

Abstract
A retrospective, observational cohort study was conducted to generate real‐world evidence in adult patients diagnosed with sarcoidosis‐associated pulmonary hypertension (SAPH) at a referral center in England between 2012 and 2019. Data from the referral center electronic medical record database were linked to the National Health Service Hospital Episode Statistics database to collect and analyze patient demographics, clinical characteristics, comorbidities, treatment patterns, health‐related quality of life (HRQoL; assessed using the EmPHasis‐10 questionnaire), healthcare resource utilization (HCRU), costs, and survival. Sixty‐two patients with SAPH were identified. At diagnosis, 84% were in WHO functional class III and presented with significant pulmonary hemodynamic impairment. Cardiovascular and respiratory comorbidities were commonly reported prediagnosis. Median EmPHasis‐10 score at diagnosis was 34, indicative of poor HRQoL. In the 1st year after diagnosis, median (Q1, Q3) per‐patient HCRU was 1 (0, 2) all‐cause inpatient hospitalizations; 3 (2, 4) same‐day hospitalizations; and 9 (6, 11) outpatient consultations. In 24 patients who were hospitalized longer than 1 day in the 1st year after diagnosis, the median duration of hospitalization was 4 days. With a median follow‐up of 1.8 years, the median overall survival was 2.9 years. In this cohort of patients with SAPH, poor HRQoL and high HCRU were observed following diagnosis. To our knowledge, this is the first study to report on HRQoL and HCRU in patients with SAPH. More research is needed on treatment options for this population with high unmet needs.
Keywords: HCRU, HRQoL, mortality, real‐world evidence, SAPH
INTRODUCTION
Sarcoidosis is an uncommon, chronic, multiorgan inflammatory disorder of unknown etiology. 1 , 2 , 3 Non‐caseating granulomas develop in multiple tissues, commonly affecting the lungs and lymphatic system and potentially causing severe morbidity. 1 , 3 , 4 , 5 Pulmonary hypertension (PH) is a well‐recognized and serious but poorly defined complication of sarcoidosis. 2 , 4 , 6 Estimates from prospective screening studies on the prevalence of PH in patients with sarcoidosis range from 3% to 6% when PH is confirmed by right heart catheterization (RHC) and from 6% to 21% for PH suspected by echocardiography. 2 , 7 , 8 , 9 , 10 , 11 , 12
Patients with sarcoidosis‐associated pulmonary hypertension (SAPH) most frequently present with persistent or unexplained dyspnea, 13 , 14 , 15 , 16 which has a negative impact on patients' health‐related quality of life (HRQoL). 2 , 17 , 18 Moreover, the development of SAPH is independently associated with a 7‐ to 10‐fold increase in the risk of death compared to patients who have sarcoidosis without PH. 2 , 19 , 20 In a study of 452 patients with sarcoidosis, PH was a major independent predictor of mortality; 10‐year survival was estimated at 65% in patients with PH, compared with 90% in patients without PH. 8
There are currently no approved drugs for the treatment of SAPH and a lack of robust data from large randomized controlled clinical trials. 2 Decisions regarding treatment are challenging and reflect in part the heterogenous nature of SAPH. Some improvement has been reported in patients with SAPH treated with pulmonary vasodilators, endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and the soluble guanylate cyclase stimulator riociguat in case series and small‐scale studies. 2 , 4 , 13 , 15 , 21 , 22 , 23 , 24 Overall, drug treatment has improved hemodynamics, but has not consistently improved exercise capacity or functional class. Single or bilateral lung transplant may be considered in selected patients. 2 , 4 , 14
To date, the economic burden of SAPH has not been studied alongside clinical outcomes for these patients. This retrospective study was conducted to generate real‐world evidence on the demographic and clinical characteristics, comorbidities, treatment patterns, HRQoL, healthcare resource utilization (HCRU), and overall survival in patients diagnosed with SAPH at a PH referral center in England between 2012 and 2019.
METHODS
Data sources
Data were collected from the electronic medical record (EMR) database used at the Sheffield Pulmonary Vascular Disease (PVD) Unit (Sheffield EMR) based at Royal Hallamshire Hospital and were linked with data from the National Health Service (NHS) Hospital Episode Statistics (HES) database, as previously described. 25 , 26
The Sheffield EMR contains clinical management and demographic data for all referred patients, including confirmed diagnoses, diagnostic procedures, further clinical tests, treatments prescribed, and specialist consultations that have taken place at the Sheffield PVD Unit. The HES database contains details on all admissions, accident and emergency attendances (i.e., emergency room visits), and outpatient appointments at all NHS hospitals in England. 27
Only aggregated, anonymised data were used in this study; researchers did not have access to personally identifiable information. HES data were linked by NHS number and date of birth to Sheffield EMR data before being deidentified and made accessible for analysis in this study. Death data in the Sheffield EMR were captured through linkage to NHS death records and were refreshed immediately before data extraction.
Categories with <7 patients in tables have been masked in line with data protection requirements to ensure that patient confidentiality was maintained (noted as “<7” in tables). 28 Where only a single count was masked as “<7” and, therefore, could be further identified, the category with the next lowest number was also suppressed (noted as “suppressed” in tables) to avoid calculation of the small value. However, no data were excluded from the analysis; small number suppression was only applied to study outputs as a final step before release. Kaplan–Meier curves were censored when the number at risk dropped to <10% of the stratum size.
Study design
Adult patients (≥18 years old at diagnosis) were included in this retrospective, observational cohort study if they were diagnosed with SAPH between January 1, 2012 and June 30, 2019. The observation period was from January 1, 2012 to September 30, 2019, from both databases. Comorbidities and HCRU post‐diagnosis were available from the HES database from January 1, 2009. For all included patients, the index date was defined as the first diagnosis date within the study period, based on Sheffield EMR records.
