

Abstract
Background
Since the beginning of the novel coronavirus (SARS-CoV-2) disease outbreak, there has been an increasing interest in discovering potential therapeutic agents for this disease. In this regard, we conducted a systematic review through an overview of drug development (in silico, in vitro, and in vivo) for treating COVID-19.
Methods
A systematic search was carried out in major databases including PubMed, Web of Science, Scopus, EMBASE, and Google Scholar from December 2019 to March 2021. A combination of the following terms was used: coronavirus, COVID-19, SARS-CoV-2, drug design, drug development, In silico, In vitro, and In vivo. A narrative synthesis was performed as a qualitative method for the data synthesis of each outcome measure.
Results
A total of 2168 articles were identified through searching databases. Finally, 315 studies (266 in silico, 34 in vitro, and 15 in vivo) were included. In studies with in silico approach, 98 article study repurposed drug and 91 studies evaluated herbal medicine on COVID-19. Among 260 drugs repurposed by the computational method, the best results were observed with saquinavir (n = 9), ritonavir (n = 8), and lopinavir (n = 6). Main protease (n = 154) following spike glycoprotein (n = 62) and other nonstructural protein of virus (n = 45) was among the most studied targets. Doxycycline, chlorpromazine, azithromycin, heparin, bepridil, and glycyrrhizic acid showed both in silico and in vitro inhibitory effects against SARS-CoV-2.
Conclusion
The preclinical studies of novel drug design for COVID-19 focused on main protease and spike glycoprotein as targets for antiviral development. From evaluated structures, saquinavir, ritonavir, eucalyptus, Tinospora cordifolia, aloe, green tea, curcumin, pyrazole, and triazole derivatives in in silico studies and doxycycline, chlorpromazine, and heparin from in vitro and human monoclonal antibodies from in vivo studies showed promised results regarding efficacy. It seems that due to the nature of COVID-19 disease, finding some drugs with multitarget antiviral actions and anti-inflammatory potential is valuable and some herbal medicines have this potential.
1. Introduction
Coronavirus disease 2019 (COVID-19), which was first identified in December 2019, and shortly after, declared a pandemic by World Health Organization (WHO) [1]. As of January 18, 2022, there have been more than 326 million confirmed cases and 5.54 million deaths globally [2]. Coronaviruses belong to the family of Coronaviridae, RNA viruses with crown-like spikes on the surface of the coronavirus particles. According to a meta-analysis of Macedo et al. [3], the mortality rate of COVID-19 was 17.1% for patients admitted to hospitals, whereas WHO estimated a fatality rate of 6.73%, which was much lower than that calculated from published studies. Among the critical cases of COVID-19, the mortality rate reaches 40% [4].
Substantial efforts have been made in the treatment of patients with COVID-19. The WHO recommendations in the treatment of COVID-19 are as follows [5]: molnupiravir (conditional), baricitinib (strong), ruxolitinib and tofacitinib (conditional), sotrovimab (conditional), casirivimab and imdevimab (conditional), IL-6 receptor blockers (tocilizumab and sarilumab) (strong), remdesivir (conditional), and systemic corticosteroids (strong). The WHO recommends not to use ivermectin, lopinavir/ritonavir, hydroxychloroquine, and convalescent plasma.
The pathogenesis of COVID-19 was explained by cytokine storm, reduction in ACE2 expression, and activation of complement pathway-induced microvascular injury and thrombosis [6]. The mechanisms of the recommended agents are focused on the mentioned pathogenesis to improve the clinical outcome of COVID-19, and antiviral therapies are missing. The anticoronaviral strategies include preventing the synthesis of viral RNA, inhibiting virus replication, blocking the virus binding to human cell receptors, or inhibiting the viruses' self-assembly process [7]. The SARS-CoV-2 contains at least four structural proteins: spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein, and 16 nonstructural proteins (NSPs). Among the translated NSPs, the main protease, also called chymotrypsin-like protease (3C-like protease), and the papain-like protease are two essential proteases for proteolytic processing of the coronavirus replicase polyprotein, therefore generating functional replication complex of the virus, whereas RNA-dependent RNA polymerase is the central enzyme for RNA synthesis. These three NSPs play crucial roles in coronavirus replication, making them attractive targets for anticoronaviral drug design [8].
The S protein, a surface-located trimeric glycoprotein of coronaviruses, promotes the attachment of viruses to host cells through binding to angiotensin-converting enzyme 2 (ACE2) and virus-cell membrane fusion during viral infection. Thus, the S protein has been considered as a major target for the development of vaccines and drug [9].
The development of a new therapeutic agent is a complex, lengthy, and expensive process, which can take 2–4 years of preclinical development and 3–6 years of clinical development and over 500 million dollar cost. There are three critical steps to develop a new drug including discovery and development, preclinical research, and clinical development [10].
Drug discovery involves screening hits, medicinal chemistry, and optimization of hits to reduce potential drug side effects. For drug discovery, two different complementary approaches can be applied: classical pharmacology, also known as phenotypic drug discovery, which is the historical basis of drug discovery, and reverse pharmacology, also known as designated target-based drug discovery. Screening methods based on phenotypic drug discovery have been used to discover new natural products mainly from the terrestrial origin [11]. These two strategies have advantages and disadvantages and promote very different screening assays. The frequent re-discovery of the same compounds, the technical difficulties associated with the isolation of compounds from extracts, and the incompatibility of natural product extracts with high-throughput screening (HTS) campaigns were the disadvantages of phenotypic drug discovery. On the other hand, natural product structures have the characteristics of high chemical diversity, biochemical specificity, and other molecular properties that make them favorable as lead structures for drug discovery, which serve to differentiate them from libraries of synthetic and combinatorial compounds [12]. Overly simplified assays, acting of drug on more than one target, the multifactorial nature of diseases, and challenges to identify a single molecular target are some limitations of target-based drug discovery. Therefore, a comprehensive screening strategy will incorporate both targeted and phenotypic assays, with one format designated as the primary screen and the other as a secondary or follow-up assay. During the spread of COVID-19 outbreak, great efforts have been made in therapeutic drug discovery against the virus. Because COVID-19 is a new, acute, severe infectious disease, the anti-SARS-CoV-2 drug development strategies are to screen existing drugs to identify potentially effective drugs, to expand indications, and to develop a vaccine [12]. The safety of conventional drugs has been mostly verified; if effective, they can be quickly applied in clinical practice (repurposing of existing drugs). The recent rises of several high transmissible strains sounded alarms for currently used vaccines and drugs. Therefore, developing broad-spectrum antiviral drugs not only to combat COVID-19 but also to provide protective arsenals against future viral outbreaks is a requirement. Scientists continue the development of broad-spectrum antiviral drugs from natural or chemical sources, which have the potential advantages of broad-spectrum therapeutic effect and insensitivity to viral evasion. Given the urgency of the SARS-CoV-2 outbreak, here we discuss the discovery and development of new therapeutics for SARS-CoV-2 infection based on the strategies from preclinical drug discovery.
2. Methods
We report this systematic review based on the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [13].
2.1. Data Sources and Searches
Studies published in PubMed, Scopus, Web of Science, EMBASE, Google Scholar, and DrugBank were searched from December 2019 to March 2021 using the following search terms: “Coronavirus,” “Covid-19,” “SARS-CoV-2,” “Drug design,” “Drug development,” “In silico,” “In vitro,” and “In vivo” alone or in combination without language restrictions. The keywords were selected using expert opinions, mesh, and related article titles.
All articles with full text or in the absence of full text with abstract are included in the screening of this study. Studies were excluded if studies were comments, editorial, letters, review, and preprints.
2.2. Data Extraction
Two researchers independently extracted data from included studies using a predefined data extraction form. All disagreement was discussed and solved after rechecking the source data with a third investigator. The data extracted, including the last name of the first author, type of the study (in silico, in vitro, in vivo), and name of the agent (chemical compound, drug, herb, etc.), studied the mechanism and efficacy of the agent according to study design that specified according to the following definition.
Computer-aided drug design can be divided into three different categories. All are based on ligands and receptors, which are briefly described [14] as follows:
Dock Receptor-Based Approach [15]. Once the three-dimensional structure of the ligand molecules and their receptor is known, the receptor-based method is a good candidate for identifying or optimizing drugs. Due to the presence of three-dimensional structures of compounds and receptors, the nature of the interaction between the ligand and the receptor and the type of structure that the ligand can have to interact with them in favorable conditions can be identified using this method. The compound is simulated on the active site of the dock (meaning anchoring) and on the interaction of the ligand with the receptor by molecular mechanics and molecular dynamics. In this method, due to the ligation of the ligand in the active position, the ligand changes in terms of conformity and changes its position in different conditions and shows interaction with the receptor in different types of situations. To determine the type of ligands that can be docked into the receptor site, the matching of the shape and the complementarity of the hydrophobic, hydrophilic, and charged parts must be considered. Various software packages such as AUTODOCK-Glide-LUDI and LigandFit are used to design the drug based on the structure of the receptor.
Ligand-Based Approach [16]. This method is used where the three-dimensional structures of the receptor are unknown and instead the structure of the ligands is known, which is one of the common methods. In this method, by indirectly studying compounds that react with biomolecules, they seek to design compounds that are pharmacologically active. In ligand-based drug design methods, in the absence of biomolecule structure, by studying specific ligands, it seeks to identify the structural and physicochemical properties of the compounds so that the desired compound can be designed based on data extracted from the study of previous compounds. This method is a kind of drug design based on pharmacophore (pharmacophore refers to the part of the drug to which the effect of the drug depends on that part of the molecule), and by studying the quantitative relationship between structure and their activity, drugs can be designed by this method. It can be said that it is a method for designing the pharmacophores of drugs.
Denovo Design-Based Approach [17]. This method is used when the structure of the ligand is unknown but the structure of the receptor is known. In this method, there is information about the structures of the receptor or quasi-receptors, but there is no structure of the main composition that can interact with the active site of the receptor. One of the functions of drug design based on this method is to suggest and present the main composition that is complementary to the active site. The basis of the method is that the database of existing 3D structures is used to find small molecules that can interact with the active site of the receptor in terms of size, geometry, and functional groups. Software packages such as GROW and LEGEND are used to design drugs by this method.
Drug design methods in the computer include quantitative structure-activity relationship (RASQ), docking, molecular dynamic simulation, and computational modeling. In these studies, the efficacy is evaluated based on the function of the drug or compound agent and the mechanism of action. Figure 1 shows the methods of computer-aided drug design (CADD).
Figure 1.
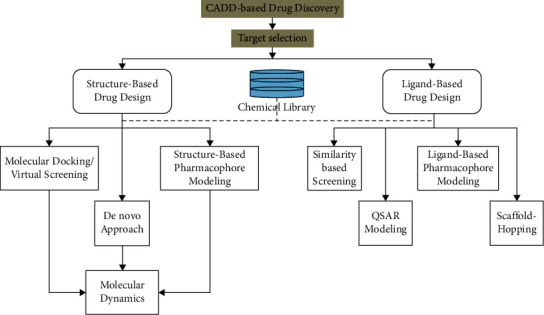
Computational methods in drug design (CADD: computer-aided drug design).
2.2.1. Computational Methods in Drug Design
Quantitative Structure-Activity Relationship (QSAR). QSAR provides studies on the relationship between chemical structure and biological activity or other biological activities that are important in selecting or removing a compound before synthesis and testing. QSAR [18] is especially important to predict the result, especially when it is not possible to experiment with a compound. Molecular descriptors, which are the most important components of QSAR, can be obtained experimentally or through mathematical formulas from various theories such as quantum mechanics, chemical graph theory, and study theories. QSAR seeks to establish a statistically significant relationship between structure and performance. It also explains the specific effect of a drug and can ultimately predict the effect of newly synthesized chemical compounds. QSAR model is also an equation that predicts a property through molecular descriptors and their coefficients. Evaluation of the effectiveness of new compounds that have been studied using this method can be reported as a percentage of enzyme inhibition if the modeling has been done and mentioned in the article.
2.2.2. Docking
In this technique, to achieve a combination with a pharmacological effect and increase the pharmacological activity of the drug, different formulations of a drug interact with the receptor, and the structure that has the best interaction with the receptor and the lowest energy level is selected for laboratory steps [19]. In this way, possible structures that have a stronger interaction with the receiver can be isolated at this stage. The same issue is considered and reported as an effectiveness measure.
2.2.3. Molecular Dynamic Simulation
In this technique, which is based on the simulation of drug-receptor interaction in the body, docking problems are solved and in fact play a complementary role in this technique [20]. Due to the time-consuming work with this technique, effective structures cannot be achieved directly through it, and the final stages of the study of drug-receptor interactions should be evaluated before starting laboratory work by having effective compounds from the previous stages. Molecular dynamic simulations produce information at the microscopic level (position and velocity of atoms). The conversion of these data into macroscopic values (pressure, energy, etc.) is done using statistical mechanics. In fact, molecular dynamics and statistical mechanics link microscopic concepts and macroscopically observable quantities. Molecular dynamic simulations are only able to predict the thermodynamic behavior and stability of the ligand binding mechanism at the active site of the target enzyme. This is reported as a criterion.
2.2.4. In Vitro
Study of drug in cell culture medium: effectiveness in these studies means inhibition of the replication of COVID-19 by the compound or drug under study [21].
The half-maximal inhibitory concentrations (IC50) are a measure of the effectiveness of a compound in inhibiting biological function [22].
In vivo studies are those in which the effects of drugs are tested on whole living organisms or cells usually animals as opposed to a tissue organism or dead organism. In vivo testing is better studied for observing the overall effects of an experiment on living subjects [23].
3. Results
In this review, we reported a significant number of articles with in silico, in vitro, and in vivo approaches for drug development of COVID-19. We retrieved a total of 2538 articles from the initial database search. After the removal of duplication and screening, 317 studies were selected for inclusion in this review. Figure 2 shows the PRISMA diagram.
Figure 2.
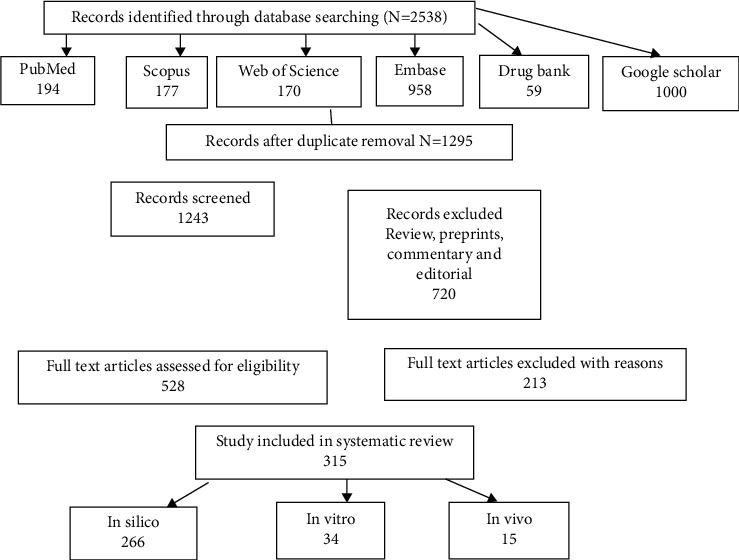
PRISMA diagram of the study.
The analysis of article contents indicated that 266 studies performed in silico approaches against viral targets; 34 studies used in vitro approaches against SARS-CoV-2; and 15 studies used in vivo (animal) models.
3.1. Results from In Silico Drug Discovery
From 267 studies used in silico approaches, 98 article studies repurposed approved drugs with a new mechanism of action and 91 studies evaluated natural products (e.g., herbal medicine) on COVID-19. The characteristics of these studies are summarized in Tables1 and 2. Also, Table 3 shows the characteristics of the remaining studies (N = 87).
Table 1.
Summary of studies with in silico method that used repurposed drugs for novel drug discovery against COVID-19.
| Author | Year | Method | Detail of method | Name of compound/drug | Target | Efficacy | Comments |
|---|---|---|---|---|---|---|---|
| Abosheasha and El-Gowily [24] | 2020 | In silico | Drug repurposing molecular docking-based virtual screening | 15 antiplatelet FDA-approved drugs | Main protease (Mpro) and spike glycoprotein (S) | Cilostazol has favorable binding interaction with Mpro (PDB ID: 6LU7) cilostazol, iloprost, epoprostenol, prasugrel, and icosapent ethyl that have a higher binding affinity on spike glycoprotein (S) | Cilostazol is a promising FDA drug against COVID-19 by inhibiting both Mpro and S protein |
| Abdul Kadhim et al. [25] | 2020 | In silico | Drug repurposing, docking | Experimental and approved drugs | Papain-like protease and RNA polymerase | Drugs that shared >70% similarity to the binding sites of those targets were reversin, pentagastrin, remdesivir, norfloxacin, and nitazoxanide against COVID-19 papain-like protease whereas benzylglutathione, lopinavir, and hydroxymethylglutathione against RNA polymerase | Antiresistance reversin showed the highest inhibitory efficacy against COVID-19 papain-like protease, and benzylglutathione is an experimental compound; however, it had the highest RNA polymerase inhibiting efficacy |
| Abu-Saleh et al. [26] | 2020 | In silico | Ligand-based/structure-based virtual screening, MD simulations, and binding energy calculations | Approved drugs and bioactive compounds listed in the DrugBank and ChEMBL databases | Main protease | Best MM/GBSA binding energy; ChEMBL275592, montelukast, ChEMBL28834. Bromocriptine and saquinavir demonstrate stability in the active site of Mpro | |
| Achilonu et al. [27] | 2020 | In silico | High-throughput virtual screening and ligand docking | FDA-approved drugs | Main protease | Isavuconazonium, a P2-P3 α-ketoamide derivative, and pentagastrina are the top three molecules (Lig13b as the benchmark) based on docking energy | |
| Aftab et al. [28] | 2020 | In silico | Repositioning/target-based virtual screening and molecular docking | Ten antiviral drugs were screened: ribavirin, remdesivir, sofosbuvir, penciclovir, nitazoxanide, nafamostat, chloroquine, galidesivir, favipiravir, and interferon | RNA-dependent RNA polymerase (RdRp) | Galidesivir and its drug-like compounds CID123624208 and CID11687749 have shown an effective attachment to the priming site of viral RdRp | CID123624208 and CID11687749 may be considered for in vitro and in vivo clinical trials |
| Ahmadi et al. [29] | 2021 | In silico | Drug repurposing study using molecular docking | Enfuvirtide, an HIV-1 fusion inhibitor peptide | SARS-CoV-2 fusion inhibitor | Enfuvirtide binding to the S2 protein of SARS-CoV-2 was remarkably stable and can act as a strong SARS-CoV-2 fusion inhibitor | |
| Ahmed et al. [30] | 2021 | In silico | Drug repurposing (high-throughput virtual screening) (HTVS) followed by re-docking with standard precision (SP) and extra-precision (XP) molecular docking | FDA-approved antiviral and anti-infection drugs | Main protease | Of 1397 potential drugs, 157 showed considerable affinity towards Mpro. High-affinity lead drugs (iodixanol, amikacin, troxerutin, and rutin) were identified. Amikacin was found to be the most potent inhibitor of main protease | Aminoglycosides may serve as a scaffold to design potent drug molecules against COVID-19 |
| Anand et al. [31] | 2021 | In silico | Molecular docking | 130 US FDA-approved drugs including hypertension, cardiovascular diseases, respiratory tract infections (RTI), antibiotics, and antiviral drugs | Structural and nonstructural proteins of SARS-CoV-2 (nsp3, nsp5, nsp10, nsp16) | 15 potent drugs exhibiting significant inhibitory potential against SARS-CoV-2 like baloxavir marboxil, danoprevir and sofosbuvir, fosinopril, moexipril, quinapril, telmisartan, azilsartan, verapamil, and doxazosin | Azithromycin, doxycycline, clarithromycin, rifamycin, and augmentin: nsp10; virginiamycin, tunicamycin, quinupristin, fidaxomicin, digoxin, and azithromycin: main protease; caspofungin, amphotericin B, ketoconazole, and micafungin: E and N proteins; virginiamycin and amphotericin B: S protein |
| Ancy et al. [32] | 2020 | In silico | Molecular docking, molecular dynamics, and binding free energy simulation study | HIV-1 protease, namely, TMB607 and TMC-310911 | Main protease | TMB607 molecule binds strongly with the SARS-CoV-2 main protease enzyme | |
| Ansari et al. [33] | 2020 | In silico | Repurposing drug molecular docking | TAT-peptide 47–57 (GRKKRRQRRRP)-conjugated repurposed drugs (i.e., lopinavir, ritonavir, favipiravir, and hydroxychloroquine) | Main protease | TP-conjugated ritonavir, lopinavir, favipiravir, and hydroxychloroquine have superior and significantly enhanced interactions with main protease | |
| Arun et al. [34] | 2020 | In silico | Repurposing drug molecular docking | Drugs available in the super DRUG2 database | Main protease | Binifibrate and bamifylline bind strongly to the enzyme active site | |
| Arya et al. [35] | 2020 | In silico | Molecular docking | FDA-approved drugs | Papain-like protease | 15 FDA-approved drugs, including chloroquine and formoterol, bind the target enzyme with significant affinity and good geometry, suggesting their potential to be utilized against the virus | |
| Baby et al. [36] | 2020 | In silico | Schrodinger's computer-aided drug discovery tools for in silico drug repurposing | FDA-approved library of drugs | RNA-dependent RNA polymerase (RdRp) | Pitavastatin, ridogrel, rosoxacin | |
| Baby et al. [36] | 2021 | In silico | Schrodinger's computer-aided drug discovery tools for in silico drug repurposing | FDA-approved library of drugs | Main protease | Tipiracil and aprepitant interacted with the main protease | |
| Baker et al. [37] | 2021 | In silico | Repurposing drug molecular docking | 50 compounds with activity against main protease | Main protease | Drugs including boceprevir, ciluprevir. narlaprevir, and telaprevir may be more potent against main protease than boceprevir and suitable for rapid repurposing | |
| Bharath et al. [38] | 2020 | In silico | Drug repurposing computer-aided drug design (CADD) | 4015 known and approved small molecules | Spike glycoprotein | Glycyrrhizic acid (GA) of plant origin may be repurposed for SARS-CoV-2 intervention | |
| Bhowmik et al. [39] | 2021 | In silico | Repurposing drugs, docking, and molecular dynamic simulation | Orientin (phytochemical) | Inhibitor of SARS-CoV-2 spike and host cell receptor GRP78 binding | Binding of orientin in the overlapping residues of GRP78 binding region of SARS-CoV-2 spike model | As a promising precautionary or therapeutic measure for COVID-19 |
| Bolelli et al. [40] | 2021 | In silico | Drug repurposing, virtual screening method | FDA-approved drugs | Main protease | Three compounds (dobutamine and its two derivatives) | |
| Cavasotto et al. [41] | 2021 | In silico | Drug repurposing, docking-based screening using a quantum mechanical scoring | FDA-approved drugs | Spike protein, main protease papain-like protease | Sovaprevir, elbasvir, danoprevir, samatasvir, Candesartan, saquinavir ritonavir, indinavir, lopinavir, brilacidin, flovagatran, aplidin, desmopressin, and felypressin listed as potential inhibitors of main protease | |
| Chandel et al. [42] | 2020 | In silico | Drug repurposing, molecular dynamics, and docking | FDA-approved drugs | Nsp9 replicase and spike proteins | Conivaptan exhibited the highest binding of the Nsp9 replicase. Tegobuvir exhibited maximum stability along with the highest binding energy at the active site of the spike proteins | |
| Chen et al. [43] | 2020 | In silico | Drug repurposing, molecular dynamics, and docking | FDA-approved drugs | Spike (S)-mediated cell entry | Cepharanthine, abemaciclib, osimertinib, trimipramine, colforsin, and ingenol | |
| Chidambaram et al. [44] | 2020 | In silico | Molecular docking | Coumarins and their analogs | Main protease | Natural coumarin analog toddacoumaquinone displayed remarkable inhibition ability. Synthetic coumarin analog (1 m) also displayed the comparable inhibition ability main protease in intricate with α-ketoamide | |
| Choudhary et al. [45] | 2020 | In silico | Drug repurposing, molecular dynamics, and docking | FDA-approved drugs | Spike glycoprotein and cellular angiotensin-converting enzyme 2 (ACE2) receptor | GR 127935 hydrochloride hydrate, GNF-5, RS504393, TNP, and eptifibatide acetate were found binding to virus binding motifs of ACE2 receptor. KT203, BMS195614, KT185, RS504393, and GSK1838705A were identified to bind at the receptor-binding site on the viral S protein | |
| Clemente et al. [46] | 2021 | In silico | Molecular docking, molecular dynamic (MD) simulations | Ibuprofen | Main protease | Racemic mixtures of the ibuprofen enantiomers might be a potential treatment for main protease | |
| Cosic et al. [47] | 2021 | In silico | Extended resonant recognition model (RRM) | Ivermectin | Spike proteins | Ivermectin could interfere with activity of spike proteins | |
| Daoud et al. [48] | 2021 | In silico | Structure-based pharmacophore approach, molecular docking, and repurposing studies | FDA-approved drugs | Main protease | Lopinavir, remdesivir, ritonavir, saquinavir, and raltegravir were successfully docked into the binding site of main protease | |
| de Oliveira et al. [49] | 2021 | In silico | Molecular modeling and virtual screening and repurposing studies | 9091 FDA-approved drugs | Spike protein | 24 best scored ligands (14 traditional herbal isolates and 10 approved drugs) as potential candidates to inhibit the S protein | Quinupristin, nilotinib, acetyldigitoxin |
| Delijewski and Haneczok [50] | 2021 | In silico | Supervised machine learning model and repurposing studies | FDA-approved drugs | Against SARS-CoV-2 | Zafirlukast as the best repurposing candidate for COVID-19 | |
