

Abstract
Many anticancer therapies (CTx) have cardiotoxic side effects that limit their therapeutic potential and cause long-term cardiovascular complications in cancer survivors. This has given rise to the field of cardio-oncology, which recognizes the need for basic, translational, and clinical research focused on understanding the complex signaling events that drive CTx-induced cardiovascular toxicity. Several CTx agents cause mitochondrial damage in the form of mitochondrial DNA deletions, mutations, and suppression of respiratory function and ATP production. In this review, we provide a brief overview of the cardiovascular complications of clinically used CTx agents and discuss current knowledge of local and systemic secondary signaling events that arise in response to mitochondrial stress/damage. Mitochondrial oxidative stress has long been recognized as a contributor to CTx-induced cardiotoxicity; thus, we focus on emerging roles for mitochondria in epigenetic regulation, innate immunity, and signaling via noncoding RNAs and mitochondrial hormones. Because data exploring mitochondrial secondary signaling in the context of cardio-oncology are limited, we also draw upon clinical and preclinical studies, which have examined these pathways in other relevant pathologies.
Keywords: cardio-oncology, cardiotoxicity, chemotherapy, DAMPs, mitochondria
INTRODUCTION
Cardiovascular disease (CVD) and cancer remain the leading causes of global morbidity and mortality, and their costs continue to rise (1). By 2030, the annual costs of CVD and cancer have been projected to rise to more than $1 trillion and $8.3 trillion, respectively (2). Although underlying etiologies of the two pathologies differ, CVD and cancer are linked in a bidirectional, causal relationship whereby each disease increases the risk of the other (3). Furthermore, some cancer treatments, such as antibiotic anthracyclines and anti-HER2 therapies, have well-known cardiotoxic effects that limit their clinical utility (4, 5).
The efficacy of anticancer therapy (CTx) is improving; thus, long-term survival of patients with cancer is at an all-time high. With longer survival, adverse cardiovascular side effects of CTx are emerging as a significant contributor to mortality and morbidity. In patients with breast cancer, cardiovascular complications now represent the leading cause of death outside of cancer reoccurrence (6, 7). Although basic cellular mechanisms that lead to CTx-induced cardiovascular toxicity have been studied for some time, most research has focused on primary damage to the heart itself. Few studies have investigated systemic factors, which may indirectly promote cardiovascular toxicity. Despite the prominent role of mitochondrial function in cardiovascular physiology, mitochondrial defects and associated signaling have only recently been explored in the context of cardio-oncology.
In this review, we highlight the contributions of emerging mitochondrial signaling mechanisms to cardiovascular complications of anticancer therapies. We briefly describe pharmacological mechanisms and cardiotoxic side effects of antineoplastic drugs, then discuss molecular and/or cellular mechanisms leading to cardiotoxicity. We focus on secondary signaling events downstream of mitochondrial damage, including changes in epigenetic signaling, innate immunity, and circulating factors such as mitochondrial nucleic acids and mitochondrial hormones.
MITOCHONDRIA AS MEDIATORS OF CARDIOTOXICITY
Mitochondria play crucial roles in energy metabolism, signaling, and cell death. Cardiomyocytes rely heavily on mitochondrial oxidative phosphorylation to meet the high energy demands of initiating contractions and maintaining ion homeostasis in the beating heart. Cardiomyocytes also have relatively low endogenous antioxidant defenses; thus, cardiomyocytes are particularly susceptible to mitochondrial damage and dysfunction. In addition to generating ATP, mitochondria are key signaling organelles that sense and respond to a variety of stressors, such as hypoxia and DNA damage. Mitochondria are major sources of reactive oxygen species (ROS), which serve important functions in signaling cascades. In the vasculature, mitochondrial ROS and mitochondrial Ca2+ dynamics regulate vascular tone and atherosclerotic processes. Mitochondria also monitor a variety of signals, such as growth factors and oxidative stress, which can promote apoptosis mediated by opening of the mitochondrial permeability transition pore (mPTP). Despite quality control processes, mitochondrial DNA (mtDNA) and proteins are susceptible to damage, and mitochondrial dysfunction has been implicated in a host of cardiovascular pathologies.
Emerging evidence suggests a role of mitochondrial damage in CTx-induced cardiotoxicity, which includes both local, intracellular effects (e.g., altered cellular metabolism and epigenetic signaling) and systemic signaling (e.g., inflammation and mitochondrial hormones). Several CTx agents promote the production of ROS, which can arise directly from redox cycling of chemotherapeutic drugs themselves, such as the formation of free radicals from anthracycline-iron complexes and semiquinone intermediates (8, 9), or indirectly through mechanisms that promote mitochondrial ROS production or suppress antioxidant mechanisms, such as mtDNA damage. Although oxidative stress has received much attention as a mechanism of CTx-induced cardiotoxicity, other cellular pathways are increasingly being investigated (10), including those involving complex mitochondrial signaling cascades (11, 12). Mitochondria are also involved in CTx-induced cardiotoxicity through their role in intrinsic apoptotic and necrotic cell pathways (12). Moreover, the influence of mitochondria on epigenetic signaling and innate immunity has recently emerged as potential contributors to adverse cardiovascular outcomes related to CTx. Thus, it is becoming clearer that mitochondrial dysfunction can contribute to cardiotoxicity through various pathways that go beyond bioenergetics and apoptosis, although much remains to be explored in the context of CTx.
CARDIOVASCULAR TOXICITY OF COMMON ANTICANCER THERAPIES
The clinical cardiovascular complications of several classes of CTx are well established, though the underlying mechanisms are incompletely understood. Approaches to combat cancer range widely and include classic, cytotoxic chemotherapies, targeted molecular therapies, and immunotherapies, among others. Although the specific mechanisms of cardiotoxicity vary between treatments, many forms of CTx have direct and indirect effects on mitochondria that impact cardiovascular health. Some CTx agents directly modulate mitochondrial function or morphology, which may impair mitochondrial respiration, promote production of mitochondrial ROS, and/or trigger mitochondria-driven apoptosis. Several agents also evoke mtDNA damage, which in turn alters mitochondrial function and promotes systemic secondary signaling mechanisms that provoke cardiovascular inflammation. Though many cardiotoxic mechanisms are treatment specific, other pathways are more generalizable. For example, tumor cell death, which occurs with most forms of CTx, promotes the release of mitochondrial fragments and cellular debris capable of eliciting cardiovascular inflammation.
Primary damage to cardiomyocytes fails to explain some of the clinical phenotypes observed in cancer survivors (e.g., increased risk of coronary artery disease and hypertension); thus, vascular toxicity has been increasingly recognized as a potential contributor to long-term adverse cardiovascular outcomes. Several classes of CTx impact endothelial control of vasomotor tone (13–15), which has implications for regulation of tissue blood flow to vital organs including the heart, brain, and kidney [for review, see Terwoord et al. (16)]. The heart relies heavily on changes in microvascular tone to ensure blood flow is adequate to meet cardiac metabolic demand, and poor microvascular function is the among the best predictors of major adverse cardiovascular events (MACE; 17). Recent evidence suggests that vasodilation mediated by endothelial production of nitric oxide (NO) is severely impaired in patients after neoadjuvant treatment with docetaxel, doxorubicin (DOX), and cyclophosphamide (15). The authors demonstrate that endothelial NO synthase (eNOS) inhibition via phosphorylation of threonine 495 augments vascular ROS production and increases the expression of the NADPH oxidases NOX2 and NOX4. In the human coronary microcirculation, NO suppresses mitochondrial ROS (18); thus, CTx-induced inhibition of NO production may unleash mitochondrial ROS production. Moreover, ROS such as superoxide scavenges NO and can cause uncoupling of eNOS promoting ROS-induced ROS release to futher reduce NO levels. In addition, high levels of ROS induce oxidative damage to mitochondrial proteins and DNA, which further exacerbates mitochondrial ROS production. In sum, CTx may promote ROS-induced ROS production in the vasculature involving mitochondrial ROS production and uncoupling of eNOS. The resulting vascular dysregulation has direct implications for cardiac blood flow in addition to altering paracrine signaling between the heart and endothelium. Some CTx agents (e.g., anthracyclines, cyclophosphamide, carmustine, and carboplatin) also modify membrane permeability and disrupt endothelial barrier function (19–21). This causes vascular damage that in turn enhances the synthesis of inflammatory cytokines, including TNF-α, IL-1β, and IL-6 (19), which are similar to the inflammatory pathways implicated in the atherosclerosis (22). Disrupted endothelial integrity also permits augmented extravasation of CTx drugs (20), which increases the exposure of underlying tissues (e.g., cardiomyocytes) to CTx agents. Together, these complex vascular effects may contribute to adverse cardiovascular outcomes associated with CTx exposure.
In the following section, we summarize key cardiotoxic mechanisms associated with several classes of CTx. The mechanisms underlying CTx-induced cardiotoxicity are complex and multifactorial, and a detailed description is beyond the scope of this review. Thus, we provide a general overview of major known mechanisms associated with cardiotoxicity and highlight mitochondrial involvement. Table 1 summarizes the mitochondrial pathophysiological signaling pathways and associated cardiotoxic outcomes for selected CTx classes. Figure 1 is a visual illustration of pathways to cardiotoxicity using examples summarized in Table 1.
Table 1.
Sample studies highlighting mitochondrial signaling pathways and anticancer therapy-induced cardiotoxicity
| Sample Studies | Antineoplastic Drugs | Mitochondrial Signaling Pathways | Cardiotoxic Outcomes |
|---|---|---|---|
| Montaigne et al. (12) | Anthracyclines and Anticancer-targeted therapies, i.e., tyrosine kinase inhibitors (TKI) | Increased mitochondrial oxidative stress and disruption intracellular Ca2+ levels leading to increase in mitochondrial Ca2+ levels The resulting Ca2+ overload pathogenic metabolic pathways lead to mitochondrial ROS production (23) | |
| Mitochondrial swelling and increased permeability of its outer membrane to apoptotic factors such as cytochrome-c | Starting with left ventricular dysfunction, multiple MACE can occur | ||
| Modulation of mitochondrial membrane permeability and other transcriptional factors that influence cell fate | |||
| Sawyer et al. (24) | Anthracyclines | DNA damage through suppressing expression and/or activity GATA4 | Disruption of cardiac sarcomeric structure starting with degradation of the titin protein, which is involved in the pathogenesis of progressive ventricular dysfunction leading to other MACE |
| Sahu et al. (19) | Alkylating agents | Vascular damage and alteration of blood-brain barrier (BBB) | Inducing inflammation and creating a cardiotoxic environment |
| Cyclophosphamide, carmustine, and carboplatin | |||
| Gorini et al. (25) | Anthracyclines (doxorubicin) | Binding to phospholipid cardiolipin in the inner mitochondrial membrane and disruption of the electron transport chain (ETC) | |
| Mitochondrial respiratory capacity impairment | Increase in ROS production, excessive oxidative stress, cell damage, cardiac muscle, and cellular membrane damage | ||
| Reduced cellular antioxidative defense [reduction of glutathione (GSH), superoxide dismutase (SOD), and catalase content or activity] leading to prolongation and/or stabilization of mitochondrial damage | |||
| Mehrotra et al. (26) | BH3 mimetics | Disruption of mitochondrial morphology and dynamics | Cardiomyopathy |
| Rasmussen et al. (27) | BH3 mimetics | Disruption of mitochondrial dynamics | Loss of viability and functionality of human cardiomyocytes |
| Manouchehri et al. (28) | Nilotinib, an oral TKI | Upregulates proatherogenic adhesion proteins on human endothelial cells | Promotion of vascular events |
| Rodríguez-Hernández et al. (29) | Ponatinib, sorafenib, and regorafenib (TKIs) | Induced mitochondrial dysfunction, uncoupling components of electron transport chain (including mitochondrial Ca2+ overload) and promoted ROS generation | General cardiovascular toxicity |
| Dempsey et al. (5) | Trastuzumab, an anti-HER2 drug | Blocking the function of neuregulin | |
| Promotion of damaging effects of oxidative stress | Myocytes attrition over time to heart failure | ||
| DNA breakage and induction of mitochondrial apoptosis | |||
| Oun et al. (30) | Cisplatin (a platinum containing agent) | Not fully understood. Either as a secondary effect from nephrotoxicity or as a direct ROS attack the heart | Arrhythmias |
| Cardiac ischemia, diastolic disturbances myocardial infarction angina | |||
| Pericarditis | |||
| Thromboembolic events | |||
| Chronic heart failure | |||
| Yuan et al. (31) | 5-fluorouracil (antimetabolite) | Disruption of RNA synthesis and inhibition of thymidylate synthase and incorporates its metabolites into RNA and DNA Vascular dysfunction with microthrombi formation |
Chest pain related to coronary vasospasm, |
| Dilated cardiomyopathy, | |||
| Vascular dysfunction with microthrombi formation | Ventricular arrhythmia, Sudden cardiac death |
MACE, major adverse cardiovascular events; ROS, reactive oxygen species.
Figure 1.
