

Abstract
The unicellular amoeba Acanthamoeba castellanii is ubiquitous in aquatic environments, where it preys on bacteria. The organism also hosts bacterial endosymbionts, some of which are parasitic, including human pathogens such as Chlamydia and Legionella spp. Here we report complete, high-quality genome sequences for two extensively studied A. castellanii strains, Neff and C3. Combining long- and short-read data with Hi-C, we generated near chromosome-level assemblies for both strains with 90% of the genome contained in 29 scaffolds for the Neff strain and 31 for the C3 strain. Comparative genomics revealed strain-specific functional enrichment, most notably genes related to signal transduction in the C3 strain and to viral replication in Neff. Furthermore, we characterized the spatial organization of the A. castellanii genome and showed that it is reorganized during infection by Legionella pneumophila. Infection-dependent chromatin loops were found to be enriched in genes for signal transduction and phosphorylation processes. In genomic regions where chromatin organization changed during Legionella infection, we found functional enrichment for genes associated with metabolism, organelle assembly, and cytoskeleton organization. Given Legionella infection is known to alter its host's cell cycle, to exploit the host's organelles, and to modulate the host's metabolism in its favor, these changes in chromatin organization may partly be related to mechanisms of host control during Legionella infection.
The first amoebae were isolated in 1913 (Pushkarew 1913), and the genus Acanthamoeba was established in 1931 by Volkonsky (1931). It comprises different species of free-living, aerobic, unicellular protozoa, present throughout the world in soil and nearly all aquatic environments (Rodríguez-Zaragoza 1994). The life cycle of Acanthamoeba includes a dormant cyst with minimal metabolic activities under harsh conditions and a motile trophozoite that can feed on small organisms and reproduce by binary fission in optimal conditions (Siddiqui and Khan 2012). Acanthamoeba is perhaps most widely known from its role as a human pathogen, acting to cause the vision-threatening eye infection Acanthamoeba keratitis, but it can also cause serious infections of the lungs, sinuses, and skin, as well as a central nervous system disease called granulomatous amoebic encephalitis (Visvesvara et al. 2007). The species Acanthamoeba castellanii was first isolated in 1930 by Castellani (1930) as a contaminant of a yeast culture.
In their natural environment, Acanthamoeba spp. are characterized by the ability to change their shape through pseudopod formation and are considered professional phagocytes as they feed on bacteria but may also phagocytose yeasts and algae. However, some bacteria are resistant to degradation and live as endosymbionts in these protozoa, and others even use the amoeba as a replication niche. Thus, Acanthamoeba are also reservoirs of microorganisms and viruses, including human pathogens, which have adapted to survive inside these cells and resist digestion, to persist, or even to replicate as intracellular parasites. At least 15 different bacterial species, two archaea, and several eukaryotes and viruses have been shown to interact with Acanthamoeba in the environment and may also coexist at the same time within the same host cell (Samba-Louaka et al. 2019).
Although it was observed early on that bacteria could resist digestion of free-living amoebae (Drozanski 1956), it was not until the discovery that Legionella pneumophila replicated in amoebae that researchers began studying the bacterium–amoeba relationship in depth (Rowbotham 1980). L. pneumophila is the agent responsible for Legionnaires’ disease, a severe pneumonia that can be fatal if not treated promptly. In addition, many species of amoebae have the ability to form highly resistant cysts in hostile environments, providing shelter for their intracellular parasites (Kilvington and Price 1990). Indeed, it is thought that L. pneumophila may survive water disinfection treatments and contaminate water distribution systems by encystation of its host (Ikedo and Yabuuchi 1986; Lasheras et al. 2006; Pagnier et al. 2008). From these contaminated water sources, L. pneumophila can reach the human lungs via aerosols contaminated with the bacteria and replicate within the alveolar macrophages, which are, like amoebae, phagocytic cells.
L. pneumophila has the ability to escape the lysosomal degradation pathway of both A. castellanii and human alveolar macrophages through the formation of a protective vacuole (the Legionella-containing vacuole [LCV]) where it multiplies to high numbers. Once the host cell has been fully exploited and nutrients become limited, L. pneumophila exits the host and infects a new cell (Mondino et al. 2020).
To establish the LCV and replicate, L. pneumophila secretes over 300 effector proteins into the host cytoplasm via a type four secretion system (T4SS) called Dot/Icm (Kubori and Nagai 2016), thereby manipulating host pathways and redirecting nutrients to the LCV (Isberg et al. 2009; Ensminger 2016). In the early stages of infection, many of these proteins target the host secretory pathway, including several small GTPases, to recruit endoplasmic reticulum–derived vesicles to the LCV (Swart et al. 2020). During the intracellular cycle, a wider range of processes, including membrane trafficking, cytoskeleton dynamics, and signal transduction pathways, is targeted by these effectors (Hubber and Roy 2010; Qiu and Luo 2017). L. pneumophila also directly alters the genome of its host by modifying epigenetic marks of the host genome in human macrophages and A. castellanii. It secretes an effector named RomA with histone methyltransferase activity that is targeted to the host's nucleus. RomA carries out genome-wide trimethylation of K14 of histone H3, leading to transcriptional changes that modulate the host response in favor of bacterial survival (Rolando et al. 2013). Concomitantly, L. pneumophila infection leads to genome-wide changes in gene expression (Li et al. 2020). In many eukaryotes, gene regulation is intertwined within the three-dimensional organization of chromosomes. The functional interplay between gene regulation and higher-order chromatin elements such as loops, self-interacting domains, and active/inactive compartments is actively being studied (Nora et al. 2012; Rennie et al. 2018). Therefore, the infection of A. castellanii by L. pneumophila provides an amenable model to investigate how an intracellular bacterial infection may affect the regulation of chromosome folding, and its consequences, in a eukaryotic host.
The investigation of genome organization and regulation of A. castellanii in response to infection requires a highly contiguous genome assembly. The reference genome sequence for A. castellanii, Neff-v1 (Clarke et al. 2013), is based on the Neff strain, isolated from soil in California in 1957 (Neff 1957). This assembly is widely used by different laboratories studying A. castellanii but is fragmented into 384 scaffolds comprising 3192 contigs, which makes chromosome-level analyses difficult, if not impossible, and basic features of the A. castellanii genome, such as the number of chromosomes and ploidy, remain undetermined. In addition, many teams investigating bacteria–amoeba interactions use the “C3” strain of A. castellanii (ATCC 50739), isolated from a drinking water reservoir in Europe in 1994 and identified as a mouse pathogen (Michel and Hauröder 1997). However, genomic information is scarce for this strain, and little is known about its similarity to the Neff strain. Notably, these two A. castellanii strains have been cultivated for several decades and were isolated from different ecological niches, but the extent of conservation between their genomes is unknown. It is difficult to investigate the factors that determine the susceptibility of different A. castellanii strains to L. pneumophila infection without proper genomic resources. These resources would also be required to apply genome-wide omics approaches.
This work intended to (1) deliver a high-quality reference genome of A. castellanii, a ubiquitous amoeba, and (2) take advantage of this assembly to explore how A. castellanii responds to L. pneumophila infection through the lens of the three-dimensional organization of its genome. We combined Illumina, Oxford Nanopore long-read, and Hi-C data to generate high-quality reference genome sequence of the C3 strain, as well as a new and improved reference assembly of the Neff genome (Neff-v2). We next analyzed the genome folding and expression changes of A. castellanii C3 strain in response to infection by L. pneumophila, providing insights into the genome structure of A. castellanii and the variations in genome folding that occur during infection.
Results
The A. castellanii Neff and C3 genome assemblies are highly contiguous and complete
We used a combination of Illumina short reads, Oxford Nanopore long reads, and Hi-C to assemble each genome to chromosome scale, with 90% of the Neff genome contained within 28 scaffolds. This is in contrast to a previous estimate of approximately 20 chromosomes inferred using pulsed-field gel electrophoresis (Rimm et al. 1988). For both the Neff and C3 strains, we first generated a raw de novo assembly using Oxford Nanopore long reads. To account for the error-prone nature of long reads, we polished the first draft assemblies with paired-end shotgun Illumina sequences using HyPo (Kundu et al. 2019). The polished assemblies were then scaffolded with long-range Hi-C contacts using our probabilistic program instaGRAAL, which exploits a Markov chain Monte Carlo algorithm to swap DNA segments until the most likely scaffolds are achieved (Marie-Nelly et al. 2014; Baudry et al. 2020a). Following the post-scaffolding polishing step of the program (Baudry et al. 2020a), the final genome assemblies displayed better contiguity (Table 1), completion, and mapping statistics than the previous versions, with the cumulative scaffold lengths quickly reaching a plateau (Fig. 1A). The assemblies of both strains are slightly longer, with a smaller number of contigs than the original Neff assembly (Neff-v1) (Fig. 1B). The BUSCO-completeness scores for both assemblies are also improved (Simão et al. 2015), with 90.6% (Neff-v2) and 91.8% (C3) complete eukaryotic universal single-copy orthologs, compared with 77.6% for Neff-v1 (Fig. 1C). An increased proportion of properly paired shotgun reads from 71% for Neff-v1 to 84% for our new Neff assembly, Neff-v2, suggested a reduced number of short misassemblies. We searched and found putative eukaryotic telomeric repeats (“TTAGG”) strongly enriched at the extremities of the Neff-v2 scaffolds and, to a lesser extent, at the extremities of C3 scaffolds (Supplemental Fig. S1), suggesting that some of these scaffolds indeed correspond to full-length chromosomes.
Table 1.
