

Significance Statement
In a mouse allograft model, the CCL8-CCR8 axis mediated early kidney inflammation through donor kidney resident macrophages. The revealed therapeutic targets could expand the transplantable donor kidney pool and promote kidney allograft longevity.
Keywords: acute allograft rejection, kidney transplantation, macrophages, inflammation, allografts
Visual Abstract

Abstract
Background
In kidney transplantation, early allograft inflammation impairs long-term allograft function. However, precise mediators of early kidney allograft inflammation are unclear, making it challenging to design therapeutic interventions.
Methods
We used an allogeneic murine kidney transplant model in which CD45.2 BALB/c kidneys were transplanted to CD45.1 C57BL/6 recipients.
Results
Donor kidney resident macrophages within the allograft expanded rapidly in the first 3 days. During this period, they were also induced to express a high level of Ccl8, which, in turn, promoted recipient monocyte graft infiltration, their differentiation to resident macrophages, and subsequent expression of Ccl8. Enhanced graft infiltration of recipient CCR8+ T cells followed, including CD4, CD8, and γδ T cells. Consequently, blocking CCL8-CCR8 or depleting donor kidney resident macrophages significantly inhibits early allograft immune cell infiltration and promotes superior short-term allograft function.
Conclusions
Targeting the CCL8-CCR8 axis is a promising measure to reduce early kidney allograft inflammation.
Kidney implantation elicits an intense immediate inflammation. Among these inflammatory factors, ischemia reperfusion injury (IRI)1 is a universal early factor, and a known risk factor for delayed graft function (DGF). DGF not only increases immediate post-transplant complications,2 but also compromises long-term kidney allograft function.3,4 Further fueling this inflammation is innate allorecognition between donor and recipient immune cells.5,6 At the center of both processes is donor kidney resident macrophages, which have recently emerged as an important player in native kidney diseases.7,8 However, their role in kidney transplant remains poorly understood.
Among macrophage-produced factors, the cys-cys (C-C) motif chemokine ligand 8 (CCL8; also known as MCP2) is implicated in various inflammatory processes.9–11 In bone marrow transplantation, circulating CCL8 levels positively correlate with severity of graft-versus-host disease.12 In solid organ transplantation, however, the role of CCL8 is less clear. In one study of murine heart and islet transplantation, CCL8’s expression was upregulated during acute rejection.13 In another study of human kidney transplantation, several CCL8 receptors were upregulated in both postperfusion and acutely rejecting kidney biopsy specimens.14
In this study, we show that donor kidney resident macrophages prominently express Ccl8 after allogeneic kidney transplantation. This, in turn, promotes recipient macrophage infiltration and expression of Ccl8, prompting subsequent allograft infiltration of CCR8-expressing T cells. Consequently, intercepting the CCL8-CCR8 axis or depleting donor kidney resident macrophages effectively reduces early allograft inflammation and improves short-term allograft function.
Methods
Mice
Male 10- to 12-week-old C57BL/6J (CD45.1+ or CD45.2+) mice were purchased from the Jackson Laboratory. BALB/c mice were bred and housed in the Division of Laboratory Animal Resources facility at Duke University. All procedures were approved by Duke Institutional Animal Care and Use Committee.
Kidney Transplantation
Kidneys from CD45.2 BALB/c (allogeneic) or CD45.2 B6 (syngeneic) mice were transplanted into bilaterally nephrectomized CD45.1 B6 recipients as we previously published.15
Depletion of Donor Kidney Resident Macrophages
We treated BALB/c or B6 donors daily with clodronate liposomes (200 μl, intravenously; Encapsula NanoSciences) for 7 days and then rested them for a week before using their kidneys for transplantation. We treated control mice similarly using PBS liposomes. FACS and immunofluorescence staining confirmed depletion of kidney resident macrophages.
Anti-CCL8 and Anti-CCR8 Antibody Treatments
Mice were treated with anti-CCL8 mAb (A16070K; 50 μg per intraperitoneal injection; BioLegend), anti-CCR8 mAb (SA214G2; 100 μg per intraperitoneal injection; BioLegend), or rat IgG2bκ (TRK4530; 50 or 100 μg per intravenous injection; BioLegend). For short-term experiments, recipients were treated daily from day −1 to +2. For longer-term experiments, recipients were treated daily from day −1 to +7, and then every other day until the end point. Day 0 was the day of kidney transplantation.
Kidney Allograft Single-Cell Ccl8 and Ccr8 Expression
We used kidneys allografts from untreated B6 recipients on day 15 post-transplantation for gene expression analysis of Ccl8 and Ccr8 at the single-cell level. We obtained single-cell RNA transcriptomics data as previously described.16 The Seurat FeaturePlot plotting function generated the Ccl8 and Ccr8 Uniform Manifold Approximation and Projections.
Flow Cytometry
At predetermined time points, animals were euthanized, and explanted kidney grafts were perfused with cold PBS and digested with collagenase IV (2 mg/ml for 45 minutes at 37°C; Worthington). Cells were stained with fluorochrome-conjugated antibodies for 45 minutes at 4°C. For intracellular staining, we first stimulated cells with PMA, ionomycin, and brefeldin A (Invitrogen) for 5–6 hours before surface staining. After surface staining, cells were fixed, permeabilized (Cytofix/Cytoperm buffers; BD), and stained for intracellular targets using specific fluorochrome-conjugated antibodies at room temperature for 1 hour. Cells were acquired on a BD Fortessa and analyzed using FlowJo version 10.8.1 (Tree Star LLC). We used the following antibodies: CD45-BUV395 (30-F11), CD45.1-BV605 (A20), CD45.2-PECy7 (104), CD11b-BUV805 (M1/70), CD11c-FITC (N418), F4/80-APC-Cy7 (BM8), CCR2-BV650 (475301), CCR8-PE (SA214G2), CX3CR1-BV785 (SA011F11), Ly6C-eFluor450 (HK1.6), MHC-II-APC (M5/114.15.2), CD3-PECy5 (145-2C11), CD4-BV650 (K129.19), CD8–Pacific Blue (53-6.7), γδTCR-BV605 (GL3), CD45.1-APC (A20), CD90.1-FITC (OX-7), TCRvβ13-BUV395 (MR12-3), IL-17-PerCP-Cy5.5 (eBio17B7), and Ki67-PE (B56) (from eBioscience, BD Bioscience, Tonbo Biosciences, or BioLegend). For CCL8 intracellular staining, purified anti-mouse CCL8 (A16070K; BioLegend) and isotype antibodies (TRK4530; BioLegend) were first conjugated with FITC using the Pierce FITC Antibody Labeling Kit (Thermo Fisher). We first treated cells with purified anti-mouse CD16/CD32 (2.4G2; Tonbo Biosciences) before staining with anti-CCL8. We excluded dead cells using Aqua LIVE/DEAD dye (Molecular Probes).
We used FACS analysis to enumerate the absolute count of a specific cell population per kidney allograft using the following formula:
Immunofluorescence Staining and Histopathology
To confirm kidney resident macrophage depletion by clodronate liposomes, frozen kidney sections from treated mice were stained using rat anti-mouse F4/80 (1:100 dilution; A3-1, BioRad) overnight at 4°C. Goat anti-rat antibody conjugated with DyLight488 (at 1:500) was used as secondary antibody for 1 hour (Novus Biologicals). Sections were mounted using medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories) and imaged using a Zeiss Axio fluorescence microscope. For histopathologic evaluation of kidney allografts, we deparaffinized and rehydrated 5-μm-thick paraffin sections and then stained with Periodic acid–Schiff (Sigma-Aldrich) and Sirius Red for morphology and fibrosis, respectively. I.H. evaluated sections stained with Periodic acid–Schiff for characteristics of allograft rejection (glomerulitis, tubulitis, peritubular capillaritis, and intimal arteritis). I.H. scored the severity of rejection features from zero (none), one (minimal), two (mild), three (moderate), to four (severe). Images were captured using a Nikon Elipse E200 microscope. Kidney fibrosis was quantified by Sirius Red–stained areas using National Institutes of Health (NIH) ImageJ software.
Kidney Function Assay
We assessed transplanted kidney allograft function by measuring serum creatinine and BUN levels using kits from Alfa Wassermann, Inc. (West Caldwell, NJ), per the manufacturer’s protocols.
