

Summary
Polymorphonuclear leucocytes (PMNs) play a protective role during Bacillus anthracis infection. However, B. anthracis is able to subvert the PMN response effectively as evidenced by the high mortality rates of anthrax. One major virulence factor produced by B. anthracis, lethal toxin (LT), is necessary for dissemination in the BSL2 model of mouse infection. While human and mouse PMNs kill vegetative B. anthracis, short in vitro half-lives of PMNs have made it difficult to determine how or if LT alters their bactericidal function. Additionally, the role of LT intoxication on PMN’s ability to migrate to inflammatory signals remains controversial. LF concentrations in both serum and major organs were determined from mice infected with B. anthracis Sterne strain at defined stages of infection to guide subsequent administration of purified toxin. Bactericidal activity of PMNs assessed using ex vivo cell culture assays showed significant defects in killing B. anthracis. In vivo PMN recruitment to inflammatory stimuli was significantly impaired at 24 h as assessed by real-time analysis of light-producing PMNs within the mouse. The observations described above suggest that LT serves dual functions; it both attenuates accumulation of PMNs at sites of inflammation and impairs PMNs bactericidal activity against vegetative B. anthracis.
Introduction
Bacillus anthracis is a Gram-positive endospore-forming bacterium that causes the disease anthrax (Mock and Fouet, 2001). The B. anthracis endospore is considered the infectious form of the bacterium, and is capable of infecting most warm blooded hosts through three distinct routes of infection: inhalation, ingestion (gastrointestinally) and cutaneous. The secreted exotoxin and capsule are responsible for much of B. anthracis virulence and are encoded on two virulence plasmids, pXO1 and pXO2 (Ivanova et al., 2003), respectively, although other virulence factors have been proposed (Mukherjee et al., 2011). The tripartite exotoxin is composed of three subunits; a receptor binding translocase, protective antigen (PA), as well as two enzymatic subunits, oedema factor (EF) and lethal factor (LF). When PA is bound to EF or LF the complex is referred to as oedema toxin (ET) and lethal toxin (LT) respectively (Kintzer et al., 2009; Moayeri and Leppla, 2009). EF is a calmodulin-activated adenylate cyclase which increases endogenous intracellular cAMP levels in the target cell, leading to host oedema, among other ramifications (Stanley and Smith, 1961; Leppla, 1982; Barth et al., 2004). LF is a metalloprotease which cleaves most mitogen-activated protein kinase kinases (MAPKK or MEK) when introduced to the host-cell cytosol by PA (Duesbery et al., 1998; Vitale et al., 1998; Agrawal et al., 2003; Moayeri et al., 2003; Glomski et al., 2007a). As the name implies, LT is capable of causing death of experimental animals and some cell types, and most importantly it is the toxin necessary for bacterial outgrowth and dissemination (Moayeri et al., 2003; Loving et al., 2009). B. anthracis capsule consists of poly-γ-d glutamic acid which is covalently linked to the cell wall and is reported to prevent phagocytosis as well as inhibit complement activation when the bacteria are exposed to serum (Makino et al., 1989; 2002; Schneerson et al., 2003). It should be noted that it has been reported that the capsule does not alter human neutrophils ability to kill B. anthracis (Mayer-Scholl et al., 2005).
Neutrophils, also known as polymorphonuclear leucocytes (PMNs), are among the first cells to arrive at sites of bacterial and fungal infections (Welkos et al., 1989; Borregaard and Cowland, 1997). PMNs are terminally differentiated cells which do not divide or synthesize large amounts of RNA or proteins (Summers et al., 2010). In addition to killing bacteria with an oxidative burst, PMNs can kill both intracellularly and extracellularly through the recruitment of granules that contain an array of antimicrobial effectors. PMNs contain three different types of granules each defined by their contents and degree of activation necessary to initiate exocytosis (Borregaard and Cowland, 1997). In the context of B. anthracis infections it is becoming increasingly clear that PMNs play an important role in bacterial clearance and delaying time to death (Cote et al., 2004; 2006; Moayeri et al., 2010). In addition to studies utilizing monoclonal antibody and chemical depletion of PMNs in mice, PMNs have been shown to surround tissues infected by B. anthracis during cutaneous infections in humans, dogs and pigs (Brachman, 1980; Meselson et al., 1994). However it is clear by high mortality rates reported for all forms of anthrax that B. anthracis has means to effectively counteract the PMN response during the course of many infections (Brookmeyer and Blades, 2002).
While there is robust literature examining the effects of B. anthracis LT on various tissue systems and cellular subtypes such as alveolar macrophages, endothelial cells, T cells and NK cells, comparatively little has been explored regarding toxins’ affects on PMNs, and what has been reported is contradictory across publications (Paccani and Baldari, 2011; Lowe and Glomski, 2012). Investigation of the ability of LT to influence PMNs bactericidal mechanisms has primarily focused on the effects of toxin components on the oxidative burst. However these studies report opposing conclusions, and did not directly address whether the changes in the oxidative burst altered PMN ability to kill B. anthracis (O’Brien et al., 1985; Crawford et al., 2006; Xu et al., 2008). However, oxidative burst was shown to not be necessary for killing of B. anthracis by human PMNs which instead kill vegetative bacilli with α-defensin (Mayer-Scholl et al., 2005). This same study suggested that the capacity of the bacteria to produce toxin components did not alter PMNs killing ability, although it should be noted that toxin concentrations in the assays were not reported. Other studies have focused on the ability of B. anthracis toxins to alter the chemotactic ability of PMNs across a chemokine gradient (Wade et al., 1985; During et al., 2005; Rossi Paccani et al., 2007; Szarowicz et al., 2009). However, results were conflicting with one having reported an increase in chemotaxis as result of PMN intoxication (Wade et al., 1985), while others reported inhibition (During et al., 2005; Szarowicz et al., 2009).
We have reported that LF can be found circulating in the bloodstream of infected mice 12 h post inoculation with subcutaneous B. anthracis Sterne strain endospores, before the bacteria have spread from the initial site of inoculation into the draining lymph node (Weiner et al., 2012). Since LT, and not ET, is necessary for B. anthracis Sterne infection to progress from the initial inoculation site into deeper tissues (Loving et al., 2009), circulating LT may thus function to dampen the host innate immune response at the systemic level. In this study we use unique and comprehensive methods to establish the levels of circulating LF throughout B. anthracis infection. We found that sublethal quantities of circulating LT (i) diminish PMNs ability to kill vegetative bacilli in vitro, (ii) hinder PMNs migration up a chemotactic gradient in vitro, and (iii) inhibit PMNs accumulation at sites of inflammation in vivo. The experiments presented here are the first to directly indicate that circulating B. anthracis LT influences PMN function remotely from an infection site, while avoiding some pit falls of in vitro intoxication studies.
Results
Circulating LT attenuates PMN ability to kill vegetative B. anthracis
Human PMNs kill vegetative bacilli through deployment of α-defensins (Mayer-Scholl et al., 2005), mice, however, lack α-defensins (Eisenhauer and Lehrer, 1992). Despite this, PMNs isolated from mice after being elicited using injections of sterile starch in vivo, were capable of phagocytosing and killing Sterne strain B. anthracis in vitro (Welkos et al., 1989). Mouse PMNs are vital for the clearance and control of B. anthracis infections since pharmacological or antibody depletion of PMNs renders mice more susceptible to infection (Cote et al., 2006; Liu et al., 2010). The ability of mouse PMNs that have not been elicited by starch to kill vegetative bacilli in vitro was determined. Stationary-phase vegetative B. anthracis which had the genes encoding the three toxin components knocked out (strain TKO), was opsonized in fresh naïve C57Bl/6 mouse serum. Opsonized TKO was then mixed with PMNs isolated from 6-week-old C57BL/6 female mice femur bone marrow. PMN killing was assessed by comparing differences in bacterial colony-forming units (cfu) over a time-course of one and a half hours between TKO incubated with live or paraformaldehyde fixed PMNs (Fig. 1). One and a half hours was the maximum time for determining differences since at later times TKO growth in the media made interpreting results difficult. At one and a half hours cfu of the TKO bacilli dropped by 65% in the presence of live PMNs, compared to samples with fixed PMNs where cfu remained steady (Fig. 1).
Fig. 1.
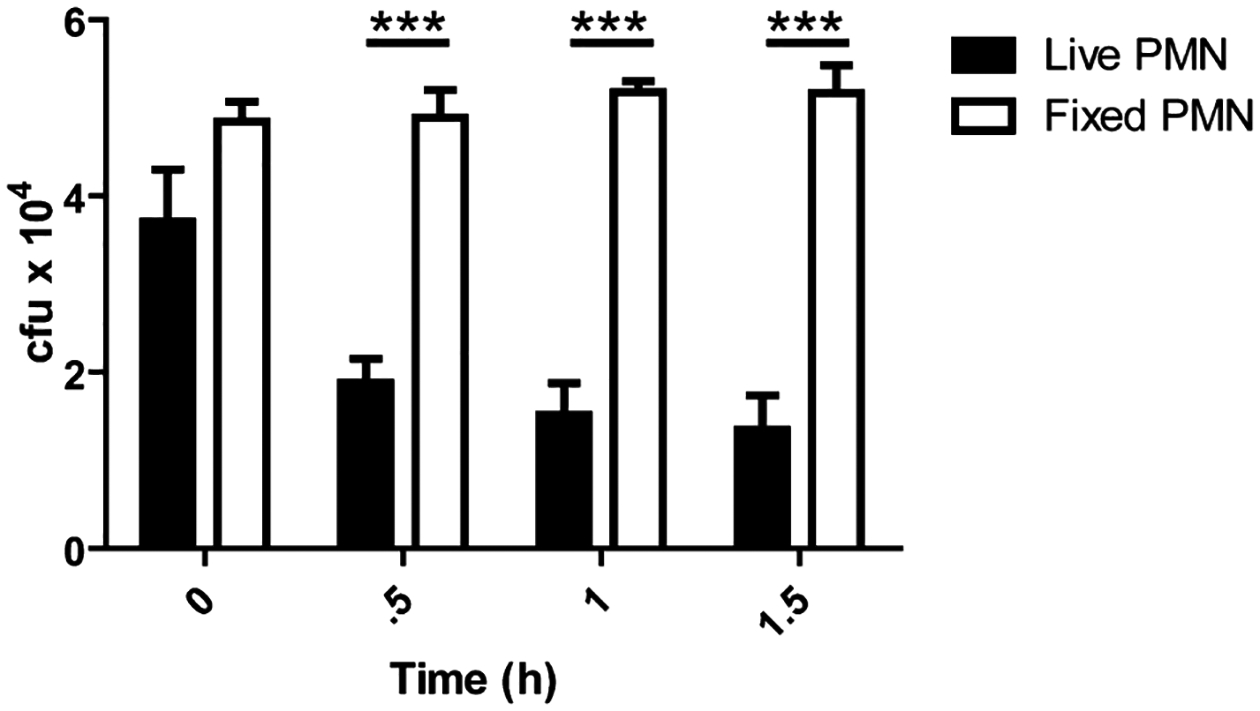
Mouse PMNs kill vegetative B. anthracis. PMNs isolated from C57B/6 mice were inoculated with vegetative B. anthracis at an moi of 1:1 and incubated at 37°C. At indicated time points samples were put on ice and mixed with 0.1% Saponin for 10 min. Samples were then serially diluted and plated for enumeration on LB agar. Comparison between samples where TKO was mixed with live versus 2% paraformaldehyde fixed PMNs with significant differences at 0.5 h (***P < 0.001), 1 h (***P < 0.001) and 1.5 h (***P < 0.001). n = 3 mice per condition.
