

Abstract
Obesity is an important independent risk factor for cardiovascular diseases, remaining an important health concern worldwide. Evidence shows that saturated fatty acid-induced inflammation in cardiomyocytes contributes to obesity-related cardiomyopathy. Dapagliflozin (Dapa), a selective SGLT2 inhibitor, exerts a favorable preventive activity in heart failure. In this study, we investigated the protective effect of Dapa against cardiomyopathy caused by high fat diet-induced obesity in vitro and in vivo. Cultured rat cardiomyocyte H9c2 cells were pretreated with Dapa (1, 2.5 μM) for 1.5 h, followed by treatment with palmitic acid (PA, 200 μM) for 24 h. We showed that Dapa pretreatment concentration-dependently attenuated PA-induced cell hypertrophy, fibrosis and apoptosis. Transcriptome analysis revealed that inhibition of PA-activated MAPK/AP-1 pathway contributed to the protective effect of Dapa in H9c2 cells, and this was confirmed by anti-p-cJUN fluorescence staining assay. Using surface plasmon resonance analysis we found the direct binding of Dapa with NHE1. Gain and loss of function experiments further demonstrated the role of NHE1 in the protection of Dapa. In vivo experiments were conducted in mice fed a high fat diet for 5 months. The mice were administered Dapa (1 mg·kg−1·d−1, i.g.) in the last 2 months. Dapa administration significantly reduced the body weight and improved the serum lipid profiles. Dapa administration also alleviated HFD-induced cardiac dysfunction and cardiac aberrant remodeling via inhibiting MAPK/AP-1 pathway and ameliorating cardiac inflammation. In conclusion, Dapa exerts a direct protective effect against saturated fatty acid-induced cardiomyocyte injury in addition to the lowering effect on serum lipids. The protective effect results from negative regulating MAPK/AP-1 pathway in a NHE1-dependent way. The current study highlights the potential of clinical use of Dapa in the prevention of obesity-related cardiac dysfunction.
Keywords: obesity, cardiomyopathy, Dapagliflozin, palmitic acid, MAPK pathway, NHE1, inflammation, rat cardiomyocyte H9c2 cell line
Introduction
Obesity, a public health problem in the general population, is an important independent risk factor for cardiovascular diseases [1]. Obesity caused chronic structural changes in the heart including left ventricular hypertrophy, further impairing the systolic/diastolic function and leading to heart failure [2]. Mechanisms underlying this progressive heart injury are complicated. Several cellular processes have been reported to participate in the progress, including cardiac inflammation, disorders in cardiac metabolism, production of reactive oxygen species (ROS), autophagy and apoptosis of cardiomyocytes [3]. Importantly, elevated free fatty acid (FFA), especially saturated fatty acid (SFA) like palmitic acid (PA), is considered as the origin of obesity-related cardiomyopathy [4]. Cumulative evidence has established that SFA is responsible for systemic and local inflammatory activation as well as immune responses initiation in circulating immune cells as well as cardiomyocytes [4, 5]. Besides, altered FA composition in obese patients results in disorders of β-oxidation metabolism, damage of mitochondria and subsequent oxidative injury in heart tissues. Furthermore, FFA-triggered inflammatory cytokines deteriorate cardiac injury via numerous pathways [6]. Targeting SFA-induced inflammation might be an effective way for preventing obesity-related cardiomyopathy.
Dapagliflozin (Dapa), known as a selective inhibitor for sodium-glucose co-transporter subtype 2 (SGLT2), has already been used clinically due to its external anti-hyperglycemic activity. Recent clinical trials have revealed exciting results that Dapa significantly reduced the risk of cardiovascular deaths and heart failure events [7, 8]. It’s worth noting that the protective effect of SGLT2 inhibitors on diabetic cardiomyopathy has also been extensively illustrated [9]. Since diabetes share a range of pathological changes with obesity, such as insulin resistance, hyperglycemia, hyperlipidemia and low-grade systemic inflammation [10], whether obese patients with cardiac dysfunction could also benefit from SGLT2 inhibition is unknown. Previous publications gave us clues that SGLT2 inhibitors might block the development of obesity systemically [11–14]. However, whether Dapa could directly benefit the heart and the potential mechanisms in obesity remains undefined.
Cardiomyocytes and heart tissue lack SGLT2 protein, hence an off-target effect might occur for SGLT2 inhibitors during their cardio-protection. Recent studies have raised that NHE1 may be the potential target for SGLT2 inhibitors. Specifically, Dapa could effectively reduce cytosolic [Ca2+] and [Na+] and increase mitochondrial [Ca2+] in primary neonatal rat cardiomyocytes, which is mediated mainly by NHE1 receptor [15]. Other studies emphasize the vital role of NHE1 in the protection of SGLT2 inhibition against cardiovascular diseases, including diabetic cardiomyopathy, myocardial infarction and heart failure [16–18]. However, no direct molecular evidence confirmed the interaction between SGLT2 inhibitors and NHE1. Besides, whether NHE1 is involved in the process of anti-SFA-induced injury in cardiomyocytes by Dapa has not been explored.
In the current study, we aimed to investigate the protective role of Dapa against high fat diet-induced obesity and cardiomyopathy. We also explored the underlying mechanism through which Dapa exerted its function in protecting cardiomyocyte. Furthermore, whether the role of NHE1 is involved in this process was also determined.
Materials and methods
Regents
PA (#408-35-5) was obtained from Sigma-Aldrich (St Louis, MO, USA). Dapa (#S1548) was obtained from Selleck (TX, USA). TUNEL apoptosis assay (#C1086) and Annexin V-FITC apoptosis detection kits (#C1062S) were obtained from Beyotime Biotech (Beijing, China). Rhodamine-phalloidin staining kit (#CA1610) was obtained from SolarBio (Beijing, China). Antibodies against p-ERK (#4370), ERK (#4695), p-JNK (#9255), JNK (#9252), p-p38 (#4511), p38 (#8690), Bcl-2 (#3498), Bax (#2772), Cleaved-Caspase3 (#9664) and GADPH (#5174) were purchased from Cell Signaling Technology (Boston, USA). Antibodies against β-MyHc (#ab50967), Col1a1 (#ab34710), TGF-β (#ab92486), TLR4 (#ab22048), MyD88 (#ab2068), MD2 (#ab24182) and p-cJUN (#ab32385) were obtained from Abcam (Cambridge, UK). Antibodies against SGLT2 (#ER64951) were obtained from HuaBio (Hangzhou, China). Antibodies against HA (#51064-2-AP) and NHE1 (#67363-1-lg) were obtained from Proteintech (Wuhan, China). Secondary antibodies were obtained from Yeasen Biotech (Shanghai, China). NHE1-fusion protein (Ag14329) was obtained from Proteintech (Wuhan, China).
