

PorX is an essential response regulator of the type IX secretion system associated with bacterial pathogenicity, gliding motility and metabolic enzyme secretion. Here, the purification, crystallization and X-ray analysis of PorX from different bacterial species are reported, representing a milestone in the structure–function studies of this essential protein.
Keywords: two-component systems, response regulators, bacterial secretion systems, bacterial pathogenesis, PorX response regulator, type IX secretion system
Abstract
Pathogenic bacteria utilize specialized macromolecular secretion systems to transport virulence factors across membrane(s) and manipulate their infected host. To date, 11 secretion systems have been identified, including the type IX secretion system (T9SS) associated with human, avian and farmed-fish diseases. As a bacterial secretion system, the T9SS also facilitates gliding motility and the degradation of different macromolecules by the secretion of metabolic enzymes in nonpathogenic bacteria. PorX is a highly conserved protein that regulates the transcription of essential T9SS components and additionally mediates the function of T9SS via direct interaction with PorL, the rotary motor protein of the T9SS. PorX is also a member of a two-component system regulatory cascade, where it serves as the response regulator that relays a signal transduced from a conserved sensor histidine kinase, PorY, to a designated sigma factor. Here, the recombinant expression and purification of PorX homologous proteins from the pathogenic bacterium Porphyromonas gingivalis and the nonpathogenic bacterium Flavobacterium johnsoniae are reported. A bioinformatical characterization of the different domains comprising the PorX protein is also provided, and the crystallization and X-ray analysis of PorX from F. johnsoniae are reported.
1. Introduction
The human oral cavity has the second largest and most diverse microbiota after the gut, harboring over 700 microbial species that form complex and dynamic biofilms (Deo & Deshmukh, 2019 ▸). The oral Gram-negative bacterium Porphyromonas gingivalis can remodel this commensal bacterial community and promote a state of dysbiosis (Gerits et al., 2017 ▸). As a result, P. gingivalis has directly been correlated with several destructive periodontal biofilm-related diseases, including periodontitis (Gerits et al., 2017 ▸). Periodontitis is a chronic inflammatory disease characterized by the destruction of the supporting structures of teeth, resulting in severe tooth loss (Gerits et al., 2017 ▸). This destructive process involves direct tissue damage from bacterial plaque products as well as indirect damage through bacterial induction of the host’s inflammatory and immune responses (Arigbede et al., 2012 ▸). In the long term, periodontitis can lead to permanent periodontal damage, an increase in systemic inflammation markers and bacteremia (Hajishengallis & Chavakis, 2021 ▸). The prevalence of periodontitis is high, with the moderate form affecting up to 46%, and the severe form up to 8.9%, of the United States population (Eke et al., 2015 ▸). In addition to the contribution of P. gingivalis to the dysbiosis in periodontal biofilms, other epidemiological and experimental studies have linked its infection to systemic conditions such as cardiovascular diseases, preterm low birth weight, rheumatoid arthritis, non-alcoholic fatty liver disease, cancer and Alzheimer’s disease (Kebschull et al., 2010 ▸; Benedyk et al., 2016 ▸; Laugisch et al., 2016 ▸; Whitmore & Lamont, 2014 ▸; Gao et al., 2016 ▸; Abbayya et al., 2015 ▸).
During infection, P. gingivalis produces an arsenal of virulence factors that are involved in tissue colonization and destruction, as well as interference with host defense systems (Lasica et al., 2017 ▸). One group of key virulence determinants are trypsin-like proteases called gingipains. These proteases are potent enzymes that are anchored to the cell surface and can be released into the extracellular milieu via the biogenesis of outer membrane vesicles (Lasica et al., 2017 ▸). Gingipain secretion is mediated by the specialized type IX secretion system (T9SS; Lasica et al., 2017 ▸). Unlike well studied secretion systems employed by acute pathogens, such as the type III secretion system, which translocate virulence factors directly into the host cell, the T9SS only translocates virulence factors across the bacterial outer membrane (Nakayama, 2015 ▸). The secretion of these virulence factors and other cargo proteins occurs in two steps (Fig. 1 ▸). In the first step, the virulence factors are translocated across the bacterial inner membrane and into the periplasm by the generalized Sec secretion system. In the second step, a designated T9SS translocon translocates these proteins across the outer membrane (Fig. 1 ▸; Lasica et al., 2017 ▸). Upon translocation across the outer membrane, the conserved targeting domain of these virulence factors is cleaved and substituted for an anionic lipopolysaccharide, to remain attached to the bacterial surface, or released into the extracellular milieu via outer membrane vesicles (Fig. 1 ▸; Veith et al., 2017 ▸). Notably, the role of the T9SS extends beyond virulence-factor secretory machinery, as it is also found in nonpathogenic species of the Fibrobacteres–Chlorobi–Bacteroidetes superphylum, where it serves additional physiological roles (McBride & Zhu, 2013 ▸). In these species, including the soil bacterium Flavobacterium johnsoniae, the T9SS supports a unique mode of gliding motility and functions to degrade macromolecules (polysaccharides and proteins) via the secretion of designated adhesins (Rhodes et al., 2011 ▸) and other digestive enzymes (Kharade & McBride, 2014 ▸, 2015 ▸), respectively.
