

Summary
Transcriptional dysregulation is a prominent feature in leukemia. Here, we systematically surveyed transcription factor (TF) vulnerabilities in leukemia and uncovered TF clusters that exhibit context-specific vulnerabilities within and between different subtypes of leukemia. Among these TF clusters, we demonstrated that acute myeloid leukemia (AML) with high IRF8 expression was addicted to MEF2D. MEF2D and IRF8 form an autoregulatory loop via direct binding to mutual enhancer elements. One important function of this circuit in AML is to sustain PU.1/MEIS1 co-regulated transcriptional outputs via stabilizing PU.1’s chromatin occupancy. We illustrated that AML could acquire dependency on this circuit through various oncogenic mechanisms that results in the activation of their enhancers. In addition to forming a circuit, MEF2D and IRF8 can also separately regulate gene expression, and dual perturbation of these two TFs leads to a more robust inhibition of AML proliferation. Collectively, our results revealed a TF circuit essential for AML survival.
Subject areas: Biological sciences, Cancer, Transcriptomics
Graphical abstract
Highlights
-
•
MEF2D is a context-specific vulnerability in IRF8hi AML
-
•
MEF2D and IRF8 form a transcriptional circuit via binding to each other’s enhancers
-
•
MEF2D-IRF8 circuit supports PU.1’s chromatin occupancy and transcriptional output
-
•
MEF2D and IRF8 can regulate separate gene expression programs alongside the circuit
Biological sciences; Cancer; Transcriptomics
Introduction
Transcription factors (TFs) encompass one or multiple direct DNA binding domains and represent one of the most important classes of proteins in interpreting eukaryotic genomes. An estimated number of ∼1,600 TFs in the human genome are likely to engage DNA in a sequence-specific manner, and many of them are biochemically and functionally obscure (Lambert et al., 2018; Vaquerizas et al., 2009). Although nearly half of these TFs are ubiquitously expressed in all types of cells, a small number of TFs, termed master regulator TFs, are expressed in a cell type specific manner and play fundamental roles in cell fate determination (Buganim et al., 2013; Davis et al., 1987; Graf and Enver, 2009). Master TFs typically form interconnected autoregulatory loops to control each other’s expression and their downstream transcriptional programs by coordinately modulating their enhancer activities (Boyer et al., 2005; Lee and Young, 2013). Such transcriptional circuitry enables rapid induction and sustained augmentation of gene expression programs to determine a cell state (Lee and Young, 2013; Young, 2011).
Malignant transformation involves transcriptional dysregulation mediated by the master TF circuitry (Bradner et al., 2017). For example, in acute myeloid leukemia (AML), an aggressive form of cancer characterized by the overproduction of immature myeloid precursor cells, recurrent oncogenic alterations occur in TFs and epigenetic regulators, such as CEBPA, RUNX1, GATA2, and KMT2A (also known as Mixed-Lineage Leukemia 1 (MLL1)), which are essential for stem cell renewal and myeloid differentiation (Döhner et al., 2015; Khwaja et al., 2016). A second category of TFs exploited in leukemia is non-oncogenic dependencies, which are not frequently mutated but required for sustaining malignant states, such as FLI1, ERG, and ZFP64 in AML (Lu et al., 2018; Roe et al., 2015; Xu et al., 2018). However, there remains an unmet need for a comprehensive understanding of TF-mediated coordination of gene regulation in cancer, which may ultimately foster the development of effective and selective therapeutic strategies. In this study, we highlight a comprehensive identification of TF clusters in a broad range of leukemias, which consequently may be utilized as predictive markers of context-specific vulnerabilities.
Myocyte Enhancer Factor (MEF2, A-D) proteins are evolutionarily conserved MADS (MCM1, AGAMOUS, DEFICIENS and SRF) TFs implicated in normal muscle, neuron, and blood system cell development, as well as in human diseases (Di Giorgio et al., 2018; Krivtsov et al., 2006; Pon and Marra, 2015; Potthoff and Olson, 2007). Among them, MEF2D is reported to be upregulated in certain types of cancer (Ma et al., 2014; Xu and Zhao, 2016), and MEF2D-fusion is a prediction marker for a poor clinical prognosis in B-cell precursor ALL (BCP-ALL) and may be involved in leukemogenesis (Gu et al., 2016a; Liu et al., 2016; Ohki et al., 2019; Suzuki et al., 2016; Tsuzuki et al., 2020). Interferon Regulatory Factor 8 (IRF8), a hematopoietic lineage-specific TF, has been implicated as a key myeloid and B lymphoid cell fate determinator (Holtschke et al., 1996; Lu et al., 2003; Tamura et al., 2015; Wang et al., 2008). Both MEF2D and IRF8 were nominated as AML essential transcriptional vulnerabilities from TF-focused CRISPR screens (Cao et al., 2021; Lu et al., 2018). Mechanistically, we found that an epigenetic reader protein ZMYND8 directly regulates IRF8 transcriptionally and IRF8 regulates MEF2D expression in AML cells. Of interest, our transcriptomic profiling of MEF2D deficient cells revealed that IRF8 could be one of its downstream targets (Cao et al., 2021). However, the essential role of MEF2D in leukemia, whether and how MEF2D maintains IRF8 expression, and why the IRF8-MEF2D network is important for AML remain to be fully investigated.
In this study, we demonstrated that genetic perturbation of MEF2D led to robust suppression of cell proliferation in a subset of AMLs with high MEF2D expression. On chromatin, we found that MEF2D regulated IRF8 transcription through occupying an AML-specific IRF8 enhancer, which eventually formed a positive feedback loop to maintain high MEF2D expression. Furthermore, we systematically identified an IRF8-bound intronic enhancer of MEF2D, which we illustrate to have high chromatin accessibility in several patient-relevant genetic alterations such as KMT2A-rearrangement. Current models depicting transcriptional circuitries in cancer focus on convergent roles of master TFs in sustaining oncogenic transcriptional programs (Bradner et al., 2017). Here, we found that MEF2D and IRF8 convergently co-regulated a set of genes, and divergently their own respective transcriptional programs to support AML. Mechanistically, the MEF2D-IRF8 circuit functionally supports the transcriptional output of PU.1, and dual perturbation of MEF2D and IRF8 displayed a more profound effect on inhibiting AML proliferation than individual gene perturbation. Together, our results highlighted the coordination between master TFs in the regulation of AML-specific transcriptional program and identified the MEF2D-IRF8 pathway as an AML-specific TF vulnerability.
Results
Integrative analysis of TF dependency map in leukemia identifies distinct TF clusters that exhibit context-specific vulnerabilities
Genome-wide CRISPR-based knockout genetic screens across a broad spectrum of cancer cell lines, such as the DepMap portal, provide rich resources for cancer type-specific vulnerability nominations (Dharia et al., 2021; Tsherniak et al., 2017). To comprehensively annotate leukemia-biased transcriptional vulnerabilities, we surveyed a total of 990 cancer cell lines including 23 AML, 7 chronic myeloid leukemia (CML), 4 T-ALL and 15 B-cell leukemia cell lines. After filtering pan-essential and non-essential genes, we established 66 TFs that displayed leukemia-biased vulnerabilities for further analysis. Unsupervised clustering of their dependency scores (copy-number-adjusted essentiality score, or CERES) (Meyers et al., 2017) nominated five distinct classes of TFs (Figure 1A). Among them, class I, II, and IV displayed the most distinguishable and cancer-type related features. Class I contained TFs that represent a class of blood-lineage specific vulnerabilities, such as MYB, ZEB2, and CBFB, that were essential for proliferation of most leukemia cell lines (Li et al., 2017; Tang et al., 2000; Zuber et al., 2011a) (Figures 1A and 1B). Class II contained TFs, such as SPI1, IRF8, MEF2D and CEBPA, that were essential for the proliferation of a subset of AML cell lines, many of which harbor a KMT2A-rearrangement or amplification (hereafter called KMT2A-r or KMT2A-amp, respectively) (Table S5) (Cao et al., 2021; Liss et al., 2021; Ohlsson et al., 2014; Zhou et al., 2014), whereas Class IV included TFs that were required for B cell leukemia proliferation (Figures 1A, 1B, and S1B–S1D). The Class III and V clustering revealed no strong exclusivity of TF dependencies in specific leukemias, but rather a broader requirement pattern across these subtypes. Notable TFs in Class III included GATA1 and STAT5B, which were both essential for AML and CML proliferation, whereas Class V included TFs such as BCL11B, TCF3, and TCF12 that were more prominently required for T-cell development/T-ALL proliferation (Ha et al., 2017; Kollmann et al., 2019; Shimizu et al., 2008; Staal et al., 2016; Wingelhofer et al., 2018; Zhan et al., 2017) (Figure 1A).
Figure 1.

Integrative analysis of TF dependency map in leukemia
(A) Heatmap (left) depicts hierarchically clustered dependency scores (CERES) of 66 TFs that are significantly enriched in AML, CML or ALL cell lines. A total of 49 leukemia cell lines with CRISPR screens available on DepMap were plotted. Pan-essential and non-essential genes were excluded. A bubble plot (right) indicates number of cell lines (by size) that are vulnerable (CERES < -0.45) to indicated TF perturbation, where color density represents significant enrichment level. AML, acute myeloid leukemia; CML, chronic myeloid leukemia; ALL, acute lymphoblastic leukemia.
(B) Violin plots of representative TF CERES effects in leukemia.
(C) Pearson correlation matrix of CERES dependency scores of AML-biased TF dependencies. A total 23 AML cell lines were used. Genetic information of cell lines used for this analysis can be found in Table S5.
We then interrogated the correlation between TF dependencies within the same subtype of leukemia. Individual hierarchical clustering of CERES in AML, CML and ALL revealed differential dependency profiles of the TF subsets (Figures S1E–S1G). In AML, HOXA10, MEIS1, MEF2C, MYB, ZFP64 and IRF8 are positively associated together. The dysregulation of these TFs are commonly reliant on certain types of AML including those harboring KMT2A-r or KMT2A-amp (Figure 1C) (Cao et al., 2021; Krivtsov et al., 2006; Lu et al., 2018; Zuber et al., 2011a,2011b). Conversely, RUNX1 and CBFB, which form a heterodimeric complex and were shown to be downregulated in KMT2A-r AML (Martinez-Soria et al., 2018; Tang et al., 2000; Zhang et al., 2003; Zhao et al., 2014), were clustered into the second subset. In ALL, B- and T-ALL biased dependencies were well separated, whereas pan-leukemic essential genes such as MYB, RUNX1 and CBFB, tend to correlate with both classes of dependencies (Figure S1E). In CML, we also observed two distinct classes of TFs (Figure S1F), implying that these TFs may be involved in different cellular pathways in the context of BCR-ABL/Ph + leukemia (Wainberg et al., 2021; Wang et al., 2017; Zhang, 2008).
We further assessed the molecular signatures that the 66 leukemia-biased TFs are involved in. Gene Ontology (GO) analysis of the TFs revealed a common signature of significant enrichment of molecular function in enhancer binding and transcription activator, and biological processes such as hematopoietic cell differentiation, T and B cell activation and cell fate commitment (Figure S1G). Using a disease-related gene prioritization tool (Jourquin et al., 2012; Liao et al., 2019), we found that these TFs were predominately enriched for various leukemia types (Figure S1H). These observations suggest that cancer cells are likely to exploit similar, if not the same, transcriptional programs that were primitively utilized by the cell of origin.
MEF2D is an AML-biased dependency
Our integrative TF analysis nominated hits that are not well studied as leukemia dependencies, including ZNF629, YBX3, ZNF98 and ZNF675 in lymphoblastic leukemia, ID3 in B-ALL, and MEF2D, ARID2 and TFAP4 in AML. We focused on MEF2D, a potent and selective vulnerability that has not yet been extensively characterized in a subset of AML (Figures 1 and2A). First, we cloned two independent sgRNAs for MEF2D and confirmed efficient depletion of MEF2D through immunoblotting (Figure S2A). To evaluate the knockout effects of MEF2D, we conducted competition-based proliferation assays in a collection of leukemia cell lines with different cytogenetics. Cas9-expressing cells were partially transduced with sgRNA expression vector linked with GFP, and relative cell growth rate was calculated by tracking the sgRNA positive (GFP positive) population over a time course. Perturbation of MEF2D strongly impaired the growth of AML cell lines, including MOLM-13, MV4; 11 and THP-1 that harbor KMT2A-r, as well as OCI-AML3 and OCI-AML5 that carry DNMT3A/NRAS/NPM1c or ASXL1/RUNX1/TET2/EZH2 mutations, respectively, but did not influence the growth of other cancer cell lines, such as K562 JURKAT, OPM1, A375 and HUH7 (Figures 2B, S2B, and S2C).
Figure 2.

MEF2D is an AML-biased dependency
(A) Violin plots of MEF2D CERES effects.
(B) Competition-based proliferation assays in MOLM-13 (AML, upper left), MV4; 11 (AML, upper right), THP1 (AML, bottom left) and K562 (an acute (erythro)blastic transformation of prior CML, bottom right) cell lines stably expressing Cas9. Plotted are relative GFP/sgRNA + population normalized based on day 3 (n = 3–5, mean ± SEM). sgNeg, negative control. sgPCNA, positive control.
(C) Scatterplot that shows a linear correlation between MEF2D’s CERE dependency scores and mRNA expression in leukemia (red) or other cancer cell lines (light gray) in the DepMap dataset. Each dot represents one cell line; the shaded regions indicate 95% confidence interval for the linear regression model.