Cell culture and transfection
The rat cardiomyocyte cell line H9c2 was purchased from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China), and cells were cultured in DMEM supplemented with 10% FBS and 100 U/mL penicillin and 100 mg/mL streptomycin. HA-tagged NHE1 plasmid was purchased from SinoBiological (Beijing, China) and NHE1 siRNA was purchased from Gene Pharma (Shanghai, China). PEI or Lipofectamine 3000 (Thermo, MA, USA) was used for cell transfection. Plasmids were mixed with PEI in a ratio of 1:3 and NHE1 siRNA was mixed with Lipofectamin 3000 in a ratio of 1:1 for 20 min, and the mixtures were added in H9c2 cells. After 8 h transfection, the medium was replaced and cells were cultured for another 24 h before other treatments.
Rhodamine-phalloidin staining
Rhodamine-phalloidin staining was used to assess cellular hypertrophy. Briefly, H9c2 cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton, and stained with rhodamine-phalloidin (1:200). DAPI (Beyotime, Shanghai, China) was then counterstained, cells were viewed and captured using Nikon 80i fluorescence microscope (Nikon, Tokyo, Japan).
Transcriptome sequencing
H9c2 cells were pretreated with Dapa for 1.5 h, and were then treated with PA for 24 h. Total mRNA was isolated using TRIzol (Takara, Kyoto, Japan), and transcriptome sequencing was conducted (LC-Biotechnology, Hangzhou, China). The differentially expressed genes (DEGs) were defined by fold change > 1.5 or < 0.67, and P < 0.05. Further analysis, including PCA analysis, drawing of Volcano plot and Venn diagram were conducted in OmicStudio (LC-Biotechnology, Hangzhou, China). Enrichment of intersecting genes was then conducted in the database of Enrichr (https://maayanlab.cloud/Enrichr/) [19]. Raw data of transcriptome sequencing were deposited in the Sequence Read Archive (SRA) in BioProject ID PRJNA784032 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA784032).
High fat diet-induced model of obesity-related cardiac dysfunction
All animal study protocols were approved by Wenzhou Medical University Animal Policy and Welfare Committee (Ethics approved number: wydw2020-0464) and adhered to the NIH guidelines (Guide for the care and use of laboratory animals). Male C57BL/6 mice with the weight of 18-22 g were obtained from Wenzhou Medical University Animal Center. Mice were acclimatized to the laboratory for at least 1 week before initiating the studies. Mice were then randomly divided into 3 groups: (1) mice fed with low fat diet (LFD, n = 8), (2) mice fed with high fat diet (HFD, n = 8), (3) mice fed with high fat diet and treated with 1 mg·kg−1·d−1 Dapa (HFD + Dapa, n = 8). High fat diet was purchased from ResearchDiets (New Jersey, USA). The model of obesity-related cardiac dysfunction was established by feeding mice with high fat diet for 5 months. Dapa was dissolved in 1% CMC-Na solution, and administered by intragastric injection for the latter 2 months. Measurements of body weight were recorded weekly. After 5 months, cardiac function of mice was examined (as described below). And then mice were sacrificed, serum samples and heart tissues were collected for further analysis. Serum lipid profiles (total cholesterol, triglyceride, low-density lipoprotein and high-density lipoprotein) and serum markers for cardiac injury (creatine kinase-MB and B-natriuretic peptide) were detected using commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer’s instructions.
Cardiac function tests
Systolic and diastolic cardiac function was assessed by non-invasive transthoracic echocardiography using a Vevo-1100 high-resolution imaging system (Fujifilm Visual Sonics, Japan), with 22–55 hMHz transducer at a frame rate of 233 Hz. Mice were anesthetized using isoflurane. Representative echocardiograph was recorded, and analysis of cardiac function, including parameters of ejection fraction (EF), fraction shortening (FS), posterior wall depth (PWTd), left ventricular diastolic dimension (LVIDd), et al. was conducted according to the echocardiograph.
Histology analysis and tissue staining
Histology analysis includes hematoxylin and eosin (H&E) staining, Masson’s staining, Sirius Red staining and immuno-staining of heart tissues. Briefly, paraffin sections of heart tissues were firstly deparaffinized, rehydrated, and then stained using H&E kit (#G1120; Solarbio, Beijing, China), Masson’s kit (#G1340; Solarbio) and Sirius red kit (#S8060; Solarbio), respectively. Sections were then sealed and viewed using Nikon microscope DM-2500 (Nikon, Tokyo, Japan). For immunochemistry staining, 5 μm heart sections were firstly deparaffinized, rehydrated and then blocked with 5% BSA. Tissue sections were then incubated with primary antibodies of anti-TNF-α (1:500) or anti-CD68 (1:500) at 4 °C overnight. After incubating with HRP-labeled secondary antibodies, chromogenic reaction was then conducted by DAB detection. For immunofluorescence staining, heart sections or cells were fixed, and then permeabilized with 0.1% Triton X-100, followed by blockage with 5% BSA. Heart sections or cells were then incubated with anti-p-cJUN antibodies (1:200) at 4 °C, overnight. After washing with PBS, sections or cells were incubated with Alexa Fluor-488 (1:500) secondary antibodies, and counterstained with DAPI. Images were acquired using Leica TCS SP8 confocal laser scanning microscope (Leica, Solms, Germany).
Apoptosis assay
Cardiomyocyte apoptosis was determined using one-step TUNEL apoptosis assay kit (#C1086, Beyotime, Shanghai, China) and Annexin V-FITC apoptosis detection kit (#C1062S, Beyotime). For TUNEL assay, cells or tissue sections were fixed, washed with PBS, and then stained using TUNEL kit. After counterstaining with DAPI, cells or slides were viewed and captured using Nikon 80i fluorescence microscope (Nikon, Tokyo, Japan). For Annexin V-FITC apoptosis detection, cells were digested and washed. Cells were then stained using Annexin V-FITC detection kit, followed by analysis using flow cytometry (FACS Calibur, BD, New Jersey, USA).
Detection of cytokines by ELISA assay
Inflammatory cytokines including IL-6 and TNF-α in the heart tissues were detected using ELISA kits (eBioscience, San Diego, CA, USA). Total amount of cytokines was calculated using the standard curve method, and the relative amount of cytokines was normalized to the protein concentration of heart tissues.
Western blot and immunoprecipitation
For Western blot, cell and tissue lysates were separated by 10% SDS-PAGE, and transferred to polyvinylidene difluoride membranes. After blocking with 5% non-fat milk for 2 h, membranes were then incubated with specific primary antibodies at 4 °C, overnight. Immunoreactive bands were detected by incubating with secondary antibodies conjugated to horseradish peroxidase and chemi-luminescence reagent (Bio-Rad, CA, USA). For immunoprecipitation assay, 600 mg cell lysates were used for IP, while 1/10 cell lysates were used for Input. Briefly, lysates were incubated with 1 μg anti-TLR4 primary antibodies at 4 °C overnight. Protein A&G beads were then added and incubated for 4 h. The precipitates were then collected as IP for further assays. Immunoblotting was performed from IP and Input to detect the formation of MD2/TLR4/MyD88.
Image J analysis software version 1.8.0 was then used for densitometric quantification of immunoblots. Relative amount of target proteins was then normalized to their respective control (GAPDH for WB and TLR4 for IP).