Figure 1.

Schematic representation of the T9SS. The secretion of virulence factors (toxins) or cargo proteins is first mediated by the generalized SEC machinery at the inner membrane (IM). Upon translocation across the IM and cleavage of the signal peptide (SP), the C-terminal domain (CTD) adopts its conserved fold and is then recognized by the T9SS designated translocon at the outer membrane (OM). Following translocation across the outer membrane, the virulence factor, still conjugated to its CTD, is shuttled to the attachment complex, where the CTD is cleaved. If an A-LPS is attached, the virulence factor or cargo proteins will remain attached to the outer membrane. Secretion through the T9SS is energized by the proton-motive force and mediated by the rotary motor complex spanning the inner and outer membranes. The regulatory cascade governing the expression of the conserved T9SS core components assembling these complexes comprises a two-component system (PorX and PorY) and an additional sigma factor (SigP). PorY serves as the sensor histidine kinase that perceives an unknown signal from the periplasm and transduces the signal, first by autophosphorylation and then by transphosphorylation to the response regulator PorX. Transphosphorylated PorX can interact with SigP and directly with the promoter regions of T9SS-associated genes to induce their translation. PorX can also interact with PorL, a conserved T9SS core component of the rotary motor complex. This figure was created with BioRender.com.
At least 18 conserved proteins are involved in the T9SS cargo translocation pathway, as confirmed by the accumulation of cargo proteins in the periplasm upon the deletion of individual T9SS components (Heath et al., 2016 ▸). These 18 proteins can be divided into four groups associated with regulation, assembly of the core T9SS structures across the inner and outer membranes, cargo translocation across the outer membrane and post-translational modification of translocated cargo (Fig. 1 ▸; Lasica et al., 2017 ▸). The expression of some of the essential T9SS components is regulated at the transcriptional level by a conserved signaling pathway composed of a two-component system (TCS) and an additional sigma factor (Kadowaki et al., 2016 ▸). TCSs are the predominant multistep signaling pathway in bacteria and their minimal composition contains an input sensor histidine kinase (SHK) and an output response regulator (RR). TCSs depend on phosphoryl-transfer reactions as a means of signal transduction and together catalyze three reactions: autophosphorylation of the SHK, transfer of the phosphoryl group to the RR and dephosphorylation of the RR (Jacob-Dubuisson et al., 2018 ▸). RRs always contain a receiver domain that undergoes phosphorylation and often harbor additional domains, including an output effector domain. The majority of these effector domains function as DNA-binding transcription factors, but can also display RNA-binding, protein-binding or even enzymatic activities (Gao et al., 2019 ▸).
In the T9SS, PorY serves as the SHK that responds to an unknown signal transmitted from the periplasm, while PorX functions as the RR but lacks the typical DNA-binding domain (Fig. 1 ▸; Kadowaki et al., 2016 ▸). To compensate for the lack of a DNA-binding domain, PorX interacts with and stabilizes the transcription factor SigP, which directly binds the promoter regions of T9SS genes (Fig. 1 ▸; Jiang et al., 2021 ▸; Yang et al., 2021 ▸; Kadowaki et al., 2016 ▸). While the precise signal triggering this pathway has yet to be identified, deletion of either porX or sigP in P. gingivalis, results in the downregulation of T9SS components and activity, and in impaired processing of gingipains, as well as decreased toxicity (Sato et al., 2010 ▸). In addition to its effect on the expression of conserved components, the regulatory cascade was found to directly modulate the rotary motor, which provides the energy for the secretion of virulence factors (Fig. 1 ▸). More specifically, PorX can directly interact with the cytoplasmic domain of the rotary core component PorL; thus, PorX is presumed to translate the regulatory signal into a mechanical output (Vincent et al., 2016 ▸).
Due to its central role in the expression and function of the T9SS, we initiated structure–function relationship studies of PorX. Here, we report the purification of PorX proteins from both P. gingivalis (PorXPG) and the nonpathogenic bacterium F. johnsoniae (PorXFJ), along with crystallization and X-ray analysis of PorXFJ. Our results represent a significant milestone in the structural determination of this critical hub protein.
2. Materials and methods
2.1. Cloning and expression
The full-length porX (UniProt B2RJJ3) and Fjoh_2906 (UniProt A5FFU4) genes were amplified by polymerase chain reaction (PCR) from P. gingivalis strain ATCC 33277 and F. johnsoniae strain UW101 (ATCC 17061) genomic DNA, respectively. The primers used for the PCR amplification were PorXFJ-FW (5′-CAGCGGCCTGGTGCCGCGCGGCAGCCATATGGATAAGATAAGAATACTTTGGGTCGATG-3′), PorXFJ-REV (5′-TTGTCGACGGAGCTCGAATTCGGATCCTTATTTAGGGTTAAATACCAAAAACGG-3′), PorXPG-FW (5′-GCAGCGGCCTGGTGCCGCGCGGCAGCCATATGGAAAAAAACATGAGACCGTATACCG-3′) and PorXPG-REV (5′-CTTGTCGACGGAGCTCGAATTCGGATCCTTACTTGGGTTGCATCGTAATTACGGGC-3′). The restriction-free cloning method (Bond & Naus, 2012 ▸) was used to insert the amplified gene between the NdeI and BamHI restriction sites in a modified pET-28a(+) vector. Each gene was fused in frame to an N-terminal His10 tag followed by a thrombin proteolytic site.
