

ABSTRACT
Introduction
Pathogenic variants in PLOD3, encoding lysyl hydroxylase‐3 (LH3), can cause a hereditary connective tissue disorder that has rarely been reported. It is a multi‐system disease, presenting with craniofacial dysmorphisms, skeletal and eye manifestations, sensorineural hearing loss, and variable skin manifestations. Severe central nervous system involvement has not been reported.
Case presentation
A 10‐month‐old girl was admitted with development delay and clustered epileptic spasms. Hypertelorism, an upturned nose, and low‐set ears were noted in physical examination. Cerebral magnetic resonance imaging showed multiple intracranial malacias and bleeding foci, extensive abnormal signals in the white matter, and obvious brain atrophy, which was consistent with cerebral small vessel disease (SVD). Electroencephalography suggested hypsarrhythmia. The vertebrae were flattened. The distal end of the metacarpal bone in the left hand was irregular. She was diagnosed with West syndrome. Whole‐exome sequencing revealed a novel homozygous variant of c.1216_1218delCTC (p.L406del) in PLOD3, which was found to be inherited from her heterozygous parents.
Conclusion
We report a patient with pathogenic PLOD3 mutation who presented with cerebral SVD. This report expands the phenotypic spectrum of LH3 deficiency.
Keywords: PLOD3, Small vessel disease, West syndrome
A patient with homozygous PLOD3 mutations presented with cerebral small vessel disease, who expands the phenotypic spectrum of lysyl hydroxylase‐3 deficiency.

INTRODUCTION
Lysyl hydroxylase‐3 (LH3) encoded by PLOD3 is an enzyme involved in the post‐translational modification of collagens. 1 LH3 also has collagen galactosyltransferase and glucosyltransferase activities. Pathogenic variants in PLOD3 have been associated with an autosomal recessive connective tissue disorder (CTD), which was first described in 2008 (MIM: 612394). 2 In 2019, Ewans et al. 3 reported another pedigree and summarized the clinical features of PLOD3‐related CTD. It is a multi‐system disease, presenting with craniofacial dysmorphisms, skeletal and eye manifestations, sensorineural hearing loss, and variable skin manifestations. Some patients show artery dissection or rupture involving large and medium‐sized vessels. 3 Severe central nervous system involvement has not been reported before. In this study, we report a patient with homozygous PLOD3 mutation who presented with West syndrome. Her cerebral magnetic resonance imaging (MRI) indicated cerebral small vessel disease (SVD). This report expands the phenotypic spectrum of LH3 deficiency.
CASE REPORT
A 10‐month‐old girl was admitted to our hospital for developmental delay and recurrent seizures. She could not sit unaided, pursue, or grip objects on admission. She was the second child of a non‐consanguineous family. She was born at full term in good condition, with a birth weight of 2.9 kg. Her brother was three years old and was healthy. Postnatal development was backward. At 3 months of age, she visited a local hospital due to psychomotor retardation. At that time, cerebral MRI indicated multiple abnormal signals in the bilateral thalamus, basal ganglia, and corona radiata. White matter volume was decreased in the left cerebral hemisphere. The left lateral ventricle was slightly dilated. At 8 months of age, she presented with clustered epileptic spasms. In physical examination, her height was 73 cm (25th–50th percentile), weight was 9.6 kg (50th percentile), and head circumference was 42 cm (3rd percentile). Dysmorphic features included hypertelorism, an upturned nose, and low‐set ears (Figure 1A). The skin was normal without bullae. The limbs could be lifted off the bed with normal muscle tone. The left limbs were more flexible than the right limbs. Cerebral MRI at this time showed multiple intracranial malacias and bleeding foci, extensive abnormal signals in the white matter, and obvious brain atrophy (Figure 1B–F). No abnormality was observed in the magnetic resonance angiography and magnetic resonance venography. Cranial CT revealed calcification in the bilateral anterior ventricle (Figure 1G). The imaging features of the patient were consistent with the diagnosis of cerebral SVD. Electroencephalography (EEG) suggested hypsarrhythmia. Auditory evoked potentials were normal. Visual evoked potentials showed moderate to a severe block of the binoculus. Myopia was detected, which was measured at −5.75D (right) and −4.50D (left). No cataracts or fundus abnormalities were observed. Metabolic investigations, including amino acids, organic acids, and acylcarnitine, were negative. Serum TORCH‐IgM was negative. Plasma DNA for cytomegalovirus was also negative. The patient was diagnosed with West syndrome based on infantile‐onset, clustered epileptic spasms, delayed development, and hypsarrhythmia on EEG. Adrenocorticotrophic hormone (2 U·kg–1·day–1) was administered for two consecutive weeks, but seizure frequency did not change. Multiple antiepileptic drugs (AEDs) were added, including topiramate (maximum dose 7 mg·kg–1·day–1), sodium valproate (32 mg·kg–1·day–1), nitrazepam (0.2 mg·kg–1·day–1), and vigabatrin (150 mg·kg–1·day–1). The seizure frequency was reduced by no more than 50%. The patient was discharged and adjustment of AED dosage continued outside the hospital.
