

SUMMARY
Sporadic pituitary adenomas occur in over 10% of the population. Hormone-secreting adenomas, including those causing Cushing’s disease (CD), cause severe morbidity and early mortality. Mechanistic studies of CD are hindered by a lack of in vitro models and control normal human pituitary glands. Here, we surgically annotate adenomas and adjacent normal glands in 25 of 34 patients. Using single-cell RNA sequencing (RNA-seq) analysis of 27594 cells, we identify CD adenoma transcriptomic signatures compared with adjacent normal cells, with validation by bulk RNA-seq, DNA methylation, qRT-PCR, and immunohistochemistry. CD adenoma cells include a subpopulation of proliferating, terminally differentiated corticotrophs. In CD adenomas, we find recurrent promoter hypomethylation and transcriptional upregulation of PMAIP1 (encoding pro-apoptotic BH3-only bcl-2 protein noxa) but paradoxical noxa downregulation. Using primary CD adenoma cell cultures and a corticotroph-enriched mouse cell line, we find that selective proteasomal inhibition with bortezomib stabilizes noxa and induces apoptosis, indicating its utility as an anti-tumor agent.
Graphical Abstract
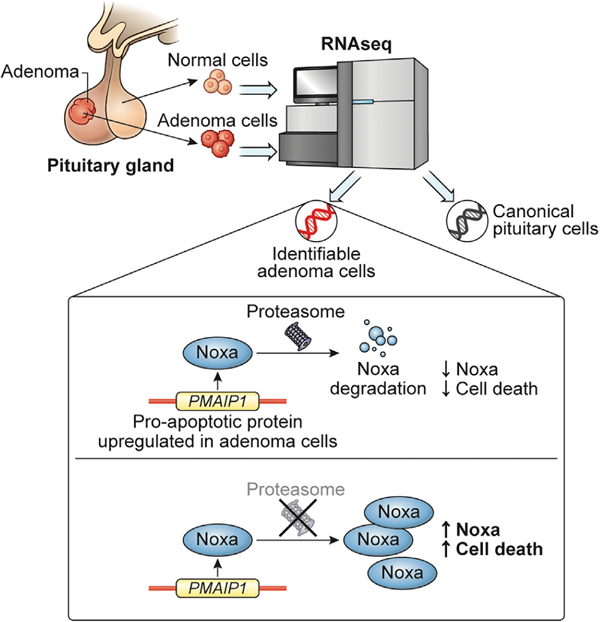
In brief
Asuzu et al. perform single-cell transcriptomic profiling in Cushing’s disease (CD) adenomas and find overexpression and DNA hypomethylation of PMAIP1, which encodes the pro-apoptotic protein noxa. Noxa is degraded by the proteasome. Proteasomal inhibition rescues noxa and induces apoptosis in CD.
INTRODUCTION
Pituitary adenomas are among the most common human central nervous system neoplasms; they are found in >10% of the population (Ezzat et al., 2004; Tjörnstrand et al., 2014) and result in disease states due to hormone oversecretion or due to compression of neural structures (Ikeda and Yoshimoto, 2009; Zaidi et al., 2017). Micro-adenomas causing Cushing’s disease (CD) cause severe morbidity and up to 2.5-fold increase in mortality despite modern treatments (Plotz et al., 1952; Ragnarsson et al., 2019). Unlike most human tumors, benign adenomas of the pituitary gland are monoclonal (Alexander et al., 1990; HERMAN et al., 1990; Schultef et al., 1991) and genetically bland, with a low mutation load (Bi et al., 2017). Point mutations in USP8 (Reincke et al., 2014; Ma et al., 2015), USP48, or BRAF (Chen et al., 2018) have been identified in up to 40% of sporadic CD adenomas. However, pathogenic mechanisms in the majority remain unexplained (Salomon et al., 2018). Proposed mechanisms for the origins of pituitary adenomas include mitotic replenishment (Gleiberman et al., 2008) or paracrine drive from pituitary stem cells; however, conclusive studies identifying and characterizing these mechanisms are lacking.
Single-cell RNA sequencing (scRNA-seq) of mammalian tissues has recently emerged as a powerful tool to study state-dependent transcriptional changes (Levine et al., 2018). Studies using mammalian pituitary glands have yielded insights into pituitary cell fates in animal models (George et al., 2018; Ho et al., 2018; Ruf-Zamojski et al., 2018; Fletcher et al., 2019) and human fetal pituitary glands (Zhang et al., 2020). However, efforts to map the post-natal human pituitary gland transcriptome have been hindered by lack of normal pituitary gland controls. Therefore, studies on the transcriptome of the pituitary gland have relied on allogenic, post-mortem pituitary glands (Jiang et al., 2011; Michaelis et al., 2011; Dorman et al., 2012).
Micro-adenomas in CD are small (<1 cm diameter), distort the pituitary gland minimally, and are often embedded deep within the normal pituitary gland. This allows for micro-neurosurgical separation of the adenoma (tumor or core cells) from adjacent normal pituitary gland (normal or margin cells) (Lu et al., 2018). Here, we used this strategy to separately procure syngeneic core and margin tissue from patients with CD for parallel transcriptional profiling of single cells. Variant analysis found no evidence of mutations at known CD point mutation loci, indicating ‘‘wild-type’’ CD adenomas. Among these cells, we identified canonical pituitary cell classes, characterized adenoma cell transcriptomes, and found a subset of proliferating cells within the CD adenoma compartment. A key CD adenoma marker gene, PMAIP1 (encoding the pro-apoptotic BH3 only Bcl-2 protein noxa), sharply demarcated adenoma cells from normal gland. Additional bulk RNA-seq, DNA methylome analysis, qRT-PCR, and immunohistochemistry buttressed these findings. The PMAIP1 locus was hypomethylated in CD adenomas compared with non-CD samples and autopsy-derived normal pituitary controls. However, despite hypomethylation and transcriptional upregulation, we found proteasomal degradation of noxa protein in CD adenomas, suggesting a mechanism for escape from apoptosis in these cells. Using the selective proteasomal inhibitor bortezomib, we rescued noxa ex vivo and in vitro, induced apoptosis, and reduced cell survival in adenoma cell lines. Our results demonstrate a mechanism of tumorigenesis with therapeutic implications for CD.
RESULTS
Human adult pituitary cell class identification
Sporadic pituitary micro-adenoma core and adjacent margin tissues were obtained from 34 patients via micro-neurosurgical dissection (Figure 1A; Video S1). Patients included 24 cases presenting with CD, one silent CD (sCD), 3 acromegaly (GH), 3 prolactinoma, and 3 non-functioning prolactinoma (NFPA) (Figure 1B; Table S1). This approach allowed for separate surgical annotation of adenomas and adjacent margin tissues for downstream analysis in 25 of 34 cases.
Figure 1. Surgical annotation captures canonical pituitary cell classes in post-natal pituitary gland.
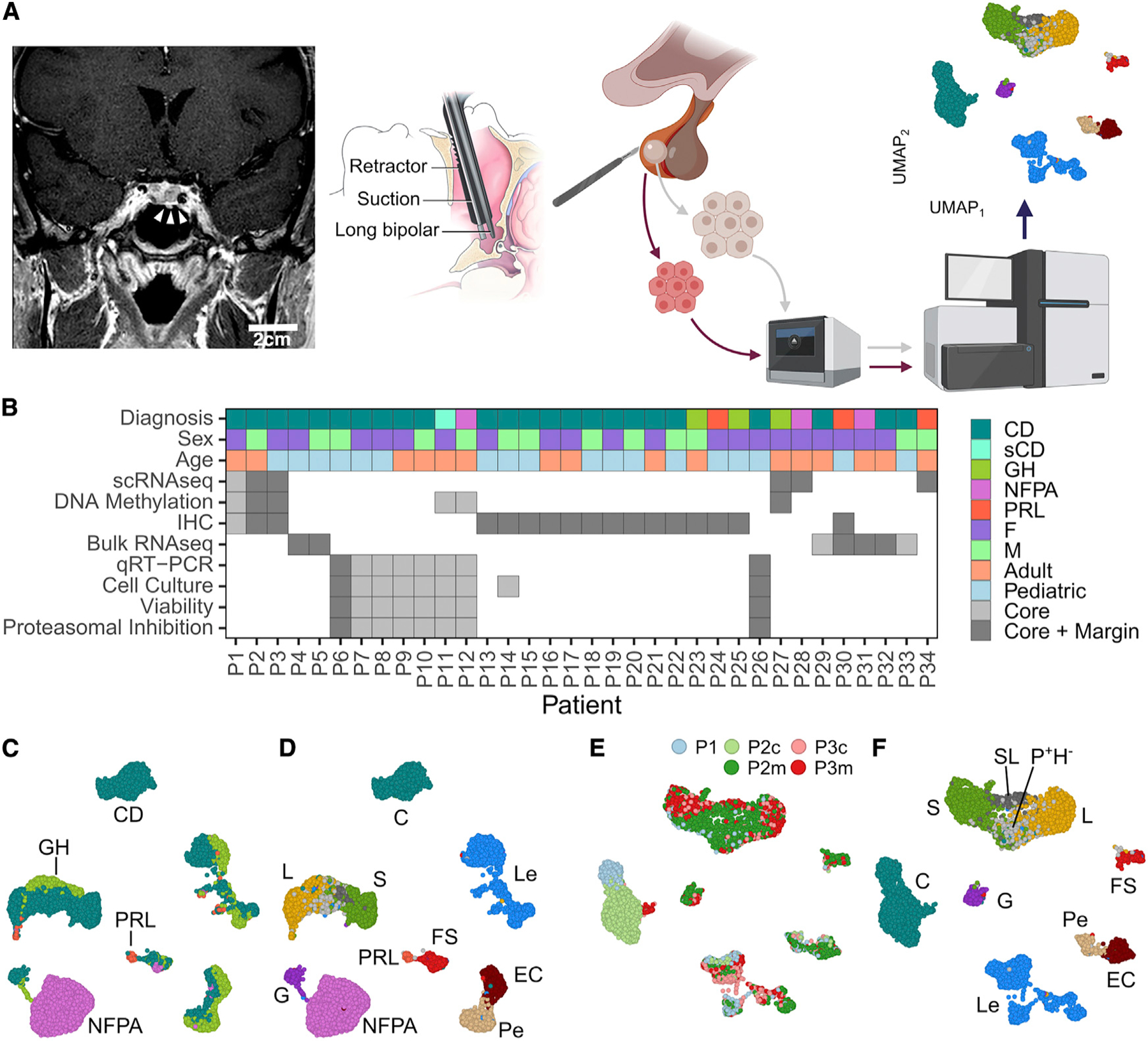
(A) A sublabial transsphenoidal approach was used for resection of microadenomas (white arrowheads) in patients with Cushing’s disease (CD). Coronal, post-gadolinium contrast enhanced magnetic resonance image from patient P1. Adenoma and adjacent normal pituitary gland were separately annotated during surgery and dissociated into a single-cell suspension, followed by GEM embedding, sequencing, and computational analysis. Scale bar: 2 cm.
(B) Summary of demographic data and analysis for patients included in the study.
(C) UMAP embedding of cells from the 6 patient samples with CD, GH, NFPA, and PRL adenomas used in scRNA-seq analysis. Cells cluster according to dominant secretory phenotype.
(D) UMAP embedding showing the cell-type identities cells from the 6 patient samples.
(E) UMAP embedding showing CD sample identity for cells from patients P1, P2, and P3.
(F) UMAP plot showing CD patient cells clustering by cell-type identity. Cell types: leukocytes (Les), endothelial cells (ECs), pericytes (Pes), folliculostellate cells (FSs), corticotrophs (Cs), gonadotroph (Gs), somatotrophs (Ss), lactotrophs (Ls), ambiguous/somato-lactotrophs (SLs), and POU1F1-positive, hormone-negative (P+H−) cells. GH, growth hormone; NFPA, non-functioning pituitary adenoma; PRL, prolactinoma.
We dissociated core and margin specimens (3 CD, P1–3, and 3 non-CD, P27, P28, and P34) into single cells for transcriptional profiling using the 10× Genomics Chromium scRNA-seq platform (Zheng et al., 2017). After running the Cell Ranger pipeline and filtering low-quality droplets (STAR Methods), we obtained a total of 27,594 cells for further analysis: 15,513 droplets from patients with CD and 12,081 from non-CD patients. Uniform manifold approximation and projection (UMAP) (McInnes et al., 2018) indicated separate clustering of CD, NFPA, and prolactinoma (PRL) adenoma cells from clusters of canonical pituitary cell types expected in margin samples (Figures 1C and 1D).
We used canonical cell-type marker genes to classify all cells from non-CD (Figure 1D) and CD samples (Figures 1E and 1F) using marker gene scores (see STAR Methods) (George et al., 2018; Ho et al., 2018; Fletcher et al., 2019). Cell identity labels closely corresponded to clusters in UMAP embeddings, supporting the classification result (Figures 1D and 1F). Canonical resident pituitary cell classes and circulating immune cells could be readily identified, including erythrocytes (Es; not shown), leukocytes (Les), endothelial cells (ECs), pericytes (Pes), folliculostellate cells (FSs), and hormone-producing cells (HPCs) including corticotrophs (Cs), gonadotrophs (Gs), somatotrophs (Ss), and lactotrophs (Ls; Table S1). PRL and NFPA adenoma cells differed sufficiently from margin cell types to warrant separate labels. Too few cells expressed thyrotroph markers to be included in further analysis, likely due to the rarity of normal thyrotrophs in the region of the pars distalis, where most adenomas occur (Baker and Jaffe, 1975).
As our primary focus was CD, we next investigated in detail tissues from three patients with sporadic CD adenomas (P1–P3; Figures 1E, 1F, and 2A) with the goal of characterizing the CD adenoma transcriptomic signature shared among them. We found no evidence in these samples of canonical CD mutations in USP8 (Reincke et al., 2014; Ma et al., 2015), USP48, or BRAF (Chen et al., 2018), using both variant calling from raw scRNAseq reads (STAR Methods; Table S1) (Robinson et al., 2011) and targeted PCR using mutation-specific primers (Figure S2).
Figure 2. Canonical pituitary cell classes are identified across samples.
