

ABSTRACT
Prescribing a ketogenic diet (KD) is a century-old dietary intervention mainly used in the context of intractable epilepsy. The classic KD and its variants regained popularity in recent decades, and they are considered potentially beneficial in a variety of neurological conditions other than epilepsy. Many patients with multiple sclerosis (MS) have attempted diet modification for better control of their disease, although evidence thus far remains insufficient to recommend a specific diet for these patients. The results of 3 pilot clinical trials of KD therapy for MS, as well as several related studies, have been reported in recent years. The preliminary findings suggest that KD is safe, feasible, and potentially neuroprotective and disease-modifying for patients with MS. Research on corresponding rodent models has also lent support to the efficacy of KD in the prevention and treatment of experimental autoimmune encephalomyelitis and toxin-induced inflammatory demyelinating conditions in the brain. Furthermore, the animal studies have yielded mechanistic insights into the molecular mechanisms of KD action in relevant situations, paving the way for precision nutrition. Herein we review and synthesize recent advances and also identify unresolved issues, such as the roles of adipokines and gut microbiota, in this field. Hopefully this panoramic view of current understanding can inform future research directions and clinical practice with regard to KD in MS and related conditions.
Keywords: β-hydroxybutyrate, decanoic acid, demyelination, experimental autoimmune encephalomyelitis, ketogenic diet, multiple sclerosis, neuroinflammation
Statement of Significance: Existing studies concerning ketogenic dietary therapy for multiple sclerosis and relevant animal models have rarely been examined systematically. This narrative review updates and synthesizes preclinical and clinical evidence regarding the effects and mechanisms of action of ketogenic diets in these contexts.
Graphical Abstract
Graphical Abstract.
Introduction
Multiple sclerosis (MS) is one of the major chronic inflammatory diseases of the central nervous system (CNS), affecting ∼2.3 million people worldwide (1). There is currently no cure for MS, and treatment remains unsatisfactory despite an increasing therapeutic armamentarium in recent decades. A substantial fraction of patients with MS expressed interest in and willingness for a trial of dietary therapy in the hope that it would ameliorate their disease (2). For instance, nearly two-thirds of the study subjects in a clinical trial of modified Atkins diet had already attempted dietary changes before they participated in that trial (3). The ketogenic diet (KD) and related dietary styles were preferred by many patients with MS. In a survey of dietary characteristics conducted in North America, these patients often adopted a low-carbohydrate, low-calorie, or Atkins diet in some periods of their disease course (4).
In contrast to the enthusiasm about diet modification on the patients’ side, evidence supporting the use of any specific diet for MS was considered to be insufficient (5–7), and KD was often not specifically addressed in review articles examining the role of dietary intervention in MS (7, 8). It has been suggested that KD might exert anti-inflammatory and neuroprotective effects in patients with various neurological disorders (9–13), including progressive MS (14). However, KD is not without potential adverse effects, and the chronic low-grade metabolic acidosis associated with KD is a cause for concern. To prevent the acidosis, it has been suggested that dietary protein, which is a source of endogenous acidic metabolites, is maintained at the level of 1–2 g/kg/d, and consumption of alkaline mineral–rich green vegetables is maximized (15). Moreover, it appears that the effects of KD on the CNS are context dependent, and KD could be detrimental in some situations. For instance, KD aggravates cognitive impairment induced by intermittent hypoxia in mice (16). This negative cognitive impact is mediated by the expansion of IFN-γ–producing T helper 1 (Th1) cells, and Th1 polarization is also implicated in the pathogenesis of experimental autoimmune encephalomyelitis (EAE) and MS (17). Nonetheless, KD has shown promise in various rodent models of dysmyelination or demyelination. In a mouse model of Pelizaeus–Merzbacher disease (a kind of inherited leukodystrophy), a KD rescued the myelination defect and clinical phenotype (18). In EAE and cuprizone-induced demyelination mouse models, KD also ameliorated disease activity in terms of neuropathology and behaviors (19, 20). Several pilot clinical trials concerning the effects of KD on patients with MS have been carried out in recent years (3, 17, 21). Therefore, this is a prime time to re-evaluate the role of KD in MS and its corresponding animal models, and to identify knowledge gaps that deserve further research.
The Effects of KD on MS (Human Studies)
A PubMed search using the search terms “ketogenic diet” AND (“multiple sclerosis” OR “experimental autoimmune encephalomyelitis”) was performed to identify relevant publications. The last search date was December 31, 2021.
Study design and execution
Three open-label clinical trials of KD therapy for MS, including 2 randomized controlled trials (class II evidence) (17, 21) and 1 single-arm pilot study (class IV evidence) (3), have been published since 2016. There were also other research articles focusing on certain aspects of KD effects in patients with MS, including the ramifications of the clinical trial conducted by Bock et al. (clinicaltrials.gov identifier: NCT01538355) (22–24). In addition, a single case report was identified (25). An overview of these studies is presented in Table 1.
TABLE 1.
Overview of studies evaluating a ketogenic diet in patients with MS: study design and execution1
| Author (year) | Study design | Dietary interventions | Supervision or coaching | Study participants | Age of study subjects, y | Other notes or comments |
|---|---|---|---|---|---|---|
| Choi et al. (2016) (17) | Randomized, parallel-group, 3-arm trial | Experimental groups: KD for 6 mo, or a single cycle of a modified FMD for 7 d followed by a Mediterranean diet for 6 mo. Comparator group: control diet | Nutritional coach during group-based workshops on 3 weekends | 60 RRMS (20 for each group; 18/18/12 in KD/FMD/control group completed study, respectively) | Adults, mean 41.3 ± 8.2 | clinicaltrials.gov identifier: NCT01538355 |
| Swidsinski et al. (2017) (24) | Interventional study (focusing on microbiome study) | KD for 6 mo | Nutritional coach during group-based workshops on 3 weekends | 10 RRMS, 14 HC | Adults, details not reported | Longitudinal evaluation of colonic microbiome change under KD, without evaluation of clinical effects; HC group was not treated with KD, so the effects of KD on MS vs. HC could not be compared |
| Bock et al. (2018) (22) | Randomized, 3-arm trial | Experimental groups: adapted (by Dr Bock) KD for 6 mo, or FMD (7-d fasting followed by usual diet). Comparator group: control diet | Nutritional coach | 24 RRMS (11 on adapted KD, 5 on FMD, 8 controls) | Adults, mean 43.1 ± 8.8 | Adapted KD and CR groups were pooled for analysis due to small sample size (because of patients' dropout and loss of blood samples) |
| Nathan et al. (2019) (25) | Single case study | KD plus CR (75%). Ketogenic ratio 2.2 | Diet diary, daily urine ketones | 1 SPMS | 60 | KD as monotherapy for SPMS |
| Benlloch et al. (2019) (27) | Prospective, mixed and quasi-experimental pilot study | Isocaloric, MCT-rich diet for 4 mo | Weekly telephone calls in which patients were asked about any problems in following the diet | 20 RRMS, 6 SPMS, 1 PPMS | Adults, mean 44.56 ± 11.27 | Not genuinely ketogenic (see text for details) |
| Brenton et al. (2019) (3) | Single-arm, open-label pilot study | Modified Atkins diet for 6 mo | The study dietitian provided a personalized educational session on initiation and maintenance of MAD, and provided contact information so that subjects could use her as a resource outside of study visits | 20 RRMS | 15–50 | Study subjects largely overweight or obese; no comparator group |
| Lee et al. (2021) (21) | Randomized, waitlist controlled, open-label study | Experimental groups: MCT-based KD for 12 wk, or modified Paleolithic diet. Comparator group: usual diet. Ketogenic ratio 0.99 ± 0.47 | Participants were taught the study diet by RD, who answered all diet-related questions throughout the study. RD made nutrition counseling calls to participants 2–3 d after visit 1, then weekly for 3 wk | 10 SPMS, 2 PPMS, 1 PRMS, 1 RRMS; 5 on MCT-based KD, 6 on modified Paleolithic diet, 4 on usual diet | 36–63 | clinicaltrials.gov identifier: NCT01915433 |
| Bock et al. (2022) (23) | Retrospective evaluation of a randomized, 3-arm trial | As described in the study of Bock et al. (2018) (22) (see above) | As described in the study of Bock et al. (2018) (22) (see above) | 40 RRMS (17 on adapted KD, 14 on FMD, 9 controls) | Adults, mean 43.06 ± 9.7 |
CR, calorie restriction; FMD, fasting mimicking diet; HC, healthy controls; KD, ketogenic diet; MAD, modified Atkins diet; MCT, medium-chain triglyceride; MS, multiple sclerosis; PPMS, primary progressive multiple sclerosis; PRMS, progressive relapsing multiple sclerosis; RD, registered dietitian; RRMS, relapsing-remitting multiple sclerosis; SPMS, secondary progressive multiple sclerosis.
