

Abstract
We investigated the hypothesis that high Ca2+ influx during traumatic brain injury induces the activation of the caspase-1 enzyme, which triggers neuroinflammation and cell apoptosis in a cell culture model of neuronal stretch injury and an in vivo model of fluid percussion injury (FPI). We first established that stretch injury causes a rapid increase in the intracellular Ca2+ level, which activates interleukin-converting enzyme caspase-1. The increase in the intracellular Ca2+ level and subsequent caspase-1 activation culminates into neuroinflammation via the maturation of IL-1β. Further, we analyzed caspase-1-mediated apoptosis by TUNEL staining and PARP western blotting. The voltage-gated sodium channel blocker, tetrodotoxin, mitigated the stretch injury-induced neuroinflammation and subsequent apoptosis by blocking Ca2+ influx during the injury. The effect of tetrodotoxin was similar to the caspase-1 inhibitor, zYVAD-fmk, in neuronal culture. To validate the in vitro results, we demonstrated an increase in caspase-1 activity, neuroinflammation and neurodegeneration in fluid percussion-injured animals. Our data suggest that neuronal injury/traumatic brain injury (TBI) can induce a high influx of Ca2+ to the cells that cause neuroinflammation and cell death by activating caspase-1, IL-1β, and intrinsic apoptotic pathways. We conclude that excess IL-1β production and cell death may contribute to neuronal dysfunction and cognitive impairment associated with TBI.
Keywords: Traumatic brain injury, Neuronal stretch injury, Fluid percussion injury, Ca2+ influx, Caspase-1, IL-1β, Neuroinflammation, Apoptosis
Introduction
Traumatic brain injury (TBI) is a complex injury with a broad spectrum of symptoms and disabilities that affects approximately 1.74 million people’s lives every year in USA [1]. TBI has been classified as primary and secondary. Primary injury is the result of mechanical forces applied to the skull and brain at the time of impact such as skull fracture, brain contusion, rupturing of blood vessels, and intracranial hemorrhage [2, 3]. Secondary injuries are characterized by a complex cascade of molecular, neurochemical, and cellular events initiated by the trauma that can lead to elevated intracranial pressure, blood-brain barrier (BBB) disruption, neuroinflammation, brain edema, cerebral hypoxia, ischemia, and delayed neurodegeneration [4-6]. Recent studies have shown evidence that neuroinflammation associated with TBI can also contribute to posttraumatic neurodegeneration [4-8].
Caspases are a family of intracellular cysteine proteases that cleave substrates after aspartic acid residues and are activated during inflammation. Caspases have been studied extensively and are essential in the initiation and implementation of cellular death [9]. Since inflammation is known to be associated with TBI, caspase-3 activity has been explored. Animal models of TBI revealed injury-induced increases in caspase-3 [10-12]. Recently, the induction of caspase-3 and apoptosis has been identified via the activation of oxidative stress and matrix metalloproteinases (MMPs) after blast TBI in rats [4]. In addition, high caspase-3 levels have been shown in the CSF and brain tissue of TBI patients [13]. Interestingly, it has been reported that serum caspase-3 levels are linked to increased mortality. In the study involving 112 TBI patients, higher caspase-3 levels were found in the serum of non-surviving than in surviving TBI patients [14].
Since caspase-1 is also linked to inflammation and apoptosis [15, 16], we hypothesize that this enzyme may also be involved in the activation of TBI-induced neuroinflammation and neurodegeneration. However, the mechanisms of neuroinflammation associated with caspase-1 and the resulting neurodegeneration in TBI is unknown. Caspase-1, formerly called IL-1β-converting enzyme (ICE), was the first caspase identified and is present in the cytosol of phagocytic cells as an inactive zymogen [17, 18]. Active caspase-1 is essential for the cleavage of prointerleukin 1 (pro-IL-1β) into their mature, biologically active forms [15, 16]. Mature IL-1β has been linked to many immune reactions including the recruitment of inflammatory cells to the site of infection [19]. In this study, we utilized a neuronal stretch injury model [20] to visualize in real time the activation of caspase-1 enzyme and the subsequent induction of apoptosis. The results obtained in the neuronal stretch injury were then validated in the fluid percussion injury (FPI) model in vivo [21-24].
This study investigates whether the influx of Ca2+ associated with stretch injury activates caspase-1 in rat brain neurons and leads to the maturation of the proinflammatory cytokine, pro-IL-1β to active IL-1β. The voltage-gated sodium channel blocker TTX and caspase-1 inhibitor (zYVAD-fmk) were used to mitigate injury-induced neuroinflammation and neurodegeneration by blocking the Ca2+ influx during the injury. We conclude that excess IL-1β production and cell death in TBI may be a risk factor for neurological disorders and cognitive impairment.
Materials and Methods
Reagents
Primary antibodies mouse anti-NeuN and rabbit anti-IL-1β were purchased from Abcam (Cambridge, MA), mouse anti-β-actin and rabbit anti-cleaved caspase-1 from Thermo scientific (Rockford, IL), and rabbit anti-PARP from BD Biosciences (San Jose, CA). All secondary Alexa Fluor-conjugated antibodies, Fluo 4AM, TUNEL kit, and DAPI were purchased from Invitrogen (Carlsbad, CA, USA). ELISA kit for IL-1β was purchased from Thermo Scientific. 3,3′-diaminobenzidine (DAB) was purchased from Sigma-Aldrich (St. Louis, MO); z-YVAD-fmk (caspase-1 inhibitor) was from Santa Cruz Biotechnology (Dallas, TX); and tetrodotoxin (TTX) was from Cayman chemicals (Ann Arbor, MI).