Due to the variable nature of B. anthracis infections in mice, injection of purified LT intraperitoneally (i.p.) was used to ensure bone marrow PMNs received consistent exposure to LT. In order to determine if in vivo LT-intoxicated PMNs displayed a defect in the ability to kill vegetative TKO, LT was injected i.p. in mice and PMN were isolated 24 h post intoxication as before. These PMN’s bactericidal activity against vegetative bacilli was then compared to non-intoxicated PMN. Significant differences in TKO cfu between un-intoxicated PMNs and LT-treated PMNs were measured using doses of 6.25 μg of LT with 84% of the bacteria surviving after 1.5 h, and 54% of the bacteria surviving when a dose of 1.56 μg LT was administered compared to 35% survival with un-intoxicated PMNs (Fig. 2A). At a dose of 0.1 μg of LT no statistically significant differences were seen from un-intoxicated control PMNs. Treatments with LF or PA alone also resulted in no significant differences when compared to untreated PMNs (Fig. 2B). We then determined that LT effects on PMNs were first seen at 6 h post injection and decreased below significance between 24 and 72 h post LT inoculation into the mouse (Fig. 2C). Purified PMNs were analysed for differences in viability by flow cytometry after 24 h in vivo incubation with 6.25 μg of LT; no differences were found when compared to non-treated control mice, indicating that the PMN viability was not affected by LT (Fig. S1).
Fig. 2.
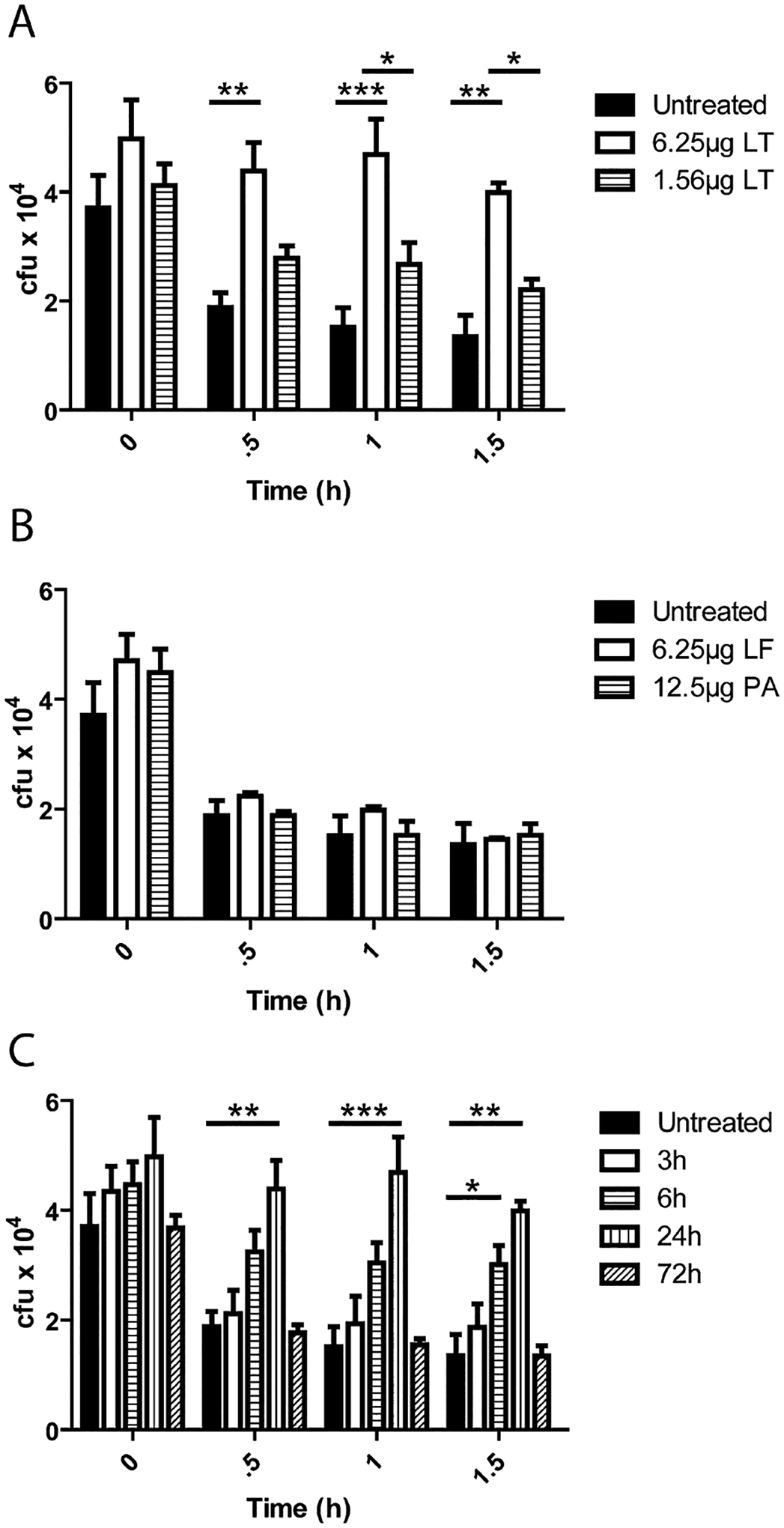
Circulating LT attenuates PMNs killing of vegetative B. anthracis. (A–C) C57Bl/6 mice were injected with toxin components (LF, PA) and PMNs were isolated at 24 h post injection. PMNs were then inoculated with TKO at an moi of 1:1 and incubated at 37°C. At indicated time points samples were put on ice and mixed with 0.1% Saponin for 10 min and vigorously mixed by pipetting. Samples were then serially diluted and plated for enumeration on LB agar.
A. Mice were injected i.p. with the indicated doses of LT, PMNs were isolated at 24 h post injection and cfu were determined at indicated time points. Significant differences in cfu over time were found between non-intoxicated and + 6.25 μg LT (0.5 h **P < 0.01, 1 h ***P < 0.001, 1.5 h **P < 0.01) and 6.25 μg LT and 1.56 μg LT (1 h *P < 0.05 and 1.5 h **P < 0.05).
B. Mice were injected i.p. with LF or PA, PMNs were isolated 24 h post injection and cfu were determined at indicated time points.
C. Mice were injected i.p. with 6.25 μg LT and PMNs were isolated from the bone marrow at 3, 6, 24 and 48 h post injection and their ability to kill TKO ex vivo was determined. Significant differences were seen between 6 h LT-treated PMNs and non-intoxicated PMNs at 1.5 h (*P < 0.05) and 24 h LT-treated PMNs and non-intoxicated PMNs at (0.5 h **P < 0.01, 1 h ***P < 0.001, 1.5 h **P < 0.01). n = 3 mice per condition.
While the concentration of injected LT sufficient to cause a defect in PMNs killing ability was low, the levels of LT ultimately conveyed to and detected in the bone marrow would presumably be much lower. In order to be able to measure the diverse amounts of LF present in serum and bone marrow at the time a defect in PMN ability to kill was seen, a mass spectrometry-based assay was used capable of detecting LF was used as previously reported (Boyer et al., 2007). It is important to note that this assay is not capable of measuring LT, only LF. However, the presence of PA83 or active PA63 does not interfere with purification of LF and measurement of its activity (Boyer et al., 2007). This indicates that the LF measured includes all LF, free and any that may be bound to PA as LT. Serum and bone marrow LF concentrations were determined in C57Bl/6 mice 24 h post injection of1.56 μg LT (1.56 μg LF + 3.12 μg PA), 6.25 μg LT (6.25 μg LF + 12.5 μg PA) and 6.25 μg LF (Table 1). The LF results from purified LT-injected mice were found to be consistent. The results indicated that a 1.56 μg injection of LT yielded circulating LF but none was detectable in the bone marrow, and circulating LF for an injected dose of 6.25 μg LT was 10-fold higher than observed for 1.56 μg LT injections. To confirm that injected LT is active in bone marrow PMNs at the times of PMN isolation, a Western blot to detect MEK3 cleavage was performed on mice which had been injected with LT, PA and LF. MEK3 cleavage was observed in PMNs 24 h after LT injection but not after PA or LF injection alone (Fig. 3). While i.p. injection of LT does not replicate an LT gradient that would be established within an active infection site, it does provide consistent measurable toxin levels in circulation and the bone marrow which would be prohibitive in the case of active infection.
Table 1.
LF concentration after injection of purified toxin components are consistent with levels found during active infection.
| Sample | 1.56 μg LTa,b | 6.25 μg LTa,c | 6.25 μg LFa,d |
|---|---|---|---|
| Serum | 0.098 ± 0.083 | 1.250 ± 0.324 | 19.43 ± 1.130 |
| Right bone marrow | < LOD | 0.041 ± 0.007 | 0.040 ± 0.006 |
| Left bone marrow | < LOD | 0.029 ± 0.011 | 0.044 ± 0.007 |
LF concentrations reported in ng ml−1 ± SE.
Three C57Bl/6 mice were injected with 1.56 μg LT (1.56 μg LF + 3.12 μg PA) in 100 μl PBS i.p. and indicated samples measured for LF concentration 24 h post intoxication.
Three C57Bl/6 mice were injected with 6.25 μg LT (6.25 μg LF + 12.5 μg PA) in 100 μl PBS i.p. and indicated samples measured for LF concentration 24 h post intoxication.
Three C57Bl/6 mice were injected with 6.25 μg LF in 100 μl PBS i.p. and indicated samples measured for LF concentration 24 h post intoxication.
Fig. 3.
