

Abstract

The viral main protease is one of the most attractive targets among all key enzymes involved in the SARS-CoV-2 life cycle. Covalent inhibition of the cysteine145 of SARS-CoV-2 MPRO with selective antiviral drugs will arrest the replication process of the virus without affecting human catalytic pathways. In this Perspective, we analyzed the in silico, in vitro, and in vivo data of the most representative examples of covalent SARS-CoV-2 MPRO inhibitors reported in the literature to date. In particular, the studied molecules were classified into eight different categories according to their reactive electrophilic warheads, highlighting the differences between their reversible/irreversible mechanism of inhibition. Furthermore, the analyses of the most recurrent pharmacophoric moieties and stereochemistry of chiral carbons were reported. The analyses of noncovalent and covalent in silico protocols, provided in this Perspective, would be useful for the scientific community to discover new and more efficient covalent SARS-CoV-2 MPRO inhibitors.
1. Introduction
The ongoing coronavirus disease-19 (COVID-19) global pandemic is etiologically caused by the highly infectious severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1,2 SARS-CoV-2 is a β-coronavirus able to infect animals and humans, causing respiratory diseases.3
In this outbreak scenario, in order to contain the infection, the scientific community suggests strong social restraint measures and active development of vaccines. More than 20 vaccines reached clinical trials, demonstrating their efficacy in the prevention of severe COVID-19 syndrome.4 Considering the rapid mutation rate of this virus family, the long-term efficacy and safety of them is currently debated; for this reason, a more specific pharmacological treatment is needed to complement vaccines both in the prophylaxis and in the treatment of COVID-19.5 Moreover, there is an urgent need to develop effective therapeutic strategies against SARS-CoV-2; in this light, drug repurposing has been an effective approach over the last 2 years, based in particular on the comparative overview between CoVs. nafamostat/camostat,6 umifenovir,7 chloroquine/hydroxychloroquine,8 lopinavir/ritonavir,9 and remdesivir,10 which are just a few examples of drugs approved for other therapeutic applications that could be used to treat COVID-19.
To date, with the goal of developing a selective antiviral drug against SARS-CoV-2, the research community worldwide evaluated several structural/nonstructural viral proteins as possible SARS-CoV-2 druggable targets. Among these, the main protease (MPRO) emerged as the most attractive target for drug design.11
1.1. SARS-CoV-2 MPRO: A Covalently Druggable Target
The main protease MPRO, also known as 3C-like protease (3CLPRO), is a key cysteinyl enzyme involved in the digestion of the polyproteins pp1a and pp1ab. It acts at 11 conserved sites with the subsequent release of 12 nonstructural proteins (Nsp, essential for the replication/transcription process), Figure 1a.
Figure 1.
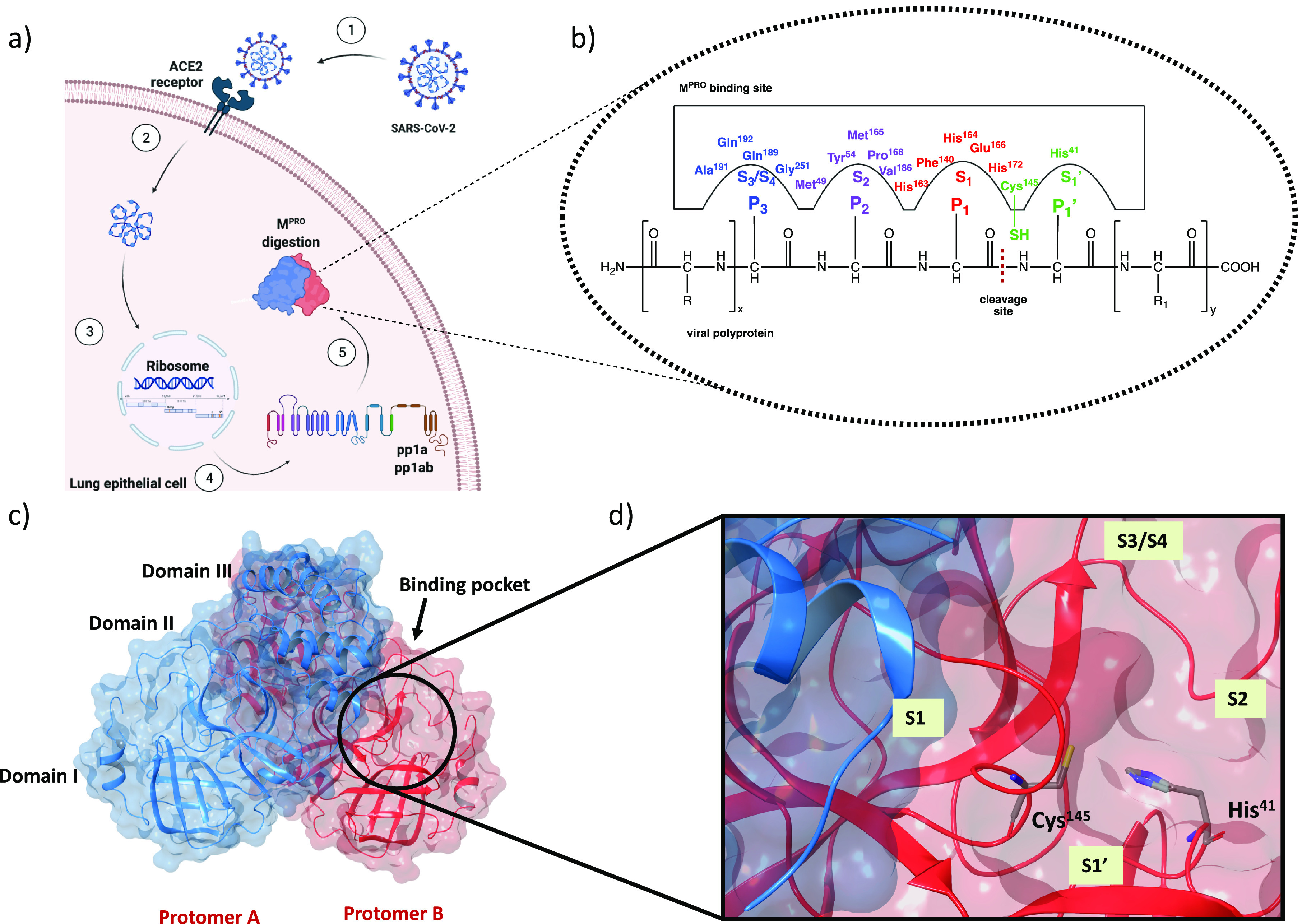
(a) Sequence of events necessary to activate the digestion process of MPRO: (1) virus entry into lung epithelial cell by interaction with ACE2 receptor, (2) release of the genomic material of SARS-CoV-2, (3 and 4) virus protein synthesis in human ribosomes, and (5) MPRO digestion. (b) SARS-CoV-2 MPRO binding site with viral polyproteins. (c) X-ray structure of dimeric SARS-CoV-2 MPRO (PDB code 6Y2F). (d) Focus on the catalytic site, with the four regions S1, S1′, S2, and S3/S4 highlighted.14
In the viral polyproteins, the amino acids from the N-terminus to the C-terminus are indicated as −P3–P2–P1↓P1′–, where P1 is generally a Gln and the arrow ↓ corresponds to the amide bond cleavage site between P1 and a small P1′ amino acid, such as serine, glycine, or alanine residues. MPRO recognizes the glutamine amino acid residues and cleaves the peptide bond between Gln and the adjacent amino acid, Figure 1b. The inhibition of its enzymatic activity could drive blockage of viral replication.12
Structurally, MPRO is a homodimer, and each protomer consists of 306 amino acids divided into 3 domains (I, II, and III): domain I (8–101) comprises 6 β-strands and an α-helix, while domain II (102–184) and domain III (201–303) include 6 β-strands and 5 α-helices, respectively. All domains are connected by long loop regions (Figure 1c).13 The binding pocket is organized into four subsites, S1′, S1, S2, and S3/S4, which are occupied by the portions P1′, P1, P2, and P3 of the viral polyproteins, respectively (Figure 1d).
In region S1′, between domains I and II, the catalytic site is located, characterized by the catalytic dyad (Cys145 and His41). During hydrolysis of the peptide bond, His41 activates the nucleophilic −SH of Cys145 by deprotonation with subsequent stabilization of the adduct by the so-called “oxyanion hole”, formed by the Gly143 and Cys145 backbones. The S1 region, characterized by the side chains of Phe140, His163, His164, Glu166, and His172, is highly specific for the glutamine residue.15 Region S2 consists of hydrophobic amino acids, such as Met49, Tyr54, Met165, Pro168, Val186, and finally, regions S3/S4, which are particularly exposed to the solvent, involve Gln189, Ala191, Gln192, and Gly251 residues. The peculiar structure of the binding pocket suggests SARS-CoV-2 MPRO as a druggable target for small molecules.5,12 In this scenario, many efforts have been devoted to identifying new selective peptidomimetic inhibitors of SARS-CoV-2 MPRO.5
An interesting approach has been provided by the covalent inhibition of the SARS-CoV-2 MPRO cysteinyl protease:16−18 the catalytic sulfur of Cys145 is covalently trapped by an electrophilic moiety P1′, a reactive warhead that mimics the amide peptide of the viral polyproteins. To be successful in the covalent inhibition, the inhibitors must meet basic structural requirements: a P1 portion, usually a cyclic glutamine analogue, capable of interacting with amino acids such as His163, Glu166, and His172 via hydrogen bonds and hydrophobic interactions; P2 and P3 moieties, projected into hydrophobic pockets (S2 and S3/S4) and characterized by substituent groups capable of favorable hydrophobic interactions with the amino acids of these sites (Figure 2a).
Figure 2.

(a) Covalent inhibition of SARS-CoV-2 MPRO. P1′, P1, P2, and P3 labels reflect the chemical analogies with the viral substrate. Warheads P1′ are in green, while fragments P1, P2, and P3 are shown in red, purple, and blue, respectively. Subregions of the binding pocket are labeled with S numbering complementary to the fragments of the inhibitor. (b) Kinetic scheme of covalent inhibition. E, I, EI, and E–I stand for enzyme, inhibitor, noncovalent enzyme–inhibitor complex, and covalent enzyme–inhibitor complex, respectively.
In this perspective, the kinetic scheme of covalent inhibition deserves special attention. As shown in Figure 2b, the process of enzymatic inhibition involves two steps: (1) the inhibitor (I) reversibly binds to the active site of the enzyme (E) and forms a noncovalent enzyme–inhibitor complex (EI); (2) due to the nucleophilic attack of the catalytic Cys145, the noncovalent complex evolves into a more stable complex (E–I), which generally irreversibly inactivates the catalytic activity. From a kinetic point of view, the first phase is correlated with the equilibrium-binding constant Ki (Ki = k2/k1), while the second stage is influenced by the inactivation constant for covalent bond formation k3 (Kinact).19
The covalent inhibition strategy has already been pursued against other proteases, including MERS and SARS-CoV-1, with remarkable results.20 Indeed, compared to traditional noncovalent agents, the electrophilic warhead inhibitors provide better efficacy, higher potency, longer residence time in the binding site of the receptor, sustained pharmacological action, and the ability to overcome resistance.16−18
In the last 2 years, the whole scientific community has invested a lot of time and resources to design new covalent SARS-CoV-2 MPRO inhibitors, and in this context, some small molecules have already reached the most advanced phases of drug development. PF-07321332, a nitrile compound developed by Pfizer as a covalent SARS-CoV-2 MPRO inhibitor, entered clinical trials in combination with ritonavir showing promising results in phase 2/3 (PAXLOVID) and was recently approved by the Committee for Medicinal Products for Human use of the EMA for the treatment of COVID-19.21−23
Considering the increasing interest of medicinal chemistry researchers in the design of new more powerful and selective covalent SARS-CoV-2 MPRO inhibitors, the main purpose of this Perspective is to provide an overview of the most representative examples reported to date in the literature in this field. In detail, the collected inhibitors were classified into different categories according to their reactive electrophilic group, underling the differences between their mechanism of inhibition.
Furthermore, with the support of the numerous available X-ray structures of these compounds in complex with the SARS-CoV-2 MPRO protease, an analysis of the most recurrent pharmacophoric portions is reported to evaluate the most suitable fragments for interaction with the different subsites of the catalytic cleft of the main protease. Biological data, such as inhibition of MPRO activity, antiviral effect in vitro, and in vivo results, are examined. This in-depth analysis aims to support the design of promising small molecules and the discovery of new, more selective SARS-CoV-2 MPRO covalent inhibitors.
2. Electrophilic Warheads in Covalent SARS-CoV-2 MPRO Inhibition
In this work, a detailed analysis of the most interesting covalent SARS-CoV-2 MPRO inhibitors is reported. According to the nature of the electrophilic warheads, the small molecules were divided into eight groups: aldehydes, ketones, α-ketoamides, Michael acceptors, α-haloacetamides, nitriles, esters, and molecules containing an electrophilic selenium/sulfur atom (Figure 3). Most of the analyzed compounds exhibited a dipeptidomimetic or tripeptidomimetic structure (such as carbonyl, α-ketoamide, Michael acceptor, α-haloacetamide, and nitrile derivatives), although some examples of nonpeptidomimetic SARS-CoV-2 MPRO inhibitors were also reported (such as activated ester derivatives, natural compounds, or ebsulfur/ebselen derivatives). The differences between their mechanisms of action and the capability to form reversible/irreversible adducts were examined. To support the discussion and analysis of the binding mechanism, the X-ray structures of the most representative ligand–protein complexes were described in detail.
Figure 3.
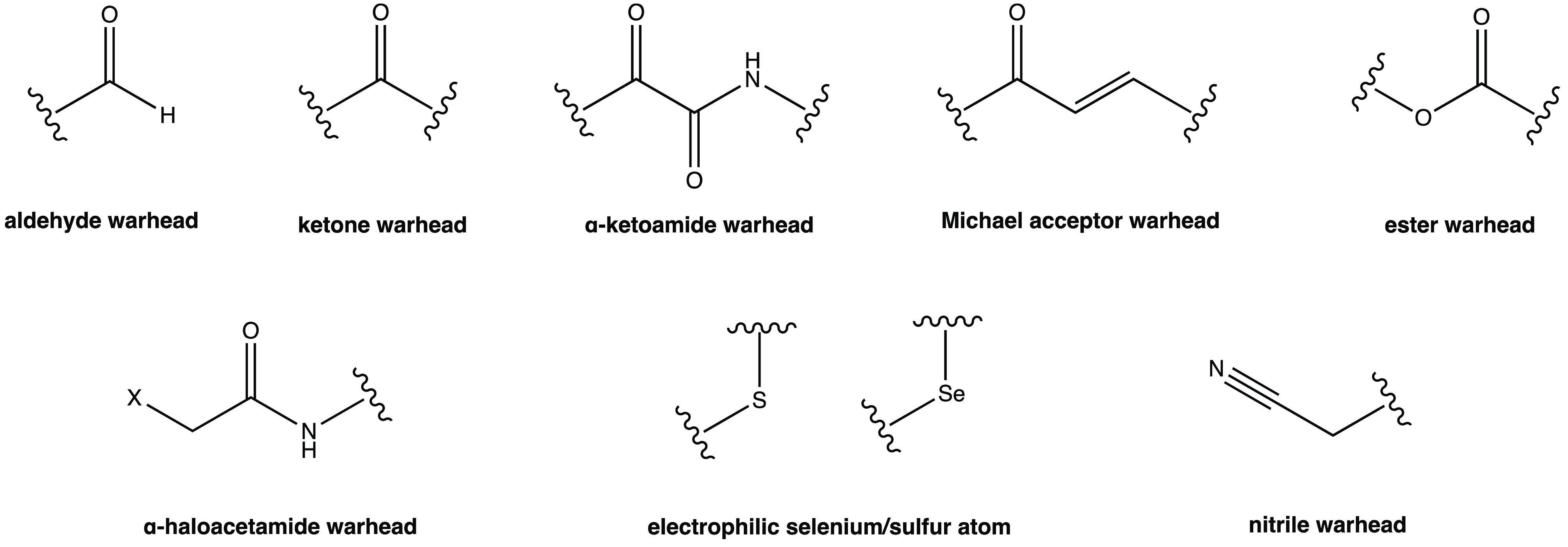
Electrophilic warheads characterizing covalent SARS-CoV-2 MPRO inhibitors.
2.1. Carbonyl Warhead
Carbonyl groups, such as aldehydes and ketones, are considered the most promising warheads in the design of new covalent inhibitors of SARS-CoV-2 Mpro. From a mechanistic point of view, their capability to form a covalent bond depends on the electrophilicity of the carbonyl carbon, which is susceptible to the nucleophilic addition of the cysteine-SH, leading to the formation of a reversible hemithioacetal adduct. The high similarity between the latter and the intermediate formed by the natural substrate during the enzymatic catalytic cycle ensures high stability of the inhibitor–protein complex and a longer residence time.24Figure 4 shows the general mechanism of covalent inhibition for this class of compounds.
Figure 4.

