTA, 1% D.O.C. (Na), 0.1% SDS, 1% Triton X-100) containing protease inhibitors (Pierce, Rockford IL). Protein concentrations were determined using BCA protein assay kit (Pierce, Rockford, IL). An equal amount of protein was loaded into SDS-PAGE gels and transferred to PVDF membranes (Millipore, MA). Membranes were blocked for 2 h with 5% non-fat milk in PBST (0.1% Tween 20 in PBS), and incubated overnight with specific primary antibody. After incubation of 2 h with horseradish peroxidase-conjugated secondary antibody, the proteins were detected by Clarity Western ECL substrate (Bio-Rad).
Cell proliferation assays
Cell proliferation was measured by the PrestoBlue Cell Viability Assay kit (A13261, Life Technologies) according to the manufacturer’s instructions. 2-4×103 cells were seeded into 96-well plates and cultured overnight. After treating with either vehicle or kinase inhibitors for 72 h, cells were collected and evaluated with an SYNERGY H1 microplate reader with Gen5 software (BioTek). In the viability assays, points and bars represent the mean of triplicates ±SEM (or four replicates).
Flow cytometric analysis of cell cycle and apoptosis
2.5x105 of cells (HCC1954, BT474 and SKOV3) were seeded in 6-cm plates. Following treating with vehicle or drugs for 48h, cells were harvested, fixed in 1% (W/V) paraformaldehyde in PBS, washed and rehydrated in PBS. DNA was stained according to the instruction of the APO-BRDU Kit, by treating the cells with DNA labeling solution, FITC labeled anti-BrdU antibody solution and PI/RNase staining buffer. Flow cytometry was analyzed using BD LSR II flow cytometer and FACSDIV 8.1 software (BD Biosciences) at the MDACC flow cytometry core facility.
mRNA sequencing
Cells (HCC1954, SKOV3 and BT474) were collected after treating with 1 μM of BETi (JQ1) for 24 h all in biological duplicates. Total RNA was isolated and purified using the RNeasy Plus Mini Kit (Qiagen). After quantification with Qubit 2.0 (Life Technologies), the samples passed through quality control steps for sample integrity and purity with Agilent 2100 and Agarose Gel Electrophoresis. After the sample QC, the cDNA library is constructed using the NEBNext Ultra II RNA Library Prep Kit (NEB) as follows: mRNA is enriched using oligo(dT) beads. The mRNA is then fragmented via sonication in fragmentation buffer, followed by cDNA synthesis using random hexamers and reverse transcriptase. After first-strand synthesis, a custom second strand synthesis buffer and enzyme mix are added to generate the second strand by nick translation. The final cDNA library is ready after a round of DNA purification, end-repair, A-tailing, ligation of sequencing adapters, size selection and PCR enrichment. For library QC, library concentration was quantified using a Qubit 2.0 fluorometer (Life Technologies) and then diluted to 1 ng/μL before checking insert size on an Agilent 2100. Libraries were then quantified to greater accuracy by quantitative PCR (Q-PCR) prior to sequencing. RNA libraries were sequenced with 20M paired-end 150bp reads on the Illumina NovaSeq 6000 platform (Novogene Inc.). Raw data was subjected to a round of QC to remove low-quality reads. All RNAseq experiments are performed in duplicates and mean values are used. The resulting mRNA sequence count data was normalized to enable cross-sample analyses.
Migration assays
The effect of drugs on cells’ migratory behavior was analyzed with the wound scratch assay. 4x105 HCC1954 and SKOV3 cells were seeded in 35 mm dishes. After 24 h, cells were treated with vehicle or kinase inhibitors for 72 h. The cells were scratched using a sterile 200-μL micropipette tip to form a straight wound. The cells were washed twice with PBS and cultured for an additional 24 h. The wound closure was measured under an EVOS FL Auto microscope (Life technologies). Images of 3 random fields were acquired at the time point of 0, 6, 12 and 24 h after wounding. The distances traveled by the cells were measured from control and experimental samples and were calculated and compared with time 0.
Membrane fluidity
Membrane fluidity was measured using the Membrane Fluidity Kit (Abcam ab189819). After a lipophilic pyrene probe incorporation into the membrane, the monomeric pyrene probe undergoes excimer formation dramatically shifting the emission spectrum of the pyrene probe to a longer red wavelength. The ratio of excimer (emission at 470 nm) to monomer (emission at 400 nm) fluorescence represents a quantitative change of the membrane fluidity. Cells were seeded in 96 well plates, treated with JQ1, SCD inhibitor and JQ1 plus SCD inhibitor for 72 h. The cells were labeled with labeling solution (15 μM of pyrenedecanoic acid (PDA), 0.08% F-127, in Perfusion Buffer) in the dark for 20 min rocking at 25°C, washed 2x with Perfusion Buffer, supplemented with culture medium and measured for fluorescence at two wavelengths (excitation at 360 nm, emission at 400 nm and 470 nm) in biological triplicates.