KD regimens used in these studies were heterogeneous, including variants such as a modified Atkins diet (3) and medium-chain triglyceride (MCT)–based KD modified from the Wahls Paleo Diet (21). The latter was considered to be the KD version of the modified Paleolithic diet. The ketogenic ratio (i.e., the ratio of grams of fat to grams of carbohydrate and protein combined) of study diets, either reported in the articles or calculated from the diet descriptions, ranged from 1 to 2.2 across studies (17, 21, 25) and was lower than that of the classic KD (with a ketogenic ratio ≤4) (26). The exception was the study published by Benlloch et al. (27), in which the ketogenic ratio was far below 1. An isocaloric KD was originally claimed as the study diet in their article, but the carbohydrate content (40%) and the degree of ketosis achieved [mean serum β-hydroxybutyrate (BHB) = 0.10 mmol/L] did not conform to KD in the general sense. Therefore, the study could not be considered a genuine KD study. Indeed, the authors admitted later that their study diet would be more appropriately termed a “coconut oil-enriched Mediterranean diet” (28).
The classic KD and MCT diet often result in different levels of nutritional ketosis, even when the ketogenic ratio is the same. They also exhibited differential clinical effects in a mouse model of hypomyelinating disease (18). Therefore, caution should be exercised in comparing and interpreting research results because different KD regimens were used across studies. Another issue that deserves attention is the differences in the calorie content of study diets. In the case study using KD as the sole therapy for secondary progressive MS, the dietary regimen was actually a combination of KD and a calorie-restricted (i.e., 75% of the recommended calorie intake) diet (25). It might be difficult to tease out the effects of KD from those of calorie restriction in this case, because intermittent calorie restriction per se might exert beneficial effects in patients with MS in the absence of significant changes in BHB concentration (29). One study made a head-to-head comparison between KD and a fasting mimicking diet (FMD) in this context (17). The results suggest that both dietary regimens are safe and feasible for patients with MS, whereas FMD leads to more significant improvement in overall quality of life, physical health, and mental health compared with KD. However, it might be difficult to determine whether the improvements seen in the FMD group can be attributed to the 7-d FMD per se or the 6-mo Mediterranean diet that followed. An ongoing study (clinicaltrials.gov identifier: NCT03508414) aims to compare KD and FMD regarding their intermediate-term (18-mo) efficacy, using change in lesion load on T2-weighted MRI of the brain as the primary outcome measure (30).
Most studies included a nutritional coach or dietitian for diet-related instruction and/or counseling during the implementation of dietary therapy (3, 17, 21, 22, 24). This was not specifically mentioned in 2 studies (25, 27). The adherence to therapeutic diet was generally assessed using food diary, urine ketones, and blood ketones measurements (3, 21, 23, 25), and the latter were also used to ensure that nutritional ketosis was achieved.
Study findings and implications
Most studies reported beneficial effects of KD on some aspects of MS-related outcomes (Table 2), including Expanded Disability Status Scale (EDSS) score (3, 17, 25), health-related quality of life (17), fatigue (3), depression (3), and anthropometric measures such as body weight, BMI, and/or total fat mass (3, 25). Two studies reported favorable effects of KD on biomarkers of glucose homeostasis (3, 21), whereas 1 study reported no significant effect in this regard (22). One study reported a significant decrease of leptin after a 3-mo period of KD (3), which is consistent with the KD-induced changes in anthropometric measures. One study found that serum neurofilament light chain, a marker of neuroaxonal damage, decreased significantly in patients treated with KD (23). By comparison, reduction of serum neurofilament light chain was not observed in patients treated with FMD or common diet. It is also notable that the temporal pattern of serum neurofilament light chain concentration was not linear in patients treated with KD, and significant reduction of this biomarker was only observed after 6-mo dietary therapy, suggesting relatively late onset of KD's neuroprotective effects. Among the KD-treated patients in the 3 clinical trials, only 1 patient experienced a clinical relapse during the study period (17); other patients did not exhibit overt disease progression. However, the clinical course of MS is usually insidious, hence further studies with a longer duration of follow-up could be needed to determine if a KD is effective in reducing disease relapse or progression.
TABLE 2.