RNA Extraction and Semiquantitative PCR
RNA was extracted from whole-kidney tissue using Trizol Reagent (Invitrogen), followed by cDNA preparation using High-Capacity RNA-to-cDNA kit (Applied Biosystems). Semiquantitative real-time PCR (ABI Prism 7500) was performed in triplicate using TaqMan master mix. We used the following TaqMan primers/probes: Ccl8 (Mm01297183_m1), Ccl2 (Mm00441242_m1), Il17a (Mm00439618_m1), Tnfa (Mm00443258_m1), Gzmb (Mm00442837_m1), Ifng (Mm01168134_m1), and Lcn2 (Mm01324470_m1). We used the ΔΔCt method to determine RNA expression, with Gapdh (Mm99999915_g1) serving as the internal control. Gene expression was normalized according to expression in naive kidney samples.
Induction and Measurements of CCL8 in Kidney Macrophages
Single-cell suspensions from BALB/c kidneys were prepared as described above. Kidney macrophages were sorted using EasySep Mouse Biotin Positive Selection Kit II and anti-mouse F4/80 biotin-labeled antibody (StemCell Technologies). Sorted cells were seeded in flat-bottomed 96-wells plates and stimulated using mouse recombinant IFN-γ (50 U/ml) for 48 hours. We determined CCL8 protein levels in culture supernatants using the Mouse MCP2 ELISA Kit (Abcam).
Alloantigen-Specific CD4 T Cell Proliferation by Donor Kidney Resident Macrophage Stimulation
We used transgenic 4C CD4 T cells were used to determine alloantigen-specific T cell response. Transgenic 4C CD4 T cells from B6 mice recognize I-Ad expressed on BALB/c cells, thus using the direct allorecognition pathway.17 We sorted 4C transgenic cells using the CD4 T cell isolation kit (StemCell Technologies), labeled the cells using the cell-proliferation dye eFluor450 (eBioscience), and cocultured them with sorted BALB/c kidney macrophages. In some experiments, macrophages were first treated with mouse recombinant IFN-γ (50 U/ml) for 48 hours before coculturing with 4C CD4 T cells. In additional experiments, at the end of IFN-γ stimulation, culture medium was replaced with control Ig (CT-Ig)– or anti-CCL8–containing medium (20 μg/ml CT-Ig or anti-CCL8) for another 24 hours before adding sorted 4C CD4 T cells for coculturing. On day 7 of cocultures, cells were harvested; stained for CD3, CD4, CD90.1, and TCRvβ13; and analyzed by FACS for eFluor450 dilution.
Donor-Specific Antibody Measurement
Donor-specific antibodies (DSAs) were measured in B6 recipients as described.15 Briefly, serum samples were incubated with BALB/c splenocytes for 1 hour on ice. Cells were then washed and stained with FITC-conjugated anti-IgG1, anti-IgG2b, and anti-IgG2c (BD). Sera from naive B6 mice without kidney transplantation were used as the negative control. DSA levels were represented as mean fluorescence intensity of the FITC channel.
Statistical Analysis
Data were presented as mean±SD. Statistical analysis was performed in GraphPad Prism 7.04 (GraphPad Inc.). Data were analyzed using the t test. P≤0.05 was considered significant.
Results
Macrophages in Kidney Allografts Show Prominent Ccl8 Expression
A diverse range of chemokines in transplant allografts have been shown to underlie allograft rejection14 and are effective targets to attenuate rejection.18 Using an unbiased approach of single-cell transcriptomics (Figure 1A) in a mouse model of allogeneic kidney transplantation, we previously identified Ccl8 as a top differentially expressed gene by macrophages in rejecting kidney allografts.16 Ccl8 expression in untransplanted donor kidneys was negligible (Figure 1B), but became conspicuously expressed on day 15 post-transplantation in the kidney allograft, with the most prominent expression in macrophage clusters (clusters 4 and 18). Scattered expression was also observed in other cell clusters, albeit at much lower levels (Supplemental Figure 1).
Figure 1.

Single-cell transcriptomic analysis of untransplanted (naive) and transplanted kidney allografts reveals distinct cell clusters and expression of Ccl8 at a single-cell level. (A) A composite Uniform Manifold Approximation and Projection (UMAP) from both untransplanted and transplanted kidneys showing 25 various unique kidney and immune cell clusters (n=2 mice per groups; four kidneys total in aggregate). A total of 17,986 cells (8552 cells from untransplanted and 9434 cells from transplanted kidneys) are represented in the UMAP. (B) Two separate UMAPs representing the expression of Ccl8 by various cell clusters in untransplanted kidneys versus transplanted kidney allografts on day 15 post-transplant (post-Tx). B-lymph, B lymphocyte; CD, collecting duct; cDC, conventional dendritic cells; DCT, distal convoluted tubule; EC, endothelial cell; LOH, Loop of Henle; Macro, macrophage; Mast/Baso, mast cell/basophil; Mono, monocyte; Neutro, neutrophil; NK, natural killer; pDC, plasmacytoid dendritic cells; PT, proximal tubule; T-lymph, T lymphocyte.
Distinct Kinetics of Donor versus Recipient Macrophages in Kidney Allografts
Transplanted kidneys initially contain donor “passenger-leukocytes,” including donor kidney resident macrophages, which are later replaced by recipient immune cells entering via circulation. We first determined the kinetics of donor versus recipient macrophage numbers after kidney transplantation.
Steady state adult kidneys contain resident macrophages derived either from the yolk sac during organogenesis or from bone marrow later on.19 Consistent with previous studies,20–22 we identified two macrophage populations in naive donor (BALB/c) kidneys: (1) CD11b+F4/80HICD11c+ cells, representing yolk sac–derived macrophages (Supplemental Figure 2A, red); and (2) CD11b+F4/80LOCD11c− cells, representing bone marrow–derived macrophages (Supplemental Figure 2A, green). As shown in Supplemental Figure 2, B and C, only yolk sac–derived macrophages expressed MHC-II. Conversely, only bone marrow–derived macrophages expressed CCR2 and Ly6C. Both macrophages expressed CX3CR1. We also observed a third myeloid cell population with a typical dendritic cell phenotype of CD11b+F4/80−CD11cHIMHC-II+ (Supplemental Figure 2A, blue). Of note, B6 kidneys harbored the same macrophage populations (data not shown).
To differentiate donor from recipient macrophages, we transplanted CD45.2 BALB/c kidneys into CD45.1 B6 recipients. As shown in Figure 2A, immediately after transplantation, the majority of persisting donor CD45.2+CD11b+ cells in the kidney allograft were yolk sac–derived F4/80HICD11c+MHC-II+ cells. Surprisingly, enumeration of these macrophages revealed they expanded in the first few days after transplantation, peaking in numbers on days 2–3. This was followed by their rapid decline and eventual disappearance by day 15 (Figure 2A, line graph). Supporting the proliferation of these macrophages, their Ki67 staining was positive on day 3 (Figure 2B). The possibility of the macrophages migrating to secondary lymphoid organs was excluded by their absence in recipient spleen or lymph nodes during the same period (data not shown). Therefore, it appears that donor macrophages were eliminated in situ in transplanted kidneys, likely by recipient alloimmunity. This notion is further supported by data from syngeneic kidney transplantation, where donor kidney resident macrophages were readily detectable on day 15 (Supplemental Figure 3).
Figure 2.
