

Abstract
Copper-containing metalloenzymes constitute a major class of proteins which catalyze a myriad of reactions in nature. Inspired by the structural and functional characteristics of this unique class of metalloenzymes, we report the conception, design, characterization, and functional studies of a de novo artificial copper peptide (ArCuP) within a trimeric self-assembled polypeptide scaffold that activates and reduces peroxide. Using a first principles approach, the ArCuP was designed to coordinate one Cu via three His residues introduced at an a site of the peptide scaffold. X-ray crystallographic, UV-vis and EPR data demonstrate that Cu binds via the Nε atoms of His forming a T2Cu environment. When reacted with hydrogen peroxide, the putative copper-hydroperoxo species is formed where a reductive priming step accelerates the rate of its formation and reduction. Mass spectrometry was used to identify specific residues undergoing oxidative modification, which showed His oxidation only in the reduced state. The redox behavior of the ArCuP was elucidated by protein film voltammetry. Detailed characterization of the electrocatalytic behavior of the ArCuP led us to determine the catalytic parameters (KM, kcat), which established the peroxidase activity of the ArCuP. Combined spectroscopic and electrochemical data showed a pH-dependence on the reactivity, which was optimum at pH 7.5.
Keywords: de novo metalloenzymes, copper proteins, self-assembly, peroxide activation, electrocatalysis
Graphical Abstract
INTRODUCTION
How the protein sequence determines the 3D structure and function of metalloenzymes remains an unsolved problem. De novo protein design allows the construction of artificial metalloproteins that are simpler yet accurate representations of the structure and function of native enzymes.1 Guided by specific sequence-to-structure determinants, the de novo designed metalloproteins provide a deep understanding of the folding and coordination properties of complex metalloenzymes. When successful, this approach produces simpler functional analogs of the native enzymes and provide a foundational understanding of the underlying chemistry. A grand challenge in the past has been the design of catalytically active de novo metalloenzymes. Recent success in this pursuit has been achieved in producing artificial de novo designed bio molecular catalysts for hydrolytic, reductive, dismutative, and oxidative chemistry.2–6 In this context, an unexplored area is to create de novo constructs whose functionality can lead to energy-relevant transformations by activation of O-O and O-H bonds. The reserve of fossil fuel is finite and not sustainable. In addition, burning of fossil fuel causes pollution and climate change. Therefore, it is necessary to find alternative forms of energy using abundant natural resources such as dioxygen and water. Development of biomolecular catalysts that catalyze energy-relevant transformations using cheap transition metals in aqueous conditions is highly desired in this regard.
Cu-containing metalloenzymes are involved in the binding, activation and reduction of O2 as part of numerous biochemical processes such as respiration and substrate oxidation.7–11 Cu is typically 3–5 coordinate in the resting state, bound to 2–3 His and 1–2 water molecules. The trigonal geometry of Cu is optimized to promote associative inner sphere binding of O2.12 The enzyme pocket tolerates the ensuing redox-induced changes in the coordination geometry to facilitate turnovers. The active sites that bind and activate substrates all have at least 1 His bound via the Nε atom, e.g. the CuB site of Cyt c Oxidase (CcO), amine oxidase and lytic polysaccharide monooxygenase (LPMO, Fig. 1A) while in others all the His are bound via the Nε atoms, such as the CuM site of peptidyl α-hydroxylating monooxygnease (PHM, Fig. 1B), tyrosinase (Ty, Fig. 1C), galactose oxidase (GO) and nitrite reductase (NiR). On the contrary, the Cu sites involved in electron transfer (ET), e.g. the blue T1Cu, purple CuA, and the T2 CuH site of PHM bind His exclusively via the Nδ atoms. It has been proposed that the reaction pathways may involve cupric superoxo (Cu(II)-O2•−), cupric hydroperxo (Cu(II)-OOH), high-valent Cu-oxyl (Cu(II)-O2•) or Cu-oxo (Cu(III)=O) species as possible intermediates.7, 13–15 Although O2 is the main substrate used by LPMOs, recent evidence has demonstrated that H2O2 can be employed as a co-substrate and that a controlled supply of H2O2 to Cu(I)-LPMO results in reaction rates that are several orders of magnitude higher compared to O2.16–18 A fundamental understanding of the coordination chemistry and the reactive intermediates are critical to not only elucidate the likely mechanisms but also as a means of energy production from biomass degradation. In addition, peroxide-based fuel cells are promising alternative energy generators which can relieve our heavy dependence on fossil fuels.19–21 One approach is the de novo design of artificial Cu peptides (ArCuPs) as functional analogs that can provide insights into the structure, function and mechanistic aspects of the native enzymes as well as to open up avenues in bioenergy production. Herein, we report the design of a tris-His coordinated ArCuP, designated as [3SCC-(I9H)3], as a functional metallopeptide for peroxide activation and its reduction to H2O (H2O2 + 2e− + 2H+ → 2H2O). A combined spectroscopic, crystallographic, mass spectrometric, and protein film electrochemistry (PFE) studies have led to the elucidation of the physical, redox, and catalytic properties of this ArCuP. The importance of reductive priming for peroxide activation has been also addressed.
Figure 1.
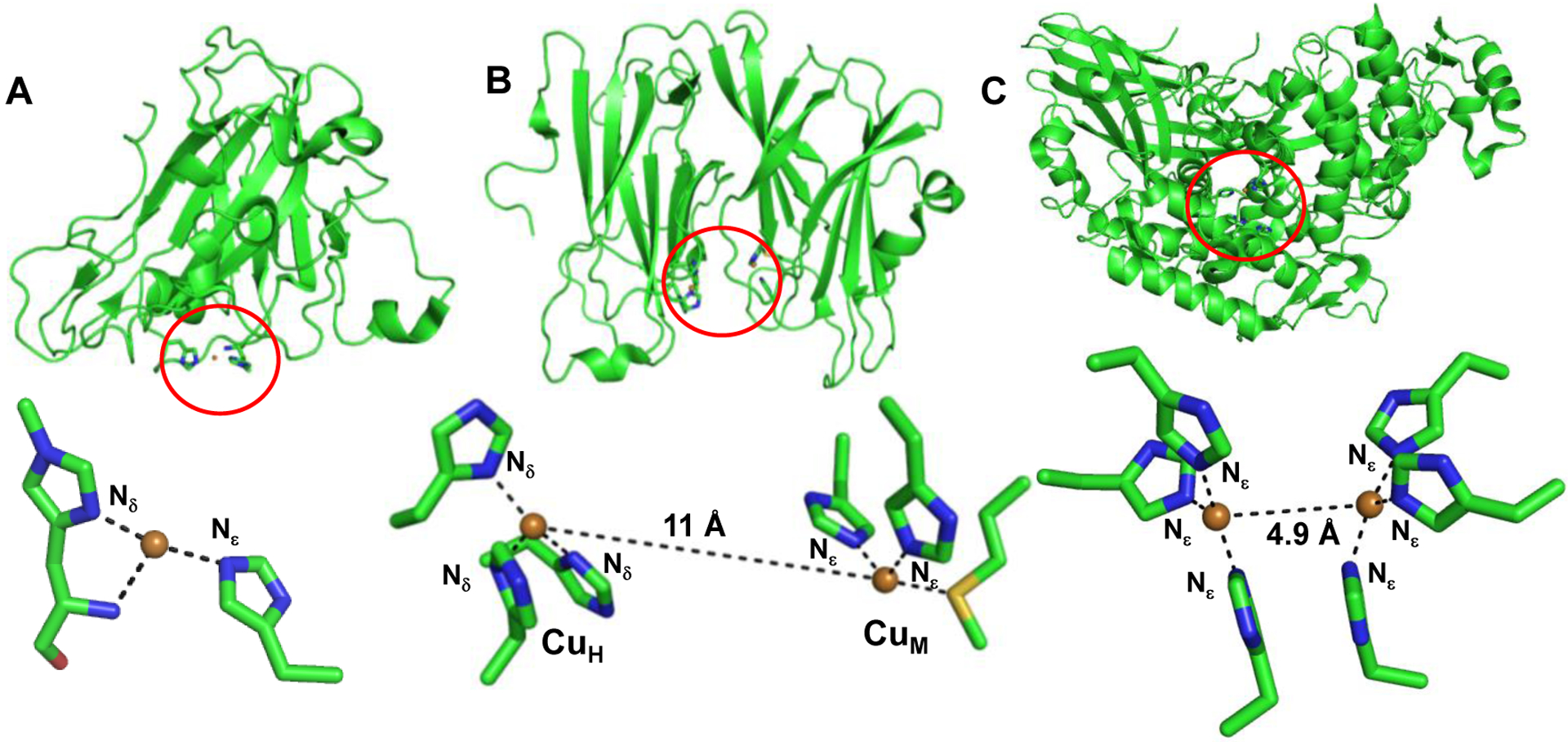
Structures of LPMO (A), PHM (B) and Ty (C). LPMO has a mixed Nδ/ Nε coordination (PDB 2YET). The CuH (ET) site of PHM (PDB 3MIH) has an all-Nδ ligation while the substrate binding CuM active site has Nε ligation. Both Cu in Ty (PDB 3HHS) bind all His via the Nε atoms.
RESULTS
Peptide design.
To create a trigonal Cu(His)3 coordination, we chose de novo designed trimeric coiled coil (3SCC) as a suitable scaffold where one His from each peptide strand ligates to Cu. The 3SCC was designed based on the heptad repeat (abcdefg)1, 22 with the general sequence (IAAIKQE)4. The Ile residues at the a and d sites direct the desired self-assembly into trimers according to the Knob-Into-Holes packing. Inter-helix ion pairs formed by the e and g site residues determine the parallel orientation of the helices. Although the identity of the rest of the heptad residues vary, it is common to place Ala at the b/c positions due its high helix-forming propensity. Polar residues are positioned at the f site to improve water solubility. The His was introduced at the a site of the 2nd heptad to form the Cu binding site. The N and C termini ends were capped with acetylated and amidated Gly, respectively. A Trp chromophore was placed at an external f site to enable quantification. The final sequence is Ac-GIAAIKQEHAAIKQEIAAIKQEIAAIKWEG-CONH2.
X-ray structure.
The ArCuP was crystallized and diffracted to 1.45Å. The overall structure of the peptide is an ordered coiled-coil assembly of parallel helices (Figs. 2, S1). The homotrimeric assemblies are formed by three copies of A, B or C chains (A3/B3/C3) where each monomer from a symmetry related molecule completes the trimer. All the amino acids occupy the preferred ϕ,ψ regions of the Ramachandran plot for a right-handed α-helix and all side chains are present in their favored rotameric conformation. Inter-helical ion pairs are formed between Glu15 and Lys20 within each peptide chain in the trimer. The orientation of the His within a trimer is symmetric with the Nε-Nε distances of 3.41Å. The His side chains are pointed towards the helical axis with both the Cβ and Nε atoms facing the N-termini. The ∠N-Cα-Cβ-Cγ dihedral angles for the His are ~ −92°. During initial refinement without added Cu, the Fo-Fc electron density map had positive density (Fig. S2A) overlapping the His residues. This residual electron density disappeared after subsequent refinement with added Cu (Fig. S2B). The Cu is bound to the Nε of all the His with Cu-Nε distance of 2.02Å (Fig. 2B,C). The Cu is lying slightly below the plane (towards C termini) of the Nε atoms.
Figure 2.