Patients were followed from diagnosis until the earliest of the following: date of death (from Sheffield EMR), date of lung transplant (from HES), date of last contact, or end of the study observation period (September 30, 2019).
For the overall survival analyses, patients were followed until death or censored at last contact according to the Sheffield EMR or HES; that is, patients were followed upon discharge from the Sheffield PVD Unit, if there was evidence in HES that they survived longer. Follow‐up for overall survival ended on September 30, 2019, in line with the observation period. For the HCRU analyses, patients were followed until the earliest of death or last contact in the HES database, or September 30, 2019.
Outcomes
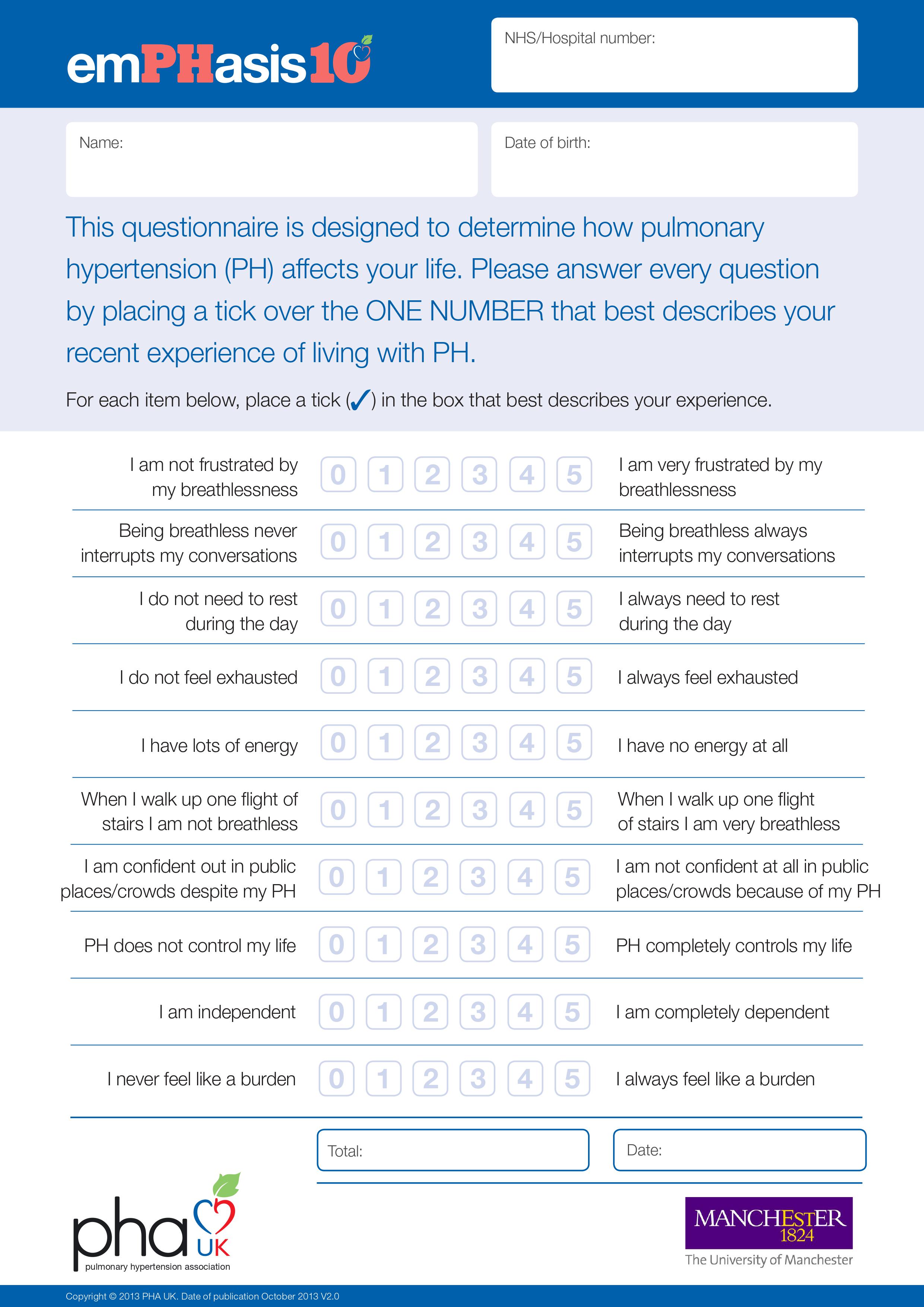
In line with the study objectives, the following outcomes were collected from the Sheffield EMR: patient demographics, clinical characteristics, survival after diagnosis, and changes in HRQoL based on EmPHasis‐10 score from diagnosis to 1 and 2 years after diagnosis. The EmPHasis‐10 is a quality‐of‐life score developed and validated specifically for the assessment of HRQoL in patients with PH (see Supporting Information). 29 , 30 Higher scores are indicative of worse HRQoL. 29
The following data were collected from HES: comorbidities identified in the 5 years before diagnosis, survival after diagnosis, and 1‐ and 3‐year HCRU and costs after diagnosis. For HCRU analyses, inpatient hospitalizations were categorized as either longer than 1 day or “same‐day spells,” meaning the discharge date was the same as the admission date. Outpatient visits were defined as any activity occurring in a hospital under an outpatient setting.