General mechanism of action of covalent carbonyl inhibitors.
In the panorama of covalent inhibitors with selective activity against SARS-CoV-2 MPRO, there are numerous examples of the carbonyl warhead, which is the most abundant in this class of small molecules.
2.1.1. Aldehyde Warhead
Among the covalent SARS-CoV-2 MPRO inhibitors with an aldehyde warhead, compounds 1 and 2 were rationally designed as two of the first peptidomimetic derivatives (Figure 5a). In vitro inhibition assays proved excellent inhibitory activity against SARS-CoV-2 MPRO (IC50 of 0.053 ± 0.005 and 0.040 ± 0.002 μM, respectively), strong anti-SARS-CoV-2 infectivity (EC50 of 0.53 ± 0.01 and 0.72 ± 0.09 μM, respectively), low cytotoxicity, and favorable pharmacokinetic and toxicity properties in vivo. Interestingly, the X-ray structures of both compounds in complex with MPRO, resolved at 1.5 Å, allowed a detailed analysis of the binding mode and mechanism of action of the proposed leads (Figure 5b and 5c shows SARS-CoV-2 MPRO in complex with 1 and 2, PDB codes 6LZE and 6M0K, respectively). To graphically illustrate the importance of each ligand moiety in stabilizing the complex, the four binding site cavities and the corresponding amino acids involved in the key interactions were highlighted. From the analysis, a similar binding mode of the two ligands was found: in both complexes, formation of the covalent bond between the aldehyde group (P1′) and the −SH of Cys145 at S1′ is enhanced by the additional H bond between the hemithioacetal group −OH and the cysteinyl backbone; the (S)-γ-lactam group (P1) is deeply inserted into the S1 site and, mimicking the glutamine residue of the natural substrates, can form three H bonds with three key amino acids of this cleft (one between His163 and the lactamic oxygen, two between Phe140 and Glu166 and the lactamic NH); the indole group (P3) is located at the surface (S3/S4 pocket) of the protein and interacts with Glu166. The only observed difference in the binding mode is represented by the P2 fragments, which differ between the two compounds (cyclohexyl and 3-fluorophenyl groups, respectively). Indeed, the 3-fluorophenyl moiety is more downrotated than the P2 in compound 1. However, despite this difference in orientation, both fragments are deeply inserted into the S2 cavity and form extensive hydrophobic interactions.25
Figure 5.
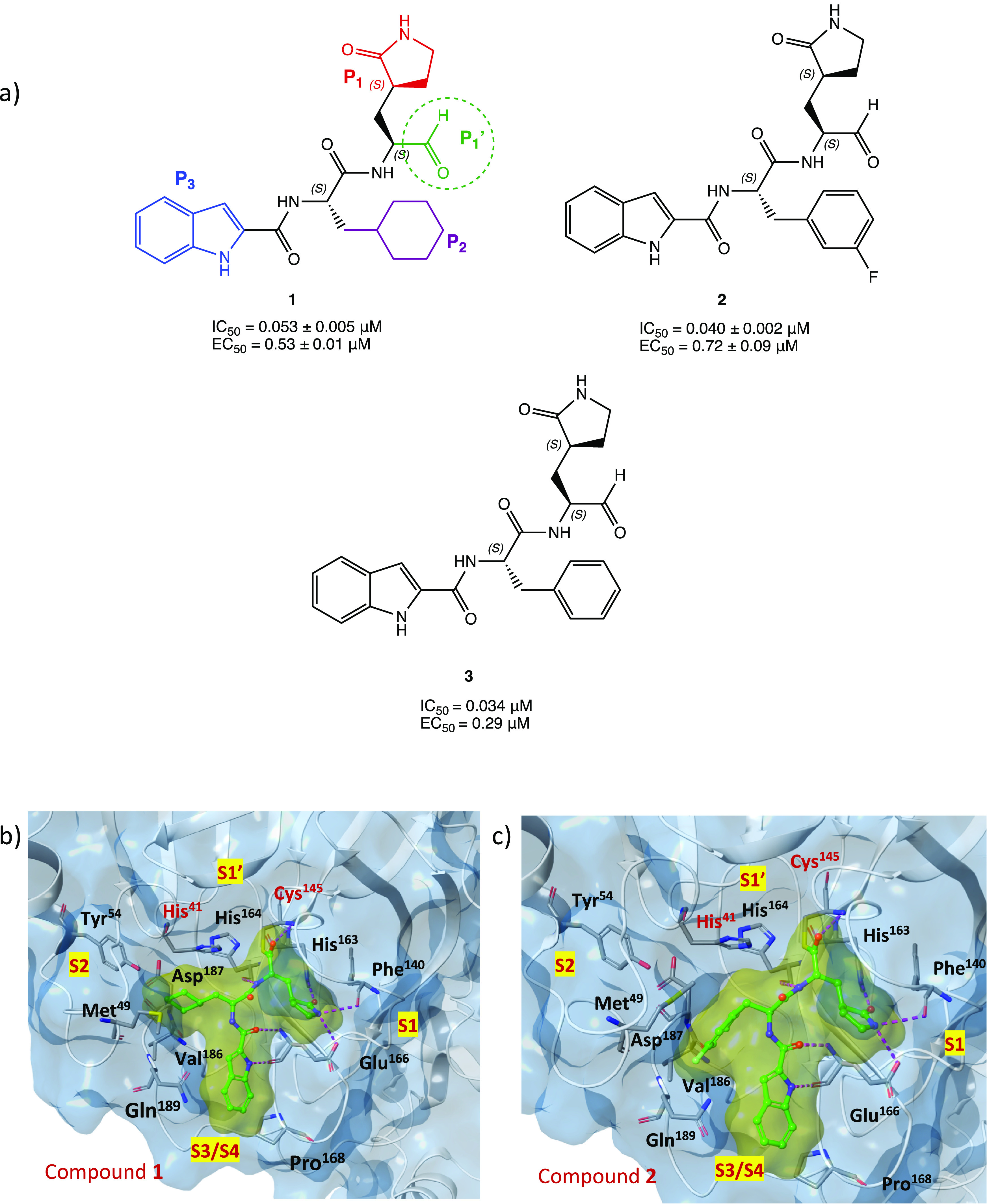
(a) Chemical structures of peptidomimetic aldehyde derivatives 1–3; for derivative 1, as an example for the whole series, the four key moieties P1′, P1, P2, and P3 for the interaction with SARS-CoV-2 MPRO are highlighted. (b) X-ray structure of SARS-CoV-2 MPRO in complex with compound 1 (PDB code 6LZE). (c) X-ray structure of SARS-CoV-2 MPRO in complex with compound 2 (PDB code 6M0K). In both crystal structures, the amino acids involved in the interaction with the ligand and the four cavities of the binding pocket (S1′, S1, S2, and S3/S4) are shown.25−27
Furthermore, in light of the interesting results, derivative 1 was chosen as a representative compound to gain insight into the mechanism of action of aldehyde inhibitors at the atomistic level using QM/MM simulations: this type of molecular simulation permitted us to elucidate the mechanism of covalent bond formation and to highlight the proton transfer processes that take place within the catalytic dyad prior to the nucleophilic attack.26
In the search of new inhibitors of rhinovirus and enterovirus proteases, the peptidomimetic derivative 3 was synthesized (Figure 5), as an analogue of 2, without the 3-F substitution on the phenyl ring. Tested against SARS-CoV-2 MPRO, compound 3 exhibited interesting inhibitory activity with an IC50 value of 0.034 μM and antiviral activity with EC50 = 0.29 μM. Structure–activity relationship (SAR) studies confirmed the importance of the γ-lactamic pentacyclic system and the indole ring: substitution with other heterocycles, such as quinoline, showed a drastic decrease in activity.27
Other examples of promising SARS-CoV-2 MPRO covalent inhibitors with an aldehyde warhead include GC-373 and the related prodrug GC-376 in which the aldehyde function is hidden as a bisulfite group to increase the solubility and that can be readily released under physiological conditions (Figure 6a).
Figure 6.
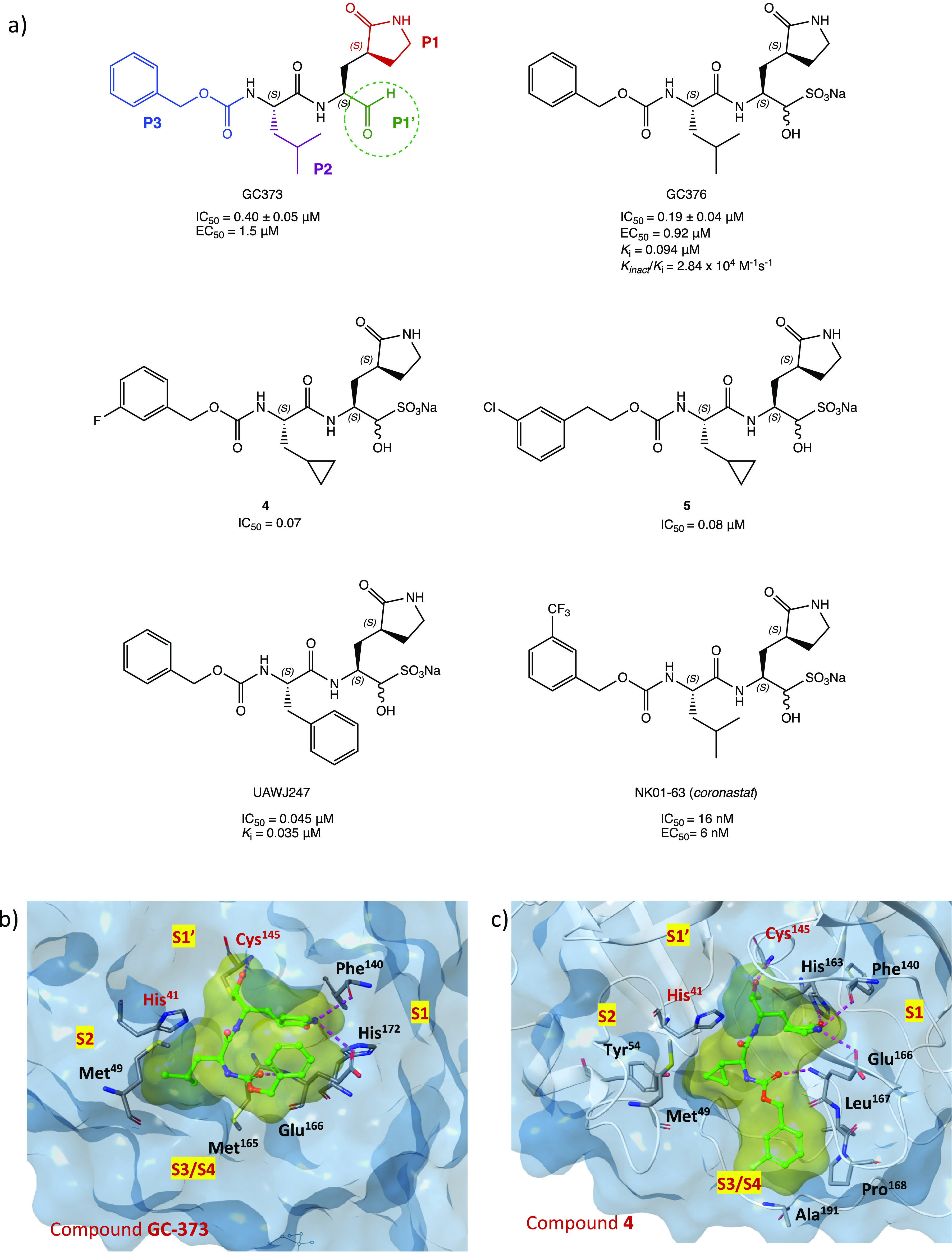
(a) Chemical structures of GC-373 (electrophilic P1′ warheads in green, P1 in red, P2 in purple, and P3 in blue), GC-376, analogues 4 and 5, UAWJ247, and NK01-63 (coronastat).28−30 (b) X-ray structure of SARS-CoV-2 MPRO in complex with GC-373 (PDB code 6WTJ(31)). (c) X-ray structure of SARS-CoV-2 MPRO in complex with 4 (PDB code 7LCO(29)).
These two compounds, initially investigated as veterinary drugs for their capability to inhibit feline coronavirus (FCoV) 3CL protease and proposed for the treatment of feline infectious peritonitis,28 were repurposed as new anti-COVID-19 agents, showing potent inhibitory activity on MPRO. Specifically, GC-373 exhibited an IC50 value against SARS-CoV-2 MPRO of 0.40 ± 0.05 μM, while the prodrug GC-376 produced slightly higher inhibition with an IC50 of 0.19 ± 0.04 μM and ki = 0.094 μM (Kinact/ki = 2.84 × 104 M–1 s–1).31,32 X-ray studies performed on both compounds in complex with SARS-CoV-2 MPRO showed an identical binding mode of the ligands (for GC-373 PDB code 6WTK; for GC-376 PDB code 6WTJ), confirming the ability of the bisulfite adduct of GC-376 to rapidly release the aldehydic function. To gain insight into the mechanism of inhibition and the binding mode of this class of compounds, the crystal structure of GC-376 in complex with the target protein is shown in Figure 6b, highlighting the key structural components responsible for the inhibition: the aldehydic warhead (P1′) forms a covalent hemithioacetal adduct with the cysteinyl −SH within the catalytic cleft in S1′; the leucine P2 inserts itself into the hydrophobic subregion S2 of the enzyme; the recurrent γ-lactame P1 of the substrate is located into the S1 site and forms multiple H bonds with polar amino acids (e.g., His163 and Glu166); the benzyl group in P3 is projected into the superficial S3/S4 site of the protein, where it forms an H bond with the Glu166 backbone.31
Further in vitro studies were performed on a model of SARS-CoV-2 infection in Vero E6 cells and confirmed the high inhibitory activity of both GC-373 and GC-376, which exhibited EC50 of 1.5 and 0.92 μM, respectively, and low cytotoxicity even at high concentration.31,32 The remarkable antiviral effect in the cellular assay (also measured in cells infected with delta and omicron SARS-CoV-2 variants) was associated with the dual effect of inhibition of the host cysteinyl protease cathepsin L, which is involved in the infection process of SARS-CoV-2 into the host cells along with other proteases such as calpains.33−35
Inspired by the in vitro results and the previous positive outcomes in animals affected by FCoV, GC-376 was examined in vivo in the K18 hACE2/SARS-CoV-2 transgenic mouse model, which expresses the human angiotensin-converting enzyme type 2 and is affected by SARS-CoV-2. The analysis of the data showed modest benefit in terms of clinical symptoms and survival but an interesting capability to reduce tissue lesions and inflammation in the tested animals.36
With the aim of improving the in vitro inhibition effect of GC-376 against MPRO, a series of analogues has been designed. From the viewpoint of SAR, the structural modifications have been directed mainly toward the P2 (leucine isopropyl) and the P3 (carboxy-benzyl) moieties; the P1 portion (γ-lactame) and the P1′ aldehyde warhead were retained in view of their pivotal role in the interaction with the target. The most representative compounds were the bisulfite prodrugs 4 and 5 (Figure 6a), which were modified at both P2 and P3 sites (cyclopropyl group and halogen-substituted phenyl, respectively). In vitro inhibition assay proved their better inhibitory activity against MPRO (IC50 of 0.07 and 0.08 μM, respectively) and their antiviral effects (EC50 of 0.57 and 0.7 μM, respectively) compared to both the lead compound GC-376 and the corresponding parent aldehydes. The interesting potency of compound 4 was confirmed by the analysis of the X-ray complex of MPRO with the desalted form of 4. As shown in Figure 6c (complex of derivative 4 with SARS-CoV-2 MPRO, PDB code 7LCO), introduction of the new structural features contributes to enhance the interaction with the target protein and achieve a better fit into the active site compared to GC-376. The more compact cyclopropyl group on the P2 unit of the ligand is able to penetrate deeper into the S2 pocket of the target; furthermore, introduction of a halogen substituent on the Cbz group (P3 fragment of the ligand) permits one to move this moiety closer to the S3/S4 cleft, while in GC-373 (Figure 6b), the unsubstituted Cbz is mainly directed toward the surface of the protease exposed to the solvent.29
To further explore the structural determinants of GC-376 for more selective inhibition of SARS-CoV-2 MPRO, UAWJ247 was developed (Figure 6a) by replacing the isopropyl moiety with a phenyl group. The in vitro inhibition assay registered an IC50 value of 0.045 μM, very close to that of the parent compound, and a ki value of 0.035 μM, confirming a better fitting of the bulkier aromatic portion into the S2 site of the MPRO binding pocket (crystal structure complex with PDB code 6XBH).30
Recently, a series of new GC-376 analogues with substantial modifications to the P2 and P3 fragments was synthesized. Among them, compound NK01-63 (coronastat) (Figure 6a) proved to be more active than the lead compound. It showed excellent inhibitory activity against SARS-CoV-2 (IC50 = 16 nM), potent antiviral effect in cell assay (EC50 = 6 nM in Huh-7ACE2 infected cells), and high selectivity against other human proteases. The crystal structure of coronastat in complex with the target protein allowed a better understanding of the drastic improvement in potency compared to the lead GC-376 (PDB code 7TIZ): the trifluoromethyl group on the benzyl moiety provides two additional H bonds with Asn142, confirming the importance of the halogen substitution in enhancing the interactions of the ligand–protein complex. These encouraging results prompted the researchers to perform further in vivo tests (mouse models) to evaluate both the activity and the pharmacokinetic profile: coronastat showed no significant toxicity, high metabolic stability, and high concentration in the plasma and lung after both oral and intraperitoneal administration.37
Maintaining the focus on the lead compound GC-376, analysis of the crystal structures of MPRO in complex with boceprevir and telaprevir, two drugs with an α-ketoamide group as a warhead (see section 2.2), allowed us to observe the importance of bicyclic proline, which can be suitably accommodated into the S2 pocket of the target protein. The merging of the pharmacophore moieties of GC-376, boceprevir, and telaprevir led to the synthesis of a new series of small molecules with an inhibition effect on SARS-CoV-2 MPRO (Figure 7a). The new derivatives were characterized by the aldehydic P1′ warhead, the γ-lactame as P1 (present in GC-376), (1R,2S,5S)-6,6-dimethyl-3-aza-bicyclo[3.1.0]hexane-2-formamide or (1S,3aR,6aS)-octahydrocyclopenta[c]-pyrrole-1-formamide as P2 (from boceprevir and telaprevir, respectively), and a hydrophobic aromatic group as P3. MI-09, MI-23, and MI-30 (Figure 7a), tested in the MPRO biochemical inhibition assay, proved to be the most interesting compounds in the series, exhibiting IC50 values in the nanomolar range (15.2, 7.6, and 17.2 nM, respectively). Using X-ray experiments, the most active inhibitor MI-23 was selected to further investigate the binding mode of this class of compounds with SARS-CoV-2 MPRO. Figure 7b shows the crystal structure of the complex MI-23/SARS-CoV-2 MPRO (PDB code 7D3I) with the relevant interactions highlighted: the aldehydic carbon interacts with the −SH of the catalytic Cys145 at the S1′ site, as expected; P1 is inserted in the S1 region and forms several hydrogen bonds, as observed for the previous compounds; the conformational constraints of the bicyclic proline structure (P2) result in a restricted trans-exo conformation of the system, which determines a deep insertion into the hydrophobic S2 site of the target; the 1-ethyl-3,5-difluorobenzene moiety at P3 occupies the S3/S4 subregion. Further biological studies were conducted on MI-09 and MI-30. Analysis of biological data showed no cytotoxicity against normal cells, remarkable antiviral activity in Vero E6-infected cells (EC50 of 0.86 and 0.54 μM, respectively), acceptable PK properties and capability to prevent SARS-CoV-2 infection in vivo (transgenic mice expressing hACE2), and ability to ameliorate lung lesion and inflammation.38 Moreover, starting from MI-09 and MI-30, a computational protocol based on QSAR and combined with molecular docking, MD simulations, and free binding energy MM/PBSA was built to design new derivatives with higher inhibition activities on SARS-CoV-2 MPRO. The virtual screening highlighted the importance of a bicyclic moiety with a three-membered ring, preferably at P2, and bulky electronegative substituents (ethyl, −CN, −F, and −Br) on the phenyl group at P3.39
Figure 7.

(a) Chemical structures of boceprevir, telaprevir, derivatives MI-09, MI-23, and MI-30, and analogues UAWJ9-36-1 and UAWJ9-36-3. (b) X-ray crystal structure of MI-23 in complex with SARS-CoV-2 MPRO (PDB code 7D3I).38,40
Similarly, retaining both the aldehyde warhead and the bicyclic fragments, two interesting compounds UAWJ9-36-1 and UAWJ9-36-3 (Figure 7a) were designed and synthesized.40
In vitro biological studies of UAWJ9-36 derivatives showed comparable inhibitory activity to GC-376 against both SARS-CoV-2 MPRO (with IC50 values of 0.051 and 0.054 μM and Kinact/ki = 8.5 × 104 and 9.3 × 104 M–1 s–1 for UAWJ9-36-1 and UAWJ9-36-3, respectively) and other known human coronaviruses MPRO. Antiviral cellular assays performed on Vero E6 and Caco2-ACE2 cell lines infected with SARS-CoV-2 demonstrated the enhanced antiviral activity of UAWJ9-36-3 with EC50 = 0.37 and 1.06 μM against the respective cell lines. Moreover, both UAWJ9-36 derivatives showed improved selectivity against host calpains/cathepsins compared to GC-376. X-ray crystal structures of the protein coresolved with both compounds (PDB codes 7LYH and 7LYI for the complexes with UAWJ9-36-1 and UAWJ9-36-3, respectively) confirmed the covalent bonding and the ability of the cyclopentyl-proline and dimethyl-cyclopropyl-proline moieties to insert in the S2 site.40
Among the SARS-CoV-2 MPRO tripeptidyl inhibitors with an aldehyde moiety as an electrophilic warhead, MPI3 and MPI8 (Figure 8) proved to be the most interesting compounds. These two derivatives feature the recurring 2-oxopyrrolidine side chain at the P1 site and a Cbz group as the N-terminal P3 cap and show remarkable IC50 values against SARS-CoV-2 MPRO (8.5 and 105 nM for MPI3 and MPI8, respectively). Despite the higher inhibition by MPI3, its activity in reducing viral infection in in vitro cell models was lower than that of MPI8, which, instead, showed complete inhibition of SARS-CoV-2 infection in Vero E6 and A549/ACE2 cell lines at concentrations of 2.5 and 0.31 μM, respectively. This discrepancy is probably due to the poor stability toward extra/intracellular proteases, which recognize natural amino acids (valine and leucine in the case of MPI3) as substrates.41 Further biological studies on MPI8 link the high potency of the tripeptidyl derivative to a dual mechanism of action: inhibition of both SARS-CoV-2 MPRO (IC50 = 105 nM) and cathepsin L (IC50 = 1.2 nM).42
Figure 8.
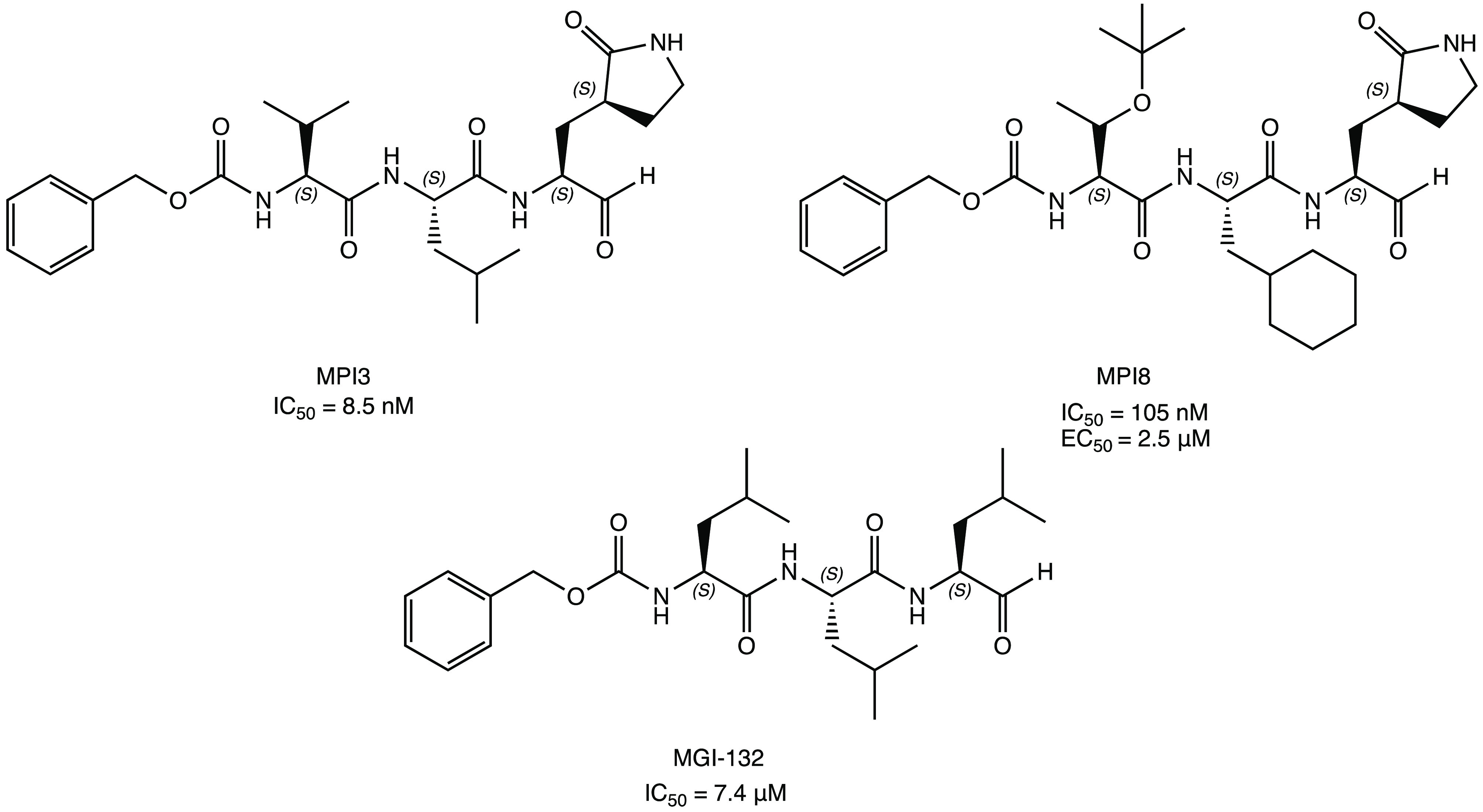
Structures of tripeptidyl inhibitors of SARS-CoV-2 MPRO: MPI3, MPI8, and MGI-132.41,43,44
Focusing on the interesting hypothesis of the dual-target inhibition of MPRO and cathepsin L, a well-known proteasome inhibitor, the trileucine peptide MGI-132 (Figure 8), was repurposed. This new compound, structurally similar to MPI3 and MPI8, showed an IC50 of 7.4 μM against MPRO and 0.15 nM against cathepsin L as well as a potent antiviral effect on Vero E6 cells infected with SARS-CoV-2.43,44
Following the same logic, the calpain/cathepsin L covalent inhibitors I and II were repositioned (Figure 9) as potential SARS-CoV-2 MPRO inhibitors through a comprehensive in vitro screening of investigational/approved inhibitors of the cysteinyl protease. In detail, the tripeptidyl compounds halted the enzymatic activity of SARS-CoV-2 MPRO in vitro at IC50 values of 0.97 ± 0.27 and 8.6 ± 1.46 μM, respectively. Calpain inhibitor II was able to block viral progression in the cell infection model (EC50 = 2.07 ± 0.76 μM in the primary CPE assay; EC50 = 3.70 ± 0.69 μM in the secondary viral yield reduction assay) with no cytotoxicity to normal cells. The analysis of the crystal structure complex of calpain inhibitor II with SARS-CoV-2 MPRO (PDB code 6XA4) confirmed the covalent mechanism of inhibition with the formation of a hemithioacetal adduct. The methionine side chain, deeply inserted into the P1 pocket, could be the starting point for the development of new inhibitors with a dual mechanism of action (MPRO and cathepsins inhibition).30,32,33
Figure 9.

Chemical structures of the repurposed calpain inhibitors I and II as new SARS-CoV-2 MPRO modulators.30,32,33
The exploration of the chemical space and structural requirements for the MPRO inhibition has led to the synthesis of a series of dipeptidomimetic compounds with a conformationally constrained cyclohexane moiety as P3. In particular, bicyclic derivative 6 (Figure 10) represented the most interesting compound with an IC50 of 0.18 ± 0.03 μM against SARS-CoV-2 MPRO and high antiviral activity (EC50 of 0.035 ± 0.001 μM in Vero E6 cells).45
Figure 10.

