

Abstract
Permissiveness of the host cell to productive infection by onco-retroviruses is cell-cycle dependent1, and nuclear localization of viral nucleoprotein preintegration complexes will occur only after cells have passed through mitosis2. In contrast, establishment of an integrated provirus after infection by the lentivirus HIV-1 is independent of host cell proliferation3–5. The ability of HIV-1 to replicate in non-dividing cells is partly accounted for by the karyophilic properties of the viral preintegration complex which, after virus infection, is actively transported to the host cell nucleus. Here we report that the gag matrix protein of HIV-1 contains a nuclear localization sequence which, when conjugated to a heterologous protein, directs its nuclear import. In addition, HIV-1 mutants containing amino-acid substitutions in this nuclear localization signal integrate and replicate within dividing but not growth-arrested cells, and thus display a phenotype more representative of an onco-retrovirus.
Nuclear localization signals (NLSs) have been identified for many karyophilic proteins of viral and cellular origin and generally resemble either the single basic domain SV40 large T-antigen NLS (PKKKRKV) or the double basic domain nucleoplasm in NLS (KRPAATKKAGQAKKKK, single-letter amino-acid code)6. Sequences within HIV-1 matrix protein with homology to the single domain motif are shown in Fig. 1a. Synthetic peptides containing putative NLSs of HIV-1 matrix were fluoresceinated and conjugated to bovine serum albumin (BSA)7. When microinjected into the cytoplasm of PtK-1 cells8, a peptide encompassing amino acids 25–33 of HIV-1 matrix directed the partial localization of BSA to cell nuclei (Fig. 1b). This nuclear import was abolished by the co-injection of wheat-germ agglutinin (WGA), an inhibitor of nuclear pore-complex-mediated import, but proceeded in the presence of WGA and its ligand, N-acetylglucosamine (GlcNAc). No nuclear localization was observed when BSA was conjugated to peptides containing four lysine to threonine substitutions (Fig. 1b) or to peptides based on a second putative NLS (amino acids 107–116 of matrix) enriched in basic amino acids (not shown).
FIG. 1.
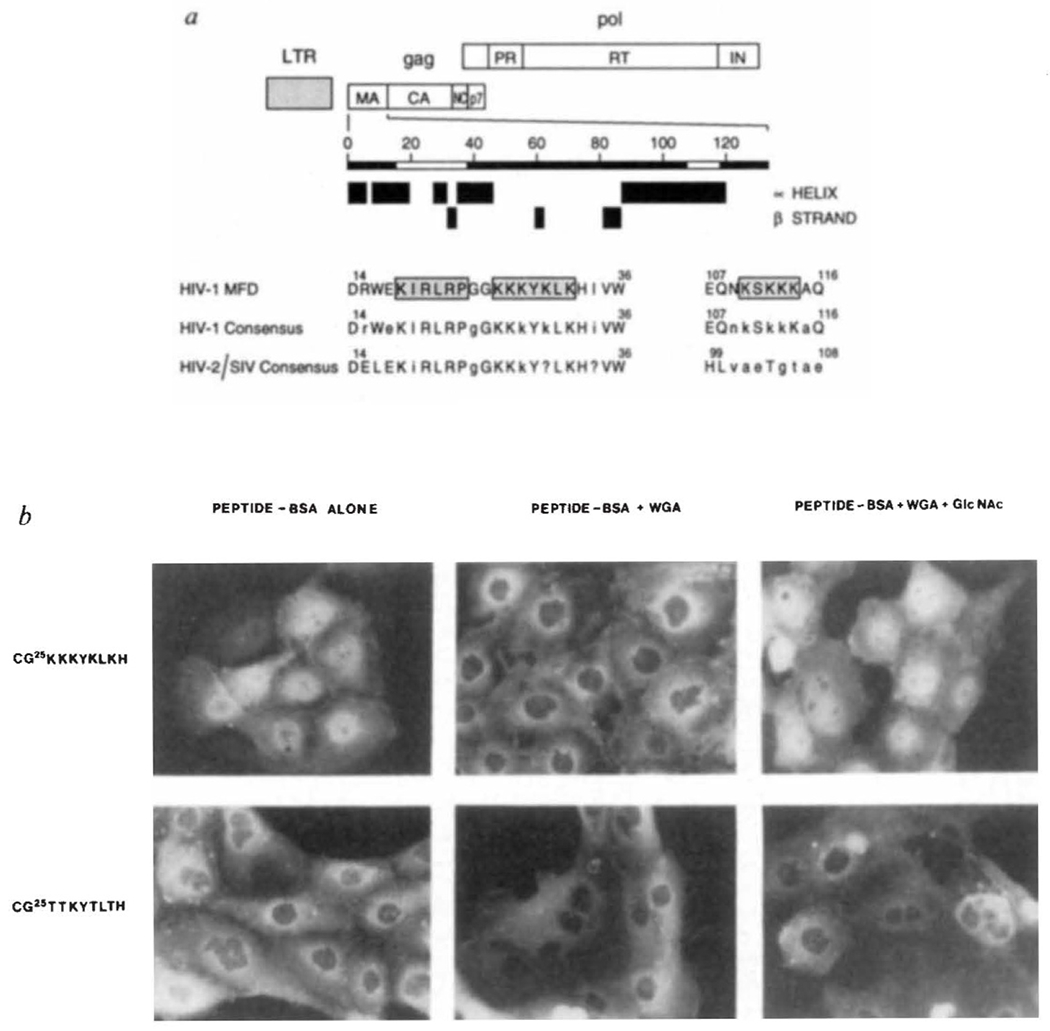
Putative nuclear targeting sequences within the HIV-1 gag matrix protein. a, Boundaries of the major products of the gag and pol polyproteins are indicated. The positions of predicted α-helices and β-strands were determined as in ref. 21. Sequences that resemble common NLS motifs6 are boxed and stippled. Amino-acid numbering, alignment and description of consensus sequences was helped by information contained within the HIV-1 sequence data base22. Upper case letters represent invariant residues whereas conserved and non-conserved amino acids are denoted by lower case letters and the question mark, respectively. b, Subcellular location of HIV-1 matrix NLS peptide–BSA conjugates after microinjection into rat epithelial cells.
