

Abstract
Heated tobacco products (HTP) are novel nicotine delivery products with limited toxicological data. HTP uses heating instead of combustion to generate aerosol (HTP-smoke). Physiologically relevant human bronchial and alveolar lung mucosa models developed at air–liquid interface were exposed to HTP-smoke to assess broad toxicological response (n = 6–7; ISO puffing regimen; compared to sham; non-parametric statistical analysis; significance: p < 0.05). Elevated levels of total cellular reactive oxygen species, stress responsive nuclear factor kappa-B, and DNA damage markers [8-hydroxy-2′-deoxyguanosine, phosphorylated histone H2AX, cleaved poly-(ADP-Ribose) polymerase] were detected in HTP-smoke exposed bronchial and/or alveolar models. RNA sequencing detected differential regulation of 724 genes in the bronchial- and 121 genes in the alveolar model following HTP-smoke exposure (cut off: p ≤ 0.01; fold change: ≥ 2). Common enriched pathways included estrogen biosynthesis, ferroptosis, superoxide radical degradation, xenobiotics, and α-tocopherol degradation. Secreted levels of interleukin (IL)1ꞵ and IL8 increased in the bronchial model whereas in the alveolar model, interferon-γ and IL4 increased and IL13 decreased following HTP-smoke exposure. Increased lipid peroxidation was detected in HTP-smoke exposed bronchial and alveolar models which was inhibited by ferrostatin-1. The findings form a basis to perform independent risk assessment studies on different flavours of HTP using different puffing topography and corresponding chemical characterization.
Subject terms: Risk factors, Respiratory tract diseases
Introduction
Heated tobacco products (HTP) are new and emerging tobacco products (first launched in 2014) that allow the user to inhale nicotine by heating the tobacco (350 °C) instead of burning it at high temperature (> 900 °C as in conventional cigarettes)1–4. HTP products are currently available in about 50 countries1–5. The total sale of HTP products is forecasted to reach nearly 68 billion USD by 2027, a seven-fold increase from 20206. HTP products are increasingly used, especially by the youth (15–24 years), current smokers and former smokers7–9. The tobacco industry claims harm reduction by HTP use compared to conventional cigarettes (CC). This remains questionable at the present state of knowledge as long-term toxicity data are missing. The operational technique (heating instead of combustion) for nicotine delivery used in HTPs also enable the tobacco industry to bypass the tobacco directives10. Most of the HTP research data currently available were obtained with tobacco heating system 2.2 which has been marketed as “IQOS” (I Quit Ordinary Smoking) by Philip Morris International (PMI)4, 5. The IQOS is operated by a battery-powered heating device with a heating blade that is inserted into a HTP specific tobacco unit (heatsticks or HTP sticks) containing processed tobacco leaf4, 5. The HTP sticks comes in different flavours (e.g. menthol, nutty, intense tobacco, mint). Upon heating, the stick releases a nicotine containing aerosol (HTP-smoke) that is inhaled by the user. The current scientific literature on HTP is dominated by tobacco industry sponsored research5, 11, 12. Till date only few independent studies on HTP have been performed and reports of short-term pathophysiological effects are reported13. Therefore, there is an urgent need of independent and comprehensive risk assessment studies on HTPs.
In general, the content of several harmful and potentially harmful constituents (HPHC) including nicotine, tobacco specific nitrosamines, tar, carbon monoxide, hydrogen cyanide, ammonia, phenol, volatile organic compounds, polycyclic aromatic hydrocarbons, aromatic amines, reactive oxygen species (ROS), carbonyls4, 14–19 are considerably lower in HTP-smoke compared to CC-smoke. However, some studies reported comparable nicotine and tar content, and percentage of free base in HTP-smoke and CC-smoke18. Highly toxic formaldehyde cyanohydrin is detected in HTP-smoke20. Almost three times higher levels of potentially carcinogenic acenapthene is also reported in HTP-smoke than in CC-smoke21. Evidence of pyrolysis products, propylene glycol and glycerin have been reported in HTP-smoke18, 19. Examination of PMI’s IQOS emission data12 on the US Food and Drug Administration’s (FDA) HPHC list revealed 56 of 93 constituents to be higher in HTP-smoke compared to CC-smoke. PMI reported levels of only 40 of 93 of these HPHC on FDA list. Among those not reported, fifteen HPHCs were two-fold or more and seven were more than ten-fold higher in HTP-smoke than in CC-smoke12. Altogether, these data show that, compared to CC-smokers, HTP-smokers inhale lower amounts of some toxicants but comparable or higher amounts of other toxicants11. Some studies have shown that the total particulate matter present in the mainstream HTP-smoke (range: 12.9–55.8 mg/heat stick) to be higher than that of CC-smoke (range: 9.8–37.7 mg/cigarette) and is dependent on the flavour as well as puffing regime14–19. The dimensional and volatility characterization of four different flavours of mainstream HTP-smoke by Pacitto et al.22 reported the median values of the total particle concentration (volatile and non-volatile) to range between 7.04 × 107 to 9.67 × 107 particles/cm3. The corresponding particle number distribution modes were about 100 nm. The volatility analysis further demonstrated that the particle number distribution mode decreases down to approximately 20 nm (at 300 °C)22. The study also estimated that the dose received by HTP-smokers in terms of non-volatile amount of particle surface area is 1–2 mm2 per puff (10–20 mm2 per heat-stick)22.
Emerging evidence from clinical studies assessing the effects on smokers and COPD patients switching to HTP are insufficient to draw any conclusions13, 23–26. However, reports on HTP use associated acute eosinophilic pneumonia are alarming27–29. Inflammatory cell recruitment, cytotoxicity, secretion of pro-inflammatory cytokines, and oxidative stress response have been shown as the primary pulmonary effects of exposure to HTP-smoke23, 30. Long-term chronic inhalation exposure of HTP-smoke in female rats resulted in significantly increased lung weight, bronchoalveolar lavage inflammatory cell recruitment, epithelial hyperplasia and metaplasia, and elevated levels of bronchoalveolar lavage inflammatory markers23, 24. Short term inhalation exposure to HTP-smoke (Health Canada Intense puffing regimen)31 in mice resulted in increased albumin in the bronchoalveolar lavage fluid indicating lung epithelial barrier leakage and infiltration of leukocytes along with increased levels of pro-inflammatory cytokines in the lung32. In vitro studies using human bronchial epithelial cells demonstrated that HTP-smoke resulted in oxidative stress and pro-inflammatory response30, 33. Higher cytotoxicity was also reported in human bronchial epithelial cells following HTP-smoke exposure compared to corresponding sham23, 30–33. Moreover, most of the HTP toxicological data was compared to CC data to showcase harm reduction. However, this does not prove the safety of HTP products, as they may cause severe adverse health effects even if the potency is lower compared to CC-smoke.
The current state of knowledge on HTP-smoke mediated pulmonary toxicity is limited. There is a lack of knowledge on the toxicological response of HTP-smoke exposure in general and particularly from different lung regions such as the conducting (bronchial) and respiratory (alveolar) zones. The cellular architecture and the corresponding molecular signature of the bronchial and alveolar of the lung are distinctly different and therefore the toxicological responses may vary as well in these lung regions. It also needs to be ascertained if and/or how HTP-smoke exposure mediated toxicity is flavour and/or puffing topography (standard or intense) dependent. In this work we hypothesized that there may be common as well as different toxicological response to HTP-smoke exposure in the bronchial and alveolar lung regions.
To address some of the above aspects, we aimed to assess the toxicological response of HTP-smoke (single flavour) using the International Organization for Standardization (ISO) puffing regimen in our established physiologically relevant bronchial (bro) and alveolar (alv) lung mucosa models developed at air–liquid interface (ALI)34, 35. The broad toxicological endpoints identified for assessment were (1) oxidative stress, (2) DNA damage, (3) altered transcript expression, and (4) secretion of pro-inflammatory cytokines. Pathway enrichment analysis of the altered gene expression was performed to identify unique as well as common candidate biological mechanisms that may drive HTP-smoke mediated toxicity in the bronchial and alveolar lung regions. The bronchial mucosa models (bro-ALI) were developed using human primary bronchial epithelial cells (PBEC) and the alveolar model (alv-ALI) was developed using representative human type II pneumocytes. Our study provides a comprehensive insight into the molecular pulmonary toxicity of HTP-smoke in two (bronchial and alveolar) different lung regions.
Methods
An overview of the experimental design, HTP-instrument, HTP stick, and puffing regimen used for performing HTP-smoke exposure on bro-ALI and alv-ALI models together with the different analyses and assays is provided in Fig. 1.
Figure 1.

Schematic presentation of the overall experimental design outlining the exposure regimen and endpoints. ALI: air–liquid interface; alv-ALI: alveolar mucosa model at ALI; bro-ALI: bronchial mucosa model at ALI; h: hours; H441: NCI-H441 (ATCC HTB-174) cell line; HTP: heated tobacco product; IFNγ: interferon gamma; IL: interleukin; IQOS: I quit ordinary smoking; LDH: lactate dehydrogenase; MDA: malondialdehyde; NFkB: nuclear factor kappa-light-chain-enhancer of activated B cells; PARP: cleaved poly [ADP-Ribose] polymerase; PBEC: human primary bronchial epithelial cells; TNFα: tumor necrosis factor alpha; γH2AX: phosphorylated histone H2AX.
HTP-smoke generation
The HTP-smoke was generated using IQOS 3 Duo HTP instrument and HEETS-Kelly Selection (flavour: intense menthol with citrus) HTP sticks. A one-day exposure experiment consisted of 6 puffing sessions conducted with 1 h intervals. Each session consisted of 10 puffs according to the International Organization for Standardization (ISO) puffing regimen (puff volume: 35 mL, puff duration: 2 s, puff frequency: 1 puff per min)36. There was 1 h (h) interval in between each session. Therefore, each bro-ALI and alv-ALI model was exposed to 60 puffs in total.