Genome statistics for the finished assemblies of Neff, C3 (this study), and the reference Neff-v1 genome
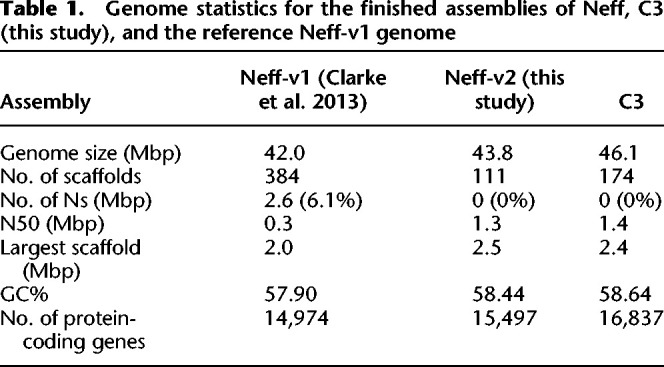
Figure 1.

Assembly statistics for A. castellanii genomes. Comparison of genome assemblies for strains C3 and Neff-v2 versus the previous Neff-v1 genome assembly (Clarke et al. 2013). (A) Cumulative length plot showing the relationship between the number of contigs (largest to smallest) and length of the assembly. (B) General continuity metrics. (C) BUSCO statistics showing the status of universal single-copy orthologs in eukaryotes for each assembly. (D) Circos plot showing syntenic blocks for all scaffolds of A. castellanii strains C3 and Neff assemblies >50 kb.
Hi-C contact maps present a convenient readout to explore large misassemblies in genome sequences (Marie-Nelly et al. 2014). Although this allowed us to manually address major unambiguous misassemblies, a number of visible misassemblies remain in complex regions such as repeated sequences near telomeres and ribosomal DNAs (rDNAs). These misassemblies could not be resolved with the data generated herein. In the C3 assembly, there are a few (at least five) inter-chromosomal misassemblies that appear to be heterozygous and cannot be resolved without a phased genome. We also found shotgun coverage to be highly heterogeneous between scaffolds, which is suggestive of aneuploidy (Supplemental Fig. S2).
The A. castellanii Neff and C3 genomes differ in size and have partly nonoverlapping gene complements
The generation of chromosome-scale genome assemblies for two different A. castellanii strains afforded us the first opportunity to compare and contrast their sizes and coding capacities. Despite their obvious relatedness (the 18S rDNA sequences of the two strains are 97% identical) (Supplemental Fig. S3), the C3 assembly was found to be 2.3 Mbp longer than the Neff-v2 assembly. We investigated this discrepancy by extracting all C3-specific regions from pairwise genome alignments with Neff sequences, which summed up to 3.2 Mbp. This comprised 5072 different sequences ranging from 200 to 22,153 bp in length, with a median of 483 bp and a mean of 637 bp. These regions were scattered across the C3 scaffolds (Supplemental Fig. S4). Using BLASTX against the NCBI Protein database (Altschul et al. 1990), we found that the majority (>90%) of these sequences had strong hits to Acanthamoeba proteins. The average gene length in the two strains (∼2.3 kb), combined with the number of additional genes in C3 relative to Neff-v2 (1307), supports the idea that most of the extra sequence in C3 relative to Neff is owing to a larger number of genes. Although the retrieved C3-specific sequence does exceed the difference in genome size between C3 and Neff-v2, the Neff-v2 genome has strain-specific genes of its own that likely account for this discrepancy. Despite the differences in gene content and genome size, the Neff and C3 genomes are highly syntenic (Fig. 1D), and the additional DNA in C3 does not appear to result in large structural rearrangements. We further tested this by aligning the Hi-C reads from C3 to the Neff genome and generating a contact map binned at 20 kb. Only three translocations were identified, in agreement with the Circos plot shown in Figure 1D (Krzywinski et al. 2009).
We used both Broccoli (Derelle et al. 2020) and OrthoFinder (Emms and Kelly 2019) for inference of orthologous groups (see Methods). A summary of the inferred orthogroups shared by, and specific to, the Neff and C3 strains of A. castellanii is presented in Figure 2, with orthogroup numbers from both orthologous clustering tools included. This figure only compares Neff against C3, irrespective of orthogroup presence or absence in outgroup taxa. In this analysis, each strain-specific gene that was not assigned to an orthogroup by either program was still considered to be a single strain-specific orthogroup in order to account for the presence of genes without any orthologs across the five species. Broccoli predicted more orthogroups overall and more strain-specific genes than OrthoFinder but predicted fewer shared orthogroups. Despite these differences, the overall trend is similar for the two outputs. The number of orthogroups shared by the two strains is roughly an order of magnitude greater than the number specific to either strain, whereas the C3 strain has a greater number of strain-specific orthogroups than the Neff strain as predicted by both programs.
Figure 2.
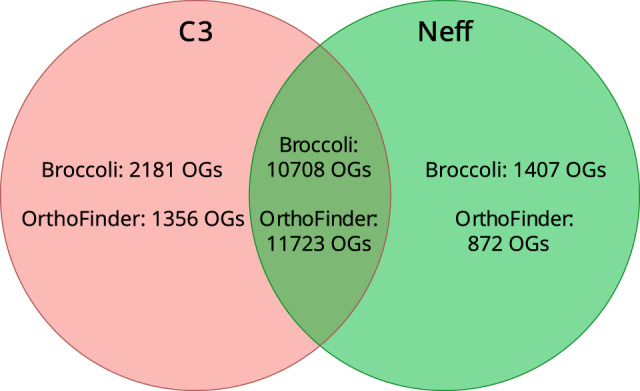
Numbers of strain-specific and shared orthologous groups in the genomes of A. castellanii strains C3 and Neff. Orthology inference was conducted with both Broccoli and OrthoFinder. Dictyostelium discoideum, Physarum polycephalum, and Vermamoeba vermiformis were used as outgroups to improve accuracy of orthogroup inference.
To investigate how similar the A. castellanii gene complement was to other members of Amoebozoa, A. castellanii orthogroups were evaluated for their presence in three outgroup species. Both Broccoli and OrthoFinder outputs were analyzed in this fashion. According to Broccoli, 43.5% of orthogroups shared by the two A. castellanii strains were not present in the other three amoebae, whereas OrthoFinder gave a figure of 48.5%. In the Neff strain, 49.3% of all orthogroups, shared or strain-specific, were not found in the three outgroup amoebae according to Broccoli compared with 51.4% as predicted by OrthoFinder. In the C3 strain, the Broccoli results indicate that 52.1% of all orthogroups are not present in the outgroup amoebae, whereas 52.8% were not found in the outgroup by OrthoFinder. This is in contrast with A. castellanii strain C3 sharing an estimated 83.1% (Broccoli) to 89.6% (OrthoFinder) of its orthogroups with the Neff strain, and the Neff strain sharing an estimated 88.3% (Broccoli) to 93.1% (OrthoFinder) of its orthogroups with the C3 strain.
A. castellanii accessory genes show strain-specific functional enrichment
In an attempt to gain insight into the functional significance of strain-specific genes in the C3 and Neff genomes, the top 30 most significantly enriched terms were identified by topGO (https://bioconductor.org/packages/topGO/ [accessed July 12, 2022]) and plotted in order of decreasing P-value for each strain/ontology combination (Supplemental Figs. S5–S9). Among C3-specific genes, five terms were found to be statistically significantly enriched in the “biological process” ontology at a 95% confidence level, whereas nine terms were enriched in the “molecular function” ontology, and two were enriched in the “cellular component” ontology. Among Neff-specific genes, five terms were significantly enriched in the “biological process” ontology at a 95% confidence level, seven were enriched in the “molecular function” ontology, and one was enriched in the “cellular component” ontology. All enriched Gene Ontology (GO) terms and the corresponding P-values can be found in Supplemental Table S2. Note that a multiple testing correction has not been applied to these P-values, consistent with the method of topGO (because of the hierarchical relationship of GO terms, the tests are not strictly independent, and the multiple testing theory does not directly apply).
For some of the enriched functional categories, the strain-specific genes contributing to the enrichment showed a relatively cohesive signal in terms of best hits when searched against the nr database with BLASTP (Altschul et al. 1990). The Neff genes annotated as “virion parts” were the same ones responsible for the enrichment in “structural molecule activity.” These genes had best BLAST hits to the major capsid protein from various nucleocytoplasmic large DNA viruses (NCLDVs). Those genes responsible for “protein homo-oligomerization” enrichment had their best BLAST hits to K+ channel tetramerization domains, whereas all those contributing to “DNA recombination” enrichment had best BLAST hits to IS607 family transposases, those contributing to “cyclic nucleotide biosynthetic process” enrichment had best BLAST hits to serine/threonine kinases, and those contributing to enrichment of both “protein-macromolecule adaptor activity” and “actin filament binding” had best BLAST hits to fascin-like proteins. In C3, there were fewer enrichment categories in which BLAST hits gave such a cohesive signal, but some examples are “amino acid transport” enrichment, in which the associated genes had best BLAST hits to serine/threonine kinases; “DNA topological change,” in which the best BLAST hits were to DNA topoisomerase 2; and “oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor,” in which the best BLAST hits of all associated genes were to Ars-J-associated glyceraldehyde-3-phosphate dehydrogenases.