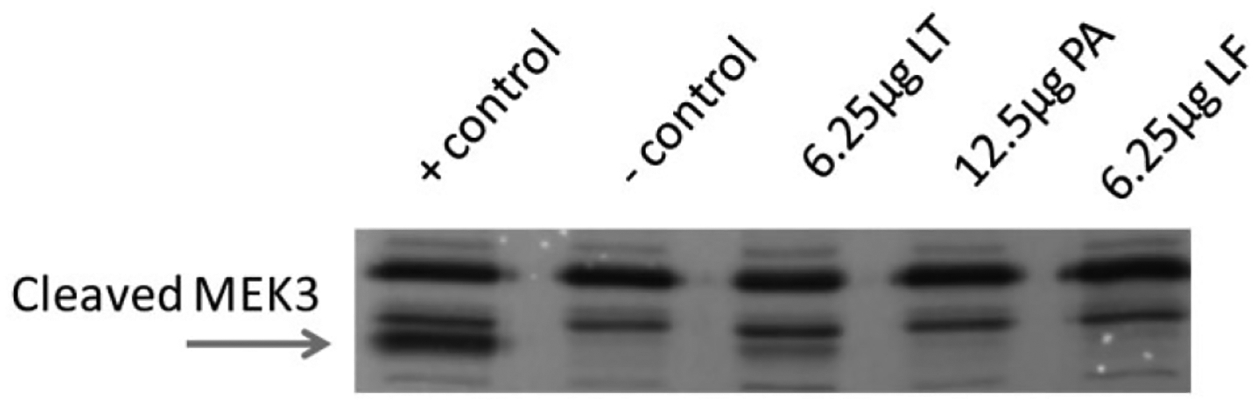
Injected LT is enzymatically active in PMNs isolated from mouse bone marrow. Western blot analysis determined LT was acting on PMNs to cleave MEK3 in vivo before cell isolation, when mice were injected with indicated amounts of LT, LF or PA. A total of 1 × 106 PMNs were isolated 24 h post injection. Positive control PMNs were treated in vitro with LT for 2 h.
LF levels increase in all major organs over time during subcutaneous B. anthracis infection
While we previously reported that LF can be found in measurable amounts in the serum of mice as early as 12 h post subcutaneous infection, the concentration of LF in circulation and in organs throughout infection has not been defined (Weiner et al., 2012). Our studies rely on the mouse model of infection, which has many advantages over other animal models, such as the availability of a wide variety of genetic tools and a BSL2 infection system using the Sterne strain of B. anthracis that lacks pX02 (Goossens, 2009). The ranges of biologically relevant levels of LF were determined in both serum and organs throughout infection in 6- to 8-week-old female A/J mice that were infected subcutaneously in the left ear with 1 × 106 spores of BIG23, a B. anthracis Sterne strain that emits light when producing toxins within a host as vegetative bacilli. Infection and dissemination were then monitored via bioluminescence. B. anthracis infection is asynchronous and time to death can vary by multiple days despite identical spore doses administered within a small timeframe (Glomski et al., 2007b; Loving et al., 2009). Because of this the stage of infection cannot be established solely by the passage of time, and must instead be defined by the stage of infection as indicated by the location of bacterial bioluminescence within the mouse. Early infection was defined as when luminescence representing vegetative bacilli can only be detected at the initial site of inoculation (Fig. S2), the mid-stage of infection was defined as when luminescence has spread to the regional draining lymph node, and late infection was defined as when luminescence can be detected in the kidneys, a time when we have determined mice are bacteraemic. At each stage of infection serum and major organs were removed for quantification of LF by a mass spectrometry-based assay. In early infection LF was found primarily in the infected ear (16.25 ± 14.32 ng g−1 tissue) associated with the vegetative bacilli, circulating in serum at 0.894 ± 0.550 ng ml−1, and at very low levels in most of the major organs where luminescence was absent, such as the spleen (≤ 0.041 ± 0.041 ng g−1 tissue) (Table 2). At mid-stage infection when luminescence spread to the draining lymph node, there was a corresponding increase in LF levels detected in the infected lymph node (547.5 ± 183.6 ng g−1 tissue), was similarly elevated in serum and the infected ears, and was detected in low levels in all other organs sampled (Table 2). In late stages of infection high levels of LF were detected in all sampled organs with the highest levels observed in serum at 1974 ± 1725 ng ml−1 (Table 2). LF was not accurately quantified in kidneys due to interference from high levels of proteases in this tissue.
Table 2.
LF levels increase in all major organs over time during B. anthracis infection.
| Sample | Early infectiona | Mid infectiona | Late infectiona |
|---|---|---|---|
| Serumc | 0.894 ± 0.550 | 27.32 ± 18.18 | 1973±1725 |
| Infected eard | 16.25 ± 14.32 | 94.25 ± 63.05 | 1201 ± 207.4 |
| Control eard | 0.099 ± 0.054 | 0.569 ± 0.336 | 28.75 ± 14.28 |
| Infected lymph noded | 0.253 ± 0.253 | 547.5 ± 183.6 | 5057 ± 2889 |
| Control lymph noded | < LODb | 2.626 ± 1.816 | 868.4 ± 575.3 |
| Heartd | 0.043 ± 0.043 | 16.70 ± 15.23 | 382.3 ± 343.7 |
| Lungsd | 0.044 ± 0.032 | 5.865 ± 5.160 | 601.1 ± 497.6 |
| Spleend | 0.041 ± 0.041 | 2.606 ± 1.717 | 1115 ± 895.9 |
| Liverd | 0.008 ± 0.004 | 9.057 ± 8.622 | 895.7 ± 611.0 |
| Right bone marrowc | < LODb | 0.145 ± 0.100 | 27.68 ± 24.42 |
| Left bone marrowc | < LODb | 0.132 ± 0.088 | 22.27 ± 17.10 |
| Braind | < LODb | 0.092 ± 0.066 | 60.25 ± 41.73 |
Injection of 1 × 105 spores of luminescent 7702 (BIG23) spores subcutaneously into the left ear of three A/J mice. Samples were collected and processed when mice reached the indicated stage of infection.
Samples where potential LF concentrations are below the limit of detection (LOD).
Samples reported in ng ml−1 ± standard error of the mean.
Samples reported in ng g−1 of tissue standard ± error of the mean.
Circulating LT caused a decrease in PMNs killing ability between a mid- and late-stage infection
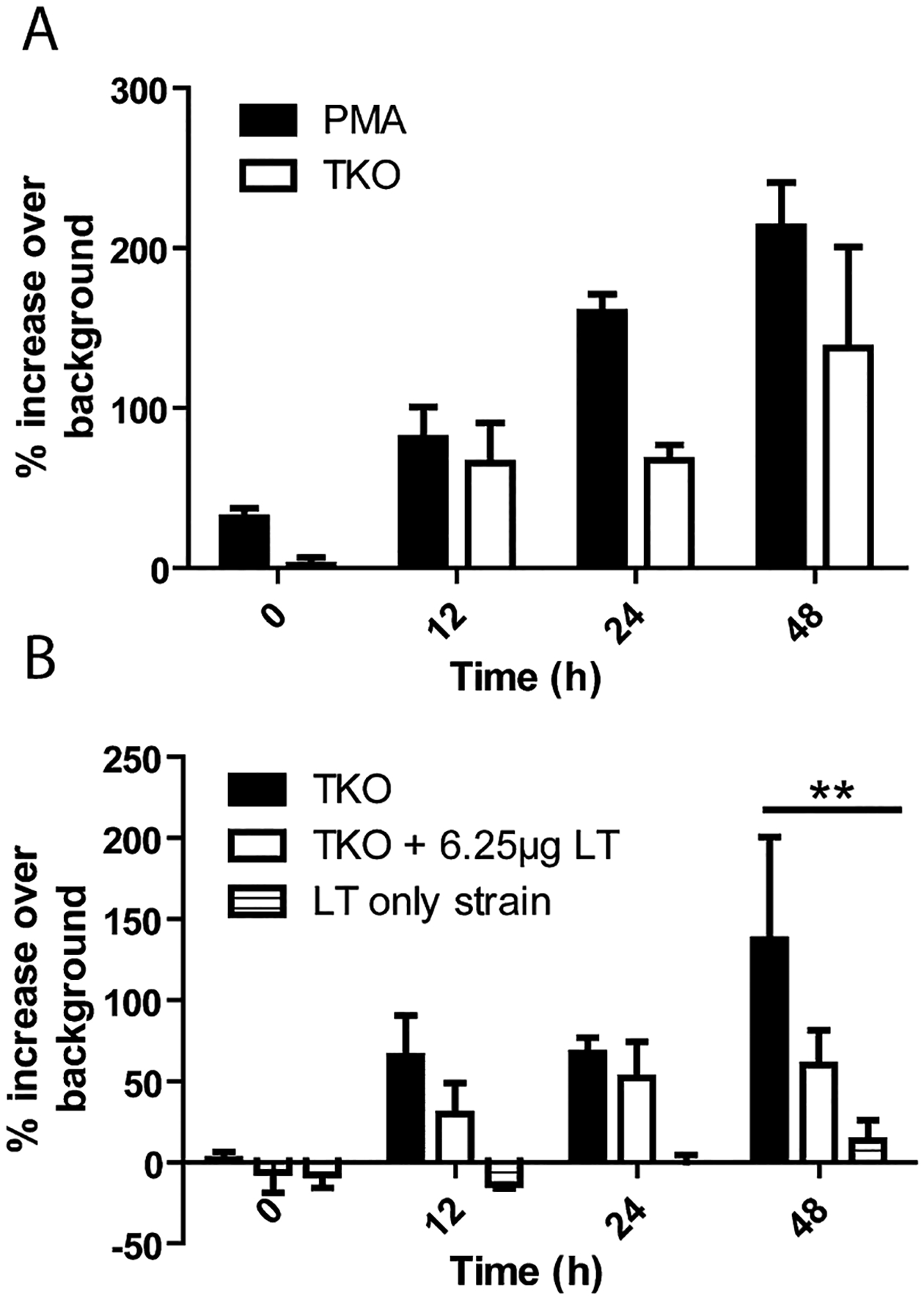
While LF can be found in circulation and in the bone marrow at very early time points, it is not clear if and when this LF is acting on bone marrow PMNs as LT. Because C57Bl/6 mice are not susceptible to the BSL2 strain of B. anthracis, A/J mice were used. We first determined if LT and not ET was responsible for dissemination in the BSL2 model of infection via the subcutaneous route of infection as was demonstrated in the inhalational route of infection (Loving et al., 2009). A/J mice were infected with 1 × 106 vegetative luminescent LF or EF knockout B. anthracis subcutaneously, and infection progression was monitored using an IVIS camera. Dissemination to the kidneys was detected in all mice infected with an EF knockout strain, while only one mouse infected with the LF knockout strain had any dissemination detected (Fig. S3). This confirms that LT is necessary for bacterial dissemination in the BSL2 mouse model of infection. The ability of A/J PMNs to kill vegetative TKO B. anthracis was determined to be equivalent to that of C57Bl/6 mice, despite A/J lack of complement protein C5 (Fig. S4). Like-wise the addition of 6.25 μg of LT attenuated A/J PMNs ability to kill vegetative TKO to a similar degree as seen in C57Bl/6 mice (Fig. S4). In order to determine if circulating LT is capable of preventing PMN function in the bone marrow during an infection, A/J mice were infected with 1 × 105 spores of BIG23 as before, and the stage of infection was determined by luminescence. At the mid-stage of infection mice were euthanized and PMNs were isolated from the bone marrow and assessed for ability to kill vegetative TKO. At the mid-stage of infection where luminescence representing toxin production was between 1 × 104 and 1 × 105 (Fig. 4A) there was no defect in PMNs ability to kill TKO (Fig. 4B). However, at a mid-stage of infection where luminescence was above 1 × 106 (Fig. 4C) there was a statistically significant defect in PMNs ability to kill at 1 h (*P < 0.05) and 1.5 h (*P < 0.05) compared to PMNs from uninfected mice (Fig. 4D). It should be noted that no spores or vegetative bacteria were detected in the bone marrow of infected mice at stages tested (data not shown). These data suggest that LF and not LT is primarily found in circulation throughout the early and mid-stages of infection.