Overview of studies evaluating a ketogenic diet in patients with MS: the results1
| Author (year) | EDSS score | Quality of life | Fatigue | Depression | Anthropometric measures | Biomarkers | Other findings or comments |
|---|---|---|---|---|---|---|---|
| Choi et al. (2016) (17) | Mildly yet significantly improved at 3 and 6 mo | Clinically meaningful improvement in MSQOL-54 at 3 mo | Not reported (assessed using MFIS)2 | Not reported (assessed using BDI)2 | Not reported2 | Slight reduction in lymphocyte and WBC counts | ▪ High compliance rate (90%)▪ Adverse events (KD/FMD/control group): respiratory tract infection (n = 12/7/9), diarrhea (n = 3/0/3), headache (n = 2/2/0), nausea (n = 2/0/0), ureteric colic (n = 1/0/0), urinary tract infection (n = 1/2/1)▪ One relapse in KD group during 6-mo study period (4 in control diet group, 3 in FMD group) |
| Swidsinski et al. (2017) (24) | Not reported | Not reported | Not reported | Not reported | Not reported | Dynamic changes in colonic microbiome | The effects of KD on colonic microbiome were biphasic. Bacterial concentrations and diversity were further reduced from baseline initially, then they started to recover at week 12 |
| Bock et al. (2018) (22) | Expression of all target genes showed statistically nonsignificant trend for positive correlation with the EDSS score | ALOX5 and COX1 expression were inversely correlated with MSQOL-54 at 6 mo | Not reported | Not reported | Improved BMI (P = 0.008) | No significant effects on fasting blood sugar and insulin | ▪ Target genes in this study were those coding for biosynthetic enzymes for proinflammatory (ALOX5, COX1, COX2) and anti-inflammatory (ALOX15) eicosanoids▪ Significant increase in serum BHB (mean 1.44 mmol/L) |
| Nathan et al. (2019) (25) | EDSS score deterioration (6.0 → 7.5) after stopping KD, and improving after resumption of KD | No formal assessment was reported | Not reported | Not reported | BW decreased from 67.1 to 65 kg and maintained (= his ideal BW) | CSF-specific OCBs remained positive | Disease activity (EDSS) apparently correlated with use/discontinuation/resumption of KD |
| Benlloch et al. (2019) (27) | Not reported | Not reported | Not reported | Not reported | Significant increase in lean mass and decrease in fat mass | ▪ Ghrelin: no significant change▪ Paraoxonase 1 (an antioxidant marker): significant increase | ▪ Significant satiating effect (decreased hunger perception)▪ Significant yet modest increase in serum BHB (mean 0.10 mmol/L after the 4-mo intervention) |
| Brenton et al. (2019) (3) | Significant decrease in EDSS score at 6 mo | Not reported | Patient-reported fatigue (MFIS) improved (P = 0.002) | Patient-reported depression scores (BDI) improved (P = 0.003) | Reductions in BMI and total fat mass (P < 0.0001) | ▪ Leptin: significantly lower at 3 mo (P < 0.0001)▪ Adiponectin: no significant change▪ Insulin and hemoglobin A1c: significantly decreased | ▪ Adherence rate: 95% at 3 mo and 75% at 6 mo▪ Reported side effects included intermittent constipation (n = 5, 25%), menstrual irregularities (n = 4, 20%), and diarrhea (n = 3, 15%)▪ No new lesion on brain MRI at 6 mo |
| Lee et al. (2021) (21) | No clinically significant change | No significant change in MSQOL-54 | No significant change in perceived fatigue (MFIS) | Not reported | Not reported2 | Reduction in fasting blood sugar and insulin | Maximal mean plasma BHB was 1.48 ± 1.10 mmol/L (at 4 wk); ketosis was maintained (>0.5 mmol/L) at 8 and 12 wk |
| Bock et al. (2022) (23) | Not reported2 | Not reported | Not reported | Not reported | Not reported2 | Significant reduction in sNfL at 6 mo | ▪ One relapse in KD group during study period (3 in control diet group, 2 in FMD group)▪ A single cycle of FMD (7-d fasting) did not affect sNfL |
ALOX, arachidonate lipoxygenase; BDI, Beck Depression Inventory; BHB, β-hydroxybutyrate; BW, body weight; COX, cyclooxygenase; CSF, cerebrospinal fluid; EDSS, Expanded Disability Status Scale; FMD, fasting mimicking diet; KD, ketogenic diet; MFIS, Modified Fatigue Impact Scale; MSQOL-54, Multiple Sclerosis Quality of Life-54 questionnaire; OCB, oligoclonal band; sNfL, serum neurofilament light chain; WBC, white blood cell.
Only baseline data were reported.
The patient characteristics varied markedly between studies. For example, patients in the clinical trial conducted by Lee et al. (21) were much more severe in terms of their EDSS score (≥4.5, as imposed by the inclusion criteria) compared with patients in Brenton et al.’s study (EDSS score 1.0–4.0) (3). In another study, the EDSS score of study subjects was not reported (27). Most studies recruited mainly patients with relapsing-remitting MS, whereas a clinical trial and a case report focused more on the effects of KD in progressive MS (21, 25). The age distribution of enrolled patients also differed between studies (Table 1), and pediatric MS has been underrepresented in existing studies. It appeared that younger patients with milder disease or a relapsing-remitting form of the disease benefited more from KD in terms of EDSS score, quality of life, and fatigue (Table 2), although further studies are clearly needed to identify the patient characteristics that lead to more favorable responses to KD intervention.
Adverse events were reported in 2 clinical trials (3, 17). The most common adverse events in KD-treated patients were respiratory tract infection [∼67% compared with 75% in control group (17)], gastrointestinal symptoms (diarrhea, constipation, nausea; 28 to 40%), and genitourinary problems (ureteric colic, urinary tract infection, menstrual irregularities; 11 to 20%). Serious adverse events were rarely encountered.
The role of gut microbiota
Gut microbiota and their metabolites are considered important players in mediating the immunological effects of dietary intervention (31). Bifidobacterium could induce intestinal Th17 cells (32), and the latter are implicated in the pathogenesis of CNS autoimmunity (33). KD has been associated with decreased abundance of Bifidobacterium in patients with obesity, which in turn results in decreased intestinal Th17 cells (34). Together, these findings suggest that KD can ameliorate MS through its microbiome-modulating effects. However, only 1 study to date reported the effects of KD on the gut microbiota of patients with MS (24). Swidsinski et al. (24) found that the overall mass and diversity of the colonic microbiome were reduced prior to KD intervention in these patients at the group level, although interindividual differences were evident. The effects of KD appeared to be dynamic during the 6-mo period of dietary intervention in these patients. The overall bacterial concentrations and diversity were further reduced initially (at ∼2 wk after the introduction of KD), then subsequently gradually recovered. An exception to this general trend was Akkermansia, which increased initially but declined later. Akkermansia has been implicated in mediating the antiseizure effect associated with KD (35). Furthermore, Akkermansia was found to be increased in patients with MS in a recent study, and the Akkermansia isolated from these patients ameliorated EAE in a mouse model (36). Taken together, these findings are broadly consistent and suggest a neuroprotective role of Akkermansia in MS and related conditions. The dynamic changes in gut microbiota, as reported by Swidsinski et al. (24), underscore the importance of longitudinal evaluation after dietary intervention. A caveat of their study was that the authors did not examine the associations between microbiome changes and clinical or paraclinical characteristics. More studies are needed to elucidate the reproducibility of their findings and the clinical significance of KD–microbiota interactions.
Limitations
The research literature reviewed has some limitations. First, these studies are largely pilot ones and exploratory in nature. The sample size is likely insufficient for clarification of KD's effects on many MS-related outcomes. In the study protocol of a 3-armed randomized controlled trial (NCT03508414) designed to compare KD and intermittent fasting, it was estimated that 111 patients needed to be enrolled to achieve a statistical power of 80%, assuming that the dropout rate is 10% (30). Adequate sample size is also required to assess potential effect modifiers. However, this is often practically difficult, as illustrated by a change of inclusion criteria due to low initial enrollment in a recent study (21). Second, the duration of treatment and follow-up was relatively short. Therefore, the impact of dietary intervention on relapse rate and long-term trajectory of disability accrual could not be assessed. Third, the ketogenic ratio of study diets was 2.2 or lower. It remains unclear whether a higher ketogenic ratio, as used in some studies of refractory epilepsy (26, 37), could bring about more therapeutic benefits in patients with MS. In addition, KD is considered particularly suitable for children with drug-resistant epilepsy because children have better dietary compliance and more efficient extraction and utilization of blood ketones (38). However, pediatric MS has been underrepresented in existing studies (Table 1), thus further research is needed to assess the role of KD in this patient population. Fourth, it has been recognized that changes in dietary macronutrient composition could rapidly alter the human gut microbiome (39), which in turn impacts health and diseases. However, KD–microbiota interaction in the context of MS was only addressed in 1 study (as reviewed above), and clinical correlations were not examined. Hopefully this issue is acknowledged and incorporated into future studies, as exemplified by an ongoing trial that listed gut microbiome change as a secondary end point (30).