Neuronal Culture
Rat brain cortices were isolated from E17 Sprague-Dawley rat embryos and stored in ice-cold HBSS containing Ca2+ and Mg2+. After rinsing with Ca2+ and Mg2+ free HBSS, the cortices were digested in 0.25 % trypsin containing EDTA (0.2 g/L) and DNase I (1.5 mg/mL) at 37 °C for 25 min. The digested tissues were neutralized with 10 % fetal bovine serum and further dissociated by trituration. The media containing the cells was filtered with a Nylon mesh to remove large pieces of tissue. Filtering was performed using a 100-μm pore sized filter followed by a 50-μm pore sized filter. Neurons were cultured (300,000 cells/well) on a poly-l-lysine (0.14 mg/mL)-coated elastic silicone membrane (Specialty Manufacturing, Saginaw, MI) in NeuroBasal media containing 2 % B-27, 1 % penicillin-streptomycin, and 0.4 mM L-glutamine at 37 °C in a CO2 (5 %) incubator [20]. Cultures were fed every 2 days. The purity of the neurons was assessed by immunostaining with anti-NeuN antibody, which normally showed 100 % enrichment of neurons.
Neuronal Stretch Injury
Neurons were allowed to grow for 8 days in vitro prior to stretch injury. On day 8, neuronal cultures were subjected to biaxial strain of 40 % at 20 s−1 strain rate using a custom stretch injury system described elsewhere [20]. For inflicting the injury, a pressure pulse was delivered to the pressure chamber and recorded by the pressure transducer for each well. The pressure pulse was given evenly for all wells to get uniform strain in all experimental conditions. The pressure pulse deforms the membrane. Since the neurons are tightly adhered to the silicone substrate, they deform along with the membrane [20, 25, 26]. For experiments with pharmacological treatments, neurons were treated with tetrodotoxin (TTX, 1 μM) or zYVAD-fmk (5 μg/mL, caspase-1 inhibitor) 30 min prior to the stretch injury. Then, the stretch injury was conducted after replacing with fresh media. At 6 and 24 h after stretch injury cells were fixed for immunostaining or proteins were extracted for western blotting and ELISA at different time points. In addition, 200-μL culture media was collected for western blotting and ELISA analysis.
Calibration of Strain
Previously, our stretch injury system has not been calibrated for use in the biaxial stretching mode [20]. Strain calibration was carried out based on the distortion of silicone membrane used for cell culture using a high-speed camera [27]. The thin elastic silicone membrane was mounted on the custom-made culture dish and exposed to pressure pulse with predefined nominal settings of pressure and time, and the elastic silicone membrane deflection was captured using a FASTCAM Mini high-speed camera (Photron, San Diego) with a resolution of 1280 × 1224 pixels at 4000 frames per second. Calibration runs were repeated at least three times. We calculated the biaxial strain based on the central region of the silicone membrane, where the strain field was relatively uniform [28], using the following simplified equation [29]:
where “α” is the radius of the silicone membrane and “w” is the displacement of the central region of the circular membrane.
Intracellular Calcium Labeling
The cortical neurons were incubated with 5 μM of Fluo-4AM (1 mM stock solution was prepared in DMSO) in culture media for 20 min at room temperature. After washing the cells in HBSS to remove excess fluorescent Fluo 4AM, the cells were subjected to stretch injury. Fluorescent microscopy was performed on a Nikon Eclipse TE-2000S inverted microscope and images were taken using a Photometrics CoolSNAP EZ CCD camera. Fluo 4AM was excited at 488 nm via a xenon light source and collected at 515 nm. The images were acquired using NIS-Elements D software. All fluorescence measurements and analysis were done in the soma using ImageJ (NIH).
We have conducted four independent stretch injury experiments. Four injury wells/condition were used from each experiment, and at least 20 neurons/well were randomly selected and counted for fluorescence analysis. Neurons that did not respond to the fluorescent staining were excluded from the selection. To reduce the effect of background noise, three areas of background absent from neurites and cell bodies were sampled and averaged. Background measurements were subtracted from the raw fluorescent data. The maximal intensity for the tested groups was compared and statistical significance was determined using a two-way analysis of variance (ANOVA).
Fluid Percussion Injury
Animals were maintained in sterile cages under pathogen-free conditions in accordance with institutional ethical guidelines for care of laboratory animals, National Institutes of Health guidelines, and the Rutgers Institutional Animal Care and Use Committee. Twenty-four hours prior to injury, 4-week-old rats were anesthetized with a ketamine/xylazine mixture (80 mg/kg)/xylene (10 mg/kg), i.p. and surgically implanted with a Luer-Lok syringe hub to the skull in a stereotaxic device. This hub surrounds a craniotomy of the same size, positioned 3.0 mm posterior from and 3.5 mm lateral from the bregma. An additional cap that surrounds the syringe hub was applied. Two screws were implanted into the skull to get additional support. Dental cement was applied around the syringe hub and between the cap to ensure fluid transmission and support.
Prior to injury, rats were anesthetized with 5 % isoflurane for 30 s to 1 min until the foot pinch reflex stopped. The animal was connected to a custom digitally controlled fluid percussion injury system (dc-FPI) [21, 24], and injury was applied to the animal at 15–18 psi with a pressure rise time of 8 ms. Rats exhibited apnea, loss of consciousness (LOC), and hyperextension of the tail and hind limbs after the injury. Similarly, control rats received the same anesthesia, surgery as injured, and connected to the dc-FPI without the injury. We used nine injured and eight control rats for this study. Animals consisting of control (uninjured) and injured with dc-FPI were euthanized after 24 hs of injury with ketamine/xylazine mixture. Brain tissues were dissected out, embedded in optimal cutting temperature (OCT) and kept frozen until we analyzed the mechanisms of injuries by immunohistochemical staining and western blotting. The sampled area for immunostaining and western blotting was just below the dc-FPI injured area in the cortex region (Fig. 3a).
Fig. 3.