The Neff strain has a divergent mannose-binding protein
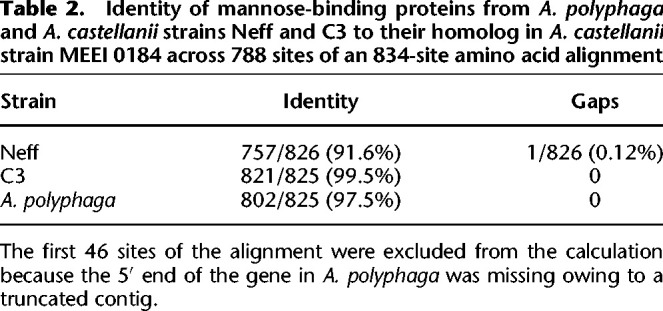
One gene of particular interest encodes a mannose-binding protein (MBP), which is known to be used as a receptor for cell entry by Legionella in some A. castellanii strains (Declerck et al. 2007). The MEEI 0184 strain of A. castellanii, an isolate from a human corneal infection, was used as a reference sequence, because it is the only strain in which the MBP was biochemically characterized (Garate et al. 2004, 2005). The orthologs from C3, Neff, and Acanthamoeba polyphaga were retrieved, and all four sequences were aligned (Supplemental Fig. S10). The percentage of identity of each sequence to the reference was calculated over the sites in the alignment in which the A. polyphaga sequence was not missing (Table 2). The C3 homolog was found to be 99.5% identical to the MEEI 0184 homolog, whereas the Neff and A. polyphaga proteins were more divergent, sharing 91.6% and 97.2% identity to MEEI 0184, respectively. Despite being of the same species as the reference, the Neff strain homolog was found to be much more divergent than the A. polyphaga sequence is from the other two A. castellanii strains.
Table 2.
Identity of mannose-binding proteins from A. polyphaga and A. castellanii strains Neff and C3 to their homolog in A. castellanii strain MEEI 0184 across 788 sites of an 834-site amino acid alignment
This observation correlates with differences in the entry of L. pneumophila upon infection between the C3 and Neff strains. Indeed, when both amoeba strains were infected with a multiplicity of infection (MOI) ratio of 0.1, we observed that L. pneumophila enters in higher numbers in C3 compared with Neff (Supplemental Fig. S11A). We then used different MOIs for infection of the Neff strain, which revealed that an MOI = 10 was required to obtain comparable numbers of bacterial entry to an MOI = 0.1 using C3. In addition, when bacterial counts are normalized to the number of bacteria that already entered the host cell, Legionella appears to replicate faster in the C3 compared with the Neff strain (Supplemental Fig. S11B). These experiments confirmed the empiric observations that led to the adoption of the C3 strain as a preferred model with respect to infection. We propose that this phenotype results partly from impaired receptor-mediated entry by Legionella into Neff cells owing to differences in the receptor encoding gene (a hypothesis that will be tested in subsequent investigations) and also from other differences in amoeba physiology between C3 and Neff strains.
Spatial organization of the A. castellanii genome
To our knowledge, no Hi-C contact maps have been published from species of Amoebozoa. Although a Hi-C library was generated on Entamoeba histolytica for the purpose of genome scaffolding (Kawano-Sugaya et al. 2020), its quality was too poor to yield a contact map of good quality. Here, the Hi-C reads we used to generate the chromosome-scale scaffolding of two A. castellanii genomes offered us the opportunity to reveal the average genome folding in a species of this clade. Hi-C reads were realigned along the new assemblies of both the C3 and Neff strains to generate genome-wide contact maps. Visualizing the Hi-C contact maps of both genomes shows that A. castellanii chromosomes are well resolved in our assemblies (Fig. 3A). In Neff, the highest intensity contacts are concentrated on the main diagonal, suggesting an absence of large-scale misassemblies. On the other hand, the C3 assembly retains a few misassembled blocks, mostly in the rDNA region where tandem repeats could not be resolved correctly with the data available to us. However, for both strains, the genome-wide contact maps reveal a grid-like pattern, with contact enrichment between chromosome extremities resulting in discrete dots. These contacts can be interpreted as a clustering of the telomeres, or subtelomeres, of the different chromosomes (Fig. 3A). Based on the presence of these inter-telomeric contact patterns, Hi-C contact maps suggest the presence of at least 35 chromosomes in both strains, ranging from ∼100 kb to 2.5 Mb in length (Supplemental Fig. S12). Additionally, we found 100 copies of 5S rDNA dispersed across most chromosomes for both strains, and 18S/28S rDNA genes show increased contacts with subtelomeres (Fig. 3A). ECC finder (Zhang et al. 2021a), a computational tool that detects extrachromosomal circular DNA elements, was run on the Oxford Nanopore long reads to verify that these rDNA sequences do not correspond to extrachromosomal circular rDNA contacting the genome at various positions. No evidence of circular DNA elements was found, suggesting that rDNA sequences in A. castellanii are interspersed within chromosomes. However, given the repetitive nature of these sequences, it is formally possible that some of the rDNA insertion sites are incorrect. In addition, the possible amplification of extrachromosomal palindromic linear rDNA, similar to that found in Dictyostelium (Sucgang et al. 2003), may also confound their proper analysis.
Figure 3.

Spatial organization of the A. castellanii genome. (A, top) Whole-genome Hi-C contact maps of the Neff (left) and C3 (right) genomes, with a magnification of the three largest scaffolds. The genomes are divided into 16-kb bins, and each pixel represents the contact intensity between a pair of bins. Each scaffold is visible as a red square on the diagonal. In both strains, there is an enrichment of interscaffold contacts toward telomeres, suggesting a spatial clustering of telomeres, as shown on the model in the right margin. (Bottom) 4C-like representation of spatial contacts between rDNA and the rest of the genome. Scaffolds are delimited by gray vertical lines. Contacts of all rDNAs are enriched toward telomeres. The genomic position of the 18S and 28S genes is highlighted with triangles on the top panel, and the occurrences of 8S rDNA sequences are shown with vertical red lines on the bottom panel. (B) High-resolution contact map for a region of the C3 genome showing chromatin loops detected by Chromosight as blue circles. (C) Size distribution of chromatin loops detected in the C3 strain.
In addition to large, interchromosomal subtelomeric contacts, we also explored the existence of intrachromosomal chromatin 3D structures in the contact maps using Chromosight, a program that detects patterns reflecting chromatin structures on Hi-C contact maps (Matthey-Doret et al. 2020). For both strains, Chromosight identified arrays of chromatin loops along chromosomes, as well as boundaries separating chromatin domains (Fig. 3B). Most chromatin loops are regularly spaced, with a typical size of 20 kb (Fig. 3C), reminiscent of the average loop size reported along metaphase chromosomes in the budding yeast Saccharomyces cerevisiae (Costantino et al. 2020; Dauban et al. 2020). The chromatin domains correspond to discrete squares along the diagonal (Supplemental Fig. S13A–C). We overlapped all predicted genes in the C3 genome with the domain borders detected from Hi-C data and measured their base expression using RNA-seq generated from that strain (see Methods). We selected the closest gene to each domain border and found that the genes overlapping domain boundaries are overall more highly expressed than those that do not (Supplemental Fig. S14C). In addition, the analysis showed that gene expression is negatively correlated with the distance to the closest domain border (Supplemental Fig. S14D). We performed the same comparison using chromatin loop anchors instead of domain borders. To a lesser extent, genes overlapping chromatin loops are also associated with higher expression (Supplemental Fig. S14A), although it is not correlated with the distance from the closest loop (Supplemental Fig. S14B). Altogether, these results suggest that the chromatin structures observed in cis are both associated with gene expression, although the association between gene expression and chromatin loop anchors is likely owing to their colocalization with domain borders (Supplemental Fig. S14E). Some microorganisms organize their chromosomes into microdomains whose boundaries correspond to highly expressed genes (e.g., budding yeasts and euryarchaeotes) (Hsieh et al. 2015; Cockram et al. 2021). Our findings in A. castellanii are therefore reminiscent of this type of organization.
L. pneumophila infection induces chromatin loop changes enriched in infection-related functions
The generation of near-complete assemblies allowed us to tackle the question of whether L. pneumophila infection impacts the 3D folding and transcription of the A. castellanii C3 strain genome. We harvested cultured A. castellanii cells before and 5 h after infection by L. pneumophila strain Paris (see Methods) (Cazalet et al. 2004). The cells were processed using Hi-C and RNA-seq (see Methods), and the resulting reads aligned against the reference C3 genome to assess changes in the genome structure and the host transcription program, respectively. RNA-seq was performed in triplicate and Hi-C in duplicate (see Methods). To measure changes in trans-chromosomal contacts, we merged the A. castellanii contact maps from our replicates and applied the serpentine adaptive binning method to improve the signal-to-noise ratio (Baudry et al. 2020b). We then computed average interactions between each pair of chromosomes before and after infection. For each pair of chromosomes, we then used the log ratio of infected over uninfected average contacts. Following infection, a global decrease in trans-subtelomeric contacts was observed, suggesting a slight declustering of chromosome ends (Fig. 4B). In addition, the scaffold bearing 18S and 28S rDNA (scaffold_29), as well as two other small scaffolds (35 and 36), displayed weaker interactions with other scaffolds during infection (Fig. 4A).
Figure 4.

Changes in trans-chromosomal contacts between A. castellanii chromosomes following L. pneumophila infection. (A) Average contact change during infection between each pair of chromosomes. Chromosome lengths are shown below the interaction matrix, with the chromosome bearing 18S and 28S rDNA highlighted in green. (B) Representative intertelomeric contacts between a pair of chromosomes (C3 scaffolds 3 and 11). The average intertelomeric contact profile generated from all pairs of chromosomes is shown as a pileup. The log ratio between the infected (I) and uninfected (U) profiles is shown as a ratio (right).