E. coli BL21 strain cells harboring pET-28a(+)-PorXFJ were cultivated in autoinduction medium (Studier, 2005 ▸) containing kanamycin (50 µg ml−1) at 37°C for 7 h. The cultivation temperature was then lowered to 22°C and expression was continued for a further 16 h. The cells were harvested by centrifugation at 6000g for 15 min at 4°C.
2.2. Recombinant protein purification
Cell pellets were resuspended in buffer A (50 mM Tris pH 8, 300 mM NaCl, 10 mM imidazole) and incubated with DNase I (10 mg ml−1) and protease-inhibitor cocktail (Calbiochem) at 4°C. The cells were then disrupted by two cycles in a French press pressure cell at 172 MPa. Cell debris was removed by high-speed centrifugation at 186 000g for 1 h at 4°C. The supernatant was applied onto a gravity Ni–NTA column (Bio-Rad Econo-Column chromatography column, Thermo Scientific HisPur Ni–NTA resin) pre-equilibrated with buffer A. The bound protein was washed with buffers B (20 mM Tris pH 8, 1 M NaCl, 20 mM imidazole) and C (20 mM Tris pH 8, 200 mM NaCl, 40 mM imidazole) and eluted with buffer D (20 mM Tris pH 8, 200 mM NaCl, 500 mM imidazole). To cleave the His tag, bovine thrombin (Prolytix) was added to the eluted protein and the mixture was then dialyzed against buffer E (10 mM Tris pH 8, 200 mM NaCl) for 16 h at 4°C. The protein was then applied onto a size-exclusion column (Superdex 200 Increase 10/300 GL, Cytivia) pre-equilibrated with buffer E (10 mM Tris pH 8, 200 mM NaCl). The purified protein was concentrated to 25 mg ml−1 and flash-frozen in liquid nitrogen. Sample purity was analyzed by SDS–PAGE and protein masses were confirmed by mass spectroscopy.
2.3. Crystallization and structure determination
PorXFJ crystals were grown using the sitting-drop vapor-diffusion method at 23°C. Each crystallization drop consisted of 0.2 µl of 5 mg ml−1 protein solution and an equal volume of precipitant solution. The screening was performed in 96-well plates (Intelli-Plate 96-3 low volume reservoir) using an NT8 robot (Formulatrix). The initial crystallization conditions were examined using commercial screening kits from Hampton Research (Index) and Molecular Dimensions (MemGold 1, MemGold2, JCSG+ and PACT).
2.4. Diffraction data collection and analysis
A native crystal was harvested from a condition consisting of 0.16 M calcium acetate, 0.08 M sodium cacodylate pH 6.4, 14.4% polyethylene glycol 8000, 25% glycerol and was flash-cooled in liquid nitrogen without the addition of any cryoprotectants.
The diffraction data set was collected on beamline 5.0.1 equipped with a PILATUS3 2M 25 Hz detector at the Lawrence Berkeley National Laboratory Advanced Light Source (ALS). Data collection was performed at 100 K; a total of 720 images were collected with an oscillation range of 0.25° and 1 s exposure per image at 0.9774 Å wavelength. The data were reduced and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▸). Crystal and data-collection characteristics are summarized in Table 1 ▸. The self-rotation function was calculated using MOLREP (Vagin & Teplyakov, 2010 ▸).
Table 1. Diffraction data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| X-ray source | Beamline 5.0.1, ALS |
| Space group | P212121 |
| No. of molecules per asymmetric unit | 2 |
| a, b, c (Å) | 84.43, 97.43. 132.99 |
| Resolution (Å) | 50.0–2.12 (2.16–2.12) |
| Reflections, total | 414342 |
| Reflections, unique | 63090 |
| R merge † (%) | 8.9 (65.6) |
| 〈I/σ(I)〉 | 22.05 (2.0) |
| Completeness (%) | 100 (99.9) |
| Multiplicity | 6.6 |
| CC* | 0.997 (0.962) |
| CC1/2 | 0.989 (0.862) |
| Wavelength (Å) | 0.9774 |
| V M (Å3 Da−1) | 2.32 |
R
merge =
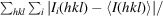
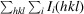 , where I
i
(hkl) is the observed intensity of an individual reflection and Ii
(hkl) is the mean intensity of that reflection.
, where I
i
(hkl) is the observed intensity of an individual reflection and Ii
(hkl) is the mean intensity of that reflection.