Fig. 4.
PMN killing ability is attenuated during active infection. (A–D) A/J mice were infected with 1 × 105 spores of BIG23 and infection was monitored by production of luminescence.
A. Black and white photograph of a single representative A/J mouse, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1, representing a mid-stage of infection where luminescence does not exceed 1 × 105 p s−1 cm−2 sr−1.
B. Comparison of PMN killing of vegetative TKO between PMNs isolated from un-infected mice and PMNs isolated from mice at a mid-stage of infection where luminescence did not exceed 1 × 105 p s−1 cm−2 sr−1. No significant differences in PMN killing ability were seen at any point.
C. Black and white photo of a single representative A/J mouse, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1, representing a mid-stage of infection where luminescence is at or exceeds 1 × 106 p s−1 cm−2 sr−1.
D. Comparison of PMN killing of vegetative TKO between PMNs isolated from un-infected mice and PMNs isolated from mice at a mid-stage of infection where luminescence was at or exceeded 1 × 106 p s−1 cm−2 sr−1. Significant differences were seen at 1 h (*P < 0.05) and 1.5 h (*P < 0.05). n = 3 mice for each experimental condition.
Circulating LT attenuates PMN accumulation at sites of inflammation
While many studies have been able to examine PMN migration and accumulation in response to a chemotactic gradient in vitro, the real-time analysis of PMN accumulation dynamics in vivo, where a complex interplay among many cell types defines these dynamics, has remained elusive. In order to analyse PMN populations in vivo, we crossed transgenic mice expressing Cre recombinase under a neutrophil elastase promoter [129-Elanetm1(cre)Roes/H mice] (Tkalcevic et al., 2000) with floxed stop luciferase mice [FVB.129S6(B6)-Gt(ROSA)26Sortm1(luc)kael/J mice] (Safran et al., 2003), resulting in progeny (NECre luc mice) that expressed luciferase in PMN only that could be detected within the mouse using a highly sensitive camera. In these mice the resulting light signal intensity is proportional to the density of PMNs in the tissue. In order to track PMNs in real time, the chemical inducer of inflammation phorbol 12-myristate 13-acetate (PMA) was initially used because it is capable of producing a strong inflammatory stimulus, with less variability than injection of bacteria. PMA was dissolved in DMSO and spotted onto the left ear of mice and luciferin was introduced i.p. Mice were then analysed using an IVIS Spectrum imaging apparatus over a time-course of 48 h (Fig. 5A). To quantify the accumulation of PMNs in the inflamed area luminescent signal was measured in both ears and at each time point the per cent increase in luminescent signal over background was calculated using the non-inflamed right ear as the background measurement. These results demonstrated that there was a statistically significant increase in the PMN-generated light signal over time in the inflamed ear, increasing from 31% to 213% over background from 0 to 48 h (Fig. 5B). PMA caused an increase in PMN accumulation compared to DMSO vehicle control at all time points (Fig. 5B). While the average luminescence in the non-inflamed control ears did not significantly vary throughout the experiment (Fig. S5A), the average luminescence in the PMA inflamed ear statistically significantly increased from 0 to 48 h (Fig. S5B). However it was noted that the light signal from PMN in each individual background ear fluctuated between time points, making it necessary to determine if both ears would fluctuate in a synchronous manner even in the absence of inflammation. Mice were injected with luciferin without induction of inflammation and imaged at multiple time points. Light signals in both ears were quantified over the 48 h time period. Independent of any inflammatory stimuli the PMN content in the ears fluctuated uniformly in both ears over time (Fig. S5C), thus validating the right ear as a proper and necessary background light-emission control.
Fig. 5.
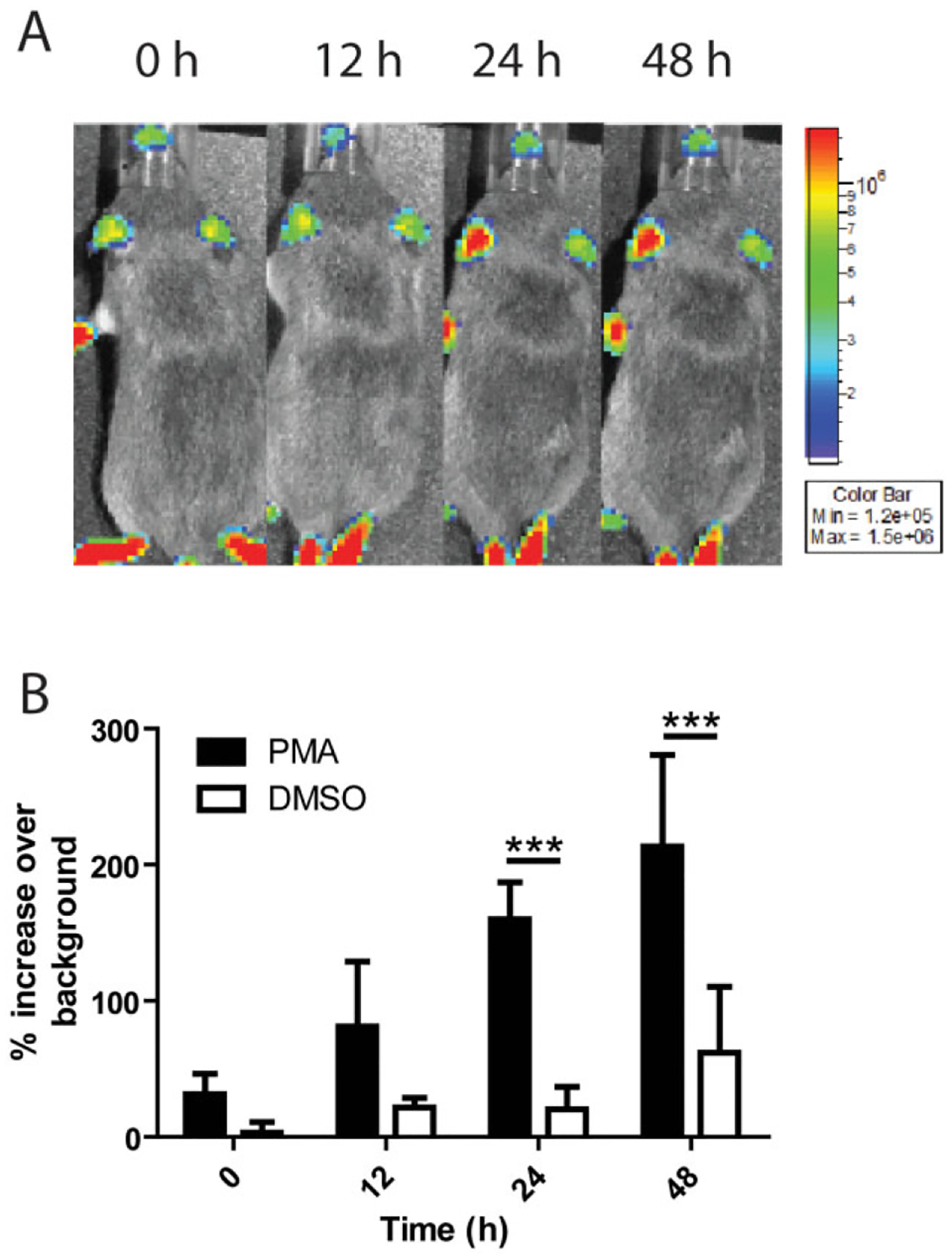
PMN accumulation at sites of inflammation can be measured in real time.
A. Black and white photos of a single representative NECre luc mouse with 2.5 μg PMA in 5 μl DMSO pipetted onto the ear, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1 over a 48 h time-course.
B. Mice were treated with either PMA or DMSO on the left ear and luminescence was quantified over time for both the treated and non-treated ear. Percentage increase in luminescence over background was then calculated, and PMN recruitment to PMA and DMSO vehicle (n = 4) control were determined where significant differences were seen at time 24 h (***P < 0.001) and 48 h(***P < 0.001). n = 6 mice per condition unless otherwise noted.
The ability of LT to prevent PMN migration in vitro has been well established (Rossi Paccani et al., 2007; Szarowicz et al., 2009). However, in vivo systems are much more complex than what can be accounted for in vitro, and the in vivo system can reflect potential LT effects on other cell types, such as endothelial and epithelial cells, as well as those on PMNs. To assess the effect that low levels of circulating LT might have on PMN accumulation at sites of inflammation in vivo, NECre luc mice were treated with increasing doses of LT and PMA was spotted onto the left ear to induce inflammation. PMA was chosen as the inflammatory stimuli because of its ability to cause a higher degree of inflammation for a longer duration than purified bacterial agonists. Light production was monitored over a time period of 48 h (Fig. 6A). It was found that mice treated with 6.25 μg of LT showed a significant decrease in light signal at both 24 and 48 h post LT injection relative to untreated controls, while a dose of 1.56 μg induced no significant differences at any time point (Fig. 6B). These results are consistent with PMN killing of vegetative bacilli experiments, where the low dose of toxin has no measurable effect, but the higher dose of 6.25 μg does. Treatment of mice with equivalent quantities of PA alone or LF alone yielded no significant difference in PMN accumulation compared to control mice (Fig. 6C). If injection of LT was causing a decrease in the net number of PMN in circulation it would be expected that there would be a decrease in luminescence over time in the non-PMA inflamed control ear. However, luminescence in the control ear of LT injected NECre luc mice did not decrease over time, indicating PMN levels in circulation were not affected by injection of LT (Fig. 6D).
