

Abstract
Phosphorylation-inducing chimeric small molecules (PHICS) can enable a kinase to act at a new cellular location or phosphorylate non-native substrates (neo-substrates)/ sites (neo-phosphorylations).[1,2] We report a modular design and high-yielding synthesis of such PHICS that endowed multiple new activities to protein kinase C (PKC). For example, while PKC is unable to downregulate the activity of a gain-of-function variant (S180A) of Bruton’s tyrosine kinase that evokes B cell malignancy phenotype, PHICS enabled PKC to induce inhibitory neo-phosphorylations on this variant. Furthermore, while PKC typically phosphorylates its membrane-associated substrates, PKC with PHICS phosphorylated multiple cytosol-based neo-substrates (e.g., BCR-ABL). Finally, a PHICS for BCR-ABL induced death of chronic myeloid leukemia cell lines. These studies show the power of synthetic chemistry to expand the chemical and functional diversity of proteins in cells using bifunctional molecules.
Keywords: Bifunctional Molecules, Kinase, PHICS, Phosphorylation, Protein Kinase C
Graphical Abstract
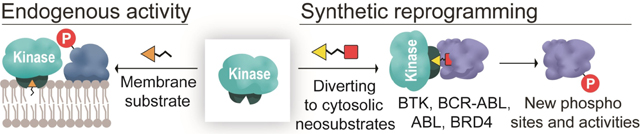
A high-yielding synthetic route to bifunctional molecules that reprogram activities of protein kinase C and engineered Abelson kinase to induce phosphorylation on neo-substrates is reported.
These phosphorylations have promising inhibitory effects on the activity of Bruton’s tyrosine kinase (BTK) and BCR-ABL-dependent cancer cells.
Nature has evolved kinase-substrate pairs for the site-, substrate-, and cellular-location-specific induction of phosphorylation. Protein kinase C (PKC) associates with the membrane through its C1 domain, wherein ligand binding to this domain induces membrane translocation and subsequent PKC-based phosphorylation of its membrane-associated targets (Scheme 1).[3] C1 ligands have two components, a polar headgroup (orange triangle, Scheme 1) that binds to the C1 domain[4] and a hydrophobic tail that inserts in the membrane. The C1 domain has a hydrophobic surface that lines a hydrophilic pocket. The binding of the polar headgroup of the ligand to the hydrophilic pocket of C1 provides a continuous hydrophobic surface, while the hydrophobic tail of the ligand facilitates the membrane translocation of PKC.
Scheme 1.

The activity of PKC in the presence of endogenous ligands (top) or PHICS (bottom).
We recently reported phosphorylation-inducing chimeric small molecules (PHICS) that promote proximity between a kinase and target protein. PHICS altered the specificity of AMP-activated protein kinase (AMPK) to induce naturally-occurring phosphorylation on Bruton’s tyrosine kinase (BTK) in cells albeit the functional consequence of such phosphorylation was unexplored.[2] PHICS that enabled PKC to phosphorylate its non-substrate BRD4 was described, but in a membrane-less, buffered solution without the competing PKC membrane translocation pathway that is observed in cells.[2] This PHICS consisted of PKC and BRD4 binders connected by a hydrophobic linker similar to the hydrophobic tail in C1 ligands (Scheme 1). To develop PKC-based PHICS that work in cells, we hypothesized that the PKC and target binders should be connected by polar linkers that will not insert in the membrane. Here, the PHICS should orient the C1 domain (and PKC) to interface with the target protein as opposed to the membrane and such reorientation may allow induction of neo-phosphorylation even on PKC substrates (Scheme 1). Furthermore, while we were able to use computational docking studies to identify linker attachment site to the benzolactam-based PKC binder,[4] the reported synthesis of benzolactam core required laborious protection/deprotection steps and the installation/removal of directing groups, which negatively affects overall yield and prevents assembly of PHICS for various targets and their subsequent biological evaluation. For example, synthesis of intermediate 6 (Figure 1A) from reported synthetic routes requires 17 steps with an overall yield of <1%.[5] Thus, the development of PKC PHICS that can work in cells requires the design of new linkers and synthetic strategies.
