

Abstract
Background
Pressure-induced constriction (PIC) is inherent to small arteries and arterioles, in which intraluminal pressure-induced vascular smooth muscle cell stretch elicits vasoconstriction. Degenerin (Deg) proteins, such as beta-epithelial Na+ channel (βENaC), have been studied in the PIC response because they are evolutionarily linked to known mechanosensors. While loss of Deg function phenotypes are plentiful, a gain-of-function phenotype has not been studied. The aim of this study was to determine if expression of exogenous βENaC in the isolated middle cerebral artery (MCA) enhances the PIC response.
Methods
Isolated MCA segments from female mice (24 weeks, n = 5) were transfected with enhanced green fluorescent protein–βENaC (EGFP–βENaC) or with EGFP alone, incubated overnight at 37 °C, then studied in a pressure myograph.
Results
Mechanical/morphological properties and vasoconstrictor responses to KCl and phenylephrine were identical in EGFP–βENaC and EGFP MCAs. In contrast, PIC responses were greater in EGFP–βENaC segments with ~2-fold greater peak myogenic tone.
Conclusions
These data confirm previous findings that βENaC is critical in the PIC response. These data provide proof-of-concept that upregulating βENaC can enhance PIC responses and lay the foundation to test the hypothesis that inflammation-mediated downregulation of βENaC contributes to cerebrovascular dysfunction.
Keywords: blood pressure, cerebrovascular, degenerin, gain-of-function, hypertension, myogenic tone, transfection
Pressure-induced constriction (PIC), also referred as myogenic constriction, is the inherent property of vascular smooth muscle cells (VSMCs) in small arteries and arterioles to constrict in response to an increase in intraluminal pressure.1,2 The response is pronounced in certain organs including the mesenteric, heart, kidney, and brain. PIC is a mechanism of blood flow autoregulation, the ability of an organ or tissue to maintain a constant blood flow in the presence of increases or decreases in perfusion pressure. Loss of PIC occurs in many inflammatory diseases including hypertension, chronic renal disease, stroke, and dementia. Loss of PIC in the cerebral circulation also occurs in preeclampsia.3,4 For instance, PIC of the middle cerebral artery (MCA) has been shown to be reduced in a rat model of preeclampsia and could be involved in the abnormal autoregulation in this disease.4
PIC is initiated by VSMC stretch. Increases in intraluminal pressure elongate VSMCs, which are wrapped circumferentially around the vessels. VSMC stretch is thought to activate a mechanoelectrical coupling event, which converts the mechanical signal at the cell membrane to signaling event into the cell plasma. Members of the degenerin/epithelial Na+ channel (Deg/ENaC) family form known mechanosensors in certain sensory neurons of the nematode Caenorhabditis elegans and fly Drosophila melanogaster.5–8 Our laboratory has shown that several members of this family are expressed in renal and cerebral VSMCs at or near the cell membrane. We and others have also shown that inhibition of vascular Deg function, using broad spectrum pharmacological inhibitors, such as amiloride and benzamil; gene-specific silencing using siRNA and dominant-negative constructs abolishes PIC responses. We have also shown that PIC is abolished in renal interlobar arteries, renal afferent arterioles, and MCAs of mice lacking normal levels of beta-epithelial Na+ channel (βENaC).9–12 While our previous loss-of-function studies demonstrate that Deg proteins mediate PIC signaling, understanding the importance of loss of Deg function in vascular disease requires a gain-of-function or enhancement of Deg expression. Since there are no pharmacological activators of βENaC, other approaches such as gene transfer by transient transfection are needed to enhance βENaC expression. However, whether transient transfection of βENaC into small artery segments actually enhances PIC has never been addressed. Therefore, the purpose of this study was to determine if transfer of exogenous βENaC gene into MCA segments enhances βENaC expression and PIC responses.
METHODS
All protocols and procedures used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Preparation of enhanced green fluorescent protein–mβENaC expression vector
Using standard cloning techniques, mouse βENaC (mβENaC) was cloned into enhanced green fluorescent protein (EGFP)-C1 expression vector at EcoR1 and BamH1 restriction enzyme sites (EGFP–mβENaC). Constructs were sequenced to ensure accuracy. EGFP fluorescence was used as indicator of gene expression. To ensure that the EGFP–mβENaC fusion protein was appropriately expressed, we examined construct expression in heterologous cells using western blotting.