(D) Immunoblotting of MEF2D in indicated whole-cell lysates. MEF2Dhi cell lines are labeled in purple. HCC, Hepatocellular carcinoma.
Moreover, we sought to assess the requirement of MEF2D in normal myeloid cell development. We transduced murine bone marrow cells constitutively expressing Cas9 with sgRNA targeting Mef2d (Figure S3D), confirmed the on-target knockout through immunoblotting (Figure S3E), and assessed the development of normal myeloid cells in colony formation assays. We did not observe a significant difference in the frequency and the absolute colony number of hematopoietic progenitor cells on Mef2d depletion (Figure S3F). This result is consistent with the notion that loss of Mef2d in a mouse model did not impede normal myeloid cell development (Pattison et al., 2020).
MEF2D has two human isoforms annotated with different Transcription Start Site (TSS). Both isoforms encode almost identical proteins, except one that excludes a small fraction of the Trans-activation Domain 2 (TAD2) through alternative splicing (Figure S2G). To verify the on-target effect of the MEF2D sgRNA, we transduced AML cells with CRISPR-resistant MEF2D-long or short cDNA and found that both isoforms were able to fully rescue the growth-arrest phenotype caused by sgRNA-mediated MEF2D inactivation (Figures S2H–S2J). This data suggested a redundant function of the two MEF2D isoforms in AML survival, although AML cell lines and primary patient samples mostly expressed the long isoform, as demonstrated by qPCR and RNA-seq (Figures S2K and S2L). In addition, inspection of cancer cell line encyclopedia (CCLE) dataset (Barretina et al., 2012) revealed a positive correlation between MEF2D’s CERES and its mRNA expression in leukemia cell lines (Figure 2C). We further confirmed by immunoblotting that high expression level of MEF2D protein was associated with MEF2D dependency in a subset of AML (Figure 2D). AML cell lines that were most reliant on MEF2D expressed high levels of MEF2D, whereas cell lines that had a basal level of MEF2D expression did not require MEF2D for their proliferation. Together, our findings validate MEF2D as a TF vulnerability in a subset of AML.
MEF2D and IRF8 form an autoregulatory loop in AML via direct binding of mutual enhancer
We set out to further explore the relationship between the leukemia-biased dependencies and their mRNA expression levels by performing a pair-wise correlation analysis. This analysis revealed that the expression levels of several TF groups were mutually positively correlated with their vulnerabilities in leukemia (Figure 3A). Evaluation of these correlated gene expression profiles may provide a rationale to guide the clinical application of TF pathway focused targeted therapy. For example, the high expression levels of TFEB, PAX5, EBF1, and FOX O 1 might be able to serve as prediction markers for EBF1 and PAX5 dependencies in B-cell leukemia (Figure 3A). Of note, we found that the high expression of a TF group (MEF2D, IRF8, HOXA10, MEIS1, SPI1, CEBPA, ZEB2, and GFI1) collectively predicted individual members’ dependencies in AML, which may imply a possible interconnection of their regulatory pathways. Among these TFs, we focused on investigating the uncharacterized function of MEF2D in AML.
Figure 3.

MEF2D-IRF8 regulatory loop in AML
(A) Similarity matrix (left) of 66 leukemia-enriched TF dependencies in their (minus) CERES effects (row) versus mRNA expressions (column) hierarchically clustered by pair-wise Pearson correlation in 49 leukemia cell lines. Heatmap (right) implicates average expression level of TFs in different subtypes of leukemia.
(B) RNA-seq rank plot of gene expression changes in MOLM-13 (up, (Cao et al., 2021)) or MV4; 11 cells 4–5 days after transduction of two independent sgNeg or sgMEF2D. Dark dots indicate IRF8-regulated genes in Figure S3A.
(C) Scatterplot of MEF2D CERES versus IRF8 CERES in leukemia cell lines.
(D and E) Scatterplot that shows a linear correlation between IRF8’s mRNA expression and MEF2D’s CERES dependency scores (D) or MEF2D’s mRNA expression and IRF8’s CERES dependency scores (E). Each dot represents one cell line; the shaded regions indicate 95% confidence interval for the linear regression model.
(F) Pearson correlation analysis of MEF2D mRNA expression with other genes in 173 AML patients. Data retrieved from TCGA dataset.
(G) Scatterplot of MEF2D and IRF8 expression in 173 AML patients.
(H) Meta-profile (top) and density plot (bottom) of MEF2D CUT&RUN and H3K27ac ChIP-seq peaks in MOLM-13 cells. Peaks are ranked by MEF2D CUT&RUN tag counts.
(I) Gene tracks of H3K27ac and MEF2D enrichment at IRF8 locus in MOLM-13 cells. Shadowed in gray is the AML-specific IRF8 enhancer (IE). (J) CUT&RUN-qPCR against MEF2D on IRF8 locus (n = 3).
We previously showed that IRF8 can directly modulate MEF2D expression in AML (Figure S3A), and MEF2D may also regulate IRF8 expression (Cao et al., 2021). We proposed that IRF8 and MEF2D could form a transcriptional circuit, but the direct downstream targets of MEF2D have not yet been investigated. To assess MEF2D-regulated transcriptional programs in AML, we analyzed RNA sequencing (RNA-seq) 4–5 days after CRISPR-mediated MEF2D depletion in 3 AML cell lines and a control cell line (K562). We observed a trend of downregulation for IRF8 and IRF8-regulated genes (defined as downregulated genes on IRF8 inactivation in 4 AML cell lines, Figure S3A) in MEF2D-addicted AML cell lines, but not in K562 cells which do not express IRF8 (Figures 3B, S3B, and S3C). Furthermore, we did not observe altered expression of other TFs that were grouped together with MEF2D and IRF8. Indeed, MEF2D and IRF8 shared a highly similar dependence pattern in leukemia (Figures 3C and 1A), and mRNA expression of one is highly correlated with the dependency score of the other in leukemia in comparison to other tumors (Figures 3D and 3E). We also retrieved AML patient transcriptomic profiling data from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network et al., 2013) and performed correlation analysis between IRF8/MEF2D and other genes. Notably, MEF2D mRNA was positively associated with IRF8 mRNA and IRF8-regulated genes in AML patients, with IRF8 as the top 1 gene correlated with MEF2D (Figures 3F, 3G, and S3D).
To further evaluate the chromatin occupancy of MEF2D and elucidate MEF2D-mediated gene regulation, we performed Cleavage Under Targets and Release Using Nuclease (CUT&RUN) (Skene et al., 2018) in parental MOLM-13 cells as well as cells transduced with either negative control and MEF2D targeting sgRNAs. This identified a total of 27,198 high confidence peaks of MEF2D. Genome-wide MEF2D-binding profiling revealed a significant overlap of MEF2D binding sites with the active histone mark H3K27ac. A large proportion of MEF2D sites were in promoters and distal elements that are likely to be cis-element (Figures 3H and S3E), consistent with an established role of MEF2D in transcriptional activation (Kim et al., 2008; Liu et al., 2007; Pon and Marra, 2015; Potthoff and Olson, 2007). Deconvolution of the MEF2D-derived sequence showed enrichment of a previously reported Mef2d motif in murine retina, as well as several hematopoietic lineage-specific TF-binding motifs, such as PU.1, FLI1, IRF8 (Figure S3F). Inspection of IRF8 locus revealed an enrichment of MEF2D signals at the previously identified AML-specific IRF8 enhancer region (IE) (Cao et al., 2021) in the parental cells or cells transduced with negative control sgRNA, but not cells with MEF2D targeting sgRNA (Figure 3I). The enrichment of MEF2D in the IRF8 IE was further validated by CUT&RUN-qPCR (Figure 3J). Furthermore, we utilized a targeted degradation system (Nabet et al., 2018) to allow rapid depletion of MEF2D to evaluate the primary transcriptional changes on MEF2D loss. We established a THP1 AML cell line with a knockout of the endogenous MEF2D and substituted with a FKBP12G36V-linked MEF2D cDNA (termed THP1-dMEF2D), which allowed us to rapidly deplete MEF2D on dTAG treatment (Figures S3G–S3I). We performed a time-course analysis of gene expression following dTAG treatment of THP1-dMEF2D cells and observed rapid loss of IRF8 transcript, but not other known AML oncogenic drivers such as FLT3 and MYC (Figure S3J). Collectively, these results support that MEF2D and IRF8 form a transcriptional circuit and regulate each other directly.
IRF8 supports PU.1’s chromatin occupancy and transcriptional output
To date, the underlying role of the MEF2D-IRF8 circuit in AML maintenance and the subsequent consequences of its activation have not yet been characterized. Previous studies reveal that IRF8 or its homologous protein IRF4 physically associates with PU.1, a master myeloid TF, on a composite DNA element (Eisenbeis et al., 1995; Tamura et al., 2015). IRF8 and PU.1 have also been suggested to play a tumor suppressive role in B cells, and that deletion of IRF8 and PU.1 led to B-ALL development in mice (Pang et al., 2016). In contrast to B-ALL, either genetic or chemical inhibition of PU.1 decreases KMT2A-r AML proliferation, partially because of the crosstalk between PU.1 and HOX/MEIS transcriptional programs (Antony-Debré et al., 2017; Zhou et al., 2014). These observations were in concert with the consequence of CRISPR-mediated PU.1 perturbation in leukemia, where PU.1 is essential for IRF8-dependent AML cell lines, but not B cell leukemia (Figure 4A and1A). Of note, PU.1 displayed a broader dependence across AML in comparison to IRF8, which may be because of PU.1’s ability to cooperate with other cell-type specific TFs, such as IRF4, CEBPα and RUNX1, for transcriptional regulation (Gu et al., 2018; Zhang et al., 1996) (Figures 1A and S1A).
Figure 4.

IRF8 supports PU.1’s chromatin occupancy and transcriptional output
(A) Scatterplot of IRF8 CERES versus PU.1 CERES in leukemia cell lines.
(B) GSEA analysis of IRF8-KO (left) and MEF2D-KO (right) RNA-seq data in MOLM-13 cells. PU.1 signature scatterplot of MEF2D CERES versus IRF8 CERES in leukemia cell lines, where the PU.1 signature is defined as the top 500 downregulated genes on PU.1 depletion. Normalized enrichment score (NES) and false discovery rate (FDR) q value are shown.
(C and D) Density plot (C) and meta-profiles (D) of PU.1, H3K27ac and H3K4me1 ChIP-seq signals at IRF8 binding sites in MOLM13-dIRF8 cells (Cao et al., 2021) on 4 h treatment of DMSO or dTAG that rapidly eliminates IRF8 protein. Peaks are ranked by IRF8 ChIP-seq tag counts (n = 3).
(E) Immunoblotting of IRF8, PU.1 and GAPDH in whole-cell lysates of MOLM13-dIRF8 cells treated with 500nM dTAG-47 for 4h.
(F and G) Gene tracks of ChIP-seq signal from Figures 4C and 4D at MEF2D (E) or CD180 (F) locus.
To evaluate whether IRF8 was functionally connected with PU.1 for gene regulation in AML, we performed transcriptomic profiling in MOLM-13 cells transduced with two independent sgRNAs targeting the DNA-binding domain of PU.1. Efficient PU.1 depletion was confirmed by immunoblotting (Figure S4B). Differential gene expression analysis revealed that loss of PU.1 resulted in the reduction of IRF8-regulated genes, but not IRF8 mRNA level (Figure S4C). A PU.1 gene signature was defined using the top 500 downregulated genes in AML with PU.1 depletion (Table S1), and gene set enrichment analysis (GSEA) showed a significant downregulation of PU.1 gene signature in both IRF8- and MEF2D-deficient AML cells, but not in K562 (Figures 4B, S4D, and S4E). Because we did not observe changes in PU.1 mRNA levels on IRF8 or MEF2D depletion, it raised a possibility that the transcriptional connection between PU.1 and the MEF2D-IRF8 transcriptional circuit might be mediated through protein-protein interaction on chromatin.
Previous studies suggest that IRF8 directs the monocyte- and dendritic cell (DC)-specific enhancer landscape establishment that are enriched for PU.1 during myeloid cell specification (Kurotaki et al., 2018; Tamura et al., 2015). Therefore, we sought to test whether IRF8 was required for PU.1 binding in AML. We utilized a previously established MOLM13-dIRF8 cell line that allowed rapid and efficient degradation of IRF8 within 1 h via the dTAG system (Cao et al., 2021). We treated the MOLM13-dIRF8 cells with 500 nM dTAG compound and performed ChIP-seq on PU.1, as well as histone markers H3K27ac and H3K4me1 that mark active cis-elements. Rapid elimination of IRF8 induced dramatic reduction of PU.1 signals at IRF8-binding sites, whereas H3K27ac, H3K4me1, and the total cellular PU.1 levels were largely unchanged (Figures 4C–4E). Furthermore, extended exposure to dTAG for 24 h only led to a very modest reduction of the H3K27ac and H3K4me1 marks (Figures S4F and S4G). Reduction of PU.1 binding was observed at the loci of MEF2D as well as other IRF8-regulated genes LY86 and CD180 (Figures 4F, 4G, S4H–S4J). Both LY86 and CD180 were implicated to be co-regulated by PU.1 and MEIS1, and were identified as leukemic stem cell markers in chemotherapy-resistant primary AML samples (Saito et al., 2010). These IRF8-dependent PU.1-enriched loci may be unique in these AML subtypes, as the PU.1 signals were stronger in the MOLM-13 and THP-1 cells in comparison to the U937 leukemia cells that harbored CALM-AF10 fusion and expressed low levels of both MEF2D and IRF8 (Figures 2D and S4H–S4J) (Minderjahn et al., 2020; Mohaghegh et al., 2019). These findings are consistent with the structural basis of PU.1/IRF/DNA ternary complex, where PU.1 and IRF4 engage on DNA in a cooperative manner, and this engagement relies on mutual protein-protein interactions (Escalante et al., 2002). To further analyze the cooperative interaction between IRF8 and PU.1, we surveyed IRF8- and PU.1-only sites and found that both these two types of regions had less active histone markers (H3K27ac and H3K4me1) in comparison to IRF8-PU.1 co-occupied regions (Figures S5A and S5B), implying that the IRF8- and PU.1-only sites tend to be less active in terms of gene transcription and enhancer function in AML. On dTAG-induced IRF8 depletion, we noted that PU.1 was preferentially lost in the IRF8-PU.1 co-occupied regions, as well as that slight loss of PU.1 in some of the PU.1-only regions (Figure S5C). We further confirmed these observations by performing ChIP-qPCR of eight representative genetic loci (Figure S5D). Collectively, these results suggest that the disassociation of PU.1 from IRF8-binding sites is an immediate event on IRF8 depletion, and loss of this interaction leads to suppression of PU.1-specific transcriptional programs required for AML proliferation.