The Effects of KD on EAE and Other CNS Demyelination Animal Models
Observational and interventional studies in human patients provide invaluable information regarding the safety, efficacy, and feasibility of KD in the treatment of MS. However, given the presence of multiple potential confounders, it has been relatively difficult to gain mechanistic understanding of dietary intervention from human studies. Animal research could play a complementary role in this regard. Several well-established animal models have been used to investigate the effects of KD on CNS demyelination. These models differ by their pathogenetic mechanisms and only recapitulate MS in some aspects. In short, the demyelinating process is immune-mediated in EAE models, whereas it is toxin-induced in cuprizone models (40). EAE is often viewed as the corresponding animal model of MS, and multiple versions of EAE have been developed (41). In the studies reviewed below, EAE was induced by myelin oligodendrocyte glycoprotein (MOG)35–55 in C57BL/6J (B6) mice. The resulting disease course was monophasic (42), in contrast to the relapsing-remitting course in the majority of patients with MS. Nonetheless, the neuropathological and immunological features of this model could mimic aspects of human MS, making it useful in dissecting the mechanisms of potential therapeutic interventions.
Table 3 summarizes the animal studies investigating the effects of KD on CNS demyelination models. Two studies examined the effects of KD, with different timing of initiation, in a MOG35–55-induced EAE mouse model. Choi et al. (17) compared the therapeutic effects of various dietary regimens. KD attenuated disease severity when initiated around the onset of EAE. However, the effect was modest compared with that of FMD. In the other study, Kim et al. (19) studied the effects of KD initiated 7 d prior to the induction of EAE (i.e., ∼2–3 wk before clinical onset of EAE). It is worth noting that spatial memory deficits in EAE manifest prior to the onset of motor disability, which is reminiscent of the early appearance of cognitive dysfunction in pediatric MS before motor handicap ensues (43). This temporal sequence allows behavioral memory testing to be performed in EAE mice without the interference of suboptimal motor performance (19). The authors found that KD-treated mice exhibited improved learning and memory and also motor performance. The pathological effects at structural and electrophysiological levels were also rescued. Together these studies suggest that both preemptive (19) and therapeutic (17) use of KD is effective in ameliorating disease activity in EAE.
TABLE 3.
Overview of studies evaluating the effects of a ketogenic diet in experimental demyelination models1
| Author (year) | Animal model and KD regimen (ketogenic ratio) | Neuropathology or morphology | Motor function | Learning/memory (behavioral paradigm) | Anxiety/depression | Body weight | Biomarkers | Other findings or comments |
|---|---|---|---|---|---|---|---|---|
| Kim et al. (2012) (19) | MOG35–55-induced EAE mouse model; KD (6.3:1) started 7 d before EAE induction and continued until the time of killing | Reduced lesion volumes on T2-weighted imaging and preserved hippocampal volume on MRI | Decreased motor disability in nadir and recovery stage (motor scale) | Sustained preservation of spatial learning and memory (MWM) | Not reported | Not reported | Reduced brain ROS (in vivo bioluminescence imaging); reduced expression of cytokines (IL-1β, IL-6, TNF-α, IL-12, IL-17) and chemokines (IFN-γ, MCP1, MIP-1α, MIP-1β) in CNS and periphery | Sustained preservation of hippocampal LTP |
| Choi et al. (2016) (17) | MOG35–55-induced EAE mouse model; KD (6.36:1) initiated at initial signs of EAE2 and continued for 30 d | Not reported | Decreased motor disability (motor scale) | Not reported | Not reported | KD ameliorated body weight loss associated with EAE | Not reported | Compared with FMD, KD had more modest effects in ameliorating EAE symptoms. FMD reversed EAE progression in some mice, whereas KD did not |
| Zhang et al. (2020) (20) | CPZ-induced demyelination mouse model; normal diet vs. KD (1.21:1) concurrent with CPZ for 5 wk | Demyelination↓; promoted oligodendrocyte maturation (inferred by enhanced Olig2 protein expression); decreased reactive astrocytes and microglia activation | Not reported | Not reported | ↓Anxiety-like behavior, ↑exploratory behavior (open field) | KD ameliorated CPZ-induced body weight loss | ↓IL-1β, TNF-α, CXCL10 expression; ↑IL-10 expression; ↓expression of HDAC3 and NLRP3 | Focus on corpus callosum; blood glucose concentrations significantly decreased in KD-fed mice |
| Liu et al. (2020) (44) | CPZ-induced demyelination mouse model; normal diet vs. KD (3.060:1) concurrent with CPZ for 5 wk | KD inhibited the activation of microglia (especially M1-like microglia) and reactive astrocytes, and enhanced the expression of mature oligodendrocytes; demyelination↓ | ↑Motor coordination (rotarod) | ↓Learning/memory deficits (MWM) | ↓Anxiety-like behavior; ↑exploratory behavior (open field) | KD ameliorated CPZ-induced body weight loss | Attenuated oxidative stress (MDA↓; GSH↑); enhanced expression of PPAR-γ and SIRT1/phospho-Akt/mTOR | Focus on hippocampus; blood glucose did not differ among the groups |
Akt/mTOR, protein kinase B/mammalian target of rapamycin; CNS, central nervous system; CPZ, cuprizone; CXCL, cysteine-X-cysteine motif chemokine ligand; EAE, experimental autoimmune encephalomyelitis; FMD, fasting mimicking diet; GSH, glutathione; HDAC, histone deacetylase; KD, ketogenic diet; LTP, long-term potentiation; MCP, monocyte chemoattractant protein; MDA, malondialdehyde; MIP, macrophage inflammatory protein; MOG, myelin oligodendrocyte glycoprotein; MWM, Morris water maze; NLRP3, nod-like receptor pyrin domain containing 3; Olig2, oligodendrocyte transcription factor 2; PPAR-γ, peroxisome proliferator-activated receptor γ; ROS, reactive oxygen species; SIRT1, sirtuin 1.
Initial signs at ∼8–14 d after immunization.
Two related studies examined the effects of KD in a cuprizone-induced demyelination mouse model in corpus callosum and hippocampus, respectively (20, 44). Both studies, with somewhat different KD regimens, reported beneficial effects of KD on neuropathological changes, including less demyelination, enhanced oligodendrocyte maturation, and reduced reactive astrocytes and microglia activation as compared with those treated with normal diet. Behavioral phenotyping showed that mice cotreated with KD exhibited better learning and memory, emotion, and motor performance than those treated with cuprizone alone. Consistent with that observed in an EAE model (17), KD also ameliorated cuprizone-induced body weight loss.