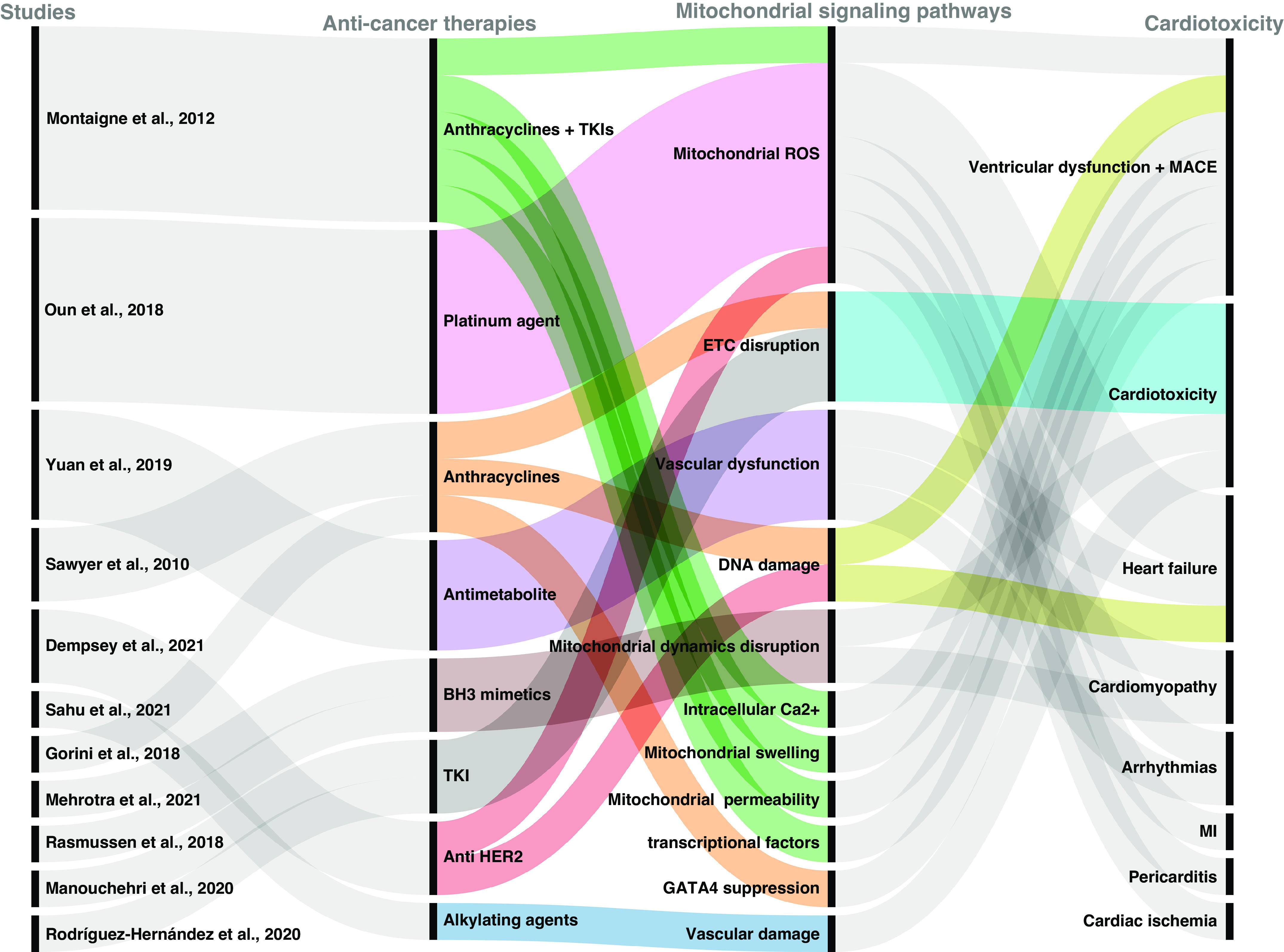
Graphical illustration of selected anticancer therapy (CTx) drugs illustrating their mitochondrial pathophysiological signaling pathways and some of the associated cardiotoxic outcomes. CTx agents work through different mechanisms and induce mitochondrial dysfunction through various pathophysiological pathways. This is graphical illustration of complex pathogenic pathways using selected examples from literature identified in our article. It is not meant to be an exhaustive depiction of all existing pathways but rather presents some key examples. Image created with Rawgraphs.io and published with permission. ETC, electron transport chain; MACE, major adverse cardiovascular events; ROS, reactive oxygen species; TKI, tyrosine kinase inhibitor.
Anthracyclines
Anthracycline antibiotics, including doxorubicin, epirubicin, and daunorubicin, are among the most effective treatments for many solid tumors, leukemias, and lymphomas. Anthracyclines intercalate into nucleic acids and inhibit the activity of topoisomerases, which disrupts DNA replication and translation. Although interfering with DNA is central to their cytotoxic and antitumor effects, anthracyclines also readily undergo redox reactions to produce free radicals (32), including nuclear and mtDNA damage. In some cells, these events can trigger apoptosis (33, 34) or cellular senescence (35), both of which can impair physiological processes and lead to organ failure (33, 34). Although anthracyclines are highly effective at combatting tumor growth, anthracycline therapy leads to irreversible, dose-dependent cardiotoxicity, which is the primary challenge that limits the cumulative dose patients receive.
Although many scholars have implicated anthracycline-induced mitochondrial ROS in cardiomyocyte death, other novel mitochondrial signaling pathways are also involved in anthracycline cardiotoxicity (11). For example, oxidative stress induced by anthracyclines also damages mtDNA (36, 37), which can further exacerbate mitochondrial ROS production and promote innate immune activation and inflammation by the release of mtDNA (discussed further in local mitochondrial signaling and systemic mitochondrial signaling below; 12). Mounting evidence suggests anthracyclines both promote and inhibit autophagic processes in a time-dependent manner (38, 39) and manipulating autophagy can ameliorate cardiotoxicity in preclinical models (40–42). These effects appear to be related to autophagic regulation of mitochondrial dynamics (i.e., fission and fusion) and respiratory function (for review, see Ref. 43). Anthracyclines also modulate mitochondrial membrane permeability and transcriptional factors that influence cell fate (12). Moreover, anthracyclines can modulate intracellular Ca2+ levels, including in the mitochondria (23). Although physiological levels of mitochondrial Ca2+ stimulate metabolic rate by increasing activity of tricarboxylic acid (TCA) cycle dehydrogenases and ATP output (44, 45), mitochondrial Ca2+ overload leads to the opening of the mPTP, release of cytochrome-c, and apoptosis (for a recent review, see Ref. 46). Ca2+-calmodulin can also promote ROS production through activation of protein kinases (12, 23), which in turn promotes peroxidation of cardiolipin, a major lipid component of mitochondrial membranes (47). Finally, mitochondrial membrane structural changes can lead to cardiomyocyte dysfunction and death, and consequently, contribute to development of MACE (12).
BH3 Mimetics
Intrinsic apoptosis is regulated by the B-cell lymphoma 2 (BCL-2) apoptotic pathway. The BCL-2 family comprises three groups of proteins regulated by different BCL-2 homology (BH) regions: prosurvival BCL-2-like proteins, proapoptotic BH3-only proteins, and proapoptotic Bcl-2-like protein 4 (BAX)/BRI1-associated receptor kinase 1 (BAK) proteins. BH3-only proteins bind to and inactive prosurvival BCL-2-like proteins to initiate apoptosis. Ultimately, BH3-only proteins promote activation of BAX/BAK, which causes mitochondrial outer membrane permeabilization and release of mitochondrial apoptotic factors such as cytochrome-c and Smac/DIABLO to induce caspase activation.
A hallmark of cancer cells is their ability to evade apoptosis by manipulating pro- and antiapoptotic genes and proteins. Many cancers overexpress prosurvival BCL-2-like proteins, thereby promoting cancer cell survival and proliferation (48). BH3 mimetics replicate the antagonistic function of BH3-only proteins on BCL-2-like proteins to promote apoptosis of cancer cells; thus, their primary mechanism of action targets mitochondrial apoptosis pathways (26, 48). Thus, it is unsurprising that BH3 mimetics disrupt mitochondrial morphology and dynamics (26).
One mechanism that has received attention as a mediator of the adverse effects of BH3 mimetics on mitochondrial morphology is inhibition of myeloid cell leukemia-1 (MCL-1), a BCL-2 family protein that is involved in mitochondrial homeostasis in diverse tissues, including the heart (49, 50). The effect of BH3 mimetics on MCL-1 may contribute to the unintended cardiotoxicity of these drugs. MCL-1 proteins are upregulated in several human hematological and solid tumors (50); indeed, MCL-1 is among the most frequently amplified genes in human cancers (51, 52). High expression of MCL-1 in tumor cells thus underlies the high efficacy of BH3 mimetics. However, MCL-1 expression in the myocardium differentially exposes the heart as tissue vulnerable to the toxic effects of BH3 mimetics. A study with cardiomyocyte-specific MCL-1 deletion found that cardiac ablation of MCL-1 in adult mice led to rapid cardiomyopathy and death (50). Mechanistically, the authors showed that myocytes from knockout animals had disorganized sarcomeres and swollen mitochondria. These changes were accompanied by reduced mitochondrial respiration and opening of the mPTP, which is consistent with increased apoptosis (50). Another independent study found that inhibition of MCL-1 by BH3 mimetics disrupted mitochondrial dynamics and led to a loss of viability and functionality in human cardiomyocytes (27). Due to the complexity of the BCL-2 apoptosis pathway and the variety of BH3 mimetics available, it is important to note that many additional mechanisms are likely involved in the mitochondrial and cardiovascular effects of BH3 mimetics.
Tyrosine Kinase Inhibitors
Tyrosine kinase inhibitors (TKIs) exert their antineoplastic effects by inhibiting kinases involved in specific signaling pathways. Kinases activate proteins through phosphorylation and are, therefore, fundamental regulators of cell signaling (53). Inhibition of specific kinases can be harnessed to combat various processes involved in tumor progression (e.g., angiogenesis). Conversely, overactivation of specific kinases can induce numerous pathologic consequences, including cancer (28); thus, TKIs which inhibit cancer-promoting kinases can be effectively targeted to combat cancer growth (54).
Kinases play a critical role in cardiac, vascular, and metabolic homeostasis, and altered kinase activity can lead to various adverse effects on the heart and the vasculature (28). The mechanism of action of some TKIs directly targets the vasculature, such as VEGF-TKIs. TKIs can also exert off-target effects on the vasculature. For example, nilotinib, an oral TKI used in the treatment of chronic myeloid leukemia (CML), impairs endothelial function, reduces nitric oxide bioavailability, and increases ROS generation in the human microcirculation (28). In human endothelial cells, nilotinib also upregulates proatherogenic adhesion proteins (28), including vascular cell adhesion protein 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), and E-selectin, all of which can recruit inflammatory cells and platelets thereby provoking vascular events (28). Other examples of potentially cardiotoxic side effects of TKIs (such as ponatinib, sorafenib, and regorafenib) include mitochondrial dysfunction, uncoupled respiration, mitochondrial Ca2+ overload, and increased ROS generation (29).
Anti-HER2 Drugs
Anti-HER2 drugs, such as the humanized monoclonal antibody trastuzumab, are targeted molecular therapies that work by blocking HER2 receptors (55). HER2 receptors have roles in cell proliferation not only in breast cancer cells but also in developing and adult hearts. Trastuzumab binds to an extracellular domain of HER2 receptors and inhibits tumor cell growth in vitro and in vivo via several mechanisms (56). Trastuzumab is the standard treatment for both early and metastatic HER2-positive breast cancer, although other novel anti-HER2 therapies are currently being used in cancer treatment (57).
Although anti-HER2 therapies are highly effective in treating cancer, they are also associated with cardiotoxic side effects that often manifest as reversible left ventricular dysfunction and heart failure (5, 55, 57). The cardiotoxic effects of trastuzumab are in part mediated by its action of blocking the function of the HER2 ligand neuregulin (5). Neuregulin is a cell-cell signaling protein secreted by coronary microvascular endothelial cells and the endocardium (58). Neuregulin is required for normal cardiac growth and maintenance and is critical in protecting cells against oxidative stress-induced cell death and promoting myocardial cell survival (5, 59). By blocking neuregulin function, trastuzumab promotes the damaging effects of oxidative stress, leading to DNA breakage and induction of mitochondrial apoptosis (5). Specifically, trastuzumab alters the balance of pro- and antiapoptotic BCL-2 proteins, which promotes mitochondrial release of cytochrome-c and initiates caspase activation and apoptosis in cardiomyocytes (60). Activation of mitochondria-mediated apoptosis occurs in conjunction with mitochondrial respiratory dysfunction, reduced ATP levels, impaired redox capacity, and loss of mitochondrial membrane potential (60). Attrition of myocytes over time is the most important mechanism leading to heart failure (59).
Platinum-Containing Agents
Platinum-based drugs such as cisplatin, carboplatin, and oxaliplatin are some of the most effective and widely used cancer chemotherapeutics (61). These drugs prevent DNA transcription and replication and induce DNA damage (62), which ultimately initiates mitochondria-mediated apoptosis (30). In addition to binding nuclear DNA, cisplatin affects mitochondrial DNA and increases mitochondrial ROS production, which is involved in the cytotoxic effects (62). Cancer cell lines that respond to cisplatin have greater mitochondrial content and higher levels of mitochondrial ROS than cisplatin-resistant cancer cells (63).
The mechanisms underlying cisplatin-induced cardiotoxicity are not completely understood, though many of the consequences are thought to be secondary to nephrotoxicity (30). There is also some evidence that cisplatin cardiotoxicity is linked to oxidative damage. Clinically, adverse cardiac outcomes associated with platinum-based therapies include silent and symptomatic arrhythmias, cardiac ischemia, diastolic disturbances without proper relaxation of ventricles before the next contraction, myocardial infarction, angina, pericarditis, thromboembolic events, and chronic heart failure (30).
Antimetabolite Agents
Antimetabolites inhibit essential biosynthetic processes or are incorporated into macromolecules, such as DNA and RNA, and inhibit their normal function (64). Antimetabolites are used in treating a variety of leukemias and solid tumors (31). Fluoropyrimidine 5-fluorouracil (5-FU) and its prodrug capecitabine are widely used antimetabolite agents. 5-FU is converted intracellularly to several active metabolites that disrupt DNA and RNA synthesis by inhibiting the action of thymidylate synthase (TS; 64). 5-FU promotes ROS production in cardiomyocytes and endothelial cells (65) and induces mitochondrial dysfunction characterized by low ATP levels, loss of mitochondrial membrane potential, and excessive mitophagy, which culminate in mitochondria-mediated apoptosis (66, 67).