Given L. pneumophila DNA was likely to have been sequenced along with the infected A. castellanii strain C3, we aligned the Hi-C reads against the L. pneumophila genome to see whether we could also track the changes in chromosome organization of the pathogen. However, the resulting contact map was extremely sparse (Supplemental Fig. S15), and the condensin-mediated secondary diagonal, which is a hallmark of most bacterial chromosome structure (Le et al. 2013; Marbouty et al. 2017), was barely visible. Therefore, the protocol must first be improved to enrich for contacts in bacterial sequence before parallel analysis of changes in host and pathogen genome organization can be considered.
We then assessed whether the behavior of cis contacts changes during infection. First, we computed the average contact frequencies according to genomic distance p(s) (see Methods), which is a convenient way to unveil variations in the compaction state of chromatin (Barbieri et al. 2012). The p(s)-curves show a global increase in long-range contacts following infection (Supplemental Fig. S16). The strengths of chromatin loops and domain borders before and 5 h after infection were quantified using Chromosight (Matthey-Doret et al. 2020). However, no significant average increase or decrease in the intensity of these structures (Supplemental Fig. S16) was identified when computed over the whole genome. To focus on infection-dependent chromatin structures, we filtered the detected patterns to retain those showing the top 20% strongest change in Chromosight score during infection (either appearing or disappearing). We performed a GO term enrichment analysis for genes associated with infection-dependent chromatin loops (see Methods). A significant enrichment for Rho GTPase and phosphorelay signal transduction, protein catabolism, and GPI biosynthesis was found (Supplemental Fig. S17A). The strongest loop changes were associated with genes encoding Rho GTPase, GOLD, and SET domains, as well as genes for proteins containing leucine-rich repeats and ankyrin repeats (Supplemental Fig. S18).
We followed the same procedure for domain borders and found that genes associated with infection-dependent domain borders were significantly enriched in “amino acid transport,” “cyclic nucleotide biosynthetic process,” “protein modification,” and “deubiquitination” (Supplemental Fig. S17B). Our results suggest that domain borders are generally associated with highly transcribed metabolic genes, consistent with previous findings showing that such borders are associated with high transcription (Le et al. 2013).
By analyzing the A. castellanii RNA-seq data after infection with L. pneumophila, we revealed that the expression of genes was globally impacted at 5 h post infection compared with uninfected cells (Supplemental Fig. S19A). This is consistent with recent results showing that transcription is globally disrupted in A. castellanii Neff following infection by L. pneumophila (Li et al. 2020). To investigate the relationship between this change in gene expression and chromatin structure, we assigned the closest domain border to each gene and compared their expression and border score changes during infection. For most of the genes, we found border intensity not to be correlated with gene expression changes (Supplemental Fig. S19B). Only genes undergoing extreme expression changes during infection corresponded to changes in associated borders (Supplemental Fig. S19C). This raises the possibility that insulation domains in A. castellanii chromosomes do not dictate gene expression programs as they do in mammals.
Recently, Li et al. (2020) investigated gene expression changes at 3, 8, 16, and 24 h after infection of A. castellanii Neff by L. pneumophila. To further validate our finding that chromatin domains are not units of regulation in A. castellanii, we used these expression results and migrated the gene annotations to our C3 assembly using Liftoff (Shumate and Salzberg 2020). This allowed us to compute coexpression between gene pairs during infection (i.e., expression correlation). We found that gene pairs within the same chromatin domain did not have a higher coexpression than gene pairs from different domains at similar genomic distances (Supplemental Fig. S13D).
Discussion
Chromosome-level assembly uncovers A. castellanii genome general organization
Generation, analysis, and comparison of the genome sequences of two A. castellanii strains revealed heterogeneous coverage across scaffolds (Supplemental Fig. S2), which is consistent with previous findings that A. castellanii has a high but variable ploidy of approximately 25n (Byers 1986). Previous estimates of the A. castellanii Neff karyotype using pulsed-field gel electrophoresis estimated 17 to 20 unique chromosomes ranging from 250 kb to just >2 Mb (Rimm et al. 1988), whereas our estimate suggests at least 35 unique chromosomes with a similar size range of 100 kb to 2.5 Mb. The discrepancy between the number of bands in the electrophoretic karyotype and our estimate may result from chromosomes of similar size comigrating on the gel, which we were able to resolve using sequence- and contact-based information.
Considering features of the nuclear biology of A. castellanii, such as suspected amitosis (Gicquaud and Tremblay 1991) and probable aneuploidy, our finding that 5S rDNA is dispersed across all chromosomes may serve to ensure a consistent copy number of 5S rDNA in daughter cells.
It was previously estimated that A. castellanii has 24 copies of rDNA genes per haploid genome (Yang et al. 1994). Our data show that both strains contain four times as many copies as originally thought. The decrease in interchromosomal contacts with rDNA-containing scaffolds during infection may reflect an alteration in the nucleolus structure, probably caused by a global increase in translational activity. This would be consistent with the global transcriptional shift observed in RNA-seq under infection conditions.
At first glance, the contact maps show a clustering of subtelomeric regions, but do not display a Rabl conformation, in which centromeres cluster to the spindle-pole body (Rabl 1885). However, the precise positions of centromeres are needed to see whether these chromosomes are acrocentric, which could lead to an overlap of the contact signal between centromeres with the contacts between subtelomeres and could mask centromere clustering.
Changes in C3 strain chromatin structure during infection likely reflect transcriptional changes
Hi-C data further revealed that A. castellanii chromosomes are (in asynchronous population) folded into arrays of chromatin loops. Infection of A. castellanii strain C3 with L. pneumophila resulted in significant changes in the positioning of these chromatin loop borders. Our analyses showed enrichment in several relevant GO terms at the sites of these infection-induced modifications, many of which correspond to known biological processes induced by L. pneumophila in amoebae and macrophages. Several enriched terms are related to cell cycle regulation, including mitotic cell cycle, cell cycle processes, and cell cycle checkpoints (Supplemental Fig. S17), consistent with recent results showing that L. pneumophila inhibits proliferation of its natural host A. castellanii (Mengue et al. 2016; Li et al. 2020). L. pneumophila–induced alterations of the host cell cycle may serve to bypass cell cycle phases that limit bacterial replication (de Jesús-Díaz et al. 2017) or serve to prevent amoebal proliferation, which has been proposed to increase the feeding efficiency of individual amoebae (Quinet et al. 2020).
Several other GO terms found enriched at infection-dependent loops or borders are related to the host cell organelles, such as assembly, microtubule cytoskeleton organization, protein localization to endoplasmic reticulum, mitochondrion organization, electron transport chain, or mitochondrial respiratory chain complexes (Supplemental Fig. S17). These are intriguing, given that it is well known that during infection, L. pneumophila hijacks host organelles such as the cytoskeleton, endoplasmic reticulum, and mitochondria in both amoebae and macrophages (Rothmeier et al. 2013; Escoll et al. 2016, 2017), and that mitochondrial respiration and electron transport chain complexes are altered in macrophages during L. pneumophila infection (Escoll et al. 2017, 2021).
Infection-dependent chromatin reorganization sites also show enrichment in functions related to changes in general host metabolism, such as biosynthetic and catabolic processes, including nucleotide and nucleoside synthesis, lipid metabolism, or amino acid and metal ion transport. To replicate intracellularly, L. pneumophila acquires all its nutrients from the cytoplasm of the host cell. Therefore, bacteria-induced modulation of the host metabolism is thought to be essential for establishing a successful infection (Escoll and Buchrieser 2019). In summary, many GO terms associated with changes in chromatin loops and borders during infection are consistent with the known biology of Legionella infection, suggesting a link between its chromatin organization and many of the changes observed in host cells during infection.
L. pneumophila infection halts host cell division and is associated with a decrease of mRNA of the A. castellanii CDC2b gene, a putative regulator of the A. castellanii cell cycle (Mengue et al. 2016). At the same time, the increase in intrachromosomal long-range contact (Supplemental Fig. S16B) is reminiscent of the variations observed throughout the S. cerevisiae cell cycle, in which an increase in compaction at the G2/M stage results in similar changes. Furthermore, the arrays of regularly spaced chromatin loops along A. castellanii chromosomes of ∼20 kb in size are similar in size to the chromatin loops observed in S. cerevisiae during the G2/M stage (Garcia-Luis et al. 2019; Dauban et al. 2020). This similarity in terms of regularity and size suggests that chromatin loops in A. castellanii may serve a similar purpose of chromosome compaction for cell division as in yeast. Our observations suggest that upon infection, the A. castellanii C3 strain population becomes enriched in cells presenting higher compaction level, consistent with an arrest in G2/M. That DNA loop anchors and domain borders overlap with highly expressed genes is also concordant with observations made in yeast and other species (Hsieh et al. 2015; Cockram et al. 2021; Zhang et al. 2021b) and could result from their role in modulating the expansion of loops by the structural maintenance of chromosome (SMC) complexes during replication (Anchimiuk et al. 2021). Unlike previously shown in Drosophila (Ramírez et al. 2018), we did not find an increase in the coexpression of genes sharing the same contact domain in A. castellanii strain C3. This suggests that the formation of chromatin domains or loops is modulated by the transcription pattern and highly expressed genes but that these structures do not act as regulatory units.