| Dey et al. [51] | 2021 | In silico | Virtual database screening, molecular docking, all-atom molecular dynamic simulation, and MM-PBSA analysis | Tretinoin, mefenamic acid, ondansetron, and artemether | Envelope (E) protein | Tretinoin as a potential SARS-CoV-2 E protein ion channel blocker and virus assembly inhibitor | |
| Durdagi [52] | 2020 | In silico | Molecular modeling approach in virtual drug screening repurposing study | FDA-approved drugs | Type 2 transmembrane serine protease (TMPRSS2) | Benzquercin as strong TMPRSS2 inhibitor | |
| Durdagi et al. [53] | 2020 | In silico | Molecular docking, MM-GBSA-based predictions, and molecular dynamic repurposing study | FDA-approved drugs | Main protease and spike receptor-binding domain bound with ACE2 COVID-19 target proteins | Pimelautide, rotigaptide, telinavir, ritonavir, pinokalant, terlakiren, cefotiam, and cefpiramide as SARS-CoV-2 main protease inhibitors. Denopamine, bometolol, naminterol, rotigaptide, and benzquercin as potential ACE2/spike protein domain inhibitors | |
| Eleftheriou et al. [54] | 2020 | In silico | Molecular docking | 34 approved and on-trial protease inhibitors | Main protease | HCV protease, DPP-4, α-thrombin, and coagulation factor Xa known inhibitors | |
| Elmezayen et al. [55] | 2021 | In silico | Molecular modeling approach in virtual drug screening repurposing study | Commercially available drugs and ZINC15 library | Main proteases | Four potential inhibitors against Mpro enzyme, two available drugs (Talampicillin and Lurasidone), and two novel drug-like compounds (ZINC000000702323 and ZINC000012481889) | |
| Encinar et al. [56] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | 9000 US Food and Drug Administration (FDA)-approved investigational and experimental drugs from the DrugBank repository | S-Adenosyl-L-methionine-binding pocket of nsp16, [2] the unique “activating surface” between nsp16 and nsp10, and [3] the RNA-binding groove of nsp16 | Tegobuvir, sonidegib, siramesine, antrafenine, bemcentinib, itacitinib, or phthalocyanine antagonism of SARS-CoV-2 RNAs lacking 20-O-methylation | |
| Farag et al. [57] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | 2000 FDA-approved drugs | Main protease | Darunavir, nelfinavir, and saquinavir bound to the central site of main protease substrate-binding pocket rosuvastatin, montelukast, and the anti-histaminic fexofenadine bound to the terminal site of main protease substrate-binding pocket | Starting point for further in vitro and in vivo testing |
| Feng et al. [58] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | FDA-approved drugs | Spike protein | Eltrombopag possesses a high binding affinity to S protein plus human ACE2 | |
| Ferraz et al. [59] | 2020 | In silico | Ligand and structure-based virtual screening, repurposing study | FDA-approved drugs | Main protease | Two oral (bedaquiline and glibenclamide) and one buccal drug (miconazole) | |
| Fischer et al. [60] | 2020 | In silico | Molecular docking approach in virtual drug screening and repurposing study | Over 606 million compounds | Main protease | 12 purchasable compounds, with binding affinity to the target protease the natural compounds (−)—taxifolin and rhamnetin as potential inhibitors of main protease | |
| Gimeno et al. [61] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | FDA-approved drugs | Main protease | Perampanel, carprofen, celecoxib, alprazolam, trovafloxacin, sarafloxacin, and ethyl biscoumacetate. Carprofen and celecoxib | Initiative for in vitro testing |
| Guo et al. [62] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study single-cell RNA sequencing | US FDA-approved drugs | Against SARS-CoV-2 | 281 FDA-approved drugs that have the potential to be effective against SARS-CoV-2 infection, 16 of which are currently undergoing clinical trials to evaluate their efficacy against COVID-19 | Including the HIV protease inhibitor lopinavir/ritonavir combination (phase 4), glucocorticoid receptor agonist dexamethasone (phase 3/4), DNA replication inhibitor niclosamide (phase 2/3), antineoplastic agent lenalidomide (phase 4), and calcineurin inhibitor tacrolimus (phase 3), ABT-737 (BCL inhibitor), brefeldin-A (protein synthesis inhibitor), indirubin (CDK inhibitor), TPCA-1 (IKK inhibitor), lopinavir (HIV protease inhibitor), GW-441756 (growth factor receptor inhibitor), treprostinil (prostacyclin analog), tyrphostin-AG-1478 (EGFR inhibitor) and epoxycholesterol (LXR agonist), fostamatinib (SYK inhibitor), VER-155008 (HSP inhibitor), KU-0063794 (MTOR inhibitor), PIK-90 (PI3K inhibitor), linsitinib (IGF-1 inhibitor), TAK-715 (p38 MAPK inhibitor), Y-27632 (Rho-associated kinase inhibitor), AZ-628 (RAF inhibitor), and lestaurtinib (FLT3 inhibitor) |
| Gupta et al. [63] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | FDA-approved drugs | Main protease | Cobicistat is the most efficient inhibitor of Mpro both in silico and in vitro | |
| Huynh et al. [64] | 2021 | In silico | Docking and molecular dynamics and repurposing study | FDA-approved drugs | Papain-like protease | The chances of drug repurposing for PLpro might be low | |
| Ibrahim et al. [65] | 2020 | In silico | Molecular dynamic simulations, molecular docking, MM-GBSA analysis, and repurposing study | DrugBank database | Main protease | DB02388 and cobicistat (DB09065) | |
| Iftikhar et al. [66] | 2020 | In silico | Molecular modeling approach in virtual drug screening and repurposing study | 4574 compounds also containing FDA-approved drugs | RdRp, main protease, and helicase | Rimantadine, bagrosin, and grazoprevir showed binding to main protease. Casopitant is a neurokinin-1 receptor that showed binding to RdRp. Meclonazepam and oxiphenisatin showed specific interactions with helicase | |
| Jain and Mujwar [67] | 2020 | In silico | Computational drug repurposing docking simulations | 2880 FDA-approved drugs | Main protease | Metocurine, dihydroergotoxine, imatinib, daunorubicin, bromocriptine, irinotecan, azelastine, gestodene, adapalene, and simvastatin | Metocurine was chosen as a safe and effective drug candidate for developing therapy against the viral Mpro enzyme of SARS-CoV-2 for the treatment of COVID-19 |
| Jarvis et al. [68] | 2020 | In silico | Tier-based scoring system repurposing study | Clinically developed drugs | Potential repurposing against COVID-19 | Four drug classes (antimalarial amino-quinolones, selective estrogen receptor modulators (SERMs), low potency tricyclic antipsychotics, and tricyclic antidepressants) as potential drug candidates for COVID-19 | The tricyclic antipsychotics and tricyclic antidepressants were further excluded based on a high adverse event profile |
| Kadioglu et al. [69] | 2021 | In silico | Repositioning/virtual drug screening, molecular docking, and supervised machine learning algorithm drug repositioning | FDA-approved drug natural compound dataset ZINC database | Spike protein, nucleocapsid protein, and 2′-o-ribose methyltransferase | Conivaptan, paritaprevir, simeprevir, dihydroergotamine, ZINC000027215482, ZINC000252515584, loniflavone, procyanidin | |
| Kandeel et al. [70] | 2020 | In silico | Drug repurposing molecular dynamic (MD) simulations followed by molecular mechanics/generalized born surface area (MM/GBSA) binding energy calculations | 1697 clinical FDA-approved drugs | Papain-like protease | Phenformin, quercetin, and ritonavir | Phenformin was more stable than quercetin and ritonavir |
| Kandwal and Fayne [71] | 2020 | In silico | Repurposing drug computational design pharmacophore features | In-development/approved drugs | Viral nucleocapsid and nonstructural proteins | Isepamicin and streptomycin (nsp3); coenzyme-I, rutin, epigallocatechin gallate-(-), and procyanidin-b-2 (nsp7/nsp8/nsp12); paromomycin (nsp10/nsp16); olomoucine, sapropterin, tetrahydrofolic acid, INS316, and adenosine phosphate (nsp15); varespladib, hexanoic acid, citric acid, OSI-027, MK-5108, stepronin, calcium gluceptate, CPP, pirenoxine, midafotel, and maltobionic acid (nucleocapsid) | |
| Khan et al. [72] | 2020 | In silico | Drugs repurposing molecular dynamic simulation | 31 FDA-approved anti-HIV drugs, and traditional Chinese medicines (TCM) database | Main protease | Saquinavir and TCM5280805 | |
| Khan et al. [73] | 2020 | In silico | Drugs repurposing molecular docking | 23 prospective drug candidates | Main protease | Epirubicin, vapreotide, and saquinavir exhibited better binding affinity | Synergistic interaction |
| Kouznetsova et al. [74] | 2020 | In silico | Drugs repurposing molecular docking | FDA-approved drugs | Papain-like protease | Inhibitors of HIV, hepatitis C, and cytomegalovirus (CMV) demonstrated some activity | |
| Krishnaprasad et al. [75] | 2020 | In silico | Drugs repurposing molecular docking | FDA-approved library of drugs | RNA-dependent RNA polymerase | Pitavastatin, ridogrel, and rosoxacin displayed superior binding with the active site | |
| Kumar et al. [76] | 2020 | In silico | Drugs repurposing docking and molecular dynamic (MD) simulations combined with molecular mechanics/generalized born surface area (MM/GBSA) | 12 FDA-approved drugs (darunavir, indinavir, saquinavir, tipranavir, diosmin, hesperidin, rutin, raltegravir, velpatasvir, ledipasvir, rosuvastatin, and bortezomib) | Main protease | Saquinavir as a potent inhibitor of dimeric main protease | |
| Kumar et al. [77] | 2020 | In silico | Drugs repurposing molecular docking molecular dynamic simulations MM/GBSA | Withaferin A (Wi-A), withanone (Wi-N) (active withanolides of ashwagandha), and caffeic acid phenethyl ester (CAPE, bioactive ingredient of propolis) | Main protease | Wi-N and CAPE possess the potential to inhibit the functional activity of main protease | |
| Kumar et al. [78] | 2020 | In silico | Drugs repurposing molecular docking | FDA-approved drugs | Main protease | Lopinavir-ritonavir, tipranavir, and raltegravir show the best molecular interaction with the main protease | |
| Kumar et al. [79] | 2020 | In silico | Drugs repurposing molecular docking molecular dynamic simulations | FDA-approved library of drugs | Main protease | Hyaluronic acid and acarbose show strong interactions with catalytic site residues of main protease | |
| Li et al. [80] | 2020 | In silico | Drug repurposing free energy perturbation-based absolute binding free energy (FEP-ABFE) predictions | Virtual screening of existing drugs | Main protease | 25 drugs were predicted, and 15 were confirmed as potent inhibitors of SARS-CoV-2 main protease. The most potent one is dipyridamole. Hydroxychloroquine (ki = 0.36 μM) and chloroquine (ki = 0.56 μM) were also found to potently inhibit main protease | |
| Liang et al. [81] | 2021 | In silico | Drug repurposing molecular docking | 2,631 FDA-approved small molecules | Multiple main proteins | 29 drugs that could actively interact with two or more target proteins, with 5 drugs (avapritinib, bictegravir, ziprasidone, capmatinib, and pexidartinib) being common candidates for all four key host proteins and 3 of them possessing the desirable molecular properties | |
| Lokhande et al. [82] | 2021 | In silico | Drugs repurposing molecular docking molecular dynamic simulations | FDA-approved drugs | Main protease | Mitoxantrone, leucovorin, birinapant, and dynasore | |
| Mahanta et al. [83] | 2020 | In silico | Drugs repurposing molecular docking molecular dynamic simulations | U.S. Food and Drug Administration-approved antimicrobial drugs | Main protease | Viomycin | |
| Mahdian et al. [84] | 2021 | In silico | Drugs repurposing molecular docking molecular dynamic simulations | FDA-approved drugs | Viral entry receptors (ACE2 and CD147) and integral enzyme of the viral polymerase (RdRp) | Ledipasvir, estradiol benzoate, and vancomycin and paritaprevir | |
| Marak et al. [85] | 2020 | In silico | Repurposing drug homology modeling molecular docking | 108 FDA-approved antiparasitic and anti-inflammatory drugs | 10 SARS-CoV-2 targets (PLpro, 3CLpro, RdRp, spike, helicase, NSP1, NSP3, NSP4, NSP9, and NSP16-NSP10) | Ivermectin, atovaquone, posaconazole, doxycycline, moxidectin, amphotericin B, chlortetracycline, spiramycin, sulfasalazine, parecoxib, and etoricoxib exhibited good binding affinities | |
| Mohapatra et al. [86] | 2020 | In silico | Repurposing drug machine learning (ML) technology | FDA-approved drugs | Against COVID-19 | 10 FDA-approved commercial drugs that can be used for repurposing amprenavir would probably be the most effective drug based on the selected criteria | |
| Molavi et al. [87] | 2021 | In silico | Repurposing drug molecular docking | 1760 FDA-approved drugs | RNA-dependent RNA polymerase (RdRp) and main protease | Nilotinib, imatinib, and dihydroergotamine for 3clpro and dexasone and raltegravir for RdRp. Raltegravir, an anti-HIV drug, was observed to be the best compound against RdRp based on docking binding energy dihydroergotamine is a suitable candidate for main protease | |
| Mulgaonkar et al. [88] | 2020 | In silico/in vitro | Repurposing drug molecular docking | FDA-approved drugs | Spike glycoprotein | BCR-ABL tyrosine kinase inhibitor, imatinib, inhibits SARS-CoV-2 | Via fusion inhibition |
| Mycroft-West et al. [89] | 2020 | In silico | Repurposing drug molecular docking molecular dynamic simulations | Heparin | Spike (S1) protein receptor-binding domain | Inhibition of viral infection arises from an overlap between the binding sites of heparin/HS on S1-RBD | Repurposing heparin and its derivatives as antiviral agents against SARS-CoV-2 |
| Nayarisseri et al. [90] | 2020 | In silico | Shape-based machine learning assisted by molecular docking and molecular dynamic simulations. ADMET studies | 31 repurposed compounds | Main protease | Remdesivir, valrubicin, aprepitant, and fulvestrant | The novel compound nCorv-EMBS herein proposed stands as a promising inhibitor to be evaluated further for COVID-19 treatment |
| Odhar et al. [91] | 2020 | In silico | Molecular docking molecular dynamic simulations | 1615 FDA-approved drugs | Main protease | Conivaptan azelastine | |
| Ortega et al. [92] | 2020 | In silico | Repurposing drug molecular docking | Famotidine | Against SARS-CoV2 | Famotidine could interact within the catalytic site of the three proteases associated with SARS-CoV2 replication | Weak binding affinity could be reached only upon intravenous administration |
| Pandey et al. [93] | 2021 | In silico | Repurposing drug molecular docking | 9 flavonoids | Spike glycoprotein | Baicalin | |
| Parveen and Alnoman [94] | 2021 | In silico | Molecular docking molecular dynamic simulation density functional theory (DFT) ADME-Tox | FDA-approved anticancer drugs (capmatinib, pemigatinib, selpercatinib, and tucatinib) | Spike glycoprotein (S1) and the main protease | Potential of selected anticancer drugs for plausible drug development to fight COVID-19 | Capmatinib, pemigatinib, selpercatinib, and tucatinib |
| Peele et al. [95] | 2020 | In silico | Molecular docking molecular dynamic simulations | USFDA-approved drugs, plant-derived natural drugs | Main protease | Lopinavir, amodiaquine, theaflavin digallate | |
| Pinzi et al. [96] | 2021 | In silico | Drug repurposing molecular docking molecular mechanic Poisson–Boltzmann surface area (MM-PBSA) | DrugBank database | Main protease | 22 candidates with putative SARS-CoV-2 Mpro inhibitory activity. Enalkiren, ethylsulfonamide-D-Trp-Gln-p-aminobenzamidine, delparantag ritonavir and lopinavir, saquinavir | Beneficial polypharmacological effects |
| Pokhrel et al. [97] | 2020 | In silico | Drug repurposing molecular dynamic simulations | US Food and Drug Administration (FDA)-approved drugs | RNA-dependent RNA polymerase | Quinupristin is particularly interesting because it is expected to bind across the RNA tunnel, blocking access from both sides | Quinupristin represents a potential anti-SARS-CoV-2 therapeutic |
| Ray et al. [98] | 2020 | In silico | Drug repurposing intramolecularly quenched fluorescence (IQF) peptide substrate | 774 FDA-approved drugs | Main protease | Ethacrynic acid, naproxen, allopurinol, butenafine hydrochloride, raloxifene hydrochloride, tranylcypromine hydrochloride, saquinavir mesylate | |
| Sachdeva et al. [99] | 2020 | In silico | Drug repurposing molecular docking | Antimalarial drugs | Spike protein and main protease | Doxycycline showed the most effective binding to the spike protein, whereas halofantrine and mefloquine bound effectively with the main protease | Doxycycline could potentially be a good candidate for repurposing for COVID-19 |
| Sang et al. [100] | 2020 | In silico | Drug repurposing molecular docking molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) | 6 approved anti-HIV drugs | Main protease | Darunavir | |
| Saxena et al. [101] | 2021 | In silico/In vitro | Drug repurposing molecular docking | FDA-approved DrugBank database | Spike protein | Ertugliflozin possesses several desired properties | Good candidate for immediate repurposing for the treatment of COVID-19 |
| Setianingsih et al. [102] | 2020 | In silico | Drug repurposing molecular docking, molecular dynamic simulations | 160 potential drugs from therapeutic target database | 13 protein targets (12 SARS-CoV-2 proteins and 1 human protein) | Suramin, the strongest binding affinity against 3 protein targets (spike protein, nucleocapsid protein, ACE2) | Suramin is the most potential to bind nucleocapsid and spike protein of SARS-CoV-2 |
| Shah et al. [103] | 2020 | In silico | Drug repurposing molecular docking | 61 molecules that are already being used in clinics or under clinical scrutiny as antiviral agents | Against the SARS-CoV-2 | 37 molecules were found to interact with >2 protein structures of COVID-19. HIV protease inhibitors and RNA-dependent RNA polymerase inhibitors showed promising features of binding to COVID-19 enzyme | Methisazone, an inhibitor of protein synthesis; CGP42112A, an angiotensin AT2 receptor agonist; and ABT450, an inhibitor of the nonstructural protein 3-4A, might become convenient treatment option as well against COVID-19 |
| Sharma and Mishra [104] | 2020 | In silico | Drug repurposing target-based virtual ligand screening | ZINC drug database and our own database of natural products | Against the SARS-CoV-2 | Antivirus drugs (ribavirin, valganciclovir, and thymidine), antibacterial drugs (cefpiramide, sulfasalazine, phenethicillin, lymecycline, demeclocycline, doxycycline, oxytetracycline, and tigecycline), anti-asthmatic drugs (montelukast, fenoterol, and reproterol), and hepatoprotective drug silybin have antiviral activity. Natural hesperidin was targeting the binding between spike RBD and human ACE2 | The natural products, such as flavonoids like neohesperidin, hesperidin, baicalin, kaempferol 3-O-rutinoside, and rutin from different sources, andrographolide, neoandrographolide, and 14-deoxy-11,12-didehydroandrographolide from A. paniculata, and a series of xanthones from the plants of Swertia genus, with antivirus, antibacteria, and anti-inflammation activity could effectively interact with these targets of SARS-CoV-2 |
| Shekhar et al. [105] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulations | 2,625 FDA-approved small molecules | Spike (S) protein fusion peptide region | Chloramphenicol succinate, imipenem, and imidurea | |
| Singh et al. [106] | 2021 | In silico | Drug repurposing molecular docking molecular dynamic simulations | 1749 FDA-approved drugs | NSP12, a RNA polymerase | 5 compounds which include 3a (paritaprevir), 3d (glecaprevir), 3h (velpatasvir), 3j (remdesivir), and 3l (ribavirin) had the best binding affinity | |
| Sinha et al. [107] | 2021 | In silico | Drug repurposing systematic pharmacokinetics, drug-likeness, basicity predictions, virtual screening, and molecular dynamic analysis | Hydroxychloroquine (HCQ), chloroquine (CQ) | Spike protein | 1-[1-(6-Chloroquinolin-4-yl) piperidin-4-yl]piperidin-3-ol and (1r,2R)-2-N-(7-chloroquinolin-4-yl)cyclohexane-1,2-diamine interact with the active site of the spike protein similar to HCQ and CQ, respectively, with augmented safety profile | |
| Soni et al. [108] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | Rifampicin | Main protease | Rifampicin docking score was −7.24 kcal·mol–1, and it can predict as a very good inhibitor of main protease | |
| Tariq et al. [109] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulations | 15 antimalarial drugs (including chloroquine) and 2413 US Food and Drug Administration-approved drugs | Main protease spike (S) protein | Paromomycin with activity against two targets spike protein and protease domain | |
| Tatar et al. [110] | 2021 | In silico | Drug repurposing molecular docking molecular dynamic simulations | 34 antiviral compounds | RNA-binding domain | Rapamycin, saracatinib, camostat, trametinib, and nafamostat were the top hit compounds | |
| Tejera et al. [111] | 2020 | In silico | Drug repurposing quantitative structure-activity relationship (QSAR) mode molecular docking molecular dynamic simulation MM-PBSA method | DrugBank database | Main protease | Levothyroxine, amobarbital, and ABP-700 | |
| Teralı et al. [112] | 2020 | In silico | Drug repurposing molecular docking | 7,173 clinically approved drug | Angiotensin-converting enzyme 2 (ACE2) | Lividomycin, burixafor, quisinostat, fluprofylline, pemetrexed, spirofylline, edotecarin, diniprofylline | |
| Trezza et al. [113] | 2020 | In silico | Drug repurposing docking simulations, with molecular dynamics (MD), supervised MD (SuMD), and steered MD (SMD) simulations | FDA-approved drugs | Spike glycoprotein | Simeprevir, lumacaftor | |
| Ugurel et al. [114] | 2020 | In silico | Drug repurposing structure-based drug design genome sequences were analyzed | FDA-approved drugs | Helicase (Nsp13) | Cangrelor, fludarabine, folic acid, and polydatin inhibit both the wild-type and mutant SARS-CoV-2 helicase | |
| Unni et al. [115] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulations | DrugBank and PubChem library | Spike protein (S protein) | Bisoxatin (DB09219) | A laxative drug |
| Vaishali et al. [116] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulation ADME properties | FDA-approved compounds | Nonstructural protein 9 (Nsp9) replicase and spike proteins | Conivaptan exhibited the highest binding energy and maximum stability of the Nsp9 replicase. Tegobuvir exhibited maximum stability along with the highest binding energy at the active site of the spike proteins | |
| Verma et al. [117] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulation MM-GBSA-based energy | FDA-approved drugs | Main protease | Top-ranked drugs including adefovir, lumefantrine, dipyridamole, dihydroergotamine, hexoprenaline, riboflavin (vitamin B2), and pantethine (vitamin B5) | |
| Wei et al. [118] | 2020 | In silico | Drug repurposing molecular docking molecular dynamic simulations | US Food and Drug Administration (FDA)-approved drugs from DrugBank and natural compounds from traditional Chinese medicine systems pharmacology (TCMSP) | Spike protein (S protein) | Digitoxin and bisindigotin in TCMSP had the highest docking scores Forsythiae fructus and Isatidis radix are components of Lianhua Qingwen, and raltegravir had relatively high binding scores | |
| Xu et al. [119] | 2021 | In silico | Drug repurposing molecular docking molecular dynamic simulations | FDA-approved drugs | Spike protein | Thymoquinone, a phytochemical compound obtained from the plant Nigella sativa, is a potential drug candidate |
Table 2.
Summary of studies with in silico method that used natural products for novel drug discovery against COVID-19.
| Author | Year | Method | Detail of method | Name of compound/drug | Target | Efficacy | Comments |
|---|---|---|---|---|---|---|---|
| Abdelli et al. [120] | 2021 | In silico | Molecular docking | Isothymol, thymol, limonene, P-cymene, and γ-terpinene derived from the essential oil of the antiviral and antimicrobial plant Ammoides verticillata (Desf.) Briq. | Inhibition of ACE2 cellular receptor | Isothymol, a major component of this plant, gives the best docking scores, as good ACE2 inhibitor | |
| Abouelela et al. [121] | 2021 | In silico | Molecular docking, dynamic simulation, and binding free energy calculation | Aloe | Main protease and spike protein | 132, 134, and 159 were the best scoring compounds against main protease, while compounds 115, 120, and 131 were the best scoring ones against spike glycoprotein. Compounds 120 and 131 were able to achieve significant stability and binding free energies during molecular dynamic simulation | |
| Adem et al. [122] | 2020 | In silico | Molecular docking | Medicinal plant-based bioactive compounds (80 flavonoid compounds) | Main protease | Hesperidin, rutin, diosmin, apiin, diacetylcurcumin, (E)-1-(2-hydroxy-4-methoxyphenyl)-3-[3-[(E)-3-(2-hydroxy-4-methoxyphenyl)-3-oxoprop-1-enyl]phenyl]prop-2-en-1-one, and beta, beta′-(4-methoxy-1,3-phenylene)bis(2′-hydroxy-4′,6′-dimethoxyacrylophenone have been found as more effective on COVID-19 than nelfinavir | |
| Allam et al. [123] | 2020 | In silico | Molecular docking, 3D shape similarity study (rapid overlay chemical similarity-ROCS) to the clinically used drugs in COVID-19 patients | 3′-Hydroxy-4′-methoxy-chroman-7-O-β-d-glucopyranoside 4, ferulic acid heptyl ester 1, naringenin 2, and 4,2′,4′-trihydroxy-6′-methoxychalcone-4′-O-β-d-glucopyranoside 3, which were isolated from peach (Prunus persica (L.) Batsch) fruits | Main protease, spike protein | Naringenin 2 and 4,2′,4′-trihydroxy-6′-methoxychalcone-4′-O-β-d-glucopyranoside 3 have a strong binding mode to a protease receptor and spike protein and also block the inflammatory storm | Recommendation of peach fruits in controlling and managing COVID-19 cases |
| Al‐Sehemi et al. [124] | 2020 | In silico | Molecular docking | 31000 natural compounds of the natural product activity and species source (NPASS) library | Spike glycoprotein | Castanospermine and karuquinone B were shown to be the best-in-class derivatives in silico able to target an essential structure of the virus and to act in the early stage of infection | |
| Attia et al. [125] | 2021 | In silico | Molecular docking | 10 phenolic antiviral | Against SARS-CoV-2 | Hesperidin showed the highest docking score | Hesperidin and its mediated ZnO nanoparticles are willing antiviral agents |
| Azim et al. [126] | 2020 | In silico | Virtual screening methods molecular docking | 27 plant metabolites | Main protease proteins (MPP), Nsp9 RNA-binding protein, spike receptor-binding domain, spike ectodomain, and HR2 domain | Asiatic acid, avicularin, guajaverin, and withaferin showed a maximum binding affinity with all key proteins in terms of lowest global binding energy | |
| Bhowmik et al. [39] | 2021 | In silico | Repurposing drugs, docking, and molecular dynamic simulation | Orientin (phytochemical) | Inhibitor of SARS-CoV-2 spike and host cell receptor GRP78 binding | Binding of orientin in the overlapping residues of GRP78 binding region of SARS-CoV-2 spike model | As a promising precautionary or therapeutic measure for COVID-19 |
| Çakır et al. [127] | 2021 | In silico | Molecular docking | Peptides derived from beta-lactoglobulin | Inhibit the host cell membrane receptors | Ala-Leu-Pro-Met-His-Ile-Arg (ALMPHIR) and Ile-Pro-Ala-Val-Phe-Lys (IPAVFK) peptides | β-Lactoglobulin (BLG) is the major whey protein of cow and sheep's milk (∼3 g/l) |
| Chatterjee et al. [128] | 2021 | In silico | Molecular docking | Hesperidin, kaempferol, quercetin, epigallocatechin | PLpro (papain-like protease), RdRp (RNA-dependent RNA polymerase), Mpro or 3cl protease, and spike protein | Hesperidin, kaempferol, quercetin, epigallocatechin | Lead to conclusive data for the treatment of polyphenols, flavonoids, and bioflavonoids against SARS-CoV-2 |
