

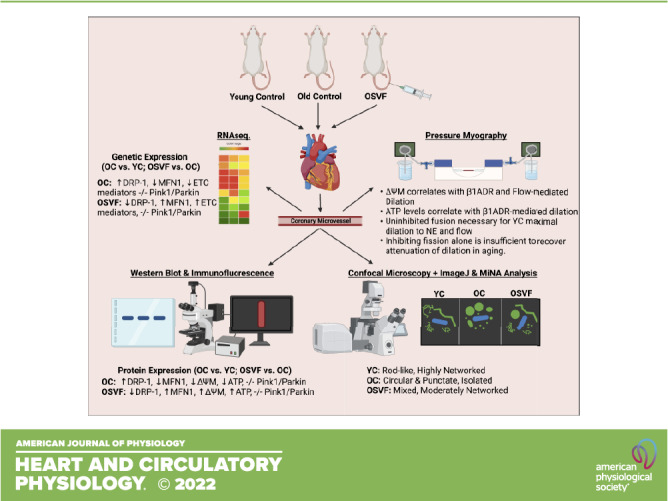
Keywords: aging, coronary, microvascular, mitochondria, stromal vascular fraction
Abstract
Aging is associated with blunted coronary microvascular vasodilatory function. Previously, systemically administered adipose stromal vascular fraction (SVF) therapy reversed aging-induced attenuation of β1-adrenergic- and flow-mediated dilation dependent on reducing mitochondrial reactive oxygen species. We hypothesized that SVF-mediated recovery of microvascular dilatory function is dependent on recovery of mitochondrial function, specifically by reducing mitochondrial hyperfission. Female Fischer-344 rats were allocated into young control, old control, and old + SVF therapy groups. Pressure myography, immunofluorescent staining, Western blot analysis, and RNA sequencing were performed to determine coronary microvascular mitochondrial dynamics and function. Gene and protein expression of fission-mediator DRP-1 was enhanced with aging but reversed by SVF therapy. SVF facilitated an increase in fusion-mediator MFN-1 gene and protein expression. Mitochondrial morphology was characterized as rod-like and densely networked in young controls, isolated circular and punctate with aging, and less circularity with partially restored mitochondrial branch density with SVF therapy. Decreased mitochondrial membrane potential and ATP bioavailability in aged animals at baseline and during flow-mediated dilation were reversed by SVF and accompanied with enhanced oxygen consumption. Dilation to norepinephrine and flow in young controls were dependent on uninhibited mitochondrial fusion, whereas inhibiting fission did not restore aged microvessel response to norepinephrine or flow. SVF-mediated recovery of β-adrenergic function was dependent on uninhibited mitochondrial fusion, whereas recovery of flow-mediated dilation was dependent on maintained mitochondrial fission. Impaired dilation in aging is mitigated by SVF therapy, which recovers mitochondrial function and fission/fusion balance.
NEW & NOTEWORTHY We elucidated the consequences of aging on coronary microvascular mitochondrial health as well as SVF’s ability to reverse these effects. Aging shifts gene/protein expression and mitochondrial morphology indicating hyperfission, alongside attenuated mitochondrial membrane potential and ATP bioavailability, all reversed using SVF therapy. Mitochondrial membrane potential and ATP levels correlated with vasodilatory efficiency. Mitochondrial dysfunction is a contributing pathological factor in aging that can be targeted by therapeutic SVF to preserve microvascular dilative function.
INTRODUCTION
Recent emphasis has been given to the role of mitochondrial signaling in the regulation of vascular tone and response to dilatory stimuli (1). These mitochondrial influences include the production of reactive oxygen species (ROS), effect of ROS on quenching nitric oxide bioavailability, mitochondrial depolarization and calcium sparks activating endothelial nitric oxide synthase (eNOS) or closing voltage-gated calcium channels, and ATP production (2–5). Adequate ATP bioavailability has recently been shown to be essential for maintenance of maximal flow- and acetylcholine-mediated vasorelaxation (6, 7). Prior work in human coronary vessels show decreased nitric oxide (NO)-mediated dilation with advanced age and loss of NO-mediated dilation with a compensatory increase in mitochondrial-derived H2O2 with the onset of coronary artery disease (8). Concomitantly, mitochondrial H2O2 levels correlate negatively with β1AR maximal dilation, whereas NO and glutathione exhibit a positive correlation (9). In support of a causative relationship, we have previously shown that aging-induced ROS directly influences the desensitization and internalization of the β1AR into endosomes, which can be reversed with therapeutic stromal vascular fraction (SVF) injection in aged female rats via a paracrine antioxidant mechanism (9). The objective of this study is to delineate the vascular mitochondrial contributions toward the progression of age-associated coronary microvascular disease (CMD) and whether SVF-mediated recovery of vascular function in CMD occurs alongside recovery of mitochondrial function.
Under homeostatic physiological conditions (healthy, youth), mitochondria function optimally exists through a balance of fission, fusion, biogenesis, and mitophagy. Dyshomeostasis toward any of these processes can lead to mitochondrial and endothelial dysfunction (10–15). An indicator of mitochondrial function is mitochondrial morphology, which is governed by fission and fusion. Dysfunctional mitochondria may undergo fission and separation into daughter mitochondria, mediated by dynamin-related protein-1 (DRP-1) or fission process 1 (Fis1). This can be followed by PTEN-induced kinase 1 (Pink1) and Parkin-mediated mitophagy in mitochondria with persistently low membrane potential to clear detrimental mitochondria, which exhibit enhanced mitochondrial ROS production (16). Dysfunctional isolated mitochondria may alternatively undergo fusion [mediated by mitofusin 1 and 2 and optic atrophy 1 (MFN1, MFN2, Opa1)] and integration with healthy mitochondrial networks to recover function (16). In particular, hyperfission is associated with greater mitochondrial ROS production, reduced NO bioavailability, aberrant respiratory complexes, and reduced capacity for angiogenesis and dilation to acetylcholine and flow (17–21). Under physiological conditions, vascular mitochondria are characterized as filamentous or rod-like and highly internet worked because of proper fusion; in contrast, aging mitochondria typically exhibit hyperfission with isolated circular or punctate morphology due to hyperfission and blunted mitophagy and biogenesis (22). Interestingly, pathological ROS overproduction correlates with mitochondrial fragmentation and mediates posttranslational modification (activation) of DRP-1 and proteolytic cleavage of Opa1 into fusion-inactive long isoforms to promote fission, which can be reversed by superoxide dismutase-2 overexpression (23).
We hypothesize that SVF-mediated recovery of coronary microvascular dilatory function is therefore dependent on recovery of mitochondrial function. Specifically, we predict SVF will reverse aging-induced mitochondrial hyperfission to restore networked mitochondrial morphology, as well as restore mitochondrial electron transport chain (ETC) function including mitochondrial membrane potential, oxygen consumption, and ATP bioavailability. Genetic and protein expression data of key mitochondrial mediators of fission, fusion, mitophagy, and oxidative phosphorylation supplement pressure myography experiments of ETC function. Finally, the contribution of fission and fusion on isolated coronary microvascular dilative response to norepinephrine (β1, α1–2-adrenergic receptor agonist) is investigated using DRP-1 (fission) inhibitor MDIVI and Opa1 (fusion) inhibitor MYLS22.
MATERIALS AND METHODS
Animal Model, Groups, and End-Point Procedures
Surgeries for all animals were carried out in accordance with approved protocols from the University of Louisville Institutional Animal Care and Use Committee and the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (24). Female Fischer-344 young control (YC, 3- to 6-mo-old virgin rats, n = 8 animals, Harlan Laboratories, Indianapolis, IN) and aged (23- to 24-mo-old virgin rats, n = 9, National Institute on Aging, Bethesda, MA) rats were housed and acclimated to the facility as previously described (25–28). The estrous cycle for young females was not monitored as our previous study showed no significant difference in coronary microvascular reactivity throughout the estrous cycle for F344 females (29). Aged rats were randomly divided into the groups: old control (OC, 24 mo, n = 9 animals) or old treated with SVF (OSVF, n = 6 animals) for which injection occurred at 23 mo of age and euthanasia at 24 mo of age. Female rats were used exclusively to match ongoing studies in our laboratory. Euthanasia was achieved by removal of the heart under deep anesthesia (lack of tactile reflex) consisting of 5% inhaled isoflurane with 1.5–2.0 L/min O2 flow in an induction chamber as previously described (25, 26, 28).
