

Abstract
The activation and differentiation of CD4+ T cells play a critical role in establishing and subsequently controlling protective adaptive immune responses. Flow cytometry is a powerful technique with which to assess the potential of xenobiotics to influence CD4+ T cell activation and differentiation. With flow cytometry, cells are stained with fluorochrome-conjugated antibodies and/or specific fluorescent probes to assess T cell activation, proliferation, effector cytokine production, and transcription factor expression. This technique allows for complex phenotypic analysis of tens to hundreds of thousands of individual cells very rapidly to assess the potential impact of a xenobiotic on CD4 effector differentiation and activation state.
Keywords: Flow cytometry, CD4+ T cells, Effector differentiation, T cell activation, TH1, TH2, TH17, TH9, TFH, Treg
1. Introduction
Since the initial discovery by Mosmann and Coffman in the late 1980s that CD4+ T cells can be divided into distinctive effector subsets (TH1 and TH2) based upon their biological functions and characteristic cytokine production [1, 2], several other CD4+ T cell effector subsets have been identified. Among these more recently identified subsets are IL-17 producing TH17 [3–5], T follicular helper cells (TFH) which take part in the germinal center reaction, IL-9 producing TH9 [6], and regulatory T cells (Treg) [7, 8] which function to suppress the activities of the other effector subsets. These subsets arise from undifferentiated naive CD4+ T cells, and their distinct effector functions play a central role in shaping the adaptive immune response. Dysregulation of effector subset differentiation can lead to immune-mediated pathologies (allergy, autoimmunity, etc.), the mounting of a nonprotective immune response toward pathogens, or to the inhibition of an immune response.
Each of these effector subsets produces characteristic effector cytokines, and the transcriptional program leading to, and enforcing, this differentiation is under the control of master transcriptional regulators (Table 1). CD4 effector subset differentiation is a complex process involving many factors including the cytokine milieu during naive CD4+ T cell activation, the specific antigen, antigen-presenting cell activation state, costimulatory molecule expression, and physiological location of activation, among others. Small perturbations of these factors can alter the ultimate differentiation of these cells. Xenobiotic exposure has the potential to alter the differentiation of CD4+ T cell and the subsequent adaptive immune response. Because dysregulation of CD4+ T cell differentiation can have significant consequences on the generation of protective adaptive immune responses, an important facet in assessing the immunotoxic potential of xenobiotics is to examine whether they alter CD4 T cell differentiation.
Table 1.
Characteristic cytokine products and master transcriptional regulators of CD4+ T cell effector subsets
Here we describe the use of flow cytometry to assess the impact of xenobiotic exposure in vitro on CD4+ T cell activation and differentiation. Cells can be stained with fluorochrome-conjugated antibodies to stain both surface and intracellular targets (Table 2). In addition, fluorescent probes can be utilized to analyze cellular processes including proliferation and DNA content for cell cycle analysis. The ability to perform simultaneous, multiparameter phenotypic analysis of surface molecules, intracellular cytokines, proliferation, and transcription factor expression rapidly for populations comprised of tens of thousands of individual cells makes flow cytometry the most appropriate technique to analyze whether xenobiotics influence CD4 differentiation.
Table 2.
Commonly stained molecules to distinguish effector subsets
| TH1 | TH2 | TH17 | TFH | TH9 | Treg | |
|---|---|---|---|---|---|---|
| Transcription factors | T-bet | GATA-3 | RORγt | Bcl-6 | PU.1 | Foxp3 |
| Surface molecules | IL-12R, CXCR3 | CCR4 | IL-23R, CCR6 | IL-21R, CXCR5 | CD25 | |
| Cytokines | IFNγ | IL-4 | IL-17A | IL-21 | IL-9, IL-10 | IL-10, TGFβ |
2. Materials
All buffers are prepared using sterile, double-deionized water. All solutions are sterile except flow cytometry staining solutions.
Hank’s Balanced Salt Solution (HBSS).
Lympholyte M (Cedarlane, Burlington, NC).
Complete RPMI culture media: RPMI 1640 supplemented with 10% FBS, 1 mM l-glutamine, 100 mg/mL sodium pyruvate, 50 μM 2-ME, essential and nonessential amino acids, 100 U/mL penicillin G, 100 U/mL streptomycin, and 50 μg/mL gentamicin.
CellTrace Violet (Life Technologies, Eugene OR).
5 mM carboxyfluorescein succinimidyl ester (CFSE) solution: resuspend dye in DMSO at 2.8 mg/mL to make 5 mM solution. Store 50 μL aliquots in −80 °C freezer or liquid nitrogen. Protect from light at all times. Warm dye to room temperature before opening. Discard unused portion of aliquot—do not refreeze.