2.5. Multiple sequence alignment
The amino-acid sequences of PorX orthologs were retrieved from the genomes of the bacterial species P. gingivalis, Tannerella forsythia, Prevotella intermedia, Flavobacterium coulminris, F. psychrophilum, F. branchiophilum and F. johnsoniae using BlastP (Johnson et al., 2008 ▸). The sequences were aligned by the Clustal Omega server with default settings (Madeira et al., 2022 ▸). The graphical representation of the aligned sequences was prepared using the ESPript 3.0 server (Robert & Gouet, 2014 ▸).
3. Results and discussion
3.1. Ortholog search and protein purification
The T9SS is a unique macromolecular machinery that is utilized by bacterial pathogens to promote infection. Each of the three major oral pathogens (P. gingivalis, Tannerella forsythia and Prevotella intermedia) associated with periodontitis and other human systemic diseases employs a T9SS (Lasica et al., 2017 ▸). Other T9SS-utilizing pathogens, including Riemerella anatipestifer, Flavobacterium coulminris, F. psychrophilum and F. branchiophilum, are responsible for major bacterial infections in birds and farmed fish, such as rainbow trout fry syndrome and cold-water disease (Li et al., 2017 ▸; Barbier et al., 2020 ▸; Good et al., 2015 ▸; Chen et al., 2022 ▸). PorX is a conserved protein that is common to all these pathogens as well as to nonpathogenic gliding bacteria (Fig. 2 ▸). As part of our efforts to purify, crystallize and determine the structure of full-length PorX, we performed an ortholog screen of PorX from the species mentioned above. The ortholog screening approach has been shown to improve the expression, purification and structural determination of challenging proteins (Savchenko et al., 2003 ▸). The eight different PorX orthologs are 514–531 residues in length and have predicted molecular weights ranging between 60 and 65 kDa. The ortholog screening revealed that only PorXFJ and PorXPG (51% sequence identity, 71% sequence similarity) were soluble under the tested conditions and thus we proceeded with their large-scale expression and purification.
Figure 2.

Multiple sequence alignment of PorX genes from pathogenic and nonpathogenic bacteria. Predicted domains are labeled, with the RR receiver domain in green, the linker/DNA-binding region in orange and the alkaline phosphatase domain in blue. Reference amino-acid numbering is according to the PorXFJ sequence.
PorXFJ and PorXPG were expressed in E. coli and purified to homogeneity, each producing an approximate yield of 13 mg purified protein per litre of culture (Fig. 3 ▸). Size-exclusion chromatography revealed that both proteins elute at a volume that corresponds to a monomer (Fig. 3 ▸). This observation stands in good agreement with the canonical mechanism reported for nonphosphorylated RRs, which exist as monomers prior to their phosphorylation and subsequent signal transduction through the cascade (Stock et al., 2000 ▸; Zschiedrich et al., 2016 ▸; Gao & Stock, 2009 ▸). As both PorX proteins were expressed in a heterologous E. coli system without their partner SHK (PorY), they were unlikely to undergo phosphorylation and therefore were predicted to exist as monomers. Mass-spectrometry results further confirmed that the purified proteins were in their nonphosphorylated forms.
Figure 3.

Purification analysis of the PorX proteins from F. johnsoniae and P. gingivalis. (a) Size-exclusion chromatography (Superdex 200 Increase) of PorXFJ (FJ) and PorXPG (PG). Both proteins elute at volumes appropriate for a monomer (∼60 kDa). Molecular markers, ferritin (440 kDa), conalbumin (75 kDa), carbonic anhydrase (29 kDa) and ribonuclease A (13.7 kDa), are indicated by a dashed gray line. (b) SDS–PAGE analysis demonstrates high purity levels of both protein homologs.
3.2. Crystallization and crystallographic data analysis
The purified proteins were extensively screened for crystallization conditions using various chemical conditions, protein concentrations and temperatures. PorXFJ crystallized under multiple conditions. However, conditions yielding single crystals resulted in poorly diffracting crystals, while other conditions displayed multilayered thin needle/plate crystals. Upon optimization of one of the original needle-forming hits (0.16 M calcium acetate, 0.08 M sodium cacodylate pH 6.5, 14.4% polyethylene glycol 8000, 20% glycerol) from the JCSG+ screen, followed by extensive rounds of seeding (both seed bead and manually with a magic wand) and reducing the protein concentration, we were able to improve the crystal morphology and resolution (Fig. 4 ▸). Next, a full diffraction data set was collected from the edge of a crystal harvested from the optimized crystallization condition (Fig. 4 ▸). We found that collecting data from the edge of the crystal reduces the effects of the thin multilayered secondary plates decorating the main crystal and therefore improved the quality of the data. Despite extensive screening attempts, conditions promoting PorXPG crystallization were not identified.
Figure 4.

(a, b) Images of PorXFJ crystals. The upper images were taken in visible light, while the lower images were taken in UV light, demonstrating that the crystals are indeed protein crystals. (a) Original crystallization condition. Scale bars are labeled in µm. (b) Optimized crystals after successive rounds of seeding. The arrow indicated the area that diffraction data were collected from. (c) Representative diffraction image.