SVF Isolation and Injection
Young (3–6 mo) rats from a green-fluorescent positive (GFP+) Fischer-344 colony (original breeders obtained from University of Missouri RRRC, Strain No. 307, colony maintained in house) were used to obtain SVF cells as previously described (26, 27, 30). SVF is a heterologous cell population containing immune cells, endothelial cells, smooth muscle cells, and mesenchymal stem cells, among others (30). Briefly, epididymal and ovarian fat pads were isolated from male or female rats, respectively. Following harvest, the fat was rinsed with sterile saline and finely minced with subsequent digestion in 0.75 mg/mL type 1 collagenase solution (Vitacyte, 011–1030) and 1 mg/mL DNAse (Sigma, 9003-96-9) in 0.1% BSA-PBS. Buoyant adipocytes following centrifugation were removed, whereas the SVF cell pellet was resuspended in 0.1% BSA-PBS as previously described and then filtered via gravity through a 20-µm mesh (26, 27, 30). The final SVF solution was collected, and cells were counted by hemocytometer, followed by reconstitution at 10 million GFP+ SVF cells in 1 mL of sterile normal saline warmed to 37°C and immediately injected as a single dose into the tail vein of rats under isoflurane anesthesia.
Isolated Coronary Arteriole Pressure Myography
Following heart excision, third-order coronary arterioles from the left anterior descending artery were isolated and transferred to a vessel chamber pressure myography system to maintain physiological temperature (37°C) and pressure (45 mmHg) being bathed in a physiological salt solution containing albumin (26, 27, 31). Vessels that achieved spontaneous tone of >20% constriction from initial diameter proceeded in the study (26, 27, 30). Initial tone achieved and maximum dilation were acquired for each vessel in the study. The drug, imaging, and flow protocols were randomized to each vessel and are described in Concentration Response to β-Adrenergic Agonism or Flow.
Flow and Fluorescence Imaging
Live isolated coronary arterioles from YC, OC, OSVF, bathed in physiological salt solution lacking albumin, were infused with fluorescent dyes for mitochondrial membrane potential dye JC-1 (2 μM, 1 h, Thermo Fisher Scientific, T3168), Biotracker ATP (5 μM, 1 h, Sigma Aldrich, SCT045), and oxygen sensing dye Biotracker 520 (10 μM, 1 h, Sigma Aldrich, SCT033). Images were captured at baseline and after 2 min at flow rates of 10 and 25 μL/min. Mean fluorescence intensity (MFI) for each image was calculated from the average of two regions of interest (ROI) of consistent size (20 × 100 µm) applied over the left and right vessel walls using NIS Elements AR Analysis software (Nikon Instruments, Melville, NY). For JC-1, carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 50 μM, 5 min, Sigma Aldrich, C2759) represents a negative control. For Biotracker ATP, oligomycin (10 μM, 1 h, Sigma Aldrich, 75351) represents the positive control, whereas camptothecin (10 μM, 1 h, Sigma Aldrich, C9911) represents the negative control. For Biotracker 520, CoCl2 represents the positive control. Analyses were performed by observers blinded to the experimental conditions.
Concentration Response to β-Adrenergic Agonism or Flow
The following experiments were randomized to each vessel. FMD (0–25 μL/min, 5 μL/min intervals, 2 min/flow rate) and concentration responses to norepinephrine (NE; α1AR, α2AR, β1AR agonist, 10−4 to 10−9 M for 2 min per dose; Sigma Aldrich, A9512) were obtained for YC, OC, and OSVF. Vasodilatory responses were recorded with and without preincubation with MDIVI (10 µM, 30 min, Tocris Bioscience, 338967-87-6, inhibits DRP-1-mediated mitochondrial fission) or with MYLS22 (50 µM, 1 h, Fisher Scientific, 50–203-0440, inhibits OPA-1-mediated mitochondrial fusion). Percent dilation responses for NE, calculated as [(luminal diameter − initial diameter)/(maximal diameter − initial diameter)] × 100, and flow were plotted against mean fluorescence intensity (MFI) values for JC-1 (mitochondrial membrane potential), Biotracker ATP, and Biotracker 520 (oxygen indicator) from paired vessels of the same animal.
Immunofluorescence Staining
Coronary arterioles measuring 750–1,000 µm in length and <250 µm in diameter were isolated and fixed in 2% paraformaldehyde for 1 h. After fixation, vessels were washed 2 × 15 min in DCF-PBS followed by 20 min in 0.5% Triton-X/DCF-PBS, all at room temperature with gentle rocking. Vessels were next placed in blocking solution containing 5% donkey serum, 0.5% BSA, and 0.5% Triton-X/DCF-PBS for 1 h, followed by incubation with primary antibodies including DRP-1 (1:100 Cell Signaling Technology, D6C7), TOM20 (1:100 Abcam, ab56783), MFN1 (1:100 Novus Biologicals, 11E9-1H12), and MFN2 (1:100 Novus Biologicals, AF7884-SP) were used in YC, OC, and OSVF. Vessels were incubated overnight at 4°C with gentle rocking. Normal rabbit serum replaced primary antibodies for negative control vessels.
The following day, the vessels were washed 3× in DCF-PBS, followed by 1-h incubation in donkey anti-rabbit IgG Alexa Fluor 594 (1:300 Invitrogen, A21207), goat anti-mouse IgG Alexa Fluor 488 (1:300 Abcam, 150113), or donkey anti-sheep Alexa Fluor 647 (1:300 Abcam, ab150179) as appropriate in blocking solution. Nuclei were stained with DAPI (Life Technologies, R37606). Vessels were placed on slides with mounting media and coverslip and then imaged with Nikon C2+ inverted microscope (Nikon Instruments, Melville, NY) with a 405- and 562-nm laser. The pixel density set for image capture was 1,024 × 1,024 with 2-µm Z-step with ×20 magnification, with 12 slices stacked together after image capture. Immunofluorescence intensity for each antibody was quantified using the Nikon NIS Elements AR Analysis software using 20 µm × 100 µm area. ROI boxes were placed on the right and left vascular walls (endothelial + smooth muscle) excluding cardiomyocytes with MFI averaged. Analyses were performed by observers blinded to the experimental conditions.
RNA Sequencing
Additional coronary arterioles were allocated for RNA sequencing via flash freezing; samples were stored at −80°C until samples from all groups were collected as previously described (28). Six isolated vessels were pooled from two animals in the same group for n = 3 biological replicates/group. RNA was harvested with RNAqueous Micro kit (AM1931, Invitrogen, Waltham, MA) and quantified on a NanoDrop (Thermo Fisher Scientific, Waltham, MA). Samples were sent to the University of Louisville Genomics Core for quality control analysis using the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA) and quantified using a Qubit fluorometric assay (Thermo Fisher Scientific, Waltham, MA) as previously described (32). Poly-A enrichments, barcoding, and Illumina Next-Gene sequencing for library preparation were performed as previously described (32, 33). Gene transcripts were analyzed by the University of Louisville Bioinformatics Core for differential expression analysis between group comparisons as previously described (34).