METHODS. PtK1 cells (rat kangaroo kidney epithelia) were grown on glass cover slips in F-12 medium supplemented with 10% fetal bovine serum. Cells were microinjected in Hank’s balanced salt solution, essentially as detailed elsewhere8. Fluoresceinated HIV-1 matrix peptide–BSA conjugates containing an average of 6 peptides per BSA molecule7 were injected with or without WGA alone (2.5 mg ml−1) or WGA and its ligand, GlcNAc(0.5 M). Microinjected cells were incubated for 30 min at 37 °C, fixed and photographed8.
The in situ microinjection approach does not take into consideration the stoichiometry of viral preintegration complex components. As shown for both strong and weak NLSs6, increasing the number of NLS motifs associated with a particle improves the rate and extent of its import. In addition to the valency effects, the size of the carrier molecule greatly influences the extent to which nuclear import of NLS peptide–carrier conjugates are imported to the nucleus. Thus, sequences sufficient to direct nuclear import of BSA (diameter 70 Å) may be incapable of directing large particles such as a retroviral preintegration complex (diameter ~ 300 Å) to the nucleus. Therefore, the effects of amino-acid substitutions in the NLS of HIV-1 matrix were examined in the context of the viral preintegration complex. Isogeneic HIV-1 variants containing amino-acid substitutions within the N-terminal NLS (residues 25–33) of HIV-1 matrix were generated by site-directed mutagenesis. A single Lys 27→Thr substitution and double Lys 26, 27→Thr substitutions within HIV-1 matrix were created and reintroduced into an infectious HIV-1 molecular clone, HIV-1 MFD9. We had previously speculated that the ability of HIV-1 to replicate in non-proliferating cells might be due to nuclear entry of the preintegration complex before mitosis4,5. Therefore, the replication of HIV-1 matrix mutants in proliferating compared with non-proliferating cells was evaluated in an integration-dependent transactivation assay10. The target cells, HeLa CD4-LTR-β-gal cells, were arrested at the G2 phase of the cell cycle by γ-ray irradiation, and the infection of non-proliferating cells relative to infection of untreated cells was measured in a single round of infection5. As previously shown5, the onco-retrovirus MuLV very poorly infects non-proliferating cells compared with HIV-1 (Fig. 2). In contrast to wild-type HIV, neither the single Lys 27→T substitution (M16), nor the double Lys 26, 27→Thr substitution (M17) was able efficiently to infect G2-arrested cells (Fig. 2). This pattern of virus replication in dividing compared with non-proliferating host cells was more representative of an onco-retrovirus phenotype.
FIG. 2.