HTP-smoke exposure
HTP-smoke exposure experiments were carried out using the exposure system previously illustrated35. Nicotine quantification (14.1 ± 1.7 µg/mL; %CV: 11.8) was performed using liquid chromatography-mass spectrometry (LC–MS/MS) analyses to inspect its distribution across all the wells in the 12 well cell culture plate containing 1 mL cell culture media in each well following 1 puffing session (“Supplementary material S1”). An air-tight pre-heated glass syringe was used to repeatedly collect 35 mL of HTP-smoke and inject it in a 3 L desiccator glass jar (37 °C and 60% humidity). The bro-ALI and alv-ALI models were exposed to HTP-smoke or clean air (sham) for 15 min (min) under identical conditions. Thereafter, the lung models were transferred to a cell incubator (37 °C, 60% humidity and 5% CO2) for 1 h until next exposure session. At the completion of repeated exposures (i.e. 6 puffing sessions), the lung models were incubated for 24 h prior to collection of basal media and cell inserts. All HTP-exposed samples were compared to their corresponding sham exposed (clean air) samples (n = 6–7 independent experiments per exposure condition). The replicates were randomly distributed in the plates and experiments performed on different days were used for both bro-ALI (developed from one donor; different vials) and alv-ALI (developed from different cell vials) for the different assays.
Bronchial and alveolar lung mucosal models
Bronchial model
The bro-ALI model was developed using PBEC harvested from macroscopically normal bronchial tissue obtained from one donor in connection with lobectomy following written and informed consent. The protocol was approved by the Swedish Ethical Review Authority (Institutional ethic committee reference number: 99–357; approved on 10th January 2000). All methods were performed in accordance with the relevant guidelines and regulations. The detailed protocol and details of cellular differentiation (Club cells, goblet cells, basal cells, ciliated cells, etc.) of the bro-ALI model has been described previously and the model has been in several studies34, 35, 37–41.
Alveolar model
The alv-ALI model was developed using NCI-H441 (ATCC HTB-174; derived from the pericardial fluid of a patient with papillary adenocarcinoma of the lung) cell line and the detailed protocol and model characteristics have been described recently35, 37. The NCI-H441 cells, representative of human type II pneumocytes, express constitutively the mRNA and protein of the major surfactant apo-protein. NCI-H441 cells were co-cultured with HULEC-5a (ATCC CRL-3244) representative of human lung microvascular endothelial cells for this purpose. The alv-ALI model characterization included light microscopy, confocal microscopy, transmission electron microscopy, and transepithelial electrical resistance measurement35, 37. Morphological characterization of the alv-ALI model demonstrated the presence of tight junction protein 1, lamellar bodies, surfactant protein C, microvilli, lipid bodies, desmosome, and tight junctions35, 37.
Assessment of HTP-smoke related pulmonary response
Cytotoxicity assessment
Membrane integrity-based cell viability assays were used for cytotoxicity assessment of bro-ALI (n = 6) and alv-ALI (n = 6) models following 24 h after the HTP-smoke exposure. Colorimetric lactate dehydrogenase (LDH; Thermo Fisher scientific Rockford, lL, US, catalog # 88953) assay and propidium iodide (PI; BD bioscience, San Jose, CA, US, catalog # 556463) staining were used according to manufacturer’s instruction and as described previously35, 37. The LDH assay was measured using BioTek 800 TS absorbance reader (Santa Clara, CA, US) and PI assay was performed using flow cytometry (BD LSRFortessa cell analyzer, BD bioscience, San Jose, CA, US). The flow cytometric data was analyzed using FlowJo software-7.6.1 (BD bioscience, San Jose, CA, US). Data are presented as percentage positive PI cells and interquartile ranges. LDH assay is shown as medians of absorbance (450 nm) and interquartile ranges. The same instruments and software were used for all subsequent enzyme linked immune sorbent assays (ELISA) and flow cytometric analysis mentioned hereafter.
Total ROS and nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB)
Total cellular ROS and NFkB was measured in both bro-ALI (n = 6) and alv-ALI (n = 6) following 2 h post HTP-smoke exposure using flow cytometry according to manufacturer’s instruction. Cells were stained with CellROX green reagent (ThermoFisher Scientific, Rockford, lL, US, Catalog #: C10444) for 30 min in incubator. Following incubation, cells were washed, collected and ROS were measured as previously described35. Expression of the stress responsive transcription factor NFkB was assessed following staining of the cells using NFkB p65 subunit kit according to manufacturer’s instruction (BD biosciences, San Jose, CA, US, catalog # 560335). Shortly, cells were stained with PE-Cy7 conjugated anti-NFkB p65 for 30 min, followed by washing, and measurement by flow cytometry. Mean fluorescent intensity (MFI) represents the level of total ROS and NFkB.
Assessment of DNA damage
DNA damage was assessed by measuring the concentration of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the basal media of HTP-smoke exposed bro-ALI (n = 6) and alv-ALI (n = 6) using a commercially available competitive enzyme linked immune sorbent assay (ELISA) kit (ThermoFisher Scientific; Rockford, lL, US, catalog # EIADNAD) according to manufacturer’s instructions. Further, cellular phosphorylated histone H2AX (γH2AX) and cleaved Poly [ADP-Ribose] Polymerase (PARP) levels as markers of DNA damage and repair were also assessed by flow cytometry in HTP-exposed bro-ALI and alv-ALI models 24 h post-exposure according to manufacturer’s instruction by flow cytometry. The cells were stained with allophycocyanin (APC) conjugated anti- γH2AX and PE conjugated anti-PARP antibodies for 30 min, and fluorescence intensity measured as described above by flow cytometry.
Transcriptomic analysis
Transcriptomic analysis was performed using the UPX 3′ RNA sequencing technology (RNAseq; Qiagen Genomic Services, Hilden, Germany) as recently described37. To determine the differentially expressed genes following HTP-smoke exposure in both bro-ALI (n = 7) and alv-ALI (n = 6), cells were collected in Qiagen RLT buffer 24 h post exposure (Qiagen, Hilden, Germany, catalog # 74104), snap frozen, and dispatched in dry ice to the service laboratory according to the service provider’s instructions. For alv-ALI, the apical layer containing type II pneumocytes were collected. A raw p value ≤ 0.01 and fold change ≥ 2 was set to select the differentially expressed genes. Gene symbols were obtained from Ensembl (Gene2ensembl; BioMart) and NCBI (Homo_sapiens.gene_info.gz). Heatmaps showing the top 25 upregulated and 25 down regulated genes (by fold change) were generated in R42. Ensembl genes without gene symbol annotation were omitted from heatmaps. RNAseq data is deposited at the Gene Expression Omnibus database at NCBI [GSE198082; https://www.ncbi.nlm.nih.gov/geo/; accessed on 14th March 2022].
Pathway and enrichment analysis
For the biological interpretation of the differentially regulated genes, canonical pathway and upstream regulator analyses were performed using the QIAGEN’s Ingenuity Pathway Analysis software (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity, content version 70750971, Release Date 2021-10-22). Significant terms were selected using Fisher`s Exact Test p-values (p < 0.05 for pathways and p < 0.01 for upstream regulators), z-scores ≥ 2 indicate activation, and z-scores ≤ − 2 indicate inhibition.
Secreted cytokine concentration
Concentrations of pro-inflammatory cytokines interferon gamma (IFNγ), interleukin (IL) 1β, IL2, IL4, IL6, IL8, IL10, IL12, IL13, and tumor necrosis factor alpha (TNFα) were measured in the basal media of the bro-ALI (n = 6) and alv-ALI (n = 6) following 24 h post-exposure as described previously35, 37. IL8 was measured using ELISA (R & D Systems, Minneapolis, MN, US, Catalog # DY208) and the remaining cytokines were measured using the V-plex immunoassay platform of Meso Scale Discovery Inc (Rockville, MD, US) at the Clinical Biomarkers facility, Science for Life Laboratory, Uppsala University, Sweden. Basal media of the samples used for RNAseq analysis were used for protein secretion analysis.
Quantitative real time polymerase chain reaction (qRT-PCR) was performed for selected pro-inflammatory genes (IFNG, IL1B, IL4, IL8, and IL13) showing significantly different secreted protein levels in the basal medium to additionally assess their transcript expression as previously described35. Actin beta (ACTB) was used as the reference control.
Lipid peroxidation
Concentration of malondialdehyde (MDA) was measured to assess lipid peroxidation43 using cell lysate of HTP-smoke exposed bro-ALI (n = 6) and alv-ALI (n = 6) models 24 h post-exposure using a commercially available colorimetric assay and according to manufacturer’s instruction. (Sigma Aldrich, St Lois, MO, US, catalog # MAK085). Further, BODIPY™ 581/591 C11 (Lipid Peroxidation Sensor) (ThermoFisher Scientific; Rockford, lL, US, Catalog #: D3861) was used to assess lipid peroxidation44 in HTP-exposed bro-ALI and alv-ALI 24 h post exposure using flow cytometry. Ferrostatin-1 (10 nM as working solution; Sigma Aldrich St Lois, MO, US, catalog # SML0583) was used as an inhibitor lipid peroxidation in this assay which in turn also acts as a ferroptosis inhibitor44, 45 The cells were incubated with 2 µM BODIPY for 30 min, washed, and fluorescence intensity was measured by flow cytometry as described above.
