

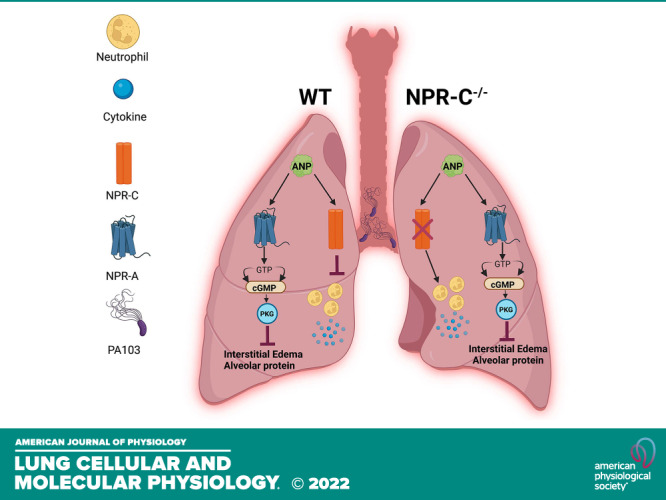
Keywords: acute lung injury, atrial natriuretic peptide, cytokines, lung inflammation, natriuretic peptide receptor C
Abstract
Atrial natriuretic peptide (ANP) protects against acute lung injury (ALI), but the receptor that mediates this effect is not known. Transgenic mice with 0 (knockout), 1 (heterozygote), or 2 (wild-type) functional copies of Npr3, the gene that encodes for natriuretic peptide receptor-C (NPR-C), were treated with intravenous infusion of ANP or saline vehicle before oropharyngeal aspiration of Pseudomonas aeruginosa (PA103) or saline vehicle. Lung injury was assessed 4 h following aspiration by measurement of lung wet/dry (W/D) weight, whole lung leukocyte and cytokine levels, and protein, leukocyte, and cytokine concentration in bronchoalveolar lavage fluid (BALF). PA103 induced acute lung injury as evidenced by increases in lung W/D ratio and protein concentration in BALF. The severity of PA103-induced lung injury did not differ between NPR-C genotypes. Treatment with intravenous ANP infusion reduced PA103-induced increases in lung W/D and BALF protein concentration in all three NPRC genotypes. PA103 increased the percentage of leukocytes that were neutrophils and cytokine levels in whole lung and BALF in NPR-C wild-type and knockout mice. This effect was blunted by ANP in wild-type mice but not in the NPR-C knockout mice. NPR-C does not mediate the protective effect of ANP on endothelial cell permeability in settings of PA103-induced injury but may mediate the effect of ANP on inhibition of the recruitment of neutrophils to the lung and thereby attenuate the release of inflammatory cytokines.
INTRODUCTION
Acute lung injury (ALI) such as the acute respiratory distress syndrome (ARDS) is a common cause of respiratory failure with an annual incidence of ∼200,000 cases in the United States and a mortality rate that exceeds 30% (1, 2). Prior to the severe acute respiratory syndrome (SARS)-2 pandemic, ∼10% of patients in intensive care units had ARDS and the majority of cases were caused by septicemia or pneumonia (3, 4). Early reports following the onset of widespread infection with SARS-associated coronavirus-2 (CoV-2) suggest that a third of patients admitted for infection from this virus also develop ARDS (5, 6). Despite the high prevalence and mortality, current management for ARDS consists of treatment of the inciting infection or lung injury and supportive care including mechanical ventilation. Although some medical therapies have been shown to improve outcome in acute respiratory failure associated with SARS-CoV-2 (7, 8), no pharmacologic therapies have been shown to reduce the severity or improve outcome in other forms of ALI or ARDS. Thus, there is an acute need to better define mechanisms responsible for modulating ALI, especially those that could represent potential therapeutic targets.
Acute respiratory failure in ARDS is caused by disruption of the alveolar-capillary membrane and increased transudation of fluid, protein, and inflammatory cells from the vascular to the alveolar space leading to decreased lung compliance and impaired gas exchange (9, 10). Endothelial cell dysfunction of the pulmonary microvasculature due to reduced cell-to-cell adherence and breakdown of adherens junctions results in the leakage of vascular components into the alveolus. A variety of inflammatory cytokines and chemokines such as thrombin, tumor necrosis factor-ɑ (TNF-ɑ), keratinocytes-derived chemokine (KC) also known as C-X-C chemokine ligand-1 (CXCL1), and macrophage inflammatory protein-2 (MIP-2) also termed C-X-C chemokine ligand-2 (CXCL2) contribute to endothelial barrier dysfunction and have been implicated in the pathogenesis of ALI/ARDS (11). These signaling proteins are released by immune cells, particularly neutrophils and macrophages that migrate to the alveolar space in response to pathogenic bacteria or cell wall components of Gram-negative pathogens, such as lipopolysaccharide (LPS) (12, 13). They are also released by other cells including endothelial cells (14).
Atrial, brain and C-type natriuretic peptide (ANP, BNP, and CNP, respectively) are the primary members of a family of small proteins characterized by a similar 17 amino acid loop. ANP and BNP are secreted mainly from the cardiac atria and ventricles, respectively, and play important physiological roles in vascular homeostasis due to their prominent effects on vascular endothelial permeability and renal sodium excretion (15). Both peptides also have vasorelaxation effects on vascular smooth muscle and inhibit the renin-angiotensin system (16). In the systemic circulation, ANP increases vascular permeability and administration of ANP causes a decrease in intravascular volume and an increase in hematocrit that are independent of its natriuretic and diuretic effects (17). In the pulmonary circulation, however, natriuretic peptides have been shown to protect against endothelial dysfunction, pulmonary edema formation, and acute lung injury (17, 18).
Despite a sizable body of evidence demonstrating the protective effects of ANP and BNP on lung injury in animal models of ARDS (17–19), the receptors that ANP and BNP might signal through to mediate protection in lung injury is not known. Natriuretic peptides interact with 3 cell surface receptors, natriuretic peptide receptor-A, -B, and -C (NPR-A, NPR-B, NPR-C, respectively). NPR-A and NPR-B possess an extracellular ligand binding domain linked to an intracellular guanylyl cyclase (GC) domain and are often referred to as particulate guanylyl cyclase receptors or guanylyl cyclase receptor-A and -B (GC-A, GC-B). NPR-A has considerably greater binding affinity for ANP and BNP than CNP, whereas the reverse is true for NPR-B. Ligand binding to NPR-A or NPR-B results in a marked increase in intracellular levels of cGMP leading to activation of cGMP-dependent kinase (PKG) that is thought to be the critical pathway leading to the biological effects of the natriuretic peptides (18). NPR-C has an extracellular ligand binding site similar to NPR-A and NPR-B, with similar affinities for ANP, BNP, and CNP, but is not connected to a GC domain and does not increase cGMP synthesis. Although initially thought to function as a clearance receptor, NPR-C was later found to be coupled with an inhibitory heterotrimeric G-protein (Gqi) composed of an α and a βγ subunit. Both subunits of the Gqi protein mediate a number of subcellular and physiological effects by either inhibiting adenyl cyclase and thus cAMP generation, or by activation of phospholipase C causing increases in phosphatidyl inositol bis-phosphate (PIP2) and diacylglycerol (DAG) (20).
In previous studies, we examined the role of ANP on lung injury in mice with genetically altered NPR-A expression. We found that ANP was nearly as effective at attenuating thrombin and LPS-induced lung injury in NPR-A knockout (KO) mice as it was in mice with normal expression of NPR-A (19). Furthermore, administration of the NPR-C-specific ligand, cANF, also blocked thrombin receptor activating peptide- (TRAP) and LPS-induced lung injury in NPR-A wild-type (WT) and KO mice (19). These findings suggested that the protective effect of ANP on ALI were not mediated by NPR-A alone. Considering, the low level of NPR-B expression on endothelial cells and low binding affinity of ANP for this receptor, we hypothesized that the beneficial effect of ANP on ALI was mediated by NPR-C. To test this hypothesis, we treated mice with 0, 1, or 2 functional copies of Npr3 (the gene that encodes for NPR-C in mice) with an infusion of saline or ANP during exposure to Pseudomonas aeruginosa strain 103 (PA103) and examined the effects on pulmonary edema formation and lung inflammatory responses.
METHODS
Animals
Male and female mice with 2 (NPR-C+/+), 1 (NPR-C+/−), or 0 (NPR-C−/−) functional copies of Npr3 were studied between the ages of 10 and 18 wk. NPR-C+/+ and NPRC+/− breeding pairs were obtained from Jackson Laboratory (Bar Harbor, ME, strain B6;129-Npr3tm1Unc/Mmnc MMRRC ID: 16). These mice were subsequently maintained and bred at the Providence VA Medical Center animal facility. Animals were given food and water ad libitum and monitored daily. DNA was extracted from mouse tails using the tissue/tail PCR genotyping kit (EZ BioResearch). NPR-C genotype was confirmed via polymerase chain reaction (PCR) using the following primers (Integrated DNA Technologies): common primer 1 Npr5′ 5′- CACAAGGACACGGAATACTC-3′ and primer 2 NprC3′ 5′- CTTGGATGTAGCGCACTATGTC-3′ for amplification of wild-type product (262 bp), primer 2, NprC3′ 5′- CTTGGATGTAGCGCACTATGTC-3′ and primer 3 Neo2 5′ ACGCGTCACCTTAATATGCG-3′ amplification of knockout product (413 bp). In addition, NPR-C KO mice have a distinct hunch-backed phenotype caused by elongation of the spine and long bones that helps to confirm the results of genotyping (13).