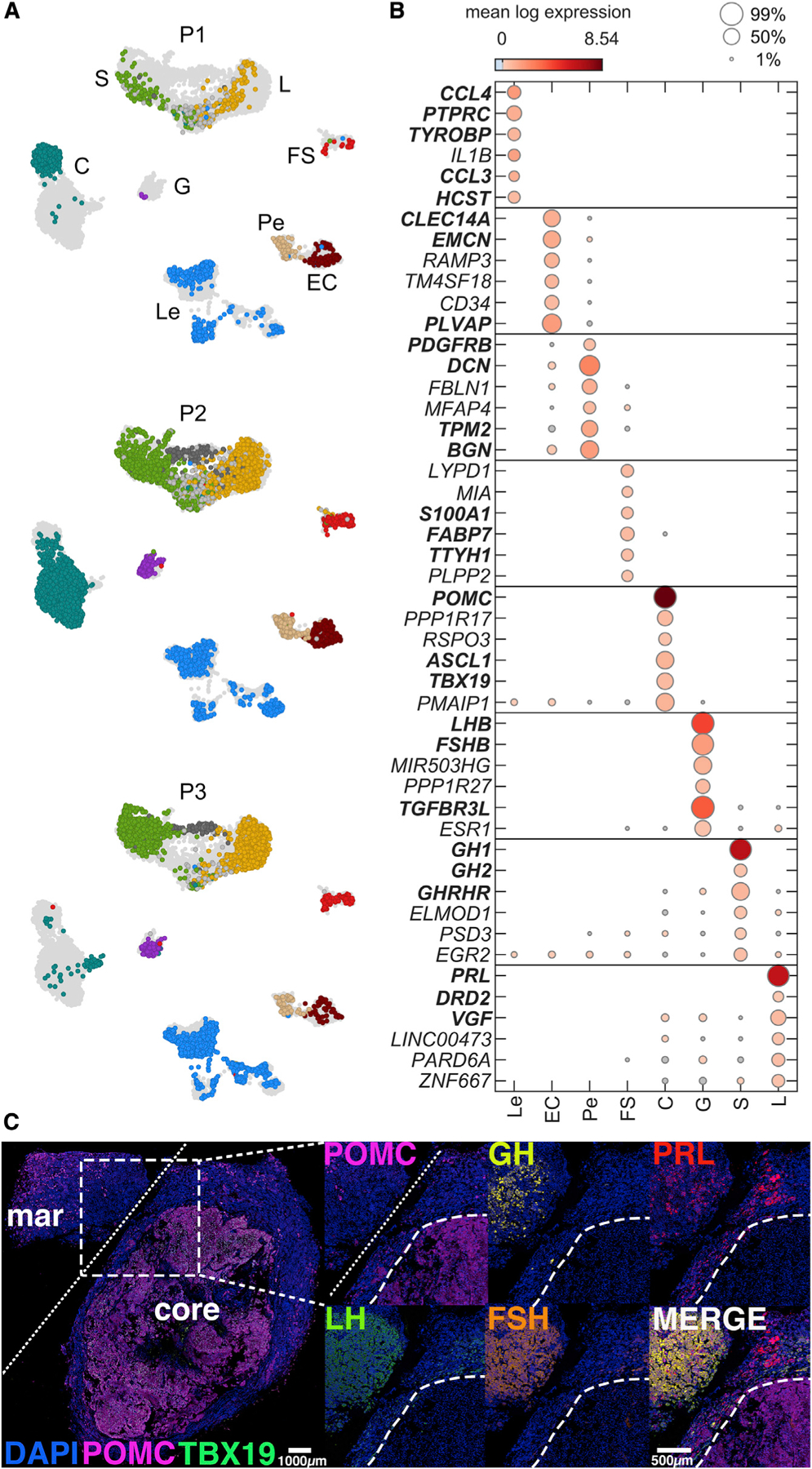
(A) The UMAP plot from Figure 1F split by patient (top, P1; middle, P2; bottom, P3), colored by cell type.
(B) Dot plot showing the expression (marker color) and percentage of cells expressing (marker size) for the top 6 cell-type upregulated genes with highest min.logFC.detected from a filtered group of robustly upregulated cell-type marker genes (STAR Methods). Genes used as a priori known classification marker genes are indicated by bold font.
(C) Multiplexed immunohistochemistry of tissue from P3, showing localization of key hormone markers in margin and core regions of the sample. White bar: 100 μm. PC, pituitary cells; HPC, hormone-producing cells; Mar, tumor margin; POMC, pro-opiomelanocortin; GH, growth hormone; PRL, prolactin; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
Pairwise differential expression analysis between cell types was performed using scran scoreMarkers with correction for patient batch (STAR Methods) to identify cell-type marker genes for human tissues (Figure 2B). Among top cell-type markers were subsets of the prior known canonical markers we used for cell classification. A complete list of identified markers is provided in Table S1. The identified cell classes in this study showed good agreement with the cell classes and gene markers in prior vertebrate studies (George et al., 2018; Ho et al., 2018; Fletcher et al., 2019; Fabian et al., 2020; Zhang et al., 2020). Immunohistochemistry of the adenoma core and surrounding margin tissue indicated uniform pro-opiomelanocortin (POMC) expression within the adenoma, while hormone immunoreactivity characteristic of non-corticotroph cells was evident in the surrounding margin tissue, consistent with scRNA-seq transcriptional profiles (Figure 2C).
Our cell classification procedure allowed some cells to remain unclassified or to be ambiguously classified as more than one canonical cell type. Most of the unclassified cells had nearest neighbors that were uniquely classified as S or L. These cells were POU1F1-positive but did not express other S or L marker genes, so we termed these POU1F1-positive hormone-negative (P+H−) cells. The most abundant subpopulation of ambiguous cells consisted of cells that expressed both S and L marker genes, termed SL, suggesting the presence of dual mammosomatrophic gene expression in our margin samples (Pasolli et al., 1994). Both SL and P+H− cells expressed several marker genes in common with S and L, including the lineage defining POU1F1 gene, the tumor suppressor MEG3-DLK1 (Gejman et al., 2008), and NLRP1 (Table S1; Figures S3A–S3B). Pseudotemporal ordering analysis (Trapnell et al., 2014) supported a single lineage determination for S, L, and P+H− cells (Figures S3C–S3D). Suppressed spontaneous and stimulated growth hormone (GH) in patients with CD has been attributed to suppressed somatotroph response to GH-releasing hormone (Magiakou et al., 1994; Borges et al., 1999). Our observation of P+H− cells, which lack key somatotroph marker gene expression, suggests transcriptional repression of the GH1 gene in a subpopulation of somatotrophs as a mechanism of GH deficiency in CD.
Consensus corticotroph-dominant genes identify adenoma cells
To identify consensus-dominant genes in adenomas causing CD, we compared corticotrophs from core and margin samples with all other cell types. Corticotrophs from core versus margin compartments were, respectively, annotated as C2c and C2m for patient P2 and C3c and C3m for patient P3. Based on surgical annotation and the large transcriptionally uniform cluster of cells that comprised P1 corticotrophs, we assigned C1 cells as core. We thus annotated C1, C2c, and C3c as core-sample corticotrophs (Ccs) and P2m and P3m as margin-sample corticotrophs (Cm). Using scoreMarkers with patient ID as batch, we computed effect sizes in gene expression separately between Cc and Cm versus every other cell type. We identified the robustly upregulated genes by selecting the union of genes with the top 20 largest effect sizes compared with all other cells for all three effect sizes reported by scoreMarkers (min.logFC.cohen, min.AUC, and min.logFC.detected), retaining only genes expressed in at least 25% of Cc or Cm, and enforced specificity to corticotroph sample by excluding genes expressed in over 25% of more than one other cell types.
We then evaluated the overlap between the identified Cc and Cm gene sets, focusing on common and Cc-specific genes (Figure 3A). Genes common to both Cc and Cm included canonical corticotroph markers (Figures 3A and 3B, blue). In addition to corticotroph lineage-restricted genes (POMC, TBX19) (Lamolet et al., 2001), we found dominant expression of genes common to corticotrophs: RAB3B, GAL, ASCL1, PCOLCE2, CALB1, and PCDH9 (Figure 3B, bottom segment; Table S1). Adenomatous corticotrophs are resistant to the hypercortisolism-induced POMC repression in CD (Neou et al., 2020). We therefore expected to find transcriptional repression of POMC in margin sample corticotrophs; POMC expression was indeed elevated in core samples relative to margin (Figure 3B). This was particularly evident in the P2 sample; with marker gene classification, we found few corticotroph-like cells in the margin sample for patient P2 (C2m, n = 19), in contrast to the abundance of corticotroph cells found in mammalian pituitary gland in the normal, non-hypercortisolemic state (Fletcher et al., 2019; Zhang et al., 2020). These cells expressed markers of terminal corticotroph differentiation such as TBX19 but were transcriptionally repressed for POMC (all but 4 were POMC–).
Figure 3. Consensus-dominant genes in adenomas causing CD.
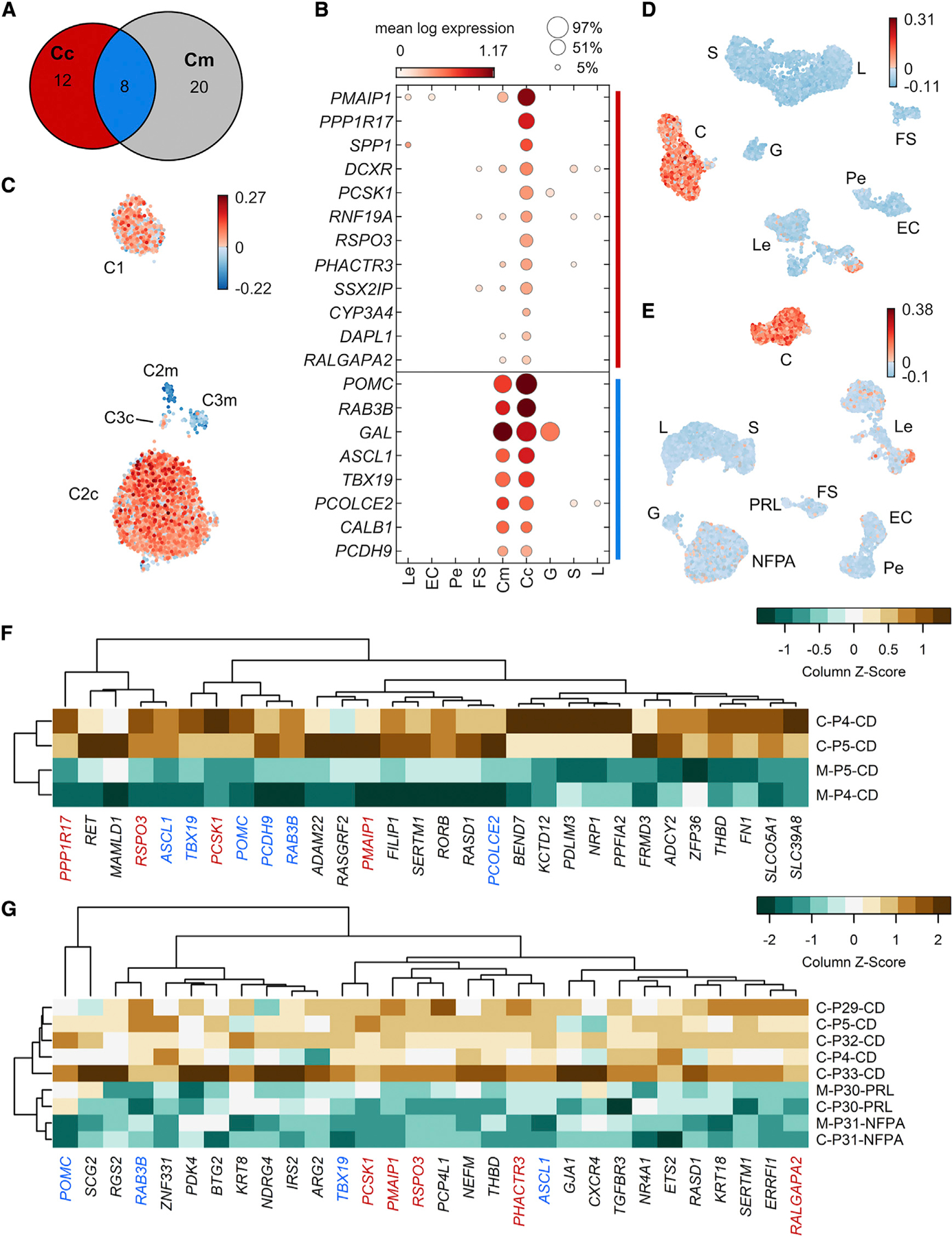
Core and margin sample corticotrophs were separately compared with all other cell types with scran’s scoreMarkers, using patient identity as a blocking factor. Stringent thresholds on effect sizes were used to identify a core set of robustly upregulated markers (STAR Methods).
(A) Venn diagram showing the number of core-specific (red), margin-specific (grey), and common (blue) marker genes identified.
(B) Dot plot showing the mean expression (marker color) and percentage (marker size) of each cell type expressing core-sample-specific genes (CD signature genes; top group, red in A) and corticotroph marker genes common to core and margin samples, which include canonical marker genes (bottom group, blue in A). Mean expression values are batch corrected across patients using scran correctSummaries. POMC expression was normalized to the second most highly expressed value to better show the levels of other genes (mean POMC: 8.29).
(C) UMAP plot of all C cells indicating the CD signature gene score per cell.
(D) UMAP plots for all cells from each patient show that cells with high CD signature scores are restricted to cell clusters classified as corticotrophs.
(E) UMAP plots for other adenoma subtypes including prolactinoma (PRL) and nonfunctioning pituitary adenoma (NFPA) verify that cells with high CD signature scores are specific to Cushing’s adenomas (C).
(F) Independent verification of overexpression of CD signature genes in pairwise bulk RNA-seq analysis of CD adenoma and adjacent normal pituitary gland (P4 and P5). Genes coidentified between bulk RNA-seq and scRNA-seq are color coded as in (B).
(G) Bulk RNA-seq comparison of CD and non-CD samples also verified upregulation of several CD signature and corticotroph marker genes.
The Cc genes that were not present in the Cm gene set were termed CD signature genes (Figures 3A, red, and 3B, top segment). Notable genes identified included phorbol-12-myristate-13-acetate-induced protein 1 gene PMAIP1 (Oda et al., 2000), protein phosphatase 1 regulatory subunit PPP1R17, secreted phosphoprotein 1 SPP1, the peptide hormone processing gene PCSK1, and the thrombospondin type 1 repeat supergene family member RSPO3. We verified the localization of CD signature genes by computing a CD signature score for all C cells, displayed on a UMAP embedding in Figure 3C. Cells with high CD signature gene scores (Cadenoma cells) were localized to P1 corticotrophs and to core samples of P2 and P3 as distinct from margin cells (Figures 3B and 3C), confirming agreement between our genetic classification methodology and anatomic demarcation at time of surgery. In all three patients, Cadenoma cells were found in cell clusters identified as corticotrophs via marker genes (Figure 3D). In integrated analysis with non-CD samples, expression of CD signature genes was not found in cell clusters representing GH-secreting adenoma (P27), non-functioning adenoma (P28), and PRL (P34) (Figure 3E).
To independently verify the overexpression of CD signature genes, we performed bulk RNA-seq in two additional syngeneic human CD adenoma-normal gland pairs (P4 and P5). We confirmed upregulation of corticotroph-restricted POMC (fold change: 7.1, false discovery rate [FDR]: 1.42E–12) and TBX19 (fold change: 5.3, FDR: 0.004) in the adenoma compartment, as well as upregulation of PMAIP1 (fold change: 5.7, FDR: 1.27E–13) in the adenoma compartment (Figure 3F). Notably, the top 30 CD upregulated genes identified by bulk RNA-seq included 6 of the corticotroph lineage-restricted genes (Figures 3F, blue labels) and 4 of the CD signature genes (red labels) identified by scRNA-seq. We also compared P4 and P5 core samples plus CD core samples from three additional patients (P29, P32, and P33) with both core and margin samples from two non-CD patients (Figure 3G; P30, PRL; P31, NFPA). Again, the scRNA-seq-identified signature genes were well represented, including PMAIP1, PCSK1, and RSPO3.