Figure 1.

A) Improved synthesis of the benzolactam core of PKC binder. B) Modular synthesis of PHICS.
PKC PHICS studies were initiated by first developing a modular and highly-yielding synthetic route (Figure 1A) to rapidly access 6 using Negishi coupling[6] between the N-Boc-3-iodoalanine methyl ester and 1-bromo-4-methoxy-2-nitrobenzene that afforded 1 in 90% yield, a substitution pattern that previously required 10 steps (intermediate 34 in ref. [5a]). DIBALH-reduction of ester 1 gave alcohol 2 in 72% yield. Hydrogenation of the nitro group in 2 and subsequent nucleophilic displacement of the triflate from (R)-benzyl-2-hydroxyisobutyrate produced 3 in 70% yield. Removal of Boc- and benzyl-protecting groups followed by lactamization resulted in formation of 4 with 58% yield. Intermediate 6 was accessed by reductive amination of 4 followed by deprotection of the phenolic group with an overall yield of 22% in 6 purification steps (9 transformations). To accommodate further functionalization, phenol 6 was alkylated with the Boc-protected para-aminomethylbenzyl group, which yielded building block 7 in 77% yield.
We leveraged our modular and facile access to the benzolactam core to synthesize PHICS for various targets, including BRD4 (Figure S2), BTK (Figure S3), Abelson kinase (ABL), BCR-ABL (Figure S4), and FKBP12 (Figure S5). To attenuate the membrane translocation of PKC, the target protein binder was appended to the benzolactam core using hydrophilic moieties (e.g., based on polyethylene glycol). We focused on protein targets for which high-quality ligands with co-crystal structures were available, selecting (S)-JQ1 (for BRD4),[7] a non-covalent variant of Ibrutinib (for BTK),[8] dihydropyrazole (for ABL and BCR-ABL),[9] and SLF (for FKBP12).[10] Our convergent and modular approach allowed the rapid assembly of PHICS against multiple targets, as the target binders can be readily functionalized with amines or acids and coupled to the Boc-deprotected free amine of benzolactam binder 7 via commercially available bis-NHS esters or ω-amino acids (Figure 1B). Using bis-NHS esters, PKC PHICS can be assembled in a single step for targets with available amine-containing binders.
Following PHICS assembly, the phosphorylation of BRD4 by PKC in vitro was investigated. BRD4 phosphorylation was detected in the presence of BRD4-targeting PHICS4 (Figure S6), indicating that the polar linkers did not inhibit PKC activity as has been proposed before.[11] We validated the role of proximity-induction in phosphorylation through the use of a chimeric molecule (iPHICS4) generated from the inactive enantiomer of (S)-JQ1 that does not bind strongly with BRD4. No significant phosphorylation was observed with this negative control.[7]
We next tested PHICS4 in cells, assessing its ability to induce the PKC interaction with neo-substrates via the C1 domain using a BRD4 construct that localizes in the cytosol.[12] HEK293T cells were co-transfected with PKC and BRD4 tagged with hemagglutinin (PKC-HA and BRD4-HA) and HA-based immunoprecipitation was performed after incubation with PHICS4 and controls. The immunoprecipitated BRD4-HA was probed with the antiphospho-PKC-substrate motif antibody and significantly higher phosphorylation levels were observed with PHICS4 over that of iPHICS4 (Figure 2A, B). These results demonstrate that PKC, which usually interacts with membranelocalized targets, can be redirected towards cytoplasmic proteins via the C1 domain and induce phosphorylation in the cellular milieu.
Figure 2.

A) Structures of PHICS4 and inactive analog iPHICS4. B) PHICS4-induced phosphorylation of cytoplasmic BRD4 by PKC or Cl-ABL in HEK293T cells. Cells were incubated with 10 μM of compound for 4 h in each treatment.