Statistical analysis
This study was descriptive in nature, without predefined hypotheses. Categorical variables are reported with frequency (n) and percentage (%). Continuous variables are reported as median (Q1, Q3) and mean (±standard deviation). Median follow‐up times were calculated. Overall survival was evaluated with Kaplan–Meier estimates with follow‐up time starting from index date and censoring at death or last contact in Sheffield EMR or HES.
For HCRU analyses, hospitalization costs were calculated from the first contact in HES until death or the last contact in HES. Costs are described using indicative tariff incomes, representative of the reimbursement received by hospitals. In England, the cost of secondary care to the health service (the payer) is calculated according to national tariff prices in 2019, based on the national average unit costs of providing each service (published as the National Schedule of Reference Costs). 31
RESULTS
Overview
Overall, 62 patients diagnosed with SAPH were identified in the database. The median time from the first record in HES to diagnosis at Sheffield PVD Unit was 4.5 years. The median follow‐up time from diagnosis was 1.8 years using Sheffield EMR data and 2.0 years using HES data. The main reasons for study exit were death in 33 (53%) patients and the last record in Sheffield EMR in 27 (44%) patients. One patient was excluded from the analysis of treatments prescribed due to data quality criteria.
Demographics
Demographics of the SAPH cohort at diagnosis are presented in Table 1. The median age at diagnosis was 63 years; 55% of patients were female, and most patients identified as white (84%).
Table 1.
Demographics and clinical characteristics at diagnosis of patients with SAPH
| Characteristic | SAPH cohort (N = 62) |
|---|---|
| Age (years), median (range) | 62.5 (33–84) |
| Sex, female, n (%) | 34 (55) |
| Ethnicity, n (%) | |
| White | 52 (84) |
| Othera | 10 (16) |
| WHO functional class, n (%) | |
| I | <7b |
| II | 0 (0) |
| III | 52 (84) |
| IV | Suppressedb |
| Exercise capacity | |
| ISWT (m) (n = 58) | 100 (40, 180) |
| RHC at diagnosis, n (%) | 54 (87) |
| Hemodynamics | |
| Heart rate, bpm (n = 51) | 85 (75, 92) |
| MAP (mmHg) (n = 50) | 102 (92, 110) |
| mRAP (mmHg) (n = 42) | 11 (6, 13) |
| mPAP (mmHg) (n = 53) | 46 (41, 55) |
| mPAWP (mmHg) (n = 47) | 12 (9, 14) |
| Cardiac index (L/min/m2) (n = 50) | 2.5 (1.9, 2.8) |
| PVR (Wood units) (n = 45) | 8.2 (6.3,11.0) |
| SvO2 (%) (n = 51) | 62.7 (58.7, 70.1) |
| Lung function | |
| FEV1/FVC (%) | 70 (58, 75) |
| DLCO (% predicted) (n = 39) | 29.3 (22.7, 41.2) |
Note: Values are expressed as median (Q1, Q3) except as noted.
Abbreviations: bpm, beats per minute; DLCO, carbon monoxide transfer factor; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; ISWT, incremental shuttle walk test; MAP, mean arterial pressure; mPAP, mean pulmonary artery pressure; mPAWP, mean pulmonary artery wedge pressure; mRAP, mean right atrial pressure; PVR, pulmonary vascular resistance; RHC, right heart catheterization; SAPH, sarcoidosis‐associated pulmonary hypertension; SvO2, mixed venous oxygen saturation; WHO, World Health Organization.
Includes patients identifying as Indian, Pakistani, Bangladeshi, or Black, and missing or unknown data.
Small numbers were suppressed in line with data protection requirements. A result of “<7” substitutes any patient count that is below 7 unique patients and, therefore, masked for data privacy reasons. “Suppressed” substitutes the next lowest number in a row or column that can be used to reverse calculate a masked value of “<7.” This means that a number that has been “Suppressed” could substitute a number >7. Values of zero or missing do not require suppression.
Clinical characteristics, comorbidities, and prescribed treatments
Clinical characteristics at diagnosis are presented in Table 1 and Supporting Information: Table 1. At diagnosis, most patients at the Sheffield PVD Unit were in WHO FC III (84%), signifying substantial pulmonary hemodynamic impairment.
Comorbidities identified in the 5 years before diagnosis are shown in Table 2. Cardiovascular and respiratory comorbidities were common.
Table 2.
Most common comorbidities (>10%) in patients with SAPH in the 5 years before diagnosis, identified from HES data
| Comorbidity | Number of patients (%) (N = 62) |
|---|---|
| Respiratory conditions | |
| Chronic obstructive pulmonary disease | 21 (34) |
| Asthma | 10 (16) |
| Other obstructive lung diseases | 23 (37) |
| Cardiovascular conditions | |
| Heart failure | 22 (36) |
| Hypertension | 22 (36) |
| Ischemic heart disease | 14 (23) |
| Valvular heart disease | 11 (18) |
| Other comorbidities of interest | |
| Diabetes | 9 (15) |
| Depression | 7 (11) |
| Pulmonary embolism | 7 (11) |
| Other conditions associated with SAPH | |
| Interstitial lung disease | 24 (39) |
| Pulmonary hypertension | |
| Primary pulmonary hypertension | 12 (19) |
| Secondary pulmonary hypertension | 41 (66) |
Note: Comorbidities were identified from the NHS Hospital Episode Statistics (HES) database based on ICD‐10 codes. Comorbidities in <7 patients were not included in the table due to data suppression.
Abbreviations: NHS, National Health Service; SAPH, sarcoidosis‐associated pulmonary hypertension.