Excessive expression of proinflammatory cytokines and chemokines in the CNS and periphery, as well as the production of reactive oxygen species, were observed in EAE mice. These were largely suppressed when KD was instituted before EAE induction (19). Qualitatively similar results were also seen in cuprizone-induced demyelination models, in which KD-treated mice had decreased expression of proinflammatory cytokines (IL-1β, TNF-α), chemokine (CXCL 10), and oxidative stress marker (malondialdehyde) (20, 44). These data were also broadly consistent with findings from patients with MS, in whom KD attenuated the mRNA expression of proinflammatory cyclooxygenase and lipoxygenase (22). Taken together, human and animal research findings suggest that the beneficial effects of KD in MS/EAE can be attributed in part to its anti-inflammatory and redox modulatory properties.
Potential Molecular Links between KD and MS/EAE
The study of KD therapy for MS/EAE to date, as reviewed above, not only suggests a promising way of dietary intervention but also yields potential mechanistic insights in relevant contexts (Figure 1). It has been increasingly recognized that the ketone body species can play dual roles. They can be transported into neurons and converted to acetyl-CoA in the mitochondria, thereby serving as energy substrates. On the other hand, the ketone bodies and related metabolites, such as decanoic acid, can also act as signaling molecules (9, 11, 45). The following is a concise review of current evidence regarding several candidate molecular players implicated in mediating the effects of KD on MS/EAE.
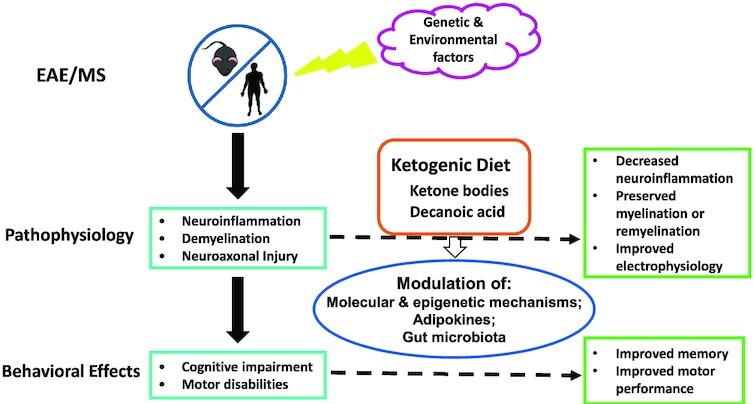
FIGURE 1.

A graphical summary of effects of a ketogenic diet in patients with MS and animal models of MS. MS is a multifactorial condition involving both genetic and environmental factors. Diet is 1 of the modifiable risk or protective factors. Specifically, the ketogenic diet could ameliorate neuroinflammation, demyelination, and their attendant behavioral effects in MS and related conditions, presumably through a variety of epigenetic and other molecular mechanisms, modulation of adipokines, and alterations of gut microbiota. EAE, experimental autoimmune encephalomyelitis; MS, multiple sclerosis.
Histone deacetylases
Histone deacetylase 3 (HDAC3), a member of class I HDACs, is involved in regulating inflammatory responses through deacetylase activity-dependent and -independent mechanisms (46). Its mRNA is expressed in significantly higher amounts in the peripheral blood mononuclear cells (PBMCs) of patients with MS (47), and it appears to be causally linked to IL-33 overexpression in these patients (48). Class I HDAC inhibition attenuates LPS-induced IL-33 expression in PBMCs derived from patients with MS (48). BHB inhibits HDAC3 in a dose-dependent manner (49), and KD ameliorates the increase in HDAC3 protein expression in the corpus callosum in a cuprizone-induced demyelination mouse model (20). HDAC1 is also involved in the pathogenesis of EAE (50), and it could also be inhibited by BHB (49).
On the other hand, sirtuin 1 (SIRT1), a member of class III HDACs, appears to have neuroprotective roles in MS and EAE (51, 52). KD is associated with enhanced SIRT1 expression in various cell types in the CNS, as well as better neurological outcomes in a cuprizone-induced demyelination mouse model (44). Taken together, these findings suggest that class I HDAC inhibition and/or SIRT1 augmentation could be potential mechanism(s) underlying the effects of KD on MS/EAE.
Acid-sensing ion channel
Acid-sensing ion channel (ASIC) subunit 1, encoded by the ASIC1 gene, is overexpressed in both acute and chronic MS lesions (53). The acidosis in the spinal cord of EAE mice is sufficient to activate ASIC1. Both genetic ablation and pharmacological blockade of ASIC1 attenuate disease activity in EAE mice (54). A pilot study showed radiological evidence of a neuroprotective effect of amiloride, an ASIC1 blocker, in a cohort of primary progressive MS patients (53). The main ketone body species, namely BHB, acetoacetate, and acetone, could inhibit ASIC-mediated currents in rat hippocampal neurons (55). Therefore, ASIC blockade might contribute to the beneficial effects of KD in patients with MS.
Forkhead box class O family
Members of the forkhead box class O (FOXO) gene family, FOXO1 and FOXO3A in particular, are implicated in the genetic susceptibility of relapsing-remitting MS (56). BHB upregulates the expression of Foxo1 (57) and Foxo3a (49) via histone β-hydroxybutyrylation and acetylation, respectively. Furthermore, FoxO1 is involved in oligodendrocyte regeneration and CNS myelination (58, 59), and FOXO3A contributes to oxidative stress resistance (49, 60). Taken together, members of the FOXO family could also be downstream mediators of KD in the context of MS/EAE.
Vesicular glutamate transporters
Vesicular glutamate transporter 1 (VGLUT1), encoded by the SLC17A7 gene, is implicated in the remyelination process (61). Its expression was downregulated in a mouse model of progressive EAE (62). KD ameliorates age-related decline in VGLUT1 expression in rat hippocampus (63). Taken together, these findings suggest that VGLUT1 might be another potential molecular player mediating the effect of KD in the context of MS/EAE. Other VGLUTs might also be relevant. For example, a 6-fold increase in VGLUT2 expression has been observed in demyelinated lesions compared with the normal-appearing white matter in people with MS, and VGLUT2 expression is presumably linked to the remyelination process in this scenario (64). A recent study showed that KD resulted in a significant increase in the abundance of VGLUT2 (encoded by Slc17a6) in the hippocampus of rats with status epilepticus (65). On the other hand, it has been demonstrated that acetoacetate and BHB could inhibit glutamate transport through VGLUT2 in an in vitro system (66). Whether and how VGLUT2 is modulated by KD, as well as its consequences, remains to be investigated in the context of MS/EAE.
NOD-, LRR- and pyrin domain–containing protein 3 inflammasome
NOD-, LRR-, and pyrin domain–containing protein 3 (NLRP3) inflammasome, the most abundant inflammasome in the CNS, has been implicated in the pathogenesis of MS, EAE, and the cuprizone-induced demyelination model (67–69). KD ameliorates the increase in NLRP3 protein expression in the corpus callosum in the cuprizone-induced demyelination mouse model (20). Ketone body species, specifically BHB, can attenuate NLRP3 activation in vitro and in vivo in the brain (70, 71). These together suggest that KD might also exert a beneficial effect through modulation of innate immune mechanisms in people with MS.