Immunofluorescence imaging experiments
SKOV3 and HCC1954 cells were seeded in coverslips overnight and treated with either vehicle or inhibitors for 48 h at 1 μM. The cells were fixed with cross-linking method. Cells were switched to fresh growth media with 4% paraformaldehyde for 5 min. After 2 times washing with PBS, the cells were incubated in 4% paraformaldehyde (in PBS) for 15 min. The fixed cells were rinsed with PBS to remove any fixation agent. Cells were then blocked with odyssey PBS for 1 h at room temperature and incubated with the anti-hERBB2/Her2 AF488 antibody (1:200, R&D systems biotechne) overnight at 4°C. After washing with PBS, coverslips were mounted on slides using Prolong gold antifade mountant with DAPI (ThermoFisher Scientific, P36935). Immunofluorescence images were acquired with a confocal microscope (Olympus).
Preparation of liposomal nanoparticles
Liposomal nanocarriers were prepared with Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and pegylated distearoyl-phosphatidyl ethanolamine (DSPE-PEG-2000) (Avanti Lipids) as previously described (Hamurcu et al., 2016). Briefly, DMPC and DSPE-PEG2000 were mixed at the ratio of (10:1) and were mixed with small molecule inhibitors at a ratio of 10:1 (w/w) and lyophilized in the presence of excess tertiary butanol. After liposomal drugs were reconstituted in PBS, the agents were systemically administered to mice.
Immunohistochemistry
The mice tissue samples were fixed in 10% neutral buffered formalin for 24 h and transferred to 70% ethanol until ready to be processed. Samples were embedded in paraffin, sectioned at 5 mm and stained routinely with hematoxylin and eosin. Unstained sections were designated for immunohistochemical staining. BCL2 (Cell Signaling. Catalog No. 15071), SCD (Cell Signaling. Catalog No. #2794), Clvd-Caspase3 (Cell Signaling. Catalog No. 9661), KI67 (Abcam. Catalog No. ab15580), BRD4 (EMD Millipore. Catalog No. ABE1391) and MCL1 (Cell Signaling. Catalog No. 39224) immunoreactivity was detected using manual staining with the R.T.U. Vectastain Universal Elite ABC kit Anti-mouse IgG/Rabbit IgG (Vector Laboratories. Catalog No. K-7100). Heat-induced antigen retrieval was done using a pH 6 Citrate-based buffer (made in house) and a tris-EDTA pH 9.0 buffer (made in house) for 30 min in the IHC-TekTM Epitope Retrieval Steamer (IHC World. Catalog No. IW-1102). The primary mouse monoclonal anti-BCL2 antibody was incubated at 1:400 dilution, the primary rabbit monoclonal anti-SCD was incubated at 1:200 dilution, the primary rabbit polyclonal anti-Clvd-Caspase3 was incubated at 1:200 dilution, the primary rabbit polyclonal anti-KI67 was incubated at 1:300 dilution, the primary rabbit polyclonal anti-BRD4 was incubated at 1:250 dilution, and the primary rabbit monoclonal anti-MCL1 at 1:100 dilution for one hour at room temperature in an incubation chamber. The anti-rabbit/mouse biotinylated universal antibody was applied for 30 min at room temperature. The Vectastain ABC reagent was incubated for 30 min at room temperature. Immunoreactivity was detected using DAB (diaminobenzidine) Peroxidase (HRP) Substrate Kit (Vector Laboratories, Catalog No. SK-4100) incubated for approximately 2 min. Slides were counterstained with Mayer’s hematoxylin (Electron Microscopy Sciences. Cat No. 26173-03) for 3 min. Hematoxylin was enhanced with 0.25% ammonium hydroxide bluing solution (made in house) for 1 min at room temperature. Sections of the experimental control group served as positive controls for validation. Negative control slides were stained by substituting the primary antibody with 2.5% normal horse serum. The stained FFPE sections from mouse models were imaged at 20X using Zeiss Axio Vision Slide Scanner.
Real-time quantitative reverse transcription PCR (RT-qPCR)
The cells were collected after indicated treatment and total RNA was extracted using the RNeasy Mini kit (QIAGE,#74104) according to manufacturer instructions. cDNA synthesis was then performed with 1 μg of total RNA by using the iScript cDNA Synthesis Kit (Bio-Rad, #1708890). QRT-PCR was performed using 2X Universal SYBR Green Fast qPCR Mix (ABclonal, #RK21203) in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) according to manufacturer instructions. All primers are synthesized from Sigma. Glyceraldehyde 3-phosphatedehydrogenase (GAPDH) was used as an internal control, and the fold change gene expression level was calculated using the 2−ΔΔCT method. The primer sequences as listed as follow. SCD (GenBank: NM_005063): Forward primer: CCTGGTTTCACTTGGAGCTGTG; Reverse primer: TGTGGTGAAGTTGATGTGCCAGC. GAPDH (GenBank: NM_002046): Forward primer: GTCTCCTCTGACTTCAACAGCG; Reverse primer: ACCACCCTGTTGCTGTAGCCAA.
Knockdown and overexpression studies
MCL1, BRD4 and control siRNA are purchased from Sigma-Aldrich: Human MCL1 siRNA (SASI HS01-00162656, SASI HS01-00162657); BRD4 (SASI_HS01_00037409, SASI_Hs01_00037410), Non-targeting siRNA Universal Negative Control (SIC001). Transfection into HCC1954, BT474, MDAMB464 and SKOV3 cells were done with DharmaFECT Duo Transfection Reagent (Dharmacon). For over expression of MCL1, cells were transfected with empty pcDNA3.1 and pcDNA3.1-V5-MCL1. After 48 h of transfection, cells were collected and analyzed for the expression of the related genes by western blotting using BRD4, MCL1 and V5-tag antibodies.