Western blot detection of βENaC in heterologous cells
To ensure the expression of EGFP and βENaC were in frame, we transfected COS-7 (African green monkey kidney fibroblasts, ATCC, Manassas, VA) and A10 cells (rat aortic/thoracic VSMCs, ATCC) with EGFP–mβENaC, EGFP, and using Lipofectamine 3000 (Lfx) (Thermo Fisher Scientific, Waltham, MA). Cells were grown to 70% confluence on 100 mm dishes and transfected with 5 µg pcDNA at 1:1.5:2 pcDNA:P3000 reagent:Lipofectamine 3000 (Thermo Fisher Scientific) in Opti-MEM Reduced Serum Medium. Cells were grown overnight, examined for EGFP fluorescence, then scraped into 1 ml KBO buffer composed of (in mM/l) 25 NaH2PO4, 300 NaCl, 20 octylglucoside, 0.5% Triton (10%), at pH ~7.4. The lysate was freeze–thawed, vortexed aggressively, centrifuged at 14,000 RPM for 30 minutes at 4 °C, and separated into soluble (S) and insoluble (I) fractions. The insoluble fraction was solubilized in 100 μl 2× Laemmli buffer, heated, and vortexed gently until in solution. Protein samples were separated using standard gel electrophoresis procedures on 4–20% gradient gels (Bio-Rad Laboratories, Hercules, CA), then transferred to nitrocellulose membranes. Membranes were first stained with Revert Red to ensure equivalent loading, then destained before blocking with Li-Cor blocking solution (LI-COR Biosciences, Lincoln, NE) for 1 hour at 4 °C. Following, membranes were exposed to primary antibodies [rabbit anti-mβENaCC-term, generated in our laboratory, 1:2,000); mouse anti-EGFPAV (JL-8, Clontech Laboratories, 1:2,000)] overnight, and rinsed then exposed to donkey anti-rabbit IR 680 and donkey anti-mouse IR 800 at 1:10,000 each, for 1 hour, then rinsed and visualized on a Li-Cor Odyssey Infrared Scanner.9,11,13,14
Isolation and transfection of mouse cerebral artery segments
To transfect cerebral artery segments, we used a modified protocol previously used by Jernigan and Drummond.13 Briefly, female, wild type mice of mixed genetic background of C57BL/6J and 129 within our mouse colony (n = 5, 24 weeks of age) were anesthetized with isoflurane, then decapitated. Brains were immediately removed and placed in ice cold physiological salt solution composed of (in mM/l) 130.0 NaCl, 4.0 KCl, 1.8 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 6.0 glucose, 4.0 NaHCO3, 10 HEPES, and 0.03 EDTA, which was equilibrated with a gas mixture of 95% O2 and 5% CO2, at pH ~7.4. Surface cerebral artery segments, including the MCA, were isolated using microsurgical instruments and a boom-stand stereo microscope (Zeiss, White Plains, NY), cut into segments, transferred to four-well glass slides (Nunc Lab-Tek, Thermo Fisher Scientific) and transiently transfected with 10 μg of expression vectors [EGFP–mβENaC, EGFP, or lipofectamine (Lfx) vehicle] in Opti-MEM containing 1% penicillin/streptomycin using Lipofectamine 3000. After 4 hours, a 1:1 ratio of Dulbecco’s Modified Eagle Medium (1×) containing 1% penicillin/streptomycin and 20% fetal bovine serum was added to the vessels. Vessels were maintained for 18 hours in a humidified incubator (95% air, 5% CO2) at 37 °C. For EGFP fluorescence and VSMC immunolabeling, surface cerebral artery segments were pooled, then randomly aliquoted to glass slide wells and transfected. The following day, whole mount segments were either imaged directly or enzymatically dissociated for immunolabeling studies as described in the following section. For vascular reactivity studies, only MCA segments were used.
Immunolabeling of enzymatically dissociated cerebral VSMCs
VSMCs were enzymatically dissociated using 78 U papain and 3 mg dithioerythritol in 4 ml Hanks Buffered Salt Solution at 37 °C for 15 minutes, followed by 6 U collagenase type II, 3 mg soybean trypsin inhibitor type II, and 3 mg elastase in 4 ml at 37 °C for 12 minutes. Vessels were collected by gentle centrifugation and washed twice in 10 ml of Hanks Buffered Salt Solution. Vessels were resuspended in 200 μl of Hanks Buffered Salt Solution. VSMCs were released by gentle trituration with a series of fire polished, plugged Pasteur pipettes in decreasing diameter, then passed over a 70-μm filter to remove any undigested debris. The collected VSMCs were fixed in 4% paraformaldehyde for 10 minutes before being pipetted onto slides, air-dried then stored at room temperature until staining.
Immunolabeling of enzymatically dissociated VSMCs
Samples were rehydrated in distilled water, rinsed in phosphate-buffered saline three times, 5 minutes each, blocked in 5% normal donkey serum for 1 hour, and then incubated with primary antibody targeted to the C-terminal region of mouse βENaC (rabbit anti-mβENaCC-term, 1:100) and mouse anti-α-smooth muscle actin (1:100, Sigma-Aldrich, St Louis, MO) overnight at 4 °C in 5% normal donkey serum. Samples were rinsed in phosphate-buffered saline three times and then incubated with secondary antibody solution of donkey anti-rabbit Alexa 546 (1:250, Thermo Fisher Scientific) and donkey anti-mouse Alexa 633 (1:250, Thermo Fisher Scientific) for 1 hour at room temperature. Following final rinses in phosphate-buffered saline, samples were cover-slipped and imaged on a Leica TCS SP8 confocal microscope using a 63× objective and 5× optical zoom. All samples comparing βENaC expression were processed side-by-side and scanned under identical conditions. Images were prepared identically for publication in Adobe Photoshop.