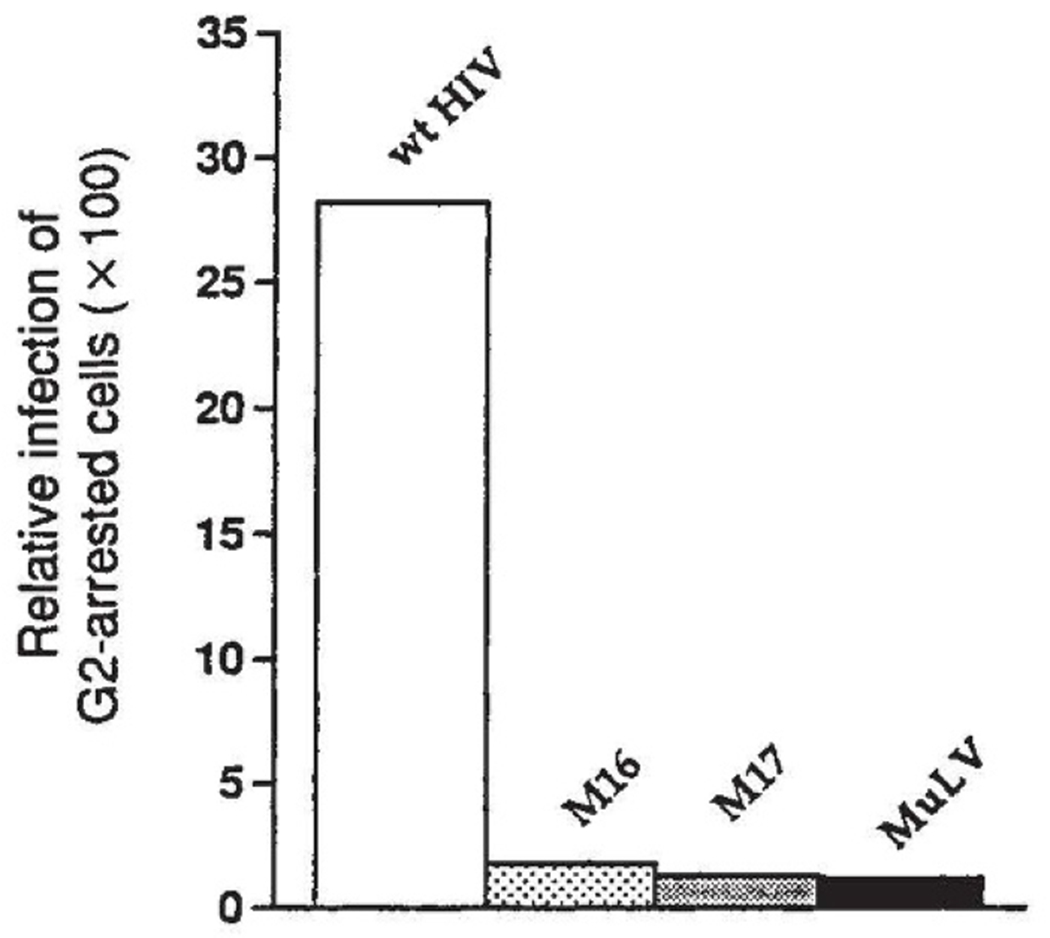
Relative infection of G2-arrested HeLa CD4 cells by HIV-1, MuLV and HIV-1 NLS matrix mutants. HeLaCD4-LTR/βgal cells were arrested in G2 by γ-irradiation, and infection was assayed by transactivation of an endogenous LTR–β-galactosidase gene as described5. In this assay, HeLa cells, which have been engineered to express high levels of the HIV receptor, CD4, contain a single copy of the bacterial β-galactosidase gene under transcriptional control of the HIV-1 LTR. On HIV-1 infection and integration β-galactosidase is produced after transactivation of the LTR by tat protein. Infected cells are scored after visualization of β-galactosidase activity in situ by staining cells with x-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). The relative infection of G2-arrested cells is the number of infected cells in the γ-irradiated (non-proliferating) culture divided by the number of infected cells in the proliferating culture for each viral infection. wt HIV is the parental MFD strain9; M16 is the single Lys 27→Thr matrix mutant of HIV-1 MFD; M17 is the double Lys 26, 27→Thr mutant of HIV-1 MFD; and MuLV is a MuLV-based retrovirus vector that encodes the HIV-1 tat gene5.