Of the 61 patients included in the analyses, 50 (81%) were prescribed pulmonary arterial hypertension (PAH)‐specific therapies within 90 days following diagnosis. Most patients (76%) were prescribed phosphodiesterase type 5 inhibitor monotherapy.
HCRU following SAPH diagnosis
A total of 42 (68%) patients had at least 1 year of observable data available after diagnosis. Total HCRU, median HCRU per patient, and median cost per patient in the first year after diagnosis of SAPH are presented in Figure 1.
Figure 1.

HCRU and associated costs over 1 year of follow‐up after SAPH diagnosis: (a) Total number of all‐cause events. (b) Median (Q1, Q3) HCRU, per patient (N = 42). (c) Median costs (Q1, Q3), per patient (N = 42). GBP, British pound sterling; HCRU, healthcare resource utilization; SAPH, sarcoidosis‐associated pulmonary hypertension.
During the 1st year after diagnosis, there were a total of 59 all‐cause inpatient hospitalizations longer than 1 day in 24 patients. Of these, 43% (25) were elective and 58% (34) were nonelective; 46% (27) of hospitalizations were PH‐specific. For these 24 patients, the median duration of hospitalization was 4 (2, 7) days. Overall, the median (Q1, Q3) number of all‐cause inpatient hospitalizations longer than 1 day was 1 (0, 2) per patient, representing a median cost of £1202 (£0, £3677) per patient. The median duration of PH‐specific hospitalizations was 2 (2, 7) days; for elective and nonelective hospitalizations, the medians were 2 (2, 3) and 7 (3, 11) days, respectively.
All‐cause same‐day spells totaled 130 in 39 patients in the year after diagnosis; of these, 80% (104) were PH‐specific. Overall, the median (Q1, Q3) number of same‐day spells in the 1st year after diagnosis was 3 (2, 4) per patient, representing a median cost of £2645 (£1660, £4140) per patient (Figure 1). The median number of outpatient consultations in the 1st year after diagnosis was 9 (6, 11) per patient, representing a median cost of £1059 (£780, £1483) per patient.
HRQoL
The median (Q1, Q3) EmPHasis‐10 score in 33 (53%) patients with available assessments at diagnosis was 34 (26, 41). The median EmPHasis‐10 score was 33 (30, 36) for the 27 patients who completed the assessment 1 year after diagnosis and 31 (11, 34) for the 18 patients who completed the assessment 2 years after diagnosis.
The median (Q1, Q3) change in EmPHasis‐10 score between diagnosis and 1 year after diagnosis for the 20 (32%) patients with available assessments at both time points was −2 (−6, 0). The median change in score between 1 year and 2 years after diagnosis was 1 (0, 7) for the 16 (26%) patients with available assessments at both time points.
A comparison with previously published HRQoL assessments in PAH and chronic thromboembolic pulmonary hypertension (CTEPH), using EmPHasis‐10, is shown in Figure 2.
Figure 2.

Median (Q1, Q3) EmPHasis‐10 scores in studies of patients with SAPH (current study), CTEPH, and PAH (30, 33, 35). CTD‐PAH, connective tissue disease pulmonary arterial hypertension; CTEPH, chronic thromboembolic pulmonary hypertension; I/D/H PAH, idiopathic/drug‐induced/heritable pulmonary arterial hypertension; PAH, pulmonary arterial hypertension; SAPH, sarcoidosis‐associated pulmonary hypertension.
Overall survival
With a median follow‐up of 1.8 years, the median (Q1, Q3) overall survival of patients with SAPH was 2.88 (1.04, not reached) years. Survival probabilities (95% CI) were 77% (67%, 88%) at 1 year and 46% (34%, 63%) at 3 years.
DISCUSSION
This retrospective study has generated pre‐ and post‐diagnosis real‐world evidence on outcomes in patients with SAPH at a large PH referral center. Patients had significant pulmonary hemodynamic impairment at diagnosis and a high prevalence of cardiovascular and respiratory comorbidities before diagnosis. Poor survival rates were observed. To our knowledge, this is the first study to report on HCRU and HRQoL in patients with SAPH. We have shown that patients with SAPH exhibit high HCRU in the first year following diagnosis. They also reported poor HRQoL at diagnosis and follow‐up.