Vascular reactivity
MCA reactivity to pressure, KCl and α-adrenergic agonists have been described previously.15 Briefly, MCA segments were cannulated onto glass cannulas in a pressure-flow chamber containing +Ca2+ physiological salt solution, and equilibrated at 0 mm Hg for 30 minutes, then at 50 mm Hg for 15 minutes at 37 °C. Inflow pressures were controlled and measured by a pressure servo-control system (Living Systems Instrumentation, St Albans City, VT). Inner and outer diameters were measured by CoolSnap color camera with a microangiometer using MetaMorph 6.1 software.
Depolarization- and α-adrenergic agonist-induced constriction responses of the isolated MCAs
Increasing concentrations of KCl (4, 20, 40, and 80 mM, 3 minutes incubation) were used to test the depolarization-induced constrictor responses at the beginning of the experiments, and α1-receptor agonist phenylephrine (PE; 10−7 to 10−4 M) was used to test the viability of the vessels at the end of the experiment at 50 mm Hg, ensuring that vessels were still viable. Vessels with a >25% constriction to 10–4 M PE at the end of the experiment were included in the analysis.
Myogenic responses of the isolated MCAs
Active diameter changes of MCAs were measured to stepwise increases in intraluminal pressure (15–90 mm Hg with 15 mm Hg increments for 5 minutes) in Ca2+-containing physiological salt solution. Following, the bath solution was exchanged for Ca2+-free physiological salt solution composed of (in mM/l) 130.0 NaCl, 4.0 KCl, 1.2 MgSO4, 1.2 KH2PO4, 6.0 glucose, 4.0 NaHCO3, 10 HEPES, 0.03 EDTA, and 2.0 EGTA, which was equilibrated with a gas mixture of 95% O2 and 5% CO2, at pH ~7.4.
Calculations and statistics
Myogenic tone (%) was calculated using the following formula: ((DP − DA)/DP) × 100, where DP is passive diameter and DA is active diameter of the vessels at a given intraluminal pressure value. Circumferential strain of the vessel wall was calculated using the following formula: (DP − D15)/D15), where DP is the passive diameter at a given intraluminal pressure and D15 is the passive diameter at 15 mm Hg under Ca2+-free conditions. Circumferential stress was calculated using the following formula: P × DP/(2 × WT), where DP is passive diameter, WT is wall thickness, and P is intraluminal pressure (where 1 mm Hg = 1.334 × 102 N/m2) under Ca2+-free conditions.
All data are expressed as mean ± standard error of the mean (SEM) and analyzed using unpaired two-tailed t test or two-way repeated-measure analysis of variance (ANOVA), where appropriate, using Prism 7.0. Differences among groups were determined using Sidak’s multiple comparison post hoc test. The specific statistical test applied is stated in figure legends. Statistical significance was considered at P < 0.05. Certain P values are provided to demonstrate confidence.
RESULTS
EGFP–mβENaC constructs are expressed as a single in-frame, fusion protein
A schematic of the EGFP–mβENaC fusion protein is shown in Figure 1a. To test fusion protein integrity, we examined expression in COS7 and A10 cells (Figure 1b,c). In COS-7 cells, EGFP is mostly expressed in the soluble or cytosolic fraction (at ~30 kDa), while the EGFP–mβENaC (~125 kDa) is expressed in the insoluble or membrane associated fraction (Figure 1b). No endogenous βENaC is found in the COS7 cells, as expected. Note that both the EGFP and mβENaC antibodies identify the same protein product in the EGFP–mβENaC transfected cells. EGFP–mβENaC protein expression appears lower than EGFP alone, likely due to its greater length. A similar expression pattern is seen in the smooth muscle cell line, A10, except overall exogenous EGFP–mβENaC expression is lower and endogenous βENaC is detected as high-molecular-weight isoform (~200 kDa) (Figure 1c). A high-molecular-weight isoform of βENaC has been detected in VSMC preparations in previous studies.16 Since the βENaC antibody is directed the extreme C-terminus of mβENaC, these findings confirm that the EGFP–mβENaC construct encodes a single, in-frame, fusion protein.
Figure 1.

EGFP–mβENaC is expressed as an in-frame fusion protein that is appropriately targeted to the membrane fraction in heterologous cells. (a) Schematic illustration of the fusion protein and the primary antibodies. The fusion protein is more than ~3-fold larger than the EGFP. The rabbit anti-mβENaC antibody targets the C-terminal of the mβENaC, and mouse anti-EGFPAV antibody targets the EGFP protein (specific antigenic site is proprietary). (b,c). Representative immunoblots showing EGFP and βENaC expression in COS7 (b) and A10 cells (c). EGFP expression is higher in soluble (S) fraction, while EGFP–mβENaC expression is higher in insoluble (I) fraction of the cell lysates. EGFP, enhanced green fluorescent protein; mβENaC, mouse beta-epithelial Na+ channel.
Expression of EGFP and EGFP–mβENaC in mouse cerebral artery segments and dissociated VSMCs
To test for fusion protein expression in VSMCs of whole mount cerebral artery segments, segments were transiently transfected with EGFP or Lfx vehicle. Representative fluorescence images show EGFP expression in dissected mouse cerebral artery segments compared with Lfx alone under identical imaging conditions (Figure 2a). The typical circumferential orientation of VSMCs is apparent indicating EGFP expression in VSMCs. Due to the reduced EGFP fluorescence with the EGFP–mβENaC fusion protein, we only examined EGFP transfected whole mount segments.
