

Abstract
BACKGROUND:
Retrospective analyses of randomized trials suggest that Black men with metastatic castration-resistant prostate cancer (mCRPC) have longer survival than White men. The authors conducted a prospective study of abiraterone acetate plus prednisone to explore outcomes by race.
METHODS:
This race-stratified, multicenter study estimated radiographic progression-free survival (rPFS) in Black and White men with mCRPC. Secondary end points included prostate-specific antigen (PSA) kinetics, overall survival (OS), and safety. Exploratory analysis included genome-wide genotyping to identify single nucleotide polymorphisms associated with progression in a model incorporating genetic ancestry. One hundred patients self-identified as White (n = 50) or Black (n = 50) were enrolled. Eligibility criteria were modified to facilitate the enrollment of individual Black patients.
RESULTS:
The median rPFS for Black and White patients was 16.6 and 16.8 months, respectively; their times to PSA progression (TTP) were 16.6 and 11.5 months, respectively; and their OS was 35.9 and 35.7 months, respectively. Estimated rates of PSA decline by ≥50% in Black and White patients were 74% and 66%, respectively; and PSA declines to <0.2 ng/mL were 26% and 10%, respectively. Rates of grade 3 and 4 hypertension, hypokalemia, and hyperglycemia were higher in Black men.
CONCLUSIONS:
Multicenter prospective studies by race are feasible in men with mCRPC but require less restrictive eligibility. Despite higher comorbidity rates, Black patients demonstrated rPFS and OS similar to those of White patients and trended toward greater TTP and PSA declines, consistent with retrospective reports. Importantly, Black men may have higher side-effect rates than White men. This exploratory genome-wide analysis of TTP identified a possible candidate marker of ancestry-dependent treatment outcomes.
Keywords: abiraterone acetate, African American, castration resistant, hormone therapy, metastatic prostate cancer, prednisone, prostate-specific antigen (PSA), race
INTRODUCTION
Compared with other common cancers in the United States, prostate cancer has 1 of the most dramatic racial disparities in incidence and outcomes.1 Evidence suggests that Black men develop prostate cancer at an earlier age, are more likely to present with advanced-stage disease, have a greater risk of early biochemical recurrence and metastasis, and have a shorter cancer-specific survival.2 Access to medical care clearly plays a key role in prostate cancer disparities.3 It is also likely that ancestry-related biologic differences contribute to prostate cancer disparities. Regardless of the causes, Black men account for roughly 30% of all prostate cancer deaths in the United States yet routinely represent ≤5% of participants in registration trials in castrate-resistant prostate cancer (CRPC), if reported at all.4–8 To improve the survival of Black Americans with prostate cancer, we have to improve access to care and, in particular, inclusion in prospective interventional trials.9
Despite more aggressive disease at presentation and progression, there are growing retrospective data to suggest that Black men with metastatic CRPC (mCRPC) may respond better than White men when both are enrolled in clinical trials. We and others first reported this in 2007, when a retrospective meta-analysis of baseline characteristics of patients with mCRPC participating in cooperative group studies revealed a modest survival advantage associated with men who self-identified as Black.10,11 We and others have subsequently confirmed this association through a much larger meta-analysis of 9 phase 3 trials of docetaxel-based regimens in men with mCRPC, demonstrating in multivariable analysis a 19% reduction in the risk of death associated with docetaxel treatment in self-described Black men.12 In addition, through a prospective observational cohort study of sipuleucel-T, a US Food and Drug Administration-approved autologous cellular immunotherapy approved for the treatment of men with mCRPC, we and others have demonstrated a clinically significant 40% improvement in survival for Black men compared with White men after adjustment for known prognostic factors.13,14 Recently, a meta-analysis of randomized controlled trials by the National Radiation Oncology Group revealed that Black men had a significantly lower hazard ratio for overall survival (OS) compared with White men on study.15 Those authors concluded that Black race did not appear to be associated with inferior stage-for-stage prostate cancer-specific mortality. A large disparity remained in all-cause mortality for Black men with nonmetastatic prostate cancer.
We hypothesized that Black men may also be more responsive to novel hormonal therapy in prostate cancer. Abiraterone acetate and prednisone (AAP) is a US Food and Drug Administration-approved treatment for men with metastatic castration-sensitive prostate cancer (mCSPC) based on the STAMPEDE trial (ClinicalTrials. gov identifier NCT00268476) and the LATITUDE study (ClinicalTrials.gov identifier NCT01715285) and for men with mCRPC based on the Cougar 302 (COU-302) study (ClinicalTrials.gov identifier NCT00887198), an international phase 3 trial of AAP versus prednisone plus placebo in men with chemotherapy-naive mCRPC that demonstrated improvements in both radiographic progression-free survival (rPFS) and OS.4 Key secondary end points, including the prostate-specific antigen (PSA) response rate and the time to PSA progression (TTP), also strongly favored patients who received AAP.4 Unfortunately, similar to other international phase 3 trials, Black men comprised of only 3.6% of the study population. Interestingly, in an exploratory retrospective analysis, we and others demonstrated that the estimated rate of a PSA decline ≥90% was higher in Black men compared with the overall study population (53.3% vs 30.8%), as was the estimated median TTP (16.6 vs 11.1 months), but there was no difference in the median rPFS (16.5 months for both).16 In an effort to further explore this trend, we performed a single-center retrospective analysis of men with mCRPC who received treatment with AAP, were selected 2:1 by race, and were controlled for prior chemotherapy.17 That study revealed higher rates of PSA decline in Black men versus White men.17 More recently, using the Veterans Health Administration Database, we demonstrated a statistically significant improvement in OS for Black men compared with White men who had mCRPC treated with AAP or enzalutamide, with the analyses adjusted for prognostic and comorbid factors (hazard ratio [HR], 0.826; 95% CI, 0.732–0.933; P = .0020).18
We performed a prospective, multicenter study of AAP treatment in 100 men who had mCRPC (50 Black men and 50 White men, based on self-identified race) to demonstrate that it is feasible to enroll an equal percentage of Black men to therapeutic trials, to report on the respective outcomes by race, and to collect on-study blood and tissue specimens for correlative science. Based on our retrospective analyses, a direct comparison of outcomes by race would require a phase 3 trial of significantly greater size. Instead, we performed a smaller multicenter pilot study to explore the clinical outcomes associated with each group in parallel to prospectively evaluate the trends observed retrospectively and to perform correlative analyses. To our knowledge, no prospective studies with prespecified race-based cohorts have been successfully completed to date in men with mCRPC, although they have been attempted.19 Here, we report here our final clinical results and the preliminary identification of a candidate single-nucleotide polymorphism (SNP) associated with outcomes of patients who received AAP in a model that included ancestry. This study is registered under Clinicaltrials.gov (identification number NCT01940276).
MATERIALS AND METHODS
Study Design
This trial was a noncomparative, open-label, multicenter pilot study of AAP in Black men and White men with mCRPC. The primary objective was to prospectively estimate rPFS in Black men and White men with mCRPC who received AAP. Patients self-identified their race and were treated until they developed evidence of clinical or radiographic disease progression, according to the Prostate Cancer Working Group 2 (PCWG2) definition,20 or until 2 years after treatment, at which point they were rolled over to the standard of care (see Supporting Fig. 1).
Study End Points
The primary end point was rPFS based on PCWG2 criteria.20 Secondary, PSA-based end points included estimation of the percentage of men who achieved PSA declines of 30%, 50%, or 90% from baseline, the time to PSA nadir, TTP, and the percentage of men who achieved PSA levels <0.1 and <0.2 ng/mL. Other secondary end points included incidence of bone flares (as previously described),16,21 safety, tolerability, and OS. In particular, we captured possible side effects associated with AAP treatment, including, but not limited to, fatigue, hypertension, peripheral edema, constipation, nausea, vomiting, anorexia, headache, hot flashes, and falls, as well as common laboratory abnormalities, including hyperglycemia, hypokalemia, hypomagnesemia, anemia, and increases in liver enzymes (according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0). An exploratory genome-wide analysis was conducted to assess the role of heritable genetic variation with respect to clinical outcomes in the study population.