Self-renewing cells are rare in human pituitary adenomas
We next asked if a population of resident tumor stem-like cells may initiate and sustain human CD adenomas. We found induction of cell-cycle genes (Tirosh and Izar, 2016) (Table S1) in a small subpopulation of Cadenoma cells, as evidenced by high cell-cycle gene scores (Figure 4A). These proliferating cells (Cpro cells) have not previously been described in sporadic adenomas. None of the Cpro cells expressed stemness markers (SOX2/SOX9). Rather, they showed robust expression of terminally differentiated corticotroph cell markers POMC and TBX19 (Figure 4B). Compared with all other corticotrophs, Cpro cells overexpressed the pituitary tumor-transforming genes PTTG1, HMGB2, TUBB, CENPF, and SMC4 (Figure 4B) in addition to histone-modifying gene H2AFZ, cell-cycle regulators CDKN2D and CDKN3, and DNA-damage repair genes BRCA2 and BARD1, among others (Table S1). These findings suggest that CD adenomas host a subpopulation of terminally differentiated, proliferating corticotrophs. Proliferating cells were found among Les, as expected due to their higher proliferative capacity, but were mostly absent from other pituitary cell types (Figure 4C). We also found proliferating cells, albeit to a lesser extent, in PRL and NFPA tumors (Figure 4C), suggesting that these cells may be involved in various pituitary adenoma subtypes.
Figure 4. Non-stem-like proliferating cells localize to the adenoma compartment.
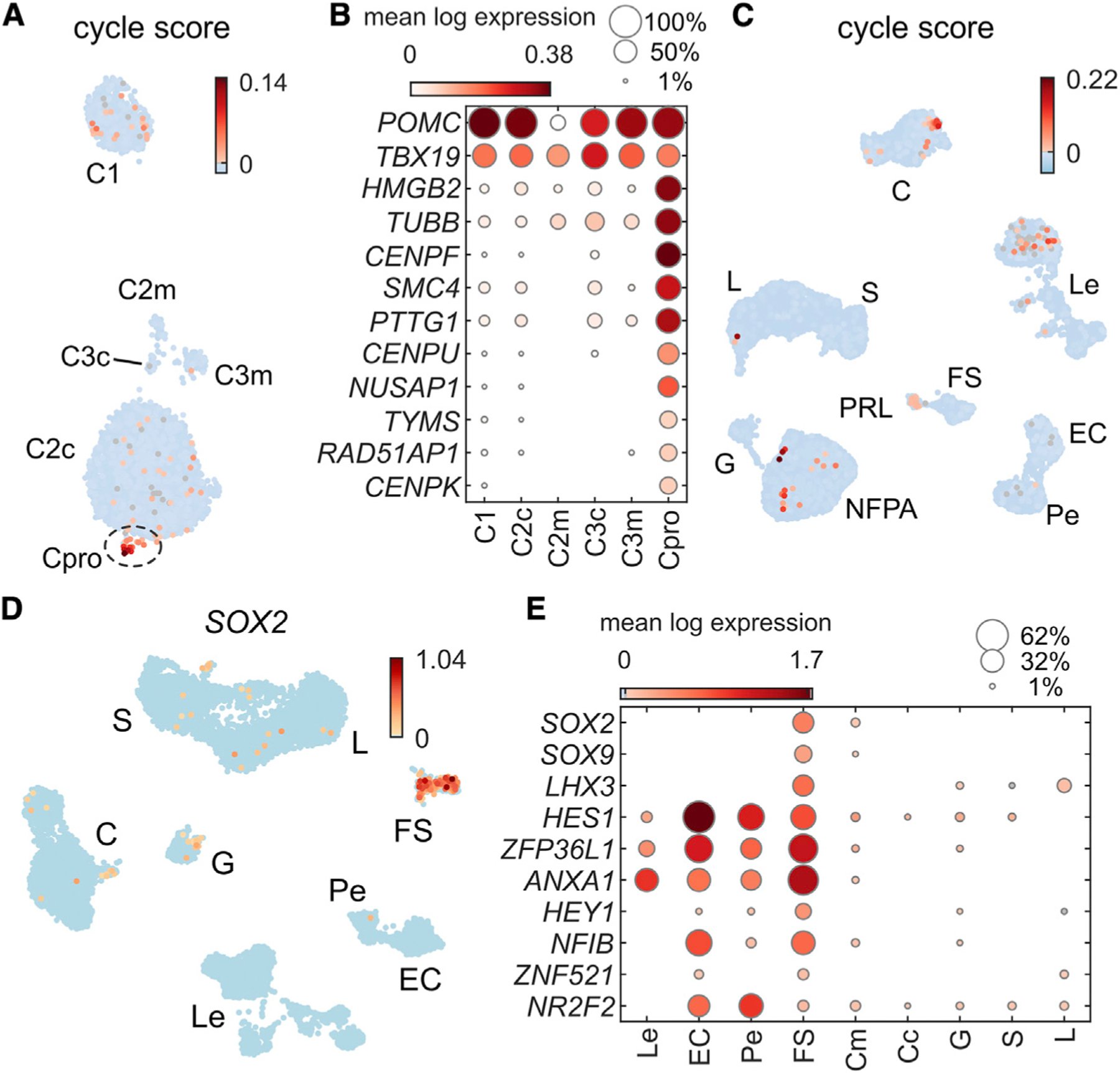
(A) UMAP plot of corticotroph cells showing cell-cycle gene scores per cell. We defined proliferating cells (Pros) for further study to have a cycle score >0.01.
(B) Dot plot showing 12 top expressed genes upregulated in Pros. These cells clearly expressed canonical corticotroph marker genes POMC and TBX19 in addition to cell-cycle genes.
(C) UMAP plot showing cell-cycle score across all six scRNA-seq patient samples, showing specificity to the C cluster and some Le cells, as well as localization to the NFPA, and PRL adenoma clusters.
(D) SOX2-positive cells are restricted to the cell cluster classified as FS cells and not expressed in the corticotroph compartment.
(E) Dot plot of select genes associated with pituitary stem cells (Cox et al., 2017; Fletcher et al., 2019) showing lack of expression in adenoma samples.
A subset of SOX2+ cells have been reported as progenitors in the mammalian pituitary gland that can self-renew and terminally differentiate into HPCs (Andoniadou et al., 2013). In human fetal pituitary glands, most SOX2+ cells do not express the FS marker S100B (Zhang et al., 2020). We found that in post-natal human pituitary samples, SOX2 was expressed in the FS compartment but not within Cadenoma cells (Figure 4D). We found very few FS cells within core samples. Patient P2 had only 3 FS cells within the core sample, and patient P3 had 45 FS cells. We also found that genes associated with self-renewing tumor stem cells (George et al., 2018) were not expressed in Cadenoma cells (Figure 4E). The paucity of stem-like cells within our adenoma samples precludes any conclusions about their potential role in CD tumorigenesis.
Discrepant PMAIP1 transcript and noxa protein expression in human CD adenomas
PMAIP1 encodes for pro-apoptotic noxa, a BH3-only Bcl2 family protein, and has been reported to be suppressed in non-functioning pituitary adenomas (Jiang et al., 2011). We confirmed that PMAIP1 overexpression was specific to CD adenomas and not found in PRL, GH adenomas, or NFPAs (Figure 5A). To investigate the unexpected upregulation of a pro-apoptotic signal, we screened for transcription factors known to bind to the PMAIP1 promoter and found upregulation of MYC (Nikiforov et al., 2007; Woloschak et al., 1994; Wang et al., 1996) overlapping with PMAIP1-expressing cells in Cadenoma (Figure 5B, yellow dots in middle panel). We verified MYC overexpression within adenoma compared with normal cells using bulk RNA-seq (P4 and P5; fold change: 4.6, FDR: 0.002) and using multiplexed immunohistochemistry (mIHC) of CD tumors compared with adjacent margin cells (Figure 5E).
Figure 5. Human adenomas causing wild-type CD have discrepant PMAIP1 transcript and noxa abundance.
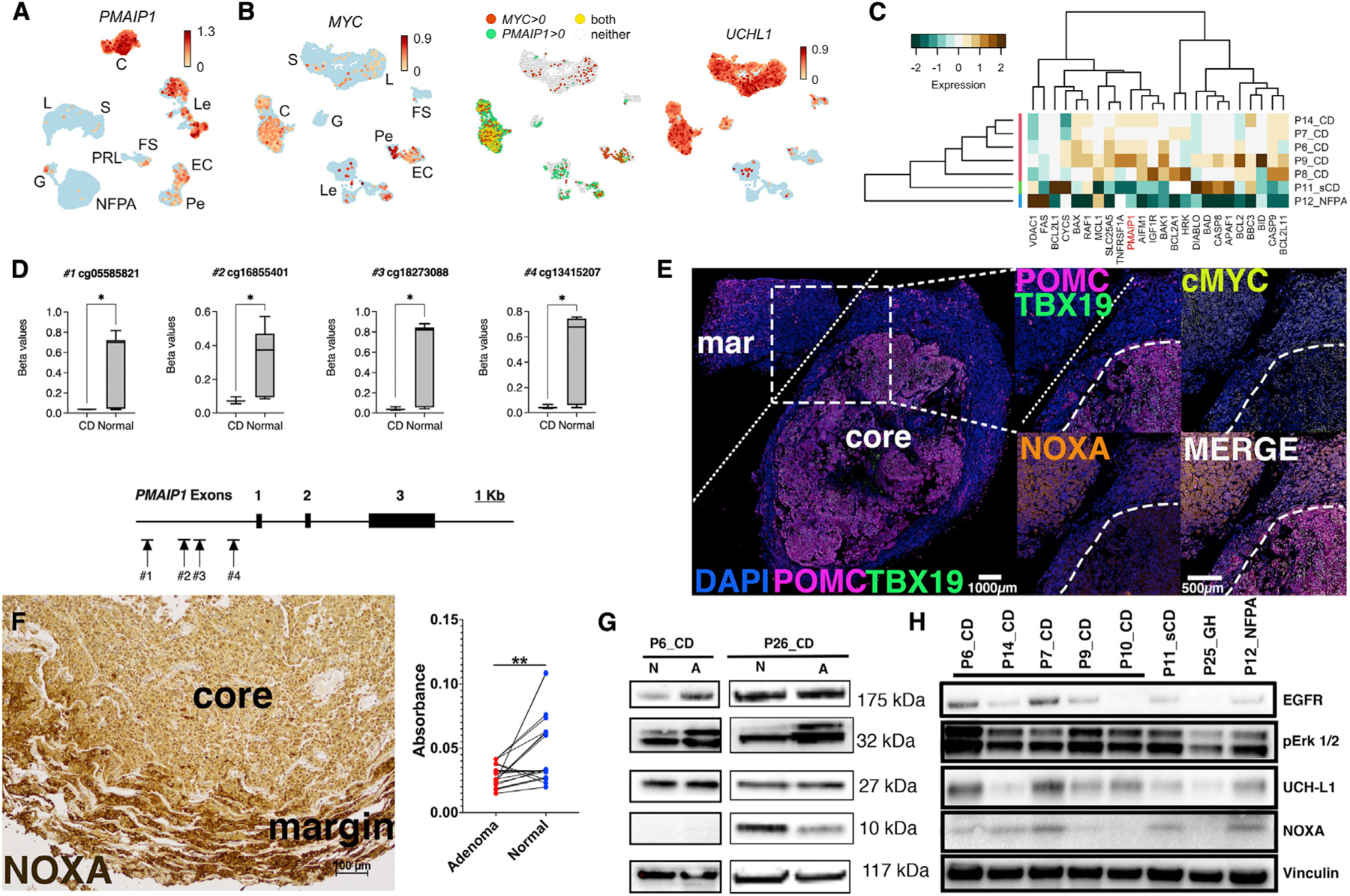
(A) UMAP plot demonstrating PMAIP1 abundance in CD adenomas (C) but not in PRL, G, or NFPA adenomas. PMAIP1 was also detected at lower levels in Les and ECs.
(B) UMAP plot showing MYC upregulation in CD corticotrophs but not other hormone-producing cells (left panel). MYC expression was also detected in Pes, ECs, and Les. Middle panel: UMAP plot showing overlap of MYC and PMAIP1 expression is mostly limited to CD corticotrophs (yellow dots). Right panel: UMAP plot showing UCHL1 abundance in most hormone-producing cell types in CD.
(C) Bulk RNA-seq of CD and non-CD samples verified overexpression of pro-apoptotic genes including PMAIP1 in CD tissues.
(D) DNA methylation levels (beta values) at CpG sites associated with the PMAIP1 promoter methylation demonstrating hypomethylation in CD (n = 3) compared with normal (autopsy-derived, n = 20) pituitary glands. *p < 0.05.
(E) Multiplex immunohistochemistry (mIHC) of 5 μm thick sections from a CD adenoma. Insets from the core-margin boundary represented by white dashed lines. White bar: 100 μm. Core-margin boundary identified by overlaying expression of POMC, TBX19, and DAPI. Compared with the margin, core adenoma cells show robust overexpression of POMC, TBX19, and MYC; however, noxa expression is decreased within the adenoma core.
(F) Representative image from noxa IHC in independent adenoma/normal pairs (n = 10). Pairwise analysis of noxa deconvoluted IHC images (absorbance = mean pixel intensity count per pixel) showing suppressed noxa signal in CD adenomas compared with normal (margin) tissues (p = 0.0013; 95% confidence interval [CI] −0.027 to −0.007). Scale bar: 100 μM.
(G) Expected epithelial growth factor (EGF) signaling upregulation and ERK1/2 phosphorylation were found in human CD adenoma primary cell lines (P6_CD and P26_CD). A, adenoma (core); N, normal (margin) pituitary gland. Noxa was undetectable or decreased in core adenomas.
(H) A survey of human primary CD adenoma cell lines revealed variable noxa expression compared with sCD adenoma, GH adenoma, and NFPA. CD, corticotroph adenoma causing Cushing’s disease; sCD, hormonally silent corticotroph adenoma; GH, growth-hormone-secreting adenoma; NFPA, non-functioning pituitary adenoma.
We further investigated the role of PMAIP1 and its encoded protein noxa in CD tumorigenesis. Using PCR arrays, we found broad transcriptional changes in apoptosis-related genes including PMAIP1 in primary cell lines derived from CD patients compared with a silent corticotroph adenoma and a non-functioning pituitary adenoma (Figure 5C). To determine the epigenomic basis for these transcriptional changes in CD adenomas (Capper et al., 2018), we performed DNA methylation analyses on three CD tumors (P1–P3) compared with 20 autopsy-derived normal pituitary glands. We found hypomethylation at the PMAIP1 promoter locus at the 5ʹ UTR and the first exon, which is critical for binding of transcription factor BMI1 (Yamashita et al., 2008), in CD adenomas compared with autopsy-derived pituitary glands (Figure 5D). In CD adenomas, CpG islands associated with PPP1R17 and RSPO3 were hypomethylated but not PCSK1 or EGLN1, indicating incomplete overlap between DNA hypomethylation and transcriptional upregulation of signature genes.
Paradoxically, with IHC, noxa protein levels were consistently depleted in CD adenomas compared with surrounding normal gland (Figure 5E). IHC analysis (Figure 5F) and subjective, blinded analysis by a trained neuropathologist confirmed that 8 of the 10 independent CD adenoma samples had decreased noxa abundance compared with adjacent normal gland (Table S1). However, noxa abundance was unchanged in GH adenoma and PRL (Figure S4). We confirmed noxa suppression in CD adenomas using western blot on whole-cell lysates from two syngeneic pairs of primary human cell lines from CD adenomas and their adjacent normal tissues (Figure 5G). We then surveyed primary cell lines of CD, hormonally silent corticotroph adenoma, GH, and non-functioning adenoma and found variable, low-noxa expression in CD adenomas but expected EGF signaling activation (Theodoropoulou et al., 2004; Figure 5H).