Mitochondrial Morphometric and Colocalization Analysis
Morphometric analysis was accomplished using ImageJ and the MiNA ImageJ tool using protocols modified from Durand et al., Merrill et al., and Valente et al. (22, 35, 36). Confocal images (×60, with ×3 optical zoom, ×180 total magnification) of microvessels stained with antibodies against mitochondrial TOM20 (described in Immunofluorescence Staining) were analyzed. Vector TrueView Autofluorescence Quenching (Vector Laboratories, SP-8400-15) was performed before DAPI counterstaining. In the MiNA Analyze Morphology window, radius was set to 3 under median filter, radius (sigma) and mask weight were set to 5 and 0.9 under unsharp mask, and block size, histogram bins, and mask slope were set to 127, 256, and 3, respectively, under enhance local contrast. Aspect ratio and form factor (inverse of circularity) were obtained by analyze particles setting size from 0.1 to infinity and circularity from 0 to 1 and selecting “display results” and “summarize.” Mitochondrial mass was determined by selecting analyze, histogram, and then list. Mitochondrial fission factor was calculated as (number of particles/mitochondrial mass). Finally, colocalization of mitochondria (TOM20) with DRP-1 and MFN-1 in these same images was determined using the EzColocalization protocol (37). Analyses were performed by observers blinded to the experimental conditions.
Colocalization Analysis
Colocalization of fluorescence signal overlap from DRP-1 with TOM20 as well as TOM20 with MFN-1 was analyzed using EzColocalization Analysis ImageJ tool (37). The reporter channels for the analysis represent TOM20 (green), DRP-1 (red), and MFN-1 (far-red), respectively. A cell identification input was not used. Threshold overlap score (TOS, linear and logarithmic), Pearson correlation coefficient (PCC), Spearman’s rank correlation coefficient (SRCC), intensity correlation quotient (ICQ), and Manders’ colocalization coefficients (M1 and M2) were reported as metrics of colocalization. PCC, SRCC, and ICQ took into account all pixels. TOS and PCC used Costes’ method for determining threshold. The average signal was also recorded. TOS (linear and logarithmic), PCC, SRCC, ICQ, M1, and M2 were reported. Analyses were performed by observers blinded to the experimental conditions.
Statistical Analysis
SigmaPlot 14 (Systat) was used for statistical analyses, with the significance level set at P < 0.05 for all analyses. Concentration- and flow-response curves were analyzed with two-way repeated-measures ANOVA with Bonferroni post hoc testing as appropriate. Mean fluorescence intensity was analyzed using one-way ANOVA with Bonferroni post hoc analysis. RNA sequences were analyzed using cufflinks-cuffdiff2 with an absolute |log2FC| ≥ 1 to determine differential expression as previously described (34). Data are presented as means ± SE as indicated. For confocal images, the images were coded and analyzed in a blind fashion with the code being broken only after all MFI quantifications were acquired. Simple linear regression analyses were performed to determine if mitochondrial membrane potential (JC-1), oxygen, or ATP significantly predicted relaxation to NE or flow.
RESULTS
Animal and Vessel Characteristics
There were no significant differences between groups for vessel wall thickness, maximum diameter, or average percent tone (Table 1). Body weight was significantly increased in OC and OSVF versus YC. There were also significant differences in total ventricular weight and left ventricular weight. The right ventricle weighed significantly more in OC versus YC with no other differences between groups.
Table 1.
Animal and vessel characteristics
| Animal and Vessel Characteristics | YC | OC | OSVF |
|---|---|---|---|
| n | 8 | 9 | 6 |
| Average age, mo | 5.6 ± 0.26 | 24.8 ± 0.10 | 24.3 ± 0.20 |
| Body weight, g | 181.63 ± 2.34* | 252.5 ± 7.80 | 252.7 ± 9.50# |
| Total ventricular weight, mg | 444.04 ± 33.08* | 658.8 ± 15.20 | 670 ± 30.0# |
| LV weight (+septum), mg | 373.71 ± 13.22* | 540.2 ± 10.10 | 491.0 ± 35.60# |
| RV weight, mg | 91.2 ± 8.23* | 145.56 ± 11.70 | 125.2 ± 16.0 |
| Vessel wall thickness, μm | 12.56 ± 0.90 | 13.15 ± 0.50 | 13.72 ± 0.69 |
| Maximum diameter, μm | 112.25 ± 3.78 | 134.4 ± 5.70 | 143.2 ± 18.10 |
| Average percent tone, % | 31.01 ± 3.31 | 30.13 ± 3.00 | 27.74 ± 4.38 |
Values are means ± SE; n, number of animals. LV, left ventricular; RV, right ventricular. Significance as P < 0.05 via one-way ANOVA with Bonferroni post hoc analysis: *young control (YC) vs. old control (OC); #old + stromal vascular fraction therapy (OSVF) vs. YC.
Aging Induces Hyperfission-Linked Gene and Protein Expression, Reversed by SVF
To examine transcriptional and translational alterations related to mitochondrial dynamics in aging and SVF therapy, we performed protein-immunofluorescence RNA sequencing. There were significant differences in pathways concerning mitochondrial fission and fusion but not mitophagy. Protein expression (immunofluorescence) of fission inducer DRP-1 was significantly upregulated with aging, reversed to YC levels by SVF (Fig. 1A). Although phosphorylated (activated) DRP-1 was not specifically measured, mitochondrially located DRP-1 (as assessed by colocalization with TOM20) was assessed. There was significantly greater logarithmic threshold overlap score [TOS(log2)] between DRP-1 and mitochondrial TOM20 in OC versus YC (Supplemental Fig. S2, A and B).
Figure 1.

Coronary microvascular genetic and protein expression related to mitochondrial function. Protein expression for fission (DRP-1), fusion (MFN-1 and MFN-2), and mitochondrial density (TOM20) with representative images. White boxes represent regions of interest placed only on the vessel wall excluding cardiomyocytes; MFI was measured only in these regions, with left and right vessel wall MFI averaged for each image (A). RNA sequencing of genetic expression associated with mitochondrial function (B). Significance determined as *P < 0.05 with one-way ANOVA and Bonferroni post hoc analysis. Scale bar = 200 µm. RNA sequencing heatmap created using Morpheus, https://software.broadinstitute.org/morpheus. MFI, mean fluorescence intensity.
There were no aging-related differences in protein expression of fusion mediators MFN-1 or MFN-2; however, SVF led to significantly enhanced MFN-1 protein expression compared with both YC and OC. MFN-1 exhibited overlap with TOM20 in each group without a significant difference between groups (Supplemental Fig. S2, A and C). Mitochondrial density marker TOM20 was not reduced in aging or following SVF treatment. Notably, gene expression of DRP-1 is significantly reduced in OSVF versus old by 33.5% (Fig. 1B, Supplemental Table S1). Mitochondrial fission protein 1 (Fis1) genetic expression exhibited no differences between groups. On the other hand, mitochondrial fission process 1 (mtfp1) was significantly upregulated in OSVF versus OC by 27.3%, with no difference between young and old.
On the contrary, MFN1 gene expression was significantly decreased in aging by 33.5% and significantly elevated in OSVF versus OC by 27.3%. There were no genetic differences between groups for MFN2. Uncoupling protein 2 (Ucp2) was significantly increased by 20.5% with aging, indicative of loss of mitochondrial membrane potential and superoxide leak. Ucp2 continued to be upregulated in OSVF versus YC. There were no genetic differences between groups for fusion mediator Opa1. There was 22.5% more expression of peroxisome proliferator-activated receptor-γ, coactivator-related 1 (Pprc1) in aging versus young, which is involved in regulation of mitochondrial biogenesis. There was no difference in Pprc1 in YC versus OSVF or OSVF versus OC.
There were additional transcriptional alterations with aging associated with mitochondrial function. Mitochondrial pyruvate carrier 1 and 2 (MPC1, -2) were significantly decreased by 29.4% and 30% in aging, respectively. Mitochondrial pyruvate carrier 1 and 2 were unchanged with SVF treatment. Bcl2-associated athanogene 2 (antiapoptotic, mitochondrial location) was significantly decreased by 33.2% in aging. Along the lines of apoptosis, caspase 12 was significantly increased by 29.6% in aging.
