

Abstract
Objective
We aim to familiarize the application status of metagenomic sequencing in diagnosing pulmonary infections, to compare metagenomic sequencing with traditional diagnostic methods, to conclude the advantages and limitations of metagenomic sequencing, and to provide some advice for clinical practice and some inspiration for associated researches.
Data Sources
The data were obtained from peer‐reviewed literature, white papers, and meeting reports.
Results
This review focused on the applications of untargeted metagenomic sequencing in lungs infected by bacteria, viruses, fungi, chlamydia pneumoniae, Mycoplasma pneumoniae , parasites, and other pathogens. Compared with conventional diagnostic methods, metagenomic sequencing is better in detecting novel, rare, and unexpected pathogens and being applied in co‐infections. Meanwhile, it can also provide more comprehensive information about pathogens. However, metagenomic sequencing still has limitations. Also, the situations that should be applied in and how the results should be interpreted are discussed in this review.
Conclusion
Metagenomic sequencing improves efficiency to identify pathogens compared with traditional diagnostic methods and can be applied in clinical diagnosis. However, the technology of metagenomic sequencing still needs to be improved. Also, clinicians should learn more about when to use metagenomic sequencing and how to interpret its results.
Keywords: application, metagenomic sequencing, pulmonary infections
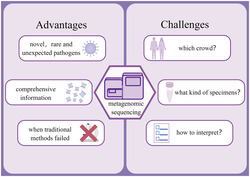
Compared with conventional diagnostic methods, metagenomic sequencing is more sensitive and better in detecting novel, rare, and unexpected pathogens. Meanwhile, it can provide more comprehensive information about pathogens. However, many challenges exist when using metagenomic sequencing. By Figdraw (www.figdraw.com).
1. BACKGROUND
Pulmonary infections remain the leading infectious cause of morbidity and mortality worldwide. 1 They can be caused by bacteria, viruses, fungi, chlamydia pneumoniae, Mycoplasma pneumoniae, parasites, and other pathogens. The conventional diagnostic progress is mainly based on the patients' clinical presentations, imaging manifestations, and laboratory examinations. The traditional laboratory tests to identify of pathogens are limited to serological assays, nucleic acid detection by specific polymerase chain reaction (PCR), and culture from respiratory samples (sputum, nasopharyngeal swabs, or tracheal aspirates). 2 , 3 As shown in Table 1, these approaches are classic but still have deficits. For instance, biomarker detection has low sensitivity, and culture processing is time‐consuming. The diagnostic yield can only reach 70%–80% even by using the most comprehensive diagnostic methods. 4 A delay or misidentification of microorganisms directly exerts a significant effect on definitive antimicrobial therapy. The condition of patients deteriorates, and mortality increases if the causative pathogens are not identified timely. Thus, a new technology that can rapidly identify pathogens with high accuracy is desperately needed.
TABLE 1.
Advantages and limitations of testing methods for diagnosing pulmonary infectious diseases
| Diagnostic test | Advantages | Limitations |
|---|---|---|
| Serological assays | Potential in diagnosing acute infection | Low sensitivity during early infection |
| Inexpensive | Low sensitivity in humoral immune deficiencies | |
| PCR | High specificity and sensitivity in detecting viruses | Hypothesis‐depended |
| Rapid | Requirement of prior sequence data for designing primers | |
| Inexpensive | Limited use in detecting unknown pathogens | |
| Culture | Gold standard in diagnosis of pathogen | Low sensitivity after using antibiotics and antifungals |
| Inexpensive | Limited use in testing fastidious organisms, unknown pathogens and virus | |
| Time consuming | ||
| Targeted NGS (e.g., 16S) | Capability in differentiating multiple species within one pathogen type | Limited use in detecting pathogens without currently available 16S sequence data |
| High sensitivity | Requirement of prior sequence data for designing primers | |
| Difficulty in identifying pathogens to the species levels when existing a high degree of complete similarity across the length of 16S for some pathogens | ||
| Expensive | ||
| Metagenomic NGS | Hypothesis‐free and unbiased | Difficulty in eliminating human host background |
| High sensitivity | Hypersensitivity leading to false‐positive results | |
| Discovery of novel, rare or unexpected pathogens | Difficulty in interpreting the results for clinicians | |
| Capability in providing comprehensive information about pathogens | Expensive |
Abbreviations: NGS, next‐generation sequencing; PCR, polymerase chain reaction.
Next‐generation sequencing (NGS), also known as high‐throughput sequencing or deep sequencing, is a deoxyribonucleic acid (DNA) sequencing technique. The release of the first massively parallel pyrosequencing platform marking the successful exploitation of NGS is in 2005. 5 Compared with the gold standard for DNA sequencing, that is, Sanger sequencing, NGS has greatly improved the throughput, and the cost has dropped dramatically. 6 The development of the technique NGS has made metagenomic sequencing a practicable approach to be applied in clinical diagnosis. Metagenomic tools can sequence all the nucleic acid from the host and pathogenic specimens. Different from targeted NGS approaches, such as 16S ribonucleic acid (RNA) gene amplification for bacteria, 7 metagenomic sequencing is untargeted and can determine pathogens, regardless of their type, and provide sufficient information about pathogens, such as their antimicrobial genes and the human host response. 8 Moreover, when aiming to use targeting antibiotics correctly, metagenomic sequencing can provide the needed results of culture and drug sensitivity reports within 24–48 h, 9 a turn‐around time shorter than the turn‐around time of conventional diagnostic methods. Thus, metagenomic sequencing has the potential advantage than the traditional approaches in detecting the pathogen in infectious diseases.
Because the first report of metagenomic next‐generation sequencing to diagnose an infectious disease was reported, which identified a neuro‐invasive pathogen of cerebrospinal fluid, 10 the applications of metagenomic sequencing in diagnosing infectious diseases such as sepsis, 11 neurological infections, 12 , 13 and pneumonia 13 gradually emerged. Here, we focus on the applications of metagenomic sequencing in lung infections, discuss its advantages and limitations compared with traditional diagnostic methods, and try to advise the applications of metagenomic sequencing in different situations.
