

Abstract
Previous genome-wide association studies (GWASs) of stroke — the second leading cause of death worldwide — were conducted predominantly in populations of European ancestry1,2. Here, in cross-ancestry GWAS meta-analyses of 110,182 patients who have had a stroke (five ancestries, 33% non-European) and 1,503,898 control individuals, we identify association signals for stroke and its subtypes at 89 (61 new) independent loci: 60 in primary inverse-variance-weighted analyses and 29 in secondary meta-regression and multitrait analyses. On the basis of internal cross-ancestry validation and an independent follow-up in 89,084 additional cases of stroke (30% non-European) and 1,013,843 control individuals, 87% of the primary stroke risk loci and 60% of the secondary stroke risk loci were replicated (P < 0.05). Effect sizes were highly correlated across ancestries. Cross-ancestry fine-mapping, in silico mutagenesis analysis3, and transcriptome-wide and proteome-wide association analyses revealed putative causal genes (such as SH3PXD2A and FURIN) and variants (such as at GRK5 and NOS3). Using a three-pronged approach4, we provide genetic evidence for putative drug effects, highlighting F11, KLKB1, PROC, GP1BA, LAMC2 and VCAM1 as possible targets, with drugs already under investigation for stroke for F11 and PROC. A polygenic score integrating cross-ancestry and ancestry-specific stroke GWASs with vascular-risk factor GWASs (integrative polygenic scores) strongly predicted ischaemic stroke in populations of European, East Asian and African ancestry5. Stroke genetic risk scores were predictive of ischaemic stroke independent of clinical risk factors in 52,600 clinical-trial participants with cardiometabolic disease. Our results provide insights to inform biology, reveal potential drug targets and derive genetic risk prediction tools across ancestries.
Subject terms: Stroke, Genome-wide association studies, Stroke, Predictive markers, Genetic markers
A cross-ancestry meta-analysis of genome-wide association studies identifies association signals for stroke and its subtypes at 89 (61 new) independent loci, reveals putative causal genes, highlighting F11, KLKB1, PROC, GP1BA, LAMC2 and VCAM1 as potential drug targets, and provides cross-ancestry integrative risk prediction.
Main
Stroke is the second leading cause of death worldwide, responsible for approximately 12% of total deaths, with an increasing burden particularly in low-income countries 6. Characterized by a neurological deficit of sudden onset, stroke is predominantly caused by cerebral ischaemia (of which the main aetiological subtypes are large-artery atherosclerotic stroke (LAS), cardioembolic stroke (CES), and small-vessel stroke (SVS)) and, less often, by intracerebral haemorrhage (ICH). The frequency of stroke subtypes differs between ancestry groups as exemplified by a higher prevalence of SVS and ICH in Asian and African populations compared with European populations. Most genetic loci associated with stroke have been identified in populations of European ancestry. The largest published GWAS meta-analysis to date (67,162 cases and 454,450 control individuals, MEGASTROKE) reported 32 stroke risk loci1. To identify new genetic associations and provide insights into stroke pathogenesis and putative drug targets, we first performed a cross-ancestry GWAS of 1,614,080 participants, including 110,182 patients who had a stroke, and followed up genome-wide significant signals in an independent dataset of 89,084 patients who had a stroke and 1,013,843 control individuals. We then characterized the identified stroke risk loci by leveraging expression and protein quantitative trait loci, cross-ancestry fine-mapping and shared genetic variation with other traits. Finally, we used a series of approaches for genomics-driven drug discovery for stroke prevention and treatment, and examined the prediction of stroke with polygenic scores (PGSs) across ancestries in the setting of both population-based studies and clinical trials.
Genetic discovery from GWASs
We performed a fixed-effect inverse-variance weighted (IVW) GWAS meta-analysis on 29 population-based cohorts or biobanks with incident stroke ascertainment and 25 clinic-based case–control studies, comprising up to 110,182 patients who had a stroke and 1,503,898 control individuals (of whom 45.5% were in longitudinal cohorts or biobanks), nearly doubling the number of cases in previous stroke GWASs (the GIGASTROKE initiative; Supplementary Table 1 and Extended Data Fig. 1). Genome-wide genotyping and imputation characteristics are described in Supplementary Table 2. The cohorts included individuals of European (66.7% of the patients who had a stroke), East Asian (24.8%), African American (3.7%), South Asian (3.3%) and Hispanic (1.4%) ancestry. Analyses were performed for any stroke (AS; comprising ischaemic stroke, ICH, and stroke of unknown or undetermined type), any ischaemic stroke regardless of subtype (AIS; n = 86,668) and ischaemic stroke subtypes (LAS, n = 9,219; CES, n = 12,790; SVS, n = 13,620). We also conducted separate GWAS analyses of incident AS and AIS (n = 32,903 and n = 16,863, respectively) in longitudinal population-based cohort studies.
Extended Data Fig. 1. GIGASTROKE study workflow.

Study workflow and rationale. EUR: European; EAS: East-Asian; AFR: African; HIS: Hispanic; SAS: South Asian; AS: any stroke; AIS: any ischaemic stroke; LAS: large artery stroke; CES: cardioembolic stroke; SVS: small vessel stroke; GWAS: genome-wide association study; IVW: inverse-variance weighted; MR-MEGA: meta-regression of multi-ethnic genetic association; COJO:conditional and joint analysis; VEGAS2:versatile gene-based association study 2; MTAG: multi-trait analysis of GWAS; TWAS: Transcriptome-wide association study; coloc: Colocalisation Test; PWAS: Proteome-wide association studies; pQTL-MR: protein quantitative trait loci Mendelian Randomization; SuSiE: sum of single effects model; MENTR: Mutation Effect prediction on Non-coding RNA TRanscription; PIP: posterior probability; FDR: false discovery rate; LDSC-COV: covariate-adjusted LD score regression; MR-Egger: Mendelian randomization-Egger; GREP: genome for REPositioning drugs; ATC: Anatomical Therapeutic Chemical; P+T: pruning and thresholding; PRScs: polygenic risk score under continuous shrinkage; BBJ: Biobank Japan; TIMI: thrombolysis in myocardial infarction; MVP: Million Veteran Program; SIREN: Stroke Investigative Research and Educational Network.
We tested up to around 7,588,359 single-nucleotide polymorphisms (SNPs) with a minor allele frequency (MAF) of ≥0.01 for association with stroke. The linkage-disequilibrium score intercepts for our ancestry-specific GWAS meta-analyses ranged from 0.91 to 1.12, suggesting that there was no systematic inflation of association statistics (Supplementary Table 3). By performing IVW GWAS meta-analyses, we identified variants associated with stroke at genome-wide significance (P < 5 × 10−8) at 60 loci, of which 33 were new (Fig. 1 and Supplementary Table 4). Lead variants at all of the new loci were common (MAF ≥ 0.05), except for low-frequency intronic variants in THAP5 (MAF = 0.02, in complete association (r2 = 1) with variants in the 5′ UTR of NRCAM) associated with cross-ancestry incident AS/AIS, and in COBL (MAF = 0.04) associated with AS/AIS in South Asian individuals. Most of the associations for these 60 loci were with AS (48 loci, 23 new) and AIS (45 loci, 18 new), and one of the AIS loci was associated only with incident AIS (Supplementary Table 4c). Although AIS subtypes were not available in some population-based cohorts (Supplementary Table 1), genome-wide significance was reached for 4 loci for LAS, 8 for CES and 7 for SVS (of which 1, 3 and 3 were new, respectively; Supplementary Table 4). Our results include a large and comprehensive description of stroke genetic risk variants in each of the five represented ancestries. In cross-ancestry meta-analyses, 53 loci (51 loci after controlling for ancestry-specific linkage-disequilibrium score intercepts) reached genome-wide significance (Supplementary Table 4), whereas 42 loci were genome-wide significant in individual ancestries (35 in Europeans, 6 in East Asians, 1 in South Asians and 2 in African Americans; Supplementary Table 4). Using conditional and joint analysis (GCTA-COJO)7, we confirmed three independent signals at PITX2 and two at SH3PXD2A1 (CES in Europeans; Supplementary Table 5). We also performed cross-ancestry gene-based association tests using VEGAS28 and MAGMA9, which revealed 267 gene-wide significant associations (P < 2.63 × 10−6) at 39 loci, of which 14 were in 8 new loci that did not reach genome-wide significance in the single-variant analyses (AGAP5/SYNPO2L/SEC24C/CHCHD1, CD96, HNRNPA0, MAMSTR, PPM1H, RALGAPA1, USP34 and USP38; Supplementary Tables 6 and 7).
Fig. 1. Identifying genetic variants that influence stroke risk.

Ideogram showing 89 genome-wide significant stroke-risk loci. The shapes correspond to ancestry: circles, cross-ancestry (CROSS-ANC); diamonds, Europeans (EUR); triangles, East Asians (EAS); squares, African Americans (AFR) or South Asians (SAS). Colours correspond to stroke types: green, AS; red, AIS; light blue, SVS; dark blue, CES; purple, LAS. The nearest genes to lead variants are displayed. Loci are characterized as follows, on the basis of replication results (Methods): bold with asterisk, high confidence; bold without asterisk, intermediate confidence; not bold, low confidence; underlined, loci identified in secondary MR-MEGA and MTAG analyses. Black and grey font indicate new and known loci, respectively. The numbers at the top indicate the chromosome.
Next, we conducted a secondary cross-ancestry GWAS meta-analysis using MR-MEGA10, which accounts for the allelic heterogeneity between ancestries. We identified three additional genome-wide significant loci for AS (all new), near TSPAN19, and in introns of DAZL and SHOC1, all showing high heterogeneity in allelic effects across ancestries (heterogeneity P < 0.01; Supplementary Table 8). To further enhance the statistical power for AIS subtypes, we conducted secondary multitrait analyses of GWASs (MTAG)11 in Europeans and East Asians, including traits correlated with specific stroke subtypes, namely (1) coronary artery disease (CAD) for LAS, both caused by atheroma; (2) atrial fibrillation for CES, as its main underlying cause; and (3) white matter hyperintensity volume (WMH, an MRI-marker of cerebral small vessel disease) for SVS (available in Europeans only). In Europeans, 11 additional loci were associated with LAS (10 new), 3 with SVS (all reported in a recent SVS GWAS2) and 5 with CES (all new; Supplementary Tables 9–11). Moreover, 18 and 15 additional genome-wide significant associations were identified (all new) for AS and AIS, respectively, using MTAG with WMH, CAD and atrial fibrillation (Supplementary Tables 12 and 13). In East Asian individuals, one locus was associated with AS (FGF5) and one with LAS (HDAC9, new in East Asians) using MTAG. This brings the number of identified stroke-risk loci from primary (IVW) and secondary (MR-MEGA and MTAG) analyses to 89 in total (61 new), of which 69 were associated with AS, 45 with AIS, 15 with LAS, 13 with CES and 10 with SVS (of these 44, 33, 11, 8 and 3 were new, respectively; Fig. 1 and Supplementary Tables 4, 8 and 9–14).
Independent follow-up of GWAS signals
We followed up genome-wide significant stroke-risk loci both internally and externally. First, we sought to replicate the 42 stroke-risk loci that reached genome-wide significance in individual ancestries in at least one other ancestry group among the discovery samples. We successfully replicated, with consistent directionality, 10 of these loci at P < 1.19 × 10−3 (accounting for the number of loci tested), of which 7 were genome-wide significant in Europeans, 1 in East Asians, and 2 in both Europeans and East Asians. An additional 15 loci showed nominal association (P < 0.05) in at least one other ancestry (Supplementary Table 15).
Second, we gathered an independent dataset of 89,084 individuals who had a stroke (AS; of which 85,546 AIS; 70.0% European, 15.6% African American, 10.1% East Asian, 4.1% Hispanic and 0.1% South Asian) and 1,013,843 control individuals, mostly from large biobanks, for external replication (the biobank setting did not allow suitable ischaemic stroke subtype analyses). Out of the 60 loci that reached genome-wide significance in the IVW meta-analyses, 48 loci (80%) replicated at P < 0.05 with consistent directionality (Extended Data Fig. 2), of which 31 (52%) replicated at P < 8.2 × 10−4 (accounting for the number of loci tested) (Supplementary Table 16). When considering both the internal and external follow-up, 52 (87%) of the 60 IVW loci replicated, of which 37 replicated with high confidence, and 15 with intermediate confidence (Methods, Fig. 1 and Supplementary Table 14). The 8 loci that did not replicate were labelled as low confidence (Methods and Supplementary Table 14). Four of these were ethnic specific and three were low-frequency variants that were monomorphic in some ancestries and were therefore probably underpowered for replication.
Extended Data Fig. 2. Graphical representation of replication results.

Shown are effect sizes and 95% confidence intervals for the 48 replicated IVW loci for (A) cross-ancestry discovery and replication association results for any stroke and (B) cross-ancestry discovery and replication association results for any ischaemic stroke. OR = odds ratio; 95% CI = 95% confidence interval.
Within the secondary analyses, none of the three MR-MEGA loci replicated, although one was borderline significant (Supplementary Table 16). Of the 26 MTAG loci, 18 (69%) replicated with AS or AIS at P < 0.05, of which 9 (35%) replicated with high confidence (P < 1.7 × 10−3, accounting for 29 secondary loci tested; Supplementary Table 16). Of the eight MTAG loci that did not replicate, seven showed a consistent directionality and four were subtype specific and were therefore underpowered to detect associations with AS or AIS.
Cross-ancestry effects and fine-mapping
For the 60 loci associated with stroke risk derived from the IVW meta-analyses, we first demonstrated the added value in terms of locus discovery of including non-European samples, showing a clear gain in power beyond sample size increase, compared with the incremental addition of European ancestry samples (Extended Data Fig. 3). We next compared the per-allele effect size across the three ancestries with the largest sample size (European, East Asian, African American). Correlations of per-allele effect sizes of index variants varied from r = 0.55 (European with African American) to r = 0.66 (European with East Asian) and r = 0.74 (East Asian with African American; Fig. 2a).
Extended Data Fig. 3. Increase in power with increase in population diversity.

The scatter plot shows the number of loci identified with incremental increase in sample size and population diversity. The diagonal line reflects the increase in number of genome-wide significant loci with increasing sample size of European ancestry only. The increase in number of loci departs from this line when adding non-European ancestry samples; EUR: European; EAS: East Asian; AFR: African; HIS: Hispanic; SAS: South Asian; BBJ: Biobank Japan; CKB: China Kadoorie Biobank. Strat0_EUR: N = 22,634 stroke cases (European population-based longitudinal cohorts); Strat1_Strat0&EUR: N = 26,253 stroke cases, adding Spanish and Estonian samples; Strat2_Strat1&EUR: N = 32,980 stroke cases, adding German and Dutch samples; Strat3_Strat2&EUR: N = 42,783, adding large European biobanks; Strat4_Strat3&EUR: N = 73,652 stroke cases, adding MEGASTROKE European; Strat5_Strat4&EAS: N = 91,303 stroke cases, adding Japanese BBJ; Strat6_Strat5&EAS: N = 99,661 stroke cases, adding Chinese CKB; Strat7_Strat6&EAS: N = 101,065 stroke cases, adding other East Asian samples; Strat8_Strat7&AFR: N = 105,026 stroke cases, adding African ancestry samples; Strat9_Strat8&HIS: N = 106,542 stroke cases, adding South American ancestry samples; Strat10_Strat9&SAS, N = 110,182 stroke cases, adding South Asian ancestry samples.