Figure 2.

Cerebral artery segments and dispersed VSMCs show EGFP and βENaC expression following transfection with EGFP or EGFP–mβENaC. (a) Representative fluorescence images of cerebral artery segments showing EGFP expression following EGFP (a, right), but not Lfx (a, left), treatment. EGFP was visualized using confocal microscopy and samples were imaged side-by-side under identical conditions. (b) Enzymatically dissociated VSMCs from EGFP control and EGFP–mβENaC transfected cerebral artery segments, immunolabeled for mβENaC and α-SM actin, then visualized using confocal microscopy. Panel columns represent EGFP (left, green), mβENaC (middle left, red), α-SM actin (middle right, blue), and merged (right) images. Single color images are shown as black and white for visual sensitivity and the merged images are presented in color. Panel rows represent EGFP control (top rows) and EGFP–mβENaC (bottom rows), no-primary antibody controls for both transfection conditions are also shown. EGFP fluorescence intensity is higher and localized to the cytoplasm in EGFP transfected (top rows), while intensity is lower and localized at or near the cell membrane with α-SM actin in EGFP–mβENaC transfected (bottom rows) VSMCs. Lfx, Lipofectamine 3000; EGFP, enhanced green fluorescent protein; VSMC, vascular smooth muscle cell; mβENaC, mouse beta-epithelial Na+ channel.
To test for upregulation of βENaC in EGFP–mβENaC transfected cerebral artery segments, we dissociated VSMCs from EGFP control and EGFP–mβENaC segments, then immunolabeled them for βENaC (red) and α-SM actin (blue). Representative images are shown in Figure 2b. Although we were not able to detect increased βENaC expression in the EGFP–mβENaC transfected VSMCs (lower panels) compared to those transfected with EGFP (upper panels), we observed differences in the intensity and localization of the EGFP signal (green) consistent with the EGFP–mβENaC fusion protein. First, the fluorescence intensity of EGFP is higher in EGFP vs. EGFP–mβENaC transfected VSMCs. Second, the EGFP fluorescence is localized to the cytoplasm in EGFP transfected VSMCs, but it is localized at or near the cell surface along with α-SM actin in EGFP–mβENaC transfected VSMCs. This would be expected since βENaC is a transmembrane protein. Thus, while overall βENaC fluorescence is not greater, which may reflect an antibody sensitivity issue, the localization pattern of EGFP fluorescence is consistent with βENaC-targeted membrane localization.
Vasoconstrictor responses to depolarizing and α-adrenergic agents are identical between EGFP and EGFP–mβENaC transfected MCAs
Changes in inner diameter in response to KCl (4–80 mM) and PE (10−7 to 10–4 M) are shown in Figure 3a,b, respectively. Vasoconstriction responses to KCl were identical between EGFP and EGFP–mβENaC transfected MCA segments (Figure 3a). A two-way repeated measures ANOVA showed an effect of KCl (P < 0.0001), but no effect of construct or interaction (P = 0.555, P = 0.335, respectively). At the highest doses of KCl (80 mM), internal diameters were 43.6 ± 2.2 vs. 46.7 ± 3.3 µm in EGFP and EGFP–mβENaC groups, respectively. Vasoconstriction responses to PE are also identical between EGFP and EGFP–mβENaC transfected MCAs (Figure 3b). A two-way repeated measures of ANOVA showed an effect of PE concentration (P < 0.0001), but no effect of construct or interaction (P = 0.729, P = 0.502, respectively). At the highest concentration of PE 10−4 M, internal diameters were 41.7 ± 2.5 vs. 40.3 ± 3.7 µm in EGFP and EGFP–mβENaC groups, respectively. These results suggest that the ability of EGFP–mβENaC segments to constrict per se is not different from EGFP controls.
Figure 3.