NADH:ubiquinone oxidoreductase core subunit V2 (Ndufv2), indicative of mitochondrial complex I function, was significantly reduced by 22% with aging. In OSVF, there were no differences in Ndufv2 or NADH:ubiquinone oxidoreductase subunit A10 (Ndufa10), also indicative of mitochondrial complex I function. Associated coenzyme 10 A (CoQ10a, itself an antioxidant) was not significantly different between groups. Coenzyme Q2 polyprenyltransferase (CoQ2) was significantly increased by 20.5% in OC versus YC. Ubiquinol-cytochrome-c reductase complex assembly factor 1 and 2 (Uqcc1 and Uqcc2, assembly factors for complex III) were not different between groups. Cytochrome-c oxidase assembly factor 20 (COX20), necessary for the assembly of mitochondrial complex IV, was reduced by 40% in aging but was significantly increased by 31.6% in YC versus OSVF with no difference between OSVF and OC. Cytochrome-c oxidase subunit 7 b (COX7b) was not changed in aging. ATP synthase membrane subunit g (ATP5mg) and ATP synthase inhibitory factor subunit 1 (ATP5if1) were unchanged between groups. Vacuolar protein-sorting 13 homolog C (Vps13c), which is a negative regulator of Parkin-mediated mitophagy in response to mitochondrial membrane depolarization was also unchanged.
Mitochondrial Morphometric Analysis of Young, Old, and Old ± SVF Microvessels
Young microvascular mitochondria were qualitatively characterized as rod-shaped and networked, whereas aged mitochondria were more isolated and punctate, with OSVF mitochondria resembling YC mitochondria (Fig. 2, A–C). Quantitatively, the aspect ratio and form factor were significantly reduced with aging and restored to YC levels by SVF treatment (Fig. 2D). Mitochondrial fission factor (particles/pixels × 1,000), also known as the mitochondrial fragmentation count, was significantly increased in aging, reversed by SVF (Fig. 2E). Mitochondrial area and footprint were significantly reduced in aging, whereas only the latter was restored with SVF (Fig. 2F). Aging significantly attenuated both individual branch length and branches per network, i.e., mitochondrial network density that was partially restored by SVF (Fig. 2G).
Figure 2.

Morphometric analysis of microvascular mitochondria in YC, aging, and SVF therapy. Qualitatively, microvascular mitochondrial morphometry was characterized as rod-like and networked in YC (A), isolated and punctate in aging (B), and less circular and more interconnected with SVF therapy (C). Aspect ratio and form factor were reduced in aging, reversed by SVF (D). Mitochondrial area (outlined areas in A–C, second from bottom images) was significantly attenuated with aging, whereas mitochondrial fission factor was significantly elevated in aging, the latter being reversed with SVF (E). Mitochondrial footprint (gray in A–C, bottom images) was significantly reduced in aging, restored with SVF (F). Mean branch length and mean number of branches per network were significantly abrogated with aging, partly restored by SVF (G). Green represents mitochondrial TOM20 and blue represents DAPI. Significance as *P < 0.05 via one-way ANOVA with Bonferroni post hoc analysis. SVF, stromal vascular fraction; YC, young control.
Mitochondrial Membrane Potential, Oxygen Consumption, and ATP
The MFI ratio of J-aggregates to JC-1 dye, i.e., mitochondrial membrane potential (ΔΨM), in live coronary microvessels is significantly abrogated with aging, restored to YC levels with SVF therapy (Fig. 3A). There was no significant difference in oxygen MFI (BioTracker 520) at baseline (Fig. 3B). Baseline ATP was not significantly altered with aging, however, but was significantly enhanced in OSVF versus OC (Fig. 3C).
Figure 3.

Baseline microvascular mitochondrial membrane potential, oxygenation, and ATP levels. Mitochondrial membrane potential (ΔΨM) was significantly attenuated with aging, restored with SVF therapy (A). There was no difference between groups in oxygen MFI (note: BioTracker 520 increases MFI with decreasing oxygen; B). ATP was not reduced with aging but was significantly enhanced with SVF therapy compared with OC (C). Significance as *P < 0.05 with one-way ANOVA and Bonferroni post hoc analysis. Scale bar = 100 μm. MFI, mean fluorescence intensity; OC, old control; SVF, stromal vascular fraction.
Intraluminal flow at 10 μL/min significantly attenuated mitochondrial membrane potential during aging only (Fig. 4A). Mitochondrial membrane potential decreased similarly between groups at 25 μL/min. Oxygen consumption was not altered significantly between YC and OC, however, but was significantly enhanced in OSVF microvessels versus OC (Fig. 4B). ATP production was held constant with ATP expenditure in YC and OSVF; ATP MFI was significantly decreased in OC compared with both YC and OSVF. Controls for JC-1 (CCCP), BioTracker 520 (CoCl2), and BioTracker ATP (oligomycin and camptothecin) enhanced or diminished MFI as appropriate (Supplemental Fig. S1).
Figure 4.

Effect of intraluminal flow on mitochondrial membrane potential, oxygen consumption, and ATP production. Mitochondrial membrane potential was significantly diminished on intraluminal flow with 10 μL/min in aging alone and was reduced in each group similarly at 25 μL/min (A). Oxygen consumption was greatest in OSVF at 10 μL/min (note: BioTracker 520 increases MFI with decreasing oxygen; B). ATP production matched ATP expenditure in YC and OSVF during intraluminal flow whereas OC lost ATP bioavailability with flow (C). Significance as *P < 0.05 with one-way ANOVA and Bonferroni post hoc analysis. MFI, mean fluorescence intensity; OC, old control; OSVF, old + stromal vascular fraction therapy; YC, young control.
Mitochondrial membrane potential (JC-1) significantly predicted percent relaxation to NE [10−4 M] and flow (15 μL/min; Fig. 5, A and D). There was no significant prediction between baseline oxygen MFI and percent relaxation to NE (Fig. 5, B and E). There was no significant prediction between baseline ATP concentration and percent relaxation to NE; similarly, no significant prediction was found with percent relaxation to flow (Fig. 5, C and F).
Figure 5.

Linear regression analysis of mitochondrial membrane potential, oxygen consumption, and ATP bioavailability with percent relaxation to norepinephrine and intraluminal flow. Mitochondrial membrane potential (JC-1) significantly predicted percent relaxation to norepinephrine [overall regression R2 = 0.193, F(1,22) = 5.015, P = 0.036; β = 16.018, P = 0.036; A] and intraluminal flow [overall regression R2 = 0.575, F(1,20) = 27.009, P < 0.001; β = 32.293, P < 0.001; D]. Oxygen consumption did not significantly predict percent relaxation to norepinephrine [overall regression R2 = 0.00078, F(1,21) = 0.0163, P = 0.9; B] or intraluminal flow [overall regression R2 = 0.0122, F(1,20) = 0.246, P = 0.625; E]. Baseline ATP levels nearly significantly predicted relaxation to norepinephrine [overall regression R2 = 0.161, F(1,21) = 4.025, P = 0.058; C], but not intraluminal flow-mediated dilation [overall regression R2 = 0.0938, F(1,20) = 2.069, P = 0.166; F]. Significance defined as P < 0.05 with linear regression analysis.
Effect of Fission and Fusion on β1AR- and Flow-Mediated Dilation
Dilation to norepinephrine was blunted in aging compared with YC and SVF therapy at 10−7 to 10−4 M doses (Fig. 6A). For YC dilation to norepinephrine, inhibition of fusion (with MYLS22) but not fission (with MDIVI) led to significantly attenuated dilation at 10−7 to 10−4 M (Fig. 6B). In OC, neither MDIVI nor MYLS22 led to significantly more robust dilation to norepinephrine (Fig. 6C). In OSVF response to norepinephrine, MYLS22 but not MDIVI led to significantly abrogated dilation at 10−7 to 10−4 M (Fig. 6D).
Figure 6.

Inhibition of fission or fusion on dilation to norepinephrine in YC, OC, and OSVF microvessels. Aging attenuated dilation to norepinephrine, reversed by SVF therapy (A). MYLS22 inhibition of fusion significantly attenuated YC dilation to norepinephrine (B). Inhibition of fission with MDIVI nearly significantly improved OC dilation to norepinephrine at early doses (C). Fusion inhibition significantly abrogated dilation to norepinephrine in OSVF vessels (D). Significance defined as P < 0.05 *[YC vs. OC (A), YC vs. MYLS22 (B), and OSVF vs. MYLS22 (D)] and #[OSVF vs OC (A), YC + MYLS22 vs. MDIVI (B), and OSVF + MYLS22 vs. MDIVI (D)] via two-way repeated-measures ANOVA with Bonferroni post hoc analysis. OC, old control; OSVF, old + stromal vascular fraction therapy; YC, young control.