Although 5-FU has been used for years in cancer treatment, it has severe side effects including cardiotoxicity (31). One potential mechanism associated with its cardiotoxicity is through vascular dysfunction with microthrombi formation (31). Cardiotoxicity caused by 5-FU often presents with chest pain related to coronary vasospasm (31). Other more serious cardiotoxic outcomes include dilated cardiomyopathy, ventricular arrhythmia, and sudden cardiac death (31).
Immunotherapies
In the past decade, immunotherapies, such as immune checkpoint inhibitors and cell-based therapies, have gained momentum as powerful anticancer tools. Under normal circumstances, immune checkpoint proteins on T cells (e.g., programmed death 1; PD-1) engage with corresponding checkpoint proteins on healthy cells (e.g., programmed death-ligand 1; PD-L1) to prevent T cells from attacking healthy cells and rein in immune responses. Thus, tumor cells that express immune checkpoint inhibitors can evade the immune system. By blocking checkpoint proteins, immune checkpoint inhibitors enable T cells to kill cancer cells and thereby promote immunogenic cell death. T cells can also be engineered to target tumor-specific antigens, which is the premise of chimeric antigen receptor (CAR) T-cell therapy.
Because of their relative novelty and the fact that latent cardiotoxicity takes years to develop, the cardiovascular consequences of immunotherapies have not been fully established. Immune-related adverse events such as myocarditis are rare but serious complications of immune checkpoint inhibitors (68). CAR-T cell therapy is associated with cytokine release syndrome, which is frequently followed by major adverse cardiovascular events and development of cardiomyopathy, arrhythmias, and/or circulatory collapse (69, 70). Preclinical evidence suggests that combining immunotherapies with other forms of CTx may potentiate cardiotoxic effects (71), though further research is needed to establish whether this occurs clinically with a range of CTx combinations. Mitochondrial respiration and dynamics are key regulators of T-cell function (72, 73), and oxidative stress generated by proinflammatory cytokines could induce mitochondrial dysfunction; however, sparse data exist examining whether mitochondria play a role in immunotherapy-induced cardiotoxicity.
LOCAL MITOCHONDRIAL SIGNALING
The following section focuses on known mechanism of intracellular communication in response to mitochondrial damage. Existing evidence, though limited, is reviewed, and put in perspective with known mechanisms of cardiovascular defects that originate in mitochondria.
Intracellular Mitochondrial Signaling
Mitochondria are no longer simply considered energy producers within the cells but have been increasingly recognized as intracellular signaling organelles. Classically, retrograde signaling reflects communication initiated from the mitochondria to the nucleus under conditions of stress (74). Nevertheless, in the past decades, it has become clear that mitochondria are constantly communicating to the nucleus, irrespective of stress, through signaling molecules. For example, the role of physiological mitochondrial reactive oxygen species (ROS) in cell proliferation, response to hypoxia and interorganellar communication is well established [for reviews, see Hamanaka and Chandel (75), Shadel and Horvath (76); Joseph et al. (77)]. More recently, the impact of metabolites primarily derived from the tricarboxylic acid (TCA) cycle or impacted by changes in its flux in remodeling the epigenome and influencing gene expression has been documented [for review, see Santos (78)]. Likewise, the ability of mitochondrial nucleic acids (DNA and RNA) to activate innate immunity and influence health outcomes has been increasingly recognized (79). In the context of cardiomyopathy, mice overexpressing a mutant mitochondrial DNA polymerase gamma (POLG) specifically in the heart showed altered nuclear DNA methylation (80). Similarly, inactivation of innate immune signaling ameliorated cardiomyopathy identified in a mouse model that expresses an exonucleolytic deficient POLG (81). Thus, it seems that mitochondrial dysfunction per se can contribute to cardiovascular outcomes through signaling mechanisms that go beyond classic stress responses.
In the below sections, we will focus on studies that provide support to the role of mitochondria in these emerging fields, namely epigenetics and innate immunity, as potential contributors to chemotherapy-associated cardiomyopathy (Fig. 2). The role of mitochondria in the context of ROS/oxidative stress and cardiotoxicity driven by cancer treatment has been reviewed elsewhere (82–86) and therefore will not be discussed here.
Figure 2.

Anticancer therapy (CTx) alters intracellular mitochondrial signaling. Several CTx agents induce mitochondrial dysfunction by promoting mitochondrial DNA (mtDNA) damage, suppressing mitochondrial metabolism, altering mitochondrial Ca2+ homeostasis, increasing production of reactive oxygen species (ROS), altering mitochondrial-nuclear communication, and/or inducing epigenetic and transcriptional remodeling. Mitochondria also play a role in CTx-induced apoptosis through release of cytochrome-c (Cyt c) and second mitochondria-derived activator of caspases (SMAC) and activation of apoptosomes. Nucleic acids in the cytosol, including mtRNA and mtDNA, activate innate immune cascades that ultimately promote inflammation. Image created with Biorender.com and published with permission. AIM2, absent-in melanoma 2; cGAS, GMP-AMP synthase; IRF3, interferon regulatory factor 3; RIG-I, retinoic acid-inducible gene I; STING, stimulator of interferon genes.
Mitochondria, Epigenetic Signaling
Anthracyclines are the best example of chemotherapy-induced cardiomyopathy, with doxorubicin (DOX) one of the most-studied drugs. Unique to this class of agents, and in particular to DOX, is not only the acute cardiac effects that present at the time of exposure but the cardiovascular outcomes that can arise years after treatment withdrawal (87). The mechanisms associated with the late-arising effects of DOX remain poorly understood, but speculation of epigenetic changes exists given the observations of chronic transcriptional remodeling detected weeks or months after DOX withdrawal (88, 89). Another long-term outcome of anthracycline treatment is mitochondrial dysfunction, which in animal models has been reported even 30 wk after cessation of DOX treatment (90). Nevertheless, it remains unclear whether mitochondrial dysfunction and transcriptional remodeling are parallel events subsequent to DOX exposure or whether mitochondrial changes actively contribute to the transcriptional outcomes through long-term epigenetic remodeling. DOX affects mitochondrial function through different mechanisms, including inhibition of complex I, mitochondrial DNA (mtDNA) damage and depletion, increased iron levels, as well as ROS (for a recent review, see Ref. 91), all of which have been shown to impinge on the status of the epigenome (92–96). Thus, it is conceivable that mitochondrial-driven epigenetic remodeling contributes to the long-term and insidious effects attributed to DOX. However, studies directly probing this connection are still emerging, and the exact cause of the long-term mitochondrial impairments observed after DOX treatment remains unclear as this drug, like all other chemotherapeutic agents, is not a specific mitochondrial inhibitor.
Short-term subchronic studies probing global epigenetic changes indicate that mitochondrial dysfunction induced by DOX might underlie epigenomic remodeling that persists after treatment ends. Ferreira and coworkers (97) treated male rats for 7 days with DOX and analyzed several biochemical and genomics parameters 2 wk after treatment ceased. At the time of analyses, the authors found persistent decrease in mtDNA content, decreased transcription of mtDNA-encoded genes and of several nuclear genes. Using biochemical assays to gauge changes in activities of enzymes associated with epigenetic modifications, the authors detected decreased histone deacetylase (HDAC) activity. Judged by the analysis of bulk 5-methyl-cytosine (5meC) levels using an ELISA assay, they also found that nuclear DNA methylation was diminished in the animals previously exposed to DOX. Previous work had demonstrated changes in levels of mitochondrial TCA cycle metabolites after DOX treatment (98), leading the authors to propose that DOX disturbed mitochondrial production of key metabolites involved in epigenetic maintenance that in turn influenced the epigenome and phenotypes of the heart (97). Notably, mtDNA depletion had been shown to alter the epigenetic landscape (92) and affect TCA cycle flux (94). Nevertheless, TCA metabolites were not directly measured in the study. Also, decreased levels of 5meC could be due to a decrease in the methyl donor s-adenosyl-methionine (SAM) and of DNA methyltransferase activity (DNMT) or reflect increased demethylation of DNA by ten eleven translocation enzymes (TETs). Alternatively, increased oxidative stress caused by DOX could directly oxidize 5meC triggering DNA repair and cytosine demethylation (99).
Despite these remaining questions, to the best of our knowledge, the work from Ferreira et al. (97) is the only to directly correlate epigenetic marks, mitochondrial and transcriptional changes after DOX withdrawal in the heart. Moving forward, it will be necessary to perform more in-depth epigenetic analysis, using deep sequencing technologies, in the hearts of animals treated with DOX, both during the treatment and after drug withdrawal. Genomics analyses that report on the epigenome status at the locus level can help to understand the relationship with gene expression. Likewise, metabolomics analyses can inform about tissue-wide changes in metabolites that can help identify contributors to the phenotype; this is particularly relevant given the wide range off-targets of DOX. It has been estimated that the half-life of mitochondria in the rodent heart is ∼14–17 days (100). Thus, allowing longer “recovery” periods after ceasing DOX treatment is necessary to assure that the protracted effects associated with DOX do not reflect leftover damaged mitochondria from the original exposure that have yet to be recycled. Finally, experiments so far involved males but including females in the analysis will help understand the relative influence, if any, of sex hormones. This is relevant as DOX-induced cardiotoxicity has been shown to be impacted by sex (101).
Mitoxantrone is a DNA topoisomerase inhibitor that causes long-term cardiomyopathy. Although an anthracycline, the cardiomyopathy associated with mitoxantrone seems to derive from cumulative mitochondrial damage (102). In one study looking at long-term cardiomyopathy associated with this exposure, rats were treated with three cycles of mitoxantrone for a total of 20 days. Animals were then evaluated 2 (D22) and 28 (D48) days after the last cycle when several parameters of mitochondria and cardiac function were followed. The authors did not observe effects on antioxidant levels in the heart at either timepoint, whereas aberrant mitochondria, changes in mitochondrial complex IV and V activities, and depletion of ATP levels were observed at D48. Plasma lactate levels, a hallmark of mitochondrial dysfunction, were also increased at this time point as was heart mass, suggesting hypertrophy likely as an effort of the muscle to compensate for bioenergetic deficit. Interestingly, these effects were not observed at D22. Instead, at this time the authors found that complex IV and V activities were increased although this did not affect ATP levels (102). Taken together, these results suggested that mitochondrial dysfunction progressed in the rat heart over time after mitoxantrone was removed. It also suggested that the initial treatment had early effects on the organelle that could not be overcome by mitochondria turnover in the 4 wk after exposure ceased. The mechanisms responsible for these protracted effects remain unclear. It is possible that the initial changes in mitochondrial function altered metabolism and established a new epigenetic landscape, which then influenced mitochondrial function after drug removal. Mitoxantrone has been shown to have a high affinity for histones in vitro (103), it is possible that it could also do so in vivo, although they would be expected to recede upon removal of the drug. More studies are needed to better understand the protracted mitochondrial and cardiovascular effects of mitoxantrone treatment.
In addition to anthracyclines, other chemotherapeutic drugs show varying degrees of cardiotoxicity, with cisplatin, trastuzumab, and sunitinib also affecting mitochondrial function (25, 104, 105). Although generally, their withdrawal eliminates the effects on the heart, cisplatin can show longer-term cardiovascular outcomes that seem to stem from its effects on kidney function (106). In renal cells, cisplatin was shown to alter TCA cycle metabolites (107), but whether it does the same in the heart and/or affects the epigenome is unknown. Many of the cardiovascular effects of cisplatin, trastuzumab, and sunitinib seem to track with previous anthracycline exposure (108), suggesting that they might precipitate but not necessarily cause the cardiac changes. Thus, if altered mitochondrial signaling plays a role in the cardiotoxicity of nonanthracycline chemotherapeutic agents, it is likely that it involves different pathways than those associated with DOX. For example, cisplatin damages nuclear and mtDNA. Previous work has shown that targeting cisplatin to mitochondria alone suffices to cause apoptosis (109). Thus, cisplatin-induced mitochondrial dysfunction may cause cardiac deficit by killing cardiomyocytes. DOX can also cause apoptosis but its high affinity for cardiolipin, a major lipid component of mitochondrial membranes, is unique to this drug (91). As cardiolipin dysfunction has broad physiological effects (110), the effects of DOX on cardiolipin may have significant consequences for long-term cardiovascular health. Interestingly, intrinsic differences in mitochondrial respiratory reserve among tissues of animals chronically treated with DOX were proposed to derive from the depletion of cardiolipin and not be associated with impaired mitochondrial respiration (111).
Decreased mitochondrial Ca2+ has been shown to alter TCA cycle metabolites, histone methylation, and the expression of genes associated with myofibroblasts differentiation (and thus fibrosis) in murine cardiac cells (112). In rats treated with cisplatin, decreased Ca2+ uptake was identified in isolated mitochondria from kidney (113). More recently, altered epigenetic status in the form of DNA methylation, the abundance of histone marks, and dysregulated expression of noncoding RNAs were associated with cisplatin exposure in renal cells (114). It is feasible to speculate that similar effects may occur in hearts exposed to cisplatin, but this remains to be determined. Interestingly, a recent study that followed testicular cancer survivors (TCS) reported mild decreased diastolic function 30 years after treatment with cisplatin (115). In a separate study, epigenetic changes were identified in the blood of an independent cohort of TCS 16 years after their treatment with cisplatin (116). Conceivably, mitochondrial changes, such as altered Ca2+ homeostasis, could drive these epigenetic outcomes in the heart or in the kidneys, or both, ultimately influencing cardiovascular health. Clearly, more studies are needed to define whether a link between mitochondrial dysfunction, epigenetic remodeling, and cisplatin-induced cardiomyopathy exist.