Chemical structure of peptidomimetic compound 6.45
2.1.2. Ketone Warhead
The ketone warhead has also been extensively explored in the design of SARS CoV-2 MPRO inhibitors. Among the initial efforts in this direction, the synthesis and biological evaluation of a series of peptidyl acyloxymethyl ketone probes with a covalent inhibition effect toward SARS-CoV-2 MPRO is of significant interest.46
Therefore, based on previous findings on a set of MPRO SARS-CoV-1 inhibitors, the α-acyloxymethyl ketone 7 and hydroxymethyl ketone (HMK) PF-00835231 (Figure 11) were synthesized as new MPRO SARS-CoV-2 covalent inhibitors.47 In particular, compound PF-00835231 exhibited an interesting Ki of 0.27 nM and an outstanding IC50 of 6.9 nM against the target protein. To gain insight into the binding mode and mechanism of action, PF-00835231 was cocrystallized in a covalent complex with SARS-CoV-2 MPRO (Figure 11b, PDB code 6XHM), highlighting the key contacts with the target: the carbonyl group of the HMK warhead (P1′) is covalently bonded to the Cys145 of the S1′ site of the binding pocket, forming a tetrahedral carbinol complex, which together with the primary −OH of the HMK group reinforces the covalent interaction by additional H bonds within the S1 cleft (His41, Gly143), the lactam P1 is positioned into the S1 pocket (as described earlier), the lipophilic leucine side chain at the P2 site of the ligand is inserted into the S2 region, surrounded by hydrophobic amino acids, and the indole moiety makes the complex more stable through van der Waals interactions with the backbones of the 189–191 amino acid residues.47 The mechanism of covalent inhibition of SARS CoV-2 MPRO was investigated by classical and QM/MM simulations, underling the importance of the hydroxymethyl group both in binding free energy increase and in the formation of the hemithioacetal adduct. Analysis of in silico data showed a significant inhibition effective of PF-00835231 against MPRO mutant forms.
Figure 11.
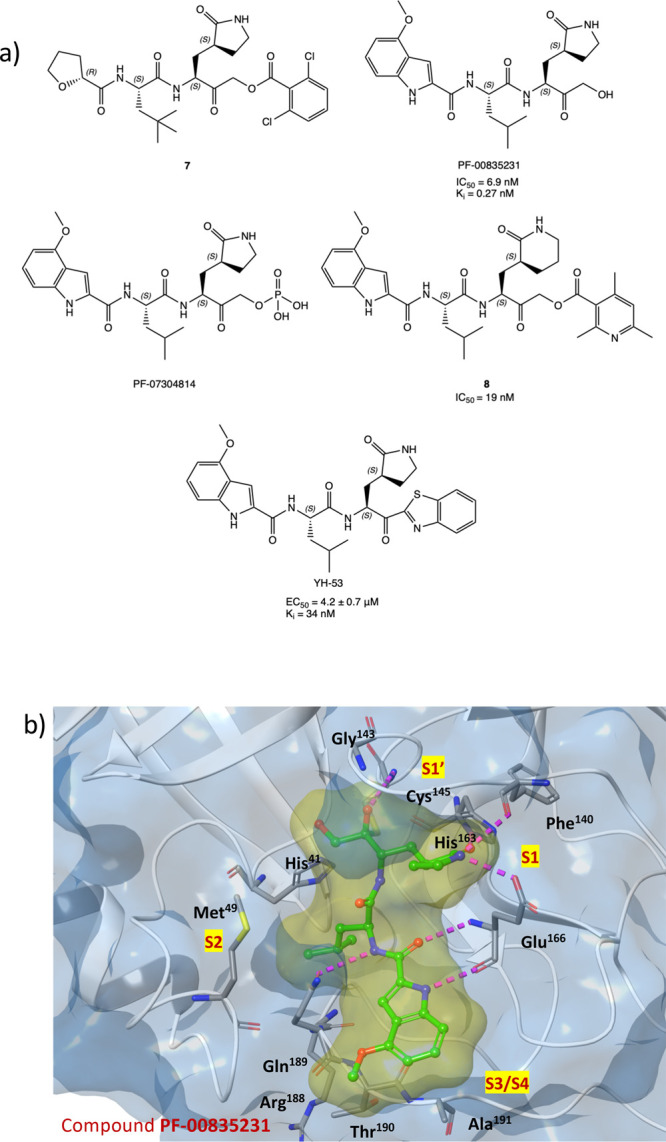
(a) Chemical structures of a series of acyloxymethyl ketone derivatives with SARS-CoV-2 MPRO inhibitory activity. (b) PF-00835231 in complex with SARS-CoV-2 MPRO (PDB code 6XHM).47,49−52
PF-00835231 was selected for in vitro/vivo biological evaluation, demonstrating appropriate physicochemical and pharmacokinetic properties for intravenous continuous infusion, similar/higher potency compared with other peptidomimetic SARS CoV-2 MPRO inhibitors (as GC-376) or other anti-SARS CoV-2 agents (remdesivir), and low cytotoxicity and elevated tolerability in an in vitro model of human airway epithelium (HAEC).47,48 Further development of PF-00835231 into the phosphate pro-drug PF-07304814 (Figure 11) demonstrated comparable antiviral and MPRO inhibitory activity but better solubility and pharmacokinetic in in vivo animal models; these more favorable features supported the progression of PF-07304814 in clinical trials (ClinicalTrials.gov Identifiers NCT04627532 and NCT04535167).49
To explore the range of substitution tolerated for maintaining MPRO inhibitory activity, the α-acycloxymethyl ketones were redesigned by replacing the pentacyclic glutamine mimic lactam with a six-membered one and inserting a heteroaromatic portion as an acyloxy group. The most interesting compound 8 (Figure 11), with a pyridyl moiety, showed a notable inhibition of MPRO (IC50 = 19 nM), excellent SARS-CoV-2 replication inhibition, and remarkable plasma and metabolic stability. The capability of 8 to covalently block the target has been confirmed by cocrystallization experiments (PDB code 7MBI).50
With a unique P1′-benzothiazolyl ketone moiety, YH-53 (Figure 11a), previously developed as a SARS-CoV-1 MPRO blocker, also proved capable of reversibly inhibiting SARS-CoV-2 MPRO with a Ki of 34 nM and to halt SARS-CoV-2 infection in vitro (EC50 = 4.2 ± 0.7 μM) with a high safety index and low cytotoxicity. X-ray experiments performed with the complex YH-53-SARS-CoV-2 MPRO (PDB code 7E18) confirmed the formation of the covalent adduct and the ability of the heteroatoms of the benzothiazolyl fragment at P1′ to form additional interactions within the catalytic binding site.
In vivo pharmacokinetic studies in animal models highlighted that the introduction of the lipophilic benzothiazole group particularly affected the cLogP value (2.37), suggesting high cell penetration in the GI tract and excellent permeability. In contrast to the almost complete absorption, the recorded bioavailability was very low (3.6%); this issue has been ascribed to the pseudopeptide structure, responsible for high first-pass metabolism in the intestine and liver after oral administration.51,52
Introduction of halogen atoms in the α position of a carbonyl atom also seemed to be an interesting way to increase the reactivity against the −SH catalytic amino acid of the viral protease. In this light, the pseudodipeptide 9 (Z-Leu-Homophe-CHF2, Figure 12) bearing an α,α-difluoromethyl ketone as an electrophilic warhead showed efficient interactions with SARS-CoV-2 MPRO in in silico simulations. In particular, the two fluoride atoms were capable to form H–halogen bonds with key residues in the catalytic pocket of S1′ (Leu141, Ser144, and Cys145).53
Figure 12.

Structure of α,α-difluoromethyl ketone 9.53
Interestingly, an example of a nonpeptidomimetic covalent MPRO inhibitor is represented by the uncommon bispidine-based ketone 10 (Figure 13), which showed noteworthy inhibitory activity against SARS-CoV-2 MPRO (IC50 of 0.75 μM). Docking simulations pointed out the central ketone group as the electrophilic center for the covalent bond with the catalytic cysteine.54
Figure 13.

Bispidine compound 10, a representative example of a nonpeptidomimetic ketone as a SARS-CoV-2 MPRO inhibitor.54
2.1.3. α-Ketoamide Warhead
Among the α-ketoamide inhibitors, the carbonyl moiety is also crucial for the formation of a reversible covalent bond with the key cysteine residue of SARS-CoV-2 MPRO. In addition to the covalent adduct with the catalytic Cys145, the α-ketoamide moiety forms additional noncovalent H bonds with the amino acids of the active site via the carbonyl oxygen and the −OH of the hemithioacetal.55 In detail, the nucleophilic attack by the cysteinyl −SH on the α-carbonyl leads to the formation of a hemithioacetal adduct. The hydroxy group of the hemithioacetal gives a hydrogen bond to His41, while the oxygen of the carboxamide moiety accepts hydrogen bonds from the main-chain amides of Gly143, Cys145, and Ser144 (Figure 14).14,56,57
Figure 14.

General mechanism of action of SARS-CoV-2 MPRO α-ketoamide inhibitors.
In accordance with the potential covalent inhibitory effect, the α-ketoamide warhead has been extensively used to develop new SARS-CoV-2 MPRO inhibitors.
In this context, a new promising series of α-ketoamide compounds with a broad spectrum of activity against the major proteases of enteroviruses, α-coronaviruses, and β-coronaviruses was developed.14,55 This class of derivatives is characterized by an α-ketoamide moiety as a warhead, a 5-membered γ-lactam ring at the P1 site (also described for aldehyde and ketone warheads), and a hydrophobic/alkylic group at the P2 site, such as cyclopropyl or cyclohexyl. Studies on viral protease inhibition and antiviral activity have shown that compound 11 (Figure 15a), a previously developed SARS-CoV-1 MPRO inhibitor (IC50 = 0.71 ± 0.36 μM), can also effectively inhibit SARS-CoV-2 MPRO (IC50 = and 0.18 ± 0.02 μM).14,55
Figure 15.
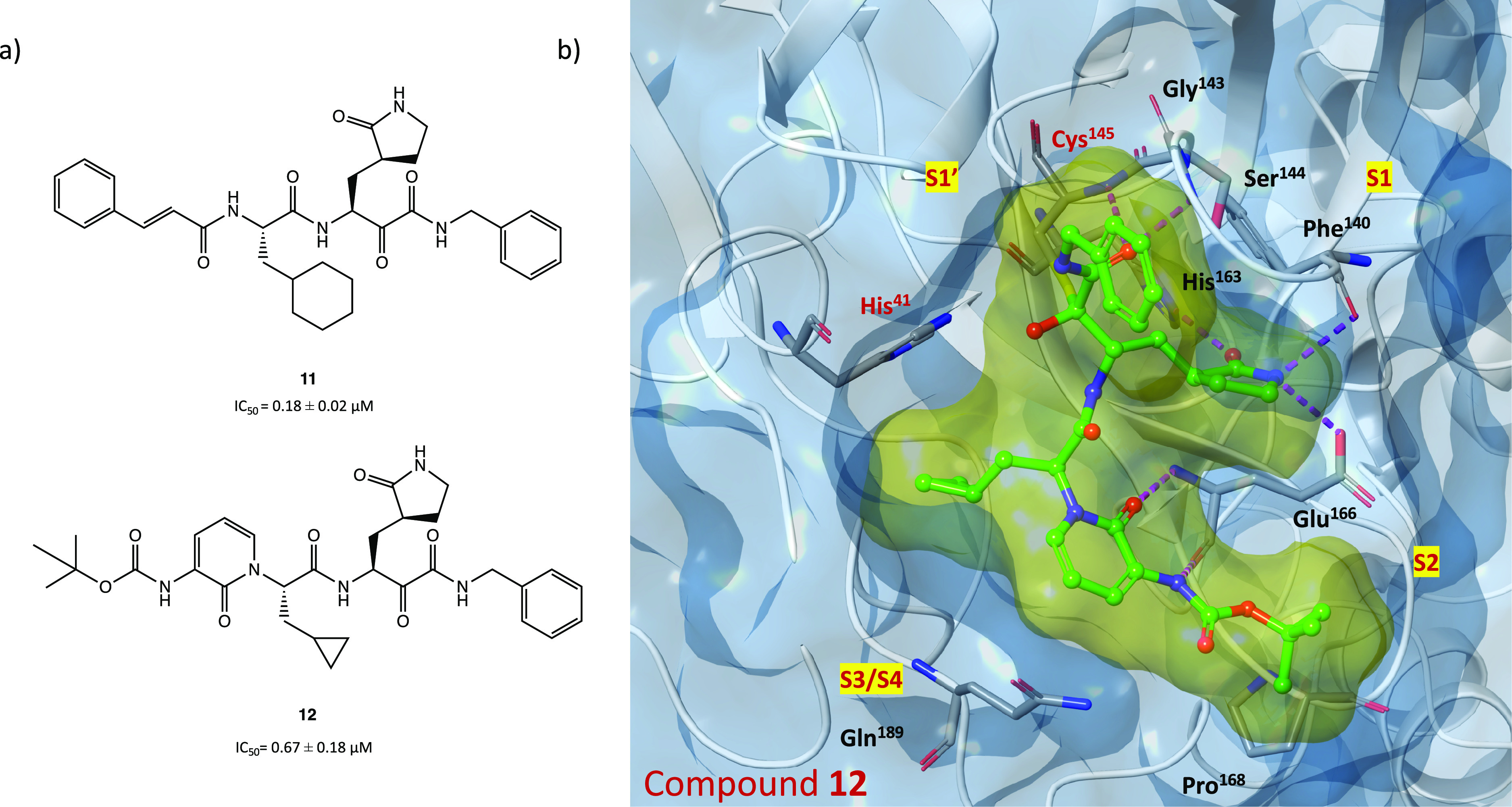
(a) Chemical structures of α-ketoamide derivatives 11 and 12. (b) X-ray structure of SARS-CoV-2 MPRO in complex with compound 12 (PDB code 6Y2F).14
Starting from the α-ketoamide 11, as a lead compound, to optimize the selective inhibition of SARS-CoV-2 MPRO, the derivative 12 was obtained (Figure 15a). With the aim of improving the pharmacokinetic parameters, a pyridone ring was inserted and the cinnamoyl and cyclohexyl moieties were replaced by a hydrophobic tert-butyloxycarbonyl group and a smaller cyclopropyl group, respectively.14 In vitro biochemical assays showed that compound 12 selectively inhibited SARS-CoV-2 MPRO with IC50 = 0.67 ± 0.18 μM and blocked SARS-CoV-2 viral infection in human Calu-3 cells with remarkable EC50 values in the range of 4–5 μM. Moreover, in vivo studies conducted in CD-1 mice highlighted favorable pharmacokinetic properties and positive tropism of the compound in the lung, the primary target of COVID-19.14
Further in silico studies (molecular docking and molecular dynamics simulations) showed that 12 is able to bind efficiently to the catalytic site of the protease. In Figure 15b (crystal structure of SARS-CoV-2 MPRO in complex with compound 12, PDB code 6Y2F), the X-ray experiment confirms the formation of the covalent adduct (Figure 14): the α-ketoamide group enhances the interaction in the S1′ cleft thanks to the H bonds with His41, Gly143, Ser144, and Cys145, the S1 site accommodates the glutamine surrogate γ-lactam ring, the key fragment amidopyridone interacts with Glu166 via two H bonds, and the tert-butyl moiety is inserted into the S2 pocket.14,58,59
In addition, starting from derivative 11, an in silico structural optimization process was carried out. The obtained compounds 13–15 were characterized by replacement of the lactam moiety in P1 by a hydantoin one, which increased the predicted binding affinity with SARS-CoV-2 MPRO through additional H-donor/acceptor bonds (Figure 16).60
Figure 16.
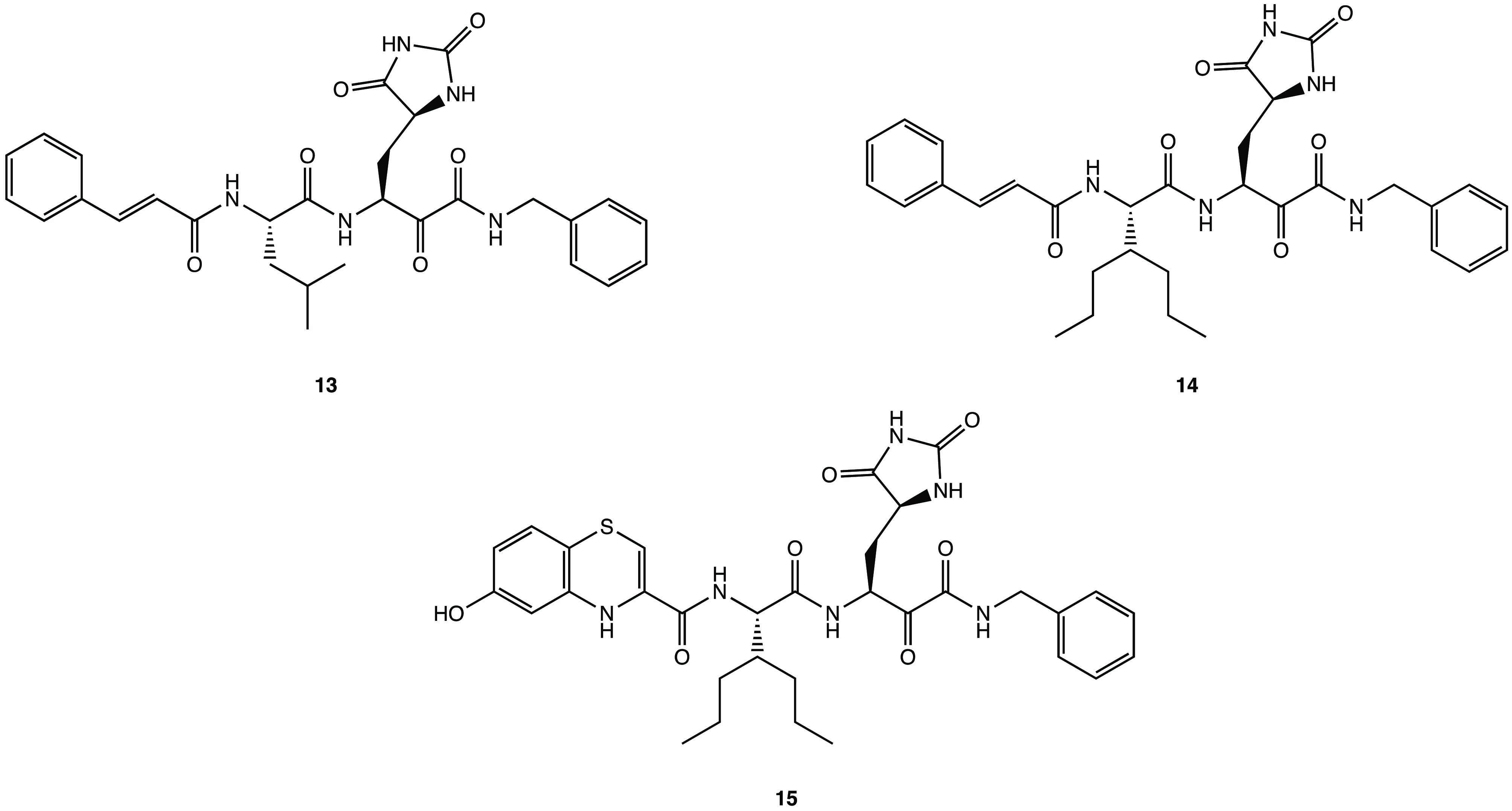
Chemical structures of hydantoin derivatives 13–15.(60)
Using GC-376 as the lead compound, the two α-ketoamide analogues UAWJ246 and UAWJ248 were developed (Figure 17) to investigate the effects of the aldehyde warhead replacement on the binding affinity with SARS-CoV-2 MPRO. Nevertheless, the biological in vitro screening showed comparable inhibitory activity against MPRO with the parent compound (IC50 = 0.045 μM and ki = 0.036 μM for UAWJ246; EC50 = 11.1 μM and ki = 0.013 μM for UAWJ248).30
Figure 17.
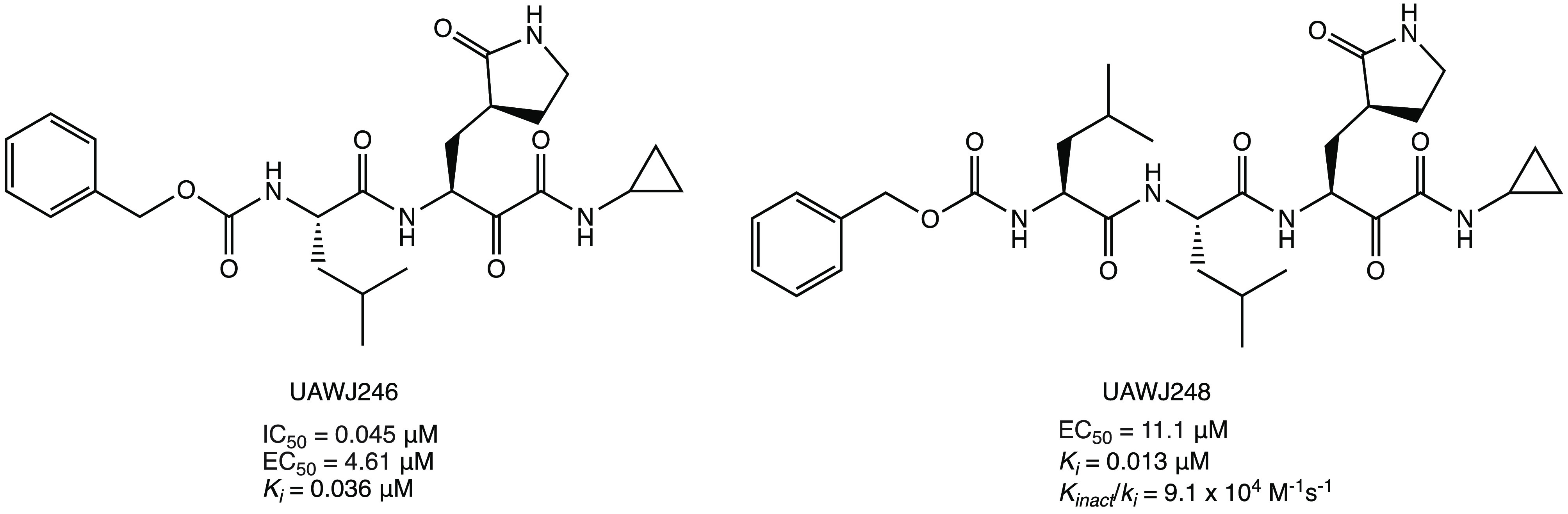
Chemical structures of GC-376 α-ketoamide analogues UAWJ246 and UAWJ248.30
As described in the previous paragraph for calpain inhibitors I and II, the calpain inhibitor XII (Figure 18) is another interesting covalent inhibitor with an α-ketoamide warhead. Compared to calpain inhibitors I and II, calpain inhibitor XII showed higher antiviral activity in the cellular model (EC50 = 0.49 ± 0.18 μM in the primary CPE assay and EC50 = 0.78 ± 0.37 μM in the secondary viral yield reduction assay) and interesting SARS-CoV-2 MPRO inhibitory activity in the submicromolar range (IC50 = 0.45 μM).30,32,40
Figure 18.

On the other hand, an in silico repurposing study of FDA-approved antiviral drugs as covalent inhibitors of SARS-CoV-2 MPRO led to the identification of boceprevir and telaprevir (Figure 19) as promising drugs with an α-ketoamide warhead. Both X-ray experiments (PDB code 6ZRU and 6ZRT) and docking studies confirmed the ability of the two drugs to form covalent adducts with Cys145.61−63 In vitro biochemical assays showed significant activity of boceprevir against SARS-CoV-2 MPRO with an IC50 of 1.59 μM.61
Figure 19.

FDA-approved α-ketoamide drugs: boceprevir and telaprevir.
Noteworthy, several in silico analyses have been carried out to design new potential inhibitors that offer intriguing ideas for future developments. The most promising of these are discussed in the following sections.
The application of various computational approaches (QSAR modeling, pharmacophore modeling, molecular docking, and molecular dynamics simulations) enabled the discovery of a novel series of α-ketoamide compounds such as derivatives 16–18 (Figure 20). Analysis of the in silico data revealed that all three molecules interacted efficiently and stably with SARS-CoV-2 MPRO (similar to the reference compound 12) and also exhibited favorable pharmacokinetic and toxicological profiles.59
Figure 20.

Chemical structures of potential SARS-CoV-2 MPRO inhibitors 16–18.(59)
Using a new advanced computational approach with a molecular covalent docking analysis, derivative 19 (Figure 21) emerged as one of the most promising molecules targeting SARS-CoV-2 MPRO. According to the binding pose, the covalent complex 19/SARS-CoV-2 MPRO was mainly stabilized by hydrogen bonds: the carbonyl group of the α-ketoamide, the triazole ring, and the oxygen in the β-lactam ring formed H bonds with Leu141, Gly143, Glu166, and His41. Moreover, to optimize the lead compound, a sulfonic group was also introduced to enhance the polarity, leading to compounds 20 and 21 (Figure 21), with improved in silico affinity for the binding pocket of the target protein.64
Figure 21.

In silico designed SARS-CoV-2 MPRO inhibitors 19–21.(64)
Among the series of novel α-ketophenylamide derivatives as anticancer agents, derivative 22 (Figure 22) was selected as a representative compound to be tested in the SARS-CoV-2 cell infection assay, and it was found to actively block viral infection in in vitro models (EC50 of 1.28 μM in the plaque reduction assay). Despite the lack of target inhibition data, the antiviral effect exhibited in the cell assay could be attributed to the inhibition of MPRO due to its structural similarity with the above-mentioned α-ketoamide derivatives.65
Figure 22.

Chemical structure of the anticancer compound 22 with activity on SARS-CoV-2.65
2.1.4. Michael Acceptor Group as a Warhead
Similar to carbonyl and α-ketoamide warheads, Michael acceptor groups have also been extensively used in the development of covalent inhibitors for cysteinyl enzymes.16−18 Michael acceptor groups (such as α,β-unsaturated carbonyl, vinyl nitriles, vinyl sulfonamides, etc.) offer potential advantages for target inhibition compared to other warheads; the Michael acceptor groups inhibit the enzymes via conjugate addition of the nucleophilic cysteinyl −SH to the electrophilic Cβ of the unsaturated system, producing a nearly irreversible and longer lasting adduct. Figure 23 shows the scheme of covalent inhibition by an α,β-unsaturated carbonyl warhead.
Figure 23.