HCRU data for patients with SAPH were collected at 1 and 3 years post‐diagnosis, although we have not presented results from the third year due to low patient numbers. The results demonstrate that SAPH incurs a high HCRU burden on a per‐patient basis in the 1st year following diagnosis when considering in‐patient hospitalizations longer than 1 day, same‐day spells, and out‐patient consultations collectively. Same‐day spells accounted for the majority of costs, and it appears that multiple same‐day admissions are common in patients with SAPH in the 1st year after diagnosis. The number of in‐patient admissions and out‐patient consultations in our study is comparable with recently published HCRU data for patients with PAH in England. 32
Patient‐reported outcomes (PROs) can improve our understanding of how patients experience SAPH. 17 HRQoL was assessed in our study by EmPHasis‐10 score. The median baseline EmPHasis‐10 score of 34 is considerably higher than the median EmPHasis‐10 scores reported for patients with PAH from a large UK multicentre study, which included patients from our center, and a large multicenter study from the United States (score range: 20–30), and for patients with CTEPH (score: 24). 30 , 33 , 34 This suggests that patients with SAPH have a worse quality of life than other forms of PH for which we have treatment interventions. EmPHasis‐10 scores have been found to correlate with WHO FC and survival in patients with PAH, with higher scores associated with worse WHO FC and a significant increase in mortality seen with scores >34. 29 , 33 , 35 Analysis of registry‐based data from the United States in patients with PAH has suggested a minimally important difference for the EmPHasis‐10 of six points (range: 5–7.6). 34 The median change in EmPHasis‐10 scores in this study was –2 in the 1st year and +1 in the 2nd year. However, it is difficult to draw firm conclusions about the change, given the low number of patients in our study with available assessments at multiple time points and the likelihood of survival bias. It is worth noting that the EmPHasis‐10 may not fully capture all aspects of HRQoL relevant to patients with SAPH and that there are currently no SAPH‐specific PROs available. 17
Published data on patients with sarcoidosis and pulmonary hypertension are relatively limited. The demographic and clinical characteristics of our cohort at diagnosis are broadly comparable to published data in patients with SAPH, although overall, the mean age in these studies was lower (range: 52–58 years vs. 63 years in our study) and our patients had a large number of comorbidities. 22 , 36 , 37 , 38 , 39 Previous European studies include a retrospective chart review conducted at the National Pulmonary Hypertension Unit of the Royal Free Hospital in London from 1999 to 2011 and an analysis of newly diagnosed patients with severe SAPH from the French Pulmonary Hypertension Registry. 22 , 36 In the UK‐based study (24 patients, mean age 58 years, 54% female), hemodynamic impairment was similar to our study. Almost 90% of patients were in WHO FC III or IV, compared with 84% in WHO FC III in our study. The French registry study included 126 patients (mean age 58 years, 52% male; 83% WHO FC III‐IV) with severe SAPH, defined as mPAP >35 mmHg or mPAP 25–35 mmHg with mean cardiac index <2.5 L/min/m2. By this definition, all patients in our cohort had severe PH at diagnosis (mean mPAP: 48 mmHg; mean mRAP 10.5 mmHg; cardiac index: 2.5 L/min/m2 (see Supporting Information: Table 1).
Studies from the United States and international registries have shown more variability in demographic and clinical characteristics than in our cohort. A similar level of hemodynamic impairment was described in a retrospective study of 95 patients with SAPH treated at the Duke Medical Center from 1990 to 2010, which included younger patients (mean age: 52 years) and a larger proportion of female (76%) and African American (86%) patients than our study. 37 International registries by Baughman et al. and Shlobin et al. reported more female patients with SAPH (71%–72%) than our study and the hemodynamic impairment in these cohorts was less severe. 38 , 39 In both studies, around 55% of patients were African American.
The clinical management of SAPH is challenging and requires individualized approaches. Treating sarcoidosis alone may not be sufficient. 40 , 41 , 42 In contrast to PAH, for which there are several approved drugs from different classes, 43 there is currently no drug approved for the treatment of SAPH, although patients treated with off‐label PAH‐specific therapies have demonstrated some improvement in small‐scale studies. 2 , 4 , 13 , 15 , 21 , 22 , 23 , 24 Most patients in this study initiated phosphodiesterase type 5 inhibitor monotherapy at diagnosis, in line with NHS clinical commissioning policy. 44 Eleven (18%) patients had no record of any PAH‐specific therapy at or within the 90 days following diagnosis. These findings reflect the lack of evidence‐based management pathways and the availability of treatment options for these patients. For patients with end‐stage SAPH, a single or bilateral lung transplant is an option. 14 Lung transplant data for our cohort were masked; however, the proportion of patients receiving this curative intervention was low (<7 patients).
Results from the present study confirm that SAPH is associated with a poor prognosis and high mortality. In our cohort, the overall survival probability was 77% at 1 year and 46% at 3 years, and the median overall survival was 2.9 years. Patients referred in this study were highly co‐morbid and with significant hemodynamic impairment. These survival figures are lower than some registry‐based studies in patients with SAPH by Boucly et al. and Shlobin et al., and may in part reflect an older and more co‐morbid population. 22 , 39
A rigorous methodology was used in our study to link two databases. Our results demonstrate that patients with SAPH present with severe hemodynamic impairment and support their poor survival prognosis. While the number of patients included in our study is small (n = 62), this represents a reasonably sized cohort for SAPH, which is a rare disease.
In conclusion, this real‐world evidence study based on 62 patients treated at one of the largest specialist PH centers in Europe adds to the limited body of evidence for SAPH and begins to address data gaps with regard to HCRU and HRQoL. Patients with SAPH have high levels of HCRU, poor quality of life, and high unmet needs, and more research is needed on the optimal medical and surgical treatment options that can reduce disease progression and improve quality of life and survival in this population.
AUTHOR CONTRIBUTIONS
Study conception: David G Kiely, Allan Lawrie, Ruvimbo Muzwidzwa, Amélie Beaudet, and Audrey Muller. Study design: David G Kiely, Fernando Exposto, Ruvimbo Muzwidzwa, Amélie Beaudet, Audrey Muller, and Rafael Sauter. Data acquisition: Allan Lawrie, Neil Hamilton, Steven Wood, and Louise Raiteri. Data analysis: Allan Lawrie, Neil Hamilton, Steven Wood, Fernando Exposto, and Louise Raiteri. Data interpretation: Allan Lawrie, David G Kiely, Neil Hamilton, Fernando Exposto, Ruvimbo Muzwidzwa, Louise Raiteri, Amélie Beaudet, Audrey Muller, Rafael Sauter, and Nadia Pillai. All authors contributed to drafting the work and revising it critically for important intellectual content. All authors gave final approval of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The authors confirm that GPP3 guidelines were followed throughout the development of the paper.