Discrepant PMAIP1 transcript and noxa abundance suggested post-translational degradation previously reported in mantle cell lymphoma (Dengler et al., 2014). To investigate this pathway, we generated a corticotroph-enriched mouse cell line (mCort) (Lu et al., 2017) with lentiviral transduction of human EGFR to simulate dysregulated EGF signaling (mCortEGFR) found in a majority of CD adenomas (Fukuoka et al., 2011a; Reincke et al., 2014; Ma et al., 2015) (Figure 6A). We confirmed expression of canonical corticotroph genes including Pomc in mCort with microarray (Figure 6B) and western blot (Figure 6C) analysis. In culture, mCort cells recapitulated pituitary cell morphology (Figure 6D). We found induction of Pomc and Pmaip1 in mCortEGFR with qRT-PCR compared with control transfected mCortEV and similar to murine corticotroph tumor cell line ATT20 (Furth et al., 1953) (Figure 6E).
Figure 6. Proteasome inhibition rescues noxa and accelerates apoptotic cell death in wild-type CD adenomas.
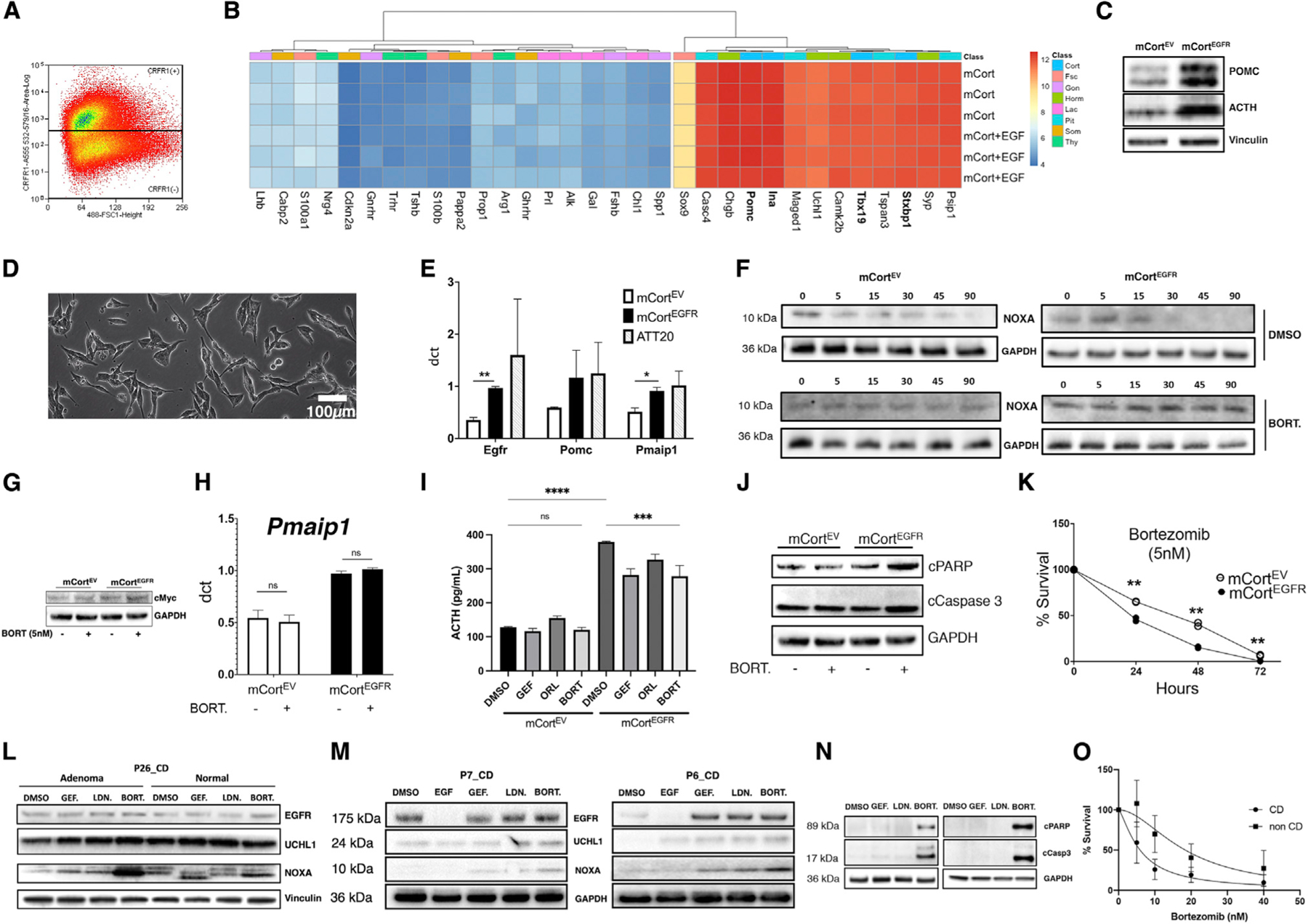
(A) Mouse pituitary cells (3 biological replicates) were flow sorted and enriched for cortricotropin-releasing hormone receptor (CRFR1).
(B) The cell line enriched for corticotrophs (mCort) demonstrated markers of corticotroph differentiation using PCR arrays (3 biological replicates per cell line). Gene expression remained stable after addition of EGF.
(C) The ACTH-producing CD phenotype was recapitulated following lentiviral human EGFR transduction (mCortEGFR) with increased POMC processing to ACTH (n = 3).
(D) Representative bright-field image showing mCort cells with elongated processes and abundant intracellular organelles.
(E) The tumorigenic sequence of CD adenomas was recapitulated with relative overexpression (qRT-PCR; ΔCT; GAPDH) of both POMC and PMAIP1 (2 experiments in technical triplicates) following lentiviral human EGFR transduction (mCortEGFR). *p < 0.05.
(F) Cycloheximide chase study (2 experiments) in murine mCort cells demonstrated post-translational degradation of noxa that was arrested with proteasome inhibitor bortezomib (5 nM, BORT.).
(G) Bortezomib (5 nM) decreased MYC protein levels in mCortEGFR but not in mCortEV cells (2 independent experiments).
(H) Bortezomib (5 nM) induced minimal transcriptional upregulation of PMAIP1 in mCort cells, suggesting a predominantly post-translational effect (3 independent experiments in technical triplicates).
(I) Supernatant from mCortEGFR cultures showed elevated ACTH production with EGFR overexpression recapitulating the adenoma phenotype (ACTH oversecretion; 3 independent experiments in technical triplicates). Additionally, bortezomib exposure reduced ACTH in supernatant from mCortEGFR cells. *p < 0.05.
(J) Bortezomib resulted in elevated cleaved PARP (cPARP) and cleaved caspase-3 (cCaspase3) in mCortEGFR cells following exposure to bortezomib (5 nM, 6 h; 2 replicates).
(K) Bortezomib exposure for 24 h decreased survival in mCortEGFR cells at lower bortezomib concentrations (IC50: 1.5 nM) compared with in mCortEV cells (IC50: 3.096 nM). **p = 0.0087, 0.0066, and 0.0080 for 24, 48, and 72 h, respectively. Data shown for dose response performed on each cell line in triplicate and timed survival in duplicate. *p < 0.05.
(L–N) Biological duplicates.
(L) Pairwise analysis of adenoma and normal cells from a patient with CD (P26_CD). Responses to DMSO control versus EGFR inhibitor gefitinib (GEF), UCHL-1 inhibitor LDN-57444 (LDN), or proteasomal inhibitor bortezomib (BORT). Protein levels of EGFR; DMSO, dimethyl sulfoxide; EGFR, epidermal growth factor receptor; UCHL-1, ubiquitin C-terminal hydrolase L1.
(M) Patient-derived CD adenoma primary cell lines P7 and P6 demonstrated elevated EGFR expression at baseline that was suppressed with EGF (100 ng, 24 h) exposure. Bortezomib (5 nM, 6 h) induced noxa at a higher level than gefitinib (10 μM, 6 h) or UCHL1 inhibitor LDN (15 μM, 6 h).
(N) Bortezomib induced markers of apoptosis with cPARP and cCaspase3 in human-derived primary CD adenoma cells.
(O) Bortezomib (24 h) decreased cumulative survival in human-derived primary cell lines from CD (n = 5; IC50: 5.929 nM) and non-CD (n = 3; IC50: 18.42 nM) cell lines (p < 0.0001); data shown in triplicates.
Since UCHL1, a specific noxa stabilizer (Brinkmann et al., 2013), is constitutively expressed in pituitary HPCs (Fletcher et al., 2019) and CD adenomas (Figure 5B, right panel), we suspected ubiquitination-independent proteasomal degradation of noxa (Dengler et al., 2014). A cycloheximide chase assay confirmed post-translational degradation of noxa in mCortEGFR (Figure 6F). We found that mCortEGFR cells overexpressed cMyc (Figure 6G) similar to CD adenomas (Figure 5B), suggesting a shared pathway in Pmaip1 expression. Proteasomal inhibition with bortezomib effectively rescued noxa within mCortEGFR cells (Figure 6F) with no transcriptional effect on the Pmaip1 mRNA (qRT-PCR; Figure 6H) (Nikiforov et al., 2007). Bortezomib treatment decreased ACTH secretion (Figure 6I), induced apoptotic markers cleaved PARP and cleaved caspase-3 (Figure 6J), and decreased cell survival specifically in mCortEGFR cells (Figure 6K).
We then compared the effect of gefitinib, a tyrosine kinase inhibitor used in CD (Fukuoka et al., 2011b), LDN-57444, an inhibitor of noxa deubiquitinase UCHL1 (Brinkmann et al., 2013), and bortezomib on noxa abundance within CD adenoma cells in primary culture. Bortezomib rescued noxa (Figures 6L–6M) and induced markers of apoptosis (cleaved PARP and cleaved caspase-3; Figure 6N), while LDN-57444 unexpectedly failed to decrease noxa. Together, these findings suggest ubiquitin-independent, proteasome-mediated noxa degradation in CD adenoma cells (Pang et al., 2014). In cell viability assays, bortezomib inhibited the growth of multiple patient-derived CD adenoma cell lines at clinically relevant doses (average half maximal inhibitory concentration [IC50] of 5.9 nM/mL), demonstrating the therapeutic potential of proteasome inhibition in CD (Figure 6O). However, the non-CD primary cell lines (sCD, NFPA, GH) required higher bortezomib concentrations (IC50 of 18.4 nM/mL) to achieve similar growth inhibition, suggesting that the CD adenoma cells were more sensitive to noxa-mediated apoptosis (Figure 6O).
DISCUSSION
scRNA-seq as a surrogate for histopathology
In this study, we performed detailed single-cell-level transcriptomic annotation of the post-natal human anterior pituitary gland in patients with CD. We also demonstrated the utility of surgical annotation of adenoma and adjacent normal pituitary gland based on transcriptomic signatures. We thus implemented a molecular histology paradigm with the potential to complement immunohistochemical analysis of tumor samples and adenoma cell identification. We confirmed cellular heterogeneity of the normal gland while finding a predominance of terminally differentiated corticotrophs in the adenoma compartment. The variable proportions of corticotrophs we identified in this study (48% in patient 1, 49% in patient 2, and 2.7% in patient 3) highlight inherent limitations in bulk-sequencing approaches to studying the pituitary gland and could partly explain the lack of mechanistic targets identified to date in CD. Syngeneic, pairwise scRNA-seq studies are ideally suited to explore these mechanisms in greater detail.
Transcriptional signatures of the plurihormonal state
Recent studies have reported production of more than one hormone type from a single anterior cell in human pituitaries (Mitrofanova et al., 2017) and in adenomas using IHC and scanning electron microscopy (Kovacs et al., 1998). Trans-differentiation has been proposed as a mechanism in these so-called plurimorphous plurihormonal adenomas (Vidal et al., 1999). This phenomenon introduces ambiguity in histology-based classification of pituitary adenomas. We identified a total of 1,453 unclassified cells and 1,200 ambiguously classified cells in our study. We were able to identify 956 of these unclassified cells whose most common neighbors were somatotrophs or lactotrophs as P+H− cells. Among the 1,200 ambiguously classified cells, 359 expressed genetic markers for both lactotrophs and somatotrophs, confirming previous reports of a plurihormonal state in the post-natal human pituitary gland.
State-dependent transcriptional changes
We found evidence of distinct transcriptomic signatures attributable to the hypercortisolemic state in CD. In our dataset, we found a very low frequency of Cnormal cells. These cell expressed corticotroph lineage-restricted TBX19 expression with absent POMC expression (Lamolet et al., 2001; Liu et al., 2001). In CD, corticotrophs in the adjacent normal gland retain significant ACTH protein (Figure S7) (McNicol, 1981; Oldfield et al., 2015). However, we found POMC transcriptional repression in margin corticotrophs likely due to the hypercortisolemic state in CD. We suspect that transcriptional repression precedes, and is uncoupled from, protein expression in Cnormal (Lee et al., 2011; Liu et al., 2016), likely due to systemic hypercortisolism. We also found evidence of hypercortisolism-related transcriptional signatures in POU1F1+ cells. Patients with CD and serum hypercortisolism have suppressed GH dynamics (Takahashi et al., 1992; Magiakou et al., 1994) and growth suppression (Yordanova et al., 2016), likely due to GH1 downregulation in the pituitary gland (Leal-Cerro et al., 1994). Our observation of the P+H− cells is suggestive of such hypercortisolism-mediated suppression of S marker genes (GH1, GHRHR, GH2) in a subset of S cells.
FS cells are rare within pituitary adenomas
SOX2+ FS cells with S100 immunoreactivity (Nakajima et al., 1980; Rinehart and Farquhar, 1953) have many proposed roles including modulation of hormone production and secretion in the anterior lobe via local paracrine actions (Allaerts et al., 1994), via direct intercellular communications (Soji et al., 1997), or via release of cytokines that influence hormone output (Lohrer et al., 2000). More recent murine work has identified a side population of SOX2+ cells within the pituitary gland that are capable of self-renewal and differentiation into HPCs (Lepore et al., 2005) (Chen et al., 2005, 2009). Whether these cells constitute adult pituitary progenitors in humans or contribute to the pathogenesis of CD has been a matter of intense study. In this study, we identified only a few cells expressing the progenitor marker SOX2, and they were confined to the FS compartment. While we did not find a tumor stem cell niche within the adenoma, paracrine signaling from SOX2+ FS cells in the tumor micro-environment cannot be excluded (Andoniadou et al., 2013; Russell et al., 2021).
Proliferating cells reside within CD adenomas
We discovered a subcluster of CProlif cells within the adenoma. These cells were more abundant within recurrent CD sample in P2. The relative abundance of Cprolif cells may reflect the greater number of adenomatous cells in P2 or may highlight the role of Cprolif cells in recurrent CD. Cprolif cells overexpress the pituitary tumor transforming gene PTTG1 which encodes securin and upregulates cell proliferation (Tfelt-Hansen, Kanuparthi and Chattopadhyay, 2006a). PTTG1 induces transformation in vitro and in vivo by inhibiting chromatid separation, causing chromosomal gain or loss (Pei and Melmed, 1997). It is abundantly expressed in more aggressive pituitary tumors (Zhang et al., 1999; Filippella et al., 2006), as well as in ovarian, esophageal, pancreatic, and colorectal tumors (Tfelt-Hansen, Kanuparthi and Chattopadhyay, 2006b), and conditional Pttg1 overexpression in the mouse pituitary gland is sufficient for ectopic generation of hormone-secreting adenomas (Abbud et al., 2005). PTTG1-expressing Cprolif cells are therefore likely tumor-initiating cells in CD.