General mechanism of inhibition conjugate systems (α,β-unsaturated carbonyl warhead is reported as an example).
N3, a compound previously evaluated for activity against SARS-CoV-1 and MERS-CoV MPRO (Figure 24a), is one of the first peptidomimetic covalent inhibitors of SARS-CoV-2 MPRO with an α,β-unsaturated Michael acceptor group as a warhead. Kinetics and X-ray studies explained the ability of the compound to irreversibly bind the thiol group of Cys145 in the MPRO catalytic site, reacting via a conjugate addition mechanism (PDB code 6LU7).15 To better understand the irreversible inhibition of N3 at the atomic level, hybrid QM/MM free energy methods were also performed. In detail, the mechanism of action of N3 consists of two steps: formation of the high-energy ion pair Cys145–/His41+ dyad and subsequent establishment of covalent bonds.66
Figure 24.

(a) Chemical structures of N3 and analogues 23–26.15,67−69 (b) X-ray crystal structure of 26 in complex with SARS-CoV-2 MPRO.69
The same in silico approaches were applied to design and computationally test three new N3 analogues 23–25 (Figure 24a), obtained by modifying the recognition portion and/or the warhead. Compared to the lead compound N3, they were predicted to possess improved thermodynamic and kinetic properties.67,68 Such an approach proves the importance of this type of in silico techniques, especially in the design of new covalent inhibitors.
The N3 analogue 26 (Figure 24a) was identified by in vitro screening of previously described SARS-CoV-1 MPRO inhibitors. It showed promising SARS-CoV-2 MPRO inhibitory activity (IC50 = 151 ± 15 nM), ability to block viral infection in Vero E6 cells (EC50 = 2.88 ± 0.23 μM), and covalent inhibition of the target protein. As illustrated by the X-ray image of the complex 26/MPRO (Figure 24b, PDB code 7JT7), the sulfur atom of Cys145 forms a covalent bond with the Cβ atom of the vinyl group (Michael addition to the α,β-unsaturated system) while the other portions (the γ-lactam ring, the isoleucine, and the phenyl) are inserted into protein regions S1, S2, and S4, respectively.69
Through a receptor-based virtual screening campaign of an in-house database of cysteine-responsive compounds, two peptidomimetic irreversible MPRO inhibitors 27 and 28 were discovered (Figure 25) with a vinyl-ketone portion as the Michael acceptor group. In vitro biochemical assays revealed promising inhibitory activity with IC50 values of 47.2 and 157.5 μM, respectively. Docking and molecular dynamics simulations confirmed the formation of a covalent adduct between the two derivatives and the reactive cysteine, highlighting the importance of the hydrophobic alkyl P2 moieties (cyclohexyl and isobutyl) and the para-substituted aromatic P3 moieties (p-F-phenyl and p-NO2-phenyl) for stable interactions with the target protein.70
Figure 25.

Chemical structures of peptidomimetic irreversible MPRO inhibitors 27 and 28.70
Recently, however, many nonpeptidomimetic compounds have also been proposed as SARS-CoV-2 MPRO inhibitors, leading to new ideas for exploring new chemical spaces and nuclei.
In an in vitro pilot screening of a highly focused compound library to identify new nonpeptidomimetic scaffolds for covalent inhibition of MPRO, the derivative SIMR-2418 emerged as one of the most active inhibitors (Figure 26) with an IC50 of 20.7 μM against SARS-CoV-2 MPRO and the most favorable ADME properties. Molecular docking and dynamics analyses showed that the unexplored core, based on the fusion between a benzo[b][1,4]oxazin-6(5H)-one and an imidazo[2,1-b]thiazole system, is crucial to achieve the desired orientation in the active site of the target protein. The presence of a cyclohexenedione fragment is essential for the covalent inhibition, while the tert-butyl group fits deeply into a hydrophobic cleft.71
Figure 26.

SIMR-2418, a promising nonpeptidomimetic SARS-CoV-2 MPRO inhibitor.71
Interestingly, covalentizer, an automated pipeline, could be an effective tool to discover new covalent SARS CoV-2 MPRO inhibitors with a Michael acceptor group. This innovative computational protocol enabled the development of a series of irreversible acrylamide agents starting from the reversible nonpeptidomimetic SARS CoV-1/2 MPRO inhibitor ML188. The most interesting compound, 29 (S enantiomer, Figure 27), showed a significant IC50 of 2.86 μM (the R enantiomer was nearly inactive with an IC50 of 86.32 μM). The X-ray crystal structure (PDB code 7NW2) provided important details about the binding mode of 29 in the catalytic pocket of SARS CoV-2 MPRO: the conjugate acrylamide moiety, which replaces the furan one, is reactive to the nucleophilic conjugate addition of the SH; the p-tert-butylphenyl moiety is projected deeply into the hydrophobic S2 pocket; the 3-F-phenetylamide fragment, absent in the lead ML-118, is essential to establish key contacts with the S4 cleft.72
Figure 27.
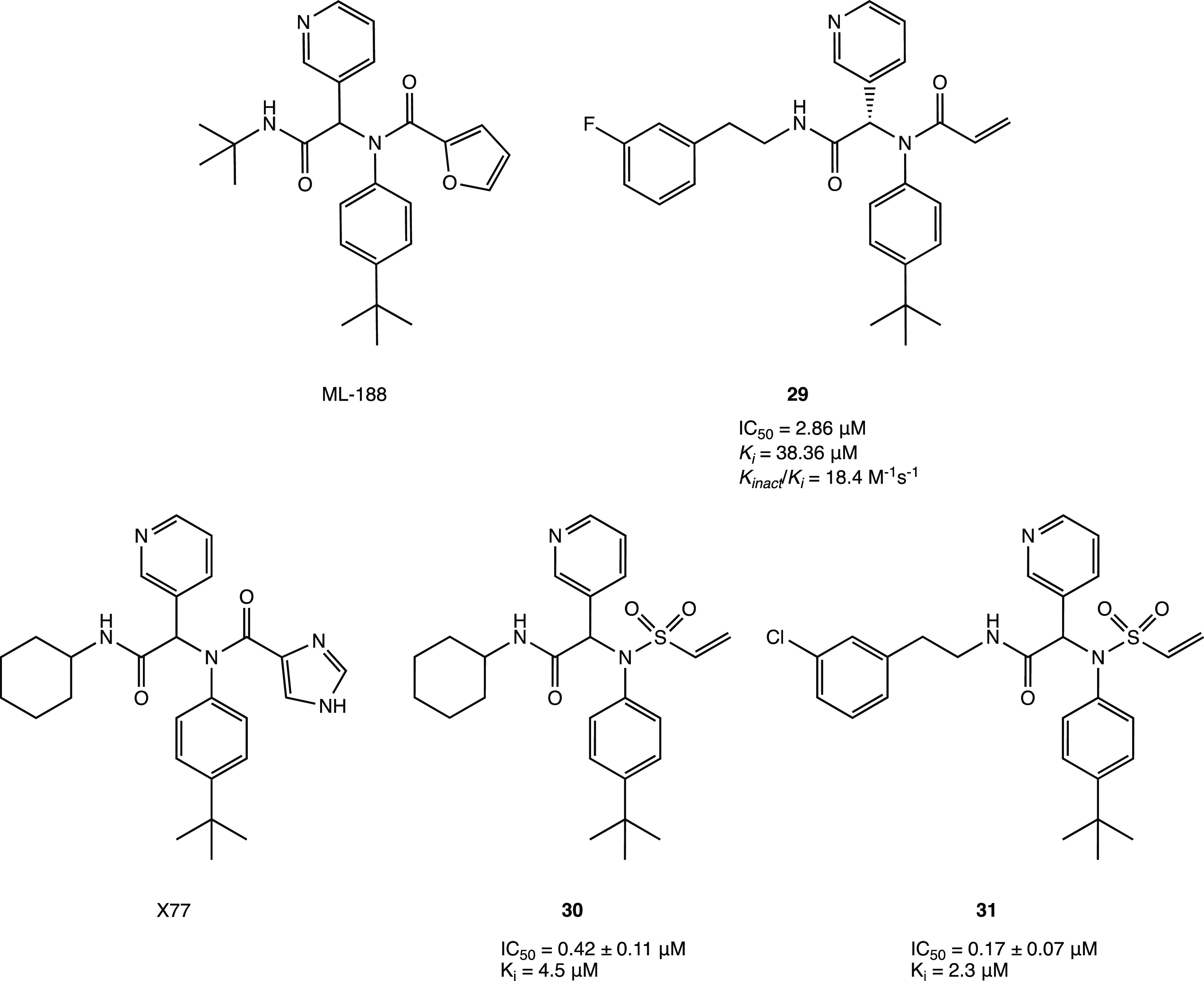
Examples of nonpeptidomimetic SARS-CoV-2 MPRO inhibitors with a covalent mechanism of action.72,73
Similarly, starting from the analysis of a previously developed noncovalent SARS CoV-2 MPRO inhibitor X77, it was possible to design new covalent agents (Figure 27). The imidazole moiety of X77, located near the catalytic Cys145 (PDB code 6W63), was replaced with several conjugated warheads. One of the most interesting compounds, 30 (Figure 27), with an unusual vinyl sulfone moiety, showed an IC50 of 0.42 ± 0.11 μM against SARS CoV-2 MPRO, an order of magnitude stronger than X77 (IC50 = 4.1 μM). The covalent inhibition mechanism was confirmed by both kinetic (ITC, isothermal titration calorimetry) and crystallographic analyses (PDB code 7MLG). Further SAR studies performed with the designed lead compounds allowed the optimization of the interaction with the target; in particular, it was found that the substitution of the cyclohexyl portion with longer chains led to compounds with higher potency compared to 30 (compound 31 showed an IC50 of 0.17 ± 0.07 μM, Figure 27).73
Among the unusual Michael acceptor warheads, the acrylonitrile moiety shows great potential. A library of more than 1400 cysteine-focused ligands from the ZINC database was investigated using covalent docking, molecular dynamics studies, and ADMET predictions. The ligands ZINC952688, ZINC2441194, ZINC2224381, and ZINC4483162 (Figure 28) were identified as the most interesting compounds, forming a covalent adduct with the MPRO via conjugate addition at the acrylic double bond.74
Figure 28.
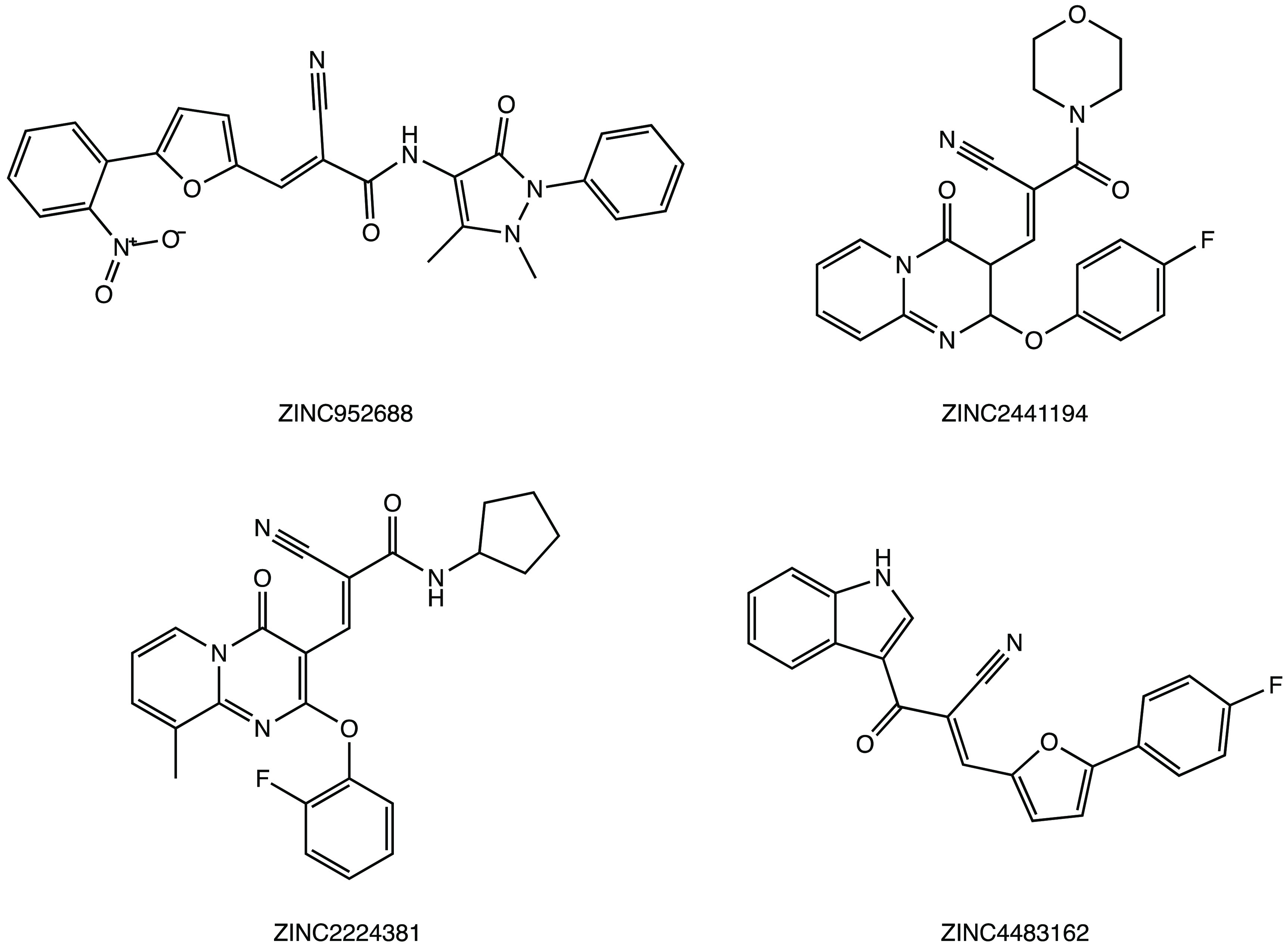
Chemical structures of ZINC database inhibitors of SARS CoV-2 MPRO.74
In addition to the above examples, several nonpeptidomimetic natural products, such as the conjugated curcuminoid and quinonoid/flavonoid systems, have also emerged as interesting lead compounds for the development of novel covalent SARS CoV-2 MPRO inhibitors in extensive in silico/in vitro screening campaigns.
Thus, curcumin (Figure 29), a natural compound known for its countless biological activities, was found to be a potential covalent inhibitor of SARS-CoV-2 MPRO in an in silico study.75 Indeed, thanks to its α,β-unsaturated carbonyl moiety, it could react with Cys145 through a Michael addition, as predicted by molecular docking and dynamics studies.75
Figure 29.

Chemical structures of natural products and analogues as potential SARS-CoV-2 MPRO inhibitors.75−77
As an example of quinonoid analogues, in a preliminary in vitro study, vitamin K3 (menadione, a synthetic form of vitamin K, Figure 29) proved to inhibit SARS-CoV-2 MPRO with an IC50 of 7.96 μM.76 Further optimization led to the development of vitamin K3 analogue 32 (5,8-dihydroxy-1,4-naphthoquinone, Figure 29), which showed a 10-fold increase in inhibitory activity compared to the parent compound (IC50 of 0.49 μM against SARS-CoV-2 MPRO). In silico and kinetic studies with both vitamin K and 32 confirmed the capability to irreversibly inhibit MPRO by forming a covalent bond via conjugate addition at the quinone core.77
Other examples of natural compounds containing Michael acceptor fragments include the flavonoids. Indeed, many virtual and in vitro screens have identified flavonoid compounds as potential covalent SARS-CoV-2 MPRO inhibitors. Myricetin, with its novel hidden electrophilic pyrogallic portion (Figure 30a), is one of the best characterized.78 The enzymatic inhibition assay confirmed its great potency with an IC50 of 0.2–0.6 μM against MPRO. The solved X-ray crystal structure in complex with SARS-CoV-2 MPRO showed that the C6′ of myricetin is covalently linked to the catalytic Cys145 (Figure 30b, PDB code 7B3E).44,79
Figure 30.
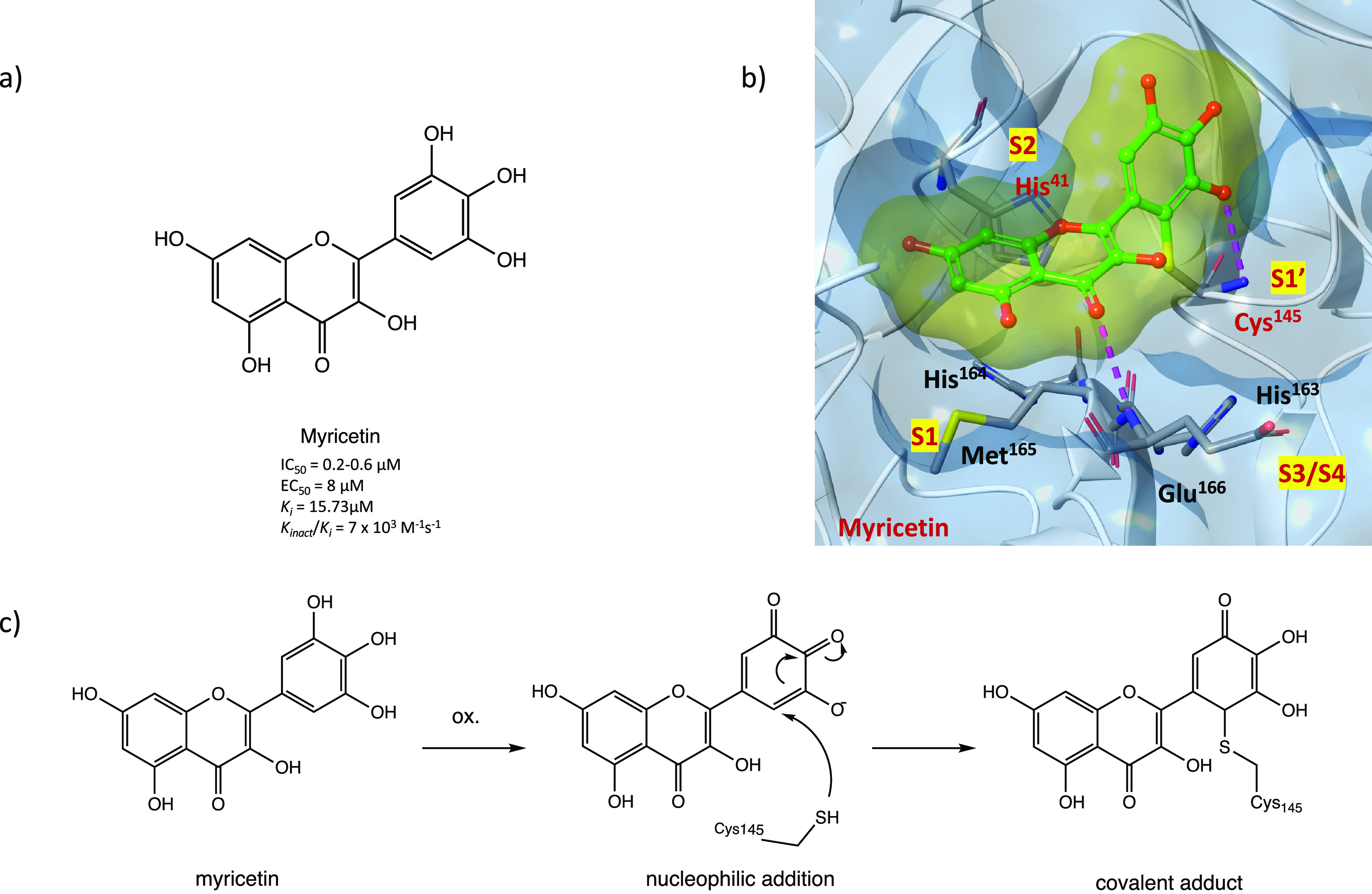
(a) Chemical structure of the flavonoid compound myricetin. (b) X-ray crystal structure of the covalent complex between myricetin and SARS-CoV-2 MPRO (PDB code 7B3E). (c) Hypothetical mechanism of action of myricetin in inhibiting SARS-CoV-2 MPRO.44,79
Considering the structure of myricetin, the irreversible inhibition could be unexplained. However, a preliminary in vivo oxidative step was proposed to explain the biological activity (Figure 30c): after the oxidation, the pyrogallol fragment is converted to an o-quinone, which could function as an α,β-unsaturated carbonyl group (as in the p-quinone group of vitamin K3, see above). The sulfur atom of Cys145 can attack o-quinone, and the resulting prototropic equilibrium could lead to the formation of the covalent adduct.79
Further in vitro cellular assays highlighted the capability of myricetin to block SARS-CoV-2 infection in Vero E6 cells with an EC50 value of 8 μM.79 Moreover, in an in vivo model of lung injury in mice it was shown to inhibit the infiltration of inflammatory cells and the production of pro-inflammatory cytokines (e.g., IL-6, TNF-α), thereby alleviating the overall inflammation.80 These findings prompted the scientific community to develop new inhibitors based on the myricetin scaffold.
Indeed, by inserting a p-OCH3 at the C7 of myricetin core, derivatives 33 and 34 (Figure 31) were obtained with improved potency compared with the parent compound (IC50 = 0.30 and 0.26 μM, respectively). In addition, insertion of a phosphonate group to the 7-OH of myricetin led to compound 35 (Figure 31), which exhibited the highest inhibitory activity against SARS-CoV-2 MPRO, demonstrating the consistency of the prodrug strategy for further development.79
Figure 31.

Chemical structures of promising myricetin analogues as covalent SARS-CoV-2 MPRO inhibitors.79
2.2. α-Haloacetamide Warhead
Alkyl halogens, especially when in the α position to a carbonyl group, have been extensively exploited to design covalent inhibitors because of their intrinsic reactivity toward nucleophiles. In this light, we report here recent efforts to develop new covalent SARS-CoV-2 MPRO inhibitors with α-haloacetamide, α,α-dihaloacetamide, and α,α,α-trihaloacetamide as reactive groups.73,81 As shown in Figure 32, the general mechanism of inhibition for this class of compounds consists of a nucleophilic attack of the cysteinyl −SH on the C–X and subsequent formation of the irreversible S–C bond.
Figure 32.