A. castellanii accessory genes may permit environmental adaptation
Among the large number of genes predicted to be strain specific in A. castellanii, several functions were found to be significantly enriched in either the Neff or C3 strain-specific gene sets. Of these, the most biologically interesting is the enrichment of the “small GTPase-mediated signal transduction” and “GTP binding” genes in C3. The enrichment of these two GO terms, along with the enrichment of protein phosphorylation, suggests that the C3 strain may have expanded its environmental sensing capabilities and associated cellular responses by expanding the gene families involved in signal transduction. Given the extensive gene repertoire of A. castellanii dedicated to cell signaling, environmental sensing, and the cellular response (Clarke et al. 2013), which is thought to help the amoeba navigate diverse habitats and identify varied prey, it seems likely that alterations of this gene repertoire in C3 may have enabled further environmental adaptations.
Another notable enrichment is the “virion parts” in the A. castellanii Neff strain. This enrichment includes a single gene with a best BLAST hit for the major capsid proteins in various NCLDVs, including a very strong hit to Mollivirus sibericum. Many NCLDVs, including Mollivirus, are known viruses of Acanthamoeba spp. (Legendre et al. 2015). Although no phylogenetic analysis has been performed to investigate the origin of this major capsid protein gene in the Neff genome, it seems plausible that it was acquired by lateral gene transfer during an NCLDV infection, perhaps by Mollivirus or a closely related virus.
Some of the remaining enriched functions seem related, but we are unable to speculate on their broader biological significance. For example, we observe enrichment of “macromolecule methylation,” “DNA topological change,” “chromatin binding,” “DNA binding,” “catalytic activity, acting on DNA,” and “DNA topoisomerase II (double-strand cut, ATP-hydrolyzing) activity” in the C3 strain, all of which appear to be related to DNA modification and maintenance. There are fewer such cases in the Neff strain: One can imagine possible connections between “protein phosphorylation,” “protein kinase activity,” and “purine ribonucleoside triphosphate binding,” as well as between “endoribonuclease activity, producing 5′-phosphomonoesters” and “nucleic acid binding” and, finally, between “actin filament binding” and “protein-macromolecule adaptor activity.” Other enrichments beyond these examples have no obvious biological significance. They could well be nonadaptive, having been generated by gene duplication, differential loss in the other amoebae strain studied, or lateral gene transfer, without conferring any significant selective advantage. An improved understanding of the cellular and molecular biology of Acanthamoeba is needed to make sense of the genetic enrichment data presented here.
Substitutions in the Neff MBP may inhibit Legionella entry
Alignment of the three A. castellanii MBPs and the A. polyphaga homolog may help explain the difference in susceptibility to Legionella infection between the Neff and C3 strains. The C3 strain MBP is highly similar to its counterpart in strain MEEI 0184, which was first to be biochemically characterized. The Neff strain MBP, however, is markedly more divergent than even the A. polyphaga MBP, which is not known to participate in Acanthamoeba–Legionella interactions (Harb et al. 1998). Infection studies comparing infection phenotypes depending on the MOI confirmed that L. pneumophila enters 10 times less into Neff cells than into C3 cells in our in vitro infection system. These results are consistent with the hypothesis that the Neff strain of A. castellanii is not a very good host for infection by Legionella partly owing to an accumulation of amino acid substitutions in its MBP, substitutions that may prevent Legionella from binding to this protein during cell entry. It is worth noting that the Neff strain has been in axenic culture since 1957, so it may be that the relaxed selective pressure on this protein, combined with repeated population bottlenecking during culture maintenance, has allowed for mutations in the Neff strain MBP gene to accumulate. At the present time, without available genome data for strains more closely related to the Neff strain, it cannot be determined whether these mutations arose in nature or in culture. However, given that the divergence of the A. polyphaga ortholog to the MEEI 0184 strain is much less than that of the Neff strain, despite all four strains having similar lifestyles in nature, evolution of the Neff strain since being deposited in the culture collection seems likely.
Methods
Strains and growth conditions
A. castellanii strains Neff and C3 were grown on amoeba culture medium (2% Bacto tryptone, 0.1% sodium citrate, 0.1% yeast extract), supplemented with 0.1 M glucose, 0.1 mM CaCl2, 2.5 mM KH2PO4, 4 mM MgSO4, 2.5 mM Na2HPO4, and 0.05 mM Fe4O21P6 at 20°C. The L. pneumophila strain Paris was grown for 3 d on N-(2-acetamido)-2-amino-ethanesulfonic acid (ACES)-buffered charcoal-yeast (BCYE) extract agar at 37°C.
Infection time course
Infection of A. castellanii C3 with L. pneumophila was performed using MOI 10 over 5 h in infection medium (0.5% sodium citrate supplemented with 0.1 mM CaCl2, 2.5 mM KH2PO4, 4 mM MgSO4, 2.5 mM Na2HPO4, 0.05 mM Fe4O21P6) at 20°C. At 5 h post infection, amoebae were collected in a 15-mL tube, pelleted by centrifugation at 300g for 10 min and washed twice in PBS, and then cross-linked in 3% formaldehyde during 20 min at room temperature (RT) with gentle shaking. Then 2.5 M glycine was added to reach a final concentration of 0.125 M over 20 min, centrifuged, and washed, and pellets were stored at −80°C until DNA extraction.
To compare bacterial entry and intracellular replication of L. pneumophila in strains Neff and C3, we infected amoeba cultures with L. pneumophila strain Paris constitutively expressing GFP. A. castellanii C3 was infected at an MOI = 0.1, and strain Neff was infected at MOI = 0.1, MOI = 1, or MOI = 10 in T25 flasks. At t = 0 h post infection (hpi) and at t = 1 hpi, 300 µL was removed from the infection; amoeba cells were lysed by vortexing the sample for 60 sec and centrifuging the sample at 14,000 rpm for 3 min; and 100 µL was spread on BCYE plates. Colony forming units (CFU) were counted to determine bacterial numbers used for infection (input, t0) and numbers of internalized bacteria (internalization, t1/t0). In parallel, at t = 0, 1, 24, 48, and 72 hpi, 300 µL of infected amoebae was removed from the infection culture and lysed as detailed before. One hundred microliters was plated in 96-well plates in duplicates, and bacterial numbers were analyzed by flow cytometry in a MACSQuant VYP flow cytometer with MACSQuantify software (Miltenyi Biotec) using a 488-nm laser and a 500–550 bandpass filter (B1 channel). For the analyses, an FSC-A/SSC-a dot plot was used to select a first gate and identify bacteria (compared with a PBS sample as negative control), and an SSC-H/SSC-A dot plot was used to select a second gate for singlets. Finally, bacterial numbers were counted as GFP-positive events in that population of single bacteria. Absolute numbers of GFP bacteria per milliliter were then calculated knowing that 50 µL of sample was acquired by the cytometer.
Hi-C
Cell pellets were suspended in 1.2 mL H2O and transferred to CK14 Precellys tubes. Cells were broken with Precellys (six cycles: 30 sec on/30 sec off) at 7500 rpm and transferred into a tube. All Hi-C libraries for A. castellanii strains C3 and Neff were prepared using the Arima kit and protocol with only the DpnII restriction enzyme. Libraries were sequenced to produce 35-bp paired-end reads on an Illumina NextSeq machine. Statistics regarding Hi-C libraries are described in Supplemental Table S1.
Short-read sequencing
Illumina libraries associated with the data submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRX12218478 and SRX12218479 were prepared from A. castellanii strains C3 and Neff genomic DNA, respectively, and sequenced by Novogene at 2 × 150 bp on an Illumina NovaSeq 6000 machine.
To generate the data associated with SRA accession number SRX4625411, a PCR-free library was prepared and sequenced by Génome Québec from purified A. castellanii strain Neff genomic DNA. The library was barcoded and run with other samples on an Illumina HiSeq X Ten instrument, producing 150-bp paired-end reads.
RNA-seq
Poly(A)-selected libraries were prepared from purified A. castellanii total RNA. A. castellanii strain C3 RNA-seq libraries were prepared using the stranded mRNA TruSeq kit from Illumina and sequenced in single-end mode at 150 bp on an Illumina NextSeq machine.
For A. castellanii strain Neff (SRA: SRX7813524), the library was prepared and sequenced by Génome Québec. The library was barcoded and run with other samples on an Illumina NovaSeq 6000 instrument, producing 300-bp paired-end reads.
Oxford Nanopore sequencing
To generate the data associated with SRA accession numbers SRX12218489 and SRX12218490, DNA was extracted from A. castellanii strains Neff and C3 using the Qiagen blood and cell culture DNA kit following the specific recommendations detailed by Oxford Nanopore Technologies in the info sheet entitled “High-molecular-weight gDNA extraction from cell lines (2018)” in order to minimize DNA fragmentation by mechanical constraints. Oxford Nanopore libraries were prepared with the ligation sequencing kit LSKQ109, flow cell model MIN106D R9. Base-calling was performed using Guppy v2.3.1-1 (https://nanoporetech.com/community).
For other libraries, genomic DNA samples were obtained from A. castellanii strain Neff using an SDS-based lysis method, followed by digestion with RNase A, then Proteinase K, and then a phenol-chloroform-based extraction. DNA samples were cleaned with Qiagen G/20 genomic clean-up columns using the manufacturer's protocol, but with double the number of wash steps. Four different libraries were prepared, using the SQK-RAD003 rapid sequencing kit (SRA: SRX4620962), the SQK-LSK308 1D2 ligation sequencing kit (SRA: SRX4620963), the SQK-RAD004 rapid sequencing kit (SRA: SRX4620964), and the SQK-LSK108 ligation sequencing kit (SRA: SRX4620965). The SQK-LSK308 and SQK-RAD003 libraries were sequenced on FLO-MIN107 flow cells, and the SQK-LSK108 and SQK-RAD004 libraries were both sequenced on a FLO-MIN106 flow cell. All four libraries were base-called with Albacore 2.1.7 (https://hub.docker.com/r/genomicpariscentre/albacore/tags), as they were sequenced before the release of Guppy. Adapters were removed from the base-called reads using Porechop v0.2.3 (https://github.com/rrwick/Porechop).
