

Introduction
The Critical Path Institute’s Polycystic Disease Outcomes Consortium, including key stakeholders (patients, medical experts, industry experts, and regulators), held a PKD Regulatory Summit in May 2021. We highlight Session 2: Advancing Drug Development for Autosomal Dominant Polycystic Kidney Disease (ADPKD).
Current Autosomal Dominant Polycystic Kidney Disease Therapy: Restricted to Adults at Risk of Rapid Progression
Diet and lifestyle clearly contribute to the risk of progression. Obesity, higher salt intake, and lower potassium intake have been linked to increased cyst growth. High water intake (1) has been suggested, but a water prescription study did not show benefit (2). Metabolic reprogramming is an important new field of study in ADPKD, and it may be that reduced caloric intake and intermittent fasting will prove to be beneficial strategies in the future.
Tolvaptan is indicated to slow kidney function decline in adults at risk of rapidly progressing ADPKD. Data regarding tolvaptan in children with ADPKD (NCT02964273) should be available soon.
Similar but varying standards for “rapidly progressing” have been adopted internationally (3). Older patients or those with smaller kidneys may still develop kidney failure but not meet the “rapidly progressing” standard. The efficacy of tolvaptan compares favorably with other renoprotective therapies (4); however, aquaretic side effects and frequent monitoring for liver injury limit full acceptance.
Prior drug approval processes were discussed. The Food and Drug Administration (FDA) agreed that a drug with a large treatment effect might be approved without labeling restrictions as there is a clear unmet need for drugs with greater therapeutic efficacy for all patients at risk of kidney failure, including those at lower risk of early progression.
Patient Selection for Trials: Risk, Monitoring, and Progression
Research is needed to define novel targets and the best timing for interventions relevant in the early disease stage before destruction of kidney parenchyma and loss of function (Figure 1) (5). These patients with ADPKD represent a crucial target for disease management. Challenges include the identification and validation of indicators, both measuring and predicting disease progression from childhood, and distinguishing slow from rapid progressors in this early-stage population.