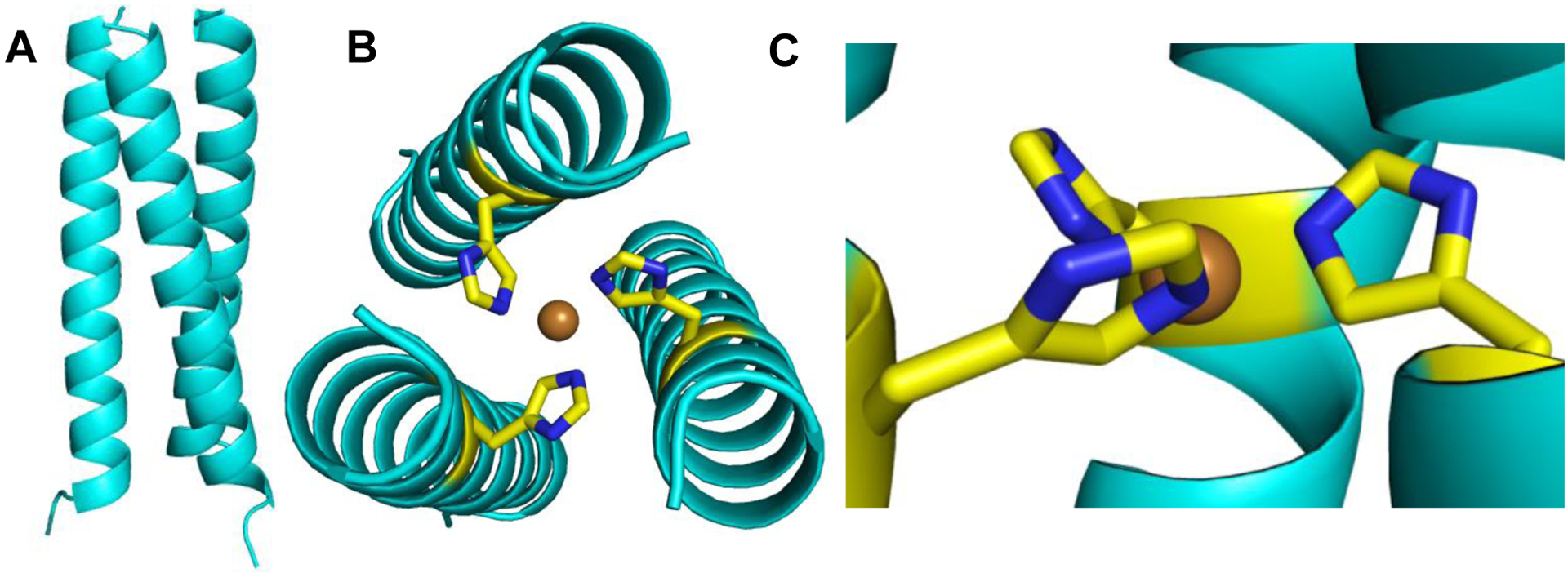
A 1.45Å X-ray structure of the ArCuP. The cartoon in A shows the parallel assembly of the peptide trimer. Views from the N-termini (B) showing the projection of the His residues towards the interior where Cu(II) binds in a an approximate trigonal planar geometry to the Nε atoms of each His. The orientation of the His residues when viewed perpendicular to the central helical axis is shown in C. The cartoon is in light cyan, His are shown as yellow and blue sticks, and Cu(II) as orange sphere. Figures generated in PyMol from the PDB code 7L33.
Electronic nature and Cu(II)/Cu(I) binding.
The UV-vis spectrum of [3SCC-Cu(I9H)3]2+ shows a broad absorption centered at ~ 590 nm with ε590=78 M−1cm−1 at pH 7.5 (Fig. 3). The λmax and the molar absorptivity are characteristic of d-d transition of Cu(II) in a Cu(His)3 environment with bound water(s).2,3 Importantly, the possibility of a Cu(His)2 ligation is eliminated, as the latter species typically absorbs at ~690 nm. Competitive binding assays using [Cu(I)(Bcs)2]3− (Bcs = Bathocuproinedisulfonate) were performed anaerobically at pH 7.5 to derive the Cu(I) binding affinity. Free ligand Bcs2− was titrated to 30 μM [3SCC-(I9H)3] bound to 15 μM Cu(I). Excess peptide was kept to minimize the amount of unbound Cu(I). A plot of the absorbance at 483 nm for [Cu(I)(Bcs)2]3− vs the equivalence of Bcs2− shows saturation at 2:1 ratio of Bcs2−:Cu(I) (Fig. S3), which suggests that 1 equivalent of Cu(I) binds to [3SCC-(I9H)3]. The Cu(I) binding affinity for [3SCC-(I9H)3] determined from equation (i) is 12 fM (Tables 1, S1). The affinity for Cu(II) determined according to the Nernst equation (eq. ii) using the redox potential (E°’) (vide infra) of the ArCuP is 184 fM (Table 1) at pH 7.5.
Figure 3.
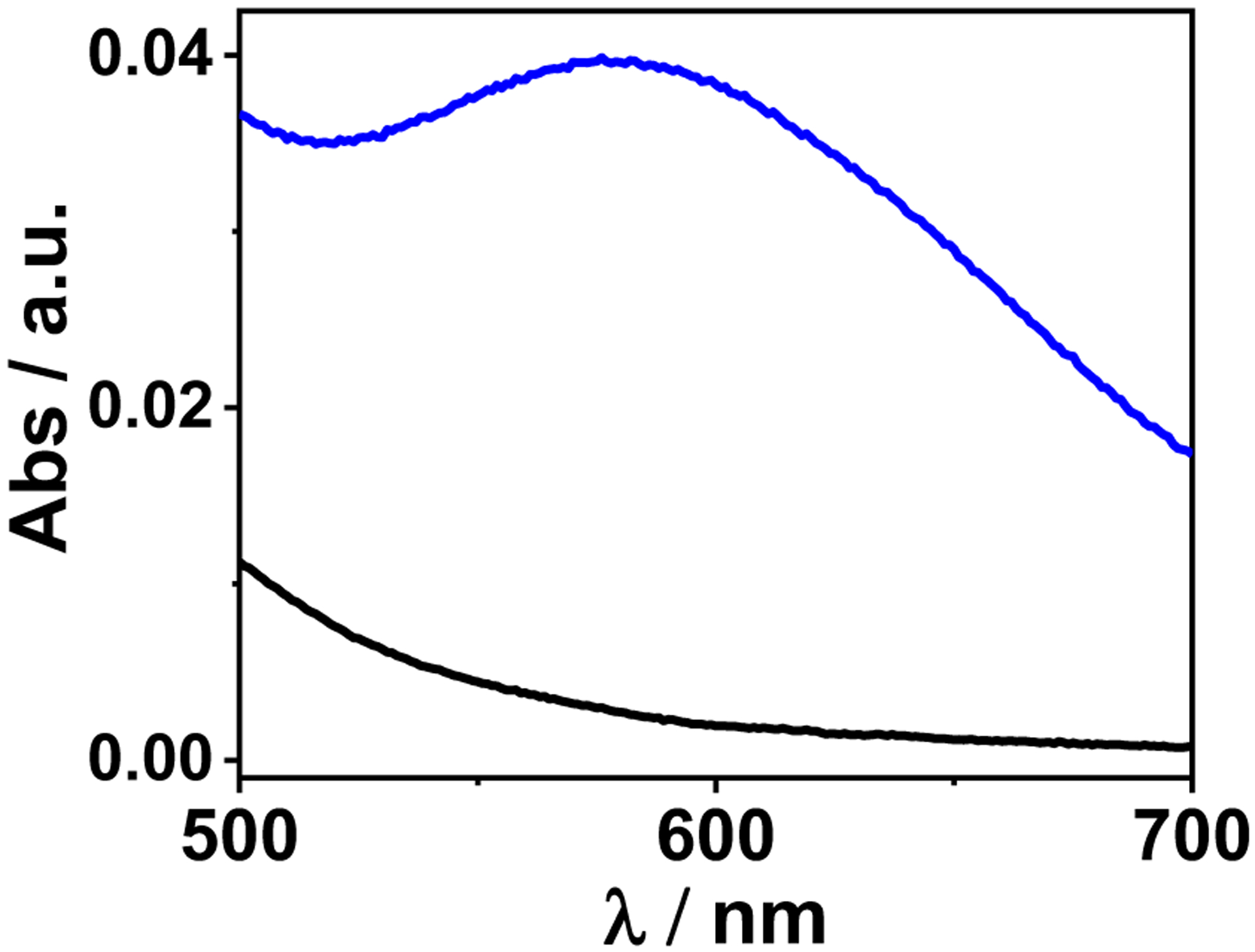
Visible spectrum of [3SCC-Cu(I9H)3]2+ showing the d-d transition (blue) versus the apo peptide (black) in 100 mM HEPES pH 7.5.
Table 1.
Relevant structural and spectroscopic parameters.
| Sample | Nε-Nε (Å) | Cu-Nε (Å) | λmax (nm) (ε / M−1cm−1) | Kd (fM) Cu(I)/Cu(II) | gx, gy | gz | Ax, Ay (MHz) | Az (MHz) |
|---|---|---|---|---|---|---|---|---|
| [3SCC-Cu(I9H)3]2+ | 3.41 | 2.03 | 590 (78) | 12 / 184 | 2.05 | 2.25 | 116 | 566 |
| [3SCC-Cu(I9H)3]2+ + H2O2 | -- | -- | 330 (6400) 387 (1400) 570 (75) |
-- | 2.06 | 2.27 | 97 | 530 |
| [3SCC-Cu(I9H)3]+ + H2O2 | -- | -- | 350 (11530) 440 (460) |
-- | 2.06 | 2.28 | 100 | 497 |
H2O2 reactivity.
Cu(II) state:
Having established that [3SCC-(I9H)3] forms a high affinity Cu(II)/(Cu(I) complex at the designed 3His site we asked whether [3SCC-Cu(I9H)3]2+/1+ will be reactive towards H2O2 in a manner that resembles the formation of Cu-OOH species of native Cu enzymes and model complexes. Addition of 12.5 mM H2O2 to 0.125 mM [3SCC-Cu(I9H)3]2+ at pH 7.5 leads to the formation of a greenish-yellow solution with new peaks at 330 and 387 nm (Fig. 4A) having ε330 = 6.4×103 M−1cm−1, and ε387 = 1.5×103 M−1cm−1. These features are typical of Cu(II)-OOH species23 assignable to the ligand to metal charge transfer transitions from the OOH− group to Cu(II).24, 25 The d-d band blue shifted to ~570 nm in the presence of H2O2 (Fig. 4A). As OOH− is a stronger field ligand than H2O, the formation of the Cu-OOH species can increase the splitting of the d-energy levels, leading to the blue shift. The new species matures within ~40 min of H2O2 addition with initial rates in the order of ~10−4 s−1 (Fig. 4B,C; Table S2), and remains stable for several hours to at least a couple of days at ambient temperature. In contrast to synthetic Cu-OOH species that are in some instances unstable26 (t1/2 of < 10 min), the stability of [3SCC-Cu(I9H)3-OOH]+ is noteworthy. A stable Cu-OOH species was formed within a hybrid Cu-streptavidin system, whose stability was attributed to the presence of H2O-mediated H-bonding of the proximal oxygen with nearby amino acids.25 It is unlikely that the stability of the Cu-OOH species in the ArCuP is due to H-bonds with nearby polar residues, as it is formed inside the hydrophobic interior. However, the possibility of H2O or backbone mediated H-bonds that stabilizes the -OOH moiety cannot be ruled out. A second possibility is that the Cu-OOH formed within the hydrophobic environment prohibits the formation of Cu-O-O-Cu type dimeric species. To test whether the observed spectral features are contributed by oxidation of aromatic side chain of Trp28, we prepared the Trp28Gln mutant. Similar absorption features are also observed when [3SCC-Cu(I9HW28Q)3]2+ is reacted with H2O2 under similar experimental conditions (Fig. S4A). This data suggests that the observed spectral features are attributable to the Cu-OOH species. Together, these results show that the 3SCC can form and stabilize the putative Cu-oxygen species.
Figure 4.