Real-time qPCR
Total mRNA prepared from H9c2 cells or heart homogenate was isolated using TRIzol. Concentrations and purity of the samples were measured in duplicate using SpectraMaxM5 Microplate Reader (Molecular Devices, San Jose, CA, USA). Reverse transcription and quantitative PCR was conducted using two-step PrimeScript RT reagent Kit (Perfect Real Time; Takara, Tokyo, Japan) and Eppendorf Mastercycler ep realplex detection system (Eppendorf, Hamburg, Germany). Primers of genes encoding MyH7, Col1a1, Tgfb1, Il1b, Il6 and Tnfa were obtained from Sangon (Shanghai, China), and the sequences were shown in Supplementary Table S1. The relative amount of each gene was normalized to β-actin.
Surface plasmon resonance (SPR) analyses
The direct binding of Dapa to NHE1 protein was detected using Biacore T200 instrument (Cytiva, Marlborough, MA, USA) with a CM7 sensor chip (Cytiva). Briefly, NHE1 protein (in acetate acid buffer) was loaded to the sensor using an Amine Coupling Kit (Cytiva). After the target sensor was placed in the instrument, different concentrations of Dapa (0, 3.125, 6.25, 12.5, 25, 50, 100 μM) were then flowed over the black and target sensors at a flow rate of 30 μL/min for a 200 s association phase, which was followed by a 60 s dissociation phase at 25 °C. The final graphs were obtained by subtracting blank sensorgrams and blank samples from the duplex. The data were analyzed with Biacore T200 software EV. The dissociation constant (KD) was calculated by global-fitting of the kinetics data from various concentrations of Dapa using a 1:1 Langmuir binding model.
Molecular docking
Molecular simulation of the binding of Dapa with NHE1 was performed using AutoDock version 4.2.6. Since no NHE1 monomer structure was reported, a predicted homology model based on Sodium/hydrogen exchanger 1 (7dsx.1.B) [20] established by Swiss-Model was used. The AutoDock Tools version 1.5.6 package was applied to generate the docking input files. A 126 × 126 × 126-point grid box with a spacing of 0.700 Å between the grid points, which could accommodate the whole NHE1 protein, was implemented. The affinity maps of NHE1 were calculated by AutoGrid. One hundred Lamarckian genetic algorithm runs with default parameter settings were processed using AutoDock 4.2.6. A series of binding forms were generated, and the lowest energy form was chosen. PyMOL 2.5 software was then used to analyze hydrogen bonds and bond lengths within the interactions.
Statistical analysis
Data presented in this study is representative of at least 3 independent experiments and is expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism 8.0 software (San Diego, CA, USA). One-way ANOVA followed by Tukey post-hoc test was used when comparing more than two groups of data. P < 0.05 was considered significant in statistics. Post-tests were run if F achieved P < 0.05 and there was no significant variance in homogeneity.
Results
Dapa exhibits direct protection in cardiomyocytes
As previously reported, SGLT2 was mainly expressed in kidney, and was not detected in cardiomyocytes and the heart [21]. Here, we first validated the expression of SGLT2 in H9c2 cells with renal tubular epithelial cells NRK-52E as the positive control. Nearly no expression of SGLT2 was detected in H9c2 cells, regardless of Dapa treatment (Supplementary Fig. S1). Despite this, a direct protection of Dapa was observed in H9c2 cardiomyocytes. As shown in Fig. 1a and Supplementary Fig. S2a–c, PA significantly increased the expression of β-MyHc, Col-1a1 and TGF-β, while pretreatment with Dapa dose-dependently downregulated these proteins related to cellular fibrosis. Consistently, qPCR assay showed a similar inhibition of PA-induced expression of these markers at the transcriptional level (Fig. 1b). To vividly visualize the protective effect of Dapa, the rhodamine-phalloidin staining on H9c2 cells was performed. Results showed that PA-induced cellular hypertrophic response was attenuated after Dapa pretreatment (Fig. 1c, Supplementary Fig. S2d). These results collectively indicated that Dapa efficiently attenuated PA-induced fibrosis and hypertrophy in H9c2 cells.
Fig. 1. Dapa directly protects cardiomyocytes against palmitic acid-induced cellular fibrosis and apoptosis.

a–c Dapa attenuated PA-induced cardiomyocyte fibrosis and hypertrophy. H9c2 cells were pretreated with Dapa, and were then stimulated with PA for 24 h. a Cell lysates were collected and proteins of β-MyHc, Col-1, and TGF-β were detected with GAPDH as loading control. b Total mRNA of H9c2 cells was isolated, and mRNA levels of fibrotic genes were detected with β-actin as the normalization control. c H9c2 cells were stained with rhodamine-phalloidin, and counterstained with DAPI. Representative images were shown (scale bar = 20 μm). d–f Dapa attenuated PA-induced cardiomyocyte apoptosis. H9c2 cells were pretreated with Dapa, and were then stimulated with PA for 24 h. d Cell lysates were collected and proteins of Bcl-2, Bax and Cleaved-Caspase-3 were detected with GAPDH as loading control. Quantifications were shown in the right panel. e H9c2 cells were stained with TUNEL kit, and counterstained with DAPI. Representative images were shown (scale bar = 25 μm). f H9c2 cells were digested and stained with Annexin V-PI apoptosis kit, cells were then detected using flow cytometry. Representative images were shown. n = 3; Means ± SEM; One-way ANOVA followed by Turkey post-hoc tests; *P < 0.05, compared with Ctrl group, #P < 0.05, compared with PA group.
Then the effects of Dapa on PA-induced apoptosis were examined in H9c2 cells. PA exposure for 24 h led to cellular apoptosis with upregulation of Bax and Cleaved-caspase3 along with downregulation of Bcl-2. This apoptotic readout was suppressed by Dapa pretreatment (Fig. 1d). Furthermore, TUNEL staining and flow cytometry assays also confirmed pretreatment with Dapa significantly mitigated PA-induced apoptosis in H9c2 cells (Fig. 1e, f, Supplementary Fig. S2e, f). Taken together, these data implicated an off-target effect of Dapa since Dapa was able to directly protect cardiomyocytes despite no SGLT2 expression was detected.