Study Sites and Patients
The study was sponsored by Duke University Health System and was deemed Investigational New Drug-exempt. The work was funded by Janssen Scientific Affairs, LLC, through an investigator-initiated contract with Duke and was approved by the Duke Institutional Review Board. This was a multicenter study that was monitored by the Duke Cancer Institute clinical research unit monitoring core.
Key eligibility included: 1) evidence of castration-resistant disease, as described in the online Supporting Methods22–29; 2) metastatic disease on conventional technetium bone scan and computed tomography imaging, 3) testosterone ≤50 ng/dL, 4) minimal hematologic, hepatic, and chemistry laboratory values (see Supporting Methods). All patients provided written informed consent. Importantly, we recognized that our Black patients may have had higher risks of diabetes mellitus and/or hypertension under suboptimal control. Therefore, we amended our eligibility from the original COU-302 phase 3 trial to include all patients who had preexisting hypertension except, for those who had uncontrolled baseline blood pressure ≥160/95. We also included all patients who had diabetes mellitus except for those poorly controlled. Other amendments made for individual Black patients who otherwise met eligibility criteria included changing tumor assessments from those with measurable disease to those with evaluable only disease, extending the window for baseline scans out to 6 weeks, and allowing for only a 2-week washout for those who received prior antiandrogen therapy. All of these changes were made in response to individual Black patients who otherwise would have been excluded from our study. In addition, we increased our number of participating centers from 3 to 10, selecting additional sites with high Black patient representation.
Genotyping
Genomic DNA from 88 consented patients, 43 white and 45 Black, was genotyped on the Illumina Infinimum Multi-Ethnic Global BeadChip. The Consolidated Standards of Reporting Trials (CONSORT) chart in Supporting Figure 2 provides additional details about the filtering process. The experimental design also included 2 patient DNA replicates and 12 controls from the International HapMap Project database. The assay was performed by the Duke Molecular Physiology Institute Molecular Genomics Shared Resource. Additional technical details are provided in the online Supporting Methods. The genotyping data and associated clinical data used in this analysis will be made available through the database of Genotypes and Phenotypes (dbGaP).
Trial Design and Data Analysis
The primary objective of this study was to estimate rPFS distribution in Black and White patients, as determined by progression based on PCWG2 criteria, symptomatic deterioration, onset of a skeletal-related event, or death. Based on the report by Ryan et al, the median time to rPFS was assumed to be 16.5 months in each group.4 With an accrual rate of 50 patients per group over a 12-month accrual period, a 24-month follow-up period, and assuming that rPFS follows an exponential distribution, based on 5000 simulations, the average width of a 2-sided 95% confidence interval (95% CI) for the median rPFS was 16 months. The expected number of rPFS events was 26 within each group.
The Kaplan-Meier product-limit approach was used to estimate the progression-free survival and OS distributions for patients within each racial group. The proportions of patients who achieved PSA declines from baseline of ≥30%, ≥50%, and ≥90% were estimated, and the exact binomial distribution was used to compute 95% CIs. Common Terminology Criteria for Adverse Events, version 4.0, was used to assess adverse events for safety end points.
Genetic analyses were conducted using a Cox model to identify SNPs for which either the genotype or a local ancestry effect was associated with TTP, using global ancestry as a covariate. The joint hypothesis of no genetic effect (ie, that neither genotype nor local ancestry is associated with TTP) was tested for each SNP using an asymptotic P value. Because these analyses were considered exploratory, they were adjusted neither to account for the variation for substituting estimates of genotypes and local and global ancestry for the actual quantities nor for multiple testing. An additional Cox analysis of rPFS for selected SNPs from the TTP model was also completed. Further technical details are provided online (see Supporting Methods).
RESULTS
Baseline Patient Characteristics
Our study enrolled patients at 10 sites between December 2013 and October 2016. Accrual of Black and White men was relatively balanced across sites (see Supporting Fig. 3). A CONSORT diagram demonstrating patient dropout is provided in Figure 1. Baseline demographics are listed in Table 1 and reveal similar characteristics between the 2 groups, including age, Gleason grade and stage at diagnosis, and patterns of metastases. Differences in performance status, median time from diagnosis to enrollment, median PSA level at enrollment, and prior treatments were not statistically significant.
Figure 1.
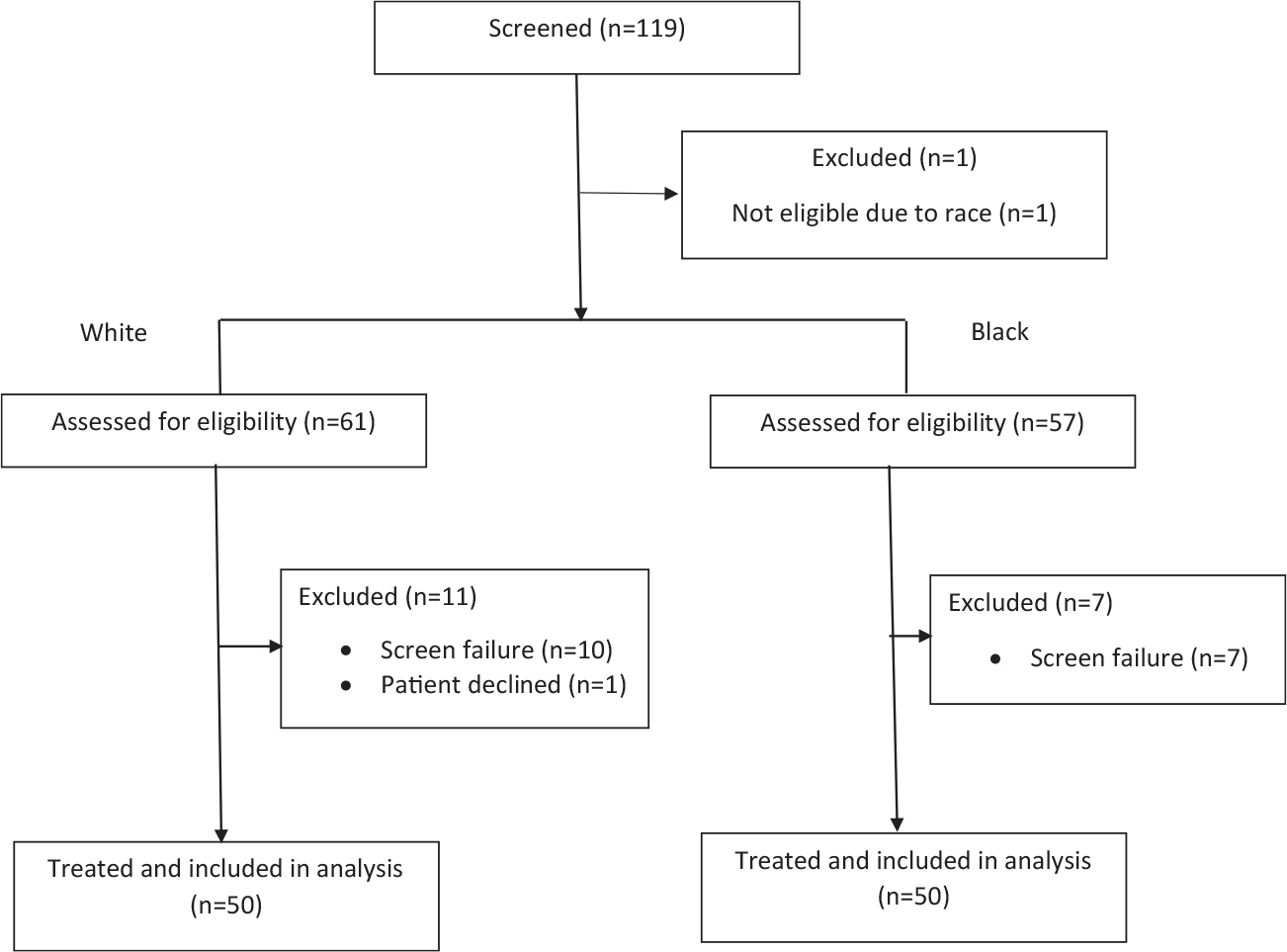
This clinical Consolidated Standards for Reporting Trials (CONSORT) diagram illustrates screen failures according to race and final intention-to-treat patient populations.