Fig. 6.
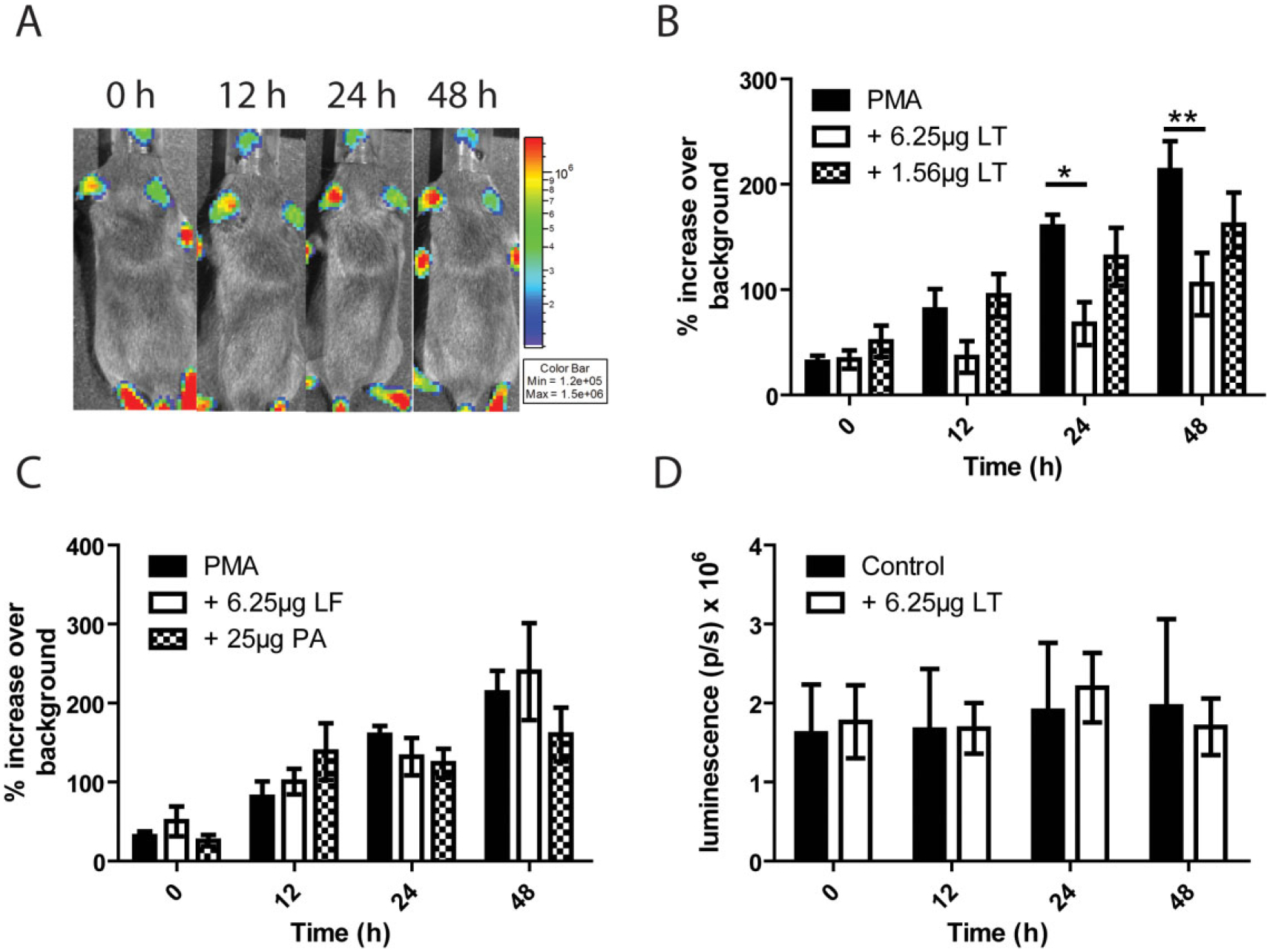
Circulating LT attenuates PMN accumulation at sites of PMA-induced inflammation.
A. Black and white photos of a single representative NECre Luc mouse with 2.5 μg PMA in 5 μl DMSO pipetted onto the ear and injected i.p. with 6.25 μg LT, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1 over a 48 h time-course.
B. Comparison of left-ear luminescence over background between mice treated with the indicated amounts of i.p. LT and non-intoxicated mice. Significant differences were found between PMA and PMA + 6.25 μg LT mice at 24 (*P < 0.05) and 48 h (**P < 0.01).
C. Comparison between mice treated with PA and LF to non-intoxicated mice. No significant differences were found.
D. Luminescence was measured in the non-PMA treated right ear in an LT-intoxicated mouse at each indicated time point and compared to luminescence in the right control ear of non-intoxicated mice. n = 6 mice per condition.
We then wanted to determine if circulating LT was capable of preventing accumulation of PMN to sites of B. anthracis infection. A total of 3 × 106 cfu of vegetative TKO were injected subcutaneously into the left ear of NECre luc mice and compared to PMN accumulation to PMA (Fig. 7A). There was no statistical difference in PMN accumulation to PMA versus TKO, however; accumulation to TKO was less consistent than was seen in PMA. Mice injected subcutaneously with TKO were then injected i.p. with 6.25 μg of LT. There was no significant difference in PMN accumulation to TKO with and without the presence of LT (Fig. 7B). NECre luc mice were then injected with 3 × 106 cfu of vegetative EF knockout B. anthracis and PMN accumulation was monitored as before. When B. anthracis is capable of producing LT locally there was statistically fewer PMNs at the site of infection at 48 h compared to TKO-infected mice (**P < 0.01) (Fig. 7B). At several time points in EF KO infected NECre luc mice there was less luminescence representing presence of PMNs in the infected ear than in the control ear (Fig. 7B). These data suggest that LT acting on its local environment may play a more vital role in preventing PMN accumulation at the site of infection than global LT.
Fig. 7.
B. anthracis LT attenuates PMN accumulation at sites of infection.
A. Comparison of left-ear luminescence over background between PMA-treated mice and mice that were injected subcutaneously with 3 × 106 cfu of vegetative TKO. No significant differences were seen at any time point.
B. Comparison between NECre luc mice which were subcutaneously injected with 3 × 106 cfu of TKO, with and withouti.p. injection of 6.25 μg LT, and NECre luc mice injected subcutaneously with 3 × 106 cfu of vegetative EF knockout B. anthracis. Significant differences were seen between TKO and EF knockout injected mice at 48 h post injection (**P < 0.05). n = 4 mice for each experimental group (A and B).
Discussion
These studies provide valuable insights into how B. anthracis utilizes LT to subvert the host PMN response to infection. While it has been shown in the mouse model of anthrax that direct intoxication of PMNs by LT was necessary for B. anthracis to disseminate and cause animal death, there have been few studies examining the functional ramifications of LT intoxication of PMNs (Loving et al., 2009; Liu et al., 2010). The reported inability of toxin-producing B. anthracis strains to alter in vitro PMN bactericidal function (Mayer-Scholl et al., 2005) may be due to insufficient concentration of LT because of too little production of LT by the bacteria during the time-course of the cell culture assay. Likewise, the time required for cells to manifest the biological effects of intoxication may also be an issue; previous reports have concluded that PMNs must be exposed to LT for at least 2 h in vitro for any measurable defects to manifest (During et al., 2005). However, due to the short lifespan of circulating PMNs of approximately 6.5 h in humans (Mauer et al., 1960), and the similar half-life of PMNs outside the host in certain conditions (Preshaw et al., 1999); the length of time needed for mouse PMNs incubation with LT in vitro and resultant biological alterations may not be feasible to assess the effect of LT on PMN function. We previously found that in the subcutaneous mouse model of B. anthracis infections, LF can be found in measurable amounts in serum after 12 h (Weiner et al., 2012), prior to circulating measureable bacteraemia. This finding leads to the possibility that circulating toxins emanating from the site of infection may be capable of manipulating the innate immune system in sites distal from areas with bacterial growth. Understanding potential roles for circulating LT might also help in understanding how residual toxins can lead to death in animals even after elimination of vegetative bacilli via antibiotics (Walsh et al., 2007).
Here we have demonstrated that circulating LT is capable of significantly impairing PMN functions crucial to resolving infections. These observations help to answer the questions of whether LT reduces PMN accumulation at inflammatory sites in vivo and prevents PMNs from killing vegetative B. anthracis. It also provides further evidence towards resolving conflicting reports in the literature regarding LT’s ability to reduce PMN migration (Wade et al., 1985; During et al., 2005; Rossi Paccani et al., 2007; Szarowicz et al., 2009). Our studies also form a connection between injected quantities of LT and biologically relevant concentrations found in an active infection. However, these results include several caveats. First, there are many variables which are difficult to take into account, such as the number of vegetative bacilli contributing to LF levels over time, and the rate of cellular uptake and clearance of LF during infection. This study also did not differentiate between LT and LF, or intra- versus extracellular toxin. It is possible that the serum toxin measurements from i.p. injected mice only had LF and no LT, due to the half-life of PA in serum being around 6 min (Moayeri et al., 2007; Liu et al., 2008). However, PA half-life in serum is not equivalent to functional half-life. It is probable that PA rapidly binds host cellular receptors and rapidly internalizes LT into target cells. These studies utilized low levels of injected LT which may be responsible for the 6–24 h in vivo incubation time needed to elicit an effect on PMNs. Presumably the saturation levels of intoxication in bone marrow PMNs is low, requiring longer periods of time for cytosolic LF to cleave PMN MEKs. Functional versus serum half-lives will have to be determined in the future. Currently no conclusions can be made about whether LF is enzymatically active within the cytosol of cells within tissues from where it was extracted and then detected enzymatically ex vivo. It is also important to differentiate local versus circulating toxin effects. Lovchik et al. reported that the toxigenic Ames strain B. anthracis could not rescue non-toxigenic B. anthracis which was co-injected into rabbits (Lovchik et al., 2012). Lovchik’s data suggest that local toxin may be more important for B. anthracis survival in its local environment. Our data demonstrate that in the subcutaneous model of infection toxin levels at the infection site are much higher than those seen in the serum and bone marrow during the early and mid-stage of infection (Table 2). The high level of LF initially found at the site of infection may play a greater role in initial survival and escape from the innate immune response than that of circulating LF. These distinctions are apparent in our data that demonstrate that during early and some parts of mid-stage infection, while comparable amounts of LF are seen in the bone marrow of infected and i.p. injected mice, there is no alteration in the ability of PMNs to kill vegetative TKO in infected mice. From these data we can speculate that while LF is constantly detected and available in circulation, PA is not. However, at some point between the mid- and late-stage of infection PA reaches a point that allows for measureable effects of intoxication on bone marrow PMNs and other cells. However, the above speculation suggests that study of the PA distribution in a kinetic fashion during infection in addition to the LF distribution is vital to our understanding of the roles of LT in anthrax. Nevertheless, LF and LT in circulation appears to play a crucial role in contributing to faster times to dissemination by attenuating peripheral PMN’s ability to respond to infection at distal sites, ensuring minimal host resistance to dissemination.