MEF2D and IRF8 are upregulated in AML carrying KMT2A-r through enhancer reactivation
Our previous study indicates that MEF2D and IRF8 are upregulated in AML patients with different cytogenetics, and an enhancer-based IRF8 transcription activation is largely mediated by IE (Cao et al., 2021). Deletion of the homologous murine IE specifically decreased Irf8 expression in myeloid cells and induced loss of DCs and accumulation of Ly6C+ monocytes (Murakami et al., 2021), suggesting that cancer cells can hijack cell-type specific enhancers to sustain their dysregulated transcriptional network (Bahr et al., 2018; Shi et al., 2013). We surveyed the epigenetic profiles at the MEF2D locus and observed a spreading of H3K27ac-enriched and IRF8-bound region to ∼10kb downstream of the MEF2D long isoform TSS in MOLM-13 and THP-1 cells, both of which were KMT2A-r AML with high IRF8 expression (Figure 5A). In contrast, we observed that H3K27ac signals were more constrained around the TSS of MEF2D in both HEL and K562 leukemia cells that were KMT2A-WT with no IRF8 expression (Figure 5A). To further confirm this observation, we performed H3K27ac CUT&RUN in primary patient samples with KMT2A-r and noted a similar H3K27ac spreading at the MEF2D locus (Figure 5B). Inspection of DNase-seq (Assi et al., 2019) and ATAC-seq (Corces et al., 2016) dataset in AML patients, we observed that a differential chromatin accessibility of the IRF8-bound MEF2D intronic enhancer in comparison with the constitutively accessible regions at the TSS of the MEF2D long isoform (Figures S6A and S6B). In normal myelopoiesis, this IRF8-bound MEF2D region was transiently activated in granulocyte monocyte progenitor (GMP) in correspondence to an elevated MEF2D expression, whereas the MEF2D TSS region was constitutively accessible across the myeloid lineages (Figures S6C and S6D). This observation implied that this cell-state-specific MEF2D regulatory element might be hijacked by certain oncogenic drivers during leukemia pathogenesis. To functionally verify the potential regulatory element of MEF2D, we employed CRISPR interference (CRISPRi) (Fulco et al., 2016; Gilbert et al., 2013) to silence the H3K27ac-enriched regions at MEF2D locus in MOLM-13 cells (Figure 5C). We found that the sgRNA#1 targeting the IRF8-bound site caused a more profound inhibition of MEF2D transcription in comparison to the sgRNAs targeting the constitutively active MEF2D TSS region (Figures 5A–5D). To further evaluate the functionality of these newly identified enhancer elements, we utilized the optimized opAsCas12a system (Gier et al., 2020) to introduce multiple guide RNAs to the flanking regions of either the IRF8-bound site at MEF2D locus or MEF2D-bound site at IRF8 locus (Figures S6E and S6F). In both scenarios, we observed a mutual and simultaneous reduction of both IRF8 and MEF2D expressions (Figure S6G).
Figure 5.

MEF2D and IRF8 are upregulated in AML carrying KMT2A-r through enhancer reactivation
(A) Gene tracks of H3K27ac and IRF8 ChIP signals in MOLM-13, THP-1, HEL or K562 cells.
(B) Gene tracks of H3K27ac CUT&RUN signals in primary AML cells, with the key oncogenic mutations labeled on the left.
(C) Schematic of CRISPRi (KRAB-dCas9)-mediated epigenomic silencing at MEF2D locus. Red bars indicate sgRNA positions.
(D) RT-qPCR analysis of mRNA expression of MEF2D in MOLM-13 cells stably expressing dCas9-KRAB and transduced with indicated sgRNAs in Figure 5B harvested on 4 days post-infection. Relative mRNA levels were normalized to GAPDH levels. Plotted are the mean ± SEM (n = 3 for negative controls, n = 2 for MEF2D sgRNAs).
(E) Volcano plots of RNA-seq data in MA4- (left) or MA9- (right) transformed human CD34+ HSPCs versus normal HSPCs isolated from BM, where KMT2A translocation was induced via CRISPR-Cas9 (Secker et al., 2020).
(F) Competition-based proliferation assays in MA9-FLT3ITD (left) or MA9-NRASG12D (right) cells, which were generated by retroviral transduction of KMT2A-AF9 followed by FLT3 FLT3ITD and NRASG12D into human CD34+ HSPC cells, respectively (Wei et al., 2008; Wunderlich et al., 2013) (n = 3, mean ± SEM).
Given the observation that the MEF2D-IRF8 circuit was frequently upregulated in AML cell lines and primary patient samples carrying KMT2A-r, we then evaluated whether KMT2A-r oncogene itself was sufficient to activate this circuit. We first inspected the public ChIP-seq/CUT&RUN dataset of KMT2A protein and its associated histone H3K79me2 mark (Krivtsov et al., 2019) in MOLM-13 cells and observed both signals at MEF2D and IRF8 loci (Figures S6H and S6I). Next, we surveyed three additional independent datasets, which profiled transcriptomic changes of KMT2A/MLL-AF9 (MA9) or KMT2A/MLL-AF4 (MA4)-transformed human CD34+ HSPCs (Horton et al., 2013; Secker et al., 2020) or model leukemias (Barabé et al., 2017), and observed consistent elevation of MEF2D and IRF8, along with other well-known KMT2A target genes HOXA, MEIS1 and PBX3 (Figure 6E, S6J, and S6K). In pediatric MA9 AML that tends to have much lower mutational burdens (Barabé et al., 2017; Dharia et al., 2021; Gröbner et al., 2018; Ma et al., 2018), MEF2D and IRF8 upregulation was also observed (Figure S6K). Furthermore, CRISPR perturbation of either IRF8 or MEF2D in two MA9-immortalized human CD34+ cell models, with either containing cooperative oncogenic FLT3ITD or NRASG12D mutations, led to a potent proliferation inhibition (Figures 5F and S6L). Of note, Flt3-ITD and Npm1c mutations were shown to synergistically rewire chromatin contacts at the Irf8 locus and upregulate Irf8 expression (Yun et al., 2021). Together, these results imply that dependency on the MEF2D-IRF8 transcriptional circuit can be attributed to different leukemia-driving events that converge on altering the epigenetic state of the MEF2D and IRF8 loci.
Figure 6.

Divergent function of MEF2D- and IRF8- regulated programs in supporting AML
(A) Competition-based proliferation assays in THP-1 cells transduced with EV or MEF2D cDNA (n = 3–4, mean ± SEM).
(B) Competition-based proliferation assays in THP-1 cells transduced with EV or IRF8 cDNA (n = 3–4, mean ± SEM).
(C–F) Exemplary tracks of genes where MEF2D and IRF8 exact overlap (C), bind in proximity with each other (D), or only IRF8 (E) or MEF2D (F) are enriched.
(G) Meta-profile (top) and density plot (bottom) of IRF8, MEF2D, SPI1, H3K27ac and H3K4me1 enrichment at IRF8-MEF2D co-bound, IRF8-only and MEF2D-regions in MOLM-13 cells. Peaks are ranked by IRF8 or MEF2D tag counts.
(H) Meta-profile (top) and density plot (bottom) depicting detailed classification of IRF8-MEF2D co-bound regions: close, IRF8 and MEF2D peaks have at least 1bp overlap; left, IRF8 binds upstream and MEF2D downstream of the chromosome; right, IRF8 binds downstream and MEF2D upstream of the chromosome. IRF8, MEF2D, SPI1, H3K27ac and H3K4me1 enrichment were plotted. Peaks are ranked by IRF8 tag counts and centered by IRF8 peaks.
(I) Competition-based proliferation assays in THP-1 cells simultaneously transduced with two indicated sgRNAs linked with GFP or mCherry, which produced a mixed population of uninfected, single, and double infected cells. Relative fluorescent (GFP+/cherry+, GFP+/cherry-, GFP-/cherry+) proportion of a single or combinatorial sgRNA was normalized with uninfected cells as an internal control, and the normalized ratios were further compared with that on day 3 to calculate the enrichment of each population (y-axis). sgPos + sgNeg group showed the genome editing was not compromised when two sgRNAs were applied, and no additive/synergistic effect was seen in this combination. (n = 3 for infection with positive and negative controls, n = 5–8 for sgIRF8+ sgMEF2D, mean ± SEM. n.s, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.001, two-tailed Welch’s unpaired t-test).
Divergent function of MEF2D- and IRF8- regulated programs in supporting AML
MEF2D and IRF8 form a transcriptional circuit to control shared downstream genes in supporting AML survival, but it remains unclear whether there are divergent functions of MEF2D and IRF8 in AML. To test this, we investigated whether forced overexpression of either MEF2D or IRF8 was sufficient to rescue each other’s deficiency-induced growth defect in AML. Notably, our results demonstrate that ectopic overexpression of either MEF2D or IRF8 in this circuit was able to partially alleviate, but not fully rescue, the growth-arrest phenotype following inactivation of the other in AML, whereas their ectopic overexpression failed to alleviate the growth-arrest phenotype induced by inactivating a pan-essential gene PCNA (Figures 6A, 6B and S7A–S7D). Collectively, these data suggested that MEF2D and IRF8 functioned convergently and divergently in supporting AML.
To investigate the divergent function of MEF2D- and IRF8- regulated programs, we examined their chromatin occupancies in AML. Closer inspection of genes enriched for MEF2D and IRF8 binding revealed co-regulated downstream targets (Figures 6C and 6D) and individually regulated genes by MEF2D and IRF8, respectively (Figures 6E and 6F). We noted that MEF2D and IRF8 co-occupied at many of the IRF8-regulated genes (Figures 6C and 6D), which suggested that MEF2D might cooperatively regulate IRF8-targeted genes. We noted that these MEF2D and IRF8 co-occupied regions displayed two distinct patterns: 1) exact overlap with IRF8 peaks, and 2) not exactly at, but in proximity with IRF8 peaks (Figures 6C and 6D). Of note, the MEF2D-only regions included CEBPE gene, which has been implicated in myeloid differentiation and immune function (Shyamsunder et al., 2019; Yamanaka et al., 1997) and were upregulated on either IRF8 or MEF2D depletion (Cao et al., 2021) (Figures 3B and S3B).
To further explore their relationship globally, we performed genome-wide co-occupancy analysis of MEF2D and IRF8. We first counted two peaks as overlap when their peak summit positions are 500 bp or less apart and identified 7,954 sites that were co-occupied by these two TFs, and 79.8% of IRF8-enriched and 69.3% of MEF2D-enriched regions were not bound by the other (Figure 6G). As expected, IRF8 was accompanied by PU.1 regardless of MEF2D binding, and unique MEF2D-enriched regions were not positively associated with high PU.1 signals. Notably, IRF8/PU.1/MEF2D co-occupied regions conferred the highest active marks. Alongside, IRF8/PU.1-only regions were weakly enriched for both H3K27ac and H3K4me1, and MEF2D-only regions correlated with low H3K4me1 but comparable H3K27ac with those bound by all the three TFs, implying that these regions may not converge as enhancers but possibly as other types of regulatory elements. Within the overlap of MEF2D-IRF8 pairs, a substantial number (around 40%) were larger than 61 bp (Figure S7E). We further classified the overlap regions based on the proximity of MEF2D peaks in accordance with whether IRF8 peaks are within (close) or over 61bp distance upstream (left) or downstream (right) (Figure S7E). Meta-profiles and density plots revealed 35.6% of the overlap regions fell into the left or right subsets. Strikingly, we also observed a correlation of the overlap regions with the H3K27ac active marks, wherein the enrichment of H3K27ac tended to enclose the MEF2D binding sites. This may suggest that the overlap between MEF2D and IRF8 shifts in correspondence to extended active marks in chromatin.
Considering that MEF2D and IRF8 not only form a transcriptional circuit to control gene expression convergently but also regulate their perspective genes expression programs divergently, we hypothesized that dual inactivation of MEF2D and IRF8 can further suppress leukemia cell proliferation in comparison to individual inactivation. To test this, we simultaneously transduced AML cells with sgMEF2D and sgIRF8 linked with different fluorescence reporter markers (GFP or mCherry), which produced a mixed population of non-infected, single, and dual color cells. Relative growth rates were monitored by the fluorescence percentage normalized to the non-infected control. Indeed, we observed a more severe growth defect in the cell population that received both MEF2D- and IRF8- targeting sgRNAs, in comparison to all the combinations with other sgRNAs (Figures 6I and S7F), which was consistent with the notion that MEF2D and IRF8 had both convergent and divergent functions that are essential for AML proliferation.