Activation of caspase-1 in fluid percussion-injured animals: a schematic representation of FPI area and the region evaluated in the cortex of rat brain. b Immunohistochemistry of cleaved caspase-1 (red) and NeuN (green) and counterstained with DAPI (blue) in rat cortex tissue sections in uninjured control and after 24 h after FPI. Yellow arrows show cleaved caspase-1 expression. Scale bar 50 μm. C Quantification of cleaved caspase-1 staining analyzed using ImageJ software. Values are mean ± SEM (n = 6); ***p < 0.001 versus control. d Western blot analysis of cleaved caspase-1 and β-actin in rat cortex tissue lysates from uninjured control and after 24 h after FPI. Densitometry measurements are mean ± SEM (n = 6); ***p < 0.001 versus control
Immunofluorescence and Microscopy
For immunofluorescence staining, the cultured neurons (on silicone membrane) and brain tissue sections (10-μm thickness) were washed with 1× PBS and fixed in 4 % paraformaldehyde for 20 min at 25 °C. After fixation, the washed membrane or tissue sections was then blocked with 3 % normal goat serum at 25 °C for 1 h in the presence of 0.1 % Triton X-100. The cell culture membranes or tissue sections were incubated overnight at 4 °C with primary antibodies to rabbit anti-cleaved caspase-1 (Thermo scientific) and mouse anti-NeuN (Abcam). All antibodies were used at the concentration of 3 μg/mL. After washing with PBS, cells or tissue sections were incubated with Alexa Fluor 488 or 594 conjugated with anti-mouse or anti-rabbit immunoglobulin G (IgG) for 1 h and mounted with immunomount containing DAPI (Invitrogen) on a slide. The silicone membrane was mounted on a cover slip and carefully cut at the edges using a scalpel blade and then again mounted on a slide using DAPI. The photographs were captured using fluorescent microscope Eclipse TE2000-U (Nikon, Melville, NY) using the NIS Elements software (Nikon, Melville, NY). Controls for the experiments consisted of the omission of primary antibodies. No staining was observed in these cases. The intensity of immunostaining was analyzed by ImageJ (NIH) software.
Western Blotting
For western blotting analysis, the proteins were extracted from ten wells of the same experiment condition and pooled to get enough proteins to run at least three western blots for each analysis. The cells were lysed with 200 μL of CelLytic M buffer (Thermo Scientific) containing a mixture of protease inhibitor (Sigma) and collected in a 1.5-mL microfuge tube and centrifuged at 10,000 rpm at 4 °C. To prepare brain cortical tissue lysates, 50 mg of cortical tissue were used. To get enough proteins for the IL-1β immunoblot, the cell culture supernatant were pooled from all ten wells of the same experiment condition and concentrated by using 30-kDa centrifugal filter tubes (EMD Millipore, Billerica, MI). Both in vitro and in vivo homogenate and cell culture supernatant protein concentrations were estimated by the bicinchoninic acid (BCA) method (Thermo Scientific, Rockford, IL). We used 20-μg protein/lane in 10 % SDS-PAGE. Molecular size-separated proteins were then transferred onto nitrocellulose membranes, blocked with superblock (Thermo Scientific), and incubated overnight with respective primary antibodies at 4 °C. In order to validate the immunocytochemical results, we used the same antibodies mentioned in the immunofluorescence and microscopy section. Then, the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies (1:1000; Fisher scientific) for 1 h at RT. The membrane was rinsed three times with 1× TBS Tween buffer for 5 min at RT. Immunoreactive bands were detected by 3,3′-diaminobenzidine (DAB) (Sigma). Data were quantified as arbitrary densitometry intensity units using the ImageJ software package.
Enzyme-Linked Immunosorbent Assay
Using commercial enzyme-linked immunosorbent assay kit (ELISA), the level of active IL-β (Thermo Scientific) was analyzed in the cell culture media, cell lysates, tissue lysates, and blood serum as per the manufacturer’s instructions.
TUNEL and PARP Analyses
Using the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL, Invitrogen) assay kit, cell apoptosis was determined in tissue sections as per manufacturer’s instructions. We used four injury wells/condition from each experiment and at least four fields/well were randomly selected and counted for TUNEL analysis. To confirm the TUNEL assay, western blotting determined the breakdown of 113-kDa poly-ADP-ribose-polymerase (PARP) to 89 and 24 kDa PARP fragments using rabbit anti-PARP primary antibody (BD Biosciences). Detection of the 89-kDa PARP fragment is an early marker for apoptosis, which is mediated by caspase-1/3 signaling pathway.
In Vitro Cell Viability Assay
After 6 and 24 h of stretch injury, the supernatant of neuronal cultures were collected and centrifuged. On the other hand, the neuronal cultures were trypsinized with 0.25 % trypsin for 5 min and neutralized with 10 % FBS, and the neurons were collected into the tubes, which contained already collected neurons from the supernatant. The neurons were washed with neurobasal media and dissociated by trituration. Ten microliters of neurons were stained with trypan blue, counted using a hemotocytometer. Both positively stained and unstained cells were counted by an observer unaware of the treatments (n = 4) [30]. All experiments were repeated at least twice. The mean ratio of dead cells to the total number of cells per field was quantified.
Data Analysis
All the results are expressed as the mean ± SEM. Statistical analyses of the data were performed using GraphPad Prism V5 (Sorrento Valley, CA). One-way ANOVA with Dunnett’s post hoc tests determined the differences between controls and experimental conditions, and a probability value of p < 0.05 was considered significant.
Results
Stretch Injury Causes Ca2+ Influx, Modulated by Tetrodotoxin
Previous studies have shown that uniaxial axonal stretch injury [31-33] and biaxial neuronal stretch injury [28] causes a high influx of calcium ions (Ca2+) that can be modulated by the voltage-gated sodium channel blocker tetrodotoxin (TTX). In this study, we confirm in our injury system that biaxial neuronal stretch increased intracellular Ca2+ immediately after injury, and the elevated levels of intracellular Ca2+ persisted over 6 and 24 h after injury when compared with the uninjured controls (Fig 1a, b). The level of intracellular Ca2+ immediately after the injury was 72.7 % higher than the uninjured cells (p < 0.001), 40.9 % higher than uninjured cells 6 h postinjury (p < 0.001), and 15.9 % higher than uninjured cells 24 h postinjury (p < 0.01). Treatment with TTX (1 μM) for 30 min reduced the influx of Ca2+ immediately after the injury and 6 and 24 h of injury when compared with the untreated injured cells (Fig 1a, b). These data support the mechanism wherein neuronal stretch injury causes high Ca2+ influx to the cells, which is blocked by TTX.
Fig. 1.