Genome assembly
Oxford Nanopore reads were filtered using filtlong v0.2.0 (rrwick/Filtlong; https://github.com/rrwick/Filtlong [accessed July 12, 2022]) with default parameters to keep the best 80% reads according to length and quality. Illumina shotgun libraries were used as reference for the filtering. A de novo assembly was generated from the raw (filtered) Oxford Nanopore long reads using flye v2.3.6 (Kolmogorov et al. 2019) with three iterations of polishing. The resulting assembly was polished using both Oxford Nanopore and Illumina reads with HyPo v1.0.1 (Kundu et al. 2019). Contigs from the polished assembly bearing >60% of their sequence or 51% identity to the mitochondrial sequence from the Neff-v1 assembly were separated from the rest of the assembly to prevent inclusion of mitochondrial contigs into the nuclear genome during scaffolding. Polished nuclear contigs were scaffolded with Hi-C reads using instaGRAAL v0.1.2 with default parameters (Baudry et al. 2020a). instaGRAAL-polish was then used to fix potential errors introduced by the scaffolding procedure. Mitotic contigs were then added at the end of the scaffolded assembly, and the final assembly was polished with the Illumina shotgun library data using two rounds of Pilon polishing. The resulting Neff-v2 assembly was edited manually to remove spurious insertion of mitochondrial contigs in the scaffold and other contaminants. The final assembly was polished again using Pilon with Rcorrector-corrected reads (Song and Florea 2015). minimap2 v2.17 (Li 2018) was used for all long read or pairwise genome alignments, and Bowtie 2 v2.3.4.1 (Langmead and Salzberg 2012) was used for short read alignments.
Genome annotation
The structural genome annotation pipeline used here was implemented similarly as described previously (Gray et al. 2020). Briefly, RNA-seq reads were mapped to the genome assembly using STAR v2.7.3a (Dobin et al. 2013), followed by both de novo and genome-guided transcriptome assembly by Trinity v2.12.0 (Grabherr et al. 2011). Both runs of Trinity were performed with Jaccard clipping to mitigate artificial transcript fusions. The resulting transcriptome assemblies were combined and aligned to the genome assembly using PASA v2.4.1 (Haas et al. 2003). Protein sequences were aligned to the genome using Spaln v2.4.2 (Gotoh 2008) to recover the most information from sequence similarity. The ab initio predictors used were AUGUSTUS v3.3.2 (Stanke et al. 2006), Snap (Korf 2004), Genemark v4.33 (Lomsadze et al. 2014), and CodingQuarry v2.0 (Testa et al. 2015). Finally, the PASA assembly, Spaln alignments, and the AUGUSTUS, Snap, and CodingQuarry gene models were combined into a single consensus with EVidenceModeler v1.1.1 (Haas et al. 2008).
Functional annotations were added using funannotate v1.5.3 (https://doi.org/10.5281/zenodo.1471785 [accessed July 12, 2022]). Repeated sequences were masked using RepeatMasker (Smit et al. 2013–2015). Predicted proteins were fed to Interproscan v5.22 (Quevillon et al. 2005), Phobius v1.7.1 (Käll et al. 2007), and Eggnog-mapper v2.0.0 (Cantalapiedra et al. 2021) to generate functional annotations. Ribosomal RNA genes were annotated separately using RNAmmer v1.2 (Lagesen et al. 2007) with HMMER 2.3.2 (Finn et al. 2011).
As described in the “Data access” section, the funannotate-based script “func_annot_from_gene_mod” used to add functional annotations to existing gene models is provided in the Zenodo record and on the associated GitHub repository.
Analysis of sequence divergence
To compute the proportion of substituted positions in aligned segments between the C3 and Neff strains, the two genomes were aligned using minimap2 with the map-ont preset and -c flag. The gap-excluded sequence divergence (mismatches/[matches + mismatches]) was then computed in each primary alignment, and the average of divergences (weighted by segment lengths) was computed. This is implemented in the script “04_compute_seq_divergence.py” available in the genome analyses repository listed at Zenodo (see “Data access”).
Orthogroup inference
Orthogroups were inferred using the predicted proteomes of both the Neff and C3 strains, with Dictyostelium discoideum, Physarum polycephalum, and Vermamoeba vermiformis as outgroups to improve the accuracy of orthogroup inference. The outgroup predicted proteomes were retrieved from PhyloFisher (Tice et al. 2021). Both Broccoli (Derelle et al. 2020) and OrthoFinder (Emms and Kelly 2019) were run with default settings for orthogroup inference.
Gene content comparison of Neff and C3 strains
Custom Python scripts were used to retrieve genes unique to each A. castellanii strain, as well as orthogroups that were shared between the two strains. Genes were only determined to be strain specific or shared if both Broccoli and OrthoFinder assigned them as such; genes were excluded from the analysis if both tools did not agree. For both strains, functional assignments for each gene ID were extracted from funannotate output and tabulated. The tabulated assignments and strain-specific gene IDs were fed into the R package topGO (https://bioconductor.org/packages/topGO/ [accessed July 12, 2022]) to analyze GO term enrichment in the strain-specific genes. A Fisher's exact test with the weight algorithm was implemented in topGO for the Neff- and C3-specific genes for each of the three ontologies (biological process, cellular component, and molecular function). When building the GOdata objects for these three ontologies, nodeSize was set to 10 for both the biological process and molecular function ontologies and to five for the cellular component ontology owing to the lower number of GO terms in this ontology. Circos plots were generated using Circos (Krzywinski et al. 2009).
MBP comparison
MBP amino acid sequences from three strains of A. castellanii (Neff, C3, and MEEI 0184) and one strain of A. polyphaga were retrieved, aligned using MAFFT-linsi (v7.475) (Katoh and Standley 2013), and visualized in Jalview (v2.11.1.3) (Waterhouse et al. 2009). The MEEI 0184 strain sequence was retrieved from the NCBI Protein database (accession number AAT37865.1), and the Neff and C3 sequences were retrieved from the predicted proteomes generated in this study with the MEEI 0184 sequence as a BLASTP (Altschul et al. 1990) query. The A. polyphaga genome does not have a publicly available predicted proteome, so its MBP protein sequence was manually extracted from several contigs in the genome sequence (NCBI accession GCA_000826345.1) using TBLASTN with the MEEI 0184 sequence as a query (the sequence encoding the first eight amino acids of the protein could not be found in the genome owing to a truncated contig).
Hi-C analyses
Reads were aligned with Bowtie 2 v2.4.1, and Hi-C matrices were generated using hicstuff v3.0.1 (https://zenodo.org/record/4066363 [accessed July 12, 2022]). For all comparative analyses, matrices were down-sampled to the same number of contacts using cooltools (https://www.github.com/mirnylab/cooltools), and balancing normalization was performed (Cournac et al. 2012; Imakaev et al. 2012). Loops and domain borders were detected using Chromosight v1.6.1 (Matthey-Doret et al. 2020) using the merged replicates at a resolution of 2 kb. We measured the intensity changes in Chromosight scores during infection using pareidolia (v0.6.1) (https://zenodo.org/record/5362241/export/json [accessed July 12, 2022]) on three pseudoreplicates generated by sampling the merged contact maps, as described previously (Yang et al. 2017). This was performed to account for contact coverage heterogeneity across replicates. The 20% threshold used to select differential patterns amounts to 1.2% false detections for loops and 2.3% for borders when comparing pseudoreplicates from the same condition.
Data access
The sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA599339 and PRJNA487265. All processed data generated in this study, as well as the assemblies and annotations used in this work, have been submitted to Zenodo (https://zenodo.org/record/5507417). Strains supporting the findings of this study are available from the corresponding authors. The code used to perform the analysis is packaged into the following Snakemake pipelines available as Supplemental Code and at GitHub: hybrid genome assembly, https://github.com/cmdoret/Acastellanii_hybrid_assembly; functional annotation of A. castellanii, https://github.com/cmdoret/Acastellanii_genome_annotation; analyses of genomic features in A. castellanii, https://github.com/cmdoret/Acastellanii_genome_analysis; and changes during infection by Legionella, https://github.com/cmdoret/Acastellanii_legionella_infection.
Supplementary Material
Acknowledgments
We thank Axel Cournac, Laura Gomez Valero, Christophe Rusniok, and Lyam Baudry for their comments on the bioinformatics analysis, Charlotte Cockram for her help with Oxford Nanopore sequencing, Olivier Espeli, and all members of the Koszul laboratory and Buchrieser laboratory for stimulating discussions. C.M.-D. is supported by the Pasteur—Paris University (PPU) International PhD Program. This research was supported by the European Research Council (ERC) under the European Union's Horizon 2020 to R.K. (ERC grant agreement 771813). The C.B. laboratory is financed by the Fondation pour la Recherche Médicale (FRM) grant no. EQU201903007847 and the Agence Nationale de la Recherche (ANR) grant no. ANR-10-LABX-62-IBEID. Research in the Archibald laboratory was supported by a grant from the Gordon and Betty Moore Foundation (GBMF5782). M.J.C. is supported by graduate student scholarships from the Natural Sciences and Engineering Research Council of Canada (NSERC) and Dalhousie University. B.F.L. and M.S. were supported by the NSERC (RGPIN-2017-05411) and by the “Fonds de Recherche Nature et Technologie,” Quebec.