General mechanism of action of an α-haloacetamide warhead.
In a comprehensive in silico and in vitro screening of newly designed covalent inhibitors with different electrophilic warheads, the α-chloroacetamide derivative 36 (Figure 33a) proved to be one of the most promising lead compounds and showed a significant inhibitory effect on SARS-CoV-2 MPRO (IC50 = 0.4 μM and ki = 16 μM) in the biochemical assay. SAR analysis highlighted that the α-chloro substitution resulted in more effective derivatives than the α-fluoro substitution due to the higher electrophilicity of C–Cl, tert-butyl had a positive effect on the activity, and replacement of the heterocyclic pyridine moiety with a carbocyclic one resulted in nearly inactive compounds. Moreover, kinetic studies and the analysis of the cocrystal structure of SARS-CoV-2 MPRO in complex with 36 (PDB code 7MLF) confirmed the binding mode of the proposed lead compound (Figure 33b): the cysteinyl −SH displaces the Cl through the covalent mechanism illustrated in Figure 32; the pyridine is inserted into the S2 pocket and forms multiple H bonds; the tert-butylphenyl is enclosed into the S4 region.73
Figure 33.
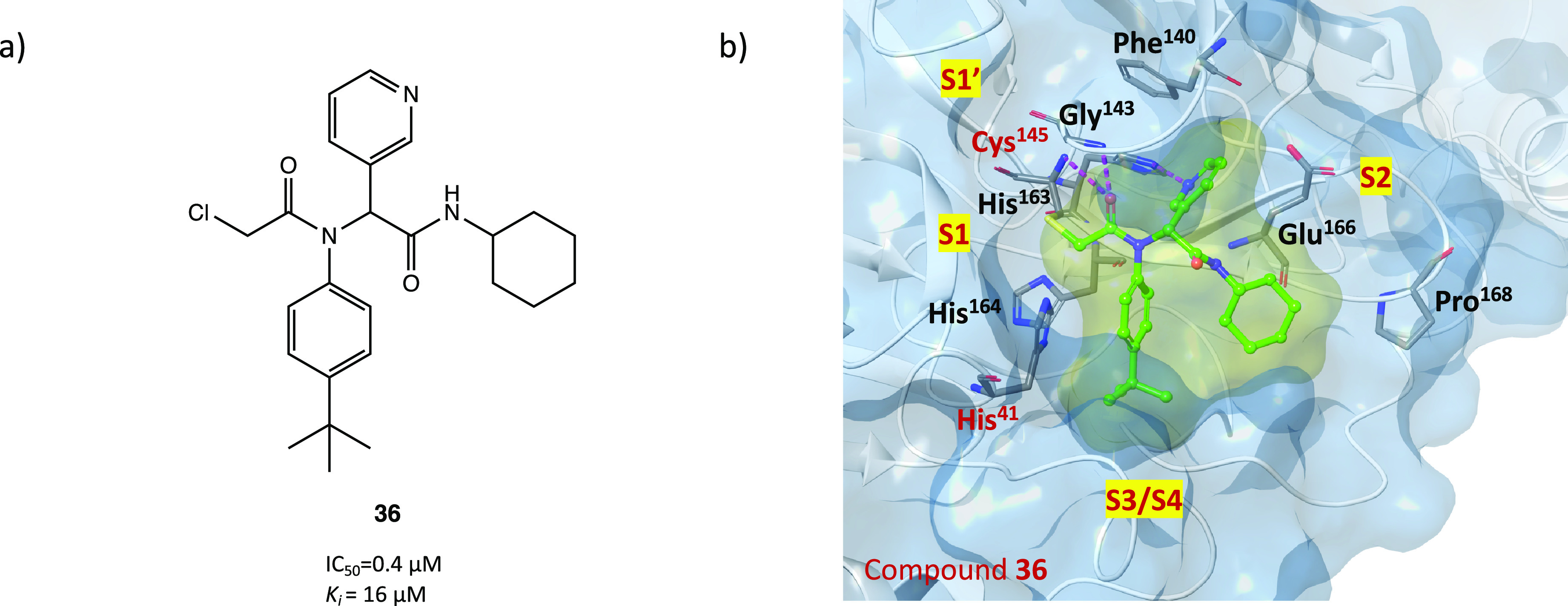
(a) Chemical structure of the α-chloroacetamide derivative 36, developed as a SARS-CoV-2 MPRO covalent inhibitor. (b) X-ray crystal structure of the covalent complex between derivative 36 and SARS-CoV-2 MPRO (PDB code 7MLF).73
Similarly, starting from the noncovalent inhibitor 37 (Figure 34), a new series of selective SARS-CoV-2 MPRO peptidomimetic covalent inhibitors was developed. The new compounds were characterized by the replacement of the furyl ring with dichloroacetamide, dibromoacetamide, tribromoacetamide, 2-bromo-2,2-dichloroacetamide, or 2-chloro-2,2-dibromoacetamide as warheads. Among all of them, derivatives 38 and 39, shown in Figure 34, were the most promising lead structures with potent and selective MPRO inhibition (IC50 of 0.43 and 0.08 μM, respectively) and outstanding efficacy in the cellular model of Caco2-hACE2 viral infection (EC50 of 2.05 and 2.15 μM, respectively). The X-ray crystal structure of the complex SARS-CoV-2 MPRO38 (PDB code 7RN1) confirmed the capability of the designed compound to covalently bind to the target protein; furthermore, the absolute R configuration of the pyridyl P1 portion appeared essential for the deep insertion into the S1 pocket.81
Figure 34.

Chemical structures of the dichloroacetamide, tribromoacetamide, and chloro-fluoro-acetamide derivatives 38 and 40 developed as SARS-CoV-2 MPRO covalent inhibitors.81
Using these derivatives as lead compounds, a new series of SARS-CoV-2 MPRO inhibitors was proposed with an interesting α-chloro-fluoroacetamide (CFA) as a warhead. Thanks to its steric and electronic features, this moiety appeared to be intrinsically selective toward cysteinyl residues and to form a pseudoreversible adduct, which could reduce the off-target labeling frequently observed for covalent inhibitors. The derivative 40 (Figure 34), with a phenyl-pentafluorosulfanyl group as P2 and a pyrimidinyl portion at P1, was the most interesting compound of the series. Remarkably, biological inhibition assays showed that the stereochemistry of the two asymmetric carbons plays a crucial role: of the four possible stereoisomers, (R,R)-cis-40 was the only one with strong inhibitory activity against SARS-CoV-2 MPRO, with an outstanding IC50 of 0.056 μM and a remarkable Kinact/ki value of 4.2 × 103 M–1 s–1. In silico simulation ascribed the higher activity of this stereoisomer to its more stable and reactive conformation within the active site.82
To discover new covalent inhibitors of SARS-CoV-2 MPRO, a virtual in silico screening of a set of commercial compounds without a peptidomimetic skeleton was performed. The computational protocol was based on a hierarchical workflow involving a first phase of filtering and large-scale noncovalent docking followed by the covalent docking of the best-ranked compounds. The most promising compounds were selected for in vitro enzyme inhibition assay, and among all of them, compound 41 (Figure 35) was the most active, with an IC50 of 8.50 μM against SARS-CoV-2 MPRO. Given the interesting and innovative nonpeptidomimetic core, the crystal structure of the ligand–protein complex was resolved to support the exploration of more potent analogues (PDB code 7VVT). In addition to the covalent bond formed between the cysteine and the chloroacetamide warhead, the other important noncovalent binding interactions involved the amino acid residues in the S2 site (His41 and Gln189) and the deeply inserted m-chlorophenyl moiety.83
Figure 35.

Chemical structure of α-chloroacetamide SARS-CoV-2 MPRO inhibitor 41.83
By means of activity-based protein profiling (ABPP) screening, several pyrazoline-based SARS-CoV-2 MPRO inhibitors with an α-chloroacetamide as a reactive group were identified. The crystal structure analysis of (R)-EN82 (IC50 value of 0.53 μM) (Figure 36) highlighted the discovery of a small protein pocket with the accommodation of the 4-phenyl substituent. Extensive SAR analyses on C-4-substituted compounds led to the isolation of the optimized trisubstituted cis derivative HW-2-010B (IC50 against the target protein in the nanomolar range, 14 nM) (Figure 36).84
Figure 36.

Chemical structures of pyrazoline-based compounds (R)-EN82, HW-2-010B, and QUB-00006-Int-07 as SARS-CoV-2 MPRO inhibitors.84,85
The application of advanced in silico techniques, such as high-resolution all-atom molecular dynamics simulations and absolute binding free energy calculations applied on SARS-CoV-2 MPRO, enabled the design of the unusual cyclic α,α-difluoro-amide QUB-00006-Int-07 as a covalent inhibitor, which showed an IC50 of 0.83 μM (Figure 36).85
2.3. Nitrile Warhead
Interestingly, the nitrile group has also been proposed as a remarkable warhead for the development of SARS-CoV-2 MPRO covalent inhibitors. Due to the difference in the electronegativity with the nitrogen atom, the carbon of nitrile is susceptible to nucleophilic addition by the catalytic cysteine of the protease with subsequent formation of the reversible thioimidate covalent adduct, as shown in Figure 37.
Figure 37.

General mechanism of SARS-CoV-2 MPRO inhibition with the nitrile warhead.
In this light, Pfizer has undertaken a comprehensive drug discovery campaign to develop new SARS-CoV-2 MPRO inhibitors for oral administration. Starting with the previously developed carbonyl compound PF-00835231 (Figure 11), which exhibited potent MPRO inhibition and anti-SARS-CoV-2 activity but no intestinal absorption, Pfizer researchers developed the first orally bioavailable anti-SARS-CoV-2 compound PF-07321332 (nirmatrelvir) (Figure 38a) with a nitrile group as the reactive warhead. In preliminary biological assays, nirmatrelvir showed potent SARS-CoV-2 MPRO inhibitory activity (Ki = 3 nM and IC50 = 19.2 nM) and anti-SARS-CoV-2 activity in cell-based assays (EC50 = 75 nM). Its less peptidomimetic structure (fewer H-bond donors and lower polarity compared to the parent compound) and the insertion of a trifluoroacetamide moiety guaranteed excellent intestinal barrier permeation and excellent oral bioavailability compared to the first in vivo evaluations.21,86
Figure 38.
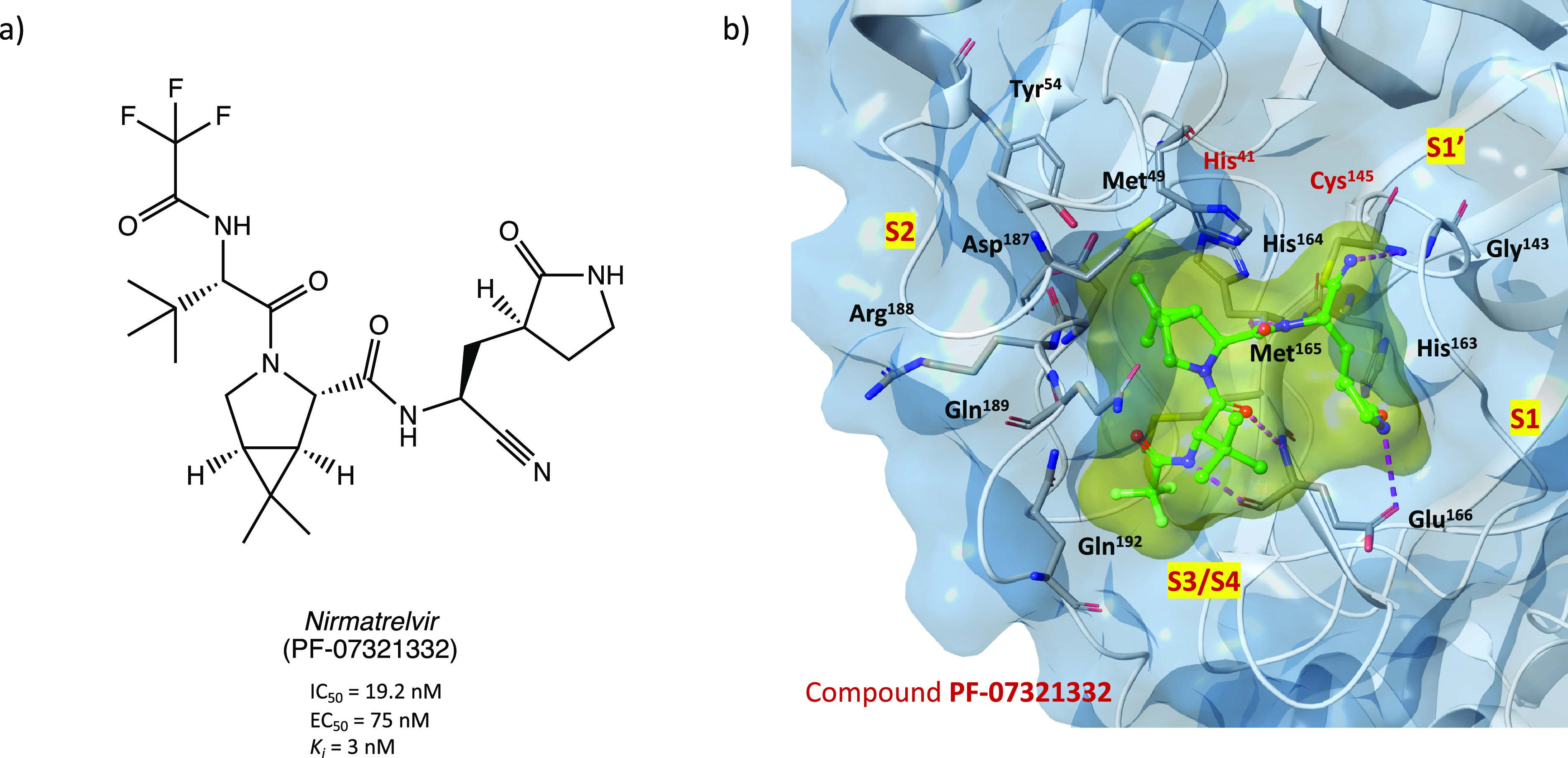
(a) Chemical structure of the orally bioavailable compound PF-07321332. (b) X-ray crystal structure of PF-07321332 in complex with SARS-CoV-2 MPRO (PDB code 7VH8).87
Thanks to its remarkable results obtained in the preclinical evaluation, it is currently under phase 3 of clinical trials in combination with ritonavir (PAXLOVID, see ClinicalTrials.gov Identifier NCT04960202), an already approved anti-HIV agent inserted in the formulation as a pharmacokinetic enhancer (it increases the systemic exposure and the half-life of nirmatrelvir thanks to its inhibitory activity on the cytochrome metabolizing enzymes). The good results already obtained prompted the European Medicines Agency (EMA) and the corresponding authorities of United States and United Kingdom to grant a conditional marketing authorization for the treatment of COVID-19.23,88,89
From a medicinal chemistry point of view, the crystal structure of SARS-CoV-2 MPRO in complex with PF-07321332 (PDB codes 7VH8 and 7VLQ) has been resolved to clarify the mechanism of action of this compound. As shown in Figure 38b, the nitrile warhead is able to form a reversible covalent thioimidate adduct with the sulfur atom of the Cys145 at S1′; furthermore, the formed imine nitrogen enhances the reversible adduct by interacting with the Gly143 and Cys145 residues of the oxyanion hole. The usual γ-lactamic ring fits into the S1 pocket and interacts with His163 and Glu166, while the dimethyl-bicycloproline group is collocated into the hydrophobic S2 pocket and is surrounded by the side chains of His41, Met49, Tyr54, Met165, and Gln189, resulting in extensive van der Waals interactions. The trifluoromethyl group, instead, is important to anchor the inhibitor at the S4 subpocket by forming a stabilizing contact with Gln192 and two ordered small molecules positioned in this site.87,90
Furthermore, given the importance of PF-07321332 as the first SARS-CoV-2 MPRO inhibitor approved for clinical use, a lot of advanced computational studies, such as steered molecular dynamics and classical-hybrid QM/MM simulations, have been conducted with the aim to clarify the mechanism of binding/inhibition and thus to guide the design of new analogues. It has been demonstrated that P1 (γ-lactamic ring) and the P2 (dimethyl-cyclopropylproline group) are essential in the ligand-binding process, contributing positively to the total binding free energy (substitution with other groups determined a drastic reduction of binding affinity). On the other hand, it has been evidenced that the P3 and P4 groups (isobutyl and trifluoromethyl) made a favorable but small contribution to the binding free energy, suggesting the possibility of modification on these sites to increase the binding strength.91,92
In addition, given the widespread distribution of SARS-CoV-2 variants of concern (beta, delta, and the rapidly spreading omicron variant), several studies are currently being conducted to investigate the efficacy of PF-07321332, providing very encouraging results. Indeed, nirmatrelvir showed capability to inhibit the most prevalent MPRO variants expressed by the most diffused SARS-CoV-2 mutant lineages in vitro93 and capability to potently block the infection of beta, delta, and omicron SARS-CoV-2 variants in both in vitro and in vivo animal models.94−98
Starting from the carbonyl derivatives GC-376 or PF-07304814, several analogues were designed with a nitrile warhead to explore the impact of this one on the inhibition activity. Among all of them, compound 42 (Figure 39) was the most active in the inhibition assay with an IC50 of 9.1 nM against the target protein and an EC50 of 2.2 μM in a plaque reduction assay conducted in Vero E6-infected cells. In general, by analyzing the results for the whole series, it appears that substitution of the carbonyl warhead with a nitrile one positively affects the activity, leading to more active and selective compounds.99
Figure 39.

Chemical structure of the nitrile derivative 42.99
2.4. Ester Warhead
Figure 40 shows the general mechanism of action of the class of inhibitor molecules with an ester warhead, which involves a nucleophilic attack of the Cys145 on the electrophilic carbonyl of the ester group followed by cleavage of the alkoxy group (−OLv) and irreversible acylation of the enzyme.
Figure 40.

Mechanism of acylation of SARS-CoV-2 MPRO mediated by ester derivatives.
The class of indole/indoline chloropyridinyl esters is one of the most frequently described in the literature as SARS CoV-2 MPRO inhibitors.52,100−102 In particular, three indole/indoline-chloropyridinyl-ester derivatives (GRL-0820, GRL-0920, and GRL-1720 in Figure 41), which have already been evaluated against SARS-CoV-1, were investigated as promising lead compounds for the design of new SARS-CoV-2 MPRO inhibitors. All compounds showed potent inhibitory activity against SARS-CoV-2 MPRO (IC50 values of 0.073, 0.25, and 0.32 μM, respectively) and anti-SARS-CoV-2 activity in Vero E6-infected cells with EC50 values of 15, 2.8, and 15 μM, respectively.52,100,101
Figure 41.

Chemical structures of chloropyridinyl esters GRL-0820, GRL-0920, and GRL-1720 repurposed as SARS-CoV-2 MPRO inhibitors.52,100,101
In silico docking analyses and kinetic studies proved that all three derivatives are able to covalently bind the catalytic Cys145 through an acyl substitution (as described in the general mechanism of action in Figure 40).100,101
Considering that the chloro-substituted pyridinyl group is a common pharmacophoric moiety, a new series of indole esters was studied (Figure 42). Compounds 43 and 44 proved to be the most potent inhibitors with IC50 values of 0.055 and 0.0342 μM, respectively. Further biological assays showed that both compounds could inhibit MPRO in HEK and A549 human lung epithelial cell lysates.102
Figure 42.

Chemical structures of indole derivatives 43 and 44.102
Interestingly, through an extensive in vitro screening of previously developed SARS-CoV-1 MPRO inhibitors, the chloropyridinyl ester MAC-5576 (Figure 43a) was selected as new covalent inhibitor of SARS-CoV-2 MPRO, exhibiting an interesting IC50 value of 81 nM. However, in the cytopathic reduction assay performed in Vero E6 cells, it did not show the desired inhibition of viral infection. Nevertheless, the X-ray crystal structure of MAC-5576 in complex with the target protein has been resolved to highlight the importance of this new nonpeptidomimetic scaffold for the design of new covalent inhibitors: as shown in Figure 43b (PDB code 7JT0), the compound is able to acylate the protease.69
Figure 43.
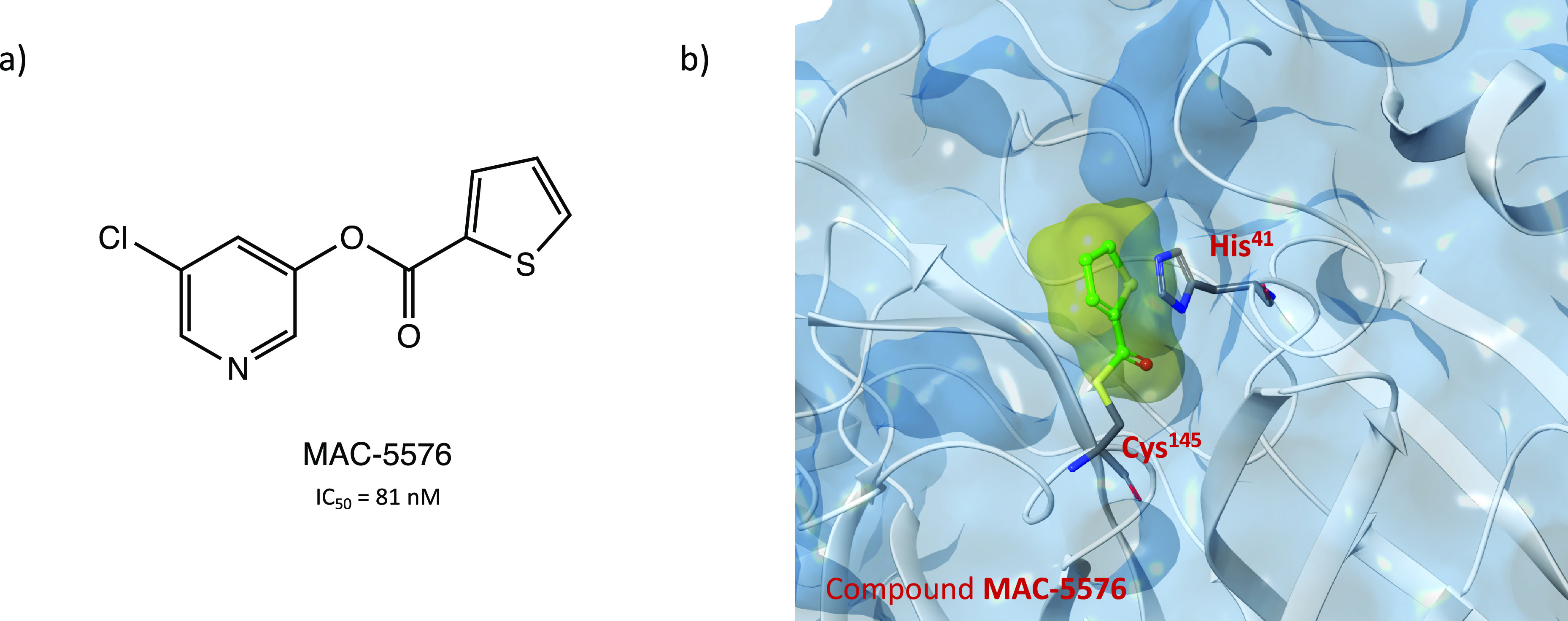
(a) Chemical structure of MAC-5576, a chloropyridinyl ester SARS-CoV-2 MPRO inhibitor. (b) Crystal structure of the covalent complex SARS-CoV-2 MPRO with MAC-5576 (PDB code 7JT0).69
As part of the SARS-CoV-2 MPRO inhibitors with an ester warhead, a series of new 5-chloropyridinyl esters of nonsteroidal anti-inflammatory drugs (NSAIDs, such as salicylic acid, ibuprofen, naproxen, and indomethacin) is of particular interest. Among all of them, (R)-naproxen derivative 45 (Figure 44) with an IC50 value of 0.16 μM was the most interesting compound. In addition, compound 46 (Figure 44), which proved less efficient in the inhibition assay (IC50 = 4.9 μM), exhibited a potent antiviral activity in cellular assays (EC50 = 24 μM in Vero E6 cells). MALDI-TOF analysis was used to demonstrate the ability of the two compounds to covalently bind SARS-CoV-2 MPRO, leading to the irreversible acylation of the enzyme.103
Figure 44.