Fig. 2. Effect-size comparison across ancestry groups of lead variants identified in stroke GWASs and cross-ancestry fine-mapping.

a, Plots showing the Pearson’s correlation coefficient (r) between the effect sizes (β) of the 60 stroke-risk alleles on AS significant after multiple-testing correction (P < 0.017) in Europeans and East Asians (left; r (95% CI) = 0.66 (0.47–0.79), P = 1 × 10−7); Europeans and African Americans (middle; r (95% CI) = 0.55 (0.33–0.71), P = 2 × 10−5); and East Asians and African Americans (right; r (95% CI) = 0.74 (0.58–0.85), P = 8 × 10−10). n = 60 independent stroke-risk variants from the IVW meta-analyses were used to compute Pearson’s correlation coefficients (r) of the effect sizes between ancestries. The nearest gene is reported for SNPs showing a difference in effect size (β, absolute value) of >0.05 between a pair of ancestries. The dots represent the effect-size (β) estimates and the bars represent the 95% CI of the estimates. Two-sided P values of the deviation of Pearson’s correlation coefficient from zero are reported. Colour corresponds to genome-wide significant association (P < 5 × 10−8) in individual ancestries: purple, European only (±cross-ancestry); green, East Asian only (±cross-ancestry); yellow, African American only (±cross-ancestry); blue, both ancestries (±cross-ancestry); red, cross-ancestry only; grey, not genome-wide significant in two plotted ancestries and in cross-ancestry. b, Locus plots of variants at SH3PXD2A in five ancestries. Fine-mapped variants are shown only in European and East Asian individuals (insufficient power for other ancestries). Variants are coloured on the basis of their linkage disequilibrium with the cross-ancestry lead variant (rs4918058), shown by the purple diamonds. In the fine-mapping plots, variants in the SuSiE 95% credible sets (CS) are shown. Shared variants between credible sets of European and East Asian participants are indicated by black circles. The red vertical lines represent the position of the lead variants in European (rs55983834) and East Asian (rs4918058) participants. The grey dashed horizontal lines represent P = 5 × 10−8. The linkage disequilibrium of each ancestry was derived from the 1000 Genomes Project.
To identify putative causal variants at stroke-risk loci identified through IVW meta-analyses, we performed multiple-causal-variant fine-mapping using SuSiE12, separately in European and East Asian participants (Methods). Across stroke types, we identified 110 and 16 95% credible set–trait pairs in European and East Asian participants, respectively, each of which having a 95% posterior probability of containing a causal variant, with multiple credible sets identified at 6 (in Europeans) and 1 (in East Asians) stroke-risk loci (Supplementary Tables 17–19). Within the credible sets identified in European participants, 17 variants were found to have a posterior inclusion probability (PIP) of >0.9. We found overlapping credible sets between European and East Asian participants at SH3PXD2A (19 overlapping variants), suggesting that there is cross-ancestry-shared genetic architecture at this locus (Fig. 2b). Two loci had credible sets with a single variant (rs10886430 at GRK5 (PIP = 0.999), associated with GRK5 platelet gene expression and thrombin-induced platelet aggregation13, and rs1549758 at NOS3, PIP = 0.995), probably representing strong targets for functional validation.
Although there were six non-synonymous variants among credible sets (rs671 (ALDH2), rs8071623 (SEPT4), rs35212307 (WDR12), rs72932557 (CARF), rs11906160 (MYH7B) and rs2501968 (CENPQ)), exonic variants for coding RNA within credible sets were few (1.2%). To detect putative causal regulatory variants, we conducted an in silico mutagenesis analysis using MENTR, a machine-learning method to precisely predict transcriptional changes caused by causal variants3. From credible sets, we obtained 78 robust predictions of variant–transcript-model sets comprising 13 variants and 19 transcripts (Supplementary Table 20), involving multiple cell types, consistent with the diversity of mechanisms that underlie stroke aetiology. For example, the G allele of rs12476527 (5′ UTR of KCNK3) is a risk allele for stroke and was predicted to increase KCNK3 expression in kidney cortex tubule cells, despite no expression quantitative trait loci (eQTL) of this variant being reported in Genotype-Tissue Expression (GTEx, v.8) or eQTLgen (2019-12-23). The same G allele has been associated with higher systolic blood pressure14. Furthermore, three variants (rs12705390 at PIK3CG, rs2282978 at CDK6 and rs2483262 at PRDM16) were predicted to affect the expression of a long non-coding RNA and enhancer RNAs, predominantly in endothelial cells, as well as other vascular cells and visceral preadipocytes, whereas a promoter variant of SH3PXD2A was predicted to modulate its expression in macrophages.
Characterizing stroke-associated loci
VEGAS2Pathway15 analysis revealed significant enrichment (P < 5.01 × 10−6) of stroke-risk loci in pathways involved in (1) carboxylation of amino-terminal glutamate residues required for the activation of proteins involved in blood clot formation and regulation; (2) negative regulation of coagulation; and (3) angiopoietin receptor Tie2-mediated signalling, involved in angiogenesis (Supplementary Table 21).
We examined shared genetic variation with 12 (in Europeans) and 10 (in East Asians) vascular risk factors and disease traits (Methods and Supplementary Methods). In Europeans, the lead variants for stroke at 57 of the 89 primary and secondary risk loci (64.0%) were associated (P < 5 × 10−8) with at least one vascular trait, most frequently blood pressure (33 loci, 37.1%; Extended Data Fig. 4 and Supplementary Table 22). After correction for multiple testing (Methods; P < 4.17 × 10−3), all of the vascular-risk traits except for low-density lipoprotein (LDL)-cholesterol showed significant genetic correlation (rg) with at least one stroke type, the strongest correlations being for CAD and LAS (rg = 0.73), atrial fibrillation and CES (rg = 0.63), and systolic blood pressure (SBP) with all stroke types (rg ranging from 0.21 for CES to 0.49 for LAS and SVS; Extended Data Fig. 5 and Supplementary Table 23). Using two-sample Mendelian randomization (MR), we found evidence for a possible causal association for every vascular-risk trait except for triglycerides with at least one stroke type (P < 4.17 × 10−3), with some subtype-specific association patterns. Genetic liability to WMH was associated with increased risk of SVS but not other stroke subtypes, whereas genetic liability to venous thromboembolism was associated with AS, AIS, CES and LAS, but not SVS (Extended Data Fig. 5 and Supplementary Table 24). Owing to a limited overlap between the European GIGASTROKE sample and cohorts included in GWASs for the exposure traits, we ran sensitivity analyses weighting our genetic instruments on the basis of a sub-sample of the UK Biobank, excluding cases included in GIGASTROKE16. The notable consistency of these with the main analyses confirmed their robustness against weak instrument bias (Supplementary Table 25). We confirmed directionality using the Steiger test (Supplementary Table 24) and ruled out reverse causation with reverse MR (Supplementary Table 26). In East Asian individuals, SBP, diastolic blood pressure (DBP), body mass index (BMI) and atrial fibrillation showed significant genetic correlation with AS (rg = 0.45, 0.39, 0.24 and 0.32 versus rg = 0.36, 0.21, 0.22 and 0.44 in Europeans) and AIS (except for BMI), with evidence for a causal association of SBP and DBP with AS, AIS and SVS; CAD with AS, AIS and LAS; and atrial fibrillation with CES (Extended Data Fig. 6 and Supplementary Tables 23 and 24). Notably, MR analyses performed with binary exposures should be interpreted with caution owing to the potential violations of the exclusion restriction assumption16.
Extended Data Fig. 4. Association of stroke risk variants with vascular risk traits.
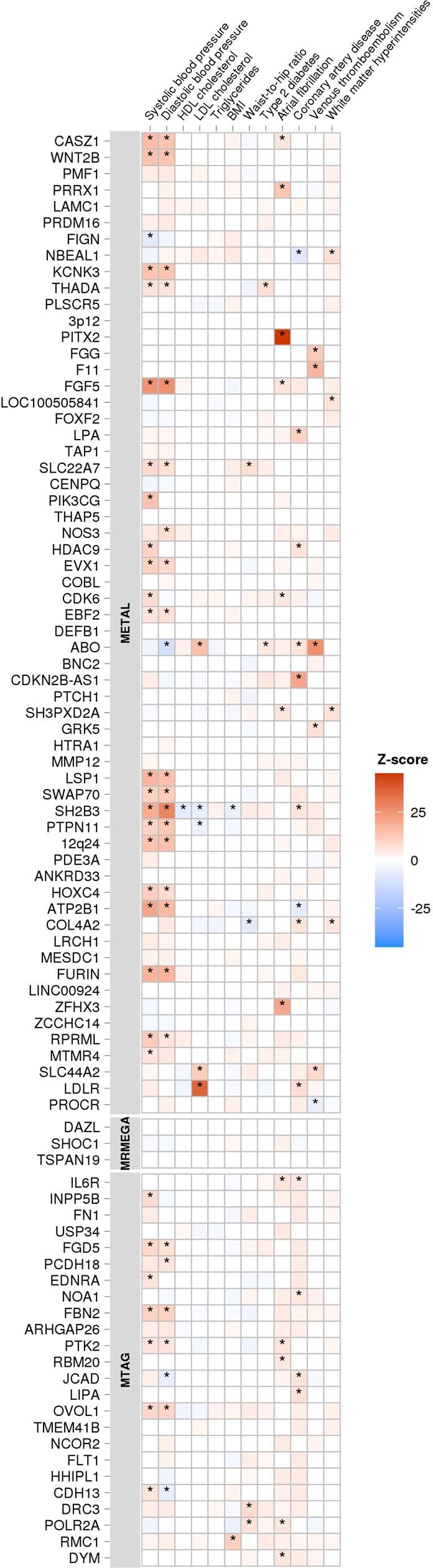
We report only associations for which the stroke lead variant of a proxy in very high LD (r2 > 0.9) showed genome-wide significant association with the vascular risk trait in a prior GWAS. Colours represent the Z-scores of association of stroke risk increasing alleles with the trait.
Extended Data Fig. 5. Genetic correlations and Mendelian randomization causal estimates of 12 vascular risk factors and disease traits with stroke (any and stroke subtypes), in European ancestry participants.

Larger squares correspond to more significant P-values, with genetic correlations or Mendelian randomization (MR) causal estimates (expressed in Z-scores) significantly different from zero at a P < 0.05 shown as a full-sized square. Genetic correlations or causal estimates that are significant after multiple testing correction (P < 4.17 × 10−3) are marked with an asterisk. Two-sided P-values were calculated using LD score regression (LDSC) for genetic correlations and inverse variance weighted analysis for MR.
Extended Data Fig. 6. Genetic correlations and Mendelian randomization causal estimates of 10 vascular risk factors and disease traits with stroke (any and stroke subtypes), in East Asian ancestry participants.

Larger squares correspond to more significant P-values, with genetic correlations or Mendelian randomization (MR) causal estimates significantly different from zero at a P < 0.05 shown as a full-sized square. Genetic correlations or causal estimates (expressed in Z-scores) that are significant after multiple testing correction (P < 5 × 10−3) are marked with an asterisk. Two-sided P-values were calculated using LD score regression (LDSC) for genetic correlations and inverse variance weighted analysis for MR. CPD: cigarettes per day.
Next, to generate hypotheses of target genes and directions of effect, we conducted transcriptome-wide association studies (TWAS) using TWAS-Fusion and eQTL based on RNA-sequencing (RNA-seq) analyses in different tissues17–20. We identified 27 genes of which the genetically regulated expression is associated with stroke and its subtypes at the transcriptome-wide level and colocalized in at least one tissue (10 genes in arteries and heart; 6 genes in brain tissue; 17 genes across tissues). Of these genes, 18 overlapped with 11 genome-wide significant stroke-risk loci (Extended Data Fig. 7 and Supplementary Table 27). For several genes of which bulk tissue expression levels showed evidence for association with stroke, human single-nucleus sequencing data of brain cells in the dorsolateral prefrontal cortex (DLPFC) showed distinct cell-specific gene expression patterns suggesting that multiple genes could be involved through different cell types21 (Extended Data Fig. 8). Overall, we observed a significant enrichment mostly in brain vascular endothelial cells and astrocytes, possibly reflecting the importance of both vascular pathology and brain response to the vascular insult in modulating stroke susceptibility (Extended Data Fig. 8 and Supplementary Tables 28 and 29). Furthermore, using proteome-wide association studies (PWAS) in DLPFC brain tissue, we found evidence for the association of ICA1L with AS and AIS through its cis-regulated protein abundance, with colocalization evidence (Extended Data Fig. 8 and Supplementary Table 30). In both TWAS and PWAS, lower ICA1L transcript or protein abundance in the DLPFC was associated with a higher risk of stroke.
Extended Data Fig. 7. Transcriptome-wide association study of stroke in multiple tissues.

Heatmap of the transcriptome-wide association studies (TWAS) of stroke (any stroke and stroke subtypes) showing transcriptome-wide significant associations with supporting evidence from colocalization; Coloured squares are TWAS significant associations based on two-sided p-values after multiple testing correction (p < 2.0 × 10−6); * Conditionally significant (p < 0.05) and COLOC PP4 ≥ 0.75; Genes are presented on the x-axis, those underlined in blue are in a stroke GWAS locus, those underlined in purple are not within a genome-wide significant stroke risk locus (Methods); Tissue types are on the y-axis (blue: cross-tissue weights; pink: arterial; orange: heart; green: brain).
Extended Data Fig. 8. Single-nucleus gene expression/enrichment analysis and proteome-wide association study (PWAS) of stroke in brain tissue.

(A) Single-nucleus gene expression data of TWAS-COLOC genes in dorsolateral prefrontal cortex (ROS-MAP study)21; Dot plot of the mean expression level in expressing cells (colour) and percent of expressing cells (circle size) of selected genes across different cell types; (B) Proteome-wide association study (PWAS) of stroke in brain tissue; Box plot showing effect estimates (odds ratio) for associations of pQTL of ICA1L in the ROS-MAP (N = 376 independent samples) and Banner (N = 152 independent samples) studies with any stroke (AS) and any ischaemic stroke (AIS), identified in PWAS after multiple testing correction. Odds ratios ± 95% CIs are shown. Dashed line indicates an odds ratio of 1. Two-sided p-values were computed using the TWAS-COLOC approach. (C) Cell-type enrichment in human and mouse single cell RNA-seq databases using STEAP; the UpSet plot displays the number of significant enrichment results, by stroke subtype (horizontally; 2 for CES, 5 for AIS, 6 for AS, and 12 for LAS) and by cell subtype (vertically; 2 cell-types show significant enrichment in LAS, AIS, and AS, 2 cell-types in AIS and AS, and 1 cell-type in LAS and AS, while 9 cell-types show significant enrichment in LAS only, 2 in CES only and 1 in AS and AIS respectively); details are displayed in Supplementary Table 29. AS: any stroke; AIS: any ischaemic stroke; LAS: large artery stroke; CES: cardioembolic stroke; VLMC: vascular and leptomeningeal cells, OPC: oligodendrocyte progenitor cells, SMC: smooth muscle cells; VSMCA: vascular smooth muscle cells, arterial.