Sterile CFSE loading buffer: 0.1% BSA fraction V in PBS.
Antibodies for T cell activation: anti-CD3 (clone 145–2C11), anti-CD28 (clone 37.52).
Fluorescence-activated cell sorting (FACS) buffer: PBS containing 2% BSA fraction V 2 mM EDTA and 0.1% sodium azide.
Fixation buffer: 4% paraformaldehyde +0.5% glutaraldehyde in PBS pH 7.2.
Fixation/permeabilization buffer: BD Cytofix/Cytoperm™ (BD Biosciences, San Jose, CA, USA).
Intracellular staining permeabilization wash buffer (BioLegend, San Diego, CA, USA) or Perm/Wash™ (BD Biosciences, San Jose, CA, USA).
Recombinant proteins for T cell polarization: IL-12, IL-2, IL-4, TGF-β1, IL-6, IL-1β, IL-21.
Antibodies for T cell polarization: anti-IL-4 (clone 11B11), anti-IFNγ (XMG1.2), anti-IL-12, anti-TGF-β1 (1D11).
5 mg/mL brefeldin A solution (1000×).
2 mM monensin solution (1000×).
1 mM PMA (phorbol 12-myristate 13-acetate) solution: powder is solubilized in DMSO (or ETOH). Used at 1 μM/L final concentration.
1 mM ionomycin calcium salt solution: used at 1 μM final concentration.
Fc Block (anti-CD16/32).
Gey’s solution A: 35 g NH4Cl, 1.85 g KCl, 0.595 g, anhydrous Na2HPO4, 0.12 KH2PO4, 5 g Glucose, 50 mg phenol red. Bring volume to 1 L in double-deionized water. Sterile filter and store at 4 °C.
Gey’s solution B: 1.05 g MgCl∙6H2O, 0.35 g MgSO4∙7H2O and 0.85 g, CaCl2 (anhydrous). Bring volume to 250 mL in double-deionized water. Sterile filter and store at 4 °C.
Gey’s solution C: 5.63 g NaHCO3. Bring volume to 250 mL in double-deionized water. Sterile filter and store at 4 °C.
Gey’s hypotonic solution (RBC Lysis Buffer): 200 mL Gey’s solution A, 50 mL Gey’s solution B and 50 mL, and Gey’s solution C. Bring volume to 700 mL in double-deionized water. Sterile filter and store at 4 °C.
3. Methods
3.1. Preparation of Spleen Cell Cultures
Euthanize mice by CO2 asphyxiation followed by cervical dislocation.
Harvest spleen(s) and place in a 15 mL conical tube containing approximately 10 mL sterile HBSS (see Note 1).
Transfer the spleen to a sterile petri dish containing 10 mL of HBSS. Rupture the collagenous spleen capsule, and generate a single spleen cell suspension by gently grinding between the coarse edges of sterile glass frosted slides in HBSS.
Place the cell suspension in a 15 mL conical centrifuge tube and centrifuge at 500 × g for 5–7 min. The large cell pellet will be brick red due to the presence of contaminating red blood cells (see Note 2).
Discard the supernatant and resuspend the pellet in 5 mL Gey’s hypotonic lysis solution to induce hypotonic lysis of RBC. Incubate 5–10 min at room temp. Add 5 mL complete RPMI and centrifuge 5–7 min at 500 × g.
Discard the supernatant. The pellet should be white/yellow with a very thin layer of red blood cells on the top of the pellet (if at all). Resuspend the cell pellet in 5 mL of complete RPMI. If the pellet is still large and red, repeat the Gey’s hypotonic lysis step.
After hypotonic lysis of red blood cells, enrich cell suspension for lymphocytes by density centrifugation using Lympholyte M. Underlay the 5 mL of cells with 5 mL room temperature Lympholyte M. Centrifuge at 1000 × g for 15 min at room temperature (see Note 3). If multiple spleens have been collected, use 1 tube per 2 spleens.
The lymphocytes will band out at the interface between the media (red) and Lympholyte M (clear). Use a filtered sterile 9 in. borosilicate pasture pipet and a rubber squeeze bulb, gently aspirate the cells from the interface, and place in another sterile 15 mL conical centrifuge tube. Bring the volume to 10 mL and centrifuge 5–7 min at 500 × g. Discard the supernatant and resuspend the cells at 6 × 106/mL in complete RPMI (see Notes 4 and 5).
3.2. Measure Cell Proliferation: CFSE and CellTrace Violet Loading
To evaluate the effects of xenobiotic treatment on cell proliferation, cells can be stained with 5-(and 6-)-carboxy-2′,7′-dichlorofluorescein diacetate, succinimidyl ester (CFSE), or CellTrace Violet on day 0 before exposure cultures are established (see Notes 6 and 7).