TABLE 1.
Baseline Characteristics at Enrollment (Unless Otherwise Stated)
| Percentage of Men |
|||
|---|---|---|---|
| Characteristic | Black Men, N = 50 | White Men, N = 50 | All Men, N = 100 |
|
| |||
| Age: Median (range), y | 69.05 (52.4–100.2) | 67.8 (49.4–87.2) | 68.5 (49.4–100.2) |
| Time from diagnosis to enrollment: Median (range), y | 3.75 (0.67–19.34) | 4.12 (0.62–23.8) | 3.99 (0.62–23.8) |
| Gleason score at diagnosis | |||
| Unknown | 6 | 4 | 5 |
| ≤7 | 38 | 40 | 39 |
| 8–10 | 56 | 56 | 56 |
| Tumor classification at diagnosis | |||
| TX | 20 | 20 | 20 |
| ≤T2 | 48 | 38 | 43 |
| T3–T4 | 32 | 42 | 37 |
| Lymph node classification | |||
| NX | 30 | 22 | 26 |
| N0 | 44 | 52 | 48 |
| N1–N3 | 26 | 26 | 26 |
| Extent of metastasis | |||
| MX | 12 | 14 | 13 |
| M0 | 52 | 54 | 53 |
| M1, M1a, M1b | 36 | 32 | 34 |
| KPS | |||
| 80–70 | 20 | 12 | 16 |
| 100–90 | 80 | 88 | 84 |
| Sites of metastases | |||
| Bone only | 44 | 38 | 41 |
| LN only | 16 | 20 | 18 |
| Bone and LN | 26 | 28 | 27 |
| Visceral | 14 | 14 | 14 |
| Median PSA (range), ng/mL | 17.73 (2.44–4194.86) | 22.35 (2.13–832.55) | 21.55 (2.13–4194.86) |
| Prior primary radiation | 48 | 58 | 53 |
| Prior prostatectomy | 34 | 46 | 40 |
| Prior bicalutamide | 66 | 72 | 69 |
| Prior sipuleucel-T | 24 | 40 | 32 |
| Prior docetaxel | 16 | 22 | 19 |
Abbreviations: KPS, Karnofsky performance status; LN, lymph node; PSA, prostate-specific antigen; y, years.
Similarly, the incidence of baseline comorbidities and symptoms varied by cohort (Table 2). Overall, Black men had modestly higher rates of hypertension (88% vs 68%), hypercholesterolemia (42% vs 30%), and obesity, defined as a body mass index ≥30 km/m2 (50% vs 44%); however, a prior history of diabetes stood out as disproportionately present in Black men compared with White men (42% vs 10%).
TABLE 2.
Common Baseline Comorbidities
| Percentage of Men |
|||
|---|---|---|---|
| Comorbidity | Black Men, N = 50 | White Men, N = 50 | All Men, N = 100 |
|
| |||
| Prior hypertension | 88 | 68 | 78 |
| Prior hypercholesterolemia | 42 | 30 | 36 |
| Prior obesity | 50 | 44 | 47 |
| Prior diabetes | 42 | 10 | 26 |
Clinical Efficacy
The median time to follow-up was 24.8 months (95% CI, 24.3–27.0 months). The primary end point for the study, rPFS, is presented in Figure 2. There were 35 rPFS events the Black cohort and 35 rPFS events in the White cohort. The estimated median rPFS distribution was 16.6 months (95% CI, 12.6–22.1 months) for the Black cohort and 16.8 months (95% CI, 11.0–33.7 months) for the White cohort.
Figure 2.
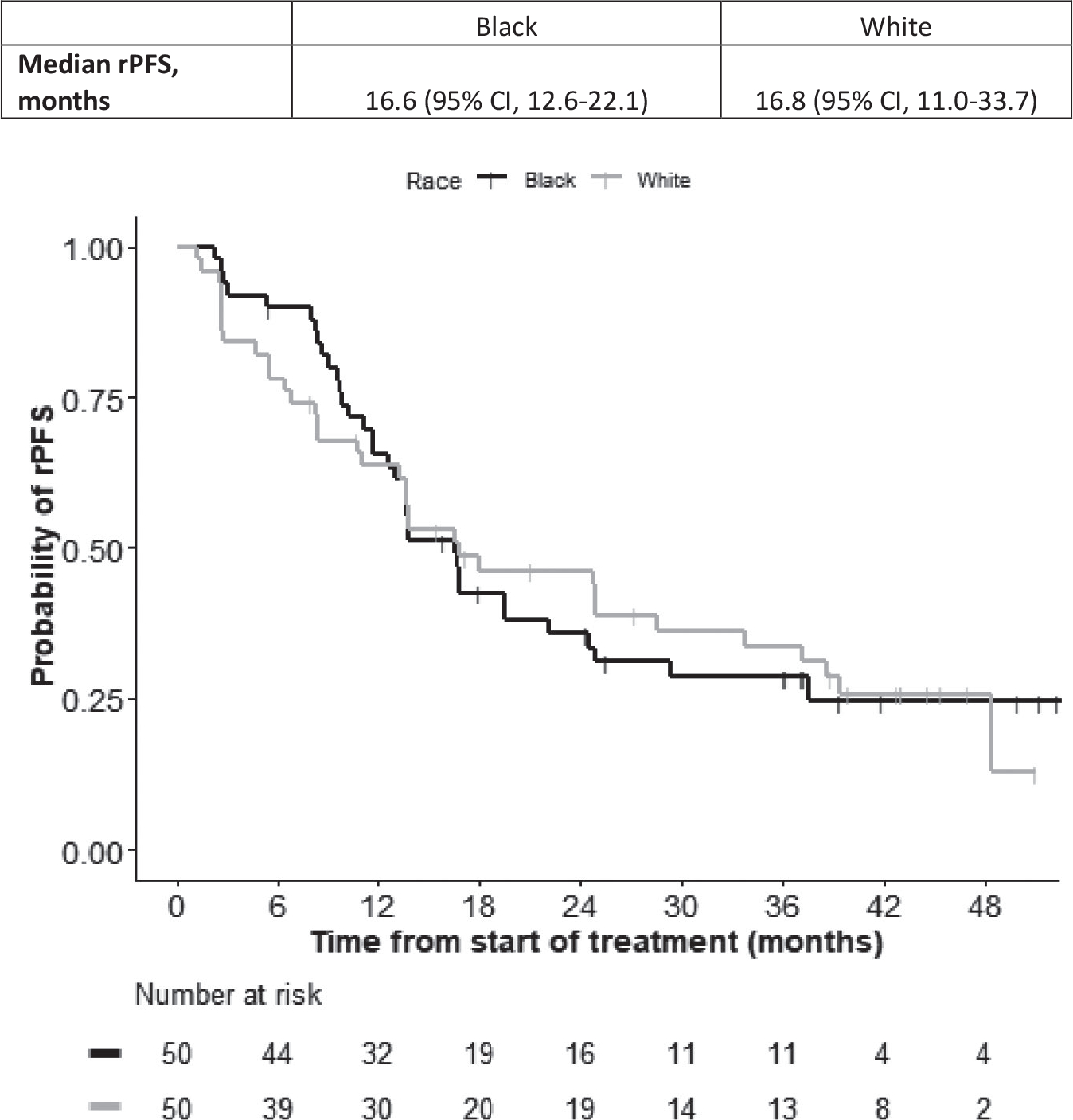
Kaplan-Meier curves illustrate radiographic progression-free survival (rPFS) in Black patients (black curve) and White patients (gray curve). Median rPFS estimates are shown with 95% CIs. This graph is for illustrative purposes only; the study was not powered for cohort comparisons. Any comparison is for hypothesis generation only.