Genomics-driven drug discovery
We used a three-pronged approach for genomics-driven discovery of drugs for the prevention or treatment of stroke4 (Methods and Fig. 3). First, using GREP22, we observed significant enrichment of stroke-associated genes (MAGMA9 or VEGAS28 false-discovery rates (FDR) < 0.05) in drug-target genes for blood and blood-forming organs (Anatomical Therapeutic Chemical Classification System B drugs, for AS, AIS and CES). This encompasses the previously described PDE3A and FGA genes1, which encode targets for cilostazol (antiplatelet agent) and alteplase (thrombolytic drug acting through plasminogen23), respectively, as well as F11, KLKB1, F2, TFPI and MUT, which encode targets for conestat alfa, ecallantide (both used for hereditary angioedema), lepirudin, dalteparin (both used to treat recurrent thromboembolism) and vitamin B12, respectively (Supplementary Table 31). Notably, the results for AS are probably driven by AIS (the vast majority of AS in the current study) and cannot be extrapolated to ICH. Second, we used Trans-Phar24 to test the negative correlations between genetically determined case–control gene expression associated with stroke (TWAS using all GTEx v.7 tissues17) and compound-regulated gene expression profiles. At FDR < 0.10, we observed significant negative correlations for BRD.A22514244 (for SVS; drug target unknown) and GR.32191 (for CES; Supplementary Table 32). GR-32191 is a thromboxane A2 receptor antagonist that has been proposed as an alternative antiplatelet therapy for stroke prevention25, and further drugs of this class are under development26. Note that one of those drugs, terutroban, was evaluated in a phase III study but did not show non-inferiority against aspirin27. Third, we used protein quantitative trait loci (pQTL) for 218 drug-target proteins as instruments for MR and found evidence for causal associations of 9 plasma proteins with stroke risk (4 cis-pQTL and 6 trans-pQTL), of which 7 were supported by colocalization analyses, with no evidence for reverse causation using the Steiger test (PROC, VCAM1, F11, KLKB1, MMP12, GP1BA and LAMC2; Supplementary Table 33). All of these replicated (at FDR < 0.05) with consistent directionality using at least one independent plasma pQTL resource and cerebrospinal fluid pQTL for PROC and KLKB1, with evidence for colocalization for PROC, F11, KLKB1 and MMP12, but not for GP1BA (for which both concordant and discordant directionality was observed) and LAMC2 (pQTL available in one replication dataset only; FDR = 0.08). Using public drug databases, we curated drugs targeting those proteins in a direction compatible with a beneficial therapeutic effect against stroke based on MR estimates and identified such drugs for VCAM1, F11, KLKB1, GP1BA, LAMC2 (inhibitors) and PROC (activators; Supplementary Table 34). Drugs targeting F11 (NCT04755283, NCT04304508, NCT03766581) and PROC (NCT02222714) are currently under investigation for stroke, and our results provide genetic support for this. Notably, F11 and KLKB1 are adjacent genes with a long-range linkage-disequilibrium pattern and complex co-regulation28, as illustrated here by the presence of a shared trans-pQTL in KNG1 (Supplementary Table 33). Additional studies are needed to disentangle causal associations and the most appropriate drug target in this region29,30. Next, for the five genes targeted by inhibitors, VCAM1, F11, KLKB1, GP1BA and LAMC2, we examined the associations of rare deleterious variants (MAF < 0.01) with stroke and stroke-related traits, applying gene-based burden tests to whole-exome sequencing data from >450,000 UK Biobank participants to support potential therapeutic targets for inhibitors31. We observed one significant protective association of rare deleterious variants in F11 with venous thromboembolism (odds ratio (OR) = 0.471, P = 2.46 × 10−4), in a direction concordant with that of MR estimates (Supplementary Table 35). To further validate the candidate drugs and estimate their potential side effects, we investigated whether the drug-target genes were associated with stroke-related phenotypes using a phenome-wide association study (PheWAS) approach. We conducted PheWAS in the Estonian Biobank (EstBB) for pQTL variants for the PROC, VCAM1, F11, KLKB1, GP1BA and LAMC2 genes. A cis-pQTL for F11, rs2289252, was associated with higher risk of venous thromboembolic disorders (P < 3.45 × 10−6), as previously described32, and showed suggestive association (P = 3.44 × 10−3) with cerebral artery occlusion with cerebral infarction (Phecode 433.21; Extended Data Fig. 9 and Supplementary Table 36). By contrast, we observed no significant association with non-stroke-related phenotypes, suggesting the safety of targeting F11. Similar profiles were observed in the UK Biobank (https://pheweb.org/UKB-SAIGE/variant/4-187207381-C-T) and FinnGen (https://r7.finngen.fi/variant/4-186286227-C-T), with no significant associations with other disorders and no overlap of subthreshold signals with side-effects reported in clinical trials33. We further confirmed the association of rs2289252 with venous thromboembolic disorders and that it has no association with other non-stroke-related phenotypes using the Phenoscanner database (Supplementary Table 37).
Fig. 3. Genomics-driven drug discovery.
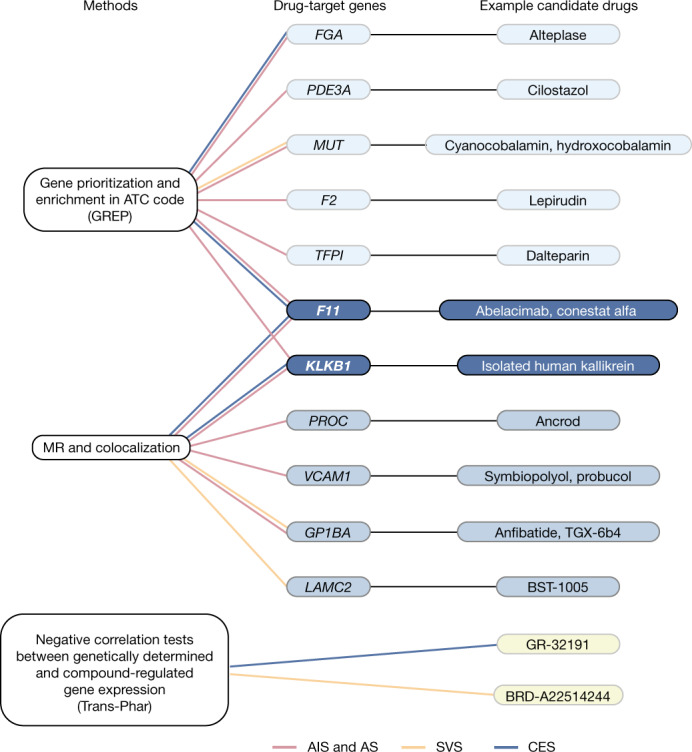
Overlap enrichment analysis using GREP22 (top). Middle, integrating MR results using cis- and trans-pQTLs as instrumental variables with data from drug databases. Bottom, negative correlation tests between compound-regulated gene expression profiles and genetically determined case–control gene expression profiles using Trans-Phar.
Extended Data Fig. 9. Drug target pQTL PheWAS.

PheWAS in Estonian biobank for rs2289252, a cis-pQTL of F11. Each triangle in the plot represents one Phecode and the direction of the triangle represents direction of effect. Two-sided P-values were calculated using logistic regression to test association between the pQTL and Phecodes (p = 3.45 × 10−6 for phenome-wide significance).
Overall, combining evidence from genomics-driven drug discovery approaches, characterization of stroke-risk loci (missense variants, TWAS, PWAS, colocalization, pathway enrichment, MR with pQTL, MENTR and PoPS34), and previous knowledge from monogenic disease models and experimental data, we found evidence for the potential functional implication of 56 genes that should be prioritized for further functional follow-up, with evidence from multiple approaches for 20 genes (Supplementary Table 38).
Integrative polygenic risk prediction
We investigated the risk prediction potential of stroke GWASs, alone and in combination with vascular-risk-trait GWASs, first in Europeans and East Asians, using ancestry-specific PGSs. PGSs were based on ancestry-specific and cross-ancestry GWAS summary statistics. We first derived single PGS (sPGS) models from single stroke GWAS summary data (Supplementary Table 39). We then constructed integrative PGS (iPGS) models, which combined multiple GWAS summary data of different traits into a PGS using elastic-net logistic regression5 (Extended Data Fig. 10). The iPGS analysis used two datasets for each ancestry for model training and evaluation, respectively. The participants in the training and evaluation datasets did not overlap and were not included in the input GWAS summary data.
Extended Data Fig. 10. Derivation and evaluation of integrative polygenic score models for Europeans and East Asians.

(A) With summary statistics of 22 GWAS (10 GIGASTROKE and 12 on vascular risk factors) and linkage disequilibrium reference data of 1000 Genomes Europeans (n = 503) and East Asians (n = 504), we computed 37 candidate PGS models using P+T, LDpred, and PRScs algorithms. For each GWAS, the best PGS model was selected based on the maximal area under the curve (AUC) values in the training dataset of Europeans (any ischaemic stroke [AIS] case-control data, Ncases/Ncontrols = 1,003/8,997) and East Asians (AIS case-control data, Ncases/Ncontrols = 577/9,232). Out of 22 selected PGS models derived from the 22 GWAS, 11 and 7 were significantly associated with AIS in the European and East Asian training dataset respectively (Bonferroni-corrected P < 0.05). (B) The significant PGS models were used as the variables for elastic-net logistic regression and the weights for the variables were trained using the model training dataset. The European iPGS model consisting of 1,213,574 variants and an East-Asian iPGS model consisting of 6,010,730 variants were constructed by combining the 11 and 7 significant PGS models using the elastic-net derived weights respectively. The European and East Asian iPGS models were evaluated in the European (a European prospective cohort data with 102,099 participants including 1,128 incident IS cases) and East-Asian (AIS case-control data, Ncases/Ncontrol = 1,470/40,459) model evaluation dataset (Methods); AS indicates any stroke; AIS, any ischaemic stroke; LAS, large artery stroke; SVS, small vessel stroke; CES, cardioembolic stroke; AF, atrial fibrillation; CAD, coronary artery disease; T2D, type 2 diabetes; SBP, systolic blood pressure; DBP, diastolic blood pressure; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triglyceride; BMI, body mass index; AUC indicates area under the curve; EUR, European; EAS, East Asian; GWAS, genome-wide association study; LD, linkage disequilibrium; PGS, polygenic score.
For Europeans, we constructed the iPGS model using 1,003 prevalent AIS cases and 8,997 controls, followed by evaluation of the model using 1,128 incident AIS cases among 102,099 participants, all from the EstBB. The improvement in predictive ability (∆C-index) was assessed over a base model including age, sex and the top 5 principal components (PCs) for population stratification. The iPGS model for Europeans incorporated 10 GIGASTROKE GWAS analyses (all stroke types, using the European and cross-ancestry analysis) and 12 vascular-risk-trait GWAS analyses (Extended Data Fig. 10 and Supplementary Table 40). The iPGS model achieved a ∆C-index of 0.027 (Supplementary Table 41), 93% higher than that for a previously constructed iPGS model for Europeans, derived from 5 MEGASTROKE GWAS analyses and similar vascular-risk-trait GWASs (∆C-index = 0.014)5. The age-, sex- and top 5 PC-adjusted hazard ratio (HR) per s.d. of the iPGS was 1.26 (95% confidence interval (CI) = 1.19–1.34, P = 2.0 × 10−15) for the GIGASTROKE-based iPGS model compared to 1.19 (95% CI = 1.12–1.26, P = 4.2 × 10−9) for the MEGASTROKE-based iPGS model. Compared with participants in the middle 10% (45–55%) of the GIGASTROKE-based iPGS model, those in the top 1% showed a >2.5-fold higher hazard of AIS (HR = 2.56, 95% CI = 1.59–4.10, P = 9.6 × 10−5; Fig. 4a and Supplementary Table 42). We further confirmed the GIGASTROKE-based European iPGS model trained on the EstBB in 403,489 European-ancestry participants of the Million Veteran Program (MVP) study, of whom 8,392 developed an AIS: HR per s.d. = 1.19 (95% CI = 1.16–1.21, P = 6.94 × 10−52), with a ∆C-index of 0.010 (Supplementary Table 43).
Fig. 4. Risk prediction in a population and trial setting.

a–d, The association of iPGS with ischaemic stroke (AIS) in European (Estonian Biobank) (a), East Asian (BioBank Japan) (b), African American (Million Veteran Program) (c) and European participants in clinical trials (d). Compared with the middle decile (45–55%) of the population as a reference group, the risk of high-iPGS groups with varying percentile thresholds was estimated using a Cox proportional hazards model for European and African American individuals and logistic regression models for East Asian individuals with adjustments for age, sex and the top five genetic principal components. e, Kaplan–Meier event rates for ischaemic stroke in European participants in five clinical trials (Methods) by tertile of GRS at 3 years (the GRS uses effect estimates of the cross-ancestry AS GWAS as weights) showing higher GRS increases risk of ischaemic stroke (Ptrend = 1.4 × 10−4). The two-sided Ptrendvalue was computed using Cox regression. Int., intermediate.
For East Asians, we derived the iPGS model using 577 cases of prevalent AIS and 9,232 control individuals, and evaluated the model using 1,470 cases of prevalent AIS and 40,459 control individuals from Biobank Japan (BBJ). A base model including age, sex and the top 5 PCs showed an area under the curve (AUC) of 0.634. The iPGS model was constructed by integrating 10 GIGASTROKE GWAS analyses and 12 vascular-risk-trait GWAS analyses (Extended Data Fig. 10 and Supplementary Table 44). The iPGS model for East Asians showed an improvement in AUC (∆AUC) of 0.019 (Supplementary Table 45). The age-, sex- and top 5 PC-adjusted odds ratio (OR) per s.d. of PGS was 1.33 (95% CI = 1.26–1.40, P = 9.9 × 10−26) for the iPGS model. The MEGASTROKE- and GIGASTROKE-based iPGS models for Europeans achieved a lower AUC improvement (∆AUC = 0.007 and 0.009, respectively) than the GIGASTROKE-based iPGS model for East Asians. While this suggests that the transferability of iPGS models from Europeans to East Asians might be limited (Supplementary Table 45), it does indicate that an ancestry-specific stroke iPGS approach yields similar improvement in predictive ability relative to their base models.
Participants in the top 1% of the iPGS showed 1.9-fold higher odds of AIS (OR = 1.90, 95% CI = 1.20–2.91, P = 0.004) compared with the middle 10% (Fig. 4b and Supplementary Table 46). We further confirmed the GIGASTROKE-based East Asian iPGS model trained on the BBJ in 1,399 cases of prevalent AIS and 86,283 controls from the Taiwan Biobank (TWB): OR per s.d. = 1.18 (95% CI = 1.12–1.25, P = 1.1 × 10−9), with a ∆AUC of 0.003 (Supplementary Table 47).
Notably, iPGS models derived from cross-ancestry stroke GWASs had a higher predictive ability compared with iPGS models derived from ancestry-specific stroke GWASs both in Europeans and East Asians (Supplementary Table 48).
