

Abstract
The tricarboxylic acid (TCA) cycle is the epicenter of cellular aerobic metabolism. TCA cycle intermediates facilitate energy production and provide anabolic precursors, but also function as intra- and extracellular metabolic signals regulating pleiotropic biological processes. Despite the importance of circulating TCA cycle metabolites as signaling molecules, the source of circulating TCA cycle intermediates remains uncertain. We observe that in mice, the concentration of TCA cycle intermediates in the portal blood exceeds that in tail blood indicating that the gut is a major contributor to circulating TCA cycle metabolites. With a focus on succinate as a representative of TCA cycle intermediate with signaling activities and using a combination of gut microbiota depletion mouse models and isotopomer tracing, we demonstrate that intestinal microbiota are not major contributors to circulating succinate. Moreover, we demonstrate that endogenous succinate production is markedly higher than intestinal succinate absorption in normal physiological conditions. Altogether, these results indicate that endogenous succinate production within the intestinal tissue is a major physiological source of circulating succinate. These results provide a foundation for investigation into the role of intestine in regulating circulating TCA cycle metabolites and their potential signaling effects in health and disease.
Introduction
Animals rely on aerobic metabolism to produce chemical energy for energy intensive physiological functions like locomotion and neuronal activity. Aerobic metabolism occurs in mitochondria where acetyl-CoA, the two-carbon metabolite derived from the catabolism of carbohydrates, proteins, or lipids, is oxidized to carbon dioxide and water producing ATP via the tricarboxylic acid cycle (TCA cycle or Krebs cycle). The TCA cycle is the epicenter of cellular aerobic metabolism and TCA cycle intermediates play essential roles in this process to facilitate cellular and organismal energy homeostasis.
While the role of TCA cycle intermediates in facilitating energy production is well defined, TCA cycle metabolites can serve other roles outside of the mitochondria. For example, TCA cycle metabolites including citrate and oxaloacetate can be exported from the mitochondria and used in synthesis of glucose and lipids (1). Additionally, the TCA cycle metabolites succinate and alpha-ketoglutarate function as natural ligands for cell surface G-protein coupled receptors (GPCRs) indicating that TCA cycle derived metabolites may serve as metabolic signals (2). Succinate signals through the succinate receptor 1 (Sucnr1), a GPCR that regulates pleiotropic biological activities including urine filtration (3), immune responses (4,5), thermogenesis (6), and liver damage and fibrosis (7,8). This succinate-Sucnr1 signaling system suggests that circulating TCA cycle intermediates have the potential to regulate interorgan communication in coordination of fuel and immune homeostasis.
Although TCA cycle intermediates are produced within the mitochondria of all cells, TCA cycle intermediates are readily detected in circulation and the major sources of circulating TCA cycle intermediates in physiological conditions are less clear. Some, but not all studies have suggested that the gut microbiota is a source of circulating TCA cycle intermediates (9,10). A study in humans with obesity and type 2 diabetes observed that circulating succinate levels are positively associated with enrichment for specific succinate-generating microbial strains and suggested that gut microbiota contribute to circulating succinate (9). In contrast, colonization of mice with succinate producing bacterial species increased succinate levels in cecal matter, but did not increase portal succinate levels (10).
These studies raise questions regarding the ability of the intestine to absorb microbiome-derived and/or dietary succinate. This is of importance as chronic supplementation of succinate in the diet or drinking water impacts metabolic homeostasis in animal models, but little evidence has been provided that such supplementation studies impact circulating succinate levels (6,10). Thus, it remains unclear whether the diet or microbiota are significant contributors to circulating levels of succinate and other TCA cycle metabolites.
In this study, we demonstrate that endogenous synthesis within the gut is a major source of circulating succinate and far exceeds the dietary contribution in normal physiological circumstances. Furthermore, using both in vivo and in vitro models, we demonstrate that intestinal tissue, and not microbiota, are the predominant sources of gut-derived succinate. These results advance our knowledge of the source of circulating succinate, interorgan metabolite communication, and will also provide guidance in investigating the physiological and/or pathophysiological functions of circulating TCA metabolite succinate in health and disease.
Methods
Materials
Sodium succinate, cysteine, norvaline and disodium succinate-2,2,3,3-D4 (D4-succinate) were purchased from Sigma-Aldrich (14170, 30089, 53721, and 293075 respectively). D-Fructose was purchased from VWR international (VWRV0226). U-C13-fructose and 2,3-C13-sodium succinate was purchased from Cambridge Isotope Labs (CLM-1553-PK, and CLM-9371-PK respectively). Dulbecco’s Modified Eagle Medium (DMEM), Advanced DMEM/F12, and PBS were purchased from Thermo Fisher (11965118, 12634010, and 10010049 respectively). Matrigel was purchased from Corning (356231).
Animals
Animal studies were conducted according to protocols approved by Duke University’s Institutional Animal Care and Use Committee (A147-19-07, A204-18-08, and A125-20-06). Wild-type conventionally reared C3H/HeJ and C57BL/6N animals were purchased from Jackson lab (000659, and 005304 respectively). All conventionally reared animals were fed a standard chow diet (LabDiet 5053, irradiated) except as otherwise noted. Conventionally reared animals were maintained in Duke’s specific pathogen free environment with corn cob bedding. Germ-free mice were obtained from the Duke Gnotobiotic Core. These mice were originally obtained from Taconic (C57BL/6NTac). All germ-free animals were fed autoclaved Envigo 2020SX diet. Germ-free animals were maintained in a flexible film isolator (Class Biologically Clean, Madison, WI) with Alpha Dri bedding. All animals were maintained at constant temperature (~ 22°C) on a 12-hour light-dark cycle (8 AM to 8 PM). Within experiments, mice were divided randomly into different treatment groups. All experimental animals used in these studies were male, wild-type C3H/HeJ mice except for the mice used in the germ-free and antibiotics induced gut microbiota depletion experiments and their controls as noted above. Except where otherwise noted with respect to specific experiments, specified treatments or euthanasia were performed at 2 PM after 5 hours food removal (9 AM to 2 PM).
Antibiotics induced gut microbiota depletion
The antibiotics-induced gut microbiota depletion protocol was adapted from a previous study (11). Specifically, mice were orally gavaged with doses of antibiotic cocktails twice daily for 5 days. The antibiotic cocktail contains 4 different antibiotics to eradicate the majority of gut microbiota: Vancomycin 50 mg/kg, Neomycin 100 mg/kg, Metronidazole 100 mg/kg, and Ampicillin 150 mg/kg. The antibiotic cocktails were administrated in a 0.2 mL volume per mouse at 7AM and 7PM. Fresh fecal contents were harvested before and after 5 days’ antibiotic treatment. The isolated fecal contents were mixed with 1 mL 0.05% cysteine supplemented PBS. A series of fecal content dilutions were generated by conducting six rounds of 1:10 dilution using the original solution from each mouse’s fecal sample. After that, a series of fecal content dilutions from the mice were plated on blood agar plates and cultured under anaerobic conditions for 48 hours. The bacterial colonies formation from diluted fecal contents serves as a surrogate indicator of gut microbiota content.
Gas chromatography-mass spectrometry (GC/MS)
Blood samples:
Plasma was prepared from collected blood by centrifugation at 2,000 g, 4°C for 15 minutes. 0.1 mmol norvaline or 0.2 mM 2-Deoxy-D-glucose (2DG) was added to 10 μL plasma samples as an internal standard for the relative quantification of TCA cycle intermediates or fructose respectively. 0.5 nmol D4-succinate was added to 10 μL plasma samples as an internal standard for the absolute quantification of succinate. To 10 μL plasma samples, 400 μL MeOH, 400 μL ddH2O, and 400 uL chloroform were added sequentially and briefly vortexed after each addition. After centrifugation at 12,000 g for 15 minutes, supernatants were collected and dried under nitrogen gas.
Culture media:
Culture media samples from intestinal organoids and cecal/fecal contents following fructose treatment were obtained and centrifuged at 12,000 g to remove cell debris. 0.5 mmol norvaline was added to 100 μL media samples as an internal standard for the analysis of TCA cycle intermediates. 400 μL MeOH, 400 μL ddH2O, and 400 μL chloroform were added sequentially to the samples and briefly vortexed after each addition. After centrifugation at 12,000 g for 15 minutes, supernatants were collected and dried under nitrogen gas.
Tissue and food samples:
Tissue samples, and food samples were powdered under liquid N2. 400 μL ice-cold 1:1 MeOH and ddH2O were added to 10 mg powdered samples. 0.1 mmol norvaline was added in methanol as an internal standard for enrichment analysis. 10 nmol D4-succinate was added in methanol as an internal standard for succinate quantification in food. These samples were homogenized (TissueLyser) for 2 minutes at 30 H and centrifuged at 12,000 g, 4°C for 20 minutes. 50 μL supernatants were combined with another 350 μL ice-cold 1:1 MeOH and ddH2O prior to adding 400 μL chloroform per sample and vortexed for 30 seconds. Samples were centrifuged at 12,000 g for 15 minutes and supernatants were collected and dried completely under nitrogen evaporator.
Relative quantification and enrichment measurements of TCA cycle intermediates:
Dried residues were derivatized with methoxylamine hydrochloride and tertbutyldimetheylchloros (TBDMS) sequentially as previously described [3]. Specifically, 25 μL of methoxylamine hydrochloride (2% (w/v) in pyridine) was added to the dried residues and incubated for 90 minutes at 40°C before the addition of 35 μL of TBDMS and incubation for 30 minutes at 60°C. The samples were then centrifuged for 2 minutes at 12,000 g and the supernatants of derivatized samples were transferred to GC vials for further analysis. GC/MS analysis was conducted as previously described using an Agilent 7890B GC system and Agilent 5977A Mass Spectrometer [3]. Specifically, 1 μL of the derivatized sample was injected into the GC column. GC temperature gradient started at 80°C for 2 minutes, increased to 280°C at the speed of 7°C per minute, and held at 280°C for until the completion of a run time of 40 minutes. Ionization was conducted by electron impact (EI) at 70 eV with Helium flow at 1 mL/min. Temperatures of the source, the MS quadropole, the interface, and the inlet were maintained at 230°C, 150°C, 280°C, and 250°C respectively. Mass spectra were recorded in mass scan mode from m/z 50 to 700.
Absolute quantification of succinate:
Dried sample residues were derivatized with methoxylamine hydrochloride and trimethylsilyl (TMS) sequentially. Specifically, 25 μL of methoxylamine hydrochloride (2% (w/v) in pyridine) was added to the dried residues and incubated for 90 minutes at 40°C before the addition of 35 μL of TMS and incubation for 30 minutes at 60°C. The samples were then centrifuged for 2 minutes at 12,000 g and the supernatants of derivatized samples were transferred to GC vials for further analysis. GC/MS analysis was conducted using Agilent 7890B GC system and Agilent 5977A Mass Spectrometer. To be specific, 1 ul of the derivatized sample was injected into the GC column. GC temperature gradient start at 80°C for 2 minutes, increased to 280°C at the speed of 7°C per minute, and held at 280°C for until the completion of a run time of 40 minutes. Ionization was conducted EI at 70 eV with Helium flow at 1 mL/min. Temperatures of the source, the MS quadropole, the interface, and the inlet were maintained at 230°C, 150°C, 280°C, and 250°C respectively. Mass spectra were recorded in mass scan mode from m/z 50 to 700.