ChIP-seq analysis
For assessment of BRD4 binding on lipid metabolism gene regulatory sites and BRD4 removal by BRD4 inhibition, we analyzed the breast cancer cell line ChIP-seq data with BRD4 and H3K27ac antibody under BETi (JQ1) treatment (GEO: GSE63581). Promoter regions are defined using the consensus of GeneHancer identified regions as well H3K27ac antibody binding sites. Relative binding of antibodies is calculated as integrated area in BET-treated normalized to DMSO-treated, after subtraction of background determined by whole cell extract control. Data is visualized in R 3.6.1 using Gviz 1.28.3 (Hahne and Ivanek, 2016) and rtracklayer 1.52.0 (Lawrence et al., 2009).
TargetScore algorithm
The algorithm is presented in detail in (Wang et al., 2021). The aim of the network-level description of adaptive responses is to better explain collective mechanisms, in contrast with conventional analysis which focuses on individual genes/proteins. The algorithm: (i) quantifies and visualizes collective adaptive responses to targeted therapy; (ii) nominates combination therapies, involving the agent that induces the adaptive response and a second agent that targets vulnerabilities induced by the adaptive response. The TargetScore algorithm nominates modules of functionally related molecular entities involved in adaptive resistance with a higher likelihood of therapeutic relevance. TargetScore also reduces the number of potential false positives as it eliminates signals from proteins with no connection to collective processes. The algorithm is formulated such that highest TargetScores correspond to potential adaptive responses (drug-induced activation of oncogenic processes or deactivation of tumor suppressors). Lowest TargetScores correspond to direct response mechanisms to targeted perturbations. TargetScore code is available as a Docker container cannin/targetscore:mcl1-analysis with instructions here: https://hub.docker.com/repository/docker/cannin/targetscore.
Input data and data quality
The molecular response data collected before and after therapy is used as the input in the analysis pipeline. The BETi RPPA response data is generated at the MDACC functional proteomics core facility. The functional proteomics core validates each antibody by analysis of western blots to observe each readout as a single or dominant band on a blot and ensure the Pearson correlation coefficient between RPPA and Western blot readouts are greater than 0.7. The dynamic range and specificity are routinely determined using peptides, phosphopeptides, growth factors, Inhibitors, RNAi, cells with wide levels of expression including 330 cell lines under multiple conditions on a single array. The intra and inter-slide reproducibility of RPPA reads are routinely monitored. In this study, BETi responses from cell lines were interrogated using 217 antibodies at time points of 24 and 48hr after drug treatment in cells cultured as 2D monolayers and 3D Matrigel cultures. The method can analyze data from both patient biopsy samples and cell lines. The method is amenable to analysis of drug response data from a single sample to detect drug-induced vulnerabilities in a sample-specific way or thousands of conditions to determine complex statistical patterns of adaptive responses across varying genomic contexts or perturbation agents. The analysis can be performed with time series (short term perturbation periods or long term acquired resistance events) or multi-dose perturbation data to monitor the evolution of adaptive response patterns in time and dose space.
Reference network construction
Network models are constructed using the signaling data stored in Pathway Commons (PC) database and manual curation/correction by experts. PC integrates detailed human pathway data from multiple public resources such as Reactome, NCI PID, PhosphoSitePlus, Panther Pathways, and HumanCyc (Cerami et al., 2011). Automated prior pathway extraction is achieved using the SignedPC module embedded in the BioPAX/PaxTools pathway analysis language and CausalPath method (Demir et al., 2010, 2013; Babur et al., 2021). In order to delineate the pathway interactions captured by the RPPA data, we identified 4 different relation types: phosphorylation, dephosphorylation, expression upregulation, and expression downregulation. The first two relations can be used to explain phospho-protein changes, and the last two can be used to explain total protein or gene expression changes. Using the BioPAX framework (Babur et al., 2014), we define a set of patterns to detect such relations and identify the signaling interactions between the phosphoproteomic entities. SignedPC resource includes 130,158 interactions, of which 107,251 define “expression up-regulation”, 20,964 define “phosphorylation”, 1942 define “expression downregulation” and 2660 define dephosphorylation events. The details of the construction of SignedPC is described in (Babur et al., 2021). The resulting interaction set extracted from the SignedPC forms the reference network.