2. APPLICATIONS OF METAGENOMIC SEQUENCING IN PULMONARY INFECTION
The metagenomic sequencing technique was introduced to clinical diagnosis and has such a short history that most research papers available are case reports with a few retrospective studies and even fewer prospective studies. The field of lung infections is no exception. Below here sketch the current application status of metagenomic sequencing in pulmonary infections.
In the last 5 years, the number of case reports using metagenomic sequencing to identify pathogens increased. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Among the 32 cases shown in Table 2, pathogens were identified in 30. Five of those 30 were bacteria, 20 were viruses, 3 were fungi, 1 was Chlamydophila psittaci, and 1 was Spirometra erinaceieuropaei. These results indicate that metagenomic sequencing is untargeted and can detect most types of pathogens. Clinicians prefer to use bronchoalveolar lavage fluid (BALF) (25/30) as a specimen, but sputum (2/30), throat swabs (2/30), blood (2/30), and pleural effusion (1/30) were also used.
TABLE 2.
Clinical applications of mNGS in pulmonary infection disease
| Ref. | Study | Case no | Specimen type | Pathogen identified | Confirmatory test of the metagenomic result |
|---|---|---|---|---|---|
| 12 | Nicole Fischer, 2014 | 1,2 | BALF | Commensal bacteria | High‐throughput,16S rRNA sequencing, PCR, and serologic analysis |
| 3 | Chlamydophila psittaci | ||||
| 13 | DagmaraW. Lewandowska, 2015 | 4 | Throat swabs and stool samples | HEV‐C104 | Whole nucleic acid high‐throughput sequencing |
| 14 | Kathryn M. Pendleton, 2017 | 5 | Mini‐BALF | Pseudomonas aeruginosa | Culture, WGS, 16S rRNA sequencing |
| 6 | Staphylococcus aureus | ||||
| 15 | Fugui Yan, 2017 | 7 | BALF | HRV‐B91 | Seroconversion of HRV‐B91 neutralizing antibodies |
| 16 | Yanpeng Li, 2018 | 8 | Pulmonary secretions | HBoV1 and HRV‐C | PCR |
| 17 | Bailu Du, 2018 | 9 | The mass at the root of the right thigh | Spirometra erinaceieuropaei | Serological tests using anti‐sparganum antibodies |
| 18 | Jian Wang, 2019 | 10 | Sputum | GkV_CN‐GZ1 | PCR |
| 19 | Bin‐Chan He, 2019 | 11 | BALF | Aspergillus fumigatus | The serum G test and the BALF GM |
| 12 | Sputum culture and the BALF GM | ||||
| 13 | The results of other tests are all negative | ||||
| 20 | Yan Lin, 2019 | 14 | Plasma | Stenotrophomonas maltophilia | / |
| 21 | Huahua Yi, 2020 | 15 | Blood, sputum, and pleural effusion | Legionella pneumophila | PCR |
| 22 | Ancong Xu, 2020 | 16 | Sputum and blood | Acinetobacter baumannii | Sputum culture |
| 23 | Liangjun Chen, 2020 | 17, 18 | BALF | SARS‐Cov‐2 | / |
| 24 | Roujian Lu, 2020 | 19–27 | BALF or cultured virus or throat swab | SARS‐Cov‐2 | PCR |
| 25 | Li‐Li Ren, 2020 | 28–32 | BALF | SARS‐Cov‐2 | Sanger sequencing and PCR |
Abbreviations: BALF, bronchoalveolar lavage fluid; HEV, human enterovirus; HRV, human rhinoviruses; HBoV, human bocavirus; GkV, gemykibivirus; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; PCR, polymerase chain reaction.
Similarly, there is an increasing trend in the number of studies, which have included multiple samples since 2017. 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 Among the 11 studies displayed in Table 3, four used children as the research subjects. BALF is obtained from an invasive operation. Other types of specimens, such as nasopharyngeal/oropharyngeal (NP/OP) swabs, sputum, blood, and endotracheal tube, are easier than BALF and safer to get. Thus, BALF is used less often. It is worth noting that a study published in 2018 used computed tomography (CT)‐guided puncture lung biopsy tissues. 31
TABLE 3.
Comparison of mNGS with traditional methods
| Ref. | Study | Specimen type | Traditional tests | Potential clinical indications | No. of positive NGS results | No. of positive other methods' results | No. of cases tested |
|---|---|---|---|---|---|---|---|
| 26 | Jian Yang, 2011 | NP aspirate samples | Conventional PCR and real‐time RT‐PCR | ALRTs | 15 | 15 | 16 |
| 27 | Xiaohui Zou, 2017 | OP swabs | Real‐time PCR | Severe pneumonia | 16 | 0 | 33 |
| 28 | Robert Schlaberg, 2017 | NP/OP swabs | Culture and PCR | CAP | 13 | 0 | 70 |
| 29 | Henan Li, 2018 | lung biopsy tissues | Culture and smear | 11 pulmonary infection, 3 pulmonary tuberculosis, 3 lung cancer, 3 pulmonary occupying lesions | 15 | 12 | 20 |
| 30 | Charles Langeliera, 2018 | BALF | Culture, Aspergillus galactomannan assay, multiplex PCR, silver stain | CAP or HAP | 13 | 6 | 22 |
| 31 | Lauge Farnaes, 2019 | Peripheral blood | Culture and PCR | CAP | 13 | 6 | 15 |
| 32 | Yun Xie, 2019 | Sputum, blood and BALF | Conventional microbial tests | Severe pneumonia | 27 | 23 | 48 |
| 33 | Tingting Pan, 2019 | BALF | Quantitative cultures, multiplex PCR | CAP | 12 | 6 | 13 |
| 34 | Yi Zhang, 2019 | 3 blood, 1 tissue sample, 4 sputum and 7 BALF | Smear and culture | PCP | 15 | 6 | 15 |
| 35 | Matt S. Zinter, 2019 | 33 BAL, 4 mini‐BAL, 4 ETA | Conventional laboratory methods | Pulmonary infection | 25 | 17 | 41 |
| 36 | Libing Yang, 2019 | ETA | Culture | VAP | 10 | 9 | 14 |
Abbreviations: ALRTs, acute lower respiratory tract infections; CAP, community‐acquired pneumonia; ETA, endotracheal aspirate; HAP, hospital‐acquired pneumonia; NP, nasopharyngeal; OP, oropharyngeal; PCP, pneumocystis pneumonia; PCR, polymerase chain reaction; RMPP, refractory mycoplasma pneumoniae pneumonia; VAP, ventilator‐associated pneumonia.