Relative quantification and enrichment measurements of fructose:
the dried residues were derivatized with methoxylamine hydrochloride and propionic anhydride sequentially adapted from a previously described method (12). 25 μL of methoxylamine hydrochloride (2% (w/v) in pyridine) was added to the dried residues and incubated for 60 minutes at 70°C. After centrifugation for 2 minutes at 12,000 g, 50 μL propionic anhydride was added and incubated for 30 minutes at 60°C prior to another round of drying under nitrogen. The dried residues were resuspended with 55ul pure ethyl acetate and transferred to GC vials for analysis. GC/MS analysis was conducted using the Agilent 7890B GC system and Agilent 5977A Mass Spectrometer. Specifically, 1 μL of the derivatized sample was injected into the GC column. GC temperature gradient started at 90°C, increased to 260°C at the speed of 9°C per minute and further increased to 290 °C at the speed of 30°C per minute. Then temperature was held at 290°C for 5 minutes with a run time of 24.9 minutes. Ionization was conducted by EI at 70 eV with Helium flow at 1 mL/min. Temperatures of the source, the MS quad, the interface, and the inlet were maintained at 230°C, 150°C, 280°C, and 250°C, respectively. Mass spectra were recorded in mass scan mode from m/z 50 to 700.
Liquid chromatography with tandem mass spectrometry (LC-MS/MS) for quantification of butyrate
Plasma samples were prepared the same way as described above. A modified LC-MS/MS method was developed to analyze the absolute quantity of butyrate as previously reported (13). Specifically, 30 μl plasma samples were mixed with 30 μl 20 μM 2,2,3,3,4,4,4-2H7-butyrate (D7-butyrate, internal standard) before adding to 1 ml acetonitrile to remove protein by precipitation. After centrifuge for 20 minutes at 1,200 g, supernatant was collected and transferred to a new Eppendorf tube, then dried completely by nitrogen gas. The dried residue was resuspended in a mixture solution consists of 50 μl HPLC water, 20 μl 3-Nitrophenylhydrazine hydrochloride (EDC, 120 mM), and 20 μl (N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (3-NPH, 200 mM) for derivatization at 40 °C for 30 minutes. After centrifuge for 10 minutes at 1,200 g in room temperature, supernatant was transferred to an LC-MS/MS vial for butyrate analysis. LC-MS/MS was conducted using a Sciex QTRAP 6500+ MS connected with a Sciex AD UHPLC. Specifically, an Agilent C18 column (Pursuit XRs C18 150 × 2.0 mm, 5 μm) was employed to generate a flow rate at 0.4 ml/min for separation. Gradient methods were conducted within two mobile phases. Mobile phase A was composed of 98% H2O and 2% acetonitrile with 0.1% formic acid. On the other hand, mobile phase B was consisting of 98% acetonitrile and 2% H2O containing 0.1% formic acid. At first, gradient started with 98% phase A and 2% phase B for 30 seconds. After 7.5 minutes, phase B was increased to 90% while phase A was decreased to 10% at 8 minutes. After maintaining at 90% phase B and 10% phase A for another 4.5 minutes, phase B was returned to initial 2% within 0.5 minutes. Finally, the column was re-equilibrated for 9 minutes with 98% phase A and 2% phase B before the next injection. The injection volume was 3 μl. MRM in negative mode was used for butyrate assay. The MS/MS parameters were set as following: curtain gas: 35 psi, source temperature: 600 °C, Gas 1: 55 psi, Gas 2: 55 psi, CAD: 10, Ion spray voltage: −4500 V, EP: −10 V, and CXP: −14.
Stable Isotope Analysis
Isotopologues that containing 0, 1, to n heavy atom(s) in a molecule were referred as M+0, M+1, to M+n respectively. The retention times of individual metabolites and their isotopomers are described in Supplementary Table 1. Isotope enrichment and labeling analysis in this study were corrected for natural isotope distribution as previously described (14,15). Specifically, the stable isotope enrichment of each metabolite in plasma, tissue, organoids, and fecal/cecal content was corrected based on the natural isotope distribution measured in the same type of samples treated without labeled molecule sources. For example, the natural isotope distribution matrix of individually measured metabolite in the blood samples was experimentally determined by assaying and averaging the blood samples from animals who had not received any treatments involving isotopically labeled sources. Similarly, the natural isotope distribution matrix of individually measured metabolites in the organoids samples was determined by assaying and averaging the blood samples from organoids that were incubated with PBS without exposure to isotopically labeled sources.
Relative quantification of the ratio endogenous production to intestinal absorption of metabolites
The crude relative quantification model used here is based on standard isotope dilution methodologies. Specifically, mice were gavaged with 1:1 isotope-labeled and unlabeled metabolites. Animals were then sacrificed over a 2-hour period with the following time points: 0 (no gavage), 10, 30, 60, and 120 minutes for the harvest of portal blood. The relative quantity and labeling pattern of metabolites in the portal blood samples were measured by GC/MS.
Metabolites in the portal blood are a mixture of metabolites transported into portal circulation following absorption as well as recirculation of metabolites from systemic circulation include metabolites derived from endogenous production which may occur in the intestine or other tissues. Thus, the labeling of absorbed isotope-labeled metabolites is diluted by the endogenously produced metabolites and recirculating metabolites which can be assessed in portal blood samples. The endogenously produced metabolites are assumed to be exclusively unlabeled because the natural isotopomer labeling of their substrates is negligible (16). One assumption made by in this model is that the redistribution of intestinal absorbed metabolites is limited during our acute oral gavage experiment. Ignoring redistribution may overestimate the absorption rate since all labeled metabolites were assumed to be from absorption. We also assumed that the metabolism of absorbed nutrients in intestinal tissues follows first-order kinetics, which is approximately true for most drugs and nutrients (17). Thus, the fraction of transported labeled metabolites following absorption will be the same as transported unlabeled metabolite from endogenous production within the intestine: (EA: Elimination of absorption; A: Absorption; EP: Elimination of production; P: Production). As we gavaged a bolus of metabolites at a 1:1 labeled to unlabeled ratio, the transported labeled metabolites are proportional to half of the total absorbed metabolite: (Tlabeled: Transported labeled metabolite). Transported unlabeled metabolites originate from both external absorption and endogenous production. Transported unlabeled metabolites are proportional to the sum of half trasnported absorption and transported intestinal production: (Tunlabeled: Transported unlabeled metabolites). Combining these equations, we crudely estimate the ratio of external absorption to endogenous production using the transported labeled and unlabeled metabolites amount: .
Average Carbon-13 Label Incorporation and Adjusted Carbon-13 Incorporation Rates
The average carbon-13 incorporation was calculated according to the equation as previously described (18). Specifically, nM+I refers to the number of C13 containing carbons in the isotopomer (M+i). For example, n = 0 for M+0, n = 1 for M+1, and n = 2 for M+2, … “A” refers to the corresponding percentage abundance of isotopomer. N refer to the total carbon number, which for glucose, N = 6.
In this study, we are comparing the carbon-13 incorporation from U-C13-fructose versus 2,3-C13-sodium succinate into glucose. Because 1 molecule of U-C13-fructose contains 6 molecules of C13 whereas 1 molecule of 2,3-C13-sodium succinate contains 2 molecules of C13, we further adjusted the carbon-13 incorporation rate by dividing by 6 for fructose and 2 for succinate respectively to calculate the adjusted carbon-13 incorporation rate.
Intestinal Organoid Culture
Intestinal crypts were isolated from 8-week-old male mice and used to grow enterocyte-enriched intestinal organoids as previously reported using ENR-growth media [Advanced DMEM/F12 supplemented with 50 ng/mL murine EGF, 1 μM N-acetylcysteine, 100 nM GSK269962, 1XB27, 2% Noggin conditional media, and 2% R-spondin-1 conditional media] (19). Briefly, small intestines were removed, incised longitudinally, and cut into 2- to 5-mm fragments before being washed with ice-cold PBS 3 times. Washed intestinal fragments were then incubated with ice-cold PBS containing 10 mM EDTA for 30 minutes on a rocker at 4 °C. Tissue was removed from solution containing EDTA and placed in ice-cold PBS and mechanically shaken by hand for 30 seconds. Crypts were enriched by filtering through a 70-uM cell strainer to remove the villi. Enriched crypts were resuspended in ENR-growth media after centrifugation at 300 g for 5 minutes. After manually counting, crypts were further diluted and embedded in Matrigel (3:1) and seeded on 12-well plates at a density of approximately 900 crypts per well and incubated with ENR-growth media. After 5 days of proliferation and differentiation, intestinal organoids were treated with either 10 mM glucose or 10 mM U-C13-fructose for overnight (5 PM to 9AM) at 37 °C. Immediately after incubation with glucose or fructose, the media were harvested for measurement of succinate labeling using GC/MS as described above.
Fecal/Cecal Content Culture
Fecal and cecal contents were freshly harvested from the anus and cecum respectively from 8-week-old male mice. For aerobic culturing, the cecal and fecal contents were incubated with or without 10 mM U-C13 fructose in 0.05% cysteine supplemented PBS and cultured in standard incubator for 30 minutes at 37 °C. For anaerobic culturing, tubes containing cecal and fecal contents were first transferred to an automated anaerobic chamber and then incubated with or without 10 mM U-C13 fructose in 0.05% cysteine supplemented PBS. After gentle mixing, the cecal and fecal contents were incubated at 37 °C within the anaerobic chamber for 30 minutes. Immediately after incubation, culture media from the fecal and cecal content samples were harvested, centrifuged at 12,000 g for 5 minutes and snap frozen for further GC/MS analysis as described above.
Statistics
All data are presented as mean ± SEM. Analysis of TCA levels between the tail and portal blood were conducted using a paired student’s T-test in GraphPad Prism. Comparisons of metabolite levels between time points following oral bolus gavage were analyzed using 1-way ANOVA and Tukey’s test. As appropriate, data were analyzed for statistical significance with GraphPad Prism using 2-way ANOVA and individual comparisons with a Tukey’s test. Statistical significance was assumed at P less than 0.05.
Data Availability:
The data that support the findings of this study are available in the supplementary material of this article or are openly available in figshare at the following URL: https://doi.org/10.6084/m9.figshare.19074176.v2.
Results
The gut contributes to circulating TCA cycle metabolites
The TCA cycle and its intermediates are essential for ATP production in mitochondria under aerobic conditions. As meal-derived nutrients are typically absorbed from the intestine and delivered to the liver prior to mixing with systemic circulation, we sampled blood from the portal vein and tail vein of mice to establish the baseline levels of the TCA cycle intermediates. We observed that relative levels of succinate, fumarate, and malate are ~ 9-fold higher in the portal blood when compared to tail blood in fasting mice (Figure 1A). Citrate levels were also higher in portal compared to tail blood, but only ~ 2-fold higher (Figure 1A). Conducting absolute quantification of succinate in the portal versus tail plasma using a labeled succinate internal standard confirmed the increase in portal blood demonstrating a succinate level of 150.4 ± 20.9 μM in portal blood compared to 51.9 ± 17.2 μM in tail blood (P < 0.001, Figure 1B). In agreement with this finding, the portal levels of succinate, malate, and citrate, were consistently higher in portal circulation compared to the tail circulation in ad-lib fed mice at both 8AM and 8PM (Figure 1C). Portal succinate levels were ~ 80 μM in the portal in contrast with ~ 40 μM in the tail circulation in ad-libitum fed mice (Figure 1D). Although the absolute levels varied with different fasted-fed conditions, the portal-peripheral gradient persisted regardless of either the fasted-fed condition or diurnal cycle indicating that circulating TCA cycle intermediates are gut-derived, and either absorbed from the intestinal lumen or generated from the intestinal tissue.