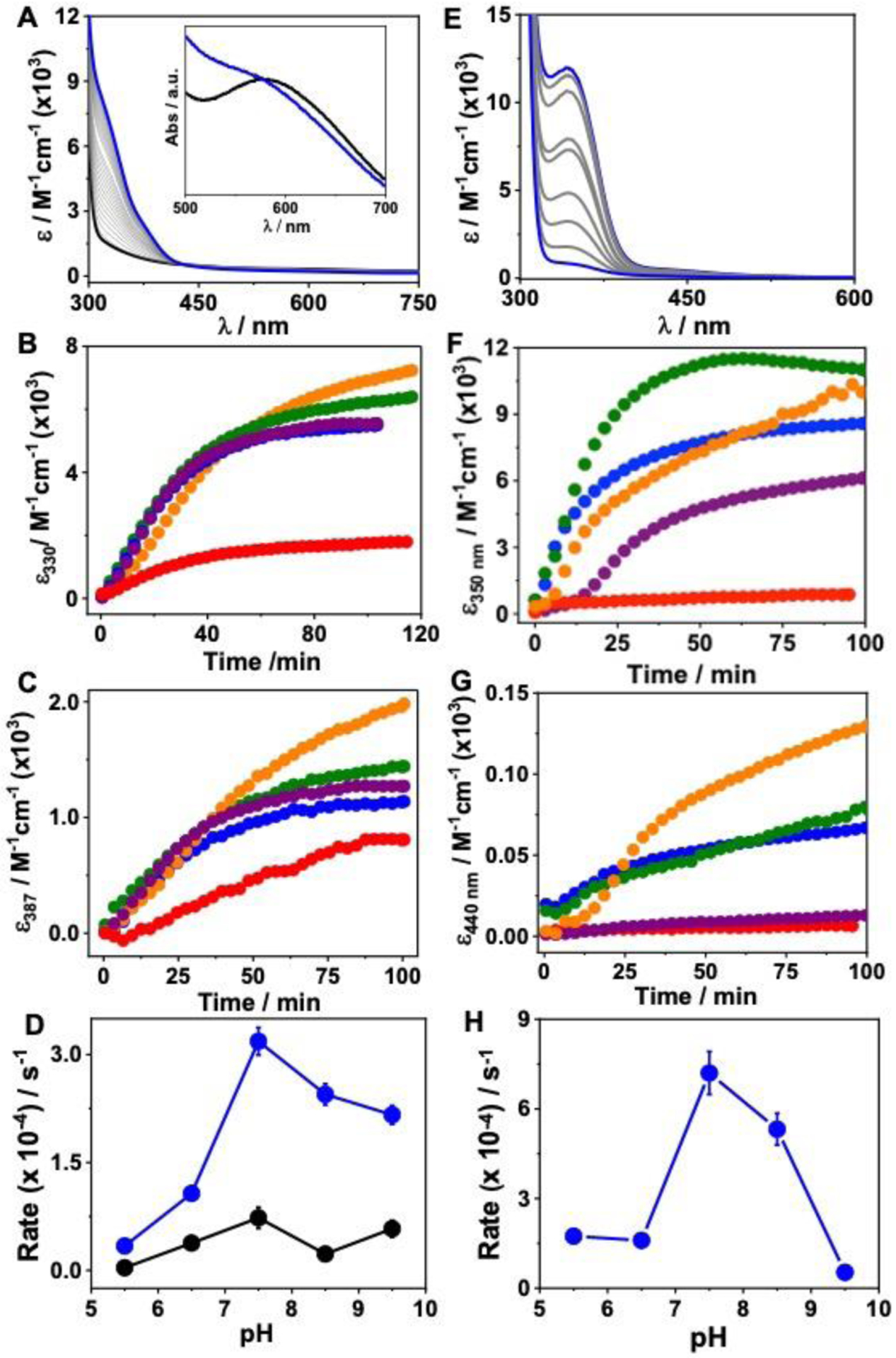
The kinetics of H2O2 reaction with [3SCC-Cu(I9H)3]2+ (A-D) and [3SCC-Cu(I9H)3]+ (E-H). A, E represent time-dependent UV-vis spectra after addition of 12.5 mM H2O2 to 0.125 mM [3SCC-Cu(I9H)3]2+ in the absence (A), and presence of 0.125 mM ASC (E) in 100 mM HEPES pH 7.5. Inset in A shows the visible region d-d band of [3SCC-Cu(I9H)3]2+ before (black) and after (blue) reaction with H2O2. Corresponding pH-dependent time traces (B, C, F, G) of the respective λmax at pH 5.5 (red), 6.5 (orange), 7.5 (olive), 8.5 (blue), and 9.5 (purple). (D, H) Plots of initial rate vs pH for 330 nm (blue), 387 nm (black) peaks in D (non-reducing) and the 350 nm peak in H (reducing). Experiments under reducing conditions were performed anaerobically. The pH-dependent assays were performed in mixed buffer at the respective pH.
pH dependence:
We next investigated whether the formation of Cu(II)-OOH is pH-dependent (Figs. 4B–D, S5). At pH 5.5, the Cu(II)-OOH species is not fully generated with an initial rate of formation that is ~10-fold slower than at pH 7.5 (Table S2). We attribute this to the fact that the His residues are not fully deprotonated at pH 5.5 which causes a weak Cu(II) binding. As a result, the subsequent binding of H2O2 is also weaker. From pH 6.5 to 9.5, the intensity for the near-UV feature remained approximately constant. A varying degree of an initial lag phase was observed at all pH except pH 7.5. The highest rate of formation of the hydroperoxo species occurs at pH 7.5.
Cu(I) state:
Cu(I) is the catalytic redox state for O2 and H2O2 dependent activity of Cu enzymes. Will the Cu(I)-form of the ArCuP have altered spectroscopic features, rates, and pH dependence for the formation of Cu-oxygen species? To address this, we performed the UV-vis studies anaerobically with [3SCC-Cu(I9H)3]+ (prepared by stoichiometric ascorbate (Asc) addition) and H2O2. A reddish-orange solution appeared with λmax of 350 nm and a shoulder at 440 nm in the pH range of 5.5–9.5 (Figs. 4E–H, S6), suggestive of some differences in the electronic nature of this species compared to that produced with the Cu(II)-form. The molar absorptivity of the Cu-OOH species and the rates of its formation are higher at all pHs (Tables 1, S2) for the Cu(I)-form. A weak d-d band at ~600 nm is visible in the energy plot (Fig. S6). The absorption features produced with Trp mutant [3SCC-Cu(I9HW28Q)3]+ and H2O2 are also similar (Fig. S4B) to that of [3SCC-Cu(I9H)3]+.
EPR spectroscopy.
The EPR spectrum (Fig. 5 blue) of [3SCC-Cu(I9H)3]2+ is representative of a T2Cu site with a dx2-y2 ground state having gz of 2.25 (Az = 566 MHz) > gx, gy of 2.05 (Ax, Ay = 116 MHz) (Table 1). The g⊥ region reveals well-resolved splitting with AN=15G arising from the interaction between the free electron on Cu(II) and the 14N nuclei of His. In the presence of H2O2, the g values shifted (Fig. 5 red) to gz of 2.27 (Az = 530 MHz), gx, gy of 2.06 (Ax, Ay = 97 MHz). The superhyperfine splitting of AN=15G remained unchanged. When H2O2 was added to [3SCC-Cu(I9H)3]+, (Fig. 5 orange) an EPR active species formed with gz of 2.28 (Az = 497 MHz), gx, gy of 2.06 (Ax, Ay = 100 MHz). Comparable EPR results are obtained for the Trp28Gln variant under analogous conditions as well (Fig. S7, Table S3).
Figure 5.
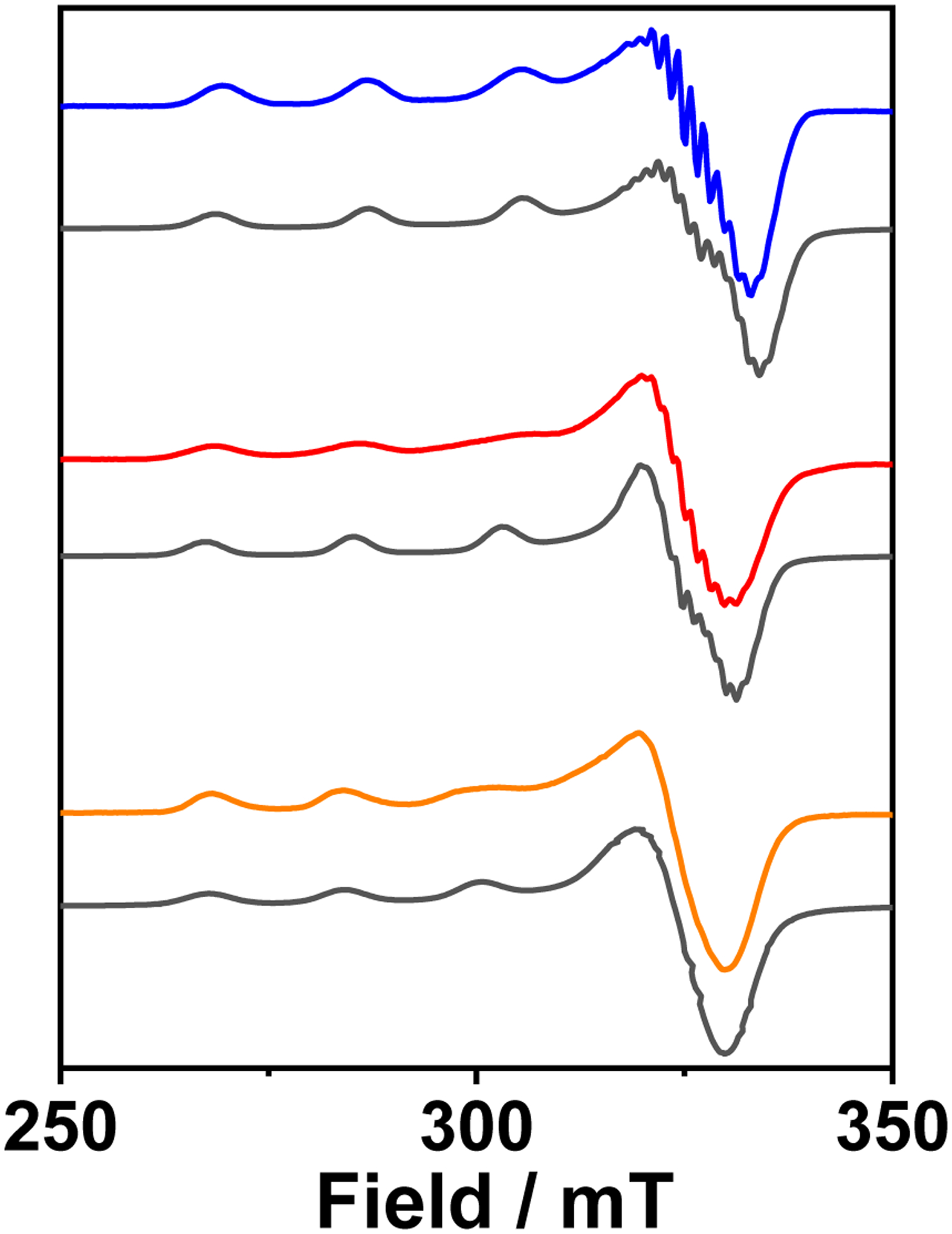
X-band EPR spectra of [3SCC-Cu(I9H)3]2+ (blue), in the presence of H2O2 (red), and the corresponding [3SCC-Cu(I9H)3]+ plus H2O2 (orange) sample in 100 mM HEPES pH 7.5. Gray traces are the simulated spectra. All data were collected at 126 K.
Monitoring peptide oxidation.
Extensive oxidative damage of LPMO in the presence of H2O2 has been observed.16, 27 We performed colorimetric assays to quantify OH• generation (Fig. S8),28 which shows that under our experimental conditions ~280 nM of 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA) is produced from hydroxylation of coumarin-3-carboxylic acid (3CCA). To test the possibility of oxidative modification of the peptide and to identify the specific amino acids undergoing oxidative modification, we performed LC-MS/MS studies upon protease digestion of the 3SCC-Cu-OOH species. These results are presented in Figs. S9, S10. The [3SCC-Cu(I9H)3]2+ sample showed ~25% oxidation with H2O2. In the Cu(I)-form the peptide level oxidation increased to ~36%. As Trp is a common site for oxidative modification, we probed whether replacement of this residue would diminish the level of oxidation. Indeed, the Trp28Gln mutant shows only 17–18% oxidation, significantly less than the parent peptide. This data clearly demonstrate that even though the Cu-OOH species is produced in Trp28Gln variant, the extent of oxidation is much less. Further analysis of the LC-MS/MS data showed that the active site His residues were oxidized only in the Cu(I)-form, while no His oxidation was observed with Cu(II)-peptide (Fig. S9 B–C), consistent with the fact that this is the site of generation of the Cu-oxygen species. In addition, several other residues were also oxidized. These include Ile2 at 10Å, Ile5 at 5Å, Lys6 at 8Å, Ile19 at 15Å, Lys20 at 17Å, Lys27 at 27Å, and Trp28 at 28Å from the Cu site.
Redox properties.