METHODS. Amino-acid substitutions within HIV-1 gag matrix were introduced by site-directed mutagenesis. A 3-kilobase (kb) Sphl fragment of the infectious molecular clone, HIV-1 MFD9 (similar in genotype to the prototypic HXB2 isolate), was inserted in M13mp19. Single (Lys 27→Thr) and double (Lys 26, 27→Thr) amino-acid substitutions were created by annealing M13 phage DNA with the mutagenic oligonucleotides, M16(5′-aggcctgggggaaagacaaaatat-3′) and M17(5′-aggcctgggggaacgacaaaatat-3′), respectively. Both oligonucleotides were designed to introduce translationally silent Stul recognition sites. DNA fragments containing the desired mutations were inserted in the HIV-1 MFD DNA backbone. Wild-type and mutant HIV-1 variants were recovered 48 h after transfection of HeLa cells and quantified by HIV-1 gag p24 ELISA. When cells are transfected with infectious DNA clones, events preceding provirus establishment are bypassed and only late events including assembly and release of virion particles are reconstituted. The amino-acid substitutions within the HIV-1 gag matrix NLS did not influence virus maturation and both quantity and morphology of virus particles produced from HeLa cells transfected with matrix mutant and wild-type molecular clones of HIV-1 were identical (not shown).
A comparison of the abilities of wild-type HIV-1 and isogeneic HIV-1 matrix mutants to elicit a spreading infection in proliferating CD4+ cells and in CD4+ cells arrested in the cell cycle by aphidicolin is shown in Fig. 3. In proliferating CD4+ cultures (−Aph), replication kinetics of both single and double HIV-1 matrix mutants was indistinguishable from that of wild-type virus. In contrast, under conditions of growth arrest (+Aph), virus antigen production in cultures infected with both HIV-1 NLS matrix mutants was not detected (Fig. 3). When the aphidicolin block was removed and cell proliferation resumed, virus replication and antigen production were detected in cultures infected with both HIV-1 matrix mutants (not shown).
FIG. 3.
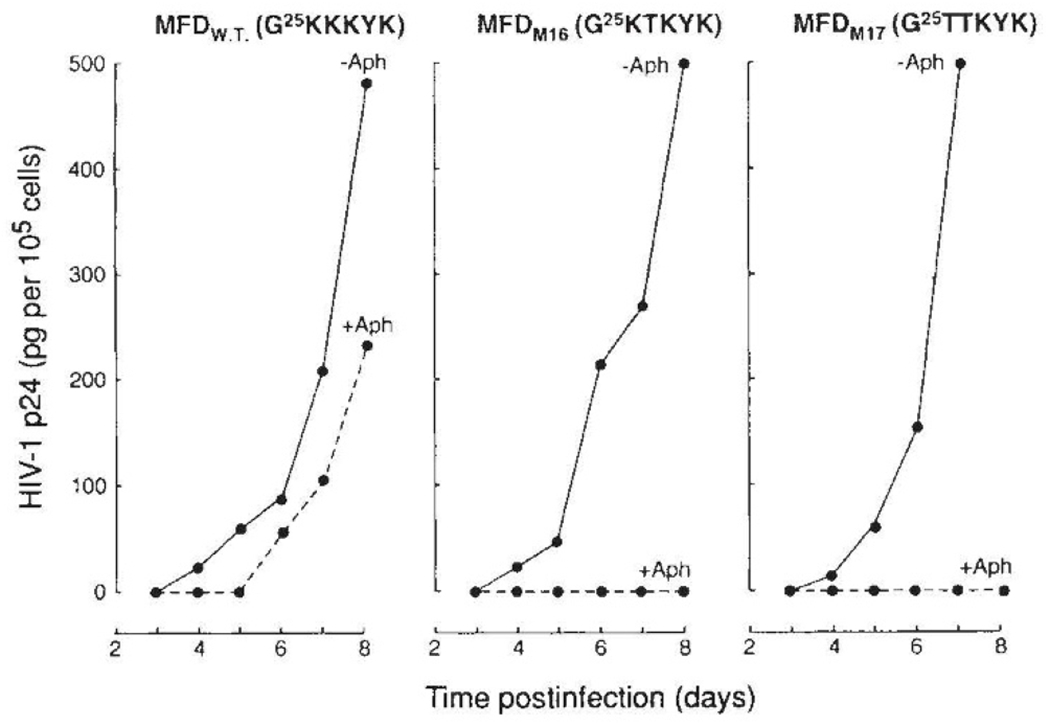
Replication of wild-type and gag matrix mutants of HIV-1 in dividing and in growth-arrested CD4+ cells. CD4+ MT-4 cell cultures were arrested at the G1/S interface of the cell cycle by the action of aphidicolin as detailed elsewhere4. Aphidicolin was removed 72 h after infection with HIV-1 (50 pg per 106 cells). At 24-h intervals, 1 × 105 cells were removed for quantification of cell-associated HIV-1 gag p24 in proliferating (solid line) and in growth-arrested (hatched line) cultures.