| Chikhale et al. [129] | 2020 | In silico | Molecular docking, dynamics | Plant Withania somnifera (Indian ginseng) | NSP15 endoribonuclease and receptor-binding domain of prefusion spike protein | Withanoside X and quercetin glucoside from W. somnifera have favorable interactions at the binding site of selected proteins | Immunomodulatory, antioxidant, and anti-inflammatory roles |
| Chikhale et al. [130] | 2021 | In silico | Molecular docking, dynamics, and network pharmacology analysis | Saikosaponins | Adjuvant therapy in the treatment of COVID-19 | Saikosaponins interact with the proteins CAT gene CAT (catalase) and checkpoint kinase 1 (CHEK1) | Possible improvement in immune response towards COVID-19 |
| Chikhalet al. [131] | 2020 | In silico | Molecular docking, dynamics | Asparagus racemosus (Willd.) | NSP15 endoribonuclease and spike receptor-binding domain | Asparoside -C and Asparoside -F have good binding with target proteins | Asparagus racemosus holds promise as SARS-CoV-2 (S) and (N) protein inhibitor |
| Chowdhury [132] | 2020 | In silico | Molecular docking, dynamics | Tinospora cordifolia (Giloy) | Main protease | Berberine can regulate main protease protein's function | |
| Dahab et al. [133] | 2020 | In silico | Molecular docking | 10 phenolic compounds of different classes (phenolic acids, flavonoids, and coumarins) | Main protease and RNA polymerase | The top 7 hits are flacourticin [3], sagerinic acid [16], hordatine a [23], hordatine B [134], N-feruloyl tyramine dimer [135], bisavenanthramides B-5 [27], and vulnibactins [38] and have better binding scores than remdesivir, the native ligand in RNA polymerase target (PDB ID: 7bV2) | Hordatines are phenolic compounds present in barley and were found to exhibit the highest binding affinity to both protease and polymerase |
| Das et al. [136] | 2020 | In silico | Molecular docking | Flavonoid-based phytochemicals of Calendula officinalis | Main protease | Rutin, isorhamnetin-3-O-β-D, calendoflaside, narcissin, calendulaglycoside B, calenduloside, and calendoflavoside have better binding energy than the native ligand | Rutin and caledoflaside showed better stability, compactness, and flexibility |
| Debnath et al. [137] | 2020 | In silico | Sequential E-pharmacophore and structure-based virtual screening (VS) | 113687 number of commercially available natural compounds | ADP-ribose phosphatase | 6 potential inhibitors having good binding affinity towards active sites | Commercially available |
| Dev and Kaur [138] | 2020 | In silico | Molecular docking | Eucalyptus essential oil | Main protease | Jensenone may represent potential treatment potential to act as main protease inhibitor | |
| Duru et al. [139] | 2021 | In silico | Molecular docking | Oil of Nigella sativa seed | Replicase polyprotein 1a, RNA-binding protein of NSP9, ADP ribose phosphatase of NSP3, 3-chymotrypsin-like protease 3CLpro, and RNA-dependent RNA polymerase RdRp, and ACE2-angiotensin-converting enzyme from the Homo sapiens | The binding affinity of caryophyllene oxide was the highest on NSP9 and RdRp targets, while α-bergamotene gave the best binding affinity on RPIA target. The binding affinity of β-bisabolene on the ACE2 was almost the same as remdesivir | |
| El-Demerdash et al. [140] | 2021 | In silico | Molecular dynamic simulations, molecular docking | 15 guanidine alkaloids | Main protease (Mpro) (PDB ID: 6lu7), spike glycoprotein (PDB ID: 6VYB), nucleocapsid phosphoprotein (PDB ID: 6VYO), membrane glycoprotein (PDB ID: 6M17), and a nonstructural protein (nsp10) (PDB ID: 6W4H) | Crambescidin 786 [5] and crambescidin 826 had the highest binding affinities. The examined 15 alkaloids especially 5 and 13 showed promising docking, ADMET, toxicity, and MD results | |
| Elekofehinti et al. [141] | 2020 | In silico | Molecular docking studies, molecular dynamics, and ADME/Tox | 50,000 natural compounds retrieved from IBS database | Papain-like protease | STOCK1N-69160 [(S)-2-((R)-4-((R)-2-amino-3-methylbutanamido)-3-(4-chlorophenyl) butanamido)propanoic acid hydrochloride] has been proposed as a novel inhibitor against COVID-19 PLpro | |
| El‐Hawary et al. [142] | 2021 | In silico | Molecular docking (a combination of metabolomics and in silico approaches) | A. terreus, the endophytic fungus associated with soybean roots | Main protease | Aspergillide B1 and 3α-hydroxy-3,5-dihydromonacolin L were found to be potent anti-COVID-19 drug candidates | |
| Emirik [143] | 2020 | In silico | Molecular docking, MM-GBSA-based predictions, and molecular dynamics | Turmeric contents | SARS-CoV-2 vital proteins | Turmeric spice has the potential to inhibit the SARS-CoV-2 vital proteins and can be used a therapeutic or protective agent against SARS-CoV-2 via inhibiting key protein of the SARS-CoV-2. Compounds 4, 23, and 6 are the most prominent inhibitor for the main protease, the spike glycoprotein, and RNA polymerase of virus, respectively | |
| Fakhar et al. [144] | 2020 | In silico | Structure-based pharmacophore modeling, virtual screening-based PHASE screen score, molecular modeling | Anthocyanin derivatives | Main protease | 6 best anthocyanin-derived natural compounds, which could be used as promising lead compounds against main protease SARS-CoV-2 | |
| Falade et al. [145] | 2021 | In silico | Molecular docking | Saponins and tannins | Main protease | Ellagic acid, arjunic acid, theasapogenol B, and euscaphic acid as potential inhibitors of SARS-CoV-2 (Mpro) with better pharmacokinetics and bioavailability compared with remdesivir | |
| Fitriani et al. [146] | 2020 | In silico | Molecular docking | Phytochemical compounds (Moringa oleifera, Allium cepa, Cocos nucifera, Psidium guajava, and Eucalyptus globulus) | Main protease | Oleanolic acid in Allium cepa, α-tocotrienol in Cocos nucifera, asiatic acid in Psidium guajava, and culinoside in Eucalyptus globulus were the most recommended compound in each medicinal plant | Oleanolic acid in Allium cepa found as a potential inhibitor of COVID-19 Mpro |
| Gangadevi et al. [147] | 2021 | In silico | Molecular dynamic simulations, molecular docking | Library of natural compounds | Host ACE2 receptor with spike RBD domain of SARS-CoV-2 | Kobophenol A, identified through docking studies, is the first compound that inhibits SARS-CoV-2 binding to cells through blocking S1-RBD to the host ACE2 receptor and thus may serve as a good lead compound against COVID-19 | |
| Gangarapu et al. [148] | 2020 | In silico | Molecular docking online pkCSM and SwissADME Web server | Phytoconstituents of Siddha official formulation kabasura kudineer and novel herbal preparation—JACOM | Spike protein | 37 compounds were screened, and of these, 9 compounds showed high binding affinity against spike protein | SNACK-V formulations could be used for effective treatment of COVID-19 |
| Ghosh et al. [149] | 2020 | In silico | Molecular dynamic simulations, molecular docking | 8 polyphenols from green tea | Main protease | 3 polyphenols (epigallocatechin gallate, epicatechin gallate, and gallocatechin-3-gallate) interact strongly with one or both catalytic residues (His41 and Cys145) of main protease | |
| Ghosh et al. [150] | 2021 | In silico | Molecular dynamic simulations, molecular docking, MM-GBSA analysis | Justicia adhatoda alkaloids | Main protease | 1 alkaloid (anisotine) had interaction with both the catalytic residues (His41 and Cys145) of Mpro and exhibited good binding affinity (−7.9 kcal/mol) | More potent Mpro inhibitor than the two previously recommended antiviral drugs (lopinavir and darunavir) |
| Gorla et al. [151] | 2020 | In silico | Molecular docking | Essential flavonoids | SARS-CoV-2 spike glycoprotein receptor-binding domain (RBD-S) and host angiotensin-converting enzyme-2 protease domain (PD-ACE2) | Biochanin A and silymarin bind significantly at the active sites of RBD-Sand PD-ACE2 | |
| Gurung et al. [152] | 2020 | In silico | Virtual screening, molecular docking | Antiviral compounds from plants | Main protease | Bonducellpin D was identified as the best lead molecule, which shows higher binding affinity | |
| Gyebi et al. [153] | 2021 | In silico | Molecular docking, ADME/Tox, and Lipinski filter analysis | African plants derived alkaloids and terpenoids | Main protease | 4 nontoxic, druggable plant-derived alkaloids (10-hydroxyusambarensine and cryptoquindoline) and terpenoids (6-oxoisoiguesterin and 22-hydroxyhopan-3-one) | |
| Elwakil et al. [154] | 2021 | In silico | Gas chromatography/mass spectrometry analysis molecular docking | Egyptian propolis | RNA-dependent RNA polymerase, spike protein S1, and main protease | Octatriacontyl pentafluoropropionate is well oriented inside the enzyme pockets, in addition to an excellent binding manner with the active site of the target macromolecules | Menoufia propolis could be a promising candidate in the combat against the pandemic COVID-19 |
| Hasan et al. [155] | 2020 | In silico | Molecular docking | Compounds present in the plant Solanum surattense | Main protease | 13 phytochemicals were studied, eight showed very strong binding affinities to main protease, and four showed moderate to strong binding affinities | |
| Hashem [156] | 2020 | In silico | Molecular docking | Honeybee and propolis | Main protease | 6 main compounds possess high binding energy with the receptor active site of the main protease | |
| Ibrahim et al. [157] | 2020 | In silico | Molecular dynamic simulations, molecular docking, MM-GBSA analysis | MolPort database that contains over 100,000 natural products | Main protease | 9 potent natural products with binding affinities (ΔG binding) >−48.0 kcal/mol four bis([1, 3]dioxolo)pyran-5-carboxamide derivatives were identified as potential drug candidates | MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646 are bis([1,3]dioxolo)pyran-5-carboxamide derivatives |
| Ibrahim et al. [158] | 2020 | In silico | Molecular dynamic simulations, molecular docking | Metabolites present in several common spices | Main protease | High potency of salvianolic acid A and curcumin as main protease inhibitors | Salvianolic acid A as an in silico natural product inhibitor against the SARS-CoV-2 main protease |
| Isa et al. [159] | 2020 | In silico | Docking and molecular dynamic (MD) simulation | Extracts of Zingiber officinale and Anacardium occidentale | Main protease | Six compounds had good binding energies. CID_9910474 and CID_10503282 had a better stability when compared to other selected phytochemicals | |
| Istifli et al. [160] | 2020 | In silico | Molecular dynamics and molecular mechanic Poisson–Boltzmann surface area (MM/PBSA) methods | 23 phytochemicals belonging to different flavonoid subgroups | Spike glycoprotein cellular proteases (transmembrane serine protease 2 (TMPRSS2), cathepsin B and L (CatB/L)). | (−)-Epicatechin gallate interacted strongly with all the proteins studied | Epicatechin gallate can be evaluated as a candidate molecule in drug development studies against 2019-nCoV since it was not the substrate of P-gp (P-glycoprotein), did not inhibit any of the cytochrome Ps, and did not show AMES toxicity or hepatotoxicity on eukaryotic cells |
| Jan et al. [161] | 2021 | In silico | Cell-based infection assay molecular modeling | 2,855 small molecules and 190 traditional herbal medicines | Main protease RNA-dependent RNA polymerase | Mefloquine, nelfinavir, and extracts of Ganoderma lucidum (RF3), Perilla frutescens, Mentha haplocalyx | |
| Jo et al. [162] | 2020 | In silico | Docking | Flavonoids | Main protease | Baicalin, herbacetin, and pectolinarin have been discovered to block the proteolytic activity. Baicalin showed an effective inhibitory activity against main protease | |
| Joshi et al. [163] | 2021 | In silico | Docking | 7100 molecules | Main protease | Several natural molecules such as δ-viniferin, myricitrin, taiwanhomoflavone A, lactucopicrin 15-oxalate, nympholide A, afzelin, biorobin, hesperidin, and phyllaemblicin B strongly binds to main protease | These molecules also showed strong binding with other potential targets of SARS-CoV-2 infection such as viral receptor human angiotensin-converting enzyme 2 (hACE2) and RNA-dependent RNA polymerase (RdRp) |
| Junior et al. [164] | 2021 | In silico | Docking and molecular dynamic (MD) simulation | Lapachol(1,4-naphthoquinone) | SARS-CoV-2 nonstructural proteins (nsps) | Lapachol derivatives VI and IX demonstrated the strongest binding | Lapachol derivatives are potential ligands for SARS-CoV-2 Nsp9 |
| Kar et al. [165] | 2020 | In silico | Molecular docking molecular dynamic simulations and analysis of MM-PBSA energy | Indian plants including Justicia adhatoda, Ocimum sanctum, and Swertia chirata | Spike protein, main protease enzyme Mpro, and RNA-dependent RNA polymerase (RdRp) | Anisotine against SARS-CoV-2 spike and Mpro proteins and amarogentin against RdRp as potential inhibitors | |
| Khalifa et al. [166] | 2020 | In silico | Molecular docking modeling structural-relationship activity | 10 anthocyanins | Main protease | Phacelianin, gentiodelphin, cyanodelphin, tecophilin | Leading molecules for further optimization and drug development process to combat COVID-19 |
| Khalifa et al. [167] | 2020 | In silico | Molecular operating environment molecular docking | 19 structural different hydrolysable tannins | Main protease | Pedunculagin, tercatain, and castalin | |
| Khan et al. [168] | 2020 | In silico | Molecular docking | Marine natural products | Main protease | C-1 (PubChem CID 11170714) exhibited good activity | |
| Kiran Raj et al. [169] | 2020 | In silico | Molecular docking | C-Phycocyanin of Spirulina platensis | Nonstructural proteins 12 | C-Phycocyanin inhibits the active site of nsp12 | |
| Krupanidhi et al. [170] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME along with toxicity analysis | Phytochemical constituents of Tinospora cordifolia | Main protease | Tinosponone | |
| Kumar et al. [171] | 2020 | In silico | Molecular docking molecular dynamic simulations and analysis of MM-PBSA energy | Novel natural metabolites | Main protease | Ursolic acid, carvacrol, and oleanolic acid could | |
| Kumar et al. [172] | 2021 | In silico | Molecular docking, ADMET, and molecular dynamic simulations | Phytoconstituents from natural herbs | Main protease | Laurolitsine | Laurolitsine, an active constituent of roots of Lindera aggregata |
| Kumar et al. [173] | 2021 | In silico | Molecular docking molecular dynamic simulations and analysis of MM-PBSA energy | Strychnos nux-vomica | Main protease | Demethoxyguiaflavine, strychnoflavine | |
| Li et al. [174] | 2021 | In silico | Network pharmacology and in vitro experiment verification molecular docking | Maxing Shigan decoction (MXSGD) | ACE2, Mpro, and RdRp | The components with strong potential affinity (top 10) with ACE2, Mpro, and RdRp are mainly from Glycyrrhiza uralensis (Chinese name: Gancao) and Semen armeniacae amarum (Chinese name: Kuxingren). Among them, amygdalin was selected as the optimal candidate component binds to all three key targets, and euchrenone, glycyrrhizin, and glycyrol also exhibited superior affinity interactions with ACE2, Mpro, and RdRp, respectively | Multicomponent, multitarget, and multi-approach intervention |
| Maiti and Banerjee [175] | 2021 | In silico | Bioinformatic molecular docking | Tea flavonoid catechin products | Spike glycoproteins | Epigallocatechin gallate and theaflavin gallate interact better than hydroxychloroquine | |
| Mahmud et al. [176] | 2021 | In silico | Molecular docking molecular dynamic simulation MM-GBSA scores | 3063 compounds from more than 200 plants from the Asian region | Main protease | Curcumin, gartanin, robinetin | |
| Mahmud et al. [177] | 2020 | In silico | Molecular docking molecular dynamic simulation MM-GBSA scores | Plant-derived natural compounds | Main protease | Carinol, albanin, myricetin | |
| Mesli et al. [178] | 2021 | In silico | Molecular docking molecular dynamic simulations | Leaves of Corchorus olitorius Linn. | Angiotensin-converting enzyme 2 | Méthyl-1,4,5-tri-O-caféoyl quinate has a stronger bond, high affinity, and gives the best docking scores compared with the co-crystallized inhibitor (PRD_002214) of the enzyme ACE2 | |
| Mohammadi et al. [179] | 2020 | In silico | Molecular docking | Marine polypeptides were isolated from the hydrolysate of Pacific oyster | Main protease | The score of Leu-Leu-Glu-Tyr-Ser-Ileu ligand was higher than remdesivir | Pacific oyster compounds may have the potency to be evolved as an anti-COVID-19 main protease |
| Murugan et al. [180] | 2020 | In silico | Molecular docking molecular dynamic simulation MM-GBSA scores | Andrographis paniculata phytochemicals | 3 nonstructural proteins (3 L main protease (3CLpro), papain-like proteinase (PLpro) and RNA-directed RNA polymerase (RdRp)), and a structural protein (spike protein (S)) | Neoandrographolide (AGP3) has shown promising binding affinity towards all the four targets | |
| Naik et al. [181] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | Natural product activity and species source (NPASS) database | Endoribonuclease exoribonuclease RNA-dependent RNA polymerase (RdRp) methyltransferase and main protease | 21 compounds showed maximum docking scores NPC214620, NPC52382, and NPC270578 are targeting five, four, and three-drug targets, respectively | Multitarget-based drug design |
| Narkhede et al. [182] | 2020 | In silico | Molecular docking molecular dynamic simulations | Natural products | Main protease | Glycyrrhizin, bicyclogermacrene, tryptanthrin, β-sitosterol, indirubin, indican, indigo, hesperetin, chrysophanic acid, rhein, berberine, and β-caryophyllene | Interactions with the COVID-19 main protease were highest in the case of glycyrrhizin and rhein |
| Nivetha et al. [183] | 2020 | In silico | Molecular docking molecular dynamic simulation MM-PBSA | Seselin purified from the leaf extracts of Aegle marmelos | Spike protein S2, main protease, and free enzyme of the SARS-CoV-2 | Seselin had inhibitory potential over multiple SARS-CoV-2 targets | |
| Ogunyemi et al. [184] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | 226 bioactive compounds from African herbs and medicinal plants | RNA-dependent RNA polymerase | Drugable alkaloids (10′-hydroxyusambarensine, cryptospirolepine, strychnopentamine) and flavonoids (usararotenoid A and 12α-epi-millettosin) | |
| Padhi et al. [185] | 2021 | In silico | Molecular docking ADME properties | 415 natural metabolites isolated from several plants, fungi, and bacteria | Spike glycoprotein (S1) and the main protease | Fusaric acid, jasmonic acid, jasmonic acid methyl ester, putaminoxin, putaminoxins B and D, and stagonolide K were predicted to have considerable (ADME) and safety indices | Jasmonic acid and putaminoxins B and D bind best to main protease |
| Pandey et al. [186] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | 10 potential naturally occurring compounds (flavonoids/non-flavonoids) | Spike glycoprotein | Fisetin, quercetin, and kaempferol consist of drug-likeness property | |
| Kumar et al. [187] | 2020 | In silico | Molecular docking | Kabasura kudineer and thonthasura kudineer are two Siddha formulations | Spike glycoprotein | Cucurbitacin B (−112.09), cardiofolioside (−111.5), apigenin (−98.84), and pyrethrin (−92.98) were observed as more effective with less bind energies | Kabasura kudineer could be a potential Siddha medicine for COVID-19 |
| Rahman et al. [188] | 2021 | In silico | Molecular docking ADMET properties | Rutin | Main protease (Mpro), RNA-dependent RNA polymerase (RdRp), papain-like protease (PLpro), and spike (S) protein | Significant binding of rutin with Mpro, RdRp, PLpro, and S proteins. Main protease exhibited the strongest binding affinity | Optimal solubility, nontoxic, and noncarcinogenic properties |
| Rahman et al. [189] | 2020 | In silico | Molecular operating environment (MOE) ligand-based pharmacophore approach and a molecular docking-based screening | Natural product activity and species source (NPASS) | Type II transmembrane serine protease (TMPRSS2) | 85 compounds with molecular docking comparable to or greater than that of the standard inhibitor (camostat mesylate) were identified. The top 12 compounds with the most favorable structural features were studied. The low-molecular-weight compound NPC306344 showed significant interaction with the active site residues of TMPRSS2 | |
| Rakib et al. [190] | 2020 | In silico | Molecular docking | Bioactive phytocompounds isolated from Tinospora crispa | Main protease | The top nine hits might serve as potential anti-SARS-CoV-2 lead molecules, with three of them exerting biological activity | |
| Ramadhan et al. [191] | 2020 | In silico | Molecular docking | Etlingera elatior plant | Main protease | Ergosterol peroxide sitostenone | |
| Rangsinth et al. [192] | 2021 | In silico | Molecular docking ADMET properties | Natural products isolated from edible and medicinal mushrooms | Main protease | 6 of 25 compounds are the best drug-like property candidates, including colossolactone VIII, colossolactone E, colossolactone G, ergosterol, heliantriol F, and velutin | |
| Rivero-Segura et al. [193] | 2021 | In silico | Molecular docking molecular dynamic simulation ADME properties | Mexican natural products | Against the SARS-CoV-2 | Quercetin, riolozatrione, and cichoriin target the key proteins of SARS-CoV-2 | Cichoriin reaches higher lung levels (100 mg/kg, IV); therefore, it may be considered in developing therapeutic tools |
| Selvaraj et al. [194] | 2020 | In silico | Homology modeling and molecular dynamic (MD) simulation MM/GBSA, MD simulations, and PCA calculations | Traditional Chinese medicine (TCM) database | Nsp 14 guanine-N7 methyltransferase (N7-MTase) | TCM 57025, TCM 3495, TCM 5376, TCM 20111, and TCM 31007 are the compounds from the TCM database, which can occupy and interact nicely with the substrate-binding site of N7-MTase | |
| Sharma and Kaur [195] | 2021 | In silico | Molecular docking, protein interaction calculator ADME studies | 12 bioactive molecules present in essential oils of eucalyptus plant leaves | Spike (S) protein | Toruatone | |
| Sharma [196] | 2020 | In silico | Molecular docking, protein interaction calculator | Eucalyptol (1,8 cineole), an essential oil component from eucalyptus oil | Main protease | Eucalyptol may represent potential treatment potential to act as main protease inhibitor | Effective binding of eucalyptol to COVID-19 proteinase |
| Sharma and Kaur [197] | 2020 | In silico | Molecular docking, protein interaction calculator | Jensenone, an essential oil component from eucalyptus oil | Main protease | Jensenone may represent potential treatment potential to act as main protease inhibitor | |
| Shawan et al. [198] | 2021 | In silico | Pharmacophore study molecular docking molecular dynamic simulation ADME properties | 43 flavonoids of 7 different classes | Against the SARS-CoV-2 | Luteolin and abyssinone II were found to develop successfully docked complex within the binding sites of target proteins | |
| Sindhu et al. [199] | 2020 | In silico | Molecular docking | Clerodendrum paniculatum leaves | Main protease | Clerodol | |
| Singh et al. [200] | 2021 | In silico | Molecular docking and structural dynamic studies | Tea (Camellia sinensis) polyphenols | Nonstructural protein 16 (NSP16) | Theaflavin compound demonstrated lower binding free energy in comparison with the standard molecule sinefungin | |
| Singh et al. [201] | 2020 | In silico | Molecular docking and structural dynamic studies molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) ADME properties | Polyphenols | RNA‐dependent RNA polymerase (RdRp) | EGCG, theaflavin (TF1), theaflavin-3′-O-gallate (TF2a), theaflavin-3′-gallate (TF2b), theaflavin 3,3′-digallate (TF3), hesperidin, quercetagetin, and myricetin strongly bind to the active site of RdRp | EGCG, TF2a, TF2b, and TF3 can inhibit RdRp and represent an effective therapy for COVID-19 |
| Srimathi et al. [202] | 2020 | In silico | Molecular docking | Traditional herbal medicine: apo-quinine, catechin, cinchonidine, cinchonine, cupreidine, epicatechin, epiprocurcumenol, epiquinine, procurcumenol, quinidine, quinine, zedoaronediol, procurcumadiol | Against the SARS-CoV-2 | Epicatechin, apo-quine | |
| Subbaiyan et al. [203] | 2020 | In silico | Molecular docking | Active constituents present in common herbs | Spike (S) protein | Epigallocatechin gallate (EGCG) was found to have the highest binding affinity with the viral S protein, followed by compounds, “F” (curcumin), “D” (apigenin), and “E” (chrysophanol) | Green tea |
| Surti et al. [204] | 2020 | In silico | Molecular docking molecular dynamic simulations | Ilimaquinone (marine sponge metabolite) | Spike receptor-binding domain, RNA-dependent RNA polymerase, Nsp10, Nsp13, Nsp14, Nsp15, Nsp16, main protease, and papain-like protease | Ilimaquinone exhibited promising inhibitory potential against all the SARS-CoV-2 target proteins, as evident from the binding energies | Most promising inhibitory candidate against the SARS-CoV-2 papain-like protease |
| Tao et al. [205] | 2020 | In silico | Network pharmacology and molecular docking. | Huashi Baidu formula (HSBDF): Chinese | Against the SARS-CoV-2 | Baicalein and quercetin were the top two compounds of HSBDF, which had high affinity with ACE2 | Regulate multiple signaling pathways through ACE2 |
| Umar et al. [206] | 2021 | In silico | Molecular docking molecular dynamic simulation ADME properties | Azadirachta indica, Mangifera indica, and Moringa oleifera: African plants | Main protease | Most of the active phytocomponents of the study plants exhibited relative inhibitory potentials against main protease and preferred pharmacological features when compared with hydroxychloroquine | Caffeic acid, chlorogenic acid, catechin, ellagic acid, gallic acid, etc. |
| Umesh et al. [207] | 2021 | In silico | Molecular docking molecular dynamic simulation ADME properties | Chemical compounds from Indian spices | Main protease | Carnosol exhibited the highest binding affinity for arjunglucoside-I and rosmanol showed a strong and stable binding affinity with favorable ADME properties | |
| Yang et al. [208] | In silico | 2020 | High-throughput virtual screening | Natural Products Research Laboratories (NPRL) | Main protease | Curcuminoid derivatives (including NPRL334, NPRL339, NPRL342, NPRL346, NPRL407, NPRL415, NPRL420, NPRL472, and NPRL473) display strong binding affinity to COVID-19 3Lpro polyprotein | NPRL-334 revealed the strongest binding affinity |
| Yu et al. [209] | 2020 | In silico | Metascape analysis protein docking molecular docking | Mongolian medicine | SARS-CoV-2 S protein RBD domain | 253 active components were predicted. Phillyrin and chlorogenic acid can effectively prevent the combination of SARS-CoV-2 S protein and ACE2 at the molecular level | |
| Zhang et al. [210] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME property network pharmacology analysis | Chinese herbal medicines | Anti-2019-nCoV activity | 13 compounds that exist in traditional Chinese medicines were found to have potential anti-2019-nCoV activity. 125 Chinese herbs were found to contain 2 or more of these 13 compounds. Of these 125 herbs, 26 are classically cataloged as treating viral respiratory infections | Regulating viral infection, immune/inflammation reactions, and hypoxia response |
Table 3.
Summary of other studies with in silico method.