In OC, flow-mediated dilation was reversed to a flow-mediated constriction, significantly different from YC at all flow rates and OSVF from 10 to 15 μL/min (Fig. 7A). In YC, MYLS22 but not MDIVI led to significantly attenuated dilation at flow rates of 15, 20, and 25 μL/min, the latter two flow rates representing flow-mediated constriction (Fig. 7B). In OC, significantly different dilation to flow could not be achieved with MYLS22 or MDIVI, albeit MDIVI did lead to dilation instead of constriction at 5 and 10 μL/min (Fig. 7C). In OSVF, flow-mediated dilation was reversed to flow-mediated constriction by MDIVI, but not MYLS22 at flow rates of 15–25 μL/min versus OSVF alone, and 20–25 μL/min versus MYLS22 (Fig. 7D).
Figure 7.

Inhibition of fission or fusion on dilation to intraluminal flow in YC, OC, and OSVF microvessels. Aging induced flow-mediated constriction, reversed by SVF therapy achieving flow-mediated dilation akin to YC (A). MYLS22 inhibition of fusion significantly attenuated YC dilation to intraluminal flow (B) OC response to flow was not significantly altered by inhibition of fusion or fission, albeit fission inhibition led to brief dilation at lower flow rates (C). Fission inhibition significantly abrogated dilation to flow in OSVF vessels (D). Significance defined as P < 0.05 via two-way repeated-measures ANOVA with Bonferroni post hoc analysis. OC, old control; OSVF, old + stromal vascular fraction therapy; YC, young control.
DISCUSSION
The major findings of this study support that hyperfission, lower mitochondrial membrane potential and ATP production, and dyshomeostatic transcriptional and translational expression of mediators of mitochondrial dynamics and ETC function are associated with aging-induced CMD. SVF can ameliorate aging-induced mitochondrial ROS coinciding with recovered microvascular function (9). As ROS production is linked to mitochondrial function and morphology, we hypothesized that aging-induced CMD is associated with mitochondrial dysfunction and that SVF-mediated recovery of microvascular function is dependent on rejuvenation of mitochondrial function. Consistent with this hypothesis, our data support that SVF therapy can target and reverse this mitochondrial dysfunction to parameters consistent with young controls. Specifically, SVF reduces DRP-1 and enhances MFN-1 and ETC mediator protein expression, partially reverses hyperfission mitochondrial morphology (to noncircular and networked), thereby increasing mitochondrial membrane potential and ATP production.
This study builds on previous work which described that a single tail-vein injection of SVF in aged rodents led to a reversal in age-related coronary flow reserve impairment compared with untreated and endothelial cell-only groups (26), and this was not due to improvements in overall cardiac function, i.e., stroke volume, cardiac output, blood pressure (9), etc. Follow-up studies to identify how SVF treatment accomplishes this improvement point to a coronary microvascular improvement in adrenergic signaling (27), increased β-adrenergic receptor expression (28), and reduced oxidative stress burden (9). To understand the mechanistic underpinnings for SVF’s ability to shift the redox landscape of aged coronary microvessels, we must first point out that the main driver of ROS generation is leakage of 1–3% of electrons from the ETC (38). At the same time, the end product of oxidative phosphorylation, ATP, is becoming increasingly recognized as necessary for optimal dilative function. Therefore, there is a trade-off in attempting to keep ROS production in check by modulating ETC efficiency, as is known to be done in aging, and maintaining maximal dilative efficiency (38). The following few paragraphs will describe SVF’s effects on ETC function as well as mitochondrial fission/fusion/mitophagy that ultimately reduce oxidative stress while restoring youthful β1AR-and flow-mediated dilative efficiency as per Fig. 8.
Figure 8.
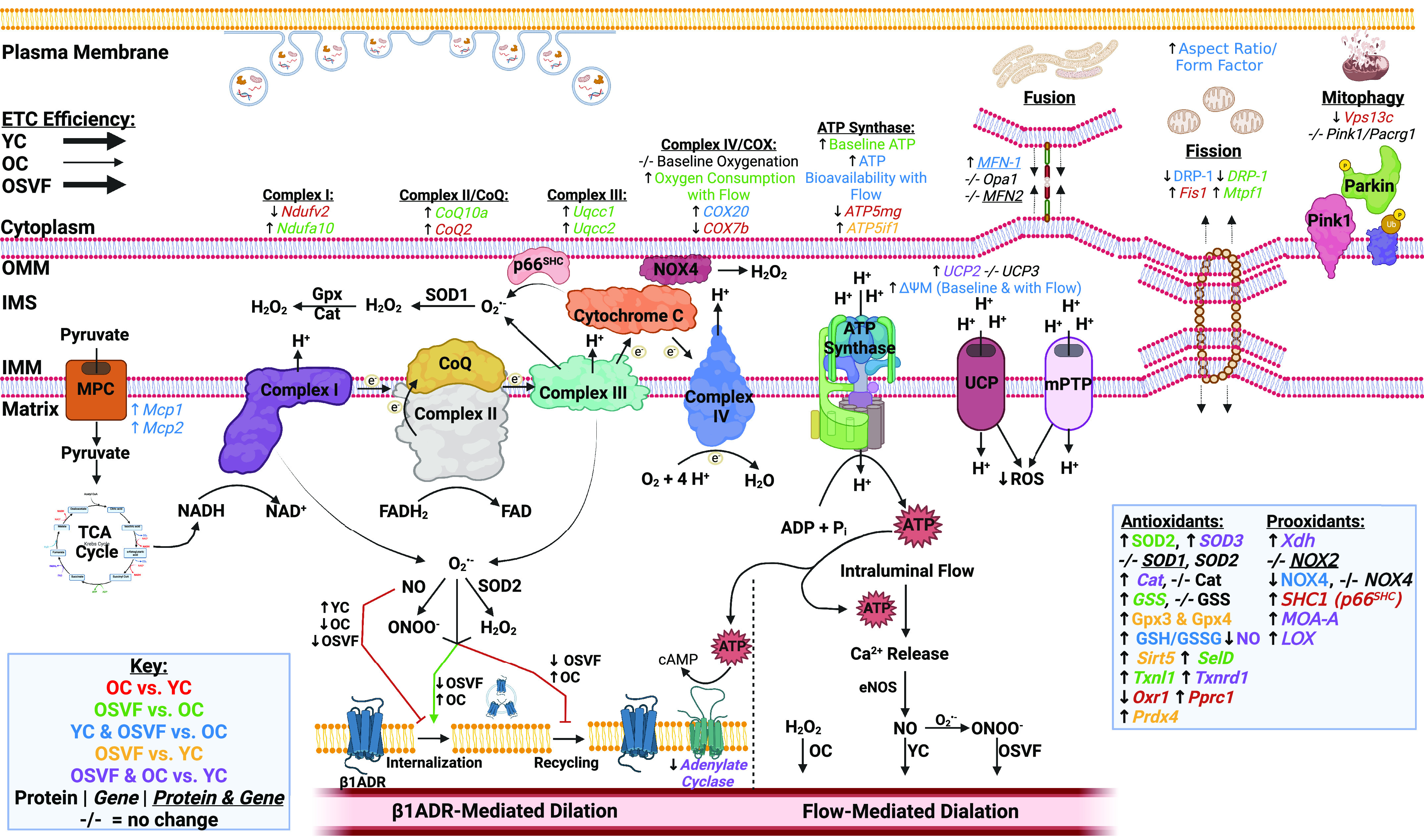
Effects of aging and SVF therapy on mitochondrial function and vasodilation. Illustration summarizes RNA sequencing data, and isolated vessel experimental results in the context of the ETC/oxidative phosphorylation, mitochondrial fission, fusion, mitophagy, and antioxidant/prooxidant enzyme balance and their influences on β1AR-mediated and flow-mediated dilation. Genes were included with significance as P < 0.1. Green arrow indicates activating whereas red line indicates inhibitory action. Note, antioxidant/prooxidant enzyme list and contributions of ROS/NO on β1AR and FMD summarize results from our preceding study (9). Image created with BioRender.com and published with permission. ROS, reactive oxygen species; SVF, stromal vascular fraction.