Author contributions: Conceptualization was done by C.M.-D., M.J.C., C.B., J.M.A., and R.K. Methodology was done by C.M.-D., M.J.C., C.B., J.M.A., and R.K. Investigation was done by M.J.C., C.M.-D., P.E., T.S., and A.T. Formal analysis was done by C.M.-D. and M.J.C. Data curation was done by C.M.-D., M.J.C., B.C., M.S., M.W.G., and B.F.L. Visualization was done by C.M.-D. and M.J.C. Writing of the original draft was done by C.M.-D., M.J.C., J.M.A., and R.K. Writing of the other drafts and editing were done by all authors. Supervision was done by J.M.A., C.B., and R.K. Funding acquisition was done by J.M.A., C.B., and R.K.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276375.121.
Competing interest statement
The authors declare no competing interests.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Anchimiuk A, Lioy VS, Bock FP, Minnen A, Boccard F, Gruber S. 2021. A low Smc flux avoids collisions and facilitates chromosome organization in Bacillus subtilis. eLife 10: e65467. 10.7554/eLife.65467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri M, Chotalia M, Fraser J, Lavitas L-M, Dostie J, Pombo A, Nicodemi M. 2012. Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci 109: 16173–16178. 10.1073/pnas.1204799109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry L, Guiglielmoni N, Marie-Nelly H, Cormier A, Marbouty M, Avia K, Mie YL, Godfroy O, Sterck L, Cock JM, et al. 2020a. instaGRAAL: chromosome-level quality scaffolding of genomes using a proximity ligation-based scaffolder. Genome Biol 21: 148. 10.1186/s13059-020-02041-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry L, Millot GA, Thierry A, Koszul R, Scolari VF. 2020b. Serpentine: a flexible 2D binning method for differential Hi-C analysis. Bioinformatics 36: 3645–3651. 10.1093/bioinformatics/btaa249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers TJ. 1986. Molecular biology of DNA in Acanthamoeba, Amoeba, Entamoeba, and Naegleria. Int Rev Cytol 99: 311–341. 10.1016/S0074-7696(08)61430-8 [DOI] [PubMed] [Google Scholar]
- Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. 2021. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38: 5825–5829. 10.1093/molbev/msab293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani A. 1930. An amoeba found in cultures of a yeast preliminary note–third note–an amoeba growing in cultures of a yeast second note–fourth note. J Trop Med Hyg 33: 160. [Google Scholar]
- Cazalet C, Rusniok C, Brüggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, et al. 2004. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet 36: 1165–1173. 10.1038/ng1447 [DOI] [PubMed] [Google Scholar]
- Clarke M, Lohan AJ, Liu B, Lagkouvardos I, Roy S, Zafar N, Bertelli C, Schilde C, Kianianmomeni A, Bürglin TR, et al. 2013. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol 14: R11. 10.1186/gb-2013-14-2-r11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockram C, Thierry A, Gorlas A, Lestini R, Koszul R. 2021. Euryarchaeal genomes are folded into SMC-dependent loops and domains, but lack transcription-mediated compartmentalization. Mol Cell 81: 459–472.e10. 10.1016/j.molcel.2020.12.013 [DOI] [PubMed] [Google Scholar]
- Costantino L, Hsieh T-HS, Lamothe R, Darzacq X, Koshland D. 2020. Cohesin residency determines chromatin loop patterns. eLife 9: e59889. 10.7554/eLife.59889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournac A, Marie-Nelly H, Marbouty M, Koszul R, Mozziconacci J. 2012. Normalization of a chromosomal contact map. BMC Genomics 13: 436. 10.1186/1471-2164-13-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauban L, Montagne R, Thierry A, Lazar-Stefanita L, Bastié N, Gadal O, Cournac A, Koszul R, Beckouët F. 2020. Regulation of cohesin-mediated chromosome folding by Eco1 and other partners. Mol Cell 77: 1279–1293.e4. 10.1016/j.molcel.2020.01.019 [DOI] [PubMed] [Google Scholar]
- Declerck P, Behets J, De Keersmaecker B, Ollevier F. 2007. Receptor-mediated uptake of Legionella pneumophila by Acanthamoeba castellanii and Naegleria lovaniensis. J Appl Microbiol 103: 2697–2703. 10.1111/j.1365-2672.2007.03530.x [DOI] [PubMed] [Google Scholar]
- de Jesús-Díaz DA, Murphy C, Sol A, Dorer M, Isberg RR. 2017. Host cell S phase restricts Legionella pneumophila intracellular replication by destabilizing the membrane-bound replication compartment. mBio 8: e02345-16. 10.1128/mBio.02345-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derelle R, Philippe H, Colbourne JK. 2020. Broccoli: combining phylogenetic and network analyses for orthology assignment. Mol Biol Evol 37: 3389–3396. 10.1093/molbev/msaa159 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozanski W. 1956. Fatal bacterial infection in soil amoebae. Acta Microbiol Pol (1952) 5: 315–317. [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20: 238. 10.1186/s13059-019-1832-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensminger AW. 2016. Legionella pneumophila, armed to the hilt: justifying the largest arsenal of effectors in the bacterial world. Curr Opin Microbiol 29: 74–80. 10.1016/j.mib.2015.11.002 [DOI] [PubMed] [Google Scholar]
- Escoll P, Buchrieser C. 2019. Metabolic reprogramming: an innate cellular defence mechanism against intracellular bacteria? Curr Opin Immunol 60: 117–123. 10.1016/j.coi.2019.05.009 [DOI] [PubMed] [Google Scholar]
- Escoll P, Mondino S, Rolando M, Buchrieser C. 2016. Targeting of host organelles by pathogenic bacteria: a sophisticated subversion strategy. Nat Rev Microbiol 14: 5–19. 10.1038/nrmicro.2015.1 [DOI] [PubMed] [Google Scholar]
- Escoll P, Song O-R, Viana F, Steiner B, Lagache T, Olivo-Marin J-C, Impens F, Brodin P, Hilbi H, Buchrieser C. 2017. Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe 22: 302–316.e7. 10.1016/j.chom.2017.07.020 [DOI] [PubMed] [Google Scholar]
- Escoll P, Platon L, Dramé M, Sahr T, Schmidt S, Rusniok C, Buchrieser C. 2021. Reverting the mode of action of the mitochondrial FOF1-ATPase by Legionella pneumophila preserves its replication niche. eLife 10: e71978. 10.7554/eLife.71978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garate M, Cao Z, Bateman E, Panjwani N. 2004. Cloning and characterization of a novel mannose-binding protein of Acanthamoeba. J Biol Chem 279: 29849–29856. 10.1074/jbc.M402334200 [DOI] [PubMed] [Google Scholar]
- Garate M, Cubillos I, Marchant J, Panjwani N. 2005. Biochemical characterization and functional studies of Acanthamoeba mannose-binding protein. Infect Immun 73: 5775–5781. 10.1128/IAI.73.9.5775-5781.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Luis J, Lazar-Stefanita L, Gutierrez-Escribano P, Thierry A, Cournac A, García A, González S, Sánchez M, Jarmuz A, Montoya A, et al. 2019. FACT mediates cohesin function on chromatin. Nat Struct Mol Biol 26: 970–979. 10.1038/s41594-019-0307-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquaud C, Tremblay N. 1991. Observations with Hoechst staining of amitosis in Acanthamoeba castellanii. J Protozool 38: 221–224. 10.1111/j.1550-7408.1991.tb04432.x [DOI] [Google Scholar]
- Gotoh O. 2008. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Res 36: 2630–2638. 10.1093/nar/gkn105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29: 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Derelle R, Klimeš V, Leger MM, Sarrasin M, Vlček Č, Roger AJ, Eliáš M, Lang BF. 2020. The draft nuclear genome sequence and predicted mitochondrial proteome of Andalucia godoyi, a protist with the most gene-rich and bacteria-like mitochondrial genome. BMC Biol 18: 22. 10.1186/s12915-020-0741-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, et al. 2003. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31: 5654–5666. 10.1093/nar/gkg770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9: R7. 10.1186/gb-2008-9-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb OS, Venkataraman C, Haack BJ, Gao LY, Kwaik YA. 1998. Heterogeneity in the attachment and uptake mechanisms of the Legionnaires’ disease bacterium, Legionella pneumophila, by protozoan hosts. Appl Environ Microbiol 64: 126–132. 10.1128/AEM.64.1.126-132.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T-HS, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. 2015. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162: 108–119. 10.1016/j.cell.2015.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol 26: 261–283. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- Ikedo M, Yabuuchi E. 1986. Ecological studies of Legionella species, I: viable counts of Legionella pneumophila in cooling tower water. Microbiol Immunol 30: 413–423. 10.1111/j.1348-0421.1986.tb02967.x [DOI] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. 2012. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9: 999–1003. 10.1038/nmeth.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol 7: 13–24. 10.1038/nrmicro1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer ELL. 2007. Advantages of combined transmembrane topology and signal peptide prediction: the Phobius web server. Nucleic Acids Res 35: W429–W432. 10.1093/nar/gkm256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano-Sugaya T, Izumiyama S, Yanagawa Y, Saito-Nakano Y, Watanabe K, Kobayashi S, Nakada-Tsukui K, Nozaki T. 2020. Near-chromosome level genome assembly reveals ploidy diversity and plasticity in the intestinal protozoan parasite Entamoeba histolytica. BMC Genomics 21: 813. 10.1186/s12864-020-07167-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilvington S, Price J. 1990. Survival of Legionella pneumophila within cysts of Acanthamoeba polyphaga following chlorine exposure. J Appl Bacteriol 68: 519–525. 10.1111/j.1365-2672.1990.tb02904.x [DOI] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540–546. 10.1038/s41587-019-0072-8 [DOI] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5: 59. 10.1186/1471-2105-5-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T, Nagai H. 2016. The type IVB secretion system: an enigmatic chimera. Curr Opin Microbiol 29: 22–29. 10.1016/j.mib.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Kundu R, Casey J, Sung W-K. 2019. HyPo: super fast & accurate polisher for long read genome assemblies. bioRxiv 10.1101/2019.12.19.882506v1 [DOI]
- Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35: 3100–3108. 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasheras A, Boulestreau H, Rogues A-M, Ohayon-Courtes C, Labadie J-C, Gachie J-P. 2006. Influence of amoebae and physical and chemical characteristics of water on presence and proliferation of Legionella species in hospital water systems. Am J Infect Control 34: 520–525. 10.1016/j.ajic.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Le TBK, Imakaev MV, Mirny LA, Laub MT. 2013. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342: 731–734. 10.1126/science.1242059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Lartigue A, Bertaux L, Jeudy S, Bartoli J, Lescot M, Alempic J-M, Ramus C, Bruley C, Labadie K, et al. 2015. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc Natl Acad Sci 112: E5327–E5335. 10.1073/pnas.1510795112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Vassiliadis D, Ong SY, Bennett-Wood V, Sugimoto C, Yamagishi J, Hartland EL, Pasricha S. 2020. Legionella pneumophila infection rewires the Acanthamoeba castellanii transcriptome, highlighting a class of sirtuin genes. Front Cell Infect Microbiol 10: 428. 10.3389/fcimb.2020.00428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze A, Burns PD, Borodovsky M. 2014. Integration of mapped RNA-seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42: e119. 10.1093/nar/gku557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbouty M, Baudry L, Cournac A, Koszul R. 2017. Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci Adv 3: e1602105. 10.1126/sciadv.1602105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie-Nelly H, Marbouty M, Cournac A, Flot J-F, Liti G, Parodi DP, Syan S, Guillén N, Margeot A, Zimmer C, et al. 2014. High-quality genome (re)assembly using chromosomal contact data. Nat Commun 5: 5695. 10.1038/ncomms6695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthey-Doret C, Baudry L, Breuer A, Montagne R, Guiglielmoni N, Scolari V, Jean E, Campeas A, Chanut PH, Oriol E, et al. 2020. Computer vision for pattern detection in chromosome contact maps. Nat Commun 11: 5795. 10.1038/s41467-020-19562-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengue L, Régnacq M, Aucher W, Portier E, Héchard Y, Samba-Louaka A. 2016. Legionella pneumophila prevents proliferation of its natural host Acanthamoeba castellanii. Sci Rep 6: 36448. 10.1038/srep36448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel R, Hauröder B. 1997. Isolation of an Acanthamoeba strain with intracellular Burkholderia pickettii infection. Zentralbl Bakteriol 285: 541–557. 10.1016/S0934-8840(97)80116-8 [DOI] [PubMed] [Google Scholar]
- Mondino S, Schmidt S, Rolando M, Escoll P, Gomez-Valero L, Buchrieser C. 2020. Legionnaires’ disease: state of the art knowledge of pathogenesis mechanisms of Legionella. Annu Rev Pathol 15: 439–466. 10.1146/annurev-pathmechdis-012419-032742 [DOI] [PubMed] [Google Scholar]
- Neff RJ. 1957. Purification, axenic cultivation, and description of a soil amoeba, Acanthamoeba sp. J Protozool 4: 176–182. 10.1111/j.1550-7408.1957.tb02505.x [DOI] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. 10.1038/nature11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnier I, Raoult D, La Scola B. 2008. Isolation and identification of amoeba-resisting bacteria from water in human environment by using an Acanthamoeba polyphaga co-culture procedure. Environ Microbiol 10: 1135–1144. 10.1111/j.1462-2920.2007.01530.x [DOI] [PubMed] [Google Scholar]
- Pushkarew BM. 1913. Über die Verbreitung der Süsswasser-protozoen durch die Luft. Arch Protistent 23: 323–362. [Google Scholar]
- Qiu J, Luo Z-Q. 2017. Legionella and Coxiella effectors: strength in diversity and activity. Nat Rev Microbiol 15: 591–605. 10.1038/nrmicro.2017.67 [DOI] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. 2005. InterProScan: protein domains identifier. Nucleic Acids Res 33: W116–W120. 10.1093/nar/gki442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet T, Samba-Louaka A, Héchard Y, Van Doninck K, Van der Henst C. 2020. Delayed cytokinesis generates multinuclearity and potential advantages in the amoeba Acanthamoeba castellanii Neff strain. Sci Rep 10: 12109. 10.1038/s41598-020-68694-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl C. 1885. Uber zelltheilung. Morphol. Jahrbuch. (ed. Gegenbauer C), pp. 214–330. Willhelm Engelmann, Leipzig, Germany.
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T. 2018. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9: 189. 10.1038/s41467-017-02525-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie S, Dalby M, van Duin L, Andersson R. 2018. Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat Commun 9: 487. 10.1038/s41467-017-02798-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimm DL, Pollard TD, Hieter P. 1988. Resolution of Acanthamoeba castellanii chromosomes by pulsed field gel electrophoresis and construction of the initial linkage map. Chromosoma 97: 219–223. 10.1007/BF00292964 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Zaragoza S. 1994. Ecology of free-living amoebae. Crit Rev Microbiol 20: 225–241. 10.3109/10408419409114556 [DOI] [PubMed] [Google Scholar]
- Rolando M, Sanulli S, Rusniok C, Gomez-Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C. 2013. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13: 395–405. 10.1016/j.chom.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, Hannemann M, Wölke S, Bausch A, Griffiths G, et al. 2013. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog 9: e1003598. 10.1371/journal.ppat.1003598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham TJ. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J Clin Pathol 33: 1179–1183. 10.1136/jcp.33.12.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samba-Louaka A, Delafont V, Rodier M-H, Cateau E, Héchard Y. 2019. Free-living amoebae and squatters in the wild: ecological and molecular features. FEMS Microbiol Rev 43: 415–434. 10.1093/femsre/fuz011 [DOI] [PubMed] [Google Scholar]
- Shumate A, Salzberg SL. 2020. Liftoff: accurate mapping of gene annotations. Bioinformatics 37: 1639–1643. 10.1093/bioinformatics/btaa1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui R, Khan NA. 2012. Biology and pathogenesis of Acanthamoeba. Parasit Vectors 5: 6. 10.1186/1756-3305-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org.
- Song L, Florea L. 2015. Rcorrector: efficient and accurate error correction for illumina RNA-seq reads. Gigascience 4: 48. 10.1186/s13742-015-0089-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7: 62. 10.1186/1471-2105-7-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucgang R, Chen G, Liu W, Lindsay R, Lu J, Muzny D, Shaulsky G, Loomis W, Gibbs R, Kuspa A. 2003. Sequence and structure of the extrachromosomal palindrome encoding the ribosomal RNA genes in Dictyostelium. Nucleic Acids Res 31: 2361–2368. 10.1093/nar/gkg348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart AL, Gomez-Valero L, Buchrieser C, Hilbi H. 2020. Evolution and function of bacterial RCC1 repeat effectors. Cell Microbiol 22: e13246. 10.1111/cmi.13246 [DOI] [PubMed] [Google Scholar]
- Testa AC, Hane JK, Ellwood SR, Oliver RP. 2015. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics 16: 170. 10.1186/s12864-015-1344-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice AK, Žihala D, Pánek T, Jones RE, Salomaki ED, Nenarokov S, Burki F, Eliáš M, Eme L, Roger AJ, et al. 2021. PhyloFisher: a phylogenomic package for resolving eukaryotic relationships. PLoS Biol 19: e3001365. 10.1371/journal.pbio.3001365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvesvara GS, Moura H, Schuster FL. 2007. Pathogenic and opportunistic free-living amoebae: Acanthamoeba spp., Balamuthia mandrillaris, Naegleria fowleri, and Sappinia diploidea. FEMS Immunol Med Microbiol 50: 1–26. 10.1111/j.1574-695X.2007.00232.x [DOI] [PubMed] [Google Scholar]
- Volkonsky M. 1931. Hartmannella castellanii Douglas et classification des Hartmannelles. Arch Zoolog Exp Gén 72: 317–319. [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zwick MG, Paule MR. 1994. Sequence organization of the Acanthamoeba rRNA intergenic spacer: identification of transcriptional enhancers. Nucleic Acids Res 22: 4798–4805. 10.1093/nar/22.22.4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang F, Yardımcı GG, Song F, Hardison RC, Noble WS, Yue F, Li Q. 2017. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res 27: 1939–1949. 10.1101/gr.220640.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Peng H, Llauro C, Bucher E, Mirouze M. 2021a. Ecc_finder: a robust and accurate tool for detecting extrachromosomal circular DNA from sequencing data. Front Plant Sci 12: 743742. 10.3389/fpls.2021.743742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Übelmesser N, Josipovic N, Forte G, Slotman JA, Chiang M, Gothe HJ, Gusmao EG, Becker C, Altmüller J, et al. 2021b. RNA polymerase II is required for spatial chromatin reorganization following exit from mitosis. Sci Adv 7: eabg8205. 10.1126/sciadv.abg8205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.