Chemical structures of 5-chloropyridinyl ester derivatives of nonsteroidal anti-inflammatory agents 45 and 46.103
2.5. Selenium/Sulfur as Electrophilic Warheads: The Case of Bselen/Ebsulfur and Analogues
The possibility of using drugs approved in therapy (well-known substances with proved efficacy and safety in humans) in the treatment of COVID-19 allows one to reduce the time and costs associated with the development of a new molecule.
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one, in Figure 45a), a heterocyclic structure with a selenium atom that has been studied as an antioxidant/anti-inflammatory agent,104,105 was one of the most interesting examples of repurposing of investigational drugs against SARS-CoV-2.
Figure 45.
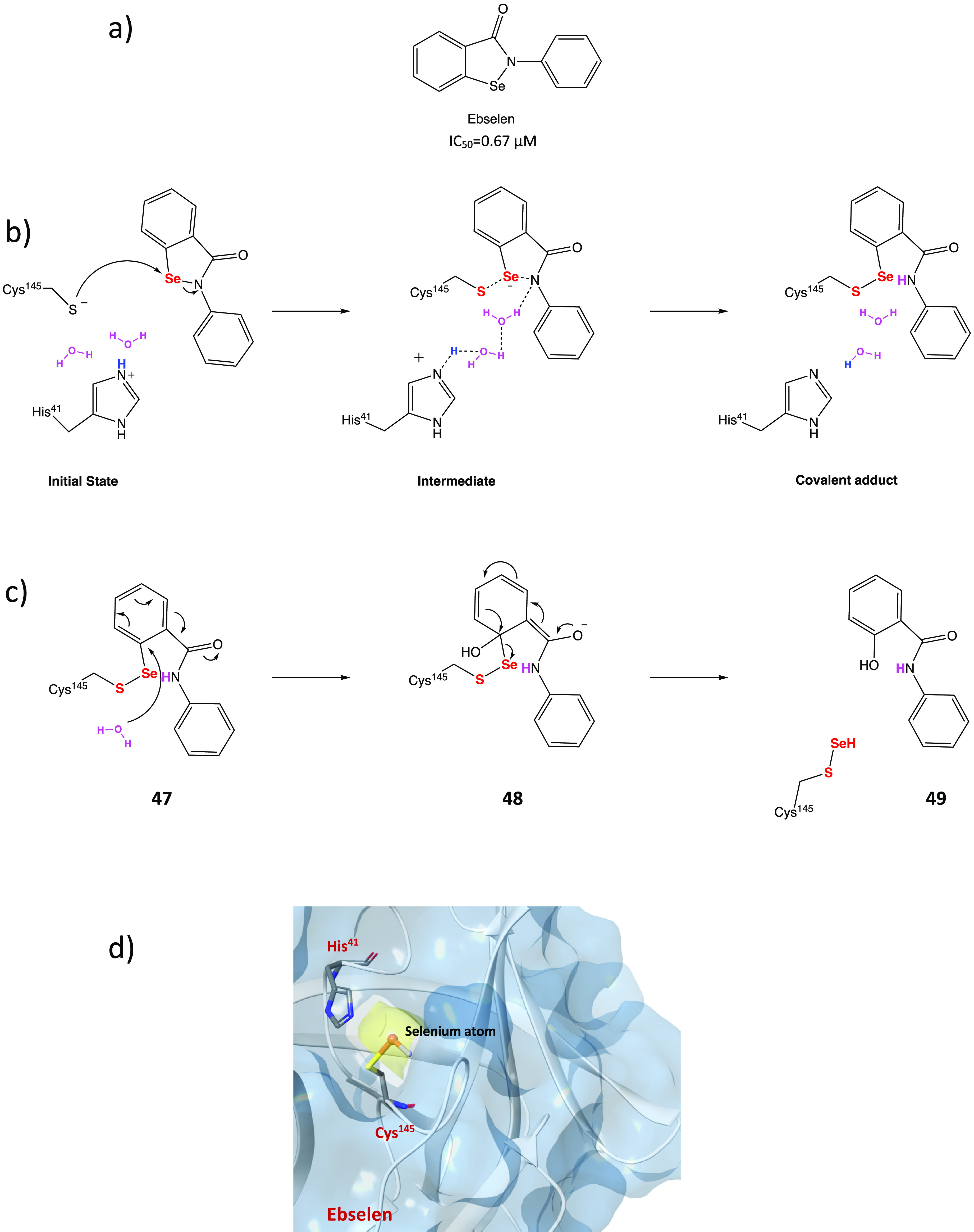
(a) Chemical structure of ebselen. (b) Proposed mechanism of action of ebselen with SARS-CoV-2 MPRO. (c) Proposed mechanism for selenylation of the catalytic site of SARS-CoV-2 MPRO. (d) Crystal structure of the selenylated cysteine145 in the SARS-CoV-2 MPRO binding site (PDB code 7BAK).107−109
Analysis of in silico and in vitro data demonstrated the strong antiviral activity of ebselen (IC50 = 0.67 μM against SARS-CoV-2 MPRO) and the capability to covalently bind the catalytic Cys145 of SARS-CoV-2 MPRO.15,106
To further explore the mechanism of covalent inhibition of this compound, a combination of docking and density functional theory (DFT) protocols clarified the ebselen ability to form a covalent adduct with Cys145 by the formation of a selenyl sulfide bond.107 In Figure 45b the mechanism of action of ebselen is shown: the first step is the activation of the thiol group of Cys145 through deprotonation mediated by His41; subsequently, the activated thiolate performs the nucleophilic attack on the electrophilic selenium atom, determining the opening of the 5-membered ring and the formation of selenyl sulfide bond, responsible for the covalent inhibition of the target. The process is mediated by a molecule of water that acts as a proton transporter.108 A mass spectrometry study suggested an additional rearrangement of the covalent adduct (Figure 45c) consisting in the hydrolysis of the ebselen-SARS-CoV-2 MPRO adduct (47) by a conserved molecule of water, forming an intermediate state (adduct 48), with subsequent selenylation of the cysteine and release of a secondary phenolic product 49. The proposed mechanism of action was confirmed by the X-ray structure shown in Figure 45d (PDB code 7BAK), where the selenium atom is covalently bonded to the cysteinyl sulfur.109
Moreover, ebselen was found to bind an allosteric site between the I and the II domains of MPRO (which is essential for the dimerization process).109,110
In view of the aforementioned data, ebselen and its analogues could represent promising lead compounds for the future development of new and more effective covalent inhibitors. In this light, many efforts have been made to analyze the SAR of the substituted N-phenyl ring.
Indeed, a collection of ebselen-related compounds has been extensively studied. Among them, 50 and 51 (4-nitro- and 5-chloro-2-fluoro derivatives, respectively; Figure 46) displayed inhibitory activity superior to that of ebselen with IC50 values of 27.95 ± 5.10 and 15.24 ± 4.58 nM, respectively.111 According to SAR analysis, the authors demonstrated that the insertion of one or two substituents on the phenyl ring can improve the inhibition activity against SARS-CoV-2 MPRO.111
Figure 46.

Ebselen derivatives 50 and 51.111
Similarly, four ebselen-based compounds 52–55 (Table 1) exhibited greater effectiveness than ebselen both against MPRO (IC50 in the submicromolar range) as well as in the viral cell model of SARS-CoV-2 replication.109
Table 1. Chemical Structures, SARS-CoV-2 MPRO Inhibitory Activity, and Inhibition of Viral Replication Data of Ebselen Derivatives 52–55(109).

To further explore the SAR for this class of compounds, a series of N-phenyl-substituted ebselen derivatives was synthesized. Compounds 56–60 (Figure 47) exhibited submicromolar IC50 values against SARS-CoV-2 MPRO (in the range 0.38–2.77 μM), suggesting that the substitution is favorable in the meta position. As for derivative 59, the most potent compound, the presence of a cyano group in the meta position improves the interaction network with an additional favorable hydrogen bond. Nevertheless, biological assays conducted in an in vitro cellular model of SARS-CoV-2 replication (Vero E6-infected cells) showed that compound 60, which was the least active in the enzymatic inhibition assay, was surprisingly the most effective in blocking viral progression (EC50 = 0.8 μM). For this reason it was selected for further studies in lung organoids, which confirmed the lack of toxicity and its ability to block viral replication.112
Figure 47.

Chemical structures of phenyl-substituted ebselen derivatives 56–60.112
The isosteric analogue of ebselen, ebsulfur, with a sulfur atom in place of selenium (Figure 48), also attracted considerable interest as a covalent SARS-CoV-2 MPRO inhibitor. Docking studies have shown that ebsulfur, in the same way as ebselen, can form a covalent S–S bond with the cysteinyl −SH of SARS-CoV-2 MPRO. On the basis of this assumption, numerous ebselen/ebsulfur analogues as SARS-CoV-2 MPRO inhibitors were designed and biologically evaluated. Among all of them, 61 and 62 were the most interesting with IC50 values of 0.074 and 0.11 μM, respectively. Docking studies proved that the furan group is essential to form additional hydrophobic interactions with Met165, Arg188, Asp187, and Met49. From this study it emerged that ebsulfur as well as ebselen could be a potential lead compound for the development of novel, broad-spectrum anticoronaviral drugs.113
Figure 48.

Chemical structures of ebsulfur and ebselen/ebsulfur analogues 61 and 62.113
The structure–activity relationships of ebsulfur were investigated focusing on three recurring components: the phenyl ring, the linker, and the benzoisothiazolone core. In particular, compound 63 (Figure 49) was found to be the best ebsulfur analogue with covalent inhibitory activity on SARS-CoV-2 MPRO. The phenyl and benzoisothiazolone rings were retained in view of their importance in the interaction with the target protein; instead, introduction of an acetamide group in the linker had a positive effect on the activity, which was confirmed by the remarkable IC50 value of 116 nM against SARS-CoV-2 MPRO.114
Figure 49.

Chemical structure of ebsulfur analogue 63.114
In the same vein, SAR investigation led to the identification of other notable ebsulfur analogues 64, 65, 66, and 67 (Figure 50) with significant IC50 values of 0.32, 0.19, 1.00, and 0.41 μM, respectively, against SARS-CoV-2 MPRO. Further structural optimization involved the introduction of a −Br in both ebsulfur and 59 to obtain the derivatives 68 and 69 (Figure 50), which exhibited an improved inhibitory activity against SARS-CoV-2 MPRO (IC50 = 0.03 and 0.32 μM, respectively) compared to ebsulfur (IC50 = 0.13 μM).115
Figure 50.
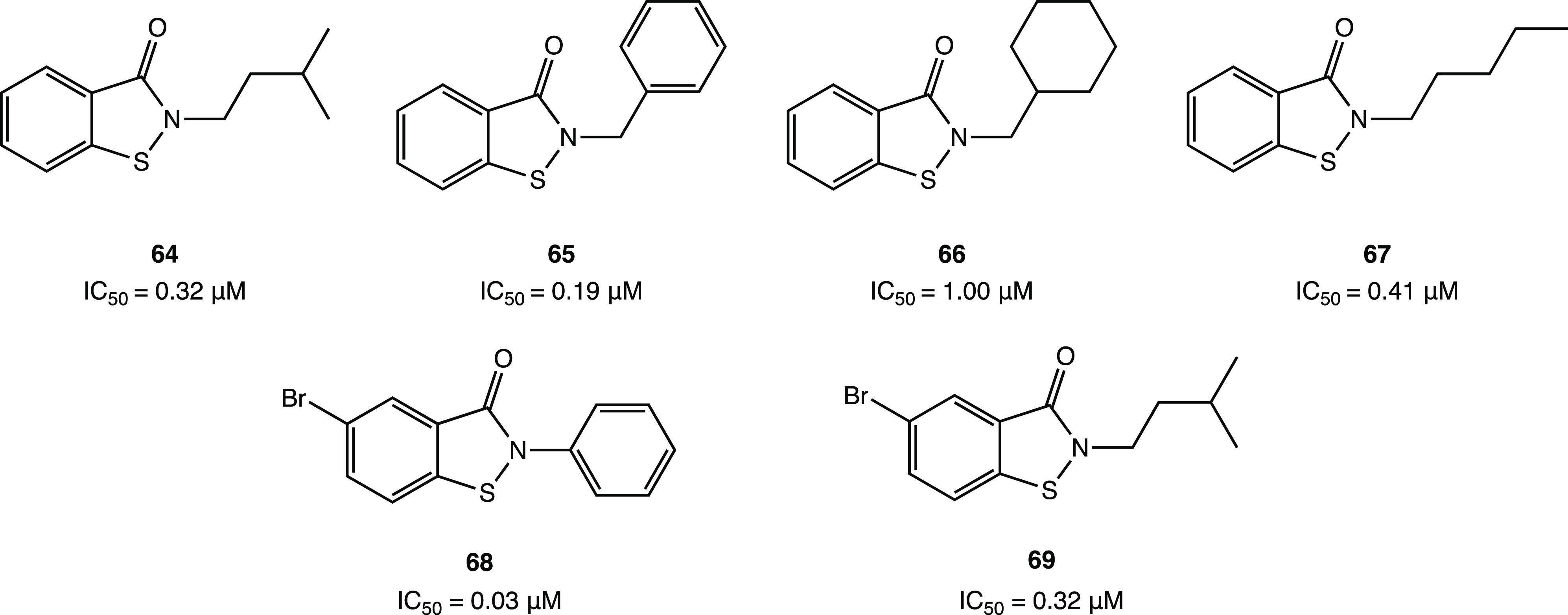
Chemical structures of the promising ebsulfur derivatives 64–69.115
3. Conclusions and Perspectives
SARS-CoV-2 MPRO is a key cysteinyl enzyme for virus replication and consequently human infection. Considering the previous findings on SARS-CoV-2 MPRO-selective inhibitors, covalent inhibition represents one of the most interesting strategies in drug discovery projects. The possibility of linking the nucleophilic cysteinyl −SH to a reactive electrophilic group (warhead) may allow more effective and durable inhibition of the targeted protein.
Given the large number of studies conducted in this field, we decided in the present review to analyze the most relevant and informative examples of covalent SARS-CoV-2 MPRO inhibitors. Specifically, our approach consisted of a primary classification of the studied compounds into eight classes depending on the nature of their electrophilic warheads: aldehyde, ketone, α-ketoamide, Michael acceptor, α-haloacetamide, nitrile, activated ester, and molecules containing an electrophilic selenium/sulfur atom. Particular attention was paid to the description of the mechanism of the catalytic inhibition, highlighting the main features and the reversibility/irreversibility of the covalent adduct formed by each warhead.
Within each class, SAR analyses evaluating the peptidomimetic and nonpeptidomimetic aspects were presented. Among these, the peptidomimetic compounds represent the largest class described in the literature to date due to their intrinsic ability to mimic the natural substrates of SARS-CoV-2 MPRO. The wide number of SARS-CoV-2 MPRO crystal structures in complex with the inhibitors (some of which have been shown in detail as examples) allowed us to highlight the most recurrent structural features and interactions with the target protein.
Table 2 lists the SARS-CoV-2 MPRO inhibitory activity and the main structural moieties (P fragments) of the most interesting compounds. Furthermore, special attention was paid to the stereochemistry of the chiral carbons, which is crucial for the arrangement of the ligands to achieve a better fit to the SARS-CoV-2 MPRO binding pocket. Following the Schechter–Berger notation, the essential structural moieties (P1′ = electrophilic warhead, P1, P2, P3) of each compound were correlated with the SARS-CoV-2 MPRO subregions (S1′, S1, S2, S3/S4). All of the analyzed inhibitors are able to form a covalent adduct with the Cys145 catalytic residue in the S1′ subregion, and several additional H bonds stabilize the ligand–protein complex with the Cys145 and Gly143 backbones and/or the His41 side chain.
Table 2. Analysis of Structural Fragments and Inhibition Data of the Most Active Covalent SARS-CoV-2 MPRO Inhibitors by Following the Schechter–Berger Notation.

Warhead masked as prodrug.
N.A.: Biological data not available.
Regarding the adjacent S1 site, which accepts the Gln residue of the native substrate, the most recurrent fragment in P1 is the five-membered γ-lactamic ring. Indeed, thanks to its capability to mimic the natural amino acid glutamine, it is capable of penetrating deeply into the pocket and forming a stabilizing interaction with key residues (Phe140, His163, His164, Glu166). The stereochemistry of the P1 five-membered γ-lactamic moiety deserves special attention: the two asymmetric carbons have generally the same configurations (S, S). By observing the X-ray structures, it is evident that this orientation within the S1 pocket guarantees an optimal fit and conformation, to be maintained in the design of new inhibitors.
The SARS-CoV-2 MPRO S2 cleft, comprising mainly hydrophobic amino acids (Met49, Tyr54, Met165, Pro168, Val186), appears to be very flexible, allowing it to bind both small and bulky aromatic/alkylic portions (P2), such as cyclohexyl,14,25,70 cyclopropyl,14,29 isoleucine,30,31,37,41,47,50,51,69,70 phenyl,25,27,30,72,73,81 and bicycloalkyl38,40,87 moieties (such as cyclopentylproline and dimethylcyclopropylproline in the analogs of boceprevir and telaprevir). As in the case of the P1 fragment, the S configuration at the asymmetric carbon is pivotal to allow a more favorable orientation within the pocket.
To interact with the hydrophobic pocket of the S3/S4 subregion, most of the covalent SARS-CoV-2 MPRO inhibitors have P3 aromatic moieties, such as indole,116 substituted phenyl (especially with halogens (fluoride,29,38,70,72,81 chloride,29,38,73 or trifluoromethyl groups37)), and an aliphatic portion, such as a tert-butyloxycarbonyl group.14 The most representative interactions occur with the amino acids Gln189, Leu167, Pro168, Gly251, and Asp187 and with the conserved water molecules on the surface of the S4 site.
Moreover, in Table 2 the biological data of the reviewed compounds are reported. Most of the SARS-CoV-2 MPRO inhibitors show IC50 values in the low micromolar/nanomolar range against MPRO and interesting antiviral activity in the cellular infection model. Among all GC-376 (with an aldehydic warhead), PF-00835231 and the corresponding phosphate prodrug PF-07304814 (ketone warheads) have already reached clinical in vivo studies and/or trials.
In particular, nirmatrelvir (PF-07321332), the first covalent SARS-CoV-2 MPRO inhibitor currently approved in the therapy for the COVID-19 treatment with a conditional marketing authorization, was obtained thanks to a lead optimization study starting from the carbonyl compound PF-00835231. Nirmatrelvir is an orally bioavailable peptidomimetic derivative with a nitrile group as an electrophilic warhead P1′, a γ-lactamic ring in P1, a dimethylbicycloproline group in P2, and a trifluoromethyl group in P3, which plays a leading role in stabilizing the covalent thioimidate adduct.
Regarding the nonpeptidomimetic covalent SARS-CoV-2 MPRO inhibitors, they are less explored in the literature but are beginning to emerge, such as the ester, natural, and ebselen/ebsulfur derivatives reported in this review.
Among the natural covalent inhibitors, the quinonoid/flavonoid systems have been extensively studied in silico/in vitro. In particular, myricetin, which exhibits high inhibitory activity (with an IC50 of 0.2–0.6 μM against MPRO and an EC50 value of 8 μM in infected Vero E6 cells) and the ability to alleviate the overall inflammation in the in vivo mice lung injury model acts as a Michael acceptor through its hidden electrophilic pyrogallic moiety. The α,β-unsaturated carbonyl group is released following the pyrogallol oxidation step. The proposed irreversible mechanism of inhibition is confirmed by the solved X-ray crystal structure in complex with SARS-CoV-2 MPRO, which shows the covalent bond between the C-6′ of myricetin and the catalytic Cys145.
Another interesting class of nonpeptidomimetic derivatives is the ester SARS-CoV-2 MPRO inhibitors, whose mechanism of action involves irreversible acylation of the enzyme following nucleophilic attack of Cys145 on the electrophilic carbonyl of the ester.
The presence of the chloro-substituted pyridinyl group is observed in all of the analyzed compounds and represents an essential pharmacophoric moiety; furthermore, maintaining the chloro-pyridinyl portion, the class of indole esters has been extensively examined in the literature as SARS CoV-2 MPRO inhibitors, Table 3.
Table 3. Analysis of Structural Fragments and Inhibition Data of the Most Active Ester SARS-CoV-2 MPRO Inhibitors.