Biaxial stretch injury causes Ca2+ influx, modulated by tetrodotoxin: a Fluo 4AM staining for intracellular Ca2+ in control, injury, and injury + TTX 24 h after stretch injury (40 % strain and 20 s−1 strain rate) of rat cortical neuronal cultures. Scale bar = 50 μm. b Quantitative analysis of calcium fluorescence at 1 min, 6 h, and 24 h after stretch injury. Values are mean ± SEM; n = 4 (20 individual neurons from each condition from each experiment. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control (first bar); #p < 0.05, ##p < 0.01, and ###p < 0.001 versus injury (in their respective time period)
Sustained Elevated Intracellular Ca2+ Causes Activation of Caspase-1 in Cultured Neurons
To test the hypothesis that stretch injury-induced increase in Ca2+ activates caspase-1, the postinjury expression of active caspase-1 was examined in cultured rat cortical neurons exposed to stretch injury with or without the treatment of TTX or zYVAD-fmk. We used anti-cleaved caspase-1 p20 fragment antibody to detect active caspase-1 by immunofluorescence and western blotting. Immunofluorescence identified that the expression level of active caspase-1 was high in injured cultures compared with uninjured cells. The expression of active caspase-1 was significantly reduced after stretch injury in the TTX-treated cultures when compared with the untreated injured cells at 6 (Fig. 2a, b) and 24 h after injury (Fig. 2c), suggesting that caspase-1 is activated by elevated calcium. The caspase-1 inhibitor, zYVAD-fmk, was used to block the maturation of procaspase-1 to active caspase-1. zYVAD-fmk pretreatment also eliminated the expression of caspase-1 in stretch-injured neurons at both 6 (data not shown) and 24 h postinjury (Fig. 2c). Figure 2d shows the intensity of cleaved caspase-1 expression by immunofluorescent staining.
Fig. 2.

Sustained elevation of intracellular Ca2+ activates caspase-1: a–c immunocytochemistry of cleaved caspase-1 (red) and NeuN (green) and counterstained with DAPI (blue) in rat cortical neuronal cultures (a) uninjured control (b) 6 h after stretch injury (40 % strain and 20 s−1 strain rate) including injury + TTX treatment condition; cleaved caspase-1 expression are shown in white arrows and c 24 h after stretch injury including injury + TTX and injury + zYVAD-fmk treatment conditions; cleaved caspase-1 expression are shown in white arrows. Scale bar= 10 μm. d Quantification of cleaved caspase-1 staining analyzed using ImageJ software. Values are mean ± SEM (n = 4); ***p < 0.001 versus control (first bar); ##p < 0.001 versus injury (6 h); and $$$p < 0.001 versus injury (24 h). e Western blot analysis of cleaved caspase-1 and β-actin 6 and 24 h after stretch injury including TTX or zYVAD-fmk pretreatment conditions. Densitometry measurements are mean± SEM (n = 4); ***p < 0.001 versus control; ###p < 0.001 versus injury (in 6 h); $$$p < 0.001 versus injury (in 24 h)
The changes in caspase-1 protein expression were validated by western blot analysis (Fig. 2e). A significant upregulation of active caspase-1 protein (~20 kDa) level was found at 6 and 24 h postinjury compared to uninjured cells (p < 0.001). In particular, the expression of active caspase-1 was higher at 24 than at 6 h after the injury. In TTX- and zYVAD-fmk-treated injured cells, the protein level of active caspase-1 was significantly reduced (p < 0.001) when compared with untreated injured cells at 6 and 24 h after injury. Taken together, these results indicate that high influx of Ca2+ to the cells due to stretch injury activates caspase-1, and the TTX ameliorates the activation of caspase-1 in stretch-injured cortical neurons.
Caspase 1 Activation in Fluid Percussion Injured Rats
To validate the in vitro results, animals were subjected to fluid percussion injury and analyzed for caspase-1 activation. In the cortex region below the site of injury (Fig. 3a), cleaved caspase-1 was identified (Fig. 3b). Colocalizing cleaved caspase-1 with NeuN staining indicated that the cleaved caspase-1 expression was primarily in neurons. The staining of cleaved caspase-1 was localized near the nuclear membrane, whereas NeuN was distributed throughout the cytoplasm. Figure 3c shows the intensity of the cleaved caspase-1 expression in rat brain cortical tissue section. FPI-induced increase in cleaved caspase-1 protein was validated by western blot in protein samples from rat cerebral cortex tissue (Fig. 3d).
Injury Induced Activation of Caspase-1 Matures IL-1β and Causes Neuroinflammation
Since caspase-1 matures interleukin 1 beta (IL-1β) [15, 16], we analyzed the level of IL-1β in injured neuronal cultures and in blood serum of FPI animals. Using an ELISA assay, the level of IL-1β was almost 4.5 times higher in stretch-injured neuronal cultures than in uninjured cells 6 h after injury and 5.5 times higher 24 h after injury. Both blocking of the calcium influx with TTX and inhibition of active caspase-1 with z-YVAD-fmk prevented the maturation and elevation of IL-1β in the cell culture (Fig. 4a).
Fig. 4.

Activation of caspase-1 matures IL-1β and causes neuroinflammation: analysis of rat cortical neuronal cell culture 6 and 24 h after stretch injury with or without TTX or zYVAD-fmk. a ELISA measurements of IL-1β in rat cortical neuronal cell culture supernatants. Values are mean ± SEM (n = 4; we used a total of 12 samples (three each) from four independent experiments), *p < 0.05 and **p < 0.01 versus control (first bar); #p < 0.05 versus injury (6 h); and $$p < 0.01 versus injury (24 h). b Western blot analysis of IL-1β in cell culture supernatants after 6 and 24 h of injury treated with or without TTX or zYVAD-fmk. The pooled and concentrated crude proteins were loaded equally for all experiment conditions. The graph shows the densitometry of the level of IL-1β. Values are mean ± SEM (n = 4; we used a total of ten samples from each independent experiment); ***p < 0.001 versus control (first bar); ###p < 0.001 versus injury (6 h); and $$$p < 0.001 versus injury (24 h). c Western blot analysis of IL-1β and β-actin in the cultured rat cortical neurons lysates after 6 and 24 h of injury treated with or without TTX or zYVAD-fmk. The graph shows the densitometry measurements of ratio of IL-1β and β-actin western blotting. Values are mean ± SEM (n = 4; we used a total of ten samples from each independent experiment); **p < 0.01 and ***p < 0.001 versus control (first bar); ##p < 0.01 versus injury (6 h); and $$$p < 0.001 versus injury (24 h).