Discussion
AML originates from immature hematopoietic cells. During leukemic transformation, cells undergo sporadic but continuous mutations which consequently lead to clonal diversification and heterogeneity (Li et al., 2016). Indeed, these mutational heterogeneities are starkly illustrated in our analysis of TF dependencies in leukemia, which highlighted differential, even opposite requirements between TFs in different subtypes of AML. Therefore, identification of the hallmarks for each subtype of AML, such as driver mutations, chromatin state, and marker gene expression, is critical for targeted therapeutic discovery. Our study mainly focuses on a subset of TFs composed of the MEF2D-IRF8 circuit and certain myeloid TFs, whose high expression levels indicate their vulnerabilities in AML, which can then be exploited for guiding future therapeutic development.
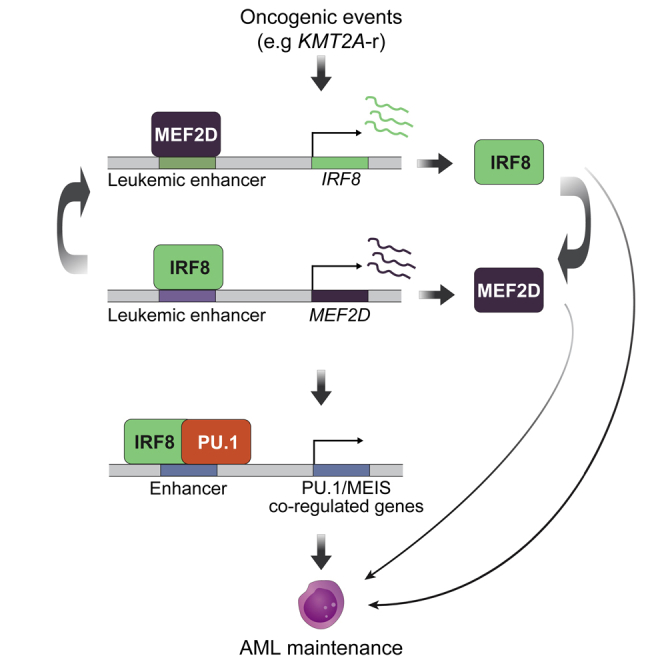
Our previous study on IRF8’s role in sustaining AML observes IRF8 directly binding to the MEF2D locus and suggests that IRF8 and MEF2D form a transcriptional circuit. Thus, our current study sought to delineate the functional role of MEF2D in this TF circuit and investigate the convergent role of this circuit, as well as whether MEF2D’s role differentiates from that of IRF8 in AML maintenance. In addition, the acquisition of dependence on this circuit and its immediate functional output were previously unknown. Here, we leveraged comprehensive approaches to extensively demonstrate the essential role of MEF2D in AML, functionally characterized a patient-relevant intronic enhancer of MEF2D and uncovered the formation of the MEF2D-IRF8 circuit via binding to mutual enhancer element. To date, recent studies revealed the essential roles and activation of MEF2D in AML carrying KMT2A-r, and different mechanisms underlying AML maintenance by MEF2D were proposed, including inhibition of CEBPE-mediated myeloid differentiation program, and direct regulation of MYC and HOXA9, which is largely redundant with its homolog TF MEF2C (Harada et al., 2022; Zhao et al., 2021). Although our model suggests the IRF8-MEF2D circuit and MYC program function in parallel in AML, we cannot entirely rule out the context- and cell type-specific roles of MEF2D. Thus, these findings can be complementary to our study and further explain the non-redundant function of MEF2D to IRF8. In line with Harada et al. (2022) highlighting the importance understanding the mechanism by which the IRF8/MEF2D TF circuit is activated, we found that AML carrying certain types of mutations, such as KMT2A-fusion and FLT3ITD and NPM1c cooperating mutations (Yun et al., 2021), can subsequently activate the MEF2D-IRF8 circuit and consequently require it for AML maintenance. To date, the concerted interaction on chromatin between IRF8 and PU.1 has not yet been shown in cellular context. We deciphered the cooperation of IRF8 with PU.1 to specifically sustain essential PU.1/MEIS1 co-regulated transcriptional programs in AML (Zhou et al., 2014). These transcriptional programs may be reinforced by both the MEF2D-IRF8 circuit and an independent MEF2D regulatory function (Figure 7), which could attribute to the enhanced proliferation arrest when MEF2D and IRF8 are simultaneously disrupted.
Figure 7.

A working model illuminating MEF2D-IRF8 circuit in AML
Oncogenic events (such as KMT2A-r and FLT3ITD + NPM1c) (Yun et al., 2021) can lead to activation and addiction to the MEF2D-IRF8 transcriptional circuit in AML. IRF8-PU.1 complex and MEF2D cooperatively maintain PU.1/MEIS1 co-regulated gene expression (Zhou et al., 2014) and AML state.
Previous studies using murine models showed that loss of Irf8 in mice causes a CML-like syndrome (Holtschke et al., 1996), and can facilitate development of acute promyelocytic leukemia (APL) in cooperation with PML-RARA fusion (Gaillard et al., 2018). Minimal reduction of PU.1 and downregulation of Irf8 are important events for preleukemic stem cell induction in mice with impaired DNA mismatch repair capacity (McKenzie et al., 2019; Will et al., 2015). In contrast to their reported tumor suppressive-like roles, we and others demonstrate that a subset of AMLs are dependent on IRF8 and PU.1 (Antony-Debré et al., 2017; Cao et al., 2021; Gozdecka et al., 2018; Liss et al., 2021; Yun et al., 2021; Zhou et al., 2014). We reason that the differential function of IRF8 and PU.1 in leukemia may be attributed to the cellular contexts and oncogenic mutations, which is supported by our leukemia dependency analysis. In addition, the anti- and pro-tumor activities in AML may also be influenced by the expression level of the TF, because precise regulation of myeloid TF concentration is critical for HSC and leukemic stem cell (LSC) development (Rosenbauer et al., 2005). This idea is supported by a recent study showing that too much or too little EWS-FLI1 oncoprotein will both abrogate tumor progression in Ewing Sarcoma (Seong et al., 2021). This principle may also be applicable in the case of IRF8 and PU.1 in leukemia, as well as other cancer dependencies, and further work is warranted to fully test these possibilities.
TFs are able to specifically modulate refined transcriptional programs and are sufficient to direct cell type specification, including differentiation, dedifferentiation and trans-differentiation (Fong and Tapscott, 2013; Henley and Koehler, 2021; Lambert et al., 2018; Takahashi and Yamanaka, 2016). It has been proposed that a TF inhibitor would theoretically confer high potency and specificity, in comparison to a signaling factor inhibitor that may impair multiple signaling pathways leading to unwanted toxicities (Bhagwat and Vakoc, 2015; Henley and Koehler, 2021). Despite obstacles in developing TF specific inhibitors (Bushweller, 2019; Darnell, 2002) and no MEF2D/IRF8 inhibitor available, advances have been made via different strategies, including protein-protein interface disruption and PROTAC/molecular glue-based degradation (Henley and Koehler, 2021), and targeting the druggable upstream regulators of the TFs (such as DOT1L, BRD4, ZMYND8 and SIK3) as an actionable strategy (Bernt et al., 2011; Cao et al., 2021; Daigle et al., 2011; Tarumoto et al., 2020; Zuber et al., 2011b). Because mouse models of Mef2d knockout showed neither apparent developmental abnormalities (Kim et al., 2008; Pon and Marra, 2015) nor defects in T, B cell or myeloid cell development (Pattison et al., 2020), we predict that therapeutic inhibition of MEF2D function or pathways may be an attractive future direction for selectively targeting AML with minimal toxicity.
Limitations of study
Although our analysis and in vitro data showed MEF2D is an AML-selective dependency, further work is needed in vivo to fully determine the role of MEF2D in leukemia progression as well as normal hematopoiesis. Our data suggest MEF2D and IRF8 may also function independently to sustain AML, but the underlying mechanism remains to be elucidated. MEF2D-IRF8 circuit can be activated through KMT2A rearrangement; however, we did not evaluate whether this circuit is required for leukemia initiation, and whether this circuit can be self-maintained or requires additional factors after activation. Another limitation of our study is that we did not evaluate the requirement of MEF2D in a more clinically relevant model, such as patient derived xenograft (PDX) or primary patient cells because of the challenge in conducting genome editing in these cells and lack of small molecule inhibitor against this circuit.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MEF2D | Abcam | Product #: ab32845 |
| Rabbit polyclonal anti-MEF2D | Bethyl Laboratories | Product #: A303-522A |
| Rabbit monoclonal anti-IRF8 | Abcam | Product #ab207418, Lot #GR3271578-1 |
| Rabbit polyclonal anti-PU.1 | Cell Signaling Technology | Product #: 2266 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse monoclonal anti-HA (clone 12CA5) | Laboratory of Gerd Blobel | N/A |
| Rabbit monoclonal anti-GAPDH | Cell Signalling Technology | Product #14C10 |
| Rabbit polyclonal anti-FKBP12 antibody | Abcam | Cat# ab24373 |
| Mouse monoclonal anti-VINCULIN | Santa Cruz Biotechnology | Product #: sc-73614 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat# ab4729; RRID: AB_211829 |
| Rabbit polyclonal anti-H3K4me1 | Abcam | Product #: ab8895 |
| PerCP anti-mouse CD45 Antibody | Biolegend | Cat# 103130 |
| IgG from rabbit serum | Sigma-Aldrich | Cat#: I8140 |
| Alexa Fluor® 680 Goat anti-mouse IgG (H + L) | Life Technologies | Product #A21058, Lot #1692967 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR | Product #926-3221, Lot #C81210-05 |
| Chemicals, peptides, and recombinant proteins | ||
| Halt Protease & Phosphatase Inhibitor Cocktail, EDTA-free (100x) | Thermo Fisher Scientific | Ref #78441, Lot #UF284419 |
| Glycogen | Roche | Ref #10901393001, Lot #11651224 |
| SuperScript II Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18064014 |
| AMPure XP | Beckman Coulter | A63880 |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 |
| Proteinase K | New England Biolabs | P8107S |
| Puromycin dihydrochloride | Sigma-Aldrich | P8833 |
| Blasticidin | Invitrogen | R21001 |
| Geneticin Selective Antibiotic (G418 Sulfate) | Thermo Fisher Scientific | 10131035 |
| Polyethylenimine, PEI | Polysciences, INC | 23966 |
| OPTI-MEM | Thermo Fisher Scientific | 31985070 |
| Hexadimethrine Bromide, Polybrene | Sigma-Aldrich | H9268 |
| Dynabeads Protein A | Thermo Fisher Scientific | Ref #10002D, Lot #00651865 |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 |
| T4 DNA polymerase | New England Biolabs | M0203L |
| T4 polynucleotide kinase | New England Biolabs | M0201L |
| Agarose, Standard, Low Electroendosmosis (EEO) | Avantor | A426-07 |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250 |
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio-Rad | 1610158 |
| 2XLaemmli Sample Buffer | Bio-Rad | 1610737 |
| Dimethyl Sulfoxide | Sigma-Aldrich | D2650 |
| Concanavalin A–coated Magnetic Beads | Bangs Laboratories | BP531 |
| Digitonin | EMD Millipore | 300410 |
| Spermidine | Sigma-Aldrich | S2501 |
| dTAG-47 | This study | N/A |
| pA-MN | This study | N/A |
| Spike-in DNA | Laboratory of Steven Henikoff | N/A |
| Roche Complete Protease Inhibitor (EDTA-free) tablets | Sigma-Aldrich | 5056489001 |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210 |
| Formaldehyde 37% Solution | Avantor | 2106-01 |
| RNase A | Thermo Fisher Scientific | EN0531 |
| Phenol/Chloroform/Isoamyl Alcohol | Thermo Fisher Scientific | BP1752I400 |
| NP-40 (Igepal CA-630) | Sigma | I8896 |
| rmIL3 | Peprotech | 213-13 |
| rmIL6 | Peprotech | 216-16 |
| rmSCF | Peprotech | 250-03 |
| Methylcellulose-based Medium with Recombinant for Mouse | STEMCELL technologies | M3434 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7570 |
| In-Fusion HD Cloning Kit | Takara Bio | 638909 |
| 2x Phusion Master Mix | Thermo Scientific | F-548 |
| Direct-zol RNA Miniprep Plus | Zymo Research | R2072 |
| QuantSeq 3′ mRNA-seq Library Prep Kit for Illumina | Lexogen | 015.96 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| Agilent High Sensitivity DNA Kit | Agilent | 5067-4626 |
| QIAquick PCR Purification Kit | QIAGEN | 28104 |
| Quick-DNA Miniprep Kit | ZYMO Research | D3025 |
| NucleoSpin Gel and PCR Clean-up Mini Kit | Macherey-Nagel | 740609.250 |
| Aligent RNA 6000 Nano Kit | Aligent | 5067-1511 |
| NEBNext® Library Quant Kit for Illumina | NEB | E7630 |
| NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® | NEB | E7770 |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | NEB | E7490 |
| Deposited data | ||
| RNA-seq, ChIP-seq and CUT&RUN data | This study | GSE186132 |
| ChIP-seq | (Tarumoto et al., 2018) | GSE109493 |
| ChIP-seq | (Mohaghegh et al., 2019) | GSE123872 |
| ChIP-seq | (Poppe et al., 2017) | GSE89212 |
| ChIP-seq and CUT&RUN | (Krivtsov et al., 2019) | GSE127508 |
| ChIP-seq | (Rathert et al., 2015) | GSE63782 |
| RNA-seq and ATAC-seq | (Corces et al., 2016) | GSE75384 |