CONFLICTS OF INTEREST
Allan Lawrie is supported by a British Heart Foundation Senior Basic Science Research Fellowship (FS/18/52/33808). Amélie Beaudet, Audrey Muller, Rafael Sauter, and Nadia Pillai are employees of Actelion Pharmaceuticals Ltd. Amélie Beaudet and Audrey Muller own stock in Johnson & Johnson. The remaining authors declare no conflict of interest.
ETHICS STATEMENT
The reported study was noninterventional, and the analysis was based on secondary data. No identifying data were prospectively collected in any of the planned approaches. Ethics approval was obtained from the Research Ethics Committee (REC) of the NHS Health Research Authority and from the Confidentiality Advisory Group (CAG) to secure s251 approval for the linkage of patient identifiable data. Following approval from the REC and CAG, an application was submitted through the NHS Digital Data Access Request Service (DARS) to access the required HES data. This application was reviewed and endorsed by the Independent Group Advising on the Release of Data (IGARD). NHS Digital and the involved entities signed a Data Sharing Agreement (DSA) to secure access to the HES data. Approval for the ASPIRE (Sheffield EMR) research database was granted by the NHS ethics committee (STH21033 IRAS 269963 REC ref 19/EM/0331).
Supporting information
Supporting information.
{kind=link}
Supporting information.
ACKNOWLEDGMENTS
Medical writing and editing support were provided by Diana Steinway, Robin Marwick, and W. Mark Roberts of Stratenym Inc., funded by Actelion Pharmaceuticals Ltd., a Janssen Pharmaceutical Company of Johnson & Johnson. Technical support for data wrangling, linkage, and pseudo‐anonymization was performed by James Allsopp, Scientific Computing, Sheffield Teaching Hospitals NHS Foundation Trust, Royal Hallamshire Hospital, Sheffield, UK. Data from the NHS Hospital Episode Statistics (HES) database is Copyright© 2022, NHS Digital. Re‐used with the permission of NHS Digital. All rights reserved.
Lawrie A, Hamilton N, Wood S, Exposto F, Muzwidzwa R, Raiteri L, Beaudet A, Muller A, Sauter R, Pillai N, Kiely DG. Healthcare resource utiliZation and quality of life in patients with sarcoidosis‐associated pulmonary hypertension. Pulm Circ 2022;12:e12136. 10.1002/pul2.12136
REFERENCES
- 1. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS), and the World Association of Sarcoidosis and (WASOG) adopted by the ATS Board of Directorsdirectors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736–55. 10.1164/ajrccm.160.2.ats4-99 [DOI] [PubMed] [Google Scholar]
- 2. Samaranayake CB, McCabe C, Wort SJ, Price LC. Sarcoidosis associated pulmonary hypertension: an update. Curr Opin Pulm Med. 2021;27:285–95. 10.1097/MCP.0000000000000793 [DOI] [PubMed] [Google Scholar]
- 3. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153–65. 10.1056/NEJMra071714 [DOI] [PubMed] [Google Scholar]
- 4. Huitema MP, Grutters JC, Rensing BJWM, Reesink HJ, Post MC. Pulmonary hypertension complicating pulmonary sarcoidosis. Netherlands Heart J. 2016;24:390–9. 10.1007/s12471-016-0847-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahmoud AR, Dahy A, Dibas M, Abbas AS, Ghozy S, El‐Qushayri AE. Association between sarcoidosis and cardiovascular comorbidity: a systematic review and meta‐analysis. Heart Lung. 2020;49:512–7. 10.1016/j.hrtlng.2020.03.013 [DOI] [PubMed] [Google Scholar]
- 6. Huitema MP, Mathijssen H, Mager JJ, Snijder RJ, Grutters JC, Post MC. Sarcoidosis‐associated pulmonary hypertension. Semin Respir Crit Care Med. 2020;41:659–72. 10.1055/s-0040-1713615 [DOI] [PubMed] [Google Scholar]
- 7. Huitema MP, Bakker ALM, Mager JJ, Rensing B, Smits F, Snijder RJ, Grutters JC, Post MC. Prevalence of pulmonary hypertension in pulmonary sarcoidosis: the first large European prospective study. Eur Respir J. 2019;54:1900897. 10.1183/13993003.00897-2019 [DOI] [PubMed] [Google Scholar]
- 8. Kirkil G, Lower EE, Baughman RP. Predictors of mortality in pulmonary sarcoidosis. Chest. 2018;153:105–13. 10.1016/j.chest.2017.07.008 [DOI] [PubMed] [Google Scholar]
- 9. Pabst S, Grohe C, Skowasch D. Prevalence of sarcoidosis‐associated pulmonary hypertension: cumulative analysis of two PULSAR studies. Eur Respir J. 2020;55:1902223. 10.1183/13993003.02223-2019 [DOI] [PubMed] [Google Scholar]
- 10. Rapti A, Kouranos V, Gialafos E, Aggeli K, Moyssakis J, Kallianos A, Kostopoulos C, Anagnostopoulou O, Sfikakis PP, Wells AU, Tzelepis GE. Elevated pulmonary arterial systolic pressure in patients with sarcoidosis: prevalence and risk factors. Lung. 2013;191:61–7. 10.1007/s00408-012-9442-4 [DOI] [PubMed] [Google Scholar]