IL-1β levels were also analyzed by western blotting cell-culture supernatants as well as cell lysates (Fig. 4b, c). Western blots showed significant elevation of IL-1β level in cell culture supernatant after 6 h (4.74 times, p < 0.001) and 24 h (5.38 times, p < 0.001) of injury when compared to uninjured controls (Fig. 4b). Similar trend of results were obtained in cell culture homogenates (Fig. 4c). In addition, treatment with TTX and z-YVAD-fmk reduced the level of IL-1β to uninjured control levels in both cell culture supernatant and cell culture homogenates.
The in vitro findings were again validated in fluid percussion injured rats. Blood was collected from control and injured animals 24 h after the injury. The levels of IL-1β in the blood serum of FPI animals was about fourfold higher than that of the uninjured control animals (p < 0.001) (Fig. 5a). Cortical tissue was collected from animals 24 h postinjury for western blot analysis. A twofold increase in IL-1β level was found compared to uninjured controls (Fig. 5b). Taken together, these data indicate that the activation of caspase-1 in stretch-injured cells and FPI in animals elevates the level of the proinflammatory cytokine, IL-1β. The attenuation of induction of caspase-1 by the treatment with TTX and caspase-1 inhibitor, z-YVAD-fmk, reduces IL-1β-dependent neuroinflammation.
Fig. 5.
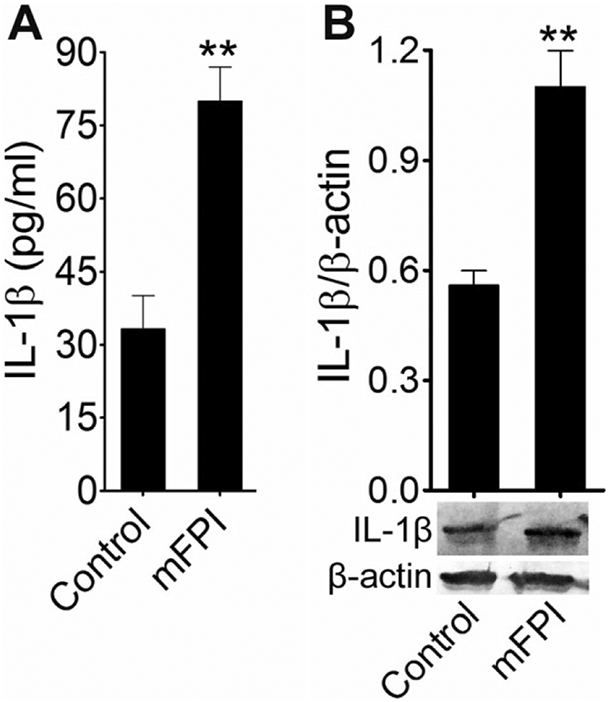
Activation of IL-1β fluid percussion-injured animals: a ELISA assay measurements of IL-1β in rat blood serum 24 h after FPI. Values are mean ± SEM (n = 6); **p < 0.01. b Western blot analysis of IL-1β and β-actin in rat cortex tissue lysates 24 h after FPI. The graph shows the densitometry of ratio of IL-1β and β-actin bands. Values are mean± SEM (n = 6); **p < 0.01
Injury Induced Activation of Caspase-1 and Neuroinflammation Leads to Cell Apoptosis
Biaxial stretch injury of neuronal cultures leads to apoptosis in some, but not all, neurons in culture. To establish the role of caspase-1 and neuroinflammation in neuronal stretch injury-associated cell death, we examined the caspase-1-mediated intrinsic apoptotic pathways in stretch-injured neuronal cultures pretreated with or without TTX or zYVAD-fmk and compared to uninjured controls. The extent of cell apoptosis was assessed by the active apoptosis markers TUNEL and poly-ADP-ribose-polymerase (PARP) and at the completion of cell death by trypan blue staining.
TUNEL labeling revealed 21.7 % positive apoptotic neurons at 6 h (p < 0.001) and 25.9 % at 24 h (p < 0.001) after injury. This value for uninjured control cultures was 5.1 % (Fig. 6a, b). In most of the injured cells, the breakage of nuclei was found that colocalized with TUNEL staining (Fig. 6a (b)). Pretreatment with TTX and zYVAD-fmk significantly decreased the number of TUNEL-positive cells at 6 h postinjury to 9.2 % (p < 0.01) and 8.1 % (p < 0.001), respectively, and to 9.8 % (p < 0.001) and 8.1 % (p < 0.001) at 24 h postinjury respectively (Fig. 6a, b).
Fig. 6.