To further validate the purported mechanism of proximity-induced phosphorylation via PHICS and expand the scope of available transformations from Ser/Thr to Tyr phosphorylation, the C1 domain was grafted onto a variant of ABL kinase (C1-ABL), and tested to determine if PHICS4 can also recruit C1-ABL to induce Tyr phosphorylation of cytosolic BRD4. Here, HEK293T cells were co-transfected with C1-ABL and BRD4 and treated with PHICS4 and control molecules (iPHICS4 and PKC binder VS1099). As expected, a significantly higher level of Tyr phosphorylation was observed in the presence of an active bifunctional PHICS4 than with inactive analog iPHICS4 or PKC binder VS1099 (Figure 2A, B), indicating that PHICS can induce phosphorylation of tyrosine residues on neo-substrates.
After inducing neo-substrate phosphorylations in cells using PKC PHICS, we focused on the known PKC substrate BTK and its hyperactive, gain-of-function variant (S180A) to determine if PHICS can expand chemical and functional diversity in cells. PKC-mediated BTK phosphorylation at Ser180 serves as an “off switch” that downregulates BTK activity, but eliminating the negative regulatory site in S180A leads to a “hyperactive phenotype” reminiscent of B cell malignancies (Figure 3D).[13] As such, PKC PHICS that can downregulate activity (e.g., autophosphorylation) of this BTK variant by installing a new off switch will have therapeutic value. We also envisioned that the PHICS-mediated ternary complex would be fundamentally different from the naturally occurring PKC-BTK complex in cells, which may induce neo-phosphorylations.
Figure 3.

A) Structures of PHICS5 and inactive analog iPHICS5. B) PHICS5-induced phosphorylation of BTK (S180A) by PKC in HEK293T cells. Cells were incubated with 1 μM of compound for 4 h in each treatment. C) Effect of Ser to Asp/Ala mutation on BTK autophosphorylation. D) PHICS expands biological activity of PKC on BTK S180A variant.
Since PKC phosphorylates membrane-associated BTK with the C1 domain buried in the lipid bilayer, PHICS-mediated BTK phosphorylation by PKC was characterized in the absence of lipid bilayer. While some phosphorylation of BTK by PKC in the absence of BTK-targeting PHICS5 was observed (quantified using PKC-motif antibody), more phosphorylation in the presence of PHICS5 was detected (Figure S8). The activity of PHICS5 was compared with its inactive analog iPHICS5, which has a pivaloyl group on the 4-aminopyrazolo[3,4-d]pyrimidine that sterically clashes in the BTK binding pocket,[2a] and iPHICS5 induced lower phosphorylation levels (Figure S8).
Next, the effect of PHICS5 on BTK’s hyperactive variant, S180A was examined. HEK293T cells were transfected with BTK-FLAG (S180A variant) and PKC-HA and a FLAG-based immunoprecipitation was performed after incubation with PHICS5 and controls (i.e., DMSO, PKC binder VS1099, and iPHICS5). As expected, a higher level of BTK (S180A) phosphorylation was observed in the presence of PHICS5 using a PKC motif antibody (Figure 3B).
Next, neo-phosphorylation sites were identified using mass spectrometry and a genetic phosphomimetic approach was used to further annotate biological activity. Several neo-phosphorylation sites (pS310, pS323, pS378, pT410) were observed on BTK obtained from cells treated with PHICS5 (Figures S9).[14] One of the modified residues (T410) is located close to the ATP binding pocket, whereas other sites (S310, S323, and S378) are on the loops of the SH2 domain in BTK. A recent study showed that the interface between the SH2 domain and kinase domain is critical for BTK activation and can be a potential site for allosteric inhibition.[15]
To explore the biological consequences of these neo-phosphorylations, a genetic approach where the phosphorylated Ser/Thr residues were mutated to Asp (phosphomimetic), and Ala (control) was adopted. In contrast to WT BTK, the S310D, S378D, and T410D variants exhibited reduced BTK autophosphorylation while the S323D variant did not have any effect as detected by western blot with a pY223 BTK-specific antibody (Figure 3C). Importantly, Ser/Thr to Ala variants showed similar levels of autophosphorylation as WT BTK, confirming that inhibitory effects of mutations arise from the introduced negative charge on Ser/Thr residues and not from the removal of the hydroxyl groups (Figure 3C). These results suggest that the activity of S180A variant of BTK can be inhibited by PHICS-induced neo-phosphorylation of BTK by PKC (Figure 3D).