Trastuzumab and sunitinib are targeted therapies that have also been shown to cause cardiotoxicity. In addition to playing a role in cell proliferation in the breast tissue, HER2 has functions in the developing and adult heart. Specifically, HER2 signaling is essential for growth and survival of cardiomyocytes under stress conditions such as mechanical strain (25, 117, 118). In addition, HER2 is expressed on the vascular endothelium, and exposure to anti-HER2 therapy could promote endothelial dysfunction as a possible contributor to the development of MACE. Thus, it is easy to envision how inhibition of Her2 in the tumor can inadvertently lead to off-target cardiovascular effects. Nevertheless, the exact mechanisms involved in trastuzumab-induced cardiotoxicity remain unclear, as this drug has been shown to induce oxidative stress, mitochondrial dysfunction, and apoptosis (119–121)—all of which contribute to myocardial dysfunction. Likewise, sunitinib has been shown to affect mitochondrial function through different mechanisms, including through inhibition of AMPK (AMP-activated protein kinase) that by virtue of sensing changes in the AMP:ATP ratio is a central regulator of energy/mitochondrial homeostasis (122). In cardiac cell culture and animal models, sunitinib was shown to inhibit mitochondrial β oxidation, impair electron transport chain (ETC) activity, increase mitochondrial ROS production, and remodel energy metabolism (123–125). Based on these findings, it would not be surprising to find that chronic but low-grade alterations in mitochondrial metabolism in the heart induced by these drugs could impact the epigenome and contribute to their cardiac effects. However, studies to address this possibility remain needed. Even if they do affect the epigenome through the mitochondria, as withdrawal of these drugs generally resolve the cardiac events, one could argue that such effects might be secondary in the context of their cardiotoxicity.
Finally, it is worth noting that chemotherapeutic drugs, by virtue of their systemic delivery, can also exert their effects by affecting mitochondrial function in immune cells (such as macrophages, monocytes, etc.) as well at vascular cells (mainly endothelial and smooth muscle cells), in turn indirectly impacting cardiovascular health. Studies have shown that epigenetic remodeling in cells from the immune system drive dysregulated overaction of those cells in the heart and vasculature (126). In macrophages, the transition from M1 to M2 polarization is driven by mitochondria (127) and epigenetic remodeling (128), dysregulation of which alters their ability to promote inflammation or an anti-inflammatory response. Recently, both DOX and ionizing radiation (IR) were shown to cause mitochondrial dysfunction in myeloid cells and a persistent proinflammatory phenotype that accelerated coronary atherosclerosis (129, 130). Likewise, proper function of a specific macrophage lineage in the heart has been shown to be necessary to remove and recycle damaged mitochondria to maintain cardiovascular health in animal models (131). Thus, conceptually mitochondrial dysfunction caused by chemotherapeutics in myeloid cells could be involved in their epigenetic remodeling and contribute to their persistent/dysregulated activation in the context of cardio-oncology. The extent to which this might contribute to the cardiac events associated with acute and/or chronic exposures to cancer treatments is unknown, and studies to specifically address this question are warranted.
Innate Immunity and Chemotherapy-Induced Cardiotoxicity: Is There a Role for Mitochondria?
Activation of innate immunity is another area in which mitochondria are now recognized players. Given the increasing evidence demonstrating the significant impact that innate immunity plays in cardiovascular outcomes (132), mitochondrial dysfunction induced by chemotherapeutics may also affect the heart through dysregulating the immune system. Seminal work from Gerald Shadel’s laboratory has demonstrated that depletion of transcription factor A mitochondrial (TFAM), a mitochondrial protein involved in mtDNA replication/transcription and nucleoid structure, leads to mtDNA leakage into the cytosol activating cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING). Together cGAS and STING are responsible for sensing cytoplasmic DNA and activating a signaling cascade that leads to a series of phosphorylation reactions that promote the dimerization and nuclear translocation of interferon regulatory factor 3 (IRF3). This in turn leads to the expression of interferon-stimulated genes (ISGs), whose products effectively handle pathogens (133). In their initial work, West and colleagues showed that activation of ISGs by depletion of TFAM potentiated type I interferon (IFN-I) responses and conferred broad viral resistance (134). More recently, they showed that genotoxin-damaged mtDNA effectively activated innate immunity. Using primary fibroblasts and cultured cancer cells, authors found that DOX treatment damaged mtDNA and engaged cGAS-STING and ISGs. They also showed that irradiation of TFAM+/− mice with IR led to enhanced nuclear DNA repair in the spleen. Given these data, these authors concluded that the release of mtDNA in the cytosol elicited a protective signaling response that enhanced nuclear DNA repair in cells and tissues. A similar conclusion was obtained by Tigano and coworkers who, by inducing double-strand breaks on the mtDNA using mitochondrial-targeted transcription activator-like effector nucleases (TALENs), also identified increased nuclear DNA repair in cultured cells exposed to IR (135). Notably, mtRNAs (and not mtDNA) induced the innate immunity cascade, which in this case involved retinoic acid-inducible gene I (RIG-I), an RNA sensing protein that also activates ISGs (135).
Together, these studies demonstrate that both mtDNA and mtRNAs, which can be released upon genotoxin exposures, engage innate immunity. This is relevant in the context of chemotherapy as many drugs used to kill cancer cells are genotoxins, which often damage the nuclear and mitochondrial genomes. Although activation of innate immunity by genotoxins might counterintuitively benefit the tumor by increasing DNA repair and thus providing a means of resistance, it might also have effects in different organs. For example, this could potentially be a means through which direct damage to the nuclear DNA of cardiomyocytes is more efficiently repaired, sparing apoptosis of these terminally differentiated cells overall protecting the heart. Conversely, hyperactivation of innate immunity upon chemotherapy could be rather detrimental to cardiovascular health. A recent study using the POLG exonucleolytic mutator mouse showed that activation of innate immunity through cGAS-STING contributed to the cardiovascular phenotypes of old animals. Specifically, they showed that elevated IFN-I signaling suppressed activation of the nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor that is a master regulator of the cellular antioxidant response. This in turn increased oxidative stress and enhanced proinflammatory cytokine responses. Ablation of the IFN-I signaling ameliorated the hyperinflammatory phenotype and cardiovascular dysfunction in aged mutator mice (81). Robust IFN-1 signaling driven by mtDNA stress and STING also appears to contribute to DOX-induced cardiotoxicity via autocrine or paracrine signaling downstream of the IFN-1 receptor (136). Mice lacking STING or the IFN-1 receptor are protected from DOX-induced left ventricular dysfunction and cardiac fibrosis, suggesting that overactivation of innate immunity driven by the release of damaged mtDNA can contribute to cardiomyopathy (136).
Whether mtDNA damage caused by chemotherapeutic agents such as DOX, cisplatin, and etoposide among others could cause overactivation of innate immunity and negatively contribute to the cardiovascular effects of these drugs remains to be explored. Nevertheless, studies using some of these agents suggest that they can activate innate immunity through mtDNA leakage in other organs, granting studies about whether similar effects are at play in the heart. For example, cisplatin treatment induced mtDNA leakage and activation of cGAS/STING in an animal model of acute kidney injury. Both knockdown of STING and removal of mtDNA ameliorated the progressive nature of the kidney injury, suggesting that mtDNA-driven engagement of cGAS-STING as critical regulator of kidney injury in the context of cisplatin exposure (137). Other chemotherapeutic agents have been shown to activate cGAS-STING (138) although it remains to be defined the extent to which such response involves sensing the mitochondrial genome in the cytosol. In this context, a high-throughput screening assay developed to identify compounds that either inhibit or bypass the Ebola virus protein VP35 IFN-antagonist function, identified five DNA intercalators as hits from a library of bioactive compounds. Most of them were anthracyclines, including the cardiotoxic drug DOX, which along the other agents triggered the DNA-sensing cGAS-STING pathway of IFN induction (139).
Another means through which mitochondria might contribute to chemotherapy-induced cardiotoxicity is through activation of the inflammasome. NLRP3 (nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing protein 3) inflammasomes are multiprotein complexes of the innate immune system that recruit pro-caspase-1 via the adaptor molecule apoptosis-associated speck-like protein (ASC). This ultimately leads to the maturation of the proinflammatory cytokines IL-1β and IL-18 (140). There is increasing evidence of the relevance of the NLRP3 inflammasome in the pathogenesis of cardiovascular diseases (CVD; 141). There are also reports that chemotherapeutic agents induce NLRP3 activation, including DOX (142) and, most notably, that the cardiotoxic effects of DOX, specifically, can be mitigated by inhibition of the NLRP3 (143, 144). Thus, although it is clear that DOX can activate NLRP3, it remains unknown whether such effects are associated with mitochondrial dysfunction or the other cellular perturbations derived from anthracycline treatment that can also activate this pathway, including modulation of ion flux, extracellular ATP, lysosomal degradation, ROS, etc. (145). Recently, oxidized mtDNA (ox-mtDNA) was shown to effectively turn on the inflammasome (146). Given that DOX can induce both increased mitochondrial ROS production and oxidation of the mtDNA (91), it is feasible that mitochondria might signal to the NLRP3 inflammasome through different mechanisms after DOX exposure. Clearly, more studies are required to determine the extent to which mitochondrial nucleic acids might signal to the immune system in the context of chemotherapy and whether these events play a role in the associated cardiovascular outcomes.
SYSTEMIC MITOCHONDRIAL SIGNALING
Many cancer therapies cause mitochondrial damage in cancer and noncancer cells alike, and the resulting mitochondrial stress can initiate secondary, systemic signaling that may contribute to cardiovascular outcomes. Nevertheless, mitochondrial-derived extracellular signaling is still an underexplored mechanism in the context of adverse cardiovascular events in patients with cancer. Secondary signaling events triggered by mitochondrial damage are well characterized in several cardiovascular conditions, including hypertension, heart failure, and endothelial dysfunction, among others [reviewed by Kadlec et al. (11) and Nakayama et al. (147)]. However, this concept has not been explored in chemotherapy-induced cardiovascular toxicity. This section provides a brief overview of mitochondria-derived signals in cardio-oncology, including circulating mitochondrial fragments, RNAs, and hormones.
mtDNA Damage, DAMPs, PAMPs
In addition to detecting bacteria and other pathogens, the innate immune system senses endogenous signals released from damaged cells and coordinates an inflammatory response to promote tissue repair. Although this is initially protective, chronic activation of inflammatory pathways has been proposed to contribute to cardiovascular dysfunction in a variety of circumstances, including cardio-oncology. Mitochondria share many evolutionarily conserved molecular and structural motifs with bacteria; thus, the innate immune system is primed to recognize and respond to mitochondrial contents released from dying cells. Cell rupture causes intracellular contents to diffuse to surrounding tissues and enter the circulation, where they serve as damage-associated molecular patterns (DAMPs) that activate innate immune signaling cascades (Fig. 3). Likewise, cellular stress, such as mtDNA damage, promotes regulated release of DAMPs (i.e., mtDNA fragments) from intact cells. Damaged mitochondria are subject to quality control processes via autophagy (i.e., mitophagy), and recent evidence suggests that perturbations to mitophagy promote extracellular release of free mitochondria (148). Several mitochondrial DAMPs are known to stimulate innate immune responses, including cell-free mtDNA (cf-mtDNA) fragments, mitochondrial membrane fractions, ATP, and formyl peptides. Circulating DAMPs are detected by a range of intracellular and cell-surface receptors on immune cells, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), formyl peptide receptors (FPRs), and purinergic receptors. Of note, mtDNA damage induced by many anticancer therapies promotes the formation and release of cf-mtDNA, which is recognized by TLRs, stimulates cGAS-STING signaling, and activates both NLRP3 and absent-in melanoma 2 (AIM2) inflammasomes (149–151). The vascular endothelium also expresses innate immune receptors and acts as an interface that senses DAMPs and orchestrates an inflammatory response by expressing adhesion molecules, releasing cytokines, and increasing vascular permeability to facilitate leukocyte migration (152, 153).
Figure 3.

Systemic mitochondrial signaling may promote cardiovascular inflammation following anticancer therapy (CTx). CTx-induced cellular stress and death promote the release of damage-associated molecular patterns (DAMPs). DAMPs released from mitochondria include cell-free mitochondrial DNA (mtDNA), ATP, formyl peptides, and mitochondrial membrane fractions, among others. Circulating mitochondrial DAMPs are recognized by pattern recognition receptors of the innate immune system, including Toll-like receptors (TLRs), purinergic receptors, formyl peptide receptors (FPRs), and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs). Activation of innate immune receptors on immune cells, cardiomyocytes, and vascular cells promotes cardiovascular inflammation that may play a role in CTx-induced cardiotoxicity. Image created with Biorender.com and published with permission.
Circulating DAMPs are elevated in patients with cancer (154), which could arise from a number of sources, including tumor growth and remodeling, tumor cell death, and carcinogenic environmental toxins (155). As the goal of most cancer therapies is to induce cancer cell death, dying tumor cells represent a major potential source of mitochondrial DAMPs, which could provoke cardiovascular inflammation. Indeed, Klee et al. (154) postulated a direct relationship of tumor size to levels of circulating DAMPs. However, nontumor cells, such as cardiomyocytes, are also susceptible to CTx-induced damage (e.g., mtDNA damage), which could lead to release of DAMPs. Thus, the origin of circulating DAMPs that trigger cardiovascular inflammation in patients with cancer undergoing therapy is not clear. In addition to DAMPs released from host cells, alterations in the microbiome and death of microbiota trigger the release of pathogen-associated molecular patterns (PAMPs; reviewed in Refs. 156, 157), which activate similar innate immune pathways and may also play a role in anticancer therapy complications.