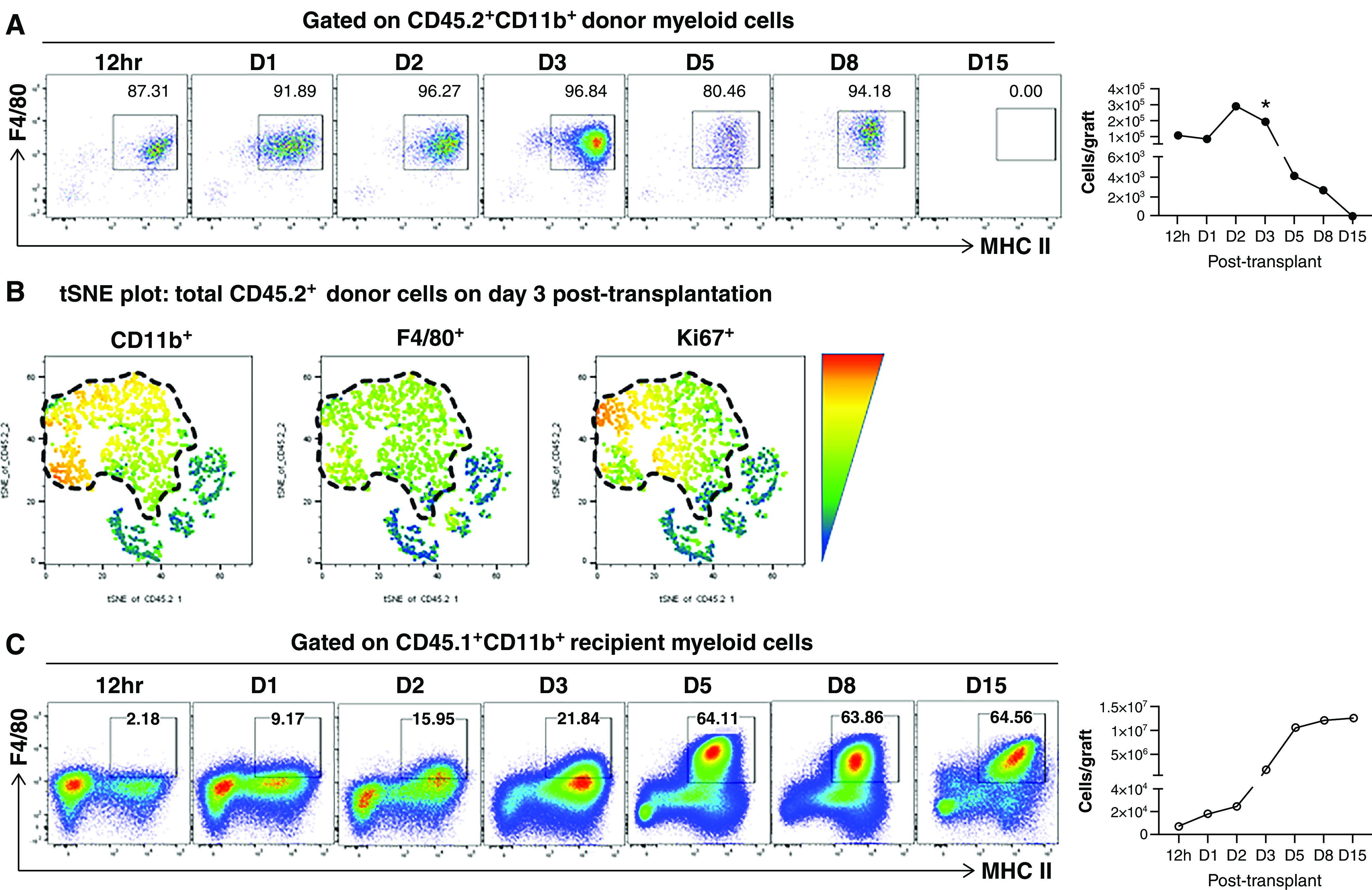
Donor and recipient kidney macrophage numbers show kinetic changes post-transplantation. (A) Representative FACS plots demonstrating the kinetics of donor kidney resident macrophages (F4/80HIMHC-IIHI) at indicated time points post-transplantation. Cells were gated on total live CD45.2+CD11b+ donor myeloid cells. The line graph on the right depicts the average absolute number of donor kidney resident macrophages present in the allograft at indicated time points (n=2–5 per time point). Data are presented as mean±SD. *P<0.05, calculated using unpaired t test by comparing with the absolute numbers of kidney resident macrophages in three age-matched untransplanted BALB/c kidneys at each time point. (B) T-distributed stochastic neighbor embedding (tSNE) plots of CD11b+, F4/80+, and Ki76+ of all donor (CD45.2+) cells in kidney allografts on day 3 post-transplantation. Cells were gated on total live CD45.2+ donor cells. Donor CD11b+ myeloid cells are outlined by the dashed line in the left tSNE plot. Their expression of F4/80 and Ki67 are outlined by the same dashed line in the next two tSNE plots. (C) Representative FACS plots demonstrating the kinetics of recipient graft-infiltrating myeloid cells at indicated time points post-transplantation. Cells were gated on total live CD45.1+CD11b+ recipient myeloid cells. The kinetics of recipient F4/80+MHC-II+ macrophage differentiation in the kidney allografts is shown by the progressive increase of the percentage of F4/80+MHC-II+ cells within this gate over time. The line graph on the right depicts the average absolute number of recipient F4/80+MHC-II+ kidney macrophages present in the allograft at indicated time points (n=2–5 per time point). Data are presented as mean±SD. D, day.
We next investigated the kinetics of recipient (CD45.1) macrophage infiltration. As shown in Figure 2C, we found that recipient CD45.1+CD11b+ cells began to infiltrate the kidney allograft as early as 12 hours post-transplantation. These cells initially did not express F4/80, suggesting they entered as circulating monocytes. In the ensuing days, they gradually upregulated F4/80 expression, which peaked on days 5–8 (Supplemental Figure 4). These F4/80HI cells also expressed a high level of MHC-II, suggesting their differentiation into a kidney resident macrophage phenotype.23 In addition to their phenotypic evolution, we observed a steep increase in their number between day 2 and 3, reaching the peak around day 5 (Figure 2C, line graph).
Collectively, these data suggest that, in unmanipulated transplant recipients, (1) kidney allograft harbors unique donor F4/80HICD11c+MHC-II+ macrophages that initially proliferate, but rapidly decline and are eventually eliminated by day 15; and (2) recipient macrophages promptly infiltrate and differentiate in the kidney allograft, and by day 15 are the only macrophages in the allograft.
CCL8 Expression in Both Donor and Recipient Kidney Macrophages Is Induced by Allogeneic Transplantation
Next, we examined CCL8 expression in both donor and recipient kidney macrophages. At baseline, kidney resident F4/80HIMHC-II+ macrophages in untransplanted donor kidneys did not express CCL8 (Figure 3A). In contrast, by day 7 post-transplantation, CCL8 was readily detectable in both donor and recipient F4/80HIMHC-II+ macrophages (Figure 3B). Ccl8 expression was also evident at earlier time points (day 1 and 3), following a pattern of gradual increase post-transplantation. Interestingly, at each time point, CCL8 expression was consistently higher in donor than in recipient macrophages (Figure 3C).
Figure 3.

Donor and recipient kidney macrophages show a kinetic expression of CCL8 post-transplantation. (A) CCL8 expression on donor kidney resident macrophages (F4/80HIMHC-IIHI cells) from naive untransplanted kidneys. (B) CCL8 expression on donor or recipient kidney macrophages 7 days post-transplantation. The left FACS plot shows intragraft donor (CD45.2+) versus recipient (CD45.1+) CD11b+ cells on day 7 post-transplantation. The middle FACS plots show the expression of F4/80 and MHC-II on CD45.2+ donor (upper plot) and CD45.1+ recipient (lower plot) myeloid cells. The right histograms show the expression of CCL8 by donor (upper histogram) and recipient (lower histogram) F4/80+MHC-II+ macrophages. (C) Quantification of CCL8 expression by donor or recipient kidney macrophages on the indicated days post-transplantation. Mean fluorescence intensity (MFI) of CCL8 by FACS normalized over isotype control was used for CCL8 quantification. n=2–3 per time point. Data are presented as mean±SD. *P≤0.05, calculated using unpaired t test.
Blocking the CCL8-CCR8 Axis Significantly Inhibits Early Allograft Inflammation and Improves Short-Term Kidney Function
In mice, CCR8 is a receptor for CCL8. This is distinct from human CCL8, which binds to CCR2.24 We first investigated CCR8 expression on various graft-infiltrating cells. As shown in Figure 4A, on day 3, CCR8 expression was readily detectable on intragraft CD4, CD8, and γδ T cells. It was also expressed on intragraft CD11b+ cells. Single-cell transcriptomics of mouse kidney allografts on day 15 confirmed Ccr8 expression by T cell clusters (Supplemental Figure 5), although expression by CD11b+ cells at that point was negative. These data suggest Ccr8 expression by CD11b+ cells is an early (e.g., day 3) event, which subsided later (e.g., day 15).