The PorXFJ crystal fulfilled the systematic absence rules of space group P212121. Further validation of the chosen space group was performed by POINTLESS (Evans, 2006 ▸), which indicated that the space-group choice was accurate, with a symmetry absence probability of 0.956. The unit-cell parameters are a = 84.44, b = 97.42, c = 132.99 Å (Table 1 ▸). We are confident that our diffraction data-set quality is adequate for structural determination, as the crystal diffracted to 2.12 Å resolution and yielded a data set with a low R merge and high multiplicity (Table 1 ▸). Although the PorXFJ protein was observed as a monomer in solution, the asymmetric unit composition suggests the presence of two monomers per asymmetric unit, with a calculated V M value and solvent content of 2.32 Å3 Da−1 and 47.00%, respectively (Matthews, 1968 ▸). Both values are within the normal range observed for soluble protein crystals. As the majority of RRs only dimerize upon phosphorylation and their dimerization is associated with signal transduction through the cascade, it is possible that the crystal packing mimics a functional dimeric state. The self-rotation plot suggests the correct assignment of the orthorhombic space group, with five clear peaks at the crystallographic axes (Fig. 5 ▸).
Figure 5.

Stereographic projection of the self-rotation function at κ = 180°. The data resolution ranged between 48 and 4 Å and the integration radius was 20 Å.
3.3. Similarities to other proteins and hints towards function
Bioinformatics analysis suggests that PorX adopts a two-domain fold: an N-terminal receiver domain (PorXFJ; residues 1–120) and a C-terminal effector domain (PorXFJ; residues 220–518) interconnected by a linker region (PorXFJ; residues 121–219) (Fig. 2 ▸). The predicted receiver domain fold shares similarity with over 200 previously determined structures of other RRs and in particular those that belong to the CheY family involved in chemotaxis (32% and 31% sequence identity and 57% and 52% sequence similarity to PorXFJ and PorXPG, respectively; Supplementary Fig. S1; Sagi et al., 2003 ▸). Upon phosphorylation, CheY binds to the C-ring of bacterial flagella, changing the rotational direction of the flagellar motor (Sagi et al., 2003 ▸). As the T9SS has been proposed to utilize the proton-motive force and a rotary apparatus to drive the secretion of virulence factors (Hennell James et al., 2020 ▸, 2021 ▸), it has been speculated that PorX might share functional similarities with CheY (Vincent et al., 2016 ▸). This hypothesis was further supported by the observed interaction of PorX with PorL, which is one of the conserved components of the T9SS rotary motor (Vincent et al., 2016 ▸).
Despite the low amino-acid sequence identity (∼10–15%), the predicted C-terminal domain of PorX shares similarities with standalone alkaline phosphatases, phosphonoacetate hydrolases and phosphodiester hydrolases (Supplementary Figs. S2 and S3). In particular, this group of enzymes coordinates two zinc ions in the active site to support the hydrolysis of organic phosphate ester bonds. Among these identified homologs, PhnA is a bacterial enzyme involved in a metabolic pathway that converts phosphonates to phosphorus and directly catalyzes the conversion of phosphonoacetate to acetate and inorganic phosphate (Supplementary Fig. S2; Agarwal et al., 2011 ▸). The other homologs belong to the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) family (Supplementary Fig. S3; Gorelik, Liu et al., 2017 ▸; Morita et al., 2016 ▸; Gorelik, Randriamihaja et al., 2017 ▸; Albright et al., 2014 ▸; Gorelik et al., 2018 ▸; Hausmann et al., 2011 ▸; Kato et al., 2012 ▸). ENPP family members (ENPP1–ENPP7) can be found in both bacteria and eukaryotes, where they participate in different cellular processes, including nucleotide hydrolysis, lipid metabolism and associated signaling transduction pathways (Zalatan et al., 2006 ▸). The conjugation of the receiver domain of PorX with an enzymatic effector domain that shares similarity to enzymes that participate in different signaling and metabolic pathways further supports a role for PorX in the signal transduction pathway that mediates the expression and function of the T9SS. Although the exact substrate of the C-terminal phosphate domain of PorX has yet to be identified, the diverse roles of the standalone phosphatase homologs in different signal transduction pathways may provide crucial clues toward its possible function.
Interestingly, previous functional studies performed in heterologous bacterial systems have demonstrated that PorX does not bind DNA promoter regions (Kadowaki et al., 2016 ▸; Vincent et al., 2016 ▸). However, a recent study performed in the presence of the native RNA polymerase from P. gingivalis suggested that nonphosphorylated, inactive, PorX can still bind directly to DNA promoter regions via the linker region interconnecting its N- and C-terminal domains (PorXFJ residues 121–219; Fig. 2 ▸; Jiang et al., 2021 ▸). Secondary-structure prediction of this PorX linker region by the HHpred server (Zimmermann et al., 2018 ▸) suggested it contains a mixture of α-helices and β-sheets, with no apparent homology to any known protein domains (DNA-binding or otherwise). However, structural prediction by AlphaFold (Jumper et al., 2021 ▸) suggested that this linker region adopts a three-helix bundle fold. The three-helix bundle is a common structural motif that can also be found in many proteins, including actin-binding proteins and DNA-binding proteins (Schneider et al., 1998 ▸).