Figure 1.
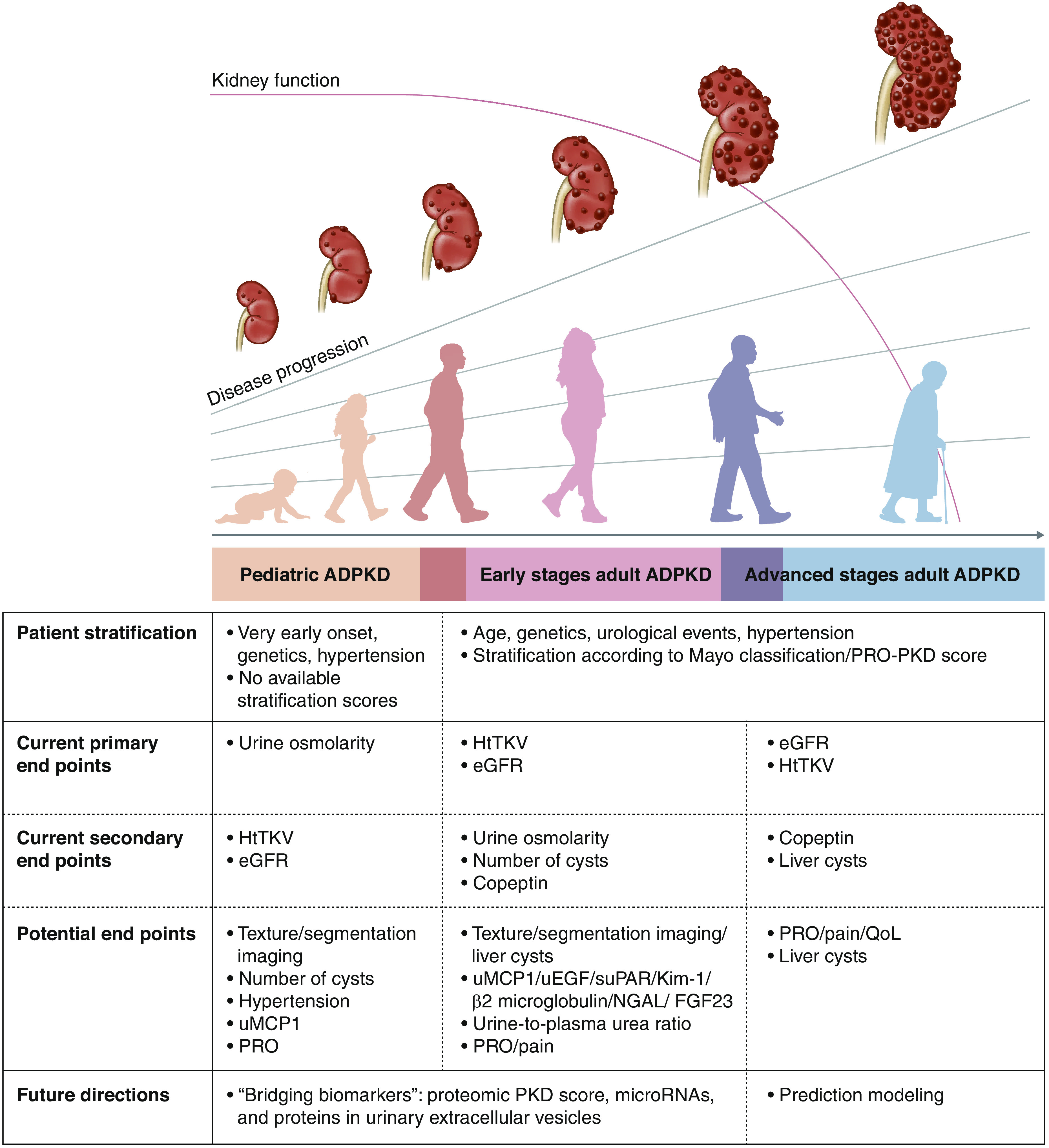
Published study end points for patients with autosomal dominant polycystic kidney disease (ADPKD) include height-adjusted total kidney volume (htTKV) and eGFR in adults and urinary osmolality in children. As GFR remains stable for years, other progression markers are needed for future clinical trials. Mayo classification (1A–1E) and/or the Predicting Renal Outcomes in Polycystic Kidney Disease (PRO-PKD) tool may be used for stratifying patients for selection in trials. Copeptin, kidney cyst number, and liver cysts are secondary end points used in previous studies, but all are not validated. Urinary monocyte chemoattractant protein-1 (uMCP1), kidney injury molecule-1 (Kim-1), β2-microglobulin, neutrophil gelatinase–associated lipocalin (NGAL), fibroblast growth factor 23 (FGF23), urinary epidermal growth factor (uEGF), soluble urokinase plasminogen activator receptor (suPAR), urine-plasma urea ratio, and patient-reported outcomes (PROs), such as pain, are potential biomarkers. Future directions in ADPKD including “bridging biomarkers,” such as microRNAs, proteomics, and proteins in urinary extracellular vesicles, are promising. Automatic segmentation using artificial intelligence/machine learning may further enhance the interpretation of kidney growth, and number and size of cysts. Combining all of these efforts will lead to the development of prediction modeling with a more precise disease progression stratification. PKD, polycystic kidney disease; QoL, quality of life.
In children, risk prediction tools are lacking. The onset of hypertension may be an important sign of high risk in this population. The ADPedKD registry (https://www.adpedkd.org/index.php?id=about) will establish the largest phenotyped pediatric ADPKD cohort, allowing for the development of risk stratification (5). ADPedKD is a global study including retrospective and prospective longitudinal data, providing the basis for the development of unified diagnostic, follow-up, and treatment practices.