Figure 4.
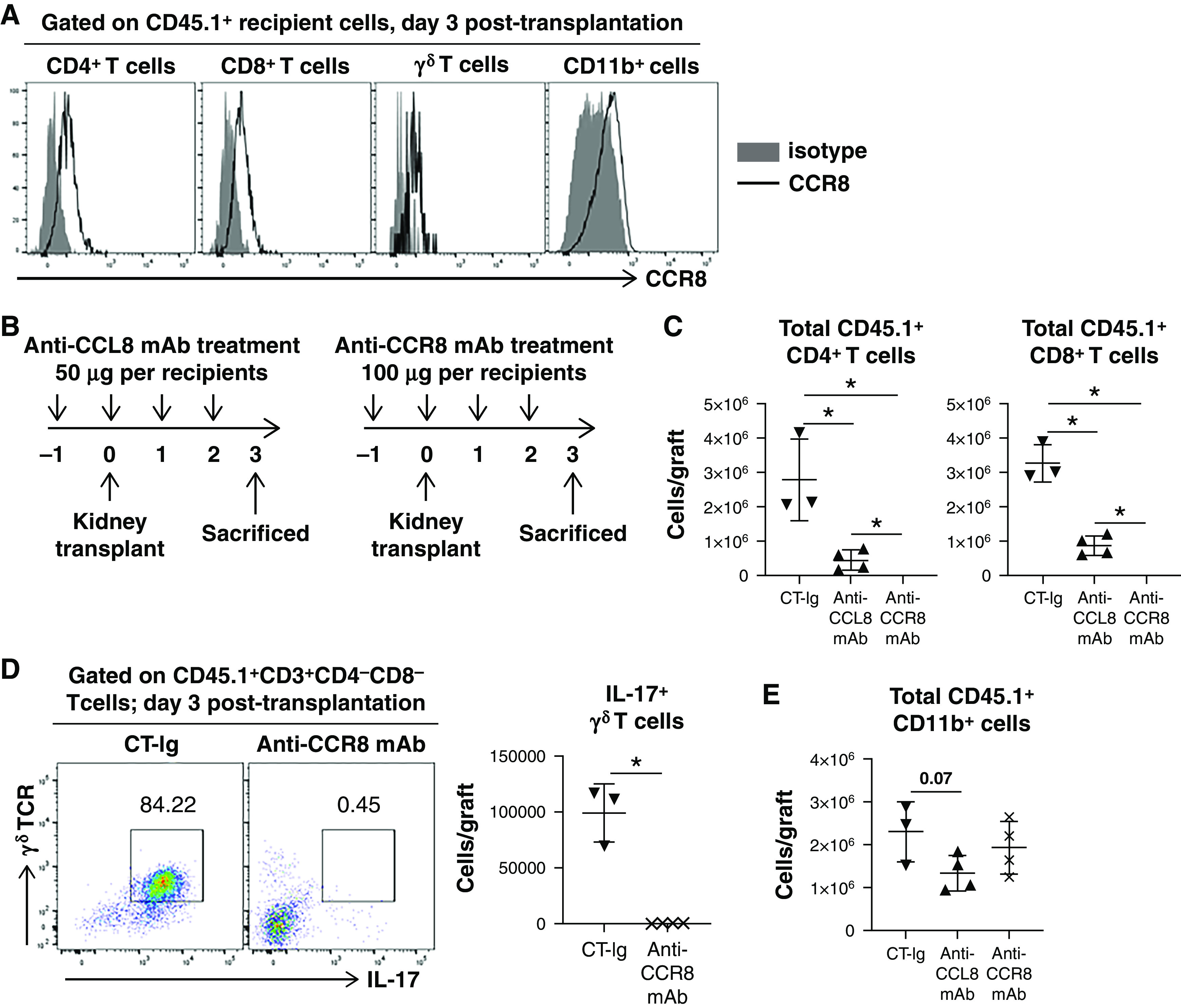
Blocking of CCL8-CCR8 axis alters graft-infiltrating immune cell number and functionality. (A) Representative histograms showing CCR8 expression by graft-infiltrating recipient (CD45.1+) CD11b+ myeloid cells, CD4 and CD8 cells, and γδ T cells on day 3 post-transplantation. (B) Treatment schemes of anti-CCL8 or anti-CCR8 mAb relative to allogeneic kidney transplantation. (C) Scatterplots showing the absolute number of graft-infiltrating recipient CD4 and CD8 T cells in the indicated groups on day 3 post-transplantation. n=3–4 per group. (D) Representative FACS plots demonstrating IL-17 expression by graft-infiltrating γδ T cells in the indicated groups on day 3 post-transplantation. Scatterplot showing the absolute number of intragraft IL-17+ γδ T cells in the indicated groups on day 3 post-transplantation. n=3–4 per group. (E) Scatterplot showing the absolute number of graft-infiltrating recipient CD11b+ cells in the indicated groups on day 3 post-transplantation. n=3–4 per group. Data are presented as mean±SD. *P≤0.05, calculated using unpaired t test.
Chemotaxis of CCR8+ cells may respond to intragraft CCL8. Therefore, we predicted that blocking CCL8-CCR8 would inhibit such chemotaxis. We tested this hypothesis by treating transplant recipients with either anti-CCL8 or anti-CCR8 mAb and comparing them with CT-Ig–treated recipients. Figure 4B shows the treatment regimens. Interestingly, both anti-CCL8 and anti-CCR8 treatment significantly reduced graft-infiltrating CD4 and CD8 T cells (Figure 4C). Moreover, anti-CCR8 also drastically reduced graft-infiltrating γδ T cells, particularly those expressing IL-17 (Figure 4D). A similar effect on γδ T cells was also observed with anti-CCL8 (data not shown). Our data suggest anti-CCR8 may be more efficacious than anti-CCL8 in blocking T cell infiltration. This could be explained by anti-CCR8 directly blocking the cell-surface receptor, whereas anti-CCL8 only indirectly blocks the axis by neutralizing the circulating ligand. In addition, anti-CCL8 treatment resulted in a trend of reduced intragraft infiltration of recipient CD11b+ cells on day 3 (Figure 4E). CCR8 blockade, on the other hand, did not reduce recipient CD11b+ cell infiltration, implicating an additional CCR8-independent chemotaxis pathway(s) used by these cells. Collectively, these data suggest the CCL8-CCR8 axis plays an important role in recipient T and myeloid cell graft infiltration early post-transplantation.
We next hypothesized that attenuation of recipient T cell infiltration by anti-CCL8 would promote superior early kidney allograft function. To test this hypothesis, B6 recipients were transplanted with BALB/c kidneys and treated with anti-CCL8 until day 27 (Figure 5A) before euthanasia on day 28. Serum creatinine was serially measured. Consistently lower serum creatinine levels were seen in anti-CCL8–treated recipients on days 3, 14, and 28 post-transplantation (Figure 5B, Supplemental Figure 6). We also observed a significant reduction in allograft fibrosis by Sirius Red staining (Figure 5C), along with a significant reduction of total intragraft CD8 T cells (Figure 5D) on day 28. Comparing data from Figure 4C (day 3) and Figure 5D (day 28), the more significant effect of CCL8-CCR8 blockade on T cell infiltration at an earlier time point suggests this axis likely plays a more prominent role early post-transplantation.
Figure 5.

CCL8 blockade improves short-term kidney transplant outcome. (A) Anti-CCL8 mAb treatment scheme. Recipients were treated with anti-CCL8 mAb (50 µg per mouse per dose; intraperitoneal), daily from day −1 to +7 and then every other day from day +9 to +27. Recipients were examined on day 28. (B) Scatterplots showing serum creatinine levels on day 28 post-transplantation. (C) Representative photomicrographs showing Sirius Red staining of kidney allografts from indicated groups. Scatterplot demonstrating quantification of allograft fibrosis determined by ImageJ as percent of Sirius Red–positive areas. (D) Total graft-infiltrating CD8 T cells on day 28 (D28) post-transplantation. n=4–9 per group. Data are presented as mean±SD. *P≤0.05, calculated using unpaired t test. Ab, antibody.