| Author | Year | Method | Detail of method | Name of compound/drug | Target | Efficacy | Comments |
|---|---|---|---|---|---|---|---|
| Abu-Melha et al. [211] | 2020 | In silico | Molecular docking combined with molecular dynamic simulation (MDS) | Hydrazones, pyrazoles, and pyrazines bearing thiazole moiety | Main protease | The average binding affinities of the compounds 3a, 3b, and 3c (−8.1 ± 0.33 kcal/mol, −8.0 ± 0.35 kcal/mol, and −8.2 ± 0.21 kcal/mol, respectively) are better than that of the positive control nelfinavir | |
| Aghaee et al. [212] | 2021 | In silico | Pharmacophore model molecular docking combined with molecular dynamic simulation (MDS), MM/PBSA, ADME studies | Pharmit website | Main protease | ML188, nelfinavir, lopinavir, ritonavir, and α-ketoamide | |
| Ahmad et al. [213] | 2021 | In silico | Structure-based virtual screening (SBVS) of ASINEX antiviral library, molecular dynamic (MD) simulations | ASINEX antiviral library | Main protease | SCHEMBL12616233, SCHEMBL18616095, and SCHEMBL20148701 compounds conformation with main protease show good stability after initial within active cavity moves, a rich intermolecular network of chemical interactions, and reliable relative and absolute binding free energies | BBB_26580140 lead and its similar analogs to be explored in vivo lead molecules |
| Ahmed et al. [214] | 2020 | In silico | Molecular docking, molecular dynamics, and structure-activity relationship | 76 prescription antiviral drugs | RNA-dependent RNA polymerase (RdRp) and main protease (Mpro) | Raltegravir, simeprevir, cobicistat, and daclatasvir have higher binding energy and strong interaction with active sites of the receptor proteins (with a precision of 85%) | |
| Alabboud and Javadmanesh [215] | 2020 | In silico | Molecular docking combined with molecular dynamic simulation | 88 conventional drugs, 16 vitamins, and 63 natural (plant) | Main protease | Various vitamins (B9, A, K, and E vitamins) exhibited a significantly strong interaction with the studied receptor. Pleconaril, adefovir dipivoxil, and stavudine in addition to plant-based compounds such as curcumin (Curcuma longa), anolignan A (Anogeissus acuminata), and phyllamyricin B (Phyllanthus myrtifolius) had strong ligand-protein interactions | |
| Alamri et al. [216] | 2020 | In silico | Structure-based virtual screening coupled with all-atom molecular dynamic (MD) simulations | Protease inhibitors database composed of ∼7,000 compounds | Papain-like protease | ADM_13083841, LMG_15521745, and SYN_15517940 showed stable conformation and interacted well with the active residues of papain-like protease | |
| Alexpandi et al. [217] | 2020 | In silico | Molecular docking | 113 quinoline drugs | Main protease, RNA-dependent RNA polymerase (RdRp) inhibitors spike-RBD-ACE2 inhibitor | Elvitegravir and oxolinic acid are able to interact with the NTP entry channel and thus interfere with the RNA-directed 5′–3′ polymerase activity of SARS-CoV-2 RdRp. Rilapladib is the only quinoline that can interrupt the spike-RBD-ACE2 complex | Quinoline, 1,2,3,4-tetrahydro-1-[(2-phenylcyclopropyl)sulfonyl]-trans-(8CI), saquinavir, elvitegravir, oxolinic acid, and rilapladib are suggested for the treatment of COVID-19 |
| Al‐Sehemi et al. [218] | 2020 | In silico | Molecular docking, MD simulation, and molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) results | Phenyl furoxan, an exogenous nitric oxide donor | Main protease | Spiro-isoquinolino-piperidine-furoxan moieties can be used as an effective ligand for main protease inhibition due to the presence of key isoquinolino-piperidine skeleton with additional NO effect | |
| Al-Shar'i [219] | 2020 | In silico | Molecular docking, MD simulation, and molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) results | Different databases | Main protease | 9 compounds with different chemotypes | |
| Badavath et al. [220] | 2020 | In silico/in vitro | Molecular docking, molecular dynamics, and structure-activity relationship studies | Screening of 118 compounds with 16 distinct heterocyclic moieties in comparison with 5 natural products and 7 repurposed drugs | Main protease | Oxidiazoles (A2 and A4) derivatives have the best docking scores. Structure-activity relationship studies showed a good comparison with a known active main protease and repurposed drug ebselen with an IC50 value of −0.67 μM | |
| Barros et al. [221] | 2020 | In silico | Molecular docking | 24 ligands | SARS-CoV-2 receptors, Nsp9 replicase, main protease (Mpro), NSP15 endoribonuclease, and spike protein (S protein) interacting with human ACE2 | Antimalarial drug metaquine and anti-HIV antiretroviral saquinavir interacted with all the studied receptors | |
| Basit et al. [222] | 2020 | In silico | Protein-protein docking and molecular dynamic simulation | Truncated version of human ACE2 (tACE2) | S Glycoprotein | tACE2 provides a high-affinity protein inhibitor for S glycoprotein | |
| Battisti et al. [223] | 2020 | In silico | Pharmacophore‐based screening, docking consensus approach (DCA), molecular dynamic simulations, common hit approach (CHA) | Aldrich Market Select (AMS) database from ChemNavigator/Sigma‐Aldrich with over 8 million unique chemical structures | Against SARS-CoV-2 | 10 compounds with high coronavirus inhibition potential | Lead molecules |
| Benítez-Cardoza and Vique-Sánchez [224] | 2020 | In silico | Molecular docking | 500,000 compounds | Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD) | 20 compounds were determined by docking focused on the region of interaction between ACE2 and RBD | |
| Cava et al. [225] | 2020 | In silico | Gene Ontology and enrichment analysis protein-protein interaction (PPI) network virtual screening method | — | Against SARS-CoV-2 | A protein-protein interaction network of 193 genes, 22 interactions, and 36 potential drugs for future treatment strategies including nimesulide, fluticasone propionate, thiabendazole, photofrin, and didanosine | Only didanosine is a real antiviral drug, while the others are mostly anti-inflammatory |
| Chen et al. [226] | 2020 | In silico | Crystal structure, virtual screening | 7173 purchasable drugs (drugs-lib), with 4574 unique compounds and their stereoisomers | Main protease | 16 candidates for consideration, ledipasvir velpatasvir | |
| Choudhary et al. [227] | 2020 | In silico | Molecular docking, MM-GBSA predictive binding energy calculations, and molecular dynamic simulation | 15,754 natural and synthetic compounds | Main protease | Compound 2 (molecular bank code AAA396) and compound 3 (molecular bank code AAD146) | |
| Chunduru et al. [228] | 2021 | In silico | Molecular docking | Novel drug-like inhibitors for COVID-19 | Main protease | Structure 61 was found to be more stable and can be further assessed for their antiviral activity to combat COVID-19 | |
| Coelho et al. [229] | 2020 | In silico | Biochemical high-throughput screening | Compound library containing known drugs, bioactive molecules, and natural products | Main protease | Organomercuric compounds thimerosal and phenylmercuric acetate, benzophenone, Evans blue, a sulfonic acid-containing dye | |
| Dai et al. [230] | 2020 | In silico | Structure-based design | — | Main protease | Designed and synthesized two lead compounds (11a and 11b) targeting main protease. Both exhibited excellent inhibitory activity | |
| de lima Menezes and da Silva [231] | 2020 | In silico | Molecular dynamic simulations, molecular docking | DrugBank database | Nonstructural protein 1 (nsp1) | Tirilazad, phthalocyanine, and Zk-806450 showed better energy score than control molecules that have in vitro activity against nsp1 from SARS-CoV-2 | Tirilazad, phthalocyanine, and Zk-806450 |
| Debnath et al. [232] | 2020 | In silico | Pharmacophores studies, structure-based virtual screening, molecular dynamic (MD) simulation | Drug molecule information retrieved from DrugBank | Main protease | DB07456 and DB13592 displayed a similar type of binding interaction with co-ligands and remdesivir, and the predicted Ki values of 2 inhibitors were found to be superior to remdesivir | |
| Di Micco et al. [233] | 2021 | In silico | Molecular docking, MM-GBSA-based predictions, and molecular dynamics | Zonulin inhibitor larazotide acetate (also called AT1001) | Main protease | AT1001, besides its well-demonstrated effect in ameliorating mucosal permeability in ALI/ARDS | |
| El Hassab et al. [234] | 2021 | In silico | Structure-based virtual screening, molecular dynamic simulation, and MM-PBSA approaches | 48 million drug-like compounds of the ZINC database | SARS-CoV-2 2′-O-methyltransferase (nsp16) | Compound 11 as the best potential nsp16 inhibitor herein identified, as it displayed a better stability and average binding free energy for the ligand-enzyme complex compared to sinefungin | |
| Elmessaoudi-Idrissi et al. [235] | 2020 | In silico | In silico screening, molecular docking, and dynamic approaches | 5000 compounds of the ZINC database | Main protease | The prominent drug-like and potent inhibitory compounds are 2-[2-(2-aminoacetyl) aminoacetyl] amino-3-(4-hydroxyphenyl)-propanamide (ZINC000004762511), 6′-fluoroaristeromycin (ZINC000001483267), and cyclo(L-histidyl-L-histidyl) (ZINC000005116916) scaffolds | |
| Feitosa et al. [236] | 2020 | In silico | Molecular docking | — | Main protease | Melatonin can have response potential in early stages for its possible effects on ACE2 and main protease, although it is also promising in more severe stages of the disease for its action against hyper-inflammation | Do not confirm antiviral activity, but can rather be used as a basis for further preclinical and clinical trials |
| Gurung et al. [237] | 2021 | In silico | Virtual screening, molecular docking | 1,36,191 molecules | Spike (S) protein receptor-binding domain (RBD) to the host cell surface receptor, angiotensin-converting enzyme 2 (ACE2) | ZINC33039472 exhibited binding free energy value lower as compared to the control (emodin) with a higher contribution by gas-phase energy and van der Waals energy to the total binding free energy | |
| Haider et al. [238] | 2020 | In silico | Computer-aided drug design (CADD) molecular docking | ZINCPharmer | Main protease | ∼200 compounds were identified as having strong interaction with Mpro (ZINC20291569, ZINC90403206, ZINC95480156) that showed the highest binding energy | |
| Hall and Ji [239] | 2020 | In silico | Homology modeling molecular docking | ZINC15 database: 3447 entries | Spike glycoprotein and main protease | Zanamivir, indinavir, saquinavir, and remdesivir are among the exciting hits of main proteinase | Flavin adenine dinucleotide (FAD) adeflavin, B2 deficiency medicine, and coenzyme A, a coenzyme may also be potentially used for the treatment of SARS-CoV-2 infections |
| Havranek and Islam [240] | 2020 | In silico | Docking and molecular dynamics | 2692 protease inhibitor compounds | Main protease | Phenyltriazolinones (PubChem ID: 104161460) and allosteric HCV NS5B polymerase thumb pocket 2 (PubChem ID: 163632044) have shown antiviral activity and also have high affinity towards the main protease | |
| Ibrahim et al. [241] | 2021 | In silico | Molecular dynamic simulations, molecular docking, MM-GBSA | 18 anti-COVID-19 drug candidates against SARS-CoV-2 main protease | Main protease | Promising binding affinities of TMC-310911 and ritonavir | |
| Jaiswal and Kumar [242] | 2020 | In silico | Docking studies and molecular dynamic simulation | Designed a protein inhibitor | Spike (S) glycoprotein | The proposed inhibitor ΔABP-D25Y | |
| Jamalan et al. [243] | 2021 | In silico | Docking and molecular dynamic (MD) simulation | Virtual screening based on GRL-0617 | Papain-like proteinase | 5-(aminomethyl)-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl]benzamide outperformed GRL-0617 in terms of binding affinity (−9.7 kcal/mol). 2-(4-fluorobenzyl)-5-nitro-1H-isoindole-1,3(2H)-dione, 3-nitro-N-[(1r)-1-phenylethyl]-5-(trifluoromethyl)benzamide, 5-acetamido-2-methyl-N-[(1S)-1-naphthalen-1-ylethyl]benzamide | |
| Jin et al. [244] | 2020 | In silico | Structure-assisted drug design, virtual drug screening, and high-throughput screening | 10,000 compounds | Main protease | Ebselen, disulfiram, tideglusib, carmofur, shikonin, PX-12 | Ebselen also exhibited promising antiviral activity in cell-based assays |
| Kanhed et al. [245] | 2021 | In silico | Systematic virtual screening approach | ASINEX BioDesign library approved drug library | Main protease | Ritonavir, nelfinavir, and saquinavir were predicted to be the most potent Mpro inhibitors. 20 molecules (pyrazoles, cyclic amides, pyrrolidine-based compounds, and miscellaneous derivatives) | |
| Kavitha et al. [246] | 2020 | In silico | Molecular docking molecular dynamic simulations | 1000 protease-inhibitor-like compounds available in the ZINC database | Main protease | 1,2,4 triazolo[1,5-a] pyrimidin-7-ones | |
| Krishnan et al. [247] | 2020 | In silico | Molecular docking | 3978 compounds with potential antiviral activity | Endoribonuclease (NSP15) | 8 compounds with good docking score and docking energy e.g., Z595015370, Z1343129850, and Z2760938911 | |
| Kumar et al. [248] | 2021 | In silico | Molecular docking molecular dynamic simulation molecular mechanic Poisson–Boltzmann surface area approaches | Million molecules and natural compound databases | Main protease | Three compounds namely ZINC14732869, ZINC19774413, and ZINC19774479 displayed better binding affinities | |
| Kumar et al. [249] | 2020 | In silico | Molecular docking molecular dynamic simulations | 13 approved antiviral drugs | Main protease | Indinavir was described as a lead drug. Indinavir possesses an important pharmacophore | Novel compound 16(hydroxyethylamine derivative) suitability as a strong candidate for therapeutic discovery against COVID-19 |
| Kwarteng et al. [250] | 2020 | In silico | Bioinformatic approach molecular docking molecular dynamic simulations | — | Nucleocapsid (N) protein | Zidovudine triphosphate, an anti-HIV agent, as a potential inhibitor of the N-terminal domain of SARS-CoV2 N protein | |
| Li et al. [251] | 2021 | In silico | Molecular docking | 21 antiviral, antifungal, and anticancer compounds | Papain-like protease | Neobavaisoflavone | |
| Mathpal et al. [252] | 2020 | In silico | Molecular docking molecular dynamic simulation MM-PBSA | 3180 FDA-approved drugs from “the ZINC database” | Main protease | ZINC03831201, ZINC08101052, ZINC01482077, and ZINC03830817 | |
| Maurya et al. [253] | 2020 | In silico | Molecular docking | Antiviral, anti-infectious, and anti-protease compounds | NSP10/NSP16 methyltransferase and main protease | Cyclocytidine hydrochloride, trifluridine adonitol, and meropenem penciclovir bound with a good docking score NSP10/NSP16 methyltransferase complexed with telbivudine, oxytetracycline dihydrate, methyl gallate, 2-deoxyglucose, and daphnetin | |
| Mohamed et al. [254] | 2020 | In silico | Molecular docking | 12 histone deacetylases (HDACs) | Main protease | Romidepsin and its active form (RedFK) | |
| Monajemi and Zain [255] | 2021 | In silico | ONIOM (own N-layered integrated molecular orbital and molecular mechanics; QM/MM) approach | N3, ebselen, disulfiram, tideglusib, carmofur, shikonin, and PX-12 | Main protease | N3, ebselen, and PX-12 inhibitors | Better inhibition from PX-12 than ebselen |
| Motiwale et al. [256] | 2020 | In silico | Molecular docking molecular dynamic simulations | Previously reported SARS-3CL protease inhibitors | Main protease | N-substituted isatin derivatives and pyrazolone | |
| Mutlu et al. [257] | 2020 | In silico | Structure-based approach molecular docking molecular dynamic simulations | FDA-approved and investigational drugs | Nsp12/Nsp8 | Two drugs, RX-3117 (fluorocyclopentenyl cytosine) and nebivolol | |
| Naidoo et al. [258] | 2020 | In silico | Molecular docking molecular dynamic simulation MM-PBSA | Cyanobacterial metabolites | Main protease (Mpro) and the papain-like protease (PLpro) | Deoxycylin, drospermopsin | Cylindrospermopsin, deoxycylindrospermopsin, carrageenan, cryptophycin 52, eucapsitrione, tjipanazole, tolyporphin, and apratoxin A exhibited promising inhibitory potential against the SARS-CoV-2 Mpro. The compounds cryptophycin 1, cryptophycin 52, and deoxycylindrospermopsin were observed to display encouraging binding energy scores with the PLpro of SARS-CoV-2 |
| Olubiy et al. [259] | 2020 | In silico | Molecular docking molecular dynamic simulations | Approved drugs, investigational drugs, natural products, and organic compounds | Main protease | Several tyrosine kinase inhibitors, which include a bioflavonoid and steroid hormones, bind best to main protease | Nilotinib, enasidenib, afatinib, ertapenem, phthalocyanine, hypericin, amrubicin, theacitrin A, theaflavin, amentoflavone, epigallocatechin gallate, glabrolide, cortisol, estradiol, testosterone |
| Özdemir et al. [260] | 2020 | In silico | Molecular docking density functional theory (DFT) ADME-Tox | 42 coumarin derivatives | Main protease | 6,7-Dihydroxy-3-phenylcoumarin derivatives gave relatively higher scores, and for all coumarins, and 4-trifluoromethylphenyl substituted coumarin had the highest score | |
| Ozdemir et al. [261] | 2020 | In silico | Molecular docking molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) | Coumarin derivatives | Spike S1 subunit, NSP5, NSP12, NSP15, and NSP16 | The highest score (−10.01 kcal/mol) in the coumarin group is 2-morpholinoethan-1-amine substituted coumarin | |
| Patel et al. [262] | 2021 | In silico | Pharmacophore studies | Drug library (having drugs and diagnostic agents, which are approved by FDA or other world authorities) and the ASINEX BioDesign library | Main protease | Ritonavir, nelfinavir, and saquinavir were predicted to be the most potent Mpro inhibitors 20 molecules categorized into four classes viz. disubstituted pyrazoles, cyclic amides, pyrrolidine-based compounds, and miscellaneous derivatives | |
| Peng et al. [263] | 2020 | In silico | Drug repositioning through virus-drug association prediction | — | Against SARS-CoV-2 | Ribavirin was predicted to be the best small molecular drug, with a higher molecular binding energy with human angiotensin-converting enzyme 2 (ACE2), followed by remdesivir, mycophenolic acid, and chloroquine (−6.29 kcal/mol) | |
| Pratama et al. [264] | 2020 | In silico | Molecular docking | Novel 5-O-benzoylpinostrobin derivatives | Main protease | Three 5-O-benzoylpinostrobin derivatives each with fluoro, tertiary butyl, and trifluoromethyl substituents at 4-position of benzoyl group showed the lowest free energy of binding value and the highest similarity of ligand-receptor interactions with co-crystallized ligands | |
| Pundir et al. [265] | 2020 | In silico | Pharmacophore-based virtual screening molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) | PubChem database | Main protease | Two compounds: PubChem3408741 and PubChem4167619 had the binding free energy of −94.02 kJ·mol−1 and −122.75 kJ·mol−1, respectively, as compared to reference X77 (−76.48 kJ·mol−1) | Lead molecules for targeting Mpro enzyme |
| Quimque et al. [266] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | 97 antiviral secondary metabolites from fungi | Papain-like protease, RNA-dependent RNA polymerase, main protease, spike glycoprotein, nonstructural protein 15 (nsp15) | Two fumiquinazoline alkaloids quinadoline B (19), scedapin C (15), and polyketide isochaetochromin D1 (8) | Quinadoline B [19] was predicted to confer favorable ADMET values, high gastrointestinal absorptive probability, and poor blood-brain barrier crossing capacities |
| Rakib et al. [267] | 2021 | In silico | Molecular docking molecular dynamic simulation ADME properties | Selenium-containing heterocyclic compounds | Main protease | Selection of the 16 most effective selenocompounds as potential anti-COVID-19 drug candidates. Ethaselen showed potential binding affinities | |
| Rane et al. [268] | 2020 | In silico | Molecular docking molecular dynamic simulations | Diarylpyrimidine analogs | Spike glycoprotein | AP-NP (2-(2-amino-5-(naphthalen-2-yl)pyrimidin-4-yl)phenol), AP-3-OMe-Ph (2-(2-amino-5-(3-methoxyphenyl)pyrimidin-4-yl)phenol), and AP-4-Me-Ph (2-(2-amino-5-(p-tolyl) pyrimidin-4-yl)phenol) from a group of diarylpyrimidine derivatives, which appears to bind at the interface of the hACE2-S complex with low binding free energy | |
| Rao et al. [269] | 2020 | In silico | Molecular docking molecular dynamic simulations | Various small molecules | Main protease | Pyranonigrin A, a secondary fungal metabolite | |
| Salman et al. [270] | 2020 | In silico | Molecular docking molecular dynamic simulation ADME properties | Library of immunomodulatory medicinal compounds with antiviral capability | SARS proteases, spike protein, and nonstructural proteins (NSP-9, 15) | 6 compounds: arzanol, ferulic acid, genistein, resveratrol, rosmanol, and thymohydroquinone | Good pharmacokinetic properties and low acute toxicity of these compounds |
| Sarma et al. [271] | 2020 | In silico | Molecular docking MM-GBSA binding free energy molecular dynamic simulations | 56,079 compounds from ASINEX and Maybridge library | RNA-binding N-terminal domain (NTD) of the N protein | ZINC00003118440 is a theophylline derivative. Pyrimidone derivatives as possible inhibitors of RNA binding to the N-terminal domain of N protein of coronavirus | Lead molecules |
| Sepay et al. [272] | 2020 | In silico | Molecular docking, bioinformatics, and molecular electrostatic potential ADME studies | Benzylidenechromanones, naturally occurring oxygen heterocyclic compounds | Main protease | (Z)-3-(4 ′-chlorobenzylidene)-thiochroman-4- | Effective pharmacological properties |
| Shehroz et al. [273] | 2020 | In silico | Pharmacophore modeling molecular docking | DrugBank, ZINC, and TIMBLE databases | Spike (S) protein | Only eight molecules fit the criteria for safe oral drugs | Lead molecules |
| Singh and Das [274] | 2021 | In silico | Molecular docking | Chloroquine (CQ) hydroxychloroquine (HCQ) azithromycin | Spike (S) protein main protease host cathepsin L (CTSL) receptor-binding domain (RBD) | Azithromycin affinity scores (ΔG) with strong interactions with ACE2, CTSL, Mpro, and RBD. CQ firm bond score with Mpro HCQ and two results (ACE2 and Mpro) were firmly bound to the receptors | |
| Stefaniu et al. [275] | 2020 | In silico | Molecular docking density functional theory (DFT) computations, drug-likeness assessment | Derivatives of benzoic acid | Main protease | 2,5-dihydroxybenzoic acid (gentisic acid) and octyl | A combination of the two compounds can be considered |
| Tachoua et al. [276] | 2020 | In silico | Molecular docking and structural dynamic study molecular mechanic Poisson–Boltzmann surface area (MM-PBSA) ADMET analysis | Chloroquine, quinine, nitazoxanide, doxycycline, lymecycline, cetirizine, mizolastine, indinavir | Main protease | Lymecycline mizolastine | |
| Uniyal et al. [277] | 2020 | In silico | Structure-based virtual screening molecular docking MM-GBSA binding free energy molecular dynamic simulations | Commercially available chemical libraries | Main protease | Compound AG-690/11203374_1 and AG-690/11203374_2 emerged as the best in silico hits based on the docking, MM-GBSA, dynamics, and ADMET studies | Lead molecules |
| Welker et al. [278] | 2020 | In silico/in vitro | Structure-activity relationships molecular docking fluorescence-based enzyme‐activity assay Vero E6 cells | A series of rationally designed competitive, noncovalent, nonpeptidic active site‐directed SARS‐CoV PLpro inhibitors | Papain-like cysteine proteases (PLpro) | R)‐5‐amino‐2‐methyl‐N‐(1‐(naphthalen‐1‐yl)ethyl) benzamide (2b), which is known to bind into the S3 and S4 pockets of the SARS‐CoV PLpro. Isoindoline as a new class of potent PLpro inhibitors | IC50 value of 2.9 ± 0.2 μM |
| Wen et al. [279] | 2021 | In silico | Structure-based screening | 8,820 compounds | Main protease | Trichostatin A | A histone deacetylase inhibitor and an antifungal compound |
| White et al. [280] | 2020 | In silico | Homology modeling and molecular dynamics approach molecular docking | ∼970,000 chemical compounds | Helicase (Nsp13) | Nilotinib and lumacaftor have significant activity in inhibiting purified recombinant SARS-CoV-2 helicase | |
| Wu et al. [281] | 2020 | In silico | Molecular docking | 11 HIV-1 protease inhibitors, 12 nucleotide-analog inhibitors, 728 approved drugs | Main protease RNA-dependent RNA polymerase | Remdesivir shows the best binding energy on RdRp and saquinavir is the best inhibitor of main protease | |
| Zaher et al. [282] | 2020 | In silico | Design, synthesis SAR study molecular docking | Newly synthesized sixteen halogenated triazole compounds | Helicase (Nsp13) | The most potent compounds were 4-(cyclopent-1-en-3-ylamino)-5-(2-(4-iodophenyl)hydrazinyl)-4H-1,2,4-triazole-3-thiol [16] and 4-(cyclopent-1-en-3-ylamino)-5-[2-(4-chlorophenyl)hydrazinyl]-4H-1,2,4-triazole-3-thiol [12] | |
| Zarezade et al. [283] | 2021 | In silico | 3D-QSAR pharmacophore modeling ADMET properties, molecular docking molecular dynamic simulation MM-PBSA, hybrid QM-MM method de novo ligand design | PubChem and ZINC databases | Human angiotensin-converting enzyme 2 and main protease | ZINC12562757 and 112,260,215 were the best potential inhibitors of the ACE2 and main protease, respectively. Evo_1 compound enjoys the highest docking energy among the designed de novo ligands | Evo_1 has a stronger potential for specific inhibition of main protease, as compared to the 112, 260, 215 compound. Lead molecules |
In silico studies used the following component of novel coronavirus as targets: main protease (N = 154), spike glycoprotein (N = 62), nonstructural protein (N = 45), RNA-dependent RNA polymerase (N = 21), papain-like protease (N = 19).
About 260 drugs were repurposed by the computational methods for COVID-19 therapy such as about 120 drugs candidate against the main protease, 52 drugs against the spike glycoprotein, 14 drugs against RNA-dependent RNA polymerase, and 28 drugs against other nonstructural proteins.
Among the studied repurposed drugs, the best results (regarding efficacy) were observed with saquinavir (N = 9 study), ritonavir (N = 8 study), lopinavir (N = 6 study), remdesivir (N = 3 study), and amikacin, danoprevir, favipiravir, and telaprevir.
Table 1 shows target-based synthesis of data for COVID-19 drug repurposing. As presented, at least two studies show the efficacy of aprepitant, cobicistat, dipyridamole, and dihydroergotamine against the main protease and tegobuvir (N = 2) against spike protein. The following list of drugs had multitarget action: avapritinib, famotidine, bictegravir, ziprasidone, capmatinib, pexidartinib, amprenavir, zafirlukast, cilostazol, paromomycin, lopinavir, and remdesivir.
A total of 91 studies used in silico methods to evaluate the effects of natural products including herbal medicine against SARS-CoV-2. Among them, 54 studies used the main protease as main target, in which eucalyptus (N = 3), Tinospora cordifolia (N = 3), and flavonoids (e.g., hesperidin, rutin, and herbacetin) were the most studied and effective. Other studied targets were as follows: spike (N = 22) and multitarget (N = 20). From the plant metabolites, oleanolic acid, hesperidin, epigallocatechin gallate, jensenone, tinosponone, and anistone show promising results in computational methods against COVID-19. Aloe, green tea, eucalyptus, curcumin, and many Chinese, Indian, and African plants were also effective on COVID-19 in silico.
The derivatives of pyrazoles, oxadiazoles, phenyltriazolinones, triazoles, benzoylpinostrobin, benzoic acid, benzylidenechromanones, coumarin, and selenium show efficacy in computational methods and could be considered as lead molecules for drug design and synthesis against COVID-19. Melatonin, ebselen, phenyl furoxan, thimerosal, isatin, romidepsin, phenyl mercurin, pleconaril, and tyrosine kinase inhibitors (such as nilotinib and imatinib) are examples of other drugs that are effective in in silico methods.
3.2. Results from In Vitro and In Vivo Studies
Screening of in vitro studies leads to finding 34 studies. Different compounds were evaluated and most studies focused on the inhibition of viral replication, which was assessed by the quantification of viral RNA by PCR and IC50 value reported. Most of the studies use the Vero E6 cell line for the assessment of replication of SARS-CoV-2 (Table 4). In particular, main protease, papain-like protease, RNA-dependent RNA polymerase, NSP-14, NSP-15, spike protein, and TMPRSS2 were evaluated in vitro as targets. Also, three studies focused on in vitro inhibition of inflammatory markers such as interleukins and the effects of the cytopathic effect on infected cell.
Table 4.
Summary of studies with in vitro method.