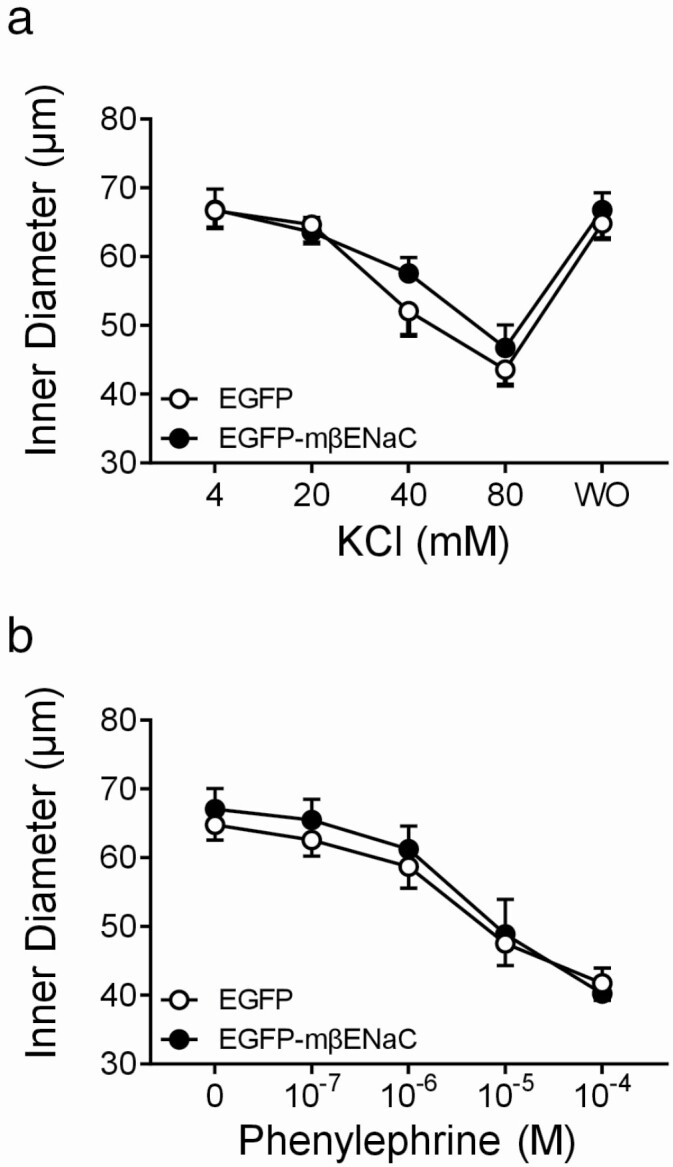
Vasoconstrictor responses of MCA segments following transfection with EGFP or EGFP–mβENaC. Vasoconstrictor responses to depolarizing agent (KCl, a) and α-adrenoceptor agonist agent (phenylephrine, PE, b) (n = 5) were not affected by expression of EGFP–mβENaC (●) compared to EGFP controls (○). Data represent internal diameters and were analyzed using a two-way repeated measures ANOVA, which showed an effect of KCl (P < 0.0001) and PE (P < 0.0001) concentration, but no effect of expression construct (P = 0.555, 0.729) or interaction (P = 0.335, 0.502). Data are presented as mean ± SEM, n = 5 in EGFP and EGFP–mβENaC groups. EGFP, enhanced green fluorescent protein; mβENaC, mouse beta-epithelial Na+ channel; MCA, middle cerebral artery; ANOVA, analysis of variance; SEM, standard error of the mean.
Transfection of MCA segments with EGFP–mβENaC enhances pressure-induced constriction
Inner diameter responses under active (Ca2+-containing) and passive (Ca2+-free) conditions, calculated myogenic tone, log-pressure vs. myogenic tone relationship, and slope of myogenic tone of MCA segments to stepwise increases in intraluminal pressure are shown in Figure 4a–e. We found the passive responses (Ca2+-free) in EGFP vs. EGFP–mβENaC are identical (Figure 4a,b). Two-way repeated measures ANOVA showed a main effect of intraluminal pressure (P < 0.0001), but no main effect of expression construct (P = 0.336) or interaction of intraluminal pressure and construct (P = 0.274) on the passive responses, suggesting that the expression of EGFP–mβENaC does not change the mechanical properties of the MCA segments compared to EGFP alone. Calculated myogenic tone for both groups is shown in Figure 4c. While EGFP and EGFP–mβENaC transfected MCA segments develop increasing myogenic tone from 15 to 90 mm Hg, tone is greater in EGFP–mβENaC segments at 45, 60, 75, and 90 mm Hg intraluminal pressure values. Moreover, peak myogenic tone is nearly 2-fold greater in EGFP–mβENaC segments (11.6 ± 0.4% vs. 6.3 ± 0.4%, at 75 mm Hg, P = 0.0006). The log-pressure vs. myogenic tone relationship and the slope of this relationship are shown in Figure 4d,e, respectively. The slope of the log-pressure vs. myogenic tone relationship is significantly higher in EGFP–mβENaC transfected MCAs at 45, 60, 75, and 90 mm Hg intraluminal pressure compared to EGFP transfected MCAs (Figure 4d). The slope of this relationship in EGFP–mβENaC is significantly higher compared to EGFP transfected MCA segments (unpaired, two-tailed t test, P = 0.0272, Figure 4e). These results suggest that transfection of the isolated MCA with EGFP–mβENaC significantly increases the PIC sensitivity.
Figure 4.