- 11. Pabst S, Hammerstingl C, Grau N, Kreuz J, Grohe C, Juergens UR, Nickenig G, Skowasch D. Pulmonary arterial hypertension in patients with sarcoidosis: the Pulsar single center experience In: Pokorski M, editor. Respiratory regulation—Clinical advances in experimental medicine and biology. 755. Dordrecht: Springer; 2013. p. 299–305. [DOI] [PubMed] [Google Scholar]
- 12. Handa T, Nagai S, Miki S, Fushimi Y, Ohta K, Mishima M, Izumi T. Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. 2006;129:1246–52. 10.1378/chest.129.5.1246 [DOI] [PubMed] [Google Scholar]
- 13. Baughman RP, Engel PJ. Sarcoidosis‐associated pulmonary hypertension: diagnosis and treatment. Adv Pulm Hypertens. 2015;14:138–44. 10.21693/1933-088X-14.3.138 [DOI] [Google Scholar]
- 14. Shino MY, Lynch Iii JP, Fishbein MC, McGraw C, Oyama J, Belperio JA, Saggar R. Sarcoidosis‐associated pulmonary hypertension and lung transplantation for sarcoidosis. Semin Respir Crit Care Med. 2014;35:362–71. 10.1055/s-0034-1376863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shlobin OA, Baughman RP. Sarcoidosis‐associated pulmonary hypertension. Semin Respir Crit Care Med. 2017;38:450–62. 10.1055/s-0037-1603767 [DOI] [PubMed] [Google Scholar]
- 16. Sulica R, Teirstein AS, Kakarla S, Nemani N, Behnegar A, Padilla ML. Distinctive clinical, radiographic, and functional characteristics of patients with sarcoidosis‐related pulmonary hypertension. Chest. 2005;128:1483–9. 10.1378/chest.128.3.1483 [DOI] [PubMed] [Google Scholar]
- 17. Currie BM, Davies EW, Beaudet A, Stassek L, Kleinman L, Baughman RP. Symptoms, impacts, and suitability of the Pulmonary Arterial Hypertension‐Symptoms and Impact (PAH‐SYMPACT) questionnaire in patients with sarcoidosis‐associated pulmonary hypertension (SAPH): a qualitative interview study. BMC Pulm Med. 2021;21:365. 10.1186/s12890-021-01694-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baughman RP, Wells A. Advanced sarcoidosis. Curr Opin Pulm Med. 2019;25:497–504. 10.1097/mcp.0000000000000612 [DOI] [PubMed] [Google Scholar]
- 19. Baughman RP, Engel PJ, Taylor L, Lower EE. Survival in sarcoidosis‐associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. 2010;138:1078–85. 10.1378/chest.09-2002 [DOI] [PubMed] [Google Scholar]
- 20. Nardi A, Brillet PY, Letoumelin P, Girard F, Brauner M, Uzunhan Y, Naccache JM, Valeyre D, Nunes H. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J. 2011;38:1368–73. 10.1183/09031936.00187410 [DOI] [PubMed] [Google Scholar]
- 21. Baughman RP, Culver DA, Cordova FC, Padilla M, Gibson KF, Lower EE, Engel PJ. Bosentan for sarcoidosis‐associated pulmonary hypertension: a double‐blind placebo‐controlled randomized trial. Chest. 2014;145:810–7. 10.1378/chest.13-1766 [DOI] [PubMed] [Google Scholar]
- 22. Boucly A, Cottin V, Nunes H, Jaïs X, Tazi A, Prévôt G, Reynaud‐Gaubert M, Dromer C, Viacroze C, Horeau‐Langlard D, Pison C, Bergot E, Traclet J, Weatherald J, Simonneau G, Valeyre D, Montani D, Humbert M, Sitbon O, Savale L. Management and long‐term outcomes of sarcoidosis‐associated pulmonary hypertension. Eur Respir J. 2017;50:1700465. 10.1183/13993003.00465-2017 [DOI] [PubMed] [Google Scholar]
- 23. Bonham CA, Oldham JM, Gomberg‐Maitland M, Vij R. Prostacyclin and oral vasodilator therapy in sarcoidosis‐associated pulmonary hypertension: a retrospective case series. Chest. 2015;148:1055–62. 10.1378/chest.14-2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baughman RP, Shlobin OA, Gupta R, Engel PJ, Stewart JI, Lower EE, Rahaghi FF, Zeigler J, Nathan SD. Riociguat for sarcoidosis‐associated pulmonary hypertension: results of a 1‐year double‐blind, placebo‐controlled trial. Chest. 2021;161:448–57. 10.1016/j.chest.2021.07.2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kiely DG, Doyle O, Drage E, Jenner H, Salvatelli V, Daniels FA, Rigg J, Schmitt C, Samyshkin Y, Lawrie A, Bergemann R. Utilising artificial intelligence to determine patients at risk of a rare disease: idiopathic pulmonary arterial hypertension. Pulm Circ. 2019;9:2045894019890549. 10.1177/2045894019890549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bergemann R, Allsopp J, Jenner H, Daniels FA, Drage E, Samyshkin Y, Schmitt C, Wood S, Kiely DG, Lawrie A. High levels of healthcare utilization prior to diagnosis in idiopathic pulmonary arterial hypertension support the feasibility of an early diagnosis algorithm: the SPHInX project. Pulm Circ. 2018;8:2045894018798613. 10.1177/2045894018798613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. NHS Digital . Hospital Episode Statistics (HES). https://digital.nhs.uk/data-and-information/data-tools-and-services/data-services/hospital-episode-statistics (2021). Accessed 18 Mar 2022.
- 28. NHS Digital . Hospital Episode Statistics (HES) analysis guide. https://digital.nhs.uk/data-and-information/data-tools-and-services/data-services/hospital-episode-statistics/users-uses-and-access-to-hospital-episode-statistics (2019). Accessed 14 Feb 2021.