Thaw aliquot of CFSE and mix just before using. If using CTV, add DMSO to the lyophilized dye to reconstitute to 2.5 μM, according to the manufacturer’s directions. Discard unused portion of CFSE or CTV aliquots.
Resuspend the cells at 107/mL in PBS + 0.1% BSA. Warm to 37°.
Add 3 μL/mL of the 5 mM dye solution to the cells. Mix and incubate for 10 min at 37°.
Add an equal volume of complete media RPMI containing 10% FBS. Spin and wash an additional time with complete media. Resuspend at 6 × 106 cells/mL in complete RPMI. The cell pellet should take on a faint neon green hue if loaded.
3.3. Exposure Treatment
Pre-coat tissue culture plasticware (6 well plates, petri dishes, etc.) with 1 μg/mL anti-CD3 (145–2C11) plus 1 μg/mL anti-CD28 (37.52) in PBS. If whole spleen is used rather than purified T cells, the anti-CD28 can be in solution. Add the antibodies to the dishes and incubate overnight at 4° or for several hours at 37°. Aspirate off the antibody solution (it can be reused once to reduce costs, if desired), and allow the wells to dry before adding the cells.
Add cells (at 6 × 106/mL in complete RPMI) to the dishes. For a six well plate, a maximum of 5 mL of cells (3.0 × 107) can be added per well.
Add the xenobiotic to be tested using a dose titration of the compound in triplicate wells for each concentration. The actual concentrations will depend on the individual compound. If the compound must be solubilized in a vehicle (e.g., EtOH or DMSO) rather than aqueous buffer, make a 1000× stock solution and dilute into the wells containing cells such that the final vehicle concentration is no more than 0.1% to prevent vehicle toxicity from confounding results. Always include vehicle-only control wells.
To allow for cell growth without media exhaustion and acidification, the media volume can be doubled on day 2, if necessary, with addition of xenobiotic to maintain the treatment concentration.
The timing of cell collection and analysis will be dependent upon the anticipated effects on the cells. Initially, samples can be stained for flow cytometry as described below and examined daily (out to day 6 or longer, depending upon the number of cells available) to evaluate whether the xenobiotic impacts the early activation and proliferation of the cells. Count viable cells upon recovery to assess cell expansion and/or death during the exposure (see Note 8).
3.4. Establishing Polarization Controls
Establishing positive controls of polarized CD4+ T cell effector subsets is necessary for analysis of data from the xenobiotic treatment cultures.
-
To prepare subset differentiation positive controls, separate T cell cultures are established at 1 × 106/mL in complete RPMI. Cells are stimulated with plate-bound anti-CD3 at 1 μg/mL and anti-CD28 at 1 μg/mL. To induce differentiation, cultures were supplemented with combinations of cytokines and anti-cytokine antibodies as below:
TH1 conditions: 5 ng/mL IL-12 and 20 μg/mL anti-IL-4 (11B11) 80 U/mL IL-2 added on day 2 [9]
TH2 conditions: 10 ng/mL rmIL-4 and 50 μg/mL anti-IFNγ, 80 U/mL IL-2 added on day 2 [9]
TH17 conditions: 10 μg/mL anti-IFNγ, 10 μg/mL anti-IL-4, 1 ng/mL TGF-β1, 100 ng/mL IL-6, and 10 ng/mL IL-1β [10]
TFH conditions: 20 μg/mL anti-IFNγ, 20 μg/mL anti-IL-4, 20 μg/mL anti-IL-12, 20 μg/mL anti-TGF-β1 (1D11), 100 ng/mL IL-6, 50 ng/mL IL-21 [11]
TH9 conditions: 10 ng/mL rmIL-4, 2 ng/mL TGF-β, and 10 μg/mL anti-IFNγ [12, 13]
TReg conditions: 5 ng/mL TGF-β1 [13]
After 4 days, cultures are supplemented with complete RPMI-1640 medium. After 5–6 days of culture, differentiated cells are restimulated for 3 days with plate-bound anti-CD3 (1 μg/mL) in polarizing conditions before flow cytometry analysis. Samples are processed and stained in parallel with xenobiotic exposed cells, as described below.
3.5. Restimulation of Cells for Intracellular Cytokine Staining (See Note 9)
When staining of intracellular cytokines is to be performed, cells are restimulated for 5 h in the presence of a protein secretion inhibitor (brefeldin A or monensin—see Notes 9 and 10) to induce maximal cytokine production and the accumulation of intracellular levels that allow for detection (see Note 11).
Remove cells from the exposure culture and centrifuge at 500 × g for 5–7 min.