3. DIAGNOSTIC YIELD
In studies targeted to identify all types of pathogens, metagenomic sequencing showed a diagnostic yield (number of positive results/number of cases tested) between 56.25% and 92.31%, whereas the conventional microbiologic tests showed 27.27%–64.29%. 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 In all studies, metagenomic sequencing detected pathogens at least as well as the conventional tests, 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 even compared with PCR which is famous for its high specificity and sensitivity in detecting viruses. 28 Furthermore, metagenomic sequencing could produce positive results in specimens previously considered by the traditional laboratory tests to be negative. 29 , 30 However, it is worth noting that assay hypersensitivity may lead to false‐positive results.
As for the capability of metagenomic sequencing in detecting different types of pathogens, a retrospective observational study conducted between 2010 and 2018 in China revealed the pathogenic bacteria positive rate of metagenomic sequencing was higher than that of traditional methods (68.7% vs. 45.4%, p = 0.006, Pearson χ 2). Meanwhile, there was no significant difference between the two methods on the pathogenic fungal positive rates. 34 However, another study showed the advantages of metagenomic sequencing to detect fungi, viruses, and opportunistic pathogens in immunocompromised hosts, whereas no significant difference existed in bacterial detection between the two techniques. 35 In another recent study, the detection rate of Pneumocystis pneumonia (PCP) was higher in high‐throughput sequencing compared with conventional methods such as Wright–Giemsa stained smear and microscopy identification. The conventional methods failed to detect Pneumocystis jirovecii in 8 of 13 samples, whereas metagenomic sequencing was successful. 35 The type pathogen that can best be detected by metagenomic sequencing is still inconclusive. More research is needed for helping clinicians achieve a better judgment in interpreting inconsistent results of metagenomic sequencing and other methods.
4. THE ADVANTAGES OF METAGENOMIC SEQUENCING
4.1. Metagenomic sequencing can detect novel, rare, and unexpected pathogens
Metagenomic sequencing is a target‐independent approach that offers a more comprehensive view of the pathogenic agents and provides a detection of common and unexpected pathogens in samples. 39 This technology does especially well in detecting rare pathogens. Chlamydophila psittaci is excluded from in standard diagnostic panels because of its rarity. However, metagenomic sequencing identified Chlamydophila psittaci successfully in a patient who had severe chlamydial pneumonia with acute respiratory distress syndrome (ARDS) in a case reported in 2014. 14 The gold standard for diagnosing a parasitic disease is to find the parasite. A rare case was reported in which metagenomic sequencing diagnosed an 11‐year‐old patient presented with pulmonary and pericardial effusion. The genomic DNA of Spirometra erinaceieuropaei was identified in a lump derived from the root of the right thigh, while sparganum was not detected in the dissected tissues. 19 Metagenomic sequencing played a decisive role in achieving a definitive diagnosis in this case. Similarly, a case reported in 2017 identified human rhinoviruses B91 (HRV‐B91), a rarely reported subtype, in BALF by deep sequencing. Here, conventional real‐time PCR assay, a conventional approach for viral identification due to its high specificity and sensitivity, produced negative results. 17 The reason for which this molecular method did not identify HRV‐B91 successfully is its target specific, which may lead to the missing of the novel, rare, and unexpected viruses.
Another merit of metagenomic sequencing is to detect a novel viral strain. A case reported in 2019 was the first to detect the Gemykibivirus (GkV) genome using metagenomic sequencing from the sputum of a patient with pneumonia. It was the first time that the GkV was found in the respiratory tract. 20 Recently, the novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS‐Cov‐2), which caused severe pneumonia in Wuhan, China, was sequenced by metagenomic sequencing. 25 , 26 , 27 Deep sequencing methods provided a great contribution in raising our awareness of a novel viral strain of this virus.
4.2. Metagenomic sequencing is more sensitive than traditional methods
Culturing is a conventional and widely used method to identify pathogens. However, culturing is time‐consuming and many pathogens that need specific culture conditions are difficult to grow. Legionella pneumophila needs a specific medium to be cultured; thus, it is harder to obtain the correct diagnosis. Metagenomic sequencing detects pathogens regardless of their taxonomy. It can identify pathogens that do not grow in culture. In a recent report, Legionella pneumophila was detected in all blood, sputum, and pleural effusion samples by metagenomic sequencing taken from a patient with community‐acquired pneumonia. 23 Many studies have revealed that metagenomic sequencing could identify more types of pathogens than the culture method. 40 , 41 , 42
Metagenomic sequencing also plays an essential role in the identification of pathogens in pulmonary fungal infections. Except for smear by microscopy, pathogen culture and PCR, serum or BALF galactomannan (GM) test, and serum 1,3‐β‐d‐glucan (G) tests help diagnose invasive pulmonary aspergillosis (IPA). 43 However, problems such as time–cost and low‐yield production still exist in traditional methods, making it difficult to diagnose IPA. In case 13, metagenomic sequencing was the only approach to detect the Aspergillus fumigatus, whereas other conventional methods failed. 21 Clinicians can consider metagenomic sequencing as a complementary method to diagnose IPA.