To demonstrate the generalizability of our approach to other targets, ABL-targeting PHICS6 (Figure 4A) was tested in vitro. As expected, PHICS6 induced higher phosphorylation of ABL relative to DMSO- or PKC-binder VS1012-treated controls (Figure S7A). Interestingly, the bifunctional molecule VS558 based on the known inhibitor dasatinib that binds to a different pocket of ABL kinase[16] still induced its phosphorylation by PKC in vitro (Figure S7B). To test if endogenous PKC can be redirected towards phosphorylation of oncogenic targets, ABL-targeting PHICS was tested in chronic myeloid leukemia cells (e.g., K562 cells) that contain oncogenic fusion gene BCR-ABL. This fusion is generated by a chromosomal translocation and codes for hyperactive tyrosine kinase that is “always-on” causing the cells to divide uncontrollably.[17]
Figure 4.

A) Structures of PHICS6 and PKC binder VS1012. B), C) PHICS6-induced phosphorylation of endogenous BCR-ABL (B) and ABL (C) in K562 cells by endogenous PKC detected with phospho-ABL (Thr735) antibody. Cells were incubated with 10 μM of compound for 4 h in each treatment. D) Effect of PHICS6 on viability of K562 and KCl22-s cells.
K562 cells were treated with PHICS6 and PKC binder VS1012 as a control and a higher degree of phosphorylation at Thr735 of BCR-ABL in the presence of PHICS6 was observed (Figure 4A, B). The enrichment of pThr735 on ABL in the PHICS6-treated sample (Figure 4C) was also detected. This phosphorylation is critical for binding to 14–33 proteins and induction of cytoplasmic sequestration of ABL.[18] To evaluate a biological effect of PHICS6, we performed a viability assay on K562 cells. PHICS6 induced dose-dependent death of K562 cells with an EC50 of 2.6 μM (Figure 4D), while PKC and ABL binders VS1012 and VS1088 did not have any significant effect at concentrations as high as 10 μM (viability>80%). Similarly, viability of another BCR-ABL-dependent cancer cell line KCl22-s was also affected by PHICS6 with an EC50 of 3.4 μM (Figure 4D). In agreement with dose-dependency in the viability assay, PHICS6 demonstrated dose-dependent increase of phosphorylation in HEK293T cells co-transfected with BCR-ABL-FLAG and PKC-HA (Figure S7C).
So far, all of the proteins successfully phosphorylated by PKC using PHICS were relatively large (>75 kDa). To test whether the kinase domain of PKC might be too far away to phosphorylate smaller proteins, the small protein FKBP12 was used. Accordingly, PHICS7 (Figure S5) was tested in vitro for the induction of FKBP12-His phosphorylation, and there was no signal observed when samples were probed with a phospho-PKC substrate motif antibody (Figure S10A). To rule out antibody bias, adenosine-5′-(γ-thio)-triphosphate (ATP-γ-S) was used for detection: this approach should provide global coverage of all the possible phosphorylations on a protein. In agreement with our previous observation, no phosphorylation of FKBP was detected (Figure S10B). Interestingly, phosphorylation on the FKBP-GST fusion was observed, and our future work will explore the utility of FKBP as a tag to induce phosphorylation on target proteins that lack high-quality chemical probes akin to development of dTAG for proteolysis targeting chimeras.[19]
