

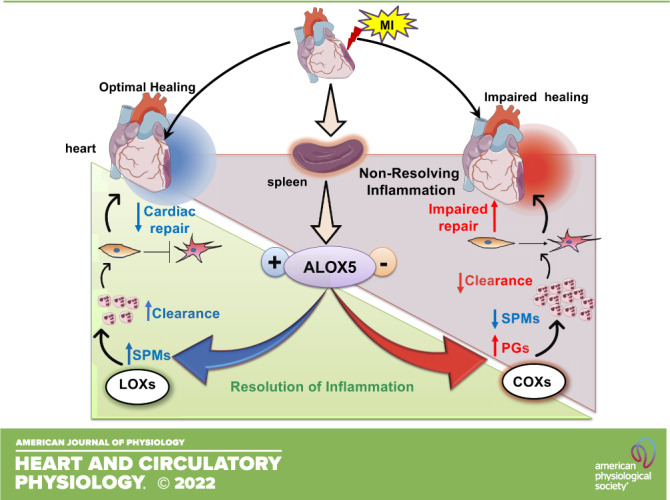
Keywords: fibroblast signaling, inflammation, non-resolving inflammation, resolution of inflammation, specialized pro-resolving mediators
Abstract
Arachidonate 5-lipoxygenase (ALOX5)-derived leukotrienes are primary signals of leukocyte activation and inflammation in response to ischemic cardiac injury (MI; myocardial infarction). Using risk-free male C57BL/6J and ALOX5-null mice (8–12 wk), we quantitated leukocytes and ALOX5-derived bioactive lipids of the infarcted left ventricle (LV) and spleen to measure the physiological inflammation and cardiac repair. Our results showed that ALOX5 endogenously generates specialized pro-resolving mediators (SPMs) that facilitate cardiac repair post-MI. Deficiency of ALOX5 leads to increase in cyclooxygenase gene expression, 6-keto prostaglandin F1α, and delayed neutrophil clearance with signs of unresolved inflammation post-MI. Consequently, ALOX5 deficiency impaired the resolution of inflammation and cardiac repair, including increased myocardium rupture post-MI in acute heart failure. On-time ALOX5 activation is critical for leukocyte clearance from the infarcted heart, indicating an essential role of ALOX5 in the resolution of inflammation. In addition, to balance the inflammatory responses, ALOX5 is also necessary for fibroblast signaling, as the ALOX5-deficient fibroblast are prone to fibroblast-to-myofibroblast differentiation leading to defective scar formation in post-MI cardiac repair. Consistent with these findings, ALOX5-null mice showed an overly inflammatory response, defective fibrotic signaling, and unresolved inflammation. These findings are indicative of a critical role of ALOX5 in myocardium healing, inflammation-resolution signaling, cardiac repair, and fibroblast pathophysiology.
NEW & NOTEWORTHY Arachidonate 5-lipoxygenase (ALOX5) is critical in synthesizing specialized pro-resolving mediators that facilitate cardiac repair after cardiac injury. Thus, ALOX5 orchestrates the overlapping phases of inflammation and resolution to facilitate myocardium healing in cardiac repair postmyocardial infarction.
INTRODUCTION
Inflammation is a universal feature of acute and chronic heart failure (CHF) following myocardial infarction (MI) (1–4); however, the physiological inflammation remains of interest to the resolution of inflammation and cardiac repair post-MI (5). After the myocardial injury, the timely initiation of an acute inflammatory response is necessary to clear injured/dead cardiomyocytes. The inflammation clearance process requires the biosynthesis of long-chain fatty acid-derived specialized pro-resolving mediators (SPMs) that act to prevent the collateral myocardium damage after ischemic injury (2, 5–7). Immune-responsive lipoxygenase family (LOX 5, 12, and 15), particularly arachidonate 5-lipoxygenase (ALOX5; synonym: 5LOX) expressed in activated leukocytes, catalyzes the endogenous biosynthesis of SPMs in the infarcted heart and spleen to clear the inflammation (8, 9). Traditionally, it was thought that the primary function of ALOX5 involved the enzymatic conversion of arachidonic acid (AA) to leukotrienes (LTs), which are potent inflammatory mediators (10–13). ALOX5-derived leukotriene B4 (LTB4) is a proinflammatory chemoattractant generated from arachidonic acid through serial activities of ALOX5-activating protein and leukotriene A4 hydrolase (12–15). Physiological inflammation is an essential signal of myocardium repair and tissue homeostasis because unresolved inflammation could drive pathological remodeling. Therefore, identifying the enzymes that catalyze the biosynthesis of resolution-promoting bioactive molecules is needed to develop a strategy for promoting resolution and delaying HF post-MI. Here, we tested whether ALOX5 is necessary to facilitate the inflammation–resolution axis during physiological healing in cardiac repair post-MI.
ALOX5, an immune-responsive metabolic enzyme expressed in neutrophils, macrophages, and dendritic cells, generates lipid mediators (LMs) during post-MI healing (14, 16). ALOX5 is highly expressed in immune cells, using a global deletion model that allowed us to assess the role of ALOX5 in spleen as immune cell reservoir, because splenic leukocyte migrates to infarct injury for the repair process after ischemic insult (6, 17). Emerging evidence indicates that ALOX5 expression is increased in certain pathological conditions such as patients infected with COVID-19 (18). It has been shown that ALOX5 promotes atherosclerosis with the enrichment of arachidonic acid in the diet, suggesting a possible nutrition-genetics interaction (19). Also, a genomic study indicates the linkage to chromosome 13q12–13 in ALOX5 that doubles the risk of MI and stroke based on the neutrophil secretive capacity to produce LTB4 (20). Our recent study confirms that post-MI acute inflammatory response coincides with resolving response with ALOX5-derived lipoxins (LXs) within 24 h (6). Clinical studies have suggested that reduced LXs promote chronic HF in subjects with marked signs of non-resolving inflammation post-MI (21). Lipoxin A4 (LXA4) and LXB4 were first identified in human leukocytes (22), and further studies identified aspirin-induced 15-epi-LXA4 and 15-epi-LXB4. LXs are produced with the collaborative effort of leukocytes, platelets as substrate donor cells, and endothelial cells as product acceptor cells (9, 23). Furthermore, in the context of HF, LTB4 is immediately biosynthesized in the infarcted area as the proinflammatory mediators; but the quantity, kinetics, and mechanisms of leukotrienes (LTs) action are not completely elucidated in HF (24, 25). The current study is designed to determine whether ALOX5-derived LTB4 is directly involved in impairing cardiac repair and non-resolving inflammation to promote HF post-MI. Our results indicate that ALOX5 directs the leukocytes-mediated generation of proinflammatory mediators (PIMs) but is also critically involved in the biosynthesis of SPMs in acute HF, supporting cardiac repair and expediting the inflammation resolution process. We found that ALOX5 simultaneously initiates the biosynthesis of SPMs, including LXs, to facilitate the resolution program in HF.
Consequently, our hypothesis that ALOX5-derived LTB4 failed to promote unresolved inflammation was not confirmed. Instead, we found that ALOX5 deficiency reduced the levels of SPMs, which in turn impaired the resolution of inflammation. Thus, ALOX5 is essential for SPMs biosynthesis and cardiac repair, and strategies that maintain or activate ALOX5 were required for the cardiac tissue repair program post-MI.
MATERIALS AND METHODS
Overall Study Design
The study aimed to define the role of ALOX5 in inflammation-resolution signaling and the cardiac repair process. The project was designed to determine that ALOX5-derived leukotrienes act as a prime downstream mediator of unresolved inflammation. We used patients and mice tissue samples to demonstrate that multiple resolving mediator’s biosynthesis is dependent on ALOX5 activation during the cardiac repair process.
Study Approval
All animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals (8th ed., 2011) and AVMA Guidelines for the Euthanasia of Animals (2013 ed.) and were approved by the Institutional Animal Care and Use Committee at the University of South Florida (Tampa, FL). Human tissue and plasma (patients and age-matched controls; demographics provided in Supplemental Tables S1 and S2; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.19293287.v3) use was approved by the Institutional Review Board, the University of Alabama at Birmingham. Cardiac patient tissue samples with active heart failure (n = 12) and control (n = 9) were obtained from the biorepository of the University of Alabama at Birmingham. The healthy tissues were taken from individuals who had accidental death with no history of heart disease or gunshot (depending on availability). The pathological human heart samples were from the apex area that was removed for placement of left ventricular assist device (LVAD). An additional set of plasma samples from control (n = 10) and patients with an ST-elevation myocardial infarction (STEMI) (n = 12) were used for lipid mediators analyses. All the patients were selected based on clinical diagnosis, ethical compliance, and laboratory information.
Mouse Coronary Ligation Surgery and Postsurgery Exclusion Criteria
Risk-free male C57Bl/6J mice (Stock No. 000664) and ALOX5−/− mice (Alox5tm1Fun, Stock No. 004155) of 8–12 wk were purchased from Jackson Laboratory and were maintained under controlled temperature (19.8°C–22.2°C). The mice were given free access to water and a standard chow diet. For initiation of an intense inflammation in the myocardium, mice were subjected to the surgical ligation of the left anterior descending coronary artery, as previously described (26, 27). Mice with postsurgery fractional shortening above 10% at day 1 were excluded from the study to maintain a consistent irreversible HF survivor cohort in wild-type (WT) and ALOX5−/− groups (28, 29).
Autopsy and Survivor Inclusion Criteria for Post-MI Survival Analysis
The mice were monitored twice daily for 56 days post-MI for survival analysis. Mice that died within 24 h of surgery were excluded from the survivor analyses and were specified as perioperative mortality (5%–8% for both genotypes). Mice died after 24 h, and before day 56, an autopsy was performed to determine the cause of death, either rupture or congestive heart failure. At autopsy, the cardiac rupture was confirmed by the presence of a clotted blood slit in the thoracic cavity, which likely occurred 3 to 6 days post-MI, and by direct microscopic identification of the left ventricle (LV) rupture site as previously described (26, 29, 30).
Heart Function Using High-Resolution Echocardiography
Heart function and geometry of WT and ALOX5−/− mice were measured by echocardiography with the parasternal long-axis and M-mode images using high-resolution ultrasound (VisualSonics Vevo 3100) as previously described (26). Pre-MI as a baseline or no-MI naïve control day 0 and postligation surgery temporal measurements were made at day 1, day 5 (acute heart failure), and day 56 (as chronic heart failure) (26). During the procedure, mice were anesthetized using isoflurane (1.5%–2%) and maintained at a physiological heart rate of 450–500 beats/min at 37 ± 1°C temperature and for still or video recordings that were subsequently analyzed in a blinded and randomized manner with respect to genotypes (26, 29).
Necropsy and Infarct Area Analysis
The infarct area was calculated as the percentage of infarct area in the total LV area, as previously reported (26, 29). As detailed in our previous reports, tissue and blood samples were collected and reserved for further molecular and cellular mass spectrometry analyses (30).
Flow Cytometry
Single mononuclear cells were isolated from no-MI controls (day 0), MI (day 1), and MI (day 5) from the LV and spleen, and quantitated as previously described (Supplemental Fig. S1; gating strategy) (6, 29, 31).
LV Multispectral Flow Cytometry (ImageStream)
Mononuclear cells were isolated from the LV as previously described in the flow cytometry protocol (30). Surface staining for macrophages was done using F4/80-PERCPCy5.5 (Thermo Fisher Scientific), and for neutrophils using Ly6G-pacific blue (eBioscience, San Diego, CA) for 30 min at 4°C and protected from light. Cells were then washed with 1× annexin-binding buffer. After the wash, cells were resuspended in 100 µL of 1× annexin-binding buffer, and 1 µL annexin V was added to the sample for 15 min at room temperature. Samples were washed with 1× annexin-binding buffer and resuspended in 100 µL of 1× annexin binding buffer containing 1 µL of propidium iodide (PI). Finally, samples were processed using ImageStream X and analyzed with IDEAS 6.2 software (Amnis) (6, 32).
LC-MS/MS Analysis from Human Plasma and Mice Spleen and Infarcted LV
Human plasma, left ventricle protein samples, mouse spleen, and left ventricle tissues were processed using solid-phase extraction methodology before mass spectrometry (30, 32, 33). Processed samples were introduced using HPLC to the electrospray ionization source of the QTrap6500+ (ABSciex, Farmingham, MA) mass analyzer in the negative ion mode as previously described (32, 34). Briefly, 100 pg of internal labeled standard mixture containing resolvin D (RvD)1-deuterated (d)5, RvD2-d5, RvD3-d5, LXA4-d5, RvE1-d4, Maresin-1-d5, 11,12-EET-d11, 14,15-EET-d11, 5-hydroxyeicosatetraenoic acid (5-HETE)-d8, 12-HETE-d8, 15-HETE-d8, prostaglandin E2 (PGE2)-d4, prostaglandin D2 (PGD2)-d9, LTB4-d5, LTC4-d5, LTE4-d5, docosahexaenoic acid (DHA)-d5, docosapentaenoic acid-d5 (DPA-d5), eicosapentaenoic acid (EPA)-d5, and AA-d5 in 1 mL of methanol were added to the previously weighed tissue samples. After homogenization, the tissue extract was kept in the dark in ice for 45 min for the protein precipitation. The tissue extract was diluted in LC-MS grade water before running through the solid phase extraction column (Strata-X Polymeric Reversed Phase). The lipid mediators were eluted in methanol and dried under a low stream of N2 before resuspending in (50:50 vol/vol) methanol-water for HPLC injection. A kinetex polar C18 LC column (100 mm × 3 mm × 2.6 µm, Phenomenex) was used for the separation, and LMs were eluted using a gradient elution of methanol-water-formic acid over an 18-min run at a flow rate of 500 µL/min. A scheduled multiple reaction monitoring (MRM) method and enhanced product ion scan (EPI) were used to profile each lipid mediator. The calibration curves for each of the LM were obtained using authentic LM standard mixtures at concentrations of 0.23, 0.69, 2.1, 6.2, 18.5, 55.6, 166.7, and 500 pg each having 100 pg of deuterated internal standard mixture. The linear calibration curve with a r2 value of 0.97–0.99 was obtained for each of the LM.