Elevated DAMPs following anticancer therapy may have important implications for cardiovascular health. Chronic elevation of DAMPs is observed in patients with a range of CVD, including hypertension, atherosclerosis, cerebrovascular disease, and heart failure (158–160). In both rodent models (161) and large patient cohorts (162), a direct correlation between DAMPs and adverse cardiovascular events has been identified. Moreover, preclinical evidence suggests that DAMPs are involved in cardiovascular disease pathogenesis (163–167). Considering the established link between high levels of DAMPs and CVD pathology, a contribution of DAMPs to elevated cardiovascular risk in patients with cancer is plausible but has not been explored to date. Genetic downregulation or pharmacological blockade of TLRs protects against vascular endothelial dysfunction and ameliorates doxorubicin-induced cardiomyopathy (159, 168, 169). Given the well-documented role of DAMPs in exacerbating inflammation in CVD, we speculate that DAMPs, irrespective of their impact on cancer treatment and progression, may facilitate cardiotoxicity during and following cancer therapy.
Although DAMPs play a key role in innate immune-mediated inflammation, they have a range of effects that can both promote and suppress inflammation (reviewed in Refs. 170, 171). In cancer, DAMPs play a dual role with both beneficial and deleterious consequences. There is growing interest in using DAMPs and associated signaling to promote immunogenic tumor cell death and improve treatment efficacy (172–174). Conversely, DAMPs are also associated with development of treatment resistance and may promote tumor progression in some instances (175–178). These paradoxical effects likely depend upon the level and type of DAMPs, mechanisms of action of antineoplastic therapies, and other systemic and local interactions. Ultimately, novel strategies should aim to limit the off-target effects of unrestrained inflammation while preserving treatment efficacy of anticancer therapies.
Mitochondria-Derived Noncoding RNAs
There is an unmet clinical need to better understand the factors that drive pathological cardiac remodeling and subsequent heart failure. Several genetic modifiers have been identified which contribute to cardiac hypertrophy; however, the precise underlying molecular mechanisms are complex, multifactorial, and incompletely understood. In addition to coding RNAs that produce proteins, long noncoding RNAs (lncRNAs) are emerging as key molecular regulators of cardiac physiology and pathophysiology. Recent studies have established the predictive value of lncRNA profile and validity for therapeutic targets (179–181; reviewed in Ref. 182). However, few studies have connected changes in lncRNA profile with cardiovascular events in relation to anticancer therapy, and to the best of our knowledge, no study has evaluated chronological changes in relation to treatment-induced cardiotoxicity in humans or in a longitudinal manner. Several regulatory lncRNAs that govern basic cellular mechanisms such as autophagy and mitochondrial metabolism have been described and represent promising treatment targets (183–185). Our group has described a critical contribution of many of these pathways to the regulation of microvascular function in health and disease as well as during acute stress (13, 14, 186–192).
The prognostic value of using noncoding RNAs (ncRNAs) and circulating microRNAs (miRNAs) as biomarkers for disease progression has been explored in CVD such as coronary syndrome (193) [reviewed by Viereck and Thum (194)], but their value as biomarkers of anticancer therapy-induced cardiotoxicity has not been investigated. The nuclear and mitochondrial genome alike encode for miRNAs that can be observed in the circulation. Mitochondria contain miRNAs, termed mitomiRs, which have been identified in several species and cell types (195). Although our knowledge of mitochondrial ncRNAs is still incomplete, they appear to be key players in the regulation of the nuclear and mitochondrial gene expression. Noncoding mitochondria-derived miRNAs have been linked to hypertension, cardiac hypertrophy, myocardial infarction, and heart failure and have been proposed as a biomarker for cardiovascular disease progression (for review, see Ref. 196). In addition, several mitochondrial miRNAs have been characterized as key players in lipid metabolism (197, 198), and hyperlipidemia is an established clinical complication in patients receiving anthracycline-based therapies that may promote CVD.
Mitochondrial dysfunction caused by disruption of mitochondrial–nuclear communication may play a role in the development of various diseases, including cardiovascular disease. The investigation of key players involved in the mitochondrial ncRNAs regulatory network may open the path for the identification and development of novel diagnostic and therapeutic approaches for mitochondrial dysfunction-related CVD (for review, see Ref. 196). However, several challenges remain to be addressed before considering the mitochondrial clinical application of ncRNAs. The biology and function of mitochondrial ncRNAs as well as their antero-retrograde translocation need to be experimentally validated and better understood.
Mitochondrial Hormones
In addition to DAMPs and ncRNAs, mitochondria produce signaling peptides that act in a paracrine and endocrine manner. Many of these mitochondria-derived peptides (MDPs) are released in response to stress and have cytoprotective effects that include preserving mitochondrial function and inhibiting initiation of apoptosis (199–201). Emerging evidence suggests that mitochondrial hormones may ameliorate CVD (for review, see Ref. 202). In a preclinical model, administration of an analog of the MDP humanin protected against DOX-induced cardiotoxicity (203). However, the cytoprotective effects of MDPs such as humanin may also promote treatment resistance and accelerate tumor growth in some circumstances (204). Further research in this developing area is warranted to investigate the role and potential therapeutic utility of MDPs in cardio-oncology.
SUMMARY AND OUTLOOK
Cardiotoxicity is a major clinical complication of anticancer therapies. In addition to direct effects of antineoplastic agents on the heart and vasculature, cardiovascular dysfunction may arise secondary to treatment-induced mitochondrial stress and associated signaling. At the intracellular level, mitochondrial damage alters epigenetic signaling, calcium dynamics, and metabolism and activates innate immune pathways. Mitochondrial damage also promotes the release of DAMPs, ncRNAs, and mitochondrial hormones to the circulation, which trigger systemic inflammation that may contribute to treatment-induced cardiotoxicity. Further research investigating the impact of cancer and its treatments on mitochondria and associated secondary signaling pathways will guide efforts to minimize the cardiovascular side effects of anticancer therapies while preserving treatment efficacy. This in turn could be applicable to other environmental exposures that also affect mitochondria, allowing better understanding and integration of knowledge to improve overall health outcomes of patients with cancer.
GRANTS
This work is funded by National Heart, Lung, and Blood Institute (NHLBI) Grant T32HL134643 (to J.D.T. and A.M.B.), American Heart Association Scientific-focused Research Network Diversity Supplement Grant 960133 (to J.C.B.), and NHLBI Grants R01HL157025 and R01HL133029.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.T. conceived and designed research; J.C.B., J.H.S., and A.M.B. prepared figures; J.C.B., J.D.T., J.H.S., and A.M.B. drafted manuscript; J.C.B., J.D.T., J.H.S., and A.M.B. edited and revised manuscript; J.C.B., J.D.T., J.H.S., and A.M.B. approved final version of manuscript.
REFERENCES
- 1. Bikomeye JC, Beyer AM, Kwarteng JL, Beyer KMM. Greenspace, inflammation, cardiovascular health, and cancer: a review and conceptual framework for greenspace in cardio-oncology research. Int J Environ Res Public Health 19: 2426, 2022. doi: 10.3390/ijerph19042426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Academies of Sciences, Engineering, and Medicine, Health and Medicine Division, Board on Global Health, Committee on Global Health and the Future of the United States. Global Health and the Future Role of the United States. Washington, DC: National Academies Press, 2017. [Google Scholar]
- 3. Bertero E, Ameri P, Maack C. Bidirectional relationship between cancer and heart failure: old and new issues in cardio-oncology. Card Fail Rev 5: 106–111, 2019. doi: 10.15420/cfr.2019.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gabani M, Castañeda D, Nguyen QM, Choi S-K, Chen C, Mapara A, Kassan A, Gonzalez AA, Khataei T, Ait-Aissa K, Kassan M. Association of cardiotoxicity with doxorubicin and trastuzumab: a double-edged sword in chemotherapy. Cureus 13: e18194, 2021. doi: 10.7759/cureus.18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dempsey N, Rosenthal A, Dabas N, Kropotova Y, Lippman M, Bishopric NH. Trastuzumab-induced cardiotoxicity: a review of clinical risk factors, pharmacologic prevention, and cardiotoxicity of other HER2-directed therapies. Breast Cancer Res Treat 188: 21–36, 2021. doi: 10.1007/s10549-021-06280-x. [DOI] [PubMed] [Google Scholar]
- 6. Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM, Meyer JE. Causes of death among cancer patients. Ann Oncol 28: 400–407, 2017. doi: 10.1093/annonc/mdw604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patnaik JL, Byers T, DiGuiseppi C, Dabelea D, Denberg TD. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: a retrospective cohort study. Breast Cancer Res 13: R64, 2011. doi: 10.1186/bcr2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simůnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 61: 154–171, 2009. doi: 10.1016/s1734-1140(09)70018-0. [DOI] [PubMed] [Google Scholar]
- 9. Nebigil CG, Désaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol 9: 1262, 2018. doi: 10.3389/fphar.2018.01262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol 41: 389–405, 2006. doi: 10.1016/j.yjmcc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 11. Kadlec AO, Beyer AM, Ait-Aissa K, Gutterman DD. Mitochondrial signaling in the vascular endothelium: beyond reactive oxygen species. Basic Res Cardiol 111: 26, 2016. doi: 10.1007/s00395-016-0546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montaigne D, Hurt C, Neviere R. Mitochondria death/survival signaling pathways in cardiotoxicity induced by anthracyclines and anti-cancer-targeted therapies. Biochem Res Int 2012: 951539, 2012. doi: 10.1155/2012/951539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hader SN, Zinkevich N, Toro LEN, Kriegel AJ, Kong A, Freed JK, Gutterman DD, Beyer AM. Detrimental effects of chemotherapy on human coronary microvascular function. Am J Physiol Heart Circ Physiol 317: H705–H710, 2019. doi: 10.1152/ajpheart.00370.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durand MJ, Hader SN, Derayunan A, Zinkevich N, McIntosh JJ, Beyer AM. BCR-ABL tyrosine kinase inhibitors promote pathological changes in dilator phenotype in the human microvasculature. Microcirculation 27: 1–5, 2020. doi: 10.1111/micc.12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szczepaniak P, Siedlinski M, Hodorowicz-Zaniewska D, Nosalski R, Mikolajczyk TP, Dobosz AM, Dikalova A, Dikalov S, Streb J, Gara K, Basta P, Krolczyk J, Sulicka-Grodzicka J, Jozefczuk E, Dziewulska A, Saju B, Laksa I, Chen W, Dormer J, Tomaszewski M, Maffia P, Czesnikiewicz-Guzik M, Crea F, Dobrzyn A, Moslehi J, Grodzicki T, Harrison DG, Guzik TJ. Breast cancer chemotherapy induces vascular dysfunction and hypertension through a NOX4-dependent mechanism. J Clin Invest 132: e149117, 2022. doi: 10.1172/JCI149117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terwoord JD, Beyer AM, Gutterman DD. Endothelial dysfunction as a complication of anti-cancer therapy. Pharmacol Ther 237: 108116, 2022. doi: 10.1016/J.PHARMTHERA.2022.108116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van de Hoef TP, van Lavieren MA, Damman P, Delewi R, Piek MA, Chamuleau SAJ, Voskuil M, Henriques JPS, Koch KT, de Winter RJ, Spaan JAE, Siebes M, Tijssen JGP, Meuwissen M, Piek JJ. Physiological basis and long-term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circ Cardiovasc Interv 7: 301–311, 2014. doi: 10.1161/CIRCINTERVENTIONS.113.001049. [DOI] [PubMed] [Google Scholar]
- 18. Beyer AM, Zinkevich N, Miller B, Liu Y, Wittenburg AL, Mitchell M, Galdieri R, Sorokin A, Gutterman DD. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol 112: 5, 2017. doi: 10.1007/s00395-016-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sahu K, Langeh U, Singh C, Singh A. Crosstalk between anticancer drugs and mitochondrial functions. Curr Res Pharmacol Drug Discov 2: 100047, 2021. doi: 10.1016/j.crphar.2021.100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffman RK, Kim B-J, Shah PD, Carver J, Ky B, Ryeom S. Damage to cardiac vasculature may be associated with breast cancer treatment-induced cardiotoxicity. Cardiooncology 7: 15, 2021. doi: 10.1186/s40959-021-00100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim Biophys Acta 1760: 267–271, 2006. doi: 10.1016/j.bbagen.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 22. Goyal A, Belur AD, Dey AK, Mehta NN. Chapter 7 - Blood inflammatory biomarkers of cardiovascular disease. In: Biomarkers in Cardiovascular Disease, edited by Nambi V. Amsterdam: Elsevier, 2019, p. 71–79. [Google Scholar]