Finally, ebselen and ebsulfur, heterocyclic structures containing a selenium/sulfur atom, represent one of the most interesting examples of repurposing of investigational compounds against SARS-CoV-2 with potent antiviral activity and capability to covalently bind the catalytic Cys145 of SARS-CoV-2 MPRO.
In view of these considerations, ebselen, ebsulfur, and their analogues could represent promising lead compounds for the future development of new and more effective covalent inhibitors.
In particular, N-phenyl ring substitutions improve the inhibition activity against SARS-CoV-2 MPRO and lead to compounds with submicromolar IC50 values against SARS-COV-2 MPRO (in the range 0.38–2.77 μM).
As highlighted in the previous sections, we reported some examples of ligand/structure-based, hybrid or nonhybrid, virtual screenings, and new computational approaches that led to biologically interesting compounds. In some cases, the integration of noncovalent and covalent docking protocols led to compounds with optimal interaction with both the catalytic cysteine and the other amino acids residues in the clefts of the binding site.117,118 Furthermore, QSAR and/or pharmacophore modeling approaches combined with molecular docking, MD simulations, and free binding energy MM/PBSA enabled the development of new derivatives with promising covalent SARS-CoV-2 MPRO inhibitory activity.26,67,68,70−72,83
In addition to the common techniques, quantum mechanics/molecular mechanics (QM/MM) calculations, although less common, are gradually becoming reliable computational approaches in the design of covalent inhibitors.24 These techniques, capable of simulating the chemical reactions occurring in enzymes at the atomistic/molecular level, are suitable for elucidating the mechanism of action, kinetics, and thermodynamics of covalent modification in a target protein.119 In particular, their application helped to clarify the covalent inhibition mechanism of SARS-COV-2 MPRO and the proton transfer process during the nucleophilic attack of the catalytic dyad.120−122
Furthermore, innovative techniques have been developed, such as the combined protocol of Advanced Deep Q-learning Network and Fragment-Based Drug Design (ADQN-FBDD), artificial intelligence (AI) with structure-based drug design (SBDD),64 and the new computational pipeline covalentizer,72 which aims to accelerate the generation of potential lead compounds and the identification of irreversible inhibitors.
In light of these considerations, we believe that an optimal integration and optimization of all of these available in silico methods could represent a crucial challenge to increasing the hit/lead compound identification rate in the current pandemic.118,123
3.1. Kinetic Covalent Inhibition
Ki values describe the exact binding affinity between the inhibitor and enzyme and provide information about the formation of the covalent complex (E–I). Determination of Ki and/or Kinact/Ki values, which reflect the binding affinity of drugs, is a prerequisite for the prediction and evaluation of drug–enzyme interactions. An important factor to consider in the development of a therapeutic protease inhibitor is the reversibility of the binding E–I complex. Irreversible protease inhibitors can achieve long-lasting effects by permanently blocking proteases and covalently modified proteins.
Table 4 lists the enzyme inhibition data (Ki or/and Kinact/Ki) of covalent SARS-CoV-2 MPRO inhibitors reported in the literature and their SARS-CoV-2 MPRO inhibitory and antiviral activity (IC50 (μM), EC50 (μM) and ratio of EC50/IC50).
Table 4. Inhibition (IC50), Antiviral (EC50) Activity, Ratio of EC50/IC50, and Equilibrium-Binding Constants Ki and Kinact/Ki (if available) of SARS-CoV-2 MPRO Covalent Inhibitorsa.
| compound | IC50 (μM) | EC50 (μM) | EC50/IC50 | Ki (μM) or/and Kinact/Ki (M–1 s–1) | ref |
|---|---|---|---|---|---|
| aldehyde warhead | |||||
| GC-376* | 0.19 | 0.92 | 4.84 | 0.094 μM | (30,31) |
| 2.84 × 104 M–1 s–1 | |||||
| UAWJ247* | 0.045 | 2.06 | 45.78 | 0.035 μM | (30) |
| UAWJ9-36-1 | 0.051 | 2.56 | 50.2 | 8.5 × 104 M–1 s–1 | (40) |
| UAWJ9-36-3 | 0.054 | 0.37 | 6.85 | 9.3 × 104 M–1 s–1 | (40) |
| ketone warhead | |||||
| PF-00835231 | 0.0069 | 2.7 × 10–4 μM | (47) | ||
| YH-53 | 4.2 | 0.034 μM | (51) | ||
| α-ketoamide warhead | |||||
| boceprevir | 1.59 | 1.90 | 1.19 | 1.18 μM | (32) |
| UAWJ246 | 0.045 | 4.61 | 102.44 | 0.036 μM | (30) |
| UAWJ248 | 11.1 | 0.013 μM | (30) | ||
| 9.1 × 104 M–1 s–1 | |||||
| Michael acceptor group as warhead | |||||
| N3 | 16.77 | 1.1 × 104 M–1 s–1 | (5,15,66) | ||
| 29 | 2.86 | 38.36 μM | (72) | ||
| 18 × 104 M–1 s–1 | |||||
| 30 | 0.42 | 4.5 μM | (73) | ||
| 31 | 0.17 | 2.3 μM | (73) | ||
| myricetin | 0.2–0.6 | 8.0 | 20 | 15.73 μM | (79) |
| 7.0 × 102 M–1 s–1 | |||||
| α-haloacetamide warhead | |||||
| 36 | 0.4 | 16 μM | (73) | ||
| 38 | 0.43 | 2.05 | 4.76 | 8.2 × 102 M–1 s–1 | (81) |
| 39 | 0.08 | 2.15 | 26.87 | 7.1 × 103 M–1 s–1 | (81) |
| 40 | 0.056 | 1.34 μM | (81,82) | ||
| 4.2 × 103 M–1 s–1 | |||||
| nitrile warhead | |||||
| nirmatrelvir | 0.019 | 0.075 | 3.94 | 0.003 μM | (88,89) |
| ester warhead | |||||
| GRL-1720 | 0.32 | 15 | 46.87 | 2.15 μM | (52,100,101) |
| 1.9 × 104 M–1 s–1 | |||||
| GRL-0820 | 0.073 | 15 | 205.47 | 0.073 μM | (52,101) |
| 44 | 0.0342 | 2.9 × 104 M–1 s–1 | (102) | ||
| selenium/sulfur as electrophilic warhead | |||||
| 61 | 0.074 | 0.031 μM | (113) | ||
| 1.13 × 105 M–1 s–1 | |||||
| 62 | 0.11 | 0.078 μM | (113) | ||
| 7.44 × 104 M–1 s–1 | |||||
Warhead masked as prodrug.
Considering α-ketoamide and carbonyl warheads, such as aldehydes and ketones, which can act as both reversible and irreversible derivatives, the enzymatic kinetic studies showed that compounds UAWJ246 (IC50 = 0.045 μM, EC50 = 4.61 μM, and ratio of EC50/IC50 = 102.44) and UAWJ247 (IC50 = 0.045 μM, EC50 = 2.06 μM, and ratio of EC50/IC50 = 45.78) reversibly bound the catalytic cysteine of SARS-CoV-2 MPRO with inhibition constant ki values of 0.036 ± 0.007 and 0.035 ± 0.008 μM, respectively. On the other hand, compounds UAWJ248 (EC50 = 11.1 μM) and GC-376 (IC50 = 0.19 μM, EC50 = 0.92 μM, and ratio of EC50/IC50 = 4.84), after an initial reversible binding complex with the enzyme, determined an irreversible inactivation with Kinact/Ki values of 9.04 × 104 and 2.84 × 104 M–1 s–1, respectively, indicating a notable potency of UAWJ248.30
Derivatives with a Michael acceptor group as a warhead are generally irreversible SARS-CoV-2 MPRO inhibitors. Kinetics data for compounds N3 (EC50 = 16.77 μM) and myricetin (IC50 = 0.2–0.6 μM, EC50 = 8.0 μM, and ratio of EC50/IC50 = 20) demonstrated an irreversible two-step mechanism for the inhibition of the SARS-CoV-2 MPRO. N3 exhibits a very potent inhibitory effect, making unachievable the Ki and Kinact measurement. Subsequently, a pseudo-second-order rate constant was determined, showing that N3 acts as a time-dependent irreversible inhibitor with Kinact/Ki values of 1.1 × 104 M–1 s–1.5,15,66 The covalent inhibitor myricetin shows a Ki value of 15.73 μM and a Kinact value of 0.011 s–1. The Ki value indicates that myricetin binds selectively to the binding pocket of the protease, providing the basis for irreversible covalent binding between myricetin and Cys145. The experimental Kinact value suggests that myricetin could react rapidly with the Cys145. Overall, considering the small size of myricetin, it is an efficient covalent binder of SARS-CoV-2 MPRO with a Kinact /Ki of 701.88 M–1 s–1.79
α-Haloacetamide derivatives are also irreversible SARS-CoV-2 MPRO inhibitors. In particular, enzymatic kinetic studies for compounds 38 (IC50 = 0.43 μM, EC50 = 2.05 μM, and ratio of EC50/IC50 = 2.76) and 39 (IC50 = 0.08 μM, EC50 = 2.15 μM, and ratio of EC50/IC50 = 26.87) proposed a two-step process for inhibition of SARS-CoV-2 MPRO: the first step is a reversible binding adduct, and the second step is an irreversible binding complex; these results were in agreement with the expected mechanism of action in which compounds 38 and 39 form a covalent bond with the catalytic Cys145.81,102 Compound 40 (IC50 = 0.056 μM) inhibited MPRO in a time-dependent manner, indicating an irreversible mode of action with a ki value of 1.34 μM and Kinact/ki value of 4167 M–1 s–1.82
Among the major covalent inhibitors of SARS-CoV-2 MPRO, nirmatrelvir, with a nitrile as the electrophilic warhead, showed remarkable biological data (IC50 = 0.019 μM, EC50 = 0.075 μM, and ratio of EC50/IC50 = 3.94) related to a notable ki value of 0.003 μM.88,89
Compounds with an electrophilic ester warhead provide an irreversible inhibitory mechanism of SARS-CoV-2 MPRO. GRL-1720 and GRL-0820 (ki values of 2.15 and 0.073 μM, respectively) exhibit potent MPRO inhibitory activity, have second-order rate inactivation constants, and demonstrate a time-dependent inhibition. The mode of action involves acylation of Cys145 in the active site, supported by the catalytic dyad.52,102,124
Ebselen and ebsulfur derivatives exhibited a concentration- and time-dependent inhibition pattern against MPRO with a biphasic character, suggesting that the rate of inactivation follows pseudo-first-order rate kinetics, implying irreversible covalent inhibition;113 among them, compounds 61 and 62 show ki values of 0.031 and 0.078 μM and Kinact/Ki values of 1.13 × 105 and 7.44 × 104 M–1 s–1, respectively.
In conclusion, in the present work, we provided a detailed analysis of the recent articles published in the literature in the last 3 years (2020–2022), focusing on the peptidomimetic and nonpeptidomimetic aspects of the covalent inhibitors against the SARS-CoV-2 main protease. As an update of previously published articles, we report only the in silico, in vitro, and in vivo biological data of the latest generation of inhibitors that have shown potent and high selectivity against the viral SARS-CoV-2 Main protease with a specific covalent inhibitory mechanism. Concentrating the attention on the electrophilic warheads, which are the pivotal moieties for the formation of the covalent adduct and the consequent reversible/irreversible mechanism of inhibition, we classified the studied molecules into eight different categories based on their reactive electrophilic groups and showed for each class the kinetic mechanism involved in the formation of the covalent adduct between the inhibitor and the catalytic Cys145 of the SARS-CoV-2 main protease. In particular, to better understand the 3D binding orientations of the ligands into the enzymatic binding pocket, we reported several X-ray crystallographic images and the corresponding list of PDB codes of the complexes. In addition, the analyses of the most recurrent pharmacophoric moieties and stereochemistry of the chiral carbons were reported to identify the most suitable molecular fragments (P1′, P1, P2, P3) for interaction with the crucial subregions (S1′, S1, S2, S3/S4) of the MPRO catalytic cleft. In Tables 2 and 3, according to the Schechter–Berger notation, we analyzed and compared the pharmacophoric moieties that are crucial for the formation of the ligand/enzyme complex during the first reversible step of the inhibition mechanism. To find a correlation with the biological data IC50 and EC50 of the most active covalent inhibitors, the available values of the equilibrium binding constants Ki and Kinact/Ki were listed in Table 4.
Acknowledgments
This work was supported by a grant from the University of Palermo, Italy (Grant PJ_RIC_FFABR_2017_161161)
Glossary
Abbreviations Used
- 3CLPRO
3C-like protease
- ABPP
activity-based protein profiling
- ADME
absorption, distribution, metabolism, and excretion
- ADQN-FBDD
advanced deep Q-learning network and fragment-based drug design
- AI
artificial intelligence
- Cbz
benzyloxycarbonyl
- CFA
α-chloro-fluoroacetamide
- cLogP
calculated LogP
- COVID-19
coronavirus disease-19
- CPE
cytopathic effect
- DFT
density functional theory
- EC50
half-maximal effective concentration
- EMA
European Medicines Agency
- FCoV
feline coronavirus
- FDA
Food and Drug Administration
- GI
gastrointestinal
- HMK
hydroxymethylketone
- IC50
half-maximum inhibitory concentration
- IL-6
interleukin 6
- Ki
inhibition constant
- MPRO
main protease
- MALDI-TOF
matrix-assisted laser desorption ionization-time of flight
- MD
molecular dynamics
- MERS-CoV
Middle East respiratory syndrome coronavirus
- MM/PBSA
molecular mechanics Poisson–Boltzmann surface area
- nM
nanomolar
- Nsp
nonstructural proteins
- PDB
Protein Data Bank
- QM/MM
quantum mechanics/molecular mechanics
- QSAR
quantitative structure–activity relationship
- SAR
structure–activity relationship
- SARS-CoV-1
severe acute respiratory syndrome coronavirus 1
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SBDD
structure-based drug design
- TNF-α
tumor necrosis factor α
Biographies
Gabriele La Monica graduated in Chemistry and Pharmaceutical Technology with full marks and honors at the University of Palermo (2019). He is a Ph.D. student in Medicinal Chemistry (University of Palermo) under the supervision of Prof. A. Lauria and A. Martorana, focusing his research on the design and synthesis of new heterocyclic small molecules endowed with biological activities. He joined the research group of Dr. Thomas Lanyon-Hogg as a visiting student at the Department of Pharmacology, University of Oxford.
Alessia Bono graduated in Chemistry and Pharmaceutical Technology with full marks and honors at the University of Palermo (2021). She is a Ph.D. student in Molecular and Biomolecular Science at the University of Palermo under the supervision of Prof. A. Lauria and A. Martorana. Her research interests are focused on the design and synthesis of new heterocyclic small molecules with biological activities.
Antonino Lauria graduated in Chemistry with full marks and honors at the University of Palermo (1994). In 1996, he became Assistant Professor, and in 2003–200,4 he joined the research group of Prof. Alan R. Katritzky at the University of Florida in Gainesville (USA), working on studies on solubility through computational chemistry. His scientific activities are mainly devoted to the design, synthesis, chemical behavior, and structure–activity relationships (SARs) of new heterocyclic compounds of biological interest. In particular, his most recent studies are devoted to the development of new classes of compounds structurally related to well-known anticancer drugs. In 2021, he became Full Professor in Medicinal Chemistry.
Annamaria Martorana graduated in Chemistry and Pharmaceutical Technology with full marks and honors at the University of Palermo (2001). In 2005, she obtained her Ph.D. degree in Medicinal Chemistry. In 2006, she became a lecturer at the University of Palermo. She joined the research group of Prof. Malcolm F. G. Stevens, Cancer Research, Centre of Biomolecular Sciences, University of Nottingham (UK) (2005–2006) and worked on the synthesis of new analogues of Temozolomide. Since 2019, she has been an associate professor at the University of Palermo, Italy. Her scientific activities focus on the design, synthesis, and biological evaluation of new small molecules for targeted therapies. Her recent studies are dedicated to the development of new classes of anticancer and antiviral agents.
Author Contributions
† G.L.M. and A.B.: Cofirst authorship.
The authors declare no competing financial interest.
References
- Wang L.; Wang Y.; Ye D.; Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta D.; Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020, 91 (1), 157–160. 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy S.; Swain B.; Verma R. S.; Gunthe S. S. SARS-CoV, MERS-CoV, and 2019-nCoV viruses: an overview of origin, evolution, and genetic variations. VirusDis. 2020, 31 (4), 411–423. 10.1007/s13337-020-00632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian L.; Gao F.; Zhang J.; He Q.; Mao Q.; Xu M.; Liang Z. Effects of SARS-CoV-2 variants on vaccine efficacy and response strategies. Expert Rev. Vaccines 2021, 20, 365–373. 10.1080/14760584.2021.1903879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella A.; Scala A.; Piperno A.; Micale N. SARS-CoV-2 Mpro: A potential target for peptidomimetics and small-molecule inhibitors. Biomolecules 2021, 11, 607. 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Du W.; Song M.; Liu Q.; Herrmann A.; Huang Q. Spontaneous binding of potential COVID-19 drugs (camostat and nafamostat) to human serine protease TMPRSS2. Comput. Struct Biotechnol J. 2021, 19, 467–476. 10.1016/j.csbj.2020.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Cao R.; Zhang H.; Liu J.; Xu M.; Hu H.; Li Y.; Zhao L.; Li W.; Sun X.; Yang X.; Shi Z.; Deng F.; Hu Z.; Zhong W.; Wang M. The anti-influenza virus drug, Arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discovery 2020, 6, 28. 10.1038/s41421-020-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juurlink D. N. Safety considerations with chloroquine, hydroxychloroquine and azithromycin in the management of SARS-CoV-2 infection. CMAJ. 2020, 192, E450–E453. 10.1503/cmaj.200528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; De Giglio M. A. R.; Roviello G. N. SARS-CoV-2: Recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. 10.2174/0929867327666200416131117. [DOI] [PubMed] [Google Scholar]
- Nguyen H. L.; Thai N. Q.; Truong D. T.; Li M. S. Remdesivir strongly binds to both RNA-dependent RNA polymerase and main protease of SARS-CoV-2: evidence from molecular simulations. J. Phys. Chem. B 2020, 124, 11337–11348. 10.1021/acs.jpcb.0c07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S.; Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi A. A. T.; Fatima K.; Mohammad T.; Fatima U.; Singh I. K.; Singh A.; Atif S. M.; Hariprasad G.; Hasan G. M.; Hassan M. I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol. Basis Dis 2020, 1866, 165878. 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E.; Rosa M.; Anjum R.; Mehmood S.; Soban M.; Mujtaba M.; Bux K.; Moin S. T.; Tanweer M.; Dantu S.; Pandini A.; Yin J.; Ma H.; Ramanathan A.; Islam B.; Mey A.; Bhowmik D.; Haider S. Dynamic profiling of β-coronavirus 3CL Mpro protease ligand-binding sites. J. Chem. Inf Model 2021, 61, 3058–3073. 10.1021/acs.jcim.1c00449. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Shindo N.; Ojida A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386. 10.1016/j.bmc.2021.116386. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Lv S.; Peng L.; Xie C.; Gao L.; Sun H.; Lin L.; Ding K.; Li Z. Development and application of novel electrophilic warheads in target identification and drug discovery. Biochem. Pharmacol. 2021, 190, 114636. 10.1016/j.bcp.2021.114636. [DOI] [PubMed] [Google Scholar]
- Gehringer M.; Laufer S. A. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J. Med. Chem. 2019, 62, 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Yang H.; Xie W.; Xue X.; Yang K.; Ma J.; Liang W.; Zhao Q.; Zhou Z.; Pei D.; Ziebuhr J.; Hilgenfeld R.; Yuen K. Y.; Wong L.; Gao G.; Chen S.; Chen Z.; Ma D.; Bartlam M.; Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Kaliya K.; Maurya S. K. Recent advances in the discovery of potent proteases inhibitors targeting the SARS coronaviruses. Curr. Top Med. Chem. 2021, 21, 307–328. 10.2174/1568026620999201111160035. [DOI] [PubMed] [Google Scholar]
- Halford B. The path to paxlovid. ACS Central Science 2022, 8, 405. 10.1021/acscentsci.2c00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfizer’s novel COVID-19 oral antiviral treatment candidate reduced risk of hospitalization or death by 89% in interim analysis of phase 2/3 EPIC-HR study; https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate.
- COVID-19: EMA recommends conditional marketing authorisation for paxlovid; https://www.ema.europa.eu/en/news/covid-19-ema-recommends-conditional-marketing-authorisation-paxlovid.
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Unraveling the SARS-CoV-2 main protease mechanism using multiscale methods. ACS Catal. 2020, 10, 12544–12554. 10.1021/acscatal.0c03420. [DOI] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X. M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.; Xu Y.; Yang H.; Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Multiscale simulations of SARS-CoV-2 3CL protease inhibition with aldehyde derivatives. Role of protein and inhibitor conformational changes in the reaction mechanism. ACS Catal. 2021, 11, 4157–4168. 10.1021/acscatal.0c05522. [DOI] [PubMed] [Google Scholar]
- Dai W.; Jochmans D.; Xie H.; Yang H.; Li J.; Su H.; Chang D.; Wang J.; Peng J.; Zhu L.; Nian Y.; Hilgenfeld R.; Jiang H.; Chen K.; Zhang L.; Xu Y.; Neyts J.; Liu H. Design, synthesis, and biological evaluation of peptidomimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65 (4), 2794–2808. 10.1021/acs.jmedchem.0c02258. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Lovell S.; Tiew K. C.; Mandadapu S. R.; Alliston K. R.; Battaile K. P.; Groutas W. C.; Chang K. O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol 2012, 86, 11754–11762. 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W.; Fischer C.; Khan M. B.; van Belkum M. J.; Lamer T.; Willoughby K. D.; Lu J.; Arutyunova E.; Joyce M. A.; Saffran H. A.; Shields J. A.; Young H. S.; Nieman J. A.; Tyrrell D. L.; Lemieux M. J.; Vederas J. C. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021, 222, 113584. 10.1016/j.ejmech.2021.113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco M. D.; Ma C.; Lagarias P.; Gao A.; Townsend J. A.; Meng X.; Dube P.; Zhang X.; Hu Y.; Kitamura N.; Hurst B.; Tarbet B.; Marty M. T.; Kolocouris A.; Xiang Y.; Chen Y.; Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, abe0751. 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W.; Khan M. B.; Fischer C.; Arutyunova E.; Lamer T.; Shields J.; Saffran H. A.; McKay R. T.; van Belkum M. J.; Joyce M. A.; Young H. S.; Tyrrell D. L.; Vederas J. C.; Lemieux M. J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ma C.; Szeto T.; Hurst B.; Tarbet B.; Wang J. Boceprevir, calpain Inhibitors II and XII, and GC-376 have broad-spectrum antiviral activity against coronaviruses. ACS Infect Dis 2021, 7, 586–597. 10.1021/acsinfecdis.0c00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G.; Gosalia D. N.; Rennekamp A. J.; Reeves J. D.; Diamond S. L.; Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11876–11881. 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Ratia K. M.; Richner J. M.; Thatcher G. R. J.; Kadam R.; Smieszek S. P.; Przychodzen B. P.; Koprivica V.; Birznieks G.; Polymeropoulos M. H.; Prabhakar B. S. Dual inhibition of cathepsin L and 3CL-Pro by GC-376 constrains SARS-CoV-2 infection Including Omicron variant. bioRxiv 2022, 479835. 10.1101/2022.02.09.479835. [DOI] [Google Scholar]