| Author | Year | Method | Detail of method | Name of compound/drug | Target | Efficacy | Comments |
|---|---|---|---|---|---|---|---|
| Agarwal et al. [284] | 2020 | In vitro | IL-1β assay using THP-1 cells, in vivo pharmacokinetic investigation in C57BL/6 mice | Alkenyl sulfonylurea derivatives | NLRP3 inhibition and reduction in the release of interleukin-1β | IL-1β inhibition IC50 of 35 nM, good oral absorption showing Cmax of 8.49 μg/mL with an AUC of 48.9 μg·h/mL and terminal half-life of 2.86 h, after oral route of administration at 3 mg/kg dose | Novel thiazolo-alkenyl sulfonylurea derivative 7 as potent, selective, and orally bioavailable NLRP3 inflammasome inhibitor |
| Akaberi et al. [285] | 2020 | In vitro | Vero E6 cells, RT-qPCR for the quantification of viral RNA, expression and purification of SARS-CoV-2 3CL protease in vitro enzymatic assay | NO-Donor S-nitroso-N-acetylpenicillamine (SNAP): nitric oxide | Main protease | Although the viral replication was not completely abolished (at 200 μM and 400 μM), SNAP delayed or completely prevented the development of viral cytopathic effect in treated cells (IC50 = 440.95 μM ± 36.15 SE) | A dose-dependent inhibition of the 3CL protease (400 μM) |
| Bernstein and Zhang [286] | 2020 | In vitro | Vero E6 cells: virus was then added at a multiplicity of infection (MOI) of 0.01, with 1 h allowed for infection. Cytotoxicity: Cell Counting Kit-8 (CCK-8) colorimetric assay | Gallium maltolate (GaM) | Inhibition of viral replication | EC50 (concentration producing 50% inhibition of viral replication) of about 14 μM (CI 95%: 8.9–22.8 μM) | No cytotoxicity was observed at concentrations up to at least 200 μM |
| Bocci et al. [287] | 2020 | In vitro | Ligand-based virtual screening/Vero E6 cells. Uninfected cells and chloroquine as control inhibition of the SARS-CoV-2-mediated CPE following infection in Vero E6 cells tissue culture infectious dose (TCID50) assay | 4000 approved drugs/glafenine, amodiaquine vorinostat, zuclopenthixol, isoxsuprine, nebivolol, ambroxol, panobinostat, and pracinostat | Inhibition of viral replication | In silico: glafenine, amodiaquine (amodiaquine), vorinostat, zuclopenthixol, isoxsuprine, nebivolol, ambroxol, panobinostat, and pracinostat. In vitro: zuclopenthixol and amodiaquine show anti-SARS-CoV-2 activity comparable with chloroquine. EC50 values were estimated as 0.13 μM for AQ, 1.35 μM for ZPX, and 2.72 μM for nebivolol | Zuclopenthixol and nebivolol exhibit in vitro antiviral activities and potencies that are comparable or better than chloroquine and hydroxychloroquine |
| Cao et al. [288] | 2020 | In vitro | African green monkey kidney Vero E6 cells were infected with virus at an MOI of 0.01 for 1 h. Immunofluorescence assay physiologically based pharmacokinetic modeling and simulations | 9 artemisinin-related compounds | Inhibition of viral replication | Arteannuin B showed the highest anti-SARS-CoV-2 potential with an EC (50) of 10.28 ± 1.12 μM. Artesunate and dihydroartemisinin showed similar EC (50) values of 12.98 ± 5.30 μM and 13.31 ± 1.24 μM, respectively | Lumefantrine could inhibit SARS-CoV-2 in vitro with an EC50 of 23.17 ± 3.22 μM. Impair viral infection by modulating host cell metabolic pathways |
| Chen et al. [289] | 2021 | In vitro | Fluorescence-based high-throughput screen, docking/pretreated with various concentrations of test compound for 1 h, followed by infection with SARS-CoV-2 (MOI of 0.0001) in the presence of test compounds, expression and purification of SARS-CoV-2 PLpro, 3CL protease from Escherichia coli enzymatic assay of SARS-CoV-2 PLpro cytotoxicity assay | 1920 natural products | Main protease, papain-like protease | Ginkgolic acid and anacardic acid were also identified as inhibitors of 3CLpro, with IC50 values of 1.79 ± 0.58 and 2.07 ± 0.35 μM, respectively, both ginkgolic acid and anacardic acid dose-dependently inhibited PLpro activity, with IC50 values of 16.30 ± 0.64 and 17.08 ± 1.30 μM, respectively | Ginkgolic acid act as an irreversible inhibitor against both PLpro and 3CLpro, suggesting it is a covalent inhibitor |
| Gendrot et al. [290] | 2020 | In vitro | Vero E6 cells, replication was estimated by RT-PCR/chloroquine as control determination of the inhibition stage | Doxycycline | Inhibition of viral replication | Median effective concentration (EC50) and 90% effective concentration (EC90) for doxycycline were 4.5 ± 2.9 μM and 23.5 ± 16.5 μM, respectively, EC50 = 4.5 μM | Interacted at both entry and postentry stages of the SARS-CoV-2 infection doxycycline has anti-inflammatory effects |
| Gupta et al. [291] | 2021 | In vitro | High-throughput virtual screening, molecular dynamic simulation, prime MM-GBSA/Vero E6 cells infected with the SARS-CoV-2 fluorescence-based biochemical assay for inhibitors of 3CLpro | FDA-approved drugs | Main protease, PLpro, RNA-directed RNA polymerase, helicase, NSP-15, NSP14, and 2′-O-methyltransferase | Troxerutin (main protease and PLpro) and bisindolylmaleimide derivatives (main protease and exon) had possible dual targets. Ivermectin has nonselective toxicity to the ATCC E6 Vero cells at ≤50 μM and 16.67 μM based on the number of nuclei counted | BIM IX specifically blocked 3CLpro in vitro enzymatic assay inhibition was observed with an IC50 value of 113.7 ± 5.2 μM a known inhibitor of protein kinase C isoforms, bisindolylmaleimide IX (BIM IX), was found to be a potent inhibitor of SARS-CoV-2 |
| Hahn et al. [292] | 2020 | In vitro | Vero B4 and 76 cells و Vero E6 cells/remdesivir RT-qPCR for the detection of extracellular SARS-CoV-2 in cell immunostaining for the detection of Intracellular SARS-CoV-2 Western blot analysis viral plaque and yield reduction assay neutral red assay (NRA) | (IMU-838) inhibitor of human dihydroorotate dehydrogenase (DHODH) | Inhibition of viral replication/immunomodulator | A virus-specific EC90 of 6.2 ± 1.9 μM was measured, with no cytotoxicity observed with drug concentrations up to 100 μM at 3 days p.i. | Active moiety of IMU-838, vidofludimus, possesses broad-spectrum antiviral activity. IMU-838 reduces T lymphocyte proliferation, cytokine production, and organ infiltration by leukocytes in various in vivo and in vitro models for autoimmunity |
| Hattori et al. [293] | 2021 | In vitro | VeroE6 cell-based assays with RNA-qPCR, cytopathic assays | Two small-molecule-compounds, named GRL-1720 and 5 h, containing an indoline and indole moiety | Main protease | EC(50) values of 15 ± 4 and 4.2 ± 0.7 μM for GRL-1720 and 5 h | Combination of 5 h and remdesivir exhibits synergism against SARS-CoV-2.5 h might serve as a lead M (pro) inhibitor |
| Jang et al. [294] | 2021 | In vitro | Vero CCL-81 cells/Calu-3 human lung epithelial cells | Gemcitabine and its analog 2′-fluoro-2′-deoxycytidine (2FdC) | Inhibition of viral replication | 50% effective concentration (EC(50)) of 1.2 μM compare to remdesivir 2FdC was marginally active (EC(50) = 175.2 μM) | Gemcitabine has a synergistic effect when combined with remdesivir |
| Jeon et al. [295] | 2020 | In vitro | Drug repositioning/chloroquine, lopinavir, and remdesivir were used as reference drugs/VeroE6 cell/immunofluorescence analysis | 48 FDA-approved drugs | Inhibition of viral replication | Niclosamide (IC50, 0.28 μM), ciclesonide (IC50, 4.33 μM) | 24 potential antiviral drug candidates against SARS-CoV-2 infection |
| Liu et al. [296] | 2020 | In vitro | Virtual screening/spiked pseudotyped lentivirus for imitating SARS-CoV-2 cell entry/ACE2-GFP expressing HEK293T 2 cells. HeLa Cells, 293T (human, kidney) cells, and Vero-E6 (African green monkey, kidney) cells | FDA-approved drugs | Angiotensin I-converting enzyme 2 (ACE2) and a spike protein | Nine potential candidates were selected and submitted to experimental studies. Three (romidepsin, saquinavir, and nelfinavir) of nine drugs possess the ability to suppress SARS-2-S pseudotyped particles to enter the ACE2 expressing cells in a concentration-dependent manner | Five clinical HDAC inhibitors including romidepsin, panobinostat, givinostat hydrochloride monohydrate, CAY10603, and sirtinol could inhibit noticeably the spike/ACE2-mediated cell entry of SARS-CoV-2 |
| Ma et al. [297] | 2020 | In vitro | Vero E6 cells using cytopathic effect and plaque reduction assay. HCoV-229E infection in Huh-7 cells. Western blot assay | Phillyrin (KD-1) ingredient of Forsythia suspensa | Inhibition of viral replication | Inhibit SARS-CoV-2. Markedly reduce the production of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, MCP-1, and IP-10) at the mRNA levels. p-NF-κB p65, NF-κB p65, and p-IκBα | KD-1 could significantly inhibit virus proliferation in vitro, the upregulated expression of pro-inflammatory cytokines induced by SARS-CoV-2 |
| Masih et al. [298] | 2020 | In vitro | LPS-stimulated RAW267.4 cells by enzyme immunoassay Western blot assay | Pyrazole derivatives | Anti-inflammatory levels of interleukin-1β, tumor necrosis factor-α, and interleukin-6 | Compound 6c as the most potent analog among the tested series. Inhibition of inhibitor kappa B-α and NF-κB | |
| Omotuyi et al. [299] | 2020 | In vitro | Kinetic assay | Aframomum melegueta. 100 secondary metabolites | Main protease, 2′-O-ribose methyltransferase (NSP16), and surface glycoprotein/ACE2 receptor interface | Diarylheptanoid (letestuianin A), phenylpropanoid (4-cinnamoyl-3-hydroxy-spiro[furan-5,2′-(1′H)-indene]-1′,2,3′(2′H,5H)-trione), flavonoids (quercetin, apigenin, and tectochrysin) have been identified as high binding compounds to SARS-CoV-2 targets in a polypharmacology manner | Di-ethyl-ether (IC50 = 0.03 mg/L), acetone (IC50 = 1.564 mg/L), ethyl-acetate (IC50 = 0.382 mg/L), and methanol (IC50 = 0.438 mg/L) fractions demonstrated the best inhibition in kinetic assay |
| Outlaw et al. [300] | 2020 | In vitro | Vero E6 cell β-Gal complementation-based fusion assay. Viral titration and plaque reduction neutralization assay quantitative RT-PCR. | A lipopeptide that is derived from the C-terminal heptad repeat (HRC) domain of SARS-CoV-2 S | Inhibition cell-cell fusion | Inhibits cell-cell fusion mediated by SARS-CoV-2 S and blocks infection by live SARS-CoV-2 in Vero E6 cell monolayers (IC50) of ∼10 nM and 90% inhibitory concentration (IC90) of ∼100 nM | |
| Pitsillou et al. [301] | 2021 | In vitro | Molecular docking, molecular dynamic simulation/papain-like protease (SARS-CoV-2) assay kit GRL-0617 as a control enzymatic inhibition assay | 300 small compounds derived predominantly from our OliveNet™ library (222 phenolics) and supplemented with synthetic and dietary compounds | Papain-like protease | Selection of 30 compounds. Hypericin possessed inhibition activity, and both rutin and cyanidin-3-O-glucoside resulted in a concentration-dependent inhibition of the PLpro | The IC50 values were not clarified in experiment |
| Pizzorno et al. [302] | 2020 | In vitro | Vero E6 cells/remdesivir, lopinavir, chloroquine as comparator | Favipiravir, ribavirin, umifenovir (arbidol), berberine, cyclosporine A, diltiazem | Inhibition of viral replication | Umifenovir, berberine, and cyclosporine A with estimated 50% inhibitory concentrations of 0.99, 5.2, 1.38, 3.5, 10.6, and 3 μM, respectively | A strong antagonism between remdesivir and berberine, in contrast with remdesivir/diltiazem, for which we describe high levels of synergy |
| Plaze et al. [303] | 2021 | In vitro | Monkey VeroE6 cells and human alveolar basal epithelial A549-ACE2 cell RT-qPCR for the presence of SARS-CoV-2 RNA | Chlorpromazine | Inhibition of viral replication | (IC50) of 8.2 μM in monkey VeroE6 cells (CC50) of 13.5 μM, and selectivity index (SI) of 1.65 IC50 of 11.3 μM, CC50 of 23.1 μM, and SI of 2.04 in human A549-ACE2 cells | |
| Reznikov et al. [304] | 2021 | In vitro | Repurposing study molecular docking/human lung A549 cells that were transfected with hACE2 (ACE2-A549) cells, Vero E6 cells | Electronic health records of over 219,000 patients with antihistamines | Inhibition of viral replication/spike protein | IC50 15.3 μg/ml for hydroxyzine, 17.4 μg/ml for diphenhydramine, and 2.24 μg/ml for azelastine | Hydroxyzine and possibly azelastine bind angiotensin-converting enzyme-2 (ACE2) and the sigma-1 receptor as off-target disease prevention |
| Sakurai et al. [305] | 2021 | In vitro | VeroE6 cells/human colon-derived Caco-2 cells/infection assay with immunofluorescence | 5-Amino levulinic acid (5-ALA) | Inhibition of viral replication | IC50 of 570 μM/IC50 of 63 μM in human Caco-2 cells. No significant cytotoxicity | 5-ALA is synthesized in most animals and plants and we are continuously consuming it in our food. Show anti-inflammation effects in humans |
| Stone et al. [306] | 2021 | In vitro | Vero E6 monkey kidney and Calu-3 human lung adenocarcinoma cells/RT-qPCR of RNA | Stenoparib poly(ADP ribose) polymerase (PARP) inhibitor | Inhibition of viral replication | Dose-dependent activity against SARS-CoV-2 at concentrations up to 30 μM with negligible cytotoxicity. Synergistic effect with remdesivir | Stenoparib impedes entry and postentry processes |
| Su et al. [307] | 2020 | In vitro | Vero E6 cells, quantitative real-time RT-PCR | Shuanghuanglian preparation, a Chinese traditional patent medicine (chlorogenic acid, phillyrin, baicalin, and baicalein) | Main protease | Dose-dependent inhibition of SARS-CoV-2 3CLpro, and the resulting IC50 values were 0.090, 0.064, and 0.076 μL/mL for three Shuanghuanglian oral liquids produced by three different pharmaceutical companies | Baicalin and baicalein, which have good drug-like properties |
| Touret et al. [308] | 2020 | In vitro | VeroE6 cells/human colon-derived Caco-2 cell real-time RT-PCR/remdesivir as control | FDA-approved chemical library | Inhibition of viral replication | 23 drugs were selected to cover the 12 different groups. 11 compounds such as macrolide antibiotics, proton pump inhibitors, antiarrhythmic agents, or CNS drugs emerged showing antiviral potency with 2 < EC50 ≤ 20 μM | Two of the highest antiviral activity were obtained for azithromycin (EC50 = 2.12 μM) and hydroxychloroquine (EC50 = 4.17 μM). Vonoprazan and sulfadoxine show a moderate activity with EC50 above 30 μM |
| Tree et al. [309] | 2021 | In vitro | Plaque inhibition assay with Vero E6 cells/differential scanning fluorimetry/ELISA assays | Unfractionated heparin (UFH), low MW heparins | Inhibition of viral replication/spike protein RBD | All the UFH preparations had potent antiviral effects, with IC50 values ranging between 25 and 41 μg·ml−1, whereas LMWHs were less inhibitory by ∼150-fold (IC50 range 3.4–7.8 mg·ml−1) | Heparin directly inhibits the binding of RBD to the human ACE2 protein receptor |
| Vatansever et al. [310] | 2021 | In vitro | Docking, screening assay/Vero E6, and A549 cell culture | 30 FDA/EMA-approved drugs | Main protease | Six FDA/EMA-approved drugs can potently inhibit Mpro with an IC50 value lower than 100 μM. Bepridil exhibited strong inhibition of SARS-CoV-2 from entry and replication inside Vero E6 and A549 cells (IC50 = 72 μM) | Bepridil indicated that it had low micromolar EC50 values |
| Wang et al. [311] | 2020 | In vitro | FRET-based enzyme activity assay of Mpro measurement of human TMPRSS2 activity by a FRET-based enzymatic assay surface plasmon resonance (SPR) analysis molecular docking African green monkey kidney cell Vero E6 | Catechin, kaempferol, quercetin, proanthocyanidins, resveratrol, and tannic acid | TMPRSS2 (transmembrane protease serine 2) main protease | Tannic acid inhibited the activities of the two proteases with an IC50 of 13.4 mM for Mpro and 2.31 mM for TMPRSS2 | A potent dual inhibitor |
| Watashi [312] | 2021 | In vitro | Virtual drug screening, docking/VeroE6 cells detecting the viral RNA by real-time RT-PCR or viral proteins by immunofluorescence | FDA-approved drugs | Main protease, PLpro, RdRp, helicase, spike, ACE2, and TMPRSS2 | Abiraterone, amodiaquine, anidulafungin, arbidol, astemizole, atazanavir, auranofin, azithromycin bazedoxifene, bexarotene, camostat IC50 values <5 μM | Cepharanthine, cetilistat, chloroquine, ciclesonide, cyclosporine A, digoxin, ivermectin, lopinavir, mefloquine, nelfinavir, niclosamide |
| Yu et al. [313] | 2021 | In vitro | Computer-aided drug design and biological verification surface plasmon resonance (SPR) assays and NanoBiT assays | Traditional Chinese medicine | Spike proteins | Glycyrrhizic acid, the most efficient and nontoxic broad-spectrum anticoronavirus molecule IC50, was 22 μM | |
| Zandi et al. [314] | 2021 | In vitro | African green monkey kidney cells (Vero CCL-81 cells) and reverse transcription-quantitative PCR (qRT-PCR). Remdesivir and β-D-N4-hydroxycytidine as positive drug controls | Nucleoside analogs/anti-HCV agents | Inhibition of viral replication | Sofosbuvir and favipiravir demonstrated no antiviral effect against COVID-2 | |
| Zhang et al. [315] | 2020 | In vitro | Hybrid virtual screening, docking, force field-based simulation/qRT-PCR assay, indirect immunofluorescence assay (IFA) and CCK-8 assay, surface plasmon resonance (SPR) assay/Vero cells | 1906 approved drugs from TargetMol-approved drug library | RNA-dependent RNA polymerase (RdRp) | Pralatrexate and azithromycin (EC50) values of 0.008 and 9.453 μM | A new therapeutic agent pralatrexate against COVID-19 by targeting RdRp |
| Zhang et al. [316] | 2020 | In vitro | African green monkey kidney Vero E6 cell line, quantitative real-time PCR (RT-PCR) analysis, immunofluorescence microscopy, Western blot analysis | Lopinavir/ritonavir, rupintrivir, and AG7404 | Inhibition of viral replication | (IC50) and half cytotoxic concentration (CC50) values of lopinavir were ∼12.01 μmol/L and 80.82 μmol/L; for ritonavir, the IC50 and CC50 values were 19.88 μmol/L and 94.71 μmol/L (SI = 4.76) | Rupintrivir inhibited SARS-CoV-2 infection only at high drug concentrations (Vero E6: IC50 = 34.08 μmol/L, AG7404: Vero E6: IC50∼195.8 μmol/L). Lopinavir/ritonavir should be stopped for clinical use due to the huge gap between in vitro IC50 and free plasma concentration |
| Zhu et al. [317] | 2021 | In vitro | Human fibroblast lung cells (MRC-5), cytopathic effect (CPE) assay, primary human airway air-liquid interface (ALI) cultures, TEER assay, CCK-8 assays | Stimulator of interferon genes (STING): dimeric amidobenzimidazole (diABZI) | Inhibition of viral replication | Potent anticoronavirus activity against both the common cold human coronavirus 229E (HCoV-229E) and SARS-CoV-2 in cell culture systems. EC50 of 120 μM |
Nitric oxide, ginkgolic acid, anacardic acid, troxerutin, bisindolylmaleimide derivatives, small molecules (GRL-172, and 5 h), baicalin and baicalein (phytochemicals), and bepridil were effective drugs in vitro against the main protease of COVID-19.
The following drugs could inhibit the spike/ACE2-mediated cell entry of COVID-19 in vitro: romidepsin, panobinostat, givinostat, sirtinol, saquinavir, lipopeptides, hydroxyzine, azelastine, heparin, and glycyrrhizic acid.
Alkenyl sulfonylurea derivatives [7], an IMU-8381 inhibitor of human dihydroorotate dehydrogenase, pyrazole derivatives, and phillyrin, regulated the expression of inflammatory cytokines (e.g., IL, TNF-α, and NF-κB) induced by SARS-CoV-2 markedly and could be considered as adjuvant treatment of COVID-19 severe disease.
Doxycycline, chlorpromazine, azithromycin, heparin, bepridil, tannic acid, and glycyrrhizic acid are well-known drugs that show both in silico and in vitro inhibitory effects against SARS-CoV-2 and should be considered for this purpose.
Also, in vitro studies show that lopinavir/ritonavir, sofosbuvir, and favipiravir have no antiviral effects against SARS-CoV-2 (huge gap between in vitro IC50 and free plasma concentration).
We included 16 in vivo studies in our final analysis. Most of them use a combination of in vitro and in vivo methods for the evaluation of novel drugs. Table 5 shows the detail of these studies.
Table 5.
Summary of studies with in vivo method.
| Author | Year | Method | Detail of method | Name of compound/drug | Target | Efficacy | Comments |
|---|---|---|---|---|---|---|---|
| Lei et al. [318] | 2020 | In vivo | BALB/c mice to determine the pharmacokinetic profiles of the fusion proteins | A recombinant protein by connecting the extracellular domain of human ACE2 to the Fc region of the human immunoglobulin IgG1 | Spike protein | The IC50 values of ACE2-Ig for SARS-CoV and SARS-CoV-2 neutralization were 0.8 and 0.1 μg·ml−1, respectively. Desirable pharmacological properties in mice | Potential applications in the diagnosis, prophylaxis, and treatment of SARS-CoV-2 |
| Rathnayake et al. [319] | 2020 | In vivo | Old male hDPP4-KI mice infected with MERSMA-CoV | Dipeptidyl and tripeptidyl series of compounds | Main protease | 7a, 6c, 7e, 7h, and 6j: EC50 values ranging from 0.15 to 0.9 μM in Vero E6 cells, IC50: 0.17 to 0.82 μM. 40% of mice treated with compound 6h survived, and all mice treated with compound 6j were alive at the end of the study | 6j resulted in the survival of MERSMA-CoV-infected hDPP4-KI mice |
| Schäfer et al. [320] | 2020 | In vivo | An immune-competent mouse model of COVID-19 by remodeling the SARS-CoV-2 S RBD at the mACE2-binding interface. Human Ace2-expressing HT1080 cells and Syrian hamster model of SARS-CoV-2 | Human mAbs (hu-mAbs) | Reduction in viral load and prevent infection | Neutralizing hu-mAbs targeting SARS-CoV-2 promotes reduction in viral load and prevent infection in macaques and hamsters effective dose: 5.3 and 16 mg/kg inhibitory concentration (IC)50/IC90 (half-maximal/90% IC) of 4.4/18 to 26/140 ng/ml | |
| Schumann et al. [321] | 2020 | In vivo | Female C57Bl/6 mice, LPS stimulation | MP1032:5-amino-2,3-dihydro-1,4-phthalazinedione sodium salt | Immune-modulating | Direct inhibitory activity on human PARP-1 was detected in the low micromolar range (IC50 = 1.55 μM). MP1032 works as a ROS scavenger, reduced the secretion of TNF-α and IL-6. MP1032 pre-treatment significantly reduced plasma cytokine levels compared with vehicle-treated mice by 50% (TNF-α) and 25% (IL-6). Significant reduction in SARS-CoV-2 replication | MP1032 could further attenuate prolonged virus replication by preventing oxidative stress or by limiting ADP ribosylation of the viral nucleocapsid protein via PARP-1 inhibition |
| Shang et al. [322] | 2021 | In vivo | African green female hACE2 transgenic mice, SARS-CoV-2 injection RNA extraction and qPCR, RNA in situ hybridization assay, histopathological examination | Endosomal acidification inhibitors, including chloroquine, bafilomycin A1, and NH4CL | Antiviral actions against SARS-CoV-2 | (40 μM), bafilomycin A1 (100 nM), and NH4CL (12.5 mM) suppressed the replication of SARS-CoV-2 in all cell types chloroquine (60 mg·kg−1) and bafilomycin A1 (0.1 mg·kg−1) markedly reduced virus yields in lung tissues. Alleviated viral pneumonia in hACE2 transgenic mice | Endosomal acidification inhibitors exhibited antiviral actions against SARS-CoV-2 |
| Sheahan et al. [323] | 2020 | In vivo | Male and female 20- to 29-week old SPF C57BL/6J (Stock 000664 Jackson Labs) mice, whole-body plethysmography 50, 150, or 500 mg/kg EIDD-2801 2 hr prior to intranasal infection with 5E + 04 PFU of mouse-adapted SARS-CoV | Ribonucleoside analog β-d-N4-hydroxycytidine | Antiviral efficacy against the three most recently emerged human CoV: SARS-CoV, MERS-CoV, and SARS-CoV-2 | Bioavailable NHC prodrug (β-d-N4-hydroxycytidine-5′-isopropyl ester), improved pulmonary function and reduced virus titer and body weight loss | Continued development of EIDD-2801 as a potent broad-spectrum antiviral |
| Tai et al. [324] | 2021 | In vivo | Sprague Dawley rats. (i) HCQ‐IV: 12 rats received a single dose of 0.590 mg HCQ sulfate per animal via IV injection; (ii) HCQ‐IT: 20 rats received a single dose of 0.590 mg HCQ sulfate per animal via intratracheal (IT) administration; and (iii) liposomal HCQ‐IT: 20 rats received a single dose of 0.284 mg liposomal HCQ sulfate per animal via IT administration 0.25, 1, 4, 24, and 72 hours postdose and for tissue/organ samples were 0.25, 4, 24, and 72 hours postdose | Inhalable liposomal hydroxychloroquine (HCQ) | Pharmacokinetic study | Compared with unformulated HCQ administered intravenously, liposomal HCQ showed higher (∼30-fold) lung exposure, longer (∼2.5-fold) half-life in lungs, but lower blood exposure with ∼20% of peak plasma concentration (Cmax) and 74% of area under the curve from 0 to 72 hours (AUC 0–72) and lower heart exposure with 23% of Cmax and 58% of AUC 0–24 (normalized for dose) | Inhalable liposomal HCQ may provide clinical benefit and serve as a potential treatment of COVID-19 |
| Tortorici et al. [325] | 2020 | In vivo | From two individuals recovering from severe COVID-19 disease. Surface plasmon resonance (SPR) and flow cytometry. Cell-cell fusion assay using VeroE6 cells/Syrian hamster model | Human neutralizing antibodies (S2E12 and S2M11) | Against SARS-CoV-2 | IC50 values were 1.2 to 6.6 ng/ml to protect hamsters against SARS-CoV-2 challenge competitively block angiotensin-converting enzyme 2 (ACE2) attachment and that S2M11 also locks the spike in a closed conformation by recognition of a quaternary epitope spanning two adjacent receptor-binding domains | Antibody cocktails for prophylaxis or therapy |
| Wahl et al. [326] | 2021 | In vivo | Coronavirus replication in human lung-only mice (LoM) and histopathologic analysis LoM were administered EIDD-2801 starting 24 h or 48 h post-SARS-CoV-2 exposure and every 12 h thereafter | Ribonucleoside analog β-D-N4-hydroxycytidine (NHC): oral prodrug EIDD-2801 (also known as molnupiravir or MK-4482) | Inhibited SARS-CoV-2 replication | EIDD-2801 pre-exposure prophylaxis significantly reduced virus titers in the human lung tissues of LoM by over 100,000-fold in two independent experiments | Prophylactic administration EIDD-2801 is highly effective |
| Wang et al. [327] | 2021 | In vivo | Male hACE2 mice inoculated intranasally with SARS-CoV-2 stock virus rhesus macaque model of SARS-CoV-2 infection | Screening the FDA-approved peptide drug library with LibDock | Against SARS-CoV-2 | Ten polypeptide drugs were selected. Dalbavancin showed the strongest inhibitory ability. (EC50) was ∼12 nM. Significant inhibition of SARS-CoV-2 pseudovirion entry into HEK293/hACE2 cells, with an IC50 of ∼53 nM in both mouse and rhesus macaque models, viral replication, and histopathological injuries caused by SARS-CoV-2 infection are significantly inhibited by dalbavancin administration | Dalbavancin directly binds to human angiotensin-converting enzyme 2 (ACE2) with high affinity, thereby blocking its interaction with the SARS-CoV-2 spike protein |
| Weston et al. [328] | 2020 | In vivo | Mice were inoculated intranasally with SARS-CoV | 20 FDA-approved drugs | Against SARS-CoV-2 | 7 of these inhibit SARS-CoV-2 at non-cytotoxic concentrations. Many of the compounds have IC50 under 10 μM, chloroquine and chlorpromazine did not inhibit viral replication in mouse lungs based on viral titers recovered at 4 dpi neither drug inhibited viral replication in the lungs, but both protected against clinical disease | |
| White et al. [329] | 2021 | In vivo | A human ACE2-expressing adenovirus to transduce the naturally resistant wild-type BALB/c mice and sensitize them to SARS-CoV-2 infection, K18-hACE2 mouse model | eEF1A inhibitor plitidepsin (aplidin): targeted the eukaryotic translation machinery | Inhibited SARS-CoV-2 replication | IC90 of 1.76 nM: Vero E6 IC90 of 0.88 Nm: human cell line significantly reduced genomic RNA content reduction of nearly 2 log units in SARS-CoV-2 viral titers in the lungs of the 0.3 mg/kg plitidepsin group, reduction of 2 log units in viral lung titers at day 3, similar to two daily 50 mg/kg doses of remdesivir | Plitidepsin as a host-targeted anti-SARS-CoV-2 agent with in vivo efficacy |
| Wu et al. [330] | 2020 | In vivo | 10 male C57BL/6 mice related to 200 mg/kg/day GB-2 everyday by oral administration. immunohistochemistry (IHC) assessment | GB-2, the formula from Tian Shang Sheng Mu of Chiayi Puzi Peitian Temple: in Taiwan: the index compound of Camellia sinensis var. assamica extract | The protein and mRNA expression of ACE2 and TMPRSS2 | No mouse death and no significant alteration in mouse activity or body weight (IHC) data revealed that the expression levels of both ACE2 and TMPRSS2 were markedly diminished in the GB-2 group compared with the control group 50 μg/mL of theaflavin could inhibit protein expression of ACE2 and TMPRSS2 | GB-2 and theaflavin could act as potential compounds for ACE2 and TMPRSS2 inhibitors |
| Yuen et al. [331] | 2021 | Ex vivo | Human lung tissues were treated with the indicated drugs overnight, followed by SARS‐CoV‐2 infection at 2 × 106 PFU/well | Small molecules targeting the ULK1/Atg1 complex involved in the induction stage of autophagy (ULK1 inhibitor SBI0206965), the ATG14/Beclin1/VPS34 complex involved in the nucleation step of autophagy (class III PI3‐kinase inhibitor VPS34‐IN1), and a widely used autophagy inhibitor that persistently inhibits class I and temporary inhibits class III PI3‐kinase (3‐MA) and a clinically approved autophagy inhibitor that suppresses autophagy by inhibiting lysosomal acidification and prevents the formation of autophagolysosome (HCQ) | Target the key cellular factors involved in key steps of the autophagy pathway | Not all the tested autophagy inhibitors suppressed SARS-CoV-2 infection inhibition of class III PI3‐kinase Vps34 downstream of ULK1, in contrast to inhibition of ULK1, which caused a significant decrease in SARS‐CoV‐2 replication (EC50) of HCQ was 19 μM, that of Vps34‐IN1 was 0.82 μM Vps34‐IN1 potently inhibited SARS‐CoV‐2 viral replication in normal ex vivo human lung tissue culture in a dose‐dependent manner | Class III PI3-kinase may be a possible target for COVID-19 therapeutic development |
| Zhang et al. [332] | 2021 | In vitro/in vivo | Preparation of MAbs from female BALB/c mice and wild-type Balb/c mice were intranasally inoculated with hACE2-encoded adenovirus 5 (Ad5-hACE2) to allow expression of the hACE2 receptor in the lung, followed by intranasal infection with live SARS-CoV-2 3 days later | Mouse anti-SARS-CoV-2-neutralizing MAbs: 2H2 and 3C1 | Spike (S) protein | (IC50) of 12 ng/mL effectively treat SARS-CoV-2-infected mice even when administered as late as 24 h post-infection |
Human monoclonal antibodies were the most evaluated drugs (N = 4), promote the reduction in viral load (in vitro), and prevent infection in animal models of SARS-CoV-2.
A prodrug of hydroxycytidine (molnupiravir) improved pulmonary function and reduced viral titer in vivo and was introduced as a potential broad-spectrum antiviral agent against SARS-CoV-2.
Chloroquine and chlorpromazine did not inhibit viral replication in mouse lungs, but protected them against clinical disease.
Dalbavancin shows significant inhibitory ability in both in vitro and in vivo models of COVID-19. The drug binds directly to ACE2 and blocks its interaction with the SARS-CoV-2 spike protein.
4. Discussion
Drug development is a multistep process, typically requiring more than five years to assure the safety and efficacy of the new compound. There are several strategies in antiviral drugs for coronaviruses including empirical testing of known antiviral drugs, large-scale phenotypic screening of compound libraries, and target-based drug discovery. To date, an increasing number of drugs have been shown to have anticoronavirus activities in vitro and in vivo, but only remdesivir and several neutralizing antibodies have been approved by the US FDA for treating COVID-19. However, remdesivir's clinical effects are controversial and new antiviral drugs are still urgently needed. Given the urgency of the SARS-CoV-2 outbreak, here we discuss the discovery and development of new therapeutics for SARS-CoV-2 infection, which have been conducted in basic research (in silico) and preclinical study (in vitro, in vivo). Our database search identified about 3000 studies, which means a global effort for drug development in the current COVID-19 pandemic. Although the chance of successful drug development is very low (less than about 10%) and till today, there are no approved drugs. Many potential candidates (at least 420 drugs) are in clinical trials.
As summarized in the results, many compounds are in the development process for COVID-19 disease. Some of these compounds are completely new and could serve as seeds (or leads) for developing antiviral drugs against COVID-19, but as we need therapeutics as soon as possible, half of the studies focused on drug repurposing (repositioning), which is a process of investigation of existing drugs for new therapeutic purposes. With the emergence of a growing COVID-19 pandemic, the drug repurposing process was being accelerated. Clinical trials using repurposed drugs may take less time and have a lower overall cost of manufacturing and could have a wide distribution of drugs. According to our results, 260 drugs repurposed by the computational methods for COVID-19, among them saquinavir, ritonavir, and lopinavir, showed the best efficacy in in silico environment. These drugs can be rapidly repurposed for clinic application for treating COVID-19 patients given their proven safety.