3.4. Phasing attempts
Based on the predicted fold similarities of the N- and C-terminal domains of PorX, we employed diverse approaches to generate an array of atomic models to recover the missing phase information by molecular-replacement experiments. These included homology models generated according to experimentally determined structures by the HHpred server and MODELLER (Šali et al., 1995 ▸; Gabler et al., 2020 ▸), the Phyre 2 server (Kelley et al., 2015 ▸) and the artificial intelligence-based AlphaFold system (Jumper et al., 2021 ▸). Molecular-replacement experiments were performed with models spanning the entire protein sequence as well as with its individual standalone domains in Phaser (McCoy et al., 2007 ▸), MrBUMP (Keegan & Winn, 2007 ▸) and BALBES (Long et al., 2008 ▸). However, all molecular-replacement attempts were unsuccessful, thus suggesting that the crystal structure and packing might differ from these predictions and therefore require full experimental phasing experiments. Accordingly, we expressed and purified a selenomethionine (SeMet) derivative of PorXFJ. Unfortunately, crystallization of SeMet-PorXFJ yielded poorly diffracting, multilayered crystals, even after optimization. As such, we are currently exploring other heavy-atom derivative soaks using the optimized wild-type crystals to retrieve the missing phase information and to determine the high-resolution structure.
4. Final remarks
Information derived from the determined structure of PorX, along with detailed biochemical studies, will allow us to answer many open questions associated with the function of PorX and to decipher the true fold of the linker domain and its associated function in DNA binding. In addition, we will be able to further investigate whether the crystal packing represents a true functional dimer and to understand the mechanisms behind signal transduction via its different domains.
Supplementary Material
Supplementary Figures. DOI: 10.1107/S2053230X22008500/ow5034sup1.pdf
Acknowledgments
We thank Professor Boaz Shaanan for his advice in data analysis. We also thank Advanced Light Source beamline 5.0.1 for support in remote data collection. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
Funding Statement
This work was funded by an Alzheimer’s Association grant to NZ (AARGD-NTF-21-851231).
References
- Abbayya, K., Puthanakar, N. Y., Naduwinmani, S. & Chidambar, Y. S. (2015). North. Am. J. Med. Sci. 7, 241–246. [DOI] [PMC free article] [PubMed]
- Agarwal, V., Borisova, S. A., Metcalf, W. W., van der Donk, W. A. & Nair, S. K. (2011). Chem. Biol. 18, 1230–1240. [DOI] [PMC free article] [PubMed]
- Albright, R. A., Ornstein, D. L., Cao, W., Chang, W. C., Robert, D., Tehan, M., Hoyer, D., Liu, L., Stabach, P., Yang, G., De La Cruz, E. M. & Braddock, D. T. (2014). J. Biol. Chem. 289, 3294–3306. [DOI] [PMC free article] [PubMed]
- Arigbede, A. O., Babatope, B. O. & Bamidele, M. K. (2012). J. Indian Soc. Periodontol. 16, 487–491. [DOI] [PMC free article] [PubMed]
- Barbier, P., Rochat, T., Mohammed, H. H., Wiens, G. D., Bernardet, J.-F., Halpern, D., Duchaud, E. & McBride, M. J. (2020). Appl. Environ. Microbiol. 86, e00799-20. [DOI] [PMC free article] [PubMed]
- Benedyk, M., Mydel, P. M., Delaleu, N., Płaza, K., Gawron, K., Milewska, A., Maresz, K., Koziel, J., Pyrc, K. & Potempa, J. (2016). J. Innate Immun. 8, 185–198. [DOI] [PMC free article] [PubMed]
- Bond, S. R. & Naus, C. C. (2012). Nucleic Acids Res. 40, W209–W213. [DOI] [PMC free article] [PubMed]
- Chen, Z., Niu, P., Ren, X., Han, W., Shen, R., Zhu, M., Yu, Y., Ding, C. & Yu, S. (2022). Appl. Environ. Microbiol. 88, e02409-21. [DOI] [PMC free article] [PubMed]
- Deo, P. N. & Deshmukh, R. (2019). J. Oral Maxillofac. Pathol. 23, 122–128. [DOI] [PMC free article] [PubMed]