Overall, we herein report a modular and highly efficient synthetic route for the rapid, combinatorial assembly of PHICS for multiple protein targets. The attachment of polar linkers to the benzolactam core recruited PKC for the phosphorylation of cytosolic targets, even though it usually operates on membrane-associated targets. These PKC-based PHICS can phosphorylate multiple neo-substrates (BRD4, ABL, and BCR-ABL), and add neo-phosphorylations to a known substrate of PKC (BTK). Using a phosphomimetic approach, it was confirmed that these neo-phosphorylations inhibit the activity of BTK, offering an avenue to regulate hyperactive BTK variants (e.g., S180A) by allowing the installation of neo-“off switches”. Furthermore, designed PHICS molecules demonstrated promising activity in the inhibition of BCR-ABL dependent cancer cells. Finally, the scope of transformations available for PHICS was expanded from Ser/Thr to Tyr using an engineered Tyr kinase that bears the C1 domain. This domain, consisting of only ≈50 amino acids, is perhaps the smallest to form small-molecule binding pocket.[3a,20] There are ≈8 proteins with C1 domains that can bind to benzolactam with high affinity.[3a] Future studies will involve optimizing of the selectivity and potency of PKC-based bifunctional molecules, explore the effect of dose and time of treatment on phosphorylation levels at different sites, investigate the mechanism by which PHICS inhibits the viability of cancer cells, and expand applications of C1 domain as a tag in protein engineering and gene therapy. Taken together, the studies presented herein broaden the scope of PHICS and demonstrate the utility of chimeric molecules for inducing unnatural phosphorylation in cells by rewiring the specificity of kinases,[21] expanding the chemical toolkit for functional diversification of proteins in cells and providing a new therapeutic strategy for the treatment of cancer.
Supplementary Material
Acknowledgements
We thank Dr. Mary O’Reilly (Pattern, Broad Institute) for contributive discussions. This work was supported by the Merkin Institute of Transformative Technologies in Healthcare, Burroughs Wellcome Fund (Career Award at the Scientific Interface), and NIH (R21AI154099). V.M.S. was supported by Damon Runyon Postdoctoral Fellowship.
Contributor Information
Dr. Veronika M. Shoba, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA); Department of Medicine, Harvard Medical School Boston, MA 02115 (USA).
Dr. Dhanushka N. P. Munkanatta Godage, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA); Department of Medicine, Harvard Medical School Boston, MA 02115 (USA).
Dr. Santosh K. Chaudhary, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA); Department of Medicine, Harvard Medical School Boston, MA 02115 (USA).
Dr. Arghya Deb, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA); Department of Medicine, Harvard Medical School Boston, MA 02115 (USA).
Dr. Sachini U. Siriwardena, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA) Department of Medicine, Harvard Medical School Boston, MA 02115 (USA).
Dr. Amit Choudhary, Chemical Biology and Therapeutics Science Program, Broad Institute of MIT and Harvard Cambridge, MA 02142 (USA); Department of Medicine, Harvard Medical School Boston, MA 02115 (USA); Divisions of Renal Medicine and Engineering Brigham and Women’s Hospital Boston, MA 02115 (USA).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- [1].a) Siriwardena SU, Munkanatta Godage DNP, Shoba VM, Lai S, Shi M, Wu P, Chaudhary SK, Schreiber SL, Choudhary A, J. Am. Chem. Soc. 2020, 142, 14052–14057; [DOI] [PubMed] [Google Scholar]; b) Modell AE, Lai S, Nguyen TM, Choudhary A, Cell Chem. Biol. 2021, 28, 1081–1089. [DOI] [PubMed] [Google Scholar]