A novel three-dimensional ultrasound method to measure the height-adjusted total kidney volume (htTKV) in children has been published (5). Three-dimensional ultrasound manual contouring volumetry outperformed the two-dimensional ultrasound ellipsoid method and was comparable with MRI volumetry in children, especially for smaller kidneys.
In adults, an average kidney length of 16.5 cm in a patient younger than 45 years is a good predictor of risk of early loss of kidney function (3). htTKV in combination with patient age and eGFR has been accepted as a prognostic biomarker by the FDA and European Medicines Agency (6).
htTKV does not fully predict the risk of progression for an individual patient as progression is sometimes nonlinear (7). Texture analysis (3) or quantification of variations in surface intensity of an image in addition to age, eGFR, and htTKV may provide better risk prediction.
Urine-plasma urea ratio reflects concentrating capacity, with lower ratios associating with rapidly progressive disease (8). Urinary monocyte chemoattractant protein-1, microRNAs, proteins in urinary exosomes, copeptin, and soluble urokinase plasminogen activator receptor levels may represent novel biomarkers.
PROPKD, a risk assessment using genotype, sex, early hypertension, or a urologic event (9), has high specificity but low sensitivity as occurrence of a urologic event before the age of 35 may be rare. Genotype may work best as part of a composite predictor tool (8).
Formal regulatory biomarker qualification procedures are useful to ensure regulatory acceptance of promising novel end points for the development of new drugs (6).
Extrarenal Manifestations: An Area of Opportunity
ADPKD is a multiorgan disorder. Liver cysts are the most common nonkidney finding, with premenopausal women over-represented as having severe hepatomegaly (10). Patient-reported symptoms increase with larger liver size, with a worse physical component score of quality of life (10). Surgical treatment includes cyst aspiration and sclerosis, fenestration, partial hepatectomy, or liver transplantation (1). Medical treatment includes the use of long-acting octreotide analogs, which is a last resort therapy for patients who are not surgical candidates.
Intracerebral aneurysms are more common and occur at an earlier age in patients with ADPKD compared with unaffected individuals. The prevalence is about 3% in the general population and three-fold higher in ADPKD. The risk of intracerebral aneurysm is 11% in ADPKD without a family history and 23% with a family history (1).
Patient Perspective
Early treatment before significant kidney damage should be a priority. End points that substitute for eGFR as a key clinical outcome and other biomarkers of progression are lacking. A recent Delphi survey of patients, caregivers, and health care professionals found that prioritized outcomes were kidney function, kidney failure, death, blood pressure, kidney growth, and cerebral aneurysm. Kidney pain was the highest-rated patient-reported outcome (PRO) (11). Pain was also found to be a significant PRO in the PKD Foundation registry.
Although not all patients with ADPKD experience pain to the same degree, it can be the most significant burden in the lives of patients who experience it.
Future Directions
Current therapies under development include Bardoxolone (FALCON/Reata), GLPG-2737 (Galapagos), and miR-17 inhibition (Regulus). Eligible patients were at high risk of progression (younger, higher Mayo imaging class, and/or some loss of kidney function). Patients with lower Mayo imaging classification who may develop kidney failure in later life or patients at highest risk with preserved eGFR are excluded from current trials. Other unmet needs include therapy targeting both liver and kidney cysts, and therapy directed at pain.
PRO measures such as pain and disease-related interference with activities of daily living were thought to be critical for future trials. The value of PROs should be strengthened by clinician- and/or observer-reported outcomes, where applicable.
Outcome measures, including composite outcomes, were considered. eGFR is a poor outcome measure for high-risk patients early in their disease course who are unlikely to have significant change in eGFR during a typical study duration. The need for better biomarkers of progression was discussed extensively. In 2016, baseline total kidney volume in combination with patient age and baseline eGFR qualified as a prognostic enrichment biomarker for ADPKD in patients at high risk of 30% decline in kidney function (6). htTKV has been accepted as a reasonably likely surrogate for accelerated but not full approval (6).