Next, we evaluated the predictive ability of the European-derived GIGASTROKE-based iPGS model in African American and indigenous African (Nigerian and Ghanaian) datasets. In 107,343 African American MVP participants, of whom 2,227 developed an AIS, the GIGASTROKE-based iPGS model showed a significant association with AIS incidence (HR per 1 s.d. = 1.11, 95% CI = 1.06–1.17, P = 1.8 × 10−5, ∆C-index = 0.003; Supplementary Table 49), although weaker than in European MVP participants (Supplementary Table 43). The participants in the top 1% of the iPGS showed 1.5-fold higher odds of AIS (HR = 1.53, 95% CI, 1.04–2.25, P = 0.03) compared with participants in the middle 10% (Fig. 4c and Supplementary Table 50). In 1,691 cases and 1,743 control participants from the indigenous African (Nigerian and Ghanaian) SIREN case–control study, the GIGASTROKE-based iPGS also showed a significant association with the odds of AIS (OR per 1 s.d. = 1.09, 95% CI = 1.02–1.17, P = 0.010, ∆AUC = 0.007; Supplementary Table 51). The GIGASTROKE-based iPGS model showed a stronger association with AIS and a larger improvement in predictive ability compared with the MEGASTROKE-based iPGS model in both MVP and SIREN (Supplementary Tables 49 and 51).
Risk prediction in clinical trials
Following up on previous work1,35, we further examined whether a genetic risk score (GRS) based on genome-wide significant risk loci from the cross-ancestry IVW AS meta-analyses could identify individuals who are at higher risk of AIS after accounting for established risk factors in five clinical trials across the spectrum of cardiometabolic disease35. The primary analysis was conducted in 51,288 European participants of whom 960 developed an incident ischaemic stroke (AIS) over a 3 year follow-up. In a Cox model adjusted for age, sex and vascular risk factors (Methods), a higher GIGASTROKE GRS was significantly associated with increased risk of AIS in Europeans (adjusted HR = 1.17, 95% CI = 1.09–1.24 per s.d. increase, P = 2 × 10−6; Supplementary Table 52). This association was substantially stronger than the association with the earlier MEGASTROKE GRS based on 32 genome-wide significant stroke-risk loci (HR = 1.07, 95% CI = 1.00–1.14, P = 0.036)1,35. Compared with patients in the lowest GIGASTROKE GRS tertile, patients in the top GRS tertile had an adjusted HR of 1.35 (95% CI = 1.16–1.58) for developing AIS, whereas those in the middle tertile had an adjusted HR of 1.13 (95% CI = 0.96–1.33, Ptrend = 1.4 × 10−4; Fig. 4e). The performance of the GRS was stronger in individuals who had not previously had a stroke (n = 44,095; adjusted HR of the top versus lowest tertile = 1.37, 95% CI = 1.14–1.65) compared with in those who previously had a stroke (n = 7,193; adjusted HR = 1.15, 95% CI = 0.87–1.54). Similar associations were observed when using effect estimates from stroke GWAS meta-analyses in Europeans or for AIS (Supplementary Table 52). In secondary analyses, we examined the association of the GIGASTROKE cross-ancestry AS GRS with incident AIS in the much smaller East Asian sample (1,312 participants of whom 27 developed an incident AIS over a 3 year follow-up), and found consistent associations (adjusted HR = 1.49, 95% CI = 1.00–2.21 per s.d. increase, P = 0.048; Supplementary Table 52), whereas the MEGASTROKE GRS was not associated with incident AIS in East Asians (adjusted HR = 0.82, 95% CI = 0.55–1.23, P = 0.34). Finally, in European trial participants (there were too few East Asian individuals for this analysis), the GIGASTROKE-based iPGS was also significantly associated with increased AIS incidence (HR per 1 s.d. increase = 1.19, 95% CI = 1.11–1.27, P = 3.2 × 10−7, ∆C-index = 0.008), performing better than the MEGASTROKE-based iPGS (Supplementary Table 53). Compared with the middle 10% of the participants, those in the top 1% had a 2.8-fold higher hazard of AIS (HR = 2.78, 95% CI = 1.67–4.61, P = 7.9 × 10−5) (Fig. 4d and Supplementary Table 54).
Discussion
Our GWAS meta-analyses, including 110,182 patients who had a stroke and 1,503,898 control participants from five different ancestries (33% of patients who had a stroke were non-European), identified 89 (61 new) risk loci for stroke and stroke subtypes (60 through primary IVW and 29 through secondary MR-MEGA and MTAG analyses). We observed substantial shared susceptibility to stroke across ancestries, with a strong correlation of effect sizes. On the basis of internal cross-ancestry validation and independent follow-up in 89,084 cases of stroke (30% non-European) and 1,013,843 control individuals, mostly from large biobanks with information on AS and AIS only, the level of confidence of these loci was intermediate or high for 87% of primary stroke-risk loci and 60% of secondary loci. Effect estimates for variants that were common across ancestries were typically similar, whereas, expectedly, variants that were rare or low frequency in one or more populations showed differences in effect size, for example, at PROCR, TAP1 or BNCZ-CNTLN (MAF ≤ 0.05 in East Asians), or at GRK5, FOXF2 or COBL (MAF ≤ 0.05 in African Americans). Ancestry-specific meta-analyses in smaller non-European populations detected fewer loci than in Europeans that were nevertheless biologically plausible, for example, 3p12 and PTCH1 for SVS in African Americans. Rare variants at 3p12 were recently shown to be associated with WMH volume36, whereas common variants at PTCH1 were associated with functional outcome after ischaemic stroke (in European individuals)37. New association signals from cross-ancestry GWASs included, for example, variants at PROCR, GRK5 and F11 (thrombosis), LPA and ATP2B1 (lipid metabolism, hypertension and atherosclerosis), SWAP70 (membrane ruffling) and LAMC1 (cerebrovascular matrisome).
Extensive bioinformatics analyses highlight genes for prioritization in functional follow-up studies (Supplementary Table 38). For example, a promoter variant of SH3PXD2A, which encodes an adaptor protein that is involved in extracellular matrix degradation through invadopodia and podosome formation, was predicted to modulate its expression in macrophages38. FURIN expression levels across tissues were associated with an increased stroke risk. FURIN has previously been implicated in CAD39 as well as in atherosclerotic lesion progression in mice40. It also has a key role in SARS-CoV-2 infectivity41, and patients with COVID-19 are at increased risk of AIS, especially LAS42; the FURIN locus was predominantly associated with LAS in our data (Supplementary Table 55).
Our results provide genetic evidence for putative drug effects using three independent approaches, with converging results from two methods (gene enrichment analysis and pQTL-based MR) for drugs targeting F11 and KLKB1. F11 and F11a inhibitors (such as abelacimab, BAY 2433334 and BMS-986177) are currently being examined in phase 2 trials for primary or secondary stroke prevention (NCT04755283, NCT04304508, NCT03766581). pQTL-based MR suggested PROC as a potential drug target for stroke. A recombinant variant of human activated protein C (encoded by PROC) was found to be safe for the treatment of acute ischaemic stroke after thrombolysis, mechanical thrombectomy or both in phase 1 and 2 trials (3K3A-APC, NCT02222714)43,44, and is poised for an upcoming phase 3 trial. 3K3A-APC is proposed as a neuroprotectant, with evidence for the protection of white matter tracts and oligodendrocytes against ischaemic injury in mice45. Weaker evidence was found for GP1BA, VCAM1 and LAMC2 as potential drug targets for stroke, with evidence for colocalization in only one pQTL dataset. Anfibatide, a GPIbα antagonist, reduced blood–brain barrier disruption after ischaemic stroke in mice46 and is being tested as an antiplatelet drug in myocardial infarction (NCT01585259). Although specific VCAM1 inhibitors are not available, probucol—a lipid lowering drug with pleiotropic effects including VCAM1 inhibition—was tested for secondary prevention against atherosclerotic events in patients with CAD (PROSPECTIVE, UMIN000003307)47.
We investigated stroke PGSs across ancestries. PGSs integrating cross-ancestry and ancestry-specific stroke GWASs with vascular-risk-factor GWASs (iPGS) analyses showed strong prediction of ischaemic stroke risk in Europeans and, importantly, in East Asians, in whom stroke incidence is highest6. These results were confirmed in several independent datasets. The iPGS performed better than stroke PGS alone and better than the previous best iPGS models in Europeans5. The transferability of European-specific iPGS models to East Asians was limited. While there were not enough African participants to generate an African-specific stroke PGS, the European iPGS showed a significant association with AIS in both African American and indigenous African participants, although expectedly weaker than in European participants. Individuals in the top 1% of the PGS distribution had a 2- to 2.5-fold risk of ischaemic stroke in East Asian and European participants compared with those in the middle 10%, whereas this risk was 1.5-fold in African American participants. Although caution is warranted when interpreting risk estimates owing to the wide CIs, these results suggest that GIGASTROKE-based iPGS models may be useful to stratify individuals exposed to genetically high risk of ischaemic stroke, especially in Europeans and East Asians. Our results highlight the importance of ancestry-specific and cross-ancestry genomic studies for the transferability of genomic risk prediction across populations, and the urgent need to substantially increase participant diversity in genomic studies, especially from the most under-represented regions such as Africa, to avoid exacerbation of health disparities in the era of precision medicine and precision public health48.
Finally, leveraging data from 5 clinical trials in 52,600 patients with cardiometabolic disease, we showed that a cross-ancestry GRS predicted ischaemic stroke, independently of clinical risk factors, and outperforming previous genetic risk evaluation35. Notably, although the trials included predominantly European participants, consistent results were observed in East Asian participants. We further confirmed the GIGASTROKE iPGS in these clinical trials.
Our study includes a considerable contribution of non-European stroke genetics resources (n = 61,528/616,014 cases/controls for the GWASs and follow-up and an additional n = 1,718/3,055 for the PGS/GRS studies). Despite substantial efforts to enhance non-European contributions to GIGASTROKE, we still had limited power for identifying shared causal variants through cross-ancestry fine-mapping. We provided independent validation of the vast majority of identified genome-wide significant associations and graded loci by level of confidence based on these findings. Despite the notable size of the follow-up study sample, with nearly 90,000 additional patients who had a stroke, this analysis remains underpowered, especially for low-frequency variants and ancestry- and subtype-specific associations, as most follow-up studies were derived from large biobanks with event ascertainment based on electronic health records and no suitable stroke subtype information. The muted risk prediction in clinical-trial participants with previous stroke history possibly points to the impact of selection or index event biases and secondary prevention therapy49.
In conclusion, our genomic findings derived from >200,000 patients who had a stroke worldwide provide critical insights to inform future biological research on stroke pathogenesis, highlight potential drug targets for intervention and provide tools for genetic risk prediction across ancestries.
Methods
All human research was approved by relevant boards and/or institutions for each study (Supplementary Table 56) and was conducted according to the Declaration of Helsinki. All of the participants provided written informed consent.
Study design and phenotypes
Information on participating studies (discovery and follow-up), study design, and definitions of stroke and stroke subtypes is provided in the Supplementary Information. Population characteristics of individual studies are provided in Supplementary Table 1.
Genotyping, imputation and GWASs
Genotyping methods, pre-imputation quality control of genotypes and imputation methods of individual cohorts (discovery and follow-up) are presented in Supplementary Table 2. High-quality samples and SNPs underwent imputation using mostly Haplotype Reference Consortium (HRC) or 1000 Genomes phase 1 or phase 3 reference panels and, less often, TOPMed, HapMap or biobank-specific reference panels. Individual studies performed a GWAS using logistic regression (or Cox regression in some longitudinal population-based cohorts) testing association of genotypes with five stroke phenotypes (AS, AIS, CES, LAS and SVS) under an additive effect model, adjusting for age, sex, principal components of population stratification and study-specific covariates when needed (Supplementary Table 2).
The R package EasyQC along with custom harmonization scripts were used to perform the quality control of individual GWAS summary results. Marker names and alleles were harmonized across studies. Meta-analyses were restricted to autosomal biallelic SNPs from the HRC panel. Duplicate markers were removed. Before the meta-analysis, we removed variants with extreme effect size values (log[OR] > 5 or log[OR] < −5), minor allele frequency (MAF) < 0.01, imputation quality scores of less than 0.50 and effective allele counts (EAC = 2 × number of cases × MAF × imputation quality score) of less than 6.
The overall analytical strategy is shown in Extended Data Fig. 1. We conducted ancestry-specific fixed-effect IVW meta-analyses in European, East Asian, African American, Hispanic and South Asian populations, followed by cross-ancestry meta-analyses using METAL50. In each meta-analysis we removed variants with heterogeneity P < 1 × 10−6 and variants available in less than one third of the total number of cases and less than one third of the total number of contributing studies. We applied the covariate adjusted linkage disequilibrium score regression (cov-LDSC) method to ancestry-specific GWAS meta-analyses without GC correction to test for genomic inflation and to compute robust SNP-heritability estimates in admixed populations51. We conducted cross-ancestry GWAS meta-analyses without genomic correction and with correction of the linkage-disequilibrium score intercept for genomic inflation observed in individual ancestry-specific GWASs. We conducted separate GWAS analyses of incident AS and AIS (n = 32,903 and n = 16,863) in longitudinal population-based cohort studies. For the meta-analysis combining both incident and prevalent stroke studies, a few incident stroke studies were removed because they were already part of a meta-analysis of stroke GWASs used as an input of the overall meta-analysis (WHI, Hisayama, REGARDS, JHS). We considered loci to be genome-wide significant for P < 5 x 10-8.
We applied the conditional and joint analysis approach7 implemented in the Genome-wide Complex Trait Analysis software52 (GCTA-COJO) to identify potentially independent signals within the same genomic region. We performed GCTA-COJO analyses on (1) European GWAS meta-analysis summary statistics using HRC imputed data of 6,489 French participants from the 3C study as in ref. 53 and (2) East Asian-ancestry-specific GWAS meta-analysis summary statistics using BBJ data as reference (Supplementary Information).
We also performed a cross-ancestry meta-regression using MR-MEGA10. Before the meta-analysis using MR-MEGA, we applied the ‘genomic inflation’ correction option to all of the input files, and removed variants with extreme effect size values (log[OR] > 5 or log[OR] < −5), MAF < 0.01, imputation quality scores of less than 0.50 and effective allele counts (EAC = 2 × number of cases × MAF × imputation quality score) of less than 6. After the meta-analysis, we considered loci to be genome-wide significant for MR-MEGA P < 5 × 10−8 and showing nominal association (P < 0.05) in at least one third of studies in any individual ancestry group (European, East Asian, African American, Hispanic and South Asian).
Multitrait association study
To identify additional stroke-risk loci we used MTAG11 in Europeans and East Asians, including traits correlated with specific stroke subtypes, namely CAD for LAS, atrial fibrillation54 for CES, and WMH55 (an MRI marker of cerebral small vessel disease, available in Europeans only) for SVS. We also ran an MTAG analysis of AS and AIS, including all three correlated traits (CAD, atrial fibrillation, WMH (European)). In European individuals, we used summary statistics of published GWAS analyses for CAD56, AF54 and WMH55. In East Asians, we used summary statistics of published GWAS analyses for CAD57 and atrial fibrillation58 (Supplementary Information). Associations were retained when the following three conditions were verified: (1) MTAG P value for stroke < 5 × 10−8; (2) P value for stroke < 0.05 in the univariate GWAS; and (3) MTAG P value for stroke less than the P value for any of the included traits in univariate GWASs.