Statistics
The results (flow cytometry, protein concentration, ELISA, qRT-PCR) were expressed as medians and interquartile ranges (25th–75th percentiles). Both bro-ALI and alv-ALI models are well-differentiated tissue-like models, which contain multiple layers of cells including different cell types of unique distribution. Hence, in this study, every model (ie. both bro-ALI and alv-ALI) are considered as a unique in vivo-like in vitro model with its own distribution of different cell types and the number of cells present might differ. Therefore, differences between treatments were examined by non-parametric statistical analysis (Wilcoxon signed rank test or Friedman test followed by Wilcoxon signed rank test, as appropriate) using GraphPad Prism (9.3.1) software (LaJolla, CA, US). A p value < 0.05 was considered as significant. The statistic method used in the RNA sequencing and pathway analysis are described in the respective sections.
Ethical approval
Institutional Review Board Statement: All procedures performed for the in vitro study were in accordance with the approval of the Swedish Ethical Review Authority (Institutional ethic committee reference number 99-357; Dated: 10.01. 2000.).
Informed consent statement
All subjects were enrolled after informed consent process for creation of the primary cell bank.
Results
Cytotoxicity
The exposure regime used in this study was not cytotoxic (> 87% cell viability) in the bro-ALI and alv-ALI models as indicated by PI staining and LDH assays (Supplementary Fig. S1). More specifically, we saw in case of both bro-ALI (0.4%; p = 0.03) and alv-ALI (7.6%; p = 0.03) a slight increase of PI positive cells following HTP-smoke exposure compared to sham. Released LDH levels were not different among HTP exposed bro-ALI compared to sham. In case of alv-ALI, released LDH levels were increased (p = 0.03) consistent to PI staining in the HTP exposed samples compared to sham. Therefore, the described exposure regime was suitable to proceed with all further experiments.
Oxidative stress
Increased total cellular ROS was detected in both bro-ALI (51%; p = 0.03; Fig. 2a) and alv-ALI (17%; p = 0.03; Fig. 2b) following HTP exposure compared to corresponding sham. as were the levels of NFkB p65 subunit in bro-ALI (61%; p = 0.03; Fig. 2c) and alv-ALI (25%; p = 06; Fig. 2d) following HTP-smoke exposure compared to sham.
Figure 2.
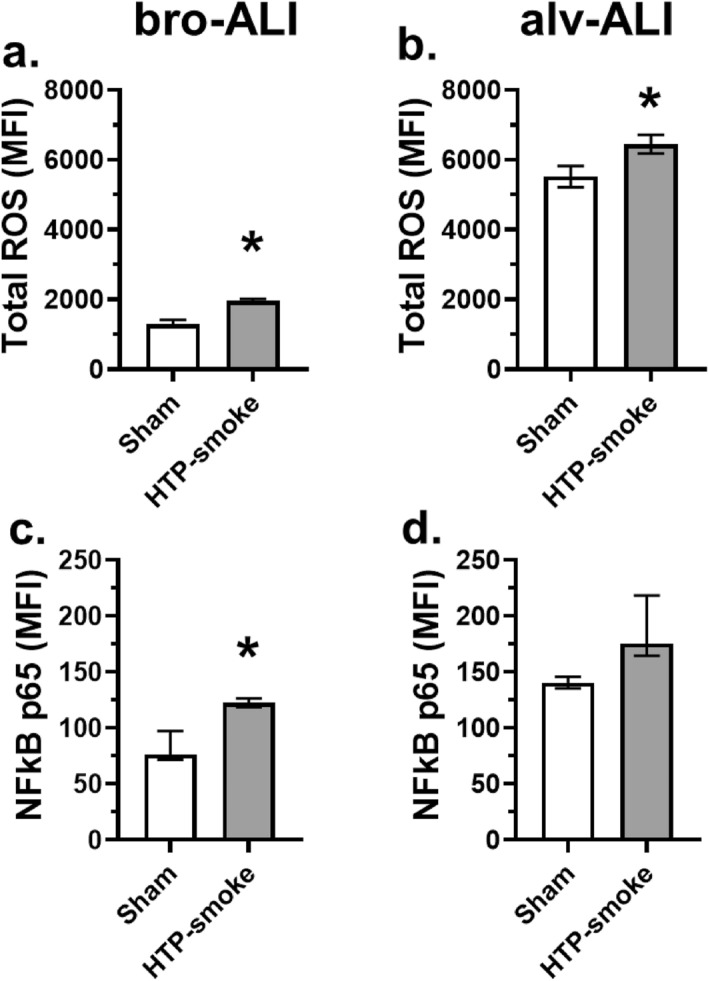
Assessment of oxidative stress response by measurement of (a, b) total cellular reactive oxygen species (ROS) and (c, d) expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) p65 subunit by flow cytometry in sham exposed and HTP-smoke exposed bro-ALI and alv-ALI models. Data are shown as medians and interquartile ranges. n = 6 per exposure condition; non-parametric statistical analysis (Wilcoxon signed rank test), *p < 0.05. ALI: air–liquid interface; alv-ALI: alveolar mucosa model at ALI; bro-ALI: bronchial mucosa model at ALI; HTP: heated tobacco product, MFI: mean fluorescent intensity.
DNA damage
The DNA damage marker 8OH-dG was significantly increased in the basal media of both models (bro-ALI: 65%; p = 0.03; Fig. 3a; and alv-ALI: 35%; p = 0.03; Fig. 3b) following HTP-smoke exposure compared to corresponding sham. The cellular expression of γH2AX was significantly increased in bro-ALI (53%; p = 0.03; Fig. 3c) but not in alv-ALI (p = 0.09; Fig. 3d) following HTP exposure. Cleaved PARP levels were significantly increased in both bro-ALI (33%; p = 0.03; Fig. 3e) and alv-ALI (44%; p = 0.03; Fig. 3f) following HTP-smoke exposure.
Figure 3.

Assessment of the levels of DNA damage markers (a, b) 8-hydroxy-2′-deoxyguanosine (8-OHdG; pg/mL), (c, d) cellular phosphorylated histone H2AX (γH2AX) and (e, f) cleaved poly [ADP-Ribose] polymerase (PARP) levels in sham exposed and HTP-smoke exposed bro-ALI and alv-ALI models. Data are shown as medians and interquartile ranges. n = 6 per exposure condition; non-parametric statistical analysis (Wilcoxon signed rank test), *p < 0.05. ALI: air–liquid interface; alv-ALI: alveolar mucosa model at ALI; bro-ALI: bronchial mucosa model at ALI; HTP: heated tobacco product; MFI: mean fluorescent intensity.
Transcriptomic alterations and proinflammatory cytokine secretion
Bronchial model
A total of 724 genes were differentially regulated (424 upregulated and 300 down regulated; n = 7; p ≤ 0.01; fold change ≥ 2) in the bro-ALI model post 24 h exposure to HTP-smoke compared to sham (Supplementary Table S1). Figure 4 shows a heat map of the top 25 upregulated and 25 down regulated genes. Enrichment analysis using the 724 differentially regulated genes identified 62 significantly enriched canonical pathways (Supplementary Table S2). Selected enriched canonical pathways are given in Table 1 and include interferon signaling, liver X receptor (LXR)/retinoid X receptor (RXR) activation, glucocorticoid receptor signaling, p38 mitogen-activated protein kinase (MAPK) signaling, IL13 signaling pathway, Toll like receptor signaling, peroxisome proliferator-activated receptors (PPAR) signaling, aryl hydrocarbon receptor (AHR) signaling, IL17 signaling, and airway pathology in chronic obstructive pulmonary disease (COPD). The secreted levels of proinflammatory cytokines (Fig. 5a–j) IL1ꞵ (p < 0.0001; Fig. 5b) and IL8 (p = 0.04; Fig. 5f) were increased in bro-ALI model (n = 6). None of the other examined cytokines was significantly altered by exposure to HTP-smoke in the bro-ALI model (Fig. 5).
Figure 4.