Many trials are performed using a combination of ritonavir-lopinavir. The results of a systematic review and meta-analysis showed that this drug combination has no more treatment effects than other therapeutic agents in COVID-19 patients and is currently not used anymore [134]. We could not find any clinical trial on saquinavir, which is the most studied drug in silico and show high potency against COVID-19. Saquinavir could be a suggestion for further clinical research.
Given that the development of synthetic chemicals for therapeutic use is a random process that might result in serendipitous discovery, many pharmaceutical companies are now focused on the development of plant-derived drugs. Natural products and their structural analogs have historically made a major contribution to pharmacotherapy, especially for cancer and infectious diseases. Nevertheless, natural products also present challenges for drug discovery, such as technical barriers to screening, isolation, characterization, and optimization. In recent years, several technological and scientific developments—including improved analytical tools, genome mining and engineering strategies, and microbial culturing advances—are addressing such challenges and opening up new opportunities. Consequently, interest in natural products as drug leads is being revitalized. Medicinal plants have attracted significant attention to treat infectious diseases. Complex molecular structures and a wide variety of natural compounds make medicinal plants an excellent biological resource for drug discovery. Our results show that various plants have potential antiviral activities and could use or be a basis for drug development against COVID-19.
Some of the studied plants are among what used by people on a daily basis, such as green tea, aloe, curcumin, and eucalyptus. A systematic review and meta-analysis of RCTs on herbal medicine in the management of COVID-19 show the significant effects of the combined therapy of herbal medicine in treating COVID-19 without any significant side effects [135].
Our results retrieved 91 in silico studies of natural products including herbal medicine. The results of this in silico approach showed that some of these studied active ingredients have a high affinity for each of the four important viral proteins compared with the inhibitors previously reported for each of these proteins. They could possibly have an inhibitory effect on the SARS-CoV-2 and COVID-19. Most evaluated chemical compounds had inhibitory effects against one or two proteins of SARS-CoV-2. In addition to having a great affinity to attach to the viral proteins, these herbal compounds have antioxidant, vasoprotective, anticarcinogenic, and antiviral properties. Thus, they can be applied as extremely safe therapeutic natural compounds and clinical assessments might have notable outcomes for controlling COVID-19.
Due to the nature of preclinical (in vitro and in vivo) studies, the number of drug development studies in this area was less than in silico studies. Again, repurposed drugs were the most studied drugs in vitro (both herbal and synthetic). Some of them such as saquinavir, heparin, glycyrrhizic acid, and chlorpromazine show efficacy in both in vitro and in silico environments. Chlorpromazine is the single agent that was found to have efficacy in in silico, in vitro, and in vivo areas.
In terms of mechanism of action, different targets from the structural and nonstructural proteins of COVID-19 were evaluated. Most of the studies focused on the main protease, papain-like protease, and spike glycoprotein. Apart from the specific protein that leads to viral replication, SARS-CoV-2 causes a surge of pro-inflammatory cytokines and chemokines, which cause damage to lung tissue and deterioration of lung function. Therefore, the design of a drug with multitarget of action against different proteins of COVID-19 and also anti-inflammatory potential could be valuable.
Currently, there is no highly efficacious and specific treatment for SARS-CoV-2. Herein, we provided data on novel compounds in therapeutic drug discovery and development. Due to the nature of SARS-CoV-2 and the rises of several high transmissible strains, repurposing existing drugs has demonstrated power by bringing several drugs to approval for treating COVID-19 patients, such as remdesivir. Our results confirmed that a large number of repurposed agents are currently being explored for treating SARS-CoV-2 infection. However, these drugs still suffer from suboptimal therapeutic effect or known strong side effect. To accelerate drug discovery and development, especially during the current pandemic, natural products capture attention again. Our results showed that various natural bioactive compounds are being investigated in the preclinical step of drug development for COVID-19. In addition to having high affinity, these herb active ingredients have antioxidant, vasoprotective, anticarcinogenic, and antiviral properties. Therefore, they can be used as extremely safe therapeutic compounds in drug design studies to control COVID-19. However, the pharmacological effects and adverse reactions of some drugs under development are still unclear, and hence, well-designed high-quality studies are needed to further study the effectiveness and safety of these potential drugs to accelerate drug development targeting SARS-CoV-2 and thus promote progress towards ending the pandemic.
Acknowledgments
This manuscript was financially supported by a grant from the Research and Technology Department of Isfahan University of Medical Sciences (grant no. 340068), Isfahan, Iran.
Data Availability
All data generated or analyzed during this study are included in this published article.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Sohrabi C., Alsafi Z., O’Neill N., et al. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19) International Journal of Surgery . 2020;76:71–76. doi: 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. WHO coronavirus (COVID-19) dashboard. 2022. https://covid19.who.int/
- 3.Macedo A., Gonçalves N., Febra C. COVID-19 fatality rates in hospitalized patients: systematic review and meta-analysis. Annals of Epidemiology . 2021;57:14–21. doi: 10.1016/j.annepidem.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim D.-H., Choe Y. J., Jeong J.-Y. Understanding and interpretation of case fatality rate of coronavirus disease 2019. Journal of Korean Medical Science . 2020;35(12) doi: 10.3346/jkms.2020.35.e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Therapeutics and COVID-19: Living Guideline . Geneva, Swizterland: World Health Organization; 2022. [PubMed] [Google Scholar]
- 6.Rothan H. A., Byrareddy S. N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. Journal of Autoimmunity . 2020;109 doi: 10.1016/j.jaut.2020.102433.102433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mei M., Tan X. Current strategies of antiviral drug discovery for COVID-19. Frontiers in Molecular Biosciences . 2021;8:p. 310. doi: 10.3389/fmolb.2021.671263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohamadian M., Chiti H., Shoghli A., Biglari S., Parsamanesh N., Esmaeilzadeh A. COVID‐19: virology, biology and novel laboratory diagnosis. The Journal of Gene Medicine . 2021;23(2) doi: 10.1002/jgm.3303.e3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momtazi-Borojeni A. A., Banach M., Reiner Ž., et al. Interaction between coronavirus S-protein and human ACE2: hints for exploring efficient therapeutic targets to treat COVID-19. Angiology . 2021;72(2):122–130. doi: 10.1177/0003319720952284. [DOI] [PubMed] [Google Scholar]
- 10.Liu C., Zhou Q., Li Y., et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases . Washington, DC, USA: ACS Publications; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lage O. M., Ramos M. C., Calisto R., Almeida E., Vasconcelos V., Vicente F. Current screening methodologies in drug discovery for selected human diseases. Marine Drugs . 2018;16(8):p. 279. doi: 10.3390/md16080279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atanasov A. G., Zotchev S. B., Zotchev V. M., Dirsch C. T., Supuran C. T. Natural products in drug discovery: advances and opportunities. Nature Reviews Drug Discovery . 2021;20(3):200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moher D., Altman D. G., Liberati A., Tetzlaff J. PRISMA statement. Epidemiology . 2011;22(1):p. 128. doi: 10.1097/ede.0b013e3181fe7825. [DOI] [PubMed] [Google Scholar]
- 14.Surabhi S., Singh B. Computer aided drug design: an overview. Journal of Drug Delivery and Therapeutics . 2018;8(5):504–509. doi: 10.22270/jddt.v8i5.1894. [DOI] [Google Scholar]
- 15.Kroemer R. T. Structure-based drug design: docking and scoring. Current Protein & Peptide Science . 2007;8(4):312–328. doi: 10.2174/138920307781369382. [DOI] [PubMed] [Google Scholar]
- 16.Stahura F. L., Bajorath J. New methodologies for ligand-based virtual screening. Current Pharmaceutical Design . 2005;11(9):1189–1202. doi: 10.2174/1381612053507549. [DOI] [PubMed] [Google Scholar]
- 17.Schneider G., Fechner U. Computer-based de novo design of drug-like molecules. Nature Reviews Drug Discovery . 2005;4(8):649–663. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 18.Lin X., Li X., Lin X. A review on applications of computational methods in drug screening and design. Molecules . 2020;25(6):p. 1375. doi: 10.3390/molecules25061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinzi L., Rastelli G. Molecular docking: shifting paradigms in drug discovery. International Journal of Molecular Sciences . 2019;20(18):p. 4331. doi: 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonso H., Bliznyuk A. A., Gready J. E. Combining docking and molecular dynamic simulations in drug design. Medicinal Research Reviews . 2006;26(5):531–568. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 21.Eddershaw P. J., Dickins M. Advances in in vitro drug metabolism screening. Pharmaceutical Science & Technology Today . 1999;2(1):13–19. doi: 10.1016/s1461-5347(98)00108-4. [DOI] [PubMed] [Google Scholar]
- 22.Georgakis N., Ioannou E., Varotsou C., Premetis G., Chronopoulou E. G., Labrou N. E. Targeting Enzymes for Pharmaceutical Development . Berlin, Germany: Springer; 2020. Determination of half-maximal inhibitory concentration of an enzyme inhibitor. [DOI] [PubMed] [Google Scholar]
- 23.Pellegatti M. Preclinicalin vivo ADME studies in drug development: a critical review. Expert Opinion on Drug Metabolism & Toxicology . 2012;8(2):161–172. doi: 10.1517/17425255.2012.652084. [DOI] [PubMed] [Google Scholar]
- 24.Abosheasha M. A., El-Gowily A. H. Superiority of cilostazol among antiplatelet FDA-approved drugs against COVID 19 Mpro and spike protein: drug repurposing approach. Drug Development Research . 2020;82 doi: 10.1002/ddr.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdul Kadhim A., Hadi N., Abdulhussein M., Abdulhussein M., Zamil S., Zamil S. Preprocessing of the candidate antiviral drugs against COVID-19 in models of SARS cov2 targets. Prensa Medica Argentina . 2020;106:p. 2. doi: 10.47275/0032-745x-220. [DOI] [Google Scholar]
- 26.Abu-Saleh A. A., Awad I. E., Yadav A., Poirier R. A. Discovery of potent inhibitors for SARS-CoV-2’s main protease by ligand-based/structure-based virtual screening, MD simulations, and binding energy calculations. Physical Chemistry Chemical Physics . 2020;22(40):23099–23106. doi: 10.1039/d0cp04326e. [DOI] [PubMed] [Google Scholar]
- 27.Achilonu I., Iwuchukwu E. A., Achilonu O. J., Fernandes M. A., Sayed Y. Targeting the SARS-CoV-2 main protease using FDA-approved Isavuconazonium, a P2-P3 α-ketoamide derivative and pentagastrin: an in-silico drug discovery approach. Journal of Molecular Graphics and Modelling . 2020;101 doi: 10.1016/j.jmgm.2020.107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aftab S. O., Ghouri M. Z., Masood M. U., et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. Journal of Translational Medicine . 2020;18(1):p. 275. doi: 10.1186/s12967-020-02439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmadi K., Farasat A., Rostamian M., Johari B., Madanchi H. Enfuvirtide, an HIV-1 fusion inhibitor peptide, can act as a potent SARS-CoV-2 fusion inhibitor: an in silico drug repurposing study. Journal of Biomolecular Structure & Dynamics . 2021;40:5566–5576. doi: 10.1080/07391102.2021.1871958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed M. Z., Zia Q., Haque A., et al. FDA-approved antiviral and anti-infection agents as potential inhibitors of SARS-CoV-2 main protease: an in silico drug repurposing study. Journal of Infection and Public Health . 2021;14:611–619. doi: 10.1016/j.jiph.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anand J., Ghildiyal T., Madhwal A., Bhatt R., Verma D., Rai N. Computational guided approach for drug repurposing against SARS-CoV-2. Future Virology . 2021;16(3) doi: 10.2217/fvl-2020-0403. [DOI] [Google Scholar]
- 32.Ancy I., Sivanandam M., Kumaradhas P. Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study. Journal of Biomolecular Structure & Dynamics . 2020;39:5368–5375. doi: 10.1080/07391102.2020.1786459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ansari M. A., Jamal Q. M. S., Rehman S., et al. TAT-peptide conjugated repurposing drug against SARS-CoV-2 main protease (3CLpro): potential therapeutic intervention to combat COVID-19. Arabian Journal of Chemistry . 2020;13(11):8069–8079. doi: 10.1016/j.arabjc.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arun K., Sharanya C., Abhithaj J., Francis D., Sadasivan C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. Journal of Biomolecular Structure & Dynamics . 2020;39:4647–4658. doi: 10.1080/07391102.2020.1779819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (COVID-19) from FDA approved drugs. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c7483f842e655654db2b42 .
- 36.Baby K., Maity S., Mehta C. H., Suresh A., Nayak U. Y., Nayak Y. Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Archives of Medical Research . 2021;52(1):38–47. doi: 10.1016/j.arcmed.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker J. D., Uhrich R. L., Kraemer G. C., Love J. E., Kraemer B. C. A drug repurposing screen identifies hepatitis C antivirals as inhibitors of the SARS-CoV2 main protease. PLoS One . 2021;16(2) doi: 10.1371/journal.pone.0245962.e0245962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bharath B. R., Damle H., Ganju S., Damle L. In silico screening of known small molecules to bind ACE2 specific RBD on spike glycoprotein of SARS-CoV-2 for repurposing against COVID-19. F1000Research . 2020;9:p. 663. doi: 10.12688/f1000research.24143.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhowmik A., Biswas S., Hajra S., Saha P. In silico validation of potent phytochemical orientin as inhibitor of SARS-CoV-2 spike and host cell receptor GRP78 binding. Heliyon . 2021;7(1) doi: 10.1016/j.heliyon.2021.e05923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolelli K., Ertan-Bolelli T., Unsalan O., Altunayar-Unsalan C. Fenoterol and dobutamine as SARS-CoV-2 main protease inhibitors: a virtual screening study. Journal of Molecular Structure . 2021;1228 doi: 10.1016/j.molstruc.2020.129449.129449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavasotto C. N., Di Filippo J. I. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Molecular Informatics . 2021;40(1) doi: 10.1002/minf.202000115.e2000115 [DOI] [PubMed] [Google Scholar]
- 42.Chandel V., Sharma P. P., Raj S., Choudhari R., Rathi B., Kumar D. Structure-based drug repurposing for targeting NSP9 replicase and spike proteins of severe acute respiratory syndrome coronavirus 2. Journal of Biomolecular Structure and Dynamics . 2020;40 doi: 10.1080/07391102.2020.1811773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C. Z., Xu M., Pradhan M., et al. Identifying SARS-CoV-2 entry inhibitors through drug repurposing screens of SARS-S and MERS-S pseudotyped particles. ACS Pharmacology & Translational Science . 2020;3(6):1165–1175. doi: 10.1021/acsptsci.0c00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chidambaram S. K., Ali D., Alarifi S., Radhakrishnan S., Akbar I. In silico molecular docking: evaluation of coumarin based derivatives against SARS-CoV-2. Journal of Infection and Public Health . 2020;13(11):1671–1677. doi: 10.1016/j.jiph.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choudhary S., Malik Y. S., Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Frontiers in Immunology . 2020;11:p. 1664. doi: 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clemente C., Freiberger M., Ravetti S., Beltramo D., Garro A. An in silico analysis of Ibuprofen enantiomers in high concentrations of sodium chloride with SARS-CoV-2 main protease. Journal of Biomolecular Structure and Dynamics . 2021;40:5653–5664. doi: 10.1080/07391102.2021.1872420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cosic I., Cosic D., Loncarevic I. Analysis of ivermectin as potential inhibitor of SARS-CoV-2 using resonant recognition model. International Journal of Sciences . 2021;10(1):1–6. doi: 10.18483/ijsci.2433. [DOI] [Google Scholar]
- 48.Daoud S., Alabed S. J., Dahabiyeh L. A. Identification of potential COVID-19 main protease inhibitors using structure-based pharmacophore approach, molecular docking and repurposing studies. Acta Pharmaceutica . 2021;71(2):163–174. doi: 10.2478/acph-2021-0016. [DOI] [PubMed] [Google Scholar]
- 49.de Oliveira O. V., Rocha G. B., Paluch A. S., Costa L. T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. Journal of Biomolecular Structure and Dynamics . 2021;39(11):3924–3933. doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delijewski M., Haneczok J. AI drug discovery screening for COVID-19 reveals zafirlukast as a repurposing candidate. Medicine in Drug Discovery . 2021;9 doi: 10.1016/j.medidd.2020.100077.100077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dey D., Borkotoky S., Banerjee M. In silico identification of tretinoin as a SARS-CoV-2 envelope (E) protein ion channel inhibitor. Computers in Biology and Medicine . 2020;127 doi: 10.1016/j.compbiomed.2020.104063.104063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durdagi S. Virtual drug repurposing study against SARS-CoV-2 TMPRSS2 target. Turkish Journal of Biology . 2020;44(SI-1):185–191. doi: 10.3906/biy-2005-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Durdagi S., Aksoydan B., Dogan B., Sahin K., Shahraki A., Birgül-İyison N. Screening of clinically approved and investigation drugs as potential inhibitors of SARS-CoV-2 main protease and spike receptor-binding domain bound with ACE2 COVID19 target proteins: a virtual drug repurposing study. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c74aa5702a9b4c1c18b2d4 .
- 54.Eleftheriou P., Amanatidou D., Petrou A., Geronikaki A. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-CoV-2 virus. Molecules . 2020;25(11):p. 2529. doi: 10.3390/molecules25112529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elmezayen A. D., Al-Obaidi A., Şahin A. T., Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics . 2021;39(8):2980–2992. doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Encinar J. A., Menendez J. A. Potential drugs targeting early innate immune evasion of SARS-coronavirus 2 via 2′-O-methylation of viral RNA. Viruses . 2020;12(5) doi: 10.3390/v12050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farag A., Wang P., Ahmed M., Sadek H. Identification of FDA approved drugs targeting COVID-19 virus by structure-based drug repositioning. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c74b2a567dfe0f38ec4ee7 .
- 58.Feng S., Luan X., Wang Y., et al. Eltrombopag is a potential target for drug intervention in SARS-CoV-2 spike protein. Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases . 2020;85 doi: 10.1016/j.meegid.2020.104419.104419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferraz W. R., Gomes R. A., Novaes A. L. S., Goulart Trossini G. H. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Medicinal Chemistry . 2020;12(20):1815–1828. doi: 10.4155/fmc-2020-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer A., Sellner M., Neranjan S., Smieško M., Lill M. A. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. International Journal of Molecular Sciences . 2020;21(10) doi: 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gimeno A., Mestres-Truyol J., Ojeda-Montes M. J., et al. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. International Journal of Molecular Sciences . 2020;21(11) doi: 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo K., Wang Z., Gao P., et al. Identification of repurposal drugs and adverse drug reactions for various courses of coronavirus disease 2019 (COVID-19) based on single-cell RNA sequencing data. 2020. https://arxiv.org/abs/2005.07856 .
- 63.Gupta A., Rani C., Pant P., et al. Structure-based virtual screening and biochemical validation to discover a potential inhibitor of the SARS-CoV-2 main protease. ACS Omega . 2020;5(51):33151–33161. doi: 10.1021/acsomega.0c04808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huynh T., Cornell W., Luan B. In silico exploration of inhibitors for SARS-CoV-2’s papain-like protease. Frontiers in Chemistry . 2021;8 doi: 10.3389/fchem.2020.624163.624163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ibrahim M. A., Abdelrahman A. H., Hegazy M.-E. F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure & Dynamics . 2020;39:5756–5767. doi: 10.1080/07391102.2020.1791958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iftikhar H., Ali H. N., Farooq S., Naveed H., Shahzad-Ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Computers in Biology and Medicine . 2020;122 doi: 10.1016/j.compbiomed.2020.103848.103848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jain R., Mujwar S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Structural Chemistry . 2020;31(6):2487–2499. doi: 10.1007/s11224-020-01605-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jarvis M. A., Hansen F. A., Rosenke K., et al. Evaluation of drugs for potential repurposing against COVID-19 using a tier-based scoring system. Antiviral Therapy . 2020;25(4):223–231. doi: 10.3851/IMP3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadioglu O., Saeed M., Greten H. J., Efferth T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Computers in Biology and Medicine . 2021;133 doi: 10.1016/j.compbiomed.2021.104359.104359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kandeel M., Abdelrahman A. H., Oh-Hashi K., et al. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. Journal of Biomolecular Structure & Dynamics . 2020;39:5129–5136. doi: 10.1080/07391102.2020.1784291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kandwal S., Fayne D. Repurposing drugs for treatment of SARS-CoV-2 infection: computational design insights into mechanisms of action. Journal of Biomolecular Structure and Dynamics . 2020;40:1316–1330. doi: 10.1080/07391102.2020.1825232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khan A., Ali S. S., Khan M. T., et al. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro) Journal of Biomolecular Structure & Dynamics . 2020;39:4659–4670. doi: 10.1080/07391102.2020.1779128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan M. A., Mahmud S., Ul Alam A., Rahman M. E., Ahmed F., Rahmatullah M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: a molecular docking study. Journal of Biomolecular Structure & Dynamics . 2020;39 doi: 10.1080/07391102.2020.1796813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kouznetsova V. L., Zhang A., Tatineni M., Miller M. A., Tsigelny I. F. Potential COVID-19 papain-like protease PLpro inhibitors: repurposing FDA-approved drugs. PeerJ . 2020;8 doi: 10.7717/peerj.9965.e9965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krishnaprasad B., Maity S., Mehta C. H., Suresh A., Nayak U. Y., Nayak Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: an in silico drug repurposing for COVID-19. F1000 Research . 2020;9:1–23. doi: 10.12688/f1000research.26359.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar D., Chandel V., Raj S., Rathi B. In silico identification of potent FDA approved drugs against coronavirus COVID-19 main protease: a drug repurposing approach. Chemical Biology Letters . 2020;7(3):166–175. [Google Scholar]
- 77.Kumar V., Dhanjal J. K., Kaul S. C., Wadhwa R., Sundar D. Withanone and caffeic acid phenethyl ester are predicted to interact with main protease (M-pro) of SARS-CoV-2 and inhibit its activity. Journal of Biomolecular Structure & Dynamics . 2020;39 doi: 10.1080/07391102.2020.1772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar Y., Singh H., Patel C. N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. Journal of Infection and Public Health . 2020;13(9):1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar P., Bhardwaj T., Kumar A., et al. Reprofiling of approved drugs against SARS-CoV-2 main protease: an in-silico study. Journal of Biomolecular Structure & Dynamics . 2020;40:3170–3184. doi: 10.1080/07391102.2020.1845976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Z., Li X., Huang Y. Y., et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proceedings of the National Academy of Sciences . 2020;117(44):27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang H., Zhao L., Gong X., Hu M., Wang H. Virtual screening FDA approved drugs against multiple targets of SARS-CoV-2. Clinical and Translational Science . 2021;14 doi: 10.1111/cts.13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lokhande K. B., Doiphode S., Vyas R., Swamy K. V. Molecular docking and simulation studies on SARS-CoV-2 Mpro reveals mitoxantrone, leucovorin, birinapant, and dynasore as potent drugs against COVID-19. Journal of Biomolecular Structure & Dynamics . 2020;39:7294–7305. doi: 10.1080/07391102.2020.1805019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mahanta S., Chowdhury P., Gogoi N., et al. Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: an in silico based approach. Journal of Biomolecular Structure & Dynamics . 2020;39:3802–3811. doi: 10.1080/07391102.2020.1768902. [DOI] [PubMed] [Google Scholar]
- 84.Mahdian S., Zarrabi M., Panahi Y., Dabbagh S. Repurposing FDA-approved drugs to fight COVID-19 using in silico methods: targeting SARS-CoV-2 RdRp enzyme and host cell receptors (ACE2, CD147) through virtual screening and molecular dynamic simulations. Informatics in Medicine Unlocked . 2021;23 doi: 10.1016/j.imu.2021.100541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marak B. N., Dowarah J., Khiangte L., Singh V. P. Step toward repurposing drug discovery for COVID-19 therapeutics through in silico approach. Drug Development Research . 2020;82(3):374–392. doi: 10.1002/ddr.21757. [DOI] [PubMed] [Google Scholar]
- 86.Mohapatra S., Nath P., Chatterjee M., et al. Repurposing therapeutics for COVID-19: rapid prediction of commercially available drugs through machine learning and docking. PLoS One . 2020;15(11) doi: 10.1371/journal.pone.0241543.e0241543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Molavi Z., Razi S., Mirmotalebisohi S. A., et al. Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomedicine & Pharmacotherapy . 2021;138 doi: 10.1016/j.biopha.2021.111544.111544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mulgaonkar N. S., Wang H., Mallawarachchi S., Ruzek D., Martina B., Fernando S. Bcr-Abl tyrosine kinase inhibitor imatinib as a potential drug for COVID-19. 2020. https://www.biorxiv.org/content/10.1101/2020.06.18.158196v2 .
- 89.Mycroft-West C. J., Su D., Pagani I., et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the spike S1 receptor-binding domain with heparin. Thrombosis and Haemostasis . 2020;120(12):1700–1715. doi: 10.1055/s-0040-1721319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nayarisseri A., Khandelwal R., Madhavi M., et al. Shape-based machine learning models for the potential novel COVID-19 protease inhibitors assisted by molecular dynamics simulation. Current Topics in Medicinal Chemistry . 2020;20(24):2146–2167. doi: 10.2174/1568026620666200704135327. [DOI] [PubMed] [Google Scholar]
- 91.Odhar H. A., Ahjel S. W., Albeer A. A. M. A., Hashim A. F., Rayshan A. M., Humadi S. S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation . 2020;16(3):236–244. doi: 10.6026/97320630016236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ortega J. T., Serrano M. L., Jastrzebska B. Class A G protein-coupled receptor antagonist famotidine as a therapeutic alternative against SARS-CoV2: an in silico analysis. Biomolecules . 2020;10(6):p. 954. doi: 10.3390/biom10060954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pandey P., Khan F., Rana A. K., Srivastava Y., Jha S. K., Jha N. K. A drug repurposing approach towards elucidating the potential of flavonoids as COVID-19 spike protein inhibitors. Biointerface Research in Applied Chemistry . 2021;11(1):8482–8501. [Google Scholar]
- 94.Parveen S., Alnoman R. B. Potential exploration of recent FDA-approved anticancer drugs against models of SARS-CoV-2’s main protease and spike glycoprotein: a computational study. Biointerface Research in Applied Chemistry . 2021;11(3):10059–10073. [Google Scholar]
- 95.Peele K. A., Potla Durthi C., Srihansa T., et al. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: a computational study. Informatics in Medicine Unlocked . 2020;19 doi: 10.1016/j.imu.2020.100345.100345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pinzi L., Tinivella A., Caporuscio F., Rastelli G. Drug repurposing and polypharmacology to fight SARS-CoV-2 through inhibition of the main protease. Frontiers in Pharmacology . 2021;12 doi: 10.3389/fphar.2021.636989.636989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pokhrel R., Chapagain P., Siltberg-Liberles J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. Journal of Medical Microbiology . 2020;69(6):864–873. doi: 10.1099/jmm.0.001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ray A. K., Gupta P. S. S., Panda S. K., Biswal S., Rana M. K. Repurposing of FDA approved drugs for the identification of potential inhibitors of SARS-CoV-2 main protease. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c74b60842e659a0edb3136 .
- 99.Sachdeva C., Wadhwa A., Kumari A., Hussain F., Jha P., Kaushik N. K. In silico potential of approved antimalarial drugs for repurposing against COVID-19. OMICS: A Journal of Integrative Biology . 2020;24(10):568–580. doi: 10.1089/omi.2020.0071. [DOI] [PubMed] [Google Scholar]
- 100.Sang P., Tian S.-H., Meng Z.-H., Yang L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Advances . 2020;10(27):15775–15783. doi: 10.1039/d0ra01899f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saxena S., Meher K., Rotella M., et al. Identification of SGLT2 inhibitor ertugliflozin as a treatment for COVID-19 using computational and experimental paradigm. 2021. https://www.biorxiv.org/content/10.1101/2021.06.18.448921v1 .