All animal protocols were approved by the Providence VA Medical Center and Brown University Institutional Animal Care and Use Committees and comply with the Health Research Extension Act and PHS policy.
RT-qPCR
RNA was isolated from homogenized whole lung tissue using the miRNeasy mini kit (Qiagen) according to the manufacturer’s protocol and quantified with NanoDrop OneC Spectrophotometer (Thermo Fisher). RNA was reverse transcribed to cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher). For qPCR, PowerUp SYBR green master mix (ThermoFisher) was used with the following primers: β-actin (forward: 5′- ACTGTCGAGTCGCGTCCACC-3′: reverse: 5′- CGATGGAGGGGAATACAGCCC-3′) and NPR3 (forward: 5′- TTCAGGAGGAGGGGTTGCAC-3′: reverse: 5′- AGTCTCCACTGGTCATGCCG-3′). qPCR was performed on the StepOnePlus Real-Time PCR system (Applied Biosystems). mRNA expression of NPR3 was normalized to housekeeping gene β-actin relative to NPRC−/−. The ΔΔCT and 2-ΔΔCT (fold change) were calculated and used for a nonparametric Mann-Whitney test.
P. aeruginosa (PA103)-Induced Lung Injury
Pseudomonas aeruginosa strain PA103 as a bacterial stock (Courtesy: Dr. Troy Stevens, University of South Alabama) was streaked on Luria broth agar plates under sterile conditions and incubated at 37°C for 18–24 h. Bacterial suspensions containing 1 × 109 CFU/mL were prepared by transferring 3–4 isolated CFUs from the plates to a sterile glass tube containing 3 mL of sterile saline solution. The bacterial suspension was quantified using Vitek Colorimeter. Mice were anaesthetized with 2.5% isoflurane (inhalation) and were held vertical while restrained on an operating board. A 100 µL of PA103 suspension ≈ 107 CFU was administered into the mouth and the nostrils were manually occluded until a clique sound confirmed aspiration of oral contents into lungs. The left external jugular vein was isolated and cannulated with a PE10 catheter (BD INTRAMEDIC). Mice received an infusion of ANP at ∼180 ng/kg/min by injecting ∼10 μL every 5 min for up to a total of 150 min starting 30 min before oropharyngeal aspiration of saline or ≈107 CFU PA103. Mice were infused through an external jugular vein via catheter secured to the dorsal area of the neck via a skin button with a Dacron patch. Mice were allowed to recover after the catheter had been placed. Mice were tethered during the infusion but allowed to move freely within their cage. Mice were monitored during the entire infusion procedure. Once the infusion was complete, mice were briefly anaesthetized with isoflurane, skin sutures were opened, and the catheter removed. The jugular vein was tied off and skin was closed again with sutures. Mice were euthanized 4 h after oropharyngeal aspiration of saline or PA103. Control mice received an equal volume of saline alone at the same infusion rate, resulting in four treatment groups: 1) intravenous saline followed by oropharyngeal aspiration of saline (Saline/Saline), 2) intravenous saline followed by oropharyngeal aspiration of PA103 (Saline/PA103), 3) intravenous ANP followed by oropharyngeal aspiration of saline (ANP/Saline), and 4) intravenous ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103).
Mice were euthanized using pentobarbitone 120 mg/kg administered by intraperitoneal injection. Bronchoalveolar lavage fluid (BALF) and plasma were collected for biochemical analysis. Experiments were performed on male and female mice in each treatment group.
Measurement of Lung Injury
Lungs were exposed via midline sternotomy and a braided silk suture was passed under the left lung hilum region and the left main bronchus was tied off. The left lung was removed, weighed immediately (wet weight), and weighed again after drying for 72 h at 80°C (dry weight) to determine the wet weight/dry weight ratio (W/D). Bronchoalveolar lavage fluid (BALF) was obtained by making a small nick in the trachea, inserting a 23 G blunt needle and injecting 300 μL of sterile phosphate-buffered saline (PBS) into right lung and aspirating it back. BALF was centrifuged at 2,000 rpm for 10 min to remove cellular debris. Protein concentration of BALF was measured using a modified Bradford assay at an absorbance of 750 nm.
Preparation of Single Cell Suspensions for Flow Cytometry
To assess changes in leukocyte content of the vascular, interstitial, and alveolar space, additional studies were performed in which 100 µL of PA103 suspension ≈ 5 × 105 CFU or saline vehicle was administered into the mouth and the nostrils were manually occluded until a clique sound confirmed aspiration of oral contents into lungs. At the time of euthanasia, the lungs were exposed via midline sternotomy and the trachea cannulated with a 23 G blunt needle. Whole lung lavage was done by repeated injection and retrieval of 500-μL aliquots of sterile chilled PBS containing 1 mM EDTA until a total of 3 mL was collected from both lungs. Following lavage, lungs were removed and placed in lung dissociation buffer (Miltenyi Biotec) and homogenized according to manufacturer’s protocol. Peripheral blood was obtained from the right ventricle and 25 µL was lysed for 10 min in 1 mL of RBC Lysis Buffer (BioLegend) and immediately washed twice in flow cytometry buffer [phosphate-buffered saline (Gibco), 1 mM EDTA (Santa Cruz), 0.5% bovine serum albumin (Sigma)]. Cell suspension of BALF was washed once in PBS.
All single-cell suspensions were fixed with 3% paraformaldehyde (Sigma) in flow cytometry buffer before flow cytometry. For flow cytometry protocol, fixed cell suspensions were washed twice in flow cytometry buffer, stained using antibodies at 1 µg/mL for 30 min on ice, and finally washed twice again in flow cytometry buffer before being analyzed using MACSQuant Analyzer 10 (Miltenyi). Fluorescent-conjugated monoclonal antibodies against CD45 [(clone 30-F11), BioLegend; Cat. No. 103125], Ly6G [(clone 1A8), BioLegend; Cat. No.127609], Ly6C [(clone HK1.4), BioLegend; Cat. No.128018], and Ly6B.2 [(clone REA115), Miltenyi; Cat. No.130-102-279] epitopes were used to identify populations of leukocytes, monocytes, and neutrophils by flow cytometry. Leukocytes were identified as CD45+ cells. Neutrophils were identified as CD45 + Ly6CmidLy6B.2+ cells and confirmed as the neutrophil population using Ly6G, which is specific to murine neutrophils. Monocytes were classified into two populations based on CD45 + Ly6ChiLy6B.2+ and CD45 + Ly6CmidLy6B.2−, which identified Ly6Chi and Ly6Cmid monocytes, respectively.
Cytokine Assay
Samples of BALF and lung homogenate were diluted in Quantikine ELISA assay-specific calibrator diluent (R&D Systems) and used to quantify secreted keratinocyte chemoattractant (KC, Cat. No. MKC00B), macrophage inhibitory protein-2 (MIP-2; Cat. No. MM200), tumor necrosis factor-α (TNF-α; Cat. No.MTA00B), and interleukin-6 (IL-6; Cat. No. M6000B). All samples were thawed on ice before dilution preparation. Protein blocking agent (assay-specific assay diluent, 50 μL) was added to each well, followed by addition of 50-μL standard, assay control, or sample dilution in duplicate to each well. Assays were performed as per the manufacturer’s protocol diluting the sample such that the sample value was on the standard curve. Cytokine concentrations were interpolated from standard curves using cubic polynomial regression analysis in GraphPad Prism.
Statistical Analysis
For all studies, data between the four treatment groups were compared using one-way ANOVA and Tukey’s multiple comparisons test. Values are expressed as means ± SD. Differences were considered significant at P < 0.05.
RESULTS
Confirmation of NPR-C Genotype
To confirm the NPR-C genotype, RNA was isolated from homogenized whole lung tissue and RT-qPCR was performed. Figure 1 depicts the level of mRNA of NPR-C (NPR-3) relative to β-actin control. NPR-C wild-type mice expressed 1.7- and 35.7-fold more NPR-3 mRNA relative to NPR-C heterozygous and knockout mice, respectively. In addition, NPR-C knockout mice were identified by their distinct hunch-backed phenotype caused by elongation of the spine and long bones due to decreased clearance of BNP (13).
Figure 1.

qPCR analysis of NPR-C mRNA (NPR-3) expression in wild-type (NPR-C+/+), heterozygous (NPR-C+/−), and knockout mice (NPR-C−/−). mRNA expression of NPR3 was normalized to housekeeping gene β-actin relative to NPRC−/−. The ΔΔCT and 2-ΔΔCT (fold change) were calculated and used for a nonparametric Mann–Whitney test (***P ≤ 0.001, ****P ≤ 0.0001; n = 15 mice NPR-C+/+, 5 mice NPR-C+/−, 12 mice NPR-C−/−. NPR-C, natriuretic peptide receptor-C.
Effect of NPR-C and ANP on PA103-Induced Lung Injury
Lung wet-to-dry weight ratios and the protein concentration of BALF fluid were measured to assess severity of acute lung injury. No differences in lung W/D weight ratio were seen between NPR-C genotypes in mice under control conditions (Saline/Saline; Fig. 2). Intravenous infusion of ANP had no effect on lung W/D in any of the NPR-C genotypes under baseline conditions (ANP/Saline). Treatment with PA103 induced significant increases in lung W/D ratio in all 3 NPR-C genotypes suggesting the occurrence of ALI (Saline/PA103), but there were no differences in lung W/D weight ratio between genotypes, suggesting that the loss of a functional NPR-C did not lead to more severe lung injury. ANP infusion reduced the PA103-induced increase in lung W/D in all 3 NPR-C genotypes (ANP/PA103; Fig. 2) and was just as effective at attenuating PA103-induced increases in lung W/D in NPR-C knockout mice as it was in wild-type mice.