The preceding step of oxidative phosphorylation is the generation of NADH, produced in the TCA cycle, which itself requires the transport of pyruvate across the inner mitochondrial membrane. The mitochondrial pyruvate carriers 1 and 2 were both genetically downregulated in aging and restored with SVF. Therefore, high pyruvate and NADH drive oxidative phosphorylation according to Le Chatelier’s principle, which is likely the case in YC and OSVF versus OC. At the same time, high NADH-to-NAD+ ratio drives matrix superoxide production from complex I (39). Aging reduced genetic expression of Ndufv2, a core subunit of complex I, whereas SVF upregulated genetic expression of Ndufa10, another core subunit. This could indicate a downregulation of complex I function in aging in an attempt to blunt superoxide production, whereas SVF therapy restores complex I function since superoxide levels were kept in check by SVF (evaluated by mitosox fluorescence) in our previous study (9). From complex I or II, electrons are shunted to coenzyme Q. CoQ10a, itself a major antioxidant, was upregulated in SVF versus OC, as were complex III assembly factors Uqcc1 and Uqcc2, again suggesting facilitation of electron transport. Complex III is another generator of superoxide, both in the mitochondrial matrix and inner mitochondrial space, so downregulation again could be an attempt by OC to halt superoxide production. Complex IV subunit COX7b and assembly factor COX20 were downregulated in aging with COX20 being restored by SVF. ATP synthase subunit ATP5mg was downregulated in aging, albeit ATP synthase inhibitory factor ATP5if1 [encodes ATP synthase inhibitory factor (IF)] was significantly upregulated in OSVF versus YC. Finally, uncoupling protein 2 (UCP2), also responsible for decoupling oxidative phosphorylation by reducing the membrane potential that drives proton flow through ATP synthase, was genetically upregulated with aging and remained so in SVF.
The culmination of these observations was that baseline ATP production was blunted in aging and significantly reinvigorated with SVF therapy. The generation of endothelial shear stress by introducing intraluminal flow facilitated greater oxygen consumption in OSVF versus OC, as well as ATP production to match demand in YC and OSVF, but not in OC. JC-1 showed that mitochondrial membrane potential, as expected by enhanced UCP2 expression, was diminished in aging as a likely compensatory mechanism to slow the respiratory rate and lower superoxide generation. On the contrary, SVF returned mitochondrial membrane potential to levels seen in YC, despite maintaining UCP2 genetic expression. It is possible that protein expression of UCP2 in SVF does not match the genetic trends. In addition, it is known that ROS activates, whereas glutathionylation of UCP2 results in inhibition (40), and we previously showed significantly increased reduced-to-oxidized glutathione ratio in OSVF versus OC (9). The mitochondrial permeability transition pore (mPTP) is also regulated by posttranslational modifications; namely, oxidized glutathione (GSSG) and peroxynitrite open the pores (41, 42), both of which are reduced in OSVF versus OC at baseline (without induced flow).
Although in this study we did not confirm protein levels of these ETC complexes and mediators, results from a recent proteomic study by Chandra et al. in aging mouse brain microvessels greatly mirrored our transcriptional findings (39, 43). Chandra et al. (43) found an aging-associated decrease in oxidative stress response proteins (antioxidant proteins), as well as an aging-associated decrease in complex I–V and ATP synthase subunits. Overall, the authors stipulated that aging initially leads to no-free radical-mediated mitochondrial dysfunction which elevates ROS in concert with a reduction in ROS scavenging ability. Together, these act as a putative event in a cyclic fashion that further drives mitochondrial dysfunction, including reduced respiratory function. The results in our study suggest the same is occurring in coronary microvasculature in aging female rats but can be mitigated by SVF therapy.
Mitochondrial health and proclivity for ROS generation are also determined by the fission and/or fusion of dysfunctional mitochondria. Morphologically, we showed mitochondria were more isolated and punctate in aging, which is known to be associated with increased ROS production. Mitochondria from OSVF microvessels exhibited greater network density and less circular shape, which is known to be associated with attenuated ROS production and similar to a YC phenotype (28). In OC, this fragmented morphology was associated with greater Fis1 and DRP-1 gene and greater DRP-1 protein expression. In addition, DRP-1 is localized to the mitochondria (where it is activated) to a greater degree in aging. DRP-1 gene and protein expression were reduced by SVF therapy, and localization to the mitochondria was nearly significantly reduced. In addition, SVF therapy conferred upregulation of fusion-mediator MFN-1, but not MFN-2 genetic and protein expression, matching a less-fragmented morphology. Inducing fusion pharmacologically has been associated with protection against ROS-induced mitochondrial fragmentation and cell death (44). It is unclear if greater fusion in OSVF is a consequence of a lower ROS environment or if activating fusion itself aids in lowering ROS levels or both.
To highlight the link between fission/fusion balance and vascular dilative function, we designed ex vivo experiments to acutely modulate fission/fusion levels to observe the dilative capacity to intraluminal flow and β1AR agonism. In YC and OVSF, both of which had more fused mitochondria in vessels at baseline, inhibition of Opa1 with MYLS22 led to significant attenuation in dilation to norepinephrine. This was not replicated with DRP-1 inhibition with MDIVI. Together, this indicates fusion confers a protective dilative effect and that blockade of fusion (leading to unbalanced fission) can blunt dilation. Thus, by adding MYLS22, we were able to make vessels from YC and OSVF behave phenotypically like OC vessels. Inhibiting fission alone was not sufficient to regenerate β1AR dilative function.
Intraluminal flow + MYLS22 significantly attenuated YC dilation, instead leading to flow-mediated constriction (with no MDIVI effect). Surprisingly, MDIVI (but not MYLS22) led to OSVF exhibiting flow-mediated constriction. The reason that blocking fission may cause flow-mediated constriction may be due to the fact that OSVF uses peroxynitrite (and hydrogen peroxide to a lesser extent) as the mediators of FMD (9). Therefore, inhibiting fission and causing hyperfusion may attenuate ROS production to the point of losing their signaling potential for dilation. This would not be the case in YC as its FMD mediator is nitric oxide. Neither MDIVI nor MYLS22 had a significant effect on OC FMD.
If mitochondrial dysfunction is severe enough, mitophagy can be instigated by Pink1 and Parkin, of which we found no genetic expression difference between groups in our study. However, microvascular vps13c, the negative regulator of Parkin, was decreased in aging. Therefore, the loss of vps13c could reflect dysfunctional ROS generating mitochondria and a compensatory response in OC to induce mitophagy. Interestingly, others have reported that loss of vps13c has been associated with reduced mitochondrial membrane potential, enhanced respiration rates (as compensation for reduced membrane potential), and fragmented mitochondrial morphology (45).
Optimal mitochondrial function could be linked to β1AR through the production of ATP. Indeed, we found a correlative link between ATP levels (and ΔΨM) and dilation to β1AR agonism. After activation of the β1AR, adenylate cyclase converts ATP into cAMP to activate PKA to inhibit smooth muscle myosin light chain kinase and block constriction. At an older age, bioavailability of ATP may be limited enough to impact this pathway (Fig. 8). Although there was no correlation in our data between ATP levels and FMD, recent investigations by others support an essential role for ATP in FMD. Wilson et al. found that blocking ATP production with oligomycin significantly blunted acetylcholine- and flow-mediated dilation through reduced IP3-mediated calcium signaling that would otherwise activate eNOS and nitric oxide production (5). This effect could be replicated by blocking mitochondrial pyruvate carriers. As nitric oxide is a necessary component of peroxynitrite, this could also potentially contribute to the loss of peroxynitrite-mediated dilation, as well as nitric oxide-mediated dilation in aging. Restored ATP with SVF therapy could therefore be important for the switch in FMD mediator to peroxynitrite after SVF therapy.