To determine whether the elimination of donor kidney resident macrophages observed in Figure 2A was also due to recipient alloimmunity, we next examined if anti-CCL8 treatment, by reducing graft T cell infiltration, also permitted longer persistence of these cells. B6 recipients were treated with daily anti-CCL8 or CT-Ig until euthanasia. As shown in Supplemental Figure 7, recipients treated with anti-CCL8 indeed harbored approximately 11-fold more donor kidney resident macrophages in the allograft on day 5 post-transplantation.
Donor Kidney Resident Macrophages Are a Key Determinant of Intragraft CCL8 Production
Because CCL8 production by donor macrophages preceded that by recipient macrophages, we hypothesized that donor kidney resident macrophages were the key instigator in orchestrating intragraft CCL8 production. If so, depleting donor kidney resident macrophages before kidney transplantation would greatly reduce intragraft CCL8 production and ensuing inflammation.
To test this hypothesis, we depleted donor kidney resident macrophages using a well-established protocol using clodronate liposome.25 As shown in Supplemental Figure 8, A and B, daily intravenous injections of clodronate liposomes in BALB/c mice (“Clodro” group) for a week effectively depleted F4/80HIMHC-II+ kidney resident macrophages (“day 8” plot), which remained depleted for at least a week (“day 14” plot). Immunofluorescence staining for F4/80 further confirmed a thorough depletion of this population in the kidneys (Supplemental Figure 8C). Control, nondepleted (“CT” group) BALB/c kidneys were from PBS-liposome treated mice (Figure 6A). As shown in Figure 6B, Ccl8 expression on day 7 was profoundly reduced in Clodro kidney allografts. Interestingly, such a differential expression was not seen with Ccl2 (data not shown), another major monocyte chemoattractant implicated in kidney transplant rejection.26 Of note, Ccl8 expression remained minimal in syngeneic transplantation, regardless of pretransplant depletion of kidney resident macrophages (Figure 6B), suggesting the induced Ccl8 expression is a consequence of allogeneicity, consistent with a previous study reporting detection of CCL8 only in allogeneic but not autologous bone marrow transplantation.12
Figure 6.

Donor kidney resident macrophages regulate post-transplant intragraft Ccl8 expression. (A) Donor (BALB/c) treatment groups. In the CT group, BALB/c mice were treated with PBS liposomes before being used as kidney donors for B6 recipients. In the Clodro group, BALB/c mice were treated with clodronate liposomes before being used as kidney donors for B6 recipients. (B) Recipients in both groups were euthanized on day 7 post-transplantation (Post-Tx) for examination. Comparisons were also made with syngeneic kidney transplantation (Syn-Tx) groups in which B6 recipients were transplanted with kidneys from B6 donors treated either with PBS liposomes (CT) or with clodronate liposomes (Clodro). Scatterplot showing the expression of Ccl8 measured by quantitative PCR in kidney grafts on day 7 post-transplantation. n=2–5 per group. (C) Expression of Ifng measured by quantitative PCR in kidney grafts retrieved on days 3 and 7 post-transplantation from allogeneic and syngeneic transplant recipients. n=2–5 per group. (D) Secretion of CCL8 by kidney macrophages upon in vitro IFN-γ stimulation. CCL8 levels were measured by ELISA in culture supernatants on indicated days. Data were collected from four independent experiments. For (B–D), data are presented as mean±SD. *P≤0.05, calculated using unpaired t test. Allo-Tx, allogeneic kidney transplantation.
We next investigated the potential upstream regulator of Ccl8 expression relevant to allogeneic transplantation. It was recently demonstrated that early recognition of allograft as nonself by innate immune cells results in intragraft IFN-γ expression,27 and IFN-γ is a known inducer of Ccl8 in macrophages.28 Therefore, we first determined Ifng expression in our model. As shown in Figure 6C, a significant expression of Ifng in kidney allografts was observed on day 3, and expression further increased by 1 log on day 7. In contrast, Ifng expression in isografts was minimal to absent (Figure 6C). We next tested if IFN-γ stimulation of sorted BALB/c kidney macrophages in vitro would induce their CCL8 production. As shown in Figure 6D, BALB/c kidney macrophages stimulated with IFN-γ indeed produced elevated levels of CCL8 in comparison with unstimulated cells. Collectively, these data support that intragraft production of CCL8 is a consequence of allogeneicity-induced IFN-γ production.
Next, we investigated if depletion of donor kidney resident macrophages would prevent early graft immune cell infiltration. As shown in Figure 7A, the Clodro group had a significant reduction of graft-infiltrating CD45.1+CD11b+ cells on day 3 in comparison with the CT group. This reduction persisted to at least day 7, as evidenced by a significant reduction of graft-infiltrating CD45.1+CD11b+F4/80+MHC-II+ macrophages in the Clodro group (Figure 7B). Furthermore, there was also a significant reduction in graft-infiltrating CD4 and CD8 T cells (Figure 7C), along with γδ T cells, including IL-17–expressing γδ T cells (Figure 7D). Il17a transcripts were also reduced in the Clodro group on day 7 (Figure 7E). Collectively, these data show that donor kidney resident macrophage depletion resulted in a drastic inhibition of Ccl8 expression in the kidney allograft and a significant reduction in recipient immune cell graft infiltration early post-transplantation.
Figure 7.

Early host immune cell allograft infiltration differs significantly in CT versus Clodro group recipients. (A) Scatterplot showing the absolute number of graft-infiltrating recipient CD11b+ cells in the indicated groups on day 3 post-transplantation. n=4–5 per group. (B) Representative FACS plots showing graft-infiltrating recipient CD11b+ cells differentiating to F4/80+MHC+ macrophages in the indicated groups on day 7 post-transplantation. Scatterplot showing the absolute number of recipient intragraft F4/80+MHC+ macrophages in the indicated groups. n=7 per group. (C) Scatterplots showing the absolute number of recipient total CD4 and CD8 T cells in the indicated groups on day 3 post-transplantation. n=4–5 per group. (D) Representative FACS plots showing IL-17+ γδ T cells in the indicated groups on day 3 post-transplantation. Cells were gated on CD45.1+CD3+ T cells. Scatterplots showing the absolute number of total γδ T cells or IL-17+ γδ T cells in the indicated groups. n=3 per group. (E) Scatterplot showing Il17a expression measured by quantitative PCR in whole-kidney grafts in the indicated groups on day 7 post-transplantation. n=2–5 per group. Data are presented as mean±SD. *P≤0.05, calculated using unpaired t test. Allo-Tx, allogeneic kidney transplantation; Syn-Tx, syngeneic kidney transplantation.
Lastly, we investigated if donor kidney resident macrophages could promote allospecific T cell proliferation. Here, we took advantage of the transgenic 4C CD4 T cells that directly recognize I-Ad expressed on BALB/c cells.17 Proliferation dye–labeled 4C T cells were cocultured with sorted BALB/c kidney macrophages (described in Methods). As shown in Supplemental Figure 9, unstimulated kidney macrophages were unable to stimulate 4C T cell proliferation. In contrast, prestimulation of kidney macrophages with IFN-γ rendered these cells capable of stimulating 4C T cell proliferation. Of note, CCL8 blockade by anti-CCL8 in this system did not block 4C T cell proliferation, suggesting the CCL8-CCR8 axis is not implicated in donor-stimulated proliferation.
Depletion of Donor Kidney Resident Macrophages Improves Short-Term Kidney Allograft Function
Aggressive early kidney allograft infiltration of immune cells has been associated with poor short- and long-term allograft function.29–34 Therefore, we investigated if reduced early graft infiltration of immune cells by pre-emptive donor kidney resident macrophage depletion would result in improved kidney allograft function. As shown in Figure 8A, serum creatinine and BUN levels were significantly lower in the Clodro group on day 28 post-transplantation. Expression of the kidney injury marker Lcn2 (NGAL) was also reduced in this group (Figure 8B). Histopathologic evaluation revealed significantly lower scores for intimal arteritis and peritubular capillaritis in the Clodro group (Figure 8C), together with reduced inflammatory markers granzyme B (Gzmb) and TNFα (Tnfa) (Figure 8D), and reduced CD8 T cell infiltration (Figure 8E). Notably, we did not observe any difference in anti-donor antibodies between the two groups on day 28 (Supplemental Figure 10).
Figure 8.