Peroxisome proliferator-activated receptor γ
Markedly decreased expression of peroxisome proliferator-activated receptor γ (PPARγ) mRNA has been found in monocytes derived from patients with relapsing-remitting MS (72). PPARγ could suppress Th17 cell differentiation, and its downregulation has been associated with exacerbation of EAE (73). Activation of PPARγ in various cell types in the brain also contributes to remyelination, as well as protection from neuroinflammation and demyelination (74–76). Decanoic acid, a major component of the MCT-based KD, is a well-known PPARγ agonist (9, 77). KD is also associated with significantly increased PPARγ protein expression in hippocampus in a cuprizone-induced demyelination model (44). Taken together, these findings suggest that KDs, particularly those enriched in decanoic acid, might confer protection against neuroinflammation in part through enhanced PPARγ action.
Adipokines
Several adipokines, both proinflammatory (leptin, fatty acid binding protein-4) and anti-inflammatory (adiponectin), have been implicated in the regulation of disease activity in MS and EAE (73, 78–80). Indeed, an animal study suggested that adiponectin could ameliorate disease activity in EAE through enhanced SIRT1/PPARγ expression and suppression of Th17 cell differentiation (73). On the other hand, PPARγ agonism also induces adiponectin expression (81, 82), suggesting a bidirectional relation between PPARγ and adiponectin. KD has been associated with increased serum adiponectin concentration in human and rodent studies (83–85). Research into the effects of KD on fasting adipokines of patients with MS, however, was scarce. One study reported significantly decreased leptin and borderline increased adiponectin concentrations after a 3-mo period of KD in MS patients who were largely obese or overweight (3). More studies are clearly needed to elucidate the role of adipokines in dietary therapy for MS.
Conclusions
Nutritional care for MS has gained wide clinical and scientific interest among patients and clinicians. Thus far, the published studies on the effects of KDs on MS are either pilot clinical trials with small sample size or anecdotal reports. Nonetheless, their findings together showed that KD is feasible and well tolerated, and it exhibits neuroprotective and disease-modifying potential in this patient population. Similarly, investigations on the effects of KDs in the corresponding rodent models of CNS demyelination yielded promising results. It has also been better recognized that major metabolites in KDs, particularly BHB and decanoic acid, are not only energy substrates but also important signaling molecules in the context of neuroinflammation. Further research in this direction is needed to elucidate the mechanisms of action and to inform evidence-based clinical practice of KDs in MS and related conditions.
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—W-SL, P-YL: designed research; W-SL, S-JL: conducted research; DS, W-SL: prepared the figure; W-SL, S-JL: wrote the first draft of the manuscript; P-YW, T-RH, DS, P-YL: reviewed and edited the manuscript for important intellectual content; W-SL: had primary responsibility for final content; and all authors: read and approved the final manuscript.
Notes
The authors reported no funding received for this study.
Author disclosures: The authors report no conflicts of interest.
W-SL and S-JL contributed equally to this work.
Abbreviations used: ASIC, acid-sensing ion channel; BHB, β-hydroxybutyrate; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; EDSS, Expanded Disability Status Scale; FMD, fasting mimicking diet; FOXO, forkhead box class O; HDAC, histone deacetylase; KD, ketogenic diet; MCT, medium-chain triglyceride; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; NLRP3, NOD-, LRR-, and pyrin domain–containing protein 3; PBMC, peripheral blood mononuclear cell; PPARγ, peroxisome proliferator-activated receptor γ; SIRT1, sirtuin 1; Th, T helper cell; VGLUT, vesicular glutamate transporter.
Contributor Information
Wei-Sheng Lin, Department of Pediatrics, Taipei Veterans General Hospital, Taipei, Taiwan; School of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan.
Shan-Ju Lin, Department of Physical Medicine and Rehabilitation, National Taiwan University Hospital Yunlin Branch, Yunlin, Taiwan.
Pei-Yin Liao, Department of Dietetics, National Taiwan University Hospital Yunlin Branch, Yunlin, Taiwan.
Divya Suresh, Department of Pediatrics, National Taiwan University Hospital Yunlin Branch, Yunlin, Taiwan.
Ting-Rong Hsu, Department of Pediatrics, Taipei Veterans General Hospital, Taipei, Taiwan; School of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan.
Pei-Yu Wang, Graduate Institute of Brain and Mind Sciences, College of Medicine, National Taiwan University, Taipei, Taiwan; Neurobiology and Cognitive Science Center, National Taiwan University, Taipei, Taiwan; Program in Translational Medicine, National Taiwan University and Academia Sinica, Taipei, Taiwan; Taiwan International Graduate Program in Interdisciplinary Neuroscience, National Taiwan University and Academia Sinica, Taipei, Taiwan; Graduate Institute of Neural Regenerative Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan.