Stretch injury activation of caspase-1 induces intrinsic apoptosis. a TUNEL staining (green) in cultured rat cortical neurons in control (a), stretch-injured neurons (b), injury + TTX (c), injury + zYVAD-fmk (d). Yellow arrows indicate TUNEL-positive cells. Nucleus was counterstained with DAPI (blue). b1, b2 shows enlarged view of a single cell with broken nucleus (DAPI) and merged with TUNEL staining. Scale bar is 100 μm in all panels except b1 and b2. b Percentage of TUNEL-positive cells in rat brain cortical neuronal culture. Values are mean ± SEM (n = 4, we used four injury wells/condition from each experiment and at least four fields/well were randomly selected and counted for TUNEL analysis. ***p < 0.001 versus control (first bar); ##p < 0.01 and ###p < 0.001 versus injury (6 h); and @@@p < 0.001 versus injury (24 h). c Changes in PARP protein levels in stretch-injured cell culture lysates. Densitometry measurements of PARP at 6 and 24 h after injury, treated with or without TTX or zYVAD-fmk. Bar graphs show the changes in relative intensity of 113- and 89-kDa fragments of PARP. Values are mean ± SEM (n = 4); **p < 0.01 and ***p < 0.001 versus 89 kDa of control (first bar); #p < 0.05 and ##p < 0.01 versus 89 kDa of injury (6 h); and @@@p < 0.001 versus 89 kDa of injury (24 h). d Percentage of dead cells measured by trypan blue at 6 and 24 h after injury treated with or without TTX or zYVAD-fmk. Values are mean ± SEM (n = 4), ***p < 0.001 versus control (first bar); ###p < 0.001 versus injury (6 h); and @@@p < 0.001 versus injury (24 h)
Apoptosis via caspase-1 was further substantiated by the cleavage of PARP. In stretch-injured neurons, western blot analysis revealed the level of cleaved fragment (89 kDa PARP) was significantly high compared to uncleaved product (113-kDa PARP) at 6 and 24 h postinjury (Fig. 6c). In uninjured control neurons, the level of the 113-kDa PARP was very high due to the normally low activity of caspase-1 enzyme. The level of the 113-kDa PARP fragment was reduced at 6 and 24 h postinjury due to the cleavage of this PARP enzyme to the 89-kDa fragment. As observed in the TUNEL study, TTX and zYVAD-fmk inhibited the cleavage of the 113-kDa PARP and significantly downregulated the level of the 89-kDa PARP at both 6 and 24 h postinjury (Fig. 6c).
Cell death associated with stretch injury was analyzed by trypan blue staining of stretch-injured neurons with or without TTX and z-YVAD-fmk pretreatment and uninjured controls (Fig. 6d). In stretch-injured neuronal cultures, the average percentages of dead cells were 16.7 and 22.7 % for 6 and 24 h of injury respectively whereas dead cells in uninjured culture were 5.3 %. As we expected, the percentages of dead cells in the TTX-pretreated injured cultures were 6.7 and 7.1 at 6 and 24 h postinjury, respectively. In zYVAD-fmk-pretreated injured cultures, the average dead cell percentages were 5.3 and 5.8 % at 6 and 24 h postinjury, respectively (Fig. 6d).
To validate the caspase-1-mediated cell apoptosis in vitro, we evaluated TUNEL and PARP western blotting from FPI animals compared to uninjured sham control animals. Analysis of TUNEL-positive cells showed numerous apoptotic neurons in cortical tissue at 24 h postinjury (Fig. 7a, b). Similarly, the 89 kDa of PARP western blot band was highly expressed in FPI animals compared to controls (p < 0.001) (Fig. 7c). Taken together, these results clearly indicate fluid percussion injury activates caspase-1, which further causes neuroinflammation and neurodegeneration in the form of cell apoptosis.
Fig. 7.

Activation of caspase-1 induces intrinsic apoptosis in fluid percussion-injured rats: a TUNEL staining (green) of rat cortex tissue sections without injury (control) and 24 h after FPI. Arrows indicate TUNEL-positive cells. The nucleus was counterstained with DAPI (blue). Scale bar 100 μm. b Percentage of TUNEL-positive cells in the adjacent area of FPI and compared with control. Values are mean ± SEM (n = 6); ***p < 0.001. c Western blotting of PARP protein levels in rat brain cortex lysates in control and 24 h after FPI. Bar graphs show the changes in relative intensity of 113- and 89-kDa fragments of PARP. Values are mean ± SEM (n = 6); ***p < 0.001
Discussion
The injury-induced elevation of intracellular Ca2+ and the activation of the proteolytic enzymes have been well established [32, 34, 35]. However, the complete downstream effects of injury-induced Ca2+ overload are not yet fully understood. As a secondary messenger, Ca2+ regulates a myriad of biochemical pathways. Here, we demonstrated that high Ca2+ influx due to neuronal stretch injury causes activation of caspase-1, which leads to neuroinflammation and cell death in cultured rat neocortical neurons. Further, using the fluid percussion injury (FPI) model in rats, we confirmed these secondary processes of neuroinflammation and cell death in vivo. To the best of our knowledge, the present findings are the first to describe the mechanisms of caspase-1-dependent neuroinflammation and neurodegeneration in the context of Ca2+ dysregulation from TBI. Figure 8 depicts a schematic presentation of our findings.
Fig. 8.

Schematic presentation of the underlying mechanisms of caspase-1-dependent neuroinflammation and neurodegeneration due to influx of Ca2+ during the brain injury
Two potential pathways involved in elevating intracellular Ca2+ following stretch injury damage the sodium channel (NaCh) causing Ca2+ dysregulation and entry via N-methyl-d-aspartate ionotropic receptor channels (NMDAr). Both pathways contribute to Ca2+ elevation after stretch injury through direct or indirect mechanisms [28, 31]. In the present study, we observed that neuronal stretch injury causes a rapid increase in Ca2+ influx. Significant attenuation of Ca2+ influx was achieved with 1 μM TTX treatment, confirming the involvement of sodium channels in the high influx of Ca2+ during injury [32]. Accordingly, TTX was used here to evaluate the role of Ca2+ overload in the activation of the caspase-1 pathway.
Several authors suggested that elevated intracellular or axonal Ca2+ levels play a pivotal role in the secondary damages to cells or axons after mechanical deformation [35-38]. Specifically, calcium-activated proteases including calpain and caspase-3 have been implicated [10, 13, 14, 34]. Here, we found that the high Ca2+ influx increased the expression of cleaved caspase-1 during neuronal stretch injury up to 24 h after the injury. The NaCh blocker TTX and caspase-1 inhibitor z-YVAD-fmk both inhibited the injury-induced expression of caspase-1, suggesting a key role of Ca2+ elevation in the activation of cleaved caspase-1. Since high concentration of zYVAD-fmk inhibits the functions of other caspases like caspase-4, we used very optimum concentration of zYVAD-fmk (5 μg/mL), and this was derived from our previous study [39].