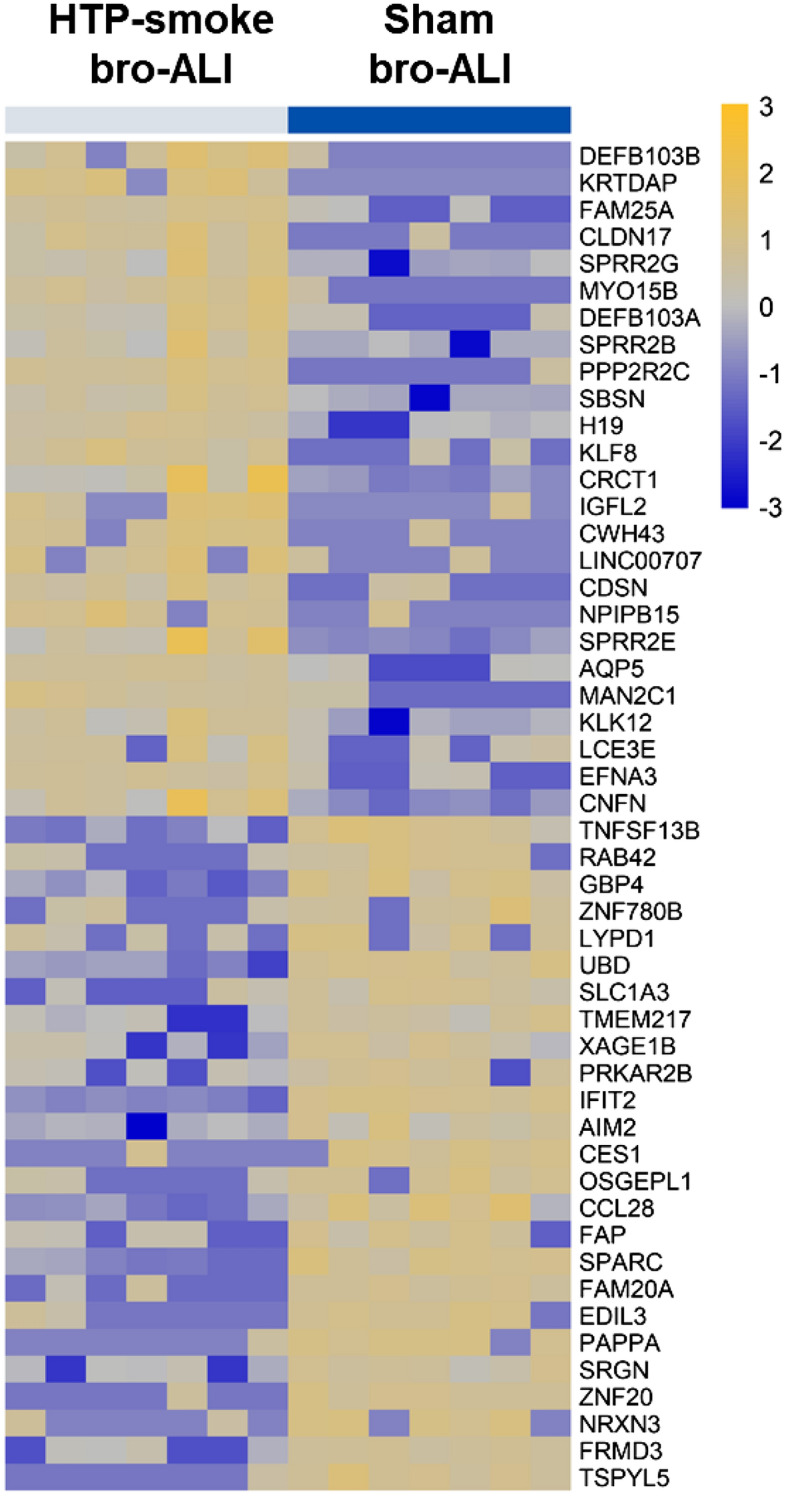
Heatmap of the top 25 up regulated and 25 down regulated genes in the bro-ALI model following exposure to HTP-smoke. n = 7 per exposure condition; significantly (raw p < 0.01) regulated genes with the highest fold changes are shown. Genes were ordered by fold-change (HTP-smoke vs Sham) and relative gene expression values are shown across samples (z-scales to mean expression per row). A complete list of the 724 differentially regulated genes is provided in Supplementary Table S1. bro-ALI: bronchial mucosa model at air–liquid interface; HTP: heated tobacco product.
Table 1.
Shown are selected enriched canonical pathways (Fisher’s Exact test p-value < 0.05) associated with the sets of differentially regulated genes in the bronchial (724 genes) and alveolar (121 genes) mucosa models developed at air liquid interface (bro-ALI and alv-ALI) post 24 h exposure to heated tobacco product (HTP)-smoke compared to sham. Terms observed with both datasets are italicized.
| Canonical pathways | p-value | Activation | Molecules |
|---|---|---|---|
| Bronchial mucosa model (bro-ALI) | |||
| Interferon signaling | 5.25E − 06 | Inhibited | IFI35, IFIT1, IFIT3, IFITM2, IRF1, ISG15, MX1, STAT2 |
| LXR/RXR activation | 8.51E − 06 | Inhibited | ABCG1, APOD, APOL1, IL1R1, IL1RN, IL33, IL36RN, PON3, S100A8, SAA1, SAA2, SAA4, TF, TLR3 |
| Glucocorticoid receptor signaling | 2.14E − 04 | – | CDKN1A, CEBPA, DUSP1, GABPB2, HLA-B, HLA-C, HLA-DPA1, HLA-F, IL18R1, IL1R1, IL1RN, KRT16, KRT40, KRT6A, KRT6B, KRT6C, KRT78, KRT80, MMP13, MT-ATP6, MT-ND2, MT-ND4L, MT-ND5, MT-ND6, PIK3R1, RARB, SERPINE1, SHC1, TAF15, TGFB3, TGFBR2, VIPR1 |
| Ferroptosis signaling pathway | 1.17E − 03 | – | ALOX15B, ALOXE3, ANGPTL4, CDKN1A, CHAC1, FTL, H2BC5, NOX1, SLC7A11, TF, TXNRD1 |
| p38 MAPK signaling | 1.95E − 03 | – | CREB3L1, DUSP1, IL1R1, IL1RN, IL33, IL36RN, IRAK3, MKNK2, TGFB3, TGFBR2 |
| IL-13 signaling pathway | 5.13E − 03 | – | ALOX15B, CXCL6, DEFB103A/DEFB103B, DEFB4A/DEFB4B, DUSP1, HBEGF, IL33, PIK3R1, TGFB3 |
| Toll-like receptor signaling | 6.03E − 03 | – | IL1RN, IL33, IL36RN, IRAK3, TICAM1, TLR3, UBD |
| PPAR signaling | 1.00E − 02 | – | CITED2, IL1R1, IL1RN, IL33, IL36RN, PDGFB, PPARD, SHC1 |
| Aryl hydrocarbon receptor signaling | 1.38E − 02 | – | ALDH3A1, ALDH3B2, CCNA1, CDKN1A, CYP1A1, CYP1B1, NCOA7, NQO1, RARB, TGFB3 |
| IL-17 signaling | 1.58E − 02 | Inhibited | CLCF1, CXCL5, DEFB103A/DEFB103B, DEFB4A/DEFB4B, IL33, MMP13, PIK3R1, TGFB3, TNFSF10, TNFSF13B, TNFSF15 |
| Superoxide radicals degradation | 1.95E − 02 | – | CYGB, NQO1 |
| α-Tocopherol degradation | 3.02E − 02 | – | CYP4F11, CYP4F3 |
| Xenobiotic metabolism signaling | 3.31E − 02 | – | ALDH3A1, ALDH3B2, CES1, CES2, CHST11, CITED2, CYP1A1, CYP1B1, FTL, NQO1, PIK3R1, PPP2R2C, SULT1E1, SULT2B1 |
| Estrogen biosynthesis | 3.63E − 02 | – | AKR1B10, AKR1C1/AKR1C2, CYP1A1, CYP1B1 |
| Airway pathology in chronic obstructive pulmonary disease (COPD) | 4.68E − 02 | – | APOD, CLCF1, IL33, TGFB3, TNFSF10, TNFSF13B, TNFSF15 |
| Alveolar mucosa model (alv-ALI) | |||
| Estrogen biosynthesis | 4.17E − 05 | – | AKR1C1/AKR1C2, AKR1C3, CYP1B1, HSD17B7 |
| Glutathione biosynthesis | 5.50E − 05 | – | GCLC, GCLM |
| Ferroptosis signaling pathway | 2.51E − 03 | – | FTH1, FTL, GCLC, H2AC20 |
| Bile acid biosynthesis, neutral pathway | 2.63E − 03 | – | AKR1C1/AKR1C2, AKR1C3 |
| NRF2-mediated oxidative stress response | 3.55E − 03 | Activated | FTH1, FTL, GCLC, GCLM, NQO1 |
| Retinoate biosynthesis I | 1.41E − 02 | – | AKR1C1/AKR1C2, AKR1C3 |
| Xenobiotic metabolism general signaling pathway | 2.34E − 02 | – | FTL, GCLC, NQO1 |
| Superoxide radicals degradation | 3.39E − 02 | – | NQO1 |
| Xenobiotic metabolism signaling | 3.63E − 02 | – | CYP1B1, FTL, GCLC, NQO1 |
| α-Tocopherol degradation | 4.17E − 02 | – | CYP4F3 |
Figure 5.

Concentration of secreted proinflammatory cytokines in the basal media of sham exposed and HTP-smoke exposed bro-ALI model. IFNγ: interferon gamma; IL: interleukin; TNFα: tumor necrosis factor alpha. Data are shown as medians and interquartile ranges. n = 6 per exposure condition; non-parametric statistical analysis (Wilcoxon signed rank test); *Significance: p < 0.05. bro-ALI: bronchial mucosa model at air–liquid interface; HTP: heated tobacco product.
Alveolar model
A total of 121 genes were differentially regulated (60 upregulated and 61 down regulated; n = 6; p < 0.01; fold change ≥ 2) in the alv-ALI model 24 h after exposure to HTP-smoke compared to sham (Supplementary Table S3). Figure 6 shows a heat map of the top 25 upregulated and 25 down regulated genes. Enrichment analysis using the 121 differentially regulated genes identified 21 significantly enriched canonical pathways (Supplementary Table S4). Selected enriched canonical pathways are provided in Table 1 and include glutathione biosynthesis, bile acid biosynthesis-neutral pathway, nuclear factor erythroid 2-related factor 2 (NRF2) mediated oxidative stress response, retinoate biosynthesis I, and xenobiotic metabolism general signaling pathway. The secretion of of pro-inflammatory cytokines (Fig. 7a–j) IFNγ (p < 0.0001; Fig. 7a) and IL4 (p = 0.01; Fig. 7d) increased whereas that of IL13 decreased (p = 0.02; Fig. 7i) following HTP-smoke exposure.
Figure 6.