4.3. Metagenomic sequencing is suitable to detect co‐infections
Metagenomic sequencing is untargeted. The capability to identify bacteria, viruses, fungi, parasites, and other pathogens at the same time in only one approach makes metagenomic sequencing widely appealing for co‐infection cases. A rare case reported in 2018 was an adult co‐infected with both human rhinovirus (HRV) and bocavirus (HBoV). 18 Almost every study covering multiple objects included samples in which a mix‐infection was detected. 28 , 29 , 30 , 31 , 32 , 33 , 35 , 36 Metagenomic sequencing can rule out co‐infections as well. A study published in 2018 reviewed 675 patients with childhood refractory Mycoplasma pneumoniae pneumonia. The culture results revealed that 18 of the 675 specimens were positive for bacteria. Another 18 specimens were picked out randomly from the remained 657 specimens to be tested by metagenomic sequencing again. Researchers used the metagenomic sequencing as a validation to assure the culture results. 44
4.4. Metagenomic sequencing provides comprehensive information about pathogens
Metagenomic sequencing allows the evaluation of phylogenetic analysis, strain‐level typing, and antigenic epitope characterization of pathogenic genomes. Phylogenetic analysis, based on sequence alignment, helps to manage endemics and outbreaks by finding the origins of viruses. For example, SARS‐CoV‐2 was classified into the genus β‐coronavirus, subgenus Sarbecovirus, and its sequence was shown to be closest with bat SARS‐like coronavirus (SL‐CoV) ZXC21 (76.5%–91.2%) and bat SL‐CoV ZC45 (76.9%–91.2%) by phylogenetic analysis. 25 , 26 , 27 The fact that SARS‐CoV‐2 probably originated from the bat was amply reported. Notably, the genetic similarity between the SARS‐CoV‐2 strains and SARS‐CoV (79%–82%) or the Middle East respiratory syndrome (MERS)‐CoV (50%–51.8%) offers a direction for managing the Corona Virus Disease 2019 (COVID‐19). 25 , 26 , 27
Metagenomic sequencing can discover novel antibiotic resistance genes in pathogens and investigate the antibiotic resistome of new strains. 45 , 46 For instance, 18 possible antibiotic resistance genes were identified from the Acinetobacter baumannii strain DMS06669, isolated from the sputum of a patient with hospital‐acquired pneumonia. Notably, 8 of 18 genes appeared in A. baumannii for the first time. 47 Based on the information in the literature, relevant databases were established. The Comprehensive Antibiotic Resistance Database (CARD; http://arpcard.mcmaster.ca) is a good example, and it renders the service for predicting an antibiotic resistance phenotype from a genome sequence. 48 , 49 , 50
4.5. Metagenomic sequencing shortens the turnaround time
Currently, the burden of multidrug‐resistant bacteria is getting heavier. We know that prolonged empirical antibiotic therapies may cause the emergence of drug‐resistant bacteria such as Achromobacter xylosoxidans and Stenotrophomonas maltophilia. 51 , 52 In numerous cases, the failure of recognizing resistant bacteria has been reported to delay the providing of appropriate antibiotic treatment for patients, resulting in severe clinical consequences. 53 , 54 Therefore, timely, accurate, and targeted antibiotic treatment is important. A study published in 2019 provided evidence that metagenomic sequencing reduced the mortality of severe pneumonia to 16.67% compared with 42.3% in control. 34 The main reason may be that using metagenomic sequencing (average of ~48 h) is faster than conventional tests (average of 3–5 days) in achieving accurate results of culture and drug sensitivity reports when a bioinformatics personnel is available. 9
As an emerging metagenomic sequencing technology, nanopore sequencing takes less time to generate the sequence. A comprehensive case report published in 2017 identified Pseudomonas aeruginosa and Staphylococcus aureus using real‐time metagenomics in mini‐BALF of two patients with bacterial pneumonia. The first application of the MinION (Oxford Nanopore Technologies, Oxford, UK), a new‐to‐market palm‐sized DNA sequencer, to identify pathogens in pneumonia revealed that real‐time metagenomics is quicker and much more convenient than culturing. It took 9 h to get the sequence from one patient's specimen, which was well aligned with the Pseudomonas aeruginosa sequence. 16 Another recent study was the first to detect pathogenic fungi using nanopore sequencing with a workflow of 10 h, and the result was consistent with routine diagnostic methods. Patients in this study were diagnosed with PCP, and nanopore sequencing confirmed the diagnosis. 55 Nanopore sequencing has top speed among all metagenomic sequencing technologies. 56
5. THE CHALLENGES IN THE APPLICATIONS OF METAGENOMIC SEQUENCING FOR CLINICIANS
5.1. The scenarios suitable for applying metagenomic sequencing
Metagenomic sequencing is good at identifying rare or novel pathogens, and it also does well in multi‐infections. To our knowledge, patients with an immune deficiency can be easily infected by opportunistic pathogens or with multiple infections. Thus, metagenomic sequencing is particularly suitable for those patients who are immunocompromised or immunodeficient. Under these conditions, when faced with infants, children, aged people with basic diseases, and repeatedly hospitalized people, clinicians could apply metagenomic sequencing to detect pathogens as soon as possible. Also, as an untargeted and comprehensive detecting method, metagenomic sequencing can be used in people suspected of being infected by specific pathogens, people with unexplained infections, people who cannot be diagnosed by repeated traditional microbial detection technology, or people who are critically ill. 16 , 57 , 58
5.2. The selection of specimens
Appropriate selection of specimens helps improve the accuracy of diagnosis. A study published in 2012 investigated the complexity and distribution of microbiota in chronic obstructive pulmonary disease (COPD) patients using a 16S ribonucleic acid (RNA) gene‐based technique to analyze sputum, bronchial aspirate, BALF, and bronchial mucosa. It revealed that the outcomes of the former two samples were similar, whereas those of the latter two were similar. This study indicated that the two most used clinical samples—sputum and BALF—were different. Sputum represented the upper bronchial tree samples, while the BALF represented the lower bronchial tree samples. 7 This study indicated that BALF was a better specimen to diagnose lung infections. The accomplishment of strict negative controls and protected bronchoscopic sampling techniques including using a catheter with a wax‐sealed tip alleviated a risk for contamination with pharyngeal microbiota when a bronchoscope went through the upper respiratory tract. 59 However, some patients may refuse invasive operation or cannot withstand the bronchoscope examination. Another study published in 2019 focused on comparing two sampling approaches (tracheal aspirate and mini‐bronchoalveolar lavage) in patients with pneumonia who were critically ill. Metagenomic sequencing was used to analyze the abundance of microbiota taken by two types of sampling, and the result showed no significant difference. This study challenged the old idea that less invasive tracheal aspirate sampling was inferior to mini‐bronchoalveolar lavage sampling because of the potential contamination from oropharyngeal microbiota. The same study demonstrated that the less invasive tracheal aspirate could replace mini‐bronchoalveolar lavage. 60 DNA sequencing of cell‐free plasma (CFPDNA) is a new commercial method that only requires blood to test, which means it is another noninvasive approach. It has a satisfactory detection rate for bacteria and fungi. However, CFPDNA sequencing has limitations in detecting RNA viruses. 33 , 61 , 62 Also, a study suggested that lung biopsy tissues also had the potential to identify pathogens. Thus, metagenomic sequencing displays many advantages in speed and sensitivity. 31 However, obtaining CT‐guided puncture lung biopsy tissues harm the patient.