In this study, stretch injury-induced Ca2+ influx was linked to the activation of caspase-1. However, caspase-1 activation also depends on the involvement of other factors such as oxidative stress or activation of inflammasomes; also linked to the high Ca2+ influx during stretch injury [40-43]. The involvement of oxidative stress in the activation of caspase-1 comes from mitochondrial perturbation with calcium overloading and opening of the mitochondrial permeability transition (MPT) pore during neuronal stretch injury (for review, see [43-46]). This is based on the release of mitochondrial cytochrome-c leading to aberrant changes in electron transport chain and hence the generation of free radicals [47, 48]. The study on the role of oxidative stress in the activation of caspase-1 during stretch injury is currently being pursued.
The present study shows that injury-induced elevation in Ca2+ is also accompanied by high levels of IL-1β due to the activation of caspase-1 in neuronal stretch injury. Caspase-1, otherwise called interleukin-converting enzyme (ICE), is involved in the maturation of proinflammatory cytokine, pro-IL-1β to IL-1β [15, 16]. Caspase-1-dependent maturation of IL-1β was supported from the treatment with caspase-1 inhibitor, zYVAD-fmk, where the level of IL-1β was reduced significantly.
Several studies have revealed the impact of proinflammatory cytokines and their significant roles in the pathophysiology of TBI. For instance, shortly after brain injury, there is mass production of IL-1β, which exacerbates damage to the brain from oxidative stress that can delay recovery [49, 50]. The mRNA and protein of these cytokines have been shown to increase markedly in the acute posttraumatic period following experimental brain trauma in rats [51-53]. These posttraumatic inflammatory cascades can also contribute to the blood-brain barrier (BBB) dysfunction and the migration of inflammatory cells from the blood to the brain [54].
Since apoptosis is a type of programmed cell death that is primarily dependent on the activity of caspases [55], they may also be involved in TBI. We and others reported the role of MMPs, which are activated in response to injury stimuli and induce apoptotic via caspases [56] [4, 57-59]. In 2012, we showed the role of caspase-1 and proinflammatory cytokine, IL-1β, in caspase-1-dependent cell death in human endothelial cells [39]. Accordingly, we concluded this study by linking the injury-induced high Ca2+ influx to caspase-1 activation and maturation of IL-1β leading to cell death via apoptosis, specifically called pyroptosis.
Here, activation of caspase-1 and release of IL-1β and eventual cell apoptosis were validated by TUNEL assay and induction of PARP in both the in vitro stretch injury and FPI models. Similar to in vitro data, the level of activated caspase-1 and IL-1β, the number of TUNEL-positive cells and the level of 89-kDa PARP fragment were significantly higher in FPI samples, providing clear evidence of the role of injury-induced influx of Ca2+. Even though other caspases especially caspase-3 has an important role in apoptosis, we have shown the evidence for the role of caspase-1 in apoptosis by treating the cells with caspase-1 inhibitor zYVAD-fmk.
In summary, the current data provides a comprehensive description on the Ca2+-mediated sequelae of pathobiological events due to neuronal stretch injury and validated the results with FPI animal model. This study focused on the signaling pathway which leads to caspase-1-dependent neuroinflammation and neurodegeneration due to influx of Ca2+ during the brain injury. These studies significantly extend our understanding of the pathobiology of TBI and provide unique insight into the rationale for the use of various therapeutic interventions targeting TBI.
Acknowledgments
This work was funded by New Jersey Commission on Brain Injury Research no. CBIR11PJT003 to Bryan J. Pfister and Vijayalakshmi Santhakumar and NIH/NINDS R01NS069861 to Vijayalakshmi Santhakumar. The authors gratefully acknowledge Mr. Brian Swenson, Dept of Biomedical Engineering, NJIT for the technical support in calibrating strain.
References
- 1.Faul M, Xu L, Wald MM, Coronado VG (2010) Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, Atlanta [Google Scholar]
- 2.Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7:728–741 [DOI] [PubMed] [Google Scholar]
- 3.Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF (2008) Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med 14:731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N et al. (2013) Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med 60:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lotocki G, de Rivero Vaccari JP, Perez ER, Sanchez-Molano J, Furones-Alonso O, Bramlett HM, Dietrich WD (2009) Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J Neurotrauma 26:1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pun PB, Lu J, Moochhala S (2009) Involvement of ROS in BBB dysfunction. Free Radic Res 43:348–364 [DOI] [PubMed] [Google Scholar]
- 7.Abdul-Muneer PM, Chandra N, Haorah J (2015) Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51:966–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Korgaonkar AA, Swietek B, Wang J, Elgammal FS, Elkabes S, Santhakumar V (2015) Toll-like receptor 4 enhancement of non-NMDA synaptic currents increases dentate excitability after brain injury. Neurobiol Dis 74:240–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 6: 1028–1042 [DOI] [PubMed] [Google Scholar]
- 10.Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE et al. (2000) Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem 74:740–753 [DOI] [PubMed] [Google Scholar]
- 11.Clausen F, Lundqvist H, Ekmark S, Lewen A, Ebendal T, Hillered L (2004) Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J Neurotrauma 21:1168–1182 [DOI] [PubMed] [Google Scholar]
- 12.Jia F, Mao Q, Liang YM, Jiang JY (2009) Effect of post-traumatic mild hypothermia on hippocampal cell death after traumatic brain injury in rats. J Neurotrauma 26:243–252 [DOI] [PubMed] [Google Scholar]
- 13.Harter L, Keel M, Hentze H, Leist M, Ertel W (2001) Caspase-3 activity is present in cerebrospinal fluid from patients with traumatic brain injury. J Neuroimmunol 121:76–78 [DOI] [PubMed] [Google Scholar]
- 14.Lorente L, Martin MM, Argueso M, Ramos L, Sole-Violan J, Riano-Ruiz M, Jimenez A, Borreguero-Leon JM (2015) Serum caspase-3 levels and mortality are associated in patients with severe traumatic brain injury. BMC Neurol 15:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller M, Ruegg A, Werner S, Beer HD (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831 [DOI] [PubMed] [Google Scholar]
- 16.Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K et al. (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7:2350–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March CJ, Kronheim SR et al. (1992) Molecular cloning of the interleukin-1 beta converting enzyme. Science 256:97–100 [DOI] [PubMed] [Google Scholar]