Isolation of Cardiac Fibroblasts
Cardiac fibroblasts from WT and ALOX5−/− mice (2–4 mo old) were isolated by enzymatic digestion with 600 U/mL of collagenase II and 60 U/mL of DNase I as previously described (35). Cells at passage 2 were plated in six-well plates (5 × 104 cells/well) and allowed to attach at 37°C overnight, then washed using DMEM/F12 media with 10% FBS and 1% antibiotics to remove unattached cells. Cardiac fibroblast was differentiated into myofibroblast by treatment with 15 ng/mL of Tgf-β (15 ng/mL) and coincubated either with 15-epi-LXA4 or LTB4 for 18 h (36).
Fibroblast-Myofibroblast Immunofluorescence
Mouse LV, human LV, and cardiac fibroblasts were fixed using 4% paraformaldehyde (PFA), permeabilized using 0.1% triton, and blocked for 1 h in 10% goat serum. Midcavity of mouse LV and cardiac fibroblasts were stained using anti-smooth muscle actin-α (SMA-α) antibody (Sigma-Aldrich) and DDR2 (Abcam) overnight at 4°C. After overnight incubation, these slides were further stained with Alexa-555 and Alexa 488 secondary antibodies for 1 h. Human LV was stained with ALOX5 antibody (Abcam, MA) overnight at 4°C and Alexa-555 respective secondary antibodies. Nuclei of both cell types were stained using Hoechst (molecular probe). Cells were mounted using anti-fade mounting media (Thermo Fisher Scientific, Grand Island, NY) and then visualized and microphotographed using Nikon A1 high-speed laser confocal microscope. Multiple LV midcavity images were merged using the Nikon A1 microscope’s large area stitching tool. Large image acquisition generates a single high magnification, wide field-of-view image (×40) by automatically stitching multiple adjacent frames from a multipoint acquisition using a motorized stage or from multiple single images captured manually (32, 36).
Real-Time Quantitative PCR for LV
For quantitative PCR, we used 2.5 μg of RNA to perform reverse transcription using SuperScript Vilo cDNA synthesis Kit (Thermo Fisher Scientific). Quantitative PCR for the ptgs1 (Mm00477214_m1), ptgs-2 (Mm00478374_m1), alox12 (Mm00545833_m1), alox15 (Mm00507789_m1), alox5 (Mm01182747_m1), and tgif (Mm01227699_m1) genes were performed using TaqMan probes as previously done. Gene expression was normalized with hypoxanthine phosphoribosyltransferase-1 (Hprt-1; Mm03024075_m1) as the housekeeping gene (32, 35, 36). The results were reported as fold change. All the experiments were duplicated with n = 5 or 6 animals per group/time point.
Fibroblast Wound Healing Assay
Fibroblasts (2 × 104) were seeded in six-well tissue culture plates in duplicates and grown for 72 h. A single scratch wound was made with a 200-mL pipette tip across the cell layer, and after rinsing with PBS, the repopulation of the denuded area was monitored by capturing images at 0, 12, and 24 h (37).
Statistical Analysis
Data are expressed as means ± SE per group. Statistical analyses were performed using GraphPad Prism 8. Analysis of variance (ANOVA), followed by Newman–Keuls post hoc test, was used for multiple comparisons of post-MI day 1, day 5, and day 56 compared with day 0 naïve control. The Kaplan–Meier test and the log-rank test were used for survival analysis. For two-group comparisons, the Student’s t test (unpaired) was applied, and P < 0.05 was considered statistically significant. In vitro experiments were done independently three times with technical replicates each time. For echocardiography and histological analysis, two individuals analyzed the data blindly for reproducibility and transparency.
RESULTS
ALOX5-Derived Bioactive Lipid Network Was Altered in Human Ischemic Myocardium
ALOX5 is a key immune-responsive enzyme in leukotriene B4 (LTB4) biosynthesis that serves as a leukocyte chemoattractant. To determine lipid mediators (LMs) levels in cardiac patients, particularly in HF (Supplemental Table S1, patient demographics), we quantitated the ALOX5-derived LMs in human ischemic myocardium using age-matched controls by an LC-MS/MS-based methodology. We determined that ALOX5-derived bioactive 5-hydroxyeicosatetraenoic acid (5-HETE); 15-epi lipoxin A4 (15-epi-LXA4), lipoxin A4 (LXA4), and resolvin D6 (RvD6) were lower in ischemic human myocardium than the healthy human myocardium (Fig. 1A). However, levels of ALOX5-derived resolvin precursor 17-hydroxy docosahexaenoic acid (17-HDHA) were higher (Fig. 1A) and 5-hydroxyeicosapentaenoate (5-HEPE) was not significantly changed in the ischemic myocardium (Fig. 1B). To assess the relationship between the quantity of ALOX5-derived LMs and the biosynthetic potential of ALOX5 activity for LMs production, we measured ALOX5 mRNA and protein expression in the ischemic and healthy control hearts. An ischemic heart displayed substantially lower ALOX5 protein and mRNA levels than were detected in control samples (Fig. 1, C–E). These results imply that the ischemic heart tissue has lower expression of ALOX5, which may correlate with its capacity to produce lower levels of SPMs, specifically in HF post-MI.
Figure 1.
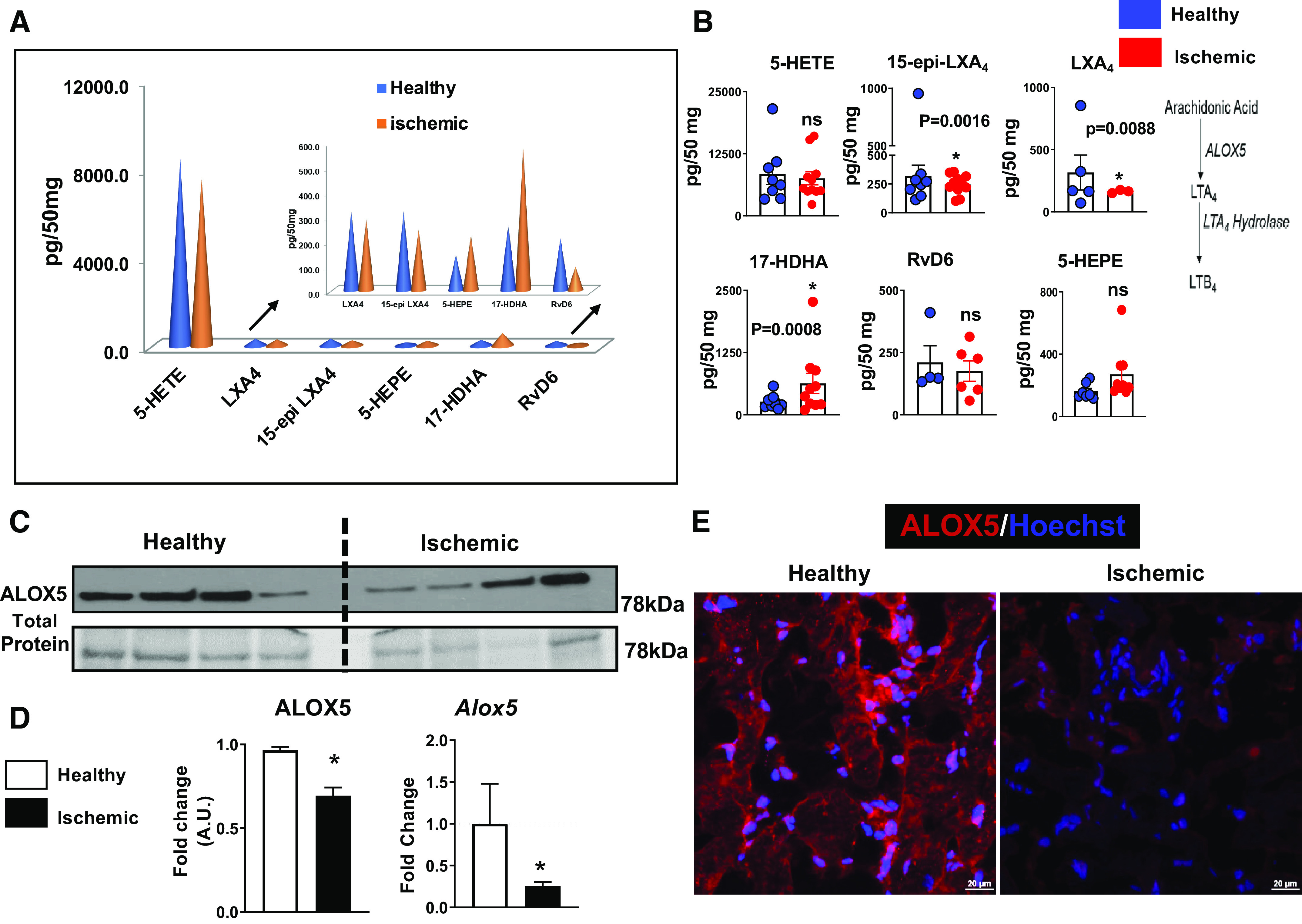
Human ischemic myocardium impairs arachidonate 5-lipoxygenase (ALOX5)-derived bioactive lipid mediators (LMs) measured using mass spectrometry. A: cone graphs displaying quantified bioactive LMs in the ischemic human heart and healthy controls. Quantification and values are pg/50 mg of left ventricle (LV) tissue (healthy human heart: n = 4–8 and ischemic human heart: n = 3–11). The detection limit was ∼1 pg. The quality controls (QC) being done for lipidomic analysis. B: bar displaying ALOX5-derived specialized pro-resolving mediators (SPMs) and LMs in the healthy and ischemic human heart (healthy human heart: n = 4–8 and ischemic human heart: n = 11; unpaired two-tailed Welch’s test was used for statistical analysis). Quantification and values are pg/50 mg of LV tissue. The detection limit was ∼1 pg. The quality controls (QC) being done for lipidomic analysis. C: immunoblot representing ALOX5 protein expression in the human healthy and ischemic control sample, n = 4 sample/group. D: bar graph displaying densitometric analysis and mRNA expression of ALOX5 protein. ALOX5 protein expression is normalized with total protein (78 kDa; n = 4); *P < 0.05 vs. healthy controls, unpaired two-tailed t test was used for statistical analysis, mRNA levels are normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT-1); n = 16–24; *P < 0.05 vs. healthy controls; unpaired two-tailed t test was used for statistical analysis. E: immunofluorescence images representing ALOX5 expression in the human heart; immunostaining was done on n = 4 independent human heart samples per group. 17-HDHA, 17-hydroxy docosahexaenoic acid; 5-HEPE, 5-hydroxyeicosapentaenoate; 5-HETE, 5-hydroxyeicosatetraenoic acid; 15-epi-LXA4, 15-epi lipoxin A4; RvD6, resolvin D6; n = no samples per group.
ALOX5-Directed Resolution Metabololipidome Dysregulated in STEMI Compared with Age-Matched Healthy Controls
In the context of ALOX5 expression and activity in the human myocardium, LTB4 was not detected in the human ischemic myocardium, and our previous reports confirmed that bioactive compounds involved in the resolution are synthesized within 24 h in mice and humans (6, 38). Therefore, to correlate the cardiac tissue LMs data to the blood plasma, we measured the plasma levels of ALOX5-derived LMs, including lipoxins and D- and E-series resolvins (collectively known as SPMs). Patients with STEMI and age-matched control subjects’ demographic characteristics are provided in Fig. 2A (Supplemental Table S2). Our assessment of plasma levels of LMs from patients with STEMI (n = 12) and age-matched controls (n = 10) revealed lower levels of ALOX5-derived intermediates and bioactive precursor 5-HEPE, with no change in 5-HETE and 17-HDHA in patients with STEMI compared with age-matched controls (Fig. 2, B–D). The SPM moiety RvD6 was undetected in STEMI plasma samples (Fig. 2E) but was readily detected in age-matched healthy controls, suggesting an activated immune response. ALOX5-derived lipoxins remained lower in STEMI plasma, indicating a failed or delayed resolution program (Fig. 2F). The quantitative plasma analysis further confirmed that multiple ALOX5-derived bioactive LMs were produced in response to ST-elevation in patients with MI compared with age-matched controls.
Figure 2.