Figure 2.
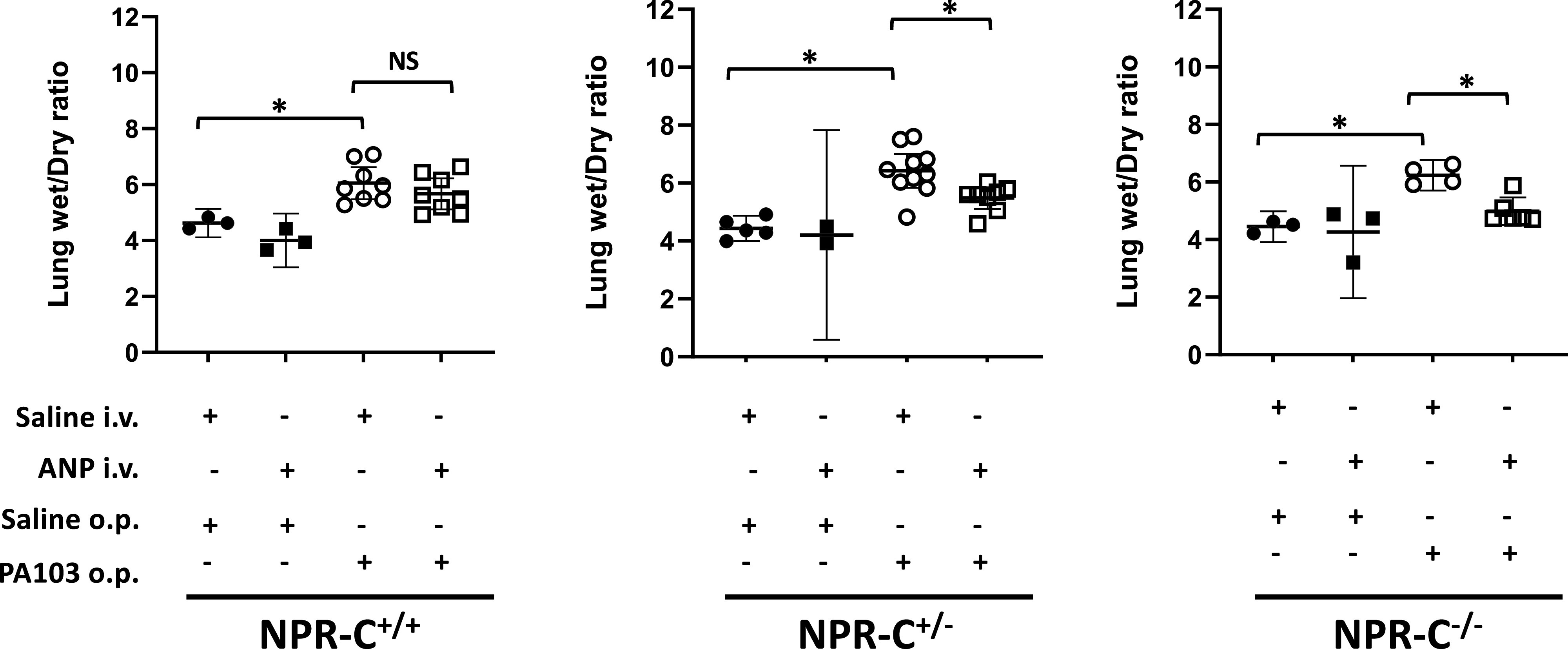
Lung wet/dry (W/D) ratio in natriuretic peptide receptor-C wild-type (NPR-C+/+), heterozygote (NPR-C+/−), and knockout (NPR-C−/−) mice treated with intravenous infusion of saline (NaCl) or atrial natriuretic peptide (ANP) and oropharyngeal aspiration of NaCl or Pseudomonas aeruginosa (PA103). n = 4–6 mice for each condition and genotype. *P < 0.05.
In addition to measurements of lung wet-to-dry ratio, severity of lung injury was also assessed by changes in protein concentration in BALF. Protein concentration of BALF did not differ between NPR-C genotypes under control conditions (Fig. 3). Intravenous infusion of ANP had no effect on BALF protein in any of the NPR-C genotypes. Treatment with PA103 induced significant increases in BALF protein in all 3 NPR-C genotypes consistent with the occurrence of ALI, although the difference did not quite reach statistical significance in the NPRC knockout mice. ANP infusion prevented a significant rise in BALF protein in PA103-treated animals in all 3 NPR-C genotypes (Fig. 3).
Figure 3.
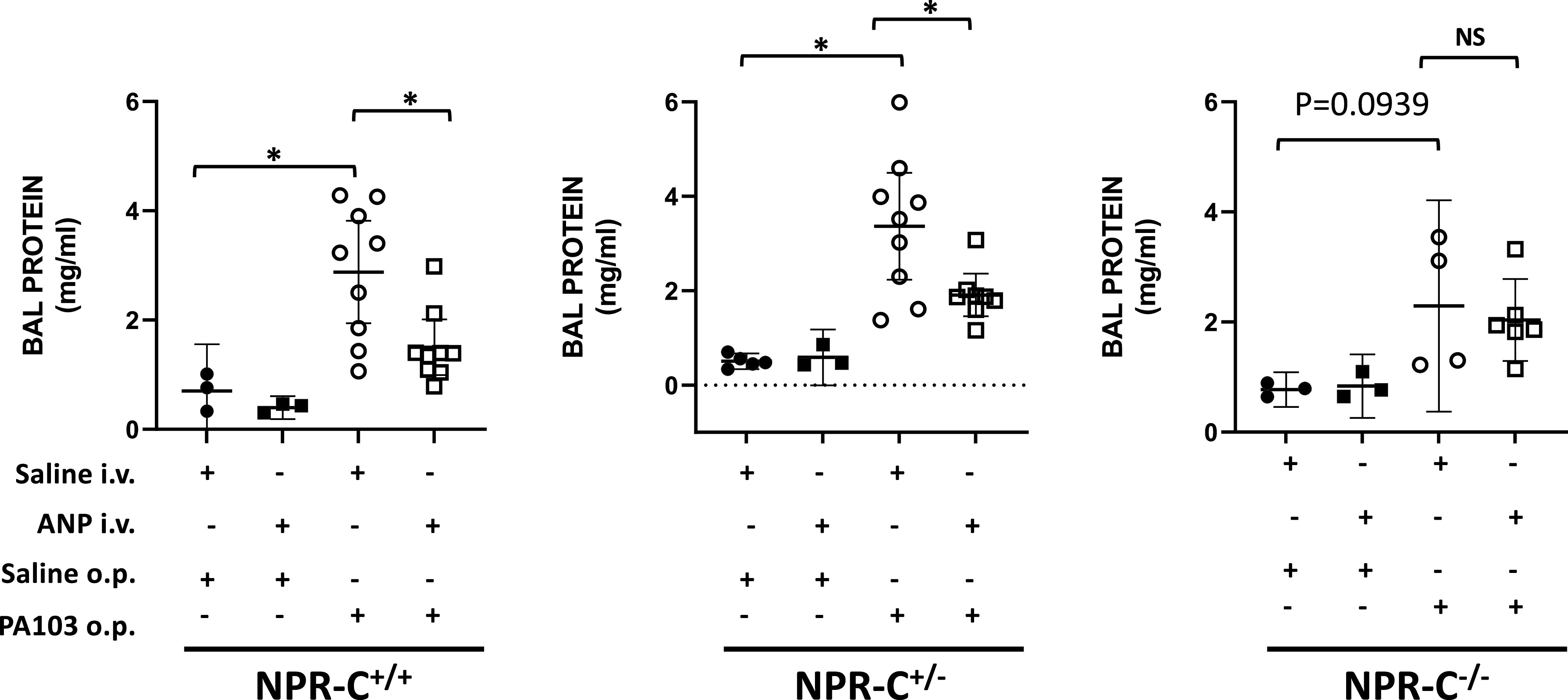
Protein concentration in bronchoalveolar lavage fluid (BALF) in natriuretic peptide receptor-C wild-type (NPR-C+/+), heterozygote (NPR-C+/−), and knockout (NPR-C−/−) mice treated with IV infusion of saline (NaCl) or atrial natriuretic peptide (ANP) and oropharyngeal aspiration of NaCl or Pseudomonas aeruginosa (PA103). n = 4–6 mice for each condition and genotype. *P < 0.05.
Effect of NPR-C and ANP on PA103-Induced Leukocyte Recruitment and Inflammatory Cytokines in Lungs
Although no difference in interstitial edema or alveolar barrier function were detected among NPR-C genotypes using lung W/D ratio and BALF protein concentration, additional experiments were done to examine the effect of NPR-C on lung inflammation by measuring differences in leukocyte populations in the blood, lung and alveolar compartments. The number of leukocytes and the percentage of leukocytes that were neutrophils in blood did not differ between NPR-C genotype and was not affected by saline infusion or aspiration of PA103 (Fig. 4, A and B). In wild-type mice, ANP infusion increased the percentage of circulating monocytes that were neutrophils in mice give saline aspiration, but not in mice given PA103. In NPR-C knockout mice, ANP infusion had no effect on the percentage of blood neutrophils in mice with saline- or PA103-aspiration (Fig. 4B). Conversely, ANP infusion increased the percentage of circulating monocytes that were Ly6Chi in NPR-C knockout mice given saline aspiration, but not PA103 and had no effect on the percentage of Ly6Chi in wild-type mice (Fig. 4C).