| RNA-seq | (Cao et al., 2021) | GSE157249 |
| ChIP-seq and CUT&RUN | (Cao et al., 2021) | GSE157636 |
| ChIP-seq | (Minderjahn et al., 2020) | GSE128834 |
| RNA-seq | (Secker et al., 2020) | GSE148714 |
| RNA-seq | (Barabé et al., 2017) | GSE71800 |
| Dnase I-seq | (Assi et al., 2019) | GSE108316 |
| Experimental models: Cell lines | ||
| Human: MOLM-13 | DSMZ | ACC-554 |
| Human: MV4-11 | ATCC | CRL-9591 |
| Human: THP1 | ATCC | TIB-202 |
| Human: HEL | ATCC | TIB-180 |
| Human: OCI-AML3 | DSMZ | ACC-582 |
| Human: U937 | ATCC | CRL-1593.2 |
| Human: K562 | ATCC | CCL-243 |
| Human: JURKAT | ATCC | TIB-152 |
| Human: REH | ATCC | CRL-8286 |
| Human: HEK293T | ATCC | CRL-3216 |
| Human: A549 | ATCC | CCL-185 |
| Human : HUH7 | JCRB | JCRB0403 |
| Human: A375 | ATCC | CRL-1619 |
| Human: OCI-AML5 | Laboratory of James D. Griffin | NA |
| Human: MA9-ITD | (Wei et al., 2008; Wunderlich et al., 2013) | NA |
| Human: MA9-RAS | (Wei et al., 2008; Wunderlich et al., 2013) | NA |
| Experimental models: Organisms/strains | ||
| Constitutive-Cas9-GFP | JAX | Stock No: 026179 |
| Oligonucleotides | ||
| sgRNA sequence see Table S2 | This study | N/A |
| qPCR primers see Table S3 | This study | N/A |
| Recombinant DNA | ||
| LentiV_neo_empty | (Tarumoto et al., 2018) | Addgene: 108101 |
| LentiV_neo_SFFV_MEF2D_short | This study | N/A |
| LentiV_neo_SFFV_MEF2D_long | This study | N/A |
| LentiV_neo_IRF8 | (Cao et al., 2021) | N/A |
| LentiV_Cas9_puro | (Tarumoto et al., 2018) | N/A |
| LRG(Lenti_sgRNA_EFS_GFP) | (Tarumoto et al., 2018) | Addgene:65656 |
| LRG2.1 | (Tarumoto et al., 2018) | Addgene:108098 |
| LRcherry2.1 | (Tarumoto et al., 2018) | Addgene:108099 |
| pSL21-mCherry | (Chen et al., 2021) | Addgene:164410 |
| pRG212 | (Gier et al., 2020) | Addgene: 149722 |
| Software and algorithms | ||
| Bowtie2 v2.3.5 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| BEDtools v2.28.0 | (Quinlan and Hall, 2010) | http://bedtools.readthedocs.io/en/latest/ |
| Samtools v1.1 | (Li et al., 2009) | http://samtools.sourceforge.net |
| HOMER v4 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| bcl2fastq Conversion Software, v2.17 | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| MACS2 v2.1 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| UCSC Genome Browser | UCSC | http://genome.ucsc.edu/ |
| deepTools | (Ramírez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
| Picard tools v1.96 | Broad Institute | https://github.com/broadinstitute/picard |
| STAR v2.5.2 | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq, htseq-count, v0.6.1pL | (Anders et al., 2015) | https://htseq.readthedocs.io/en/release_0.11.1/ |
| R Bioconductor DESeq2 package v1.14.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MSigDB v6.1 | (Liberzon et al., 2015) | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| Cufflinks | (Trapnell et al., 2013) | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ |
| deepTools | (Ramírez et al., 2016) | https://deeptools.readthedocs.io/en/develop/index.html |
| IGVtools, 2.4.10 | Broad Institute | https://software.broadinstitute.org/software/igv/igvtools |
| GraphPad Prism 7 | GraphPad Software | N/A |
Resource availability
Lead contact
Further information and reasonable requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Junwei Shi (jushi@upenn.edu).
Materials availability
All plasmids will be deposited to Addgene for public requests (Addgene numbers in the key resources table).
Data and code availability
The accession number for the RNA-seq, ChIP-seq and CUT&RUN, generated in this study is: GSE186132. Data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not generate original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
Experimental model and subject details
Cell lines
HEK293T, A375 and HUH7 cell lines were cultured in DMEM (Corning) supplemented with 10% Bovine Calf Serum (FCS) and 1% Penicillin/Streptomycin. MOLM-13, MV4-11, THP-1, OCI-AML3, K562, U937, HEL, REH, OPM1 and JURKAT cell lines were cultured in RPMI-1640 (Gibco) supplemented with 10% FCS and 1% Penicillin/Streptomycin. MA9-ITD and MA9-RAS cell lines were cultured in RPMI-1640 supplemented with 10% Bovine Fetal Serum (FBS) and 1% Penicillin/Streptomycin. The OCI-AML5 cell line was cultured in alpha-MEM supplemented with 20% FBS, 1% Penicillin/Streptomycin and 10 ng/mL GM-CSF. All cells were maintained at 37°C with 5% CO2.
Primary AML samples
The protocols used for this study were approved by the University Pennsylvania’s institutional review board. The primary AML samples were obtained from the Stem Cell and Xenograft Core Facility at The Perelman School of Medicine at the University of Pennsylvania. Specimens were used after informed consent in accordance with the Declaration of Helsinki. The primary AML samples were frozen in FCS and 10% DMSO in liquid nitrogen until used. Patient clinical info is included in Table S4.
Mouse models
Around 12–16 weekold male Gt(ROSA)26Sortm1.1(CAG-cas9∗,-EGFP)Fezh mice (constitutively expressing Cas9-GFP mice) were used in this study and purchased from the Jackson Laboratory. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia.
Method details
sgRNA and plasmid cloning
Cancer cell lines with stable Cas9 expression used in this study were generated via lentiviral transduction of spCas9 expression vector (Addgene: 108,100) and MOLM-13 cell line with stable Cas12a expression were generated via transduction of the AsCas12a lentiviral expression vector (Addgene: 149,723), followed by 1–2 μg/mL puromycin selection for 1 week. For dCas9-KRAB-mediated MEF2D regulatory element inhibition, sgRNAs were designed to target the TSS of MEF2D (NM_005920) and the proximal H3K27ac-enriched regions. For the AsCas12a-mediated deletion of the MEF2D and IRF8 regulatory elements, sgRNAs were designed to delete the region surrounding the identified enhancer elements. The sense and antisense DNA oligos of all the human sgRNAs were annealed and phosphorylated using T4 PNK (NEB M0201S). For human Cas9 sgRNAs, the oligos were ligated with T4 DNA ligase (NEB B0202S) to either BsmBI digested LRG2.1 plasmid (Addgene: 108,098) or LRCherry2.1 (Addgene: 108,099) backbones co-expressing a GFP or Cherry fluorescent tag, respectively. For murine Cas9 sgRNAs, the oligos were ligated into the BbsI digested pSL21-mCherry (Addgene:164,410) vector. The AsCas12a crRNAs were ligated with T4 DNA ligase (NEB B0202S) to an AsCas12a crRNA vector co-expressing GFP and neomycin selection gene (Addgene: 149,722). All sgRNA/crRNA sequences are listed in Table S2.
MEF2D cDNA (NM_001271629 and NM_005920) were obtained from the MOLM-13 cDNA and subcloned into a modified LentiV_Neo lentiviral expression vector (Addgene: 108,101) with an EFS promoter swapped to SFFV using the In-Fusion cloning system (Takara Bio). PCR mutagenesis and In-Fusion cloning were used to introduce synonymous mutations CGA → AGG (R), ATC → ATT (I), ACC → ACT (T), GAC → GAT (D), GAG → GAA (E) and CGG → CGT (R) into the MEF2D cDNA to generate a sgRNA-resistant construct.
Virus production and transduction
For lentivirus production, HEK293T cells were co-transfected with the plasmid DNA of interest, lentiviral packaging plasmids pPAX2 and VSVG, and the cationic polymer reagent Polyethyleneimine (PEI). To produce lentivirus in 6-well plates, 5 μg of the DNA, 2.5 μg of VSVG, 3.5 μg pPAX2, 1 mg/mL PEI and 500 μL OPTI-MEM were used. To produce lentivirus in 10-cm plates, 10 μg of the DNA, 5 μg of VSVG, 7.5 μg pPAX2, 1 mg/mL PEI and 1 mL OPTI-MEM were used. The components were incubated for 18 min at room temperature and transfected into >95% confluent HEK293T cells. The HEK293T cells were incubated at 37°C with 5% CO2 for ∼6–8 h before aspiration of theold media and replenishment with fresh DMEM. Supernatant containing lentivirus was collected at 24, 48 and 72h post-transfection and filtered using a 0.45 μm PVDF membrane (Millex-HV). For retrovirus production, retroviruses were produced in 10-cm plates using 10 μg plasmid, 2 μg pCL-Eco, 2 μg VSVG, 40 μL PEI and 1 mL OPTI-MEM. Virus was collected at 24 and 48 h post-transfection, pooled together and filtered by the 0.45μm filter.
To lentivirally/retrovirally transduce the cells, the cells were combined with 4 μL/mL polybrene (2 mg/mL) and virus, followed by spin-infection at 650 x g for 25 min at room temperature. Media was replaced on 12 h postinfection, administered with appropriate antibiotic selection if needed (1 mg/mL G418).
Competition-based cell proliferation assay
To validate the effect of knockout on proliferation, cancer cell lines stably expressing spCas9 were transduced with LRG2.1 or LRcherry2.1 sgRNAs. The transduction of the sgRNAs were measured based on the fluorescence detected using Guava Easycyte HT Instrument (Millipore). For single gene validation, percentage of the GFP + or mCherry + transduced cells were measured on Day 3 post-infection and measured every 3 days for 15–21 days. The GFP% was normalized based on Day 3. For dual CRISPR-based disruption, cells were co-transduced with GFP and mCherry-tagged gRNAs and measured using the Guava Easycyte HT Instrument (Millipore) from day 3 post-infection. The percentage of transduction efficiency of non-transduced, GFP+, Cherry+, GFP+/Cherry+ were measured based on their ratios in the mixed population of cells.
Immunoblotting
Cells were counted using the Guava Easycyte HT Instrument (Millipore) and washed with 1X PBS. Proteins were lysed using a 26-gauge x 5/8-inch needle and 1 mL syringe (BD) in Laemmli sample buffer (Bio-Rad) containing 3% β-Mercaptoethanol. Extracted protein-containing lysates were boiled at 95°C for 7 min. Extracts were loaded onto 10% polyacrylamide gels and transferred to 0.45 μm nitrocellulose membranes. The membrane was blocked with 5% milk in TBST at room temperature in a shaking platform, followed by 1° antibody (MEF2D: Bethyl #A303-522A, PU.1: CST #2266, Vinculin: Santa Cruz #sc-73614, GAPDH: CST #5014, IRF8: Abcam #ab207418) diluted in 5% milk in TBST overnight incubation in a shaking platform at 4°C. Membrane was washed with PBST for 3 × 5 min. The 2° antibody solution was prepared with 1% secondary antibody and 1X LICOR blocking buffer in PBST. The membrane was incubated in the dark with the 2° antibody solution for 1h and imaged using the Odyssey CLx (LI-COR) imager. The image was quantified in Image™ Studio Lite.
Colony formation assay
Bone marrow cells were isolated from the femurs of Cas9-expressing mice (JAX). ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) was used to lyse the erythroid cells, and the remaining BM cells were cultured in IMDM media supplemented with 15% FBS, 1% Penicillin/Streptomycin, 10 ng/mL rmIL-6, 10 ng/mL rmIL-3 and 20 ng/mL rmSCF. BM cells were transduced with sgRNA in pSL21-mCherry retroviral vectors as described above. On day 2 post-infection, GFP+/mCherry + double-positive cells were sorted, and 25,000 cells were seeded into MethoCult GF M3434 media containing murine cytokines. Following 10 days of incubation, differentiated colonies were counted according to the manufacturer’s protocols, including burst-forming unit-erythroid (BFU-E), macrophage (CFU-GM), CFU-granulocyte and CFU-granulocyte, erythrocyte, macrophage, megakaryocyte (CFU-GEMM).
RNA-seq
MOLM-13 cells were harvested on day 5 post-infection of sgRNA, MV4; 11 and K562 cells were harvested on day 4 post-infection, and THP-1 cells were harvested on day 10 post-infection. Approximately 1–5 million cells transduced with sgRNAs were collected and lysed in Trizol (Invitrogen). Total RNA was isolated using the Direct-zol RNA Miniprep Plus kit (ZYMO) with DNAse I treatment, followed by RNA quality check using the RNA Nano 6000 Bioanalyzer kit (Agilent). RNA-seq libraries were prepared using the QuantSeq 3′ FWD mRNA-seq Library Prep Kit (Lexogen) with 0.5–2 μg of total RNA inputs following the manufacturer’s protocol. Library quality was assessed using the High Sensitivity Bioanalyzer kit (Agilent) and libraries were sequenced on the Nextseq 500 platform with 75 bp single-end reads.