Identification of MAPK signaling as a functioning pathway
To explore the off-target effect of Dapa and the potential mechanism mediating its protection in cardiomyocytes, a transcriptome sequencing was performed. PCA analysis revealed that 3 groups of cells were well divided (Fig. 2a). Gene expressions with fold change more than 1.5 or less than 0.67, and P value less than 0.05 were defined as significantly changed. Totally, 3432 and 124 DEGs were obtained in PA and PA + Dapa groups, respectively (Supplementary Fig. S3a). Of these DEGs, 70 intersecting genes were significantly changed in both the two comparisons, and might be responsible for the protective effect of Dapa (Fig. 2b). Therefore, these 70 intersecting genes were selected for further pathway enrichment analysis using an online tool Enrichr [19]. Panther pathway enrichment analysis indicated p38-MAPK pathway was the most enriched (Fig. 2c). Pathway enrichment analysis based on NCI_Nature or BioCarta also confirmed the result that p38-MAPK or ERK1/2-MAPK signaling contributed to the protective effect of Dapa (Supplementary Fig. S3b, c). Besides, the hub-protein analysis showed that inflammation-associated proteins were essential in the process. Specifically, MAPK1 (ERK2) and MAPK3 (ERK1) ranked frontally, indicating that MAPK signaling was critically involved in modulating the protective function of Dapa (Fig. 2d). To fully understand the potential mechanisms, the transcription factors prediction analysis was further conducted. Interestingly, components of AP-1 [22], including Jun and JunD, ranked frontally among the top 10 transcription factors (Fig. 2e). These results prompted us that Dapa directly protected cardiomyocytes via regulating MAPK/AP-1 signaling.
Fig. 2. MAPK signaling contributes to the direct cardiomyocyte protection of Dapa.

H9c2 cells were pretreated with Dapa, and were then stimulated with PA for 24 h. Samples were isolated using TRIzol, and transcriptome sequencing was conducted. a PCA analysis of the sequencing results. b Venn diagram of differential genes in Ctrl VS PA, and PA VS PA + Dapa. c Panther pathway enrichment analysis of 70 intersecting genes. d PPI hub-proteins analysis of 70 intersecting genes. e TRRUST transcription analysis of 70 intersecting genes. Dotted lines represented P = 0.05. Black column showed significant changed items.
Verification of the negative regulation of MAPK/AP-1 pathway by Dapa in cardiomyocytes
As transcriptome sequencing indicated, MAPK/AP-1 pathway was the most probable regulator for these intersecting genes and might be the potential pathway mediating the protective effect of Dapa. Since the activation of MAPK/AP-1 pathway often relies on the phosphorylation of the key kinases. We then validated the hypothesis by checking Dapa’s effect on the phosphorylation of MAPK/AP-1 pathway in H9c2 cells. Using immunofluorescence staining of phosphorylated-cJUN (p-cJUN), we found that PA was able to activate cJUN transcription via increasing p-cJUN in the nucleus. As expected, a dose-dependent decrease of p-cJUN in the nucleus was observed after pretreatment with Dapa (Fig. 3a, Supplementary Fig. S4a). Similarly, Western blotting analysis demonstrated that PA-induced excessive activation of MAPK pathway was also diminished by Dapa (Fig. 3b, Fig. S4b–d). Furthermore, PA-induced over-expression of pro-inflammatory cytokines in H9c2 cells was also restrained by Dapa pretreatment (Fig. 3c).
Fig. 3. Dapa inhibits MAPK signaling and inflammation in cardiomyocytes.

H9c2 cells were pretreated with Dapa, and were then stimulated with PA for 1 h. a H9c2 cells were fixed and stained with anti-p-cJUN antibody, and counterstained with DAPI. Representative images were shown (scale bar = 25 μm). b Cell lysates were collected and proteins of p-ERK, p-JNK and p-p38 were detected with total proteins as loading control. c Cells were pretreated with Dapa, and were then stimulated with PA for 8 h. Total mRNA was isolated using TRIzol, and mRNA levels of pro-inflammatory genes were detected with β-actin as the normalization control. n = 3; Means ± SEM; One-way ANOVA followed by Turkey post-hoc tests; *P < 0.05, compared with Ctrl group, #P < 0.05, compared with PA group.
Dapa targets NHE1 hence regulating MAPK/AP-1 pathway to protect cardiomyocytes
Subsequently, we aimed to understand how Dapa regulated PA-induced activation of MAPK/AP-1 pathway. PA was reported to directly bind to MD2, the accessory protein of TLR4, and resulted in the activation of TLR4/MyD88 dependent MAPK signaling [23]. Therefore, whether Dapa could influence the formation of MD2/TLR4/MyD88 complex was firstly examined. Surprisingly, Western blotting analysis failed to observe any significant change after pre-treatment with Dapa (Supplementary Fig. S5). This suggested that Dapa may regulate MAPK/AP-1 pathway in a TLR4/MyD88 independent way.
Recent studies [17, 18, 24] demonstrated that NHE1 might be one of the direct targets of SGLT2 inhibitors in cardiovascular diseases. Interestingly, NHE1 was also reported to be associated with MAPK signaling [25]. Hence, it is hypothesized that NHE1 participated in the protective function of Dapa in cardiomyocytes. Since no direct evidence showed the binding of NHE1 and Dapa, we firstly conducted SPR assay and confirmed the direct binding of Dapa to NHE1 protein with a KD value of 1.09 μM (Fig. 4a). Similarly, molecular docking showed that Dapa bound to NHE1 structure with a docking score of −5.86 kcal/mol (Fig. 4b). Overexpression and knockdown of NHE1 was then performed. Transfection of NHE1 siRNA significantly decreased the expression of NHE1 while transfection of HA-NHE1 plasmid significantly elevated NHE1 expression (Supplementary Fig. S6a, b). Furthermore, knockdown of NHE1 was already to inhibit phosphorylation of MAPK pathway while addition of Dapa did not show better effect (Fig. 4c, Supplementary Fig. S6c). However, overexpression of NHE1 worsen the effect and abolished the inhibitory effect of Dapa on MAPK pathway, probably due to that Dapa was not enough in this saturating condition (Fig. 4d, Supplementary Fig. S6d). Consistently, cellular hypertrophy and apoptosis was prevented in NHE1-knockdown group with comparative effect in NHE1-knockdown plus Dapa group (Fig. 4e, f, Supplementary Fig. S6e, f). Moreover this was aggravated in NHE1-overexpressed group and the protective effect of Dapa was also abolished after NHE1 overexpression (Fig. 4g, h, Supplementary Fig. S6g, h). Taken together, these results indicated that Dapa targets NHE1 hence regulating MAPK/AP-1 pathway to protect cardiomyocytes against PA-induced cellular injury.
Fig. 4. Dapa targets NHE1 hence regulating MAPK/AP-1 pathway to protect cardiomyocytes.

a Direct binding of Dapa and NHE1 protein was shown by SPR assay. Ka binding constant, Kd dissociation constant, KD equilibrium dissociation constant. b Molecular docking of Dapa with NHE1. c–h H9c2 cells were transfected with NHE1 siRNA or HA-NHE1 plasmid, and were then treated with Dapa and PA. Cell lysates from knockdown (c) or overexpression (d) assay were collected and proteins of p-ERK, p-JNK, and p-p38 were detected with total proteins as loading control. H9c2 cells after knockdown of NHE1 were stained with rhodamine-phalloidin (e) or TUNEL kit (f), and counterstained with DAPI. Representative images were shown. g, h H9c2 cells after overexpression of NHE1 were stained with TUNEL kit, and counterstained with DAPI (e, g, scale bar = 50 μm; f, h, scale bar = 100 μm). Representative images were shown. n = 3; Means ± SEM; One-way ANOVA followed by Turkey post-hoc tests.