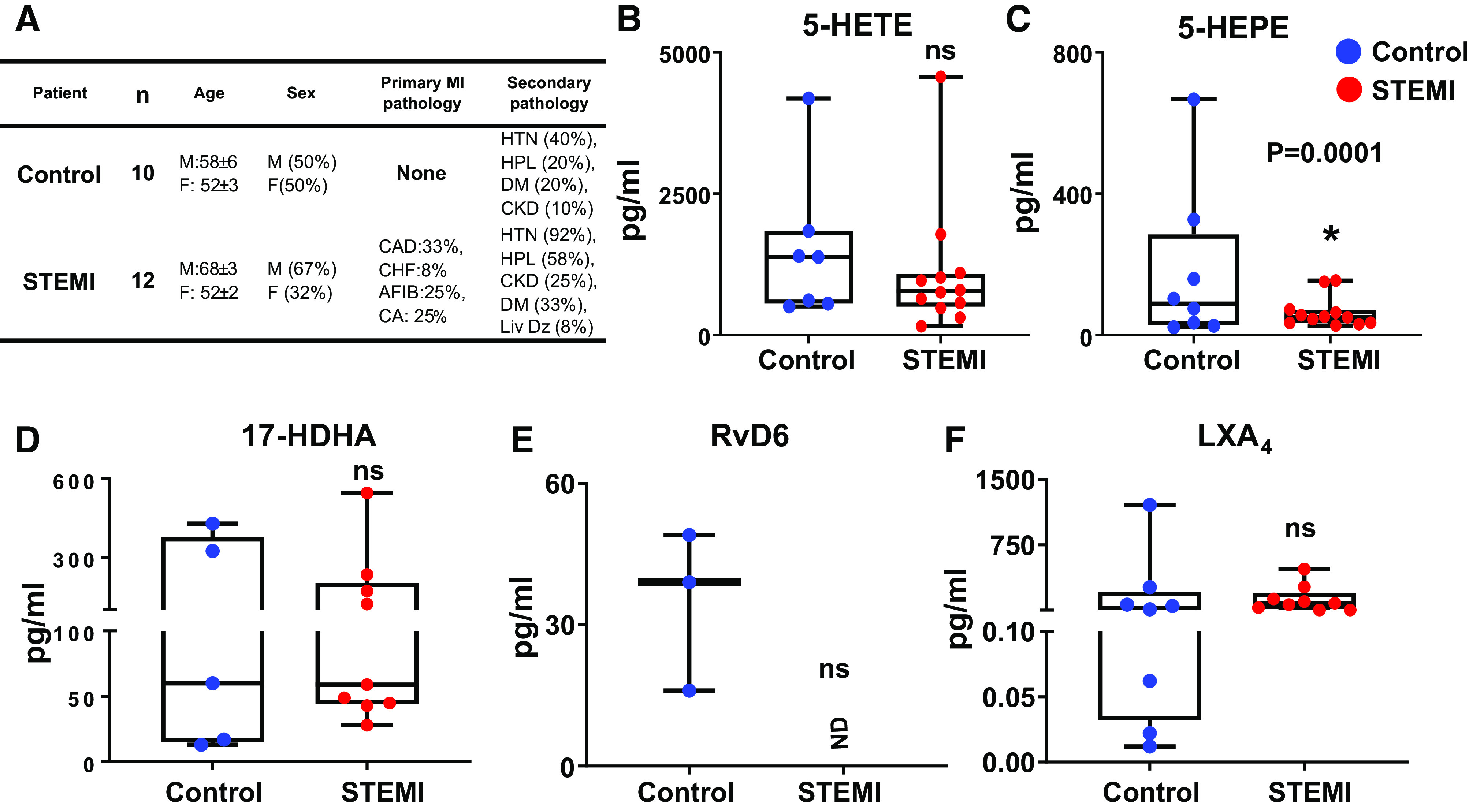
Dysregulation of arachidonate 5-lipoxygenase (ALOX5) directed resolution metabololipidome in STEMI, compared with healthy controls. Human plasma samples were harvested within 24 h post-myocardial infarction (MI) with confirmed ST-elevation and biomarker validation. A: table displaying patient demographics and age-matched controls. Whisker plots representing 5-hydroxyeicosatetraenoic acid (5-HETE, B), 5-hydroxyeicosapentaenoate (5-HEPE, C), 17-hydroxy docosahexaenoic acid (17-HDHA, D), resolvin D6 (RvD6, E), and lipoxin A4 (LXA4, F) levels in patients with elevated STEMI compared with age-matched controls; ND = not detectable; n = 3–12 patients with MI or age-matched controls/group; *P = 0.0001 vs. control, unpaired two-tailed t test was used for statistical analysis. STEMI group composed of heterogeneous patients (coronary artery disease, congestive heart failure, hypertension, cardiac arrest, chronic kidney disease, diabetes mellitus, liver disease, hyperlipidemia, etc.).
ALOX5−/− Mice Diminished SPMs Biosynthesis in the Spleen and Infarcted Myocardium post-MI
Given that biopsy of human myocardium immediately after MI and at various times was unfeasible; therefore, we expanded the further investigation to determine the role of ALOX5 in inflammation-resolution signaling using the HF mouse model. We applied an LC-MS/MS quantitative approach that determined comprehensive bioactive LMs in the spleen and infarcted LV from both WT and ALOX5−/− mice post-MI (day 1 and day 5) with respective to naïve controls (day 0) (Supplemental Tables S3, S4, and S5). In naïve WT mice, the SPMs concentration is the negligible to maximum peak value of 80 pg/50 mg in WT, with limited change in ALOX5−/− mice heart (left ventricle: LV). In response to cardiac injury, SPMs biosynthesis peaked at day 1 with a marked increase of resolution markers [LXA4, macrophage-derived mediator of inflammation resolution (MaR1), protectin D1 (PD1), and RvD5) in the infarcted tissue in WT however, there was no change in the levels of SPMs post-MI (day 1 and day 5) in ALOX5−/− infarcted LV (Fig. 3A). The pie chart shows a similar trend, with a lower level of SPMs in ALOX5−/− mice (25%) compared with WT mice (75%) at day 1 post-MI, indicative of a defective resolution phase. Furthermore, at day 5 post-MI, the SPMs were diminished in the injured heart in WT mice suggesting termination of the inflammation-resolution phase (Fig. 3B). Spleen is a major leukocyte reservoir, thus inherits a higher concentration of SPMs than LV (6). Similar to LV, SPMs concentration increased post-MI day 1 in WT-spleen, which diminished at day 5 post-MI. However, the SPMs level in ALOX5−/− spleen was further diminished post-MI day 1 and day 5 (Fig. 3C). The pie chart reflected the overall percent changes in SPMs of WT versus ALOX5−/− in spleen (Fig. 3D). The MS spectra of PD1 in infarcted LV post-MI day 1 are provided in Fig. 3E and overall concentration of SPMs is provided in Supplemental Tables S3, S4, and S5. These quantitative outcomes of resolution mediators confirmed that ALOX5 is essential for the biosynthesis of SPMs and inflammation-resolution signaling that plays a crucial role in cardiac repair post-MI.
Figure 3.
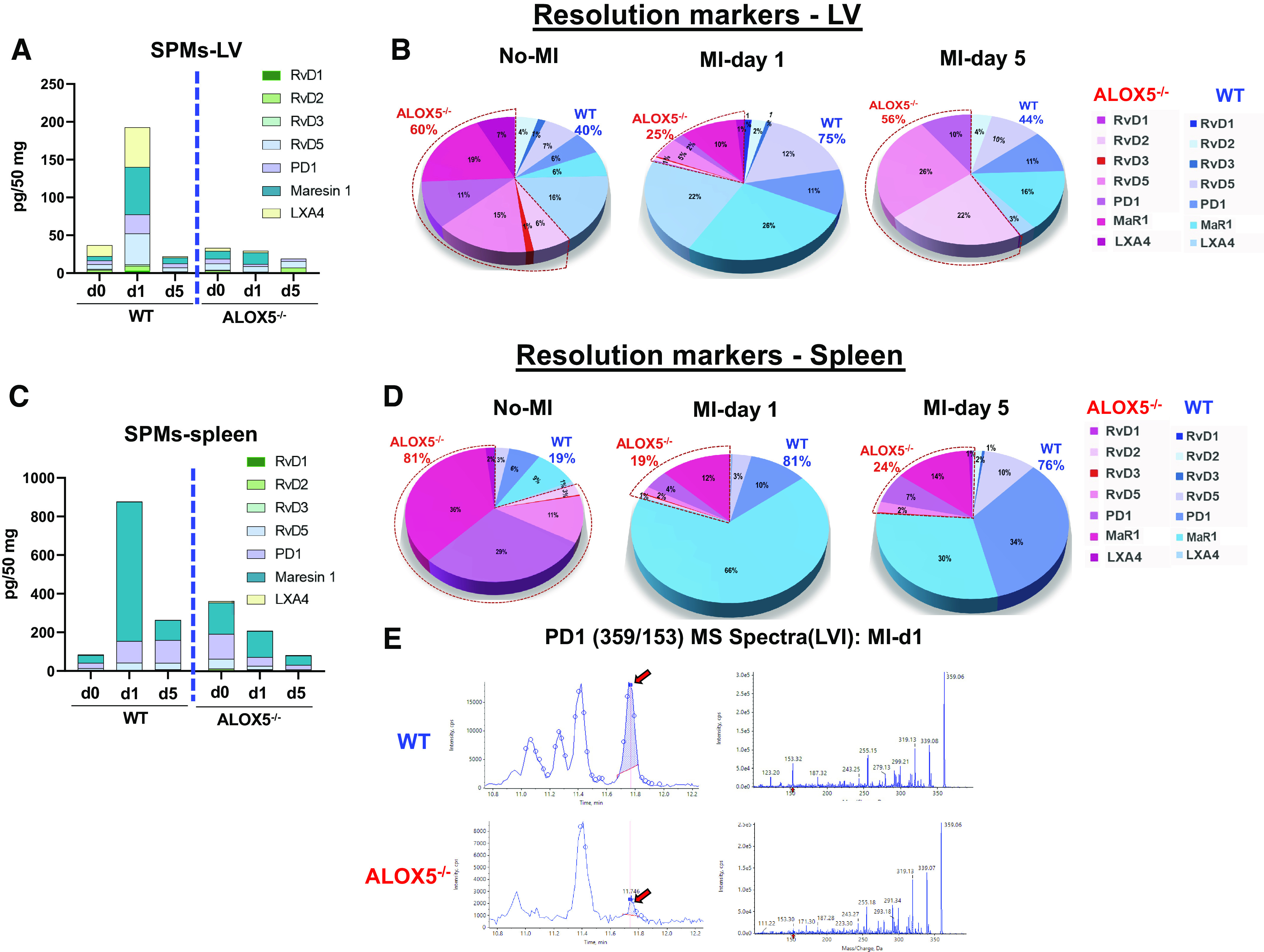
Arachidonate 5-lipoxygenase (ALOX5) deficiency lowered specialized pro-resolving mediators (SPMs) in infarcted left ventricle (LV) and spleen post-myocardial infarction (MI). Left ventricles and spleen were collected after coronary ligation-induced MI-operated mice, and myocardium tissue was harvested from infarct zones and quantitated lipid mediators (LMs) compared with naïve controls (no-MI). Interleaved graphs of LV SPMs (A), LV SPMs pie chart (B), interleaved graphs of spleen SPMs (C), and spleen SPMs pie chart (D) represent the percent composition (averaged) of resolvin D (RvD)1, RvD2, RvD3, RvD4, RvD5, protectin D1 (PD1), macrophage-derived mediator of inflammation resolution (MaR1), lipoxin A4 (LXA4) at day 0 naïve control, day 1, and day 5 post-MI in wild-type (WT) and ALOX5−/− mice. E: MS spectra of PD in infarcted LV at MI-day 1 in WT and ALOX5−/− mice. Quantification and values are pg/50 mg of LV tissue. The detection limit was ∼1 pg determined using mass spectrometry; n = 5 per group/day for both genotypes; Values are the average of each SPMs.
ALOX5 Deletion Resulted in Compensatory Biosynthesis of Proinflammatory Mediators in LV post-MI
Inflammation-resolution mediators biosynthesis is an overlapping process of cardiac repair (6), thus, both SPMs and proinflammatory mediators (PIMs) are quantitated and organized in a segregated manner. The increase in LTB4 is an obvious change, and the WT-LV showed an increase in LTB4 at day 1 post-MI, however, instead of complete abrogation of LTB4 in ALOX5−/− LV, lower levels were noted, might be due to nonenzymatic biosynthesis of LTB4 in ALOX5−/− mice. Spleen LTB4 content of ALOX5−/− mice was higher in the absence of injury or infection, suggestive of immune cell-specific biosynthesis (Supplemental Table S3). Instead, there was a dramatic increase in the levels of prostaglandins species (PGD2, PGE2, PGF2a, and 6 K-PGF1a) in LV post-MI from day 1 to day 5 in ALOX5−/− mice infarcted LV compared with the respective WT mice (Fig. 4A). The pie chart showed amplified PGs in ALOX5−/− mice (67%) at day 1 and further amplified feed-forward at day 5 compared with WT (33%), suggestive of amplified PIMs milieu at the site of injury post-MI (Fig. 4B). Likewise, PGs were amplified in the spleen at day 1 in ALOX5 mice compared with WT, suggestive of intensified inflammation (Fig. 4, C and D). The MS spectra of PGE2 in infarcted LV post-MI day 1 are provided in Fig. 4E, and the overall concentration of PIMs is provided in Supplemental Tables S3, S4, and S5. Thus, genetic deficiency of ALOX5 in mice increased PIMs (PGD2, PGE2, PGF2a, and 6 K-PGF1a) suggestive of increased proinflammatory response in the spleen (day 1) and heart (day 1 and day 5) post-MI, thereby impaired cardiac repair.