Figure 1. The gut is a contributor to circulating TCA cycle intermediates.
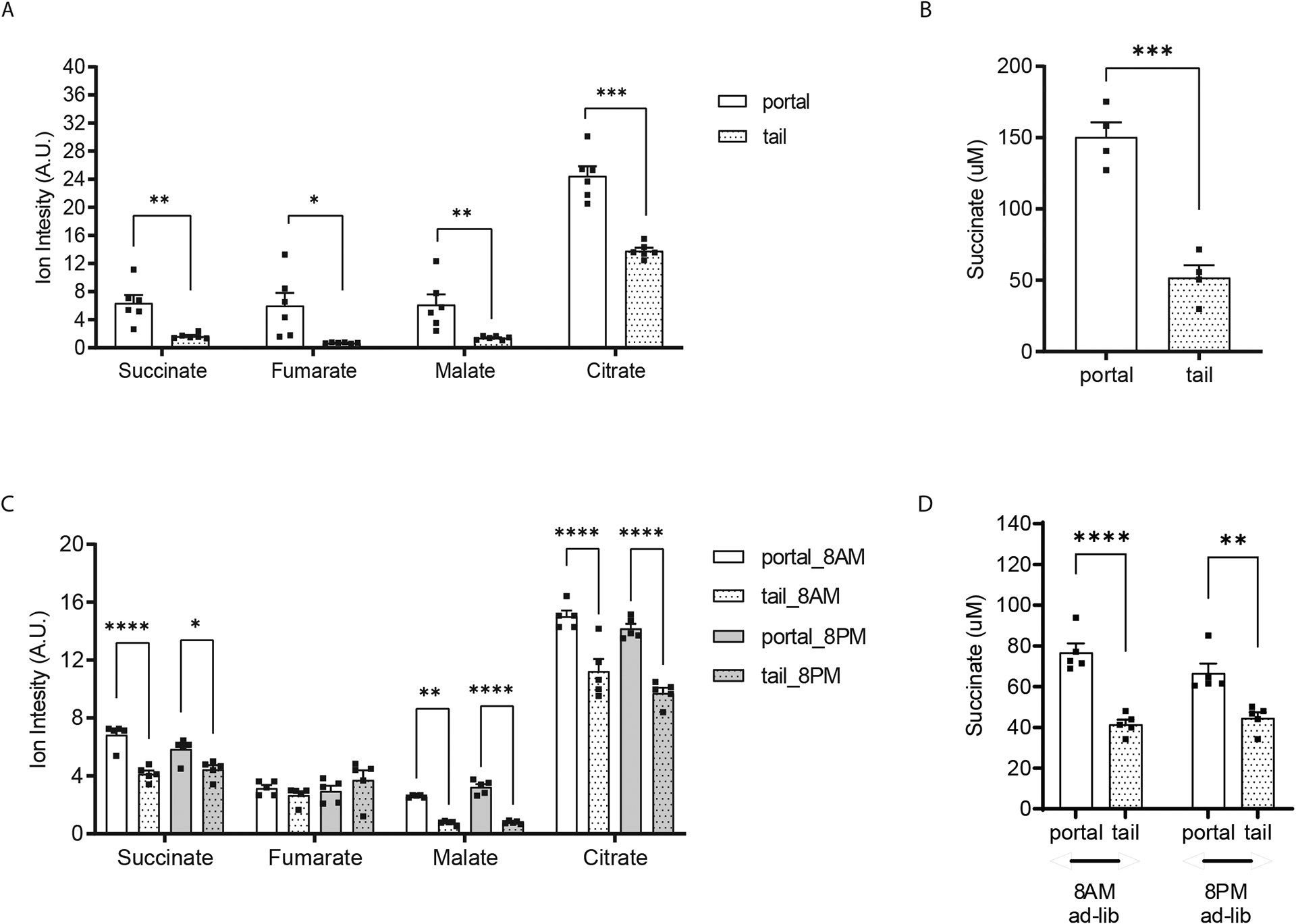
Relative quantification of TCA cycle metabolites (A) and absolute quantification of succinate (B) in portal versus tail plasma samples from 8-weeks old, male mice after 5 hours food removal. Relative quantification of TCA cycle metabolites (C) and absolute quantification of succinate (D) in portal versus tail plasma samples from ad-lib fed 8-weeks old, male mice on either 8AM or 8PM. Norvaline and D4-sodium succinate were used as internal standards for relative and absolute quantification measurement by GC/MS, respectively (n=6 per group in panel A and B; n=5 per group in panel C and D). * p<0.05; ** p<0.01; *** p<0.001. Analysis in A and B performed via paired student t-test. Analysis in panel C and D performed via two-way ANOVA Bonferroni analysis for post hoc comparisons.
To examine the contribution of gut carbohydrate metabolism to portal TCA cycle intermediates we orally gavaged mice with U-C13-fructose (0.48 g/kg) as fructose can be metabolized at high rates by both microbiota and enterocytes in the gut (20,21). The relative quantification and enrichment of TCA cycle intermediates in the portal blood were assessed by GC/MS at intervals for 2 hours following gavage. The rapid appearance of labeled carbons in portal TCA cycle intermediates supports the hypothesis that orally administered fructose can be readily metabolized into TCA cycle intermediates in gut (Figure 2A, B, C). While fructose-derived carbons were readily incorporated into portal TCA cycle intermediates, the quantity of the TCA cycle intermediates (succinate, malate, and citrate) in the portal vein remained relatively constant over this two-hour period (Figure 2D, E, F). The increase in isotope enrichment without changes in TCA cycle intermediate levels indicate that TCA cycle intermediates are constitutively secreted from the gut into circulation although the source of substrate for TCA cycle intermediate production is variable. As TCA cycle metabolites derived directly from U-C13-fructose will predominantly be M+2, we traced the percentage of enrichment of M+2 TCA metabolites at baseline, 10 minutes and 30 minutes after gavage in the portal versus tail circulation (Figure 2G–I). The enrichment is modestly, but consistently higher in the portal versus tail blood for all measured intermediates and timepoints. Additionally, the kinetics of M+2 TCA cycle metabolite enrichment are very similar in the portal and tail circulation (Figure 2G to I). Together, these data indicate that the labeled TCA cycle metabolites in the systemic circulation are predominantly derived from portal circulation and less likely to be derived from the other tissues like the liver.
Figure 2. Circulating levels of TCA cycle intermediates tend to remain relatively constant.
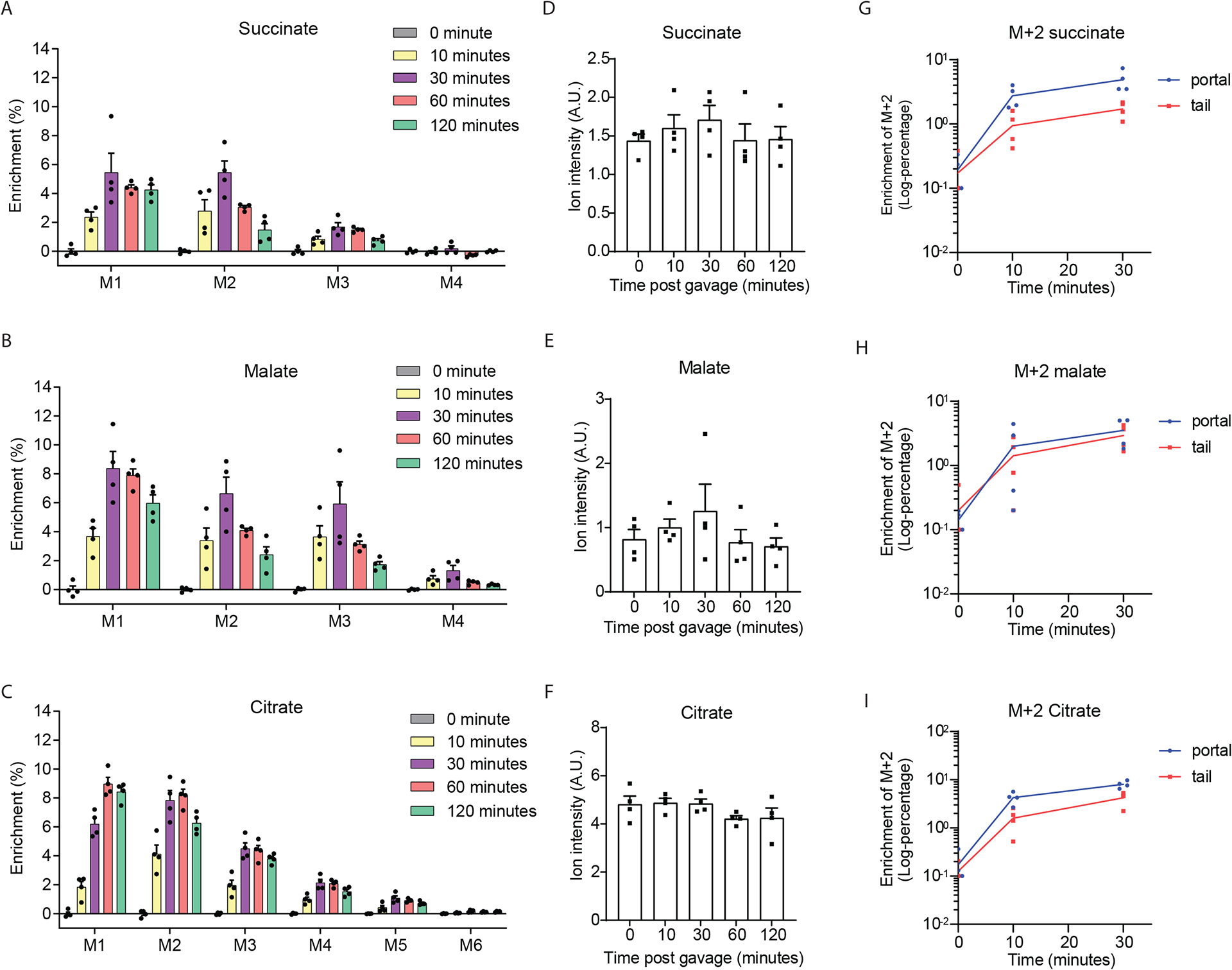
Enrichment (A, B, C) and relative quantification (D, E, F) of portal TCA cycle intermediates after oral-gavage in 8-weeks old male mice with U-C13-fructose (0.48 g/Kg body weight, n=4 per group). Enrichment of the M+2 TCA cycle intermediates (D, E, F) in the plasma from the portal vein versus tail vein from the same group. Time to log-transformed enrichment of the M+2 TCA cycle intermediates plots of the first 3 time points (D, E, F). Every number at each time points was added with 0.05 in panel D to F to avoid log-transform 0 which will lead to mathematic error. Data represent means ± SEM. Analysis performed via one-way ANOVA and Fisher’s LSD for post-hoc analysis comparing to baseline (0 minutes).