Administration of Dapa reduces HFD-induced cardiac dysfunction and remodeling
We then explored whether Dapa could improve obesity-related cardiac dysfunction in vivo. Therefore, a mouse model of HFD-induced cardiac dysfunction was established. After successively feeding with HFD for 3 months, mice were treated with 1 mg·kg−1·d−1 Dapa in gavage for the lasting 2 months. Administration of Dapa decreased the body weight of mice (Supplementary Fig. S7a), and improved HFD-induced disorders of serum lipid profiles (Supplementary Fig. S7b), including total cholesterol (TC), triglyceride (TG), low density lipoprotein (LDL) and high-density lipoprotein (HDL). These data were consistent with the results from type 2 diabetic patients enrolled in a clinical trial [26].
To confirm HFD-induced cardiac dysfunction and evaluate the effect of Dapa, echocardiography analysis was utilized. As shown in Table 1 and Supplementary Fig. S8, HFD mice exerted severe cardiac dysfunction, including the decrease of ejection fraction (EF), fraction shortening (FS). Administration of Dapa significantly improved the situation. Moreover, Dapa significantly reduced cardiac hypertrophy and remodeling, evidenced by the decrease of systolic left ventricular internal dimension (LVIDs), diastolic anterior wall thickness (AWTd), heart weight tibia length ratio (HW/TL) and diastolic posterior wall thickness (PWTd, P = 0.1). However, Dapa had little effect on LVIDd. The most used serologic tests for the diagnosis of dysfunctional heart, serum CK-MB and BNP were also in line with the echocardiography findings (Table 1).
Table 1.
Cardiac echocardiographic parameters and systemic and organ parameters of the experimental mice.
| Parameters | LFD (n = 8) | HFD (n = 8) | HFD + Dapa (n = 8) |
|---|---|---|---|
| EF (%) | 68.88 ± 1.46 | 52.26 ± 1.09* | 63.12 ± 1.93# |
| FS (%) | 38.21 ± 1.20 | 26.57 ± 0.72* | 34.00 ± 1.42# |
| LVIDs (mm) | 2.45 ± 0.04 | 3.08 ± 0.06* | 2.75 ± 0.06# |
| LVIDd (mm) | 3.97 ± 0.09 | 4.20 ± 0.10* | 4.18 ± 0.10ns |
| PWTd (mm) | 0.73 ± 0.03 | 0.86 ± 0.05* | 0.78 ± 0.01ns |
| AWTd (mm) | 0.99 ± 0.04 | 1.22 ± 0.04* | 1.07 ± 0.05# |
| Weight (g) | 26.8 ± 0.66 | 48.3 ± 3.44* | 37.3 ± 3.55# |
| HW/TL (mg/mm) | 6.49 ± 0.51 | 8.33 ± 0.71* | 7.29 ± 0.82# |
| CK-MB (U/L) | 165.48 ± 33.79 | 273.37 ± 40.73* | 211.66 ± 49.59# |
| BNP (ng/L) | 111.59 ± 23.36 | 291.8 ± 20.52* | 142.55 ± 26.86# |
*P < 0.05, compared with LFD mice; #P < 0.05, nsP > 0.05, compared with HFD mice.
Histological analysis further confirmed the echocardiography findings. As shown in Fig. 5a, heart size was increased in HFD mice while treatment with Dapa restored heart size. H&E staining showed that Dapa significantly reduced disorganized myofibers (longitudinal section, Fig. 5b) and increased cross-sectional cardiomyocyte area of myofibers (transverse section, Fig. 5c, Supplementary Fig. S9a). Masson’s stained heart sections demonstrated that cardiac fibrosis was significantly enhanced with more connective tissues in the interstitial area in HFD mice, which was reduced after Dapa treatment (Fig. 5d, Supplementary Fig. S9b). Sirius staining further confirmed the anti-fibrotic effect of Dapa, with decreased collagen deposition in both interstitial and perivascular areas (Fig. 5e, f, Supplementary Fig. S9c, d). Consistently, the expression levels of fibrosis and hypertrophy markers (β-MyHc, Col1a1, TGF-β) were also significantly decreased by Dapa at both transcriptional and translational levels (Fig. 5g, h, Supplementary Fig. S9e).
Fig. 5. Dapa attenuates HFD-induced cardiac fibrosis, remodeling and cardiomyocytes apoptosis.

Mice were randomly divided into 3 groups, including LFD, HFD and HFD + Dapa. After 3 months of high fat diet, mice were treated with 1 mg·kg−1·d−1 Dapa for another 2 months in HFD + Dapa group. a Representative macrographs of hearts from different groups of mice. Hematoxylin-eosin staining of heart sections. Representative images of longitudinal views (b, scale bar = 30 μm) and transverse views (c, scale bar = 10 μm). d Representative images of Masson’s staining of heart sections (scale bar = 50 μm). Sirius Red staining of heart sections. Representative images of interstitial views (e, scale bar = 50 μm) and perivascular views (f, scale bar = 30 μm). g Western blot analysis of fibrotic and hypertrophy protein markers in heart tissue. GAPDH was used as loading control. h Quantitative PCR analysis of fibro-genic genes in heart tissue, and β-actin was used as normalization control. i Western blot analysis of apoptotic protein markers in heart tissue. GAPDH was used as loading control. j Representative images of TUNEL staining of heart sections. White arrows showed positive TUNEL staining cells in heart tissue (scale bar = 25 μm). n = 8 (a-f, h, j), n = 6 (g, i); Means ± SEM; One-way ANOVA followed by Turkey post-hoc tests; *P < 0.05, compared with LFD group, #P < 0.05, compared with HFD group.
Apoptosis of cardiomyocytes results in the loss of contractile tissue, hence playing an important role in the development of obesity-related cardiac dysfunction and remodeling [27]. In consistent with our in vitro results, an anti-apoptosis effect of Dapa was observed by immunoblotting. Downregulation of Bcl-2, and upregulation of Bax and cleaved-caspase 3 in heart tissues of HFD mice were significantly reversed in HFD + Dapa group (Fig. 5i, Supplementary Fig. S9f). This was further confirmed by TUNEL analysis of heart sections (Fig. 5j, Supplementary Fig. S9g). Collectively, these data demonstrated that Dapa showed active cardio-protection, with improved cardiac dysfunction and remodeling, despite the positive influence on lipid metabolism.
Dapa reduces cardiac inflammation via inhibiting MAPK/AP-1 pathway
We next aimed to investigate whether the function of Dapa in reducing inflammation was also attributed to MAPK/AP-1 pathway inhibition in vivo. Western blotting results found that HFD induced excessive phosphorylation of ERK, JNK and p38 in the heart, while Dapa reduced the activation of MAPK signaling (Fig. 6a, Supplementary Fig. S10a). The activation of transcription factor AP-1 was further detected by immunofluorescence staining of phosphorylated cJUN in heart sections. Conversely, the enhanced transcriptional activity of AP-1 in HFD-fed mice was inhibited by Dapa treatment (Fig. 6b, Supplementary Fig. S10b).