Figure 4.
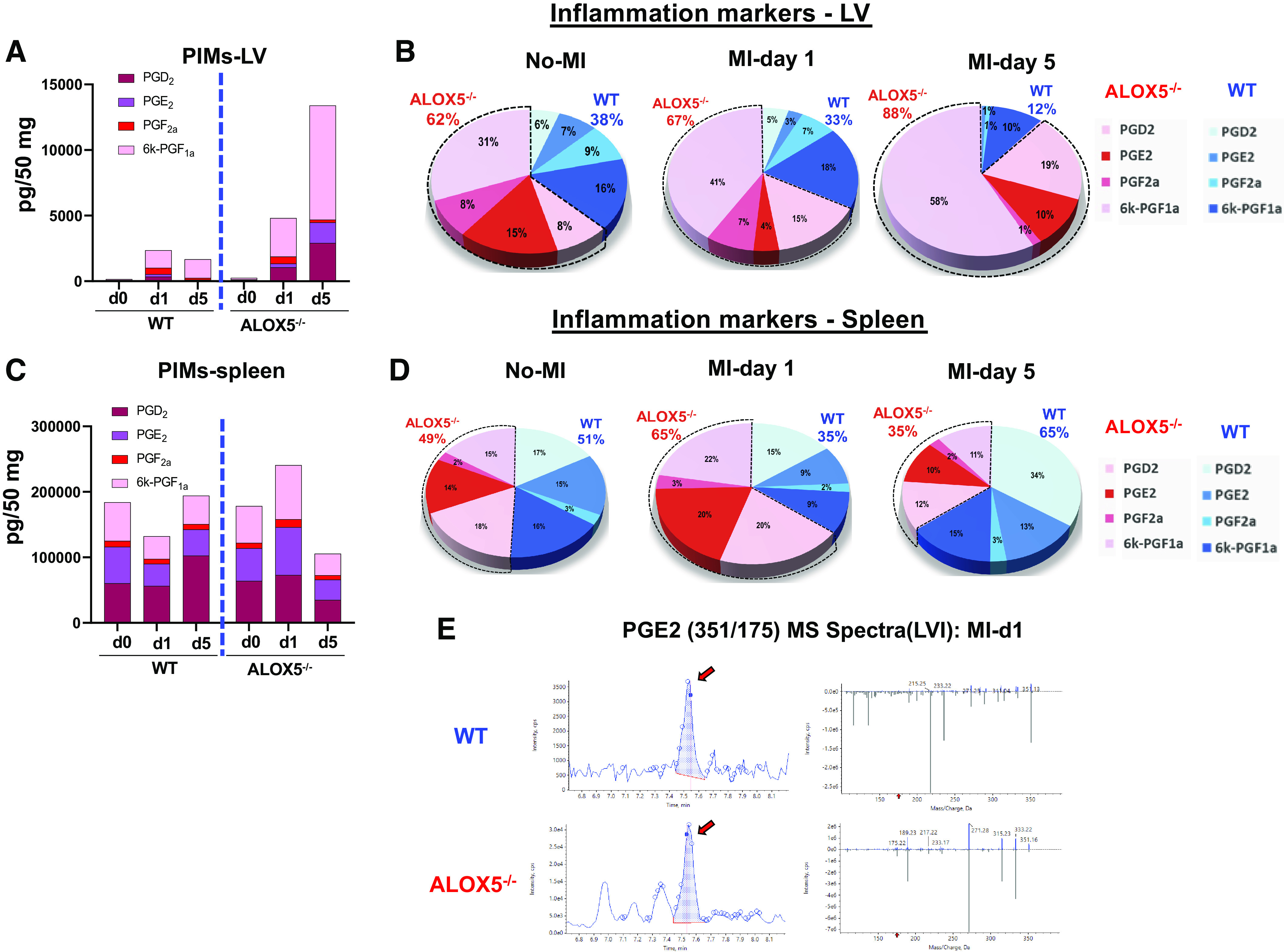
Arachidonate 5-lipoxygenase (ALOX5) deficiency amplified proinflammatory mediators (PIMs) in infarcted left ventricle (LV) and spleen post-myocardial infarction (MI). Spleen was collected after coronary ligation-induced MI-operated mice and quantitated lipid mediators (LMs) compared with naïve controls (no-MI). Interleaved graphs of LV PIMs (A), PIMs pie chart of LV (B), interleaved graphs of spleen PIMs (C), and PIMs pie chart of spleen represent percent composition (averaged) (D) of prostaglandin (PGs) (PGD2, PGE2, PGF2a, and 6 K-PGF1a) at day 0 naïve control, day 1, and day 5 post-MI indicative of amplified inflammation at day 1 in ALOX5−/− mice compared with wild-type (WT) mice and no-MI naïve controls. E: MS spectra of prostaglandin E2 (PGE2) in infarcted LV (LVI) at MI-day 1 in WT and ALOX5−/− mice. Quantification and values are pg/50 mg of spleen tissue. The detection limit was ∼1 pg determined using mass spectrometry; n = 5 per group/day for both genotypes; values are the average of each PGs.
ALOX5 and SPM Deficiency Reduced Survival and Increased the Propensity for Infarcted Myocardial Rupture and Profound Cardiac Remodeling in Acute and Chronic HF
The ALOX5 deletion in mice resulted in lower SPMs essential for myocardium healing (6). We monitored post-MI survival in ALOX5−/− mice and compared this with WT during the transition from acute HF (post-MI day 1 and day 5) to CHF (post-MI day 56). ALOX5−/− mice displayed reduced post-MI survival (31%) than did WT (56%; P < 0.001), and ALOX5−/− also had higher myocardial rupture rates than in the WT mice (Fig. 5A). Specifically, the overall rupture rate was 65% in ALOX5−/− mice compared with 28% in WT mice (Fig. 5A). LV-free wall rupture occurred within 3–6 days after MI, most commonly at days 4 and 5 post-MI. Infarcted LV rupture was determined based on the presence of a slit-like site on the myocardium with a blood-filled chest cavity. To compare the differences in injured myocardium between WT and ALOX5−/− groups, we determined the infarcted area after MI (Table 1). The echocardiography and gravimetric analysis revealed extensive cardiac remodeling in ALOX5−/− and WT mice during acute and chronic HF, with a similarly infarcted area indicative of successful coronary ligation (Fig. 4B and Table 1). Mice LVs from both genotypes in acute HF (MI days 1 and 5) and chronic HF (MI day 56) were stained with hematoxylin and eosin to analyze the myocardial structure following cardiac injury. We observed infarct expansion in ALOX5−/− mice compared with WT, suggestive of advanced cardiac remodeling (Fig. 5C). ALOX5−/− mice displayed signs of impaired cardiac repair with earlier development of progressive end-stage acute HF and defective scar formation, supporting the high mortality post-MI (Fig. 5C). Our results indicated that ALOX5 deletion increased the propensity of ruptures and reduced survival with profound pathological remodeling.
Figure 5.
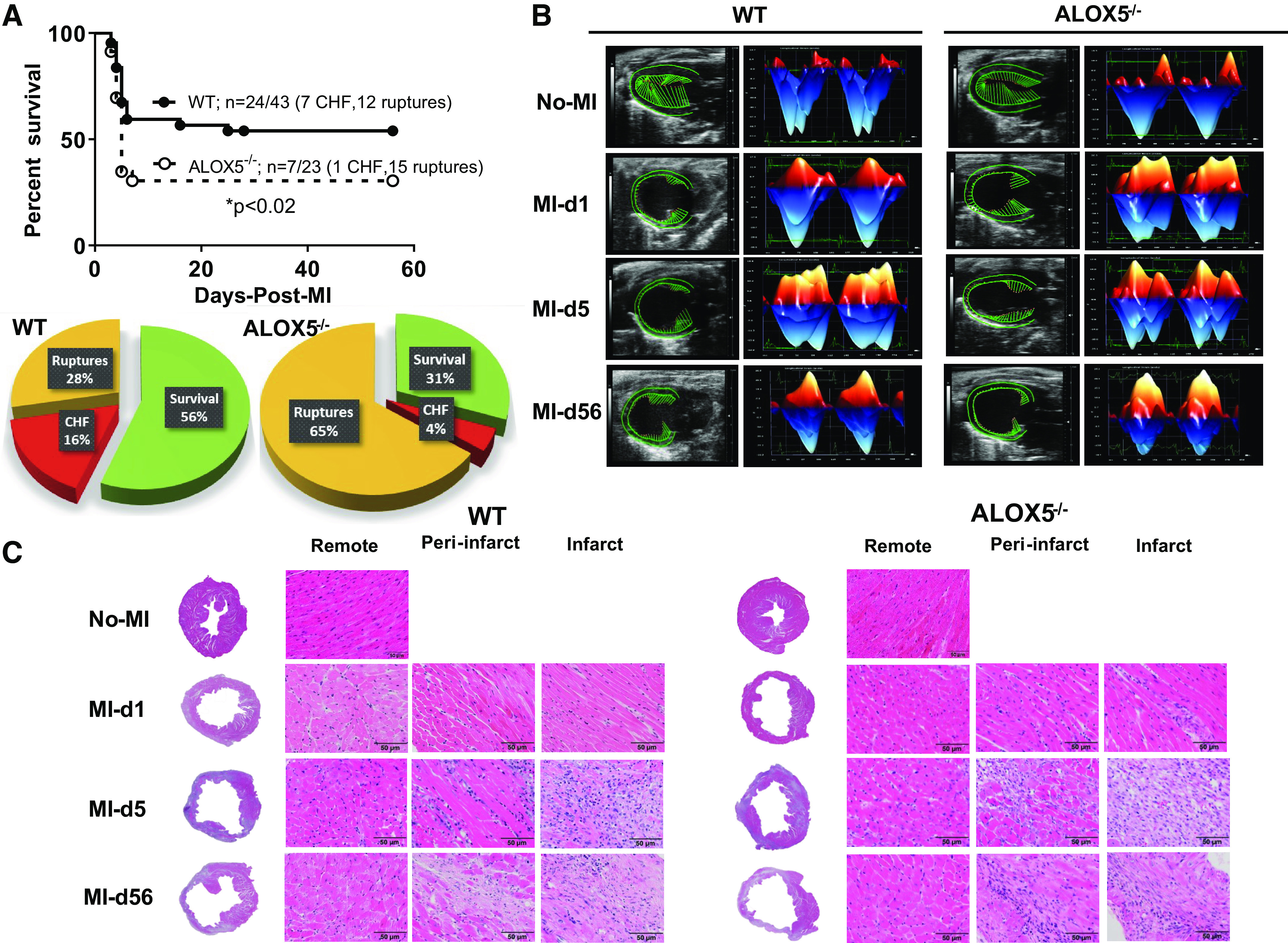
Deletion of arachidonate 5-lipoxygenase (ALOX5) decreased post-myocardial infarction (MI) survival, worsened cardiac function, and structure in acute and chronic heart failure (HF). A: Kaplan–Meier survival curve compared by log-rank test representing decreased survival in ALOX5−/− male mice post-MI during cardiac healing from acute HF (day 1 and day 5-post-MI) to chronic HF (day 56 post-MI). *P < 0.02 vs. wild-type (WT), n = 43 (male) WT and n = 23 (male) ALOX5−/−. Pie charts representing percent survivor (green), percent mortality due to congestive heart failure (red), and percent ruptures (orange) in the transition of acute HF (day 1 and day 5 post-MI) to chronic HF (day 56 post-MI) in WT and ALOX5−/− mice. B: echocardiographic representation of speckle-tracking analysis in the long-axis B-mode; the left ventricle (LV) is in mid-systole and longitudinal three-dimensional strain in WT and ALOX5−/− mice in acute HF (day 1 and day 5 post-MI) and chronic HF (day 56 post-MI), WT: No-MI (n = 27); MI = day 1 (n = 15); MI = day 5 (n = 14); MI-day 56 (n = 10), ALOX5−/− (n): No-MI (n = 13); MI = day 1 (n = 11); MI = day 5 (n = 7); MI-day 56 (n = 7). C: hematoxylin and eosin (H&E)-stained LV dilation indicative of structural remodeling in the infarct, peri-infarct, and remote areas in acute to chronic HF transition in WT and ALOX5−/− mice post-MI compared with naïve controls. Complete LV image in ×1.25. Remote, peri-infarct and infarct images are ×40. n = 5 or 6 mice/group, scale bar = 50 μm.
Table 1.