Microbiota are not essential to maintain circulating levels of TCA cycle metabolites
TCA cycle intermediates found in portal blood appear to be constitutively produced in the gut. The cellular origin of these TCA cycle intermediates is uncertain as both the intestinal tissue and microbiota residing in the gut have the capacity to produce them. Given interest in succinate as a metabolic signal (22), we focused on succinate as a representative TCA cycle intermediate in the remainder of this study. We measured the relative levels of portal TCA cycle intermediates and absolute levels of succinate in blood samples of conventionally reared mice to age- and strain-matched mice that were reared in a germ-free environment. Due to the fact that the germ-free mice need to be maintained in restricted germ-free insulators and regulated with special bedding and diet, the conventionally reared mice and germ-free mice were actually fed on different rodent chow diets, Lab Diet 5053 for conventionally reared mice and Envigo 2020SX for germ-free mice. Despite differences in amino acid and fiber content between the two diets, the succinate content is comparable (Figure 3A). Moreover, the circulating succinate levels were similar in conventionally reared wildtype male mice fed either the 5053 Diet or the Envigo diet for one week (Figure 3B). These data indicate that dietary differences were not a major determinant of circulating succinate levels in the conventionally reared versus germ-free mice. Butyrate is a well-established circulating metabolite produced largely by microbiota (23). As expected, portal butyrate levels are 143-fold higher in the portal blood of the conventionally reared mice compared to the germ-free mice (Figure 3C). In contrast, succinate levels were similar in the germ-free mice and conventionally reared mice in blood samples harvested from both portal vein and IVC (Figure 3D). Actually, levels of the majority of TCA cycle metabolites (succinate, fumarate and citrate) in the portal blood were similar, and portal malate levels were even higher in the germ-free mice than the conventionally reared mice (Figure 3E). These data indicate that intestinal microbiota are not necessary to maintain portal TCA cycle metabolite levels.
Figure 3. Intestinal microbiota are not a major source of circulating succinate.
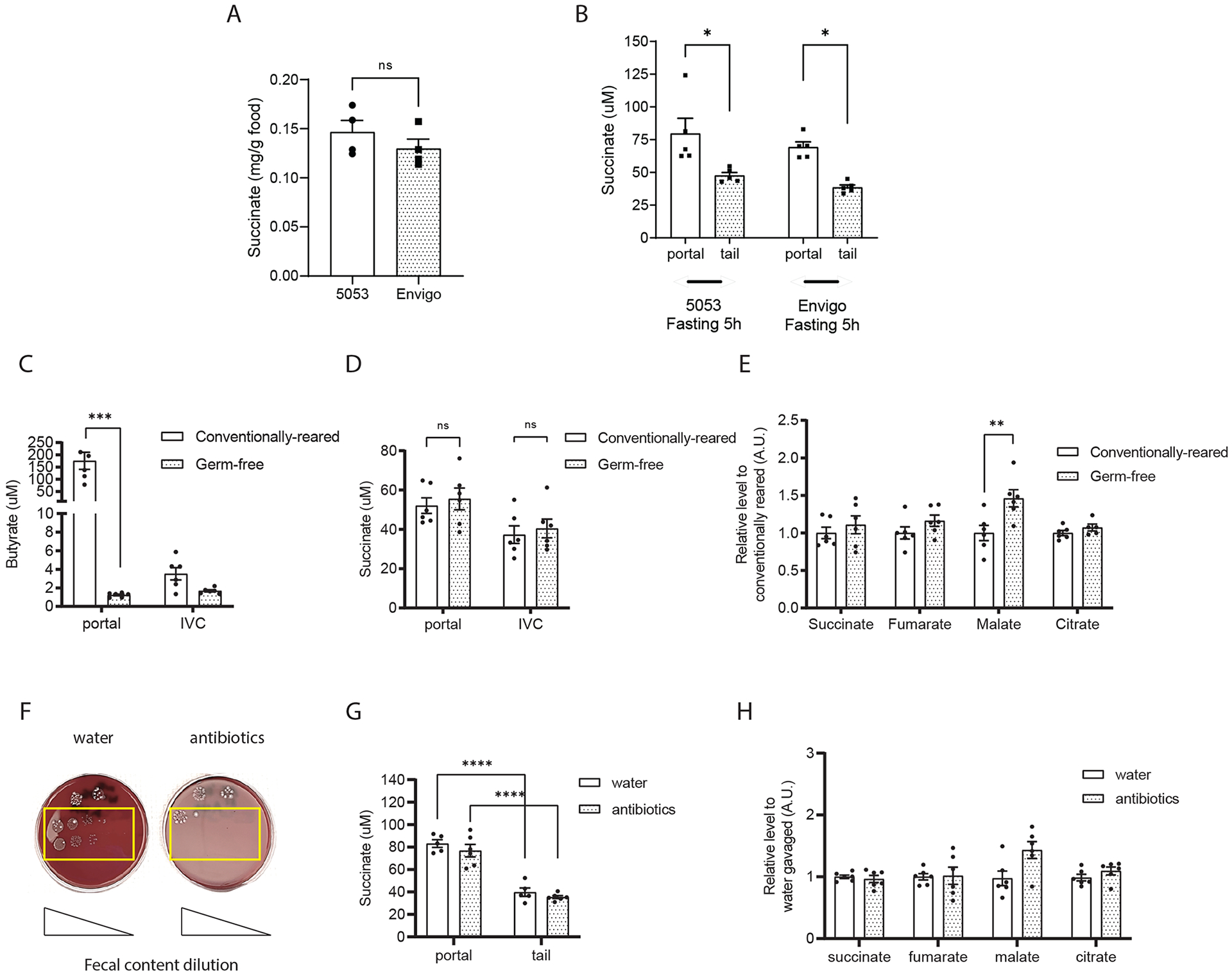
(A) Succinate content in normal chow diets: Lab Diet 5053 (conventionally reared mice) and Envigo 2020SX (germ-free mice). (B) Succinate levels in the plasma from portal and tail vein of wildtype 8-week male mice fed on either Lab Diet 5053 and Envigo 2020SX for a week. Absolute quantification of butyrate (C) and succinate (D) in plasma from the portal vein versus inferior vena cava of 12-week-old germ-free male mice versus their conventionally reared mice. Relative quantification of portal TCA cycle intermediates levels in the germ-free and conventionally reared mice (E). Colony formation on the blood agar from fecal content harvested from 8-weeks old, male mice after treated with either water or antibiotic cocktails for 5 days (F). Absolute levels of portal versus tail succinate levels (G) and relative levels of portal TCA cycle intermediates levels (H) in the water versus antibiotic cocktails treated mice. Butyrate levels were measured by LC-MS/MS using D7-butyrate as internal standard. TCA cycle metabolites and absolute succinate levels were measured by GC/MS using norvaline and/or D4-succinate as internal standard (n=6 per group). Data represent means ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Analysis in A and B performed via paired student t-test. Analysis in C to H were analyzed by two-way ANOVA and Bonferroni analysis for post hoc comparisons.
As germ-free mice may have distinct interorgan transport of metabolites including TCA cycle metabolites (24), we have also compared the circulating TCA cycle metabolites levels in wildtype mice with or without antibiotic-induced gut microbiota depletion. Specifically, after oral gavage of an antibiotic cocktail (Vancomycin, Neomycin, Metronidazole, plus Ampicillin) for 5 days, the gut microbiota were largely depleted (Figure 3F). As in the germ-free mice, there was no reduction in portal succinate or other TCA cycle intermediate levels after antibiotic treatment and the portal-tail gradient persisted (Figure 3G and 3H). Altogether, results from both the germ-free and gut-microbiota depletion experiments show that intestinal microbiota are not a major source of circulating TCA cycle metabolites.
Labeled precursors produce distinct succinate labeling patterns when exposed to intestinal organoids versus microbiota
Although intestinal microbiota are not essential for intestinal succinate production, this does not exclude the possibility that microbiota can contribute to circulating succinate in conventionally bred animals, and that other undefined mechanisms maintain portal succinate levels. Whereas eukaryotes generate succinate from carbohydrate precursors exclusively via the TCA cycle, microbiota could generate succinate from carbohydrate precursors via the glyoxylate shunt independent from the TCA cycle (25) (Figure 4A). Thus, the labeling pattern of succinate produced from C-13-labeled carbohydrate-derived three-carbon substrates are likely to be different when produced in mammalian cells versus microbiota (25). Specifically, mammalian cells using predominantly the TCA cycle will generate M+2 succinate in the first turn of the cycle and M+1 succinate in the second turn of the cycle, leading to predominant enrichment in M+1 and M+2 succinate (Figure 4A). In contrast, microbiota use a highly active PEP carboxykinase which converts PEP into oxaloacetate and then condenses acetyl-CoA and oxaloacetate via isocitrate lyase into isocitrate which is used to generate glyoxylate and succinate (25). The succinate generated in this pathway from C-13-labeled carbohydrate-derived three-carbon substrates will be enriched at M+3 and M+4 in addition to M+1 and M+2 (Figure 4A).
Figure 4. Intestinal organoids compared with cecal and fecal contents generate distinct succinate labeling patterns from labeled precursors.
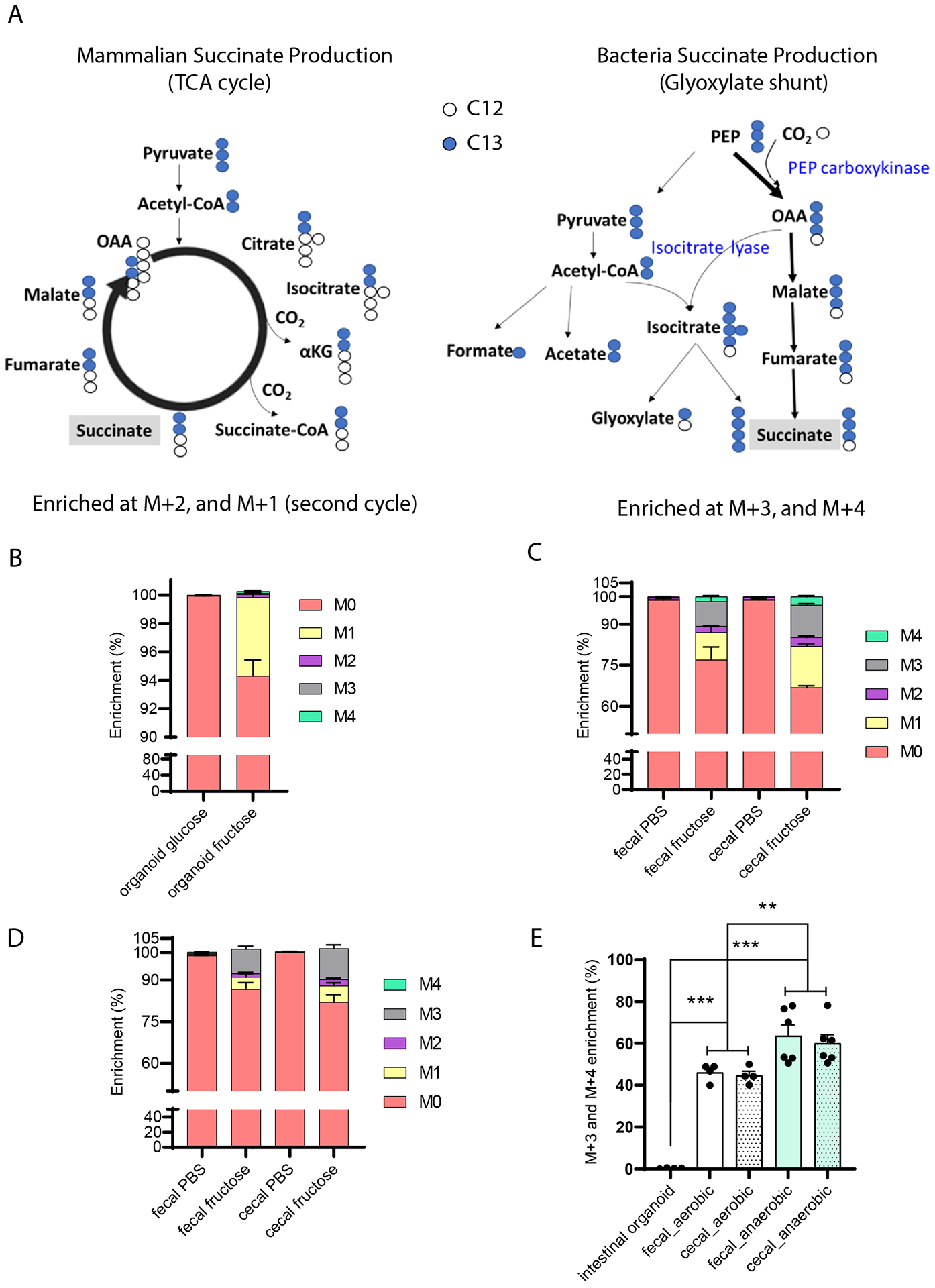
(A) Theoretical isotope-labeling patterns of succinate produced from C13-labeled three carbon substrates in mammalian cells versus bacteria. Measured labeling pattern in (B) mammalian cells and (C, D) microbial enriched samples. Intestinal crypts and fresh cecal and fecal contents were harvested from 8-weeks-old male mice. Intestinal organoids were differentiated into enterocytes prior to treatment with 100 mM U-C13-fructose for 24 hours (n=3 per group). Freshly harvested cecal and fecal contents were cultured with 100 mM U-C13-fructose for 30 minutes in aerobic (C, n=4 per group) or anaerobic condition (D, n=6 per group). (E) Quantification of the ratio of M+3 and M+4 succinate to total labeled succinate in cultured intestinal organoids and cecal/fecal contents described in (B) and (C, D). Data represent means ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Data were analyzed by two-way ANOVA and Bonferroni analysis for post hoc comparisons.