Our studies also present several methodological advantages over those previously published. The first advantage is that PMNs from intoxicated mice were used instead of directly intoxicating PMNs in vitro. Because PMNs have a short half-life and it has been demonstrated that different intoxication times can lead to different biological results, differences in in vitro intoxication protocols could have led to the disagreements in the literature (Szarowicz et al., 2009). Intoxication in vivo allows for circulating and bone marrow PMNs to interact with LT in a similar physiological context as they would during an active infection. The second advantage is the development of an in vivo PMN accumulation assay, which represents a major advance in understanding factors that influence how PMNs act in vitro. Current in vitro PMN migration assays do not reflect many of the factors influencing PMN function within the highly integrated immune response that arises in response to infection. These factors include failing to account for multiple chemoattractant gradients occurring simultaneously and that the use of diffusion-based assays leads to temporary and unstable gradients (meaning the slope of chemoattractant gradient will change as cells migrate across the gradient) (Levchenko and Iglesias, 2002; Lin et al., 2004). In vitro chemotaxis and migration assays also do not take into account potential effects of LT on the cellular epithelium and endothelium. This is why it is important to differentiate between circulating LT effects on chemotaxis and the ability to accumulate at sites of inflammation. In vivo, it is possible that circulating LT is not directly acting on PMNs, but could be acting on the endothelium to repress expression of cellular adhesins or expression of chemokines. This possibility is supported by Fig. 7B, where EF knockout B. anthracis-infected mice show almost no increase in PMN accumulation over time compared to TKO-infected mice injected with LT. During the time-course of the in vivo PMN accumulation assay, very little LF would be found in circulation, meaning most LF and LT could be acting locally on resident dendritic and endothelial cells, and not globally on PMNs. Regardless, in the end the results are the same: fewer PMNs accumulate within the site of inflammation. In addition previous chemotaxis assays relied on treatment of PMNs in vitro, where it has been shown that short incubation times (< 2 h) with LT can lead to different results in the assay (Rossi Paccani et al., 2007; Szarowicz et al., 2009). Importantly, data acquired with our novel in vivo PMN accumulation assay are consistent with our in vitro migration assays which together support the interpretation that LT inhibits PMN migration into inflammatory sites (Levchenko and Iglesias, 2002).
Understanding the mechanisms by which LT secretion by B. anthracis promotes bacterial growth and dissemination in a host is vital to developing means to reduce anthrax mortality. We have focused our research on the effects of LT on PMN function because of the critical role PMNs play in resolving and controlling B. anthracis infections, and since LT and not ET is necessary for dissemination in the Sterne strain mouse model of infection (Cote et al., 2004; 2006; Loving et al., 2009; Liu et al., 2010). Our work demonstrates that LT freely circulating in the bloodstream independently of vegetative bacteria is capable of having dramatic effects at sites distal to the infection. However, gaps in knowledge still remain regarding the role that circulating LT might play in the context of active infection. It will be important for future studies to implement new methods that differentiate between total-LF and LT and intra- versus extracellular toxin in various organs during the course of B. anthracis infection.
This information would result in a much greater understanding of how B. anthracis is so effective at causing lethal infection. It will also be necessary to determine what cellular mechanisms within both PMNs themselves and other cell types that are being disrupted by LT to inhibit both PMN migration and bactericidal activity. B. anthracis capsule may also play a supporting role in modulation of the immune system. Capsule components shed during infection can be found in circulation and may function to activate the complement cascade at sites remote from infection disrupting the chemoattractant gradient (Makino et al., 2002). Circulating capsule may also increase the efficiency of cellular intoxication by stabilizing intracellular PA pore formation (Kintzer et al., 2012).
The majority of studies focusing on the effects of bacterial products on components of the immune system focus on immune cells which have been directly exposed to toxins in vitro, or immune cells which have been isolated directly from the infected sites. Our studies demonstrate that circulating LT can affect PMNs’ ability to respond to infection by direct intoxication, in the case of inhibiting PMN killing ability, and indirectly through intoxicating the local cellular environment, as in the case of PMN accumulation at the infection site. While exotoxins produced by other pathogens such as Clostridium species and Staphylococcus aureus are reported to cause effects remote from infection sites, these phenotypes are generally reported to be at late stages of disease progression or at concentrations which result in outward symptoms (Otto, 2012; Stevens et al., 2012). Our data suggest that minute concentrations of circulating exotoxin have the potential to greatly affect the host immune response to infection. The degree of early intoxication of host cells may play a greater role in determining a lethal versus non-lethal infection than previously thought. By further study of B. anthracis toxin kinetics during the course of anthrax, it will be possible to determine the thresholds of LT intoxication needed for infection to progress beyond the initial site of infection. These studies and techniques can be applied broadly to other exotoxin secreting pathogens to better understand how initial slight differences in toxin concentrations can precipitate differences in patient outcomes.
Experimental procedures
Ethics statement
All mouse husbandry and manipulation were performed following protocols approved by the University of Virginia Animal Care and Use Committee (protocol #3671) conforming to AAALAC International accreditation guidelines. When at all possible we have strived to replace the use of animals in our studies with in vitro or non-invasive assays, reduced the number of animals utilized, and refined our use of animals to minimize their suffering and maximize the data extracted from each experiment.
Mice
All Mouse strains used were bred in specific pathogen-free conditions within vivaria at University of Virginia All experiments used female mice between 6 and 8 weeks of age. Strains used were C57Bl/6 mice (Jackson labs, Bar Harbor, Maine), A/J mice (Jackson labs), FVB.129S6(B6)-Gt(ROSA)26Sortm1(luc)kael/J mice (Jackson labs), 129-Elanetm1(cre)Roes/H mice (Medical Research Council, London, UK), and NECre luc mice bred at University of Virginia.
Bacterial strains and growth conditions
Bacillus anthracis Sterne strain 7702 was obtained from BEI Resources (Manassas, VA). The B. anthracis Sterne 7702 strain with deletion of the pagA, lef and cya genes (referred to in the text as the triple exotoxin knockout or TKO) was graciously provided by Dr Scott Stibitz from the Center for Biologics Evaluation and Research, Food and Drug Administration (Janes and Stibitz, 2006). BIG23 was constructed by integrating plasmid pIG6–19, a kind gift from Michele Mock at the Pasteur Institute, into B. anthracis strain 7702 using previously described conjugative methods (Glomski et al., 2007b). The resultant BIG23 expressed the luxABCDE genes from Photorhabdus luminescens under the control of the protective antigen promoter; therefore the vegetative cells of this strain are luminescent when growing with the host. Cultures of TKO B. anthracis were grown in Luria–Bertani (LB) broth overnight and then separated into 500 μl aliquots with 15% glycerol and stored at −80°C. For recovery of experimental aliquots from −80°C, samples were thawed on ice and then 200 μl was added to 800 μl of LB broth and allowed to shake for 1 h at 37°C.
Bacterial toxin components
The following reagents were obtained through BEI Resources, NIAID, NIH: Anthrax Protective Antigen (PA), Recombinant from Bacillus anthracis, NR-140 and Anthrax Lethal Factor (LF-HMA), Recombinant from Bacillus anthracis, NR-723. Toxin components were resuspended in PBS + 1 mg ml−1 of bovine serum albumin to a concentration of 1 mg ml−1 and stored at −80°C.
Mouse infection
A/J mice were anaesthetized with 3% isofluorane (Piramal Healthcare, Andhra Pradesh, India) mixed with oxygen using an Isotec 5 vaporizer (Absolute Anesthesia, Piney River, VA). Purified BIG23 B. anthracis spores at a dose of 1 × 105 spores in 10 μl PBS (Invitrogen, Carlsbad, CA) were injected subcutaneously in the left ear using a 0.5 cc insulin syringe as previously published (Glomski et al., 2007b).
Infection monitoring and luminescent imaging
In order to monitor the progression of infections using luminescent BIG23, mice were anaesthetized using 3% isoflurane mixed with oxygen from the XGI-8 gas anaesthesia system supplied with a Xenogen IVIS Spectrum. Images were acquired as previously reported and analysed using Living Image software (version 2.50.1, Xenogen) (Glomski et al., 2007b). Once any detectible luminescent signal was detected in a vital organ, or a mouse appeared moribund, mice were euthanized per animal use protocol.
Measurement of lethal factor concentration in tissue samples
Organs from infected A/J or LT intoxicated C57Bl/6 mice were removed and placed into 100–1000 μl of a cell lysis cocktail, depending on the organ, consisting of 0.2% Triton X-100 (Promega), 0.56 mg ml−1 Pefablock (AppliChem, Darmstadt, Germany), 3.125 mg ml−1 6-aminohexanoic acid (Spectrum Chemical, Brunswick, NJ), 0.3125 mg ml−1 Antipain (VWR, West Chester, PA) and 22 μg ml−1 of E64-D (Cayman Chemical, Ann Arbor, MI) in PBS. Samples were then homogenized using kontes pellet pestles (Fisher Scientific, Waltham, MA). Samples then sat on ice for 1 h were then centrifuged and supernatants were removed and stored at −80°C until later use. Blood samples were allowed to clot for 20 min then spun down to collect serum, which was then stored at −80°C until later use. Quantification of LF was performed using matrix assisted laser desorption/ionization (MALDI) time of flight (TOF) mass spectrometry (MS) to detect synthetic peptide cleavage products generated by LF with a limit of detection (LOD) of 0.005 ng ml−1 as previously described (Boyer et al., 2007). LF levels in serum were reported in ng ml−1.
Opsonization of B. anthracis
Blood was collected from mice by cardiac puncture and allowed to clot at room temperature for 10 min. Clotted blood was centrifuged at 1800 g for 15 min at room temperature and serum was collected. Ninety-five microlitres of B. anthracis from aliquots stored at −80°C which was diluted 1:5 in fresh LB broth and shaken for 1 h at 37°C, was then added to 500 μl of serum and allowed to incubate 37°C shaking for 30 min.