Temporal echocardiography and necropsy parameters in acute and chronic heart failure in WT and ALOX5−/− mice
| No-MI Controls |
MI (Day 1) |
MI (Day 5) |
MI (Day 56) |
|||||
|---|---|---|---|---|---|---|---|---|
| WT | ALOX5−/− | WT | ALOX5−/− | WT | ALOX5−/− | WT | ALOX5−/− | |
| Echo parameters | ||||||||
| n | 27 | 13 | 15 | 11 | 14 | 7 | 10 | 7 |
| Heart rate, beats/min | 462 ± 11 | 468 ± 15 | 456 ± 9 | 475 ± 11 | 454 ± 11 | 448 ± 11 | 488 ± 12 | 435 ± 9 |
| EDD, mm | 3.7 ± 0.1 | 3.9 ± 0.1 | 4.7 ± 0.1* | 4.5 ± 0.1* | 5.5 ± 0.1* | 5.2 ± 0.2* | 5.8 ± 0.2* | 5.7 ± 0.2* |
| ESD, mm | 2.4 ± 0.1 | 2.7 ± 0.1* | 4.3 ± 0.1* | 4.1 ± 0.1* | 5.0 ± 0.2* | 4.9 ± 0.2* | 5.4 ± 0.4* | 5.1 ± 0.2* |
| Fractional shortening, % | 34 ± 1.7 | 32 ± 1.5 | 9 ± 0.97* | 10 ± 0.91* | 10 ± 2.7* | 7 ± 0.6*$ | 9 ± 0.02* | 10 ± 1.2* |
| IVSd, mm | 0.73 ± 0.03 | 0.70 ± 0.03 | 0.61 ± 0.04* | 0.48 ± 0.03* | 0.54 ± 0.03* | 0.53 ± 0.7* | 0.54 ± 0.14* | 0.46 ± 0.05* |
| PWTd, mm | 0.67 ± 0.04 | 0.71 ± 0.04 | 0.54 ± 0.04* | 0.58 ± 0.03* | 0.49 ± 0.04* | 0.47 ± 0.04* | 0.52 ± 0.10* | 0.50 ± 0.05* |
| IVSs, mm | 1.10 ± 0.03 | 1.03 ± 0.03 | 0.74 ± 0.05* | 0.58 ± 0.03* | 0.63 ± 0.05* | 0.64 ± 0.08* | 0.65 ± 0.17* | 0.60 ± 0.06* |
| PWTs, mm | 1.06 ± 0.03 | 1.09 ± 0.05 | 0.60 ± 0.04* | 0.64 ± 0.04* | 0.57 ± 0.04* | 0.54 ± 0.04* | 0.61 ± 0.13* | 0.53 ± 0.04* |
| GLS | −19 ± 0.7 | −16 ± 0.7* | −4 ± 1.1* | −4 ± 0.5* | −6 ± 1.2* | −4 ± 0.08* | −7 ± 0.4* | −5 ± 1.9* |
| Necropsy parameters | ||||||||
| n | 11 | 11 | 9 | 12 | 10 | 14 | 23 | 9 |
| Body weight, g | 28 ± 1 | 25 ± 0.8 | 23 ± 0.4 | 26 ± 0.9 | 23 ± 0.6 | 24 ± 0.9 | 30 ± 0.4 | 28 ± 0.8 |
| LV, mg | 94 ± 3 | 78 ± 2* | 89 ± 4* | 86 ± 3* | 105 ± 5* | 103 ± 2* | 111 ± 3* | 102 ± 6* |
| LV/BW, mg/g | 3.4 ± 0.1 | 3.3 ± 0.09 | 4.0 ± 0.1* | 3.4 ± 0.09*$ | 4.6 ± 0.29* | 4.4 ± 0.24* | 3.7 ± 0.12* | 3.7 ± 0.19* |
| Right ventricle, mg | 22 ± 1 | 20 ± 0.5 | 21 ± 1 | 20 ± 0.9 | 21 ± 1.1 | 19 ± 1.2 | 24 ± 0.8 | 24 ± 2.5 |
| Spleen, mg | 78 ± 5 | 69 ± 3* | 56 ± 6* | 69 ± 8* | 84 ± 4* | 78 ± 2* | 88 ± 5* | 68 ± 3* |
| RV mass/BW, mg/mg | 0.77 ± 0.03 | 0.78 ± 0.03 | 0.95 ± 0.05* | 0.82 ± 0.05* | 0.92 ± 0.07* | 0.80 ± 0.06* | 0.81 ± 0.03* | 0.87 ± 0.08* |
| Lung mass (wet)/BW, mg/mg | 5 ± 0.22 | 6 ± 0.53 | 7 ± 0.60 | 6 ± 0.33 | 7 ± 0.85 | 7 ± 0.77 | 5 ± 0.17 | 5 ± 0.61 |
| Lung mass (dry)/BW, mg/mg | 1 ± 0.05 | 1 ± 0.05 | 1 ± 0.06 | 1 ± 0.04 | 1 ± 0.09 | 2 ± 0.02 | 1 ± 0.04 | 1 ± 0.07 |
| Tibia, mm | 17 ± 0.1 | 17 ± 0.2 | 17 ± 0.01 | 17 ± 0.09 | 17 ± 0.06 | 17 ± 0.17 | 17 ± 0.09 | 17 ± 0.12 |
| Infarct area, % | ND | ND | 47 ± 1* | 47 ± 4* | 50 ± 3* | 53 ± 2* | 55 ± 4* | 55 ± 5* |
Values are means ± SE; n, mice sample size. ALOX5−/−, arachidonate 5-lipoxygenase-null mice; BW, body weight; EDD, end-diastolic dimension; ESD, end-systolic dimension; GLS, global longitudinal strain; IVSd, intraventricular septum diastole; IVSs, intraventricular septum systole; LV, left ventricle; MI, myocardial infarction; ND, not determined; PWTd, posterior wall thickness diastole; PWTs, posterior wall thickness systole; RV, right ventricle; WT, wild-type.
P < 0.05 vs. no-MI naïve controls; $P < 0.05 vs. respective WT post-MI.
ALOX5 Deficiency Primed Immature Neutrophils with Delayed Clearance post-MI
After the cardiac injury, the splenic leukocytes mobilize to the heart to facilitate healing of the injured myocardium (6). During the early signs of inflammation, PGs and LTB4 facilitate vascular leakage that enables the infiltration of neutrophils (39). Furthermore, SPMs are biosynthesized to counterregulate leukocyte infiltration and stimulate clearance, cardiac repair, and resolution of inflammation (6, 9, 13). To measure clearance, we checked the influence of the ALOX5 deletion on leukocyte kinetics. The flow cytometric analyses revealed an increase in Ly6G+ cells (i.e., mature neutrophils) in the infarcted LV of ALOX5−/− mice compared with WT mice at day 1 post-MI (8.2 ± 1.4 vs. 3.2 ± 1.3%). Compared with WT (1.2 ± 0.5%), the neutrophils (Ly6G+) were higher in ALOX5−/− mice (4.1 ± 1%) at day 5 post-MI, which suggests that neutrophil clearance was delayed because of insufficient SPMs. Similar kinetics of neutrophils were observed at splenic sites (Fig. 6, A, C, and D). Compared with WT mice, both splenic and LV neutrophils at post-MI-day 1 displayed lower Ly6G expression in ALOX5−/− mice, indicating the neutrophils are not fully mature or functional (Fig. 6B). The LV and splenic CD11b+/F4/80+ population in ALOX5−/− and WT mice were quantitatively similar, but the phenotypic classification of macrophages revealed a skew toward the splenic proinflammatory F4/80+/Ly6Chi population in ALOX5−/− compared with WT (Supplemental Fig. S2, A–F). Together, these results suggest that the ALOX5 deletion impaired neutrophil clearance without altering the initiation of inflammation post-MI, implying non-resolving inflammation. We used high-resolution ImageStream flow cytometry to determine leukocyte function (Fig. 7A). Representative images contained bright-field (BF), annexin+ (apoptotic-green), F4/80+ (macrophages-red) and Ly6G+ (neutrophils-purple), and PI (propidium iodide-brown). Mononuclear cells were isolated from the LV of WT and ALOX5−/− mice from no-MI controls, post-MI day 1, and day 5. Post-MI day 1 ImageStream analysis in WT mice revealed that macrophages phagocytosed annexin+Ly6G+ (Fig. 7B, right) whereas ALOX5−/− mice displayed an annexin marker on macrophages, which is indicative of impaired and delayed phagocytosis (Fig. 7C, right). Flow cytometry and high-resolution ImageStream microscopy-based analyses suggested that ALOX5−/− mice have impaired macrophage-directed phagocytic neutrophil clearance, leading to non-resolving inflammation and failure of cardiac repair.
Figure 6.
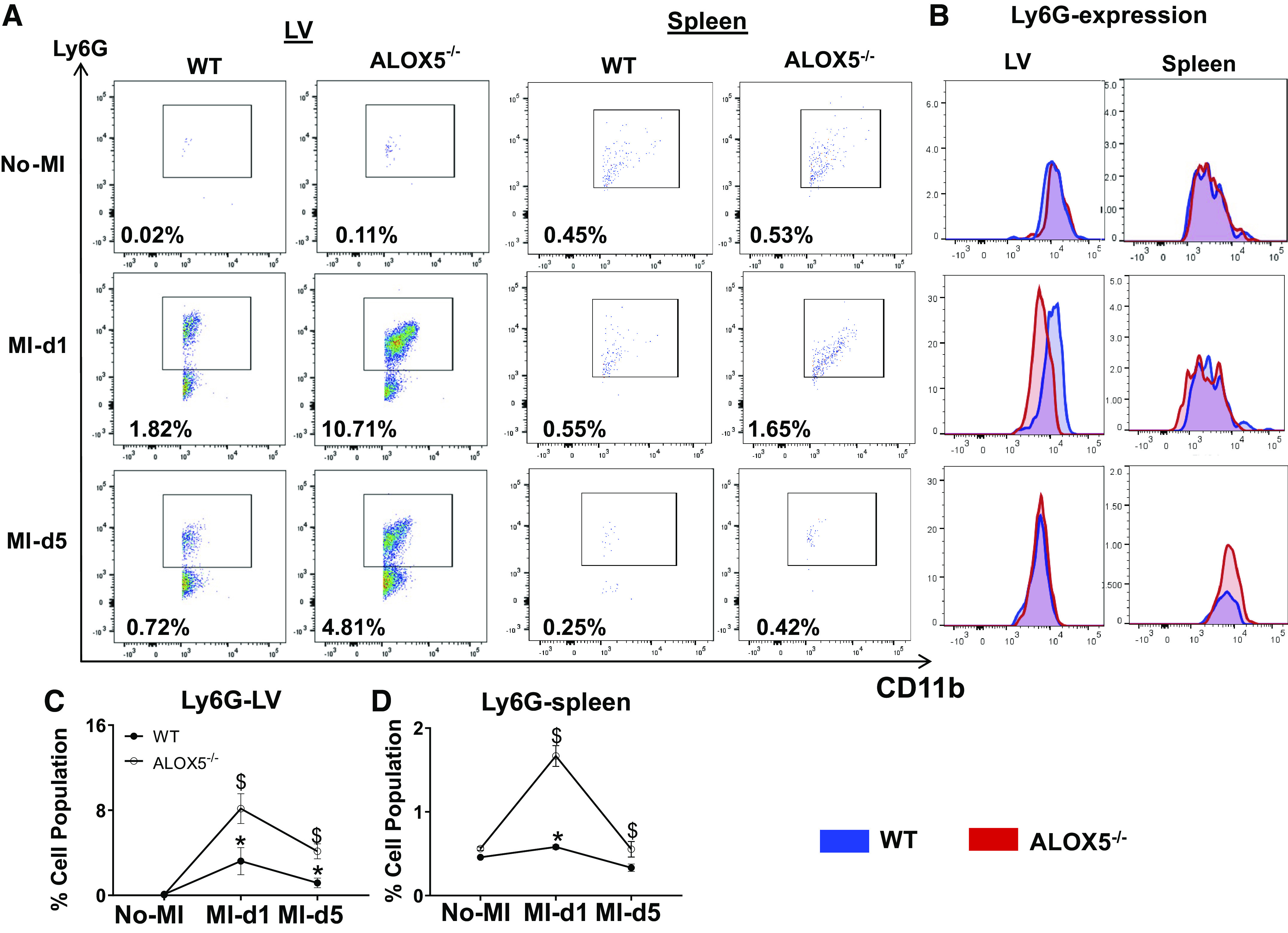
Arachidonate 5-lipoxygenase (ALOX5) deficiency primed immature neutrophils with delayed clearance. A: representative flow cytometry (FACS) dot plots showing the neutrophil population (CD11b+/Ly6G+) in left ventricle (LV, left) and spleen (right) from wild type (WT) and ALOX5−/− mice at day 1 and day 5 post-myocardial infarction (MI) compared with no-MI naïve controls. B: representative LV and spleen stacked histograms displaying Ly6G expression in WT and ALOX5−/− mice at day 1 and day 5 post-MI compared with naïve controls no-MI. The line graph presents the percentage of the Ly6G+ population in LV (C) and spleen (D) in WT and ALOX5−/− mice at day 1 and day 5 post-MI compared with naïve controls. n = 5 mice/group/daytime point. *P < 0.05 compared with no-MI naïve control, and $P < 0.05 compared with ALOX5−/− at respective time points by two-way analyses of variance (ANOVA).
Figure 7.