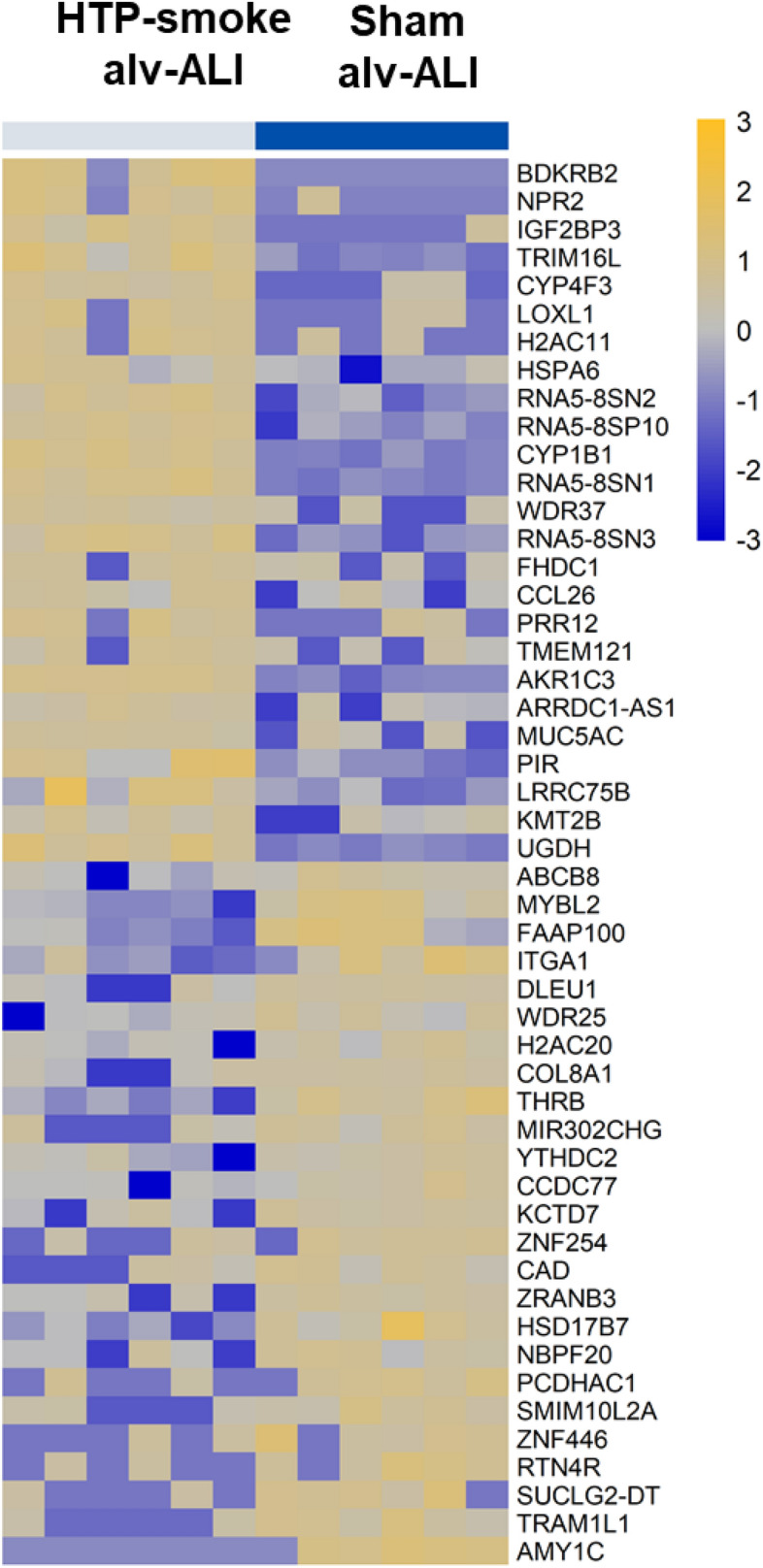
Heatmap of the top 25 up regulated and 25 down regulated genes in the alv-ALI model following exposure to HTP-smoke. n = 6 per exposure condition; significantly (raw p < 0.01) regulated genes with the highest fold changes are shown Genes were ordered by fold-change (HTP-smoke vs Sham) and relative gene expression values are shown across samples (z-scales to mean expression per row). A complete list of the 121 differentially regulated genes is provided in Supplementary Table S3. alv-ALI: alveolar mucosa model at air–liquid interface; HTP: heated tobacco product.
Figure 7.

Concentration of secreted proinflammatory cytokines in the basal media of sham exposed and HTP-smoke exposed alv-ALI model. IFNγ: interferon gamma; IL: interleukin; TNFα: tumor necrosis factor alpha. Data are shown as medians and interquartile ranges. n = 6 per exposure condition; non-parametric statistical analysis (Wilcoxon signed rank test); *Significance: p < 0.05. alv-ALI: alveolar mucosa model at air–liquid interface; HTP: heated tobacco product.
Consistent with the findings of RNAseq analysis, transcript expression of IFNG, IL1B, IL4, IL8, and IL13 as detected by qRT-PCR remained unaltered in both bro-ALI and alv-ALI exposed to HTP-smoke compared to the corresponding sham (Supplementary Fig. S2).
Bronchial and alveolar models
Significantly enriched pathways common to both bro-ALI and alv-ALI included estrogen biosynthesis, ferroptosis signaling pathway, superoxide radicals degradation, xenobiotic metabolism signaling, and α-tocopherol degradation (Table 1). Upstream regulator analysis in bro-ALI predicted the majority of the cytokines (24 out of 28) to be inhibited (Supplementary Table S5). As for example, IL1RN, IL4, IL5, and IL10 were predicted to be activated in bro-ALI but only TNFα for alv-ALI (Supplementary Table S6).
Lipid peroxidation
Significantly increased MDA levels were detected in both models (bro-ALI: p = 0.03; Fig. 8a; and alv-ALI: p = 0.03; Fig. 8b) following HTP exposure. MDA levels were undetectable in the sham exposed bro-ALI and alv-ALI models. The lipid peroxidation assay revealed significant increase in both models (bro-ALI: 64%; p = 0.03; Fig. 8c; and alv-ALI: 61%; p = 0.03; Fig. 8d) after HTP-smoke exposure. Addition of ferrostatin-1 significantly reduced the lipid peroxidation in both HTP-exposed models (bro-ALI: − 18%; p = 0.03; and alv-ALI: − 24%; p = 0.03) compared ferrostatin-1 untreated samples. The lipid peroxidation was also significantly reduced by ferrostatin-1 treatment sham samples in alv-ALI model (− 12%; p = 0.03) but not in the bro-ALI model. Although reduced by ferrostatin-1, the lipid peroxidation levels after HTP-smoke exposure remained higher in both models (bro-ALI: (34%; p = 0.03) and alv-ALI (23%; p < 0.03) compared to sham.
Figure 8.
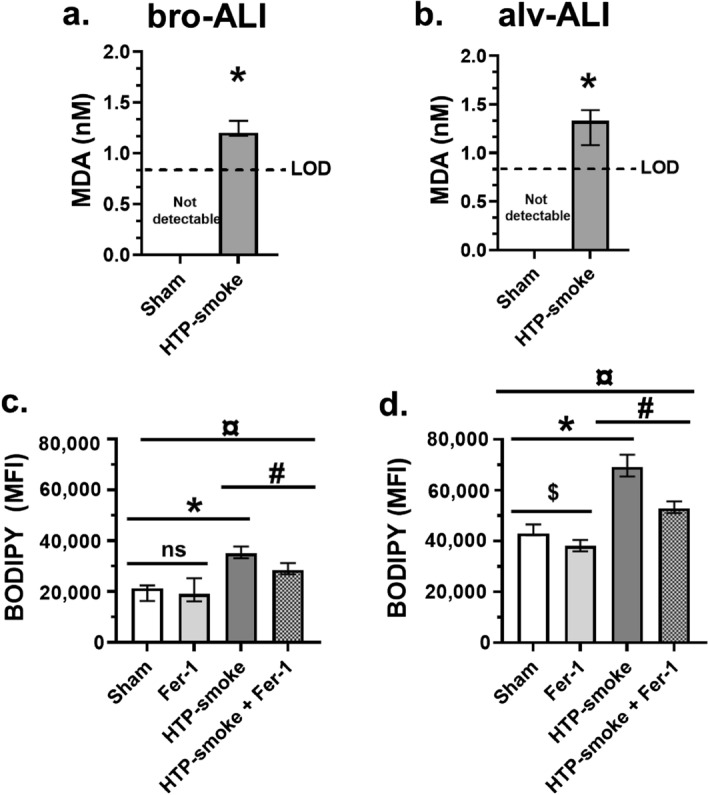
Assessment of lipid peroxidation levels in the bro-ALI and alv-ALI models due to sham exposure and HTP-smoke exposure. (a, b) Colorimetric malondialdehyde (MDA) assay and (c, d) BODIPY 581/591 C11 (Lipid Peroxidation Sensor) assay using ferrostatin-1 by flow cytometry. Data are shown as medians and interquartile ranges. n = 6 per exposure condition; non-parametric statistical analysis (Wilcoxon signed rank test or Friedman test followed by Wilcoxon signed rank test, as appropriate); ns: not significant; #,¤,$,*p < 0.05. MDA levels were below the limit of detection (LOD) in both bro-ALI and alv-ALI sham samples. Therefore, a value just below the LOD (0.85 nM) of MDA is assigned to all the sham samples for statistical analysis. ALI: air–liquid interface; alv-ALI: alveolar mucosa model at ALI; bro-ALI: bronchial mucosa model at ALI; Fer-1: ferrostatin-1; HTP: heated tobacco product; MFI: mean fluorescent intensity.