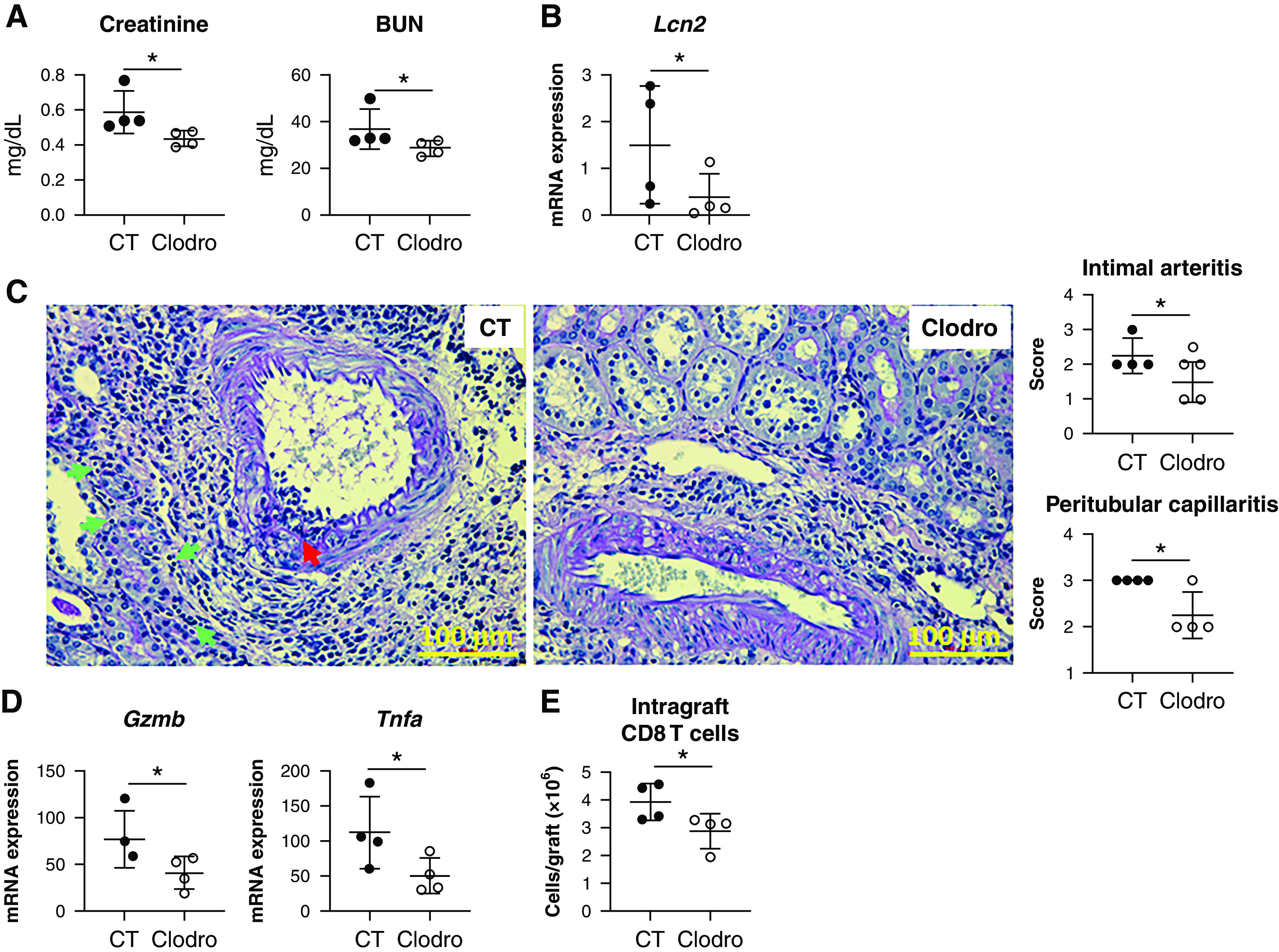
Kidney allograft function in Clodro recipients on day 28 post-transplantation is significantly better than that in CT recipients. (A) Scatterplots showing serum creatinine and BUN levels in the indicated groups on day 28 post-transplantation. (B) Scatterplot showing the expression level of the kidney injury maker gene Lcn2 measured by quantitative PCR in the indicated groups on day 28 post-transplantation. (C) Representative PAS-stained sections of kidney allografts from CT versus Clodro recipients. Red arrow, intimal arteritis; green arrows, peritubular capillaritis. Images are representative of n=4 per group. Scatterplots showing averaged pathologic scores for intimal arteritis and peritubular capillaritis in the indicated groups. (D) Scatterplots showing the expression level of inflammatory marker genes Gzmb and Tnfa measured by quantitative PCR in the indicated groups. (E) Scatterplot showing the absolute number of graft-infiltrating CD8 T cells in the indicated groups. CD8 T cell numbers were calculated from FACS analysis of individual kidney allografts. For (A), (B), (D), and (E), n=4 per group. Data are presented as mean±SD. *P≤0.05, calculated using unpaired t test.
Collectively, these data suggest depletion of donor kidney resident macrophages before transplantation significantly improves short-term kidney allograft function associated with a reduction of early inflammation.
Discussion
Our previous single-cell transcriptomics of mouse kidney allografts revealed that Ccl8 is prominently expressed by allograft macrophages.16 This study made a series of novel observations examining the cellular origin and kinetics of Ccl8 expression after kidney transplantation, and its role in mediating early allograft inflammation. We show that donor kidney resident macrophages initially expand after transplantation and are the primary source of early CCL8 production. This, in turn, promotes recipient monocyte infiltration, their differentiation, and production of CCL8. Through its cognate receptor CCR8, CCL8 promotes recipient T cell infiltration and allograft inflammation. Consequently, peritransplant blockade of CCL8 or CCR8, or pretransplant depletion of donor kidney resident macrophages, attenuates early graft inflammation and improves short-term kidney allograft function. These data thus reveal a previously unrecognized role of CCL8-CCR8 in mediating the effects of donor kidney resident macrophages on early allograft inflammation.
An interesting observation here is that intragraft Ccl8 expression is induced only by allogeneic, but not syngeneic, kidney transplantation. Mechanistic investigation revealed that CCL8 production in kidney macrophages can be induced upon IFN-γ stimulation. Indeed, we observed that Ifng is expressed in allogeneic, but not syngeneic, kidney transplantation. These results support the idea that the observed intragraft CCL8 induction is a consequence of intragraft IFN-γ production in response to allogeneic kidney transplantation. It was previously shown that expression of intragraft IFN-γ is promoted by recipient innate immune cells as a result of recognition of allograft nonself27 via PIR-α-MHC-I and CD47-SIRP-α interactions.5,6 Such innate alloimmune recognition may be directly upstream of IFN-γ induction, or it may be further potentiated by a nonspecific inflammatory milieu precipitated by IRI and production of the damage-associated molecular patterns commonly associated with kidney implantation.35 Indeed, kidney resident macrophages respond to IRI by secreting inflammatory cytokines and chemokines.36 Ongoing studies aim to elucidate specific signaling requirements for these events to identify potential therapeutic targets.
CCL8, or MCP-2, is a member of the conserved CC chemokine family that promotes leukocyte recruitment. In contrast to CCL2/MCP-1, which has been extensively investigated in kidney transplantation,37 the role of CCL8 in transplantation is entirely unknown. CCL8 binds to multiple receptors on leukocytes, including CCR1, CCR2, CCR5, and CCR8,24,38 suggesting CCL8 may have the capacity to recruit a broad range of leukocytes. Interestingly, blockade of CCR1 and/or CCR5 inhibits graft infiltration of CD4 and CD8 cells and monocyte/macrophages, and prolongs allograft survival.39,40 These studies, however, did not investigate the role of CCL8 in mediating the effect of CCR1 and/or CCR5. CCL8 also induces expression of adhesion molecules41 and enhances endothelial permeability,42 both of which can further facilitate T cell migration.43–45 Indeed, we show that blocking CCL8-CCR8 dramatically inhibits graft infiltration of immune cells early post-transplantation. Notably, T cells under homeostasis do not express cell surface CCR8. Instead, the intracellularly stored CCR8 can be rapidly transported to the cell surface upon stimulation.46 Here, we show that graft-infiltrating αβ and γδ T cells both express surface CCR8, and that blocking the CCL8-CCR8 axis severely impairs recruitment of these cells to the kidney allograft. The finding of the effect of CCL8 on γδ T cell infiltration is unexpected and not previously reported. It is known that γδ TCR confers these cells with the ability to respond to antigens in an MHC-independent manner.47 Coupled with the finding that the majority of γδ T cells in the allograft also expressed IL-17, our data suggest the chemotaxis and function of these cells are likely also MHC independent. Collectively, our data provide strong evidence that the CCL8-CCR8 axis is an important linchpin between donor kidney resident macrophages and early immune cell infiltration and therefore represents a promising therapeutic target.