- Eke, P. I., Dye, B. A., Wei, L., Slade, G. D., Thornton-Evans, G. O., Borgnakke, W. S., Taylor, G. W., Page, R. C., Beck, J. D. & Genco, R. J. (2015). J. Periodontol. 86, 611–622. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gabler, F., Nam, S. Z., Till, S., Mirdita, M., Steinegger, M., Söding, J., Lupas, A. N. & Alva, V. (2020). Curr. Protoc. Bioinform. 72, e108. [DOI] [PubMed]
- Gao, R., Bouillet, S. & Stock, A. M. (2019). Annu. Rev. Microbiol. 73, 175–197. [DOI] [PubMed]
- Gao, R. & Stock, A. M. (2009). Annu. Rev. Microbiol. 63, 133–154. [DOI] [PMC free article] [PubMed]
- Gao, S., Li, S., Ma, Z., Liang, S., Shan, T., Zhang, M., Zhu, X., Zhang, P., Liu, G., Zhou, F., Yuan, X., Jia, R., Potempa, J., Scott, D. A., Lamont, R. J., Wang, H. & Feng, X. (2016). Infect Agents Cancer, 11, 3. [DOI] [PMC free article] [PubMed]
- Gerits, E., Verstraeten, N. & Michiels, J. (2017). J. Oral Microbiol. 9, 1300366. [DOI] [PMC free article] [PubMed]
- Good, C., Davidson, J., Wiens, G. D., Welch, T. J. & Summerfelt, S. (2015). J. Fish Dis. 38, 409–413. [DOI] [PMC free article] [PubMed]
- Gorelik, A., Liu, F., Illes, K. & Nagar, B. (2017). J. Biol. Chem. 292, 7087–7094. [DOI] [PMC free article] [PubMed]
- Gorelik, A., Randriamihaja, A., Illes, K. & Nagar, B. (2017). FEBS J. 284, 3718–3726. [DOI] [PubMed]
- Gorelik, A., Randriamihaja, A., Illes, K. & Nagar, B. (2018). FEBS J. 285, 2481–2494. [DOI] [PubMed]
- Hajishengallis, G. & Chavakis, T. (2021). Nat. Rev. Immunol. 21, 426–440. [DOI] [PMC free article] [PubMed]
- Hausmann, J., Kamtekar, S., Christodoulou, E., Day, J. E., Wu, T., Fulkerson, Z., Albers, H. M. H. G., van Meeteren, L. A., Houben, A. J. S., van Zeijl, L., Jansen, S., Andries, M., Hall, T., Pegg, L. E., Benson, T. E., Kasiem, M., Harlos, K., Kooi, C. W. V., Smyth, S. S., Ovaa, H., Bollen, M., Morris, A. J., Moolenaar, W. H. & Perrakis, A. (2011). Nat. Struct. Mol. Biol. 18, 198–204. [DOI] [PMC free article] [PubMed]
- Heath, J. E., Seers, C. A., Veith, P. D., Butler, C. A., Nor Muhammad, N. A., Chen, Y. Y., Slakeski, N., Peng, B., Zhang, L., Dashper, S. G., Cross, K. J., Cleal, S. M., Moore, C. & Reynolds, E. C. (2016). PLoS One, 11, e0164313. [DOI] [PMC free article] [PubMed]
- Hennell James, R., Deme, J., Kjæer, A., Alcock, F., Silale, A., Lauber, F., Berks, B. & Lea, S. M. (2020). bioRxiv, 2020.05.11.089193.
- Hennell James, R., Deme, J. C., Kjær, A., Alcock, F., Silale, A., Lauber, F., Johnson, S., Berks, B. C. & Lea, S. M. (2021). Nat. Microbiol. 6, 221–233. [DOI] [PMC free article] [PubMed]
- Jacob-Dubuisson, F., Mechaly, A., Betton, J. & Antoine, R. (2018). Nat. Rev. Microbiol. 16, 585–593. [DOI] [PubMed]
- Jiang, C., Yang, D., Hua, T., Hua, Z., Kong, W. & Shi, Y. (2021). mSphere, 6, e00428-21. [DOI] [PMC free article] [PubMed]
- Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S. & Madden, T. L. (2008). Nucleic Acids Res. 36, W5–W9. [DOI] [PMC free article] [PubMed]
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., Petersen, S., Reiman, D., Clancy, E., Zielinski, M., Steinegger, M., Pacholska, M., Berghammer, T., Bodenstein, S., Silver, D., Vinyals, O., Senior, A. W., Kavukcuoglu, K., Kohli, P. & Hassabis, D. (2021). Nature, 596, 583–589. [DOI] [PMC free article] [PubMed]
- Kadowaki, T., Yukitake, H., Naito, M., Sato, K., Kikuchi, Y., Kondo, Y., Shoji, M. & Nakayama, K. (2016). Sci. Rep. 6, 23288. [DOI] [PMC free article] [PubMed]
- Kato, K., Nishimasu, H., Okudaira, S., Mihara, E., Ishitani, R., Takagi, J., Aoki, J. & Nureki, O. (2012). Proc. Natl Acad. Sci. USA, 109, 16876–16881. [DOI] [PMC free article] [PubMed]
- Kebschull, M., Demmer, R. T. & Papapanou, P. N. (2010). J. Dent. Res. 89, 879–902. [DOI] [PMC free article] [PubMed]