The TargetScore and target detection
A TargetScore that quantifies the adaptive pathway responses to a perturbation as a sum of the response from each individual protein and its pathway neighborhood, is calculated for each entity on the reference network. The calculation combines the cell type-specific drug response data with signaling network information extracted from signaling databases. Positive TargetScore identifies proteins involved in adaptive response (e.g., upregulation of receptor tyrosine kinase expression by MEK inhibitor via a feedback loop) and negative TargetScore corresponds to the immediate impact of the drug (e.g., inhibition of ERK phosphorylation by MEK inhibitor). The mathematical formulation of the TargetScore is.
| (Equation 1) |
where fsi represents the functional score (see below) of the node of interest, Δxi is the proteomic response, log normalized with respect to the unperturbed (or pretreatment) level. The σ is the standard deviation of Δxi over all perturbation conditions and all drug doses for a sample. fsi scores ensure high positive TargetScore correspond to adaptive responses. The division of Δx by σ normalizes a readout with respect to the dynamic range of the corresponding antibody. Node j is a node in the pathway neighborhood of the node i, with readout Δxj and standard deviation. pij is the pathway distance between nodes i and j. Wij represents the signaling interaction between nodes i and j extracted algorithmically from databases (i.e., reference network). The cumulative TargetScore over multiple doses can be calculated as the area under the curve of the TargetScore at each dose versus drug doses.
A functional score (fs) is assigned to each of the proteomic entities measured in the MDACC RPPA and DFCI/MSKCC Zeptosens proteomics platforms (Tibes et al., 2006). We used manual curation, resources such as Phosphosite database, COSMIC Cancer Gene Census and TUSON tumor suppressor/oncogene resource (Davoli et al., 2013). We assigned (+1) for total levels or activating phosphorylation of oncoproteins and inhibitory phosphorylation of tumor suppressors. Similarly, a functional score of (−1) is assigned to total levels or activating phosphorylation of tumor suppressors and inhibitory phosphorylation of oncoproteins.
Determination of response pathways
The resistance and response (drug activity) pathways are detected by the reanalysis of the TargetScore and the underlying network models. Proteins that are connected within the first neighborhood of each other in the underlying network and have the highest scores (20 top entities) represent the potential adaptive response pathway(s) and those with low TargetScore represent the potential response/drug activity pathway. The pathway relations are extracted from the reference network model and visualized using the Cytoscape software (Shannon et al., 2003).
Analysis of patient and cell line genomics and proteomics data
The correlative analyses of tumors of patients with breast and ovarian cancer are performed using the TCGA breast invasive carcinoma (N = 1108) and ovarian serous cystadenocarcinoma (N = 606) data stored in the cBioPortal. The patients in the basal breast cancer subtype are selected based on the annotation in (Sanchez-Vega et al., 2018). The HER2 amplified breast cancer cases are selected based on the ERBB2 gene GISTIC copy-number assignments stored in the cBioPortal. The correlations between molecular entities are computed using the TCGA microarray data for mRNA and RPPA data for phosphoproteomics. The Spearman’s correlations are computed using the cBioPortal analysis and visualization suits. The cell line genomics analyses are performed using the data from the CCLE resource (Ghandi et al., 2019; Barretina et al., 2012). The cell line mutation data is based on WGS and WES followed by a mutation calling for SNVs and InDels, and filtering out germline variants performed at the Broad Institute as described in (Ghandi et al., 2019; Barretina et al., 2012). The copy-number data is based on SNP arrays processed with circular binary segmentation of copy-number levels and GISTIC copy-number calls as described in (Barretina et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of protein expression based on IHC experiments
Cleaved Caspase3, KI67, BCL2, MCL1 and SCD1 expressions were assessed within the tumor populated areas by counting the number of positively stained cells. On each slide across all antibodies and conditions, three independent ROIs populated by tumor cells are selected randomly. In each ROI, the number of positively stained cells per unit area are counted. Cells positive for Cleaved Caspase3 (nuclear/cytoplasmic), KI67(nuclear), BCL2 (nuclear/cytoplasmic), MCL1(cytoplasmic) and SCD(cytoplasmic) were included when differential staining was observed. The scenes were assessed and quantified by three independent observers and a consensus count was reached. The mean and standard deviation value of positive counts from different regions were calculated. The statistical significance between different treatment arms were assessed with an unpaired t test.
Quantification of band intensities based on western blot experiments
The Western blot images were quantified using Adobe PhotoShop (Version: 22.5.0) by calculation of mean pixel intensities on inverted black and white images after subtraction of background intensities. Equal areas were selected for each band across conditions enabling cross comparison. The band intensities are normalized with respect to the beta-actinin controls and in relevant cases for phosphoproteins, normalized with respect to total protein levels.
Statistical evaluation of TargetScores
The statistical assessment detects and eliminates the TargetScores that are likely driven solely by the network structure with no significant impact from the cell type-specific data. For this purpose, the probability of observing a given TargetScore is calculated over a null distribution of TargetScore values generated with randomized drug response data and the fixed network structure. A random sampling of proteomic responses (randomized antibody label mixing) at each drug dose generates randomized datasets over all antibody readouts. The TargetScore is calculated for 1000 independent random datasets using Equation 1, and the null score distribution is constructed. Next, the FDR-adjusted p value is calculated for the TargetScore value from actual data. For increased statistical power, the p values across biologically similar or highly overlapping conditions (e.g., similar BETis) that lead to similar TargetScores are merged using the Stouffer’s method with the formula (Stouffer et al., 1949).
| (Equation 2) |
Z scores are obtained through p-value-to-Z conversion and k is the total number of conditions merged. Resulting Z-value is back converted to p-values and FDR-adjusted using the Benjamini-Hochberg method.