References
- 1. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O, Multiple sclerosis. Lancet. 2018;391:1622–36. [DOI] [PubMed] [Google Scholar]
- 2. Brenton JN, Goldman MD. A study of dietary modification: perceptions and attitudes of patients with multiple sclerosis. Mult Scler Relat Disord. 2016;8:54–7. [DOI] [PubMed] [Google Scholar]
- 3. Brenton JN, Banwell B, Bergqvist AGC, Lehner-Gulotta D, Gampper L, Leytham Eet al. . Pilot study of a ketogenic diet in relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm. 2019;6:e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fitzgerald KC, Tyry T, Salter A, Cofield SS, Cutter G, Fox RJet al. . A survey of dietary characteristics in a large population of people with multiple sclerosis. Mult Scler Relat Disord. 2018;22:12–18. [DOI] [PubMed] [Google Scholar]
- 5. Burgos R, Breton I, Cereda E, Desport JC, Dziewas R, Genton Let al. . ESPEN guideline clinical nutrition in neurology. Clin Nutr. 2018;37:354–96. [DOI] [PubMed] [Google Scholar]
- 6. Evans E, Levasseur V, Cross AH, Piccio L. An overview of the current state of evidence for the role of specific diets in multiple sclerosis. Mult Scler Relat Disord. 2019;36:101393. [DOI] [PubMed] [Google Scholar]
- 7. Parks NE, Jackson-Tarlton CS, Vacchi L, Merdad R, Johnston BC. Dietary interventions for multiple sclerosis-related outcomes. Cochrane Database Syst Rev. 2020;5:CD004192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bagur MJ, Murcia MA, Jiménez-Monreal AM, Tur JA, Bibiloni MM, Alonso GLet al. . Influence of diet in multiple sclerosis: a systematic review. Adv Nutr. 2017;8:463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JHet al. . Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018;17:84–93. [DOI] [PubMed] [Google Scholar]
- 10. Koh S, Dupuis N, Auvin S. Ketogenic diet and neuroinflammation. Epilepsy Res. 2020;167:106454. [DOI] [PubMed] [Google Scholar]
- 11. Offermanns S, Schwaninger M. Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol Med. 2015;21:245–55. [DOI] [PubMed] [Google Scholar]
- 12. Pinto A, Bonucci A, Maggi E, Corsi M, Businaro R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer's disease. Antioxidants (Basel). 2018;7(5):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Storoni M, Plant GT. The therapeutic potential of the ketogenic diet in treating progressive multiple sclerosis. Mult Scler Int. 2015;2015:681289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuen AWC, Walcutt IA, Sander JW. An acidosis-sparing ketogenic (ASK) diet to improve efficacy and reduce adverse effects in the treatment of refractory epilepsy. Epilepsy Behav. 2017;74:15–21. [DOI] [PubMed] [Google Scholar]
- 16. Olson CA, Iniguez AJ, Yang GE, Fang P, Pronovost GN, Jameson KGet al. . Alterations in the gut microbiota contribute to cognitive impairment induced by the ketogenic diet and hypoxia. Cell Host Microbe. 2021;29(9):1378–92.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi IY, Piccio L, Childress P, Bollman B, Ghosh A, Brandhorst Set al. . A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. 2016;15:2136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stumpf SK, Berghoff SA, Trevisiol A, Spieth L, Duking T, Schneider LVet al. . Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus-Merzbacher disease. Acta Neuropathol. 2019;138:147–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim DY, Hao J, Liu R, Turner G, Shi FD, Rho JM. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS One. 2012;7:e35476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang N, Liu C, Zhang R, Jin L, Yin X, Zheng Xet al. . Amelioration of clinical course and demyelination in the cuprizone mouse model in relation to ketogenic diet. Food Funct. 2020;11:5647–63. [DOI] [PubMed] [Google Scholar]
- 21. Lee JE, Titcomb TJ, Bisht B, Rubenstein LM, Louison R, Wahls TL. A modified MCT-based ketogenic diet increases plasma β-hydroxybutyrate but has less effect on fatigue and quality of life in people with multiple sclerosis compared to a modified paleolithic diet: a waitlist-controlled, randomized pilot study. J Am Coll Nutr. 2021;40:13–25. [DOI] [PubMed] [Google Scholar]
- 22. Bock M, Karber M, Kuhn H. Ketogenic diets attenuate cyclooxygenase and lipoxygenase gene expression in multiple sclerosis. EBioMedicine. 2018;36:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bock M, Steffen F, Zipp F, Bittner S. Impact of dietary intervention on serum neurofilament light chain in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2022;9(1):e1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swidsinski A, Dörffel Y, Loening-Baucke V, Gille C, Göktas Ö, Reißhauer Aet al. . Reduced mass and diversity of the colonic microbiome in patients with multiple sclerosis and their improvement with ketogenic diet. Front Microbiol. 2017;8:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nathan J, Khedekar Kale D, Naik VD, Thakker F, Bailur S. Dietary therapy in secondary progressive multiple sclerosis: a case report. Cureus. 2019;11:e5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford Ret al. . Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3:175–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benlloch M, López-Rodríguez MM, Cuerda-Ballester M, Drehmer E, Carrera S, Ceron JJet al. . Satiating effect of a ketogenic diet and its impact on muscle improvement and oxidation state in multiple sclerosis patients. Nutrients. 2019;11(5):1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benlloch M, López-Rodríguez MM, Cuerda-Ballester M, Drehmer E, Carrera S, Ceron JJet al. . Reply to “When is a ketogenic diet ketogenic? Comment on satiating effect of a ketogenic diet and its impact on muscle improvement and oxidation state in multiple sclerosis patients. Nutrients 2019, 11, 1156.”. Nutrients. 2019;11(8):1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cignarella F, Cantoni C, Ghezzi L, Salter A, Dorsett Y, Chen Let al. . Intermittent fasting confers protection in CNS autoimmunity by altering the gut microbiota. Cell Metab. 2018;27:1222–35.e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bahr LS, Bock M, Liebscher D, Bellmann-Strobl J, Franz L, Prüß Aet al. . Ketogenic diet and fasting diet as nutritional approaches in multiple sclerosis (NAMS): protocol of a randomized controlled study. Trials. 2020;21:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexander M, Turnbaugh PJ. Deconstructing mechanisms of diet-microbiome-immune interactions. Immunity. 2020;53:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng Fet al. . Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. 2016;113:E8141–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BMLet al. . Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell. 2021;184:6281–98.e6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ang QY, Alexander M, Newman JC, Tian Y, Cai J, Upadhyay Vet al. . Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell. 2020;181:1263–75.e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell. 2018;173:1728–41.e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cox LM, Maghzi AH, Liu S, Tankou SK, Dhang FH, Willocq Vet al. . Gut microbiome in progressive multiple sclerosis. Ann Neurol. 2021;89:1195–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seo JH, Lee YM, Lee JS, Kang HC, Kim HD. Efficacy and tolerability of the ketogenic diet according to lipid:nonlipid ratios—comparison of 3:1 with 4:1 diet. Epilepsia. 2007;48:801–5. [DOI] [PubMed] [Google Scholar]
- 38. Masino SA, Rho JM. Mechanisms of ketogenic diet action. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. 4th ed. New York: Oxford University Press; 2012. p. 1003–24. [Google Scholar]
- 39. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BEet al. . Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118:723–36. [DOI] [PubMed] [Google Scholar]
- 41. Rangachari M, Kuchroo VK. Using EAE to better understand principles of immune function and autoimmune pathology. J Autoimmun. 2013;45:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15:1074–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin WS, Lin SJ, Hsu TR. Cognitive assessment and rehabilitation for pediatric-onset multiple sclerosis: a scoping review. Children (Basel). 2020;7(10):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu C, Zhang N, Zhang R, Jin L, Petridis AK, Loers Get al. . Cuprizone-induced demyelination in mouse hippocampus is alleviated by ketogenic diet. J Agric Food Chem. 2020;68:11215–28. [DOI] [PubMed] [Google Scholar]