Noxa degradation in CD
The PMAIP1 promoter that encodes noxa spans about 4 kb and contains regulatory regions for over 40 transcription factors and coactivators (Guikema, Amiot and Eldering, 2017a). PMAIP1 is upregulated by p53 and p73 in response to DNA damage, by the polycomb repressive complex 1 (PRC1) member Bmi1 (Yamashita et al., 2008; Teshima et al., 2014; Li et al., 2018) as well as PRC2 (Xie et al., 2014), by DNA methyltransferase Dnmt1-induced CpG methylation, by ER stress due to accumulation of misfolded proteins, by a Hif1α-mediated response to hypoxia, and by elevated MTOR/AKT signaling in response to unregulated growth signaling (Guikema, Amiot and Eldering, 2017b). PMAIP1 activation therefore represents a common pathway for induction of apoptosis in response to cellular stress or unregulated proliferation.
MYC-driven PMAIP1 overexpression induces apoptosis in tumors (Wirth et al., 2014). However, despite MYC/PMAIP1 coexpression, corticotroph adenomas escape apoptosis by post-translational degradation of noxa protein. Mantle cell lymphoma cells similarly evade apoptosis by extensive proteasomal degradation of noxa (Dengler et al., 2013a). Inhibition of this pathway using the proteasome inhibitor bortezomib has been shown to induce the apoptotic program in mantle cell lymphoma (Dengler et al., 2013b). UCHL-1 is a noxa deubiquitinase (Brinkmann et al., 2013) and is also specifically expressed in hormone-producing pituitary cells (Fletcher et al., 2019). We found that specific inhibition of UCHL-1 did not further destabilize noxa in CD adenomas, while bortezomib was able to rescue noxa effectively. These data suggest that noxa deregulation may be ubiquitin independent and that proteasomal inhibition may lead to noxa rescue even with deubiquitinase (e.g., USP8) activating mutations (Ma et al., 2015; Reincke et al., 2015).
Limitations of the study
Our study was limited by a modest number of single-cell libraries from limited patients, which may have precluded in-depth assessment of normal thyrotrophs in the present study. These limitations highlight inherent difficulties acquiring fresh normal adult pituitary gland tissues, which are only accessible during en route dissections for pituitary adenectomies. There may also exist histone modifications that underly our results or post-transcriptional changes that further contribute to the pathogenesis of wild-type CD that were not captured by our single-cell or DNA methylation approaches. Future studies involving comprehensive surveys of the chromatin landscape and proteome in CD may yield additional targetable disease mechanisms.
Conclusions
We generated a transcriptomic map of the post-natal human pituitary gland and defined signature genes for CD adenomas. Our study uncovered a previously unrecognized population of proliferating cells within the adenoma that may drive tumorigenesis. In addition, we identified apoptosis escape via proteasome-mediated noxa degradation as an oncogenic mechanism. Selective proteasomal inhibition rescued noxa and triggered apoptosis in human primary cells from CD, suggesting its clinical utility for human CD.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be addressed to and will be fulfilled by the lead contact, Prashant Chittiboina, MD, MPH (prashant.chittiboina@nih.gov).
Materials availability
The human pLV-V5-EGFR plasmid generated in this study will be deposited with Addgene (addgene.org). Original Western blot images will be deposited at Mendeley by the time of publication. All additional information required to reanalyze the data reported in this paper is available from the lead author upon request.
Data and code availability
Single-cell RNA-seq and whole RNA-seq data has been uploaded to GEO and will be publicly available by the date of publication. Whole microscopy data reported in this paper will be shared by the lead author upon request.
No original code was generated in this study
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Annotated surgical samples were obtained from non-consecutive patients (n = 34) that underwent surgery by author PC between 2014 and 2019 (clinicaltrials.gov identifier NCT00060541) (Table S1). Their clinical diagnoses included CD (n = 24), silent-CD (n = 1), non-functioning pituitary adenoma (n = 1), prolactinoma (n = 1), and GH-secreting adenoma (n = 2). The study was conducted at the National Institutes of Health (NIH) Clinical Center in Bethesda, MD and approved by the Combined Neuroscience Institutional Review Board (IRB) of the National Institute of Neurological Disorders and Stroke, Bethesda, MD. Written informed consent was obtained from each patient for research study participation, and the study was conducted according to the standards set by the IRB.
Corticotroph cells were harvested from female, BALB/c mice (aged 6–8 weeks; Taconic Biosciences, USA). Mice were euthanized according to IACUC standards of humane and ethical practices and following institutional protocols. Cells were digested and homogenized in 1 mg/mL collagenase (Sigma-Aldrich, USA) for 15 min and cultured in DMEM (Thermo Fischer Scientific, USA) with 10% FBS (Thermo Fischer Scientific, USA) and 1% penicillin-streptomycin (Gibco, USA) in 5% CO2 at 37°C.
METHOD DETAILS
Patients
Annotated surgical samples were obtained from non-consecutive patients (n = 34) that underwent surgery by author PC between 2014 and 2019 (clinicaltrials.gov identifier NCT00060541) (Table S1). Their clinical diagnoses included CD (n = 24), silent-CD (n = 1), non-functioning pituitary adenoma (n = 3), prolactinoma (n = 3), and GH-secreting adenoma (n = 3). Pituitary core-margin pairs for syngeneic, pairwise transcriptomic analysis were obtained from 13 non-consecutive patients. All hormone secreting adenomas (CD, GH, PRL) had evidence of hormonally active pituitary microadenomas and biochemical evidence of endocrinopathy (96).
Hypercortisolism in CD patients was diagnosed as previously described (Asuzu et al., 2017) based on elevated late night salivary cortisol (chemiluminescent enzyme immunoassay, Siemens Immulite 1000 analyzer, NIH Department of Laboratory Medicine, Bethesda MD; normal range <100 ng/dL), elevated 24-h urine free cortisol (high performance liquid chromatography/tandem mass spectrometry, NIH Department of Laboratory Medicine, Bethesda, MD; normal range: 1.4–20 μg/24H for 3–8 year-olds, 2.6–37 for 9–12 year-olds, 4.0–56 μg/24H for 13–17 year-olds and 3.5–45.0 μg/24H for ≥18 year-olds) or low-dose dexamethasone suppression testing (1mg overnight or 2mg over 48 h) (Lonser et al., 2017; Nieman et al., 2008). Supplementary midnight serum cortisol (normal range <7.5 mcg/dL) and ACTH (Nichols Advantage Immunochemiluminometric assay or chemiluminescence immunoassay, Siemens Immulite 2500 analyzer, NIH Department of Laboratory Medicine, Bethesda MD, normal range 5–46 pg/mL) (Papanicolaou et al., 2016), and 24-h urinary 17- hydroxycorticosteroids (17-OHS/Cr) excretion (Quest Diagnostics Nichols Institute, Chantilly VA; normal range 3–10 mg/24H for males and 2–6 mg/24H for females) (Hsiao et al., 2009) were used as needed. Pituitary ACTH production was verified using early morning ACTH levels, 8mg overnight dexamethasone suppression testing (DST) and ovine corticotropin-releasing hormone (CRH) stimulation testing (Dichek et al., 1994; Günes et al., 2013; Nieman et al., 1993). Average post-CRH or post-DDAVP stimulated levels were determined preoperatively in all CD patients by averaging serum cortisol at 30 and 45 min after stimulation, and for ACTH by averaging serum levels at 15 and 30 min after stimulation.
All patients underwent a sub-labial trans-sphenoidal approach for microscope-assisted selective adenectomy. Adenoma was identified by visual inspection and removed by micro-surgical dissection along a circumferential pseudo-capsular plane (97). Pituitary adenoma was removed en-bloc within its pseudocapsule (Video) and sent for histopathological examination following formalin fixation and paraffin embedding (FFPE). The en-route or adjacent non-viable adjacent normal pituitary gland was removed separately and annotated for analysis (98).
Research specimens were labeled separately and sent for bulk RNAseq, scRNAseq analysis, immunohistochemistry, or were dissociated for primary cell culture. The study was conducted at the National Institutes of Health (NIH) Clinical Center in Bethesda, MD and approved by the Combined Neuroscience Institutional Review Board (IRB) of the National Institute of Neurological Disorders and Stroke, Bethesda, MD. Written informed consent was obtained from each patient for research study participation, and the study was conducted according to the standards set by the IRB.
Single cell RNA sequencing
Human pituitary adenomas and adjacent normal pituitary glands were stored and transported in 0.9% NaCl or RPMI 1640 on ice. Samples were chopped into ~2–4mm3 sized pieces and dissociated using the Human Tumor Dissociation Kit in gentleMACS C tubes using the gentleMACS dissociator (Bergisch Gladbach, North Rhine-Westphalia, Germany). Samples were briefly centrifuged at 300 rpm for 30 s and filtered through a 70-nm strainer. The single cell suspensions were washed with 10 mL of RPMI 1640 and re-centrifuged at 2100 rpm for 5 min at 4° Celsius. Single-cell suspensions then underwent red blood cell (RBC) lysis using the RBC Lysis Solution 10X (Bergisch Gladbach, North Rhine-Westphalia, Germany). Following RBC lysis, non-viable cells were removed using the Dead Cell Removal Kit (Bergisch Gladbach, North Rhine-Westphalia, Germany). Single cell suspension quality, number and viability were assessed with a dual fluorescence cell counter Luna-FL (Logos Biosystems, Gyeonggi-do, South Korea). A total of 8,000–10,000 cells from each normal or adenoma cell suspension were washed twice with PBS+0.04% BSA and resuspended to a final concentration of 1000 cells per μL.
10X Genomics’ Chromium instrument and Single Cell 3ʹ Reagent kit (V3 and 3.1) were used to prepare the individually barcoded scRNAseq libraries following the manufacturer’s protocol. Library quality was assessed by BioAnalyzer traces (Agilent BioAnalyzer High Sensitivity Kit) and quantitated using the Qubit system. Sequencing was performed on the Illumina NextSeq machine, using the 150-cycle High Output kit with 28 bp read 1, 8 bp sample index, and 91 bp read 2. Following sequencing, the bcl files were demultiplexed into a FASTQ, aligned to human transcriptome GRCh38, and single-cell 3ʹ gene counting was performed using the standard 10X Genomics’s CellRanger software pipeline (mkfastq, count, aggr; V3.0.2). In total, 48900 droplets were recovered from six patient samples: 28,724 from CD samples and 20176 from non-CD samples.
scRNAseq data dimensionality reduction and visualization
Linear dimensionality reduction of gene-space was performed using principal component analysis (PCA) computed on a set of highly variable genes (100). Genes were clustered into 30 bins based on mean expression. Normalized dispersion was computed and the 2,000 genes with highest normalized dispersion were retained. Prior to PCA, the expression values for each gene were standardized and clipped to a maximum of 10 standard deviations from the mean. The top principal components (PCs) were then computed using MATLAB’s svds function, and the number of PCs retained was determined using a permutation test (17). The number of significant PCs was determined by comparing the data’s true PC singular values to a null distribution generated by reshuffling the counts among cells for each gene independently. Significant PCs had singular values larger than the null distribution’s max at p < 0.01, resulting in 41 significant PCs for the full N = 28,724 cells. PCA scores were used as inputs for downstream analysis, k-nearest neighbor classification (knnsearch, MATLAB R2021b), and further nonlinear dimensionality reduction using Uniform Manifold Approximation and Projection (UMAP) (101) for visualization (umap-learn, v0.5.2) (Figure 1D).
Cell classification, quality control filtering, and ambient RNA removal
Cell classification and quality control were performed in parallel so that cell type-specific thresholds for common cell quality control metrics could be applied. This procedure was performed separately for each data from each patient. Cells were first classified based on expression of known marker genes. Erythrocytes expressed at least one of HBB, HBA1, or HBA2; leukocytes expressed at least two of AIF1, C1QA, CCL3, CCL4, CCL4L2, CCL5, CD3D, CD52, CD53, CST7, CTSS, FCER1A, FCER1G, GPR183, HCST, IL7R, ITGB2, LYZ, NKG7, PTPRC, S100A8, S100A9, TRBC2, or TYROBP; pericytes, two of PDGFRB, DCN, TPM2, TAGLN, BGN, or ACTA2; endothelial cells, two of PLVAP, EMCN, CAVIN2, KDR, CLEC14A, PECAM1; and folliculostellate cells, two of SOX2, S100B, S100A1, S100A11, FABP7, SOX9, TTYH1, LYPD1, RFX4, MGST1, CCDC80, or HSD11B1. HPCs expressed one of SCG2, SCG3, CHGB, SEZ6L2, SYP, SNAP25, or UCHL1, as well as the following cell type-specific markers: C expressed one of POMC, TBX19, RAB3B, or ASCL1; G expressed two of CGA, LHB, FSHB, GNRHR, NR5A1, or TGFBR3L; S expressed two of POU1F1, GH1, GH2, GHRHR, or PAPPA2; and L expressed two of POU1F1, PRL, DRD2, or VGF. Based on the distribution of expression levels across all cells from a given patient’s samples, an expression threshold was computed to define whether a cell expressed the marker gene using Otsu’s optimal thresholding algorithm (99), as previously described (17).
Cell type scores, defined as the number of cell type markers expressed, were computed for every cell type in each cell. Three outcomes are possible for each cell: a cell could be uniquely classified (satisfying exactly one cell type condition), ambiguously classified (satisfying two or more cell type conditions), or unclassified (satisfying none of the cell type conditions). Dropouts in marker genes could lead to unclassified cells, so we attempted to classify these cells using K-nearest neighbor classification, as previously described (17). We examined the K = 30 neighbors of each unclassified cell and assigned a cell type only if the majority of their neighbors were uniquely classified as that type. The classification of cell types resulting from this procedure matched well with the visually obvious clusters in the 2-dimensional UMAP embeddings.
Cell type-specific thresholds were used to identify low quality droplets for exclusion from further study. Outliers in fraction of mitochondrial genes were identified using a threshold of 2.5 median absolute deviations (MAD) above or 3.5 MAD below the cell type-specific median for a total of 3,325 droplets. Similarly, we identified 3,344 outlier droplets in terms of library complexity with >2 MAD below or >3.5 MAD above the cell type-specific median number of genes expressed. Outlier thresholds were also applied to unclassified cells, for which the threshold was defined as the cell type count-weighted average of thresholds from other cell types. Finally, erythrocytes were excluded. A total of 11,891 cells were removed during quality control, leaving 16,833 cells including unclassified and ambiguous cells for further analysis.
The final UMAP embeddings and cell classification were obtained after cell filtering using a new round of dimensionality reduction following the same procedure outlined above. The top 2,000 highly variable genes were identified among remaining cells, PCA was performed, and 51 significant PCs (permutation test, p < 0.01) were retained. A fresh K-nearest neighbor classification of unclassified cells was performed using the new PCA coordinates to obtain the final cell classes. A total of 1,435 cells remained unclassified, while 1,200 cells were ambiguously classified.