Discussion
To comprehensively assess the adverse effects of HTP-smoke, we exposed physiologically relevant human bronchial and alveolar lung mucosa models at ALI to HTP-smoke using an ISO puffing regimen. The bro-ALI and alv-ALI models represents the conducting and respiratory zones of the lung respectively thereby allowing us to assess the adverse effects of HTP exposure at two different levels of the lung. The conducting and respiratory zones of the lung have their unique cellular architecture and molecular signature many of which are present in the physiologically relevant bronchial and alveolar models used in this study and mentioned earlier. Increased oxidative stress due to HTP-smoke exposure in both bro-ALI and alv-ALI was detected by elevated cellular total ROS and NFkB levels. Pathways involved in oxidative stress or down-stream of oxidative stress are enriched in the transcriptomic profiles of bronchial and alveolar models exposed to HTP-smoke. This indicates oxidative stress as the main upstream event to HTP-smoke exposure in the bronchial and alveolar models. This is consistent with other studies on HTP-smoke which demonstrated HTP-smoke exposure resulted in oxidative stress23, 30, 32, 33. Similar effects have also been widely reported in case of CC-smoke46–48. However, the extent of adverse effects was in general lower in case of HTP-smoke compared to CC-smoke23, 30, 32, 33. DNA damage on bro-ALI and alv-ALI are indicated by increased levels of 8-OH-dG, γH2AX, and cleaved PARP levels upon HTP-smoke exposure. This is also consistent with reported effects of CC-smoke exposure on oxidative DNA damage49–52. Elevated levels of 8-OH-dG, γH2AX, and cleaved PARP levels have been reported in case of CC-smoke exposure, COPD, and lung cancer43, 53. 8-OH-dG is a widely studied DNA damage marker43, 53. γH2AX functions as a coordinator of DNA damage response by recruiting specific proteins and providing a binding site for the downstream signaling molecules54. Several studies have linked exposure to CC-smoke and γ-H2AX formation in vitro and in vivo54, 55. PARPs are multidomain proteins that are DNA repair factors which are activated to repair DNA lesions56. PARPs are also involved in cellular stress response as well as maintaining cellular homeostasis56. Therefore, the findings of this study indicate oxidative DNA damage in both bronchial and alveolar models as another adverse consequence of HTP-smoke exposure.
While interpreting the response of the bronchial and alveolar model to HTP-smoke exposure, it is important to consider the intrinsic characteristics of the two models. First, the bronchial model is developed from human primary bronchial epithelial cells collected from macroscopically normal bronchial tissue whereas the alveolar model is developed from immortalized lung adenocarcinoma cell line exhibiting characteristics of type II pneumocytes. Thus, there are inherent differences between the two cell types used. This is also supported by the higher basal levels of LDH, % PI positive cells, ROS, NFkB, 8-OHdG, γH2AX, and BODIPY in the alveolar model compared to the bronchial model. Further, the bronchial model differentiates into Club cells, goblet cells, basal cells, ciliated cells etc. and each have their unique cellular and molecular characteristics. On the other hand, presence of tight junction protein 1, lamellar bodies, surfactant protein C, microvilli, lipid bodies, desmosome, and tight junctions are features of the alveolar model. The role of the surfactant layer serves as a protective physical barrier in alveoli has been also demonstrated in cell culture57. The above reviewed information highlights the fact that each lung model has its own characteristics. Therefore, in this study the data generated from the two models have been dealt separately However, while performing pathway enrichment analysis for biological relevance of the toxicological response, few common enriched pathways (ferroptosis, superoxide radical degradation, xenobiotic metabolism, estrogen biosynthesis, α-tocopherol) were detected among bronchial and alveolar models. Identification of common pathways may provide important clue regarding common toxicological mechanisms at both lung regions apart from lung region specific effects. Based on the finding of ferroptosis as a common enriched pathway, lipid peroxidation assay using ferrostatin-1 inhibitor was performed to identify a plausible biochemical mechanism of HTP-smoke toxicity. Therefore, ferroptosis as one of the important candidate toxicological pathway of HTP-smoke exposure warrant detailed mechanistic evaluation.
In the following section we have briefly addressed some of the enriched canonical pathways in bro-ALI and alv-ALI that are mentioned in Table 1 to outline the plausible mechanisms of HTP-smoke exposure related pulmonary toxicity in each lung region. It is important to note that Supplementary Tables S2 and Tables S4 includes several other pathways which contributes to the toxicity of HTP-smoke specifically for the bronchial and alveolar regions. Enrichment of AHR signaling pathway, glutathione biosynthesis, NRF2-mediated oxidative stress response, superoxide radicals degradation, xenobiotic metabolism signaling following transcriptomic analysis in bro-ALI and/ or alv-ALI in our exposure experiments are consistent with the CC-smoke associated oxidative stress and inflammatory response43, 58. It is well established that these pathways act in a cascading manner. Recent studies suggest, AHR suppresses pulmonary oxidative response and inflammation in response to CC-smoke as a protective mechanism59, 60. The role of AHR receptor in maintaining lung Club cell homeostasis have been also reported61. As described previously, our multicellular bro-ALI models consist of Club cells following differentiation of the PBEC at ALI34. We detected only an increased levels of IL1ꞵ and IL8 in the bro-ALI, whereas in alv-ALI, IFNγ and IL4 levels increased and IL13 decreased following HTP-smoke exposure. However, alteration of secreted levels of other proinflammatory cytokines were not detected. It has been demonstrated that CC-smoke exposure in human bronchial epithelial cell lines induce the release of neutrophil chemoattractant IL1ꞵ and IL862. Alteration of IFNγ, IL4 and IL13 is also consistent with reports demonstrating the differential effects of CC-smoke on the regulation these cytokines in human airway epithelial cells62, 63. IL17 pathway is widely implicated in CC-smoke exposure including a positive correlation of IL17 concentration and sputum neutrophil count in COPD patients64, 65. Elevated levels of DNA damage response and IL17 have been also reported following CC-smoke exposure64. CC-smoke along with other environmental agents activates p38 MAPK pathway in the lung and is involved in pulmonary inflammation in asthma and COPD62, 66. However, the physiological relevance of the slight changes in the levels of secreted proinflammatory cytokines detected in this study cannot be ascertained. It is not uncommon to observe a difference between transcript expression profile and secreted levels of cytokines due to the time lag among transcriptional, translational, and secretory processes as also observed in other studies35, 37. A time course analysis of the transcript expression and cytokine secretion following HTP-smoke exposure may reveal the kinetics more appropriately.
Dysregulation of estrogen metabolism due to CC-smoke exposure have been detected via upregulation of a key cytochrome P450 gene that can metabolize estrogens such as beta-estradiol to potentially carcinogenic catechol and quinine forms67. Glucocorticoid signaling is ubiquitously present in the various organ systems including lung and demonstrate anti-inflammatory and anti-proliferative roles68. Alpha-tocopherol is the most bioavailable vitamin E isomer in human tissues and is considered as one of the most potent antioxidant vitamin E which attenuate inflammation specifically via suppressing NFkB mediated cytokine production69. LXR is a nuclear hormone receptor with anti-inflammatory properties and are known for triggering “reverse cholesterol transport”70, 71. PPARγ represents a disease-relevant pathophysiological and pharmacological target in COPD72. PPARγ activation state likely contributes to NF-κB-p65 dependent, CC-smoke induced chemokine-mediated regulation of inflammatory cell accumulation72. Exposure to CC-smoke also activates Toll like receptor signaling58. NRF2 is a key transcription factor for cell survival during oxidative stress by activating the expression of detoxification and antioxidant genes58.
Ferroptosis is a non-apoptotic and oxidative damage-related regulated cell death73, 74. Lipid peroxidation plays a central role in mediating ferroptosis44, 75. Increased peroxidized phospholipids has been reported in several ferroptosis models44. Smoking induced increase in lipid peroxidation can lead to DNA lesions43. Elevated levels of MDA have been reported in CC-smoke associated cancers43. Measurement of BODIPY-C11 is used to measure lipid peroxidation in ferroptosis44, 45. Elevated levels of MDA and BODIPY-C11 were detected in HTP-smoke exposed bro-ALI and alv-ALI. Ferrostatin-1, a synthetic antioxidant, inhibits lipid peroxidation and is also used as a specific inhibitor of ferroptosis45, 76. Our findings demonstrate the reduction of lipid peroxidation following ferrostatin-1 treatment in HTP-smoke exposed bro-ALI and alv-ALI models. These findings are consistent with a recent report of CC-smoke induced lipid peroxidation and ferroptosis77. This supports the fact that lipid peroxidation and ferroptosis are two important toxicological mechanisms associated HTP-smoke exposure down stream of oxidative stress.
This study provides a broad and comprehensive base to investigate HTP-smoke induced toxicity yet there are some limitations. Regarding the exposure set up, a direct way of exposing the bronchial and alveolar models to HTP-smoke would be preferable compared to the indirect method used in this study. However, due to the operational restrictions of the HTP device, it was difficult to perform direct exposures. In this context, it is also important to note that translation of the dosimetry from our in vitro exposure system to the lung in vivo is difficult. Though the particle number count as well as the particle size distribution is likely to differ between experimental puffing system and real-world puffing and inhalation and deposition across different lung regions yet measurement of the same would have been informative. Possibility to include multiple donors to develop the bro-ALI model for performing the experiments would have been ideal. Moreover, assessment of the HTP-smoke exposure using PBEC from smokers, COPD subjects, asthmatics, and chronic bronchitis patients would have added more dimensions to the study. The NCI-H441 cell line used to develop the alveolar model is an immortalized lung adenocarcinoma cell line which are inherently different compared to normal type II pneumocytes. On the other hand, though primary bronchial epithelial cells were obtained from macroscopically normal regions of the lung in connection with lobectomy, yet the molecular status of those apparently normal lung regions are unknown. Inclusion of immune effector cells such as macrophages in the bro-ALI and alv-ALI would have further added the physiological relevance of the lung mucosa models. This can affect the cell–cell interaction and cross talk that can in turn influence cytokine secretion and release. In this exposure set up, we have restricted ourselves to only one flavour (intense menthol with citrus) of heat-stick. Since flavours corresponds to additives which gives rise to toxic products upon thermal degradation, assessment of multiple flavours together with physical and chemical characterization of HTP-smoke would be necessary in future. Precise measurement of the dose (e.g. total particulate mass) deposited over each transwell membrane will make the exposure methodology more robust. However, similar nicotine concentration was detected in all the wells with low dispersion suggesting comparable exposure dose in all the cell culture plate wells. In this study we have used an ISO puffing regimen and an acute-repeated exposure model. It remains to be seen how the toxicity changes in the event of an intense puffing regimen (e.g. Health Canada Intense)31 and chronic exposure settings. The current study mainly focusses on transcriptomic data and corresponding biological pathways. It would be valuable to evaluate the significant pathways at protein level for better functional relevance.