Independent follow-up of GWAS signals
First, we sought to replicate internally the 42 stroke-risk loci reaching genome-wide significance in IVW meta-analyses within individual ancestries, in at least one other ancestry group among the discovery samples, considering both nominal replication levels (P < 0.05) and multiple-testing corrected significance at P < 1.19 × 10−3 (0.05/42). Second, we gathered independent datasets totalling 89,084 AS (including 85,546 AIS; and 70.0% European, 15.6% African American, 10.1% East Asian, 4.1% Hispanic and 0.1% South Asian) and 1,013,843 controls for external replication of associations with AS and AIS (Supplementary Tables 1 and 2). These comprised eight biobanks (82,263 cases, 930,988 controls) and four hospital-based cohorts (6,821 cases, 82,855 controls). We considered both nominal replication levels (P < 0.05) and multiple-testing corrected significance at P < 8.2 × 10−4 (0.05/60) and P < 1.3 × 10−3 (0.05/29) for follow-up of genome-wide significant loci from the IVW and the MR-MEGA/MTAG meta-analyses, respectively (two-sided P values were used for both discovery and replication analyses). We considered stroke-risk loci as high confidence in the case of significant internal inter-ancestry and/or external replication after accounting for the number of loci tested, nominally significant replication in both internal and external replication analyses, or evidence of involvement in monogenic stroke; intermediate confidence in the case of nominal significance in either internal inter-ancestry or external replication analyses but not both; and low confidence in the absence of formal replication.
Gene-based analyses
We performed gene-based tests of common variant associations using VEGAS28 and MAGMA9. Both VEGAS2 and MAGMA considered variants in the gene or within 10 kb on either side of a gene’s transcription site to compute a gene-based P value. We performed MAGMA tests using the default parameters, whereas the VEGAS2 analyses were performed using the ‘-top 10’ parameter that tests enrichment of the top 10% variants assigned to a gene accounting for the linkage disequilibrium between variants and the total number of variants within a gene. We used 1000 Genomes phase 3 continental reference samples of European, East Asian, African, South Asian and South American (for our Hispanic samples) ancestry and to compute the linkage disequilibrium between variants for respective ancestry-specific gene-based analyses. We then meta-analysed ancestry-specific gene-based results, using Stouffer’s method for sample-size-weighted combination of P values. Gene-wide significance was defined as P < 2.72 × 10−6, correcting for 18,371 autosomal protein-coding genes tested.
Pathway-based analyses
We used the ancestry-specific gene-based association P values generated using VEGAS2 to perform pathway analyses for individual ancestry groups, testing enrichment of gene-based P values in Biosystems pathways with VEGAS2Pathway8,15. For each stroke phenotype, we meta-analysed the ancestry-specific pathway association P values using Stouffer’s method considering the number of cases in each ancestry-specific GWAS; for example, for AS, we considered 73,652, 27,413, 3,961, 1,516 and 3,640 cases in European-, East Asian-, African American-, Hispanic- and South Asian-specific GWAS analyses to combine the respective ancestry-specific pathway association P values. Pathway-wide significance was defined at P < 5.01 × 10−6 correcting for 9,977 Biosystems pathways tested.
Shared genetic variation
We examined shared genetic variation with 12 vascular risk factors and related disease traits in Europeans using summary statistics of GWASs on SBP59, DBP59, BMI and waist-to-hip ratio60, high density lipoprotein (HDL) cholesterol61, LDL cholesterol61, triglycerides61, type 2 diabetes62, WMH volume55, atrial fibrillation54, CAD56 and venous thromboembolism32. We extracted sentinel stroke-risk variants (or a proxy (r2 > 0.9)) that showed genome-wide significant association (P < 5 × 10−8) with the aforementioned vascular-risk traits.
We then systematically examined genetic correlations and potentially causal associations between vascular-risk traits and risk of stroke using linkage-disequilibrium score regression (LDSC) and MR analyses, with 12 (in Europeans) and 6 (in East Asians) vascular-risk traits. In individuals of European ancestry, we used summary statistics of the aforementioned GWASs32,54–56,59–62. For the analysis in East Asians, we used unpublished GWAS analyses for SBP, DBP, LDL and HDL cholesterol, triglycerides and BMI in up to 53,323 participants of the independent Tohoku Medical Megabank Project (Supplementary Information).
We used cov-LDSC to compute genetic correlations between stroke and vascular-risk traits, using European and East Asian GWAS summary files and 1000Gp3v5 reference data of respective continental ancestries (considering the recommended subset of high-quality HapMap3 SNPs only).
For MR analyses, we constructed genetic instruments for each vascular-risk trait based on genome-wide significant associations (P < 5 × 10−8) after clumping for linkage disequilibrium at r2 < 0.01 (based on European and East Asian 1000 Genomes reference panels). We applied two-sample MR analyses in the GIGASTROKE summary statistics separately for individuals of European and East Asian ancestry based on variant associations derived from the aforementioned sources. After extraction of the association estimates and harmonization of their direction-of-effect alleles, we computed MR estimates with fixed-effect IVW analyses63. As a measure of pleiotropy, we assessed heterogeneity across the MR estimates for each instrument in the IVW MR analyses with Cochran’s Q statistic (P < 0.05 was considered to be significant)64. We further applied alternative MR methods that are more robust to the use of pleiotropic instruments: the weighted median estimator enables the use of invalid instruments under the assumption that at least half of the instruments used in the MR analysis are valid65; MR-Egger regression allows for the estimation of an intercept term, provides less precise estimates and relies on the assumption that the strengths of potential pleiotropic instruments are independent of their direct associations with the outcome66. The intercept obtained from MR-Egger regression was used as a measure of directional pleiotropy (P < 0.05 indicated significance)66. MR analyses were performed in R v.4.1.1 using the Mendelian Randomization package.
For all genetic correlation and MR analyses, we set statistical significance at Bonferroni-corrected P < 4.17 × 10−3 in Europeans (correcting for 12 vascular-risk traits) and P <8.33 × 10−3 in East Asians (correcting for 6 vascular-risk traits).
Cross-ancestry fine mapping
Fine-mapping was performed separately for Europeans and East Asians using susieR v.0.9.112 on all variants within 3 Mb of the lead variant of each genomic risk locus (60 loci reached genome-wide significance in the IVW meta-analysis). Unrelated individuals from the UK Biobank (n = 420,000) and BBJ (n = 170,000) were used as ancestry-matched linkage-disequilibrium reference panels that fulfil the sample size requirement67. After extracting variants present in the linkage disequilibrium reference panel, the default settings of susieR were used while allowing for a maximum of 10 putative causal variants in each locus. The fine-mapping results were checked for potential false-positive findings using a diagnostic procedure implemented in SuSiE. In brief, we compared observed and expected z-scores for each variant at a given locus and removed the variant if the difference between the observed and expected z-score was too high after manual inspection. We compared the variants in credible sets of the same loci between Europeans and East Asians.
To detect putative causal regulatory variants, we conducted an in silico mutagenesis analysis using MENTR (mutation effect prediction on non-coding RNA transcription; https://github.com/koido/MENTR), a machine-learning method to precisely predict transcriptional changes induced by causal variants3,68. The in silico mutations predicted to have strong effects are highly concordant with the observed effects of known variants in a cell-type-dependent manner. Furthermore, MENTR does not use population datasets and is therefore less susceptible to linkage-disequilibrium-dependent association signals, enabling precise prediction of the effects of causal variants on transcriptional changes. From 1,274 variants in the credible sets from the European and East Asian fine-mapping, we searched FANTOM5 promoters and enhancers, obtained by cap analysis of gene expression, within ±100 kb from each variant. As a result, we found 37,878 variant–transcript pairs comprising 1,270 variants and 2,350 transcripts. We used MENTR with the pretrained FANTOM5 347 cell/tissue models + LCL models69–72 and extracted reliable predictions using the predetermined robust threshold (absolute in silico mutation effects ≥ 0.1, achieving >90% concordance for predicting effects on expression).
TWAS and PWAS
We performed TWAS using TWAS-Fusion19 to identify genes of which the expression is significantly associated with stroke risk. We restricted the analysis to tissues considered to be relevant for cerebrovascular disease, and used precomputed functional weights from 21 publicly available eQTL reference panels from blood (Netherlands Twin Registry; Young Finns Study)19,20, arterial and heart (GTEx v.7))17 and brain tissues (GTEx v.7, CommonMind Consortium)17,18. Moreover, we used the newly developed cross-tissue weights generated in GTEx v.8 using sparse canonical correlation analysis (sCCA) across 49 tissues available on the TWAS-Fusion website, including gene expression models for the first three canonical vectors (sCCA1–3), which were shown to capture most of the gene expression signal73. TWAS-Fusion was then used to estimate the TWAS association statistics between predicted gene expression and stroke by integrating information from expression reference panels (SNP-expression weights), GWAS summary statistics (SNP-stroke effect estimates) and linkage disequilibrium reference panels (SNP correlation matrix)19. Transcriptome-wide significant genes (eGenes) and the corresponding eQTLs were determined using Bonferroni correction, based on the average number of features (5005.8 genes) tested across all reference panels and correcting for the 5 stroke phenotypes (P < 2.0 × 10−6). eGenes were then tested in conditional analysis as implemented using the Fusion software19. To ensure that the observed associations did not reflect random correlation between gene expression and non-causal variants associated with stroke, we performed a colocalization analysis on the conditionally significant genes (P < 0.05) to estimate the posterior probability of a shared causal variant between the gene expression and trait association (PP4)74. We used a prior probability of P < 2.0 × 10−6 for the stroke association. Genes presenting a PP4 ≥ 0.75, for which eQTLs did not reach genome-wide significance in association with stroke, and were not in linkage disequilibrium (r2 < 0.01) with any of the lead SNPs of genome-wide significant risk loci for stroke, were considered to be new, i.e. not within a genome-wide significant stroke risk locus.
Using similar parameters in TWAS-Fusion19, we also performed a proteome-wide association study. For this analysis, we used the precomputed weights for protein expression in DLPFC75 from the ROS/MAP study (n = 376 individuals, n = 1,475 proteins)76 and the Banner Sun Health Institute study (n = 152 individuals, n = 1,145 proteins)77. Proteome-wide significant genes and the corresponding pQTLs were determined using Bonferroni correction, on the number of proteins tested across the reference panel and correcting for the 5 stroke phenotypes (P < 1.7 × 10−4 for ROS/MAP and P < 2.2 × 10−8 for the Banner Sun Health Institute study). We then followed the same method as described for the TWAS.
Brain single-cell expression analyses
Single-nucleus RNA-sequencing data of the DLPFC region of 24 ageing individuals chosen to represent the range of pathologic and clinical diagnoses of AD dementia, from the ROS/MAP cohorts, was obtained21. RNA profiles of cells annotated as endothelial, pericytes or smooth muscle cells and vascular leptomeningeal cells (VLMC) were used, and a pseudobulk RNA profile was generated for each cell type by averaging the expression of all genes across the cells. Average expression levels and the percentage of expressed genes were calculated for genes of interest using the DotPlot function from the Seurat package v.4.0.4 in R v.4.1.1.
We also conducted a cell-type enrichment analysis using the STEAP pipeline (https://github.com/ComPopBio/STEAP). This is an extension of CELLECT and uses S-LDSC78, MAGMA9 and H-MAGMA79 for enrichment analysis. Stroke GWAS summary statistics were first munged. Expression specificity profiles were then calculated using human and mouse single-cell RNA-seq databases (Supplementary Table 28). Cell-type enrichment was calculated using three models: MAGMA, H-MAGMA (incorporating chromatin interaction profiles from human brain tissues in MAGMA) and stratified linkage-disequilibrium score regression. P values were corrected for the number of independent cell types in each database (Bonferroni correction).
Genomics-driven drug discovery
We used three methodologies for in-depth genomics-driven drug discovery as described previously4: (1) an overlap enrichment analysis of disease-risk genes in drug-target genes in medication categories; (2) negative correlation tests between genetically determined case–control gene expression profiles and compound-regulated gene expression profiles; and (3) endophenotype MR. Details of the methods are described in the following sections. For the overlap enrichment analysis and the endophenotype MR-nominated drug targets, we curated drug candidates from four major drug databases: DrugBank23, Therapeutic Target Database (TTD)80, PharmGKB81 and Open Target Platform82. As for the endophenotype MR, we curated drugs with opposite effects against the signs of the MR effect estimates. By contrast, the negative correlation tests directly prioritized candidate compounds. We manually curated supporting evidence for candidate drugs and compounds.
Overlap enrichment analysis of disease-risk genes in drug-target genes in medication categories
We ran MAGMA9 and VEGAS28 to summarize variant-level P values into gene level and used the genes with FDR < 0.05 in either MAGMA or VEGAS2 as the disease-risk genes. We then used GREP22 to perform a series of Fisher’s exact tests for the enrichment of the disease-risk genes in the drug-target genes involved in the drug indication categories, Anatomical Therapeutic Chemical Classification System codes.
Negative correlation tests between genetically determined and compound-regulated gene expression profiles
We nominated the compounds with inverse effects on gene expression against genetically determined gene expression by using Trans-Phar24. In brief, genetically determined case–control gene expression was inferred for 44 tissues in the Genotype-Tissue Expression project (v.7)17 with FOCUS83, and the genes in the top decile for the absolute value of the z-score were used for the following correlation analysis. The Library of Integrated Network-based Cellular Signatures project (LINCS) CMAP L1000 library data84 were used for the compound library. After matching the tissues in GTEx with the cell lines in the LINCS L1000 library, we performed a series of Spearman’s rank correlation tests for 308,872 pairs of genetically determined and compound-perturbed tissue- or cell-type specific gene expression profiles. We prioritized compounds with FDR < 0.1, as we previously showed that the compounds with FDR < 0.1 contained plausible therapeutic targets with literature supports4.
Endophenotype MR
To pin-point the disease-causing proteins that were targeted by existing drugs, we performed MR analyses (specifically, a Wald ratio test) by using lead variants in pQTL as instrumental variables and five stroke phenotypes as outcomes: AS, AIS, CES, LAS and SVS. We used the tier 1 lead variants defined in ref. 85 to avoid confounding by horizontal pleiotropy. The tier 1 variants, summarized from five pQTL studies (n = 997 to 6,861)86–90, did not include variants with heterogeneous effect sizes among the studies or with a number of associated proteins of larger than five. We restricted the lead variants to the variants associated with drug-target proteins. For the lead variants of pQTLs that were missing in the stroke GWAS summary statistics, the proxy variants with the largest r2 were used if the r2 was greater than 0.8 (1000 Genomes, European). In total, we used 277 lead variants for 218 drug-target proteins for MR and considered FDR < 0.05 as the threshold to identify significant associations. We used the TwoSampleMR R package91 for MR analysis. As post-MR quality controls, we performed (1) a directionality check of causal relationships by Steiger filtering92 and (2) colocalization analysis for the proteins with FDR < 0.05. To examine colocalization assuming multiple causal variants per locus, coloc74 was applied to the decomposed signals by SuSiE12 for the variants within 500 kb upstream and downstream of the lead variants (coloc + SuSiE)93. If SuSiE did not converge after 10,000 iterations, coloc was used instead. coloc + SuSiE and coloc were run with their respective default parameters. For the two pQTL studies without public summary statistics86,90, we compared the r2 between the lead variants of the pQTL study and the stroke GWAS. We considered that colocalization occurred when the maximum posterior probability (that is, PP.H4) was greater than 0.75 or r2 was greater than 0.8.