Ambient RNA was detected for highly expressed genes, including HBB, POMC, GH1, and PRL. For final visualization and differential expression analysis, we removed contaminating counts from each sample using the R package SoupX. Default parameters were used, with cell type classes provided as cluster IDs.
Identification of POU1F1 lineage cells
Most unclassified cells remaining appeared in UMAP embeddings near cells uniquely classified as S or L. These cells were retained for further analysis, identified as unclassified cells whose most common uniquely classified neighbors were either S or L. These accounted for 956 of the 1,435 unclassified cells. These cells were POU1F1-positive but did not express S or L marker genes sufficiently and were termed P+H− cells. Similarly, we retained the most commonly occurring set of ambiguously classified cells, which satisfied classification for both S and L and termed these SL. These cells accounted for 359 of the 1,200 ambiguous cells. The remaining unclassified and ambiguous cells were excluded from further analysis, leaving a final total of 15,513 cells. Final number of cells per type in each sample are outlined in Table S3. Pseudotemporal analysis of the POU1F1 lineage cells was performed using R packages Seurat and Monocle3 (PMID: 24658644).
Identification of cell type-upregulated genes
Marker genes were identified for cell contrasts of interest using the scoreMarkers function from scran, using patient ID as the batch variable. For every pairwise contrast between cell types of interest, this function computes batch-corrected estimates of three gene expression effect sizes logFC.cohen (standardized log-fold-change in expression), AUC (area under the curve, closely related to the Wilcoxon rank sum test statistic), and logFC.detected (log-fold-change in proportion of cells with nonzero expression). We considered upregulated genes to be the set of genes simultaneously satisfying min.logFC.cohen>0, min.AUC>0.5, and min.logFC.detected>0, indicating that by all three effect size metrics the gene was upregulated in a given cell type compared to all other cell types of interest. Further filtering on upregulated genes was performed as indicated in the text and figure legends, by filtering on proportion of cells expressing the gene and selecting the union of genes with the top values of each effect size.
Variant calling
Bam files produced per sample by Cell Ranger were imported into the CLCbio Genomics Workbench v20 (qiagen.com) and processed through a workflow consisting of the following supported tools in order: ‘‘Extract Reads’’, ‘‘Map Reads to Reference’’, ‘‘Local Realignment’’, ‘‘Low Frequency Variant Detection’’, ‘‘Annotate with Exon Numbers’’, ‘‘Annotate from Known Variants’’, ‘‘Predict Splice Site Effect’’, ‘‘Amino Acid Changes’’, ‘‘Export VCF’’. Per genome reference and annotations, those for hg38 (ftp.ensembl.org) were used in conjunction with known variants reported by and available from ClinVar (20200419), HapMap (phase 3, v97), the 1000 Genomes Project (phase 3, v99), and dbSNP (build 151). Local realigned mapping files and variant call files were exported from the Workbench and samtools (www.htslib.org) used to subsequently sort and index the mapping files.
Bulk RNA sequencing
Paired human adenoma and normal pituitary gland were procured from surgery and immediately flash frozen in 1 mL of Qiazol (Qiagen, Germany) within Lysing Matrix E tubes (MP Biomedicals, Santa Ana, CA) in dry ice. We thawed the contents and homogenized the tissues using a Fast Prep-24 Homogenizer, (MP Biomedicals) for 20 s at 4.5 m/s. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Germany). RNA quantity and quality were measured by NanoDrop ND-1000, and integrity assessed by agarose gel electrophoresis. PolyA+ mRNA isolation, size fragmentation, cDNA synthesis, size selection, and next gen sequencing were performed at the National Intramural Sequencing Center, Bethesda, MD according to their standard protocols, as described previously (102). RNA-Seq libraries were constructed from 1 μg total RNA after rRNA depletion using Ribo-Zero GOLD (Illumina). The Illumina TruSeq RNA Sample Prep V2 Kit was used according to manufacturer’s instructions.
Paired-end sequence files (.fastq) per sample were quality inspected using the FastQC tool 0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) then adaptor clipped (TruSeq3-PE-2.fa:2:30:10) and trimmed to remove 5ʹ nucleotide bias (HEAD-CROP:12) and low quality calls (TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:15) using the Trimmomatic tool 0.39 (http://www.usadellab.org/cms/?page=trimmomatic). Surviving intact pairs of reads per sample were then imported into the CLCbio Genomics Workbench v11 (https://www.qiagenbioinformatics.com/) and reference mapped by sample in stranded fashion against the current instance of the human genome (GRCh38) using the “RNA-Seq Analysis” tool supported therein under default parameters. Expression in counts per known annotated gene (Homo_sapiens.GRCh38.96.chr.gff3) were then exported from the tool.
Analysis of bulk RNA sequencing data for paired core-margin samples was performed using the edgeR package on total exon counts per gene. A paired analysis was performed with GLM-based approach was used with design matrix including patient and sample type. Genes were filtered by expression with filterByExpr, dispersion estimated with estimateDisp, and the GLM fit with glmFit. The log ratio test was used to detect differences between core vs margin or CD vs nonCD samples using glmLRT, and significant genes were recorded at FDR<0.01 and log fold-change > 1.5.
Multiplex fluorescence immunohistochemistry and multispectral imaging
Multiplex fluorescence immunohistochemistry (mIHC) was performed on 5 μm-thick paraffin sections of pituitary tissue samples using select antibody panels targeting relevant biomarkers of pituitary adenoma signaling pathways. Briefly, sections were first deparaffinized and treated using a standard antigen unmasking step in 10 mM Tris/EDTA buffer pH 9.0. Sections were then blocked with Human BD Fc Blocking solution (BD Biosciences) and treated with True Black Reagent (Biotium) to quench intrinsic tissue autofluorescence. The sections were then immunoreacted for 1 h at RT using 1 μg/mL cocktail mixture of immunocompatible antibodies targeting EGFR (GeneTex Inc, USA), phosphorylated EGFR Tyr992 (Cell Signaling Technologies, USA), POMC (LifeSpan Biosciences, USA), c-Myc (Santa Cruz, USA), TBX19 (ThermoFisher, USA), NOXA (Novus Biologicals, USA), TSH (Abcam, USA), GH (Novus Biologicals, USA), LH (Novus Biologicals, USA), prolactin (Novus Biologicals, USA) or FSH (Novus Biologicals, USA). Primary antibodies were either directly conjugated or indirectly labelled with secondary antibodies using the following spectrally compatible fluorophores: Alexa Fluor 430, Alexa Fluor 488, Alexa Fluor 546, Alexa Fluor 594, Alexa Fluor 647, IRDye 800CW. After washing off excess antibodies, sections were counterstained using 1 μg/mL DAPI (Thermo Fisher Scientific) for visualization of cell nuclei. Slides were then mounted using Immu-Mount medium (Thermo Fisher Scientific) and imaged using an Axio Imager.Z2 slide scanning epifluorescence microscope (Zeiss) equipped with a 20X/0.8 Plan-Apochromat (Phase-2) non-immersion objective (Zeiss), a high-resolution ORCA-Flash4.0 sCMOS digital camera (Hamamatsu), a 200W X-Cite 200DC broad band lamp source (Excelitas Technologies), and 7 customized filter sets (Semrock). Image tiles (600 × 600 μm viewing area) were individually captured at 0.325 micron/pixel spatial resolution, and tiles seamlessly stitched into whole specimen images using the ZEN 2 image acquisition and analysis software program (Zeiss). Pseudocolored stitched images were then exported to Adobe Photoshop and overlaid as individual layers to create multicolored merged composites.
Immunohistochemistry
Surgically derived tissue specimens were fixed in formalin and embedded in paraffin. Tissue blocks were then sectioned (5 μm) and stained with an automated immunostainer (Bond-Max, Leica). Tissues were probed using noxa antibodies (ThermoFisher, USA) after being validated with positive control tissue samples and a high pH (EDTA) epitope retrieval. Slides were reviewed for relative noxa abundance by a blinded, board-certified neuropathologist (ARC). Digital IHC images were converted to grayscale and the auto-threshold function was used to delineate antibody positive tissue. The percent area of antibody positive tissue was compared to the number of positive objects identified to determine the relative protein expression. Analysis of immunohistochemistry was completed using ImageJ v1.53e.
Primary human corticotroph cell culture
Primary human corticotroph tumor and normal pituitary cells were digested and homogenized in 1 mg/mL Clostridium hemolyticum collagenase (Sigma-Aldrich, USA) for 15 min and cultured in RPMI 1640 (Thermo Fischer Scientific, USA) with 10% fetal bovine serum (FBS) (Thermo Fischer Scientific, USA) and 1% penicillin-streptomycin (Gibco, USA) in 5% CO2 at 37°C. Cells incubated with human epidermal growth factor (hEGF) (Sigma-Aldrich, USA) were serum starved using 0% FBS media for two-hours prior to incubating with 100ng of hEGF for 24-h prior to whole-cell lysis.
Corticotroph enriched mouse corticotroph cells
Pooled corticotroph cells were harvested (<30 min post-euthanasia) from female, BALB/c mice (aged 6–8 weeks; Taconic Biosciences, USA), digested and homogenized in 1 mg/mL collagenase (Sigma-Aldrich, USA) for 15 min and cultured in DMEM (Thermo Fischer Scientific, USA) with 10% FBS (Thermo Fischer Scientific, USA) and 1% penicillin-streptomycin (Gibco, USA) in 5% CO2 at 37°C. Following dissociation and culture of pooled mouse (n = 10) pituitary cells, corticotrophs were isolated by flow cytometry using anti – corticotropin-releasing hormone receptor 1 (anti-CRHR1) antibody (Invitrogen, USA) followed by Alexa Fluor-555 conjugated antibody (Thermo Fisher, USA).(46). Cell sorting and analysis were carried out using a MoFlo Astrios cell sorter and Summit acquisition and analysis software (Beckman Coulter, USA) to identify mouse corticotroph cells only (mCort). We confirmed the gene expression profile of mCort cells with microarray analysis (Clariom S Mouse, Thermo Fisher, USA) and analyzed with R packages (affy, limma and pheatmap). Corticotroph phenotype preservation was confirmed by the presence of secreted ACTH at each generation in the cell culture media by enzyme-linked immunosorbent assay (ELISA), using the ACTH ELISA kit (MDBio products, USA) according to the manufacturer’s instructions. All animals were euthanized in accordance with the standards and guidelines of the Institutional Animal Care and Use Committee of the National Institutes of Health.
ATT-20 D16:16 Mouse Corticotroph Tumor Cells
ATT-20 D16:16 murine pituitary corticotroph tumor cells were provided as a generous gift from Dr. Steven L. Sabol at the National Heart, Lung and Blood Institute. ATT-20 cells were cultured in DMEM (Thermo Fischer Scientific, USA) with 10% FBS (Thermo Fischer Scientific, USA) and 1% penicillin-streptomycin (Gibco, USA) in 5% CO2 at 37°C. ACTH was measured to ensure preservation of phenotype (as performed in corticotroph enriched mouse corticotroph cells).
Lentiviral construct and transduction of mouse corticotrophs
The human V5-EGFR construct was generated using a lentiviral (pLV) expression system containing a neomycin resistance gene on a pCMV backbone. V5-EGFR was inserted into the pLV backbone using the BsrGI-SbfI restriction sites. Virus particles were generated using the Dharmacon™ Trans-Lentiviral packaging kits (TLP5916, Horizon Discovery, UK) following co-transfection into HEK293T producer cells, as per the manufacturer’s instructions. The corticotroph enriched mouse corticotrophs (mCort) were used as our target cell line and killing curve was generated against G418 Geneticin (Gibco, USA) before viral transduction. Transduction for both empty vector (pLV) and recombinant vector (pLV- V5-EGFR) were performed as per the manufacturer’s instructions. Mouse corticotroph cells were collected from cultures grown in absence of penicillin/streptomycin and suspended at 50,000 cells/mL in complete DMEM media. Virus particles were diluted at 5, 10, 50, 100 and 500-fold using DMEM complete media containing polybrene 10 μg/mL and 0.5 mL was distributed among 5 wells in a 6 well culture plate, as well as one with only 0.5 mL of media. Cell suspension (1mL) was added to each well and incubated with the viral particles for 48–72 h in 5% CO2 at 37°C. Using neomycin, mouse corticotrophs with the empty vector (MCEV) and those with the V5-EGFR (mCortEGFR) were selected to establish stable cell lines.
RNA extraction and quantitative Real Time-PCR (qRT-PCR)
RNA was extracted from both human and mouse cells using the RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. Complementary DNA (cDNA) libraries were constructed using SuperScript III for qRT-PCR (Invitrogen Life Technologies, USA). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the Sso Universal IT SYBR green supermix (BioRad, USA). Gene expression in human primary cell cultures were determined after template amplification using the Prime PCR assay H384 plates for Mitochondrial Apoptosis and EGFR Signaling Pathways (BioRad, USA) according to the manufacturer’s instructions. Gene expression in the mouse corticotroph cells and AtT-20 cells were determined using primers designed for epidermal growth factor receptor (EGFR), pro-opiomelanocortin (POMC), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) (Table S11). Relative gene expression was calculated using the ΔCt method with GAPDH as the housekeeping gene.
Western immunoblotting
Cells were washed with ice-cold PBS (Invitrogen Life Technologies, USA) and whole-cell lysates were collected on ice using a radio-immunoprecipitation assay (RIPA) based lysis buffer (Thermo Scientific, USA) containing a protease inhibitor cocktail (Halt protease inhibitor, Sigma-Aldrich, USA). Whole-cell lysates were quantified using the bicinchoninic acid (BCA) protein assay (Thermo Scientific, USA). Proteins were then electrophoretically separated on 4%–12% NuPAGE Bis-Tris gels (Invitrogen, USA) and electroblotted onto polyvinylidene difluoride membranes (PVDF) with the Trans-Blot Transfer Turbo System (Bio-Rad, USA). After blocking membranes for 1 h in 5% bovine serum albumin (BSA) in 0.05% TBS-Tween at room temperature, they were incubated overnight at 4°C with primary antibodies. Primary antibodies include anti-EGFR A10 (Santa Cruz, USA) at 1:1000, anti-Vinculin 7F9 (Santa Cruz, USA) at 1:2000, anti-Erk1/2 (Cell Signaling Technologies, USA) at 1:1000, anti-phospho-Erk1/2 (Cell Signaling Technologies, USA) at 1:2000, anti-UCH-L1 (Santa Cruz, USA) at 1:1000, anti-NOXA D8L7U (Cell Signaling Technologies, USA) at 1:1000, anti-cleaved PARP (Cell Signaling Technologies, USA) at 1:1000, and anti-caspase 3 (Cell Signaling Technologies, USA) at 1:1000. Membranes were then washed with TBS-Tween and incubated with peroxidase-conjugated secondary antibody for 1 h at room temperature while rocking. Membranes were developed with ECL (Super Signal West Femto, Thermo Fisher, USA). Immunoreactive signal was detected and imaged using the ChemiDoc MP Imaging System (Bio-Rad, USA) with subsequent densitometric quantification performed using ImageJ v1.53e.
Post-translational degradation assay
Protein half-life in murine mCort cells were measured after treatment with Bortezomib (5nM, 6 h) using 20ug/mL of cycloheximide (CHX) (Sigma-Aldrich, USA). Cells were harvested at the indicated times following CHX treatment (0, 5, 15, 30, 45, 90 min) and cells were lysed prior to western immunoblotting as previously mentioned.