FIGURE 1.
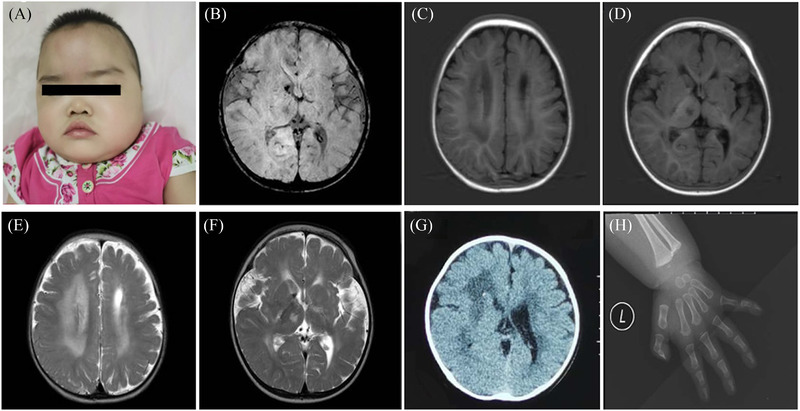
Clinical features and images of the patient with PLOD3 mutation. (A) The patient showed hypertelorism, an upturned nose, and low‐set ears. (B–F) Cerebral magnetic resonance imaging (MRI) of the patient revealed abnormal signals in the bilateral thalamus, basal ganglia, and corona radiata, with multiple intracranial malacias and bleeding foci. White matter volume was decreased in the left cerebral hemisphere. (G) Cranial CT revealed calcification in the anterior ventricle. (H) X‐ray of the left hand showed the distal end of the metacarpal bone was irregular.
Whole‐exome sequencing data from the patient, her brother, and her parents were analyzed using a trio‐based analysis. The patient carried a homozygous variant of c.1216_1218delCTC (p.L406del) in PLOD3, which was inherited from her heterozygous parents. Her brother also carried the heterozygous variant (Figure 2A,C). The variant has never been reported with minor allelic frequency <0.01. The Leu406 residue is extremely conserved in various species. In order to further clarify the pathogenicity of this variant, we compared LH3 synthesized by fibroblasts established from the skin of the patient and two healthy children with normal mental and motor development after we got the approval from the Ethics Committee of Beijing Children's Hospital (2022‐E‐081‐R). Total cellular protein from dermal fibroblasts was collected and subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Subsequently, proteins were transferred to a nitrocellulose membrane and incubated with a rabbit polyclonal PLOD3 antibody (Proteintech Group, Manchester, UK) and glyceraldehyde‐3‐phosphate dehydrogenase antibody (Proteintech Group) respectively. Western blotting showed markedly reduced levels of LH3 in the patient (Figure 2B). Then the patient was followed up for 2 months. We found that the patient also had bone abnormalities. The vertebrae were flattened, and the lower and upper edges were slightly concave. The distal end of the metacarpal bone in the left hand was irregular (Figure 1H). No abnormalities were found on coronary and macrovascular ultrasounds. Based on these findings, this variant was considered to be pathogenic. Unfortunately, urine pyridinoline compounds level and LH3 activity were not measured because of condition limitations, which could further clarify the pathogenicity of the mutation. The last follow‐up was at 1 year and 2 months of age. The patient still had epileptic spasms with no improvement in cognitive and motor skills. We suggested the parents try a ketogenic diet.