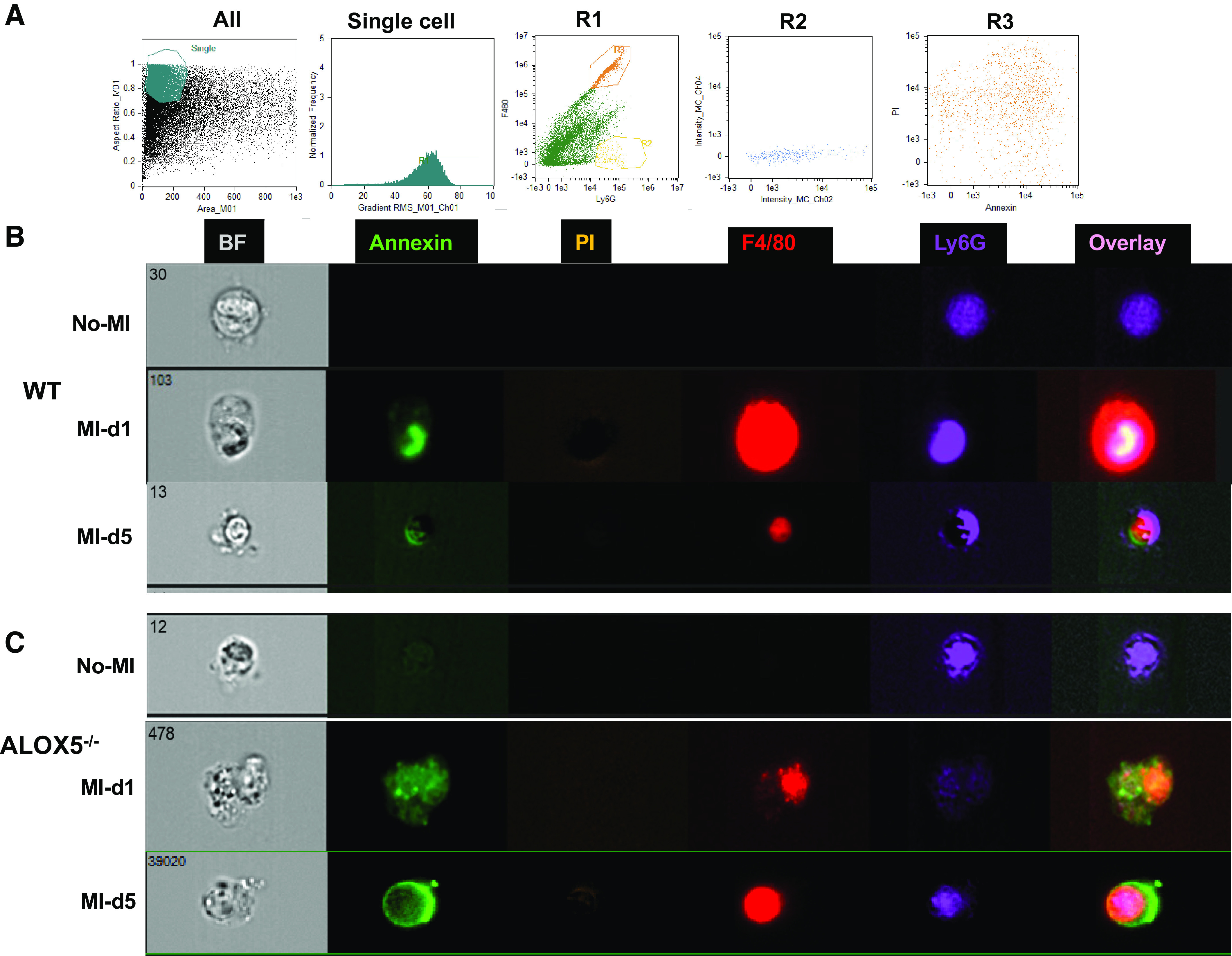
Arachidonate 5-lipoxygenase (ALOX5) deficiency delayed macrophage-directed phagocytosis post-myocardial infarction (MI). A: wild-type (WT) and ALOX5−/− mice were operated on to ligate the left anterior descending artery (MI). After 24 h (day 1) and day 5 of MI surgery, ventricular tissue was collected from infarct areas, processed for mononuclear cell suspension, and compared with naïve controls. B and C: representative images from ImageStream analysis selected for focus, size, and aspect ratio labeled for the phenotypes analyzed: annexin (apoptotic cells; green), dead cells (orange), F4/80+ (macrophages, red), or Ly6G+ (neutrophil; purple), and all channels merged with and without brightfield (BF) in LV of WT and ALOX5−/− mice at day 1 and day 5 post-MI compared with naïve controls. n = 3 mice/group/day time point. For the Image stream, nearly 1 million events were acquired for each analysis.
ALOX5 Deletion Overactivated COXs with Increased PGs and Defective TGF-β Signaling in the Infarcted Myocardium post-MI
Activated leukocytes biosynthesize SPMs that overlap with LTB4 as initial signs of inflammation and resolution (6). The ALOX5-derived chemotactic signal indicator LTB4 directs leukocytes toward an ischemic injury post-MI. Expression of the LOX-encoding genes is higher in infarcted LV and is associated with SPM generation at the ischemic site (6). Furthermore, we analyzed the mRNA expression of the LOXs (-15, -12, and -5) in the transmural area of the infarcted LV (LVI). Compared with WT, ALOX5−/− mice were devoid of Alox5 expression in LVI tissues from day 0 to day 56 post-MI (Fig. 8A). Similarly, the Alox12 level remained lower in ALOX5−/− LVI than in WT (Fig. 8B). However, the Alox15 mRNA expression was significantly increased in ALOX5−/− LVI at day 5 post-MI compared with WT mice and decreased at day 56 post-MI (Fig. 8C). In contrast, both cyclooxygenases (COXs, COX-1 and -2) mRNA expression remains elevated in the infarcted LV in ALOX5−/− mice, from no-MI control to day 56 post-MI, which is indicative of sustained inflammation marked with higher levels of PGs (PGD2, PGE2, PGF2a, and 6 K-PGF1a) in the infarcted area (Fig. 8, D and E).
Figure 8.
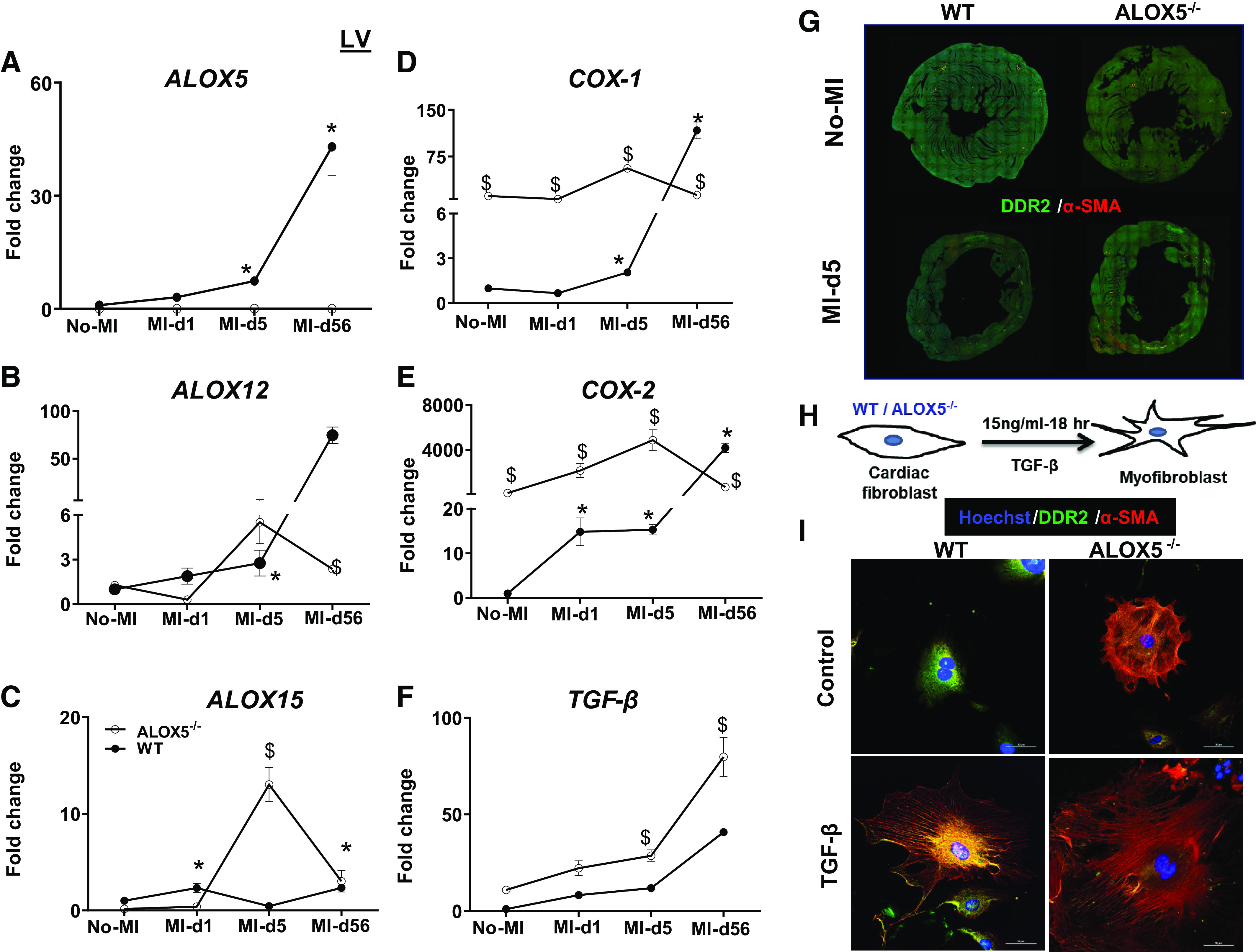
Deletion of arachidonate 5-lipoxygenase (ALOX5) upregulated cyclooxygenases (COXs) and dysregulated fibroblast. Temporal kinetics of ALOXs (-15, -12, and -5) (A–C), COXs (-1 and -2) (D and E), and TGF-β (F) expression in the infarcted tissue of the left ventricle from wild-type (WT) and ALOX5−/− mice in the transition of acute (day 1 and day 5) to chronic (day 56) HF compared with naïve controls; n = 6/group/day. *P < 0.05 compared with no-myocardial infarction (MI) naïve control; $P < 0.05 compared with ALOX5−/− at respective time points by two-way analysis of variance (ANOVA). G: DDR2 (green) and α-smooth muscle actin (α-SMA) (red) expression in LV-middle section of WT and ALOX5−/− mice reconstructed using image stitching tool in Nikon A1 microscope. H: in vitro study design showing cardiac fibroblast isolated from male C57BL/6 WT and ALOX5−/− mice (6–8 wk of age) treated with TGF-β (15 ng/mL) for myofibroblast differentiation. I: immunofluorescence representative images showing DDR2 (green) and α-SMA (red) expression in nontreated and TGF-β (15 ng/mL)-treated cardiac fibroblast. The nuclei are stained blue with Hoechst. Data are representative of n = 3 technical replicates with three independent repeats done on a different day. Images (×40) were acquired using Nikon A1 confocal microscope. Scale bar = 50 μm.
After the cardiac injury, TGF-β plays an essential role in matrix remodeling and mature scar formation for optimal cardiac repair (40). Therefore, we measured TGF-β levels in ALOX5−/− mice. TGF-β mRNA levels remained elevated in ALOX5−/− LVI compared with WT from no-MI control to day 56 post-MI, which is characteristic of overactivated and defective TGF-β signaling (Fig. 8F). Thus, ALOX5 deletion led to the upregulation of COX-1 and -2 with a marked increase in PGs levels and defective overactivation of TGF-β that triggered a proinflammatory milieu of non-resolving inflammation in acute and chronic HF.
Non-Resolving Inflammation and Impaired Cardiac Repair in ALOX5−/− Mice Are Marked by Overactivation of Fibroblasts
After MI, leukocyte clearance and scar formation are apparent responses. ALOX5−/− mice are non-responsive to healing postinjury and display higher TGF-β expression, suggestive of fibroblasts preactivation. We studied the fibroblast-myofibroblast axis to compare post-MI matrix remodeling in ALOX5−/− and WT mice. Analysis of picrosirius red (PSR) staining of the LV revealed higher collagen levels in the border zone of ALOX5−/− mice compared with WT mice at day 5 and day 56 post-MI (Supplemental Figs. S3 and S4). We further evaluated the fibroblast-myofibroblast transition, marked by an increase of α-smooth muscle actin (α-SMA) expression. When compared with WT, ALOX5−/− mice had higher α-SMA expression at day 5 post-MI in the infarcted zone (Fig. 8G).
We analyzed the fibroblast phenotype from WT and ALOX5−/− mice to determine the transition of fibroblast to secretory myofibroblast. Cardiac fibroblasts were differentiated to myofibroblast using TGF-β, which allowed us to distinguish the fibroblast-myofibroblast morphology in WT and ALOX5−/− mice (Fig. 8H). TGF-β induces fibroblast hypertrophy within 18 h and shows myofibroblast signatures. Cardiac fibroblasts isolated from ALOX5−/− mice, when treated with TGF-β, grew as round and large cells with increased α-SMA expression, suggestive of a fibroblast-to-myofibroblast transition (Fig. 8I; expanded in Supplemental Fig. S5). By contrast, WT fibroblast treated with TGF-β displayed typical spindle-enriched myofibroblast morphology with an increase in α-SMA expression compared with the naïve state. ALOX5−/− fibroblasts treated with TGF-β displayed hypertrophic cardiac myofibroblast morphology and an increase in α-SMA expression indicative of overactivated TGF-β signaling in ALOX5−/− mice compared with WT mice. Thus, ALOX5−/− mice fibroblasts are prone to fibroblast-to-myofibroblast differentiation, which suggests the critical role of ALOX5 in fibroblast physiology.