Primary human samples were first washed with the Dead Cell Removal Kit (MACS) to remove dead cells. Total RNA was isolated from 1 to 3 million cells using Direct-zol RNA Miniprep Plus kit (ZYMO), followed by construction of the RNA-seq library using NEBNext Ultra II RNA Library Prep Kit for Illumina. One μg of total RNA was poly-A selected followed by fragmentation using the NEBNext PolyA(A) mRNA Magnetic Isolation module. The selected RNAs were subjected to a first and second strand cDNA synthesis. The libraries were end-repaired and linked with Illumina adaptors. The libraries were PCR amplified using 8 cycles, followed by sequencing using the Nextseq 500 platform with 75bp single-end reads or 2 × 42 paired-end reads. Sequencing data information was provided in Table S6.
ChIP-sequencing and ChIP-qPCR
For targeted degradation, MOLM13-dIRF8 (cells derived from MOLM13 expressing degradable IRF8 via dTAG system with endogenous IRF8 depletion) cells (Cao et al., 2021) were treated with either DMSO or 500 nM dTAG-47 compound and collected at 4 and 24h timepoints. For all the experiments, cells were collected in culture media, and crosslinked in 1% formaldehyde for 20 min at room temperature, and the reaction was sequestered with 0.125M Glycine in PBS for 10 min. The crosslinked cells were spun down, washed with PBS and frozen in −80°C until use. Ten million cells were used for H3K27ac and H3K4me1, and 50 million cells for the SPI1, respectively. Cell pellets were lysed in cell lysis buffer (10mM Tris pH 8.0, 10 mM NaCl, 0.2% NP-40) with protease inhibitor (Roche) and incubated for 15 min on ice. The cells were spun down for 30 s at 4,200 RPM and the buffer was removed. The cells were resuspended in 1mL nuclear lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS) per 10 million cells, followed by a 10 min incubation on ice. The cells were sonicated with a bioruptor for 5 min at 4°C and spun down at max speed for 15 min at 4°C. Supernatants containing fragmented chromatins were collected, diluted with 7mL IP dilution buffer (20 mM Tris pH 8.0, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.01% SDS) per 10 million cells, and applied with respective antibodies (PU.1: 10 μg, CST#2266; H3K27ac: 2 μg, #ab4729, H3K4me1: 2μg, #ab8895). For each antibody, 30 μL of protein A magnetic beads (Invitrogen) were applied after washing with 0.5% BSA in TBS for 3 times and blocked at 4°C on a rotor for 2h. The mixture was incubated overnight at 4°C in rotation. The beads were washed once with IP wash I buffer (20 mM Tris pH 8.0, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.01% SDS), twice with high salt buffer (20 mM Tris pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.01% SDS), once with IP wash 2 buffer (10 mM Tris pH 8.0, 1 mM EDTA, 0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate), and twice with 1xTE. The beads were incubated with 200 μL elution buffer (50mM Tris 8.0.10mM EDTA, 1%SDS), vortexed 3x for 5s, followed by shaking at 950 RPM at 65°C for 15 min. Supernatants were collected and reverse crosslinking was performed in 0.25M NaCl at 65°C overnight. non-DNA content was eliminated with 2 μL RNase A for 1hat 37°C, followed by 0.2 mg/mL Proteinase K for 2hat 42°C. ChIP DNA was purified using QIAquick PCR purification kit according to manufacturer’s instructions, and eluted with 40 μL water. ChIP-seq libraries were constructed using NEBNext Ultra II DNA Library Prep Kit for Illumina (E7645L) and the optimal cycle numbers were determined by NEBNext Library Quant Kit for Illumina (E7630L). Library quality was assessed using the high sensitivity Bioanalyzer kit (Agilent) and libraries were pooled and single-end sequenced for 75 bp on the Nextseq 500 platform. For ChIP-qPCR, PU.1 peak loci with or without IRF8 binding, and H3K27ac/H3K4me1-enriched regions obtained from ChIP-seq were selected. ChIP DNA was quantified using the ViiA 7 Real-Time PCR System (Applied Biosystems) with Power SYBR Green Master Mix (Thermo Fisher). A series of input dilution was used for normalization. qPCR Primer sequences and their chromosomal coordinates used for ChIP-qPCR are included in Table S3. Sequencing data information was provided in Table S6.
CUT&RUN and CUT&RUN qPCR
CUT&RUN experiments were slightly modified from a previous protocol (Cao et al., 2021; Skene et al., 2018). Concanavalin A-coated beads were washed with Binding buffer (20 mM HEPES-KOH, 10 mM KCl, 1 mM NaCl and 1 mM MnCl2) prior to use. For MOLM-13 cells, 2 × 106 live cells were collected, and an extra 2 × 106 cells were harvested and saved as input in CUT&RUN qPCR. For primary patient cells, 5 × 105 cells were purified from the Dead Cell Removal kit (MACS) before use. Cells were washed twice in Wash buffer (150 mM NaCl, 20 mM HEPES-KOH pH 7.9, 0.5 mM Spermidine and protease inhibitor cocktail), and incubated in 1mL of Wash buffer containing 20 μL of Concanavalin A-coated beads for 5–10 min with rotation at room temperature. Cell-attached beads were then resuspended with 300 μL Antibody buffer (Wash buffer with 0.05% Digitonin and 2mM EDTA) in combination with 20 μL MEF2D antibody (Abcam, #ab32845), 2μL of H3K27ac antibody (Abcam #ab4729) or 2 μL IgG overnight at 4°C with rotation. Based were washed once with Dig-wash buffer (Wash buffer with 0.05% Digitonin) and resuspended in 300 μL Dig-wash buffer with 700 ng/mL pA-MN purified protein at 4°C for 1 h with rotation. Beads were then washed twice with Dig-wash buffer, resuspended in Dig-wash buffer and chilled on a heat block on ice. Chromatin digestion was conducted in the presence of CaCl2 for 30 min and sequestered with an equal volume of 2x STOP solution (340 mM NaCl, 20 mM EDTA, 50 μg/mL RNase A, 4 mM EGTA, 0.02% Digitonin, 50 μg/mL Glycogen and 2 pg/mL heterogeneous spike-in DNA) at 37°C for 10 min. The supernatant was collected, and DNA and the input samples were subjected to Phenol-Chloro-form extraction. For CUT&RUN qPCR, the MEF2D peak loci at the IRF8 enhancer region were used for qPCR analysis. Sequencing libraries were constructed with NEBNext Ultra II DNA Library Prep Kit for Illumina following a previously described method (Liu et al., 2018) for TF or the default method for histone marks. Adapters were diluted 50x and 10–12 cycles of PCR amplification were applied. Library quality was assessed using the high sensitivity Bioanalyzer kit (Agilent) and libraries were pooled and paired-end sequenced for 2 × 42bp on the Nextseq 500 platform. qPCR Primer sequences and their chromosomal coordinates used for CUT&RUN-qPCR are included in Table S3. Sequencing data information was provided in Table S6.
RT-PCR
Total RNA was isolated using the Direct-zol RNA Miniprep Plus Kit (ZYMO) with DNAse I treatment manufacturer protocol. Reverse transcription of isolated RNA to cDNA was performed using RevertAid Reverse Transcriptase (Thermo) supplemented with 20 U/μL Riboblock. RT-PCR of cDNA was performed with PowerSYBR Green PCR Master Mix (Thermo #4367659) on the ABI 7900HT standard real-time PCR machine. All ΔCt values were normalized based on GAPDH expression. All the RT primers are listed in Table S3.
Deletion of regulatory elements
To delete the genomic regions at potential MEF2D or IRF8 enhancer elements bound by IRF8 or MEF2D, respectively, we utilized the Cas12a system (Gier et al., 2020) to induce simultaneous double-strand breaks flanking the TF binding sites. To enhance deletion efficiency, we designed four crRNAs on CRISPick (Doench et al., 2016), two of which target the upstream and two for the downstream of the element, with at least a 40bp gap between each crRNA. The combinatorial crRNAs targeting the IRF8 or MEF2D enhancer will result in a 540-680bp or 850-950bp deletion, respectively. A quadrupole crRNA combination for the ROSA locus was used as a negative control. The total 4 crRNAs were cloned in a crRNA array of the pRG212 lentiviral expression vector (Addgene: 149,722). MOLM-13 cells stably expressing AsCas12a were transduced with the designated array, followed by 4-day neomycin selection. Measurement of the GFP fluorescent tag using the Guava Easycyte HT Instrument (Millipore) was used for further validation of crRNA transduction and measurement of selection efficiency. To measure the efficiency of the enhancer deletion, gDNA was isolated using the Quick-DNA Miniprep Plus Kit (ZYMO) and genotyped. PCR products were run in 1% agarose gel and band intensities were measured using Image Lab (BioRad) with value normalized by product size. Percentage of enhancer deletion was calculated using normalized enhancer-deleted band intensity divided by sum of normalized enhancer-deleted and intact band intensity, multiplied by 100. To measure transcriptional changes upon enhancer deletion, total RNA was isolated using the Direct-zol RNA Miniprep Plus Kit (ZYMO), followed by RT-PCR. Genotyping primers were listed in Table S3.
Identification of leukemia-selective TF dependencies
Genome-wide “drop-out” CRISPR screen data were retrieved and analyzed on the DepMap portal (https://depmap.org/portal/) using the 21Q2 Public + Score CERES dataset containing perturbation effects of 17,645 genes in 990 cell lines. Briefly, CERES was calculated and scaled as previously described (Meyers et al., 2017), such that the median non-essential and common essential gene perturbation effects are 0 and -1, respectively. The pan-dependent genes were defined as those whose ranks in 90% of cell lines were above a given dependency cut-off. The cut-off value is determined by the central minimum in the 90th percentile lowest dependency line in the gene rank histogram (Dempster et al., 2019).
To identify potential leukemia-specific dependencies, CERES in 23 AML cell lines, 14 ALL cell lines and 7 CML cell lines (termed “in-group”) were compared with the remaining cell lines (term “out-group”) as previously described (Dharia et al., 2021). Briefly, t-statistics and log-odd ratios of differential CERES were calculated based on a fitted linear regression model. Effect size was determined by the difference between the CERES in the in-group and the out-group. Cancer subtype-biased dependencies were identified as those with a negative t-statistics and p value lower than 0.05. Common essential and non-essential genes were filtered out, and genes categorized as TF based on previous studies (Lambert et al., 2018; Lu et al., 2018; Vaquerizas et al., 2009) were used for further analysis. For analysis on TF dependencies in combination with their mRNA expression, RNA-seq datasets (Expression 21Q2 Public) in leukemia cell lines were downloaded on the DepMap portal, and pairwise Pearson correlation between CERES of one gene and mRNA expression (normalized to log2(TPM+1)) of the others were conducted in R. Hierarchical clustering on TF dependencies was performed with the pheatmap (https://rdrr.io/cran/pheatmap/) or ComplexHeatmap (Gu et al., 2016b) function in R.
RNA-seq analysis
Primary AML samples were aligned to hg19 using STAR Aligner (Dobin et al., 2013). For cell lines, RNA-seq analysis was performed on the Quantseq FWD 2.3.6 pipeline (Lexogen). Briefly, sequencing reads were aligned to hg38 using STAR Aligner (Dobin et al., 2013) with low quality reads and adaptor contamination removed. RPM-normalized track files (bigwig) were generated with the bedGraphToBigWig tool. Raw read counts were generated using HTseq-count (Anders et al., 2015). For CRISPR-mediated disruption samples, raw read counts >10 were retained. Differential gene expression analysis was performed using DE-Seq 1.30.1 (Love et al., 2014). GO and ORA analysis was performed on WebGestalt 2019 (Liao et al., 2019). GSEA analysis was performed in accord with the GSEA instruction (https://www.gsea-msigdb.org/gsea/index.jsp) with Molecular Signature Database v6.1 (MSigDB) (Liberzon et al., 2015) and custom gene signatures that are listed on Table S1.
ChIP-seq and CUT&RUN processing
For ChIP-seq and CUT&RUN analysis, reads were aligned to hg19 using Bowtie2 v2.3.5 (Langmead and Salzberg, 2012), following parameters as previously described (Skene et al., 2018):--local--very-sensitive-local--no-unal--no-mixed--no-discordant--phred33-I 10-X 700-k1-N1. Picard tools v1.96 (Broad Institute) was applied to displace presumed PCR duplicates using the MarkDuplicates command. Bam files with uniquely mapped reads retained were generated using Samtools v1.1 (Li et al., 2009). Fragments between 40 and 1000 bp were kept. Blacklist regions defined by ENCODE, random chromosomes, and mitochondria were removed, and filtered bam files were used for further analysis. Reads per kilobase per million mapped reads (RPKM)-normalized bigwig files were created using deeptools bamCoverage (Ramírez et al., 2016) with parameters-bs 10--smoothLength 50--normalizedUsing RPKM--skipNAs in cell lines, or default parameters in primary patient cells to visualize binding signals. Bigwig files of replicates were pooled together using WiggleTools mean setting. Tracks were loaded to UCSC genome browser for visualization. Pearson correlation of replicates were calculated using deepTools multiBigwigSummary bins with binning size as 10,000. Correlation heatmap were plotted using deepTools plotCorrelation with default hierarchical clustering.