Resuspend cells at 1 × 107 cells/mL (or a minimum of 100 μL per sample), in complete RPMI containing a final concentration of 1 μg/mL PMA, 1 μg/mL ionomycin salt, and 5 μg/mL brefeldin A or 2 μg/mL monensin (see Note 12).
Incubate cells at 37 °C 5% CO2 for 4–6 h (5 h is recommended).
Add 3 mL complete c × g for 5–7 min. Wash cells, pellet cells, by spinning 500 × g.
Discard supernatant and resuspend the cells in 3–4 mL complete RPMI and centrifuge 5–7 min at 500 × g.
Discard supernatant and proceed to cell surface staining.
3.6. Staining Surface Markers for Flow Cytometry Analysis
Cells are recovered from the cultures, counted, and resuspended at 107 cells/mL in FACS buffer. Place 100 μL of the cell suspension per staining tube (106/tube).
To prevent nonspecific binding of staining antibodies to Fc Receptors, the cells are incubated with 10 μg/mL Fc Block (anti-CD16/32) for 15 min at 4 °C. For surface staining, 100 μL of the antibody cocktail can be added to the cells without removal of the Fc Block.
While the cells are being incubated with Fc Block, dilute antibodies for the cell surface staining in FACS buffer to create desired antibody staining cocktail. Generally, the staining cocktail is 2× the recommended final staining concentrations. Each lab may wish to titrate antibodies to determine optimal staining.
Ensure cells are in suspension before adding staining antibodies by flicking with the finger or very briefly vortexing tubes, and add then 100 μL antibody staining cocktail per 106 cells. A list of possible targets and suggested antibody clones relevant to CD4 T cell differentiation is provided in Table 3. Gently mix cells and antibody cocktail by flicking tube or gently pipetting up and down two or three times.
Incubate the cells in the dark at 4 °C or on wet ice for 20–30 min.
Add 2–3 mL FACS buffer per tube and centrifuge at 500 × g for 5–7 min. Discard the supernatant. Repeat this step to insure all unbound antibody is washed out.
After two washes in FACS buffer, cells can be stained for 30 min with secondary reagents in FACS buffer following steps 4–6, when necessary.
(a) After the final centrifugation step, discard supernatant. Samples can be resuspended in 500 μL FACS buffer and analyzed immediately or stored on ice protected from light for several hours before analysis.
-
(b) Alternatively, after the final wash, the pellet can be dislodged by brief vortexing and the cells fixed by addition of ice-cold fixative (4% paraformaldehyde and 0.5% glutaraldehyde) followed by a 45-min incubation at room temperature.
After 45 min in the dark, wash out fixative by repeating step 6 and resuspend in 0.5 mL FACS buffer. Store at 4 °C in the dark until analyzed (up to 3 days).
(c) If intracellular staining of cytokines and/or transcription factors, proceed to the intracellular staining section.
Table 3.
Suggested antibody clones for flow cytometric analysis of CD4+ differentiation
| Specificity | Clone | Specificity | Clone | Specificity | Clone |
|---|---|---|---|---|---|
| BCL-6 | K112–91 | CD62L | MEL-14 | IL-17A | ICFC |
| CCR4 | 2G12 | CD69 | H1.2F3 | IL-21 | B25168 or IL-21R-Fc chimeric protein |
| CCR6 | 29–2L17 | Fc Block | 2.4G2 | IL-21R | 4A9 |
| CXCR3 | CXCR3–173 | Foxp3 | 150D | IL-23R | 12B2B64 |
| CXCR5 | 2G8 | GATA-3 | 16E10A23 | RORγt | Q31–378 or B2D |
| CD3 | 145–2C11 | IFNγ | XMG1.2 | Pu.1 | 9G7 or ab88082 |
| CD4 | RM4.5 or GK 1.5 | IL-4 | 11B11 | T-bet | 4B10 |
| CD25 | PC61 | IL-9 | RM9A4 | TGFβ | TW7–16B4 |
| CD28 | 37.51 | IL-10 | JES5–16E3 | ||
| CD44 | IL-12R | 114 |
3.7. Flow Cytometry Controls
To accurately set up the flow cytometer and to confirm that detected signals are from specific staining and not from nonspecific interactions between cells and the staining reagents, several controls must be prepared and analyzed along with the experimental samples.
Unstained sample: cells that are processed alongside the stained samples, but no staining reagents are included. These must not have been stained with CFSE or CTV, either.
Single color controls: cells stained with only one of the fluorophores used in the experiment. Along with the unstained sample, these will be used to determine spectral overlap and to perform compensation on the samples (see Notes 13 and 14).