To conclude, we have assessed pulmonary toxicity of HTP-smoke using a broad spectrum of endpoints. This study identified oxidative stress, DNA damage, lipid peroxidation, and ferroptosis as key features of HTP-smoke mediated pulmonary toxicity via a complex regulation of molecular cascade. The findings presented here shows that HTP-smoke exposure is toxic by itself, and the adverse effects are consistent with those reported for CC-smoke. The findings indicate that similar as well as differential toxicological response may drive the effects in different lung regions. Overall, the safety profile of HTP remains questionable. More independent studies are necessary to identify specific mechanisms of HTP use associated toxicity along with studies addressing the long-term adverse effects. This will aid towards evidence-based policy making decisions.
Supplementary Information
Acknowledgements
The study was supported by Grants from the Swedish Heart Lung Foundation (KG: 20200776; SU: 20210548; LP: 20210211), Swedish Research Council (LP: 2018-03233), and the Swedish Fund for Research without Animal Experiments. The authors would like to acknowledge support of the Clinical biomarker facility at SciLifeLab (Science for Life Laboratory, Uppsala University, SE-751 85 Uppsala Sweden) for providing assistance in protein analyses. We acknowledge the Biomedicum Small Molecule Mass Spectrometry Core facility (Karolinska Institutet, Stockholm, Sweden), supported by KI/SLL, for nicotine quantification.
Author contributions
M.R., S.U., L.P., G.J., and K.G., conceived and designed the project. M.R., Ma.I., Mi.I., S.U., and K.G., performed experiments and/or analyzed the data. M.R., Ma.I., J.B., L.P., G.J., S.U., and K.G. wrote the manuscript. S.U. and K.G. contributed equally. All authors have read and approved the manuscript.
Funding
Open access funding provided by Karolinska Institute. Swedish Heart Lung Foundation, Swedish Research Council, and Forska Utan DjurFörsök/Do Research without animal.
Data availability
All data presented in the study are available on request from the corresponding authors and are also included in the supplementary section. RNA sequencing data is deposited at the Gene Expression Omnibus database at NCBI [GSE198082; use token (ahkpuammjlerlgp) to access].
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Swapna Upadhyay and Koustav Ganguly.
Contributor Information
Swapna Upadhyay, Email: swapna.upadhyay@ki.se.
Koustav Ganguly, Email: koustav.ganguly@ki.se.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-20657-y.
References
- 1.https://www.euro.who.int/__data/assets/pdf_file/0008/443663/Heated-tobacco-products-brief-eng.pdf. (2020). [Accessed 14.02.2022].
- 2.Heated tobacco products: Information sheet, 2nd Edn. https://www.who.int/publications/i/item/WHO-HEP-HPR-2020.2 (2020). [Accessed 14.02.2022].
- 3.Heated tobacco products. https://www.cdc.gov/tobacco/basic_information/heated-tobacco-products/ (2020). [Accessed 14.02.2022].
- 4.Bitzer ZT, Goel R, Trushin N, Muscat J, Richie JP., Jr Free radical production and characterization of heat-not-burn cigarettes in comparison to conventional and electronic cigarettes. Chem. Res. Toxicol. 2020;33:1882–1887. doi: 10.1021/acs.chemrestox.0c00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonavicius E, McNeill A, Shahab L, Brose LS. Heat-not-burn tobacco products: A systematic literature review. Tob. Control. 2019;28:582–594. doi: 10.1136/tobaccocontrol-2018-054419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.https://www.grandviewresearch.com/industry-analysis/heat-not-burn-market (2020) [Accessed 14.02.2022].
- 7.Laverty AA, Vardavas CI, Filippidis FT. Prevalence and reasons for use of Heated Tobacco Products (HTP) in Europe: An analysis of Eurobarometer data in 28 countries. Lancet Reg. Health Eur. 2021;8:100159. doi: 10.1016/j.lanepe.2021.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallus S, et al. Use and awareness of heated tobacco products in Europe. J. Epidemiol. 2021;32(3):139–144. doi: 10.2188/jea.JE20200248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czoli CD, White CM, Reid JL, O’Connor RJ, Hammond D. Awareness and interest in IQOS heated tobacco products among youth in Canada, England and the USA. Tob. Control. 2020;29(1):89–95. doi: 10.1136/tobaccocontrol-2018-054654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Directive 2014/40/EU of the European Parliament and of the Council. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32014L0040 (2014) [Accessed 14.02.2022].
- 11.Glantz SA. Heated tobacco products: The example of IQOS. Tob. Control. 2018;27(Suppl 1):s1–s6. doi: 10.1136/tobaccocontrol-2018-054601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.St Helen G, Jacob IP, Nardone N, Benowitz NL. IQOS: Examination of Philip Morris International's claim of reduced exposure. Tob. Control. 2018;27(1):s30–s36. doi: 10.1136/tobaccocontrol-2018-054321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Znyk M, Jurewicz J, Kaleta D. Exposure to Heated tobacco products and adverse health effects, a systematic review. Int. J. Environ. Res. Public Health. 2021;18(12):6651. doi: 10.3390/ijerph18126651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bekki K, Inaba Y, Uchiyama S, Kunugita N. Comparison of chemicals in mainstream smoke in heat-not-burn tobacco and combustion cigarettes. J. UOEH. 2017;39(3):201–207. doi: 10.7888/juoeh.39.201. [DOI] [PubMed] [Google Scholar]
- 15.Farsalinos KE, Yannovits N, Sarri T, Voudris V, Poulas K. Nicotine delivery to the aerosol of a heat-not-burn tobacco product: Comparison with a tobacco cigarette and E-cigarettes. Nicotine Tob. Res. 2018;20:1004–1009. doi: 10.1093/ntr/ntx138. [DOI] [PubMed] [Google Scholar]
- 16.Farsalinos KE, et al. Carbonyl emissions from a novel heated tobacco product (IQOS): Comparison with an e-cigarette and a tobacco cigarette. Addiction. 2018;113(11):2099–2106. doi: 10.1111/add.14365. [DOI] [PubMed] [Google Scholar]
- 17.Leigh NJ, Palumbo MN, Marino AM, O'Connor RJ, Goniewicz ML. Tobacco-specific nitrosamines (TSNA) in heated tobacco product IQOS. Tob. Control. 2018;27(Suppl 1):s37–s38. doi: 10.1136/tobaccocontrol-2018-054318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, et al. Chemical analysis and simulated pyrolysis of tobacco heating system 2.2 compared to conventional cigarettes. Nicotine Tob. Res. 2019;21(1):111–118. doi: 10.1093/ntr/nty005. [DOI] [PubMed] [Google Scholar]
- 19.Salman R, et al. Free-base and total nicotine, reactive oxygen species, and carbonyl emissions from IQOS, a heated tobacco product. Icotine Tob. Res. 2019;21(9):1285–1288. doi: 10.1093/ntr/nty235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis B, Williams M, Talbot P. iQOS: Evidence of pyrolysis and release of a toxicant from plastic. Tob. Control. 2019;28(1):34–41. doi: 10.1136/tobaccocontrol-2017-054104. [DOI] [PubMed] [Google Scholar]
- 21.Auer R, Concha-Lozano N, Jacot-Sadowski I, Cornuz J, Berthet A. Heat-not-burn tobacco cigarettes: Smoke by any other name. JAMA Intern. Med. 2017;177(7):1050–1052. doi: 10.1001/jamainternmed.2017.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pacitto A, Stabile L, Scungio M, Rizza V, Buonanno G. Characterization of airborne particles emitted by an electrically heated tobacco smoking system. Environ. Pollut. 2018;240:248–254. doi: 10.1016/j.envpol.2018.04.137. [DOI] [PubMed] [Google Scholar]
- 23.Kopa PN, Pawliczak R. IQOS—A heat-not-burn (HnB) tobacco product—Chemical composition and possible impact on oxidative stress and inflammatory response. A systematic review. Toxicol. Mech. Methods. 2020;30(2):81–87. doi: 10.1080/15376516.2019.1669245. [DOI] [PubMed] [Google Scholar]
- 24.Moazed F, Chun L, Matthay MA, Calfee CS, Gotts J. Assessment of industry data on pulmonary and immunosuppressive effects of IQOS. Tob. Control. 2018;27(1):s20–s25. doi: 10.1136/tobaccocontrol-2018-054296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polosa R, Morjaria JB, Prosperini U, Busà B, Pennisi A, Gussoni G, Rust S, Maglia M, Caponnetto P. Health outcomes in COPD smokers using heated tobacco products: A 3-year follow-up. Intern. Emerg. Med. 2021;16(3):687–696. doi: 10.1007/s11739-021-02674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pataka A, et al. Acute effects of a heat-not-burn tobacco product on pulmonary function. Medicina (Kaunas). 2020;56(6):292. doi: 10.3390/medicina56060292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aokage T, et al. Heat-not-burn cigarettes induce fulminant acute eosinophilic pneumonia requiring extracorporeal membrane oxygenation. Respir. Med. Case Rep. 2018;26:87–90. doi: 10.1016/j.rmcr.2018.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaaban T. Acute eosinophilic pneumonia associated with non-cigarette smoking products: A systematic review. Adv. Respir. Med. 2020;88(2):142–146. doi: 10.5603/ARM.2020.0088. [DOI] [PubMed] [Google Scholar]
- 29.Kamada T, Yamashita Y, Tomioka H. Acute eosinophilic pneumonia following heat-not-burn cigarette smoking. Respirol Case Rep. 2016;4:e00190. doi: 10.1002/rcr2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaur G, Muthumalage T, Rahman I. Mechanisms of toxicity and biomarkers of flavoring and flavor enhancing chemicals in emerging tobacco and non-tobacco products. Toxicol. Lett. 2018;288:143–155. doi: 10.1016/j.toxlet.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ISO20778. "Cigarettes—Routine analytical cigarette smoking machine—Definitions and standard conditions with an intense smoking regime. https://www.iso.org/standard/69065.html (2018). Accessed 14 Feb 2022.