We examined succinate enrichment within fecal and cecal contents in vitro and in cultured intestinal organoids after incubation with U-C13-fructose. As expected, isotope-labeled succinate was identified in both the intestinal organoids and fecal and cecal contents when cultured with U-C13-fructose. In the intestinal organoid experiment, the M+1 and M+2 succinate accounts for 83.7% of the labeled succinate (Figure 4B), consistent with the hypothesis that mammalian cells predominantly use the TCA cycle to generate succinate from carbohydrate precursors. In contrast, fecal and cecal contents generated a much higher proportion of M+3 and M+4 labeled succinate in both aerobic and anaerobic culture conditions, accounting for nearly 45% and 60% of the labeled succinate, respectively (Figure 4C–E). These substantial differences indicate that the contribution of microbiota versus mammalian tissue to succinate production can be distinguished based on labeling patterns following administration of labeled carbohydrate precursors.
Portal succinate is produced from intestinal tissue and not the microbiota
We next compared the succinate labeling pattern in portal blood, intestinal tissue, and cecal contents in mice after providing large amounts of labeled precursors in the form of U-C13-fructose. The fructose dose, 4 g/kg, is higher than the amount that mice can fully absorb in the small intestine, thus providing adequate labeling in both intestinal tissue and the bulk of intestinal bacteria present in the cecum and colon (20,26). Portal blood, intestinal tissue, and cecal contents were harvested from animals that were not gavaged or at either 10 minutes or 2 hours after gavage. The two time points were chosen based on the liquid transition speed in typical mice. Specifically, 10 minutes is sufficient for liquid to transition to the small intestine, but not cecum, whereas 2 hours is aimed at ensuring cecal fructose metabolism (20,26). The portal succinate labeling pattern and specifically the proportion of M+1 and M+2 labeled succinate consistent with production in the eukaryotic TCA cycle were similar in portal blood and intestinal tissue, and distinct from the pattern in the cecal contents at both time points (Figure 5A–B). These data demonstrate that portal succinate is predominantly produced by intestinal tissue and not microbiota.
Figure 5. The succinate labelling pattern in portal plasma is similar to that in intestinal tissue while distinct from that in the cecal contents following gavage with U-C13-fructose.
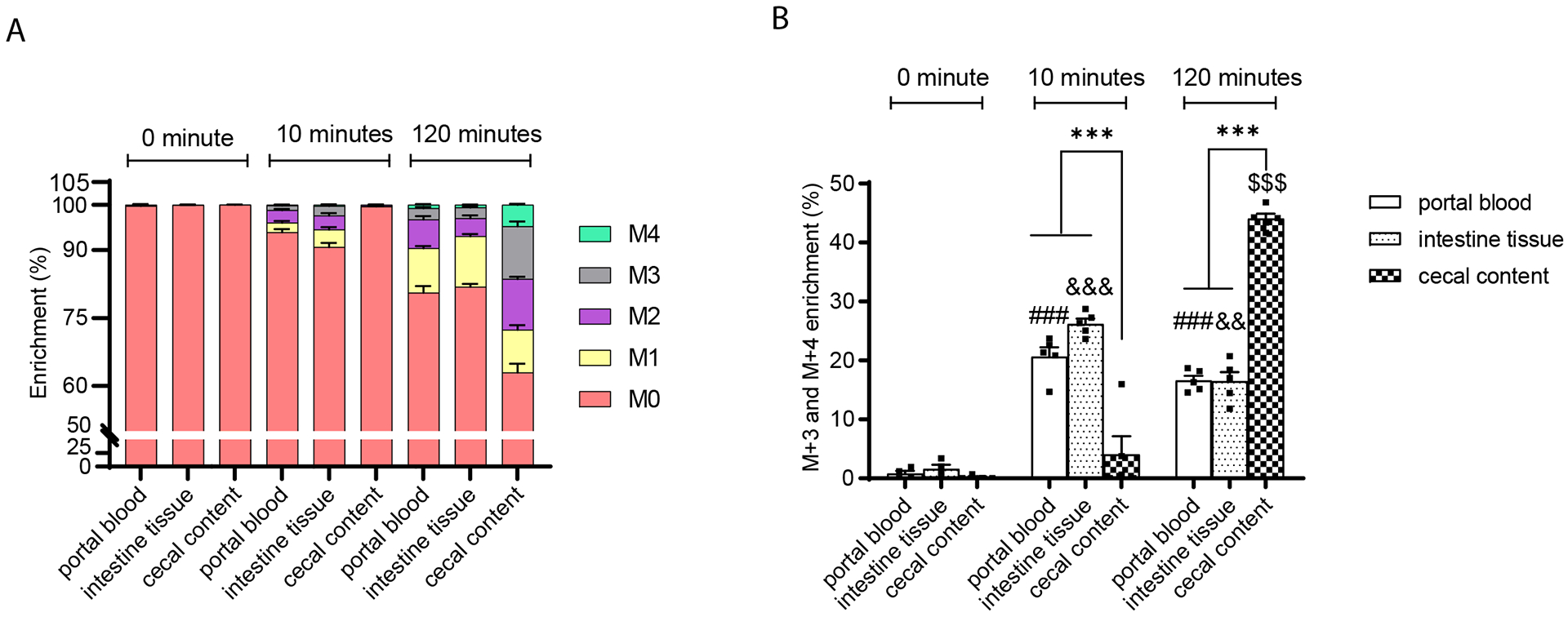
(A) The enrichment and (B) the ratio of M+3 plus M+4 to total labeled succinate in the portal plasma, intestinal tissue, and cecal contents after gavage of 8-weeks-old male mice with U-C13-fructose (4g/Kg body weight) (n=4 per group at each time point). Data represent means ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Data were analyzed by two-way ANOVA and Tukey’s test was applied for post hoc comparisons. # portal plasma comparisons across time points, & intestinal tissue comparisons across time points, $ cecal content comparisons across time points, * comparisons between tissues within time points.
Compared to endogenous production, intestinal succinate absorption capacity is limited.
Prior studies have suggested that dietary succinate supplementation can regulate multiple systemic biological activities through the effects of absorbed succinate on the Sucnr1 receptor in diverse tissues (6,27,28). However, there is limited data demonstrating that oral or dietary succinate supplementation increases circulating succinate levels (29). Our data indicates that succinate produced by microbiota in the large intestine is not readily absorbed, but this does not exclude the possibility that dietary succinate could be absorbed in the small intestine. Thus, we sought to examine the intestinal capacity for absorption of orally administered succinate and compare this to the rate of endogenous intestinal succinate production.
Portal succinate levels are a mixture of succinate absorbed from the intestinal lumen, succinate produced endogenously within intestinal tissue, and succinate carried into the portal circulation via systemic recirculation or via production in other tissues. The content of succinate in typical human or mouse diets is not well defined. Succinate may be added to foods as a flavor enhancer at levels not to exceed 0.08% (g/g) in condiments and less than 0.006% (g/g) in meat products per FDA guidelines (21CFR184, Sec. 184.1091). As indicated by GC/MS measurement in Supplementary 1A, the succinate content of standard mouse chow diets LabDiet 5053 and Envigo 2020 SX, are 146.5 μg/g and 113.8 μg/g comprising less than 0.015% of food by weight.
To define the upper limits of intestinal succinate absorption and to compare this to the rate of endogenous gut succinate production, we established a crude relative quantification model (Figure 6A and B). We first tested this model using fructose, taking advantage of the fact that fructose is quickly absorbed and readily metabolized in the intestine and that endogenous fructose production is likely small relative to fructose absorption in mammals (30). We measured the relative quantification and enrichment of portal fructose after oral-gavage with 1:1 U-C13-fructose and unlabeled fructose (0.48 g/Kg body weight) (n=4 per time point). As expected, within 10 minutes of fructose gavage, both portal and tail fructose levels rapidly and robustly increased approximately 50-fold and 20-fold above their baseline levels, respectively (Figure 6C). The relative abundance of M+6 fructose was similar to that of M+0 fructose at the 10-minute time point and comparable to the 1:1 ratio of labeled and unlabeled fructose that was provided by gavage (Figure 6D). The lack of dilution of M+6 fructose with M+0 fructose indicates that endogenously produced fructose was not a major contributor to portal fructose at this time point. The gradual decline in M+6 compared to M+0 fructose from 30 minutes on is indicative of the dilution of the gavaged mixture of labeled and unlabeled fructose with endogenously produced fructose (Figure 6D). Based on the relative quantification model, we estimate that the rate of intestinal fructose absorption is approximately 25-fold greater than the rate of endogenously produced fructose production at the 10-minute time point (Figure 6E). These findings are consistent known aspects of fructose metabolism in mice (31) and illustrate that this simplified relative quantification model is useful in estimating the relative amounts of absorbed versus endogenously produced substrates in vivo.
Figure 6. Relative quantification of the ratio of intestinal fructose absorption to endogenous fructose production.