In a nutshell, when selecting specimens, the patient's will, physical condition, and representativeness should all be taken into consideration.
5.3. The interpretation of metagenomic sequencing results
The detection of microorganisms by metagenomic sequencing can prove their existence but not their pathogenicity because it is difficult to distinguish normal microbiota, colonized microbiota, contamination, or bona fide pathogens. 8 The lung is considered to be sterile, but this view has been overturned nowadays. Microbial communities exist in the lungs of healthy humans. 63 Also, the composition of lung microbiota in those affected with chronic pulmonary diseases differs from healthy people. 64 , 65 , 66 Nucleic acid contamination may occur in several procedures during the workflow. As for clinicians, they should be prudent to avoid contamination when collecting specimens. Notably, in theory, contamination with commensal oral flora organisms seems to be inescapable. 67
Without a standardized protocol, it is difficult for clinicians to interpret the results of metagenomic sequencing. Databases that consist of potentially contaminating genera help clinicians recognize normal microbiota, contamination, and bona fide pathogens. 68 , 69 Negative controls that monitor external contamination are recommended to be sequenced simultaneously. 56 , 68 Quantitative and semiquantitative statistical analyses have been reported to distinguish colonization from infection. 16 , 32 However, an acknowledged approach is required. Clinicians should also concentrate on the clinical manifestations of patients and outcomes of other traditional diagnostic methods. For instance, whether opportunistic pathogens caused disease depends on the patients' presentation. 56 Remarkably, the lung pathogenicity of a microbe identified by metagenomic sequencing should have been evidenced by the existing literature. 32 Furthermore, negative metagenomic sequencing outcomes may be due to very low quantities for sequencing 42 and do not exclude the existence of causative agents. In summary, clinicians should be well‐rounded when interpreting metagenomic sequencing reports. Cooperation within a group consisting of experts from medical microbiology, computational biology, and clinicians to interpret results of metagenomic sequencing is advised. 9
6. CONCLUSION
The mortality associated with pulmonary infections is higher than that of other infectious diseases. 1 Up to 60% of pulmonary infection cases were treated without gathering evidence for the presence of pathogens, 4 , 13 as traditional clinical diagnostic methods have low sensitivity and are time‐consuming. It is important to identify pathogens as early as possible because this information promotes the management of diseases and consequently improves the prognosis of the patients.
NGS has become a research hotspot in recent years. However, this technology still needs improvement to narrow the gap between academic research and clinical applications. Importantly, several obstacles should be overcome to improve specificity of the technology. The first is to isolate pathogens from complicated clinical samples reliably. Human sequences need to be removed as much as possible. A study compared three DNA extraction methods by eliminating the nucleic acid of hosts and found that the one with Bensonase could obtain a higher yield of microbial DNA. 70 Reference sequence databases should be improved by verifying the accuracy of the sequences and the databases should also be updated periodically. The specification operation of clinicians to collect specimens which can avoid contamination is also an approach to improve the specificity. On the other hand, the cost should be cut down because it is still much higher than that of routine diagnostic methods. 71
As for the applications of metagenomic sequencing in lung infections, many challenges exist. When a patient has a pulmonary infection, clinical samples are commonly taken from sputum, NP/OP swabs, BALF, and lung puncture biopsy tissues. The location, shape, and size of lung lesions along with sputum quality and invasive operation tolerance of patients should be in consideration to determine which type of sample is the most appropriate for detecting pathogens in lung infections using metagenomic sequencing. Commonly, BALF is the first choice as it is taken from lower respiratory tract directly. Sputum expectoration and induced sputum can be alternative if patients reject invasive fiberoptic bronchoscopy. Patients with viral pneumonia usually do not have obvious expectoration. For those suspected of viral lower respiratory tract infection, NP/OP swabs can be collected. For peripheral pulmonary lesions, localized lesions, or mediastinal/hilar lymphadenopathy, invasive operation should be selected individually according to the characteristics of the lesions to obtain tissues for examination. Another challenge is to interpret the results. Clinicians should combine results from all tests with patients' manifestations and interpret them from all possible angles. Whether microorganisms detected are pathogenic bacteria, colonizing bacteria or background bacteria should be carefully discriminated. And the detection results should be cross verified by other traditional microbial technologies.
In summary, metagenomic sequencing is a diagnostic technology that complements current diagnostic methods for pathogen detection in pulmonary infections. In addition to the diagnostic role, metagenomic sequencing also informs genotyping, predicting drug resistance, and guiding the medication. This technology still needs to be improved, and clinicians must enhance their understanding of its results. Metagenomic sequencing will be an important supplement for the targeted diagnostic methods for pathogen detection.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of the article.