CUT&RUN and ChIP-Seq enriched signals were called using MACS v2.1 (Zhang et al., 2008) with adjusted p value threshold 0.01 unless indicated. Genes in proximity with peaks were annotated against the hg19 genome using annotatePeaks.pl from HOMER v4 (Heinz et al., 2010).
Peak overlap and shifting analysis
The Venn diagram of comparisons of the ChIP/CUT&RUN peaks was generated using Bioconductor package ChIPpeakAnno (Zhu et al., 2010) function findOverlappingPeaks with mapped gap equal to 500bp. To identify shifts of IRF8 and MEF2D overlapped binding regions, we first calculated genomic distance (with orientation) and absolute distance between IRF8 and MEF2D peak summits. The distance distribution follows an approximate normal distribution. We then defined lower 20% of the peaks as IRF8 left-shifted peaks and upper 20% as IRF8 right-shifted peaks over MEF2D. Heatmaps and metaplots were generated using deepTools plotHeatmap (Ramírez et al., 2016).
Peak overlap and differential analysis
The Venn diagram of the IRF8 and PU.1 peaks comparison was generated using Bioconductor package ChIPpeakAnno (Zhu et al., 2010) function findOverlappingPeaks with mapped gap equal to 500bp. Metaplot and signal heatmap of the unique and overlapped enriched regions were generated using deepTools computeMatrix and plotHeatmap. We further quantified PU.1 read counts under IRF8 and PU.1 enriched regions using deepTools multiBigwigSummary BED-file setting to quantify RPKM normalized bigwig files. Using the same method, we then extended 1000bp on both sides of the narrow peaks, and quantified RPKM read counts of histone marks H3K27ac and H3K4me1. Differential analysis between DMSO and dTAG were performed using DESeq2. Boxplot of log2FC results from DESeq2 were plotted using ggplot2.
Quantification and statistical analysis
Statistical analysis was performed on either Graphpad Prism 9 or R. For linear regression, statistical analysis (p value) was calculated using a two-tailed F-test. For RNA-seq, ChIP-seq, RT-PCR and proliferation assay, statistical parameters including p value, biological replicates, SEM and FDR are indicated directly on the figures or in the corresponding figure legends.
Acknowledgments
We thank James D. Griffin for OCI-AML5 cell line, James C. Mulloy for retroviral human leukemia lines, Will Bailis for sharing Cas9+ murine BM cells, Mitchell J. Weiss and Ruopeng Feng for helpful suggestions, Yeqiao Zhou and Robert B. Faryabi for enhancer analysis, and Krista A. Budinich for helping with the sgRNA cloning and immunoblotting. This work was supported by NIH grant R01 CA258904 (to J.S.) and NIH training grant T32 DK07780 (to B.P.)
Author contributions
Z.C. and J.S. conceived of and designed the study. B.Y.P. and Z.C. performed most experiments and analyzed data; F.A. contributed to the murine colony formation assay; Q.C. established the Cas12a-mediated deletion system; Z.Z. contributed to the CUT&RUN experiments. H.H and Z.C. analyzed and interpreted the NGS data. J.Q. provided the dTAG-47 compound. K.M.B, S.L.B., Z.C., and J.S. supervised the research. B.Y.P., Z.C., and J.S. wrote the manuscript with inputs from all authors.
Declaration of interests
The authors declare no conflict of interest.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105139.
Contributor Information
Zhendong Cao, Email: zhendon@pennmedicine.upenn.edu.
Junwei Shi, Email: jushi@upenn.edu.
Supplemental information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony-Debré I., Paul A., Leite J., Mitchell K., Kim H.M., Carvajal L.A., Todorova T.I., Huang K., Kumar A., Farahat A.A., et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Invest. 2017;127:4297–4313. doi: 10.1172/JCI92504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assi S.A., Imperato M.R., Coleman D.J.L., Pickin A., Potluri S., Ptasinska A., Chin P.S., Blair H., Cauchy P., James S.R., et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 2019;51:151–162. doi: 10.1038/s41588-018-0270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C., von Paleske L., Uslu V.V., Remeseiro S., Takayama N., Ng S.W., Murison A., Langenfeld K., Petretich M., Scognamiglio R., et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515–520. doi: 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- Barabé F., Gil L., Celton M., Bergeron A., Lamontagne V., Roques É., Lagacé K., Forest A., Johnson R., Pécheux L., et al. Modeling human MLL-AF9 translocated acute myeloid leukemia from single donors reveals RET as a potential therapeutic target. Leukemia. 2017;31:1166–1176. doi: 10.1038/leu.2016.302. [DOI] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt K.M., Zhu N., Sinha A.U., Vempati S., Faber J., Krivtsov A.V., Feng Z., Punt N., Daigle A., Bullinger L., et al. MLL-Rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat A.S., Vakoc C.R. Targeting transcription factors in cancer. Trends Cancer. 2015;1:53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J.E., Hnisz D., Young R.A. Transcriptional addiction in cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushweller J.H. Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer. 2019;19:611–624. doi: 10.1038/s41568-019-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A.G., Hoadley K., Triche T.J., Laird P.W., et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Budinich K.A., Huang H., Ren D., Lu B., Zhang Z., Chen Q., Zhou Y., Huang Y.-H., Alikarami F., et al. ZMYND8-regulated IRF8 transcription axis is an acute myeloid leukemia dependency. Mol. Cell. 2021;81:3604–3622.e10. doi: 10.1016/j.molcel.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Arai E., Khan O., Zhang Z., Ngiow S.F., He Y., Huang H., Manne S., Cao Z., Baxter A.E., et al. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell. 2021;184:1262–1280.e22. doi: 10.1016/j.cell.2021.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S.R., Olhava E.J., Therkelsen C.A., Majer C.R., Sneeringer C.J., Song J., Johnston L.D., Scott M.P., Smith J.J., Xiao Y., et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Davis R.L., Weintraub H., Lassar A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Dempster J.M., Rossen J., Kazachkova M., Pan J., Kugener G., Root D.E., Tsherniak A. Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. Preprint at bioRxiv. 2019 doi: 10.1101/720243. [DOI] [Google Scholar]
- Dharia N.V., Kugener G., Guenther L.M., Malone C.F., Durbin A.D., Hong A.L., Howard T.P., Bandopadhayay P., Wechsler C.S., Fung I., et al. A first-generation pediatric cancer dependency map. Nat. Genet. 2021;53:529–538. doi: 10.1038/s41588-021-00819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio E., Hancock W.W., Brancolini C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta. Rev. Cancer. 2018;1870:261–273. doi: 10.1016/j.bbcan.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H., Weisdorf D.J., Bloomfield C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- Eisenbeis C.F., Singh H., Storb U. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 1995;9:1377–1387. doi: 10.1101/gad.9.11.1377. [DOI] [PubMed] [Google Scholar]
- Escalante C.R., Brass A.L., Pongubala J.M.R., Shatova E., Shen L., Singh H., Aggarwal A.K. Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol. Cell. 2002;10:1097–1105. doi: 10.1016/S1097-2765(02)00703-7. [DOI] [PubMed] [Google Scholar]
- Fong A.P., Tapscott S.J. Skeletal muscle programming and re-programming. Curr. Opin. Genet. Dev. 2013;23:568–573. doi: 10.1016/j.gde.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco C.P., Munschauer M., Anyoha R., Munson G., Grossman S.R., Perez E.M., Kane M., Cleary B., Lander E.S., Engreitz J.M. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard C., Surianarayanan S., Bentley T., Warr M.R., Fitch B., Geng H., Passegué E., de Thé H., Kogan S.C. Identification of IRF8 as a potent tumor suppressor in murine acute promyelocytic leukemia. Blood Adv. 2018;2:2462–2466. doi: 10.1182/bloodadvances.2018018929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gier R.A., Budinich K.A., Evitt N.H., Cao Z., Freilich E.S., Chen Q., Qi J., Lan Y., Kohli R.M., Shi J. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-17209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozdecka M., Meduri E., Mazan M., Tzelepis K., Dudek M., Knights A.J., Pardo M., Yu L., Choudhary J.S., Metzakopian E., et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018;50:883–894. doi: 10.1038/s41588-018-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T., Enver T. Forcing cells to change lineages. Nature. 2009;462:587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- Gröbner S.N., Worst B.C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V.A., Johann P.D., Balasubramanian G.P., Segura-Wang M., Brabetz S., et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- Gu X., Ebrahem Q., Mahfouz R.Z., Hasipek M., Enane F., Radivoyevitch T., Rapin N., Przychodzen B., Hu Z., Balusu R., et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Invest. 2018;128:4260–4279. doi: 10.1172/JCI97117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Churchman M., Roberts K., Li Y., Liu Y., Harvey R.C., McCastlain K., Reshmi S.C., Payne-Turner D., Iacobucci I., et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016;7 doi: 10.1038/ncomms13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Ha V.L., Luong A., Li F., Casero D., Malvar J., Kim Y.M., Bhatia R., Crooks G.M., Parekh C. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia. 2017;31:2503–2514. doi: 10.1038/leu.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T., Heshmati Y., Kalfon J., Perez M.W., Xavier Ferrucio J., Ewers J., Hubbell Engler B., Kossenkov A., Ellegast J.M., Yi J.S., et al. A distinct core regulatory module enforces oncogene expression in KMT2A-rearranged leukemia. Genes Dev. 2022;36:368–389. doi: 10.1101/gad.349284.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley M.J., Koehler A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021;20:669–688. doi: 10.1038/s41573-021-00199-0. [DOI] [PubMed] [Google Scholar]
- Holtschke T., Löhler J., Kanno Y., Fehr T., Giese N., Rosenbauer F., Lou J., Knobeloch K.-P., Gabriele L., Waring J.F., et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/S0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- Horton S.J., Jaques J., Woolthuis C., van Dijk J., Mesuraca M., Huls G., Morrone G., Vellenga E., Schuringa J.J. MLL-AF9-mediated immortalization of human hematopoietic cells along different lineages changes during ontogeny. Leukemia. 2013;27:1116–1126. doi: 10.1038/leu.2012.343. [DOI] [PubMed] [Google Scholar]
- Jourquin J., Duncan D., Shi Z., Zhang B. GLAD4U: deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012;13(Suppl 8):S20. doi: 10.1186/1471-2164-13-S8-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A., Bjorkholm M., Gale R.E., Levine R.L., Jordan C.T., Ehninger G., Bloomfield C.D., Estey E., Burnett A., Cornelissen J.J., et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers. 2016;2 doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- Kim Y., Phan D., van Rooij E., Wang D.-Z., McAnally J., Qi X., Richardson J.A., Hill J.A., Bassel-Duby R., Olson E.N. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Invest. 2008;118:124–132. doi: 10.1172/JCI33255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann S., Grundschober E., Maurer B., Warsch W., Grausenburger R., Edlinger L., Huuhtanen J., Lagger S., Hennighausen L., Valent P., et al. Twins with different personalities: STAT5B—but not STAT5A—has a key role in BCR/ABL-induced leukemia. Leukemia. 2019;33:1583–1597. doi: 10.1038/s41375-018-0369-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A.V., Twomey D., Feng Z., Stubbs M.C., Wang Y., Faber J., Levine J.E., Wang J., Hahn W.C., Gilliland D.G., et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Krivtsov A.V., Evans K., Gadrey J.Y., Eschle B.K., Hatton C., Uckelmann H.J., Ross K.N., Perner F., Olsen S.N., Pritchard T., et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 2019;36:660–673.e11. doi: 10.1016/j.ccell.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki D., Nakabayashi J., Nishiyama A., Sasaki H., Kawase W., Kaneko N., Ochiai K., Igarashi K., Ozato K., Suzuki Y., Tamura T. Transcription factor IRF8 governs enhancer landscape dynamics in mononuclear phagocyte progenitors. Cell Rep. 2018;22:2628–2641. doi: 10.1016/j.celrep.2018.02.048. [DOI] [PubMed] [Google Scholar]
- Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Young R.A. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Mar B.G., Zhang H., Puram R.V., Vazquez F., Weir B.A., Hahn W.C., Ebert B., Pellman D. The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood. 2017;129:497–508. doi: 10.1182/blood-2016-05-714493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Mason C.E., Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 2016;36:100–106. doi: 10.1016/j.gde.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J., Tamayo P. The molecular signatures Database hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss F., Frech M., Wang Y., Giel G., Fischer S., Simon C., Weber L.M., Nist A., Stiewe T., Neubauer A., et al. IRF8 is an AML-specific susceptibility factor that regulates signaling pathways and proliferation of AML cells. Cancers. 2021;13:764. doi: 10.3390/cancers13040764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Williams A.H., Kim Y., McAnally J., Bezprozvannaya S., Sutherland L.B., Richardson J.A., Bassel-Duby R., Olson E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA. 2007;104:20844–20849. doi: 10.1073/pnas.0710558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R., et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-F., Wang B.-Y., Zhang W.-N., Huang J.-Y., Li B.-S., Zhang M., Jiang L., Li J.-F., Wang M.-J., Dai Y.-J., et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–183. doi: 10.1016/j.ebiom.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B., Klingbeil O., Tarumoto Y., Somerville T.D.D., Huang Y.-H., Wei Y., Wai D.C., Low J.K.K., Milazzo J.P., Wu X.S., et al. A transcription factor addiction in leukemia imposed by the MLL promoter sequence. Cancer Cell. 2018;34:970–981.e8. doi: 10.1016/j.ccell.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Medina K.L., Lancki D.W., Singh H. IRF-4, 8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003;17:1703–1708. doi: 10.1101/gad.1104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Liu J., Liu L., Duan G., Wang Q., Xu Y., Xia F., Shan J., Shen J., Yang Z., et al. Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res. 2014;74:1452–1462. doi: 10.1158/0008-5472.CAN-13-2171. [DOI] [PubMed] [Google Scholar]