Pressure-induced vasoconstriction (PIC) responses of MCA segments following transfection with EGFP or EGFP–mβENaC. (a,b) Intraluminal diameters of MCA segments transfected with EGFP control (a) or EGFP–mβENaC (b) in response to stepwise increases in intraluminal pressure under active (Ca2+-containing, ●) and under passive (Ca2+-free, ○) conditions. The passive responses (Ca2+-free) in EGFP vs. EGFP–mβENaC were compared using two-way repeated measures ANOVA, which showed a main effect of intraluminal pressure (P < 0.0001), but no main effect of expression construct (P = 0.336) or interaction of intraluminal pressure and construct (P = 0.274) on the response. (c) Calculated myogenic tone of isolated MCAs following transfection with EGFP (○) or EGFP–mβENaC (●). Two-way repeated measures ANOVA showed an effect of intraluminal pressure (P < 0.0001) and EGFP–mβENaC expression (P = 0.0001) and an interaction between genotype and intraluminal pressure (P < 0.0451) on intraluminal pressure vs. % myogenic tone. Sidak’s multiple comparison post hoc test showed a significant effect of expression of EGFP–mβENaC on myogenic tone at 45 (P = 0.0170), 60 (P = 0.0156), 75 (P = 0.0006), and 90 (P = 0.0124) mm Hg intraluminal pressure values. (d) Pressure and myogenic tone relationship was linearized by plotting pressure log vs. tone to obtain the slope or estimate of PIC sensitivity. (e) Slope of the relationship between log of intraluminal pressure vs. myogenic tone of the MCA following transfection with EGFP or EGFP–mβENaC. Individual data points are shown. PIC sensitivity was significantly increased in MCA segments following transfection with EGFP–mβENaC compared to that of EGFP (P = 0.0272, unpaired two-tailed t test). Data are presented as mean ± SEM, n = 5 in each group. *Significantly different from control at P < 0.05. †Significantly different from control at P < 0.001. EGFP, enhanced green fluorescent protein; mβENaC, mouse beta-epithelial Na+ channel; MCA, middle cerebral artery; ANOVA, analysis of variance; SEM, standard error of the mean.
Mechanical and morphological properties of EGFP and EGFP–mβENaC transfected MCAs are similar
To determine whether differences in mechanical and morphological properties might contribute to the increased PIC responses in EGFP–mβENaC transfected MCA segments, we calculated the circumferential wall strain and stress under Ca2+-free conditions (Figure 5a,b). The circumferential strain (0.25 ± 0.02 vs. 0.27 ± 0.01, P = 0.82, Figure 5a) and stress (4.1 ± 0.5 vs. 4.6 ± 0.09, P = 0.95, Figure 5b), at 90 mm Hg, were identical in EGFP and EGFP–mβENaC MCAs, respectively. These findings suggest that mechanical properties do not account for the enhanced PIC responses in the EGFP–mβENaC transfected MCA segments. We also found that the wall thickness (12.6 ± 1.07 vs. 13.6 ± 0.98 μm, P = 0.51) and wall-to-lumen ratio (0.19 ± 0.02 vs. 0.20 ± 0.01, P = 0.56) are identical between EGFP and EGFP–mβENaC transfected MCA segments, respectively, suggesting that morphological changes do not contribute to the enhanced PIC responses in EGFP–mβENaC transfected MCA segments.
Figure 5.
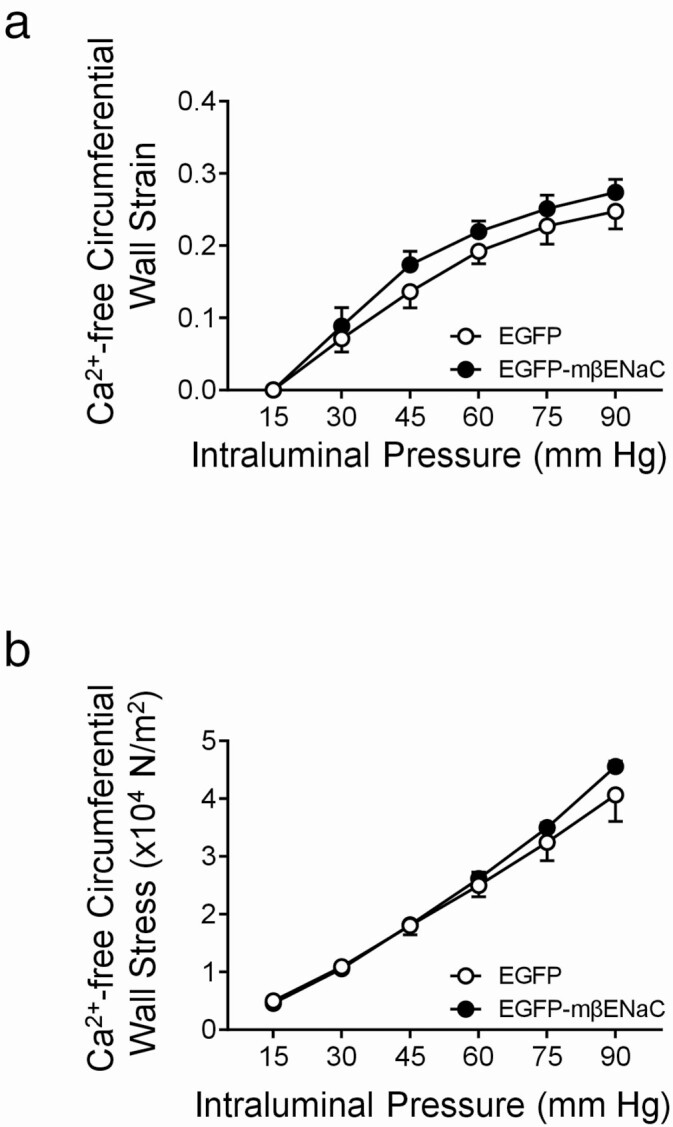
Mechanical properties of MCA segments following transfection with EGFP and EGFP–mβENaC. Data were analyzed using a two-way repeated measures ANOVA, which showed an effect of intraluminal pressure (P < 0.0001), but no effect of expression construct (P = 0.0892) or interaction between pressure and construct (P = 0.2133). Data are presented as mean ± SEM, n = 5 in EGFP (○) and EGFP–mβENaC (●) groups. Sidak’s multiple comparison post hoc test showed that circumferential wall strain (a) and stress (b) under Ca2+-free conditions are identical between EGFP and EGFP–mβENaC MCAs. Data are presented as mean ± SEM, n = 5 in each group. EGFP, enhanced green fluorescent protein; mβENaC, mouse beta-epithelial Na+ channel; MCA, middle cerebral artery; ANOVA, analysis of variance; SEM, standard error of the mean.