Statistical evaluation and quantification of drug responses in cell lines
The drug response is quantified as area under the dose-response curves by treating the area as a sum of the trapezoids generated by the responses to consecutive drug doses. The maximum drug dose is quantified as Amax = 1-Vmax, where A is the effect at maximum dose and V is the observed viability at maximum dose. The drug synergy is quantified using the Bliss score with the formula; CI= (EA + EB-EAEB)/EAB, where E is the effect of drugs A, B and AB (the combination of drugs A and B) on viability (i.e., fraction of cells killed). The differences are assessed with an unpaired t test for two-group comparisons (Figures 4B, 4C, 4F, 5A, 5D, 5E). We used ANOVA test for statistical assessment of multi-group comparisons (Figures 3H and 6C).
Statistical evaluation of drug responses in xenograft models
We used 9 animals/arm for control, 8 animals/arm for treatment cohorts. The statistical differences were assessed with an Mann-Whitney U test and error bars represent the +/−SEM of the responses across the animals in each arm.
Processing and differential analysis of transcriptomic responses
The raw RNA sequencing reads were aligned using STAR with the reference genome GRCh38.d1.vd1.fa and annotation file gencode.v22.annotation.gtf. The quality controls of raw reads and aligned reads were assessed using FastQC and RSeQC, respectively. The gene counts are quantified using htseq-count from the aligned RNA reads in BAM files. The normalized read counts are calculated using DESeq2. The significant mRNA fold changes upon drug treatment were detected as FDR adjusted Q-values < 0.05. To eliminate noise from rare transcripts, we included the mRNA species with an abundance over a minimal threshold (minimum count >200, in at least one condition – i.e., before or after treatment in at least one cell line). The significant differential responses are visualized as a heatmap and quantified to demonstrate fold differences of mRNA species that change in opposite directions in resistant vs. sensitive lines.
Go-term enrichment
We downloaded GO-term gene associations from -associations/goa_human.gaf.gz, and tested the enrichment of the terms in differentially expressed 127 genes using Fisher’s exact test. We used the Benjamini-Hochberg method to select the significance threshold to get a result set with 0.1 FDR.
Differential analysis of proteomic responses
The proteomic response was quantified by normalizing the median normalized proteomic readouts from drug treated samples with respect to the matched untreated samples. The proteomic differential response (Figure 5) is quantified as ST,I, = ΣD[(ΔXT,DHCC1954,I-ΔXT,DSKOV3,I)+(ΔXT,DBT474,I-ΔXT,DSKOV3,I)], where Δx is the phosphoproteomic response for entity i, t is the time point (24 or 48h), and d represents BETi (JQ1, iBET151, iBET726, or iBET762). The statistical significance is quantified using a t test followed by a Bonferroni multiple hypothesis correction with the null hypothesis Ho: (Xt,dHCC1954,i) = μ(Xt,dSKOV3,i).
Supplementary Material
Highlights.
MCL1-driven apoptosis evasion is an adaptive response to BET inhibition (BETi)
Co-targeting MCL1 and BET is highly synergistic in breast cancer cells
MCL1 copy-number alterations can predict responses to BET and MCL1 targeting
Activities of fatty acid synthesis and RTK pathways link BETi to apoptosis evasion
ACKNOWLEDGMENTS
This work is supported by grants from OCRA Collaborative Research Award (A.K. and G.B.M.); US NCI grants U24CA210950 (G.B.M.), P50CA217685 (G.B.M.), P30 CA016672 (MDACC Support Grant, the Bioinformatics Shared Resource; A.K.), and U01CA217842 (A.K. and G.B.M.); ICI Fund (A.K.); NIGMS P41 GM103504 (C.S.); CPRIT High-Impact/High-Risk Award (RP170640; A.K.); a kind gift from the Miriam and Sheldon Adelson Medical Research Foundation (G.B.M.); SAC110052 from the Susan G. Komen Foundation (G.B.M.); and BCRF-18-110 from the Komen Foundation (PDF17483544; D.M.). Confocal microscopy experiments are performed at the MDACC Advanced Microscopy Facility funded by NIH S10 RR029552. N.S. is a CPRIT Scholar in Cancer Research with funding from the Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant RR160021 and a recipient of Early Career Award by Ovarian Cancer Research Alliance (grant 649968).
Footnotes
DECLARATION OF INTERESTS
G.B.M., SAB/consultant: AstraZeneca, Chrysallis Biotechnology, ImmunoMET, Ionis, Lilly, Nuevolution, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda; stock/options/financial: Catena Pharmaceuticals, ImmunoMet, SignalChem, Tarveda; licensed technology HRD assay to Myriad Genetics; DSP patent with Nanostring. Z.D.: shareholder in Vivoz Biolabs, LLC. M.C.: paid employee of Axcella Health.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111304.