Differences in PSA kinetics were observed across various time points and thresholds by race. Black men had a median TTP of 16.6 months (95% CI, 11.6 months to not reached), whereas White men had a median TTP of 11.5 months (95% CI, 8.5–19.3 months) (Fig. 3), similar to prior retrospective reports. Among Black men, the estimated rates of PSA decline from baseline by ≥30%, ≥50%, and ≥90% were 82%, 74%, and 48%, respectively, and, among White men, the rates were 78%, 66%, and 38%, respectively (Table 3). Black men also had a lower rate of no PSA decline as the best response (4%; 95% CI, 0.004–0.14) compared with White men (10%; 95% CI, 0.03–0.21). The best PSA decline to <0.1 ng/mL was observed in 18% of Black men, and the best PSA decline to <0.2 ng/mL was observed in 26% of Black men (Table 3). Among White men, the best PSA decline to <0.1 ng/mL was observed in 8%, and the PSA decline to <0.2 ng/mL was observed in 10%.
Figure 3.
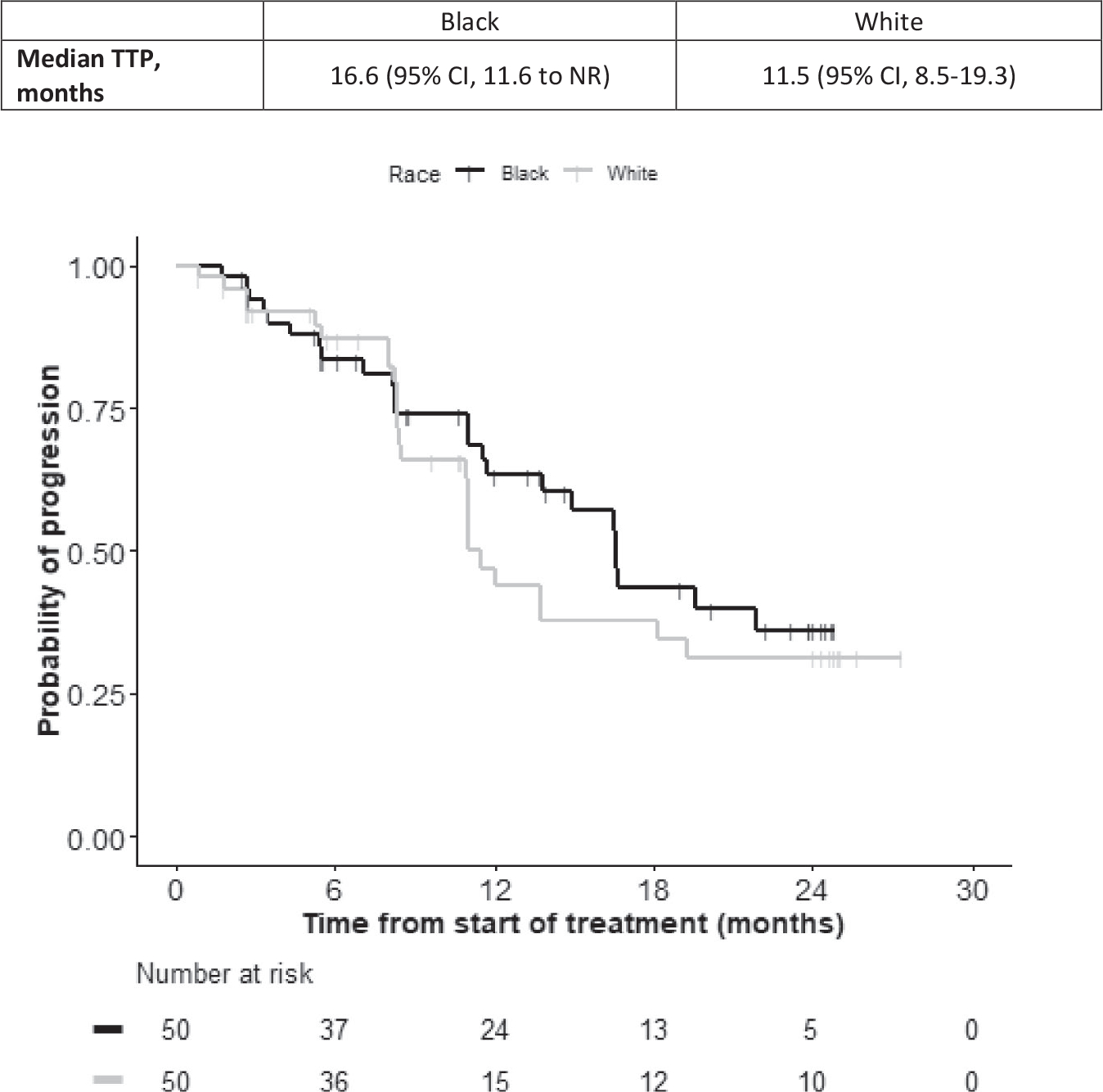
Kaplan-Meier curves illustrate the time to prostate-specific antigen progression (TTP) in Black patients (black curve) and White patients (gray curve) who had metastatic, castrate-resistant prostate cancer and received treatment with abiraterone acetate plus prednisone. Median TTP estimates are shown with 95% CIs. This graph is for illustrative purposes only; the study was not powered for cohort comparisons. Any comparison is for hypothesis generation only. NR indicates not reached.
TABLE 3.
Proportion of Prostate-Specific Antigen Decline From Baseline for Black Men and White Men Who Had Metastatic, Castrate-Resistant Prostate Carcinoma Treated With Abiraterone Acetate Plus Prednisone
| Percentage of Men (95% CI) |
||
|---|---|---|
| PSA Status | Black Men | White Men |
|
| ||
| Decline ≥30% | 82 (0.69–0.91) | 78 (0.64–0.88) |
| Decline ≥50% | 74 (0.60–0.85) | 66 (0.51–0.79) |
| Decline ≥90% | 48 (0.34–0.63) | 38 (0.25–0.53) |
| No PSA decline | 4 (0.004–0.14) | 10 (0.03–0.21) |
| PSA < 0.1 ng/mL | 18 (0.1–0.3) | 8 (0.02–0.2) |
| PSA < 0.2 ng/mL | 26 (0.14–0.4) | 10 (0.03–0.21) |
Abbreviation: PSA, prostate-specific antigen.
Black men and White men had the same number of deaths (27 each). The median OS was 35.9 months (95% CI, 24.5–43.0 months) in Black patients and 35.7 months (95% CI, 27.1 to not reached) in White patients (Fig. 4). The observed rate of bone scan flare among Black men was 16% in their first on-study imaging, similar to previous observations with enzalutamide, whereas the bone scan flare rate among White men was only 4%.26
Figure 4.