Discussion
PIC involves multiple signaling molecules including extracellular matrix proteins, integrins, G-proteins, cytoskeletal proteins, voltage gated Ca2+ channels, and mechanically gated ion channels.17,18 Previous studies that have blocked Deg proteins, including the βENaC, support the concept that βENaC mediates the PIC response. For example, pharmacological (e.g., amiloride, benzamil), gene-silencing (e.g., dominant-negative cDNA or siRNA against ENaC), or genetic (βENaC loss-of-function mutation) inhibition of βENaC in isolated small renal and cerebral arteries resulted in nearly abolished PIC in these vessels.9,13,19 While there is an abundance of studies that demonstrate the importance of ENaC proteins, including βENaC, using a loss-of-function approach, there are no studies that support the role of ENaC in PIC using a gain-of-function approach. In the present study, we found that expression of exogenous βENaC using transient transfection increased PIC responsiveness in MCA segments nearly 2-fold, without altering overall vascular contractility and mechanical/morphological properties.
While inhibitory approaches are useful in demonstrating the importance of βENaC to the PIC response, they are insufficient to gain understanding the importance of loss of βENaC on cerebrovascular dysfunction in disease. Determining the importance of VSMC βENaC inhibition in disease requires a “rescue,” or enhancement, of βENaC expression. However, whether or not enhancement of βENaC expression can improve the PIC response has not been established. Therefore, the purpose of this investigation was to provide proof-of-concept evidence that transfection of βENaC in MCA segments could enhance the PIC response without changing other vascular properties. The major finding of this study is that transient expression of βENaC doubles PIC responsiveness independent of vascular contractile or mechanical properties.
Emerging evidence suggests that a loss of PIC responsiveness may be linked to inflammation in certain diseases, one example is preeclampsia. Preeclampsia is the development of new onset hypertension in the last trimester and affects 5–8% of pregnancies. Preeclampsia and animal models of placental ischemia are characterized by elevated serum cytokines and cerebrovascular dysfunction, including loss of PIC and autoregulation of cerebral blood flow.4,20–23 Previous findings from our laboratory group demonstrated the downregulation of cerebrovascular βENaC expression in a rat placental ischemic model of preeclampsia.4,24 Follow-up studies showed that exposure to elevated levels of proinflammatory cytokine tumor necrosis factor α, in vivo and in vitro, inhibits vascular βENaC expression.24 These findings suggest that tumor necrosis factor α induced inhibition of VSMC βENaC may underlie the cerebrovascular dysfunction in placental ischemia. However, the importance of βENaC in a disease model where it is downregulated cannot be discerned by using βENaC inhibition. Thus, understanding the importance of cytokine-mediated βENaC inhibition in preeclampsia/placental ischemia will require an enhancement of βENaC expression and/or function.
To address this gap, we tested whether exogenous expression of βENaC in the isolated MCA would enhance PIC. Since pharmacological activation of ENaC channels is unavailable, we used transient transfection to upregulate its expression, an approach used previously by our laboratory group. Female mice were utilized for two reasons. First, our laboratory group is interested in understanding the role of βENaC in vascular dysfunction in preeclampsia, a disease specific to females. Second, we rationalized that females might have a greater likelihood of benefiting from βENaC because this group tends to have weaker PIC responses than males.25–27 Potential inhibitory effects of estrogen on basal βENaC expression were not a major consideration.28,29 Whether or not this approach is effective at increasing ENaC in males was not addressed.
The results of the present study provide gain-of-function evidence that βENaC is an important mediator of PIC in the MCA. Our findings also provide proof-of-concept that expression of exogenous βENaC increases MCA PIC responsiveness without altering overall contractility. This approach may be useful to assess the importance of specific proteins downregulated in disease models where PIC responsiveness is impaired, such as βENaC in preeclampsia.
Acknowledgments
We acknowledge the technical support of Emily Hildebrandt. This work was supported by National Institutes of Health Grants P20GM104357, P20GM121334, P20121334, P01HL051971, and R01HL1136684.
Disclosure
The authors declared no conflict of interest.