- 102.Setianingsih H., Yatmasari E., Ilmawan M. F., Zahroh H., Lestari E. D. P. Targeting multiple SARS-CoV-2 and human proteins: in silico approach for COVID-19 drug repurposing. International Journal of Pharmaceutical Research . 2020;12(2):3184–3192. [Google Scholar]
- 103.Shah B., Modi P., Sagar S. R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sciences . 2020;252 doi: 10.1016/j.lfs.2020.117652.117652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sharma S. K., Mishra A. K. Analysis of therapeutic targets for COVID 19-nCoV/SARS-CoV-2 and discovery by virtual screening methods. TANG (Humanitas Medicine) . 2020;10(2):p. 14. [Google Scholar]
- 105.Shekhar N., Sarma P., Prajapat M., et al. In silico structure-based repositioning of approved drugs for spike glycoprotein S2 domain fusion peptide of SARS-CoV-2: rationale from molecular dynamics and binding free energy calculations. mSystems . 2020;5(5) doi: 10.1128/msystems.00382-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Singh S. K., Upadhyay A. K., Reddy M. S. Screening of potent drug inhibitors against SARS-CoV-2 RNA polymerase: an in silico approach. 3 Biotech . 2021;11(2):1–13. doi: 10.1007/s13205-020-02610-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sinha M., Gupta A., Gupta S., et al. Analogue discovery of safer alternatives to HCQ and CQ drugs for SAR-CoV-2 by computational design. Computers in Biology and Medicine . 2021;130 doi: 10.1016/j.compbiomed.2021.104222.104222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soni H., Gautam D., Sharma S., Malik J. Rifampicin as potent inhibitor of COVID-19 main protease: in-silico docking approach. Saudi Journal of Medical and Pharmaceutical Sciences . 2020;6(9):588–593. doi: 10.36348/sjmps.2020.v06i09.001. [DOI] [Google Scholar]
- 109.Tariq A., Mateen R. M., Afzal M. S., Saleem M. Paromomycin: a potential dual targeted drug effectively inhibits both spike (S1) and main protease of COVID-19. International Journal of Infectious Diseases . 2020;98:166–175. doi: 10.1016/j.ijid.2020.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tatar G., Ozyurt E., Turhan K. Computational drug repurposing study of the RNA binding domain of SARS‐CoV‐2 nucleocapsid protein with antiviral agents. Biotechnology Progress . 2021;37(2) doi: 10.1002/btpr.3110.e3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tejera E., Munteanu C. R., López-Cortés A., Cabrera-Andrade A., Pérez-Castillo Y. Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules . 2020;25(21) doi: 10.3390/molecules25215172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Teralı K., Baddal B., Gülcan H. O. Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: insights from a molecular mechanics-assisted structure-based virtual screening experiment. Journal of Molecular Graphics and Modelling . 2020;100 doi: 10.1016/j.jmgm.2020.107697.107697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Trezza A., Iovinelli D., Santucci A., Prischi F., Spiga O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Scientific Reports . 2020;10(1) doi: 10.1038/s41598-020-70863-9.13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ugurel O. M., Mutlu O., Sariyer E., et al. Evaluation of the potency of FDA-approved drugs on wild type and mutant SARS-CoV-2 helicase (NSP13) International Journal of Biological Macromolecules . 2020;163:1687–1696. doi: 10.1016/j.ijbiomac.2020.09.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Unni S., Aouti S., Thiyagarajan S., Padmanabhan B. Identification of a repurposed drug as an inhibitor of spike protein of human coronavirus SARS-CoV-2 by computational methods. Journal of Biosciences . 2020;45(1) doi: 10.1007/s12038-020-00102-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vaishali C., Prakash P., Sharma S., Raj B., Rathi D. Structure based drug repurposing through targeting NSP9 replicase and spike proteins of SARS-CoV-2. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c74b26337d6c3f76e27a44 . [DOI] [PMC free article] [PubMed]
- 117.Verma D., Kapoor S., Das S., Thakur K. Potential inhibitors of SARS-CoV-2 main protease (M Pro) identified from the library of FDA approved drugs using molecular docking studies. 2020. https://www.preprints.org/manuscript/202004.0149/v1 . [DOI] [PMC free article] [PubMed]
- 118.Wei T. Z., Wang H., Wu X. Q., et al. In silico screening of potential spike glycoprotein inhibitors of SARS-CoV-2 with drug repurposing strategy. Chinese Journal of Integrative Medicine . 2020;26(9):663–669. doi: 10.1007/s11655-020-3427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu H., Liu B., Xiao Z., et al. Computational and experimental studies reveal that thymoquinone blocks the entry of coronaviruses into in vitro cells. Infectious Diseases and Therapy . 2021;10(1):483–494. doi: 10.1007/s40121-021-00400-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abdelli I., Hassani F., Bekkel Brikci S., Ghalem S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. Journal of Biomolecular Structure and Dynamics . 2021;39(9):3263–3276. doi: 10.1080/07391102.2020.1718553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abouelela M. E., Assaf H. K., Abdelhamid R. A., et al. Identification of potential SARS-CoV-2 main protease and spike protein inhibitors from the genus aloe: an in silico study for drug development. Molecules . 2021;26(6):p. 1767. doi: 10.3390/molecules26061767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Adem S., Eyupoglu V., Sarfraz I., Rasul A., Ali M. Identification of potent COVID-19 main protease (Mpro) inhibitors from natural polyphenols: an in silico strategy unveils a hope against CORONA. 2020. https://www.preprints.org/manuscript/202003.0333/v1 . [DOI] [PMC free article] [PubMed]
- 123.Allam A. E., Assaf H. K., Hassan H. A., Shimizu K., Elshaier Y. A. M. M. Anin silicoperception for newly isolated flavonoids from peach fruit as privileged avenue for a countermeasure outbreak of COVID-19. RSC Advances . 2020;10(50):29983–29998. doi: 10.1039/d0ra05265e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Al‐Sehemi A. G., Olotu F. A., Sanal Dev D., et al. Natural products database screening for the discovery of naturally occurring SARS‐cov‐2 spike glycoprotein blockers. ChemistrySelect . 2020;5(42) doi: 10.1002/slct.202003349.13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Attia G. H., Moemen Y. S., Youns M., Ibrahim A. M., Abdou R., El Raey M. A. Antiviral zinc oxide nanoparticles mediated by hesperidin and in silico comparison study between antiviral phenolics as anti-SARS-CoV-2. Colloids and Surfaces B: Biointerfaces . 2021;203 doi: 10.1016/j.colsurfb.2021.111724.111724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Azim K. F., Ahmed S. R., Banik A., Khan M. M. R., Deb A., Somana S. R. Screening and druggability analysis of some plant metabolites against SARS-CoV-2: an integrative computational approach. Informatics in Medicine Unlocked . 2020;20 doi: 10.1016/j.imu.2020.100367.100367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Çakır B., Okuyan B., Şener G., Tunali-Akbay T. Investigation of beta-lactoglobulin derived bioactive peptides against SARS-CoV-2 (COVID-19): in silico analysis. European Journal of Pharmacology . 2021;891 doi: 10.1016/j.ejphar.2020.173781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chatterjee S., Maiti S., Banerjee A., Kanwar M. In-silico study of hesperidin, epigallocatechin (EGCG), kaempferol and quercetin. 2021. https://assets.researchsquare.com/files/rs-525091/v1_covered.pdf?c=1631867322 .
- 129.Chikhale R. V., Gurav S. S., Patil R. B., et al. SARS-CoV-2 host entry and replication inhibitors from Indian ginseng: an in-silico approach. Journal of Biomolecular Structure & Dynamics . 2020;39:4510–4521. doi: 10.1080/07391102.2020.1778539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chikhale R., Sinha S. K., Wanjari M., et al. Computational assessment of saikosaponins as adjuvant treatment for COVID-19: molecular docking, dynamics, and network pharmacology analysis. Molecular Diversity . 2021;25:1889–1904. doi: 10.1007/s11030-021-10183-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chikhale R. V., Sinha S. K., Patil R. B., et al. In-silico investigation of phytochemicals from Asparagus racemosus as plausible antiviral agent in COVID-19. Journal of Biomolecular Structure and Dynamics . 2020;39(14) doi: 10.1080/07391102.2020.1784289. [DOI] [PubMed] [Google Scholar]
- 132.Chowdhury P. In silico investigation of phytoconstituents from Indian medicinal herb “Tinospora cordifolia (giloy)” against SARS-CoV-2 (COVID-19) by molecular dynamics approach. Journal of Biomolecular Structure and Dynamics . 2020;39:6792–6809. doi: 10.1080/07391102.2020.1803968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dahab M. A., Hegazy M. M., Abbass H. S. Hordatines as a potential inhibitor of COVID-19 main protease and RNA polymerase: an in-silico approach. Natural Products and Bioprospecting . 2020;10(6):453–462. doi: 10.1007/s13659-020-00275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Alhumaid S., Al Mutair A., Al Alawi Z., Alhmeed N., Zaidi A. R. Z, Tobaiqy M. Efficacy and safety of lopinavir/ritonavir for treatment of COVID-19: a systematic review and meta-analysis. Tropical Medicine and Infectious Disease . 2020;5(4):p. 180. doi: 10.3390/tropicalmed5040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ang L., Song E., Lee H. W., Lee M. S. Herbal medicine for the treatment of coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis of randomized controlled trials. Journal of Clinical Medicine . 2020;9(5):p. 1583. doi: 10.3390/jcm9051583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Das P., Majumder R., Mandal M., Basak P. In-silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. Journal of Biomolecular Structure & Dynamics . 2020;39(16):1–16. doi: 10.1080/07391102.2020.1796799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Debnath P., Debnath B., Bhaumik S., Debnath S. In silico identification of potential inhibitors of ADP-ribose phosphatase of SARS-CoV-2 NSP3 by combining E-pharmacophore- and receptor-based virtual screening of database. ChemistrySelect . 2020;5(30):9388–9398. doi: 10.1002/slct.202001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dev S. A., Kaur I. Bioactive molecules from eucalyptus essential oil as potential inhibitors of COVID 19 corona virus infection by molecular docking studies. Kragujevac Journal of Science . 2020;42:29–43. doi: 10.5937/kgjsci2042029d. [DOI] [Google Scholar]
- 139.Duru C. E., Duru I. A., Adegboyega A. E. In silico identification of compounds from Nigella sativa seed oil as potential inhibitors of SARS-CoV-2 targets. Bulletin of the National Research Centre . 2021;45(1):1–13. doi: 10.1186/s42269-021-00517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.El-Demerdash A., Metwaly A. M., Hassan A., et al. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against sars-cov-2 (Covid-19) Biomolecules . 2021;11(3):1–25. doi: 10.3390/biom11030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Elekofehinti O. O., Iwaloye O., Josiah S. S., Lawal A. O., Akinjiyan M. O., Ariyo E. O. Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Molecular Diversity . 2020;25:1761–1773. doi: 10.1007/s11030-020-10151-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.El‐Hawary S. S., Mohammed R., Bahr H. S., et al. Soybean‐associated endophytic fungi as potential source for anti‐COVID‐19 metabolites supported by docking analysis. Journal of Applied Microbiology . 2021;131(3):1193–1211. doi: 10.1111/jam.15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Emirik M. Potential therapeutic effect of turmeric contents against SARS-CoV-2 compared with experimental COVID-19 therapies: in silico study. Journal of Biomolecular Structure & Dynamics . 2020;40:2024–2037. doi: 10.1080/07391102.2020.1835719. [DOI] [PubMed] [Google Scholar]
- 144.Fakhar Z., Faramarzi B., Pacifico S., Faramarzi S. Anthocyanin derivatives as potent inhibitors of SARS-CoV-2 main protease: an in-silico perspective of therapeutic targets against COVID-19 pandemic. Journal of Biomolecular Structure & Dynamics . 2020;39:6171–6183. doi: 10.1080/07391102.2020.1801510. [DOI] [PubMed] [Google Scholar]
- 145.Falade V. A., Adelusi T. I., Adedotun I. O., Abdul-Hammed M., Lawal T. A., Agboluaje S. A. In silico investigation of saponins and tannins as potential inhibitors of SARS-CoV-2 main protease (Mpro) Silico Pharmacology . 2021;9(1) doi: 10.1007/s40203-020-00071-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fitriani I. N., Utami W., Zikri A. T., Santoso P. In silico approach of potential phytochemical inhibitor from Moringa oleifera, Cocos nucifera, Allium cepa, Psidium guajava, and Eucalyptus globulus for the treatment of COVID-19 by molecular docking. 2020. https://www.scienceopen.com/document?vid=45020f08-2651-475b-b2bc-eec16a177515 .
- 147.Gangadevi S., Badavath V. N., Thakur A., et al. Kobophenol A inhibits binding of host ACE2 receptor with spike rbd domain of SARS-CoV-2, a lead compound for blocking COVID-19. The journal of physical chemistry letters . 2021;12(7):1793–1802. doi: 10.1021/acs.jpclett.0c03119. [DOI] [PubMed] [Google Scholar]
- 148.Gangarapu K., Sathiyarajeswaran P., Kanakavalli K., et al. In silico computational screening of kabasura kudineer-official siddha formulation and JACOM-novel herbal coded formulation against SARS-CoV-2 spike protein. 2020. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3574412 . [DOI] [PMC free article] [PubMed]
- 149.Ghosh R., Chakraborty A., Biswas A., Chowdhuri S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors—an in silico docking and molecular dynamics simulation study. Journal of Biomolecular Structure and Dynamics . 2020;39(12) doi: 10.1080/07391102.2020.1779818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghosh R., Chakraborty A., Biswas A., Chowdhuri S. Identification of alkaloids from Justicia adhatoda as potent SARS CoV-2 main protease inhibitors: an in silico perspective. Journal of Molecular Structure . 2021;1229 doi: 10.1016/j.molstruc.2020.129489.129489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gorla U. S., Rao G. K., Kulandaivelu U. S., Alavala R. R., Panda S. P. Lead finding from selected flavonoids with antiviral (SARS-CoV-2) potentials against COVID-19: an in-silico evaluation. Combinatorial Chemistry & High Throughput Screening . 2020;24 doi: 10.2174/1386207323999200818162706. [DOI] [PubMed] [Google Scholar]
- 152.Gurung A. B., Ali M. A., Lee J., Farah M. A., Al-Anazi K. M. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sciences . 2020;255 doi: 10.1016/j.lfs.2020.117831.117831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gyebi G. A., Ogunro O. B., Adegunloye A. P., Ogunyemi O. M., Afolabi S. O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics . 2021;39(9):3396–3408. doi: 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Elwakil B., Shaaban M. M., Bekhit A. A., El-Naggar M. Y., Olama Z. A. Potential anti-COVID-19 activity of Egyptian propolis using computational modeling. Future Virology . 2021;16(2):107–116. [Google Scholar]
- 155.Hasan A., Al Mahamud R., Jannat K., et al. Phytochemicals from Solanum surattense Burm. f. have high binding affinities for C-3 like main protease of COVID-19 (SARS-CoV-2) Journal of Medicinal Plants Studies . 2020;8(4):20–26. [Google Scholar]
- 156.Hashem H. In silico approach of some selected honey constituents as SARS-CoV-2 main protease (COVID-19) inhibitors. 2020. https://chemrxiv.org/engage/chemrxiv/article-details/60c749e1842e651579db2e34 .
- 157.Ibrahim M. A. A., Abdeljawaad K. A. A., Abdelrahman A. H. M., Hegazy M. E. F. Natural-like products as potential SARS-CoV-2 Mpro inhibitors: in-silico drug discovery. Journal of Biomolecular Structure & Dynamics . 2020;39:5722–5734. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ibrahim M. A. A., Abdelrahman A. H. M., Hussien T. A., et al. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Computers in Biology and Medicine . 2020;126 doi: 10.1016/j.compbiomed.2020.104046.104046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Isa M. A., Mustapha A., Qazi S., et al. In silico molecular docking and molecular dynamic simulation of potential inhibitors of 3C-like main proteinase (3CLpro) from severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) using selected African medicinal plants. Advances in Traditional Medicine . 2020;22 doi: 10.1007/s13596-020-00523-w. [DOI] [Google Scholar]
- 160.Istifli E. S., Netz P. A., Sihoglu Tepe A., Husunet M. T., Sarikurkcu C., Tepe B. In silico analysis of the interactions of certain flavonoids with the receptor-binding domain of 2019 novel coronavirus and cellular proteases and their pharmacokinetic properties. Journal of Biomolecular Structure and Dynamics . 2020;40 doi: 10.1080/07391102.2020.1840444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jan J. T., Cheng T. R., Juang Y. P., et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proceedings of the National Academy of Sciences of the United States of America . 2021;118(5) doi: 10.1073/pnas.2021579118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jo S., Kim S., Kim D. Y., Kim M. S., Shin D. H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. Journal of Enzyme Inhibition and Medicinal Chemistry . 2020;35(1):1539–1544. doi: 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Joshi R. S., Jagdale S. S., Bansode S. B., et al. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure & Dynamics . 2021;39(9):3099–3114. doi: 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Junior N. N., Santos I. A., Meireles B. A., et al. In silico evaluation of lapachol derivatives binding to the NSP9 of SARS-CoV-2. Journal of Biomolecular Structure & Dynamics . 2021;40 doi: 10.1080/07391102.2021.1875050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kar P., Kumar V., Vellingiri B., et al. Anisotine and amarogentin as promising inhibitory candidates against SARS-CoV-2 proteins: a computational investigation. Journal of Biomolecular Structure & Dynamics . 2020;40:4532–4542. doi: 10.1080/07391102.2020.1860133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khalifa I., Nawaz A., Sobhy R., Althawb S. A., Barakat H. Polyacylated anthocyanins constructively network with catalytic dyad residues of 3CLpro of 2019-nCoV than monomeric anthocyanins: a structural-relationship activity study with 10 anthocyanins using in-silico approaches. Journal of Molecular Graphics and Modelling . 2020;100 doi: 10.1016/j.jmgm.2020.107690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Khalifa I., Zhu W., Mohammed H. H. H., Dutta K., Li C. M. Tannins inhibit SARS-CoV-2 through binding with catalytic dyad residues of 3CL (pro): an in silico approach with 19 structural different hydrolysable tannins. Journal of Food Biochemistry . 2020;44(10) doi: 10.1111/jfbc.13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Khan M. T., Ali A., Wang Q., et al. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2—a molecular dynamic study. Journal of Biomolecular Structure and Dynamics . 2020;39:3627–3637. doi: 10.1080/07391102.2020.1769733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kiran Raj T., Ranjithkumar R., Kanthesh B. M., Gopenath T. S. C-phycocyanin of spirulina plantesis inhibits NSP12 required for replication of sars-COV-2: a novel finding in-silico. International Journal of Pharmaceutical Sciences and Research . 2020;11(9):4271–4278. [Google Scholar]
- 170.Krupanidhi S., Abraham Peele K., Venkateswarulu T., et al. Screening of phytochemical compounds of Tinospora cordifolia for their inhibitory activity on SARS-CoV-2: an in silico study. Journal of Biomolecular Structure & Dynamics . 2020;39(15):1–5. doi: 10.1080/07391102.2020.1787226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kumar A., Choudhir G., Shukla S. K., et al. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. Journal of Biomolecular Structure & Dynamics . 2020;39:3760–3770. doi: 10.1080/07391102.2020.1772112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kumar N., Singh A., Gulati H. K., et al. Phytoconstituents from ten natural herbs as potent inhibitors of main protease enzyme of SARS-COV-2: in silico study. Phytomedicine . 2021;1(4) doi: 10.1016/j.phyplu.2021.100083.100083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kumar B., Parasuraman P., Murthy T. P. K., Murahari M., Chandramohan V. In silico screening of therapeutic potentials fromStrychnos nux-vomica against the dimeric main protease (Mpro) structure of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics . 2021;40:1–19. doi: 10.1080/07391102.2021.1902394. [DOI] [PubMed] [Google Scholar]
- 174.Li Y., Chu F., Li P., et al. Potential effect of Maxing Shigan decoction against coronavirus disease 2019 (COVID-19) revealed by network pharmacology and experimental verification. Journal of Ethnopharmacology . 2021;271 doi: 10.1016/j.jep.2021.113854.113854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Maiti S., Banerjee A. Epigallocatechin gallate and theaflavin gallate interaction in SARS-CoV -2 spike-protein central channel with reference to the hydroxychloroquine interaction: bioinformatics and molecular docking study. Drug Development Research . 2021;82(1):86–96. doi: 10.1002/ddr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mahmud S., Uddin M. A. R., Paul G. K., et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Briefings in Bioinformatics . 2021;22(2):1402–1414. doi: 10.1093/bib/bbaa428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mahmud S., Uddin M. A. R., Zaman M., et al. Molecular docking and dynamics study of natural compound for potential inhibition of main protease of SARS-CoV-2. Journal of Biomolecular Structure & Dynamics . 2020;39(16):1–9. doi: 10.1080/07391102.2020.1796808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mesli F., Ghalem M., Daoud I., Ghalem S. Potential inhibitors of angiotensin converting enzyme 2 receptor of COVID-19 by Corchorus olitorius Linn using docking, molecular dynamics, conceptual DFT investigation and pharmacophore mapping. Journal of Biomolecular Structure and Dynamics . 2021;40(16) doi: 10.1080/07391102.2021.1896389. [DOI] [PubMed] [Google Scholar]
- 179.Mohammadi M., Rajabi S., Piroozmand A., Mirhosseini S. A. In silico study of pacific oyster antiviral polypeptides as potential inhibitory compounds for SARS-CoV-2 main protease. Jentashapir Journal of Cellular and Molecular Biology . 2020;11(3) doi: 10.5812/jjcmb.108932. [DOI] [Google Scholar]
- 180.Murugan N. A., Pandian C. J., Jeyakanthan J. Computational investigation on Andrographis paniculata phytochemicals to evaluate their potency against SARS-CoV-2 in comparison to known antiviral compounds in drug trials. Journal of biomolecular structure & dynamics . 2020;39:4415–4426. doi: 10.1080/07391102.2020.1777901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Naik B., Gupta N., Ojha R., Singh S., Prajapati V. K., Prusty D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. International Journal of Biological Macromolecules . 2020;40:1–17. doi: 10.1016/j.ijbiomac.2020.05.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Narkhede R. R., Pise A. V., Cheke R. S., Shinde S. D. Recognition of natural products as potential inhibitors of COVID-19 main protease (Mpro): in-silico evidences. Natural Products and Bioprospecting . 2020;10(5):297–306. doi: 10.1007/s13659-020-00253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nivetha R., Bhuvaragavan S., Muthu Kumar T., Ramanathan K., Janarthanan S. Inhibition of multiple SARS-CoV-2 proteins by an antiviral biomolecule, seselin from Aegle marmelos deciphered using molecular docking analysis. Journal of Biomolecular Structure & Dynamics . 2020;40 doi: 10.1080/07391102.2021.1955009. [DOI] [PubMed] [Google Scholar]
- 184.Ogunyemi O. M., Gyebi G. A., Elfiky A. A., et al. Alkaloids and flavonoids from African phytochemicals as potential inhibitors of SARS-Cov-2 RNA-dependent RNA polymerase: an in silico perspective. Antiviral Chemistry and Chemotherapy . 2020;28 doi: 10.1177/2040206620984076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Padhi S., Masi M., Chourasia R., Rajashekar Y., Rai A. K., Evidente A. ADMET profile and virtual screening of plant and microbial natural metabolites as SARS-CoV-2 S1 glycoprotein receptor binding domain and main protease inhibitors. European Journal of Pharmacology . 2021;890 doi: 10.1016/j.ejphar.2020.173648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pandey P., Rane J. S., Chatterjee A., et al. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. Journal of Biomolecular Structure & Dynamics . 2020;39:6306–6316. doi: 10.1080/07391102.2020.1796811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kumar M., Sundaram K., Ramasamy M. S. Coronavirus spike (S) glycoprotein (2019-Ncov) targeted siddha medicines kabasura kudineer and Thonthasura kudineer – in silico evidence for corona viral drug. Asian Journal of Pharmaceutical Research and Health Care . 2020;12(1):20–27. doi: 10.18311/ajprhc/2020/25103. [DOI] [Google Scholar]
- 188.Rahman F., Tabrez S., Ali R., Alqahtani A. S., Ahmed M. Z., Rub A. Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. Journal of Traditional and Complementary Medicine . 2021;11(2):173–179. doi: 10.1016/j.jtcme.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rahman N., Basharat Z., Yousuf M., Castaldo G., Rastrelli L., Khan H. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2) Molecules . 2020;25(10):p. 2271. doi: 10.3390/molecules25102271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rakib A., Paul A., Chy M., et al. Biochemical and computational approach of selected phytocompounds from Tinospora crispa in the management of COVID-19. Molecules . 2020;25(17):p. 3936. doi: 10.3390/molecules25173936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ramadhan D. S. F., Fakih T. M., Arfan A. Activity prediction of bioactive compounds contained in etlingera elatior against the SARS-CoV-2 main protease: an in silico approach. Borneo Journal of Pharmacy . 2020;3(4):235–242. doi: 10.33084/bjop.v3i4.1634. [DOI] [Google Scholar]
- 192.Rangsinth P., Sillapachaiyaporn C., Nilkhet S., Tencomnao T., Ung A. T., Chuchawankul S. Mushroom-derived bioactive compounds potentially serve as the inhibitors of SARS-CoV-2 main protease: an in silico approach. Journal of Traditional and Complementary Medicine . 2021;11(2):158–172. doi: 10.1016/j.jtcme.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rivero-Segura N., Gomez-Verjan J. In silico screening of natural products isolated from Mexican herbal medicines against COVID-19. Biomolecules . 2021;11(2):p. 216. doi: 10.3390/biom11020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Selvaraj C., Dinesh D. C., Panwar U., Abhirami R., Boura E., Singh S. K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 guanine-N7 methyltransferase (NSP14) for identifying antiviral inhibitors against COVID-19. Journal of biomolecular structure & dynamics . 2020;39:4582–4593. doi: 10.1080/07391102.2020.1778535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sharma A. D., Kaur I. Eucalyptus essential oil bioactive molecules from against SARS-CoV-2 spike protein: insights from computational studies. 2021. https://europepmc.org/article/ppr/ppr260910 .
- 196.Sharma A. D. Eucalyptol (1, 8 cineole) from eucalyptus essential oil a potential inhibitor of COVID 19 corona virus infection by molecular docking studies. 2020. https://www.preprints.org/manuscript/202003.0455/v1 .
- 197.Sharma A. D., Kaur I. Molecular docking studies on jensenone from eucalyptus essential oil as a potential inhibitor of COVID 19 corona virus infection. 2020. https://arxiv.org/abs/2004.00217 .