ETHICS STATEMENT
No ethics approval was needed, no human subjects were involved in this review paper, and no consent to participate and publish was needed.
AUTHOR CONTRIBUTIONS
YC and JFX were the major contributors to the writing of the manuscript. LCF and YHC revised the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Fund for Distinguished Young Scholars to JFX (81925001), Shanghai Leading Talent Program (No. 2016036 to JFX), the National Natural Science Foundation of China (81800063 to LCF).
Chen Y, Fan L‐C, Chai Y‐H, Xu J‐F. Advantages and challenges of metagenomic sequencing for the diagnosis of pulmonary infectious diseases. Clin Respir J. 2022;16(10):646‐656. doi: 10.1111/crj.13538
Yan Chen and Li‐Chao Fan contributed equally.
Funding information This work was supported by the National Natural Science Fund for Distinguished Young Scholars to JFX (81925001) and Shanghai Leading Talent Program (No. 2016036 to JFX). LCF was supported by the National Natural Science Foundation of China (81800063).
Funding information National Natural Science Foundation of China, Grant/Award Numbers: 81925001, 81800063; Shanghai Leading Talent Program, Grant/Award Number: 2016036
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Hansen V, Oren E, Dennis LK, Brown HE. Infectious disease mortality trends in the United States, 1980–2014. Jama. Nov 22, 2016;316(20):2149‐2151. doi: 10.1001/jama.2016.12423 [DOI] [PubMed] [Google Scholar]
- 2. Loens K, Van Heirstraeten L, Malhotra‐Kumar S, Goossens H, Ieven M. Optimal sampling sites and methods for detection of pathogens possibly causing community‐acquired lower respiratory tract infections. J Clin Microbiol. Jan 2009;47(1):21‐31. doi: 10.1128/JCM.02037-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajapaksha P, Elbourne A, Gangadoo S, Brown R, Cozzolino D, Chapman J. A review of methods for the detection of pathogenic microorganisms. Analyst. Jan 14, 2019;144(2):396‐411. doi: 10.1039/c8an01488d [DOI] [PubMed] [Google Scholar]
- 4. Ewig S, Torres A, Angeles Marcos M, et al. Factors associated with unknown aetiology in patients with community‐acquired pneumonia. Eur Respir J. Nov 2002;20(5):1254‐1262. doi: 10.1183/09031936.02.01942001 [DOI] [PubMed] [Google Scholar]
- 5. Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high‐density picolitre reactors. Nature. Sep 15 2005;437(7057):376‐380. doi: 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voelkerding KV, Dames SA, Durtschi JD. Next‐generation sequencing: from basic research to diagnostics. Clin Chem. Apr 2009;55(4):641‐658. doi: 10.1373/clinchem.2008.112789 [DOI] [PubMed] [Google Scholar]
- 7. Cabrera‐Rubio R, Garcia‐Nunez M, Seto L, et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. Nov 2012;50(11):3562‐3568. doi: 10.1128/JCM.00767-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. Jun 2019;20(6):341‐355. doi: 10.1038/s41576-019-0113-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next‐generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. Feb 10, 2018;66(5):778‐788. doi: 10.1093/cid/cix881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. Jun 19, 2014;370(25):2408‐2417. doi: 10.1056/NEJMoa1401268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell‐free DNA sequencing test for infectious disease. Nat Microbiol. Apr 2019;4(4):663‐674. doi: 10.1038/s41564-018-0349-6 [DOI] [PubMed] [Google Scholar]
- 12. Miller S, Naccache SN, Samayoa E, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. May 2019;29(5):831‐842. doi: 10.1101/gr.238170.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next‐generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. Jun 2017;141(6):776‐786. doi: 10.5858/arpa.2016-0539-RA [DOI] [PubMed] [Google Scholar]
- 14. Fischer N, Rohde H, Indenbirken D, et al. Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia. Emerg Infect Dis. Jun 2014;20(6):1072‐1075. doi: 10.3201/eid2006.131526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewandowska DW, Zagordi O, Zbinden A, et al. Unbiased metagenomic sequencing complements specific routine diagnostic methods and increases chances to detect rare viral strains. Diagn Microbiol Infect Dis. Oct 2015;83(2):133‐138. doi: 10.1016/j.diagmicrobio.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pendleton KM, Erb‐Downward JR, Bao Y, et al. Rapid pathogen identification in bacterial pneumonia using real‐time metagenomics. Am J Respir Crit Care Med. Dec 15, 2017;196(12):1610‐1612. doi: 10.1164/rccm.201703-0537LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yan F, Xiao Y, Li M, et al. Metagenomic analysis identified human rhinovirus B91 infection in an adult suffering from severe pneumonia. Am J Respir Crit Care Med. Jun 1, 2017;195(11):1535‐1536. doi: 10.1164/rccm.201609-1908LE [DOI] [PubMed] [Google Scholar]
- 18. Li Y, Deng X, Hu F, et al. Metagenomic analysis identified co‐infection with human rhinovirus C and bocavirus 1 in an adult suffering from severe pneumonia. J Infect. Mar 2018;76(3):311‐313. doi: 10.1016/j.jinf.2017.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du B, Tao Y, Ma J, et al. Identification of sparganosis based on next‐generation sequencing. Infect Genet Evol. Dec 2018;66:256‐261. doi: 10.1016/j.meegid.2018.10.005 [DOI] [PubMed] [Google Scholar]
- 20. Wang J, Li Y, He X, et al. Gemykibivirus genome in lower respiratory tract of elderly woman with unexplained acute respiratory distress syndrome. Clin Infect Dis. Aug 16, 2019;69(5):861‐864. doi: 10.1093/cid/ciz072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He BC, Liu LL, Chen BL, Zhang F, Su X. The application of next‐generation sequencing in diagnosing invasive pulmonary aspergillosis: three case reports. American Journal of Translational Research. 2019;11(4):2532‐2539. [PMC free article] [PubMed] [Google Scholar]