Although the CCL8-CCR8 axis is the focus of this study, it is likely that donor kidney resident macrophages use additional mechanisms to promote graft inflammation. Published literature in native kidney diseases demonstrate these cells efficiently clear small immune complexes, sense TLR agonists, and recruit monocytes and neutrophils.20 On the other hand, these cells can also stimulate proangiogenic and reparative pathways.21 Indeed, in allogeneic kidney transplantation, donor kidney resident macrophages may persist long after transplantation, and their longevity may correlate with rejection-free graft survival.48 Whether this indicates an active immune regulatory role of these cells remains to be determined. Future studies are also needed to identify the switch between proinflammatory and proreparative programs in these cells that can be precisely targeted to benefit allograft outcome.
A clear understanding of the pathways implicated in donor kidney resident macrophage–mediated early allograft inflammation will have two immediate clinical implications. The first is the potential to expand the usage of “marginal donor” kidneys, particularly those with a prolonged cold ischemia time (CIT), which are commonly discarded.49 Notably, prolonged CIT promotes aggressive early recipient macrophage infiltration,50 an event that, in our model, could be effectively attenuated by blocking the CCL8-CCR8 axis or by depleting donor kidney resident macrophages. Therefore, targeting these steps may mitigate the negative effect of CIT and, consequently, expand our transplantable deceased donor kidney pool. The second is the potential to reduce peritransplant inflammation to facilitate transplant tolerance.51 All current approaches of transplant tolerance induction require pre-emptive recipient conditioning for days to weeks before transplantation to circumvent challenges brought by peritransplant inflammation.52 Consequently, these approaches are only feasible for living donor kidney transplantation. Blocking the CCL8-CCR8 axis or depleting donor kidney resident macrophages may sufficiently reduce peritransplant inflammation so that pre-emptive recipient conditioning is no longer necessary, and tolerance induction can be safely delayed to the post-transplant period. If proven true, this would significantly extend current tolerance approaches to deceased donor kidney transplantation.
In summary, our study elucidates the role of CCL8-CCR8 originating from donor kidney resident macrophages in early kidney allograft inflammation. Therapeutic targets revealed by these mechanistic studies will have the potential to expand the transplantable donor kidney pool and to promote kidney allograft longevity.
Disclosures
J. Kwun reports receiving research funding from Alexion, eGenesis, and MorphoSys. X. Luo reports serving on the editorial boards of American Journal of Transplantation and Transplantation; having patents or royalties with Diabetes-Free Inc.; receiving research funding from NIH; receiving honoraria from NIH, University of Illinois Chicago, and Virginia Tech; and having consultancy agreements with ViaCyte. X. Shen reports having ownership interest in AccuBeing and Xilis; having patents or royalties with Duke University and Xilis Inc.; serving as chief scientific officer of the Terasaki Institute; and being employed by Terasaki Institute and Xilis Inc. All remaining authors have nothing to disclose.
Funding
This work was supported by the Henry E. Haller Jr. Foundation (A. Dangi, X. Luo); NIH grants R01DK132889, R01HL139812, and R01AI114824 (all to A. Dangi, X. Luo); and the American Society of Nephrology Ben J Lipps Research fellowship (I. Husain).
Supplementary Material
Acknowledgments
We would like to thank the Duke Core for Microsurgery and Surgical Models in Small Animals for their technical assistance in murine kidney transplantation.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Author Contributions
A. Dangi, I. Husain, C.Z. Jordan, N. Natesh, X. Shen, and S. Yu were responsible for data curation and formal analysis; A. Dangi, I. Husain, J. Kwun, X. Luo, N. Natesh, X. Shen, and S. Yu were responsible for investigation; A. Dangi, I. Husain, N. Natesh, and X. Shen were responsible for methodology; A. Dangi, J. Kwun, and X. Luo conceptualized the study; A. Dangi and X. Luo wrote the original draft and were responsible for project administration; J. Kwun and X. Luo provided supervision; X. Luo reviewed and edited the manuscript and was responsible for funding acquisition; N. Natesh and X. Shen were responsible applying software packages to analysis of single cell RNA sequencing data; and S. Yu was responsible for validation.
Data Sharing Statement
Data will be made available upon request to the corresponding author.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2022020139/-/DCSupplemental.
Supplemental Figure 1. Violin plot depicting Ccl8 expression by various immune and non-immune cell clusters from rejection kidney allografts.
Supplemental Figure 2. Identification and phenotypic characterization of kidney resident macrophages.
Supplemental Figure 3. Donor kidney-resident and recipient graft-infiltrating macrophages co-exist in syngeneic kidney transplant grafts on day 15 post-transplantation.
Supplemental Figure 4. Up-regulation of F4/80 mean fluorescent intensity (MFI) on graft-infiltrating recipient CD11b+ cells post allogeneic kidney transplantation.
Supplemental Figure 5. UMAPs of un-transplanted kidneys or transplanted kidneys on day 15 post-transplantation (in allogeneic recipients) showing Ccr8 expression in distinct cell clusters.
Supplemental Figure 6. Recipient serum creatinine levels at various time points post-transplantation in CT-Ig and anti-CCL8 Ab treated recipients.
Supplemental Figure 7. Anti-CCL8 treatment permits longer survival of donor kidney resident macrophages post-transplantation.
Supplemental Figure 8. Depletion of kidney resident macrophages in kidney donors.
Supplemental Figure 9. Alloantigen-specific CD4 T cell proliferation stimulated by donor kidney resident macrophages.
Supplemental Figure 10. Donor-specific alloantibodies on day 28 post-transplantation.