Fig. 6. Dapa attenuates MAPK/AP1 cascades and cardiac inflammation in HFD mice.

Mice were randomly divided into 3 groups, including LFD, HFD and HFD + Dapa. After 3 months of high fat diet, mice were treated with 1 mg·kg−1·d−1 Dapa for another 2 months. a Western blot analysis of proteins in MAPK pathway in heart tissue. GAPDH was used as loading control. b Representative images of anti-p-cJUN immunofluorescence staining in heart sections (scale bar = 25 μm). c Quantitative analysis of inflammatory genes in heart tissue, and β-actin was used as normalization control. d ELISA assay of inflammatory cytokines in heart tissues. e, f Representative images of anti-TNF-α and anti-CD68 immunochemistry staining in heart sections (scale bar = 30 μm). n = 8; Means ± SEM; One-way ANOVA followed by Turkey post-hoc tests; *P < 0.05, compared with LFD group, #P < 0.05, compared with HFD group.
Activation of MAPK/AP-1 pathway induced transcription and secretion of inflammatory cytokines, thereby triggering chronic inflammation [28]. Based on previous findings, inflammatory cytokines in transcript and protein levels were detected. As expected, a significant increase of inflammatory cytokines was observed in HFD mice, while treatment with Dapa successfully prevented the over-expression of these cytokines mRNA (Fig. 6c) and secretion (Fig. 6d). An immunochemistry staining of anti-TNF-α in heart tissues further confirmed that Dapa shifted overall milieu towards low cardiac inflammation (Fig. 6e). Since cardiac inflammation was strongly associated with macrophages infiltration, we then stained heart sections with CD68 antibodies, a specific markers for macrophages. Similarly, Dapa reduced infiltrated macrophages amount in HFD mice (Fig. 6f). Taken together, these data demonstrated that Dapa inhibited MAPK/AP-1 pathway and reduced cardiac inflammation in vivo.
Discussion
Disorders of lipid profiles, especially the excessive SFA often accompanied and further promoted cardiac dysfunction and remodeling via activating several detrimental signaling pathways [29]. In the current study, we found that Dapa provided direct cardio-protection against SFA-induced apoptosis, deleterious remodeling and consequent cardiac dysfunction through inhibiting NHE1/MAPK/AP-1 pathway-mediated inflammation in cardiomyocytes. A summary of our main findings is schemed in Fig. 7.
Fig. 7. Schematic illustration of the main findings.
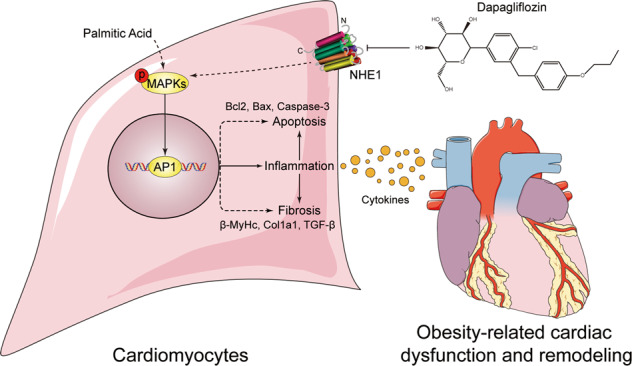
Dapa directly inhibits saturated fatty acid (PA)-induced MAPK signaling and inflammation, thus attenuating cell apoptosis and fibrosis in cardiomyocytes, which might be associated with the function of NHE1 receptor.
Emerging evidence have emphasized the role of chronic inflammation in the development of obesity-related cardiac dysfunction [30, 31]. Adiponectin secreted from expanded adipocyte tissue as well as the overwhelming circulating lipids create systemic inflammation. This reaction leads to subsequent dysregulation of insulin action in peripheral tissues, such as skeletal and cardiac muscle [32]. Specifically, the excessive lipid exerts cardiac lipotoxicity and triggers cardiac inflammation. Lipid overloads the circulating monocytes and induces the formation of monocytes into pro-inflammatory macrophages. The formed macrophages then infiltrates into cardiac tissue and in turn aggravates cardiac and systemic inflammation by secreting inflammatory cytokines [33, 34]. On the other hand, FFA such as PA, could directly activate detrimental pathways and induce the apoptosis and inflammatory response in cardiomyocytes [35, 36]. It is worth noting that these harmful effects of PA are attributed to the activation of MD2/TLR4/MyD88/NF-κB signaling [23, 37]. Our study also observed a significant cardiac inflammation characterized by macrophages infiltration, secretion of inflammatory cytokines in HFD-fed mice, which were mitigated by treatment with Dapa. However, we found that Dapa could not influence the enhanced interaction of MD2/TLR4/MyD88 complex, which indicated that Dapa may regulate cardiac inflammation in other ways.
MAPK signaling has been reported to be critically involved in regulating inflammation, insulin resistance, ROS production and cardiac injury. Numerous studies have demonstrated that inhibition of MAPK signaling attenuated obesity-related multiple injuries, such as airway damage [38], cardiac dysfunction [39], and renal dysfunction [40]. The vital role of JNK/MAPK has been raised recently. Activation of JNK results in the inflammatory response via promoting macrophages polarization. Besides, phosphorylation of IRS by JNK in cardiomyocytes leads to insulin resistance and disrupts normal cardiac metabolism, producing excessive ROS and further aggravating cardiac injury [41]. In our study, we conducted a transcriptome sequencing analysis and demonstrated that the protective effect of Dapa in cardiomyocytes was attributed to the inhibition of MAPK signaling and transcription of AP-1. A significant decrease of MAPK/AP-1 activation was also observed in HFD mice treated with Dapa.
The rare expression of SGLT2 in cardiomyocytes and hearts prompted us to seek other receptors mediating the function of Dapa. Recent studies indicated that Na+/H+ exchanger 1 (NHE1) might be the non-classical target of SGLT2 inhibitors. Jiang et al. reported that empagliflozin directly inhibited the activity of NHE1 in cardiomyocytes to regulate disrupted autosis, therapy leading to the reduced infarct size and improved cardiac function [17]. Another study carried out by Uthman et al. demonstrated that activity of NHE1 was inhibited by class of SGLT2 inhibitors, (including canagliflozin, Dapa and empagliflozin) with a significant decrease of [Na+]c [42]. However, Chung et al. found that neither empagliflozin nor other SGLT2i had effects on [Na+]i over a wide range of concentrations in primary rat ventricular cardiomyocytes [24]. In our study, we confirmed the direct binding of Dapa with NHE1. Using NHE1 overexpression and knockdown system, we further examined its role in the protection of Dapa. Interestingly, the protective effect of Dapa against PA-induced apoptosis and hypertrophy was all blunted after NHE1 overexpression or knockdown. We could even observe a worsened phenotype in cardiomyocytes when NHE1 was overexpressed, which was consistent with previous findings [17]. These results partially indicated that Dapa’s direct cardiac protection might be mediated by its inhibition of NHE1. Indeed, NHE1 was also regarded as a pivotal target in many cardiovascular diseases, including ischemia-reperfusion injury, maladaptive cardiac hypertrophy and heart failure [43]. Our results highlighted its function in saturated fatty acid-induced cardiomyopathy as well.