REFERENCES
- Akbani R, Ng PKS, Werner HMJ, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. (2014). A pan-cancer proteomic perspective on the Cancer Genome Atlas. Nat. Commun 5, 3887. 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci C, D’Alessio A, Iacopino F, Proietti G, Di Leone A, Masetti R, and Sica G (2018). Pivotal role of human stearoyl-CoA desaturases (SCD1 and 5) in breast cancer progression: oleic acid-based effect of SCD1 on cell migration and a novel pro-cell survival role for SCD5. Oncotarget 9, 24364–24380. 10.18632/oncotarget.25273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur Ö, Aksoy BA, Rodchenkov I, Sümer SO, Sander C, and Demir E (2014). Pattern search in BioPAX models. Bioinformatics 30, 139–140. 10.1093/bioinformatics/btt539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur Ö, Luna A, Korkut A, Durupinar F, Siper MC, Dogrusoz U, Vaca Jacome AS, Peckner R, Christianson KE, Jaffe JD, et al. (2021). Causal interactions from proteomic profiles: molecular data meet pathway knowledge. Patterns 2, 100257. 10.10167/j.patter.2021.100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancerdrug sensitivity. Nature 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al. (2018). A comprehensive pan-cancer molecular study of Gynecologic and breast cancers. Cancer Cell 33, 690–705.e9. 10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, Dickson A, Ryan KM, Kalna G, Edwards J, et al. (2018). MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 9,19. 10.1038/s41419-017-0035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD, and Sander C (2011). Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 39, D685–D690. 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir E, Babur O, Rodchenkov I, Aksoy BA, Fukuda KI, Gross B, Sümer OS, Bader GD, and Sander C (2013). Using biological pathway data with paxtools. PLoS Comput. Biol 9, e1003194. 10.1371/journal.pcbi.1003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir E, Cary MP, Paley S, Fukuda K, Lemer C, Vastrik I, Wu G, D’Eustachio P, Schaefer C, Luciano J, et al. (2010). The BioPAX community standard for pathway data sharing. Nat. Biotechnol 28, 935–942. 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroshow DB, Eder JP, and LoRusso PM (2017). BET inhibitors: a novel epigenetic approach. Ann. Oncol 28, 1776–1787. 10.1093/annonc/mdx157. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028. 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gry M, Rimini R, Strömberg S, Asplund A, Pontén F, Uhlén M, and Nilsson P (2009). Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genom. 10, 365. 10.1186/1471-2164-10-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Statistical genomics: methods and protocols. In Visualizing Genomic Data Using Gviz and Bioconductor, Mathé E and Davis S, eds. (Springer New York; ), pp. 335–351. [DOI] [PubMed] [Google Scholar]
- Hamurcu Z, Ashour A, Kahraman N, and Ozpolat B (2016). FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 7, 16619–16635. 10.18632/oncotarget.7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, and Johnston PG (2013). Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Hölscher AS, Schulz WA, Pinkerneil M, Niegisch G, and Hoffmann MJ (2018). Combined inhibition of BET proteins and class I HDACs synergistically induces apoptosis in urothelial carcinoma cell lines. Clin. Epigenetics 10, 1. 10.1186/s13148-017-0434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. (2011). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109. 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, and Spiegelman BM (1996). ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 10, 1096–1107. 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- Kim TK, Herbst RS, and Chen L (2018). Defining and understanding adaptive resistance in cancer immunotherapy. Trends Immunol. 39, 624–631. 10.1016/j.it.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut A, Wang W, Demir E, Aksoy BA, Jing X, Molinelli EJ, Babur Ö, Bemis DL, Onur Sumer S, Solit DB, et al. (2015). Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. Elife 4, e04640. 10.7554/eLife.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- Latif AL, Newcombe A, Li S, Gilroy K, Robertson NA, Lei X, Stewart HJS, Cole J, Terradas MT, Rishi L, et al. (2021). BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat. Commun 12, 241. 10.1038/s41467-020-20378-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842. 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550. 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, and Green DR (2006). Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760. 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Meric-Bernstam F, Frampton GM, Ferrer-Lozano J, Yelensky R, Pérez-Fidalgo JA, Wang Y, Palmer GA, Ross JS, Miller VA, Su X, et al. (2014). Concordance of genomic alterations between primary and recurrent breast cancer. Mol. Cancer Ther 13, 1382–1389. 10.1158/1535-7163.MCT-13-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, and Brugge JS (2012). Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239. 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashed M, Chisholm JW, and Igal RA (2012). Stearoyl-CoA desaturase activity modulates the activation of epidermal growth factor receptor in human lung cancer cells. Exp. Biol. Med 237, 1007–1017. 10.1258/ebm.2012.012126. [DOI] [PubMed] [Google Scholar]