- Ma X., Liu Y., Liu Y., Alexandrov L.B., Edmonson M.N., Gawad C., Zhou X., Li Y., Rusch M.C., Easton J., et al. Pan-cancer genome and transcriptome analyses of 1, 699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Soria N., McKenzie L., Draper J., Ptasinska A., Issa H., Potluri S., Blair H.J., Pickin A., Isa A., Chin P.S., et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34:626–642.e8. doi: 10.1016/j.ccell.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie M.D., Ghisi M., Oxley E.P., Ngo S., Cimmino L., Esnault C., Liu R., Salmon J.M., Bell C.C., Ahmed N., et al. Interconversion between tumorigenic and differentiated states in acute myeloid leukemia. Cell Stem Cell. 2019;25:258–272.e9. doi: 10.1016/j.stem.2019.07.001. [DOI] [PubMed] [Google Scholar]
- Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S., et al. Computational correction of copy-number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minderjahn J., Schmidt A., Fuchs A., Schill R., Raithel J., Babina M., Schmidl C., Gebhard C., Schmidhofer S., Mendes K., et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 2020;11:402. doi: 10.1038/s41467-019-13960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh N., Bray D., Keenan J., Penvose A., Andrilenas K.K., Ramlall V., Siggers T. NextPBM: a platform to study cell-specific transcription factor binding and cooperativity. Nucleic Acids Res. 2019;47:e31. doi: 10.1093/nar/gkz020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K., Sasaki H., Nishiyama A., Kurotaki D., Kawase W., Ban T., Nakabayashi J., Kanzaki S., Sekita Y., Nakajima H., et al. A RUNX–CBFβ-driven enhancer directs the Irf8 dose-dependent lineage choice between DCs and monocytes. Nat. Immunol. 2021;22:301–311. doi: 10.1038/s41590-021-00871-y. [DOI] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A., et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki K., Kiyokawa N., Saito Y., Hirabayashi S., Nakabayashi K., Ichikawa H., Momozawa Y., Okamura K., Yoshimi A., Ogata-Kawata H., et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica. 2019;104:128–137. doi: 10.3324/haematol.2017.186320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson E., Hasemann M.S., Willer A., Lauridsen F.K.B., Rapin N., Jendholm J., Porse B.T. Initiation of MLL-rearranged AML is dependent on C/EBPα. J. Exp. Med. 2014;211:5–13. doi: 10.1084/jem.20130932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang S.H.M., Minnich M., Gangatirkar P., Zheng Z., Ebert A., Song G., Dickins R.A., Corcoran L.M., Mullighan C.G., Busslinger M., et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia. 2016;30:1375–1387. doi: 10.1038/leu.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison M.J., Naik R.J., Reyskens K.M.S.E., Arthur J.S.C. Loss of Mef2D function enhances TLR induced IL-10 production in macrophages. Biosci. Rep. 2020;40 doi: 10.1042/BSR20201859. BSR20201859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon J.R., Marra M.A. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget. 2016;7:2297–2312. doi: 10.18632/oncotarget.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe M., Wittig S., Jurida L., Bartkuhn M., Wilhelm J., Müller H., Beuerlein K., Karl N., Bhuju S., Ziebuhr J., et al. The NF-κB-dependent and -independent transcriptome and chromatin landscapes of human coronavirus 229E-infected cells. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff M.J., Olson E.N. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P., Roth M., Neumann T., Muerdter F., Roe J.-S., Muhar M., Deswal S., Cerny-Reiterer S., Peter B., Jude J., et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525:543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J.-S., Mercan F., Rivera K., Pappin D.J., Vakoc C.R. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell. 2015;58:1028–1039. doi: 10.1016/j.molcel.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F., Koschmieder S., Steidl U., Tenen D.G. Effect of transcription-factor concentrations on leukemic stem cells. Blood. 2005;106:1519–1524. doi: 10.1182/blood-2005-02-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y., Kitamura H., Hijikata A., Tomizawa-Murasawa M., Tanaka S., Takagi S., Uchida N., Suzuki N., Sone A., Najima Y., et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secker K.-A., Bruns L., Keppeler H., Jeong J., Hentrich T., Schulze-Hentrich J.M., Mankel B., Fend F., Schneidawind D., Schneidawind C. Only hematopoietic stem and progenitor cells from cord blood are susceptible to malignant transformation by MLL-AF4 translocations. Cancers. 2020;12 doi: 10.3390/cancers12061487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong B.K.A., Dharia N.V., Lin S., Donovan K.A., Chong S., Robichaud A., Conway A., Hamze A., Ross L., Alexe G., et al. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell. 2021;39:1262–1278.e7. doi: 10.1016/j.ccell.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Whyte W.A., Zepeda-Mendoza C.J., Milazzo J.P., Shen C., Roe J.-S., Minder J.L., Mercan F., Wang E., Eckersley-Maslin M.A., et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu R., Engel J.D., Yamamoto M. GATA1-related leukaemias. Nat. Rev. Cancer. 2008;8:279–287. doi: 10.1038/nrc2348. [DOI] [PubMed] [Google Scholar]
- Shyamsunder P., Shanmugasundaram M., Mayakonda A., Dakle P., Teoh W.W., Han L., Kanojia D., Lim M.C., Fullwood M., An O., et al. Identification of a novel enhancer of CEBPE essential for granulocytic differentiation. Blood. 2019;133:2507–2517. doi: 10.1182/blood.2018886077. [DOI] [PubMed] [Google Scholar]
- Skene P.J., Henikoff J.G., Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018;13:1006–1019. doi: 10.1038/nprot.2018.015. [DOI] [PubMed] [Google Scholar]
- Staal F.J.T., Famili F., Garcia Perez L., Pike-Overzet K. Aberrant Wnt signaling in leukemia. Cancers. 2016;8:78. doi: 10.3390/cancers8090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Okuno Y., Kawashima N., Muramatsu H., Okuno T., Wang X., Kataoka S., Sekiya Y., Hamada M., Murakami N., et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J. Clin. Oncol. 2016;34:3451–3459. doi: 10.1200/JCO.2016.66.5547. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Tamura T., Kurotaki D., Koizumi S.i. Regulation of myelopoiesis by the transcription factor IRF8. Int. J. Hematol. 2015;101:342–351. doi: 10.1007/s12185-015-1761-9. [DOI] [PubMed] [Google Scholar]
- Tang Y.Y., Shi J., Zhang L., Davis A., Bravo J., Warren A.J., Speck N.A., Bushweller J.H. Energetic and functional contribution of residues in the core binding factor beta (CBFbeta ) subunit to heterodimerization with CBFalpha. J. Biol. Chem. 2000;275:39579–39588. doi: 10.1074/jbc.M007350200. [DOI] [PubMed] [Google Scholar]
- Tarumoto Y., Lu B., Somerville T.D.D., Huang Y.-H., Milazzo J.P., Wu X.S., Klingbeil O., El Demerdash O., Shi J., Vakoc C.R. LKB1, Salt-Inducible Kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol. Cell. 2018;69:1017–1027.e6. doi: 10.1016/j.molcel.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarumoto Y., Lin S., Wang J., Milazzo J.P., Xu Y., Lu B., Yang Z., Wei Y., Polyanskaya S., Wunderlich M., et al. Salt-inducible kinase inhibition suppresses acute myeloid leukemia progression in vivo. Blood. 2020;135:56–70. doi: 10.1182/blood.2019001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M., et al. Defining a cancer dependency map. Cell. 2017;170:564–576.e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki S., Yasuda T., Kojima S., Kawazu M., Akahane K., Inukai T., Imaizumi M., Morishita T., Miyamura K., Ueno T., et al. Targeting MEF2D-fusion oncogenic transcriptional circuitries in B-cell precursor acute lymphoblastic leukemia. Blood Cancer Discov. 2020;1:82–95. doi: 10.1158/2643-3230.BCD-19-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Wainberg M., Kamber R.A., Balsubramani A., Meyers R.M., Sinnott-Armstrong N., Hornburg D., Jiang L., Chan J., Jian R., Gu M., et al. A genome-wide atlas of co-essential modules assigns function to uncharacterized genes. Nat. Genet. 2021;53:638–649. doi: 10.1038/s41588-021-00840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Lee C.H., Qi C., Tailor P., Feng J., Abbasi S., Atsumi T., Morse H.C. IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood. 2008;112:4028–4038. doi: 10.1182/blood-2008-01-129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Yu H., Hughes N.W., Liu B., Kendirli A., Klein K., Chen W.W., Lander E.S., Sabatini D.M. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 2017;168:890–903.e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Wunderlich M., Fox C., Alvarez S., Cigudosa J.C., Wilhelm J.S., Zheng Y., Cancelas J.A., Gu Y., Jansen M., et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B., Vogler T.O., Narayanagari S., Bartholdy B., Todorova T.I., da Silva Ferreira M., Chen J., Yu Y., Mayer J., Barreyro L., et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med. 2015;21:1172–1181. doi: 10.1038/nm.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingelhofer B., Maurer B., Heyes E.C., Cumaraswamy A.A., Berger-Becvar A., de Araujo E.D., Orlova A., Freund P., Ruge F., Park J., et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32:1135–1146. doi: 10.1038/s41375-017-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M., Mizukawa B., Chou F.-S., Sexton C., Shrestha M., Saunthararajah Y., Mulloy J.C. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–e97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Zhao Y.-C. MEF2D/Wnt/β-catenin pathway regulates the proliferation of gastric cancer cells and is regulated by microRNA-19. Tumour Biol. 2016;37:9059–9069. doi: 10.1007/s13277-015-4766-3. [DOI] [PubMed] [Google Scholar]
- Xu Y., Milazzo J.P., Somerville T.D.D., Tarumoto Y., Huang Y.-H., Ostrander E.L., Wilkinson J.E., Challen G.A., Vakoc C.R. A TFIID-SAGA perturbation that targets MYB and suppresses acute myeloid leukemia. Cancer Cell. 2018;33:13–28.e8. doi: 10.1016/j.ccell.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka R., Barlow C., Lekstrom-Himes J., Castilla L.H., Liu P.P., Eckhaus M., Decker T., Wynshaw-Boris A., Xanthopoulos K.G. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:13187–13192. doi: 10.1073/pnas.94.24.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R.A. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun H., Narayan N., Vohra S., Giotopoulos G., Mupo A., Madrigal P., Sasca D., Lara-Astiaso D., Horton S.J., Agrawal-Singh S., et al. Mutational synergy during leukemia induction remodels chromatin accessibility, histone modifications and three-dimensional DNA topology to alter gene expression. Nat. Genet. 2021;53:1443–1455. doi: 10.1038/s41588-021-00925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T., Rindtorff N., Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. The role of aberrant transcription factor in the progression of chronic myeloid leukemia. Leuk. Lymphoma. 2008;49:1463–1469. doi: 10.1080/10428190802163305. [DOI] [PubMed] [Google Scholar]
- Zhang D.E., Hohaus S., Voso M.T., Chen H.M., Smith L.T., Hetherington C.J., Tenen D.G. Function of PU.1 (Spi-1), C/EBP, and AML1 in early myelopoiesis: regulation of multiple myeloid CSF receptor promoters. Curr. Top. Microbiol. Immunol. 1996;211:137–147. doi: 10.1007/978-3-642-85232-9_14. [DOI] [PubMed] [Google Scholar]
- Zhang L., Li Z., Yan J., Pradhan P., Corpora T., Cheney M.D., Bravo J., Warren A.J., Bushweller J.H., Speck N.A. Mutagenesis of the Runt domain defines two energetic hot spots for heterodimerization with the core binding factor beta subunit. J. Biol. Chem. 2003;278:33097–33104. doi: 10.1074/jbc.M303972200. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9 doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Zhang P., Galbo P.M., Zhou X., Aryal S., Qiu S., Zhang H., Zhou Y., Li C., Zheng D., et al. Transcription factor MEF2D is required for the maintenance of MLL-rearranged acute myeloid leukemia. Blood Adv. 2021;5:4727–4740. doi: 10.1182/bloodadvances.2021004469. 2021004469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Chen A., Yan X., Zhang Y., He F., Hayashi Y., Dong Y., Rao Y., Li B., Conway R.M., et al. Downregulation of RUNX1/CBFβ by MLL fusion proteins enhances hematopoietic stem cell self-renewal. Blood. 2014;123:1729–1738. doi: 10.1182/blood-2013-03-489575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Wu J., Li B., Liu D., Yu J., Yan X., Zheng S., Wang J., Zhang L., Zhang L., et al. PU.1 is essential for MLL leukemia partially via crosstalk with the MEIS/HOX pathway. Leukemia. 2014;28:1436–1448. doi: 10.1038/leu.2013.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.J., Gazin C., Lawson N.D., Pagès H., Lin S.M., Lapointe D.S., Green M.R. ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinf. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Rappaport A.R., Luo W., Wang E., Chen C., Vaseva A.V., Shi J., Weissmueller S., Fellmann C., Fellman C., et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M., et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq, ChIP-seq and CUT&RUN, generated in this study is: GSE186132. Data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not generate original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.