- 45. Newman JC, Verdin E. β-Hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nguyen HCB, Adlanmerini M, Hauck AK, Lazar MA. Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature. 2020;584:286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang F, Shi Y, Wang L, Sriram S. Role of HDAC3 on p53 expression and apoptosis in T cells of patients with multiple sclerosis. PLoS One. 2011;6:e16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang F, Tossberg JT, Spurlock CF, Yao SY, Aune TM, Sriram S. Expression of IL-33 and its epigenetic regulation in multiple sclerosis. Ann Clin Transl Neurol. 2014;1:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan Net al. . Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Göschl L, Preglej T, Hamminger P, Bonelli M, Andersen L, Boucheron Net al. . A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis. J Autoimmun. 2018;86:51–61. [DOI] [PubMed] [Google Scholar]
- 51. Martin A, Tegla CA, Cudrici CD, Kruszewski AM, Azimzadeh P, Boodhoo Det al. . Role of SIRT1 in autoimmune demyelination and neurodegeneration. Immunol Res. 2015;61:187–97. [DOI] [PubMed] [Google Scholar]
- 52. Tegla CA, Azimzadeh P, Andrian-Albescu M, Martin A, Cudrici CD, Trippe R 3rdet al. . SIRT1 is decreased during relapses in patients with multiple sclerosis. Exp Mol Pathol. 2014;96:139–48. [DOI] [PubMed] [Google Scholar]
- 53. Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MIet al. . Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain. 2013;136:106–15. [DOI] [PubMed] [Google Scholar]
- 54. Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJet al. . Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–9. [DOI] [PubMed] [Google Scholar]
- 55. Zhu F, Shan W, Xu Q, Guo A, Wu J, Wang Q. Ketone bodies inhibit the opening of acid-sensing ion channels (ASICs) in rat hippocampal excitatory neurons in vitro. Front Neurol. 2019;10:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gökdoğan Edgünlü T, Ünal Y, Karakaş Çelik S, Genç Ö, Emre U, Kutlu G. The effect of FOXO gene family variants and global DNA methylation on RRMS disease. Gene. 2020;726:144172. [DOI] [PubMed] [Google Scholar]
- 57. Zhang H, Tang K, Ma J, Zhou L, Liu J, Zeng Let al. . Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8(+) T-cell memory development. Nat Cell Biol. 2020;22:18–25. [DOI] [PubMed] [Google Scholar]
- 58. Jablonska B, Scafidi J, Aguirre A, Vaccarino F, Nguyen V, Borok Eet al. . Oligodendrocyte regeneration after neonatal hypoxia requires FOXO1-mediated p27Kip1 expression. J Neurosci. 2012;32:14775–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Palazuelos J, Klingener M, Aguirre A. TGFβ signaling regulates the timing of CNS myelination by modulating oligodendrocyte progenitor cell cycle exit through SMAD3/4/FoxO1/Sp1. J Neurosci. 2014;34:7917–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJet al. . Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–21. [DOI] [PubMed] [Google Scholar]
- 61. Pfeiffer F, Frommer-Kaestle G, Fallier-Becker P. Structural adaption of axons during de- and remyelination in the cuprizone mouse model. Brain Pathol. 2019;29:675–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sevastou I, Pryce G, Baker D, Selwood DL. Characterisation of transcriptional changes in the spinal cord of the progressive experimental autoimmune encephalomyelitis biozzi ABH mouse model by RNA sequencing. PLoS One. 2016;11:e0157754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hernandez AR, Hernandez CM, Campos KT, Truckenbrod LM, Sakarya Y, McQuail JAet al. . The antiepileptic ketogenic diet alters hippocampal transporter levels and reduces adiposity in aged rats. J Gerontol A Biol Sci Med Sci. 2018;73:450–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard Iet al. . Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun. 2015;6:8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zheng YQ, Jin MF, Suo GH, Wu YJ, Sun YX, Ni H. Proteomics for studying the effects of ketogenic diet against lithium chloride/pilocarpine induced epilepsy in rats. Front Neurosci. 2020;14:562853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara Cet al. . Metabolic control of vesicular glutamate transport and release. Neuron. 2010;68:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Aryanpour R, Pasbakhsh P, Zibara K, Namjoo Z, Beigi Boroujeni F, Shahbeigi Set al. . Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int Immunopharmacol. 2017;51:131–9. [DOI] [PubMed] [Google Scholar]
- 68. Hou B, Zhang Y, Liang P, He Y, Peng B, Liu Wet al. . Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020;11:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schuh E, Lohse P, Ertl-Wagner B, Witt M, Krumbholz M, Frankenberger Met al. . Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm. 2015;2:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shippy DC, Wilhelm C, Viharkumar PA, Raife TJ, Ulland TK. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer's disease pathology. J Neuroinflammation. 2020;17:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim Det al. . The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wouters E, Grajchen E, Jorissen W, Dierckx T, Wetzels S, Loix Met al. . Altered PPARγ expression promotes myelin-induced foam cell formation in macrophages in multiple sclerosis. Int J Mol Sci. 2020;21:9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang K, Guo Y, Ge Z, Zhang Z, Da Yet al. . Adiponectin suppresses T helper 17 cell differentiation and limits autoimmune CNS inflammation via the SIRT1/PPARγ/RORγt pathway. Mol Neurobiol. 2017;54:4908–20. [DOI] [PubMed] [Google Scholar]
- 74. Duvanel CB, Honegger P, Pershadsingh H, Feinstein D, Matthieu JM. Inhibition of glial cell proinflammatory activities by peroxisome proliferator-activated receptor gamma agonist confers partial protection during antimyelin oligodendrocyte glycoprotein demyelination in vitro. J Neurosci Res. 2003;71:246–55. [DOI] [PubMed] [Google Scholar]
- 75. Klotz L, Diehl L, Dani I, Neumann H, von Oppen N, Dolf Aet al. . Brain endothelial PPAR-gamma controls inflammation-induced CD4+ T cell adhesion and transmigration in vitro. J Neuroimmunol. 2007;190:34–43. [DOI] [PubMed] [Google Scholar]
- 76. Kanakasabai S, Pestereva E, Chearwae W, Gupta SK, Ansari S, Bright JJ. PPARγ agonists promote oligodendrocyte differentiation of neural stem cells by modulating stemness and differentiation genes. PLoS One. 2012;7:e50500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell Met al. . The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem. 2014;129:426–33. [DOI] [PubMed] [Google Scholar]
- 78. Bove R, Healy BC, Musallam A, Soltany P, Diaz-Cruz C, Sattarnezhad Net al. . Fatty acid binding protein-4 is associated with disability in multiple sclerosis patients. Mult Scler. 2019;25:344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marrodan M, Farez MF, Balbuena Aguirre ME, Correale J. Obesity and the risk of multiple sclerosis. The role of leptin. Ann Clin Transl Neurol. 2021;8:406–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sanna V, Di Giacomo A, La Cava A, Lechler RI, Fontana S, Zappacosta Set al. . Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. 2003;111:241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Iwaki M, Matsuda M, Maeda N, Funahashi T, Matsuzawa Y, Makishima Met al. . Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52:1655–63. [DOI] [PubMed] [Google Scholar]
- 82. Banga A, Unal R, Tripathi P, Pokrovskaya I, Owens RJ, Kern PAet al. . Adiponectin translation is increased by the PPARγ agonists pioglitazone and omega-3 fatty acids. Am J Physiol Endocrinol Metab. 2009;296:E480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rosenbaum M, Hall KD, Guo J, Ravussin E, Mayer LS, Reitman MLet al. . Glucose and lipid homeostasis and inflammation in humans following an isocaloric ketogenic diet. Obesity (Silver Spring). 2019;27:971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Partsalaki I, Karvela A, Spiliotis BE. Metabolic impact of a ketogenic diet compared to a hypocaloric diet in obese children and adolescents. J Pediatr Endocrinol Metab. 2012;25:697–704. [DOI] [PubMed] [Google Scholar]
- 85. Widiatmaja DM, Lutvyani A, Sari DR, Kurniasari H, Meiliana ID, Fasitasari Met al. . The effect of long-term ketogenic diet on serum adiponectin and insulin-like growth factor-1 levels in mice. J Basic Clin Physiol Pharmacol. [Internet] 2021. doi: 10.1515/jbcpp-2021-0287. [DOI] [PubMed] [Google Scholar]