- 23. Peng T-I, Jou M-J. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201: 183–188, 2010. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 24. Sawyer DB, Peng X, Chen B, Pentassuglia L, Lim CC. Mechanisms of anthracycline cardiac injury: can we identify strategies for cardioprotection? Prog Cardiovasc Dis 53: 105–113, 2010. doi: 10.1016/j.pcad.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorini S, De Angelis A, Berrino L, Malara N, Rosano G, Ferraro E. Chemotherapeutic drugs and mitochondrial dysfunction: focus on doxorubicin, trastuzumab, and sunitinib. Oxid Med Cell Longev 2018: 7582730, 2018. [Erratum in Oxid Med Cell Longev 2019: 9601435, 2019]. doi: 10.1155/2018/7582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mehrotra N, Kharbanda S, Singh H. BH3 mimetics in cancer therapy and their future perspectives with nanodelivery. Nanomedicine (Lond) 16: 1067–1070, 2021. doi: 10.2217/nnm-2021-0059. [DOI] [PubMed] [Google Scholar]
- 27. Rasmussen ML, Taneja N, Neininger AC, Wang L, Robertson GL, Riffle SN, Shi L, Knollmann BC, Burnette DT, Gama V. MCL-1 inhibition by selective BH3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. iScience 23: 101015, 2020. doi: 10.1016/j.isci.2020.101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manouchehri A, Kanu E, Mauro MJ, Aday AW, Lindner JR, Moslehi J. Tyrosine kinase inhibitors in leukemia and cardiovascular events: from mechanism to patient care. Arterioscler Thromb Vasc Biol 40: 301–308, 2020. doi: 10.1161/ATVBAHA.119.313353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodríguez-Hernández MA, de la Cruz-Ojeda P, López-Grueso MJ, Navarro-Villarán E, Requejo-Aguilar R, Castejón-Vega B, Negrete M, Gallego P, Vega-Ochoa Á, Victor VM, Cordero MD, Del Campo JA, Bárcena JA, Padilla CA, Muntané J. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol 36: 101510, 2020. doi: 10.1016/j.redox.2020.101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton transactions 47: 6645–6653, 2018. [Erratum in Dalton Trans 47: 7848, 2018]. doi: 10.1039/c8dt00838h. [DOI] [PubMed] [Google Scholar]
- 31. Yuan C, Parekh H, Allegra C, George TJ, Starr JS. 5-FU induced cardiotoxicity: case series and review of the literature. Cardiooncology 5: 13, 2019. doi: 10.1186/s40959-019-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vásquez-Vivar J, Martasek P, Hogg N, Masters BSS, Pritchard KA, Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 36: 11293–11297, 1997. doi: 10.1021/bi971475e. [DOI] [PubMed] [Google Scholar]
- 33. Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem 275: 33585–33592, 2000. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 34. Kalivendi S. V, Kotamraju S, Zhao H, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase: effect of antiapoptotic antioxidants and calcium. J Biol Chem 276: 47266–47276, 2001. doi: 10.1074/jbc.M106829200. [DOI] [PubMed] [Google Scholar]
- 35. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7: 165–176, 2017. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Serrano J, Palmeira CM, Kuehl DW, Wallace KB. Cardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administration. Biochim Biophys Acta 1411: 201–205, 1999. doi: 10.1016/S0005-2728(99)00011-0. [DOI] [PubMed] [Google Scholar]
- 37. Adachi K, Fujiura Y, Mayumi F, Nozuhara A, Sugiu Y, Sakanashi T, Hidaka T, Toshima H. A deletion of mitochondrial DNA in murine doxorubicin-induced cardiotoxicity. Biochem Biophys Res Commun 195: 945–951, 1993. doi: 10.1006/BBRC.1993.2135. [DOI] [PubMed] [Google Scholar]
- 38. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, Pattillo CB, Orr AW, Sadoshima J, Hill JA, Bhuiyan MS. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep 9: 2002, 2019. doi: 10.1038/s41598-018-37862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li DL, Wang Z. V, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, Hill JA. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133: 1668–1687, 2016. doi: 10.1161/CIRCULATIONAHA.115.017443/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y, Lu X, Wang X, Qiu Q, Zhu P, Ma L, Ma X, Herrmann J, Lin X, Wang W, Xu X. Atg7-based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circ Res 129: e166–e182, 2021. doi: 10.1161/CIRCRESAHA.121.319104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res 157: 104846, 2020. doi: 10.1016/J.PHRS.2020.104846. [DOI] [PubMed] [Google Scholar]
- 42. Li S, Wang W, Niu T, Wang H, Li B, Shao L, Lai Y, Li H, Janicki JS, Wang XL, Tang D, Cui T. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev 2014: 748524, 2014. doi: 10.1155/2014/748524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 8: 46663–46680, 2017. doi: 10.18632/ONCOTARGET.16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nicholls DG. Mitochondria and calcium signaling. Cell Calcium 38: 311–317, 2005. doi: 10.1016/J.CECA.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 45. Wescott AP, Kao JPY, Lederer WJ, Boyman L. Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat Metab 1: 975–984, 2019. [Erratum in Nat Metab 1: 1168, 2019]. doi: 10.1038/S42255-019-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Márta K, Hasan P, Rodríguez-Prados M, Paillard M, Hajnóczky G. Pharmacological inhibition of the mitochondrial Ca 2+ uniporter: relevance for pathophysiology and human therapy. J Mol Cell Cardiol 151: 135–144, 2021. doi: 10.1016/J.YJMCC.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45: 643–650, 2009. doi: 10.1016/J.CECA.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 48. Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell 34: 879–891, 2018. doi: 10.1016/j.ccell.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 49. Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 27: 1351–1364, 2013. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson ÅB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 27: 1365–1377, 2013. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ 22: 2098–2106, 2015. doi: 10.1038/cdd.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R, Golub TR. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell 21: 547–562, 2012. doi: 10.1016/j.ccr.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roskoski RJ. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res 100: 1–23, 2015. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 54. Traxler P. Tyrosine kinase inhibitors in cancer treatment (Part II). Expert Opin Ther Pat 8: 1599–1625, 1998. doi: 10.1517/13543776.8.12.1599. [DOI] [Google Scholar]
- 55. Babar T, Blomberg C, Hoffner E, Yan X. Anti-HER2 cancer therapy and cardiotoxicity. Curr Pharm Des 20: 4911–4919, 2014. doi: 10.2174/1381612820666140604145037. [DOI] [PubMed] [Google Scholar]
- 56. Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 27: 5838–5847, 2009. doi: 10.1200/JCO.2009.22.1507. [DOI] [PubMed] [Google Scholar]
- 57. Sendur MAN, Aksoy S, Altundag K. Cardiotoxicity of novel HER2-targeted therapies. Curr Med Res Opin 29: 1015–1024, 2013. doi: 10.1185/03007995.2013.807232. [DOI] [PubMed] [Google Scholar]
- 58. Lemmens K, Doggen K, De Keulenaer GW. Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease: implications for therapy of heart failure. Circulation 116: 954–960, 2007. doi: 10.1161/CIRCULATIONAHA.107.690487. [DOI] [PubMed] [Google Scholar]
- 59. Sandoo A, Kitas GD, Carmichael AR. Breast cancer therapy and cardiovascular risk: focus on trastuzumab. Vasc Health Risk Manag 11: 223–228, 2015. doi: 10.2147/VHRM.S69641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grazette LP, Boecker W, Matsui T, Semigran M, Force TL, Hajjar RJ, Rosenzweig A. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J Am Coll Cardiol 44: 2231–2238, 2004. doi: 10.1016/J.JACC.2004.08.066. [DOI] [PubMed] [Google Scholar]
- 61. Bruno PM, Liu Y, Park GY, Murai J, Koch CE, Eisen TJ, Pritchard JR, Pommier Y, Lippard SJ, Hemann MT. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med 23: 461–471, 2017. doi: 10.1038/nm.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One 8: e81162, 2013. doi: 10.1371/JOURNAL.PONE.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, Aulitzky WE, Essmann F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis 10: 1–12, 2019. doi: 10.1038/s41419-019-2081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3: 330–338, 2003. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 65. Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, Bucci EO, Finzi G, Sessa F, Noonan DM, Albini A. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One 10: e0115686, 2015. doi: 10.1371/JOURNAL.PONE.0115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eskandari MR, Moghaddam F, Shahraki J, Pourahmad J. A comparison of cardiomyocyte cytotoxic mechanisms for 5-fluorouracil and its pro-drug capecitabine. Xenobiotica 45: 79–87, 2015. doi: 10.3109/00498254.2014.942809. [DOI] [PubMed] [Google Scholar]
- 67. Li Y, Zhang Y, Zhou X, Lei X, Li X, Wei L. Dynamic observation of 5-fluorouracil-induced myocardial injury and mitochondrial autophagy in aging rats. Exp Ther Med 22: 1451, 2021. doi: 10.3892/ETM.2021.10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mahmood SS, Fradley MG, Cohen J. V, Nohria A, Reynolds KL, Heinzerling LM, Sullivan RJ, Damrongwatanasuk R, Chen CL, Gupta D, Kirchberger MC, Awadalla M, Hassan MZO, Moslehi JJ, Shah SP, Ganatra S, Thavendiranathan P, Lawrence DP, Groarke JD, Neilan TG. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol 71: 1755–1764, 2018. doi: 10.1016/j.jacc.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lefebvre B, Kang Y, Smith AM, Frey N. V, Carver JR, Scherrer-Crosbie M. Cardiovascular effects of CAR T cell therapy: a retrospective study. JACC CardioOncol 2: 193–203, 2020. doi: 10.1016/J.JACCAO.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, Lee DH, Zlotoff DA, Zhang L, Drobni ZD, Hassan MZO, Bassily E, Rhea I, Ismail-Khan R, Mulligan CP, Banerji D, Lazaryan A, Shah BD, Rokicki A, Raje N, Chavez JC, Abramson J, Locke FL, Neilan TG. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J Am Coll Cardiol 74: 3099–3108, 2019. doi: 10.1016/j.jacc.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Du S, Zhou L, Alexander GS, Park K, Yang L, Wang N, Zaorsky NG, Ma X, Wang Y, Dicker AP, Lu B. PD-1 modulates radiation-induced cardiac toxicity through cytotoxic T lymphocytes. J Thorac Oncol 13: 510–520, 2018. doi: 10.1016/j.jtho.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van der Windt GJW, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8 + T cell memory development. Immunity 36: 68–78, 2012. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang C-H, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, Chen Q, Huang SC-C, O'Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166: 63–76, 2016. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jazwinski SM. The retrograde response: a conserved compensatory reaction to damage from within and from without. Prog Mol Biol Transl Sci 127: 133–154, 2014. doi: 10.1016/B978-0-12-394625-6.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35: 505–513, 2010. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell 163: 560–569, 2015. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Joseph SK, Booth DM, Young MP, Hajnóczky G. Redox regulation of ER and mitochondrial Ca2+ signaling in cell survival and death. Cell Calcium 79: 89–97, 2019. doi: 10.1016/j.ceca.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Santos JH. Mitochondria signaling to the epigenome: a novel role for an old organelle. Free Radic Biol Med 170: 59–69, 2021. doi: 10.1016/j.freeradbiomed.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu Z, Sainz AG, Shadel GS. Mitochondrial DNA: cellular genotoxic stress sentinel. Trends Biochem Sci 46: 812–821, 2021. doi: 10.1016/J.TIBS.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Koczor CA, Ludlow I, Fields E, Jiao Z, Ludaway T, Russ R, Lewis W. Mitochondrial polymerase gamma dysfunction and aging cause cardiac nuclear DNA methylation changes. Physiol Genomics 48: 274–280, 2016. doi: 10.1152/physiolgenomics.00099.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lei Y, Martinez CG, Torres-Odio S, Bell SL, Birdwell CE, Bryant JD, Tong CW, Watson RO, West LC, Phillip West A. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci Adv 7: 7548–7574, 2021. doi: 10.1126/sciadv.abe7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brown S-A, Sandhu N, Herrmann J. Systems biology approaches to adverse drug effects: the example of cardio-oncology. Nat Rev Clin Oncol 12: 718–731, 2015. doi: 10.1038/nrclinonc.2015.168. [DOI] [PubMed] [Google Scholar]
- 83. Quryshi N, Norwood Toro LE, Ait-Aissa K, Kong A, Beyer AM. Chemotherapeutic-induced cardiovascular dysfunction: physiological effects, early detection—the role of telomerase to counteract mitochondrial defects and oxidative stress. Int J Mol Sci 19: 797, 2018. doi: 10.3390/ijms19030797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Varricchi G, Ameri P, Cadeddu C, Ghigo A, Madonna R, Marone G, Mercurio V, Monte I, Novo G, Parrella P, Pirozzi F, Pecoraro A, Spallarossa P, Zito C, Mercuro G, Pagliaro P, Tocchetti CG. Antineoplastic drug-induced cardiotoxicity: a redox perspective. Front Physiol 9: 167, 2018. doi: 10.3389/fphys.2018.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fu X, Tang J, Wen P, Huang Z, Najafi M. Redox interactions-induced cardiac toxicity in cancer therapy. Arch Biochem Biophys 708: 108952, 2021. doi: 10.1016/j.abb.2021.108952. [DOI] [PubMed] [Google Scholar]
- 86. Dominic A, Hamilton D, Abe J-I. Mitochondria and chronic effects of cancer therapeutics: the clinical implications. J Thromb Thrombolysis 51: 884–889, 2021. doi: 10.1007/s11239-020-02313-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Steinherz L, Steinherz P. Delayed cardiac toxicity from anthracycline therapy. Pediatrician 18: 49–52, 1991. [PubMed] [Google Scholar]
- 88. Berthiaume JM, Wallace KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic doxorubicin treatment. Cardiovasc Toxicol 7: 178–191, 2007. doi: 10.1007/S12012-007-0026-0/TABLES/6. [DOI] [PubMed] [Google Scholar]