To provide further support for our findings, we conducted MR analyses with two additional recent independent pQTL datasets, using the same methodology and significance thresholds (FDR < 0.05 for MR and PP.H4 > 0.75 for colocalization) as above: one study comprised both plasma (n = 529) and cerebrospinal fluid (n = 835) pQTL datasets94, the second is one of the largest plasma pQTL studies conducted in 35,559 Icelandic individuals95.
Protective rare variants
For the five genes targeted by inhibitors—VCAM1, F11, KLKB1, LAMC2 and GP1BA—we extracted the associations of rare deleterious variants (MAF < 0.01) with stroke and stroke-related traits from the gene-based burden tests in the whole-exome sequencing data of >450,000 UK Biobank participants31. As stroke and stroke-related traits, we extracted 30 traits belonging to 9 vascular risk factor and disease categories (Supplementary Table 35). We applied Bonferroni correction and the corrected P-value threshold was 0.05/5/30 = 3.33 × 10−4 (5 and 30 represent the number of tested genes and traits, respectively).
PheWAS
PheWAS analysis was performed using R (v.4.0.3). We used the PheWAS R package96 (https://github.com/PheWAS/PheWAS) function createPhenotypes to translate ICD10 diagnosis codes into phecodes for the PheWAS analysis. We tested the associations between phecodes and genetic variants using logistic regression and adjusting for sex, birth year and ten genotype PCs. We applied Bonferroni correction to select statistically significant associations (number of tested phecodes: 1,809; number of tested SNPs: 8; corrected P-value threshold: 0.05/(1,809 × 8) = 3.45 × 10−6). The results were visualized using the PheWAS library. To further characterize the associations of the genetic variants with other phenotypes, we searched for all eight SNPs in the PhenoScanner database97,98.
Polygenic risk prediction
We constructed iPGS models for stroke in European and East Asian individuals (Extended Data Fig. 10). For each ancestry, independent datasets were used for model training and evaluation. We used as input summary statistics data of multiple GWAS analyses for stroke outcomes and vascular-risk traits to derive iPGS models. We denote the number of input GWASs as N. For each of the N GWAS summary data, 37 candidate single-trait polygenic score (sPGS) models were generated using the P+T99,100, LDpred101 and PRScs102 algorithms with an ancestry-specific linkage-disequilibrium reference panel from the 1000 Genomes Project103 (Supplementary Methods). The plink (v.1.90b6.8)104, LDpred (v.1.0.11)101 and PRScs.py (5 June 2021)102 programs were used to compute the P+T, LDpred and PRScs models, respectively. Subsequently, among the 37 candidate models, the best sPGS model, which was defined as the model that showed a maximal improvement in AUC over a base model (age, sex and top five PCs were included in the base model), was selected using the model training dataset5,100. Then, N best sPGS models were selected from the N input GWASs. Among the N best sPGS models, we retained models that were significantly associated with AIS in the model-training dataset (Bonferroni-corrected P < 0.05).
Then, each retained best sPGS was z-transformed (zero mean and unit s.d.) over the model-training dataset, followed by elastic-net logistic regression105 to model the associations between the N sPGS variables and AIS with the adjustments for age, sex and top five genetic PCs. Two regularization parameters (α and λ) were optimized using tenfold cross-validation. Coefficients (weights) for the retained sPGS models were then determined by elastic-net logistic regression with the optimal regularization parameters, followed by integration of the sPGS models into a single iPGS model according to a formula presented previously5. Elastic-net regression was performed using the glmnet R package106.
The predictive ability of the iPGS model was estimated using the model-evaluation dataset, whereby we evaluated the improvement in C-index for a prospective cohort dataset or AUC for a case-control dataset over a base model that includes age, sex and top five genetic PCs.
We used EstBB data for the model training and evaluation of iPGS model in Europeans. The model-training dataset was composed of 1,003 cases of prevalent AIS at the baseline and 8,997 control individuals. The control individuals were randomly selected among EstBB participants who had no history of AS at the baseline and who did not develop AS during the follow-up. The remaining 102,099 EstBB participants were used for the model evaluation (mean ± s.d. age at the baseline, 44.0 ± 15.7 years; 37.8% men). Among the participants in the model-evaluation dataset, 1,128 cases of incident AIS were observed during 4.6 ± 4.8 years. To derive the European iPGS model, we incorporated 5 ancestry-specific and 5 cross-ancestry stroke GWAS analyses (AS, AIS, LAS, SVS and CES) from the GIGASTROKE project, and 12 GWAS analyses of vascular-risk traits from other groups (Extended Data Fig. 10). To avoid the overlap of participants across datasets, the GWAS summary statistics for stroke outcomes were recalculated for the iPGS analysis by excluding the EstBB from the meta-analysis of GIGASTROKE studies. To enable comparison with a previous European iPGS model based on the MEGASTROKE GWAS5, we incorporated 12 GWAS analyses of vascular-risk traits (atrial fibrillation, CAD, T2D, SBP, DBP, TC, LDL-C, HDL-C, TG, BMI, height and smoking)54,56,59–61,107,108 into the GIGASTROKE-based iPGS model. The iPGS model for Europeans was further evaluated in two external cohorts of European ancestry (MVP and pooled data of clinical trials) as well as in two studies of participants with African ancestry (MVP and SIREN).
For the East Asian iPGS model, we used BBJ data for the model training and evaluation. The model-training dataset was composed of 577 cases of AIS and 9,232 control individuals, whereas there were 1,470 cases of AIS and 40,459 control individuals in the model-evaluation dataset. The mean ± s.d. of age at recruitment was 69.2 ± 10.8 years for cases and 66.5 ± 12.5 years for controls in the model evaluation dataset. The percentage of male participants was 70.0% for cases and 53.1% for controls. The two case–control datasets were not included in the meta-analysis of GIGASTROKE studies and, therefore, the overlap of participants across datasets was avoided. To derive the East Asian iPGS model, we incorporated 5 ancestry-specific and 5 cross-ancestry stroke GWAS analyses (AS, AIS, LAS, SVS and CES) from the GIGASTROKE project, and 12 GWAS analyses of vascular-risk traits (Extended Data Fig. 10). The iPGS model for East Asian individuals was further evaluated in an external study of East Asian ancestry (TWB).
GRS in clinical trials
Participants who had consented for genetic testing and who were of European ancestry from the ENGAGE AF-TIMI 48 (effective anticoagulation with factor Xa next generation in atrial fibrillation)109, SOLID-TIMI 52 (stabilization of plaques using darapladib)110, SAVOR-TIMI 53 (saxagliptin assessment of vascular outcomes recorded in patients with diabetes mellitus)111, PEGASUS-TIMI 54 (prevention of cardiovascular events in patients with prior heart attack using ticagrelor compared to placebo on a background of aspirin)112 and FOURIER (further cardiovascular outcomes research with PCSK9 inhibition in patients with elevated risk)113 trials were included in this analysis. Methods for genotyping and imputation have previously been published35,114 and are summarized in Supplementary Table 2. A set of 58 sentinel variants at stroke-risk loci identified in the IVW meta-analysis was used to calculate a GRS for each trial participant and identify tertiles of genetic risk (Supplementary Table 57). A Cox model was used to estimate HRs for ischaemic stroke associated with the quantitative GRS and across genetic risk groups, adjusted for clinical risk factors (age, sex, hypertension, hyperlipidaemia, diabetes, smoking, CAD, atrial fibrillation and congestive heart failure) and the first five principal components of population stratification. Analyses were conducted primarily in participants of European ancestry (n = 51,288, with 960 incident AIS)—with secondary analyses in the much smaller East Asian (n = 1,312, with 27 incident AIS) ancestry subset—using the AS cross-ancestry IVW meta-analysis effect estimates as weights for the primary analysis and ancestry-specific, as well as AIS effect estimates for secondary analyses. We also looked separately at associations with incident stroke in participants with and without previous stroke.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-05165-3.
Supplementary information
Supplementary Methods, including a description of the GWAS of stroke-risk factors in Tohuku Medical Megabank and Calculation of candidate polygenic score models; description of study populations in the GIGASTROKE initiative; study-specific acknowledgements; information on other members of participating consortia; and Supplementary References.
Supplementary Tables 1–56.
Acknowledgements
Detailed acknowledgements are provided in the Supplementary Information. We thank the participants and staff of contributing studies. We also thank Michèle M. Sale, who passed away in 2018, for her important contributions to some of the studies included in this manuscript, as principal investigator of one of the grants that funded “The Sea Islands Genetics Network (SIGNET)” (R01 DK084350). She contributed specifically to SIGNET-REGARDS from the REasons for Geographic And Racial Differences in Stroke (REGARDS, U01 NS041588) and served as a key contributor to SiGN, COMPASS and METASTROKE.
Extended data figures and tables
Author contributions
S. Debette, M.D., Y.K., L.M., J.E.H., M.O.O. and C.T.F. jointly supervised the research. S. Debette and M.D. designed and conceived the study. D.C., M.F., M.N., S.N., T. Konuma, Y.O., J.Q.T., R.F.-S., S. Trompet, J.B., T.B., K.W., M.R., Y.-H.J., B.W., S.B., H.L., M.A.N., C.Y., A. Mishra, S.R., J.I.R., M.C., F.K., T.H., Y.S., A.S., G.C., A.K., D. Strbian, Q.Y., F.V., J.L., A.C., N.H., T.J., K.K., K. Lepik, J.C.-M., N.P.T.-A., R.M., M.G., J.E.H., E.Y.-D., M. Shi, Y.H., M. Koido, A. Mishra, Q.L.-G., I.C., M.V.V., R.W., K. Lin, M.J.K., A.L., D.P., G.-V.R., H.-J.B., H.T., J.E.H., J. He, K.-J.L., L.T., L.B., V. Srinivasasainagendra, Y.J.K. and Y.-C.L. contributed to bioinformatics analyses. Y.R., M.B., C.A., D.W., P.R., T. Meitinger, K. Cho, K. Christensen, Yi-Ching Liaw, Yung-Po Liaw, B.N., A.T.-H., R.F.-S., J.W.J., M.E., T.B., K.W., M.J., F.-E.d.L., P.L., M.R., L.F., P.F., C.J., K.L.K., H.H., Y.-H.J., C.E.J., J.K., R.V., B.W., S.B., E.C.S., J.-A.Z., H.S.M., N.G., M.C., G.P., M.O’D., N.M., F.K., M. Sasaki, C.R., K.T., M.S.S., K.P., D. Strbian, J.C.H., S.S., A.H., L.L., V.G., N.S., D.-A.T., R.S., T.R., H.A., M.A.I., P.H., K.-G.H., F.M., V.A., R.Z., S.W.-S., M.A.I., S.K., B.M., H.X., J.C., C.-O.S., L.M., J.R., D. Saleheen, R.D.C., J. Hata, J.M.M.H., T.N., T.A., M. Koido, T. Kitazono, S. Tiedt, M.D., C.G., A.P., T. Morisaki, T. Meitinger, M. Kamouchi, Y.K., S. Debette, I.R., M.I., N.A., I.M., A.L., A.K.H., C.C., D.P., D.B., J. He, K.S., L.T., L.B., M.O., M.I.G., N.C.O.-M., P.W., P.D.-J., R.J., R.A., S. Damrauer, S.V., T.T., V. Salomaa, Y.-L.H., P.-H.C., K. Lee and Y.-P.L. contributed samples and phenotyping. S. Debette, M.D., Y.K., L.M., J.E.H., M.O.O., C.T.F., A. Mishra, R.M., T.H., T.J., Y.H., M. Koido, M. Shi, K.K. and S.N. wrote and edited the manuscript. All of the authors provided critical revision.
Peer review
Peer review information
Nature thanks Paul Timmers and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Summary statistics generated by the GIGASTROKE consortium across ancestries and stroke subtypes are available in the GWAS Catalog (GCST90104534–GCST90104563). The integrated polygenic risk score models of stroke in Europeans and East Asians are available in the PGS Catalog (PGS002724 and PGS002725). Individual level data can be requested directly from the authors of the contributing studies, listed in Supplementary Table 1. Single-nucleus RNA-seq data have been deposited in the SYNAPSE database as part of the Religious Orders Study and Memory and Aging Project (ROSMAP) (https://www.synapse.org) and through the RADC Resource Sharing Hub (https://www.radc.rush.edu). We used publicly available data from GTEx (https://gtexportal.org/home/), the Gusev laboratory (http://gusevlab.org/projects/fusion/), the FinnGen Freeze 7 cohort (https://www.finngen.fi/en/access_results), PhenoScanner v.2 database (http://www.phenoscanner.medschl.cam.ac.uk), pQTL summary statistics (10.1038/s41588-020-0682-6, http://www.phpc.cam.ac.uk/ceu/proteins/, http://metabolomics.helmholtz-muenchen.de/pgwas/index.php, https://zenodo.org/record/264128), deCODE genetics (https://www.decode.com/summarydata/) and summary statistics using the UK Biobank whole-exome sequencing (10.1038/s41586-021-04103-z).
Code availability
The code for computation of the integrated polygenic risk score of stroke are available at GitHub (https://github.com/hacchy1983/iPGS-construction). The drug discovery analysis was conducted using the following publicly available tools: GREP (https://github.com/saorisakaue/GREP), Trans-Phar (https://github.com/konumat/Trans-Phar), and the TwoSampleMR (https://mrcieu.github.io/TwoSampleMR/), coloc (https://chr1swallace.github.io/coloc/) and susieR (https://stephenslab.github.io/susieR/index.html) R packages.