Major complementary DNA products of HIV-1 reverse transcription in the cytoplasm and nucleus of acutely infected cells are illustrated in Fig. 4a. Viral cDNA synthesis in dividing compared with growth-arrested cells infected with wild-type HIV-1 and an HIV-1 matrix mutant (Lys 26, 27→Thr) was equivalent (Fig. 4b). Nuclear localization of viral DNA, shown by abundance of nucleus-specific two long terminal repeat (LTR) circle forms of viral DNA, was equivalent in dividing and growth-arrested cells infected with wild-type HIV-1. In contrast, after infection with the matrix mutant, formation of two LTR circle forms was evident in dividing cells but not growth-arrested cells. Synthesis and nuclear import of viral DNA in proliferating and growth-arrested cells infected with a single (Lys 27→Thr) HIV-1 matrix mutant was identical to that for the double matrix mutant (not shown). Taken together, these results suggest that under conditions of growth arrest, amino-acid substitutions within HIV-1 matrix antigen interrupt the ability of viral nucleic acids to enter the nucleus and as a consequence restricts the ability of HIV-1 to elicit a spreading infection in non-proliferating host cells.
FIG. 4.
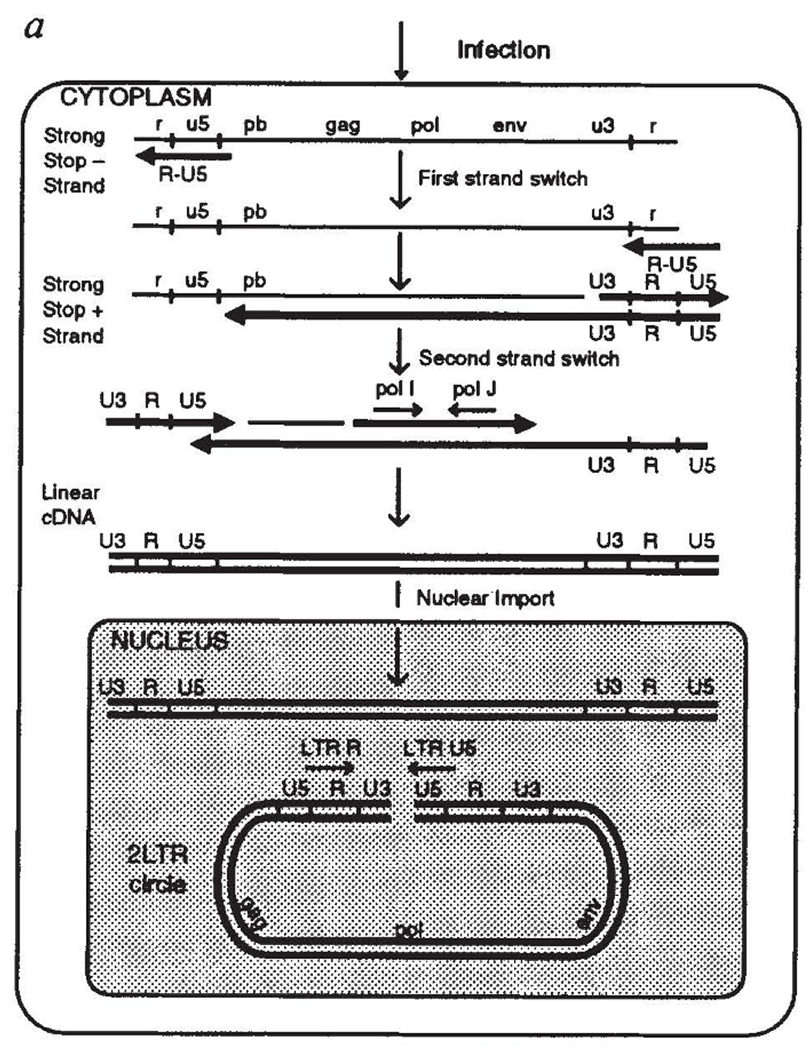
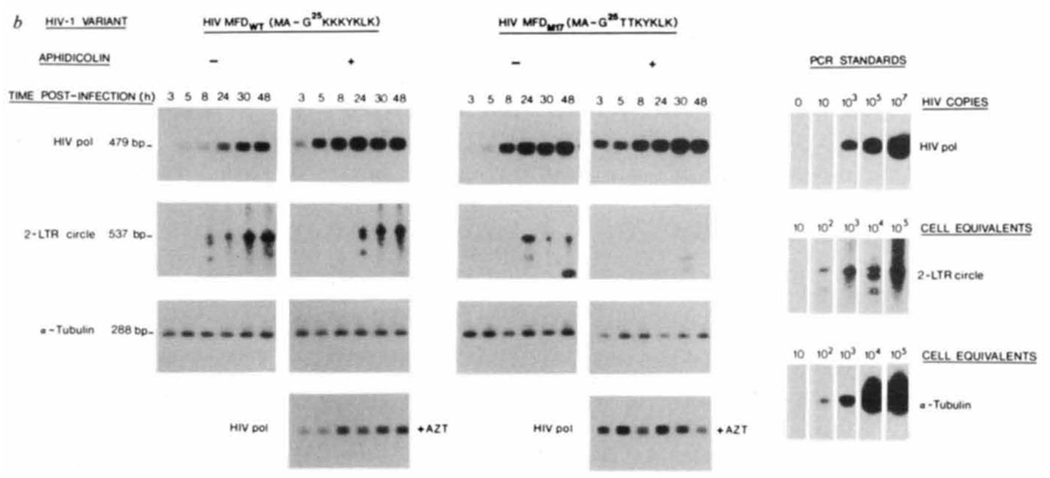
Synthesis and nuclear import of viral DNA by wild-type and gag matrix mutants of HIV-1 in proliferating and growth-arrested CD4+ T-cell cultures. a, Major reverse transcription products in cytoplasm and nucleus of HIV-1-infected cells. After reverse transcription is initiated, two strand-switching events result in a full-length cDNA containing duplicated U3, R and U5 regions. After transport to the nucleus, some