- 89. Richard C, Ghibu S, Delemasure-Chalumeau S, Guilland JC, Des Rosiers C, Zeller M, Cottin Y, Rochette L, Vergely C. Oxidative stress and myocardial gene alterations associated with doxorubicin-induced cardiotoxicity in rats persist for 2 months after treatment cessation. J Pharmacol Exp Ther 339: 807–814, 2011. doi: 10.1124/JPET.111.185892. [DOI] [PubMed] [Google Scholar]
- 90. Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 108: 2423–2429, 2003. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- 91. Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res 126: 926–941, 2020. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modifications in the nucleus. Cancer Biol Ther 7: 1182–1190, 2008. doi: 10.4161/cbt.7.8.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Case AJ, Madsen JM, Motto DG, Meyerholz DK, Domann FE. Manganese superoxide dismutase depletion in murine hematopoietic stem cells perturbs iron homeostasis, globin switching, and epigenetic control in erythrocyte precursorcells. Free Radic Biol Med 56: 17–27, 2013. doi: 10.1016/j.freeradbiomed.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lozoya OA, Martinez-Reyes I, Wang T, Grenet D, Bushel P, Li J, Chandel N, Woychik RP, Santos JH. Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol 16: e2005707, 2018. doi: 10.1371/JOURNAL.PBIO.2005707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lozoya OA, Wang T, Grenet D, Wolfgang TC, Sobhany M, Da Silva DG, Riadi G, Chandel N, Woychik RP, Santos JH. Mitochondrial acetyl-CoA reversibly regulates locus-specific histone acetylation and gene expression. Life Sci Alliance 2: e201800228, 2019. doi: 10.26508/LSA.201800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lozoya OA, Xu F, Grenet D, Wang T, Grimm SA, Godfrey V, Waidyanatha S, Woychik RP, Santos JH. Single nucleotide resolution analysis reveals pervasive, long-lasting DNA methylation changes by developmental exposure to a mitochondrial toxicant. Cell Rep 32: 108131, 2020. doi: 10.1016/J.CELREP.2020.108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ferreira A, Cunha-Oliveira T, Simões RF, Carvalho FS, Burgeiro A, Nordgren K, Wallace KB, Oliveira PJ. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology 390: 63–73, 2017. doi: 10.1016/J.TOX.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 98. Carvalho RA, Sousa RPB, Cadete VJJ, Lopaschuk GD, Palmeira CMM, Bjork JA, Wallace KB. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology 270: 92–98, 2010. doi: 10.1016/J.TOX.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 99. Gong Z, Zhu JK. Active DNA demethylation by oxidation and repair. Cell Res 21: 1649–1651, 2011. doi: 10.1038/cr.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 246: 2425–2429, 1971. doi: 10.1016/S0021-9258(18)62305-1. [DOI] [PubMed] [Google Scholar]
- 101. Moulin M, Piquereau J, Mateo P, Fortin D, Rucker-Martin C, Gressette M, Lefebvre F, Gresikova M, Solgadi A, Veksler V, Garnier A, Ventura-Clapier R. Sexual dimorphism of doxorubicin-mediated cardiotoxicity potential role of energy metabolism remodeling. Circ Heart Fail 8: 98–108, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001180. [DOI] [PubMed] [Google Scholar]
- 102. Rossato LG, Costa VM, Dallegrave E, Arbo M, Silva R, Ferreira R, Amado F, Dinis-Oliveira RJ, Duarte JA, De Lourdes Bastos M, Palmeira C, Remião F. Mitochondrial cumulative damage induced by mitoxantrone: Late onset cardiac energetic impairment. Cardiovasc Toxicol 14: 30–40, 2014. doi: 10.1007/S12012-013-9230-2. [DOI] [PubMed] [Google Scholar]
- 103. Hajihassan Z, Rabbani-Chadegani A. Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins. J Biomed Sci 16: 31, 2009. doi: 10.1186/1423-0127-16-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Finsterer J, Ohnsorge P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul Toxicol Pharmacol 67: 434–445, 2013. doi: 10.1016/j.yrtph.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 105. Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol 309: H1453–H1467, 2015. doi: 10.1152/ajpheart.00554.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dugbartey GJ, Peppone LJ, de Graaf IAM. An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 371: 58–66, 2016. doi: 10.1016/j.tox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Choi YM, Kim HK, Shim W, Anwar MA, Kwon JW, Kwon HK, Kim HJ, Jeong H, Kim HM, Hwang D, Kim HS, Choi S. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One 10: e0135083, 2015. doi: 10.1371/journal.pone.0135083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yeh ETH, Tong AT, Lenihan DJ, Yusuf SW, Swafford J, Champion C, Durand JB, Gibbs H, Zafarmand AA, Ewer MS. Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation 109: 3122–3131, 2004. doi: 10.1161/01.CIR.0000133187.74800.B9. [DOI] [PubMed] [Google Scholar]
- 109. Wisnovsky SP, Wilson JJ, Radford RJ, Pereira MP, Chan MR, Laposa RR, Lippard SJ, Kelley SO. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem Biol 20: 1323–1328, 2013. doi: 10.1016/J.CHEMBIOL.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dudek J, Hartmann M, Rehling P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim Biophys Acta Mol Basis Dis 1865: 810–821, 2019. doi: 10.1016/J.BBADIS.2018.08.025. [DOI] [PubMed] [Google Scholar]
- 111. Pereira GC, Pereira SP, Tavares LC, Carvalho FS, Magalhães-Novais S, Barbosa IA, Santos MS, Bjork J, Moreno AJ, Wallace KB, Oliveira PJ. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion 30: 95–104, 2016. doi: 10.1016/j.mito.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 112. Lombardi AA, Gibb AA, Arif E, Kolmetzky DW, Tomar D, Luongo TS, Jadiya P, Murray EK, Lorkiewicz PK, Hajnóczky G, Murphy E, Arany ZP, Kelly DP, Margulies KB, Hill BG, Elrod JW. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun 10: 4509, 2019. doi: 10.1038/s41467-019-12103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gemba M, Nakatani E, Teramoto M, Nakano S. Effect of cisplatin on calcium uptake by rat kidney cortical mitochondria. Toxicol Lett 38: 291–297, 1987. doi: 10.1016/0378-4274(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 114. Loren P, Saavedra N, Saavedra K, Zambrano T, Moriel P, Salazar LA. Epigenetic mechanisms involved in cisplatin-induced nephrotoxicity: an update. Pharmaceuticals (Basel) 14: 491–491, 2021. doi: 10.3390/PH14060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bjerring AW, Fosså SD, Haugnes HS, Nome R, Stokke TM, Haugaa KH, Kiserud CE, Edvardsen T, Sarvari SI. The cardiac impact of cisplatin-based chemotherapy in survivors of testicular cancer: a 30-year follow-up. Eur Heart J Cardiovasc Imaging 22: 443–450, 2021. doi: 10.1093/ehjci/jeaa289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bucher-Johannessen C, Page CM, Haugen TB, Wojewodzic MW, Fosså SD, Grotmol T, Haugnes HS, Rounge TB. Cisplatin treatment of testicular cancer patients introduces long-term changes in the epigenome. Clin Epigenetics 11: 179, 2019. doi: 10.1186/S13148-019-0764-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Negro A, Brar BK, Gu Y, Peterson KL, Vale W, Lee KF. erbB2 is required for G protein-coupled receptor signaling in the heart. Proc Natl Acad Sci USA 103: 15889–15893, 2006. doi: 10.1073/PNAS.0607499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol 17: 627–638, 2015. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 119. ElZarrad MK, Mukhopadhyay P, Mohan N, Hao E, Dokmanovic M, Hirsch DS, Shen Y, Pacher P, Wu WJ. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS One 8: e79543, 2013. [Erratum inPLoS One9: 10.1371/annotation/3ba18ef8-8c9c-45ab-9bc5-ad571a54a28c]. doi: 10.1371/journal.pone.0079543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kitani T, Ong SG, Lam CK, Rhee JW, Zhang JZ, Oikonomopoulos A, Ma N, Tian L, Lee J, Telli ML, Witteles RM, Sharma A, Sayed N, Wu JC. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation 139: 2451–2465, 2019. doi: 10.1161/CIRCULATIONAHA.118.037357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pecoraro M, Pinto A, Popolo A. Trastuzumab-induced cardiotoxicity and role of mitochondrial connexin43 in the adaptive response. Toxicol In Vitro 67: 104926, 2020. doi: 10.1016/j.tiv.2020.104926. [DOI] [PubMed] [Google Scholar]
- 122. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135, 2018. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, Renault G, Tharaux PL, Dhaun N, Tavitian B. Cardiac metabolic deregulation induced by the tyrosine kinase receptor inhibitor sunitinib is rescued by endothelin receptor antagonism. Theranostics 7: 2757–2774, 2017. doi: 10.7150/THNO.19551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Stuhlmiller TJ, Zawistowski JS, Chen X, Sciaky N, Angus SP, Hicks ST, Parry TL, Huang W, Beak JY, Willis MS, Johnson GL, Jensen BC. Kinome and transcriptome profiling reveal broad and distinct activities of erlotinib, sunitinib, and sorafenib in the mouse heart and suggest cardiotoxicity from combined signal transducer and activator of transcription and epidermal growth factor receptor inhibition. J Am Heart Assoc 6: e006635, 2017. doi: 10.1161/JAHA.117.006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bouitbir J, Alshaikhali A, Panajatovic MV, Abegg VF, Paech F, Krähenbühl S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 426: 152281, 2019. doi: 10.1016/j.tox.2019.152281. [DOI] [PubMed] [Google Scholar]
- 126. Zarzour A, Kim HW, Weintraub NL. Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol 39: 984–990, 2019. doi: 10.1161/ATVBAHA.119.312193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol 10: 1462, 2019. doi: 10.3389/FIMMU.2019.01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol 34: 216–223, 2013. doi: 10.1016/J.IT.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kotla S, Zhang A, Imanishi M, Ko KA, Lin SH, Gi YJ, Moczygemba M, Isgandarova S, Schadler KL, Chung C, Milgrom SA, Banchs J, Yusuf SW, Amaya DN, Guo H, Thomas TN, Shen YH, Deswal A, Herrmann J, Kleinerman ES, Entman ML, Cooke JP, Schifitto G, Maggirwar SB, McBeath E, Gupte AA, Krishnan S, Patel ZS, Yoon Y, Burks JK, Fujiwara K, Brookes PS, Le NT, Hamilton DJ, Abe JI. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox Biol 47: 102132, 2021. doi: 10.1016/J.REDOX.2021.102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ortiz de Choudens S, Sparapani R, Narayanan J, Lohr N, Gao F, Fish BL, Zielonka M, Gasperetti T, Veley D, Beyer A, Olson J, Jacobs ER, Medhora M. Lisinopril mitigates radiation-induced mitochondrial defects in rat heart and blood cells. Front Oncol 12: 828177, 2022. doi: 10.3389/FONC.2022.828177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, et al.. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183: 94–109.e23, 2020. doi: 10.1016/J.CELL.2020.08.031. [DOI] [PubMed] [Google Scholar]
- 132. Koc A, Cagavi E. Cardiac immunology: a new era for immune cells in the heart. Adv Exp Med Biol 1312: 75–95, 2021. doi: 10.1007/5584_2020_576. [DOI] [PubMed] [Google Scholar]
- 133. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545, 2014. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557, 2015. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591: 477–481, 2021. doi: 10.1038/s41586-021-03269-w. [DOI] [PubMed] [Google Scholar]
- 136. Lei Y, VanPortfliet JJ, Chen Y-F, Bryant JD, Ragan KB, Tong CW, West LC, Bosenberg MW, Li P, Shadel GS, Upton JW, West AP. ZBP1 sequesters cGAS in the cytoplasm and sustains type I interferon responses to mitochondrial DNA (Preprint). bioRxiv PPR499779, 2022. doi: 10.1101/2022.05.30.493783. [DOI]
- 137. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Tanaka Y, Hirokawa N, Nangaku M, Inagi R. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 29: 1261–1273.e6, 2019. doi: 10.1016/J.CELREP.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 138. Yum S, Li M, Chen ZJ. Old dogs, new trick: classic cancer therapies activate cGAS. Cell Res 30: 639–648, 2020. doi: 10.1038/s41422-020-0346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, Morlock LK, García-Sastre A, Leung DW, Williams NS, Fernandez-Sesma A, Bukreyev A, Basler CF. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing Ebola virus immune evasion. mBio 8: e00368-17, 2017. doi: 10.1128/MBIO.00368-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Loppnow H, Werdan K, Werner C. The enhanced plasma levels of soluble tumor necrosis factor receptors (sTNF-R1; sTNF-R2) and interleukin-10 (IL-10) in patients suffering from chronic heart failure are reversed in patients treated with beta-adrenoceptor antagonist. Auton Autacoid Pharmacol 22: 83–92, 2002. doi: 10.1046/J.1474-8673.2002.00245.X. [DOI] [PubMed] [Google Scholar]
- 141. Pellegrini C, Martelli A, Antonioli L, Fornai M, Blandizzi C, Calderone V. NLRP3 inflammasome in cardiovascular diseases: pathophysiological and pharmacological implications. Med Res Rev 41: 1890–1926, 2021. doi: 10.1002/MED.21781. [DOI] [PubMed] [Google Scholar]
- 142. Wei S, Ma W, Li X, Jiang C, Sun T, Li Y, Zhang B, Li W. Involvement of ROS/NLRP3 inflammasome signaling pathway in doxorubicin-induced cardiotoxicity. Cardiovasc Toxicol 20: 507–519, 2020. doi: 10.1007/S12012-020-09576-4. [DOI] [PubMed] [Google Scholar]
- 143. Sun Z, Lu W, Lin N, Lin H, Zhang J, Ni T, Meng L, Zhang C, Guo H. Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochem Pharmacol 175: 113888, 2020. doi: 10.1016/J.BCP.2020.113888. [DOI] [PubMed] [Google Scholar]
- 144. Zhenzhu S, Xingxiao H, Na L, Wenqiang L, Hangyuan G. Dihydromyricin alleviates doxorubicin-induced myocardial injury by inhibiting NLRP3 inflammasome in rats. Zhonghua Bing Li Xue Za Zhi 49: 1046–1051, 2020. doi: 10.3760/CMA.J.CN112151-20200108-00020. [DOI] [PubMed] [Google Scholar]