Isotype controls: these are fluorescently conjugated isotype-matched antibodies that will not stain molecules on the target cell population. They are often myeloma proteins. Any staining with these reagents is not antigen-specific and is due to nonspecific interactions with the cell. The intensity of the actual staining antibody is compared to the isotype controls to confirm that staining is antigen-specific and above the background isotype control value.
Fluorescence minus one (FMO) controls: with FMO controls, individual samples are stained with the entire staining panel with one of the stains left out. One FMO control is stained for each of the various fluorophores to be used in the experiment (see Notes 15 and 16).
3.8. Intracellular Staining
After the final wash (step 6 above), briefly vortex to break up the cell pellet and add 50 μL ice-cold fixation/permeabilization buffer per 106 cells. Gently mix by pipetting up and down or brief vortexing.
Incubate cells in fixation/permeabilization buffer on ice for 20 min, protected from light.
Centrifuge at 500 × g for 5–7 min and discard the supernatant.
Resuspend the cells in 500 μL permeabilization buffer.
Repeat steps 3 and 4 twice. During these washes, prepare intracellular staining cocktail.
Dilute antibodies against cytokines and/or transcription factors (all intracellular targets) at appropriate dilution in intracellular staining permeabilization buffer. A list of possible targets and suggested antibody clones relevant to CD4 T cell differentiation is provided in Table 3.
After the final centrifugation step, discard the supernatant and briefly vortex to break up cell pellet. Add 100 μL of intracellular staining solution to the cells.
Incubate on ice for 1 h or overnight at 4 °C degrees in the dark.
After incubation, add 2–3 mL in the intracellular staining permeabilization buffer and centrifuge at 500 × g for 5–7 min. Discard supernatant and repeat this step (see Note 17).
After the final centrifugation, resuspend cells at 3 × 106 cells/mL in FACS buffer. Cells can be analyzed immediately or stored in the dark at 4 °C for up to 3 days before analysis.
3.9. Staining Panel Design
Designing staining panels for flow cytometry is essential for the successful performance of the experiment. Unlike in the past, there are now a multitude of fluorescent dyes and fluorescent proteins that can be used for flow cytometry. Choosing the most appropriate staining reagents and their associated fluorophores is essential.
-
The first step in designing a successful panel is to determine the capabilities of the flow cytometer that will be used. Essentially, you must know what laser excitation wavelengths are available and what emission wavelengths can be detected and their detection efficiency.
There are some basic rules for panel design that must be followed for a successful flow cytometry experiment.- Insure that there is only one target molecule for a particular wavelength (see Note 18).
- The longer the emission wavelength of the fluorophore, the lower the energy, so far-red wavelengths should be used to stain for relatively abundant targets. Higher-energy fluorophores, such as yellow and green, should be used for more rare targets, when possible (see Note 19).
- If possible, choose fluorophores that are spectrally distinct to minimize potential spectral overlap. Fluorescent spectra viewers available online from vendors such as Molecular Probes, BD Biosciences, and BioLegend can be of assistance with this.
- If secondary reagents are used to detect unlabeled antibodies binding to target molecules, insure that the intended target is the only antibody that will react with the secondary. For example, if two mouse IgG primary staining antibodies are used, they must be directly conjugated to the fluorophore because a secondary will react with both. Such data is meaningless because it is impossible to distinguish which primary antibody is being detected.
3.10. Data Analysis
As with any experiment, the critical component of this experimental approach is to correctly analyze and interpret collected data. An excellent primer is available to assist in this process [14]. One of the most powerful components of flow cytometry data analysis is the ability to electronically distinguish cells of interest to be analyzed further. This process is termed “gating.”
Once data is collected on a flow cytometer, .fcs data files can be analyzed by use of third-party software package such as ImmPort (http://Immport.niaid.nih.gov) FlowJo (Tree Star, Inc.), FCS Express (DeNovo Software), or Flowing Software (http://www.uskonaskel.fi).
Before data analysis can take place, the spectral overlap between fluorophores must be corrected by a process called “compensation.” This is the purpose of the unstained population and the single-color controls that were analyzed. Compensation is used to subtract the overlap of one fluorochrome into another channel [15]. This can be performed during data collection on the cytometer or can be done in post-acquisition data analysis.
The first step in data analysis is to define the cell population of interest. Typically, a 2D dot or density plot is displayed of the forward scatter (cell size) versus side scatter (granularity).
The viable lymphocyte populations can be gated, as shown in Fig. 1a. Smaller and/or more granular particles that are detected by the cytometer may represent subcellular debris, dead cells, and/or populations not of interest in this setting (e.g., granulocytes) (see Note 8).
Once the viable lymphocyte population has been gated on, the next step is to define the CD4+ T cell population for further analysis. Display the side scatter versus CD4 intensities on a 2D dot or density display. The CD4+ T cells can be gated as shown in Fig. 1b.