ALOX5-Derived SPMs, Including LXs, Are Essential for Scar Maturation and Fibroblast Migration during Wound Healing
ALOX5 deletion decreases the formation of both LTB4 and SPMs. Lack of ALOX5 in cardiac fibroblast displayed hypertrophic morphology. Therefore, to define the molecular mechanism, we determined whether supplementation of LTB4 or specific SPM 15-epi-LXA4 in media could limit the TGF-β-induced hypertrophy in cardiac fibroblasts isolated from WT and ALOX5−/− mice (Fig. 9A). Cardiac fibroblasts of ALOX5−/− mice treated with TGF-β + LTB4 displayed a more hypertrophic morphology with higher α-SMA expression (red) and lower DDR2 (green) compared with TGF-β-only treated ALOX5−/− fibroblasts, suggesting that LTB4 further enhances the myofibroblast transition (Fig. 9B; expanded in Supplemental Fig. S6). Of note, 15-epi-LXA4 treated WT cardiac fibroblasts displayed lower α-SMA expression and limited myofibroblast growth with an increase in DDR2 expression. By contrast, ALOX5−/− cardiac fibroblasts remained hypertrophic with TGF-β + LTB4 treatment with higher α-SMA expression. TGF-β + 15-epi-LXA4-treated cardiac fibroblasts displayed limited hypertrophy with an increase in DDR2 expression, but the α-SMA expression remained unchanged compared with WT cardiac fibroblasts (Fig. 9B).
Figure 9.
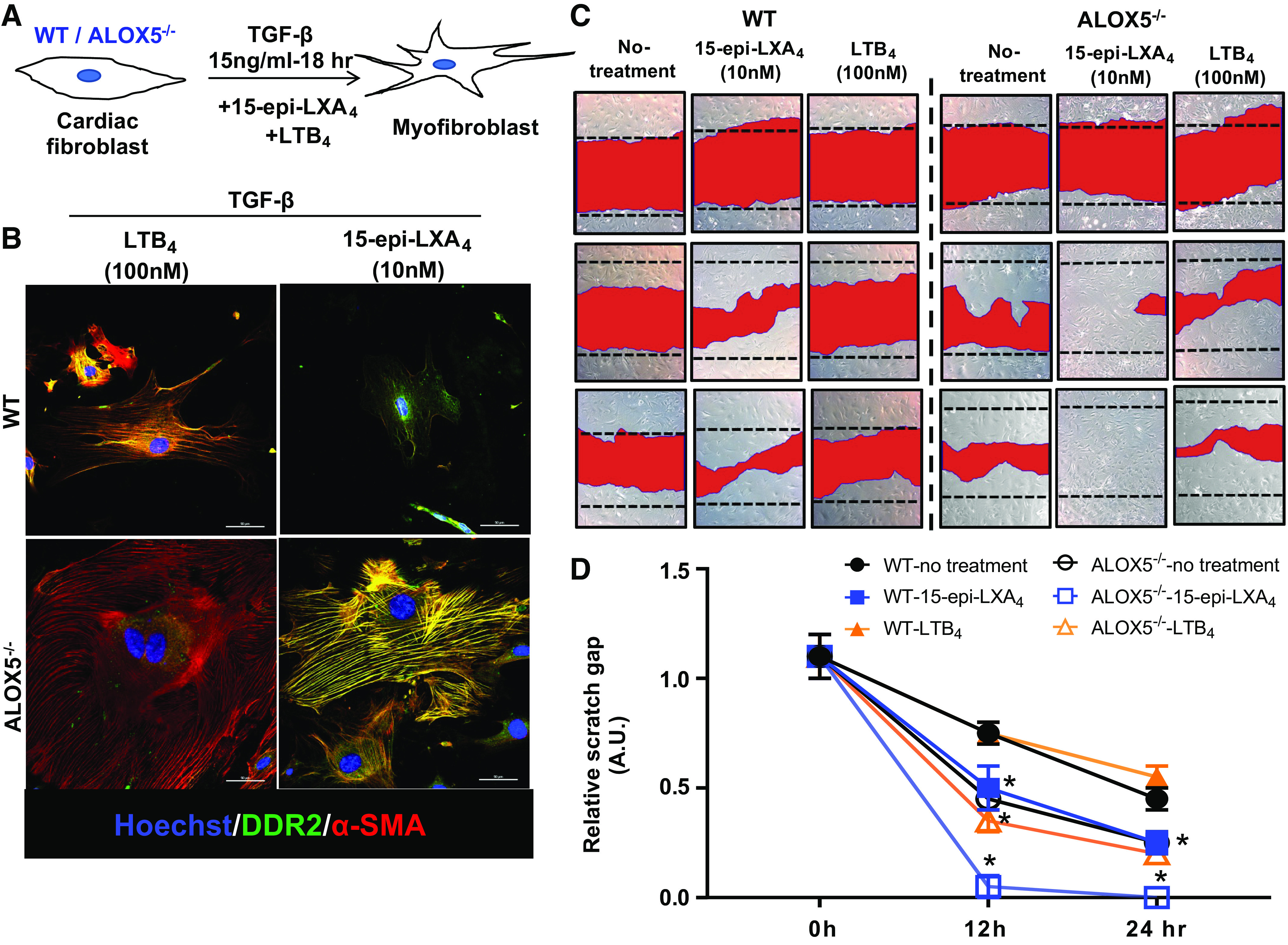
Arachidonate 5-lipoxygenase (ALOX5)-derived leukotrienes-lipoxins are essential for fibroblast migration to prevent rupture. A: in vitro fibroblast signaling study design showing cardiac fibroblast isolated from male C57BL/6 WT and ALOX5−/− mice treated with TGF-β + leukotriene B4 (LTB4) or +15-epi-lipoxin A4 (LXA4) for myofibroblast differentiation. B: representative immunofluorescence images showing DDR2 (green) and α-smooth muscle actin (α-SMA, red) expression in LTB4 (100 nM)- and 15-epi-LXA4 (10 nM)-treated cardiac fibroblast from wild-type (WT) and ALOX5−/− mice. The nuclei are stained blue with Hoechst. Data are representative of n = 3 technical replicates with three independent repeats done at different days. Images are taken on ×40 of Nikon A1 confocal microscope. Scale bar = 50 μm. C: representative images from scratch assay using cardiac fibroblast (CF) from WT and ALOX5−/− mice. CF (WT/ALOX5−/−) were treated with either ALOX5-derived LTB4, or 15-epi-LXA4 to depict cell migration. Data are representative of n = 3 technical replicates with three independent repeats done on a different day. D: line graph representing relative scratch gap (on time cell migration) of WT and ALOX5−/− treated either with LTB4 (100 nM) or 15-epi-LXA4 (10 nM). *P < 0.05 compared with respective WT-control at respective time point by two-way analysis of variance (ANOVA).
These results confirmed that ALOX5-mediated LXs are essential for cardiac repair, optimizing the fibroblast-to-myofibroblast transition and limiting the overactivation of cardiac fibroblasts. We further confirmed LXs (SPM) mediated cardiac repair using wound healing scratch assay. The ALOX5−/− fibroblasts showed higher migratory capacity than WT control fibroblasts. Stimulation of cardiac fibroblasts (WT and ALOX5−/−) with 15-epi-LXA4 led to a significant increase in migratory capacity compared with untreated controls. No difference was observed between respective untreated and LTB4-treated cardiac fibroblasts. However, the migratory capacity was higher in all ALOX5−/− cardiac fibroblast groups (Fig. 9, C and D). The results pointed to the critical role of ALOX5 in fibroblast-myofibroblast transition and migration signaling.
DISCUSSION
We revealed that activated leukocytes expressed ALOX5 generated the inflammatory mediator LTB4 and facilitates the biosynthesis of SPMs, such as lipoxins, MaR1, PD1, and RvD5 in the cardiac repair process. In this study, we revisited the inflammation-resolution signaling and attempted to redefine the role of ALOX5 in the biosynthesis of SPMs in acute HF. Our results suggest that ALOX5 1) is indispensable for biosynthesis of SPMs (lipoxins, MaR1, PD1, and RvD5) in the infarcted heart; 2) is inactivated in humans, leading to dysregulation of bioactive LMs in the plasma and ischemic myocardium; and 3) deficiency of ALOX5 promotes defective TGF-β signaling with marked fibroblasts disposition to myofibroblasts. In the clinical setting, HF is secondary to the myocardial injury in which unresolved inflammation is a prime contributor to pathological remodeling, leading to morbidity and mortality, which begins with atherogenesis (41–43). Traditionally, ALOX5 is considered an initiator of inflammatory responses because ALOX5-derived LTB4 serves as a chemoattractant for leukocytes (neutrophils and monocytes/macrophages) (10, 13, 14, 44). Furthermore, the leukocytes have classically been thought to be key initiator of the short-lived proinflammatory phase. This phase usually lasts from a few hours to a few days depending on the magnitude of the injury, followed by the reparative phase essential for physiological healing (45). The following four integrative mechanisms suggest an obligatory role of ALOX5 in initiating the inflammation-resolution program for both SPMs biosynthesis and cardiac healing.
First, post-MI mouse survival and delayed HF progression are readily definable endpoints that provide the ultimate therapeutic efficacy test. In mice, deletion of 12/15LOX improved survival to 89% compared with 56% in controls. The 12/15LOX knockout animals display elevated biosynthesis of epoxyeicosatrienoic acids (EETs) in the plasma, spleen, and LV, leading to a reduction in rupture and activation of reparative CD206+ macrophages and neutrophils post-MI (30, 34, 46). In contrast, deficiency of ALOX5 in mice causes higher rupture rates and lower survival (31% vs. 56% in controls) associated with lower levels of LTB4 and SPMs in the infarcted LV post-MI. In humans, ischemic myocardium contains substantial amounts of 5-HETE and shows lower levels of ALOX5 mRNA and protein expression in ischemic tissue than controls. ALOX5-derived lipoxin A4, and 15-epi-lipoxin A4, are reduced with lower expression of ALOX5 mRNA and protein in human ischemic myocardium, which is suggestive of unresolved inflammation in a failing heart. ALOX5 deletion displayed lower levels of LTB4 and SPMs post-MI in infarcted LV and spleen post-MI day 1, blunting the essential initial inflammatory and resolving responses required for cardiac healing. The recent LTIMI (Leukotrienes and Thromboxane in Myocardial Infarction) trial confirmed that leukotrienes are initiators in an acute setting but independent of major adverse cardiovascular events (MACEs) within 1-year post-MI, suggestive of a transient increase during the initiation of inflammation (47, 48). In patients with STEMI, SPMs are biosynthesized before the peak of troponin T levels compared with healthy controls and stable patients with CAD (49). The targeted analyses of SPMs within 24 h post-MI confirm sex- and race-specific differences in SPM biosynthesis, while comprehensive lipidome measurement indicates that monohydroxy arachidonate fatty acids mediators were decreased with a reduced level of lipoxins and RvD6, suggesting SPMs as prognostic markers of MI event (50, 51).
Second, after cardiac injury, elevated levels of PIMs, such as LTB4 and PGs, are key markers of an initial inflammatory response through the action of ALOX5 and COXs, respectively (2). Biosynthesis of LTB4 directs first-responder neutrophil entry, which is essential for removing dead cardiomyocytes and directly activates monocytes/macrophages for cardiac repair (25, 52). Depletion of neutrophils impairs cardiac healing, suggesting the role of activated neutrophils in resolving post-MI inflammation (52). Infiltrated leukocytes, such as neutrophils and monocytes/macrophages, are essential for the biosynthesis of lipoxins (lipoxin A4, lipoxins B4, and 15-epi-lipoxin A4) in coordination with platelets, endothelial cells, and macrophages (53). Lipoxins are trihydroxytetraene-containing bioactive LMs actively biosynthesized during the initial phase of cardiac repair using two main pathways: 1) via activated leukocytes and 2) through the interaction of platelet-leukocyte and leukocyte-epithelial cell interactions (6, 53). Depleting either neutrophils or macrophages impairs cardiac healing and biosynthesis of SPMs (6, 52). The outcome presented herein shows impaired cardiac repair and higher rupture rates in ALOX5−/− mice possibly due to the lower synthesis of SPMs, which indicates ALOX5 is not only essential in SPMs biosynthesis and but also for ECM remodeling in HF.
Third, resolution of inflammation with the biosynthesis of SPMs at the site of atherosclerotic plaques, cardiac injury, or infection is essential for the resolution program. Therefore, targeted enzymes would focus on developing novel therapeutic strategies to delay chronic HF (6, 21, 31, 42, 54). For LTB4-induced inflammation, ALOX5 enzyme activity and protein expression are associated with disease progression of respiratory and cardiovascular diseases, including pathogenesis of aortic valve stenosis, but ALOX5 single-nucleotide polymorphisms are unrelated to these disease outcomes (55, 56). Precise, quantitative, and temporal analyses of targeted ALOX5 products confirmed that SPMs and PIMs are biosynthesized after leukocyte entry at the site of infarction. Not surprisingly, ALOX5−/− mice failed to generate inflammation-clearance signals and therefore showed impaired cardiac repair; however, amplified PIMs are indicative of unresolved inflammation. Our previous reports align with the present outcome, given that ALOX5-derived SPMs, particularly RvD1 and 15-epi-lipoxinA4, are responsible for neutrophil clearance and alter macrophage plasticity toward a reparative phenotype. ALOX5-derived RvD1 and 15-epi-lipoxin A4 SPMs are activators of formyl peptide receptor 2 in the spleen and infarcted LV that facilitate the resolution of inflammation (33, 36).