Competing interests
C.D.A. has received sponsored research support from Bayer, and has consulted for ApoPharma; T. Konuma is an employee of JAPAN TOBACCO; M. E. reports grants from Bayer and fees paid to the Charité from Abbot, AstraZeneca, Bayer, Boehringer Ingelheim, BMS, Daiichi Sankyo, Amgen, GSK, Sanofi, Covidien, Novartis and Pfizer, all outside the submitted work; B.M.P. serves on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson; P.A. works with Fondation Alzheimer (nonprofit foundation) and Genoscreen (biotech company); H.L.L.’s participation in this project was part of a competitive contract awarded to Data Tecnica International by the National Institutes of Health to support open science research; M.A.N.’s participation in this project was part of a competitive contract awarded to Data Tecnica International by the National Institutes of Health to support open science research, he also currently serves on the scientific advisory board for Clover Therapeutics and is an advisor to Neuron23; N.A.M. declares institutional research grants to the TIMI Study Group at Brigham and Women’s Hospital from Amgen, Pfizer, Ionis, Novartis, AstraZeneca and NIH. The TIMI Study Group has received institutional research grant support through Brigham and Women’s Hospital from Abbott, Amgen, Anthos Therapeutics, ARCA Biopharma, AstraZeneca, Daiichi-Sankyo, Eisai, Intarcia, Ionis Pharmaceuticals, MedImmune, Merck, Novartis, Pfizer, Regeneron Pharmaceuticals, Roche, The Medicines Company, Zora Biosciences, Janssen Research and Development, Siemens Healthcare Diagnostics and Softcell Medical; F.K.K. declares that the TIMI Study Group has received institutional research grant support through Brigham and Women’s Hospital from Abbott, Amgen, Anthos Therapeutics, ARCA Biopharma, AstraZeneca, Daiichi-Sankyo, Eisai, Intarcia, Ionis Pharmaceuticals, MedImmune, Merck, Novartis, Pfizer, Regeneron Pharmaceuticals, Roche, The Medicines Company and Zora Biosciences; M.S.S. has consultancies with Althera, Amgen, Anthos Therapeutics, AstraZeneca, Beren Therapeutics, Bristol-Myers Squibb, DalCor, Dr. Reddy’s Laboratories, Fibrogen, IFM Therapeutics, Intarcia, Merck, Moderna, Novo Nordisk and Silence Therapeutics, and research grant support through Brigham and Women’s Hospital from Abbott, Amgen, Anthos Therapeutics, AstraZeneca, Bayer, Daiichi-Sankyo, Eisai, Intarcia, Ionis, Medicines Company, MedImmune, Merck, Novartis, Pfizer, Quark Pharmaceuticals; C.T.R. has consultancies with Anthos, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Daiichi Sankyo, Janssen and Pfizer, institutional research grant to the TIMI Study Group at Brigham and Women’s Hospital from Anthos, AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, Janssen, National Institutes of Health and Novartis, and consultancies with Anthos, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Daiichi Sankyo, Janssen and Pfizer. T.H. receives personal fees from Genome Analytics Japan; J.C.H. is supported by a personal fellowship from the British Heart Foundation (FS/14/55/30806), and acknowledges additional support from the Nuffield Department of Population Health (NDPH), University of Oxford, the British Heart Foundation Centre for Research Excellence, Oxford, and the Oxford Biomedical Research Centre. J.C.H. holds steering committee and Data and Safety Monitoring Board (DSMB) positions for various cardiovascular randomized controlled trials, and is a principal investigator/co-principal investigator of research grants from industry related to cardiovascular clinical trials and observational studies that are governed by University of Oxford contracts that protect personal independence. NDP.H also has a staff policy of not taking personal payments from industry (further details can be found online; https://www.ndph.ox.ac.uk/files/about/ndph-independence-of-research-policy-jun-20.pdf/@@download); S.S. has consultancies with Biogen; P.U.H. reports grants from German Ministry of Research and Education, during the conduct of the study, research grants from the German Ministry of Research and Education, European Union, Charité–Universitätsmedizin Berlin, Berlin Chamber of Physicians, German Parkinson Society, University Hospital Würzburg, Robert Koch Institute, German Heart Foundation, Federal Joint Committee (G-BA) within the Innovationfond, German Research Foundation, Bavarian State (ministry for science and the arts), German Cancer Aid, Charité–Universitätsmedizin Berlin (within Mondafis; supported by an unrestricted research grant to the Charité from Bayer), University Göttingen (within FIND-AF randomized; supported by an unrestricted research grant to the University Göttingen from Boehringer- Ingelheim), University Hospital Heidelberg (within RASUNOA-prime; supported by an unrestricted research grant to the University Hospital Heidelberg from Bayer, BMS, Boehringer-Ingelheim and Daiichi Sankyo), outside the submitted work; K.G.H. reports a study grant by Bayer, lecture fees/advisory board fees from Abbott, Alexion, AMARIN, AstraZeneca, Bayer, Biotronik, Boehringer Ingelheim, Bristol-Myers-Squibb, Daiichi Sankyo, Edwards Lifesciences, Medtronic, Pfizer, Premier Research, SUN Pharma and W. L. Gore & Associates; H.J.G. has received travel grants and speakers honoraria from Fresenius Medical Care, Neuraxpharm, Servier and Janssen Cilag as well as research funding from Fresenius Medical Care; J.M.M.H. is full time employee of Novo Nordisk; E.Y.-D. is full-time employee of Novo Nordisk. S. Damrauer receives research support from RenalytixAI and personal consulting fees from Calico Labs, outside the scope of the current research. H.B. reports grants from AstraZeneca, AstraZeneca Korea, Bayer Korea, Boehringer Ingelheim Korea, Boryung Pharmaceutical, Bristol Myers Squibb, Bristol Myers Squibb Korea, Chong Gun Dang Pharmaceutical, Daiichi Sankyo, Daiichi Sankyo Korea, Dong-A ST, Esai, Jeil Pharmaceutical, JLK, Korean Drug, SAMJIN Pharm., Servier Korea, Shinpoong Pharm., Shire International and Yuhan Corporation, and personal fees from Amgen Korea, Esai Korea, Otsuka Korea, Takeda Korea and Viatris Korea outside the submitted work. C.C. has received research support from GSK. The funders of the study had no role in the collection, analysis or interpretation of data, in the writing of the report or in the decision to submit the paper for publication. C.C. is a member of the advisory board of Vivid genetics; F.A.M. is supported by the German Research Foundation (Deutsche Forschungsgemeinschaft (DFG) within the UNION-CVD Clinician-Scientist Programme (project number 413657723) and has been previously supported by a MD/PhD Fellowship of the Interdisciplinary Center for Clinical Research, University Hospital Würzburg. A. Mishra, R.M., T.J., S.N., D.C.P., M. Koido, Q.L.G., M. Shi, Y.H., M.K.G., I.C., K.K., Yi-Ching Liaw, F.C.V., K. Lin., B.S.W., V. Srinivasasainagendra, L.P., G.C., M.R.C., L.T., R.A., G.V.R., N.H., Y.H.J., J.Q.T., V.A., J.C., M.N., C.Y., E.Y., M.J.K., A.J.L., R.L., T.A., N.D.A., M.K.B., T.M.B., D.A.B., J.C.B., C.B., S.B., A.C., P.M.R., K. Cho, Z.C., J.W.C., P.L.d., R.d.C., M.E., L.E.F., M.I.G., N.C.G., V.G., J. Hata, J. He., A.K.H., Y. Ho., A.S.H., H.I.H., M.I., M.A.J., C.E.J., C.J., M. Kamouchi, K.L.K., T. Kitazono, S.J.K., A.K., P.L., L.J.L., K. Lee, K. Lepik, J.L., L.L., A. Manichaikul, H.S.M., T. Meitinger, B.D.M., T. Morisaki, T.H.M., B.G.N., M.J.O., Y.O., N.C.O., B.O., A.P., S.S.R., J.R., M.S.S., R.L.S., D. Saleheen, E.C.S., V. Salomaa, M. Sargurupremraj, M. Sasaki, C.L.S., C.O.S., A.S., N.L.S., K.S., Y.S., Y.V.S., K.T., S. Tiedt, T.T., N.P.T., H.K.T., D.T., S. Trompet, A.M.T., A.T., M.v.V., R.V., S.S.V., K.L.W., P.W., D.W., P.W.W., H.X., Q.Y., K.Y., I.Y.M., C.G., T.N., J.W.J., I.L.R., D. Strbian, Y.J.K., P.C., E.M., M.R.I., H.A., S.W., K. Christensen, M.A.I., T.R., B.B.W., G.M.L., M.R., E.M.S., J.K., P.H.F., R.Z., K.P., R.F., F.d.L., T.L., Y.M.R., W.T.L., K.J.J., L.B., G.P., D.I.C., J.I.R., J.Z., T.J.N., M.F., Yung-Po Liaw, I.F., R.G.W., M.O.O., J.E.H., L.M., Y.K., M.D. and S. Debette declare no competing interests.
Footnotes
A list of authors and their affiliations appears online.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Aniket Mishra, Rainer Malik, Tsuyoshi Hachiya, Tuuli Jürgenson, Shinichi Namba, Daniel C. Posner
These authors jointly supervised this work: Christian T. Ruff, Mayowa O. Owolabi, Jennifer E. Huffman, Lili Milani, Yoichiro Kamatani, Martin Dichgans, Stephanie Debette
A list of authors and their affiliations appear online
Change history
11/14/2022
A Correction to this paper has been published: 10.1038/s41586-022-05492-5
Contributor Information
Martin Dichgans, Email: Martin.Dichgans@med.uni-muenchen.de.
Stephanie Debette, Email: stephanie.debette@u-bordeaux.fr.
The COMPASS Consortium:
Joshua C. Bis, Jin-Moo Lee, Yu-Ching Cheng, James F. Meschia, Wei Min Chen, Michèle M. Sale, Alan B. Zonderman, Michele K. Evans, James G. Wilson, Adolfo Correa, Matthew Traylor, Cathryn M. Lewis, Cara L. Carty, Alexander Reiner, Jeffrey Haessler, Carl D. Langefeld, Rebecca F. Gottesman, Kristine Yaffe, Yong Mei Liu, Charles Kooperberg, Leslie A. Lange, Karen L. Furie, Donna K. Arnett, Oscar R. Benavente, Raji P. Grewal, and Leema Reddy Peddareddygari
The INVENT Consortium:
Charles Kooperberg, Kristian Hveem, Sara Lindstrom, Lu Wang, Erin N. Smith, William Gordon, Astrid van Hylckama Vlieg, Mariza de Andrade, Jennifer A. Brody, Jack W. Pattee, Jeffrey Haessler, Ben M. Brumpton, Pierre Suchon, Ming-Huei Chen, Kelly A. Frazer, Constance Turman, Marine Germain, James MacDonald, Sigrid K. Braekkan, Sebastian M. Armasu, Nathan Pankratz, Rebecca D. Jackson, Jonas B. Nielsen, Franco Giulianini, Marja K. Puurunen, Manal Ibrahim, Susan R. Heckbert, Theo K. Bammler, Bryan M. McCauley, Kent D. Taylor, James S. Pankow, Alexander P. Reiner, Maiken E. Gabrielsen, Jean-François Deleuze, Chris J. O’Donnell, Jihye Kim, Barbara McKnight, Peter Kraft, John-Bjarne Hansen, Frits R. Rosendaal, John A. Heit, Weihong Tang, Pierre-Emmanuel Morange, Andrew D. Johnson, and Christopher Kabrhel
The Dutch Parelsnoer Initiative (PSI) Cerebrovascular Disease Study Group:
Ewoud J. van Dijk, Peter J. Koudstaal, Gert-Jan Luijckx, Paul J. Nederkoorn, Robert J. van Oostenbrugge, Marieke C. Visser, Marieke J. H. Wermer, and L. Jaap Kappelle
The Estonian Biobank:
Tõnu Esko, Andres Metspalu, Reedik Mägi, and Mari Nelis
The NINDS Stroke Genetics Network (SiGN):
Marguerite R. Irvin, Frank-Erik de Leeuw, Christopher R. Levi, Jane Maguire, Jordi Jiménez-Conde, Pankaj Sharma, Cathie L. M. Sudlow, Kristiina Rannikmäe, Reinhold Schmidt, Agnieszka Slowik, Joanna Pera, Vincent N. S. Thijs, Arne G. Lindgren, Andreea Ilinca, Olle Melander, Gunnar Engström, Kathryn M. Rexrode, Peter M. Rothwell, Tara M. Stanne, Julie A. Johnson, John Danesh, Adam S. Butterworth, Laura Heitsch, Giorgio B. Boncoraglio, Michiaki Kubo, Alessandro Pezzini, Arndt Rolfs, Anne-Katrin Giese, David Weir, Rebecca D. Jackson, Owen A. Ross, Robin Lemmons, Martin Soderholm, Mary Cushman, Katarina Jood, Caitrin W. McDonough, Steven Bell, Birgit Linkohr, Tsong-Hai Lee, and Jukka Putaala
The MEGASTROKE Consortium:
Christopher D. Anderson, Oscar L. Lopez, Xueqiu Jian, Ulf Schminke, Natalia Cullell, Pilar Delgado, Laura Ibañez, Jerzy Krupinski, Vasileios Lioutas, Koichi Matsuda, Joan Montaner, Elena Muiño, Jaume Roquer, Chloe Sarnowski, Naveed Sattar, Gerli Sibolt, Alexander Teumer, Loes Rutten-Jacobs, Masahiro Kanai, Anne-Katrin Giese, Solveig Gretarsdottir, Natalia S. Rost, Salim Yusuf, Peter Almgren, Hakan Ay, Steve Bevan, Robert D. Brown, Jr, Caty Carrera, Julie E. Buring, Wei-Min Chen, Ioana Cotlarciuc, Paul I. W. de Bakker, Anita L. DeStefano, Marcel den Hoed, Qing Duan, Stefan T. Engelter, Guido J. Falcone, Rebecca F. Gottesman, Stefan Gustafsson, Ahamad Hassan, Elizabeth G. Holliday, George Howard, Fang-Chi Hsu, Erik Ingelsson, Tamara B. Harris, Brett M. Kissela, Dawn O. Kleindorfer, Claudia Langenberg, Robin Lemmens, Didier Leys, Wei-Yu Lin, Erik Lorentzen, Patrik K. Magnusson, Patrick F. McArdle, Sara L. Pulit, Kenneth Rice, Saori Sakaue, Bishwa R. Sapkota, Christian Tanislav, Gudmar Thorleifsson, Unnur Thorsteinsdottir, Christophe Tzourio, Cornelia M. van Duijn, Matthew Walters, Nicholas J. Wareham, Najaf Amin, Hugo J. Aparicio, John Attia, Alexa S. Beiser, Claudine Berr, Mariana Bustamante, Valeria Caso, Seung Hoan Choi, Ayesha Chowhan, Jean-François Dartigues, Hossein Delavaran, Marcus Dörr, Ian Ford, Wander S. Gurpreet, Anders Hamsten, Atsushi Hozawa, Martin Ingelsson, Motoki Iwasaki, Sara Kaffashian, Lalit Kalra, Olafur Kjartansson, Manja Kloss, Daniel L. Labovitz, Cathy C. Laurie, Linxin Li, Lars Lind, Cecilia M. Lindgren, Hirata Makoto, Naoko Minegishi, Andrew P. Morris, Martina Müller-Nurasyid, Bo Norrving, Soichi Ogishima, Eugenio A. Parati, Nancy L. Pedersen, Markus Perola, Pekka Jousilahti, Silvana Pileggi, Raquel Rabionet, Iolanda Riba-Llena, Marta Ribasés, Jose R. Romero, Anthony G. Rudd, Antti-Pekka Sarin, Ralhan Sarju, Mamoru Satoh, Norie Sawada, Ásgeir Sigurdsson, Albert Smith, O. Colin Stine, David J. Stott, Konstantin Strauch, Takako Takai, Hideo Tanaka, Emmanuel Touze, Shoichiro Tsugane, Andre G. Uitterlinden, Einar M. Valdimarsson, Sven J. van der Lee, Kenji Wakai, Stephen R. Williams, Charles D. A. Wolfe, Quenna Wong, Taiki Yamaji, Dharambir K. Sanghera, Kari Stefansson, Kent D. Taylor, Nicolas Martinez-Majander, Kenji Sobue, Carolina Soriano-Tárraga, and Henry Völzke
The SIREN Consortium:
Onoja Akpa, Fred S. Sarfo, Albert Akpalu, Reginald Obiako, Kolawole Wahab, Godwin Osaigbovo, Lukman Owolabi, Morenikeji Komolafe, Carolyn Jenkins, Oyedunni Arulogun, Godwin Ogbole, Abiodun M. Adeoye, Joshua Akinyemi, Atinuke Agunloye, Adekunle G. Fakunle, Ezinne Uvere, Abimbola Olalere, and Olayinka J. Adebajo
The China Kadoorie Biobank Collaborative Group:
Junshi Chen, Robert Clarke, Rory Collins, Yu Guo, Chen Wang, Jun Lv, Richard Peto, Yiping Chen, Zammy Fairhurst-Hunter, Michael Hill, Alfred Pozarickij, Dan Schmidt, Becky Stevens, Iain Turnbull, and Canqing Yu
The International Stroke Genetics Consortium (ISGC):
Quentin Le Grand and Leslie E. Ferreira
The Biobank Japan:
Akiko Nagai and Yoishinori Murakami
The CHARGE Consortium:
Mirjam I. Geerlings, Natalie C. Gasca, Vilmundur Gudnason, Marion van Vugt, Rebecca F. Gottesman, Eric J. Shiroma, Sigurdur Sigurdsson, Mohsen Ghanbari, Eric Boerwinkle, Alexa S. Beiser, Bernard Fongang, Ruiqi Wang, Mohammad K. Ikram, and Uwe Völker
The GIGASTROKE Consortium:
Phil L. de Jager, Rafael de Cid, Børge G. Nordestgaard, Muralidharan Sargurupremraj, and Shefali S. Verma
Extended data
is available for this paper at 10.1038/s41586-022-05165-3.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-05165-3.