- 22. Lin Y, Wang BX, Zhang NN, et al. Metagenomic analysis identified Stenotrophomonas maltophilia pneumonia in an infant suffering from unexplained very severe pneumonia. Front Pediatr. 2019;7:380‐386. doi: 10.3389/fped.2019.00380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yi H, Fang J, Huang J, Liu B, Qu J, Zhou M. Legionella pneumophila as cause of severe community‐acquired pneumonia. China Emerg Infect Dis. Jan 2020;26(1):160‐162. doi: 10.3201/eid2601.190655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu A, Zhu H, Gao B, et al. Diagnosis of severe community‐acquired pneumonia caused by Acinetobacter baumannii through next‐generation sequencing: a case report. BMC Infect Dis. Jan 15, 2020;20(1):45‐51. doi: 10.1186/s12879-019-4733-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L, Liu W, Zhang Q, et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg Microbes Infect. Dec 2020;9(1):313‐319. doi: 10.1080/22221751.2020.1725399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England). Feb 22, 2020;395(10224):565‐574. doi: 10.1016/s0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ren LL, Wang YM, Wu ZQ, et al. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin Med J (Engl). Feb 11, 2020:1015‐1024. doi: 10.1097/CM9.0000000000000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang J, Yang F, Ren L, et al. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J Clin Microbiol. Oct 2011;49(10):3463‐3469. doi: 10.1128/JCM.00273-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou X, Tang G, Zhao X, et al. Simultaneous virus identification and characterization of severe unexplained pneumonia cases using a metagenomics sequencing technique. Sci China Life Sci. Mar 2017;60(3):279‐286. doi: 10.1007/s11427-016-0244-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schlaberg R, Queen K, Simmon K, et al. Viral pathogen detection by metagenomics and pan‐viral group polymerase chain reaction in children with pneumonia lacking identifiable etiology. J Infect Dis. May 1, 2017;215(9):1407‐1415. doi: 10.1093/infdis/jix148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H, Gao H, Meng H, et al. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next‐generation sequencing. Front Cell Infect Microbiol. 2018;8:205‐215. doi: 10.3389/fcimb.2018.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langelier C, Zinter MS, Kalantar K, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am J Respir Crit Care Med. Feb 15, 2018;197(4):524‐528. doi: 10.1164/rccm.201706-1097LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farnaes L, Wilke J, Ryan Loker K, et al. Community‐acquired pneumonia in children: cell‐free plasma sequencing for diagnosis and management. Diagn Microbiol Infect Dis. Jun 2019;94(2):188‐191. doi: 10.1016/j.diagmicrobio.2018.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Y, Du J, Jin W, et al. Next generation sequencing for diagnosis of severe pneumonia: China, 2010‐2018. J Infect. Feb 2019;78(2):158‐169. doi: 10.1016/j.jinf.2018.09.004 [DOI] [PubMed] [Google Scholar]
- 35. Pan T, Tan R, Qu H, et al. Next‐generation sequencing of the BALF in the diagnosis of community‐acquired pneumonia in immunocompromised patients. J Infect. Jul 2019;79(1):61‐74. doi: 10.1016/j.jinf.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Ai JW, Cui P, Zhang WH, Wu HL, Ye MZ. A cluster of cases of pneumocystis pneumonia identified by shotgun metagenomics approach. J Infect. Feb 2019;78(2):158‐169. doi: 10.1016/j.jinf.2018.08.013 [DOI] [PubMed] [Google Scholar]
- 37. Zinter MS, Dvorak CC, Mayday MY, et al. Pulmonary metagenomic sequencing suggests missed infections in immunocompromised children. Clin Infect Dis. May 17, 2019;68(11):1847‐1855. doi: 10.1093/cid/ciy802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang L, Haidar G, Zia H, et al. Metagenomic identification of severe pneumonia pathogens in mechanically‐ventilated patients: a feasibility and clinical validity study. Respir Res. Nov 27, 2019;20(1):265‐276. doi: 10.1186/s12931-019-1218-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palacios G, Druce J, Du L, et al. A new arenavirus in a cluster of fatal transplant‐associated diseases. N Engl J Med. Mar 6, 2008;358(10):991‐998. doi: 10.1056/NEJMoa073785 [DOI] [PubMed] [Google Scholar]
- 40. Toma I, Siegel MO, Keiser J, et al. Single‐molecule long‐read 16S sequencing to characterize the lung microbiome from mechanically ventilated patients with suspected pneumonia. J Clin Microbiol. Nov 2014;52(11):3913‐3921. doi: 10.1128/JCM.01678-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tunney MM, Klem ER, Fodor AA, et al. Use of culture and molecular analysis to determine the effect of antibiotic treatment on microbial community diversity and abundance during exacerbation in patients with cystic fibrosis. Thorax. Jul 2011;66(7):579‐584. doi: 10.1136/thx.2010.137281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sung JY, Hwang Y, Shin MH, et al. Utility of conventional culture and MALDI‐TOF MS for identification of microbial communities in bronchoalveolar lavage fluid in comparison with the GS junior next generation sequencing system. Ann Lab Med. Mar 2018;38(2):110‐118. doi: 10.3343/alm.2018.38.2.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alanio A, Bretagne S. Challenges in microbiological diagnosis of invasive aspergillus infections. F1000Res. 2017;6(F1000 Faculty Rev):157‐166. doi: 10.12688/f1000research.10216.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu JR, Lu J, Dong F, et al. Low bacterial co‐infection invalidates the early use of non‐anti‐mycoplasma pneumoniae antibiotics in pediatric refractory mycoplasma pneumoniae pneumonia patients. Front Pediatr. 2018;6:296‐305. doi: 10.3389/fped.2018.00296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bugrysheva JV, Sue D, Gee JE, et al. Antibiotic resistance markers in Burkholderia pseudomallei strain Bp1651 identified by genome sequence analysis. Antimicrob Agents Chemother. Jun 2017;61(6):e00010‐17. doi: 10.1128/aac.00010-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dunne WM Jr, Jaillard M, Rochas O, Van Belkum A. Microbial genomics and antimicrobial susceptibility testing. Expert Rev Mol Diagn. Mar 2017;17(3):257‐269. doi: 10.1080/14737159.2017.1283220 [DOI] [PubMed] [Google Scholar]