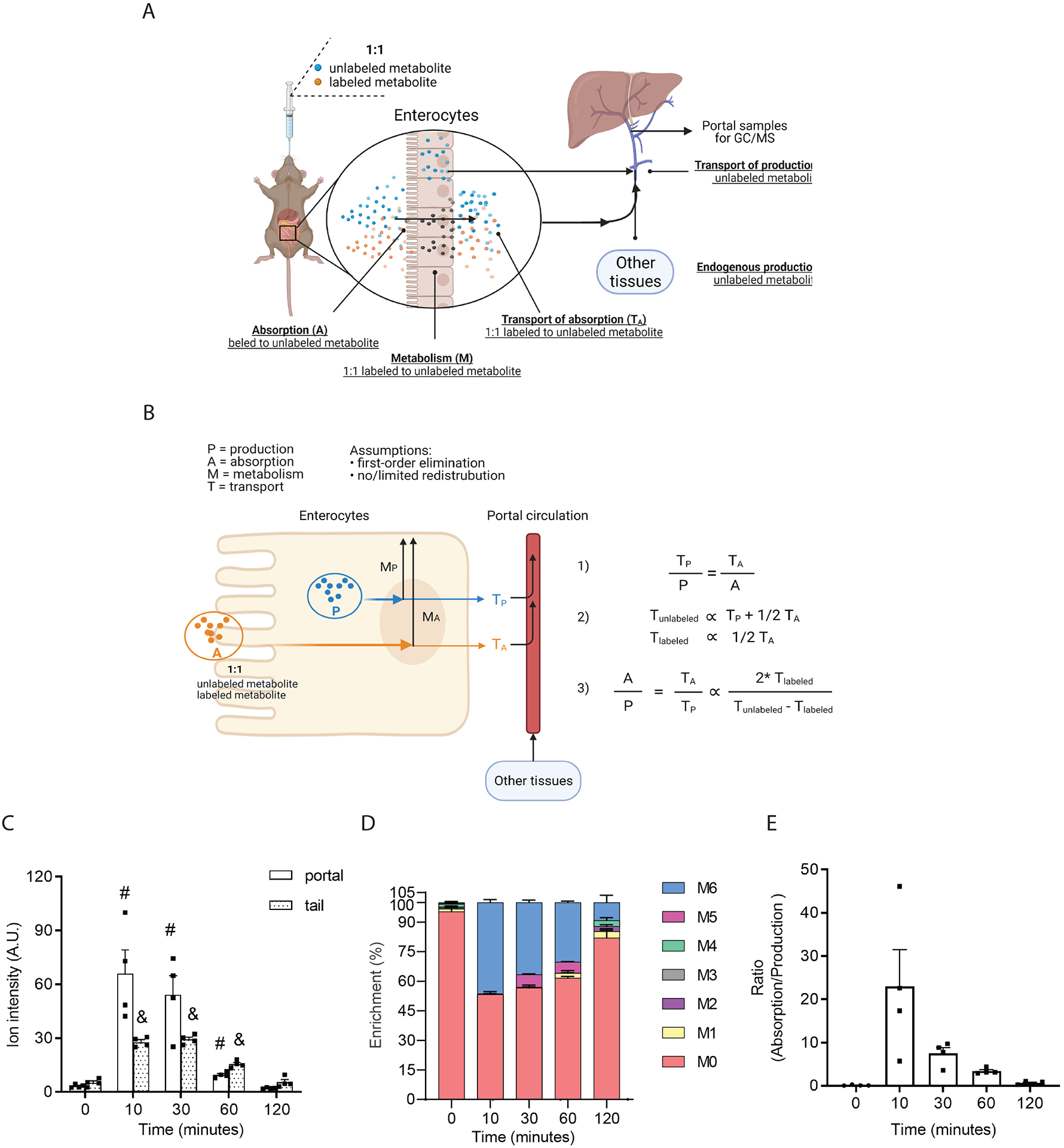
Diagram of the method (A) and model (B) for estimating the ratio of fructose absorption to fructose production. Quantification of fructose (C) and its enrichment (D) in portal plasma after gavage of 1:1 U-C13 fructose and unlabeled fructose (0.48 g/Kg body weight, n=4 per group). 2DG was used as internal standard. (E) The estimated ratio of intestinal fructose absorption to endogenous fructose production. Data represent means ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Data in panel A were analyzed by two-way ANOVA with time and location as co-variable Bonferroni analysis for post hoc comparisons; # portal comparisons between time points, & tail comparisons between time points, * comparisons between tail and portal. Data in panel C were analyzed by one-way ANOVA and Fisher’s LSD for post-hoc analysis of the ratios of M+6 to M+0 fructose between baseline (0 minutes) and time points post fructose administration (10, 30, 60, 120 minutes).
Next, we gavaged mice with 1:1 disodium succinate-2,2,3,3-D4 and unlabeled disodium succinate at a dose of 1.46 g/kg which is close to the solubility of succinate in water to compare the capability of intestinal succinate absorption to endogenous production. This amount of succinate (~ 36.5 mg for a 25g mouse) is more than 60-fold higher than the amount of succinate consumed by a mouse on a standard chow diet in a day (~ 0.6 mg succinate for a mouse that eats 4g food per day). After gavage of this supraphysiological succinate bolus, total portal succinate levels increased within 10 minutes and declined to baseline levels after ~ 1 hour (Figure 7A). This suggests that succinate can be absorbed from the intestinal lumen. Interestingly, at the 10 minute time point, the increase in magnitude for M+0 succinate (from 3.35 ± 1.32 to 8.70 ± 5.10 A.U.) is higher than for M+4 succinate (from 0.41 ± 0.08 to 3.44 ± 2.99 A.U.), suggesting that the osmolar stress of the supraphysiological succinate bolus may promote the secretion of endogenous gut-derived succinate (Figure 7B). During the 2 hour time period, the maximum proportion of M+4 succinate with respect to total succinate ranges from 20 to 25% (Figure 7C). This indicates that the absorbed exogenous 1:1 labeled to unlabeled succinate was substantially diluted by endogenous unlabeled succinate (Figure 7D). Using our model (Figure 6), we estimated that at peak absorption of this supraphysiological succinate bolus, the contribution of endogenous succinate and absorbed succinate are approximately equal (Figure 7D). These results indicate that in normal physiological circumstances, where succinate is present in ingested foods in limited amounts, the vast majority of circulating succinate is produced endogenously, and that succinate absorption likely contributes negligibly to portal succinate levels.
Figure 7. The intestinal absorption of succinate is limited.
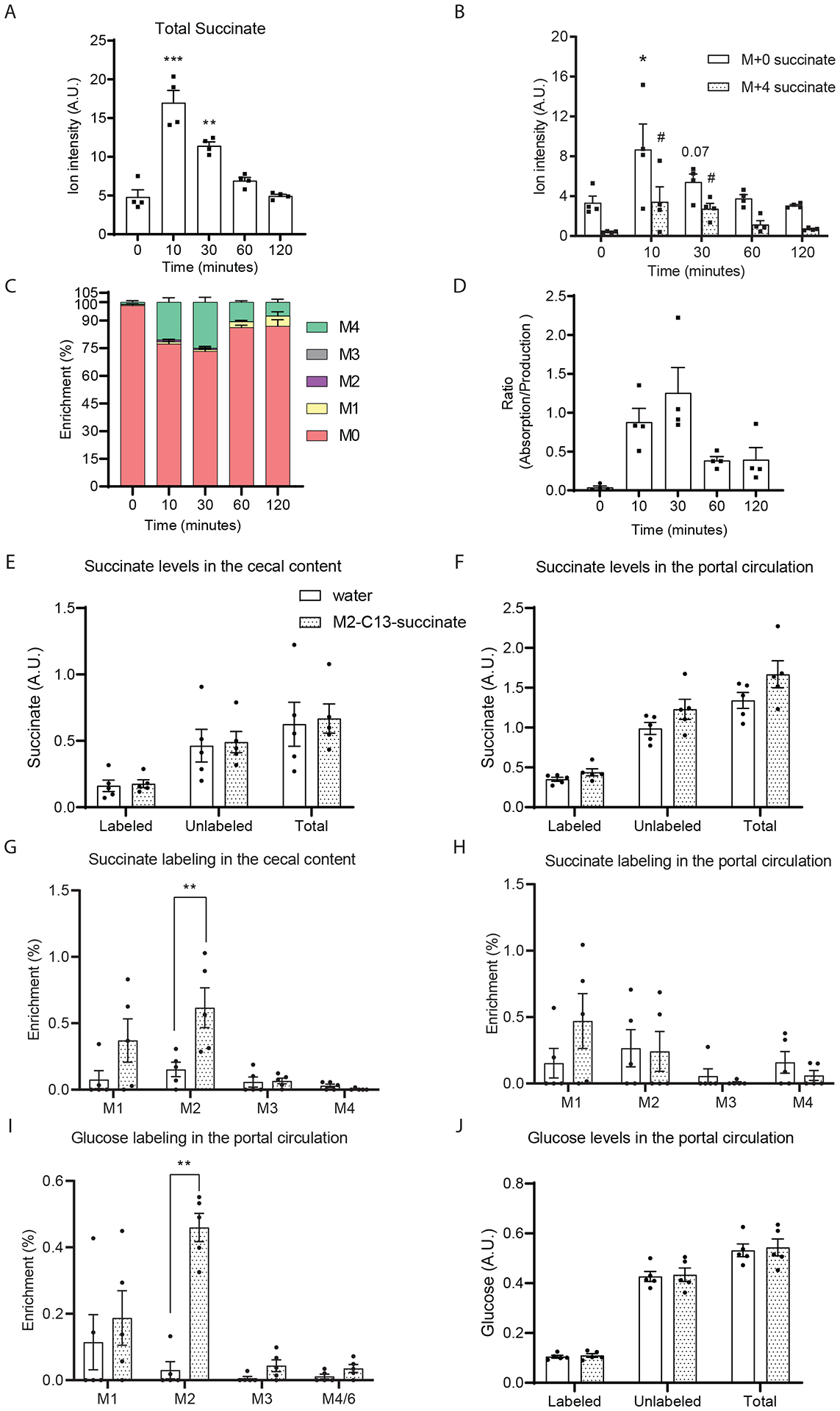
(A) Total succinate ion counts and (B) M+0 and M+4 isotopomers ion counts and (C) enrichment in portal plasma after gavage of 1:1 D-4 succinate and unlabeled succinate (1.46 g sodium succinate/Kg body weight). Norvaline was used as internal standard. (D) The estimated ratio of intestinal absorption to endogenous production of succinate. (n=4 at each time point). Measured succinate levels (E, F) and labeling (G, H) in the cecal content and portal circulation after supplementing drinking water with 0.5% 2,3-C13-sodium succinate for 3 days. Glucose labeling (I) and quantification (J) were also measured by GC/MS. Data represent means ± SEM. * or # p<0.05; ** or ## p<0.01; *** or ### p<0.001. In panel A to D, data were analyzed by one-way ANOVA and Fisher’s LSD for post-hoc analysis between baseline (0 minutes) and time points post D4-succinate administration; In panel B, * indicates the comparison of M+0 succinate and # indicates the comparison of M+4 succinate with the zero-time point. In panel E to F, data were analyzed by two-way ANOVA and Tukey’s test was applied for post hoc comparison.
As this finding seems contradictory to the view suggested in prior studies that dietary succinate supplementation impacts systemic succinate levels and systemic metabolic function, we further examined this by treating wild-type mice with 2,3-C13-sodium succinate supplemented drinking water (0.5% gram/ml) ad-libitum for 3 days. This dose of succinate supplementation was suggested by a prior study to significantly increase circulating succinate levels (29). It is estimated that an adult mouse consumes ~ 5.8 mL water per day (32). Supplementing 0.5% sodium succinate in drinking water is equivalent to supplementing ~ 0.026 g succinate per day. This amount of succinate is ~ 43-fold higher than the amount contained in a mouse’s typical daily diet and is close to the supraphysiological dose we administered in previous experiments (1.46 g/kg). Interestingly, despite supplementing a large amount of M+2 succinate in the drinking water, the total measured succinate remained stable in both the cecal content and portal circulation (Figure 7E, F). This indicates that the endogenous succinate production rate by both microbiota and intestinal tissue far exceeds the succinate that was delivered by oral supplementation. Note that there is a modest increase in M+2 succinate and a trend for increased M+1 succinate in cecal contents (Figure 7G), indicating that the oral delivery of M+2 succinate was successful. For the portal succinate, neither the labeling nor the quantity increased after C13-succinate supplementation (Figure 6H). Interestingly, there is a slight but statistically significant increase of M+2 glucose in the portal circulation, although this does not alter the total glucose levels (Figure 7I, J). This suggests that absorbed M+2 succinate can be converted into glucose in very limited amounts. This data confirms that endogenous succinate production is the major source of circulating succinate.