Figure 4.
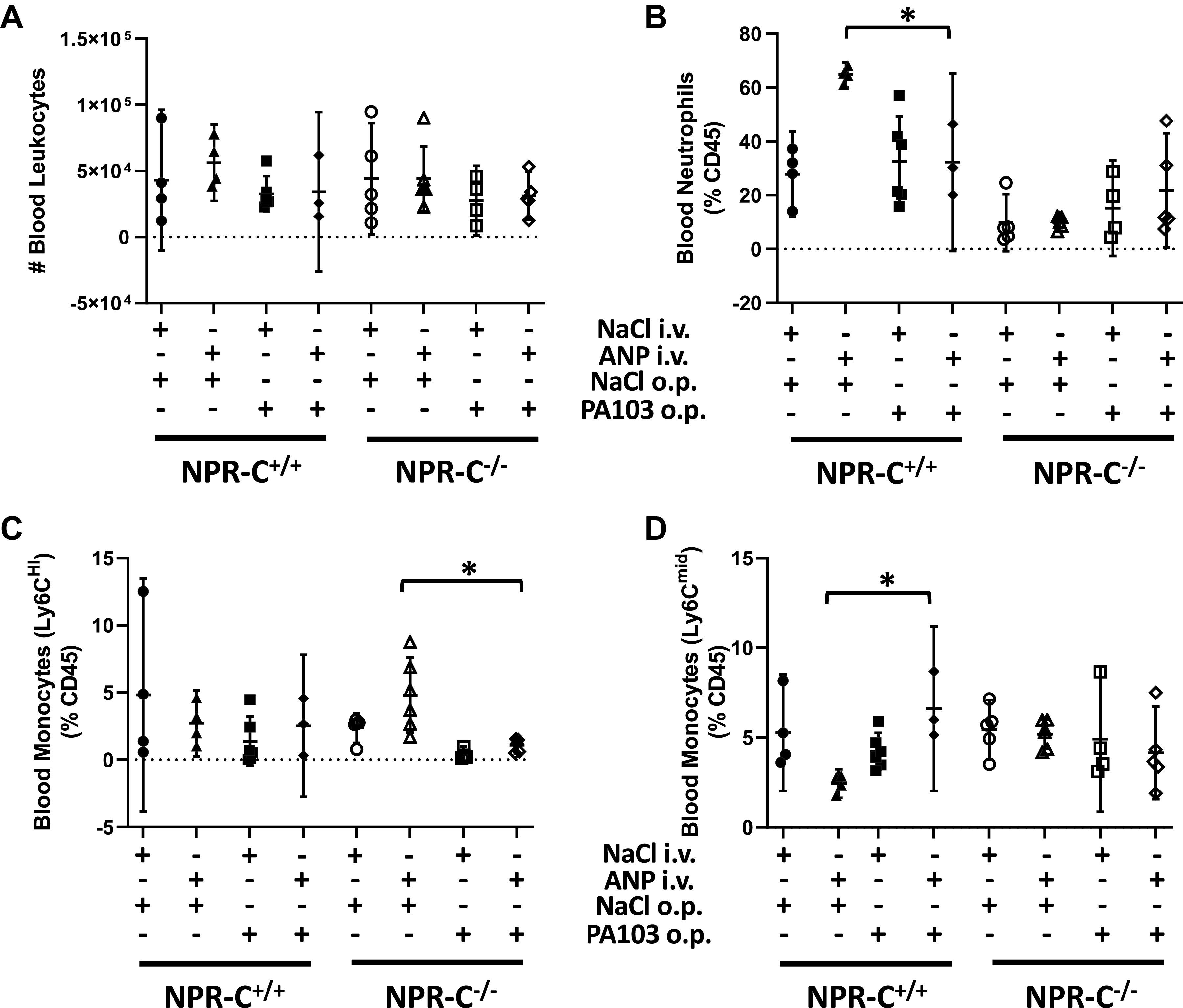
Total leukocyte counts (A) and percentage of neutrophils (B), Ly6Chi monocytes (C), and Ly6Cmid monocytes (D) in blood of natriuretic peptide receptor-C wild-type (NPR-C+/+) and knockout (NPR-C−/−) mice treated with infusion of saline followed by oropharyngeal aspiration of saline (NaCl/NaCl), infusion of saline and oropharyngeal aspiration of Pseudomonas aeruginosa (PA103; NaCl/PA103), infusion of atrial natriuretic peptide (ANP) followed by oropharyngeal aspiration of saline (ANP/NaCl), or infusion of ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103). n = 6 mice for each condition and genotype. *P < 0.05.
Total leukocyte counts from lung homogenates and the percentage of leukocytes that were Ly6Chi monocytes did not differ between by NPR-C genotypes and were not affected by ANP or PA103 (Fig. 5, A and C) although there was a strong trend toward an increase in total lung leukocytes levels in NPR-C knockout mice treated with PA103 that was not seen in wild-type mice (Fig. 5A). PA103 markedly increased the percentage of lung leukocytes that were neutrophils and decreased the percentage of leukocytes that were Ly6Cmid in NPR-C knockout mice, but not in wild-type mice. This effect was not altered by treatment with ANP (Fig. 5, B and D).
Figure 5.
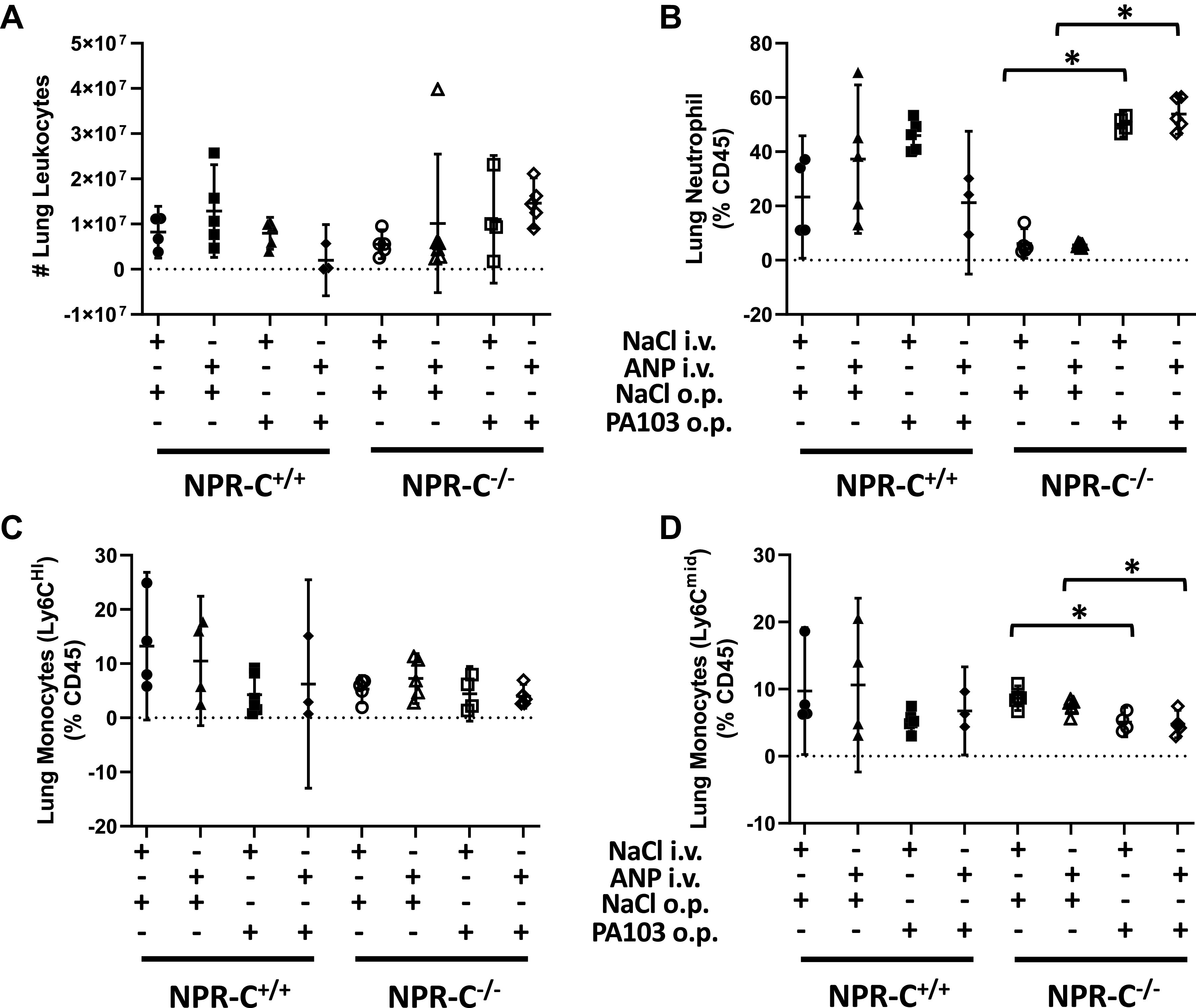
Total leukocyte counts (A) and percentage of neutrophils (B), Ly6Chi monocytes (C), and Ly6Cmid monocytes (D) in whole lung homogenates obtained from natriuretic peptide receptor-C wild-type (NPR-C+/+) and knockout (NPR-C−/−) mice treated with infusion of saline followed by oropharyngeal aspiration of saline (NaCl/NaCl), infusion of saline and oropharyngeal aspiration of Pseudomonas aeruginosa (PA103; NaCl/PA103), infusion of atrial natriuretic peptide (ANP) followed by oropharyngeal aspiration of saline (ANP/NaCl), or infusion of ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103). n = 6 mice for each condition and genotype. *P < 0.05.