- 198.Shawan M. M. A. K., Halder S. K., Hasan M. A. Luteolin and abyssinone II as potential inhibitors of SARS-CoV-2: an in silico molecular modeling approach in battling the COVID-19 outbreak. Bulletin of the National Research Centre . 2021;45(1):27–21. doi: 10.1186/s42269-020-00479-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sindhu T., Arathi K., Akhilesh A., et al. Antiviral screening of Clerodol derivatives as COV 2 main protease inhibitor in novel corona virus disease: in silico approaches. Asian Journal of Pharmacy and Technology . 2020;10(2):60–64. doi: 10.5958/2231-5713.2020.00012.4. [DOI] [Google Scholar]
- 200.Singh R., Bhardwaj V. K., Sharma J., Purohit R., Kumar S. In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. Journal of Traditional and Complementary Medicine . 2021;12 doi: 10.1016/j.jtcme.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Singh S., Fulbabu M., Sonawane A., Kar P., Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA‐dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. Journal of Biomolecular Structure and Dynamics . 2020;39 doi: 10.1080/07391102.2020.1796810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Srimathi R., Mohan Maruga Raja M. K., Kathiravan M. K. In silico screening of traditional herbal medicine derived chemical constituents for possible potential inhibition against sars-cov-2. Journal of Natural Remedies . 2020;20(2):79–88. doi: 10.18311/jnr/2020/25278. [DOI] [Google Scholar]
- 203.Subbaiyan A., Ravichandran K., Singh S. V., et al. In silico molecular docking analysis targeting SARS-CoV-2 Spike protein and selected herbal constituents. Journal of Pure and Applied Microbiology . 2020;14(1):989–998. doi: 10.22207/jpam.14.spl1.37. [DOI] [Google Scholar]
- 204.Surti M., Patel M., Adnan M., et al. Ilimaquinone (marine sponge metabolite) as a novel inhibitor of SARS-CoV-2 key target proteins in comparison with suggested COVID-19 drugs: designing, docking and molecular dynamics simulation study. RSC Advances . 2020;10(62):37707–37720. doi: 10.1039/d0ra06379g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tao Q., Du J., Li X., et al. Network pharmacology and molecular docking analysis on molecular targets and mechanisms of Huashi Baidu formula in the treatment of COVID-19. Drug Development and Industrial Pharmacy . 2020;46(8):1345–1353. doi: 10.1080/03639045.2020.1788070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Umar H. I., Josiah S. S., Saliu T. P., Jimoh T. O., Ajayi A., Danjuma J. B. In-silico analysis of the inhibition of the SARS-CoV-2 main protease by some active compounds from selected African plants. Journal of Taibah University Medical Sciences . 2021;16(2):162–176. doi: 10.1016/j.jtumed.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Umesh K. D., Selvaraj C., Singh S. K., Dubey V. K. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure & Dynamics . 2021;39(9):3428–3434. doi: 10.1080/07391102.2020.1763202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang J. S., Chiang J. H., Tsai S., et al. In silico de novo curcuminoid derivatives from the compound library of natural products research laboratories inhibit COVID-19 3CLpro activity. Natural Product Communications . 2020;15(9) doi: 10.1177/1934578x20953262. [DOI] [Google Scholar]
- 209.Yu J.-w., Wang L., Bao L.-d. Exploring the active compounds of traditional Mongolian medicine in intervention of novel coronavirus (COVID-19) based on molecular docking method. Journal of Functional Foods . 2020;71 doi: 10.1016/j.jff.2020.104016.104016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang D.-h., Wu K.-l., Zhang X., Deng S.-q., Peng B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. Journal of Integrative Medicine . 2020;18(2):152–158. doi: 10.1016/j.joim.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Abu-Melha S., Edrees M. M., Riyadh S. M., Abdelaziz M. R., Elfiky A. A., Gomha S. M. Clean grinding technique: a facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing Thiazole moiety against SARS-CoV-2 main protease (Mpro) Molecules . 2020;25(19) doi: 10.3390/molecules25194565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Aghaee E., Ghodrati M., Ghasemi J. B. In silico exploration of novel protease inhibitors against coronavirus 2019 (COVID-19) Informatics in Medicine Unlocked . 2021;23 doi: 10.1016/j.imu.2021.100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ahmad S., Waheed Y., Ismail S., Najmi M. H., Ansari J. K. Rational design of potent anti-COVID-19 main protease drugs: an extensive multi-spectrum in silico approach. Journal of Molecular Liquids . 2021;330 doi: 10.1016/j.molliq.2021.115636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ahmed S., Mahtarin R., Ahmed S. S., et al. Investigating the binding affinity, interaction, and structure-activity-relationship of 76 prescription antiviral drugs targeting RdRp and Mpro of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics . 2020;39 doi: 10.1080/07391102.2020.1796804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Alabboud M., Javadmanesh A. In silico study of various antiviral drugs, vitamins, and natural substances as potential binding compounds with SARS-CoV-2 main protease. DYSONA-Life Science . 2020;1(2):44–63. [Google Scholar]
- 216.Alamri M. A., Tahir ul Qamar M., Mirza M. U., Alqahtani S. M., Froeyen M., Chen L. L. Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. Journal of Pharmaceutical Analysis . 2020;10(6):546–559. doi: 10.1016/j.jpha.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Alexpandi R., De Mesquita J. F., Pandian S. K., Ravi A. V. Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and Spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: an in silico analysis. Frontiers in Microbiology . 2020;11:p. 1796. doi: 10.3389/fmicb.2020.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Al-Sehemi A. G., Pannipara M., Parulekar R. S., et al. Potential of NO donor furoxan as SARS-CoV-2 main protease (Mpro) inhibitors: in silico analysis. Journal of Biomolecular Structure and Dynamics . 2020;39(15):1–15. doi: 10.1080/07391102.2020.1790038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Al-Shar’i N. A. Tackling COVID-19: identification of potential main protease inhibitors via structural analysis, virtual screening, molecular docking and MM-PBSA calculations. Journal of Biomolecular Structure and Dynamics . 2020;39:6689–6704. doi: 10.1080/07391102.2020.1800514. [DOI] [PubMed] [Google Scholar]
- 220.Badavath V. N., Kumar A., Samanta P. K., et al. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. Journal of Biomolecular Structure & Dynamics . 2020;40 doi: 10.1080/07391102.2020.1845800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Barros R. O., Junior F. L. C. C., Pereira W. S., Oliveira N. M. N., Ramos R. M. Interaction of drug candidates with various SARS-CoV-2 receptors: an in silico study to combat COVID-19. Journal of Proteome Research . 2020;19(11):4567–4575. doi: 10.1021/acs.jproteome.0c00327. [DOI] [PubMed] [Google Scholar]
- 222.Basit A., Ali T., Rehman S. U. Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. Journal of Biomolecular Structure & Dynamics . 2020;39:3605–3614. doi: 10.1080/07391102.2020.1768150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Battisti V., Wieder O., Garon A., Seidel T., Urban E., Langer T. A computational approach to identify potential novel inhibitors against the coronavirus SARS-CoV-2. Molecular Informatics . 2020;39(10) doi: 10.1002/minf.202000090.e2000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Benítez-Cardoza C. G., Vique-Sánchez J. L. Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD), to develop a drug. Life Sciences . 2020;256 doi: 10.1016/j.lfs.2020.117970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cava C., Bertoli G., Castiglioni I. In silico discovery of candidate drugs against COVID-19. Viruses . 2020;12(4) doi: 10.3390/v12040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen Y. W., Yiu C.-P. B., Wong K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research . 2020;9:p. 129. doi: 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Choudhary M. I., Shaikh M., Tul-Wahab W., Ur-Rahman R. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One . 2020;15(7) doi: 10.1371/journal.pone.0235030.e0235030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chunduru K., Sankhe R., Begum F., et al. In silico study to evaluate the antiviral activity of novel structures against 3c-like protease of novel coronavirus (COVID-19) and SARS-CoV. Medicinal Chemistry . 2021;17(4):380–395. doi: 10.2174/1573396316999200727125522. [DOI] [PubMed] [Google Scholar]
- 229.Coelho C., Gallo G., Campos C. B., Hardy L., Würtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One . 2020;15(10) doi: 10.1371/journal.pone.0240079.e0240079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Dai W., Zhang B., Jiang X. M., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science . 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.de Lima Menezes G., da Silva R. A. Identification of potential drugs against SARS-CoV-2 non-structural protein 1 (NSP1) Journal of Biomolecular Structure and Dynamics . 2020;39(15):1–11. doi: 10.1080/07391102.2020.1792992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Debnath B., Saha A. K., Bhaumik S., Debnath S. In silico identification of potential inhibitors of the main protease of SARS-CoV-2 using combined ligand-based and structure-based drug design approach. Eurasian Journal of Medicine and Oncology . 2020;4(4):336–348. [Google Scholar]
- 233.Di Micco S., Musella S., Scala M. C., et al. In silico analysis revealed potential anti-SARS-CoV-2 main protease activity by the Zonulin inhibitor larazotide acetate. Frontiers in Chemistry . 2021;8:p. 1271. doi: 10.3389/fchem.2020.628609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.El Hassab M. A., Ibrahim T. M., Al-Rashood S. T., Alharbi A., Eskandrani R. O., Eldehna W. M. In silico identification of novel SARS-COV-2 2′-O-methyltransferase (NSP16) inhibitors: structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. Journal of Enzyme Inhibition and Medicinal Chemistry . 2021;36(1):727–736. doi: 10.1080/14756366.2021.1885396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Elmessaoudi-Idrissi M., Tsukiyama-Kohara K., Nourlil J., et al. Structure-guided discovery approach identifies potential lead compounds targeting Mpro of SARS-CoV-2. VirusDisease . 2020;31(4):549–553. doi: 10.1007/s13337-020-00627-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Feitosa E. L., Júnior F. T. D. S. S., Nery Neto J. A. O. N., et al. Covid-19: rational discovery of the therapeutic potential of melatonin as a sars-cov-2 main protease inhibitor. International Journal of Medical Sciences . 2020;17(14):2133–2146. doi: 10.7150/ijms.48053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gurung A. B., Ali M. A., Lee J., Farah M. A., Al-Anazi K. M. Identification of potential SARS-CoV-2 entry inhibitors by targeting the interface region between the spike RBD and human ACE2. Journal of Infection and Public Health . 2021;14(2):227–237. doi: 10.1016/j.jiph.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Haider Z., Subhani M. M., Farooq M. A., et al. In-silico pharmacophoric and molecular docking-based drug discovery against the main protease (Mpro) of SARS-CoV-2, a causative agent COVID-19. Pakistan Journal of Pharmaceutical Sciences . 2020;33(6):2697–2705. [PubMed] [Google Scholar]
- 239.Hall D. C., Ji H. F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Medicine and Infectious Disease . 2020;35 doi: 10.1016/j.tmaid.2020.101646.101646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Havranek B., Islam S. M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. Journal of Biomolecular Structure & Dynamics . 2020;39:4304–4315. doi: 10.1080/07391102.2020.1776158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ibrahim M. A., Abdelrahman A. H., Allemailem K. S., Almatroudi A., Moustafa M. F., Hegazy M.-E. F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. The Protein Journal . 2021;40:296–309. doi: 10.1007/s10930-020-09945-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Jaiswal G., Kumar V. In-silico design of a potential inhibitor of SARS-CoV-2 S protein. PLoS One . 2020;15(10) doi: 10.1371/journal.pone.0240004.e0240004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jamalan M., Barzegari E., Gholami-Borujeni F. Structure-based screening to discover new inhibitors for papain-like proteinase of SARS-CoV-2: an in silico study. Journal of Proteome Research . 2020;20(1):1015–1026. doi: 10.1021/acs.jproteome.0c00836. [DOI] [PubMed] [Google Scholar]
- 244.Jin Z., Du X., Xu Y., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature . 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 245.Kanhed A. M., Patel D. V., Teli D. M., Patel N. R., Chhabria M. T., Yadav M. R. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Molecular Diversity . 2021;25(1):383–401. doi: 10.1007/s11030-020-10130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kavitha K., Sivakumar S., Ramesh B. 1, 2, 4 triazolo[1, 5-a] pyrimidin-7-ones as novel SARS-CoV-2 main protease inhibitors: in silico screening and molecular dynamics simulation of potential COVID-19 drug candidates. Biophysical Chemistry . 2020;267 doi: 10.1016/j.bpc.2020.106478.106478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Krishnan D. A., Sangeetha G., Vajravijayan S., Nandhagopal N., Gunasekaran K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Informatics in Medicine Unlocked . 2020;20 doi: 10.1016/j.imu.2020.100392.100392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kumar R., Kumar R., Tanwar P. In silico drug designing for COVID-19: an approach of high-throughput virtual screening, molecular, and essential dynamics simulations. Journal of Biomolecular Structure and Dynamics . 2021;40(16) doi: 10.1080/07391102.2021.1897681. [DOI] [PubMed] [Google Scholar]
- 249.Kumar S., Sharma P. P., Shankar U., et al. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: molecular docking, molecular dynamics simulation, and structure-activity relationship studies. Journal of Chemical Information and Modeling . 2020;60(12):5754–5770. doi: 10.1021/acs.jcim.0c00326. [DOI] [PubMed] [Google Scholar]
- 250.Kwarteng A., Asiedu E., Sakyi S. A., Asiedu S. O. Targeting the SARS-CoV2 nucleocapsid protein for potential therapeutics using immuno-informatics and structure-based drug discovery techniques. Biomedicine and Pharmacotherapy . 2020;132 doi: 10.1016/j.biopha.2020.110914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Li D., Luan J., Zhang L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochemical and Biophysical Research Communications . 2021;538:72–79. doi: 10.1016/j.bbrc.2020.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Mathpal S., Joshi T., Sharma P., et al. A dynamic simulation study of FDA drug from zinc database against COVID-19 main protease receptor. Journal of Biomolecular Structure and Dynamics . 2020;40 doi: 10.1080/07391102.2020.1821785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Maurya S. K., Maurya A. K., Mishra N., Siddique H. R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. Journal of Receptors and Signal Transduction . 2020;40(6):605–612. doi: 10.1080/10799893.2020.1772298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Mohamed M. F. A., Abuo-Rahma G. E. D. A., Hayallah A. M., Aziz M. A., Nafady A., Samir E. Molecular docking study reveals the potential repurposing of histone deacetylase inhibitors against Covid-19. International Journal of Pharmaceutical Sciences and Research . 2020;11(9):4261–4270. [Google Scholar]
- 255.Monajemi H., Zain S. M. Strong inhibition of M-protease activity of coronavirus by using PX-12 inhibitor based on ab initio ONIOM calculations. Journal of Chemical Research . 2021;45(1-2):136–140. doi: 10.1177/1747519820938025. [DOI] [Google Scholar]
- 256.Motiwale M., Yadav N. S., Kumar S., et al. Finding potent inhibitors for COVID-19 main protease (M-pro): an in silico approach using SARS-CoV-3CL protease inhibitors for combating CORONA. Journal of Biomolecular Structure & Dynamics . 2020;40 doi: 10.1080/07391102.2020.1829501. [DOI] [PubMed] [Google Scholar]
- 257.Mutlu O., Ugurel O. M., Sariyer E., et al. Targeting SARS-CoV-2 NSP12/NSP8 interaction interface with approved and investigational drugs: an in silico structure-based approach. Journal of Biomolecular Structure and Dynamics . 2020;40 doi: 10.1080/07391102.2020.1819882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Naidoo D., Roy A., Kar P., Mutanda T., Anandraj A. Cyanobacterial metabolites as promising drug leads against the Mpro and PLpro of SARS-CoV-2: an in silico analysis. Journal of Biomolecular Structure & Dynamics . 2020;39:6218–6230. doi: 10.1080/07391102.2020.1794972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Olubiy O. O., Olagunju M., Keutmann M., Loschwitz J., Strodel B. High throughput virtual screening to discover inhibitors of the main protease of the coronavirus SARS-CoV-2. Molecules . 2020;25(14) doi: 10.3390/molecules25143193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Özdemir M., Köksoy B., Ceyhan D., Bulut M., Yalçın B. In silico, 6lu7 protein inhibition using dihydroxy-3-phenyl coumarin derivatives for SARS-CoV-2. Journal of the Turkish Chemical Society, Section A: Chemistry . 2020;7(3):691–712. [Google Scholar]
- 261.Ozdemir M., Koksoy B., Ceyhan D., et al. Design and in silico study of the novel coumarin derivatives against SARS-CoV-2 main enzymes. Journal of Biomolecular Structure & Dynamics . 2020;40 doi: 10.1080/07391102.2020.1863263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Patel D. V., Patel D. M., Patel A. M., et al. Carbazole-based semicarbazones and hydrazones as multifunctional anti-Alzheimer agents. Journal of Biomolecular Structure and Dynamics . 2021;6(2):1–22. doi: 10.1080/07391102.2021.1942212. [DOI] [PubMed] [Google Scholar]
- 263.Peng L., Tian X., Shen L., et al. Identifying effective antiviral drugs against SARS-CoV-2 by drug repositioning through virus-drug association prediction. Frontiers in Genetics . 2020;11:p. 1072. doi: 10.3389/fgene.2020.577387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Pratama M. R. F., Poerwono H., Siswodihardjo S. Molecular docking of novel 5-O-benzoylpinostrobin derivatives as SARS-CoV-2 main protease inhibitors. Pharmaceutical Sciences . 2020;26:S63–S77. doi: 10.34172/ps.2020.57. [DOI] [Google Scholar]
- 265.Pundir H., Joshi T., Joshi T., et al. Using Chou’s 5-steps rule to study pharmacophore-based virtual screening of SARS-CoV-2 Mpro inhibitors. Molecular Diversity . 2020;25 doi: 10.1007/s11030-020-10148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Quimque M. T. J., Notarte K. I. R., Fernandez R. A. T., et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. Journal of Biomolecular Structure and Dynamics . 2020;39 doi: 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rakib A., Nain Z., Sami S. A., et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: an in silico investigation. Briefings in Bioinformatics . 2021;22(2):1476–1498. doi: 10.1093/bib/bbab045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Rane J. S., Pandey P., Chatterjee A., et al. Targeting virus-host interaction by novel pyrimidine derivative: an in silico approach towards discovery of potential drug against COVID-19. Journal of Biomolecular Structure and Dynamics . 2020;39:5768–5778. doi: 10.1080/07391102.2020.1794969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Rao P., Shukla A., Parmar P., et al. Reckoning a fungal metabolite, pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophysical Chemistry . 2020;264 doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Salman S., Shah F. H., Idrees J., et al. Virtual screening of immunomodulatory medicinal compounds as promising anti-SARS-COV-2 inhibitors. Future Virology . 2020;15(5):267–275. doi: 10.2217/fvl-2020-0079. [DOI] [Google Scholar]
- 271.Sarma P., Shekhar N., Prajapat M., et al. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain) Journal of Biomolecular Structure and Dynamics . 2020;39 doi: 10.1080/07391102.2020.1753580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Sepay N., Sepay N., Al Hoque A., Mondal R., Halder U. C., Muddassir M. In silico fight against novel coronavirus by finding chromone derivatives as inhibitor of coronavirus main proteases enzyme. Structural Chemistry . 2020;31(5):1831–1840. doi: 10.1007/s11224-020-01537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Shehroz M., Zaheer T., Hussain T. Computer-aided drug design against spike glycoprotein of SARS-CoV-2 to aid COVID-19 treatment. Heliyon . 2020;6(10) doi: 10.1016/j.heliyon.2020.e05278.e05278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Singh B., Das A. In silico drug repositioning of hydroxychloroquine (HCQ) for the treatment of viral diseases. International Journal of Progressive Research in Science and Engineering . 2021;2(3):13–15. [Google Scholar]
- 275.Stefaniu A., Pirvu L., Albu B., Pintilie L. Molecular docking study on several benzoic acid derivatives against SARS-CoV-2. Molecules . 2020;25(24) doi: 10.3390/molecules25245828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Tachoua W., Kabrine M., Mushtaq M., Ul-Haq Z. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. Journal of Molecular Graphics and Modelling . 2020;101 doi: 10.1016/j.jmgm.2020.107758.107758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Uniyal A., Mahapatra M. K., Tiwari V., Sandhir R., Kumar R. Targeting SARS-CoV-2 main protease: structure based virtual screening, in silico ADMET studies and molecular dynamics simulation for identification of potential inhibitors. Journal of Biomolecular Structure & Dynamics . 2020;40 doi: 10.1080/07391102.2020.1848636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Welker A., Kersten C., Müller C., et al. Structure-activity relationships of benzamides and isoindolines designed as SARS-CoV protease inhibitors effective against SARS-CoV-2. ChemMedChem . 2020;16 doi: 10.1002/cmdc.202000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wen L., Tang K., Chik K. K.-H., et al. In silico structure-based discovery of a SARS-CoV-2 main protease inhibitor. International Journal of Biological Sciences . 2021;17(6):1555–1564. doi: 10.7150/ijbs.59191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.White M. A., Lin W., Cheng X. Discovery of COVID-19 inhibitors targeting the SARS-CoV-2 NSP13 helicase. The Journal of Physical Chemistry Letters . 2020;11(21):9144–9151. doi: 10.1021/acs.jpclett.0c02421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Wu Y., Chang K. Y., Lou L., Edwards L. G., Doma B. K., Xie Z. R. In silico identification of drug candidates against COVID-19. Informatics in Medicine Unlocked . 2020;21 doi: 10.1016/j.imu.2020.100461.100461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Zaher N. H., Mostafa M. I., Altaher A. Y. Design, synthesis and molecular docking of novel triazole derivatives as potential CoV helicase inhibitors. Acta Pharmaceutica . 2020;70(2):145–159. doi: 10.2478/acph-2020-0024. [DOI] [PubMed] [Google Scholar]
- 283.Zarezade V., Rezaei H., Shakerinezhad G., et al. The identification of novel inhibitors of human angiotensin-converting enzyme 2 and main protease of SARS-CoV-2: a combination of in silico methods for treatment of COVID-19. Journal of Molecular Structure . 2021;1237 doi: 10.1016/j.molstruc.2021.130409.130409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Agarwal S., Pethani J. P., Shah H. A., et al. Identification of a novel orally bioavailable NLRP3 inflammasome inhibitor. Bioorganic & Medicinal Chemistry Letters . 2020;30(21) doi: 10.1016/j.bmcl.2020.127571.127571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Akaberi D., Krambrich J., Ling J., et al. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biology . 2020;37 doi: 10.1016/j.redox.2020.101734.101734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bernstein L. R., Zhang L. Gallium maltolate has in vitro antiviral activity against SARS-CoV-2 and is a potential treatment for COVID-19. Antiviral Chemistry & Chemotherapy . 2020;28 doi: 10.1177/2040206620983780.2040206620983780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bocci G., Bradfute S. B., Ye C., et al. Virtual and in vitro antiviral screening revive therapeutic drugs for COVID-19. ACS Pharmacology & Translational Science . 2020;3(6):1278–1292. doi: 10.1021/acsptsci.0c00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Cao R., Hu H., Li Y., et al. Anti-SARS-CoV-2 potential of artemisinins in vitro. ACS Infectious Diseases . 2020;6(9):2524–2531. doi: 10.1021/acsinfecdis.0c00522. [DOI] [PubMed] [Google Scholar]
- 289.Chen Z., Cui Q., Cooper L., et al. Ginkgolic acid and anacardic acid are specific covalent inhibitors of SARS-CoV-2 cysteine proteases. Cell and Bioscience . 2021;11(1) doi: 10.1186/s13578-021-00564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Gendrot M., Andreani J., Jardot P., et al. In vitro antiviral activity of doxycycline against SARS-CoV-2. Molecules . 2020;25(21) doi: 10.3390/molecules25215064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Gupta Y., Maciorowski D., Zak S. E., et al. Bisindolylmaleimide IX: a novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in-vitro validation assays. Methods . 2021;195 doi: 10.1016/j.ymeth.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hahn F., Wangen C., Häge S., et al. IMU-838, a developmental DHODH inhibitor in phase II for autoimmune disease, shows anti-SARS-CoV-2 and broad-spectrum antiviral efficacy in vitro. Viruses . 2020;12(12) doi: 10.3390/v12121394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hattori S. I., Higashi-Kuwata N., Hayashi H., et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nature Communications . 2021;12(1):p. 668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Jang Y., Shin J. S., Lee M. K., et al. Comparison of antiviral activity of gemcitabine with 2′-fluoro-2′-deoxycytidine and combination therapy with remdesivir against SARS-CoV-2. International Journal of Molecular Sciences . 2021;22(4) doi: 10.3390/ijms22041581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Jeon S., Ko M., Lee J., et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrobial Agents and Chemotherapy . 2020;64(7) doi: 10.1128/AAC.00819-20.e00819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Liu K., Zou R., Cui W., et al. Clinical HDAC inhibitors are effective drugs to prevent the entry of SARS-CoV2. ACS Pharmacology & Translational Science . 2020;3(6):1361–1370. doi: 10.1021/acsptsci.0c00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ma Q., Li R., Pan W., et al. Phillyrin (KD-1) exerts anti-viral and anti-inflammatory activities against novel coronavirus (SARS-CoV-2) and human coronavirus 229E (HCoV-229E) by suppressing the nuclear factor kappa B (NF-κB) signaling pathway. Phytomedicine . 2020;78 doi: 10.1016/j.phymed.2020.153296.153296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Masih A., Agnihotri A. K., Srivastava J. K., Pandey N., Bhat H. R., Singh U. P. Discovery of novel pyrazole derivatives as a potent anti-inflammatory agent in RAW264.7 cells via inhibition of NF-ĸB for possible benefit against SARS-CoV-2. Journal of Biochemical and Molecular Toxicology . 2020;35 doi: 10.1002/jbt.22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Omotuyi I. O., Nash O., Ajiboye B. O., et al. Aframomum melegueta secondary metabolites exhibit polypharmacology against SARS-CoV-2 drug targets: in vitro validation of furin inhibition. Phytotherapy Research . 2020;35 doi: 10.1002/ptr.6843. [DOI] [PubMed] [Google Scholar]
- 300.Outlaw V. K., Bovier F. T., Mears M. C., et al. Inhibition of coronavirus entry in vitro and ex vivo by a lipid-conjugated peptide derived from the SARS-CoV-2 spike glycoprotein HRC domain. mBio . 2020;11(5) doi: 10.1128/mBio.01935-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Pitsillou E., Liang J., Ververis K., Hung A., Karagiannis T. C. Interaction of small molecules with the SARS-CoV-2 papain-like protease: in silico studies and in vitro validation of protease activity inhibition using an enzymatic inhibition assay. Journal of Molecular Graphics and Modelling . 2021;104 doi: 10.1016/j.jmgm.2021.107851.107851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Pizzorno A., Padey B., Dubois J., et al. In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antiviral Research . 2020;181 doi: 10.1016/j.antiviral.2020.104878.104878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Plaze M., Attali D., Prot M., et al. Inhibition of the replication of SARS-CoV-2 in human cells by the FDA-approved drug chlorpromazine. International Journal of Antimicrobial Agents . 2021;57(3) doi: 10.1016/j.ijantimicag.2020.106274.106274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Reznikov L. R., Norris M. H., Vashisht R., et al. Identification of antiviral antihistamines for COVID-19 repurposing. Biochemical and biophysical research communications . 2021;538:173–179. doi: 10.1016/j.bbrc.2020.11.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Sakurai Y., Ngwe Tun M. M., Kurosaki Y., et al. 5-amino levulinic acid inhibits SARS-CoV-2 infection in vitro. Biochemical and Biophysical Research Communications . 2021;545:203–207. doi: 10.1016/j.bbrc.2021.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Stone N. E., Jaramillo S. A., Jones A. N., et al. Stenoparib, an inhibitor of cellular poly(ADP-ribose) polymerase, blocks replication of the SARS-CoV-2 and HCoV-NL63 human coronaviruses in vitro. mBio . 2021;12(1) doi: 10.1128/mbio.03495-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Su H. X., Yao S., Zhao W. F., et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacologica Sinica . 2020;41(9):1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Touret F., Gilles M., Barral K., et al. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Scientific Reports . 2020;10(1) doi: 10.1038/s41598-020-70143-6.13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Tree J. A., Turnbull J. E., Buttigieg K. R., et al. Unfractionated heparin inhibits live wild type SARS-CoV-2 cell infectivity at therapeutically relevant concentrations. British Journal of Pharmacology . 2021;178(3):626–635. doi: 10.1111/bph.15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Vatansever E. C., Yang K. S., Drelich A. K., et al. Bepridil is potent against SARS-CoV-2 in vitro. Proceedings of the National Academy of Sciences . 2021;118(10) doi: 10.1073/pnas.2012201118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wang S. C., Chen Y., Wang Y. C., et al. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. American Journal of Cancer Research . 2020;10(12):4538–4546. [PMC free article] [PubMed] [Google Scholar]
- 312.Watashi K. Identifying and repurposing antiviral drugs against severe acute respiratory syndrome coronavirus 2 with in silico and in vitro approaches. Biochemical and Biophysical Research Communications . 2021;538:137–144. doi: 10.1016/j.bbrc.2020.10.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Yu S., Zhu Y., Xu J., et al. Glycyrrhizic acid exerts inhibitory activity against the spike protein of SARS-CoV-2. Phytomedicine . 2021;85 doi: 10.1016/j.phymed.2020.153364.153364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zandi K., Amblard F., Musall K., et al. Repurposing nucleoside analogs for human coronaviruses. Antimicrobial Agents and Chemotherapy . 2021;65(1) doi: 10.1128/AAC.01652-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Zhang H., Yang Y., Li J., et al. A novel virtual screening procedure identifies pralatrexate as inhibitor of SARS-CoV-2 RdRp and it reduces viral replication in vitro. PLoS Computational Biology . 2020;16(12) doi: 10.1371/journal.pcbi.1008489.e1008489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Zhang L., Liu J., Cao R., et al. Comparative antiviral efficacy of viral protease inhibitors against the novel SARS-CoV-2 in vitro. Virologica Sinica . 2020;35(6):776–784. doi: 10.1007/s12250-020-00288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Zhu Q., Zhang Y., Wang L., et al. Inhibition of coronavirus infection by a synthetic STING agonist in primary human airway system. Antiviral Research . 2021;187 doi: 10.1016/j.antiviral.2021.105015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Lei C., Qian K., Li T., et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nature Communications . 2020;11(1):p. 2070. doi: 10.1038/s41467-020-16048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Rathnayake A. D., Zheng J., Kim Y., et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Science Translational Medicine . 2020;12(557) doi: 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Schäfer A., Muecksch F., Lorenzi J. C. C., et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-cov-2 infection in vivo. Journal of Experimental Medicine . 2020;218(3) doi: 10.1084/jem.20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Schumann S., Kaiser A., Nicoletti F., et al. Immune-modulating drug MP1032 with SARS-CoV-2 antiviral activity in vitro: a potential multi-target approach for prevention and early intervention treatment of COVID-19. International Journal of Molecular Sciences . 2020;21(22) doi: 10.3390/ijms21228803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Shang C., Zhuang X., Zhang H., et al. Inhibitors of endosomal acidification suppress SARS-CoV-2 replication and relieve viral pneumonia in hACE2 transgenic mice. Virology Journal . 2021;18(1):p. 46. doi: 10.1186/s12985-021-01515-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Sheahan T. P., Sims A. C., Zhou S., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Science Translational Medicine . 2020;12(541) doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Tai T. T., Wu T. J., Wu H. D., et al. A strategy to treat COVID-19 disease with targeted delivery of inhalable liposomal hydroxychloroquine: a preclinical pharmacokinetic study. Clinical and Translational Science . 2021;14(1):132–136. doi: 10.1111/cts.12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Tortorici M. A., Beltramello M., Lempp F. A., et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science . 2020;370(6519):950–957. doi: 10.1126/science.abe3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Wahl A., Gralinski L. E., Johnson C. E., et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature . 2021;591 doi: 10.1038/s41586-021-03312-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wang G., Yang M. L., Duan Z. L., et al. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Research . 2021;31(1):17–24. doi: 10.1038/s41422-020-00450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Weston S., Coleman C. M., Haupt R., et al. Broad anti-coronavirus activity of food and drug administration-approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo. Journal of Virology . 2020;94(21) doi: 10.1128/JVI.01218-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.White K. M., Rosales R., Yildiz S., et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science . 2021;371(6532):926–931. doi: 10.1126/science.abf4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Wu C. Y., Lin Y. S., Yang Y. H., Shu L. H., Cheng Y. C., Liu H. T. GB-2 inhibits ACE2 and TMPRSS2 expression: in vivo and in vitro studies. Biomedicine and Pharmacotherapy . 2020;132 doi: 10.1016/j.biopha.2020.110816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Yuen C. K., Wong W. M., Mak L. F., et al. Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. Journal of Medical Virology . 2021;93(4):2076–2083. doi: 10.1002/jmv.26583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zhang C., Wang Y., Zhu Y., et al. Development and structural basis of a two-MAb cocktail for treating SARS-CoV-2 infections. Nature Communications . 2021;12(1):p. 264. doi: 10.1038/s41467-020-20465-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.