- Nifoussi SK, Vrana JA, Domina AM, De Biasio A, Gui J, Gregory MA, Hann SR, and Craig RW (2012). Thr163 phosphorylation causes Mcl-1 stabilization when degradation is independent of the adjacent GSK3-targeted phosphodegron, promoting drug resistance in cancer. PLoS One 7, e47060. 10.1371/journal.pone.0047060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. (2006). mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508. 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri C, Tosoni D, Comai R, Rabellino A, Segat D, Caneva F, Luzzi P, Di Fiore PP, and Tacchetti C (2005). Relationships between EGFR signaling--competent and endocytosis-competent membrane microdomains. Mol. Biol. Cell 16, 2704–2718. 10.1091/mbc.e04-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen GW, Yao Y, Zhang J, Dulak A, Castriotta L, Jacques K, Zhao W, Gharahdaghi F, Hattersley MM, Lyne PD, et al. (2018). BRD4 amplification facilitates an oncogenic gene expression program in high-grade serous ovarian cancer and confers sensitivity to BET inhibitors. PLoS One 13, e0200826. 10.1371/journal.pone.0200826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, Alvarez MJ, Kendsersky ND, Pelz CR, Johnson-Camacho K, et al. (2018). Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun 9, 3815. 10.1038/s41467-018-05729-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhrig F, and Schulze A (2016).The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16, 732–749. 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley BJ, Yang WP, Zhu J, Madireddi MT, Lawrence RM, et al. (2011). Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res 9, 1551–1561. 10.1158/1541-7786.MCR-11-0126. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic signaling pathways in the cancer genome Atlas. Cell 173, 321–337.e10. 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. (2016). Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Hu G, Jia J, Yao M, Wang X, Lu W, Hutchins AP, Chen J, Ozato K, and Yao H (2020). DNA damage induces dynamic associations of BRD4/P-TEFb with chromatin and modulates gene transcription in a BRD4-dependent and -independent manner. Front. Mol. Biosci 7, 618088. 10.3389/fmolb.2020.618088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer SA, Suchman EA, DeVinney LC, Star SA, and Williams RM Jr. (1949). The American Soldier. In Vol.1: Adjustment during Army Life (Princeton University Press; ). [Google Scholar]
- Stubbs CD, and Smith AD (1984). The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim. Biophys. Acta 779, 89–137. 10.1016/0304-4157(84)90005-4. [DOI] [PubMed] [Google Scholar]
- Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PKS, et al. (2017). Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med 9, eaal5148. 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. (2018). BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell 33, 401–416.e8. 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KN, and Schlaepfer DD (2018). Adaptive resistance to chemotherapy, A multi-FAK-torial linkage. Mol. Cancer Ther 17, 719–723. 10.1158/1535-7163.MCT-17-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther 5, 2512–2521. 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Turk HF, Barhoumi R, and Chapkin RS (2012).Alteration of EGFR spatio-temporal dynamics suppresses signal transduction. PLoS One 7, e39682. 10.1371/journal.pone.0039682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Luna A, Yan G, Li X, Babur O, Mills GB, Sander C, and Korkut A (2021). Targeting adaptation to cancer treatment by drug combinations. Preprint at bioRxiv. 10.1101/2021.04.14.439861. [DOI] [Google Scholar]
- Wang R, Xia L, Gabrilove J, Waxman S, and Jing Y (2013). Downregulation of Mcl-1 through GSK-3b activation contributes to arsenic trioxide-induced apoptosis in acute myeloid leukemia cells. Leukemia 27, 315–324. 10.1038/leu.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, and Chiang CM (2006). Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20, 2383–2396. 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Vakoc CR (2017). Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med 7, a026674. 10.1101/cshperspect.a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB (2019). Why geneticists stole cancer research even though cancer is primarily a signaling disease. Sci. Signal 12, eaaw3483. 10.1126/scisignal.aaw3483. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, et al. (2018). BRD4 facilitates replication stress-induced DNA damage response. Oncogene 37, 3763–3777. 10.1038/s41388-018-0194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO (GSE209934) and are publicly available as of the date of publication. The accession code is available in the key resources table. Proteomics data has been deposited to MDACC RPPA repository. The link to proteomics data is available in the key resources table. Microscopy and western blot data reported in this paper will be shared by the lead contact upon request.
The TargetScore code is shared as open source. The link is provided in the key resources table.
Any additional information, software and scripts required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MCL1 | Cell Signaling Technology | Cat#4572,RRID:AB_2281980 |
| MCL1 | Cell Signaling Technology | Cat# 39224,RRID:AB_2799149 |
| BCL2 | Cell Signaling Technology | Cat# 4223, RRID:AB_1903909 |
| BCL2 | Cell Signaling Technology | Cat# 15071,RRID:AB_2744528 |
| BRD4 | Cell Signaling Technology | Cat# 13440,RRID:AB_2687578 |