- 47. Si‐Tuan N, Ngoc HM, Hang PTT, Nguyen C, Van PH, Huong NT. New eight genes identified at the clinical multidrug‐resistant Acinetobacter baumannii DMS06669 strain in a Vietnam hospital. Ann Clin Microbiol Antimicrob. Nov 14, 2017;16(1):74‐80. doi: 10.1186/s12941-017-0250-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McArthur AG, Waglechner N, Nizam F, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. Jul 2013;57(7):3348‐3357. doi: 10.1128/AAC.00419-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia B, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model‐centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. Jan 4, 2017;45(D1):D566‐D573. doi: 10.1093/nar/gkw1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. Jan 8, 2020;48(D1):D517‐D525. doi: 10.1093/nar/gkz935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next‐generation sequencing when applied to clinical practice. Clin Infect Dis. Nov 13, 2018;67(suppl_2):S231‐S240. doi: 10.1093/cid/ciy693 [DOI] [PubMed] [Google Scholar]
- 52. Feigelman R, Kahlert CR, Baty F, et al. Sputum DNA sequencing in cystic fibrosis: non‐invasive access to the lung microbiome and to pathogen details. Microbiome. Feb 10, 2017;5(1):20‐33. doi: 10.1186/s40168-017-0234-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. Feb 1999;115(2):462‐474. doi: 10.1378/chest.115.2.462 [DOI] [PubMed] [Google Scholar]
- 54. Piskin N, Aydemir H, Oztoprak N, et al. Inadequate treatment of ventilator‐associated and hospital‐acquired pneumonia: risk factors and impact on outcomes. BMC Infect Dis. Oct 24, 2012;12:268‐276. doi: 10.1186/1471-2334-12-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Irinyi L, Hu Y, Hoang MTV, et al. Long‐read sequencing based clinical metagenomics for the detection and confirmation of Pneumocystis jirovecii directly from clinical specimens: a paradigm shift in mycological diagnostics. Med Mycol. Nov 23, 2019:1‐11. doi: 10.1093/mmy/myz109 [DOI] [PubMed] [Google Scholar]
- 56. Gu W, Miller S, Chiu CY. Clinical metagenomic next‐generation sequencing for pathogen detection. Annu Rev Pathol. Jan 24, 2019;14:319‐338. doi: 10.1146/annurev-pathmechdis-012418-012751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parize P, Muth E, Richaud C, et al. Untargeted next‐generation sequencing‐based first‐line diagnosis of infection in immunocompromised adults: a multicentre, blinded, prospective study. Clin Microbiol Infect. Aug 2017;23(8):574 e1‐574 e6. doi: 10.1016/j.cmi.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 58. Ruppe E, Cherkaoui A, Lazarevic V, Emonet S, Schrenzel J. Establishing genotype‐to‐phenotype relationships in bacteria causing hospital‐acquired pneumonia: a prelude to the application of clinical metagenomics. Antibiotics (Basel). Nov 29, 2017;6(4):30‐44. doi: 10.3390/antibiotics6040030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol. Oct 2019;20(10):1279‐1290. doi: 10.1038/s41590-019-0451-9 [DOI] [PubMed] [Google Scholar]
- 60. Kalantar KL, Moazed F, Christenson SC, et al. Metagenomic comparison of tracheal aspirate and mini‐bronchial alveolar lavage for assessment of respiratory microbiota. Am J Physiol Lung Cell Mol Physiol. Mar 1, 2019;316(3):L578‐L584. doi: 10.1152/ajplung.00476.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. De Vlaminck I, Martin L, Kertesz M, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci U S a. Oct 27, 2015;112(43):13336‐13341. doi: 10.1073/pnas.1517494112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell‐free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn Microbiol Infect Dis. Nov 2018;92(3):210‐213. doi: 10.1016/j.diagmicrobio.2018.06.009 [DOI] [PubMed] [Google Scholar]
- 63. Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. Oct 2012;160(4):258‐266. doi: 10.1016/j.trsl.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu D, Hou C, Li Y, et al. Analysis of the bacterial community in chronic obstructive pulmonary disease sputum samples by denaturing gradient gel electrophoresis and real‐time PCR. BMC Pulmonary Med. Nov 18, 2014;14:179‐185. doi: 10.1186/1471-2466-14-179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma‐associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. Feb 2013;131(2):346‐52.e1–3. doi: 10.1016/j.jaci.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Molyneaux PL, Cox MJ, Willis‐Owen SA, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. Oct 15, 2014;190(8):906‐913. doi: 10.1164/rccm.201403-0541OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O'Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. Jun 15, 2016;196(12):4839‐4847. doi: 10.4049/jimmunol.1600279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC biology. Nov 12, 2014;12:87‐98. doi: 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Strong MJ, Xu G, Morici L, et al. Microbial contamination in next generation sequencing: implications for sequence‐based analysis of clinical samples. PLoS Pathog. Nov 2014;10(11):e1004437. doi: 10.1371/journal.ppat.1004437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wen Y, Xiao F, Wang C, Wang Z. The impact of different methods of DNA extraction on microbial community measures of BALF samples based on metagenomic data. American Journal of Translational Research. 2016;8(3):1412‐1425. [PMC free article] [PubMed] [Google Scholar]
- 71. Balloux F, Bronstad Brynildsrud O, van Dorp L, et al. From theory to practice: translating whole‐genome sequencing (WGS) into the clinic. Trends Microbiol. Dec 2018;26(12):1035‐1048. doi: 10.1016/j.tim.2018.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.