Compared to fructose, succinate is a poor substrate for intestinal glucose production in vivo
Although we demonstrated that intestinal succinate absorption is limited compared to endogenous succinate production, it remains possible that absorbed succinate could be metabolized within intestinal tissue. We observed a modest but statistically significant increase in M+2 glucose in the portal circulation after administration of drinking water supplemented with M+2 succinate. Indeed, a prior study suggested that microbiome-derived succinate contributes to intestinal gluconeogenesis without altering circulating succinate levels (33). However, the contribution of succinate to intestinal glucose production was not directly assessed in vivo. To address this question, we compared the enrichment of label in circulating glucose after oral gavage with C13-labeled fructose versus an equimolar amount of C13-labeled succinate. Note that because the normal consumption of dietary succinate is very limited, when we orally gavage the mice with the same molar concentration (0.89M) of succinate and fructose, we are providing a physiological amount of dietary U-C13-fructose (1.6 g/Kg), but a supraphysiological amount of 2,3-C13-disodium succinate (1.46 g/Kg). This is the same amount of succinate that we gavaged in Figure 6, and which is ~ 60-fold higher than the amount consumed in a typical rodent diet per day. As indicated in Figure 8A, 30 minutes after gavage which represents the peak of absorption, nearly 50% of the portal glucose was labeled in the fructose gavaged animals whereas only ~ 10% of glucose was labeled following succinate gavage. The average incorporation of carbon from fructose into glucose is ~ 9-fold greater than the rate of incorporation of carbon from succinate into glucose in the portal circulation (Figure 8B). After correcting for the differences in the number of C13 carbons contained in the U-C13-fructose versus 2,3-C13-disodium succinate tracers, the adjusted carbon incorporation rate from fructose is still more than 3-fold higher from fructose than from succinate (Figure 8C). Thus, compared to fructose, succinate is a poor substrate for intestinal glucose production in vivo. Moreover, this data suggests that even when we provided the gut with a massive, supraphysiological amount of succinate, the contribution of succinate to circulating glucose is limited.
Figure 8. Succinate is a poor substrate for intestinal glucose production.

8-week old wildtype mice were oral gavage with the same molar concentration of water, U-C13-fructose or 2,3-C13-sodium succinate after 5 hours food removal. (A) Glucose labeling in the plasma samples from portal vein or tail vein was measured by GC/MS. Average carbon 13 incorporation and adjusted carbon 13 incorporation were calculated. Data represent means ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Data were analyzed by two-way ANOVA and Tukey’s test was applied for post hoc comparison.
Discussion
Recent studies illustrate important roles for TCA cycle intermediates outside of their role in energy production including important roles for circulating TCA cycle intermediates as metabolic signals (2,22). This field has advanced with the discovery of cell-surface receptors sensitive to specific TCA cycle metabolites including succinate and alpha-ketoglutarate (34). While the role of metabolites like succinate in regulating cellular function and organismal homeostasis is rapidly advancing, few studies have assessed the source of circulating TCA cycle intermediates.
We observed that the levels of TCA cycle intermediates are markedly higher in the portal blood than the peripheral circulation indicating the gut is likely a major source of circulating TCA cycle intermediates (Figure 1A, B). In support of this, when treated mice with C13-tracers, the slopes of isotope-labeled TCA cycle metabolites are very similar in the portal and tail circulation, suggesting that peripheral TCA cycle intermediates are predominantly derived from portal circulation (Figure 2G to I). Recently Jang and colleagues used steady-state stable isotopic tracer infusions to assess the relative production and consumption of a wide range of metabolites including TCA cycle metabolites within different organs/tissues in pigs (35). Based on sampling of the pancreaticoduodenal vein, they concluded that TCA cycle metabolites are the most highly exported metabolites from pancreas. However, as this vein carries venous blood from both the pancreas and duodenum, these investigators could not discriminate whether the duodenum versus pancreas is the actual source. Moreover, in this study, blood obtained from the inferior and superior mesenteric veins which drain the gut, but not that from splenic vein which drains the pancreatic tail also exported a significant amount of TCA cycle metabolites. Hence, data from this study may support our findings that the gut is a major contributor to circulating TCA cycle metabolites.
Fructose is a dietary sugar that is preferentially metabolized in selected cell types including enterocytes in the intestine, hepatocytes in the liver, and proximal tubule cells in the kidney (30,31). Fructose is not readily metabolized in the pancreas (31). Gavaging mice with labeled fructose rapidly and robustly labels TCA cycle intermediates in the portal vein and to comparable degrees within duodenal tissue (Fig 2 A–F and 7 A). Although multiple intestinal epithelial cell types (enterocytes, goblet cells, and Paneth cells) may express the Glut5 luminal fructose transporter and fructolytic enzymes (36), these are expressed at high levels in absorptive enterocytes which are thought to mediate the bulk of intestinal fructose metabolism (31,37). Altogether, these results indicate that enterocytes in the proximal intestine are major contributors to gut-derived circulating TCA cycle metabolites.
According to studies conducted by us and others, the physiological circulating succinate levels range from 0 to 50 μM in humans (9), and 0 to 100 μM in rodents (27). This range of succinate coincides with the pharmacological range (EC50) of succinate receptor (Sucnr1) activation - 20 to 50 μM in human and 10 to 100 μM in rodents (2). Thus, changes in the circulating succinate level within the physiological range have potential to affect the activity of Sucnr1 and its downstream signaling. This supports succinate’s potential role as an important circulating metabolic signal.
While our results indicate that enterocytes are major sources of circulating succinate in physiological circumstances, other tissues may also play important roles. As previously noted, Jang et al and others have observed TCA cycle production in range of normal tissues (35,38). Additionally, acute cellular hypoxia such as myocardial infarction, ischemia of the kidney, or liver ischemia are known to result in succinate production, and this may increase circulating succinate levels (39–41). Exercise-induced hypoxia in skeletal muscles can produce substantial amounts of succinate which can transiently increase circulating succinate levels (42). Chronic, mild increases in circulating succinate are associated with cardiometabolic diseases including obesity, hypertension, and type-2 diabetes and the source of this succinate is unclear (9,43). Additional studies will be required to determine whether intestinal TCA cycle metabolite production is a contributor to changes in circulating succinate associated with pathological conditions.
Intestinal microbiota are increasingly recognized as a major source of bioactive metabolites in systemic circulation which may have pleiotropic physiological effects (44). As examples, microbiota-derived short-chain fatty acids and acetate both play important roles in hepatic gluconeogenesis and de novo lipogenesis participating in diet-induced metabolic disease (21,45). Serena et al. observed an enrichment of succinate-generating microbiota in association with circulating succinate levels in patients with metabolic diseases and hypothesized that the microbiota contributes to this increase (9). De Vadder et al. subsequently transplanted one of these succinate-generating microbial strains, P. copri, into conventional mice and observed significantly increased succinate in cecal contents in association with improved glucose tolerance (10). However, this was not associated with increased circulating succinate indicating that increased levels of succinate in the intestinal lumen are not sufficient to increase circulating succinate levels (10). De Vadder et al. interpreted this data to suggest that microbially-derived succinate is absorbed and consumed within the intestine as gluconeogenic substrate although they did not directly assess intestinal glucose production from succinate. Moreover, the expression of glucose-6-phosphatase, the terminal step in glucose production is largely restricted to the small intestine whereas succinate production from microbiota occurs predominantly in the colon (46). In this study, we have demonstrated that the intestinal capacity for glucose production from orally delivered succinate is limited as the incorporation of labeled succinate into glucose is markedly less than that from the equimolar amount of orally delivered fructose (Figure 8). Moreover, labeled circulating glucose derived drinking water supplemented with a large amount of labeled succinate contributes to less than 1% circulating glucose (Figure 7I). Furthermore, these data indicate that the modest increases in circulating succinate levels after external supplementation is not likely due to the metabolite exchange and interconversion as exogenous succinate is diluted by the great amount of endogenous production in the intestinal tissue. In further support of this, we showed that portal succinate levels are comparable in conventional mice, germ-free mice, and mice with gut microbiota depletion following antibiotic treatment, demonstrating that gut microbiota are not essential for maintaining circulating succinate levels. Moreover, by comparing portal succinate labeling patterns after oral administration of labeled substrate, we demonstrate that intestinal tissue rather than microbiota are the predominant contributor to portal succinate in conventional mice (Figure 5A, B). Altogether, we and others have demonstrated that although microbiota can generate substantial amounts of succinate in the intestinal lumen, this microbiota-produced succinate is distinct from that produced endogenously in the small intestine. Thus, microbiota are not a major contributor to circulating succinate. These results raise questions as to the causal nature of the improvements in glucose homeostasis after transplantation of succinate-generating microbiota. At a minimum, the improvements in metabolism associated with this transplant cannot be attributed changes in circulating succinate as a metabolic signal.
These studies also raise questions about the capacity of the intestine to absorb succinate from the lumen. This question is important as several studies have suggested that dietary supplementation of succinate can increase circulating succinate and regulate biological functions through activation of SUCNR1 in multiple organs including the liver and adipose tissue (6,27,47). However, few studies assessed the impact of dietary succinate supplementation on circulating succinate levels. Here, we examined intestinal succinate absorption capacity and determine the relative importance of succinate absorption compared to endogenous production.
To approach this problem, we first sought literature regarding the succinate content of typical diets and were surprised to find no clear reference for this in typical human or animal diets. Hence, we measured succinate levels in two standard mouse chow diets and found that succinate contributes no more than 0.015% g/g (Figure 3A and B). We elected to gavage mice with a quantity of labeled succinate that far exceeds the typical daily dietary content. At an extreme, supraphysiological dose, succinate absorption transiently approached the levels of endogenous succinate production (Figure 7A–D). These results suggest that the modest amounts of succinate present in typical diets contribute negligibly to gut-derived circulating succinate levels. Moreover, supplementing a large dose of succinate by drinking water for 3 days did not significantly alter the succinate levels in either cecal contents or portal circulation. This confirms the extensive rate of endogenous succinate production compared with typical exogenous supplementation.
While the results from this study support the conclusion that the intestine tissue is a major source of circulating TCA cycle metabolites, there are several limitations. First, we encountered some technical limitations when assessing the levels of TCA cycle metabolites. For example, we have not included α-ketoglutarate measurements in this manuscript as metabolites generated through derivatization of α-ketoglutarate in our GC/MS method are unstable resulting in highly variable results within and across cohorts. Also, we noticed some variability in absolute quantification of circulating succinate levels within and across cohorts. Although we added D4-succinate as an internal standard to all of our samples to minimize batch effects, some technical variability remains in this analysis. Another and likely the more important potential explanation for the variability of absolute succinate levels across experiments is that we have noted that there are substantial differences in circulating TCA cycle metabolite levels across different mouse strains which were used in this study (data not shown).