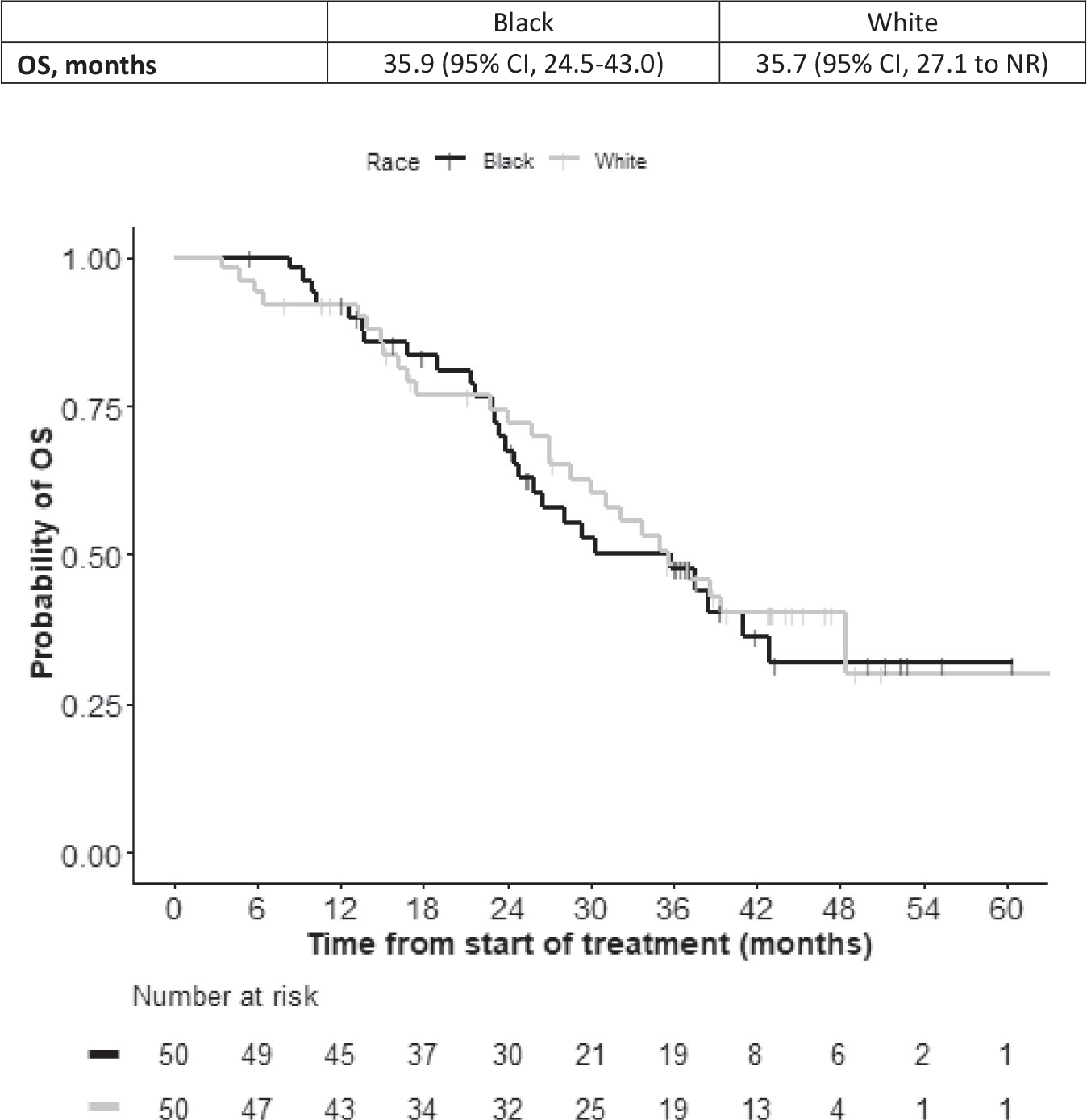
Kaplan-Meier curves illustrate overall survival (OS) in Black patients (black curve) and White patients (gray curve) who had metastatic, castrate-resistant prostate cancer and received treatment with abiraterone acetate plus prednisone. Median OS estimates are shown with 95% CIs. The graph is for illustrative purposes only; the study was not powered for cohort comparisons. Any comparison is for hypothesis generation only. NR indicates not reached.
Safety
The most common adverse events (≥10%) reported on study are listed in Table 4. Overall, the toxicity profile was similar to that in prior reports; however, the incidence of some toxicities appeared to vary by cohort. Black men and White men, respectively, reported different rates of fatigue (26% and 42%, respectively), cough (18% and 30%), headache (6% and 18%), falls (8% and 14%), vomiting (6% and 16%), peripheral edema (26% and 16%), constipation (28% and 10%), anorexia (20% and 4%), hot flashes (20% and 2%), and dyspepsia (14% and 6%).
TABLE 4.
Adverse Event Profile in Black Men and White Men
| All Grades, % |
Grades 3 and 4, % |
|||||
|---|---|---|---|---|---|---|
| Adverse Event | Black Men | White Men | Total | Black Men | White Men | Total |
|
| ||||||
| Fatigue | 26 | 42 | 34 | 4 | 8 | 6 |
| Hypertension | 46 | 40 | 43 | 24 | 16 | 20 |
| Cough | 18 | 30 | 24 | 0 | 0 | 0 |
| Pain | 24 | 24 | 24 | 2 | 6 | 4 |
| Edema, limbs | 26 | 16 | 21 | 0 | 0 | 0 |
| Constipation | 28 | 10 | 19 | 0 | 0 | 0 |
| Pain in extremity | 14 | 18 | 16 | 2 | 2 | 2 |
| Nausea | 16 | 16 | 16 | 0 | 0 | 0 |
| Dyspnea | 18 | 14 | 16 | 0 | 0 | 0 |
| Anorexia | 20 | 4 | 12 | 0 | 0 | 0 |
| Headache | 6 | 18 | 12 | 0 | 0 | 0 |
| Hot flashes | 20 | 2 | 11 | 0 | 0 | 0 |
| Urinary tract infection | 12 | 10 | 11 | 4 | 4 | 4 |
| Fall | 8 | 14 | 11 | 0 | 0 | 0 |
| Vomiting | 6 | 16 | 11 | 0 | 0 | 0 |
| Dyspepsia | 14 | 6 | 10 | 0 | 0 | 0 |
| Urinary frequency | 10 | 10 | 10 | 0 | 0 | 0 |
| Laboratory adverse events | ||||||
| Hyperglycemia | 26 | 14 | 20 | 10 | 4 | 7 |
| Hypokalemia | 34 | 20 | 27 | 12 | 4 | 8 |
| Anemia | 16 | 14 | 15 | 6 | 2 | 4 |
| Hypomagnesemia | 16 | 8 | 12 | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 10 | 10 | 10 | 2 | 0 | 1 |
| Alanine aminotransferase increased | 10 | 10 | 10 | 0 | 2 | 1 |
| Alkaline phosphatase increased | 10 | 4 | 7 | 4 | 0 | 2 |
Laboratory adverse events were similar to those reported previously,4 although they varied in incidence by cohort. Estimated rates differed in Black men and White men for both all-grade hyperglycemia (26% and 14%) and grade 3 and 4 hyperglycemia (10% and 4%), respectively. Estimated rates of hypokalemia (34% and 20%) and hypomagnesemia (16% and 8%) also differed in Black men and White men, respectively. In particular, grade 3 and 4 rates of hypokalemia were more frequent among in Black men compared with White men (12% and 4%, respectively). There was no evidence of higher baseline screening rates of electrolyte abnormalities, and diuretic antihypertensive medications were not commonly used in either population. Glucocorticoid use (prednisone) was mandated on study (5 mg twice daily), although dose adjustments were allowed at the discretion of the treating physician.
Genetic Analyses
A genome-wide analysis was conducted on the basis of data from 83 patients across 934,674 SNPs. In Table 5, we present results for the top genic SNPs, ranked according to their corresponding unadjusted P values (≤1.0e-07), for the association with TTP. Of the top genic SNPs, 6 of 9 had a greater prevalence in White patients. In particular, we identified a missense variant in SPHK1 interactor, AKAP domain containing (SPHKAP) (reference SNP rs16824283) with high ancestral variation, with a minor allelic frequency of 0.28 for White patients and 0.06 for Black patients (see Supporting Table 1). Three other SNPs in the SPHKAP gene were identified from our top ranking SNPs, and all demonstrated higher prevalence in our White patients.
TABLE 5.