Figure 6.
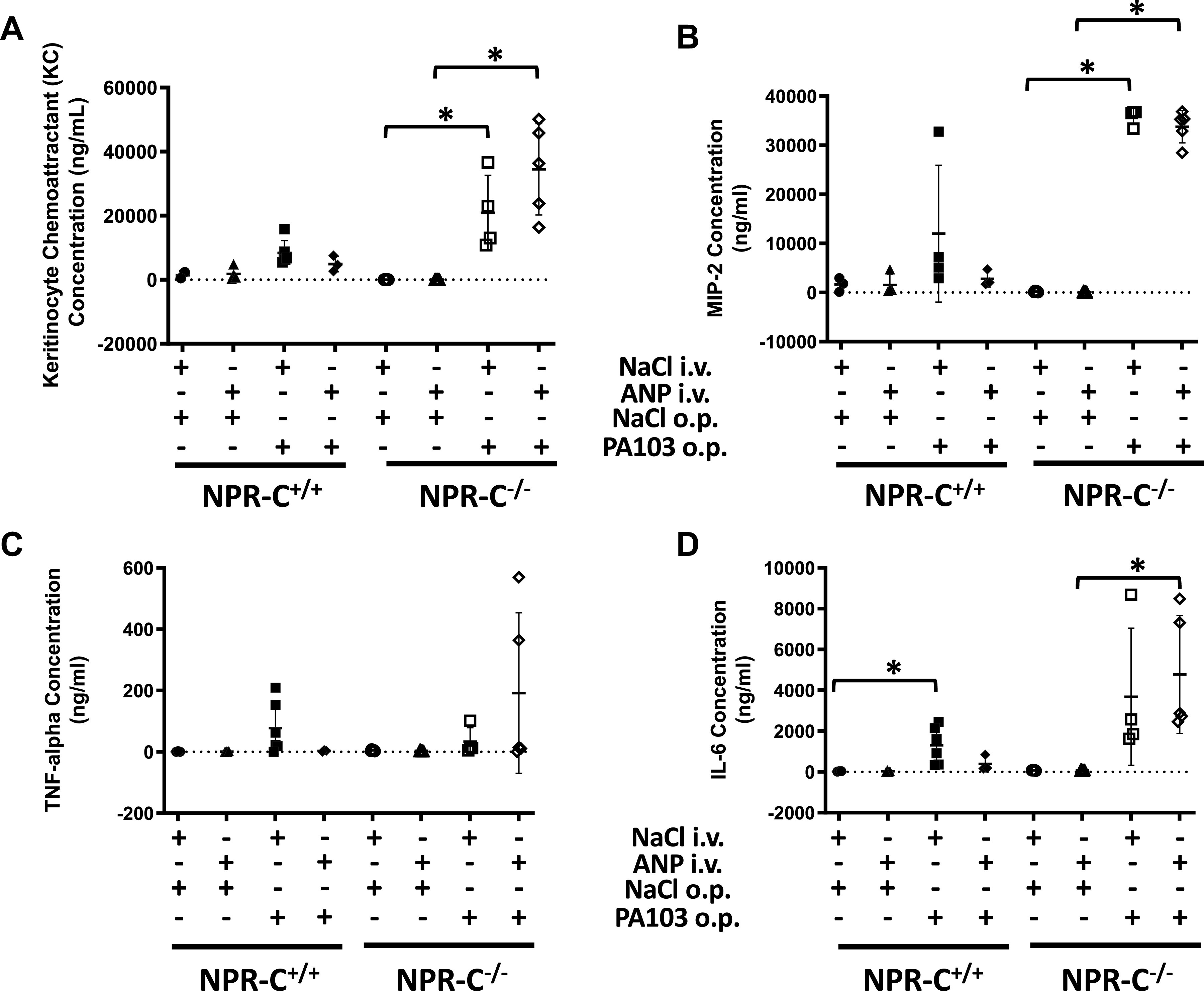
Concentration of CXCL1/KC (A), CXCL2/MIP2 (B), tumor necrosis factor-α (TNFα; C), and interleukin-6 (IL-6; D) in whole lung homogenates obtained from natriuretic peptide receptor-C wild-type (NPR-C+/+) and knockout (NPR-C−/−) mice treated with infusion of saline followed by oropharyngeal aspiration of saline (NaCl/NaCl), infusion of saline and oropharyngeal aspiration of Pseudomonas aeruginosa (PA103; NaCl/PA103), infusion of atrial natriuretic peptide (ANP) followed by oropharyngeal aspiration of saline (ANP/NaCl), or infusion of ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103). n = 6 mice for each condition and genotype. *P < 0.05.
The concentration of inflammatory cytokines in lung homogenates did not differ between by NPR-C genotypes and were not affected by infusion of ANP under baseline conditions (Fig. 6). In wild-type mice, PA103 tended to increase cytokine concentration (although the increase reached statistical significance only for IL-6) and the effect of PA103 on cytokine levels was prevented by ANP infusion (Fig. 6). In knockout mice, PA103 increased KC and MIP with a strong trend toward an increase in IL-6. The effect of PA103 on cytokine levels was not affected by ANP (Fig. 6). PA103-induced increases in KC and MIP-2 were several-fold greater in knockout than in wild-type mice.
In BALF, total leukocyte counts were greater in NPR-C knockout than in wild-type mice at baseline, but were not affected by ANP or PA103 (Fig. 7A). PA103 increased the percentage of BALF leukocytes that were neutrophils and Ly6Chi monocytes in both genotypes but the increase was only significant in NPR-C knockout mice. Treatment with ANP prevented the PA103-induced increase in Ly6Chi monocytes, but not the increase in neutrophils in NPR-C knockout mice (Fig. 7, B and C).
Figure 7.
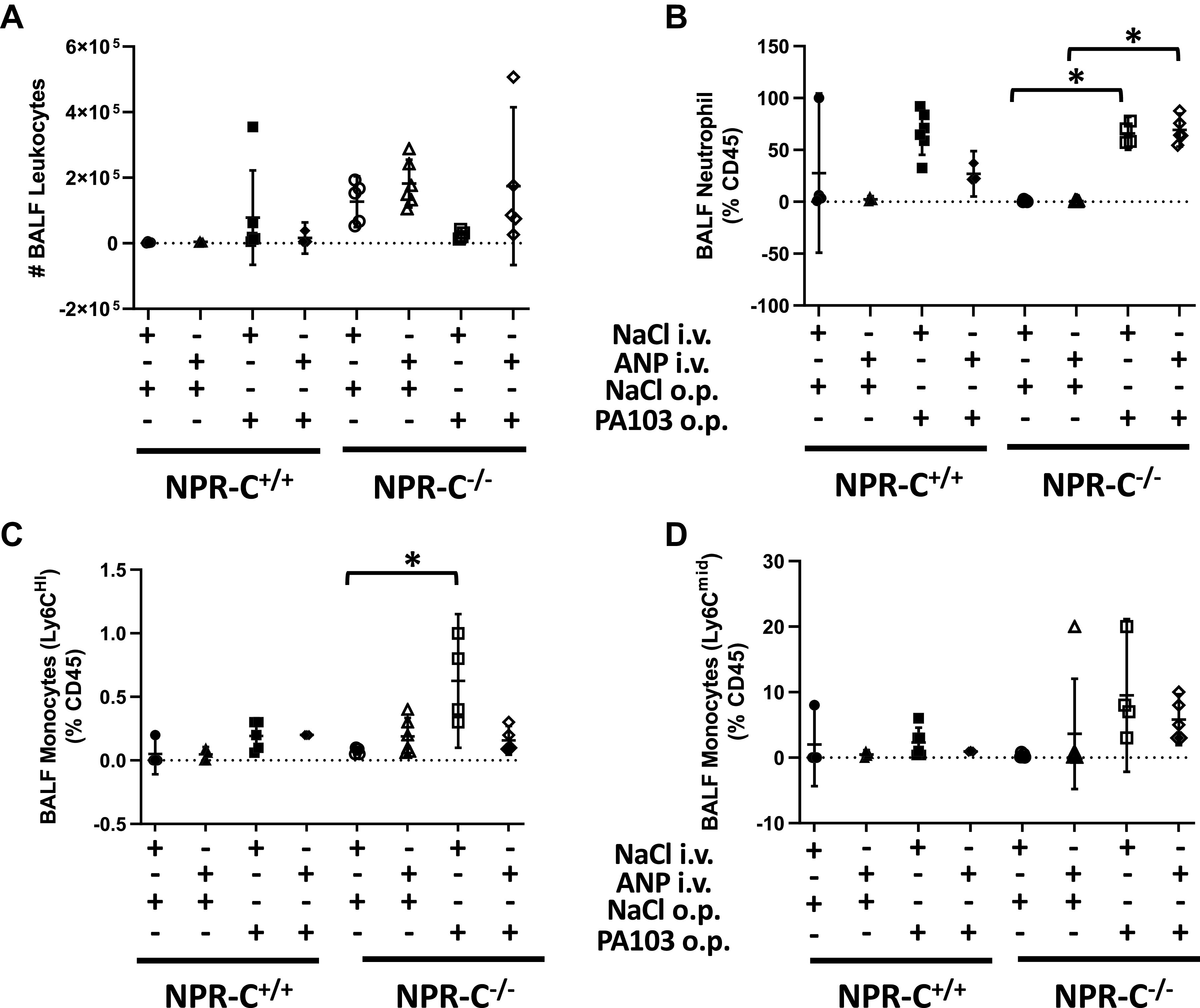
Total leukocyte counts (A) and percentage of neutrophils (B), Ly6Chi monocytes (C), and Ly6Cmid monocytes (D) in bronchoalveolar lavage fluid (BALF) obtained from natriuretic peptide receptor-C wild-type (NPR-C+/+) and knockout (NPR-C−/−) mice treated with infusion of saline followed by oropharyngeal aspiration of saline (NaCl/NaCl), infusion of saline and oropharyngeal aspiration of Pseudomonas aeruginosa (PA103; NaCl/PA103), infusion of atrial natriuretic peptide (ANP) followed by oropharyngeal aspiration of saline (ANP/NaCl), or infusion of ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103). n = 6 mice for each condition and genotype. *P < 0.05.