- Cáceres C. J.; Cardenas-Garcia S.; Carnaccini S.; Seibert B.; Rajao D. S.; Wang J.; Perez D. R. Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci. Rep 2021, 11, 9609. 10.1038/s41598-021-89013-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Iketani S.; Zask A.; Khanizeman N.; Bednarova E.; Forouhar F.; Fowler B.; Hong S. J.; Mohri H.; Nair M. S.; Huang Y.; Tay N. E. S.; Lee S.; Karan C.; Resnick S. J.; Quinn C.; Li W.; Shion H.; Xia X.; Daniels J. D.; Bartolo-Cruz M.; Farina M.; Rajbhandari P.; Jurtschenko C.; Lauber M. A.; McDonald T.; Stokes M. E.; Hurst B. L.; Rovis T.; Chavez A.; Ho D. D.; Stockwell B. R. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. 10.1038/s41467-022-29413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.; Li Y. S.; Zeng R.; Liu F. L.; Luo R. H.; Huang C.; Wang Y. F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z.; Zou Q.; Zhang H.; Qu W.; Long Y.; Li M.; Yang R.; Liu X.; You J.; Zhou Y.; Yao R.; Li W.; Liu J.; Chen P.; Liu Y.; Lin G.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G.; Li W.; Wei Y.; Zheng Y.; Lei J.; Yang S. SARS-CoV-2 M pro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D.; Tong J. B.; Zhang X.; Xiao X. C.; Bian S. Computational strategies towards developing novel SARS-CoV-2 Mpro inhibitors against COVID-19. J. Mol. Struct. 2022, 1247, 131378. 10.1016/j.molstruc.2021.131378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z.; Sacco M.; Hu Y.; Ma C.; Meng X.; Zhang F.; Szeto T.; Xiang Y.; Chen Y.; Wang J. Rational design of hybrid SARS-CoV-2 main protease inhibitors guided by the superimposed cocrystal structures with the peptidomimetic inhibitors GC-376, telaprevir, and boceprevir. ACS Pharmacol Transl Sci. 2021, 4, 1408–1421. 10.1021/acsptsci.1c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Ma X. R.; Ma Y.; Alugubelli Y. R.; Scott D. A.; Vatansever E. C.; Drelich A. K.; Sankaran B.; Geng Z. Z.; Blankenship L. R.; Ward H. E.; Sheng Y. J.; Hsu J. C.; Kratch K. C.; Zhao B.; Hayatshahi H. S.; Liu J.; Li P.; Fierke C. A.; Tseng C.-T. K.; Xu S.; Liu W. R. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors. ChemMedChem 2021, 16, 942–948. 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X. R.; Alugubelli Y. R.; Ma Y.; Vatansever E. C.; Scott D. A.; Qiao Y.; Yu G.; Xu S.; Liu W. R. MPI8 is potent against SARS-CoV-2 by inhibiting dually and selectively the SARS-CoV-2 main protease and the host cathepsin L. ChemMedChem. 2022, 17, e202100456. 10.1002/cmdc.202100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi E.; Kuzikov M.; Esposito F.; Albani S.; Demitri N.; Giabbai B.; Camasta M.; Tramontano E.; Rossetti G.; Zaliani A.; Storici P. Structural and biochemical analysis of the dual inhibition of MG-132 against SARS-CoV-2 main protease (Mpro/3CLpro) and human cathepsin-L. Int. J. Mol. Sci. 2021, 22, 11779. 10.3390/ijms222111779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzikov M.; Costanzi E.; Reinshagen J.; Esposito F.; Vangeel L.; Wolf M.; Ellinger B.; Claussen C.; Geisslinger G.; Corona A.; Iaconis D.; Talarico C.; Manelfi C.; Cannalire R.; Rossetti G.; Gossen J.; Albani S.; Musiani F.; Herzog K.; Ye Y.; Giabbai B.; Demitri N.; Jochmans D.; Jonghe S. D.; Rymenants J.; Summa V.; Tramontano E.; Beccari A. R.; Leyssen P.; Storici P.; Neyts J.; Gribbon P.; Zaliani A. Identification of inhibitors of SARS-CoV-2 3CL-Pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. 10.1021/acsptsci.0c00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Kim Y.; Bickmeier N.; Rathnayake A. D.; Nguyen H. N.; Zheng J.; Kashipathy M. M.; Baird M. A.; Battaile K. P.; Lovell S.; Perlman S.; Chang K.; Groutas W. C. Structure-guided design of conformationally constrained cyclohexane inhibitors of severe acute respiratory syndrome coronavirus-2 3CL protease. J. Med. Chem. 2021, 64, 10047–10058. 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Plassche M. A. T.; Barniol-Xicota M.; Verhelst S. H. L. Peptidyl acyloxymethyl ketones as activity-based probes for the main protease of SARS-CoV-2. Chembiochem 2020, 21, 3383–3388. 10.1002/cbic.202000371. [DOI] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J. Jr; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J. 3rd; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries M.; Mohamed A. S.; Prescott R. A.; Valero-Jimenez A. M.; Desvignes L.; O’Connor R.; Steppan C.; Devlin J. C.; Ivanova E.; Herrera A.; Schinlever A.; Loose P.; Ruggles K.; Koralov S. B.; Anderson A. S.; Binder J.; Dittmann M. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CL pro inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol 2021, 95 (10), e01819-20. 10.1128/JVI.01819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boras B.; Jones R. M.; Anson B. J.; Arenson D.; Aschenbrenner L.; Bakowski M. A.; Beutler N.; Binder J.; Chen E.; Eng H.; Hammond H.; Hammond J.; Haupt R. E.; Hoffman R.; Kadar E. P.; Kania R.; Kimoto E.; Kirkpatrick M. G.; Lanyon L.; Lendy E. K.; Lillis J. R.; Logue J.; Luthra S. A.; Ma C.; Mason S. W.; McGrath M. E.; Noell S.; Obach R. S.; O’Brien M. N.; O’Connor R.; Ogilvie K.; Owen D.; Pettersson M.; Reese M. R.; Rogers T. F.; Rosales R.; Rossulek M. I.; Sathish J. G.; Shirai N.; Steppan C.; Ticehurst M.; Updyke L. W.; Weston S.; Zhu Y.; White K. M.; García-Sastre A.; Wang J.; Chatterjee A. K.; Mesecar A. D.; Frieman M. B.; Anderson A. S.; Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID-19. Nat. Commun. 2021, 12, 6055. 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.; Belovodskiy A.; Hena M.; Kandadai A. S.; Joyce M. A.; Saffran H. A.; Shields J. A.; Khan M. B.; Arutyunova E.; Lu J.; Bajwa S. K.; Hockman D.; Fischer C.; Lamer T.; Vuong W.; van Belkum M. J.; Gu Z.; Lin F.; Du Y.; Xu J.; Rahim M.; Young H. S.; Vederas J. C.; Tyrrell D. L.; Lemieux M. J.; Nieman J. A. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2022, 65 (4), 2905–2925. 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S.; Kobayashi K.; Senda M.; Funai Y.; Seki Y.; Tamai I.; Schäkel L.; Sakata K.; Pillaiyar T.; Taguchi A.; Taniguchi A.; Gütschow M.; Müller C. E.; Takeuchi K.; Hirohama M.; Kawaguchi A.; Kojima M.; Senda T.; Shirasaka Y.; Kamitani W.; Hayashi Y. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022, 65 (4), 2926–2939. 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- Hattori S. I.; Higashi-Kuwata N.; Hayashi H.; Allu S. R.; Raghavaiah J.; Bulut H.; Das D.; Anson B. J.; Lendy E. K.; Takamatsu Y.; Takamune N.; Kishimoto N.; Murayama K.; Hasegawa K.; Li M.; Davis D. A.; Kodama E. N.; Yarchoan R.; Wlodawer A.; Misumi S.; Mesecar A. D.; Ghosh A. K.; Mitsuya H. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella A.; Gentile D.; Rescifina A.; Piperno A.; Mognetti B.; Gribaudo G.; Sciortino M. T.; Holzer W.; Pace V.; Micale N. Pseudo-dipeptide bearing α,α-difluoromethyl ketone moiety as electrophilic warhead with activity against coronaviruses. Int. J. Mol. Sci. 2021, 22, 1398. 10.3390/ijms22031398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov D.; Baev D.; Kalinin M.; Dalinger A.; Chirkova V.; Belenkaya S.; Khvostov A.; Krut’Ko D.; Medved’Ko A.; Volosnikova E.; Sharlaeva E.; Shanshin D.; Tolstikova T.; Yarovaya O.; Maksyutov R.; Salakhutdinov N.; Vatsadze S. Design and evaluation of bispidine-based SARS-CoV-2 main protease inhibitors. ACS Med. Chem. Lett. 2022, 13 (1), 140–147. 10.1021/acsmedchemlett.1c00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Kusov Y.; Nian Y.; Ma Q.; Wang J.; von Brunn A.; Leyssen P.; Lanko K.; Neyts J.; de Wilde A.; Snijder E. J.; Liu H.; Hilgenfeld R. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Banerjee S. An insight into the interaction between α-ketoamide-based inhibitor and coronavirus main protease: a detailed in silico study. Biophys Chem. 2021, 269, 106510. 10.1016/j.bpc.2020.106510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal D.; Warshel A. Exploring the mechanism of covalent inhibition: simulating the binding free energy of α-ketoamide inhibitors of the main protease of SARS-CoV-2. Biochemistry 2020, 59, 4601–4608. 10.1021/acs.biochem.0c00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Pitsillou E.; Karagiannis C.; Darmawan K. K.; Ng K.; Hung A.; Karagiannis T. C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. 10.1016/j.compbiolchem.2020.107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oubahmane M.; Hdoufane I.; Bjij I.; Jerves C.; Villemin D.; Cherqaoui D. COVID-19: in silico identification of potent α-ketoamide inhibitors targeting the main protease of the SARS-CoV-2. J. Mol. Struct. 2021, 1244, 130897. 10.1016/j.molstruc.2021.130897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frecer V.; Miertus S. Antiviral agents against COVID-19: structure-based design of specific peptidomimetic inhibitors of SARS-CoV-2 main protease. RSC Adv. 2020, 10, 40244–40263. 10.1039/D0RA08304F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R.; Ruiz-Moreno A. J.; Cong Y.; Dinesh Kumar N.; Velasco-Velazquez M. A.; Neochoritis C. G.; Smith J.; Reggiori F.; Groves M. R.; Dömling A. Repurposing the HCV NS3–4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 2021, 12, 370–379. 10.1039/D0MD00367K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzupir A. O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020, 1222, 128878. 10.1016/j.molstruc.2020.128878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Phillips G.; Weiss K. L.; Zhang Q.; Coates L.; Kovalevsky A. Direct observation of protonation state modulation in SARS-CoV-2 main protease upon inhibitor binding with neutron crystallography. J. Med. Chem. 2021, 64, 4991–5000. 10.1021/acs.jmedchem.1c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B.; He F.; Liu D.; Fang M.; Wu Z.; Xu D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020, 972133. 10.1101/2020.03.03.972133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Liang B.; Chen Y.; Fuk-Woo Chan J.; Yuan S.; Ye H.; Nie L.; Zhou J.; Wu Y.; Wu M.; Huang L. S.; An J.; Warshel A.; Yuen K.-Y.; Ciechanover A.; Huang Z.; Xu Y. A new class of α-ketoamide derivatives with potent anticancer and anti-SARS-CoV-2 activities. Eur. J. Med. Chem. 2021, 215, 113267. 10.1016/j.ejmech.2021.113267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. A microscopic description of SARS-CoV-2 main protease inhibition with Michael acceptors. Strategies for improving inhibitor design. Chem. Sci. 2021, 12, 3489–3496. 10.1039/D0SC04978F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafet K.; Serrano-Aparicio N.; Lodola A.; Mulholland A. J.; González F. V.; Świderek K.; Moliner V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. 10.1039/D0SC06195F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí S.; Arafet K.; Lodola A.; Mulholland A. J.; Świderek K.; Moliner V. Impact of warhead modulations on the covalent inhibition of SARS-CoV-2 Mpro explored by QM/MM simulations. ACS Catal. 2022, 12, 698–708. 10.1021/acscatal.1c04661. [DOI] [PubMed] [Google Scholar]
- Iketani S.; Forouhar F.; Liu H.; Hong S. J.; Lin F. Y.; Nair M. S.; Zask A.; Huang Y.; Xing L.; Stockwell B. R.; Chavez A.; Ho D. D. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021, 12, 2016. 10.1038/s41467-021-22362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola G.; Ettari R.; Previti S.; Di Chio C.; Messere A.; Di Maro S.; Hammerschmidt S. J.; Zimmer C.; Zimmermann R. A.; Schirmeister T.; Zappalà M.; Cosconati S. Lead discovery of SARS-CoV-2 main protease inhibitors through covalent docking-based virtual screening. J. Chem. Inf Model 2021, 61, 2062–2073. 10.1021/acs.jcim.1c00184. [DOI] [PubMed] [Google Scholar]
- Manandhar A.; Srinivasulu V.; Hamad M.; Tarazi H.; Omar H.; Colussi D. J.; Gordon J.; Childers W.; Klein M. L.; Al-Tel T. H.; Abou-Gharbia M.; Elokely K. M. Discovery of novel small-molecule inhibitors of SARS-CoV-2 main protease as potential leads for COVID-19 treatment. J. Chem. Inf Model 2021, 61, 4745–4757. 10.1021/acs.jcim.1c00684. [DOI] [PubMed] [Google Scholar]
- Zaidman D.; Gehrtz P.; Filep M.; Fearon D.; Gabizon R.; Douangamath A.; Prilusky J.; Duberstein S.; Cohen G.; Owen C. D.; Resnick E.; Strain-Damerell C.; Lukacik P.; Barr H.; Walsh M. A.; von Delft F.; London N. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806. 10.1016/j.chembiol.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stille J. K.; Tjutrins J.; Wang G.; Venegas F. A.; Hennecker C.; Rueda A. M.; Sharon I.; Blaine N.; Miron C. E.; Pinus S.; Labarre A.; Plescia J.; Burai Patrascu M.; Zhang X.; Wahba A. S.; Vlaho D.; Huot M. J.; Schmeing T. M.; Mittermaier A. K.; Moitessier N. Design, synthesis and in vitro evaluation of novel SARS-CoV-2 3CLpro covalent inhibitors. Eur. J. Med. Chem. 2022, 229, 114046. 10.1016/j.ejmech.2021.114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A. S.; Islam R.; Parves M. R.; Mamun A. A.; Shahriar I.; Hossain M. I.; Hossain M. N.; Ali M. A.; Halim M. A. Cysteine focused covalent inhibitors against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40 (4), 1639–1658. 10.1080/07391102.2020.1831610. [DOI] [PubMed] [Google Scholar]
- Teli D. M.; Shah M. B.; Chhabria M. T. Screening of natural compounds as potential inhibitors of SARS-CoV-2 main protease and spike RBD: targets for COVID-19. Front Mol. Biosci 2021, 7, 599079. 10.3389/fmolb.2020.599079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z.; Zhao W.; Niu W.; Gao X.; Gao X.; Gong Y.; Gao X. Molecules inhibit the enzyme activity of 3-chymotrypsin-like cysteine protease of SARS-CoV-2 virus: the experimental and theory studies. bioRxiv 2020, 120642. 10.1101/2020.05.28.120642. [DOI] [Google Scholar]
- Wang R.; Hu Q.; Wang H.; Zhu G.; Wang M.; Zhang Q.; Zhao Y.; Li C.; Zhang Y.; Ge G.; Chen H.; Chen L. Identification of vitamin K3 and its analogues as covalent inhibitors of SARS-CoV-2 3CLpro. Int. J. Biol. Macromol. 2021, 183, 182–192. 10.1016/j.ijbiomac.2021.04.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen J.; Albani S.; Hanke A.; Joseph B. P.; Bergh C.; Kuzikov M.; Costanzi E.; Manelfi C.; Storici P.; Gribbon P.; Beccari A. R.; Talarico J. C.; Spyrakis J. F.; Lindahl K. E.; Zaliani A.; Carloni P.; Wade R. C.; Musiani F.; Kokh D. B.; Rossetti G. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS Pharmacol Transl Sci. 2021, 4, 1079–1095. 10.1021/acsptsci.0c00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H.; Yao S.; Zhao W.; Zhang Y.; Liu J.; Shao Q.; Wang Q.; Li M.; Xie H.; Shang W.; Ke C.; Feng L.; Jiang X.; Shen J.; Xiao G.; Jiang H.; Zhang L.; Ye Y.; Xu Y. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. 10.1038/s41467-021-23751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T.; Cui M.; Zheng C.; Wang M.; Sun R.; Gao D.; Bao J.; Ren S.; Yang B.; Lin J.; Li X.; Li D.; Yang C.; Zhou H. Myricetin inhibits SARS-CoV-2 viral replication by targeting Mpro and ameliorates pulmonary inflammation. Front Pharmacol 2021, 12, 669642. 10.3389/fphar.2021.669642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Xia Z.; Sacco M. D.; Hu Y.; Townsend J. A.; Meng X.; Choza J.; Tan H.; Jang J.; Gongora M. V.; Zhang X.; Zhang F.; Xiang Y.; Marty M. T.; Chen Y.; Wang J. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021, 143, 20697–20709. 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane D.; Onitsuka S.; Re S.; Isogai H.; Hamada R.; Hiramoto T.; Kawanishi E.; Mizuguchi K.; Shindo N.; Ojida A. Selective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide. Chemical Science 2022, 13, 3027–3034. 10.1039/D1SC06596C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M.; Nie T.; Shao Q.; Li M.; Su H.; Xu Y. In silico screening-based discovery of novel covalent inhibitors of the SARS-CoV-2 3CL protease. Eur. J. Med. Chem. 2022, 231, 114130. 10.1016/j.ejmech.2022.114130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon P.; Boike L.; Dovala D.; Henning N. J.; Knapp M.; Spradlin J. N.; Ward C. C.; Wolleb H.; Zammit C. M.; Fuller D.; Blake G.; Murphy J. P.; Wang F.; Lu Y.; Moquin S. A.; Tandeske L.; Hesse M. J.; McKenna J. M.; Tallarico J. A.; Schirle M. F. D.; Nomura D. K. Discovery of potent pyrazoline-based covalent SARS-CoV-2 main protease inhibitors. bioRxiv 2022, 483025. 10.1101/2022.03.05.483025. [DOI] [PubMed] [Google Scholar]
- El Khoury L.; Jing Z.; Cuzzolin A.; Deplano A.; Loco D.; Sattarov B.; Hedin F.; Wendeborn S.; Ho C.; El Ahdab D.; Jaffrelot Inizan T.; Sturlese M.; Sosic A.; Volpiana M.; Lugato A.; Barone M.; Gatto B.; Macchia M. L.; Bellanda M.; Battistutta R.; Salata C.; Kondratov I.; Iminov R.; Khairulin A.; Mykhalonok Y.; Pochepko A.; Chashka-Ratushnyi V.; Kos I.; Moro S.; Montes M.; Ren P.; Ponder J. W.; Lagardere L.; Piquemal J.-P.; Sabbadin D. Computationally driven discovery of SARS-CoV-2 Mpro inhibitors: from design to experimental validation. Chem. Sci. 2022, 13, 3674. 10.1039/D1SC05892D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia C. S. B. Novel nitrile peptidomimetics for treating COVID-19. ACS Med. Chem. Lett. 2022, 13, 330–331. 10.1021/acsmedchemlett.2c00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Fang C.; Zhang Q.; Zhang R.; Zhao X.; Duan Y.; Wang H.; Zhu Y.; Feng L.; Zhao J.; Shao M.; Yang X.; Zhang L.; Peng C.; Yang K.; Ma D.; Rao Z.; Yang H. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2022, 13 (9), 689–693. 10.1007/s13238-021-00883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar P. E.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Lamb Y. N. Nirmatrelvir plus ritonavir: first approval. Drugs 2022, 82, 585. 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Lin C.; Zhou X.; Zhong F.; Zeng P.; Yang Y.; Zhang Y.; Yu B.; Fan X.; McCormick P. J.; Fu R.; Fu Y.; Jiang H.; Zhang J. Structural basis of the main proteases of coronavirus bound to drug candidate PF-07321332. J. Virol 2022, 96, e0201321 10.1128/jvi.02013-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Computational simulations on the binding and reactivity of a nitrile inhibitor of the SARS-CoV-2 main protease. Chem. Commun. (Camb) 2021, 57, 9096–9099. 10.1039/D1CC03953A. [DOI] [PubMed] [Google Scholar]
- Ngo S. T.; Nguyen T. H.; Tung N. T.; Mai B. K. Insights into the binding and covalent inhibition mechanism of PF-07321332 to SARS-CoV-2 Mpro. RSC Adv. 2022, 12, 3729–3737. 10.1039/D1RA08752E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S.; Ekanayake K. B.; Otting G.; Nitsche C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. 10.1016/j.bmcl.2022.128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Foo C. S.; Jochmans D.; Vangeel L.; De Jonghe S.; Augustijns P.; Mols R.; Weynand B.; Wattanakul T.; Hoglund R. M.; Tarning J.; Mowbray C. E.; Sjö P.; Escudié F.; Scandale I.; Chatelain E.; Neyts J. The oral protease inhibitor (PF-07321332) protects syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. 10.1038/s41467-022-28354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Wang Y.; Lavrijsen M.; Lamers M. M.; de Vries A. C.; Rottier R. J.; Bruno M. J.; Peppelenbosch M. P.; Haagmans B. L.; Pan Q. SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022, 32, 322–324. 10.1038/s41422-022-00618-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai D. K.; Yurgelonis I.; McMonagle P.; Rothan H. A.; Hao L.; Gribenko A.; Titova E.; Kreiswirth B.; White K. M.; Zhu Y.; Anderson A. S.; Cardin R. D. Nirmatrelvir, an orally active Mpro inhibitor, is a potent inhibitor of SARS-CoV-2 variants of concern. bioRxiv 2022, 476644. 10.1101/2022.01.17.476644. [DOI] [Google Scholar]
- Greasley S. E.; Noell S.; Plotnikova O.; Ferre R.; Liu W.; Bolanos B.; Fennell K.; Nicki J.; Craig T.; Zhu Y.; Stewart A. E.; Steppan C. M. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J. Biol. Chem. 2022, 298 (6), 101972. 10.1016/j.jbc.2022.101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales R.; McGovern B. L.; Rodriguez M. L.; Rai D. K.; Cardin R. D.; Anderson A. S.; group P.S.P.s.; Sordillo E. M.; van Bakel H.; Simon V.; García-Sastre A.; White K. M. Nirmatrelvir, molnupiravir, and remdesivir maintain potent in vitro activity against the SARS-CoV-2 Omicron variant. bioRxiv 2022, 476685. 10.1101/2022.01.17.476685. [DOI] [Google Scholar]
- Bai B.; Arutyunova E.; Khan M. B.; Lu J.; Joyce M. A.; Saffran H. A.; Shields J. A.; Kandadai A. S.; Belovodskiy A.; Hena M.; Vuong W.; Lamer T.; Young H. S.; Vederas J. C.; Tyrrell D. L.; Lemieux J.; nieman J. A. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021, 12, 1722–1730. 10.1039/D1MD00247C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori S. I.; Higshi-Kuwata N.; Raghavaiah J.; Das D.; Bulut H.; Davis D. A.; Takamatsu Y.; Matsuda K.; Takamune N.; Kishimoto N.; Okamura T.; Misumi S.; Yarchoan R.; Maeda K.; Ghosh A. K.; Mitsuya H. GRL-0920, an indole chloropyridinyl ester, completely blocks SARS-CoV-2 infection. mBio 2020, 11, e01833-20. 10.1128/mBio.01833-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Raghavaiah J.; Shahabi D.; Yadav M.; Anson B. J.; Lendy E. K.; Hattori S. I.; Higashi-Kuwata N.; Mitsuya H.; Mesecar A. D. Indole chloropyridinyl ester-derived SARS-CoV-2 3CLpro inhibitors: enzyme inhibition, antiviral efficacy, structure-activity relationship, and X-ray structural studies. J. Med. Chem. 2021, 64, 14702–14714. 10.1021/acs.jmedchem.1c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach J.; Lemke C.; Pillaiyar T.; Schakel L.; Al Hamwi G.; Diett M.; Gedschold R.; Geiger N.; Lopez V.; Mirza S.; Namasivayam V.; Schiedel A. C.; Sylvester K.; Thimm D.; Vielmuth C.; Phuong Vu L.; Zyulina M.; Bodem J.; Gutschow M.; Muller C. E. Targeting the main protease of SARS-CoV-2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew. Chem., Int. Ed. Engl. 2021, 60, 10423–10429. 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Shahabi D.; Yadav M.; Kovela S.; Anson B. J.; Lendy E. K.; Bonham C.; Sirohi D.; Brito-Sierra C. A.; Hattori S. I.; Kuhn R.; Mitsuya H.; Mesecar A. D. Chloropyridinyl esters of nonsteroidal anti-inflammatory agents and related derivatives as potent SARS-CoV-2 3CL protease inhibitors. Molecules 2021, 26, 5782. 10.3390/molecules26195782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y.; Feng Q.; Kumagai T.; Torikai K.; Ohigashi H.; Osawa T.; Noguchi N.; Niki E.; Uchida K. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J. Biol. Chem. 2002, 277, 2687–2694. 10.1074/jbc.M109641200. [DOI] [PubMed] [Google Scholar]
- Sakurai T.; Kanayama M.; Shibata T.; Itoh K.; Kobayashi A.; Yamamoto M.; Uchida K. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem. Res. Toxicol. 2006, 19, 1196–1204. 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- Monajemi H.; Zain S. M. Strong inhibition of M-protease activity of coronavirus by using PX-12 inhibitor based on ab initio ONIOM calculations. Journal of Chemical Research 2021, 45, 136–140. 10.1177/1747519820938025. [DOI] [Google Scholar]
- Madabeni A.; Nogara P. A.; Omage F. B.; Rocha J. B. T.; Orian L. Mechanistic insight into SARS-CoV-2 mpro inhibition by organoselenides: the ebselen case study. Appl. Sci. 2021, 11 (14), 6291. 10.3390/app11146291. [DOI] [Google Scholar]
- Parise A.; Romeo I.; Russo N.; Marino T. The Se-S bond formation in the covalent inhibition mechanism of SARS-CoV-2 main protease by ebselen-like inhibitors: a computational study. Int. J. Mol. Sci. 2021, 22, 9792. 10.3390/ijms22189792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amporndanai K.; Meng X.; Shang W.; Jin Z.; Rogers M.; Zhao Y.; Rao Z.; Liu Z.-J.; Yang H.; Zhang L.; O’Neill P. M.; Samar Hasnain S. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. 10.1038/s41467-021-23313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez C. A.; Byléhn F.; Perez-Lemus G. R.; Alvarado W.; de Pablo J. J. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 2020, 6, abd0345. 10.1126/sciadv.abd0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmudzinski M.; Rut W.; Olech K.; Granda J.; Giurg M.; Burda-Grabowska M.; Zhang L.; Sun X.; Lv Z.; Nayak D.; Kesik-Brodacka M.; Olsen S. K.; Hilgenfeld R.; Drag M. Ebselen derivatives are very potent dual inhibitors of SARS-CoV-2 proteases - PLpro and Mpro in vitro studies. bioRxiv 2020, 273979. 10.1101/2020.08.30.273979. [DOI] [Google Scholar]
- Huff S.; Kummetha I. R.; Tiwari S. K.; Huante M. B.; Clark A. E.; Wang S.; Bray W.; Smith D.; Carlin A. F.; Endsley M.; Rana T. M. Discovery and mechanism of SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022, 65 (4), 2866–2879. 10.1021/acs.jmedchem.1c00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Y.; Chen C.; Su J.; Li J. Q.; Jiang Z.; Gao H.; Chigan J. Z.; Ding H. H.; Zhai L.; Yang K. W. Ebsulfur and ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg Chem. 2021, 112, 104889. 10.1016/j.bioorg.2021.104889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Feng B.; Han S.; Wang P.; Chen W.; Zang Y.; Li J.; Hu Y. Discovery of highly potent SARS-CoV-2 M pro inhibitors based on benzoisothiazolone scaffold. Bioorg. Med. Chem. Lett. 2022, 58, 128526. 10.1016/j.bmcl.2022.128526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thun-Hohenstein S. T. D.; Suits T. F.; Malla T. R.; Tumber A.; Brewitz L.; Choudhry H.; Salah E.; Schofield C. J. Structure-activity studies reveal scope for optimization of ebselen-type inhibition of SARS-CoV-2 main protease. ChemMedChem 2021, 17 (4), e202100582. 10.1002/cmdc.202100582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehri B.; Kohnehpoushi M.; Ghavami R. High predictive QSAR models for predicting the SARS coronavirus main protease inhibition activity of ketone-based covalent inhibitors. J. Iran. Chem. Soc. 2022, 19 (5), 1865–1876. 10.1007/s13738-021-02426-2. [DOI] [Google Scholar]
- De Cesco S.; Kurian J.; Dufresne C.; Mittermaier A. K.; Moitessier N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- Luo Y. L. Mechanism-based and computational-driven covalent drug design. J. Chem. Inf Model 2021, 61, 5307–5311. 10.1021/acs.jcim.1c01278. [DOI] [PubMed] [Google Scholar]
- Fernandes H. S.; Sousa S. F.; Cerqueira N. M. F. S. New insights into the catalytic mechanism of the SARS-CoV-2 main protease: an ONIOM QM/MM approach. Mol. Divers 2022, 26, 1373–1381. 10.1007/s11030-021-10259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodola A.; Callegari D.; Scalvini L.; Rivara S.; Mor M. Design and SAR analysis of covalent inhibitors driven by hybrid QM/MM simulations. Methods Mol. Biol. 2020, 2114, 307–337. 10.1007/978-1-0716-0282-9_19. [DOI] [PubMed] [Google Scholar]
- Awoonor-Williams E.; Walsh A. G.; Rowley C. N. Modeling covalent-modifier drugs. Biochim Biophys Acta Proteins Proteom 2017, 1865, 1664–1675. 10.1016/j.bbapap.2017.05.009. [DOI] [PubMed] [Google Scholar]
- Świderek K.; Moliner V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 MPRO. Chem. Sci. 2020, 11, 10626–10630. 10.1039/D0SC02823A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zev S.; Raz K.; Schwartz R.; Tarabeh R.; Gupta P. K.; Major D. T. Benchmarking the ability of common docking programs to correctly reproduce and score binding modes in SARS-CoV-2 protease Mpro. J. Chem. Inf Model 2021, 61, 2957–2966. 10.1021/acs.jcim.1c00263. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Gong G.; Grum-Tokars V.; Mulhearn D. C.; Baker S. C.; Coughlin M.; Prabhakar B. S.; Sleeman K.; Johnson M. E.; Mesecar A. D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]