Single-Nucleotide Polymorphism Association With the Time to Prostate-Specific Antigen Progression, Ranked by Unadjusted P Values
| MAFa |
||||||
|---|---|---|---|---|---|---|
| SNP | Gene | P TOT | P GT | HRGT (95% CI) | White Men | Black Men |
|
| ||||||
| rs80275835 | SPHKAP | 9.8e-08 | 2.4e-05 | 7.68 (2.98–19.80) | 0.07 | 0.01 |
| rs75639111 | TM4SF20 | 2.7e-07 | 4.4e-04 | 9.95 (2.76–35.85) | 0.03 | 0.01 |
| rs6733782 | C2orf83 | 3.6e-07 | 1.4e-03 | 3.90 (1.69–8.98) | 0.06 | 0.06 |
| rs10502901 | ACAA2 | 4.9e-07 | 7.4e-08 | 19.94 (6.70–59.30) | 0.04 | 0.08 |
| rs6760875 | SPHKAP | 5.1e-07 | 2.0e-03 | 2.11 (1.31–3.38) | 0.45 | 0.28 |
| rs10177414 | MFF | 6.1e-07 | 7.3e-03 | 1.97 (1.20–3.22) | 0.45 | 0.14 |
| rs16824283 | SPHKAP | 6.8e-07 | 3.8e-02 | 2.15 (1.04–4.43) | 0.28 | 0.06 |
| rs4530360 | SPHKAP | 7.2e-07 | 1.2e-03 | 2.47 (1.43–4.27) | 0.28 | 0.17 |
| rs73102745 | NGEF | 8.8e-07 | 1.1e-03 | 2.62 (1.47–4.67) | 0.12 | 0.14 |
Abbreviations: GT, genotype; HR, hazard ratio; MAF, minor allelic frequency; PTOT, unadjusted P value for the association with the time to prostate-specific antigen progression; SNP, single-nucleotide polymorphism.
MAFs represent those observed in the analysis population.
Next, we cross-referenced these SNPs with the large HumanVar and HumanDiv Polymorphism Phenotyping, version 2 (Polyphen-2) databases, which are used to predict for pathogenicity. The rs16824283 SNP revealed scores of 0.78 and 0.96 in HumanVar and HumanDiv Polyphen-2, respectively, predicting that this SNP is possibly or probably deleterious and worthy of further independent evaluation.30 Similar results were seen compared with the rPFS and OS results, although there were fewer events with these end points. Additional information on these SNPs is provided in Supporting Table 1.
DISCUSSION
To our knowledge, our study is the first interventional trial in mCRPC to prespecify parallel patient treatment groups by self-identified race and to evaluate clinical efficacy and safety outcomes prospectively by race. Our study was designed to enroll equal numbers of Black men and White men with mCRPC, although White men made up a greater percentage of men with mCRPC who received care at many of our sites. In addition, we allowed a more liberal eligibility criteria for common comorbidities like hypertension or diabetes and amended our study several times to broaden eligibility in response to individual Black patients who otherwise would have been excluded for a single factor. Consequently, we might have expected worse tolerance and efficacy among Black men with baseline higher rates of comorbidities; however, such outcomes were not observed.
Our study was not designed to compare outcomes by race; however, as with other retrospective studies, we observed that Black men who had mCRPC and received AAP treatment demonstrated higher rates of unconfirmed PSA declines and a greater magnitude of PSA decline, including men who reached undetectable PSA levels, and a longer TTP compared with White men who had mCRPC. These clinical outcomes are consistent with our prior retrospective analyses from the phase 3 COU-302 trial and our multi-institutional analyses.4,12,31 The median TTP in the COU-302 overall study population was 11.1 months, similar to that of our White cohort (11.5 months); whereas, in a retrospective analysis of Black men from the COU-302 trial, the median TTP (16.5 months) was similar to that in our Black cohort (16.6 months).4,16 In practice, many practitioners will continue to treat men on abiraterone who have PSA progression after a nadir and will wait for radiographic progression, as was done in the COU-302 study. However, if Black men are more likely to develop radiographic progression close to the TTP, this may warrant a different practice pattern. More prospective data are needed to confirm these patterns and the timing of progression.
Biologically, because PSA expression is transcriptionally regulated by androgen receptor signaling, we hypothesize that differences in hormonal signaling by race in men with mCRPC could alter PSA response and progression. Finally, in our study, the longer term outcomes of OS and rPFS were similar by race, providing encouraging data to support intentional broadening of eligibility criteria to improve the inclusion of disproportionately affected Black men with mCRPC into prospective trials.
We report clinical differences in adverse events by race, which may be clinically important, specifically including greater rates and greater severity of adverse events related to adrenal hormone suppression (ie, hypertension, hypokalemia, and hypomagnesemia) in Black men compared with White men, which has not previously been recognized. If confirmed, these results could support different thresholds for monitoring and managing these important adverse events by race. For instance, AAP has also been studied using a lower starting dose of prednisone (5 mg daily instead of 5 mg twice daily).32 Although adverse event rates were similar with a lower prednisone dose, Black men were underrepresented in the study. We also observed differences in symptom reporting on patients by race. The extent to which this was driven by differences in culture, education, or trust is unknown in our study but warrants further prospective investigation.
In the current prospective study, we were able to collect DNA to perform genome-wide genotyping and initially assess the role of heritable genetic variation with respect to outcomes in this mCPRC patient population stratified by race. In addition to assessing the prevalence of genetic variation caused by genotype, ie, the number of copies (0, 1, or 2) of allelic alterations inherited from one’s parents, we also assessed the role of ancestry-dependent variation quantified by local ancestry, ie, the number of copies of DNA inherited from the African ancestor. We adjusted the results further based on the estimated average proportion of African DNA, or global ancestry, as a continuous variable. This exploratory analysis identified a missense variant, rs16824283, as 1 of the top SNPs associated with TTP, unadjusted P values < 6.8e-07, and HR estimates of 2.15 for genotype (95% CI, 1.04–4.43) and 12.37 for local ancestry (95% CI, 2.18–7 0.06). The SNP rs16824283 is located in SPHKAP, which functions as a protein coding gene. SPHK1 has high predicted pathogenicity based on HumanVar and HumanDiv Polyphen-2 scores and has an allelic frequency that may vary considerably across ancestral populations. HumanVar and HumanDiv Polyphen-2 scores of 0.78 and 0.96, respectively, predict that this SNP is probably or likely functionally damaging,30 whereas SIFT scores from 0.013 to 0.031 (Ion Reporter Software) predict that this SNP is deleterious.33 Cross-referencing this SNP in the Single Nucleotide Polymorphism Database (dbSNP), another large population-based database for SNPs, the minor allelic frequency of rs16824283 in European populations ranged from 0.26 to 0.30, compared with an estimated minor allelic frequency for African or African American populations in the range from 0.03 to 0.06.34–36
The biologic significance of SPHK1 and its downstream protein, sphingosine-1-phosphate (S1P) are well characterized in prostate cancer. SPHK1 has been associated with PSA, tumor volume, positive margins, surgical and treatment failure, Gleason grade, and survival and with driving prostate cancer growth, migration, invasion, and metastasis.37–40 In addition, circulating S1P levels have been associated with the time to PSA and radiographic progression and the time to metastasis.41 In the context of prostate cancer treatment, SPHK1 expression modulates chemotherapy and radiotherapy efficacy.42–47 Moreover, in the metastatic bone microenvironment, S1P promotes proliferation and chemotherapeutic and radiotherapy resistance.48 SPHK1 also modulates hypoxia.49–53 Therefore, the biologic and predictive significance of SPHKAP/SPHK1 as a predictor of clinical benefit from AAP based on ancestry is worthy of further investigation.
Our study has several limitations. As mentioned above, this was a noncomparative study to evaluate clinical outcomes for each group in parallel. Therefore, the cross-population comparisons should be interpreted as exploratory and hypothesis-generating/supportive, but not conclusive. Similarly, based on our small sample size, our SNP analyses should be viewed as exploratory, with a potential for measurement error in the local and global ancestry estimates or associations with other imbalances in comorbidities.54,55 We are currently conducting a second, race-stratified study of abiraterone acetate, prednisone, and apalutamide (ClinicalTrials.gov identifier NCT03098836) to independently assess these associations and other ancestry-related biologic determinants of outcome in this patient population.