References
- 1. Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol 1902; 28:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 1999; 79:387–423. [DOI] [PubMed] [Google Scholar]
- 3. Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, Montezano AC. Vascular smooth muscle contraction in hypertension. Cardiovasc Res 2018; 114:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ryan MJ, Gilbert EL, Glover PH, George EM, Masterson CW, McLemore GR Jr, LaMarca B, Granger JP, Drummond HA. Placental ischemia impairs middle cerebral artery myogenic responses in the pregnant rat. Hypertension 2011; 58:1126–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Voglis G, Tavernarakis N. Mechanotransduction in the nematode Caenorhabditis elegans. In Kamkin A, Kiseleva I (eds), Mechanosensitivity in Cells and Tissues. Moscow, Academia Publishing House Ltd; 2005. [PubMed] [Google Scholar]
- 6. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 2002; 82:735–767. [DOI] [PubMed] [Google Scholar]
- 7. Syntichaki P, Tavernarakis N. Genetic models of mechanotransduction: the nematode Caenorhabditis elegans. Physiol Rev 2004; 84:1097–1153. [DOI] [PubMed] [Google Scholar]
- 8. Zhong L, Hwang RY, Tracey WD. Pickpocket is a DEG/ENaC protein required for mechanical nociception in Drosophila larvae. Curr Biol 2010; 20:429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. VanLandingham LG, Gannon KP, Drummond HA. Pressure-induced constriction is inhibited in a mouse model of reduced betaENaC. Am J Physiol Regul Integr Comp Physiol 2009; 297:R723–R728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim EC, Choi SK, Lim M, Yeon SI, Lee YH. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS One 2013; 8:e84194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge Y, Gannon K, Gousset M, Liu R, Murphey B, Drummond HA. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced βENaC expression. Am J Physiol Renal Physiol 2012; 302:F1486–F1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grifoni SC, Chiposi R, McKey SE, Ryan MJ, Drummond HA. Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-ENaC. Am J Physiol Renal Physiol 2010; 298:F285–F292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jernigan NL, Drummond HA. Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol 2006; 291:F1184–F1191. [DOI] [PubMed] [Google Scholar]
- 14. Grifoni SC, Gannon KP, Stec DE, Drummond HA. ENaC proteins contribute to VSMC migration. Am J Physiol Heart Circ Physiol 2006; 291:H3076–H3086. [DOI] [PubMed] [Google Scholar]
- 15. Nemeth Z, Hildebrandt E, Ryan MJ, Granger JP, Drummond HA. Pressure-induced constriction of the middle cerebral artery is abolished in TrpC6 knockout mice. Am J Physiol Heart Circ Physiol 2020; 319:H42–H50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jernigan NL, LaMarca B, Speed J, Galmiche L, Granger JP, Drummond HA. Dietary salt enhances benzamil-sensitive component of myogenic constriction in mesenteric arteries. Am J Physiol Heart Circ Physiol 2008; 294:H409–H420. [DOI] [PubMed] [Google Scholar]
- 17. Martino F, Perestrelo AR, Vinarský V, Pagliari S, Forte G. Cellular mechanotransduction: from tension to function. Front Physiol 2018; 9:824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drummond HA. βENaC is a molecular component of a VSMC mechanotransducer that contributes to renal blood flow regulation, protection from renal injury, and hypertension. Front Physiol 2012; 3:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jernigan NL, Drummond HA. Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 2005; 289:F891–F901. [DOI] [PubMed] [Google Scholar]
- 20. LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep 2007; 9:480–485. [DOI] [PubMed] [Google Scholar]
- 21. Sharma A, Satyam A, Sharma JB. Leptin, IL-10 and inflammatory markers (TNF-alpha, IL-6 and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am J Reprod Immunol 2007; 58:21–30. [DOI] [PubMed] [Google Scholar]
- 22. Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension 2006; 48:711–716. [DOI] [PubMed] [Google Scholar]
- 23. LaMarca BB, Bennett WA, Alexander BT, Cockrell K, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-alpha. Hypertension 2005; 46:1022–1025. [DOI] [PubMed] [Google Scholar]
- 24. Duncan JW, Younes ST, Hildebrandt E, Ryan MJ, Granger JP, Drummond HA. Tumor necrosis factor-α impairs cerebral blood flow in pregnant rats: role of vascular β-epithelial Na+ channel. Am J Physiol Heart Circ Physiol 2020; 318:H1018–H1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gros R, Van Wert R, You X, Thorin E, Husain M. Effects of age, gender, and blood pressure on myogenic responses of mesenteric arteries from C57BL/6 mice. Am J Physiol Heart Circ Physiol 2002; 282:H380–H388. [DOI] [PubMed] [Google Scholar]
- 26. Wang S, Zhang H, Liu Y, Li L, Guo Y, Jiao F, Fang X, Jefferson JR, Li M, Gao W, Gonzalez-Fernandez E, Maranon RO, Pabbidi MR, Liu R, Alexander BT, Roman RJ, Fan F. Sex differences in the structure and function of rat middle cerebral arteries. Am J Physiol Heart Circ Physiol 2020; 318:H1219–H1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geary GG, Krause DN, Duckles SP. Estrogen reduces myogenic tone through a nitric oxide-dependent mechanism in rat cerebral arteries. Am J Physiol 1998; 275:H292–H300. [DOI] [PubMed] [Google Scholar]
- 28. Yusef YR, Thomas W, Harvey BJ. Estrogen increases ENaC activity via PKCdelta signaling in renal cortical collecting duct cells. Physiol Rep 2014; 2:e12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matalon S, Bartoszewski R, Collawn JF. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol 2015; 309:L1229–L1238. [DOI] [PMC free article] [PubMed] [Google Scholar]