- 18.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM et al. (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356:768–774 [DOI] [PubMed] [Google Scholar]
- 19.Arend WP, Palmer G, Gabay C (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38 [DOI] [PubMed] [Google Scholar]
- 20.Magou GC, Guo Y, Choudhury M, Chen L, Hususan N, Masotti S, Pfister BJ (2011) Engineering a high throughput axon injury system. J Neurotrauma 28:2203–2218 [DOI] [PubMed] [Google Scholar]
- 21.Wahab RA, Neuberger EJ, Lyeth BG, Santhakumar V, Pfister BJ (2015) Fluid percussion injury device for the precise control of injury parameters. J Neurosci Methods 248:16–26 [DOI] [PubMed] [Google Scholar]
- 22.Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, Longhi L, Laurer H et al. (2005) Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience 136:971–989 [DOI] [PubMed] [Google Scholar]
- 23.Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK (2005) Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma 22:42–75 [DOI] [PubMed] [Google Scholar]
- 24.Neuberger EJ, Abdul Wahab R, Jayakumar A, Pfister BJ, Santhakumar V (2014) Distinct effect of impact rise times on immediate and early neuropathology after brain injury in juvenile rats. J Neurosci Res 92:1350–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrison B 3rd, Saatman KE, Meaney DF, McIntosh TK (1998) In vitro central nervous system models of mechanically induced trauma: a review. J Neurotrauma 15:911–928 [DOI] [PubMed] [Google Scholar]
- 26.Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, Satin LS (2007) Injury-induced alterations in CNS electrophysiology. Prog Brain Res 161:143–169 [DOI] [PubMed] [Google Scholar]
- 27.Skotak M, Wang F, Chandra N (2012) An in vitro injury model for SH-SY5Y neuroblastoma cells: effect of strain and strain rate. J Neurosci Methods 205:159–168 [DOI] [PubMed] [Google Scholar]
- 28.Geddes-Klein DM, Schiffman KB, Meaney DF (2006) Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. J Neurotrauma 23:193–204 [DOI] [PubMed] [Google Scholar]
- 29.Winston FK, Macarak EJ, Gorfien SF, Thibault LE (1989) A system to reproduce and quantify the biomechanical environment of the cell. J Appl Physiol 67:397–405 [DOI] [PubMed] [Google Scholar]
- 30.Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM et al. (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci 6:1064–1071 [DOI] [PubMed] [Google Scholar]
- 31.Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci Off J Soc Neurosci 21:1923–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH (2004) Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci Off J Soc Neurosci 24: 4605–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuen TJ, Browne KD, Iwata A, Smith DH (2009) Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res 87:3620–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Reyn CR, Mott RE, Siman R, Smith DH, Meaney DF (2012) Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. J Neurochem 121:793–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber JT (2012) Altered calcium signaling following traumatic brain injury. Front Pharmacol 3:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Povlishock JT, Erb DE, Astruc J (1992) Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity. J Neurotrauma 9(Suppl 1):S189–200 [PubMed] [Google Scholar]
- 37.Gitler D, Spira ME (1998) Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron 20:1123–1135 [DOI] [PubMed] [Google Scholar]
- 38.Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58:365–375 [DOI] [PubMed] [Google Scholar]
- 39.Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J (2012) The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler Thromb Vasc Biol 32:1167–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL et al. (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492:123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schoneberg T et al. (2012) Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun 3:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byrne AM, Lemasters JJ, Nieminen AL (1999) Contribution of increased mitochondrial free Ca2+ to the mitochondrial permeability transition induced by tert-butylhydroperoxide in rat hepatocytes. Hepatology 29:1523–1531 [DOI] [PubMed] [Google Scholar]
- 43.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE et al. (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366:177–196 [DOI] [PubMed] [Google Scholar]
- 44.Cai J, Yang J, Jones DP (1998) Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta 1366:139–149 [DOI] [PubMed] [Google Scholar]
- 45.Montal M (1998) Mitochondria, glutamate neurotoxicity and the death cascade. Biochim Biophys Acta 1366:113–126 [DOI] [PubMed] [Google Scholar]
- 46.Siesjo BK, Hu B, Kristian T (1999) Is the cell death pathway triggered by the mitochondrion or the endoplasmic reticulum? J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 19:19–26 [DOI] [PubMed] [Google Scholar]
- 47.Reed JC (1998) Bcl-2 family proteins. Oncogene 17:3225–3236 [DOI] [PubMed] [Google Scholar]
- 48.Saikumar P, Dong Z, Weinberg JM, Venkatachalam MA (1998) Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene 17:3341–3349 [DOI] [PubMed] [Google Scholar]
- 49.Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99:4–9 [DOI] [PubMed] [Google Scholar]
- 50.Lucas SM, Rothwell NJ, Gibson RM (2006) The role of inflammation in CNS injury and disease. Br J Pharmacol 147(Suppl 1):S232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hans VH, Kossmann T, Joller H, Otto V, Morganti-Kossmann MC (1999) Interleukin-6 and its soluble receptor in serum and cerebrospinal fluid after cerebral trauma. Neuroreport 10:409–412 [DOI] [PubMed] [Google Scholar]
- 52.Shohami E, Bass R, Wallach D, Yamin A, Gallily R (1996) Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 16:378–384 [DOI] [PubMed] [Google Scholar]
- 53.Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, McIntosh TK (1995) Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res Mol Brain Res 30:125–130 [DOI] [PubMed] [Google Scholar]
- 54.Soares HD, Hicks RR, Smith D, McIntosh TK (1995) Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci Off J Soc Neurosci 15:8223–8233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462 [DOI] [PubMed] [Google Scholar]
- 56.Abdul-Muneer PM, Pfister BJ, Haorah J, Chandra N. 2015. Role of matrix metalloproteinases in the pathogenesis of traumatic brain injury. Mol Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mannello F, Luchetti F, Falcieri E, Papa S (2005) Multiple roles of matrix metalloproteinases during apoptosis. Apoptosis 10:19–24 [DOI] [PubMed] [Google Scholar]
- 58.Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y (2007) Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem 101:566–576 [DOI] [PubMed] [Google Scholar]
- 59.Haorah J, Schall K, Ramirez SH, Persidsky Y (2008) Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: novel mechanism for neurodegeneration associated with alcohol abuse. Glia 56:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]