Finally, this study demonstrates the feasibility of prospective race-based studies inclusive of a high percentage of Black men. With proper representation in treatment trials, more insights into how Black patients tolerate and respond to therapy based on genetic and cultural factors can be gained. Ultimately, larger prospective randomized studies, proportionately inclusive of Black men, will be necessary to investigate fully the biologic determinants associated with ancestry and outcomes.
Supplementary Material
Acknowledgments
We acknowledge Janssen Scientific Affairs, LLC, for supporting this trial. The infrastructure to complete this trial was supported by 2 consecutive Department of Defense Congressionally Directed Medical Research Program Prostate Cancer Clinical Trials Consortium grants (W81XWH-09-1-0152 and W81XWH-14-2-0198). We also acknowledge support from a National Institutes of Health/National Cancer Institute Feasibility Studies to Build Collaborative Partnerships in Cancer Research (P20) Award (1P20-CA202925-01A1) and a Prostate Cancer Foundation Movember Challenge Award. In addition, we acknowledge the assistance of the Duke Molecular Physiology Institute Molecular Genomics Core for generating the genotyping data. We also thank our colleagues and coordinators for passionately championing this study and, most of all, the patients and families who participated in this work to bring these results to light.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Daniel J. George reports institutional research support from Acerta, Astellas, Bristol-Myers Squibb (BMS), Bayer, Calithera, Exelixis, Janssen Pharma, Myovant, Pfizer, Novartis, and Sanofi-Aventis; and personal fees from the AACR, Astellas, AstraZeneca, Bayer H/C, BMS, Capio Biosciences, Constellation Pharma, EMD Serono, Exelixis, Flatiron, Ipsen, Janssen Pharma, Merck, Michael J Hennessey Associates, Millennium Medical Publishing, Modra Pharma, Myovant Sciences, NCI Genitourinary, Nektar Therapeutics, Pfizer, Physician Education Resource, RevHealth LLC, Propella Therapeutics, Sanofi, UroGPO, UroToday, and Vizuri Health Sciences, outside the submitted work. Susan Halabi serves on the on the Data and Safety Monitoring Boards at Bayer, Eisai, and Ferring and receives funding through the American Society for Clinical Oncology as a statistician on the TAPUR trial. A. Oliver Sartor reports grants from Innocrin, Invitae, Merck, and Sotio; grants and personal fees from Advanced Accelerator Applications, AstraZeneca, Bayer, BMS, Clarity Pharmaceuticals, Clovis Oncology, Constellation, Dendreon, EMD Serono, Endocyte, Fusion, Janssen, Myovant, Myriad, Noria Therapeutics Inc, Novartis, Noxopharm, Pfizer, Progenics, POINT Biopharma, Sanofi, Tenebio, Telix, and Theragnostics; personal fees from Astellas, Blue Earth Diagnostics, Bavarian Nordic, Celgene, and Fusion; and other support from the National Cancer Institute and NRG Oncology, outside the submitted work. Guru P. Sonpavde reports institutional research funding from AstraZeneca, Bayer, Boehringer-Ingelheim, Merck, Pfizer, and Sanofi; grants and personal fees from Gilead, Janssen, and Sanofi; personal fees from Amgen, AstraZeneca, Bicycle Therapeutics, BMS, Dava Oncology, Eisai, Elsevier Practice Update, EMD Serono/Pfizer, Exelixis, G1 Therapeutics, Genentech, Janssen, Medscape, Merck, Novartis, Sanofi, Scholar Rock, and Seattle Genetics/Astellas; personal fees from Clinical Care Options, Onclive, Physicians Education Resource, Research to Practice, and UpToDate; service on steering committees of trials for Bavarian Nordic, BMS, QED, and Seattle Genetics (all unpaid) as well as AstraZeneca and Debiopharm; and other support from Bavarian Nordic and Debiopharm, outside the submitted work. Matthew I. Milowsky reports institutional research funding from Acerta, Amgen, Arvinas, Astelllas, BMS, Clovis Oncology, Constellation, Genentech, Incyte, Innocrin, Inovio, Johnson & Johnson, Merck, Mirati, Pfizer, Regeneron, Roche, Seagen, Syndax, and X4Pharmaceuticals, outside the submitted work. Michael Goodman reports institutional research funding from Janssen, outside the submitted work. Michael R. Harrison reports institutional research funding from Argos, Bayer, BMS, Exelixis, Merck, Pfizer, and Seattle Genetics; and personal fees from BMS, Exelixis, Genentech/Roche, and Janssen, outside the submitted work. Megan McNamara reports institutional research funding from Acerta Pharma, Astellas Pharma, AstraZeneca, Boehringer-Ingelheim, BMS, Cerulean Pharma, Clovis Oncology, Constellation Pharmaceuticals, Incyte, Innocrin Pharma, Inovio Pharmaceuticals, Jounce Therapeutics, MedImmune, Merck, Mirati Therapeutics, Roche/Genentech, Seattle Genetics, Syndax, V Foundation, and X4 Pharmaceuticals; personal fees from Bayer and BioClin Therapeutics; and other institutional support from Asieris. Dadong Zhang reports institutional research funding from AbbVie, Acerta, Astellas, Janssen, Merck, Merrimack, Mirati, Novartis, OmniSeq, Pfizer, PGDx, and Regeneron; personal fees from Amgen, AstraZeneca, Bayer, BMS, Calithera, Dendreon, Exelixis, Foundation Medicine, Genentech/Roche, Genomic Health, IQVIA, Janssen, Larvol, Merck, MUH Associates, Nanorobotics, Pacific Genuity, Pfizer, Pharmacyclics, QED Therapeutics, Sanofi-Aventis, and Seattle Genetics; service on the American Society for Clinical Oncology Clinical Guidelines Committee for Treatment of Metastatic Renal Cell Carcinoma; service on the Aravive Data Safety Monitoring Board; and stock ownership/employment (spouse) from Archimmune Therapeutics and Capio Biosciences, outside the submitted work. Andrew J. Armstrong reports grants from Janssen during the course of the study; institutional research funding from Astellas, AstraZeneca, Bayer, Beigene; BMS, Constellation, Dendreon, Genentech/Roche, Janssen, Merck, Novartis, and Pfizer; personal fees from Astellas/Pfizer, AstraZeneca, Bayer, BMS, Clovis Oncology, Dendreon, Genentech/Roche, and Merck; and personal fees from Clovis Oncology and Janssen, outside the submitted work. The remaining authors made no disclosures.
Additional supporting information may be found in the online version of this article.