| Cleaved PARP | Cell Signaling Technology | Cat# 9541, RRID:AB_331426 |
| p-AKT(T308) | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| p-AKT (S473) | Cell Signaling Technology | Cat# 9271, RRID:AB_329825 |
| p-PKC | Cell Signaling Technology | Cat# 9375, RRID:AB_2284224 |
| P-MEK1/2 | Cell Signaling Technology | Cat# 9121, RRID:AB_331648 |
| p-GSK3 | Cell Signaling Technology | Cat# 9331, RRID:AB_329830 |
| p-HER2(T1248)/p-EGFR (Tyr1173) | Cell Signaling Technology | Cat# 2244, RRID:AB_331705 |
| p-P38 | Cell Signaling Technology | Cat# 4511, RRID:AB_2139682 |
| SCD | Cell Signaling Technology | Cat# 2794, RRID:AB_2183099 |
| BRD4 (IHC) | EMD Millipore. | Catalog No. ABE1391 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| β-actin | Sigma-Aldrich | Cat# A5316, RRID:AB_476743 |
| hERBB2/Her2 AF488 | R and D Systems | Cat# FAB9589G, RRID:AB_2800468 |
| Ki67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| V5-Tag (D3H8Q) | Cell signaling | Cat #13202, RRID:AB_2687461 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#:11668027 |
| PrestoBlue Cell Viability Assay kit | Life Technologies | Cat#:A13261 |
| Cellbrite orange | Biotium | Cat#:30022 |
| Prolong gold antifade mountant with DAPI | ThermoFisher Scientific | Cat#:P36935 |
| APO-BRDU™ Kit | BD Pharmingen | Cat#:556405 |
| Membrane Fluidity Kit | abcam | Cat#:Ab189819 |
| RNeasy Plus Mini Kit | Qiagen | Cat#:74134 |
| DharmaFECT Duo Transfection Reagent (Dharmacon) | Horizon Discovery | Cat#:T-2010-03 |
| RNeasy Mini kit | QIAGEN | Cat#: 74104 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat#:1708890 |
| Universal SYBR Green Fast qPCR Mix | ABclonal, | Cat#: #RK21203 |
| R.T.U. Vectastain Universal Elite ABC kit Anti-mouse IgG/Rabbit IgG | Vector Laboratories | Cat#: K-7100 |
| DAB (diaminobenzidine) Peroxidase (HRP) Substrate Kit | Vector Laboratories | Cat#: SK-4100 |
| Mayer’s hematoxylin | Electron Microscopy Sciences. | Cat#: 26173-03 |
| JQ1 | Selleckchem | Cat#: S7110 |
| SCD-inhibitor | MedChemExpress | Cat#: A939572 |
| MCL1 inhibitor-S63845 | Chemietek | Cat#: CT-S63845 |
| EGFR inhibitor-Lapatinib | Selleckchem | Cat#: S2111 |
| AKT inhibitor- MK-2206 2HCl | Selleckchem | Cat#: S1078 |
| PKC inhibitor- Go 6983 | Selleckchem | Cat#: S2911 |
| P38 MAPK inhibitor- Doramapimod (BIRB 796) | Selleckchem | Cat#: S1574 |
| MEK inhibitor- Trametinib (GSK1120212) | Selleckchem | Cat#: S2673 |
| Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) | Avanti Lipids | Cat#: 850345 |
| pegylated distearoyl-phosphatidyl ethanolamine (DSPE-PEG-2000) | Avanti Lipids | Cat#: 880128 |
| Deposited data | ||
| Gene expression changes occurring in response to treatment with JQ1 in breast cancer cell lines | This study | GEO: GSE209934 |
| Response and resistance to BET bromodomain inhibitors in triple negative breast cancer [ChIP-Seq] | Shu et al. (2016) | GEO: GSE63581 |
| Protein expression changes occurring in response to treatment with JQ1 in breast cancer cell lines | This study | https://tcpaportal.org/mclp/#/download |
| Gene expression and mRNA profiles in CCLE | CCLE | https://portals.broadinstitute.org/ccle/data/browseData?conversationPropagation=begin |
| mRNA and protein expression data in patients with breast cancer | TCGA |
https://www.cbioportal.org/study/summary?id=brca_tcga
https://www.cbioportal.org/study/summary?id=ov_tcga |
| Experimental models: Cell lines | ||
| BT474 | MD Anderson Cancer Center Cell Line Repository | N/A |
| BT20 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1954 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1937 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC1419 | MD Anderson Cancer Center Cell Line Repository | N/A |
| HCC70 | MD Anderson Cancer Center Cell Line Repository | N/A |
| MDAMB468 | MD Anderson Cancer Center Cell Line Repository | N/A |
| MCF7 | MD Anderson Cancer Center Cell Line Repository | N/A |
| SKBR3 | MD Anderson Cancer Center Cell Line Repository | N/A |
| SKOV3 | MD Anderson Cancer Center Cell Line Repository | N/A |
| Experimental models: Organisms/strains | ||
| Nude athymic NCr female mice | Department of Experimental Radiation Oncology, MD Anderson Cancer Center | N/A |
| Oligonucleotides | ||
| SCD1 (NM_005063): Forward primer: CCTGGTTTCACTTGGAGCTGTG | Sigma | N/A |
| SCD1 (NM_005063):Reverse primer: TGTGGTGAAGTTGATGTGCCAGC | Sigma | N/A |
| GAPDH (NM_002046): Forward primer: GTCTCCTCTGACTTCAACAGCG | Sigma | N/A |
| GAPDH (NM_002046): Reverse primer: ACCACCCTGTTGCTGTAGCCAA | Sigma | N/A |
| Human MCL1 siRNA | Sigma Aldrich | SASI HS01-00162656, SASI HS01-00162657 |
| Human BRD4 siRNA | Sigma Aldrich | SASI_HS01_00037409, SASI_Hs01_00037410 |
| Non-targeting siRNA Universal Negative Control | Sigma Aldrich | SIC001 |
| Recombinant DNA | ||
| pcDNA3.1 | Invitrogene | V79020 |
| Software and algorithms | ||
| Prism | GraphPad Software | N/A |
| Microsoft Excel | Microsoft | N/A |
| ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| TargetScore | Wang et al. (2021). https://doi.org/10.1101/2021.04.14.439861 | https://hub.docker.com/repository/docker/cannin/targetscore |
| rtracklayer 1.52.0 | Lawrence et al. (2009). | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html |
| Gviz 1.28.3 | Hahne and Ivanek (2016) | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| FACSDIV8.1 software | MDACC flow cytometry core facility | BD Biosciences (https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software) |