Inhibitor compounds
Human and mouse cells treated with inhibitors were incubated for 6 h prior to whole cell lysis. Compounds used were the EGFR inhibitor, Gefitinib ZD1839 (SelleckChem, USA) at 10 μM or 20 μM, the proteasomal inhibitor, Bortezomib (Millipore, USA) at 5nM or 10nM, and the UCHL-1 inhibitor, LDN-57444 (Sigma-Aldrich, USA) at 15uM. All were solubilized using DMSO (ThermoFisher, USA) as the vehicle and controls were incubated with DMSO only, at the relative concentration of the corresponding inhibitor drug counterpart.
Cell viability assays
Cell cultures were seeded in 96-well plates at a density of 5,000–10,000 cells per well in 100uL of media and allowed to grow for 24 h prior to treatment. Cells were treated with increasing concentrations of Gefitinib (5um, 10um, 20um, 40um) and Bortezomib (5nM, 10nM, 15nM, 20nM) for 24 h prior to viability assay. DMSO controls were incubated with the highest concentration of DMSO used corresponding to the comparative drug treatments. Prior to analysis, plates were equilibrated to room temperature and cell viability assay performed using CellTiterGlo (Promega Corporation, USA) according to the manufacturer’s instructions. Plates were incubated for 30 min at room temperature prior to recording luminescence on the Synergy Neo2 microplate reader (BioTek, USA).
Illustrative figures
Figure 1A diagram was created on Biorender.com.
QUANTIFICATION AND STATISTICAL ANALYSIS
DNA methylation beta values (Figure 5E) were compared using two-sample T tests after testing for variance equality, and using Welch’s approximation for degrees of freedom. Quantification of noxa immunohistochemistry (Figure 5H) in tumor-margin samples was performed using paired T-tests. EGFR and PMAIP1 gene expression (Figures 6E and 6H), and mean cell survival (Figure 6K) were compared between mCortEV and mCortEGFR cell lines using paired T-tests. ACTH ELISA results (Figure 6I) were compared between mCortEV and mCortEGFR cell lines, and between DMSO versus bortezomib treatment in mCortEGFR cells using paired T-tests. p values <0.05 two-tailed were considered statistically significant. Statistical analyses were performed using STATA 14/IC (StataCorp LP, College Station, Texas) or GraphPad Prism 9.0 (GraphPad Software, La Jolla, California).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGFR | GeneTex Inc | Cat #GTX20231; RRID:AB_368210 |
| Phospho-EGFR Tyr992 | Cell signaling | Cat #2235s; RRID:AB_331708 |
| POMC | LifeSpan Biosciences | Cat #LS-C340619–100 |
| c-Myc | Santa Cruz | Cat #sc-40 AF546; RRID:AB_627268 |
| TBX19 | Thermo Fisher | Cat #MA5–31431; RRID:AB_2787067 |
| Noxa | Novus Biologicals | Cat #NB600–1159AF594; RRID:AB_10001419 |
| TSH | Abcam | Cat #ab64378; RRID:AB_1143452 |
| GH1 | Novus Biologicals | Cat #NBP2–53262 |
| LH | Novus Biologicals | Cat #NBP2–47733AF700 |
| Prolactin | Novus Biologicals | Cat #NBP3–08245AF488 |
| FSH | Novus Biologicals | Cat #NBP2–47734AF532 |
| DAPI | Thermo Fisher | Cat #D1306; RRID:AB_2629482 |
| CRHR1 | Invitrogen | Cat #720290 |
| EGFR A10 | Santa Cruz | Cat #sc-373746; RRID:AB_10920395 |
| Vinculin 7F9 | Santa Cruz | Cat #sc-73614; RRID:AB_1131294 |
| Erk 1/2 | Cell Signaling | Cat #4695; RRID:AB_390779 |
| Phospho Erk 1/2 Thr202/Tyr204 | Cell Signaling | Cat #4370; RRID:AB_2315112 |
| UCH-L1 | Santa Cruz | Cat #sc-271639; RRID:AB_10714950 |
| Noxa D8L7U | Cell Signaling | Cat #14766; RRID:AB_2798602 |
| Cleaved PARP | Cell Signaling | Cat #5625; RRID:AB_10699459 |
| Caspase 3 | Cell Signaling | Cat # 9662; RRID:AB_331439 |
|
Chemicals, peptides, and recombinant proteins | ||
| Clostridium histolyticum collagenase | Sigma-Aldrich | Cat #9001–12-1 |
| RPMI 1640 | Thermo Fisher | Cat #11875093 |
| FBS | Thermo Fisher | Cat #26140 |
| 1% penicillin-streptomycin | Gibco | Cat #15070063 |
| Human epidermal growth factor | Sigma-Aldrich | Cat #11376454001 |
| DMEM | Thermo Fisher | Cat #11965084 |
| Bortezomib | Millipore | Cat #CAS 179324–69-7 |
| Gefitinib | SelleckChem | Cat #ZD1839 |
| LDN-57444 | Sigma-Aldrich | Cat # 668467–91-2 |
| DMSO | Thermo Fisher | Cat #85190 |
|
Critical commercial assays | ||
| ACTH ELISA | MDBio products | Cat #M046006 |
| Prime PCR assay | BioRad | Cat #H384 |
| Cyclohexamide assay | Sigma-Aldrich | Cat #66–81-9 |
| CellTiterGlo | Promega | Cat #G9241 |
|
Deposited data | ||
| Bulk RNAseq and single-cell RNAseq data | GEO | GSE208112 |
|
Experimental models: Cell lines | ||
| Mouse: mCortEGFR: BALB/c | This paper | N/A |
| Mouse: mCortEV: BALB/c | This paper | N/A |
| Mouse: ATT-20 D16:6 | Dr. Steven Sabol, NHLBI | N/A |
|
Oligonucleotides | ||
| EGFR for 5’CCCATGCGGAACTTACAGGAA’3 | Sigma | Cat #VC00021 |
| EGFR rev 5’TTGGATCACATTTGGGGCAAC’3 | Sigma | Cat #VC00021 |
| POMC for 5’ATGCCGAGATTCTGCTACAGT’3 | Sigma | Cat #VC00021 |
| POMC rev 5’CCACACATCTATGGAGGTCTGAA’3 | Sigma | Cat #VC00021 |
| PMAIP1 for 5’GCAGAGCTACCACCTGAGTTC’3 | Sigma | Cat #VC00021 |
| PMAIP1 rev 5’CTTTTGCGACTTCCCAGGCA’3 | Sigma | Cat #VC00021 |
|
Other | ||
| Chromium instrument | 10× genetics | N/A |
| NextSeq sequencing machine | Illumina | N/A |
| TruSeq sequencing machine | Illumina | N/A |
| Infinium MethylationEPIC chip | Illumina | Cat #20042130 |
Highlights.
scRNA-seq indicates transcriptomic signatures of the human adult pituitary gland
CD adenomas overexpress PMAIP1, which encodes the pro-apoptotic protein noxa
Noxa is degraded in CD adenomas to evade apoptosis
Bortezomib proteasomal inhibition stabilizes Noxa and induces apoptosis
ACKNOWLEDGMENTS
This study was supported by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke, Eunice Kennedy Shriver National Institute for Child Health, Human Development, and the National Institute of Diabetes and Digestive Kidney Diseases and National Human Genome Research Institute, Bethesda, MD, USA.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111223.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abbud RA, Takumi I, Barker EM, Ren SG, Chen DY, Wawrowsky K, and Melmed S (2005). Early multipotential pituitary focal hyperplasia in the α-subunit of glycoprotein hormone-driven pituitary tumor-transforming gene transgenic mice. Mol. Endocrinol 19, 1383–1391. 10.1210/me.2004-0403. [DOI] [PubMed] [Google Scholar]
- Alexander JM, Biller BM, Bikkal H, Zervas NT, Arnold A, and Klibanski A (1990). Clinically nonfunctioning pituitary tumors are monoclonal in origin. J. Clin. Invest 86, 336–340. 10.1172/JCI114705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaerts W, Tijssen AM, Jeucken PH, Drexhage HA, and de Koning J (1994). Influence of folliculo-stellate cells on biphasic luteinizing hormone secretion response to gonadotropin-releasing hormone in rat pituitary cell aggregates. Eur. J. Endocrinol 130, 530–539. 10.1530/eje.0.1300530. [DOI] [PubMed] [Google Scholar]
- Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, Gaston-Massuet C, Mollard P, Jacques TS, Le Tissier P, et al. (2013). Sox2+ stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell 13, 433–445. 10.1016/j.stem.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Asuzu D, Chatain GP, Hayes C, Benzo S, McGlotten R, Keil M, Beri A, Sharma ST, Nieman L, Lodish M, et al. (2017). Normalized early postoperative cortisol and ACTH values predict nonremission after surgery for Cushing disease. J. Clin. Endocrinol. Metab 102, 2179–2187. 10.1210/jc.2016-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BL, and Jaffe RB (1975). The genesis of cell types in the adenohypophysis of the human fetus as observed with immunocytochemistry. Am. J. Anat 143, 137–161. 10.1002/aja.1001430202. [DOI] [PubMed] [Google Scholar]
- Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, Mei Y, Schumacher SE, Ben-David U, Chevalier A, et al. (2017). Landscape of genomic alterations in pituitary adenomas. Clin. Cancer Res 23, 1841–1851. 10.1158/1078-0432.CCR-16-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges MHS, Pinto AC, DiNinno FB, Camacho-Hübner C, Grossman A, Kater CE, and Lengyel AM (1999). IGF-I levels rise and GH responses to GHRH decrease during long-term prednisone treatment in man. J. Endocrinol. Invest 22, 12–17. 10.1007/BF03345472. [DOI] [PubMed] [Google Scholar]
- Brinkmann K, Zigrino P, Witt A, Schell M, Ackermann L, Broxtermann P, Schüll S, Andree M, Coutelle O, Yazdanpanah B, et al. (2013). Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep 3, 881–891. 10.1016/j.celrep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, et al. (2018). DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hersmus N, Van Duppen V, Caesens P, Denef C, and Vankelecom H (2005). The adult pituitary contains a cell population displaying stem/progenitor cell and early embryonic characteristics. Endocrinology 146, 3985–3998. 10.1210/en.2005-0185. [DOI] [PubMed] [Google Scholar]
- Chen J, Gremeaux L, Fu Q, Liekens D, Van Laere S, and Vankelecom H (2009). Pituitary progenitor cells tracked down by side population dissection. Stem Cell 27, 1182–1195. 10.1002/stem.51. [DOI] [PubMed] [Google Scholar]
- Chen J, Jian X, Deng S, Ma Z, Shou X, Shen Y, Zhang Q, Song Z, Li Z, Peng H, et al. (2018). Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun 9, 3171–3179. 10.1038/s41467-018-05275-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B, Roose H, Vennekens A, and Vankelecom H (2017). Pituitary stem cell regulation: who is pulling the strings? J. Endocrinol 234, R135–R158. 10.1530/joe-17-0083. [DOI] [PubMed] [Google Scholar]
- Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Horn H, Ott G, van der Kuip H, and Aulitzky WE (2013a). The phenotype high noxa mRNA/low NOXA protein levels constitutes a critical achilles heel of mantle cell lymphoma (MCL) cells. Blood 122, 644. 10.1182/blood.V122.21.644.644. [DOI] [Google Scholar]
- Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Horn H, Ott G, van der Kuip H, and Aulitzky WE (2013b). The phenotype high noxa mRNA/low NOXA protein levels constitutes a critical achilles heel of mantle cell lymphoma (MCL) cells. Blood 122, 644. 10.1182/blood.V122.21.644.644. [DOI] [Google Scholar]
- Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Vöhringer MC, Horn H, Ott G, Aulitzky WE, and van der Kuip H (2014). Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: a potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis 5, e1013–10. 10.1038/cddis.2013.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichek HL, Nieman LK, Oldfield EH, Pass HI, Malley JD, and Cutler GB Jr. (1994). A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab 78, 418–422. [DOI] [PubMed] [Google Scholar]
- Dorman K, Shen Z, Yang C, Ezzat S, and Asa SL (2012). CtBP1 interacts with ikaros and modulates pituitary tumor cell survival and response to hypoxia. Mol. Endocrinol 26, 447–457. 10.1210/me.2011-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, and McCutcheon IE (2004). The prevalence of pituitary adenomas: a systematic review. Cancer 101, 613–619. 10.1002/cncr.20412. [DOI] [PubMed] [Google Scholar]
- Fabian P, Tseng KC, Smeeton J, Lancman JJ, Dong PDS, Cerny R, and Crump JG (2020). Lineage analysis reveals an endodermal contribution to the vertebrate pituitary. Science 370, 463–467. 10.1126/science.aba4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippella M, Galland F, Kujas M, Young J, Faggiano A, Lombardi G, Colao A, Meduri G, and Chanson P (2006). Pituitary tumour transforming gene (PTTG) expression correlates with the proliferative activity and recurrence status of pituitary adenomas: a clinical and immunohistochemical study. Clin. Endocrinol 65, 536–543. 10.1111/j.1365-2265.2006.02630.x. [DOI] [PubMed] [Google Scholar]
- Fletcher PA, Smiljanic K, Maso Prévide R, Iben JR, Li T, Rokic MB, Sherman A, Coon SL, and Stojilkovic SS (2019). Cell type- and sex-dependent transcriptome profiles of rat anterior pituitary cells. Front. Endocrinol 10, 623–625. 10.3389/fendo.2019.00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, and Melmed S (2011a). EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Invest 121, 4712–4721. 10.1172/JCI60417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, and Melmed S (2011b). EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Invest 121, 4712–4721. 10.1172/JCI60417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth J, Gadsen EL, and Upton AC (1953). Acth secreting transplantable pituitary tumors. PSEBM (Proc. Soc. Exp. Biol. Med.) 84, 253–254, Society for Experimental Biology. 10.3181/00379727-84-20607. [DOI] [PubMed] [Google Scholar]
- Gejman R, Batista DL, Zhong Y, Zhou Y, Zhang X, Swearingen B, Stratakis CA, Hedley-Whyte ET, and Klibanski A (2008). Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab 93, 4119–4125. 10.1210/jc.2007-2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Akima S., George AS, McGee SR, Daly AZ, Brinkmeier ML, Ellsworth BS, and Camper SA (2018). Single-cell RNA sequencing reveals novel markers of male pituitary stem cells and hormone-producing cell-types. Endocrinology 159, 3910–3924. 10.1210/en.2018-00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleiberman AS, Michurina T, Encinas JM, Roig JL, Krasnov P, Balordi F, Fishell G, Rosenfeld MG, and Enikolopov G (2008). Genetic approaches identify adult pituitary stem cells. Proc. Natl. Acad. Sci. USA 105, 6332–6337. 10.1073/pnas.0801644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guikema JE, Amiot M, and Eldering E (2017a). Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer. Expert Opin. Ther. Targets 21, 767–779. 10.1080/14728222.2017.1349754. [DOI] [PubMed] [Google Scholar]
- Guikema JE, Amiot M, and Eldering E (2017b). Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer. Expert Opin. Ther. Targets 21, 767–779. 10.1080/14728222.2017.1349754. [DOI] [PubMed] [Google Scholar]
- Günes M, Celik O, and Kadioglu P (2013). Reliability of the diagnostic tests for Cushing’s syndrome performed in a tertiary referral center. Pituitary 16, 139–145. [DOI] [PubMed] [Google Scholar]
- Herman V, Fagin J, Gonsky R, Kovacs K, and Melmed S (1990). Clonal origin of pituitary adenomas. J. Clin. Endocrinol. Metab 71, 1427–1433. 10.1210/jcem-71-6-1427. [DOI] [PubMed] [Google Scholar]
- Ho H, Hu P, Peel MT, Chen S, Camara PG, Epstein DJ, Wu H, and Liebhaber SA (2018). Single cell transcriptomic analysis of the adult mouse pituitary reveals a novel multi-hormone cell cluster and physiologic demand-induced lineage plasticity. Preprint at bioRxiv Genomics 10.1101/475558. [DOI] [PMC free article] [PubMed]
- Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, Robinson-White AJ, Nesterova M, Lacroix A, and Stratakis CA (2009). Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J. Clin. Endocrinol. Metab 94, 2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, and Yoshimoto T (2009). Visual disturbances in patients with pituitary adenoma. Acta Neurol. Scand 92, 157–160. 10.1111/j.1600-0404.1995.tb01031.x. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Gui S, and Zhang Y (2011). Analysis of differential gene expression by bead-based fiber-optic array in nonfunctioning pituitary adenomas. Hormone and metabolic research = Hormonund Stoffwechselforschung = Hormones et metabolisme 43, 325–330. 10.1055/s-0031-1271748. [DOI] [PubMed] [Google Scholar]
- Kovacs K, Horvath E, Stefaneanu L, Bilbao J, Singer W, Muller PJ, Thapar K, and Stone E (1998). Pituitary adenoma producing growth hormone and adrenocorticotropin: a histological, immunocytochemical, electron microscopic, and in situ hybridization study. J. Neurosurg 88, 1111–1115. 10.3171/jns.1998.88.6.1111. [DOI] [PubMed] [Google Scholar]
- Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, and Drouin J (2001). A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell 104, 849–859. 10.1016/S0092-8674(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Leal-Cerro A, Pumar A, Garcia-Garcia E, Dieguez C, and Casanueva FF (1994). Inhibition of growth hormone release after the combined administration of GHRH and GHRP-6 in patients with Cushing’s syndrome. Clin. Endocrinol 41, 649–654. 10.1111/j.1365-2265.1994.tb01831.x. [DOI] [PubMed] [Google Scholar]
- Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ, and Gasch AP (2011). A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol 7, 514. 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore DA, Roeszler K, Wagner J, Ross SA, Bauer K, and Thomas PQ (2005). Identification and enrichment of colony-forming cells from the adult murine pituitary. Exp. Cell Res 308, 166–176. 10.1016/j.yexcr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, and Levine AJ (2018). Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 22, 2216–2225. 10.1016/j.celrep.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu X, Xia Z, Yu X, Xie L, Ma X, Zhou H, Liu L, Wang J, Yang Y, and Liu H (2018). Repression of Noxa by Bmi1 contributes to deguelin-induced apoptosis in non-small cell lung cancer cells. J. Cell Mol. Med 22, 6213–6227. 10.1111/jcmm.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lin C, Gleiberman A, Ohgi KA, Herman T, Huang HP, Tsai MJ, and Rosenfeld MG (2001). Tbx19, a tissue-selective regulator of POMC gene expression. Proc. Natl. Acad. Sci. USA 98, 8674–8679. 10.1073/pnas.141234898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550. 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Lohrer P, Gloddek J, Nagashima AC, Korali Z, Hopfner U, Pereda MP, Arzt E, Stalla GK, and Renner U (2000). Lipopolysaccharide directly stimulates the intrapituitary interleukin-6 production by folliculostellate cells via specific receptors and the p38α mitogen-activated protein kinase/nuclear factor-κb Pathway**Supported by a grant from the DFG: sta 285/7–3. Endocrinology 141, 4457–4465. 10.1210/endo.141.12.7811. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Nieman L, and Oldfield EH (2017). Cushing’s disease: Pathobiology, diagnosis, and management. J. Neurosurg 126, 404–417. [DOI] [PubMed] [Google Scholar]
- Lu J, Chatain GP, Bugarini A, Wang X, Maric D, Walbridge S, Zhuang Z, and Chittiboina P (2017). Histone deacetylase inhibitor SAHA is a promising treatment of Cushing disease. J. Clin. Endocrinol. Metab 102, 2825–2835. 10.1210/jc.2017-00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Montgomery BK, Chatain GP, Bugarini A, Zhang Q, Wang X, Edwards NA, Ray-Chaudhury A, Merrill MJ, Lonser RR, and Chittiboina P (2018). Corticotropin releasing hormone can selectively stimulate glucose uptake in corticotropinoma via glucose transporter 1. Mol. Cell. Endocrinol 470, 105–114. 10.1016/j.mce.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z-Y, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, Mao Y, Li YM, Hu RG, Zhang ZY, et al. (2015). Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res 25, 306–317. 10.1038/cr.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiakou MA, Mastorakos G, Gomez MT, Rose SR, and Chrousos GP (1994). Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing’s disease before and after surgical cure. J. Clin. Endocrinol. Metab 78, 131–137. 10.1210/jcem.78.1.7507118. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: uniform Manifold approximation and projection for dimension reduction http://arxiv.org/abs/1802.03426.
- McNicol AM (1981). Patterns of corticotropic cells in the adult human pituitary in Cushing’s disease. Diagn. Histopathol 4, 335–341. [PubMed] [Google Scholar]
- Michaelis KA, Knox AJ, Xu M, Kiseljak-Vassiliades K, Edwards MG, Geraci M, Kleinschmidt-DeMasters BK, Lillehei KO, and Wierman ME (2011). Identification of growth arrest and DNA-damage-inducible gene β (GADD45β) as a novel tumor suppressor in pituitary gonadotrope tumors. Endocrinology 152, 3603–3613. 10.1210/en.2011-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrofanova LB, Konovalov PV, Krylova JS, Polyakova VO, and Kvetnoy IM (2017). Plurihormonal cells of normal anterior pituitary: facts and conclusions. Oncotarget 8, 29282–29299. 10.18632/oncotarget.16502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Yamaguchi H, and Takahashi K (1980). S100 protein in folliculostellate cells of the rat pituitary anterior lobe. Brain Res 191, 523–531. 10.1016/0006-8993(80)91300-1. [DOI] [PubMed] [Google Scholar]
- Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, Letourneur F, Diry S, Diedisheim M, Izac B, et al. (2020). Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 37, 123–134.e5. 10.1016/j.ccell.2019.11.002. [DOI] [PubMed] [Google Scholar]
- Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, and Montori VM (2008). The diagnosis of Cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab 93, 1526–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, and Cutler GB Jr. (1993). A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab 77, 1308–1312. [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Riblett M, Tang WH, Gratchouck V, Zhuang D, Fernandez Y, Verhaegen M, Varambally S, Chinnaiyan AM, Jakubowiak AJ, and Soengas MS (2007). Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA 104, 19488–19493. 10.1073/pnas.0708380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, and Tanaka N (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058. 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Oldfield EH, Vance ML, Louis RG, Pledger CL, Jane JA Jr., and Lopes MBS (2015). Crooke’s changes in cushing’s syndrome depends on degree of hypercortisolism and individual susceptibility. J. Clin. Endocrinol. Metab 100, 3165–3171. 10.1210/JC.2015-2493. [DOI] [PubMed] [Google Scholar]
- Pang X, Zhang J, Lopez H, Wang Y, Li W, O’Neill KL, Evans JJD, George NM, Long J, Chen Y, and Luo X (2014). The carboxyl-terminal tail of Noxa protein regulates the stability of Noxa and Mcl-1. J. Biol. Chem 289, 17802–17811. 10.1074/jbc.M114.548172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou DA, Yanovski JA, Cutler GB, Chrousos GP, and Nieman LK (2016). A single midnight serum cortisol measurement distinguishes Cushing’s syndrome from pseudo-Cushing. J. Clin. Endocrinol. Metab 83, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Pasolli HA, Torres AI, and Aoki A (1994). The mammosomatotroph: a transitional cell between growth hormone and prolactin producing cells? An immunocytochemical study. Histochemistry 102, 287–296. 10.1007/BF00269165. [DOI] [PubMed] [Google Scholar]
- Pei L, and Melmed S (1997). Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol. Endocrinol 11, 433–441. 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- Plotz CM, Knowlton AI, and Ragan C (1952). The natural history of cushing’s syndrome. Am. J. Med 13, 597–614. 10.1016/0002-9343(52)90027-2. [DOI] [PubMed] [Google Scholar]
- Ragnarsson O, Olsson DS, Papakokkinou E, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, Berinder K, Bensing S, et al. (2019). Overall and disease-specific mortality in patients with cushing disease: a Swedish nationwide study. J. Clin. Endocrinol. Metab 104, 2375–2384. 10.1210/jc.2018-02524. [DOI] [PubMed] [Google Scholar]
- Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, et al. (2015). Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet 47, 31–38. 10.1038/ng.3166. [DOI] [PubMed] [Google Scholar]
- Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, et al. (2015). Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet 47, 31–38. 10.1038/ng.3166. [DOI] [PubMed] [Google Scholar]
- Rinehart JF, and Farquhar MG (1953). Electron microscopic studies of the anterior pituitary gland. J. Histochem. Cytochem 1, 93–113. 10.1177/1.2.93. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf-Zamojski F, Ge Y, Nair V, Zamojski M, Pincas H, Toufaily C, Tome-Carcia J, Stoeckius M, Stephenson W, Smith GR, Bernard DJ, et al. (2018). Single-cell Stabilization Method Identifies Gonadotrope Transcriptional Dynamics and Pituitary Cell Type Heterogeneity. Nucleic Acids Res, 1–11. 10.1093/nar/gky991. [DOI] [PMC free article] [PubMed]
- Russell JP, Lim X, Santambrogio A, Yianni V, Kemkem Y, Wang B, Fish M, Haston S, Grabek A, Hallang S, et al. (2021). Pituitary stem cells produce paracrine WNT signals to control the expansion of their descendant progenitor cells. Elife 10, e59142. 10.7554/eLife.59142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, Matsuba C, Takasumi Y, Ballesteros-Merino C, Fox BA, et al. (2018). The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, Cushing’s disease and endocrine-inactive subtypes. Clin. Cancer Res 24, 4126–4136. 10.1158/1078-0432.CCR-17-2206. [DOI] [PubMed] [Google Scholar]
- Schulte HM, Oldfield EH, Allolio B, Katz DA, Berkman RA, and Ali IU (1991). Clonal composition of pituitary adenomas in patients with cushing’s disease: determination by X-chromosome inactivation analysis. J. Clin. Endocrinol. Metab 73, 1302–1308. 10.1210/jcem-73-6-1302. [DOI] [PubMed] [Google Scholar]
- Soji T, Mabuchi Y, Kurono C, and Herbert DC (1997). Folliculo-stellate cells and intercellular communication within the rat anterior pituitary gland. Microsc. Res. Tech 39, 138–149. . [DOI] [PubMed] [Google Scholar]
- Takahashi H, Bando H, Zhang C, Yamasaki R, and Saito S (1992). Mechanism of impaired growth hormone secretion in patients with Cushing’s disease. Acta Endocrinol 127, 13–17. 10.1530/acta.0.1270013. [DOI] [PubMed] [Google Scholar]
- Teshima K, Nara M, Watanabe A, Ito M, Ikeda S, Hatano Y, Oshima K, Seto M, Sawada K, and Tagawa H (2014). Dysregulation of BMI1 and microRNA-16 collaborate to enhance an anti-apoptotic potential in the side population of refractory mantle cell lymphoma. Oncogene 33, 2191–2203. 10.1038/onc.2013.177. [DOI] [PubMed] [Google Scholar]
- Tfelt-Hansen J, Kanuparthi D, and Chattopadhyay N (2006a). The emerging role of pituitary tumor transforming gene in tumorigenesis. Clin. Med. Res 4, 130–137. 10.3121/cmr.4.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tfelt-Hansen J, Kanuparthi D, and Chattopadhyay N (2006b). The emerging role of pituitary tumor transforming gene in tumorigenesis. Clin. Med. Res 4, 130–137. 10.3121/cmr.4.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoropoulou M, Arzberger T, Gruebler Y, Jaffrain-Rea ML, Schlegel J, Schaaf L, Petrangeli E, Losa M, Stalla GK, and Pagotto U (2004). Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J. Endocrinol 183, 385–394. 10.1677/joe.1.05616. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjörnstrand A, Gunnarsson K, Evert M, Holmberg E, Ragnarsson O, Rosén T, and Filipsson Nyström H (2014). The incidence rate of pituitary adenomas in western Sweden for the period 2001–2011. Eur. J. Endocrinol 171, 519–526. 10.1530/EJE-14-0144. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Luis Syro EH, Horvath E, Uribe H, and Kovacs K (1999). Ultra-structural and immunoelectron microscopic study of three unusual plurihormonal pituitary adenomas. Ultrastruct. Pathol 23, 141–148. 10.1080/019131299281635. [DOI] [PubMed] [Google Scholar]
- Wang DG, Johnston CF, Atkinson AB, Heaney AP, Mirakhur M, and Buchanan KD (1996). Expression of bcl-2 oncoprotein in pituitary tumours: comparison with c-myc. J. Clin. Pathol 49, 795–797. 10.1136/jcp.49.10.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Stojanovic N, Christian J, Paul MC, Stauber RH, Schmid RM, Häcker G, Krämer OH, Saur D, and Schneider G (2014). MYC and EGR1 synergize to trigger tumor cell death by controlling NOXA and BIM transcription upon treatment with the proteasome inhibitor bortezomib. Nucleic Acids Res 42, 10433–10447. 10.1093/nar/gku763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloschak M, Roberts JL, and Post K (1994). c-myc, c-fos, and c-myb gene expression in human pituitary adenomas. J. Clin. Endocrinol. Metab 79, 253–257. 10.1210/jcem.79.1.8027238. [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, and Orkin SH (2014). Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80. 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, Hasegawa A, Motohashi S, Iwama A, and Nakayama T (2008). Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J. Exp. Med 205, 1109–1120. 10.1084/jem.20072000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordanova G, Martin L, Afshar F, Sabin I, Alusi G, Plowman NP, Riddoch F, Evanson J, Matson M, Grossman AB, et al. (2016). Long-term outcomes of children treated for Cushing’s disease: a single center experience. Pituitary 19, 612–624. 10.1007/s11102-016-0756-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi HA, Cote DJ, Castlen JP, Burke WT, Liu YH, Smith TR, and Laws ER Jr. (2017). Time course of resolution of hyperprolactinemia after transsphenoidal surgery among patients presenting with pituitary stalk compression. World Neurosurg 97, 2–7. 10.1016/j.wneu.2016.09.066. [DOI] [PubMed] [Google Scholar]
- Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, and Melmed S (1999). Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J. Clin. Endocrinol. Metab 84, 761–767. 10.1210/jcem.84.2.5432. [DOI] [PubMed] [Google Scholar]
- Zhang S, Cui Y, Ma X, Yong J, Yan L, Yang M, Ren J, Tang F, Wen L, and Qiao J (2020). Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat. Commun 11, 5275. 10.1038/s41467-020-19012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq and whole RNA-seq data has been uploaded to GEO and will be publicly available by the date of publication. Whole microscopy data reported in this paper will be shared by the lead author upon request.
No original code was generated in this study
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request