Cytokine levels in BALF did not differ between NPR-C genotypes at baseline and were not affected by treatment with ANP (Fig. 8). PA103-induced increase in KC and TNF-α in both genotypes and increased IL-6 in wild-type mice (Fig. 8). Administration of ANP completely blocked the PA103-induced increases in BALF cytokine levels for all four cytokines in wild-type mice, but had no effect in NPR-C knockout mice.
Figure 8.
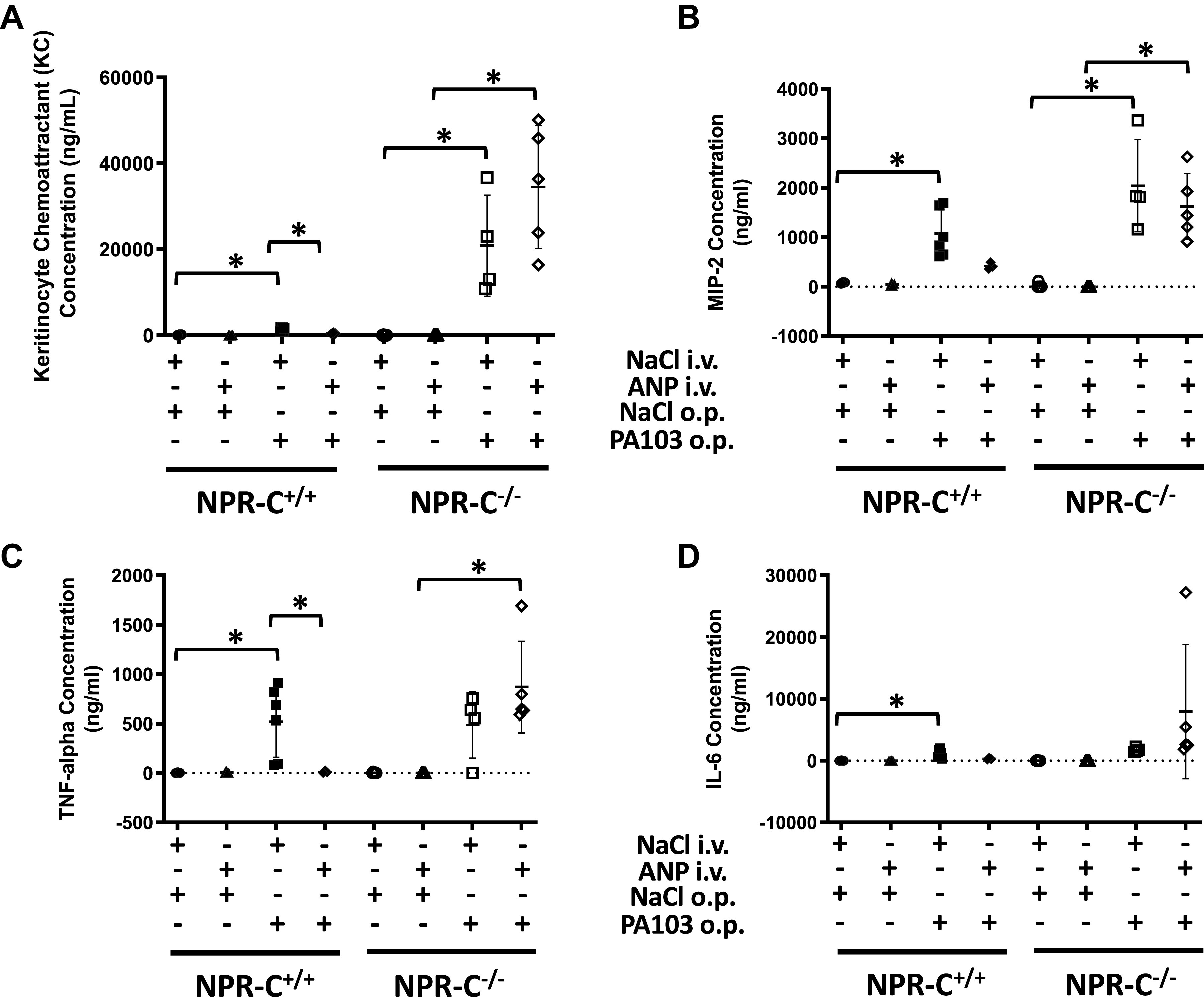
Concentration of CXCL1/KC (A), CXCL2/MIP2 (B), tumor necrosis factor-α (TNFα; C), and interleukin-6 (IL-6; D) in bronchoalveolar lavage fluid (BALF) obtained from natriuretic peptide receptor-C wild-type (NPR-C+/+) and knockout (NPR-C−/−) mice treated with infusion of saline followed by oropharyngeal aspiration of saline (NaCl/NaCl), infusion of saline and oropharyngeal aspiration of Pseudomonas aeruginosa (PA103; NaCl/PA103), infusion of atrial natriuretic peptide (ANP) followed by oropharyngeal aspiration of saline (ANP/NaCl), or infusion of ANP followed by oropharyngeal aspiration of PA103 (ANP/PA103). n = 6 mice for each condition and genotype. *P < 0.05.
DISCUSSION
ANP has been shown to attenuate increased permeability of lung vascular endothelial cells and protect against the development of acute lung injury in a variety of animal models, but the receptor that mediates this effect is not clear. In an earlier study (19), we found that acute lung injury induced by LPS was greater in mice with disrupted expression of the particulate guanylyl cyclase-liked receptor NPR-A than in wild-type mice, suggesting that NPR-A mediated the protective effects of endogenous ANP against lung injury in these mice. However, we also found that administration of exogenous ANP was nearly as effective at reducing lung injury in mice that were homozygous or heterozygous for a disrupted NPR-A gene as it was in attenuating lung injury in wild-type mice. We therefore hypothesized, that in the NPR-A knockout mice, ANP was acting via its other receptor, the non-guanylyl cyclase-linked receptor, NPR-C. In the present study, we examined the effects of acute lung injury and ANP administration in mice with genetically disrupted expression of NPR-C. Under baseline conditions, we found no evidence of increased pulmonary edema as assessed by differences in lung W/D weight ratio or BALF protein concentration between mice with 0, 1, or 2 copies of a functional NPR-C gene. Intratracheal administration of PA103 caused acute lung injury as assessed by increases in lung W/D weight and BALF protein concentration, but we saw no difference in the severity of PA103-induced lung injury between NPR-C genotypes. Intravenous infusion of ANP blunted PA103-induced increases in lung W/D weight ratio and BALF protein concentrations in all 3 genotypes and appeared to be equally effective at reducing the severity of lung injury in both wild-type and knockout mice. Thus, the ability of ANP to attenuate PA103-induced lung injury was not diminished by partial or absent expression of NPR-C.
The ability of ANP to attenuate PA103-induced lung injury in NPR-C heterozygote and KO mice argues against our primary hypothesis that the protective effects of ANP on lung injury are mediated via NPR-C. These findings along with our earlier studies (19) in the NPR-A mice suggest that neither NPR-A or NPR-C are solely responsible for the protective effects of ANP and that ANP may act via both receptors to achieve its beneficial effects. Although it is possible that ANP could be acting via NPR-B or another unknown receptor, this is unlikely due to the very low binding affinity of ANP for NPR-B and the lack of any other known binding sites (21). In our earlier study (19) expression of NPR-B or NPR-C in whole lung and in pulmonary microvascular endothelial cells were not increased in NPR-A knockout mice. Thus, it is unlikely that changes in expression of one receptor affects expression of the others.
Although we were unable to detect any difference in the effect of ANP administration on lung W/D weight or BALF protein concentration between NPR-C genotypes treated with PA103, NPR-C KO mice appeared to have a significantly greater degree of neutrophil recruitment to the lung and higher levels of lung inflammatory cytokines in response to PA103. Furthermore, ANP infusion reduced the PA103-induced increases in percentage of neutrophils and cytokine levels in the lung and BALF of wild-type mice, but had no effect in NPR-C knockout mice.
Whether or not the higher cytokine levels found in the lungs of the knockout mice is the cause or the result of greater neutrophil counts in the lung is uncertain. Although neutrophils are capable of expressing KC and MIP-2 (22), macrophages are the primary source of these cytokines that are potent chemotactic agents for polymorphonuclear (PMN) leukocytes. (23–25). In the present study, we did not see greater increases in lung Ly6Chi or LyC6mid monocyte in the lungs of NPR-C KO compared to WT mice. Thus, it appears that the higher cytokine levels in the KO mice is due to greater numbers of neutrophils or greater release of these cytokines from neutrophils.