Protein film voltammetry (PFV) was used to study the kinetics of ET processes. The peptide films adsorbed on pyrrolytic graphite electrode (PGE) demonstrate a well-developed redox wave with an E1/2 of 256 mV (Epc = 239 mV, Epa = 273 mV) vs SHE, Ic of −7.1 μA and Ia of 6.2 μA at pH 6.5 (Fig. 6, Table 2) in N2-saturated buffer. A relatively small peak-to-peak separation of 34 mV is suggestive of a quasi-reversible redox process. The overall peak shape is characteristic of a homogenous protein film. Repeated CV scans showed that the peptide remains stably adsorbed on the electrode (Fig. S11). The cathodic peak is symmetric with a full width at half height (ΔE1/2c) of 92 mV, which is close to the theoretical value of 91 mV for an ideal and reversible 1e− redox process. The anodic peak deviates from symmetry with ΔE1/2a of 132 mV, suggesting that dispersion and electrostatic effects influence the oxidation process.29–31 The peak current of the redox wave shows a linear dependence with scan rate, diagnostic of a surface controlled electrochemical process (Fig. S12). The slopes obtained from the current vs scan rate plot were used to determine the surface coverage (Γ) according to equation (iii), which was found to be 88±8 pmoles cm−2. A clear pH-dependence of the redox process was observed where an increase in pH from 5.5 to 9.5 caused a gradual cathodic potential shift from an E1/2 of 289 mV at pH 5.5 to 176 mV at pH 9.5 (Fig. S13A, Table 2). This result indicates that the reduction of Cu(II) to Cu(I) is more favorable in acidic conditions. The positive charge of His at lower pH stabilizes electron-rich Cu(I) preferentially over Cu(II). A similar trend has been observed for a heme protein.32 The plots of peak potentials vs pH (Fig. S13B) are linear with slopes of 33 mV/pH and 24 mV/pH for anodic and cathodic peaks, respectively. However, since these slopes are less than the theoretical 59 mV/pH for a PCET process, it appears that the redox process of the ArCuP is not associated with the chemical event of H+ transfer.
Figure 6.
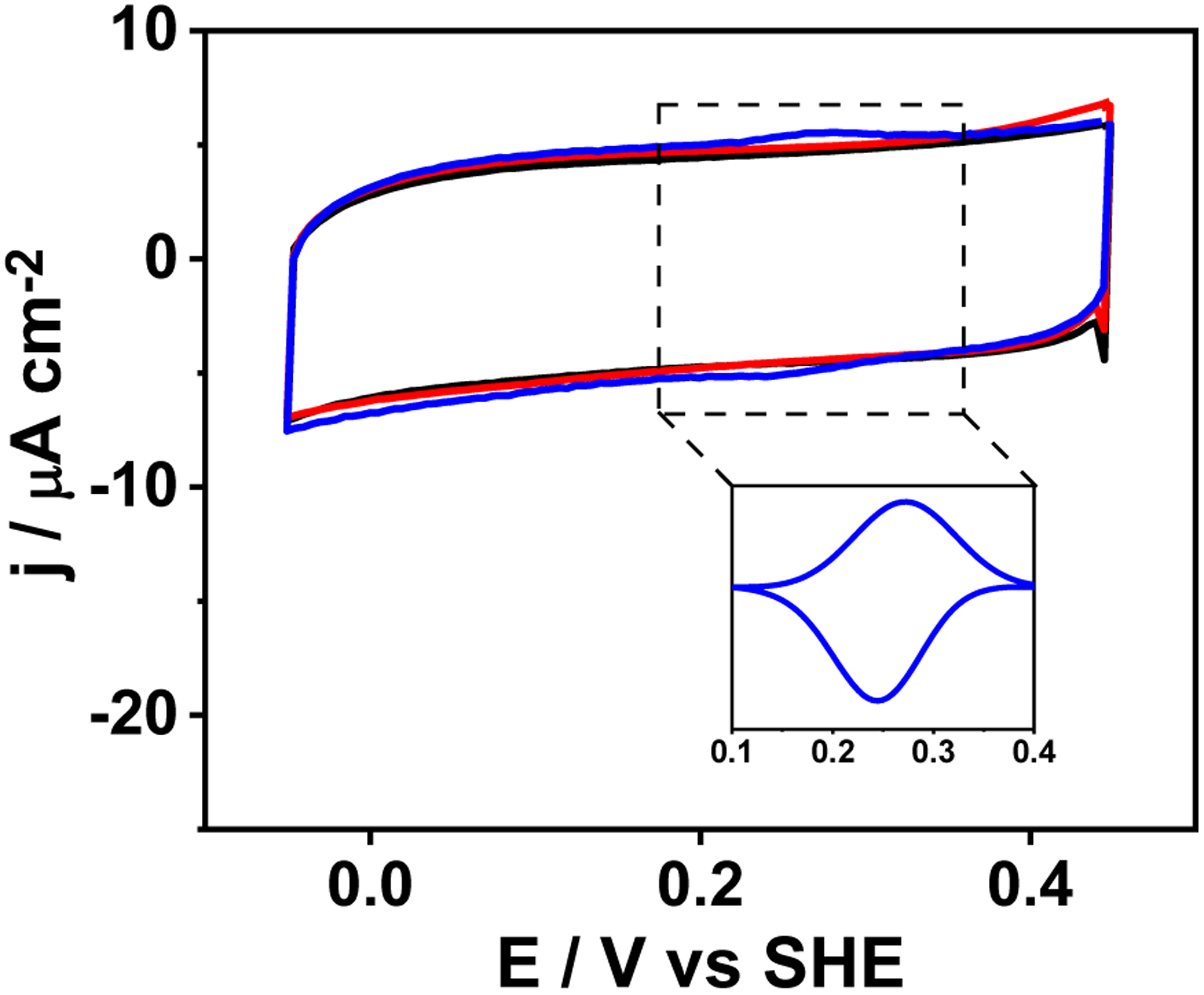
PFV scans of PGE (black trace), [3SCC-(I9H)3] (red trace), [3SCC-Cu(I9H)3]2+ (blue trace) at 50 mVs−1 in N2-saturated 80 mM mixed buffer at pH 6.5. The inset shows background corrected peak for [3SCC-Cu(I9H)3]2/1+. Peptide samples were dropped on PGE and dried under N2 prior to data collection. Data were collected against Ag/AgCl reference electrode and converted to SHE.
Table 2.
Redox and catalytic properties of the ArCuP.
| pH | Epc/mV | Epa/mV | E1/2/mV | ▲Ep/mV | ▲E1/2c/mV | ▲El/2a/mV | Ic/μA | Ia/μA | kcat /s−1 |
|---|---|---|---|---|---|---|---|---|---|
| 5.5 | 279 (±8) | 298 (±8) | 289 | 19 | 126 | 94 | −6.9 (±0.2) | 6.1 (±0.1) | 0.45 (±0.01) |
| 6.5 | 239 (±6) | 273 (±5) | 256 | 34 | 92 | 132 | −7.1 (±0.1) | 6.2 (±0.1) | 0.54 (±0.02) |
| 7.5 | 210 (±4) | 248 (±3) | 229 | 38 | 98 | 144 | −6.7 (±0.1) | 6.2 (±0.1) | 0.70 (±0.02) |
| 8.5 | 160 (±7) | 218 (±5) | 189 | 58 | 110 | 130 | −6.8 (±0.1) | 6.3 (±0.1) | 0.65 (±0.02) |
| 9.5 | 149 (±10) | 203 (±8) | 176 | 54 | 100 | 140 | −6.7 (±0.1) | 6.5 (±0.1) | 0.62 (±0.02) |
Electrocatalytic H2O2 reduction.
The electrocatalytic H2O2 reduction activity of the ArCuP was established by linear sweep voltammograms (LSVs) collected anaerobically with increasing H2O2 concentrations. To avoid complications due to electrodeposition of Cu, the cathodic potential was restricted to −300 mV. In the presence of 1 mM H2O2, an electrocatalytic current was observed with an onset potential (Eonset) of ~250 mV (Fig. 7A, blue trace).The blank PGE in the absence (Fig. 7A black trace) and presence of 1 mM H2O2 (Fig. 7A wine trace) does not show catalytic response, neither does free Cu(II) (Fig. 7A gray trace) or the apo peptide (Fig. 7A, cyan trace) in the presence of H2O2. These results indicate electrocatalytic reduction of H2O2 by the ArCuP. Moreover, the Eonset is similar to the redox peak of Cu(II)/(I) (Fig. 7A inset) suggesting that the catalytic process is coupled to the reduction of Cu(II) to Cu(I). A comparable current was observed for Trp28Gln mutant also (Fig. S14). LSVs recorded after rinse tests established that the activity was attributable to [3SCC-Cu(I9H)3]2+ and not electrodeposited Cu, as no current was observed after the rinse test (Fig. 7A orange trace). LSVs recorded with increasing H2O2 (2–15 mM) showed that the current increased concurrently (Fig. 7A purple, green, red traces) suggesting that the catalytic process is dependent on H2O2.
Figure 7.
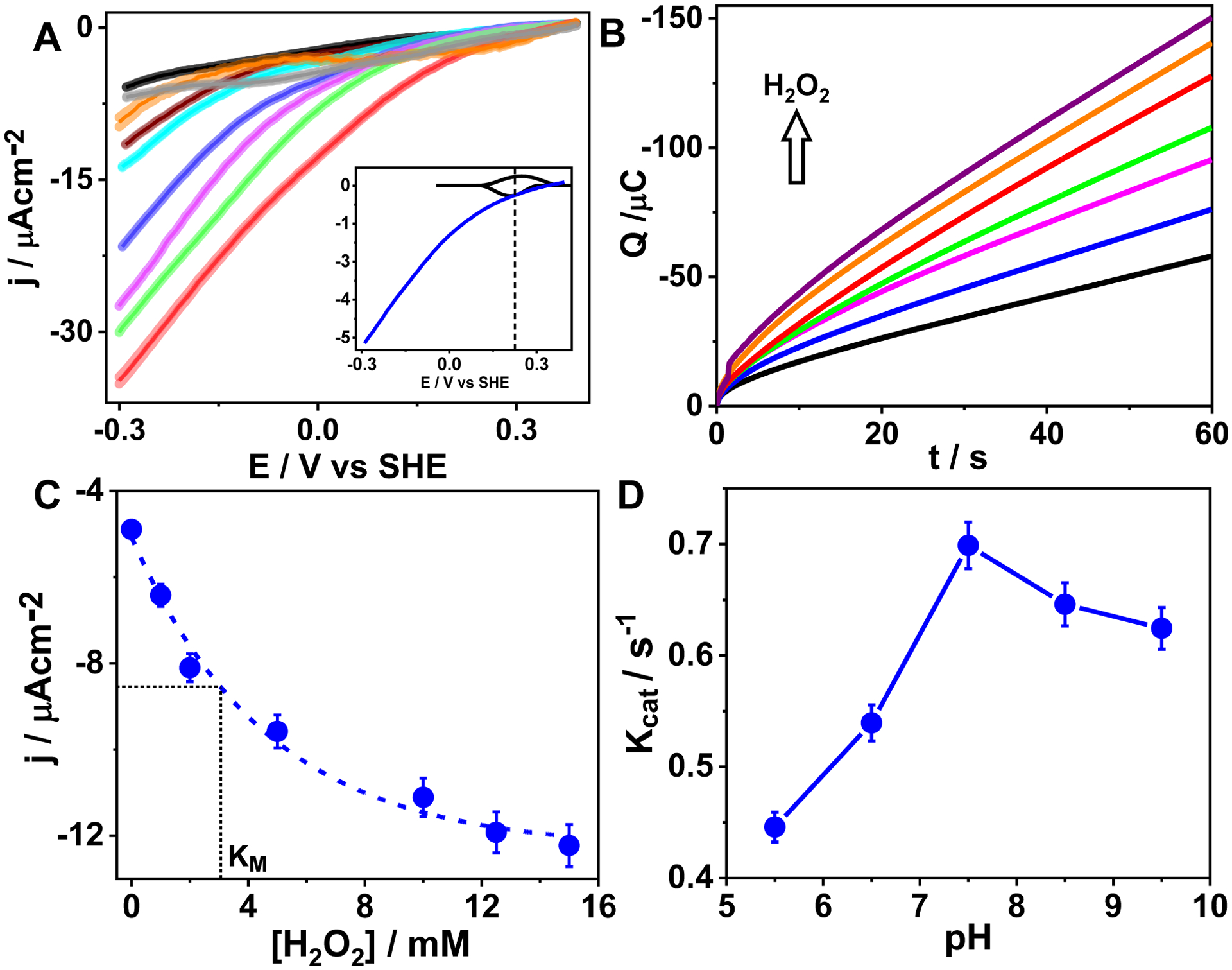
A) LSVs of blank PGE in the absence (black trace) and presence of 1 mM H2O2 (wine trace), apo [3SCC-(I9H)3] (cyan trace), free Cu(II) with 1 mM H2O2 (gray trace), and [3SCC-Cu(I9H)3]2+ films on PGE (blue trace) in presence of 1 mM H2O2 in N2-saturated 80 mM mixed buffer at pH 7.5. Magenta, green, and red traces represent LSVs of [3SCC-Cu(I9H)3]2+ in the presence of 2 mM, 5 mM, and 10 mM H2O2. The orange trace is the rinse test LSV of [3SCC-Cu(I9H)3]2+. The inset is an overlay of the non-catalytic peaks and the corresponding LSV with 1 mM H2O2 shown at 4-fold decreased scale to demonstrate that the catalytic H2O2 reduction is coupled to the reduction of Cu(II) to Cu(I). ν = 100 mV s−1. The shaded regions represent error bars from 3 separate experiments of fresh protein films at each condition. B) Charge vs time traces from chronoamperometry experiments of [3SCC-Cu(I9H)3]2+ films on PGE at −0.3 V vs SHE in the presence of 0–15 mM H2O2 in 80 mM N2-saturated mixed buffer at pH 7.5. (C) Plot of the corresponding catalytic current densities vs [H2O2] showing a KM of ~3 mM. D) Plot of kcat vs pH at 12.5 mM H2O2. Error bars are from 2–3 independent measurements.