- 145. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10: 128, 2019. doi: 10.1038/s41419-019-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhang Z, Zhang H, Li D, Zhou X, Qin Q, Zhang Q. Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J Cell Mol Med 25: 8159–8168, 2021. doi: 10.1111/JCMM.16574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J 475: 839–852, 2018. doi: 10.1042/BCJ20170714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Choong C-J, Okuno T, Ikenaka K, Baba K, Hayakawa H, Koike M, Yokota M, Doi J, Kakuda K, Takeuchi T, Kuma A, Nakamura S, Nagai Y, Nagano S, Yoshimori T, Mochizuki H. Alternative mitochondrial quality control mediated by extracellular release. Autophagy 17: 2962–2974, 2021. doi: 10.1080/15548627.2020.1848130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sharma BR, Karki R, Kanneganti TD. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur J Immunol 49: 1998–2011, 2019. doi: 10.1002/eji.201848070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, Ren J. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis 12: 673, 2021. doi: 10.1038/s41419-021-03961-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, Aung LHH, Li P. F, Yu T, Chu X. M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis 11: 776, 2020. doi: 10.1038/s41419-020-02985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, Drummer C, Johnson C, Xu K, Jiang X, Wang H, Yang X. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol 40: e138–e152, 2020. doi: 10.1161/ATVBAHA.120.314330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Klee NS, McCarthy CG, Martinez-Quinones P, Webb RC. Out of the frying pan and into the fire: damage-associated molecular patterns and cardiovascular toxicity following cancer therapy. Ther Adv Cardiovasc Dis 11: 297–317, 2017. doi: 10.1177/1753944717729141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Heijink IH, Pouwels SD, Leijendekker C, De Bruin HG, Zijlstra GJ, Van Der Vaart H, Ten Hacken NHT, Van Oosterhout AJM, Nawijn MC, Van Der Toorn M. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 52: 554–562, 2015. [Erratum in Am J Respir Cell Mol Biol 54: 597–599, 2016]. doi: 10.1165/RCMB.2013-0505OC. [DOI] [PubMed] [Google Scholar]
- 156. De Vos WM, Tilg H, Hul M, Van Cani PD. Gut microbiome and health: mechanistic insights. Gut 71: 1020–1032, 2022. doi: 10.1136/gutjnl-2021-326789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res 30: 492–506, 2020. doi: 10.1038/s41422-020-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 306: H184–H196, 2014. doi: 10.1152/ajpheart.00328.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107: 119–130, 2015. doi: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. [Erratum in Nature 490: 292, 2012]. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, Olsson AK. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res 75: 2653–2662, 2015. doi: 10.1158/0008-5472.CAN-14-3299. [DOI] [PubMed] [Google Scholar]
- 162. Guan T, Zhang H, Yang J, Lin W, Wang K, Su M, Peng W, Li Y, Lai Y, Liu C. Increased risk of cardiovascular death in breast cancer patients without chemotherapy or (and) radiotherapy: a large population-based study. Front Oncol 10: 619622, 2021. doi: 10.3389/FONC.2020.619622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep 3: 1077, 2013. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hosseini H, Li Y, Kanellakis P, Tay C, Cao A, Liu E, Peter K, Tipping P, Toh BH, Bobik A, Kyaw T. Toll-like receptor (TLR)4 and MyD88 are essential for atheroprotection by peritoneal B1a B cells. J Am Heart Assoc 5: e002947, 2016. doi: 10.1161/JAHA.115.002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, Hashiguchi T, Maruyama I. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost 5: 109–116, 2007. doi: 10.1111/j.1538-7836.2006.02255.x. [DOI] [PubMed] [Google Scholar]
- 166. Ishikawa T, Abe K, Takana-Ishikawa M, Yoshida K, Watanabe T, Imakiire S, Hosokawa K, Hirano M, Hirano K, Tsutsui H. Chronic inhibition of toll-like receptor 9 ameliorates pulmonary hypertension in rats. J Am Heart Assoc 10: e019247, 2021. doi: 10.1161/JAHA.120.019247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ueda H, Yamaguchi O, Taneike M, Akazawa Y, Wada-Kobayashi H, Sugihara R, Yorifuji H, Nakayama H, Omiya S, Murakawa T, Sakata Y, Otsu K. Administration of a TLR9 inhibitor attenuates the development and progression of heart failure in mice. JACC Basic Transl Sci 4: 348–363, 2019. doi: 10.1016/j.jacbts.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Liang CF, Liu JT, Wang Y, Xu A, Vanhoutte PM. Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arterioscler Thromb Vasc Biol 33: 777–784, 2013. doi: 10.1161/ATVBAHA.112.301087. [DOI] [PubMed] [Google Scholar]
- 169. McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, Webb RC. Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. Am J Hypertens 30: 173–181, 2017. doi: 10.1093/ajh/hpw113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35: 5931–5941, 2016. doi: 10.1038/onc.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12: 860–875, 2012. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 172. Hayashi K, Nikolos F, Lee YC, Jain A, Tsouko E, Gao H, Kasabyan A, Leung HE, Osipov A, Jung SY, Kurtova AV, Chan KS. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat Commun 11: 6299, 2020. doi: 10.1038/s41467-020-19970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13: 54–61, 2007. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 174. González-Reyes S, Fernández JM, González LO, Aguirre A, Suárez A, González JM, Escaff S, Vizoso FJ. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol Immunother 60: 217–226, 2011. doi: 10.1007/s00262-010-0931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29: 5299–5310, 2010. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 17: 666–676, 2010. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Rajput S, Volk-Draper LD, Ran S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther 12: 1676–1687, 2013. doi: 10.1158/1535-7163.MCT-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhou J, Chen X, Gilvary DL, Tejera MM, Eksioglu EA, Wei S, Djeu JY. HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Sci Rep 5: 15085, 2015. doi: 10.1038/srep15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Xia W, Chen H, Xie C, Hou M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging (Albany NY) 12: 8241–8260, 2020. doi: 10.18632/AGING.103136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Xuan L, Sun L, Zhang Y, Huang Y, Hou Y, Li Q, Guo Y, Feng B, Cui L, Wang X, Wang Z, Tian Y, Yu B, Wang S, Xu C, Zhang M, Du Z, Lu Y, Yang BF. Circulating long non-coding RNAs NRON and MHRT as novel predictive biomarkers of heart failure. J Cell Mol Med 21: 1803–1814, 2017. doi: 10.1111/JCMM.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Pandal P, Shulkin A, Vazquez E, Cortez-Toledo E, Ramos D, Vargas CR, Wilson M, Durbin-Johnson BP, Britton M, Settles ML, López JE. Early expression of circulating long non-coding RNA correlates with the risk of 1-year mortality after acute myocardial infarction. J Am Coll Cardiol 75: 186, 2020. doi: 10.1016/s0735-1097(20)30813-5. [DOI] [Google Scholar]
- 182. Gomes CPC, Schroen B, Kuster GM, Robinson EL, Ford K, Squire IB, Heymans S, Martelli F, Emanueli C, Devaux Y; EU-CardioRNA COST Action (CA17129). Regulatory RNAs in heart failure. Circulation 141: 313–328, 2020. doi: 10.1161/CIRCULATIONAHA.119.042474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Nukala SB, Jousma J, Cho Y, Lee WH, Ong SG. Long non-coding RNAs and microRNAs as crucial regulators in cardio-oncology. Cell and Bioscience 12: 24, 2022. doi: 10.1186/S13578-022-00757-Y/TABLES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Rhee JW, Ky B, Armenian SH, Yancy CW, Wu JC. Primer on biomarker discovery in cardio-oncology: application of omics technologies. JACC CardioOncol 2: 379–384, 2020. doi: 10.1016/J.JACCAO.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Bär C, Chatterjee S, Pires IF, Rodrigues P, Sluijter JPG, Boon RA, Nevado RM, Andrés V, Sansonetti M, de Windt L, Ciccarelli M, Hamdani N, Heymans S, Videira RF, Tocchetti CG, Giacca M, Zacchigna S, Engelhardt S, Dimmeler S, Madonna R, Thum T. Non-coding RNAs: update on mechanisms and therapeutic targets from the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc Res 116: 1805–1819, 2020. [Erratum in Cardiovasc Res 117: 177, 2021]. doi: 10.1093/CVR/CVAA195. [DOI] [PubMed] [Google Scholar]
- 186. Ma C, Beyer AM, Durand M, Clough AV, Zhu D, Toro LN, Terashvili M, Ebben JD, Hill RB, Audi SH, Medhora M, Jacobs ER. Hyperoxia causes mitochondrial fragmentation in pulmonary endothelial cells by increasing expression of pro-fission proteins. Arterioscler Thromb Vasc Biol 38: 622–635, 2018. doi: 10.1161/ATVBAHA.117.310605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Chabowski DS, Kadlec AO, Ait-Aissa K, Hockenberry JC, Pearson PJ, Beyer AM, Gutterman DD. Lysophosphatidic acid acts on LPA1 receptor to increase H2O2 during flow-induced dilation in human adipose arterioles. Br J Pharmacol 175: 4266–4280, 2018. doi: 10.1111/BPH.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Kadlec AO, Chabowski DS, Ait-Aissa K, Hockenberry JC, Otterson MF, Durand MJ, Freed JK, Beyer AM, Gutterman DD. PGC-1α overexpression in coronary artery disease recruits nitric oxide and hydrogen peroxide during flow-mediated dilation and protects against increased intraluminal pressure. Hypertension 70: 166–173, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ait-Aissa K, Kadlec AO, Hockenberry J, Gutterman DD, Beyer AM. Telomerase reverse transcriptase protects against angiotensin ii-induced microvascular endothelial dysfunction. Am J Physiol Heart Circ Physiol 314: H1053–H1060, 2018. doi: 10.1152/ajpheart.00472.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Durand MJ, Zinkevich NS, Riedel M, Gutterman DD, Nasci VL, Salato VK, Hijjawi JB, Reuben CF, North PE, Beyer AM. Vascular actions of angiotensin 1-7 in the human microcirculation. Arterioscler Thromb Vasc Biol 36: 1254–1262, 2016. doi: 10.1161/ATVBAHA.116.307518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Beyer AM, Freed JK, Durand MJ, Riedel M, Ait-Aissa K, Green P, Hockenberry JC, Garret Morgan R, Donato AJ, Peleg R, Gasparri M, Rokkas CK, Santos JH, Priel E, Gutterman DD. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res 118: 856–866, 2016. doi: 10.1161/CIRCRESAHA.115.307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Freed JK, Beyer AM, Logiudice JA, Hockenberry JC, Gutterman DD. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res 115: 525–532, 2014. doi: 10.1161/CIRCRESAHA.115.303881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Biener M, Giannitsis E, Thum T, Bär C, Stoyanov KM, Salbach C, de Gonzalo-Calvo D, Frey N, Mueller-Hennessen M. Prognostic value of circulating microRNAs compared to high-sensitivity troponin T in patients presenting with suspected acute coronary syndrome to the emergency department. Clin Biochem 99: 9–16, 2022. doi: 10.1016/j.clinbiochem.2021.09.009. [DOI] [PubMed] [Google Scholar]
- 194. Viereck J, Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ Res 120: 381–399, 2017. doi: 10.1161/CIRCRESAHA.116.308434. [DOI] [PubMed] [Google Scholar]
- 195. Bandiera S, Rüberg S, Girard M, Cagnard N, Hanein S, Chrétien D, Munnich A, Lyonnet S, Henrion-Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One 6: e20746, 2011. doi: 10.1371/journal.pone.0020746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Jusic A, Devaux Y; EU-CardioRNA COST Action (CA17129). Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res Cardiol 115: 23, 2020. doi: 10.1007/s00395-020-0783-5. [DOI] [PubMed] [Google Scholar]
- 197. Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LOF, Moore KJ, Suárez Y, Lai EC, Fernández-Hernando C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA 108: 9232–9237, 2011. doi: 10.1073/PNAS.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wang T, Li M, Guan J, Li P, Wang H, Guo Y, Shuai S, Li X. MicroRNAs miR-27a and miR-143 regulate porcine adipocyte lipid metabolism. Int J Mol Sci 12: 7950–7959, 2011. doi: 10.3390/IJMS12117950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Thummasorn S, Apaijai N, Kerdphoo S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc Ther 34: 404–414, 2016. doi: 10.1111/1755-5922.12210. [DOI] [PubMed] [Google Scholar]
- 200. Thummasorn S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. High-dose Humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc Ther 35: e12289, 2017. doi: 10.1111/1755-5922.12289. [DOI] [PubMed] [Google Scholar]
- 201. Jia Y, Lue YH, Swerdloff R, Lee KW, Cobb LJ, Cohen P, Wang C. The cytoprotective peptide humanin is induced and neutralizes Bax after pro-apoptotic stress in the rat testis. Andrology 1: 651–659, 2013. doi: 10.1111/J.2047-2927.2013.00091.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Yang Y, Gao H, Zhou H, Liu Q, Qi Z, Zhang Y, Zhang J. The role of mitochondria-derived peptides in cardiovascular disease: recent updates. Biomed Pharmacother 117: 109075, 2019. doi: 10.1016/j.biopha.2019.109075. [DOI] [PubMed] [Google Scholar]
- 203. Lue Y, Gao C, Swerdloff R, Hoang J, Avetisyan R, Jia Y, Rao M, Ren S, Atienza V, Yu J, Zhang Y, Chen M, Song Y, Wang Y, Wang C. Humanin analog enhances the protective effect of dexrazoxane against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol 315: H634–H643, 2018. doi: 10.1152/ajpheart.00155.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Moreno Ayala MA, Gottardo MF, Zuccato CF, Pidre ML, Nicola Candia AJ, Asad AS, Imsen M, Romanowski V, Creton A, Isla Larrain M, Seilicovich A, Candolfi M. Humanin promotes tumor progression in experimental triple negative breast cancer. Sci Rep 10: 8542, 2020. doi: 10.1038/s41598-020-65381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]