- [2].a) Stanton BZ, Chory EJ, Crabtree GR, Science 2018, 359, eaao5902; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gerry CJ, Schreiber SL, Nat. Chem. Biol. 2020, 16, 369–378; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nalawansha DA, Crews CM, Cell Chem. Biol. 2020, 27, 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Das J, Rahman GM, Chem. Rev. 2014, 114, 12108–12131; [DOI] [PubMed] [Google Scholar]; b) Blumberg PM, Kedei N, Lewin NE, Yang D, Czifra G, Pu Y, Peach ML, Marquez VE, Curr. Drug Targets 2008, 9, 641–652; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Newton AC, Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang G, Kazanietz MG, Blumberg PM, Hurley JH, Cell 1995, 81, 917–924. [DOI] [PubMed] [Google Scholar]
- [5].a) Ma D, Tang W, Kozikowski AP, Lewin NE, Blumberg PM, J. Org. Chem. 1999, 64, 6366–6373; [Google Scholar]; b) Kozikowski P, Chen Y, Subhasish T, Lewin NE, Blumberg PM, Zhong Z, D’Annibale MA, Wang WL, Shen Y, Langley B, ChemMedChem 2009, 4, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tuttle JB, Azzarelli JM, Bechle BM, Dounay AB, Evrard E, Gan X, Ghosh S, Henderson J, Kim J-Y, Parikh VD, Verhoest PR, Tetrahedron Lett. 2011, 52, 5211–5213. [Google Scholar]
- [7].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Johnson AR, Kohli PB, Katewa A, Gogol E, Belmont LD, Choy R, Penuel E, Burton L, Eigenbrot C, Yu C, Ortwine DF, Bowman K, Franke Y, Tam C, Estevez A, Mortara K, Wu J, Li H, Lin M, Bergeron P, Crawford JJ, Young WB, ACS Chem. Biol. 2016, 11, 2897–2907. [DOI] [PubMed] [Google Scholar]
- [9].Simpson GL, Bertrand SM, Borthwick JA, Campobasso N, Chabanet J, Chen S, Coggins J, Cottom J, Christensen SB, Dawson HC, Evans HL, Hobbs AN, Hong X, Mangatt B, Munoz-Muriedas J, Oliff A, Qin D, Scott-Stevens P, Ward P, Washio Y, Yang J, Young RJ, J. Med. Chem. 2019, 62, 2154–2171. [DOI] [PubMed] [Google Scholar]
- [10].Holt DA, Luengo JI, Yamashita DS, Oh HJ, Konialian AL, Yen HK, Rozamus LW, Brandt M, Bossard MJ, Levy MA, Eggleston DS, Liang J, Schultz LW, Stout TJ, Clardy J, J. Am. Chem. Soc. 1993, 115, 9925–9938. [Google Scholar]
- [11].Wada R, Suto Y, Kanai M, Shibasaki M, J. Am. Chem. Soc. 2002, 124, 10658–10659. [DOI] [PubMed] [Google Scholar]
- [12].Fukazawa H, Masumi A, Biol. Pharm. Bull. 2012, 35, 2064–2068. [DOI] [PubMed] [Google Scholar]
- [13].Kang SW, Wahl MI, Chu J, Kitaura J, Kawakami Y, Kato RM, Tabuchi R, Tarakhovsky A, Kawakami T, Turck CW, Witte ON, Rawlings DJ, EMBO J. 2001, 20, 5692–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E, Nucleic Acids Res. 2015, 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Duarte DP, Lamontanara AJ, La Sala G, Jeong S, Sohn YK, Panjkovich A, Georgeon S, Kükenshöner T, Marcaida MJ, Pojer F, De Vivo M, Svergun D, Kim HS, Dal Peraro M, Hantschel O, Nat. Commun. 2020, 11, 2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, Xie D, Zhang Y, Klei HE, Cancer Res. 2006, 66, 5790–5797. [DOI] [PubMed] [Google Scholar]
- [17].Ren R, Nat. Rev. Cancer 2005, 5, 172–183. [DOI] [PubMed] [Google Scholar]
- [18].Nihira K, Taira N, Miki Y, Yoshida K, Oncogene 2008, 27, 7285–7295. [DOI] [PubMed] [Google Scholar]
- [19].Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, Scott TG, Vittori S, Perry JA, Qi J, Winter GE, Wong KK, Gray NS, Bradner JE, Nat. Chem. Biol. 2018, 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vandemoortele G, Eyckerman S, Gevaert K, Trends Biotechnol. 2019, 37, 1078–1090. [DOI] [PubMed] [Google Scholar]
- [21].McCormick JW, Pincus D, Resnekov O, Reynolds KA, Trends Biochem. Sci. 2020, 45, 259–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.