- 32.Bhat TA, et al. Acute effects of heated tobacco product (IQOS) aerosol inhalation on lung tissue damage and inflammatory changes in the lungs. Nicotine Tob. Res. 2021;23(7):1160–1167. doi: 10.1093/ntr/ntaa267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leigh NJ, Tran PL, O'Connor RJ, Goniewicz ML. Cytotoxic effects of heated tobacco products (HTP) on human bronchial epithelial cells. Tob. Control. 2018;27(1):s26–s29. doi: 10.1136/tobaccocontrol-2018-054317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji J, et al. Development of combining of human bronchial mucosa models with XposeALI for exposure of air pollution nanoparticles. PLoS ONE. 2017;12:e0170428. doi: 10.1371/journal.pone.0170428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganguly K, et al. Addressing the challenges of E-cigarette safety profiling by assessment of pulmonary toxicological response in bronchial and alveolar mucosa models. Sci. Rep. 2020;10:20460. doi: 10.1038/s41598-020-77452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ISO 3308: 2000-04-15: Routine Analytical Cigarette-smoking Machine—Definition and Standard Conditions. International Organization for Standardization. https://www.iso.org/standard/60404.html#:~:text=ISO%203308%3A2012%3A,complying%20with%20the%20standard%20conditions (2000). Accessed 14 Feb 2022.
- 37.Rahman M, et al. Differential effect of SARS-CoV-2 Spike glycoprotein 1 on human bronchial and alveolar lung mucosa models: Implications for pathogenicity. Viruses. 2021;13(12):2537. doi: 10.3390/v13122537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji J, et al. Multi-cellular human bronchial models exposed to diesel exhaust particles: Assessment of inflammation, oxidative stress and macrophage polarization. Part Fibre Toxicol. 2018 doi: 10.1186/s12989-018-0256-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji J, et al. Exposure of normal and chronic bronchitis-like mucosa models to aerosolized carbon nanoparticles: Comparison of pro-inflammatory oxidative stress and tissue injury/repair responses. Nanotoxicology. 2019;13(10):1362–1379. doi: 10.1080/17435390.2019.1655600. [DOI] [PubMed] [Google Scholar]
- 40.Thimraj TA, et al. Evaluation of diacetyl mediated pulmonary effects in physiologically relevant air–liquid interface models of human primary bronchial epithelial cells. Toxicol. In Vitro. 2019;61:104617. doi: 10.1016/j.tiv.2019.104617. [DOI] [PubMed] [Google Scholar]
- 41.Johanson G, et al. Analysis of acrolein exposure induced pulmonary response in seven inbred mouse strains and human primary bronchial epithelial cells cultured at air–liquid interface. Biomed. Res. Int. 2020 doi: 10.1155/2020/3259723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Team, R. C., A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/index.html (2020). Accessed 14 Feb 2022.
- 43.Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat. Res. Rev. Mutat. Res. 2021;787:108365. doi: 10.1016/j.mrrev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan HF, et al. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target Ther. 2021;6(1):49. doi: 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez AM, Kim A, Yang WS. Detection of ferroptosis by BODIPY™ 581/591 C11. Methods Mol. Biol. 2020;2108:125–130. doi: 10.1007/978-1-0716-0247-8_11. [DOI] [PubMed] [Google Scholar]
- 46.Barr J, et al. Nicotine induces oxidative stress and activates nuclear transcription factor kappa B in rat mesencephalic cells. Mol. Cell Biochem. 2007;297(1–2):93–99. doi: 10.1007/s11010-006-9333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin AH, et al. Inflammatory effects of menthol vs non-menthol cigarette smoke extract on human lung epithelial cells: A double-hit on TRPM8 by reactive oxygen species and menthol. Front. Physiol. 2017;8:263. doi: 10.3389/fphys.2017.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol. 2011;254(2):72–85. doi: 10.1016/j.taap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leanderson P, Tagesson C. Cigarette smoke-induced DNA damage in cultured human lung cells: Role of hydroxyl radicals and endonuclease activation. Chem. Biol. Interact. 1992;81(1–2):197–208. doi: 10.1016/0009-2797(92)90034-I. [DOI] [PubMed] [Google Scholar]
- 50.Chen Z, et al. Oxidative DNA damage is involved in cigarette smoke-induced lung injury in rats. Environ. Health Prev. Med. 2015;20(5):318–324. doi: 10.1007/s12199-015-0469-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toyooka T, Ibuki Y. Cigarette sidestream smoke induces phosphorylated histone H2AX. Mutat. Res. 2009;676(1–2):34–40. doi: 10.1016/j.mrgentox.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Künzi L, Holt GE. Cigarette smoke activates the parthanatos pathway of cell death in human bronchial epithelial cells. Cell Death Discov. 2019;5:127. doi: 10.1038/s41420-019-0205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sears CR. DNA repair as an emerging target for COPD-lung cancer overlap. Respir. Investig. 2019;57(2):111–121. doi: 10.1016/j.resinv.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 54.Ishida M, et al. Smoking cessation reverses DNA double-strand breaks in human mononuclear cells. PLoS ONE. 2014;9:e103993. doi: 10.1371/journal.pone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Albino AP, et al. gammaH2AX: A potential DNA damage response biomarker for assessing toxicological risk of tobacco products. Mutat. Res. 2009;678(1):43–52. doi: 10.1016/j.mrgentox.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bai P. Biology of poly (ADP-Ribose) polymerases: The factotums of cell maintenance. Mol. Cell. 2015;58(6):947–958. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- 57.Radiom M, et al. Pulmonary surfactant inhibition of nanoparticle uptake by alveolar epithelial cells. Sci Rep. 2020;10:19436. doi: 10.1038/s41598-020-76332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuo L, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell Mol. Physiol. 2014;307(3):L205–L218. doi: 10.1152/ajplung.00330.2013. [DOI] [PubMed] [Google Scholar]
- 59.Guerrina N, Traboulsi H, Eidelman DH, Baglole CJ. The aryl hydrocarbon receptor suppresses chronic smoke-induced pulmonary inflammation. Front. Toxicol. 2021;3:653569. doi: 10.3389/ftox. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guerrina N, et al. Aryl hydrocarbon receptor deficiency causes the development of chronic obstructive pulmonary disease through the integration of multiple pathogenic mechanisms. FASEB J. 2021 doi: 10.1096/fj.202002350R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu KY, et al. Aryl hydrocarbon receptor is essential in the control of lung club cell homeostasis. J. Inflamm. Res. 2021;14:299–311. doi: 10.2147/JIR.S284800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: A mechanistic review. Int. J. Environ. Res. Public Health. 2018;15:1033. doi: 10.3390/ijerph15051033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mertens TCJ, et al. Cigarette smoke differentially affects IL-13-induced gene expression in human airway epithelial cells. Physiol. Rep. 2017;5:e13347. doi: 10.14814/phy2.13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao C, et al. IL-17-mediated inflammation promotes cigarette smoke-induced genomic instability. Cells. 2021;10:1173. doi: 10.3390/cells10051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, et al. Increased expression of CD4+IL-17+ cells in the lung tissue of patients with stable chronic obstructive pulmonary disease (COPD) and smokers. Int. Immunopharmacol. 2013;15(1):58–66. doi: 10.1016/j.intimp.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 66.Pelaia C, Vatrella A, Gallelli L, Lombardo N, Sciacqua A, et al. Role of p38 mitogen-activated protein kinase in asthma and COPD: Pathogenic aspects and potential targeted therapies. Drug Des. Devel. Ther. 2021;15:1275–1284. doi: 10.2147/DDDT.S300988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siegfried JM. Early changes in pulmonary gene expression following tobacco exposure shed light on the role of estrogen metabolism in lung carcinogenesis. Cancer Prev. Res. (Phila). 2010;3(6):692–695. doi: 10.1158/1940-6207.CAPR-10-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013;34(9):518–530. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ji X, Yao H, Meister M, Gardenhire DS, Huanbiao M. Dietary supplements for chronic obstructive pulmonary disease. Antioxidants (Basel). 2021;10:883. doi: 10.3390/antiox10060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smoak K, et al. Effects of liver X receptor agonist treatment on pulmonary inflammation and host defense. J. Immunol. 2008;180(5):3305–3312. doi: 10.4049/jimmunol.180.5.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Higham, A. et al. Respir. Res. 14, 106. 10.1186/1465-9921-14-106. (2013).
- 72.Solleti SK, et al. Airway epithelial cell PPARγ modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;309:L293–304. doi: 10.1152/ajplung.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dixon SJ, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang D, Kroemer G. Ferroptosis. Curr. Biol. 2020;30(21):R1292–R1297. doi: 10.1016/j.cub.2020.09.068. [DOI] [PubMed] [Google Scholar]
- 75.Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miotto G, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328. doi: 10.1016/j.redox. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshida M, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019;10:3145. doi: 10.1038/s41467-019-10991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in the study are available on request from the corresponding authors and are also included in the supplementary section. RNA sequencing data is deposited at the Gene Expression Omnibus database at NCBI [GSE198082; use token (ahkpuammjlerlgp) to access].