FIGURE 2.
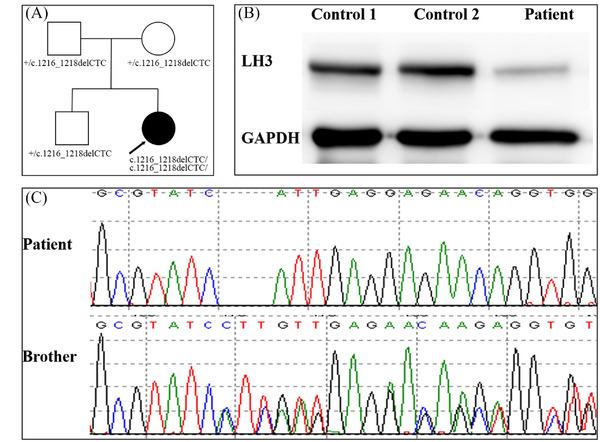
Family pedigree and effect of the mutation on LH3 expression. (A) Family pedigree. The arrow indicates the proband. (B) LH3 protein level was analyzed by western blotting analysis from fibroblasts cells of the patient and the two healthy controls. (C) Sanger sequencing shows the c.1216_1218delCTC homozygous variant in PLOD3 in the patient and the heterozygous variant in her brother.
DISCUSSION
Pathogenic variants in PLOD3 can cause hereditary CTD. Nine patients with PLOD3 mutations have been reported in the literature. 2 , 3 , 4 , 5 These patients all had multi‐system involvement. Almost all the joints showed mixed hypermobility and contracture. Seven of the patients presented craniofacial dysmorphisms including prominent eyes, malar or midface hypoplasia, upturned nose, and low‐set ears. Six patients had sensorineural deafness. Five patients had ocular disorders such as early cataracts, myopia, flat retinas, and optically empty vitreous. Skin blistering was recorded in two patients. Two patients had large and medium‐sized vessel involvement, one with coronary artery dissection, and the other with spontaneous cerebral arterial hemorrhage and right popliteal aneurysm. Our patient had craniofacial dysmorphisms and skeletal and eye manifestations, which were similar to the reported cases. However, our patient presented with West syndrome, and her cerebral MRI revealed small vessel involvement. Epileptic encephalopathy and cerebral SVD have not been reported associated with PLOD3 mutations. So this study expands the phenotypic spectrum of LH3 deficiency.
The term cerebral SVD encompasses all pathological processes that affect the small vessels of the brain, including small arteries and arterioles, but also capillaries and small veins. 6 Patients with cerebral SVD present with stroke, dementia, gait deterioration, and other symptoms. MRI often shows confluent, bilateral, and symmetrical cerebral white matter lesions, lacunes of presumed vascular origin, cerebral microbleeds, dilated perivascular spaces, and total cerebral atrophy. 7 Because brain biopsies are invasive tests, we usually make a preliminary diagnosis of cerebral SVD according to brain MRI changes. Our patient's MRIs showed confluent white matter lesions, intracranial malacia, and bleeding foci, which were in accordance with the features of cerebral SVD. A group of inherited cerebral SVDs has been reported. 8 , 9 However, except for COL4A1 and COL4A2‐related diseases, other monogenic SVDs are not present in infants. COL4A1 and COL4A2‐related diseases are systemic diseases, including a broad spectrum of central nervous system lesions: periventricular leukoencephalopathy, porencephaly, and cerebral calcification; lesions of the eyes causing cataracts, retinal arterial tortuosity, and strabismus; lesions of the kidneys leading to renal cysts, nephrosis, and haematuria; lesions of the heart causing arrhythmia; and lesions of the skeletal muscles causing dystrophic changes, weakness, and myoglobinuria. 10 Compared to PLOD3‐related diseases, both have a central nervous system and eye involvement. However, visceral involvement, such as the heart and kidneys, was not reported in PLOD3‐related disease.