REFERENCES
- 1.Surveillance, Epidemiology, and End Results (SEER) Program; National Cancer Institute. Overview of the SEER Program. Accessed January 4, 2021. https://seer.cancer.gov/index.html
- 2.Pietro GD, Chornokur G, Kumar NB, Davis C, Park JY. Racial differences in the diagnosis and treatment of prostate cancer. Int Neurourol J. 2016;20(suppl 2):S112–S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halabi S, Vogelzang NJ, Kornblith AB, et al. Pain predicts overall survival in men with metastatic castration-refractory prostate cancer. J Clin Oncol. 2008;26:2544–2549. [DOI] [PubMed] [Google Scholar]
- 4.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–223. [DOI] [PubMed] [Google Scholar]
- 7.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. [DOI] [PubMed] [Google Scholar]
- 8.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. [DOI] [PubMed] [Google Scholar]
- 9.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. [DOI] [PubMed] [Google Scholar]
- 10.Halabi S, Small EJ, Vogelzang NJ, Barrier RC Jr, George SL, Gilligan TD. Impact of race on survival in men with metastatic hormone-refractory prostate cancer. Urology. 2004;64:212–217. [DOI] [PubMed] [Google Scholar]
- 11.Halabi S, Vogelzang NJ, Ou SS, Kelly WK, Small EJ. Clinical outcomes by age in men with hormone refractory prostate cancer: a pooled analysis of 8 Cancer and Leukemia Group B (CALGB) studies. J Urol. 2006;176:81–86. [DOI] [PubMed] [Google Scholar]
- 12.Halabi S, Dutta S, Tangen CM, et al. Overall survival of black and white men with metastatic castration-resistant prostate cancer treated with docetaxel. J Clin Oncol. 2019;37:403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higano CS, Armstrong AJ, Sartor AO, et al. Real-world outcomes of sipuleucel-T treatment in PROCEED, a prospective registry of men with metastatic castration-resistant prostate cancer. Cancer. 2019;125:4172–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sartor O, Armstrong AJ, Ahaghotu C, et al. Survival of African-American and Caucasian men after sipuleucel-T immunotherapy: outcomes from the PROCEED registry. Prostate Cancer Prostatic Dis. 2020;23:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dess RT, Hartman HE, Mahal BA, et al. Association of black race with prostate cancer-specific and other-cause mortality. JAMA Oncol. 2019;5:975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efstathiou E, Deshpande H, George D, et al. Abstract CT313: an exploratory analysis of efficacy and safety of abiraterone acetate (AA) in black patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) without prior chemotherapy (ctx). Cancer Res. 2014;74(19 suppl):CT313. [Google Scholar]
- 17.Ramalingam S, Humeniuk MS, Hu R, et al. Prostate-specific antigen response in black and white patients treated with abiraterone acetate for metastatic castrate-resistant prostate cancer. Urol Oncol. 2017;35:418–424. [DOI] [PubMed] [Google Scholar]
- 18.McNamara MA, George DJ, Ramaswamy K, et al. Overall survival by race in chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC) patients treated with abiraterone acetate or enzalutamide [abstract]. J Clin Oncol. 2019;37(7 suppl):212. [Google Scholar]
- 19.Tsao CK, Sfakianos J, Liaw B, et al. Phase II trial of abiraterone acetate plus prednisone in black men with metastatic prostate cancer. Oncologist. 2016;21:1414–e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong AJ, Al-Adhami M, Lin P, et al. Association between new unconfirmed bone lesions and outcomes in men with metastatic castration-resistant prostate cancer treated with enzalutamide: secondary analysis of the PREVAIL and AFFIRM randomized clinical trials. JAMA Oncol. 2020;6:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan Y Detecting structure of haplotypes and local ancestry. Genetics. 2014;196:625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. [DOI] [PubMed] [Google Scholar]
- 24.International HapMap 3 Consortium; Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redden DT, Divers J, Vaughan LK, et al. Regional admixture mapping and structured association testing: conceptual unification and an extensible general linear model. PLoS Genet. 2006;2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szulc P, Bogdan M, Frommlet F, Tang H. Joint genotype- and ancestry-based genome-wide association studies in admixed populations. Genet Epidemiol. 2017;41:555–566. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Zhu X, Qin H, et al. Adjustment for local ancestry in genetic association analysis of admixed populations. Bioinformatics. 2011;27:670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Therneau T A Package for Survival Analysis in R. Accessed January 4, 2021. https://rdrr.io/cran/survival/f/inst/doc/survival.pdf
- 29.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. Springer; 2000. [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halabi S, Dutta S, Tangen CM, et al. Clinical outcomes in men of diverse ethnic backgrounds with metastatic castration-resistant prostate cancer. Ann Oncol. 2020;31:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. 2017;377:352–360. [DOI] [PubMed] [Google Scholar]
- 33.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- 34.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke L, Fairley S, Zheng-Bradley X, et al. The International Genome Sample Resource (IGSR): a worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017;45(D1):D854–D859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dayon A, Brizuela L, Martin C, et al. Sphingosine kinase-1 is central to androgen-regulated prostate cancer growth and survival. PLoS One. 2009;4:e8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malavaud B, Pchejetski D, Mazerolles C, et al. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur J Cancer. 2010;46:3417–3424. [DOI] [PubMed] [Google Scholar]
- 39.Brizuela L, Ader I, Mazerolles C, Bocquet M, Malavaud B, Cuvillier O. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: implication for resistance to therapeutics in prostate cancer. Mol Cancer Ther. 2012;11:1841–1851. [DOI] [PubMed] [Google Scholar]
- 40.Lee CF, Dang A, Hernandez E, et al. Activation of sphingosine kinase by lipopolysaccharide promotes prostate cancer cell invasion and metastasis via SphK1/S1PR4/matriptase. Oncogene. 2019;38:5580–5598. [DOI] [PubMed] [Google Scholar]
- 41.Nunes J, Naymark M, Sauer L, et al. Circulating sphingosine-1-phosphate and erythrocyte sphingosine kinase-1 activity as novel biomarkers for early prostate cancer detection. Br J Cancer. 2012;106:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pchejetski D, Golzio M, Bonhoure E, et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005;65:11667–11675. [DOI] [PubMed] [Google Scholar]
- 43.Akao Y, Banno Y, Nakagawa Y, et al. High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun. 2006;342:1284–1290. [DOI] [PubMed] [Google Scholar]
- 44.Pchejetski D, Doumerc N, Golzio M, et al. Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol Cancer Ther. 2008;7:1836–1845. [DOI] [PubMed] [Google Scholar]
- 45.Sauer L, Nunes J, Salunkhe V, et al. Sphingosine kinase 1 inhibition sensitizes hormone-resistant prostate cancer to docetaxel. Int J Cancer. 2009;125:2728–2736. [DOI] [PubMed] [Google Scholar]
- 46.Aoyama Y, Sobue S, Mizutani N, et al. Modulation of the sphingolipid rheostat is involved in paclitaxel resistance of the human prostate cancer cell line PC3-PR. Biochem Biophys Res Commun. 2017;486:551–557. [DOI] [PubMed] [Google Scholar]
- 47.Pchejetski D, Bohler T, Brizuela L, et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010;70:8651–8661. [DOI] [PubMed] [Google Scholar]
- 48.Brizuela L, Martin C, Jeannot P, et al. Osteoblast-derived sphingosine 1-phosphate to induce proliferation and confer resistance to therapeutics to bone metastasis-derived prostate cancer cells. Mol Oncol. 2014;8:1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O. Sphingosine kinase 1: a new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008;68:8635–8642. [DOI] [PubMed] [Google Scholar]
- 50.Cho SY, Lee HJ, Jeong SJ, et al. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1 alpha inactivation in hypoxic PC-3 prostate cancer cells. J Pineal Res. 2011;51:87–93. [DOI] [PubMed] [Google Scholar]
- 51.Cho SY, Cho S, Park E, et al. Coumestrol suppresses hypoxia inducible factor 1alpha by inhibiting ROS mediated sphingosine kinase 1 in hypoxic PC-3 prostate cancer cells. Bioorg Med Chem Lett. 2014;24:2560–2564. [DOI] [PubMed] [Google Scholar]
- 52.Ader I, Gstalder C, Bouquerel P, et al. Neutralizing S1P inhibits intratumoral hypoxia, induces vascular remodelling and sensitizes to chemotherapy in prostate cancer. Oncotarget. 2015;6:13803–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee MS, Lee SO, Kim KR, Lee HJ. Sphingosine kinase-1 involves the inhibitory action of HIF-1alpha by chlorogenic acid in hypoxic DU145 cells. Int J Mol Sci. 2017;18:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Harashima SI, Liu Y, Usui R, Inagaki N. Sphingosine kinase 1-interacting protein is a novel regulator of glucose-stimulated insulin secretion. Sci Rep. 2017;7:779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Harashima SI, Wang Y, et al. Sphingosine kinase 1-interacting protein is a dual regulator of insulin and incretin secretion. FASEB J. 2019;33:6239–6253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.