Isolation of PMNs
Mice were euthanized by cervical dislocation while under anaesthesia and femurs were removed. Bone marrow was removed from femur using a 1 ml syringe and a 26-gauge half inch needle with 500 μl of 1× Hanks balanced salt solution (HBSS). Once collected 1 ml of bone marrow suspension was carefully layered over 1.5 ml of Lympholyte Mammal (Cedar lane laboratories, Ontario, Canada) and centrifuged for 20 min at 800 g at room temperature. After centrifugation the pellet consisting of red blood cells (RBCs) and PMNs were resuspended in 4.5 ml of dH2O on ice, and after 30 s 0.5 ml of 10× HBSS was added. PMNs were centrifuged at 4°C for 5 min at 500 g, supernatant was discarded and PMNs were resuspended in 1 ml of 1× HBSS and counted by haemocytometer. Purity and viability of PMNs was established using flow cytometry.
Assessment of PMNs killing of vegetative bacteria
Heat-treated mouse serum was made by collecting blood from C57Bl/6 mice by cardiac puncture and allowing blood to clot at room temperature for 10 min. After clotting blood was centrifuged at 1800 g for 15 min and serum was collected. Serum was then heated at 55°C for 20 min before being frozen at −20°C for later use. Purified PMNs and opsonized bacteria were mixed at an moi of 1:1 in a master mix of 1× HBSS and 10% heat-inactivated mouse serum. Two hundred microlitres of the master mix were pipetted in triplicate for each time point into U-bottomed 96-well tissue culture plates (Corning, Corning, New York). Plates were centrifuged at 450 g for 10 min at 4°C, and then placed at 37°C + 10% CO2 for the times indicated in the figures. At designated time points PMNs were lysed by addition of 0.5% Saponin and vigorous pipetting with subsequent incubation on ice for 10 min. After lysis samples were diluted appropriately onto LB agar plates and incubated at 37°C overnight to enumerate cfu.
Western blotting for MEK3 cleavage
PMNs were harvested from mice with and without pre-treatment with LT. As a positive control 1 × 106 PMNs were treated with 100 ng of LT (100 ng LF, 200 ng PA) for 1 h at 37°C with 10% CO2. For each condition 1 × 106 PMNs were lysed using SDS running buffer with β-mercaptoethanol and heated to 95°C for 4 min. Samples were loaded onto a SDS 10% polyacrylamide gel along with 10 μl of a pre-stained protein ladder (Bio-Rad, Hercules, CA). The gel was run at 161 volts and transferred to a PVDF transfer membrane (Biotechnology systems, Boston, MA) at 100 volts for 1 h. Membrane was first treated with rabbit anti-mouse MEK3 (C-19, Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:500 dilution in 1% milk (0.4 μg ml−1), following with goat anti-rabbit alkaline phosphatase conjugate (Millipore, Billerica, MA) at 1:1000 (1 μg ml−1). Blot was visualized using SigmaFast BCIP/NBT tablets (Sigma-Aldrich, St. Louis, MO).
PMN accumulation monitoring and luminescent imaging
Mice were anaesthetized using 3% isoflurane mixed with oxygen from the XGI-8 gas anaesthesia system supplied with a Xenogen IVIS Spectrum. To induce inflammation 5 μl of a 0.5 μg μl−1 phorbol 12-myristate 13-acetate (PMA) suspended in dimethyl sulfoxide (DMSO) was pipetted onto the left ear of mice. At indicated time points 150 μl of luciferin at a concentration of 30 mg ml−1 suspended in DPBS (Perkin Elmer, Waltham, MA) was injected i.p. into mice and 8–10 min were allowed to elapse for diffusion throughout the mouse. Images were acquired as previously reported and analysed using Living Image software (version 2.50.1, Xenogen) (Glomski et al., 2007b).
Flow cytometry staining
Single cell suspensions of bone marrow isolated PMNs were resuspended in 100 μl flow cytometry staining buffer (1× PBS 3% FBS 0.05% sodium azide). Fc receptor-blocking antibody (CD16/32, eBioscience, San Diego, CA) was added and incubated on ice for 30 min. Samples were centrifuged and resuspended in 100 μl flow staining buffer. Antibodies against specific cell surface markers were added, incubated for 1 h on ice, and then washed. Samples were then fixed overnight with 4% paraformaldehyde at 4°C. Antibodies used were: anti-Ly6C FITC (RB6–8C5, BD Biosciences), anti-CD86 PE (PO3.1, eBioscience, San Diego, CA), anti-CD11b APC (M1/70, eBioscience), anti-MHC II PacBlue (M5/114.15.2, eBiosciences), anti-CD80 PE Cy5 (16–10A1, eBioscience), anti-CD69 PE Cy7 (H1.2F3, eBioscience) and anti-CD45 Alexa780 (30-F11, eBioscience). Live/Dead staining was performed with fixable Aqua Dead Cell Stain kit (Invitrogen).
Statistical analysis
All statistical analysis and was performed using GraphPad Prism software (version 5, GraphPad Software, San Diego, CA). Unless otherwise noted all statistical values reported were determined using a two-way ANOVA and graphed to display mean with the standard error of the mean.
Supplementary Material
Fig. S1. Circulating LT does not decrease viability of bone marrow PMNs. C57Bl/6 mice were injected with 6.25 μg of LT and PMNs were isolated from the bone marrow of intoxicated and non-intoxicated mice 24 h later. Cells were then stained for flow cytometry. PMNs were gated for by Ly-6G and side scatter, the bar graph represents % viability of cells as determined by fixable Aqua Dead Cell Stain in that subset. n = 3 mice per condition.
Fig. S2. Three defined stages of B. anthracis infection. (A–C) Black and white photos of a single A/J mouse injected subcutaneously in the left ear with 1 × 105 luminescent B. anthracis Sterne strain (BIG23) spores, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1 at different stages of infection.
A. Early stage of infection defined by vegetative bacilli, represented by luminescence, being confined to the initial site of spore inoculation.
B. Mid-stage of infection defined by vegetative bacilli spread to the regional draining lymph node.
C. Late-stage infection defined by bacterial spread to the kidneys.
Fig. S3. Ability to produce LT makes B. anthracis more virulent. A/J mice were infected with 1 × 106 cfu of vegetative LF or EF knockout BIG23. Once light was detected in the kidneys, representing dissemination, mice were euthanized according to ACUC protocol. There was a significant defect in ability to disseminate in LF knockout. B. anthracis significance determined by log rank test (**P < 0.01). n = 4 mice for each experimental condition.
Fig. S4. A/J PMNs kill vegetative TKO similarly to C57Bl/6 PMNs. (A–C) PMNs isolated from A/J and C57Bl/6 mice were inoculated with vegetative B. anthracis at an moi of 1:1 and incubated at 37°C. At indicated time points samples were put on ice and mixed with 0.1% Saponin for 10 min. Samples were then serially diluted and plated for enumeration on LB agar.
A. Comparison between PMN ability to kill vegetative TKO between C57Bl/6 and A/J PMNs. No statistical difference was seen at any time.
B. Effects of LT on ability of A/J PMN to kill vegetative TKO. A/J mice were i.p. injected with 6.25 μg of LT and PMNs were isolated 24 h later. Significant differences were seen between LT intoxicated and non-intoxicated PMNs at 0.5 h (*P < 0.05), 1 h (**P < 0.01) and 1.5 h (**P < 0.01).
C. A/J and C57Bl/6 PMN ability to kill vegetative TKO was determined after i.p. injection of 6.25 μg LT 24 h prior to PMN isolation. No significant differences in killing ability were seen at any time point. n = 3 mice for each experimental group.
Fig. S5. PMN accumulation at sites of inflammation can be measured in real time.
Aand B. NECre Luc mouse with 2.5 μg PMAin 5 μl DMSO pipetted onto the ear had net luminescence measured over the course of 48 h in both the non-inflamed right ear (A) and the inflamed left ear(B), where a significant increase was found from time zero to 48 h (*P = 0.0213) where n = 6 mice per condition.
C. Luminescence was measured in both ears of a NECre Luc mouse over the course of 48 h. Results are representative of three separate mice.
Acknowledgements
We would like to express our thanks to Alice Kenney and the staff in the University of Virginia mouse vivaria for their highly professional husbandry of our mice. Additionally, we must recognize the outstanding training and aid that was provided by Joanne Lannigan and her staff at the University of Virginia Flow Cytometry Core. We would like to thank Dr Borna Mehrad and Marie Burdick for allowing use of lab space and materials to perform the in vitro PMN migration assay. Recognition is also due to Amanda House and Kelly Smucker for providing consistent laboratory support that made these studies possible. This manuscript also benefitted immensely from the editorial critiques and aid of Dr Erik Hewlett, Dr Heath Damron, David E. Lowe and Amanda L. House.