The relationship between phenotype and genotype of the PLOD3‐related disease was not clear now. However, there may be a correlation between the phenotype and protein structure. 5 The LH3 protein contains three domains (GT domain, AC domain, and LH domain). 1 The individuals with severe skin disease all had at least one PLOD3 variant in the LH domain. 2 , 4 The variant of our patient was in the AC domain, which was different from the previous cases. This may be the reason for the special phenotype of this patient.
In clinical practice, patients similar to our case are often misdiagnosed with perinatal brain injury or congenital viral infection. We can distinguish it from perinatal brain injury with a detailed perinatal history. Congenital viral infection is often associated with jaundice, liver and spleen enlargement, thrombocytopenia, and retinopathy. White matter lesions are often patchy and asymmetric. Some present with cortical developmental malformations. 11 In laboratory examinations, pathogen tests are often positive. These characteristics can help differentiate diagnosis.
Here, we report a patient with a pathogenic PLOD3 variant who presented with cerebral SVD. This report expands the phenotypic spectrum of LH3 deficiency. It also reminds us that if the MRI of an infant shows certain characteristics, including intracranial confluent and symmetric white matter lesions, microbleeding, encephalomalacia foci, and porencephaly, monogenic SVDs should be considered after excluding secondary factors.
CONSENT FOR PUBLICATION
Consent was obtained from the patient's guardian.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Zhou J, Feng W, Zhuo X, Lu W, Wang J, Fang F, et al. Cerebral small vessel disease caused by PLOD3 mutation: Expanding the phenotypic spectrum of lysyl hydroxylase‐3 deficiency. Pediatr Investig. 2022;6:219–223. 10.1002/ped4.12328
REFERENCES
- 1. Myllylä R, Wang C, Heikkinen J, Juffer A, Lampela O, Risteli M, et al. Expanding the lysyl hydroxylase toolbox: new insights into the localization and activities of lysyl hydroxylase 3 (LH3). J Cell Physiol. 2007;212:323‐329. DOI: 10.1002/jcp.21036 [DOI] [PubMed] [Google Scholar]
- 2. Salo AM, Cox H, Farndon P, Moss C, Grindulis H, Risteli M, et al. A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene. Am J Hum Genet. 2008;83:495‐503. DOI: 10.1016/j.ajhg.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maddirevula S, Alzahrani F, Al‐Owain M, Al Muhaizea MA, Kayyali HR, AlHashem A, et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet Med. 2019;21:736‐742. DOI: 10.1038/s41436-018-0138-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Pajouhanfar S, Baghdadi T, et al. Mutations in PLOD3, encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa‐like blistering phenotype with abnormal anchoring fibrils and type VII collagen deficiency. Matrix Biol. 2019;81:91‐106. DOI: 10.1016/j.matbio.2018.11.006 [DOI] [PubMed] [Google Scholar]
- 5. Ewans LJ, Colley A, Gaston‐Massuet C, Gualtieri A, Cowley MJ, McCabe MJ, et al. Pathogenic variants in PLOD3 result in a Stickler syndrome‐like connective tissue disorder with vascular complications. J Med Genet. 2019;56:629‐638. DOI: 10.1136/jmedgenet-2019-106019 [DOI] [PubMed] [Google Scholar]
- 6. Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18:684‐696. DOI: 10.1016/S1474-4422(19)30079-1 [DOI] [PubMed] [Google Scholar]
- 7. Sargurupremraj M, Suzuki H, Jian X, Sarnowski C, Evans TE, Bis JC, et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat Commun. 2020;11:6285. DOI: 10.1038/s41467-020-19111-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi JC. Genetics of cerebral small vessel disease. J Stroke. 2015;17:7‐16. DOI: 10.5853/jos.2015.17.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marini S, Anderson CD, Rosand J. Genetics of cerebral small vessel disease. Stroke. 2020;51:12‐20. DOI: 10.1161/STROKEAHA.119.024151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meuwissen ME, Halley DJ, Smit LS, Lequin MH, Cobben JM, de Coo R, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med. 2015;17:843‐853. DOI: 10.1038/gim.2014.210 [DOI] [PubMed] [Google Scholar]
- 11. Livingston JH, Stivaros S, Warren D, Crow YJ. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol. 2014;56:612‐626. DOI: 10.1111/dmcn.12359 [DOI] [PubMed] [Google Scholar]