References
- 1.Malik R, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traylor, M. et al. Genetic basis of lacunar stroke: a pooled analysis of individual patient data and genome-wide association studies. Lancet Neurol.10.1016/s1474-4422(21)00031-4 (2021). [DOI] [PMC free article] [PubMed]
- 3.Koido, M. et al. Predicting cell-type-specific non-coding RNA transcription from genome sequence. Preprint at bioRxiv10.1101/2020.03.29.011205 (2020).
- 4.Namba, S. et al. A practical guideline of genomics-driven drug discovery in the era of global biobank meta-analysis. Preprint at medRxiv10.1101/2021.12.03.21267280 (2021). [DOI] [PMC free article] [PubMed]
- 5.Abraham G, et al. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat. Commun. 2019;10:5819. doi: 10.1038/s41467-019-13848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol.20, 795–820 (2021). [DOI] [PMC free article] [PubMed]
- 7.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishra A, Macgregor S. VEGAS2: software for more flexible gene-based testing. Twin Res. Hum. Genet. 2015;18:86–91. doi: 10.1017/thg.2014.79. [DOI] [PubMed] [Google Scholar]
- 9.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magi R, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet. 2017;26:3639–3650. doi: 10.1093/hmg/ddx280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turley P, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 2018;50:229–237. doi: 10.1038/s41588-017-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G, Sarkar A, Carbonetto P, Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez BAT, et al. A platelet function modulator of thrombin activation is causally linked to cardiovascular disease and affects PAR4 receptor signaling. Am. J. Hum. Genet. 2020;107:211–221. doi: 10.1016/j.ajhg.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giri A, et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019;51:51–62. doi: 10.1038/s41588-018-0303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra A, MacGregor S. A novel approach for pathway analysis of GWAS data highlights role of BMP signaling and muscle cell differentiation in colorectal cancer susceptibility. Twin Res. Hum. Genet. 2017;20:1–9. doi: 10.1017/thg.2016.100. [DOI] [PubMed] [Google Scholar]
- 16.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 2016;40:597–608. doi: 10.1002/gepi.21998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fromer M, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gusev A, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright FA, et al. Heritability and genomics of gene expression in peripheral blood. Nat. Genet. 2014;46:430–437. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cain, A. et al. Multi-cellular communities are perturbed in the aging human brain and with Alzheimer’s disease. Preprint at bioRxiv10.1101/2020.12.22.424084 (2022).
- 22.Sakaue S, Okada Y. GREP: genome for REPositioning drugs. Bioinformatics. 2019;35:3821–3823. doi: 10.1093/bioinformatics/btz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wishart DS, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konuma T, Ogawa K, Okada Y. Integration of genetically regulated gene expression and pharmacological library provides therapeutic drug candidates. Hum. Mol. Genet. 2021;30:294–304. doi: 10.1093/hmg/ddab049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chamorro A. TP receptor antagonism: a new concept in atherothrombosis and stroke prevention. Cerebrovasc. Dis. 2009;27:20–27. doi: 10.1159/000209262. [DOI] [PubMed] [Google Scholar]
- 26.Yan A, et al. Thromboxane A2 receptor antagonist SQ29548 reduces ischemic stroke-induced microglia/macrophages activation and enrichment, and ameliorates brain injury. Sci. Rep. 2016;6:35885. doi: 10.1038/srep35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bousser MG, et al. Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet. 2011;377:2013–2022. doi: 10.1016/S0140-6736(11)60600-4. [DOI] [PubMed] [Google Scholar]
- 28.Safdar H, et al. Regulation of the F11, Klkb1, Cyp4v3 gene cluster in livers of metabolically challenged mice. PLoS ONE. 2013;8:e74637. doi: 10.1371/journal.pone.0074637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Haan HG, et al. Targeted sequencing to identify novel genetic risk factors for deep vein thrombosis: a study of 734 genes. J. Thromb. Haemost. 2018;16:2432–2441. doi: 10.1111/jth.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohmann JL, et al. Genetic determinants of activity and antigen levels of contact system factors. J. Thromb. Haemost. 2019;17:157–168. doi: 10.1111/jth.14307. [DOI] [PubMed] [Google Scholar]
- 31.Backman JD, et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindstrom S, et al. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood. 2019;134:1645–1657. doi: 10.1182/blood.2019000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verhamme P, et al. Abelacimab for prevention of venous thromboembolism. N. Engl. J. Med. 2021;385:609–617. doi: 10.1056/NEJMoa2105872. [DOI] [PubMed] [Google Scholar]
- 34.Weeks E. M. et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. Preprint at medRxiv10.1101/2020.09.08.20190561 (2020). [DOI] [PMC free article] [PubMed]
- 35.Marston NA, et al. Clinical application of a novel genetic risk score for ischemic stroke in patients with cardiometabolic disease. Circulation. 2021;143:470–478. doi: 10.1161/CIRCULATIONAHA.120.051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malik R, et al. Whole-exome sequencing reveals a role of HTRA1 and EGFL8 in brain white matter hyperintensities. Brain. 2021;144:2670–2682. doi: 10.1093/brain/awab253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Söderholm M, et al. Genome-wide association meta-analysis of functional outcome after ischemic stroke. Neurology. 2019;92:e1271–e1283. doi: 10.1212/WNL.0000000000007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma D, et al. Inhibition of KLF5-Myo9b-RhoA pathway-mediated podosome formation in macrophages ameliorates abdominal aortic aneurysm. Circ. Res. 2017;120:799–815. doi: 10.1161/CIRCRESAHA.116.310367. [DOI] [PubMed] [Google Scholar]
- 39.Yang X, et al. FURIN expression in vascular endothelial cells is modulated by a coronary artery disease-associated genetic variant and influences monocyte transendothelial migration. J. Am. Heart Assoc. 2020;9:e014333. doi: 10.1161/JAHA.119.014333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yakala GK, et al. FURIN inhibition reduces vascular remodeling and atherosclerotic lesion progression in mice. Arterioscler. Thromb. Vasc. Biol. 2019;39:387–401. doi: 10.1161/ATVBAHA.118.311903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantuti-Castelvetri L, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nannoni S, de Groot R, Bell S, Markus HS. Stroke in COVID-19: a systematic review and meta-analysis. Int. J. Stroke. 2021;16:137–149. doi: 10.1177/1747493020972922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyden P, et al. Phase 1 safety, tolerability and pharmacokinetics of 3K3A-APC in healthy adult volunteers. Curr. Pharm. Des. 2013;19:7479–7485. doi: 10.2174/1381612819666131230131454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyden P, et al. Final results of the RHAPSODY trial: a multi-center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A-APC, a recombinant variant of human activated protein C, in Combination with Tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann. Neurol. 2019;85:125–136. doi: 10.1002/ana.25383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huuskonen MT, et al. Protection of ischemic white matter and oligodendrocytes in mice by 3K3A-activated protein C. J. Exp. Med. 2022;219:e20211372. doi: 10.1084/jem.20211372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu W, et al. Blockade of platelet glycoprotein receptor Ib ameliorates blood-brain barrier disruption following ischemic stroke via Epac pathway. Biomed. Pharmacother. 2021;140:111698. doi: 10.1016/j.biopha.2021.111698. [DOI] [PubMed] [Google Scholar]
- 47.Yamashita S, et al. Probucol trial for secondary prevention of atherosclerotic events in patients with coronary heart disease (PROSPECTIVE) J. Atheroscler. Thromb. 2021;28:103–123. doi: 10.5551/jat.55327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben-Eghan C, et al. Don’t ignore genetic data from minority populations. Nature. 2020;585:184–186. doi: 10.1038/d41586-020-02547-3. [DOI] [PubMed] [Google Scholar]
- 49.Dudbridge F, et al. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat. Commun. 2019;10:1561. doi: 10.1038/s41467-019-09381-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo Y, et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum. Mol. Genet. 2021;30:1521–1534. doi: 10.1093/hmg/ddab130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.3C Study Group. Vascular factors and risk of dementia: design of the Three-City Study and baseline characteristics of the study population. Neuroepidemiology. 2003;22:316–325. doi: 10.1159/000072920. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen JB, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018;50:1234–1239. doi: 10.1038/s41588-018-0171-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sargurupremraj M, et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat. Commun. 2020;11:6285. doi: 10.1038/s41467-020-19111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikpay M, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishigaki K, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020;52:669–679. doi: 10.1038/s41588-020-0640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Low SK, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat. Genet. 2017;49:953–958. doi: 10.1038/ng.3842. [DOI] [PubMed] [Google Scholar]
- 59.Evangelou E, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pulit SL, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019;28:166–174. doi: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xue A, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018;9:2941. doi: 10.1038/s41467-018-04951-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bowden J, Hemani G, Davey Smith G. Invited Commentary: detecting individual and global horizontal pleiotropy in Mendelian randomization—a job for the humble heterogeneity statistic? Am. J. Epidemiol. 2018;187:2681–2685. doi: 10.1093/aje/kwy185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017;46:1985–1998. doi: 10.1093/ije/dyx102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benner C, et al. Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet. 2017;101:539–551. doi: 10.1016/j.ajhg.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou J, et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018;50:1171–1179. doi: 10.1038/s41588-018-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forrest AR, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hon CC, et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204. doi: 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garieri M, et al. The effect of genetic variation on promoter usage and enhancer activity. Nat. Commun. 2017;8:1358. doi: 10.1038/s41467-017-01467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feng H, et al. Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies. PLoS Genet. 2021;17:e1008973. doi: 10.1371/journal.pgen.1008973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giambartolomei C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wingo AP, et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet. 2021;53:143–146. doi: 10.1038/s41588-020-00773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bennett DA, et al. Religious orders study and rush memory and aging project. J. Alzheimers Dis. 2018;64:S161–S189. doi: 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beach TG, et al. Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology. 2015;35:354–389. doi: 10.1111/neup.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sey NYA, et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 2020;23:583–593. doi: 10.1038/s41593-020-0603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen X, Ji ZL, Chen YZ. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whirl-Carrillo M, et al. An evidence-based framework for evaluating pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2021;110:563–572. doi: 10.1002/cpt.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ochoa D, et al. Open Targets Platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 2021;49:D1302–D1310. doi: 10.1093/nar/gkaa1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mancuso N, et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet. 2019;51:675–682. doi: 10.1038/s41588-019-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Subramanian A, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng J, et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020;52:1122–1131. doi: 10.1038/s41588-020-0682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Emilsson V, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. doi: 10.1126/science.aaq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Folkersen L, et al. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet. 2017;13:e1006706. doi: 10.1371/journal.pgen.1006706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suhre K, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017;8:14357. doi: 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun BB, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao C, et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 2018;9:3268. doi: 10.1038/s41467-018-05512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hemani G, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13:e1007081. doi: 10.1371/journal.pgen.1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17:e1009440. doi: 10.1371/journal.pgen.1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang C, et al. Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat. Neurosci. 2021;24:1302–1312. doi: 10.1038/s41593-021-00886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferkingstad E, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 2021;53:1712–1721. doi: 10.1038/s41588-021-00978-w. [DOI] [PubMed] [Google Scholar]
- 96.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–2376. doi: 10.1093/bioinformatics/btu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Staley JR, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32:3207–3209. doi: 10.1093/bioinformatics/btw373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kamat MA, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. doi: 10.1093/bioinformatics/btz469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khera AV, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vilhjálmsson BJ, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 2015;97:576–592. doi: 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zou H, Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. B. 2005;67:301–320. [Google Scholar]
- 106.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 107.Mahajan A, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat. Genet.42, 441–447 (2010). [DOI] [PMC free article] [PubMed]
- 109.Giugliano RP, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2013;369:2093–2104. doi: 10.1056/NEJMoa1310907. [DOI] [PubMed] [Google Scholar]
- 110.O’Donoghue ML, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–1015. doi: 10.1001/jama.2014.11061. [DOI] [PubMed] [Google Scholar]
- 111.Scirica BM, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 112.Bonaca MP, et al. Long-term use of ticagrelor in patients with prior myocardial infarction. N. Engl. J. Med. 2015;372:1791–1800. doi: 10.1056/NEJMoa1500857. [DOI] [PubMed] [Google Scholar]
- 113.Sabatine MS, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 114.Marston NA, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation. 2020;141:616–623. doi: 10.1161/CIRCULATIONAHA.119.043805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, including a description of the GWAS of stroke-risk factors in Tohuku Medical Megabank and Calculation of candidate polygenic score models; description of study populations in the GIGASTROKE initiative; study-specific acknowledgements; information on other members of participating consortia; and Supplementary References.
Supplementary Tables 1–56.
Data Availability Statement
Summary statistics generated by the GIGASTROKE consortium across ancestries and stroke subtypes are available in the GWAS Catalog (GCST90104534–GCST90104563). The integrated polygenic risk score models of stroke in Europeans and East Asians are available in the PGS Catalog (PGS002724 and PGS002725). Individual level data can be requested directly from the authors of the contributing studies, listed in Supplementary Table 1. Single-nucleus RNA-seq data have been deposited in the SYNAPSE database as part of the Religious Orders Study and Memory and Aging Project (ROSMAP) (https://www.synapse.org) and through the RADC Resource Sharing Hub (https://www.radc.rush.edu). We used publicly available data from GTEx (https://gtexportal.org/home/), the Gusev laboratory (http://gusevlab.org/projects/fusion/), the FinnGen Freeze 7 cohort (https://www.finngen.fi/en/access_results), PhenoScanner v.2 database (http://www.phenoscanner.medschl.cam.ac.uk), pQTL summary statistics (10.1038/s41588-020-0682-6, http://www.phpc.cam.ac.uk/ceu/proteins/, http://metabolomics.helmholtz-muenchen.de/pgwas/index.php, https://zenodo.org/record/264128), deCODE genetics (https://www.decode.com/summarydata/) and summary statistics using the UK Biobank whole-exome sequencing (10.1038/s41586-021-04103-z).
The code for computation of the integrated polygenic risk score of stroke are available at GitHub (https://github.com/hacchy1983/iPGS-construction). The drug discovery analysis was conducted using the following publicly available tools: GREP (https://github.com/saorisakaue/GREP), Trans-Phar (https://github.com/konumat/Trans-Phar), and the TwoSampleMR (https://mrcieu.github.io/TwoSampleMR/), coloc (https://chr1swallace.github.io/coloc/) and susieR (https://stephenslab.github.io/susieR/index.html) R packages.