Conclusions
Age-related microvascular dysfunction and hyperconstriction can be ameliorated by targeting mitochondrial dysfunction, including ROS-associated hyperfission and ETC dysfunction. SVF therefore has significant translational potential for patients with CMD and could be theoretically superior to current clinical standards (e.g., β-blockers) that treat symptoms but do not address the root pathological cause of microvascular dysfunction and hyperconstriction (and may even potentiate root pathological cause through blockade of microvascular β-adrenergic-mediated dilation). Future directions should confirm these findings in human coronary microvessels for enhanced clinical translatability. Overall, SVF is a feasible therapeutic alternative for aging-induced CMD that acts to reduce oxidative stress, restore dilation to FMD, and β1AR agonism with mechanistic underpinnings of rejuvenated mitochondrial function and dynamics.
LIMITATIONS
Due to the unique size of isolated microvessels in this study (∼100 µm in diameter), certain experiments that were attempted were not successful such as mitochondrial respiratory analysis (even with pooled microvessels). Although larger vessels or aorta could be used in such an experiment, we felt it may not be an accurate reflection of microvasculature dynamics and therefore could be misleading in the context of this study. To our knowledge, this is the first RNA-seq analysis performed on coronary microvessels from young and old F344 rats, but the data do not provide information on protein and protein quality, and future experiments would need to be performed to confirm these findings. For the mitochondrial dynamics (e.g., membrane potential, oxygenation, and ATP), it is important to note that these are not distinguishable between endothelial and smooth muscle cell layers without the addition of live cell-specific markers, which were not included in the present study. We must acknowledge MDIVI and MYLS22 inhibitors of DRP-1 and Opa1 are known to have limitations in specificity with some off-target effects. Specifically, MDIVI can inhibit mitochondrial complex I (46) and Opa1 also has a role in angiogenesis via limiting NF-κB signaling (47). Activation of DRP-1 via phosphorylation was not assessed, albeit we did assess DRP-1/TOM20 colocalization (greater DRP-1 activation in the mitochondria; Supplemental Fig. S2). Lastly, we did not evaluate changes in autophagy regulation downstream of mitophagy such as LC3B or p62.
DATA AVAILABILITY
RNA sequencing data have been uploaded to the Gene Expression Omnibus with accession number GSE184674 and can be accessed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184674 with reviewer access token mvqfqcwjvyrfsn.
SUPPLEMENTAL DATA
Supplemental Figs. S1 and S2: http://doi.org/10.6084/m9.figshare.20657466.
GRANTS
A.J.L. was funded by National Institutes of Health (NIH) Grants R01 AG053585, R01 DE030103, and P30 ES030283 and a grant from the Gheens Foundation. A.B. was funded by NIH Grants R01 HL133029 and R01 HL157025-01 and by Advancing a Healthier Wisconsin Endowment.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Amanda LeBlanc is an editor of American Journal of Physiology-Heart and Circulatory Physiology and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article.
AUTHOR CONTRIBUTIONS
E.P.T., A.B., and A.J.L. conceived and designed research; E.P.T., R.N., G.R., J.E.B., and A.J.L. performed experiments; E.P.T. and R.N. analyzed data; E.P.T., A.B., and A.J.L. interpreted results of experiments; E.P.T. prepared figures; E.P.T. drafted manuscript; E.P.T., G.R., J.E.B., A.B., and A.J.L. edited and revised manuscript; E.P.T., R.N., G.R., J.E.B., A.B., and A.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Eric Rouchka and Dr. Xiaohong Li of the University of Louisville Bioinformatics Core for assistance with RNA sequencing.
This work was orally presented at the 2022 Experimental Biology Conference and was Published in the Accompanying FASEB Journal as a conference abstract (see https://doi.org/10.1096/fasebj.2022.36.S1.R1930).
Online graphical abstract was created with Biorender.com and published with permission.
REFERENCES
- 1. Tracy EP, Stielberg V, Rowe G, Benson D, Nunes SS, Hoying JB, Murfee WL, LeBlanc AJ. State of the field: cellular and exosomal therapeutic approaches in vascular regeneration. Am J Physiol Heart Circ Physiol 322: H647–H680, 2022. doi: 10.1152/ajpheart.00674.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–H503, 2014. doi: 10.1152/ajpheart.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gáspár T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013. doi: 10.1161/ATVBAHA.112.300560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park SY, Kwon OS, Andtbacka RHI, Hyngstrom JR, Reese V, Murphy MP, Richardson RS. Age-related endothelial dysfunction in human skeletal muscle feed arteries: the role of free radicals derived from mitochondria in the vasculature. Acta Physiol (Oxf) 222: e12893, 2018. doi: 10.1111/apha.12893. [DOI] [PubMed] [Google Scholar]
- 5. Wilson C, Lee MD, Heathcote HR, Zhang X, Buckley C, Girkin JM, Saunter CD, McCarron JG. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J Biol Chem 294: 737–758, 2019. doi: 10.1074/jbc.RA118.005913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bharath LP, Mueller R, Li Y, Ruan T, Kunz D, Goodrich R, Mills T, Deeter L, Sargsyan A, Anandh Babu PV, Graham TE, Symons JD. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can J Physiol Pharmacol 92: 605–612, 2014. doi: 10.1139/cjpp-2014-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park SK, La Salle DT, Cerbie J, Cho JM, Bledsoe A, Nelson A, Morgan DE, Richardson RS, Shiu YT, Boudina S, Trinity JD, Symons JD. Elevated arterial shear rate increases indexes of endothelial cell autophagy and nitric oxide synthase activation in humans. Am J Physiol Heart Circ Physiol 316: H106–H112, 2019. doi: 10.1152/ajpheart.00561.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beyer AM, Zinkevich N, Miller B, Liu Y, Wittenburg AL, Mitchell M, Galdieri R, Sorokin A, Gutterman DD. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol 112: 5, 2017. doi: 10.1007/s00395-016-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tracy EP, Dukes M, Beare J, Rowe G, Nair R, LeBlanc AJ. Stromal vascular fraction restores vasodilatory function by reducing oxidative stress in aging-induced coronary microvascular disease. Antioxid Redox Signal, 2022. doi: 10.1089/ars.2021.0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J 35: e21620, 2021. doi: 10.1096/fj.202100067R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan Y, Cheng Z, Mao L, Xu G, Li N, Zhang M, Weng P, Zheng L, Dong X, Hu S, Wang B, Qin X, Jiang X, Chen C, Zhang J, Zou Z. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles. J Nanobiotechnology 20: 149, 2022. doi: 10.1186/s12951-022-01338-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forrester SJ, Preston KJ, Cooper HA, Boyer MJ, Escoto KM, Poltronetti AJ, Elliott KJ, Kuroda R, Miyao M, Sesaki H, Akiyama T, Kimura Y, Rizzo V, Scalia R, Eguchi S. Mitochondrial fission mediates endothelial inflammation. Hypertension 76: 267–276, 2020. doi: 10.1161/HYPERTENSIONAHA.120.14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res 112: 1171–1188, 2013. doi: 10.1161/CIRCRESAHA.111.300233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu X, Cao K, Lv W, Feng Z, Liu J, Gao J, Li H, Zang W, Liu J. Punicalagin attenuates endothelial dysfunction by activating FoxO1, a pivotal regulating switch of mitochondrial biogenesis. Free Radic Biol Med 135: 251–260, 2019. doi: 10.1016/j.freeradbiomed.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 15. Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, Cui Z, Cui T, Wang XL, Shen YH. PINK1-parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One 10: e0132499, 2015. doi: 10.1371/journal.pone.0132499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5: a011072, 2013. doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chehaitly A, Guihot A-L, Proux C, Grimaud L, Aurrière J, Legouriellec B, Rivron J, Vessieres E, Tétaud C, Zorzano A, Procaccio V, Joubaud F, Reynier P, Lenaers G, Loufrani L, Henrion D. Altered mitochondrial Opa1-related fusion in mouse promotes endothelial cell dysfunction and atherosclerosis. Antioxidants (Basel) 11: 1078, 2022. doi: 10.3390/antiox11061078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hung CH, Cheng SS, Cheung YT, Wuwongse S, Zhang NQ, Ho YS, Lee SM, Chang RC. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol 14: 7–19, 2018. doi: 10.1016/j.redox.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T, Ushio-Fukai M. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep 23: 3565–3578, 2018. doi: 10.1016/j.celrep.2018.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubramaniam V, Watson PA, Reusch JE. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol 304: H1624–H1633, 2013. doi: 10.1152/ajpheart.00987.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song SB, Hwang ES. A rise in ATP, ROS, and mitochondrial content upon glucose withdrawal correlates with a dysregulated mitochondria turnover mediated by the activation of the protein deacetylase SIRT1. Cells 8: 11, 2018. doi: 10.3390/cells8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durand MJ, Ait-Aissa K, Levchenko V, Staruschenko A, Gutterman DD, Beyer AM. Visualization and quantification of mitochondrial structure in the endothelium of intact arteries. Cardiovasc Res 115: 1546–1556, 2019. doi: 10.1093/cvr/cvy294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res 122: 58–73, 2018. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Council NR. Guide for the Care and Use of Laboratory Animals (8th edition). Washington, DC: The National Academies Press, 2011, p. 246. [Google Scholar]
- 25. Kelm NQ, Beare JE, Weber GJ, LeBlanc AJ. Thrombospondin-1 mediates Drp-1 signaling following ischemia reperfusion in the aging heart. FASEB Bioadv 2: 304–314, 2020. doi: 10.1096/fba.2019-00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelm NQ, Beare JE, Yuan F, George M, Shofner CM, Keller BB, Hoying JB, LeBlanc AJ. Adipose-derived cells improve left ventricular diastolic function and increase microvascular perfusion in advanced age. PLoS One 13: e0202934, 2018. doi: 10.1371/journal.pone.0202934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowe G, Kelm NQ, Beare JE, Tracy E, Yuan F, LeBlanc AJ. Enhanced beta-1 adrenergic receptor responsiveness in coronary arterioles following intravenous stromal vascular fraction therapy in aged rats. Aging (Albany NY) 11: 4561–4578, 2019. doi: 10.18632/aging.102069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rowe G, Tracy E, Beare JE, LeBlanc AJ. Cell therapy rescues aging-induced beta-1 adrenergic receptor and GRK2 dysfunction in the coronary microcirculation. Geroscience 44: 329–348, 2022. doi: 10.1007/s11357-021-00455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. LeBlanc AJ, Reyes R, Kang LS, Dailey RA, Stallone JN, Moningka NC, Muller-Delp JM. Estrogen replacement restores flow-induced vasodilation in coronary arterioles of aged and ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 297: R1713–R1723, 2009. [Erratum in Am J Physiol Regul Integr Comp Physiol 298: R1446, 2010]. doi: 10.1152/ajpregu.00178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leblanc AJ, Touroo JS, Hoying JB, Williams SK. Adipose stromal vascular fraction cell construct sustains coronary microvascular function after acute myocardial infarction. Am J Physiol Heart Circ Physiol 302: H973–H982, 2012. doi: 10.1152/ajpheart.00735.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chilian WM, Eastham CL, Marcus ML. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am J Physiol Heart Circ Physiol 251: H779–H788, 1986. doi: 10.1152/ajpheart.1986.251.4.H779. [DOI] [PubMed] [Google Scholar]
- 32. Mboge MY, Chen Z, Khokhar D, Wolff A, Ai L, Heldermon CD, Bozdag M, Carta F, Supuran CT, Brown KD, McKenna R, Frost CJ, Frost SC. A non-catalytic function of carbonic anhydrase IX contributes to the glycolytic phenotype and pH regulation in human breast cancer cells. Biochem J 476: 1497–1513, 2019. doi: 10.1042/BCJ20190177. [DOI] [PubMed] [Google Scholar]
- 33. Lanceta L, O’Neill C, Lypova N, Li X, Rouchka E, Waigel S, Gomez-Gutierrez JG, Chesney J, Imbert-Fernandez Y. Transcriptomic profiling identifies differentially expressed genes in palbociclib-resistant ER+ MCF7 breast cancer cells. Genes (Basel) 11: 467, 2020. doi: 10.3390/genes11040467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miralda I, Vashishta A, Rogers MN, Rouchka EC, Li X, Waigel S, Lamont RJ, Uriarte SM. Whole transcriptome analysis reveals that Filifactor alocis modulates TNFα-stimulated MAPK activation in human neutrophils. Front Immunol 11: 497, 2020. doi: 10.3389/fimmu.2020.00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merrill R, Filippo K, Strack S. Measuring mitochondrial shape with ImageJ. In: Techniques to Investigate Mitochondrial Function in Neurons, edited by Strack S, Usachev Y.. Humana Press, 2017, p. 31–48. [Google Scholar]
- 36. Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem 119: 315–326, 2017. doi: 10.1016/j.acthis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 37. Stauffer W, Sheng H, Lim HN. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep 8: 15764, 2018. doi: 10.1038/s41598-018-33592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tracy EP, Hughes W, Beare J, Rowe G, Beyer AM, LeBlanc AJ. Aging induced impairment of vascular function - mitochondrial redox contributions and physiological/clinical implications. Antioxid Redox Signal 35: 974–1015, 2021. doi: 10.1089/ars.2021.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ait-Aissa K, Blaszak SC, Beutner G, Tsaih SW, Morgan G, Santos JH, Flister MJ, Joyce DL, Camara AKS, Gutterman DD, Donato AJ, Porter GA Jr, Beyer AM. Mitochondrial oxidative phosphorylation defect in the heart of subjects with coronary artery disease. Sci Rep 9: 7623, 2019. doi: 10.1038/s41598-019-43761-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem 286: 21865–21875, 2011. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem 238: 623–630, 1996. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- 42. Imaizumi N, Aniya Y. The role of a membrane-bound glutathione transferase in the peroxynitrite-induced mitochondrial permeability transition pore: formation of a disulfide-linked protein complex. Arch Biochem Biophys 516: 160–172, 2011. doi: 10.1016/j.abb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 43. Chandra PK, Cikic S, Rutkai I, Guidry JJ, Katakam PVG, Mostany R, Busija DW. Effects of aging on protein expression in mice brain microvessels: ROS scavengers, mRNA/protein stability, glycolytic enzymes, mitochondrial complexes, and basement membrane components. Geroscience 44: 371–388, 2022. doi: 10.1007/s11357-021-00468-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szabo A, Sumegi K, Fekete K, Hocsak E, Debreceni B, Setalo G Jr, Kovacs K, Deres L, Kengyel A, Kovacs D, Mandl J, Nyitrai M, Febbraio MA, Gallyas F Jr, Sumegi B. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem Pharmacol 150: 86–96, 2018. doi: 10.1016/j.bcp.2018.01.038. [DOI] [PubMed] [Google Scholar]
- 45. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet 98: 500–513, 2016. doi: 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40: 583–594.e6, 2017. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Herkenne S, Ek O, Zamberlan M, Pellattiero A, Chergova M, Chivite I, Novotná E, Rigoni G, Fonseca TB, Samardzic D, Agnellini A, Bean C, Di Benedetto G, Tiso N, Argenton F, Viola A, Soriano ME, Giacomello M, Ziviani E, Sales G, Claret M, Graupera M, Scorrano L. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab 31: 987–1003.e8, 2020. doi: 10.1016/j.cmet.2020.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1 and S2: http://doi.org/10.6084/m9.figshare.20657466.
Data Availability Statement
RNA sequencing data have been uploaded to the Gene Expression Omnibus with accession number GSE184674 and can be accessed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184674 with reviewer access token mvqfqcwjvyrfsn.