The above findings suggest that NPR-C may play an important role in limiting recruitment of neutrophils to the lung or suppressing the release of inflammatory cytokines. ANP has been shown to reduce leukocyte/endothelial interactions. For example, Morikis et al. (26) demonstrated that ANP reduced accumulation of neutrophils in a mouse skin wound after treatment with thrombin (26). In that study, ANP was found to reduce the deformability of PMNs and thereby limit the area of adhesive contact with the endothelium, resulting in decreased rolling, adhesion, and transendothelial migration (26). Whether or not the effect of ANP on leukocyte recruitment is mediated by NPR-C is not known.
To our knowledge, the role of NPR-C in mediating cytokine levels in the lung during acute lung injury has not been studied. The natriuretic peptides have long been associated with immune-modulatory properties (27) and several recent publications speak to the therapeutic potential of ANP due to this effect (28–30). Although ANP has been shown to induce the production of reactive oxygen species (ROS) in polymorphonuclear (PMN) cells and stimulate their mobilization (31–33), most immunomodulatory effects of ANP have been described in macrophages. ANP has been shown to blunt LPS-induced increases in nitric oxide (NO) production and prostaglandin E2 (PGE2) release from macrophages and to decrease secretion of IL-6 in adipose-derived macrophages (34, 35). ANP has also been shown to inhibit TNFα production in macrophages by impeding the activation of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) (36, 37). The receptors that mediate these myriad effects of ANP on macrophage function have not been fully identified, but macrophages have been shown to express all 3 NPRs (38) and it appears that both NPR-A and NPR-C are involved. For example, the ability of ANP to blunt NO production and TNF-α secretion in macrophages has been attributed to NPR-A (35–37), whereas its effect on PGE2 release from murine macrophages is thought to occur via NPR-C (34).
The effect of ANP on modulating cytokine release and PMN homing to the lung in response to acute lung injury is unclear. Zhu et al. (39) found that ANP infusion blunted the increase in serum and lung levels of interleukin-1β (IL-1β), IL-6, interleukin-10 (IL-10), and TNF-α in rats with oleic acid-induced lung injury. The findings of the present study suggest that ANP may play an important role in limiting recruitment of neutrophils to the lung by suppressing secretion of KC, MIP-2, TNFα, and IL-6 and that this effect may be mediated via NPR-C. Although the present study was limited to the effect of ANP on acute lung injury, other investigators (4, 40) have found that C-type natriuretic peptide (CNP), which has vascular anti-inflammatory properties that are mediated via NPR-C (41–43), also inhibits the release of chemokines such as MCP-1 and IL-6 as well as S100A8, a member of the S100 family of Ca2+-binding proteins that has been shown to amplify macrophage production of proinflammatory cytokines (44). CNP has also been shown to limit leukocyte rolling in murine mesenteric postcapillary venules and inhibit platelet-leukocyte interactions via selective attenuation of leukocyte P-selectin expression (43). Thus, it is possible that the ability of NPR-C to limit cytokine production and neutrophil recruitment to the lung is due to its interactions with both ANP and CNP.
The mitigating effects of ANP on PA103-induced lung injury observed in the present study agree with other studies that found a protective effect of ANP on lung injury induced by thrombin receptor activator, LPS, Staphylococcus aureus, and oleic acid (19, 39, 45, 46). Determining how the different NPRs mediate the protective effects of ANP on lung edema formation is vital to advancing the natriuretic peptides as a potential therapeutic target for treating ARDS. The findings of the present study, refute the hypothesis that NPR-C is the primary receptor involved in controlling fluid exudation from the vascular space to the alveoli, but suggest that NPR-C has a significant effect on modulating cytokine release and recruitment of neutrophils to the lung during PA103-induced injury. In the present study, higher neutrophil counts and inflammatory cytokine levels in the NPR-C knockout mice did not translate into a higher degree of lung injury as assessed by accumulation of fluid in the lung or protein in the alveolar space, but other measures of lung injury such as ventilatory function, gas exchange, time to recovery, or survival were not assessed. It is possible that the mechanisms involved in pulmonary edema formation and inflammation are regulated by different NPRs and that both NPR-A and NPR-C play significant roles in limiting different types of lung injury.
Due to the limited treatment options available for ARDS and other types of acute lung injury, the protective effect of ANP offers a new potential therapeutic approach. Plasma ANP levels increase markedly in patients with septic shock (38, 47), but it is unclear if this response protects against worsening lung injury and if further increases in plasma ANP levels would be beneficial. Clinical studies with ANP have shown significant improvement in lung injury scores and gas exchange in patients with acute lung injury (48). However, ANP infusion has been associated with hypotension in clinical trials of heart failure, likely due to its effect on decreasing intravascular volume (49). Considering that the diuretic and vasorelaxant effects of ANP are mediated via NPR-A (17), the development of a selective NPR-C agonist could potentially provide the benefits of limiting recruitment of inflammatory cells and cytokine release during acute lung injury without affecting intravascular tone or volume.
In conclusion, data from the present study do not support the hypothesis that NPR-C mediates the protective effects of ANP on alveolar fluid accumulation that occurs during lung injury. However, functional NPR-C expression appears to play a significant role in limiting neutrophil recruitment to the lung and suppression of inflammatory cytokines. Whether or not this effect plays an important role in reducing lung injury will require further study, but data from the present study adds to the growing body of literature supporting a protective role of the natriuretic peptides and their receptors in acute lung injury and points to NPR-C as a potential therapeutic target for new treatments of ARDS.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This material is the result of work supported with resources and the use of facilities at the Providence VA Medical Center and supported with National Institutes of Health Grants R01 HL123965 (to J.R.K. and E.O.H.), P20 GM103652 (to E.O.H.), R03 AI130528 and R35 GM124911 (to C.T.L.), and T32 HL134625 (to B.G.).
DISCLOSURES
J.R.K. is a consultant for Palatin. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
E.O.H., C.T.L., and J.R.K. conceived and designed research; A.K., V.L., Z.S.W., B.G., and J.B. performed experiments; E.O.H., V.L., Z.S.W., B.G., J.B., C.T.L., and J.R.K. analyzed data; E.O.H., A.K., Z.S.W., B.G., C.T.L., and J.R.K. interpreted results of experiments; E.O.H., Z.S.W., and B.G. prepared figures; E.O.H., C.T.L., and J.R.K. drafted manuscript; E.O.H., Z.S.W., and J.R.K. edited and revised manuscript; E.O.H., A.K., V.L., Z.S.W., J.B., C.T.L., and J.R.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Huetran Duong for data collection and analysis and Jason Machan for assistance with statistical analysis of part of the data. Graphical abstract image created with BioRender and published with permission.
REFERENCES
- 1. Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R, Brochard L, Brower R, Esteban A, Gattinoni L, Rhodes A, Slutsky AS, Vincent JL, Rubenfeld GD, Thompson BT, Ranieri VM. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med 38: 1573–1582, 2012. [Erratum in Intensive Care Med 38: 1731–1732, 2012]. doi: 10.1007/s00134-012-2682-1. [DOI] [PubMed] [Google Scholar]
- 2. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3. Laffey JG, Bellani G, Pham T, Fan E, Madotto F, Bajwa EK, Brochard L, Clarkson K, Esteban A, Gattinoni L, van Haren F, Heunks LM, Kurahashi K, Laake JH, Larsson A, McAuley DF, McNamee L, Nin N, Qiu H, Ranieri M, Rubenfeld GD, Thompson BT, Wrigge H, Slutsky AS, Pesenti A; The LUNG SAFE Investigators and the ESICM Trials Group. Potentially modifiable factors contributing to outcome from acute respiratory distress syndrome: the LUNG SAFE study. Intensive Care Med 42: 1865–1876, 2016. doi: 10.1007/s00134-016-4571-5. [DOI] [PubMed] [Google Scholar]
- 4. Osawa H, Yamabe H, Kaizuka M, Tamura N, Tsunoda S, Baba Y, Shirato K, Tateyama F, Okumura K. C-Type natriuretic peptide inhibits proliferation and monocyte chemoattractant protein-1 secretion in cultured human mesangial cells. Nephron 86: 467–472, 2000. doi: 10.1159/000045836. [DOI] [PubMed] [Google Scholar]
- 5. Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit Care 24: 516, 2020. doi: 10.1186/s13054-020-03240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, Wu Y, Zhang L, Yu Z, Fang M, Yu T, Wang Y, Pan S, Zou X, Yuan S, Shang Y. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med 8: 475–481, 2020. [Erratum in Lancet Respir Med 8: e26, 2020]. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.REMAP-CAP Investigators; Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med 384: 1491–1502, 2021. doi: 10.1056/NEJMoa2100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.RECOVERY Collaborative Group; Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 384: 693–704, 2021. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6: 147–163, 2011. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM; Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shanley TP, Schmal H, Warner RL, Schmid E, Friedl HP, Ward PA. Requirement for C-X-C chemokines (macrophage inflammatory protein-2 and cytokine-induced neutrophil chemoattractant) in IgG immune complex-induced lung injury. J Immunol 158: 3439–3448, 1997. [PubMed] [Google Scholar]