To further elucidate the kinetics of H2O2 reduction, anaerobic chronoamperometry (CA) scans were collected at −0.3V for 60s. Similar to the LSVs, a higher charge passed with an increase in H2O2 (Fig. 7B). The KM for H2O2 reduction was found to be ~3 mM (Fig. 7C). The current saturated at ~12.5–15 mM H2O2 with a maximum current density of ~ −12 μAcm−2. From these results, a kcat of 0.72 s−1 at pH 7.5 was calculated according to equation (iv) assuming a 2e− reduction of H2O2 to H2O. The maximum kcat was obtained at pH 7.5 (Figs. 7D, S15). Due to uncertainty in quantifying the amount of H2O formed, the Faradaic efficiency (FE) of the ArCuP could not be reliably obtained.
DISCUSSION
Several mononuclear Cu complexes with N-ligands have been synthesized and their reactivity studies with peroxide have been reported.24, 33–38 These studies have served as benchmark models for establishing the spectroscopic and reactivity features of Cu-OOH species. However, these complexes are studied in organic media, which makes a direct correlation to the enzyme’s redox and reactivity patterns challenging. Hybrid systems prepared by incorporation of synthetic Cu complexes into streptavidin cavity have been characterized.25 X-ray structures of Cu-OOH species established that H-bond between the proximal O and a nearby amino acid side chain stabilized the Cu-OOH species while deletion of this H-bonding makes this species reactive towards substrates. Here, all the coordinating N atoms to Cu are provided by an external ligand scaffold. We sought out to de novo design an ArCuP where all three endogenous His ligands are provided by the peptide scaffold itself, akin to the native Cu enzymes, and to characterize its spectroscopic, redox and catalytic properties towards H2O2 both in solution as well as adsorbed on electrode surface.
Design considerations of the ArCuP.
To create the intended Cu site, we used the rules of de novo peptide design where an Ile at both the a and d site of the heptad produces trimeric assemblies. We replaced an a site Ile from the second heptad with His to produce the trishistidine environment for Cu. The rationale for choosing the a site vs d site stems from the fact that the a site residues are known to point towards the helical interior while the d site residues point toward the helical interface.3, 39, 40 Therefore, we surmised that all 3His in the a site could be properly oriented for Cu coordination. The X-ray structures of the ArCuPs validated the general design principles. The binding of Cu(II) via all Nε atoms of His is consistent with the active sites of Cu enzymes involved in substrate binding and activation (vide supra). This coordination mode therefore bears significance for the ability of the ArCuP to bind, activate, and catalyze H2O2 reduction.
ArCuP binds Cu tightly and forms a T2Cu site.
From competitive titrations and E°’, the Cu(I) and Cu(II) affinity were determined to be ~15 fM and 184 fM, respectively. To compare, the corresponding affinity for azurin is 0.03 fM and 25 fM,41 which indicate that Cu(I) and Cu(II) bind azurin 400-fold and 8-fold tighter than to [3SCC-(I9H)3]. The Cu(I) binding affinity for the Cu transporter Ctr1c, the Cu pump Ccc2n and the Cu chaperone Atx1 are even tighter (~0.14–0.73 aM).42, 43 Compared to related 3SCC TRI peptide systems (KdCu(I) = 0.2 pM; KdCu(II) = 8.7 nM;), the Cu affinity in the ArCuP is stronger.2 In other examples, our values are comparable to the α3D variants designed to bind Cu.44 The EPR spectrum of [3SCC-Cu(I9H)3]2+ (Fig. 5) is representative of Cu bound to typically N ligands and 1–2 water molecule(s). For example, the Cu-streptavidin system with a CuN3(OH2)2 coordination had two types of species with gz of 2.22, 2.26 and Az = 492, 514 MHz.25 LMPO has a slightly larger gz of 2.27 and a smaller Az of 456 MHz.45 The T2 CuH site of PHM has a gz of 2.29 and a large Az of 471 MHz.46 In processed amine oxidases (AO) with Cu(His)3(OH2)1–2 ligation, the gz values are found in the range of 2.28–2.32 while the Az values vary between 480 and 561 MHz.7, 47–49 Furthermore, superhyperfine splitting in the g⊥ region of AO47–49 and LPMO45, 50 have been observed with AN of 13–14 G. These values are comparable to the [3SCC-Cu(I9H)3]2+. The lack of water ligation in the X-ray structure is intriguing and it most likely indicates cryoreduction of Cu(II) to Cu(I) causing the water(s) to dissociate upon irradiation. The cryoreduction of Cu(II) has been widely observed in Cu proteins.51–53 In solution studies, the reduction does not happen, which produces UV-vis and EPR parameters characteristic of water(s) bound Cu(II) system.
The E°’ of the ArCuP aligns with functional T2Cu sites.
The E°’ of the ArCuP (Table 2) is similar to what has been reported for His-coordinated Cu sites in many enzymes. For example, the E°’ for LPMO and the CuH site of PHM at pH 6 are 273 mV and 270 mV, respectively.54, 55 In the T2Cu site of NiR, the E°’ is in the range of 218–310 mV depending on pH.56, 57 These data indicate that irrespective of the role of T2Cu sites, may it be substrate activation (LPMO, NiR) or ET (CuH site of PHM), the E°’ for CuN3 sites in native metalloenzymes are tightly regulated within a narrow range. This is in contrast to the T1Cu sites, which solely participate in ET, where the E°’ span across a wide range of ~600 mV (183–800 mV).10 Furthermore, in engineered Az, the E°’ for the T1Cu site has been tuned to encompass as high as a 2V range.55
Nature of the Cu-oxygen species.
The relatively high E°’ of [3SCC-Cu(I9H)3]2+/1+ suggested that the ArCuP can be reduced using Asc (E° ~ 100 mV).58 Furthermore, a high E°’ for the 2e− reduction of H2O2 to H2O or OH− in acidic (1.77 V) or basic (0.88 V) conditions should make its reduction by [3SCC-Cu(I9H)3]1+ thermodynamically accessible. The differences in the absorption maxima for the reaction of the ArCuP with H2O2 in the Cu(II) vs Cu(I) form indicate that the electronic nature for the resultant species are different from one another, while both are representative of a Cu(II)-oxygen species. The near-UV features at 330 nm and 387 nm along with the absorptivity values for the species generated with the Cu(II)-peptide, lead us to assign this as the Cu(II)-OOH. The species produced with Cu(I) form has red shifted features at 350 nm and 440 nm (Figs. 4, S6) with a higher absorptivity for the 350 nm peak but lower for the 440 nm peak compared to those formed with Cu(II)-peptide. The most noteworthy difference is the rate at which the Cu-oxygen species are generated in these conditions. A ~3-fold higher rate of formation with Cu(I) suggests that the reduced form is more favorable for H2O2 reaction than with Cu(II). Reduction of the Cu(II) to Cu(I) is implied to prime the active site of LPMO for H2O2 binding.16–18 An analogous process could occur in the activation of H2O2 by the ArCuP, accounting for the faster rate of reaction with Cu(I). The reductive priming process facilitates the electrocatalytic H2O2 reduction of the metallopeptide. In synthetic systems it has been demonstrated that Cu(II)-OOH species can be generated from Cu(I) and H2O2.37, 59 Similar process likely produces the Cu(II)-OOH species in our system as well. The Trp28Gln mutant also produced similar spectral features to the parent peptide upon H2O2 reaction under identical conditions. This result provided further evidence that the optical features are indeed arising from the Cu-OOH species within the ArCuP and not from the oxidation of aromatic residues such as Trp. Additional control experiments (Fig. S16) proved that the presence of H2O2, Cu and peptide are all necessary to produce the UV features for the Cu-oxygen species seen in Fig. 4.
The changes in the EPR parameters upon H2O2 reaction is consistent with what has been observed for synthetic and biomolecular systems producing Cu-OOH species as well.24, 25 The overall shapes of the EPR spectra of the parent peptide and the Trp mutant are similar when the Cu-oxygen species are generated. (Figs. 5, S7). Spin integration for all the data in Fig. 5 showed that the relative spin concentrations of Cu(II) are 1 : 1.4 : 1 in [3SCC-Cu(I9H)3]2+ : [3SCC-Cu(I9H)3]2+ + H2O2 : [3SCC-Cu(I9H)3]+ + H2O2, indicating a similar relative amount of Cu(II) present in these samples. The differences in the Az and the loss of superhyperfine features, along with changes in the UV-vis spectral features for the Cu-oxygen species generated with Cu(II) or Cu(I) peptides indicate differences in the electronic nature of the final species. What is the origin of this difference? The LC-MS/MS data (Fig. S9 B–C) shows a critical distinction in the oxidation of active site His between the Cu(II) and Cu(I) forms. While no His oxidation was observed in the Cu(II) form, the Cu(I)-peptide showed oxidation of the active site His. Therefore, the most likely explanation for the differences in the spectral properties is borne out by the fact that the Cu(II)-OOH species generated from Cu(II)-peptide features unoxidized His (Fig. 8B), while the Cu(II)-OOH species generated from the Cu(I)-peptide is bound to oxidized 2-oxo His (Fig. 8C), giving rise to different electronics of these species.
Fig. 8.