Once this population has been defined, it is then possible to assess surface molecule expression (Fig. 1c) along with any intracellular targets for which cells have been stained (Fig. 2).
Fig. 1.
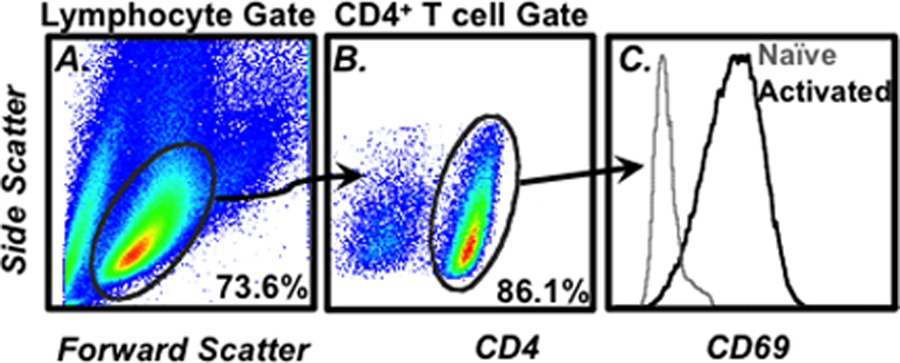
Example of flow cytometry gating strategy. (a) The cell size (forward scatter) vs. cell granularity (side scatter) is plotted on a 2D density plot. The lymphocytes can be identified by characteristic size and scatter profiles and then gated on for further analysis. (b) To identify CD4+ T cells in the gated lymphocytes, a 2D density plot of CD4 expression vs. side scatter is prepared. The CD4+ cells can then be gated upon for additional analysis such as (c) CD69 surface expression. The numbers in A and B represent the frequency of cells within the indicated gate
Fig. 2.
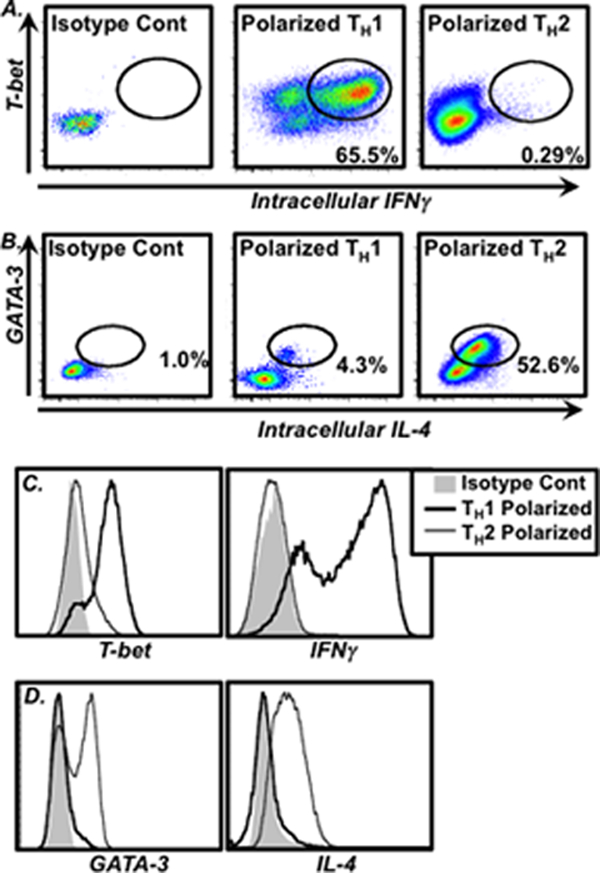
Example of intracellular analysis of TH1 and TH2 cells. Cells were in vitro polarized and stained as described above. Example results of intracellular staining of (a) T-bet and IFNγ (identifying TH1 cells) or (b) GATA-3 and IL-4 (identifying TH2 cells) in CD4+ gated lymphocytes is shown. The frequency of cells within specific gates is shown in the plots. (c and d) Histogram overlays comparing expression levels of these molecules by polarized TH1 (thick black line) and TH2 cells (thin black like) are shown. Isotype control staining is shown for reference (shaded histogram)
4. Notes
To collect the spleen after mice are euthanized, flood the mouse surface with 70% EtOH. Place the mouse on its right side (if viewed from the back), and make an incision in the skin on the left side over the rib cage and extending down the side below the ribs. Cut through the body wall to expose the internal organs. The spleen is a small, thin brick-red organ that is visible under the ribs. It is easily distinguished from the kidney (which is round) or the liver (which spans the peritoneal cavity and is much larger than the spleen). Using a pair of forceps, remove the spleen and then cut the small connection to completely remove it from the body. It should come out as an intact organ. If it is significantly enlarged and more pink in color or it has a significant portion that is black, this indicates there is some pathology in the mouse (e.g., lymphoma) and these cells should not be used. A typical spleen from a wild-type mouse will have 0.5–1.5 × 108 cells.