linear molecules undergo circularization, yielding 2 LTR circle forms. Although such circle forms of viral DNA do not appear to be precursors of the provirus23, they are formed only after synthesis of full-length viral cDNA and its transport to the nucleus1,23 and thus represent a specific marker with which to follow nuclear localization of viral DNA. HIV-1 pol I and J primers amplify products of reverse transcription which follow the second strand switch24, whereas LTR R and U5 primers amplify 2 LTR circle forms of extrachromosomal viral DNA. Viral RNA and cDNA are represented by thin lines/lower-case letters and thick lines/uppercase letters, respectively. pb, Primer binding site. b, Proliferating (− Aph) and growth-arrested ( +Aph) CD4+ MT-4 cells were infected with HIV-1 as outlined in Fig. 3. At the indicated times postinfection, cell aliquots were withdrawn (2–3 × 105 cells) for isolation of total cellular DNA as detailed elsewhere25. HIV-1 pol-specific amplification products obtained in the presence of AZT (1 μm) represent incomplete reverse transcription products contained within the virion and not de novo synthesis of viral DNA26,27.
METHODS. PCR conditions for amplification of linear and circle forms of viral DNA were exactly as detailed elsewhere4,25. One and two rounds of PCR were used for pol and LTR circle primers, respectively. PCR amplification products were visualized after Southern blot transfer and hybridization with [α-32P]-labelled oligonucleotide probes. HIV-1 pol standards were generated by PCR on log dilutions of cloned HIV-1 MF DNA whereas standards for circle viral DNA forms and cellular DNA generated by PCR on log dilutions of HIV-1-infected MT-4 cells.