It is important to note that mutual regulation exists in the relationship between NHE1 and MAPK signaling. On the one hand, ERK, p38-MAPK, and JNK have all been implicated in the acute regulation of NHE1 activity after various stimuli [44, 45]. NHE1 was also reported to be directly phosphorylated by ERK effector p90 ribosomal S6 kinase (p90RSK) and p38-MAPK [46, 47]. On the other hand, NHE1 plays a central role in the regulation of MAPK activity after certain stimuli. Specific NHE1 inhibitor was able to inhibit the activation of ERK [48] and phosphorylation of p38-MAPK [49, 50]. The mechanisms underlying the NHE1 regulating MAPK might be associated with [Ca2+]i and subsequent ROS production [49, 50]. In our study, we also investigated the relationship between NHE1 and MAPK signaling. In consistent with the previous reports, overexpression of NHE1 resulted in a significant activation of MAPK signaling, especially for ERK1/2 pathway, while knockdown of NHE1 decreased the phosphorylation of MAPK signaling.
Despite the fact that Dapa directly protects cardiomyocytes in a NHE1/MAPK signaling, we observed an evident lowering lipid effect in vivo, which was also confirmed by Wang et al.’s findings [11]. Although we did not explore how Dapa affecting lipid metabolism and body weight, its potential function in liver, kidney and adipose tissue might be the reason. Using a western diet-induced obesity mouse model, Wang et al. found that Dapa was able to decrease lipid accumulation in hepatic and renal tissue, hence restoring normal organ function [11]. Xu et al. reported that SGLT2 inhibition by empagliflozin was able to enhance energy expenditure via upregulation of uncoupling protein 1 in brown fat and white adipose tissue, therapy producing more heat and promoting weight loss in HFD induce obese mice [12]. Another study carried out by Yokono et al. demonstrated that selective SGLT2 inhibitor ipragliflozin reduced the body fat mass by increasing fatty acid oxidation and upregulating energy expenditure through AMPK/SIRT1 pathway [13, 14]. Besides, JNK1/2 was reported to regulate lipid metabolism in hepatocyte in a PPAR-α/FGF-21 dependent way. Considering that Dapa could inhibit PA-induced JNK activation (current data) in cardiomyocytes, we proposed that it may also improve lipid metabolism in a JNK dependent way. However, all these hypotheses need further experiments to accurately validate.
There are also some limitations in our study. First of all, it is hard to estimate whether the lowering lipid effect or inhibition of cardiac inflammation by Dapa contributed more to the improvement of cardiac function in vivo. An exact lowering lipid drug, such as atorvastatin used as positive a control in the mouse study might be the solution and will be our future exploration. Besides, combined application of Dapa with NHE1 specific inhibitor or administration of Dapa in NHE1 knockout mice would further convince the role of NHE1 in the cardio-protection of Dapa. Future studies focused on combinatorial therapies would certainly help.
In summary, our studies provide evidence that Dapa significantly protects against obesity-related cardiac dysfunction and remodeling. The underlying mechanism might be at least associated with its direct protection in cardiomyocytes via a NHE1/MAPK/AP-1 pathway. Our data clearly highlights the potential clinical utility of SGLT2 inhibitors in the prevention of obesity-related cardiac dysfunction.
Supplementary information
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81600341), the Medical Health Science and Technology Project of Zhejiang Province (2021RC091), the Natural Science Foundation of Zhejiang Province (LQ15H020005), Wenzhou Science Technology Bureau Foundation (Y20190616), and Zhejiang Provincial Science and Technology Innovation Program (New Young Talent Program) for College Students (2021R413084).
Author contributions
PRS and WJH contributed to the literature search and study design. KL, NY and CXY carried out the cellular experiments. KL and WL carried out the analysis for RNA-Sequence and molecular docking. KL and JFQ carried out the experiments on gain and loss of NHE1. KL, NY and DYX carried out the in vivo experiments. KL, CXY and WL contributed to the statistical analysis. KL drafted the article. GL, PRS and WJH revised the article before submission. NY and WWZ carried out the experiments in revision stage. KL, SJY, GL, WJH and PRS revised the article in the revision stage.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Ke Lin, Na Yang.
Contributor Information
Wei-jian Huang, Email: weijianhuang69@126.com.
Pei-ren Shan, Email: prshan@126.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-022-00885-8.
References
- 1.Horwich TB, Fonarow GC, Clark AL. Obesity and the obesity paradox in heart failure. Prog Cardiovasc Dis. 2018;61:151–6. doi: 10.1016/j.pcad.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiwari S, Ndisang JF. The role of obesity in cardiomyopathy and nephropathy. Curr Pharmacol Des. 2014;20:1409–17. doi: 10.2174/13816128113199990562. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009;139:1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- 5.Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126:233–45. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol. 2020;598:2977–93. doi: 10.1113/JP276747. [DOI] [PubMed] [Google Scholar]
- 7.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383:1413–24. doi: 10.1056/NEJMoa2022190. [DOI] [PubMed] [Google Scholar]
- 8.McMurray JJV, DeMets DL, Inzucchi SE, Køber L, Kosiborod MN, Langkilde AM, et al. A trial to evaluate the effect of the sodium-glucose co-transporter 2 inhibitor dapagliflozin on morbidity and mortality in patients with heart failure and reduced left ventricular ejection fraction (DAPA-HF) Eur J Heart Fail. 2019;21:665–75. doi: 10.1002/ejhf.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N, Zhou H. SGLT2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy. Drug Des Devel Ther. 2020;14:4775–88. doi: 10.2147/DDDT.S269514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–6. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Luo Y, Wang X, Orlicky DJ, Myakala K, Yang P, et al. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents renal and liver disease in western diet induced obesity mice. Int J Mol Sci. 2018;19:137. doi: 10.3390/ijms19010137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, et al. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. 2017;20:137–49. doi: 10.1016/j.ebiom.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JY, Lee M, Lee JY, Bae J, Shin E, Lee YH, et al. Ipragliflozin, an SGLT2 Inhibitor, ameliorates high-fat diet-induced metabolic changes by upregulating energy expenditure through activation of the AMPK/SIRT1 pathway. Diabetes Metab J. 2021;45:921–32. doi: 10.4093/dmj.2020.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi: 10.1016/j.ejphar.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Uthman L, Baartscheer A, Schumacher CA, Fiolet JWT, Kuschma MC, Hollmann MW, et al. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front Physiol. 2018;9:1575. doi: 10.3389/fphys.2018.01575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Uthman L, Bakker D, Sari S, Chen S, Hollmann MW, et al. Empagliflozin decreases lactate generation in an nhe-1 dependent fashion and increases α-ketoglutarate synthesis from palmitate in type ii diabetic mouse hearts. Front Cardiovasc Med. 2020;7:592233. doi: 10.3389/fcvm.2020.592233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang K, Xu Y, Wang D, Chen F, Tu Z, Qian J, et al. Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell. 2021. 10.1007/s13238-020-00809-4. [DOI] [PMC free article] [PubMed]