Second, the models used to compare the relative rates of succinate absorption versus endogenous production relies on several assumptions which include: 1) the assumption that the metabolism of absorbed and endogenously produced succinate follows first-order kinetics, and 2) the assumption that recirculation of labeled succinate is negligible. First, although most of metabolites follow first-order elimination kinetics (48), it is possible that succinate elimination actually follows zero-order elimination rule. However, the rapid peak and return to baseline within 1 hour of gavage is inconsistent with a zero-order elimination model. Second, although we disregard the recirculation of intestinal absorbed labeled metabolites in our model, this assumption could lead to an overestimation of the absorption of succinate. Thus, if recirculation is substantial, the actual intestinal absorption rate would be even smaller than what we estimated here and would further support our conclusion that intestinal absorption of succinate makes a minor contribution. Third, our conclusions are limited to mice. While gut physiology in mice certainly has many similarities to that of other animals, there are well-defined differences. For example, the small intestinal length normalized to surface and the small intestine normalized to colon length are much smaller in mice than human (49). Additionally, the mouse cecum size, gut microbiota composition, and responses to dietary interventions are also different from that in human (50). Additional studies will be required to determine whether our observations regarding intestinal TCA cycle production extend to humans and other animals.
In summary, murine intestinal tissue is a major source of circulating TCA cycle metabolites. The role of intestinally-derived circulating TCA cycle intermediates as metabolic signals remains to be investigated in future studies.
Supplementary Material
Acknowledgements:
This work was supported by NIH grants 5R01 DK121710 (MAH). Metabolomics support was provided by the North Carolina Diabetes Research Center (NCDRC) (P30 DK124723).
Conflict of interest statement:
The authors state that there are no conflicts of interest in connection with this article. Mark A. Herman receives research support from Eli Lilly, and Co.
Reference
- 1.Owen OE, Kalhan SC, and Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. The Journal of biological chemistry 277, 30409–30412 [DOI] [PubMed] [Google Scholar]
- 2.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, and Ling L (2004) Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–193 [DOI] [PubMed] [Google Scholar]
- 3.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, and Peti-Peterdi J (2008) Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. The Journal of clinical investigation 118, 2526–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, and Artyomov MN (2016) Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell metabolism 24, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwärzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, and Carballido JM (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nature immunology 9, 1261–1269 [DOI] [PubMed] [Google Scholar]
- 6.Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, Banks AS, Haigis MC, Kajimura S, Murphy MP, Gygi SP, Clish CB, and Chouchani ET (2018) Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho EH (2018) Succinate as a Regulator of Hepatic Stellate Cells in Liver Fibrosis. Front Endocrinol (Lausanne) 9, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Correa PR, Kruglov EA, Thompson M, Leite MF, Dranoff JA, and Nathanson MH (2007) Succinate is a paracrine signal for liver damage. Journal of hepatology 47, 262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serena C, Ceperuelo-Mallafre V, Keiran N, Queipo-Ortuno MI, Bernal R, Gomez-Huelgas R, Urpi-Sarda M, Sabater M, Perez-Brocal V, Andres-Lacueva C, Moya A, Tinahones FJ, Fernandez-Real JM, Vendrell J, and Fernandez-Veledo S (2018) Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. The ISME journal 12, 1642–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Backhed F, and Mithieux G (2016) Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell metabolism 24, 151–157 [DOI] [PubMed] [Google Scholar]
- 11.Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, Littman DR, and Diehl GE (2018) Critical Role for the Microbiota in CX(3)CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49, 151–163.e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N, Ai Y, Wang L, Xi R, Astapova I, Han Y, Li J, Barth BB, Lu M, Gao Z, Mines R, Zhang L, Herman M, Hsu D, Zhang GF, and Shen X (2018) Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell metabolism 27, 1249–1262.e1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He W, Wang Y, Xie EJ, Barry MA, and Zhang GF (2021) Metabolic perturbations mediated by propionyl-CoA accumulation in organs of mouse model of propionic acidemia. Molecular genetics and metabolism 134, 257–266 [DOI] [PubMed] [Google Scholar]
- 14.Fernandez CA, Des Rosiers C, Previs SF, David F, and Brunengraber H (1996) Correction of 13C mass isotopomer distributions for natural stable isotope abundance. Journal of mass spectrometry : JMS 31, 255–262 [DOI] [PubMed] [Google Scholar]
- 15.Zhang GF, Jensen MV, Gray SM, El K, Wang Y, Lu D, Becker TC, Campbell JE, and Newgard CB (2020) Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell metabolism [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilsson R (2020) Validity of natural isotope abundance correction for metabolic flux analysis. Mathematical biosciences 330, 108481. [DOI] [PubMed] [Google Scholar]
- 17.Borowy CS, and Ashurst JV (2021) Physiology, Zero and First Order Kinetics. in StatPearls, StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC., Treasure Island (FL). pp [PubMed] [Google Scholar]
- 18.Shree M, and S KM (2018) Intracellular Fate of Universally Labelled (13)C Isotopic Tracers of Glucose and Xylose in Central Metabolic Pathways of Xanthomonas oryzae. Metabolites 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 [DOI] [PubMed] [Google Scholar]
- 20.Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, and Rabinowitz JD (2018) The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell metabolism 27, 351–361.e353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, Trefely S, Fernandez S, Carrer A, Miller KD, Schug ZT, Snyder NW, Gade TP, Titchenell PM, Rabinowitz JD, and Wellen KE (2020) Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mills E, and O’Neill LA (2014) Succinate: a metabolic signal in inflammation. Trends in cell biology 24, 313–320 [DOI] [PubMed] [Google Scholar]
- 23.Macfarlane S, and Macfarlane GT (2006) Composition and metabolic activities of bacterial biofilms colonizing food residues in the human gut. Applied and environmental microbiology 72, 6204–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai Y, Liu CW, Yang Y, Hsiao YC, Ru H, and Lu K (2021) High-coverage metabolomics uncovers microbiota-driven biochemical landscape of interorgan transport and gut-brain communication in mice. Nature communications 12, 6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn S, Jung J, Jang IA, Madsen EL, and Park W (2016) Role of Glyoxylate Shunt in Oxidative Stress Response. The Journal of biological chemistry 291, 11928–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woting A, and Blaut M (2018) Small Intestinal Permeability and Gut-Transit Time Determined with Low and High Molecular Weight Fluorescein Isothiocyanate-Dextrans in C3H Mice. Nutrients 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Chen Y, Zhao L, Tian Q, deAvila JM, Zhu MJ, and Du M (2021) Dietary succinate supplementation to maternal mice improves fetal brown adipose tissue development and thermogenesis of female offspring. The Journal of nutritional biochemistry, 108908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, Erle DJ, Anderson MS, Locksley RM, Raftery D, and von Moltke J (2018) Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41.e37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Xu YQ, Yuan YX, Xu PW, Zhang C, Li F, Wang LN, Yin C, Zhang L, Cai XC, Zhu CJ, Xu JR, Liang BQ, Schaul S, Xie PP, Yue D, Liao ZR, Yu LL, Luo L, Zhou G, Yang JP, He ZH, Du M, Zhou YP, Deng BC, Wang SB, Gao P, Zhu XT, Xi QY, Zhang YL, Shu G, and Jiang QY (2021) Succinate induces skeletal muscle fiber remodeling via SUCNR1 signaling. EMBO reports 22, e53027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hannou SA, Haslam DE, McKeown NM, and Herman MA (2018) Fructose metabolism and metabolic disease. The Journal of clinical investigation 128, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herman MA, and Birnbaum MJ (2021) Molecular aspects of fructose metabolism and metabolic disease. Cell metabolism [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmanov AA, Reed DR, Beauchamp GK, and Tordoff MG (2002) Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behavior genetics 32, 435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Bäckhed F, and Mithieux G (2016) Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell metabolism 24, 151–157 [DOI] [PubMed] [Google Scholar]
- 34.Tretter L, Patocs A, and Chinopoulos C (2016) Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochimica et biophysica acta 1857, 1086–1101 [DOI] [PubMed] [Google Scholar]
- 35.Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, Morscher RJ, Reyes J, Frezza C, Hwang HY, Imai A, Saito Y, Okamoto K, Vaspoli C, Kasprenski L, Zsido GA 2nd, Gorman JH 3rd, Gorman RC, and Rabinowitz JD (2019) Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell metabolism 30, 594–606.e593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kishida K, Pearce SC, Yu S, Gao N, and Ferraris RP (2017) Nutrient sensing by absorptive and secretory progenies of small intestinal stem cells. American journal of physiology. Gastrointestinal and liver physiology 312, G592–g605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Jawadi A, Patel CR, Shiarella RJ, Romelus E, Auvinen M, Guardia J, Pearce SC, Kishida K, Yu S, Gao N, and Ferraris RP (2020) Cell-Type-Specific, Ketohexokinase-Dependent Induction by Fructose of Lipogenic Gene Expression in Mouse Small Intestine. The Journal of nutrition 150, 1722–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS, and Arany Z (2020) Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science (New York, N.Y.) 370, 364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, and Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wijermars LG, Schaapherder AF, Kostidis S, Wüst RC, and Lindeman JH (2016) Succinate Accumulation and Ischemia-Reperfusion Injury: Of Mice but Not Men, a Study in Renal Ischemia-Reperfusion. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 16, 2741–2746 [DOI] [PubMed] [Google Scholar]
- 41.Chan TS, Cassim S, Raymond VA, Gottschalk S, Merlen G, Zwingmann C, Lapierre P, Darby P, Mazer CD, and Bilodeau M (2018) Upregulation of Krebs cycle and anaerobic glycolysis activity early after onset of liver ischemia. PloS one 13, e0199177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, Paschini M, Paulo JA, Garrity R, Laznik-Bogoslavski D, Ferreira JCB, Carl CS, Sjøberg KA, Wojtaszewski JFP, Jeppesen JF, Kiens B, Gygi SP, Richter EA, Mathis D, and Chouchani ET (2020) pH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 183, 62–75.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y, and Tones MA (2007) Circulating succinate is elevated in rodent models of hypertension and metabolic disease. American journal of hypertension 20, 1209–1215 [DOI] [PubMed] [Google Scholar]
- 44.Tang WH, Kitai T, and Hazen SL (2017) Gut Microbiota in Cardiovascular Health and Disease. Circulation research 120, 1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, and Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajas F, Bruni N, Montano S, Zitoun C, and Mithieux G (1999) The glucose-6 phosphatase gene is expressed in human and rat small intestine: regulation of expression in fasted and diabetic rats. Gastroenterology 117, 132–139 [DOI] [PubMed] [Google Scholar]
- 47.Mills EL, Harmon C, Jedrychowski MP, Xiao H, Garrity R, Tran NV, Bradshaw GA, Fu A, Szpyt J, Reddy A, Prendeville H, Danial NN, Gygi SP, Lynch L, and Chouchani ET (2021) UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nature metabolism 3, 604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Hui S, Mirek ET, Jonsson WO, Anthony TG, Lee WD, Zeng X, Jang C, and Rabinowitz JD (2022) Circulating metabolite homeostasis achieved through mass action. Nature metabolism 4, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casteleyn C, Rekecki A, Van der Aa A, Simoens P, and Van den Broeck W (2010) Surface area assessment of the murine intestinal tract as a prerequisite for oral dose translation from mouse to man. Laboratory animals 44, 176–183 [DOI] [PubMed] [Google Scholar]
- 50.Nguyen TL, Vieira-Silva S, Liston A, and Raes J (2015) How informative is the mouse for human gut microbiota research? Disease models & mechanisms 8, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article or are openly available in figshare at the following URL: https://doi.org/10.6084/m9.figshare.19074176.v2.