The study outlined here is pertinent to the understanding of determinants which govern the ability of HIV-1 to replicate within non-proliferating cells. This pattern of HIV-1 replication is supported by in vitro studies demonstrating provirus establishment in G2-arrested CD4+ HeLa cells5 and primary macrophages3,11 and is reflected by distribution of the virus in non-T-cell compartments of the infected host, such as terminally differentiated and non-proliferating cells of macrophage or microglial phenotype12–15. The propensity for replication in non-dividing cells distinguishes HIV-1, and perhaps lentiviruses in general16, from the animal onco-retroviruses1,2. Although retroviruses such as MuLV will infect non-proliferating cells and initiate viral DNA synthesis17 within the context of a preintegration complex, the large physical size of retroviral preintegration complexes18 precludes them from passing through the aqueous channel of nuclear pores, requiring instead that they enter the nuclear compartment after breakdown of the nuclear membrane during mitosis2. The requirement for mitosis at cell division is obviated by the karyophilic properties of the preintegration complex of HIV-1, which undergoes active nuclear import after acute virus infection of non-proliferating cells4. Determinants which govern nuclear import characteristics of HIV-1 DNA are contained within the virus core because MuLV pseudotypes containing gag/pol proteins of HIV-1 will integrate within non-proliferating cells5. Of the gag/pol proteins, integrase19,20 and matrix20 proteins of gag and reverse transcriptase protein of pol20 have been identified in association with viral nucleic acids in the context of the viral preintegration complex. We have shown here that the HIV-1 gag matrix protein, by virtue of a nuclear localization sequence at its N terminus, contributes to the karyophilic properties of the viral preintegration complex and influences the ability of the virus to replicate within non-proliferating cells. □
ACKNOWLEDGEMENTS:
We thank T. Pushkarakya for technical assistance, H. Varmus and S. Rhode for discussion and C. Bowers for manuscript preparation. S.H. is a scholar of the American foundation for AIDS research, AmFAR. This work was supported by the NIH (M.S., D.G., M.E.) and by AmFAR (M.B.).
References
- 1.Varmus HE & Swanstrom R in RNA Tumor Viruses (eds Weiss R, Teich N, Varmus HE & Coffin J) 369–512 (Cold Spring Harbor Laboratory Press, New York, 1984). [Google Scholar]
- 2.Roe T, Reynolds TC, Yu G & Brown PO EMBO J. 12, 2099–2108 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinberg JB, Mathews TJ, Cullen BR & Malim MH J. exp. Med 174, 1477–1482 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bukrinsky MI et al. Proc. natn. Acad. Sci. U.S.A 89, 6580–6584 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis P Hensel M &, Emerman M EMBO. J 11, 3053–3068 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forbes DJ A. Rev. Cell Biol 8, 495–529 (1992). [DOI] [PubMed] [Google Scholar]
- 7.Goldfarb DS, Gariepi J, Schoolnik G & Kornberg RD Nature 322, 641–644 (1986). [DOI] [PubMed] [Google Scholar]
- 8.Breeuwer M & Goldfarb DS Cell 60, 999–1008 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Stevenson M et al. J. Virol 64, 3792–3803 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimpton J & Emerman M J. Virol 66, 2232–2239 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gendelman HE et al. J. Virol 58, 67–74 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw GM et al. Science 226, 1165–1170 (1984). [DOI] [PubMed] [Google Scholar]
- 13.Koenig S et al. Science 223, 1089–1093 (1986). [Google Scholar]
- 14.Wiley CA, Schrier RD, Nelson JA, Lampert PW & Oldstone MBA Proc. natn. Acad. Sci. U.S.A 83, 7089–7093 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price RW et al. Science 239, 586–592 (1988). [DOI] [PubMed] [Google Scholar]
- 16.Haase AT et al. Virology 119, 399–410 (1982). [DOI] [PubMed] [Google Scholar]
- 17.Miller DG, Adam MA & Miller AD Molec. cell. Biol 10, 4239–4242 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowerman B, Brown PO, Bishop JM & Varmus HE Genes Dev. 3, 469–478 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Farnet CM & Haseltine WA J. Virol 65, 1990–1915 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukrinsky MI et al. Proc. natn. Acad. Sci. U.S.A 90, 6125–6129 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eliopoulos EE et al. Int. J. biol. Macromol 4, 263–276 (1982). [Google Scholar]
- 22.Meyers G et al. Human Retroviruses and AIDS (Los Alamos National Laboratory, New Mexico, 1992). [Google Scholar]
- 23.Brown PO, Bowerman B, Varmus HE & Bishop JM Cell 49, 347–356 (1987). [DOI] [PubMed] [Google Scholar]
- 24.Hsu W-S & Temin HM Science 250, 1227–1232 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Bukrinsky MI, Stanwick TL, Dempsey MP & Stevenson M Science 254, 423–427 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lori F et al. J. Virol 66, 5067–5074 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trono D J. Virol 66, 4893–4900 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]