- 18.Bayes-Genis A, Iborra-Egea O, Spitaleri G, Domingo M, Revuelta-López E, Codina P, et al. Decoding empagliflozin’s molecular mechanism of action in heart failure with preserved ejection fraction using artificial intelligence. Sci Rep. 2021;11:12025. doi: 10.1038/s41598-021-91546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene set knowledge discovery with Enrichr. Curr Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Y, Gao Y, Ilie A, Kim D, Boucher A, Li B, et al. Structure and mechanism of the human NHE1-CHP1 complex. Nat Commun. 2021;12:3474. doi: 10.1038/s41467-021-23496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyani CN, Plastira I, Sourij H, Hallström S, Schmidt A, Rainer PP, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. 2020;158:104870. doi: 10.1016/j.phrs.2020.104870. [DOI] [PubMed] [Google Scholar]
- 22.Nicolaides NC, Correa I, Casadevall C, Travali S, Soprano KJ, Calabretta B. The Jun family members, c-Jun and JunD, transactivate the human c-myb promoter via an Ap1-like element. J Biol Chem. 1992;267:19665–72. doi: 10.1016/S0021-9258(18)41826-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Qian Y, Fang Q, Zhong P, Li W, Wang L, et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat Commun. 2017;8:13997. doi: 10.1038/ncomms13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. Off-target effects of SGLT2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc Res. 2021;12:2794–806. doi: 10.1093/cvr/cvaa323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen SF, Darborg BV, Rentsch ML, Rasmussen M. Regulation of mitogen-activated protein kinase pathways by the plasma membrane Na+/H+ exchanger, NHE1. Arch Biochem Biophys. 2007;462:195–201. doi: 10.1016/j.abb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi T, Fukui T, Nakanishi N, Yamamoto S, Tomoyasu M, Osamura A, et al. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: comparison with sitagliptin. Cardiovasc Diabetol. 2017;16:8. doi: 10.1186/s12933-016-0491-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M, et al. Blockade of interleukin-6 signaling augments regulatory T-cell reconstitution and attenuates the severity of graft-versus-host disease. Blood. 2009;114:891–900. doi: 10.1182/blood-2009-01-197178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 29.Ebbesson SO, Voruganti VS, Higgins PB, Fabsitz RR, Ebbesson LO, Laston S, et al. Fatty acids linked to cardiovascular mortality are associated with risk factors. Int J Circumpolar Health. 2015;74:28055. doi: 10.3402/ijch.v74.28055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. 2016;118:1786–807. doi: 10.1161/CIRCRESAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen TS, Jansen KM. Impact of obesity-related inflammation on cardiac metabolism and function. J Lipid Atheroscler. 2021;10:8–23. doi: 10.12997/jla.2021.10.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:139239. doi: 10.1155/2013/139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ res. 2020;126:789–806. doi: 10.1161/CIRCRESAHA.119.312321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prieur X, Roszer T, Ricote M. Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim Biophys Acta. 2010;1801:327–37. doi: 10.1016/j.bbalip.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 35.He Y, Zhou L, Fan Z, Liu S, Fang W. Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by N‑acetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell Death Dis. 2018;9:568. doi: 10.1038/s41419-018-0593-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mangali S, Bhat A, Udumula MP, Dhar I, Sriram D, Dhar A. Inhibition of protein kinase R protects against palmitic acid-induced inflammation, oxidative stress, and apoptosis through the JNK/NF-kB/NLRP3 pathway in cultured H9c2 cardiomyocytes. J Cell Biochem. 2019;120:3651–63. doi: 10.1002/jcb.27643. [DOI] [PubMed] [Google Scholar]
- 37.Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012;53:2002–13. doi: 10.1194/jlr.D029546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal AK, Makhija S, Stahr N, Sandey M, Suryawanshi A, Saxena A, et al. Dendritic cell-restricted progenitors contribute to obesity-associated airway inflammation via ADAM17-p38 MAPK-dependent pathway. Front Immunol. 2020;11:363. doi: 10.3389/fimmu.2020.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S, Gu J, Xu Z, Zhang Z, Bai T, Xu J, et al. Zinc rescues obesity-induced cardiac hypertrophy via stimulating metallothionein to suppress oxidative stress-activated BCL10/CARD9/p38 MAPK pathway. J Cell Mol Med. 2017;21:1182–92. doi: 10.1111/jcmm.13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo M, Luo P, Zhang Z, Payne K, Watson S, Wu H, et al. Zinc delays the progression of obesity-related glomerulopathy in mice via down-regulating P38 MAPK-mediated inflammation. Obesity (Silver Spring) 2016;24:1244–56. doi: 10.1002/oby.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6:174–84. doi: 10.1016/j.molmet.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia. 2018;61:722–6. doi: 10.1007/s00125-017-4509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeves AM, Ennis IL. Na+/H+ exchanger and cardiac hypertrophy. Hipertens Riesgo Vasc. 2020;37:22–32. doi: 10.1016/j.hipert.2019.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Goss GG, Jiang L, Vandorpe DH, Kieller D, Chernova MN, Robertson M, et al. Role of JNK in hypertonic activation of Cl−-dependent Na+/H+ exchange in Xenopus oocytes. Am J Physiol Cell Physiol. 2001;281:C1978–90. doi: 10.1152/ajpcell.2001.281.6.C1978. [DOI] [PubMed] [Google Scholar]
- 45.Pederson SF, Varming C, Christensen ST, Hoffmann EK. Mechanisms of activation of NHE by cell shrinkage and by calyculin A in Ehrlich ascites tumor cells. J Membr Biol. 2002;189:67–81. doi: 10.1007/s00232-001-0190-2. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, et al. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem. 1999;274:20206–14. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- 47.Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K, et al. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol. 2001;21:7545–57. doi: 10.1128/MCB.21.22.7545-7557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamazaki T, Komuro I, Kudoh S, Zou Y, Nagai R, Aikawa R, et al. Role of ion channels and exchangers in mechanical stretch-induced cardiomyocyte hypertrophy. Circ Res. 1998;82:430–7. doi: 10.1161/01.RES.82.4.430. [DOI] [PubMed] [Google Scholar]
- 49.Aker S, Snabaitis AK, Konietzka I, Van De Sand A, Böngler K, Avkiran M, et al. Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc Res. 2004;63:273–82. doi: 10.1016/j.cardiores.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Javadov S, Baetz D, Rajapurohitam V, Zeidan A, Kirshenbaum LA, Karmazyn M. Anti-hypertrophic effect of Na+/H+ exchanger isoform 1 inhibition is mediated by reduced mitogen-activated protein kinase activation secondary to improved mitochondrial integrity and decreased generation of mitochondrial-derived reactive oxygen species. J Pharmacol Exp Ther. 2006;317:1036–43. doi: 10.1124/jpet.105.100107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.