- 29. Yorke J, Corris P, Gaine S, Gibbs JS, Kiely DG, Harries C, Pollock V, Armstrong I. emPHasis‐10: development of a health‐related quality of life measure in pulmonary hypertension. Eur Respir J. 2014;43:1106–13. 10.1183/09031936.00127113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hendriks PM, van Thor MCJ, Wapenaar M, Chandoesing P, van den Toorn LM, van den Bosch AE, Post MC, Boomars KA. The longitudinal use of EmPHasis‐10 and CAMPHOR questionnaire health‐related quality of life scores in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Respir Med. 2021;186:106525. 10.1016/j.rmed.2021.106525 [DOI] [PubMed] [Google Scholar]
- 31. NHS . Past national tariffs: documents and policies. https://www.england.nhs.uk/publication/past-national-tariffs-documents-and-policies/ (2021). Accessed 18 Mar 2022.
- 32. Exposto F, Hermans R, Nordgren Å, Taylor L, Sikander Rehman S, Ogley R, Davies E, Yesufu‐Udechuku A, Beaudet A. Burden of pulmonary arterial hypertension in England: retrospective HES database analysis. Ther Adv Respir Dis. 2021;15:1753466621995040. 10.1177/1753466621995040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis RA, Armstrong I, Bergbaum C, Brewis MJ, Cannon J, Charalampopoulos A, Church AC, Coghlan JG, Davies RJ, Dimopoulos K, Elliot C, Gibbs J, Gin‐Sing W, Haji G, Hameed AG, Howard LS, Johnson MK, Kempny A, Kiely DG, Lo Giudice F, McCabe C, Peacock AJ, Peleyeju O, Pepke‐Zaba J, Polwarth G, Price L, Sabroe I, Schreiber BE, Sheares K, Taboada D, Thompson A, Toshner MR, Wanjiku I, Wort SJ, Yorke J, Condliffe R. EmPHasis‐10 health‐related quality of life score predicts outcomes in patients with idiopathic and connective tissue disease‐associated pulmonary arterial hypertension: results from a UK multicentre study. Eur Respir J. 2021;57:2000124. 10.1183/13993003.00124-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borgese M, Badesch D, Bull T, Chakinala M, DeMarco T, Feldman J, Ford HJ, Grinnan D, Klinger JR, Bolivar L, Shlobin OA, Frantz RP, Sager JS, Mathai SC, Kawut S, Leary PJ, Gray MP, Popat RA, Zamanian RT. EmPHasis‐10 as a measure of health‐related quality of life in pulmonary arterial hypertension: data from PHAR. Eur Respir J. 2020;57:2000414. 10.1183/13993003.00414-2020 [DOI] [PubMed] [Google Scholar]
- 35. Guillevin L, Armstrong I, Aldrighetti R, Howard LS, Ryftenius H, Fischer A, Lombardi S, Studer S, Ferrari P. Understanding the impact of pulmonary arterial hypertension on patients' and carers' lives. Euro Respir Rev. 2013;22:535–42. 10.1183/09059180.00005713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dobarro D, Schreiber BE, Handler C, Beynon H, Denton CP, Coghlan JG. Clinical characteristics, haemodynamics and treatment of pulmonary hypertension in sarcoidosis in a single centre, and meta‐analysis of the published data. Am J Cardiol. 2013;111:278–85. 10.1016/j.amjcard.2012.09.031 [DOI] [PubMed] [Google Scholar]
- 37. Parikh KS, Dahhan T, Nicholl L, Ruopp N, Pomann GM, Fortin T, Tapson VF, Rajagopal S. Clinical features and outcomes of patients with sarcoidosis‐associated pulmonary hypertension. Sci Rep. 2019;9:4061. 10.1038/s41598-019-40030-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baughman RP, Shlobin OA, Wells AU, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Birring SS, Kouranos V, O'Hare L, Baran JM, Cal JG, Lower EE, Engel PJ, Nathan SD. Clinical features of sarcoidosis associated pulmonary hypertension: results of a multi‐national registry. Respir Med. 2018;139:72–8. 10.1016/j.rmed.2018.04.015 [DOI] [PubMed] [Google Scholar]
- 39. Shlobin OA, Kouranos V, Barnett SD, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Lower EE, Engel PJ, Wort J, Price L, Wells AU, Nathan SD, Baughman RP. Physiological predictors of survival in patients with sarcoidosis‐associated pulmonary hypertension: results from an international registry. Eur Respir J. 2020;55:1901747. 10.1183/13993003.01747-2019 [DOI] [PubMed] [Google Scholar]
- 40. Wang J, Brusca S, Sharp M, Kolb TM, Hassoun PM, Damico RL, Mathai SC. Subtypes and outcomes of sarcoidosis‐associated pulmonary hypertension. Am J Respir Crit Care Med. 2019;199:A2514. 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A2514 [DOI] [Google Scholar]
- 41. Duong H, Bonham CA. Sarcoidosis‐associated pulmonary hypertension: pathophysiology, diagnosis, and treatment. Clin Pulm Med. 2018;25:52–60. 10.1097/CPM.0000000000000252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerke AK. Treatment of sarcoidosis: a multidisciplinary approach. Front Immunol. 2020;11:545413. 10.3389/fimmu.2020.545413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galiè N, Channick RN, Frantz RP, Grünig E, Jing ZC, Moiseeva O, Preston IR, Pulido T, Safdar Z, Tamura Y, McLaughlin VV. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53:1801889. 10.1183/13993003.01889-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. NHS England Specialised Commissioning Team . A11/P/c: trgeted therapies for use in pulmonary hypertension in adults. https://www.england.nhs.uk/wp-content/uploads/2018/07/Targeted-therapies-for-use-in-pulmonary-hypertension-in-adults.pdf (2015). Accessed 18 Mar 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.