References
- 1.Thurman JM: Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol 123: 7–13, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schröppel B, Legendre C: Delayed kidney graft function: From mechanism to translation. Kidney Int 86: 251–258, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Zhao H, Alam A, Soo AP, George AJT, Ma D: Ischemia-reperfusion injury reduces long term renal graft survival: Mechanism and beyond. EBioMedicine 28: 31–42, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuquay R, Renner B, Kulik L, McCullough JW, Amura C, Strassheim D, et al. : Renal ischemia-reperfusion injury amplifies the humoral immune response. J Am Soc Nephrol 24: 1063–1072, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai H, Lan P, Zhao D, Abou-Daya K, Liu W, Chen W, et al. : PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science 368: 1122–1127, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai H, Friday AJ, Abou-Daya KI, Williams AL, Mortin-Toth S, Nicotra ML, et al. : Donor SIRPα polymorphism modulates the innate immune response to allogeneic grafts. Sci Immunol 2: eaam6202, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JG, Na M, Kim MG, Park SH, Lee HJ, Kim DK, et al. : Immune cell composition in normal human kidneys. Sci Rep 10: 15678, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conway BR, O’Sullivan ED, Cairns C, O’Sullivan J, Simpson DJ, Salzano A, et al. : Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J Am Soc Nephrol 31: 2833–2854, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilgelm AE, Richmond A: Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front Immunol 10: 333, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto M, Ota A, Hori T, Imai S, Sohma H, Suzuki N, et al. : Early expression of plasma CCL8 closely correlates with survival rate of acute graft-vs.-host disease in mice. Exp Hematol 39: 1101–1112, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Klementowicz JE, Mahne AE, Spence A, Nguyen V, Satpathy AT, Murphy KM, et al. : Cutting Edge: Origins, recruitment, and regulation of CD11c(+) cells in inflamed islets of autoimmune diabetes mice. J Immunol 199: 27–32, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hori T, Naishiro Y, Sohma H, Suzuki N, Hatakeyama N, Yamamoto M, et al. : CCL8 is a potential molecular candidate for the diagnosis of graft-versus-host disease. Blood 111: 4403–4412, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdi R, Means TK, Ito T, Smith RN, Najafian N, Jurewicz M, et al. : Differential role of CCR2 in islet and heart allograft rejection: Tissue specificity of chemokine/chemokine receptor function in vivo. J Immunol 172: 767–775, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Lo DJ, Weaver TA, Kleiner DE, Mannon RB, Jacobson LM, Becker BN, et al. : Chemokines and their receptors in human renal allotransplantation. Transplantation 91: 70–77, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dangi A, Yu S, Lee FT, Burnette M, Wang JJ, Kanwar YS, et al. : Murine cytomegalovirus dissemination but not reactivation in donor-positive/recipient-negative allogeneic kidney transplantation can be effectively prevented by transplant immune tolerance. Kidney Int 98: 147–158, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dangi A, Natesh NR, Husain I, Ji Z, Barisoni L, Kwun J, et al. : Single cell transcriptomics of mouse kidney transplants reveals a myeloid cell pathway for transplant rejection. JCI Insight 5: e141321, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brennan TV, Hoang V, Garrod KR, Liu FC, Hayden T, Kim J, et al. : A new T-cell receptor transgenic model of the CD4+ direct pathway: Level of priming determines acute versus chronic rejection. Transplantation 85: 247–255, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Schenk, AD, Rosenblum, JM, Fairchild, RL: Chemokine-directed strategies to attenuate allograft rejection. Clin Lab Med 28: 441–454, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. : Specification of tissue-resident macrophages during organogenesis. Science 353: aaf4238, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamatiades EG, Tremblay ME, Bohm M, Crozet L, Bisht K, Kao D, et al. : Immune monitoring of trans-endothelial transport by kidney-resident macrophages. Cell 166: 991–1003, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puranik AS, Leaf IA, Jensen MA, Hedayat AF, Saad A, Kim KW, et al. : Kidney-resident macrophages promote a proangiogenic environment in the normal and chronically ischemic mouse kidney. Sci Rep 8: 13948, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al. : Single-Cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol 30: 767–781, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirsch S, Austyn JM, Gordon S: Expression of the macrophage-specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J Exp Med 154: 713–725, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. : Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol 12: 167–177, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno SG: Depleting macrophages in vivo with clodronate-liposomes. Methods Mol Biol 1784: 259–262, 2018 [DOI] [PubMed] [Google Scholar]
- 26.Ho J, Rush DN, Nickerson PW: Urinary biomarkers of renal transplant outcome. Curr Opin Organ Transplant 20: 476–481, 2015 [DOI] [PubMed] [Google Scholar]
- 27.Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, et al. : Non-self recognition by monocytes initiates allograft rejection. J Clin Invest 124: 3579–3589, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Struyf S, Proost P, Vandercappellen J, Dempe S, Noyens B, Nelissen S, et al. : Synergistic up-regulation of MCP-2/CCL8 activity is counteracted by chemokine cleavage, limiting its inflammatory and anti-tumoral effects. Eur J Immunol 39: 843–857, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Guillén-Gómez E, Dasilva I, Silva I, Arce Y, Facundo C, Ars E, et al. : Early macrophage infiltration and sustained inflammation in kidneys from deceased donors are associated with long-term renal function. Am J Transplant 17: 733–743, 2017 [DOI] [PubMed] [Google Scholar]
- 30.Bräsen JH, Khalifa A, Schmitz J, Dai W, Einecke G, Schwarz A, et al. : Macrophage density in early surveillance biopsies predicts future renal transplant function. Kidney Int 92: 479–489, 2017 [DOI] [PubMed] [Google Scholar]
- 31.Magil AB, Tinckam K: Monocytes and peritubular capillary C4d deposition in acute renal allograft rejection. Kidney Int 63: 1888–1893, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Bergler T, Jung B, Bourier F, Kühne L, Banas MC, Rümmele P, et al. : Infiltration of macrophages correlates with severity of allograft rejection and outcome in human kidney transplantation. PLoS One 11: e0156900, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girlanda R, Kleiner DE, Duan Z, Ford EA, Wright EC, Mannon RB, et al. : Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant 8: 600–607, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyairi S, Ueda D, Yagisawa T, Okada D, Keslar KS, Tanabe K, et al. : Recipient myeloperoxidase-producing cells regulate antibody-mediated acute versus chronic kidney allograft rejection. JCI Insight 6: e148747, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aiello S, Podestà MA, Rodriguez-Ordonez PY, Pezzuto F, Azzollini N, Solini S, et al. : Transplantation-induced ischemia-reperfusion injury modulates antigen presentation by donor renal CD11c+F4/80+ macrophages through IL-1R8 regulation. J Am Soc Nephrol 31: 517–531, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD: Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int 71: 619–628, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Viedt C, Orth SR: Monocyte chemoattractant protein-1 (MCP-1) in the kidney: Does it more than simply attract monocytes? Nephrol Dial Transplant 17: 2043–2047, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Ruffing N, Sullivan N, Sharmeen L, Sodroski J, Wu L: CCR5 has an expanded ligand-binding repertoire and is the primary receptor used by MCP-2 on activated T cells. Cell Immunol 189: 160–168, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Yun JJ, Whiting D, Fischbein MP, Banerji A, Irie Y, Stein D, et al. : Combined blockade of the chemokine receptors CCR1 and CCR5 attenuates chronic rejection. Circulation 109: 932–937, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Bedke J, Kiss E, Schaefer L, Behnes CL, Bonrouhi M, Gretz N, et al. : Beneficial effects of CCR1 blockade on the progression of chronic renal allograft damage. Am J Transplant 7: 527–537, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Orejudo M, Rodrigues-Diez RR, Garcia-Redondo A, Alique M, Egido J, Selgas R, et al. : SP081 MCP-2/CCR8 axis is activated in experimental renal and vascular inflammation. Nephrol Dial Transplant 30: iii405–iii406, 2015 [Google Scholar]
- 42.Xue S, Tang H, Zhao G, Fang C, Shen Y, Yan D, et al. : C-C motif ligand 8 promotes atherosclerosis via NADPH oxidase 2/reactive oxygen species-induced endothelial permeability increase. Free Radic Biol Med 167: 181–192, 2021 [DOI] [PubMed] [Google Scholar]
- 43.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B: Monocyte chemotactic proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J 8: 1055–1060, 1994 [DOI] [PubMed] [Google Scholar]
- 44.Roth SJ, Carr MW, Springer TA: C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol 25: 3482–3488, 1995 [DOI] [PubMed] [Google Scholar]
- 45.Taub DD, Proost P, Murphy WJ, Anver M, Longo DL, van Damme J, et al. : Monocyte chemotactic protein-1 (MCP-1), -2, and -3 are chemotactic for human T lymphocytes. J Clin Invest 95: 1370–1376, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gombert M, Dieu-Nosjean MC, Winterberg F, Bünemann E, Kubitza RC, Da Cunha L, et al. : CCL1-CCR8 interactions: An axis mediating the recruitment of T cells and Langerhans-type dendritic cells to sites of atopic skin inflammation. J Immunol 174: 5082–5091, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Ribot JC, Lopes N, Silva-Santos B: γδ T cells in tissue physiology and surveillance. Nat Rev Immunol 21: 221–232, 2021 [DOI] [PubMed] [Google Scholar]
- 48.Malone AF, Wu H, Fronick C, Fulton R, Gaut JP, Humphreys BD: Harnessing expressed single nucleotide variation and single cell RNA sequencing to define immune cell chimerism in the rejecting kidney transplant. J Am Soc Nephrol 31: 1977–1986, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helanterä I, Ibrahim HN, Lempinen M, Finne P: Donor age, cold ischemia time, and delayed graft function. Clin J Am Soc Nephrol 15: 813–821, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorbacheva V, Fan R, Beavers A, Fairchild RL, Baldwin WM 3rd, Valujskikh A: Anti-donor MHC class II alloantibody induces glomerular injury in mouse renal allografts subjected to prolonged cold ischemia. J Am Soc Nephrol 30: 2413–2425, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chong AS, Alegre ML: Transplantation tolerance and its outcome during infections and inflammation. Immunol Rev 258: 80–101, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Issa F, Strober S, Leventhal JR, Kawai T, Kaufman DB, Levitsky J, et al. : The Fourth International Workshop on Clinical Transplant Tolerance. Am J Transplant 21: 21–31, 2021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.