- 12. Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 46: 566–572, 2012. doi: 10.1165/rcmb.2011-0392TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, Smithies O. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci USA 96: 7403–7408, 1999. doi: 10.1073/pnas.96.13.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol 6: 61, 2013. doi: 10.1186/1756-8722-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wijeyaratne CN, Moult PJ. The effect of alpha human atrial natriuretic peptide on plasma volume and vascular permeability in normotensive subjects. J Clin Endocrinol Metab 76: 343–346, 1993. doi: 10.1210/jcem.76.2.8432776. [DOI] [PubMed] [Google Scholar]
- 16. Clinkingbeard C, Sessions C, Shenker Y. The physiological role of atrial natriuretic hormone in the regulation of aldosterone and salt and water metabolism. J Clin Endocrinol Metab 70: 582–589, 1990. doi: 10.1210/jcem-70-3-582. [DOI] [PubMed] [Google Scholar]
- 17. Sabrane K, Kruse MN, Fabritz L, Zetsche B, Mitko D, Skryabin BV, Zwiener M, Baba HA, Yanagisawa M, Kuhn M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest 115: 1666–1674, 2005. doi: 10.1172/JCI23360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klinger JR, Warburton R, Carino GP, Murray J, Murphy C, Napier M, Harrington EO. Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp Cell Res 312: 401–410, 2006. doi: 10.1016/j.yexcr.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 19. Klinger JR, Tsai SW, Green S, Grinnell KL, Machan JT, Harrington EO. Atrial natriuretic peptide attenuates agonist-induced pulmonary edema in mice with targeted disruption of the gene for natriuretic peptide receptor-A. J Appl Physiol (1985) 114: 307–315, 2013. doi: 10.1152/japplphysiol.01249.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anand-Srivastava MB. Natriuretic peptide receptor-C signaling and regulation. Peptides 26: 1044–1059, 2005. doi: 10.1016/j.peptides.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 21. Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H, Goeddel DV. Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science 252: 120–123, 1991. doi: 10.1126/science.1672777. [DOI] [PubMed] [Google Scholar]
- 22. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol 5: 508, 2014. doi: 10.3389/fimmu.2014.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moser B, Clark-Lewis I, Zwahlen R, Baggiolini M. Neutrophil-activating properties of the melanoma growth-stimulatory activity. J Exp Med 171: 1797–1802, 1990. doi: 10.1084/jem.171.5.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schumacher C, Clark-Lewis I, Baggiolini M, Moser B. High- and low-affinity binding of GRO alpha and neutrophil-activating peptide 2 to interleukin 8 receptors on human neutrophils. Proc Natl Acad Sci USA 89: 10542–10546, 1992. doi: 10.1073/pnas.89.21.10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci USA 86: 612–616, 1989. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morikis VA, Radecke C, Jiang Y, Heinrich V, Curry FR, Simon SI. Atrial natriuretic peptide down-regulates neutrophil recruitment on inflamed endothelium by reducing cell deformability and resistance to detachment force. Biorheology 52: 447–463, 2015. doi: 10.3233/BIR-15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vollmar AM. The role of atrial natriuretic peptide in the immune system. Peptides 26: 1086–1094, 2005. doi: 10.1016/j.peptides.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 28. Bae CR, Hino J, Hosoda H, Miyazato M, Kangawa K. C-type natriuretic peptide (CNP) in endothelial cells attenuates hepatic fibrosis and inflammation in non-alcoholic steatohepatitis. Life Sci 209: 349–356, 2018. doi: 10.1016/j.lfs.2018.08.031. [DOI] [PubMed] [Google Scholar]
- 29. Liu S, Chirkov YY, Horowitz JD. Neutrophil-initiated myocardial inflammation and its modulation by B-type natriuretic peptide: a potential therapeutic target. Int J Mol Sci 20: 129, 2018. doi: 10.3390/ijms20010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Najenson AC, Courreges AP, Perazzo JC, Rubio MF, Vatta MS, Bianciotti LG. Atrial natriuretic peptide reduces inflammation and enhances apoptosis in rat acute pancreatitis. Acta Physiol 222: e12992, 2018. doi: 10.1111/apha.12992. [DOI] [PubMed] [Google Scholar]
- 31. Biselli R, Farrace S, De Simone C, Fattorossi A. Potentiation of human polymorphonuclear leukocyte activation by atrial natriuretic peptide. Inhibitory effect of carnitine congeners. Inflammation 20: 33–42, 1996. doi: 10.1007/BF01487743. [DOI] [PubMed] [Google Scholar]
- 32. Vollmar AM, Förster R, Schulz R. Effects of atrial natriuretic peptide on phagocytosis and respiratory burst in murine macrophages. Eur J Pharmacol 319: 279–285, 1997. doi: 10.1016/s0014-2999(96)00859-x. [DOI] [PubMed] [Google Scholar]
- 33. Wiedermann CJ, Niedermühlbichler M, Braunsteiner H, Widermann CJ. Priming of polymorphonuclear neutrophils by atrial natriuretic peptide in vitro. J Clin Invest 89: 1580–1586, 1992. doi: 10.1172/JCI115752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiemer AK, Lehner MD, Hartung T, Vollmar AM. Inhibition of cyclooxygenase-2 by natriuretic peptides. Endocrinology 143: 846–852, 2002. doi: 10.1210/endo.143.3.8680. [DOI] [PubMed] [Google Scholar]
- 35. Kiemer AK, Vollmar AM. Autocrine regulation of inducible nitric-oxide synthase in macrophages by atrial natriuretic peptide. J Biol Chem 273: 13444–13451, 1998. doi: 10.1074/jbc.273.22.13444. [DOI] [PubMed] [Google Scholar]
- 36. Kiemer AK, Hartung T, Vollmar AM. cGMP-mediated inhibition of TNF-alpha production by the atrial natriuretic peptide in murine macrophages. J Immunol 165: 175–181, 2000. doi: 10.4049/jimmunol.165.1.175. [DOI] [PubMed] [Google Scholar]
- 37. Tsukagoshi H, Shimizu Y, Kawata T, Hisada T, Shimizu Y, Iwamae S, Ishizuka T, Iizuka K, Dobashi K, Mori M. Atrial natriuretic peptide inhibits tumor necrosis factor-alpha production by interferon-gamma-activated macrophages via suppression of p38 mitogen-activated protein kinase and nuclear factor-kappa B activation. Regul Pept 99: 21–29, 2001. doi: 10.1016/s0167-0115(01)00218-x. [DOI] [PubMed] [Google Scholar]
- 38. Beishuizen A, Götz JM, Kip L, Haanen C, Vermes I. Elevated plasma levels of endothelin are associated with the severity of sepsis and presence of shock in contrast to the levels of atrial natriuretic peptide. Mediators Inflamm 1: 419–423, 1992. doi: 10.1155/S0962935192000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu YB, Zhang YB, Liu DH, Li XF, Liu AJ, Fan XM, Qiao CH, Ling F, Liu YL. Atrial natriuretic peptide attenuates inflammatory responses on oleic acid-induced acute lung injury model in rats. Chin Med J (Engl) 126: 747–750, 2013. [PubMed] [Google Scholar]
- 40. Kimura T, Nojiri T, Hosoda H, Ishikane S, Shintani Y, Inoue M, Miyazato M, Okumura M, Kangawa K. C-type natriuretic peptide attenuates lipopolysaccharide-induced acute lung injury in mice. J Surg Res 194: 631–637, 2015. doi: 10.1016/j.jss.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 41. Blier AS, Veron W, Bazire A, Gerault E, Taupin L, Vieillard J, Rehel K, Dufour A, Le Derf F, Orange N, Hulen C, Feuilloley MGJ, Lesouhaitier O. C-type natriuretic peptide modulates quorum sensing molecule and toxin production in Pseudomonas aeruginosa. Microbiology (Reading) 157: 1929–1944, 2011. doi: 10.1099/mic.0.046755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khambata RS, Panayiotou CM, Hobbs AJ. Natriuretic peptide receptor-3 underpins the disparate regulation of endothelial and vascular smooth muscle cell proliferation by C-type natriuretic peptide. Br J Pharmacol 164: 584–597, 2011. doi: 10.1111/j.1476-5381.2011.01400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scotland RS, Cohen M, Foster P, Lovell M, Mathur A, Ahluwalia A, Hobbs AJ. C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc Natl Acad Sci USA 102: 14452–14457, 2005. doi: 10.1073/pnas.0504961102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, Chazin WJ, Nakatani Y, Yui S, Makino H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther 8: R69, 2006. doi: 10.1186/ar1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Birukova AA, Xing J, Fu P, Yakubov B, Dubrovskyi O, Fortune JA, Klibanov AM, Birukov KG. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol 299: L652–663, 2010. doi: 10.1152/ajplung.00202.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xing J, Moldobaeva N, Birukova AA. Atrial natriuretic peptide protects against Staphylococcus aureus-induced lung injury and endothelial barrier dysfunction. J Appl Physiol (1985) 110: 213–224, 2011. doi: 10.1152/japplphysiol.00284.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hartemink KJ, Groeneveld AB, de Groot MC, Strack van Schijndel RJ, van Kamp G, Thijs LG. Alpha-atrial natriuretic peptide, cyclic guanosine monophosphate, and endothelin in plasma as markers of myocardial depression in human septic shock. Crit Care Med 29: 80–87, 2001. doi: 10.1097/00003246-200101000-00019. [DOI] [PubMed] [Google Scholar]
- 48. Mitaka C, Hirata Y, Nagura T, Tsunoda Y, Amaha K. Beneficial effect of atrial natriuretic peptide on pulmonary gas exchange in patients with acute lung injury. Chest 114: 223–228, 1998. doi: 10.1378/chest.114.1.223. [DOI] [PubMed] [Google Scholar]
- 49. Fifer MA, Molina CR, Quiroz AC, Giles TD, Herrmann HC, De Scheerder IR, Clement DL, Kubo S, Cody RJ, Cohn JN. Hemodynamic and renal effects of atrial natriuretic peptide in congestive heart failure. Am J Cardiol 65: 211–216, 1990. doi: 10.1016/0002-9149(90)90087-h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.