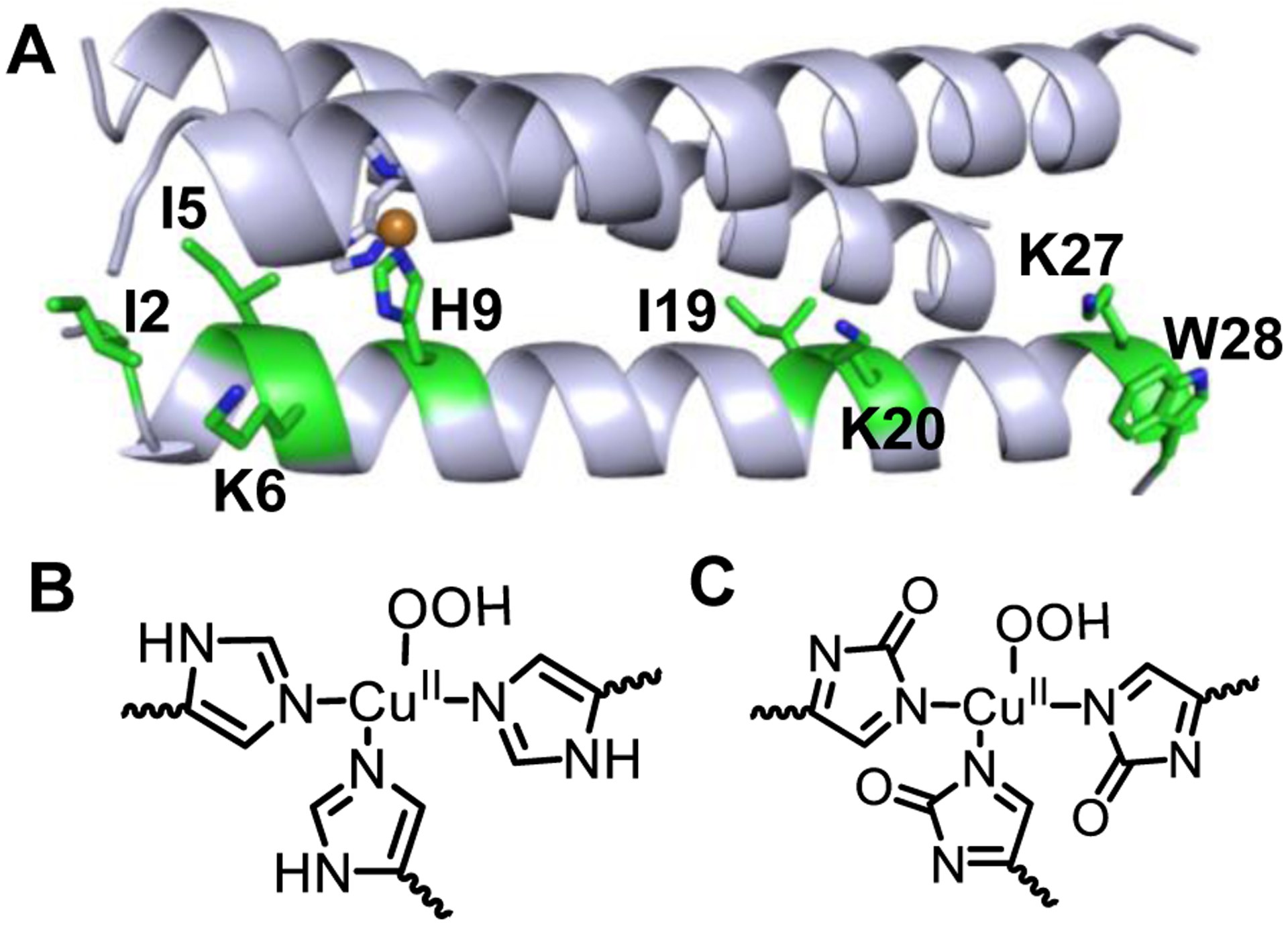
A) Amino acid oxidation sites detected in LC-MS/MS shown as green. Only one strand is highlighted for clarity. His oxidation is observed only in the Cu(I)-form, while the Cu(II)-form does not show His oxidation. Proposed structures of the CuII-OOH species generated with Cu(II)-peptide having unoxidized His (B) and with Cu(I)-peptide (C) bound to 2-oxo His. The structural differences between B and C likely dictate the spectral features of the CuII-OOH species generated with Cu(II) vs Cu(I)-forms of the peptide.
Oxidative modification of specific side chains and the role of Trp28.
Oxidative modification of the peptide was observed towards both the N-termini and C-termini residues (Fig. 8). Ile, Lys, and Trp are the primary residues that are preferentially oxidized over others. The specific oxidation sites span across a wide distance range of 5–28Å from the Cu site. The oxidation of the active site His residues with the Cu(I)-form is commensurate with the fact that this is the site of generation of the reactive Cu-oxygen species. Under reducing conditions, an ~11% increase in the total peptide oxidation was observed, which bolsters the notion that Cu(I)-peptide is more reactive to H2O2 than the Cu(II) form. Importantly, replacement of the aromatic Trp28 with Gln on the peptide exterior caused a significant decrease in the total peptide-level oxidation. This observation is not unexpected, as extensive oxidation of amino acid residues has been also observed in LPMO in the presence of H2O2, where His (including the active site His), Trp, Tyr, and Met residues are oxidized located up to ~19Å from the Cu site.16, 27 For LMPO, this could be a strategy for the enzyme to scavenge the reactive species at these aromatic sites.
Catalytic parameters for H2O2 reduction.
The catalytic nature of [3SCC-Cu(I9H)3]2+ for H2O2 reduction was established using voltammetry and constant potential amperometry experiments. The catalytic onset occurs at the same potential to that of the Cu(II)/Cu(I) redox process. This further supports that Cu(I) is beneficial for the initial binding and reduction of H2O2. Both the catalytic currents and open circuit potentials (Figs. 7B–C, S17) increased with H2O2, before reaching saturation at 12.5–15 mM of H2O2. The KM of 3 mM H2O2 is approximately twice than what has been reported for the decaheme cytochrome MtrC from Shewanella oneidensis MR-1 immobilized on indium tin oxide (ITO) electrodes,60 suggesting that MtrC can reach vmax at half the H2O2 concentration than the ArCuP. The benchmark enzyme horseradish peroxidase (HRP) supported on pyrene has an even lower KM of 0.2 mM.61 The kcat of 0.7 s−1 at pH 7.5 is ~7-fold lower than that of MtrC/ITO electrodes for H2O2 reduction.60 The pH dependence of kcat from the electrocatalytic studies follows a similar trend with the relative rates obtained in UV-vis studies where a maximum rate is obtained at pH 7.5.
CONCLUSIONS
In conclusion, a de novo trimeric ArCuP has been designed and characterized by X-ray crystallography and spectroscopy. A T2Cu coordination environment is formed with an all-Nε ligation of the His, akin to several functional Cu enzymes. Activation of H2O2 occurs at a faster rate by reduced Cu(I) as opposed to the Cu(II) form. UV-vis and EPR data suggested the formation of Cu-hydroperoxo species. The ArCuP can be immobilized stably on PGE electrode, which allowed us to characterize the redox behavior of the ArCuP in detail. The EPR and redox properties of the ArCuP are similar to related Cu enzymes and artificial biomolecular systems. Oxidative modification is observed at Ile, Lys, His and Trp residues. The origin of spectral differences of the Cu(II)-OOH species generated from Cu(II) vs Cu(I)-forms of the peptide is attributed to the presence of oxidized His, which is observed only with Cu(I)-peptide. Direct ET between the electrode and the Cu site of the immobilized peptide facilitated the electrocatalytic reduction of H2O2. Determination of catalytic parameters (kcat, KM) provided insights into the peroxide reduction activity of the ArCuP. Our results pave way to a new artificial metalloenzyme where endogenous amino acids host a transition metal active site for energy-relevant electrocatalytic transformations.
EXPERIMENTAL SECTION
Materials and Methods.
All chemicals and reagents were of analytical grade and used without further purification. All the glassware and plasticware were immersed in a 10 mM ethylenediaminetetraacetic acid bath overnight, followed by soaking in 10% and 1% nitric acid baths, respectively. All buffer solutions were prepared in Milli-Q (Millipore) water and treated with Chelex 100 overnight to remove trace metals. Tris(hydroxymethyl) aminomethane (Tris, Frontier Scientific), 4- (2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 3-morpholinopropane-1-sulfonic acid (MOPS), 2-(N-morpholino) ethanesulfonic acid (MES), sodium acetate, NaCl and HCl were purchased from Fisher Scientific. The mixed buffers contained 20 mM each of MES, sodium acetate, HEPES and TRIS. For electrochemistry experiments 0.1 M KCl was added as supporting electrolyte to the mixed buffers. CuCl2 (Alfa Aesar), ascorbic acid (VWR), NaOH (VWR), and H2O2 (VWR) stock solutions were made in water. Coumarin-3-carboxylic (3-CCA) acid and 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA) were purchased from Acros Organics and VWR, respectively. Potassium phosphate, LC-MS grade formic acid, acetonitrile, and water were obtained from Fisher Scientific. Sequencing grade Glu-C was purchased from Promega.
Peptide synthesis and purification.
Peptides were synthesized by an automated microwave peptide synthesizer (Liberty Blue, CEM) on a 0.1 mmol scale using Rink Amide ProTide Resin (CEM) with a poly(ethylene glycol) polystyrene backbone. All coupling steps were performed at 90°C for 4 min. The amino acids (Advanced Chemtech) were weighed in concentrations of 0.2M and dissolved in DMF (VWR). Oxyma (1M; CEM) and N, N’-diisopropylcarbodiimide (DIC) (0.5M; Advanced Chemtech) in DMF were used as the activator base and activator, respectively. 20 % piperidine (Sigma) in DMF was used as the deprotection solution. All His residues were coupled at 50°C for 10 min to avoid racemization. Cleavage of the peptides from the resin and side chain deprotection were achieved by 92.5% trifluoroacetic acid (TFA; Advanced Chemtech), 2.5% triisopropylsilane (TIS; Sigma–Aldrich), 2.5% ethane-1,2-dithiol (EDT; Sigma–Aldrich), and 2.5 % water in 10 mL volume for 2h at RT with stirring. After cleavage, the crude peptides were filtered and excess TFA was evaporated under a gentle stream of N2 gas. The crude peptides were then precipitated, washed with cold diethyl ether, dissolved in water, and lyophilized. The peptides were again dissolved in 10% acetic acid and purified by using a C18-semi preparative column using an Agilent 1260 Infinity II HPLC system. A linear gradient from solvent A (0.1% TFA in H2O) to solvent B (90% acetonitrile, 10% H2O, 0.1 % TFA) was used. The peptide was injected at 1 mL/min and run with 10% B/90% A for 5 min, followed by a linear gradient to 70% B over 25min at a flow rate of 3 mL/min. The peptide eluted at 50–60% solvent B (Fig. S18A). The chromatogram was monitored at 214 and 280 nm. The purified peptide was lyophilized and stored at −20°C. Peptide mass and purity were analyzed by ESI-MS and MALDI-MS (Fig. S18B).
MALDI-MS.
The identity and purity of the purified peptides were confirmed by MALDI-TOF MS (Bruker Voyager) with sinapinic acid as the matrix (Sigma, 10mg/mL stock in ACN/H2O with 0.1 % TFA). The instrument was externally calibrated by using a ProteoMass calibration kit (Sigma–Aldrich).
X-ray crystallography.
Solutions for Cu(II)-bound peptide were prepared by adding 1 equivalent of CuCl2 to 40 mg/mL apo peptide dissolved in water. This was then mixed with equal volume of the well solution containing 0.1 M Tris pH 8.5, 0.2 M MgCl2, and 30% PEG 4000. Crystals were grown by hanging drop method at RT, which appeared within 7–20 days. Crystals were harvested and soaked with 30% PEG 400 as cryoprotectant prior to freezing in liquid N2.
X-ray diffraction data were collected at the Life Sciences Collaborative Access Team beamline 21-ID-G at the Advanced Photon Source of the Argonne National Laboratory. Diffraction images were indexed and integrated using XDS62 followed by data reduction and analysis using Aimless63 in the CCP4i264 interface of the CCP4 suite.65 Data quality assessment and twinning analysis was performed by phenix.xtriage that indicated no twinning was present.66 The structures were solved using Phaser-MR67 in Phenix68 by molecular replacement using 4DZL69 as the search model using correction for translational non-crystallographic symmetry in the data set, followed by automatic model building in Buccaneer70 in CCP4 or AutoBuild71 in Phenix. Refinement was performed in phenix.refine72 with iterative model building and density fitting in Coot.73 The A3 and C3 trimers were refined with 100% occupancy of Cu, while the B3 trimer was refined with partial Cu occupancy of 16%. Waters were automatically added during refinement in Phenix. Data validation was performed at wwPDB OneDep System. The structure factors and coordinate files have been deposited with PDB code 7L33. Data collection statistics are in Table S4.
UV-Vis spectroscopy.
The UV-vis spectra were recorded on an Agilent 8454 or Cary 5000 spectrophotometer. Peptide samples were prepared in either MES, MOPS, HEPES, TRIS or CHES buffer as needed. All the kinetics experiments were performed with 0.125 mM of [3SCC-Cu(I9H)3]2+ in the appropriate buffer after addition 12.5 mM H2O2 and the kinetics was monitored for 100 min with 60s interval between scans. For reactions with the Cu(I)-form, [3SCC-Cu(I9H)3]+ was generated by addition of 1 equivalent Asc before H2O2 addition. All components were degassed and the kinetics was monitored anaerobically in a septa-capped cuvette (Starna Cells). The initial rates were calculated from the first 300s of the kinetic run in each case.