There will be larger aggregates plus the ruptured spleen capsule in the petri dish with the single-cell suspension. Care should be used not to transfer these into the 15 mL centrifuge tube with single-cell suspension, as they will lead to larger aggregate formation and significantly reduce the number of cells in the single-cell suspension available for the experiment. The easiest way to avoid this is to carefully rinse the petri dish with the HBSS cell suspension. Avoid pulling larger chunks into the pipet. Once the cell suspension is in the 10 mL pipette, hold the pipette vertically for 30 s to a minute. Aggregates will settle out (they will be visible as small red clumps) due to gravity, and a drop or two is then expelled from the pipette to dispense out the aggregates before the suspension is transferred to the new centrifuge tube.
If multiple spleens have been collected, only place the cells from two spleens per Lympholyte M tube. Use multiple tubes, if necessary. Cell yield and purity will be reduced if there are too many cells in the tube with Lympholyte M .
Take care not to transfer much of the Lympholyte M into the new tube. If enough Lympholyte is transferred to the new tube and mixed with the cell suspension and media, the density of the cell suspension could increase to the point that the cells will not pellet during subsequent centrifugation steps.
This protocol will examine whether the xenobiotic influences CD4+ differentiation. This could be due to direct interactions with the T cells and or indirectly by influencing the APCs in the culture. To determine if any observed effect is due to a direct interaction between the xenobiotic and the T cells, CD4+CD62L+ naive cells can be purified from the splenocyte cell suspension and used in parallel experiments.
When designing flow cytometry panels, it is important to keep in mind that use of CFSE will occupy the FITC/AF488 channel and CTV will occupy the DAPI/Pacific Blue/BV421 channel.
If CTV or CFSE are being used, take an aliquot of cells to stain for the pre-/day 0 stain before labeling with those reagents. Immediately after loading, the fluorescence intensity of those dyes will make compensation very difficult. To collect a control population of undivided cells, we routinely wait 24 h.
If there are significant numbers of dead cells in the culture, it is very useful to stain the cells with a fixable live/dead indicator to allow gating out of dead cells, which have a tendency to bind staining antibodies nonspecifically. Examples of such live/dead viability stains are Zombie dyes from BioLegend or the LIVE/DEAD fixable dead cell stains from Molecular Probes. Choose a fluorochrome compatible with the other staining reagents you are using (i.e., do not choose a viability stain that fluoresces in the same wavelengths as another of your stains). Dyes such as propidium iodide can be used, but their very broad emission spectra lead to their overlapping with other fluorophores and make data interpretation much more difficult.
The restimulation step is only required if staining intracellular cytokines. Surface molecule and transcription factor expression can be assessed without restimulation.
When using protein-transport inhibitors upon restimulation of cells, make sure to check recommended inhibitors for targeted cytokines. For example, IL-4 and IL-10 are more readily detected when brefeldin A is used as an inhibitor compared to monensin [16, 17].
A control without PMA + ionomycin stimulation, but with brefeldin A or monensin, should also be included.
Surface staining of cells should be performed prior to fixation to enhance staining fixation-sensitive targets as they may become inaccessible for antibody binding upon fixation.
Because some of the staining targets will vary due to activation state, treatment, etc., it is often difficult to use the antibody staining reagent for single-color controls. It is acceptable to stain single-color controls with any antibody that conjugated to the same fluorochrome. The only exception is with the use of tandem dyes (PE-Cy7, APC-Alexa Fluor 750, etc.). Tandem dyes rely on a FRET signal and vary from lot to lot, so the best practice is to use the staining reagent on the single-color controls, as well.
Antibody capture beads can be used in place of single-color controls. With the beads, the staining antibody is mixed with the beads and antibodies are bound by anti-Fc antibodies bound to the bead. These have the advantage that they can be prepared and used repeatedly in experiments over a couple of weeks, they are uniform in size, and they are very bright. However, they will not display an autofluorescence signal like single-stained cells, and the very high fluorescence intensity can lead to overcompensation, influencing data analysis. Both are valid controls and can be used to set compensation.
The FMO controls are functionally analogous to isotype controls to confirm that staining with a reagent is antigen specific. They have the added advantage that they can assess changes in autoflourescence and spectral overlap on a specific fluorophore’s signal in the context of the other staining antibodies. They can also help assess signal spread (or digital spread) in the detection of the samples on digital flow cytometers [15]. This is not an issue for older analog machines like the BD FACSCalibur.