Effectiveness of a drug in adults can provide evidence in support of efficacy in pediatric patients (i.e., extrapolation of efficacy) if (1) the disease course is similar in adult and pediatric patients and (2) the response to the drug is similar in adult and pediatric patients. Assuming that these criteria are met, a biomarker pertinent to disease pathophysiology and the mechanism of action of the drug, which correlates with clinical outcomes in adult and pediatric patients with ADPKD, could be used to evaluate or “bridge” the efficacy of a specific drug in adults to pediatric patients with ADPKD (i.e., using a “bridging biomarker” approach).
Future studies targeting kidney-based efficacy end points should assess the effects on extrarenal disease manifestations as part of safety assessments (e.g., effects on the liver) and vice versa. The potential risks and benefits of composite kidney and liver efficacy end points were considered.
Disclosures
N.K. Dahl serves as a principal investigator for clinical trials sponsored by Allena, Kadmon, Reata, Regulus, and Sanofi; serves on the unbranded speakers bureau for Otsuka; reports consultancy agreements with Otsuka Pharmaceutical and Vertex; reports honoraria from AstraZeneca, the National Kidney Foundation, and Otsuka Pharmaceutical; reports advisory or leadership roles for the Natera Scientific Advisory Board and the PKD Foundation; and reports other interests or relationships with the Medical Advisory Board of the National Kidney Foundation serving Connecticut Chapter. D. Mekahli serves on advisory boards for Otsuka Pharmaceutical, Reata, and Sanofi Genzyme as a representative of the University Hospital of Leuven and has received educational grants from Otsuka Pharmaceutical paid to the University Hospital of Leuven, all outside the submitted work. The remaining author has nothing to disclose.
Funding
The 2021 PKD Regulatory Summit was funded/supported by Otsuka Pharmaceutical and the PKD Foundation. Critical Path Institute is supported by the Food and Drug Administration (FDA) of the US Department of Health and Human Services (HHS), and it is 54.2% funded by FDA/HHS, totaling $13,239,950, and 45.8% funded by nongovernment source(s), totaling $11,196,634.
Acknowledgments
Participants in the 2021 PKD Regulatory Summit Session 2 included Arlene Chapman (University of Chicago); N.K. Dahl (Yale University); Karen Edwards (patient speaker); Lainie Esquivel (patient speaker); Ali Hariri (Sanofi); Pravin Jadhav (Otsuka Pharmaceuticals); Romaldas Maciulaitis (European Medicines Agency); D. Mekahli (Universitair Ziekenhuis Leuven); Kirtida Mistry, Norman Stockbridge, and Joseph Toerner (FDA); Craig Ostroff (Goldfinch Bio); Chris Rusconi (PKD Foundation); Roser Torra (Fundacio Puigvert); and H. Womack (patient speaker).
The contents are those of the authors and do not necessarily represent the official views of nor an endorsement by FDA/Department of Health and Human Services or the US Government.
The content of this article reflects the personal experience and views of the author(s) and should not be considered medical advice or recommendation. The content does not reflect the views or opinions of the American Society of Nephrology (ASN) or CJASN. Responsibility for the information and views expressed herein lies entirely with the author(s).
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
See related Perspectives, “Drug Development for Cystic Kidney Diseases,” on pages 1549–1550; “Perspectives on Drug Development in Autosomal Recessive Polycystic Kidney Disease,” on pages 1551–1554; and “Current Challenges and Perspectives on Developing a Clinical Trial Design for ADPKD,” on pages 1559–1562.
Author Contributions
N.K. Dahl and D. Mekahli conceptualized the study; N.K. Dahl, D. Mekahli, and H. Womack wrote the original draft; and N.K. Dahl and D. Mekahli reviewed and edited the manuscript.