Competitive titration with Bcs:
All assays were performed using degassed solutions using a septa-capped cuvette. For both forward and reverse titrations excess ligand was taken to ensure no free Cu(I) remains in solution at the beginning of each titration. In the forward titration, a 30 μM [3SCC-(I9H)3] solution was mixed with 15 μM of Cu(II) and 3 mM Asc in 100 mM MOPS pH 7.4 to form [3SCC-Cu(I9H)3]+. Bcs2− was then gradually titrated into the solution and stirred for 10–15 min, which led to an increase in absorbance at 483 nm due to the formation of [Cu(Bcs)2]3− (ε483= 13,300 M−1cm−1). For the reverse titration a 15 μM solution of Cu(II) plus 60 μM Bcs2− were taken and Asc was added to form [Cu(Bcs)2]3−. Aliquots of [3SCC-(I9H)3] were then titrated into this solution. A decrease in the absorbance at 483 nm was observed with each addition, indicating the formation of [3SCC-Cu(I9H)3]+. The formation or disappearance of [Cu(Bcs)2]3− was plotted against equivalents of Bcs2− or [3SCC-(I9H)3] to obtain the stoichiometry of Cu(I) to [3SCC-(I9H)3]. The plots of [Cu(Bcs)2]3− vs the ratio of total [3SCC-(I9H)3] and [3SCC-Cu(I9H)3]+ (1/θ) were made to determine the binding affinity (Kd) of Cu(I) for the peptide according to equation (i):
| (i) |
where β2 = 1019.8M−2 for [Cu(Bcs)2]3−.
EPR spectroscopy.
300 μL solutions containing 0.5 mM [3SCC-Cu(I9H)3]2+/1+ were prepared in 100 mM HEPES buffer pH 7.5 and 100-fold H2O2 was added as needed. After 40 minutes, 30% glycerol (final volume) was added, loaded into 3 mM thin wall 6.25” long EPR tubes (Wilmad) prior to freezing in liquid N2. The EPR spectra were acquired on a Bruker EMX spectrometer controlled with a Bruker ER 041 X G microwave bridge operating at X-band (~9.35 GHz) equipped with a precision temperature controller. The EPR spectra were recorded at 126 K and 1.26 mW microwave power, with a 600 mT field sweep in 167.77 s (two scans), and 0.25 mT field modulation amplitude. Simulations were executed with Matlab and the EasySpin74 computational package as S = ½ species with axial g tensors.
Coumarin-3-carboxylic acid assay (3-CCA).
The oxidation of 3-CCA to fluorescent 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA) by OH• produced by HRP, catalase, and [3SCC-Cu(I9H)3]2+ in the presence of H2O2 was monitored on a PTI fluorimeter (λex = 395 nm, λem = 450 nm,) following a known procedure.28 The samples contained metal bound catalysts (0.25 mM), 3-CCA (5 mM), and H2O2 (25 mM) in 100 mM HEPES pH 7.5. The concentration of 7-OH-CCA produced in the assays was calculated from calibration curve using known amounts of 7-OH-CCA.
Mass spectrometry.
Peptide Glu-C digestion:
For digestion experiments, 0.125 mM of [3SCC-Cu(I9H)3]+/2+ was reacted with 12.5 mM H2O2 in 100 mM HEPES pH 7.5 for 40 min to form the Cu-OOH species according to the UV kinetics. The reaction was terminated with 0.1% TFA. Then, 10 μM of samples were incubated in 100 mM phosphate buffer, pH 8.0 at 95°C for 15 minutes. The samples were cooled to RT and a 1:20 Glu-C/peptide ratio was added to the samples for overnight digestion at 37°C with sample mixing. Digestion was terminated by heating the samples to 95°C for 10 min and the addition of 0.1% formic acid.
LC-MS/MS analysis:
All samples were analyzed on a Thermo Scientific Orbitrap Fusion Tribrid (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Ultimate 3000 Nano UHPLC system (Dionex, Sunnyvale, CA, USA) using a 150 × 0.075 mm PepMap 100 C18, 2 μm particle size, analytical column (Thermo Fisher Scientific) in trapping mode with a C18 trap cartridge. The peptides were separated using mobile phase A (water with 0.1% formic acid) and mobile phase B (acetonitrile with 0.1% formic acid) at a flow rate of 0.3 μL/min. The peptide separation gradient consisted of 2 to 30% solvent B over 4 min, ramped to 95% solvent B over 2 min, held for 4 min, and then returned to 2% solvent B over 1 min and held for 6 min. Peptides were eluted directly into the nanospray source of an Orbitrap Fusion instrument controlled with Xcalibur version 3.1 (Thermo Fisher, San Jose, CA) using a conductive nanospray emitter. The spray voltage was set to 2400 V and ion transfer tube was set to 300°C. The MS1 data were collected at a resolution of 60000 with the scan range of 250–2000 m/z. In CID mode, full MS scans were followed by eight subsequent MS/MS scans on the top eight most abundant peptide ions. Collision energy was set to 35%.
Data analysis:
The percent oxidation of the intact peptide was calculated by summing the area of the selected ion chromatogram peaks of the all the oxidized peptide peaks and then divided by the sum of the area of the unoxidized plus oxidized peptides. CID MS/MS fragmentation was used to identify the major sites of oxidation within the peptide obtained after Glu-C digestion. The presence or absence of unoxidized or oxidized b and y ions were used to identify the site of oxidation.
Electrochemistry.
All experiments were performed at RT under a N2 atmosphere using a Wave Driver 20 bipotentiostat (Pine Instruments) with pyrolytic graphite electrode (PGE) as the working electrode, coiled platinum wire as the counter electrode, and Ag/AgCl (in saturated KCl) as the reference electrode. 10 μL of 250 μM [3SCC-Cu(I9H)3]2+ samples were drop-cast on PGE followed by 6 μL of 0.1 M Polymyxin B to facilitate electrostatic interaction between the enzyme and the electrode. The drop was dried under a gentle stream of N2 gas, and voltammograms were recorded in N2-saturated 80 mM mixed buffer solutions with 100 mM KCl as a supporting electrolyte. Before each run, PGE was polished using slurry of 0.1 μm alumina solution. The potential values were referenced against SHE. The Cu(II) binding affinity was determined according to the Nernst equation (ii)
| (ii) |
where:
The electrochemical surface coverage of the metallopeptide was determined using eq. (iii)
| (iii) |
where ip is the peak current, n is the number of electrons involved, A is the surface area of the electrode, Γ is the surface coverage, ν is the scan rate, and F, R, T represent standard symbols.
The catalytic rate constant kcat was determined from eq. (iv)
| (iv) |
where jcat is the catalytic current density and n is the number of electrons (n = 2 in this case).
Supplementary Material
ACKNOWLEDGEMENTS
S.C. thanks the University of Mississippi for support. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The LC-MS/MS work was performed at the Glycoscience Center of Research Excellence supported by the National Institutes of Health grant P20GM103460.
Footnotes
Supporting Information
The Supporting Information is available free of charge. Relevant data tables, structures, UV-vis, EPR, LC/MS-MS, electrochemistry, HPLC, and MALDI data.
The authors declare no competing financial interest.
REFERENCES
- [1].Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, and Pecoraro VL Protein Design: Toward Functional Metalloenzymes, Chem. Rev, 2014, 114, 3495–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, and Pecoraro VL Designing a Functional Type 2 Copper Center That Has Nitrite Reductase Activity within α-Helical Coiled Coils, Proc. Natl. Acad. Sci., USA, 2012, 109, 21234–21239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zastrow ML, Peacock AFA, Stuckey JA, and Pecoraro VL Hydrolytic Catalysis and Structural Stabilization in a Designed Metalloprotein, Nat. Chem, 2012, 4, 118–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mathieu E, Tolbert AE, Koebke KJ, Tard C, Iranzo O, Penner-Hahn JE, Policar C, and Pecoraro V Rational De Novo Design of a Cu Metalloenzyme for Superoxide Dismutation, Chem. Eur. J, 2020, 26, 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Faiella M, Andreozzi C, De Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, and Lombardi A An Artificial Di-Iron Oxo-Protein with Phenol Oxidase Activity, Nat. Chem. Biol, 2009, 5, 882–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, and DeGrado WF Alteration of the Oxygen-Dependent Reactivity of De Novo Due Ferri Proteins, Nat. Chem, 2012, 4, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, and Tian L Copper Active Sites in Biology, Chem. Rev, 2014, 114, 3659–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaim W, and Rall J Copper—a “Modern” Bioelement, Angew. Chem. Int. Ed, 1996, 35, 43–60 [Google Scholar]
- [9].Mirica LM, Ottenwaelder X, and Stack TDP Structure and Spectroscopy of Copper− Dioxygen Complexes, Chem. Rev, 2004, 104, 1013–1046 [DOI] [PubMed] [Google Scholar]
- [10].Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, and Lu Y Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers, Chem. Rev, 2014, 114, 4366–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, and Eijsink VG An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides, Science, 2010, 330, 219–222 [DOI] [PubMed] [Google Scholar]
- [12].Astruc D, and Astruc D (1995) Electron Transfer and Radical Processes in Transition-Metal Chemistry, Vol. 4, VCH New York. [Google Scholar]
- [13].Bertini L, Breglia R, Lambrughi M, Fantucci P, De Gioia L, Borsari M, Sola M, Bortolotti CA, and Bruschi M Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations, Inorg. Chem, 2018, 57, 86–97 [DOI] [PubMed] [Google Scholar]
- [14].Wang B, Johnston EM, Li P, Shaik S, Davies GJ, Walton PH, and Rovira C QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate, ACS Catal., 2018, 8, 1346–1351 [Google Scholar]
- [15].Chylenski P, Bissaro B, Sørlie M, Røhr ÅK, Várnai A, Horn SJ, and Eijsink VGH Lytic Polysaccharide Monooxygenases in Enzymatic Processing of Lignocellulosic Biomass, ACS Catal., 2019, 9, 4970–4991 [Google Scholar]
- [16].Bissaro B, Røhr ÅK, Müller G, Chylenski P, Skaugen M, Forsberg Z, Horn SJ, Vaaje-Kolstad G, and Eijsink VG Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2, Nat. Chem. Biol, 2017, 13, 1123. [DOI] [PubMed] [Google Scholar]
- [17].Bissaro B, Røhr ÅK, Skaugen M, Forsberg Z, Horn SJ, Vaaje-Kolstad G, and Eijsink VG Fenton-Type Chemistry by a Copper Enzyme: Molecular Mechanism of Polysaccharide Oxidative Cleavage, bioRxiv, 2016, 097022 [Google Scholar]
- [18].Bissaro B, Streit B, Isaksen I, Eijsink VGH, Beckham GT, DuBois JL, and Røhr ÅK Molecular Mechanism of the Chitinolytic Peroxygenase Reaction, Proc. Natl. Acad. Sci., USA, 2020, 117, 1504–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mazurenko I, de Poulpiquet A, and Lojou E Recent Developments in High Surface Area Bioelectrodes for Enzymatic Fuel Cells, Curr. Opin. Electrochem, 2017, 5, 74–84 [Google Scholar]
- [20].Fukuzumi S, Yamada Y, and Karlin KD Hydrogen Peroxide as a Sustainable Energy Carrier: Electrocatalytic Production of Hydrogen Peroxide and the Fuel Cell, Electrochim. Acta, 2012, 82, 493–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miglbauer E, Wójcik PJ, and Głowacki ED Single-Compartment Hydrogen Peroxide Fuel Cells with Poly(3,4-Ethylenedioxythiophene) Cathodes, Chem. Commun, 2018, 54, 11873–11876 [DOI] [PubMed] [Google Scholar]
- [22].Chakraborty S, Hosseinzadeh P, and Lu Y Metalloprotein Design and Engineering, Encyclepedia of Inorganic and Bioinorganic Chemistry, 2014, 1–51 [Google Scholar]
- [23].Efforts to characterize the Cu-oxygen species by resonance Raman has been unsuccessful due to strong background signals from the peptide.