It is unnecessary to use isotype and FMO controls in the same experiment. It is best to choose one type of control and use it in all experiments to allow for easier experiment—to experiment comparisons.
During washes post-intracellular cytokine/transcription factor staining, allowing cells to sit in permeabilization buffer for 3–5 min before spinning down may help reduce background of nonspecific antibody staining.
For example, because they are detected in the same channel, you can’t use GFP with Alexa Fluor 488 or FITC, nor can you use FITC and Alexa Fluor 488 together. These will be detected by the same detector and can’t be compensated.
The goal is to collect similar fluorescent intensity values for all of the fluorophores. If there is a large difference in intensities between overlapping fluorophores, compensation is much more difficult to perform and is generally less reliable. The general rule of thumb is dimmer targets (fewer molecules) get the brighter dyes (higher-energy yellow and green), while brighter targets (more molecules) get the dimmer dyes (far red and near IR).
References
- 1.Mosmann TR, Coffman RL (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145–173. 10.1146/annurev.iy.07.040189.001045 [DOI] [PubMed] [Google Scholar]
- 2.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL (1986) Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136(7):2348–2357 [PubMed] [Google Scholar]
- 3.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278(3):1910–1914. 10.1074/jbc.M207577200 [DOI] [PubMed] [Google Scholar]
- 4.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM (2006) Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24(6):677–688. 10.1016/j.immuni.2006.06.002 [DOI] [PubMed] [Google Scholar]
- 5.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421(6924):744–748. 10.1038/nature01355 [DOI] [PubMed] [Google Scholar]
- 6.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK (2008) IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat Immunol 9(12):1347–1355. 10.1038/ni.1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asano M, Toda M, Sakaguchi N, Sakaguchi S (1996) Autoimmune disease as a consequence of developmental abnormality of a T cell sub-population. J Exp Med 184(2):387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155(3):1151–1164 [PubMed] [Google Scholar]
- 9.Thauland TJ, Koguchi Y, Wetzel SA, Dustin ML, Parker DC (2008) Th1 and Th2 cells form morphologically distinct immunological synapses. J Immunol 181(1):393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stritesky GL, Yeh N, Kaplan MH (2008) IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol 181(9):5948–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu KT, Kanno Y, Cannons JL, Handon R, Bible P, Elkahloun AG, Anderson SM, Wei L, Sun H, O’Shea JJ, Schwartzberg PL (2011) Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity 35(4):622–632. 10.1016/j.immuni.2011.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, Jabeen R, McKinley C, Ahyi AN, Han L, Nguyen ET, Robertson MJ, Perumal NB, Tepper RS, Nutt SL, Kaplan MH (2010) The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol 11(6):527–534. 10.1038/ni.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fantini MC, Dominitzki S, Rizzo A, Neurath MF, Becker C (2007) In vitro generation of CD4+ CD25+ regulatory cells from murine naive T cells. Nat Protoc 2(7):1789–1794. 10.1038/nprot.2007.258 [DOI] [PubMed] [Google Scholar]
- 14.Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR (2006) Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol 7(7):681–685. 10.1038/ni0706-681 [DOI] [PubMed] [Google Scholar]
- 15.Roederer M (2001) Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry 45(3):194–205 [DOI] [PubMed] [Google Scholar]
- 16.Vicetti Miguel RD, Maryak SA, Cherpes TL (2012) Brefeldin A, but not monensin, enables flow cytometric detection of interleukin-4 within peripheral T cells responding to ex vivo stimulation with Chlamydia trachomatis. J Immunol Methods 384(1–2):191–195. 10.1016/j.jim.2012.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muris AH, Damoiseaux J, Smolders J, Cohen Tervaert JW, Hupperts R, Thewissen M (2012) Intracellular IL-10 detection in T cells by flowcytometry: the use of protein transport inhibitors revisited. J Immunol Methods 381(1–2):59–65. 10.1016/j.jim.2012.04.011 [DOI] [PubMed] [Google Scholar]
- 18.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100(6):655–669 [DOI] [PubMed] [Google Scholar]
- 19.Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89(4):587–596 [DOI] [PubMed] [Google Scholar]
- 20.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6):1121–1133. 10.1016/j.cell.2006.07.035 [DOI] [PubMed] [Google Scholar]
- 21.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, Vinuesa CG (2009) The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31(3):457–468. 10.1016/j.immuni.2009.07.002 [DOI] [PubMed] [Google Scholar]
- 22.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY (2005) Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22(3):329–341 [DOI] [PubMed] [Google Scholar]
- 23.Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299(5609):1057–1061 [DOI] [PubMed] [Google Scholar]