- Keegan, R. M. & Winn, M. D. (2007). Acta Cryst. D63, 447–457. [DOI] [PubMed]
- Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. (2015). Nat. Protoc. 10, 845–858. [DOI] [PMC free article] [PubMed]
- Kharade, S. S. & McBride, M. J. (2014). J. Bacteriol. 196, 961–970. [DOI] [PMC free article] [PubMed]
- Kharade, S. S. & McBride, M. J. (2015). J. Bacteriol. 197, 147–158. [DOI] [PMC free article] [PubMed]
- Lasica, A. M., Ksiazek, M., Madej, M. & Potempa, J. (2017). Front. Cell. Infect. Microbiol. 7, 215. [DOI] [PMC free article] [PubMed]
- Laugisch, O., Wong, A., Sroka, A., Kantyka, T., Koziel, J., Neuhaus, K., Sculean, A., Venables, P. J., Potempa, J., Möller, B. & Eick, S. (2016). Clin. Oral Invest, 20, 675–683. [DOI] [PMC free article] [PubMed]
- Li, N., Zhu, Y., LaFrentz, B. R., Evenhuis, J. P., Hunnicutt, D. W., Conrad, R. A., Barbier, P., Gullstrand, C. W., Roets, J. E., Powers, J. L., Kulkarni, S. S., Erbes, D. H., García, J. C., Nie, P. & McBride, M. J. (2017). Appl. Environ. Microbiol. 83, e01769-17. [DOI] [PMC free article] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Madeira, F., Pearce, M., Tivey, A. R. N., Basutkar, P., Lee, J., Edbali, O., Madhusoodanan, N., Kolesnikov, A. & Lopez, R. (2022). Nucleic Acids Res. 50, W276–W279. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McBride, M. J. & Zhu, Y. (2013). J. Bacteriol. 195, 270–278. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Morita, J., Kano, K., Kato, K., Takita, H., Sakagami, H., Yamamoto, Y., Mihara, E., Ueda, H., Sato, T., Tokuyama, H., Arai, H., Asou, H., Takagi, J., Ishitani, R., Nishimasu, H., Nureki, O. & Aoki, J. (2016). Sci. Rep. 6, 20995. [DOI] [PMC free article] [PubMed]
- Nakayama, K. (2015). J. Periodont Res. 50, 1–8. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rhodes, R. G., Nelson, S. S., Pochiraju, S. & McBride, M. J. (2011). J. Bacteriol. 193, 599–610. [DOI] [PMC free article] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Sagi, Y., Khan, S. & Eisenbach, M. (2003). J. Biol. Chem. 278, 25867–25871. [DOI] [PubMed]
- Šali, A., Potterton, L., Yuan, F., van Vlijmen, H. & Karplus, M. (1995). Proteins, 23, 318–326. [DOI] [PubMed]
- Sato, K., Naito, M., Yukitake, H., Hirakawa, H., Shoji, M., McBride, M. J., Rhodes, R. G. & Nakayama, K. (2010). Proc. Natl Acad. Sci. USA, 107, 276–281. [DOI] [PMC free article] [PubMed]
- Savchenko, A., Yee, A., Khachatryan, A., Skarina, T., Evdokimova, E., Pavlova, M., Semesi, A., Northey, J., Beasley, S., Lan, N., Das, R., Gerstein, M., Arrowmith, C. H. & Edwards, A. M. (2003). Proteins, 50, 392–399. [DOI] [PubMed]
- Schneider, J. P., Lombardi, A. & DeGrado, W. F. (1998). Fold. Des. 3, R29–R40. [DOI] [PubMed]
- Stock, A. M., Robinson, V. L. & Goudreau, P. N. (2000). Annu. Rev. Biochem. 69, 183–215. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Veith, P. D., Glew, M. D., Gorasia, D. G. & Reynolds, E. C. (2017). Mol. Microbiol. 106, 35–53. [DOI] [PubMed]
- Vincent, M. S., Durand, E. & Cascales, E. (2016). Front. Cell. Infect. Microbiol. 6, 96. [DOI] [PMC free article] [PubMed]
- Whitmore, S. E. & Lamont, R. J. (2014). PLoS Pathog. 10, e1003933. [DOI] [PMC free article] [PubMed]
- Yang, D., Jiang, C., Ning, B., Kong, W. & Shi, Y. (2021). J. Biol. Chem. 296, 100574. [DOI] [PMC free article] [PubMed]
- Zalatan, J. G., Fenn, T. D., Brunger, A. T. & Herschlag, D. (2006). Biochemistry, 45, 9788–9803. [DOI] [PubMed]
- Zimmermann, L., Stephens, A., Nam, S. Z., Rau, D., Kübler, J., Lozajic, M., Gabler, F., Söding, J., Lupas, A. N. & Alva, V. (2018). J. Mol. Biol. 430, 2237–2243. [DOI] [PubMed]
- Zschiedrich, C. P., Keidel, V. & Szurmant, H. (2016). J. Mol. Biol. 428, 3752–3775. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures. DOI: 10.1107/S2053230X22008500/ow5034sup1.pdf