The findings and conclusions in this publication are those of the author(s) and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Footnotes
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
References
- Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, and Pulendran B (2003) Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 424: 329–334. [DOI] [PubMed] [Google Scholar]
- Barth H, Aktories K, Popoff MR, and Stiles BG (2004) Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev 68: 373–402, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borregaard N, and Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89: 3503–3521. [PubMed] [Google Scholar]
- Boyer AE, Quinn CP, Woolfitt AR, Pirkle JL, McWilliams LG, Stamey KL, et al. (2007) Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal Chem 79: 8463–8470. [DOI] [PubMed] [Google Scholar]
- Brachman PS (1980) Inhalation anthrax. Ann N Y Acad Sci 353: 83–93. [DOI] [PubMed] [Google Scholar]
- Brookmeyer R, and Blades N (2002) Prevention of inhalational anthrax in the U.S. outbreak. Science 295: 1861. [DOI] [PubMed] [Google Scholar]
- Cote CK, Rea KM, Norris SL, van Rooijen N, and Welkos SL (2004) The use of a model of in vivo macrophage depletion to study the role of macrophages during infection with Bacillus anthracis spores. Microb Pathog 37: 169–175. [DOI] [PubMed] [Google Scholar]
- Cote CK, Van Rooijen N, and Welkos SL (2006) Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect Immun 74: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MA, Aylott CV, Bourdeau RW, and Bokoch GM (2006) Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J Immunol 176: 7557–7565. [DOI] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280: 734–737. [DOI] [PubMed] [Google Scholar]
- During RL, Li W, Hao B, Koenig JM, Stephens DS, Quinn CP, and Southwick FS (2005) Anthrax lethal toxin paralyzes neutrophil actin-based motility. J Infect Dis 192: 837–845. [DOI] [PubMed] [Google Scholar]
- Eisenhauer PB, and Lehrer RI (1992) Mouse neutrophils lack defensins. Infect Immun 60: 3446–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski IJ, Fritz JH, Keppler SJ, Balloy V, Chignard M, Mock M, and Goossens PL (2007a) Murine splenocytes produce inflammatory cytokines in a MyD88-dependent response to Bacillus anthracis spores. Cell Microbiol 9: 502–513. [DOI] [PubMed] [Google Scholar]
- Glomski IJ, Piris-Gimenez A, Huerre M, Mock M, and Goossens PL (2007b) Primary involvement of pharynx and Peyer’s patch in inhalational and intestinal anthrax. PLoS Pathog 3: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens PL (2009) Animal models of human anthrax: the Quest for the Holy Grail. Mol Aspects Med 30: 467–480. [DOI] [PubMed] [Google Scholar]
- Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, et al. (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423: 87–91. [DOI] [PubMed] [Google Scholar]
- Janes BK, and Stibitz S (2006) Routine markerless gene replacement in Bacillus anthracis. Infect Immun 74: 1949–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, et al. (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol 392: 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer AF, Tang II, Schawel AK, Brown MJ, and Krantz BA (2012) Anthrax toxin protective antigen integrates poly-gamma-D-glutamate and pH signals to sense the optimal environment for channel formation. Proc Natl Acad Sci USA 109: 18378–18383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla SH (1982) Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA 79: 3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko A, and Iglesias PA (2002) Models of eukaryotic gradient sensing: application to chemotaxis of amoebae and neutrophils. Biophys J 82: 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Nguyen CM, Wang SJ, Saadi W, Gross SP, and Jeon NL (2004) Effective neutrophil chemotaxis is strongly influenced by mean IL-8 concentration. Biochem Biophys Res Commun 319: 576–581. [DOI] [PubMed] [Google Scholar]
- Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, et al. (2008) Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem 283: 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Miller-Randolph S, Crown D, Moayeri M, Sastalla I, Okugawa S, and Leppla SH (2010) Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe 8: 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovchik JA, Drysdale M, Koehler TM, Hutt JA, and Lyons CR (2012) Expression of either lethal toxin or edema toxin by Bacillus anthracis is sufficient for virulence in a rabbit model of inhalational anthrax. Infect Immun 80: 2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving CL, Khurana T, Osorio M, Lee GM, Kelly VK, Stibitz S, and Merkel TJ (2009) Role of anthrax toxins in dissemination, disease progression, and induction of protective adaptive immunity in the mouse aerosol challenge model. Infect Immun 77: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DE, and Glomski IJ (2012) Cellular and physiological effects of anthrax exotoxin and its relevance to disease. Front Cell Infect Microbiol 2: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S, Uchida I, Terakado N, Sasakawa C, and Yoshikawa M (1989) Molecular characterization and protein analysis of the cap region, which is essential for encapsulation in Bacillus anthracis. J Bacteriol 171: 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S, Watarai M, Cheun HI, Shirahata T, and Uchida I (2002) Effect of the lower molecular capsule released from the cell surface of Bacillus anthracis on the pathogenesis of anthrax. J Infect Dis 186: 227–233. [DOI] [PubMed] [Google Scholar]
- Mauer AM, Athens JW, Ashenbrucker H, Cartwright GE, and Wintrobe MM (1960) Leukokinetic studies. Ii. A method for labeling granulocytes in vitro with radioactive Diisopropylfluorophosphate (Dfp). J Clin Invest 39: 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Scholl A, Hurwitz R, Brinkmann V, Schmid M, Jungblut P, Weinrauch Y, and Zychlinsky A (2005) Human neutrophils kill Bacillus anthracis. PLoS Pathog 1: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M, Guillemin J, Hugh-Jones M, Langmuir A, Popova I, Shelokov A, and Yampolskaya O (1994) The Sverdlovsk anthrax outbreak of 1979. Science 266: 1202–1208. [DOI] [PubMed] [Google Scholar]
- Moayeri M, and Leppla SH (2009) Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med 30: 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri M, Haines D, Young HA, and Leppla SH (2003) Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest 112: 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri M, Wiggins JF, and Leppla SH (2007) Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun 75: 5175–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, et al. (2010) Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog 6: e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock M, and Fouet A (2001) Anthrax. Annu Rev Microbiol 55: 647–671. [DOI] [PubMed] [Google Scholar]
- Mukherjee DV, Tonry JH, Kim KS, Ramarao N, Popova TG, Bailey C, et al. (2011) Bacillus anthracis protease InhA increases blood-brain barrier permeability and contributes to cerebral hemorrhages. PLoS ONE 6: e17921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J, Friedlander A, Dreier T, Ezzell J, and Leppla S (1985) Effects of anthrax toxin components on human neutrophils. Infect Immun 47: 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2012) MRSA virulence and spread. Cell Microbiol 14: 1513–1521. doi: 10.1111/j.1462-5822.2012.01832.x. Epub 17 July 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccani SR, and Baldari CT (2011) T cell targeting by anthrax toxins: two faces of the same coin. Toxins (Basel) 3: 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preshaw PM, Schifferle RE, and Walters JD (1999) Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodontal Res 34: 197–202. [DOI] [PubMed] [Google Scholar]
- Rossi Paccani S, Tonello F, Patrussi L, Capitani N, Simonato M, Montecucco C, and Baldari CT (2007) Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell Microbiol 9: 924–929. [DOI] [PubMed] [Google Scholar]
- Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, and Kaelin WG Jr (2003) Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol Imaging 2: 297–302. [DOI] [PubMed] [Google Scholar]
- Schneerson R, Kubler-Kielb J, Liu TY, Dai ZD, Leppla SH, Yergey A, et al. (2003) Poly(gamma-D-glutamic acid) protein conjugates induce IgG antibodies in mice to the capsule of Bacillus anthracis: a potential addition to the anthrax vaccine. Proc Natl Acad Sci USA 100: 8945–8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley JL, and Smith H (1961) Purification of factor I and recognition of a third factor of anthrax toxin. J Gen Microbiol 282: 49–66. [DOI] [PubMed] [Google Scholar]
- Stevens DL, Aldape MJ, and Bryant AE (2012) Life-threatening clostridial infections. Anaerobe 18: 254–259. doi: 10.1016/j.anaerobe.2011.11.001. Epub 20 November 2011. [DOI] [PubMed] [Google Scholar]
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, and Chilvers ER (2010) Neutrophil kinetics in health and disease. Trends Immunol 31: 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarowicz SE, During RL, Li W, Quinn CP, Tang WJ, and Southwick FS (2009) Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect Immun 77: 2455–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, and Roes J (2000) Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 12: 201–210. [DOI] [PubMed] [Google Scholar]
- Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, and Montecucco C (1998) Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun 248: 706–711. [DOI] [PubMed] [Google Scholar]
- Wade BH, Wright GG, Hewlett EL, Leppla SH, and Mandell GL (1985) Anthrax toxin components stimulate chemotaxis of human polymorphonuclear neutrophils. Proc Soc Exp Biol Med 179: 159–162. [DOI] [PubMed] [Google Scholar]
- Walsh JJ, Pesik N, Quinn CP, Urdaneta V, Dykewicz CA, Boyer AE, et al. (2007) A case of naturally acquired inhalation anthrax: clinical care and analyses of anti-protective antigen immunoglobulin G and lethal factor. Clin Infect Dis 44: 968–971. [DOI] [PubMed] [Google Scholar]
- Weiner ZP, Boyer AE, Gallegos-Candela M, Cardani AN, Barr JR, and Glomski IJ (2012) Debridement increases survival in a mouse model of subcutaneous anthrax. PLoS ONE 7: e30201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welkos SL, Trotter RW, Becker DM, and Nelson GO (1989) Resistance to the Sterne strain of B. anthracis: phagocytic cell responses of resistant and susceptible mice. Microb Pathog 7: 15–35. [DOI] [PubMed] [Google Scholar]
- Xu L, Fang H, and Frucht DM (2008) Anthrax lethal toxin increases superoxide production in murine neutrophils via differential effects on MAPK signaling pathways. J Immunol 180: 4139–4147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Circulating LT does not decrease viability of bone marrow PMNs. C57Bl/6 mice were injected with 6.25 μg of LT and PMNs were isolated from the bone marrow of intoxicated and non-intoxicated mice 24 h later. Cells were then stained for flow cytometry. PMNs were gated for by Ly-6G and side scatter, the bar graph represents % viability of cells as determined by fixable Aqua Dead Cell Stain in that subset. n = 3 mice per condition.
Fig. S2. Three defined stages of B. anthracis infection. (A–C) Black and white photos of a single A/J mouse injected subcutaneously in the left ear with 1 × 105 luminescent B. anthracis Sterne strain (BIG23) spores, overlaid with a false colour representation of photon emission intensity as indicated by the scale on the right in p s−1 cm−2 sr−1 at different stages of infection.
A. Early stage of infection defined by vegetative bacilli, represented by luminescence, being confined to the initial site of spore inoculation.
B. Mid-stage of infection defined by vegetative bacilli spread to the regional draining lymph node.
C. Late-stage infection defined by bacterial spread to the kidneys.
Fig. S3. Ability to produce LT makes B. anthracis more virulent. A/J mice were infected with 1 × 106 cfu of vegetative LF or EF knockout BIG23. Once light was detected in the kidneys, representing dissemination, mice were euthanized according to ACUC protocol. There was a significant defect in ability to disseminate in LF knockout. B. anthracis significance determined by log rank test (**P < 0.01). n = 4 mice for each experimental condition.
Fig. S4. A/J PMNs kill vegetative TKO similarly to C57Bl/6 PMNs. (A–C) PMNs isolated from A/J and C57Bl/6 mice were inoculated with vegetative B. anthracis at an moi of 1:1 and incubated at 37°C. At indicated time points samples were put on ice and mixed with 0.1% Saponin for 10 min. Samples were then serially diluted and plated for enumeration on LB agar.
A. Comparison between PMN ability to kill vegetative TKO between C57Bl/6 and A/J PMNs. No statistical difference was seen at any time.
B. Effects of LT on ability of A/J PMN to kill vegetative TKO. A/J mice were i.p. injected with 6.25 μg of LT and PMNs were isolated 24 h later. Significant differences were seen between LT intoxicated and non-intoxicated PMNs at 0.5 h (*P < 0.05), 1 h (**P < 0.01) and 1.5 h (**P < 0.01).
C. A/J and C57Bl/6 PMN ability to kill vegetative TKO was determined after i.p. injection of 6.25 μg LT 24 h prior to PMN isolation. No significant differences in killing ability were seen at any time point. n = 3 mice for each experimental group.
Fig. S5. PMN accumulation at sites of inflammation can be measured in real time.
Aand B. NECre Luc mouse with 2.5 μg PMAin 5 μl DMSO pipetted onto the ear had net luminescence measured over the course of 48 h in both the non-inflamed right ear (A) and the inflamed left ear(B), where a significant increase was found from time zero to 48 h (*P = 0.0213) where n = 6 mice per condition.
C. Luminescence was measured in both ears of a NECre Luc mouse over the course of 48 h. Results are representative of three separate mice.