References
- 1.Chebib FT, Torres VE: Autosomal dominant polycystic kidney disease: Core curriculum 2016. Am J Kidney Dis 67: 792–810, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rangan GK, Wong ATY, Munt A, Zhang JQJ, Saravanabavan S, Louw S, Allman-Farinelli M, Badve SV, Boudville N, Chan J, Coolican H, Coulshed S, Edwards ME, Erickson BJ, Fernando M, Foster S, Gregory AV, Haloob I, Hawley CM, Holt J, Howard K, Howell M, Johnson DW, Kline TL, Kumar K, Lee VW, Lonergan M, Mai J, McCloud P, Pascoe E, Biostat M, Peduto A, Rangan A, Roger SD, Sherfan J, Sud K, Torres VE, Vilayur E, Harris DCH: Prescribed water intake in autosomal dominant polycystic kidney disease. NEJM Evid 1: EVIDoa2100021, 1–13, 2021. 10.1056/EVIDoa2100021 [DOI] [PubMed] [Google Scholar]
- 3.Chebib FT, Torres VE: Assessing risk of rapid progression in autosomal dominant polycystic kidney disease and special considerations for disease-modifying therapy. Am J Kidney Dis 78: 282–292, 2021 [DOI] [PubMed] [Google Scholar]
- 4.Meijer E, Gansevoort RT: Vasopressin V2 receptor antagonists in autosomal dominant polycystic kidney disease: Efficacy, safety, and tolerability. Kidney Int 98: 289–293, 2020 [DOI] [PubMed] [Google Scholar]
- 5.Gimpel C, Bergmann C, Mekahli D: The wind of change in the management of autosomal dominant polycystic kidney disease in childhood. Pediatr Nephrol 37: 473–487, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith KA, Thompson AM, Baron DA, Broadbent ST, Lundstrom GH, Perrone RD: Addressing the need for clinical trial end points in autosomal dominant polycystic kidney disease: A report from the Polycystic Kidney Disease Outcomes Consortium (PKDOC). Am J Kidney Dis 73: 533–541, 2019 [DOI] [PubMed] [Google Scholar]
- 7.Brosnahan GM, Abebe KZ, Moore CG, Rahbari-Oskoui FF, Bae KT, Grantham JJ, Schrier RW, Braun WE, Chapman AB, Flessner MF, Harris PC, Hogan MC, Perrone RD, Miskulin DC, Steinman TI, Torres VE; HALT-PKD Trial Investigators : Patterns of kidney function decline in autosomal dominant polycystic kidney disease: A post hoc analysis from the HALT-PKD trials. Am J Kidney Dis 71: 666–676, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heida JE, Gansevoort RT, Messchendorp AL, Meijer E, Casteleijn NF, Boertien WE, Zittema D; DIPAK Consortium : Use of the urine-to-plasma urea ratio to predict ADPKD progression. Clin J Am Soc Nephrol 16: 204–212, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornec-Le Gall E, Audrézet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, Morin MP, Moal MC, Dantal J, Wehbe B, Perrichot R, Frouget T, Vigneau C, Potier J, Jousset P, Guillodo MP, Siohan P, Terki N, Sawadogo T, Legrand D, Menoyo-Calonge V, Benarbia S, Besnier D, Longuet H, Férec C, Le Meur Y: The PROPKD score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 942–951, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neijenhuis MK, Kievit W, Verheesen SM, D’Agnolo HM, Gevers TJ, Drenth JP: Impact of liver volume on polycystic liver disease-related symptoms and quality of life. United European Gastroenterol J 6: 81–88, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho Y, Rangan G, Logeman C, Ryu H, Sautenet B, Perrone RD, Nadeau-Fredette AC, Mustafa RA, Htay H, Chonchol M, Harris T, Gutman T, Craig JC, Ong ACM, Chapman A, Ahn C, Coolican H, Kao JT, Gansevoort RT, Torres V, Pei Y, Johnson DW, Viecelli AK, Teixeira-Pinto A, Howell M, Ju A, Manera KE, Tong A: Core outcome domains for trials in autosomal dominant polycystic kidney disease: An international Delphi survey. Am J Kidney Dis 76: 361–373, 2020 [DOI] [PubMed] [Google Scholar]