- [24].Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, and Karlin KD Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry, J. Am. Chem. Soc, 2015, 137, 2867–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mann SI, Heinisch T, Ward TR, and Borovik A Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins, J. Am. Chem. Soc, 2017, 139, 17289–17292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhu Q, Lian Y, Thyagarajan S, Rokita SE, Karlin KD, and Blough NV Hydrogen Peroxide and Dioxygen Activation by Dinuclear Copper Complexes in Aqueous Solution: Hydroxyl Radical Production Initiated by Internal Electron Transfer, J. Am. Chem. Soc, 2008, 130, 6304–6305 [DOI] [PubMed] [Google Scholar]
- [27].Paradisi A, Johnston EM, Tovborg M, Nicoll CR, Ciano L, Dowle A, McMaster J, Hancock Y, Davies GJ, and Walton PH Formation of a Copper (II)–Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by H2O2, J. Am. Chem. Soc, 2019, 141, 18585–18599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mitra S, Prasad P, and Chakraborty S A Unified View of Assessing the Pro-Oxidant Versus Antioxidant Nature of Amyloid Beta Conformers, ChemBioChem, 2018, 19, 2360–2371 [DOI] [PubMed] [Google Scholar]
- [29].Hirst J Elucidating the Mechanisms of Coupled Electron Transfer and Catalytic Reactions by Protein Film Voltammetry, Biochim. Biophys. Acta, 2006, 1757, 225–239 [DOI] [PubMed] [Google Scholar]
- [30].Brown AP, and Anson FC Cyclic and Differential Pulse Voltammetric Behavior of Reactants Confined to the Electrode Surface, Anal. Chem, 1977, 49, 1589–1595 [Google Scholar]
- [31].Allen JB, and Larry RF (2001) Electrochemical Methods Fundamentals and Applications, John Wiley & Sons. [Google Scholar]
- [32].Nazemi Z, Prasad P, and Chakraborty S Kinetics of Oxygen Reduction by a Beta Barrel Heme Protein on Hyrid Bioelectrodes, ChemElectroChem, 2020, 7, 1029–1037 [Google Scholar]
- [33].Kakuda S, Peterson RL, Ohkubo K, Karlin KD, and Fukuzumi S Enhanced Catalytic Four-Electron Dioxygen (O2) and Two-Electron Hydrogen Peroxide (H2O2) Reduction with a Copper (II) Complex Possessing a Pendant Ligand Pivalamido Group, J. Am. Chem. Soc, 2013, 135, 6513–6522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim B, Jeong D, and Cho J Nucleophilic Reactivity of Copper (II)–Alkylperoxo Complexes, Chem. Commun, 2017, 53, 9328–9331 [DOI] [PubMed] [Google Scholar]
- [35].Maiti D, Narducci Sarjeant AA, and Karlin KD Copper− Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases, Inorg. Chem, 2008, 47, 8736–8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Paria S, Ohta T, Morimoto Y, Ogura T, Sugimoto H, Fujieda N, Goto K, Asano K, Suzuki T, and Itoh S Generation, Characterization, and Reactivity of a CuII–Alkylperoxide/Anilino Radical Complex: Insight into the O–O Bond Cleavage Mechanism, J. Am. Chem. Soc, 2015, 137, 10870–10873 [DOI] [PubMed] [Google Scholar]
- [37].Park GY, Lee JY, Himes RA, Thomas GS, Blackburn NJ, and Karlin KD Copper–Peptide Complex Structure and Reactivity When Found in Conserved His-Xaa-His Sequences, J. Am. Chem. Soc, 2014, 136, 12532–12535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tsuji T, Zaoputra AA, Hitomi Y, Mieda K, Ogura T, Shiota Y, Yoshizawa K, Sato H, and Kodera M Specific Enhancement of Catalytic Activity by a Dicopper Core: Selective Hydroxylation of Benzene to Phenol with Hydrogen Peroxide, Angew. Chem. Int. Ed, 2017, 56, 7779–7782 [DOI] [PubMed] [Google Scholar]
- [39].Touw DS, Nordman CE, Stuckey JA, and Pecoraro VL Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chakraborty S, Touw DS, Peacock AF, Stuckey J, and Pecoraro VL Structural Comparisons of Apo- and Metalated Three-Stranded Coiled Coils Clarify Metal Binding Determinants in Thiolate Containing Designed Peptides, J. Am. Chem. Soc, 2010, 132, 13240–13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Marks J, Pozdnyakova I, Guidry J, and Wittung-Stafshede P Methionine-121 Coordination Determines Metal Specificity in Unfolded Pseudomonas Aeruginosa Azurin, J. Biol. Inorg. Chem, 2004, 9, 281–288 [DOI] [PubMed] [Google Scholar]
- [42].Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S, and Wedd AG Unification of the Copper (I) Binding Affinities of the Metallo-Chaperones Atx1, Atox1, and Related Proteins Detection Probes and Affinity Standards, J. Biol. Chem, 2011, 286, 11047–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xiao Z, Loughlin F, George GN, Howlett GJ, and Wedd AG C-Terminal Domain of the Membrane Copper Transporter Ctr1 from Saccharomyces Cerevisiae Binds Four Cu(I) Ions as a Cuprous-Thiolate Polynuclear Cluster: Sub-Femtomolar Cu(I) Affinity of Three Proteins Involved in Copper Trafficking, J. Am. Chem. Soc, 2004, 126, 3081–3090 [DOI] [PubMed] [Google Scholar]
- [44].Plegaria JS, Duca M, Tard C. d., Friedlander TJ, Deb A, Penner-Hahn JE, and Pecoraro VL De Novo Design and Characterization of Copper Metallopeptides Inspired by Native Cupredoxins, Inorg. Chem, 2015, 54, 9470–9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Borisova AS, Isaksen T, Dimarogona M, Kognole AA, Mathiesen G, Várnai A, Røhr ÅK, Payne CM, Sørlie M, Sandgren M, and Eijsink VGH Structural and Functional Characterization of a Lytic Polysaccharide Monooxygenase with Broad Substrate Specificity, J. Biol. Chem, 2015, 290, 22955–22969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen P, Bell J, Eipper BA, and Solomon EI Oxygen Activation by the Noncoupled Binuclear Copper Site in Peptidylglycine α-Hydroxylating Monooxygenase. Spectroscopic Definition of the Resting Sites and the Putative CuIIM−OOH Intermediate, Biochemistry, 2004, 43, 5735–5747 [DOI] [PubMed] [Google Scholar]
- [47].Suzuki S, Sakurai T, Nakahara A, Oda O, Manabe T, and Okuyama T Spectroscopic Aspects of Copper Binding Site in Bovine Serum Amine Oxidase, FEBS Lett, 1980, 116, 17–20 [DOI] [PubMed] [Google Scholar]
- [48].Adelson CN, Johnston EM, Hilmer KM, Watts H, Dey SG, Brown DE, Broderick JB, Shepard EM, Dooley DM, and Solomon EI Characterization of the Preprocessed Copper Site Equilibrium in Amine Oxidase and Assignment of the Reactive Copper Site in Topaquinone Biogenesis, J. Am. Chem. Soc, 2019, 141, 8877–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ruggiero CE, Smith JA, Tanizawa K, and Dooley DM Mechanistic Studies of Topa Quinone Biogenesis in Phenylethylamine Oxidase, Biochemistry, 1997, 36, 1953–1959 [DOI] [PubMed] [Google Scholar]
- [50].Courtade G, Ciano L, Paradisi A, Lindley PJ, Forsberg Z, Sørlie M, Wimmer R, Davies GJ, Eijsink VGH, Walton PH, and Aachmann FL Mechanistic Basis of Substrate-O(2) Coupling within a Chitin-Active Lytic Polysaccharide Monooxygenase: An Integrated NMR/EPR Study, Proc. Natl. Acad. Sci, 2020, 117, 19178–19189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hemsworth GR, Taylor EJ, Kim RQ, Gregory RC, Lewis SJ, Turkenburg JP, Parkin A, Davies GJ, and Walton PH The Copper Active Site of CBM33 Polysaccharide Oxygenases, J. Am. Chem. Soc, 2013, 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Macedo S, Pechlaner M, Schmid W, Weik M, Sato K, Dennison C, and Djinović-Carugo K Can Soaked-in Scavengers Protect Metalloprotein Active Sites from Reduction During Data Collection?, J. Synchrotron Rad, 2009, 16, 191–204 [DOI] [PubMed] [Google Scholar]
- [53].Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, Hodgson KO, Hedman B, and Solomon EI Spectroscopic and Computational Insight into the Activation of O2 by the Mononuclear Cu Center in Polysaccharide Monooxygenases, Proc. Natl. Acad. Sci., USA, 2014, 111, 8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Aachmann FL, Sørlie M, Skjåk-Bræk G, Eijsink VG, and Vaaje-Kolstad G NMR Structure of a Lytic Polysaccharide Monooxygenase Provides Insight into Copper Binding, Protein Dynamics, and Substrate Interactions, Proc. Natl. Acad. Sci, 2012, 109, 18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chauhan S, Hosseinzadeh P, Lu Y, and Blackburn NJ Stopped-Flow Studies of the Reduction of the Copper Centers Suggest a Bifurcated Electron Transfer Pathway in Peptidylglycine Monooxygenase, Biochemistry, 2016, 55, 2008–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jacobson F, Pistorius A, Farkas D, De Grip W, Hansson Ö, Sjölin L, and Neutze R Ph Dependence of Copper Geometry, Reduction Potential, and Nitrite Affinity in Nitrite Reductase, J. Biol. Chem, 2007, 282, 6347–6355 [DOI] [PubMed] [Google Scholar]
- [57].Suzuki S, Yamaguchi K, Kataoka K, Kobayashi K, Tagawa S, Kohzuma T, Shidara S, and Iwasaki H Spectroscopic Characterization and Intramolecular Electron Transfer Processes of Native and Type 2 Cu-Depleted Nitrite Reductases, J. Biol. Inorg. Chem, 1997, 2, 265–274 [Google Scholar]
- [58].Borsook H, and Keighley G Oxidation-Reduction Potential of Ascorbic Acid (Vitamin C), Proc. Natl. Acad. Sci, 1933, 19, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim S, Saracini C, Siegler MA, Drichko N, and Karlin KD Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent, Inorg. Chem, 2012, 51, 12603–12605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Reuillard B, Ly KH, Hildebrandt P, Jeuken LJ, Butt JN, and Reisner E High Performance Reduction of H2O2 with an Electron Transport Decaheme Cytochrome on a Porous ITO Electrode, J. Am. Chem. Soc, 2017, 139, 3324–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Reuillard B, Le Goff A, Holzinger M, and Cosnier S Non-Covalent Functionalization of Carbon Nanotubes with Boronic Acids for the Wiring of Glycosylated Redox Enzymes in Oxygen-Reducing Biocathodes, J. Mater. Chem. B, 2014, 2, 2228–2232 [DOI] [PubMed] [Google Scholar]
- [62].Kabsch W XDS, Acta Crystallogr. D, 2010, 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Evans PR, and Murshudov GN How Good Are My Data and What Is the Resolution?, Acta Crystallogr. D, 2013, 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Potterton L, Agirre J, Ballard C, Cowtan K, Dodson E, Evans PR, Jenkins HT, Keegan R, Krissinel E, and Stevenson K CCP4i2: The New Graphical User Interface to the CCP4 Program Suite, Acta Crystallogr. D, 2018, 74, 68–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, and McCoy A Overview of the CCP4 Suite and Current Developments, Acta Crystallogr. D, 2011, 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zwart PH, Grosse-Kunstleve RW, and Adams PD Phenix.Xtriage, CCP4 Newsl. 43,, 2005, contribution 7, [Google Scholar]
- [67].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ Phaser Crystallographic Software, J. Appl. Crystallogr, 2007, 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, and McCoy AJ Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix, Acta Crystallogr. D, 2019, 75, 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fletcher JM, Boyle AL, Bruning M, Bartlett GJ, Vincent TL, Zaccai NR, Armstrong CT, Bromley EHC, Booth PJ, Brady RL, Thomson AR, and Woolfson DN A Basis Set of De Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology, ACS Synth. Biol, 2012, 1, 240–250 [DOI] [PubMed] [Google Scholar]
- [70].Cowtan K The Buccaneer Software for Automated Model Building. 1. Tracing Protein Chains, Acta Crystallogr. D, 2006, 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- [71].Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung L-W, Read RJ, and Adams PD Iterative Model Building, Structure Refinement and Density Modification with the Phenix Autobuild Wizard, Acta Crystallogr. D, 2008, 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD Towards Automated Crystallographic Structure Refinement with Phenix. Refine, Acta Crystallogr. D, 2012, 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Emsley P, and Cowtan K Coot: Model-Building Tools for Molecular Graphics, Acta Crystallogr. D, 2004, 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- [74].Stoll S, and Schweiger A Easyspin, a Comprehensive Software Package for Spectral Simulation and Analysis in Epr, J. Magn. Reson, 2006, 178, 42–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.