Finally, we determined that ALOX5 deletion increased fibroblast proliferation and signs of hypertrophy in vitro. This was consistent with higher TGF-β expression post-MI in ALOX5−/− infarcted LV. Fibroblasts of ALOX5−/− mice were primed with a smooth muscle actin expression in the naïve state because of the anomalous increase of COX-1 and COX-2 marked with high levels of PIMs. Given that PGE2 is known to support unresolved inflammation and fibrotic signaling therefore, post-MI, overactivation of COX-2 outweighed the biosynthesis of PGs (PGD2, PGE2, PGF2a, and 6 K-PGF1a) in acute cardiac repair, promoting non-resolving inflammation in ALOX5−/− mice (57). In contrast, the ALOX5 derived 15-epi-LXA4 limited the process of fibroblast-to-myofibroblast hypertrophy and differentiation of these studies consisted of in vitro data where 15-epi-LXA4 limited hypertrophy in ALOX5−/− fibroblast (36). ALOX5 is also expressed in multiple cells (neutrophils, monocytes/macrophages, lymphocytes, endothelial cells) (18, 58) therefore, future studies evaluating whether MI influences specific cell types are warranted.
In conclusion, these results support the critical role of ALOX5 in cardiac repair and SPMs biosynthesis within the injured myocardium. The leukotriene biosynthesizing ALOX5 enzyme not only produced LTB4 but also catalyzed the synthesis of SPMs, which are critical to turn-off inflammation and turn-on resolution needed for cardiac repair. The American Heart Association has recommended the consumption of marine sourced oils since 2000 for primary prevention of cardiovascular disease, and indeed, the SPMs biosynthesis is ALOX5 enzyme- and omega-3 fatty acid-dependent (6, 59). Thus, the studies show a critical finding that the inflammatory phase and safe clearance of inflammation (resolution) phase begins simultaneously during cardiac repair, and ALOX5 is essential to delay HF. Future studies are needed to confirm the role of the ALOX5 enzyme network target and SPMs signature in patients with acute and chronic HF.
Limitations and Perspective
Multiomic analyses of human infarcts suggest the diversified role of multiple cells in cardiac pathophysiology (60). Here, we focused on ALOX5 immune-responsive enzyme. ALOX5 is a complex enzyme essential in the biosynthesis of PIMs and SPMs; therefore, more studies are warranted to determine the cell-specific ALOX5 role in cardiac repair. The current study used risk-free young WT and ALOX5−/− mice; however, the use of cardiometabolic risk as a diseased model (obesity, diabetes, and/or hypertension) likely will have different outcome. In clinical settings, future studies are warranted considering sex, age, race, and time points of ischemic insult to clarify the role of ALOX5 in cardiac pathophysiology.
SUPPLEMENTAL DATA
Supplemental Tables S1–S5 and Figs. S1–S6: https://doi.org/10.6084/m9.figshare.19293287.v3.
GRANTS
This work was supported, in part, by National Heart, Lung, and Blood Institute Grants HL132989 and HL144788 (to G. V. Halade).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.V.H. and P.A. conceived and designed research; G.V.H., V.K., S.H., and V.P. performed experiments; G.V.H., V.K., S.H., V.P., N.A.L., and P.A. analyzed data; G.V.H., V.K., and P.A. interpreted results of experiments; G.V.H. and V.K. prepared figures; G.V.H., V.K., N.A.L., and P.A. drafted manuscript; G.V.H. edited and revised manuscript; G.V.H., V.K., S.H., N.A.L., and P.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Wayne State Lipidomics Core Director Dr. Krishna Rao Maddipati for analyses of bioactive lipid mediators in human plasma and tissue. We acknowledge Servier Medical Art gallery (Creative Commons license) that was used to illustrate the online graphical abstract.
REFERENCES
- 1. Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res 119: 159–176, 2016. doi: 10.1161/CIRCRESAHA.116.308030. [DOI] [PubMed] [Google Scholar]
- 2. Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol 109: 444, 2014. doi: 10.1007/s00395-014-0444-7. [DOI] [PubMed] [Google Scholar]
- 3. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116: 1254–1268, 2015. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 128: 2657–2669, 2018. doi: 10.1172/JCI97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 79: 103992, 2022. doi: 10.1016/j.ebiom.2022.103992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Halade GV, Norris PC, Kain V, Serhan CN, Ingle KA. Splenic leukocytes define the resolution of inflammation in heart failure. Sci Signal 11: eaao1818, 2018. doi: 10.1126/scisignal.aao1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123: 92–100, 2013. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jadapalli JK, Halade GV. Unified nexus of macrophages and maresins in cardiac reparative mechanisms. FASEB J 32: 5227–5237, 2018. doi: 10.1096/fj.201800254R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 510: 92–101, 2014. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 274: 23679–23682, 1999. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 11. Funk CD. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat Rev Drug Discov 4: 664–672, 2005. doi: 10.1038/nrd1796. [DOI] [PubMed] [Google Scholar]
- 12. Peters-Golden M, Henderson WR Jr.. Leukotrienes. N Engl J Med 357: 1841–1854, 2007. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 13. Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220: 568–575, 1983. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 14. Haeggstrom JZ. Leukotriene biosynthetic enzymes as therapeutic targets. J Clin Invest 128: 2680–2690, 2018. doi: 10.1172/JCI97945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237: 1171–1176, 1987. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 16. Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, Frey A, Bayer B, Vogel B, Ertl G, Bauersachs J, Frantz S. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol 108: 367, 2013. doi: 10.1007/s00395-013-0367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325: 612–616, 2009. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwarz B, Sharma L, Roberts L, Peng X, Bermejo S, Leighton I, Casanovas-Massana A, Minasyan M, Farhadian S, Ko AI, Dela Cruz CS, Bosio CM, Yale IMPACT Team. Cutting edge: severe SARS-CoV-2 infection in humans is defined by a shift in the serum lipidome, resulting in dysregulation of eicosanoid immune mediators. J Immunol 206: 329–334, 2021. doi: 10.4049/jimmunol.2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R, Lusis AJ, Mehrabian M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med 350: 29–37, 2004. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- 20. Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, Samani NJ, Gudmundsson G, Grant SF, Thorgeirsson G, Sveinbjornsdottir S, Valdimarsson EM, Matthiasson SE, Johannsson H, Gudmundsdottir O, Gurney ME, Sainz J, Thorhallsdottir M, Andresdottir M, Frigge ML, Topol EJ, Kong A, Gudnason V, Hakonarson H, Gulcher JR, Stefansson K. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet 36: 233–239, 2004. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 21. Reina-Couto M, Carvalho J, Valente MJ, Vale L, Afonso J, Carvalho F, Bettencourt P, Sousa T, Albino-Teixeira A. Impaired resolution of inflammation in human chronic heart failure. Eur J Clin Invest 44: 527–538, 2014. doi: 10.1111/eci.12265. [DOI] [PubMed] [Google Scholar]
- 22. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA 81: 5335–5339, 1984. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA 92: 9475–9479, 1995. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poeckel D, Funk CD. The 5-lipoxygenase/leukotriene pathway in preclinical models of cardiovascular disease. Cardiovasc Res 86: 243–253, 2010. doi: 10.1093/cvr/cvq016. [DOI] [PubMed] [Google Scholar]
- 25. Sasaki K, Ueno A, Katori M, Kikawada R. Detection of leukotriene B4 in cardiac tissue and its role in infarct extension through leucocyte migration. Cardiovasc Res 22: 142–148, 1988. doi: 10.1093/cvr/22.2.142. [DOI] [PubMed] [Google Scholar]
- 26. Halade GV, Kain V, Ingle KA. Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. Am J Physiol Heart Circ Physiol 314: H255–H267, 2018. doi: 10.1152/ajpheart.00528.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindsey ML, Brunt KR, Kirk JA, Kleinbongard P, Calvert JW, de Castro Brás LE, DeLeon-Pennell KY, Del Re DP, Frangogiannis NG, Frantz S, Gumina RJ, Halade GV, Jones SP, Ritchie RH, Spinale FG, Thorp EB, Ripplinger CM, Kassiri Z. Guidelines for in vivo mouse models of myocardial infarction. Am J Physiol Heart Circ Physiol 321: H1056–H1073, 2021. doi: 10.1152/ajpheart.00459.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res 112: 675–688, 2013. doi: 10.1161/CIRCRESAHA.111.300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pullen AB, Kain V, Serhan CN, Halade GV. Molecular and cellular differences in cardiac repair of male and female mice. J Am Heart Assoc 9: e015672, 2020. doi: 10.1161/JAHA.119.015672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kain V, Ingle KA, Kabarowski J, Barnes S, Limdi NA, Prabhu SD, Halade GV. Genetic deletion of 12/15 lipoxygenase promotes effective resolution of inflammation following myocardial infarction. J Mol Cell Cardiol 118: 70–80, 2018. doi: 10.1016/j.yjmcc.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Halade GV, Kain V, Serhan CN. Immune responsive resolvin D1 programs myocardial infarction-induced cardiorenal syndrome in heart failure. FASEB J 32: 3717–3729, 2018. doi: 10.1096/fj.201701173RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halade GV, Kain V, Black LM, Prabhu SD, Ingle KA. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging (Albany NY) 8: 2611–2634, 2016. doi: 10.18632/aging.101077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, Halade GV. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 84: 24–35, 2015. doi: 10.1016/j.yjmcc.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Halade GV, Kain V, Ingle KA, Prabhu SD. Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. Am J Physiol Heart Circ Physiol 313: H89–H102, 2017. doi: 10.1152/ajpheart.00040.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kain V, Halade GV. Immune responsive resolvin D1 programs peritoneal macrophages and cardiac fibroblast phenotypes in diversified metabolic microenvironment. J Cell Physiol 234: 3910–3920, 2019. doi: 10.1002/jcp.27165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kain V, Liu F, Kozlovskaya V, Ingle KA, Bolisetty S, Agarwal A, Khedkar S, Prabhu SD, Kharlampieva E, Halade GV. Resolution agonist 15-epi-lipoxin A4 programs early activation of resolving phase in post-myocardial infarction healing. Sci Rep 7: 9999, 2017. doi: 10.1038/s41598-017-10441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, Zhu ZX, Cai WH, Zhu YT, Wei TM, Ye HB, Cong WT, Jin LT. High-glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PLoS One 9: e108182, 2014. doi: 10.1371/journal.pone.0108182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Halade GV, Kain V, Dillion C, Beasley M, Dudenbostel T, Oparil S, Limdi NA. Race-based and sex-based differences in bioactive lipid mediators after myocardial infarction. ESC Heart Fail 7: 1700–1710, 2020. doi: 10.1002/ehf2.12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tourki B, Halade G. Leukocyte diversity in resolving and nonresolving mechanisms of cardiac remodeling. FASEB J 31: 4226–4239, 2017. doi: 10.1096/fj.201700109R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frangogiannis NG. Transforming growth factor-β in myocardial disease. Nat Rev Cardiol 19: 435–455, 2022. doi: 10.1038/s41569-021-00646-w. [DOI] [PubMed] [Google Scholar]
- 41. Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16: 389–406, 2019. doi: 10.1038/s41569-019-0169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 7: 12859, 2016. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tourki B, Halade GV. Heart failure syndrome with preserved ejection fraction is a metabolic cluster of non-resolving inflammation in obesity. Front Cardiovasc Med 8: 695952, 2021. doi: 10.3389/fcvm.2021.695952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, Taguchi S, Lu M, Olefsky JM. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 21: 239–247, 2015. doi: 10.1038/nm.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48: 504–511, 2010. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Halade GV, Kain V, Tourki B, Jadapalli JK. Lipoxygenase drives lipidomic and metabolic reprogramming in ischemic heart failure. Metabolism 96: 22–32, 2019. doi: 10.1016/j.metabol.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stodolkiewicz E, Rewerska B, Rzeszutko M, Tomala M, Chrustowicz A, Zmudka K, Sanak M, Szczeklik W. Leukotriene biosynthesis in coronary artery disease. Results of the Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) study. Pol Arch Intern Med 128: 43–51, 2018. doi: 10.20452/pamw.4140. [DOI] [PubMed] [Google Scholar]
- 48. Szczeklik W, Stodolkiewicz E, Rzeszutko M, Tomala M, Chrustowicz A, Zmudka K, Sanak M. Urinary 11-dehydro-thromboxane B2 as a predictor of acute myocardial infarction outcomes: results of leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study. J Am Heart Assoc 5: e003702, 2016. doi: 10.1161/JAHA.116.003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fosshaug LE, Colas RA, Anstensrud AK, Gregersen I, Nymo S, Sagen EL, Michelsen A, Vinge LE, Øie E, Gullestad L, Halvorsen B, Hansen TV, Aukrust P, Dalli J, Yndestad A. Early increase of specialized pro-resolving lipid mediators in patients with ST-elevation myocardial infarction. EBioMedicine 46: 264–273, 2019. doi: 10.1016/j.ebiom.2019.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halade GV, Kain V, Dillion C, Beasley M, Dudenbostel T, Oparil S, Limdi NA. Race-based and sex-based differences in bioactive lipid mediators after myocardial infarction. ESC Heart Fail 7: 1700–1710, 2020. doi: 10.1002/ehf2.12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jove M, Mate I, Naudi A, Mota-Martorell N, Portero-Otin M, De la Fuente M, Pamplona R. Human aging is a metabolome-related matter of gender. J Gerontol A Biol Sci Med Sci 71: 578–585, 2016. doi: 10.1093/gerona/glv074. [DOI] [PubMed] [Google Scholar]
- 52. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38: 187–197, 2017. doi: 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
- 53. Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat 68–69: 433–455, 2002. doi: 10.1016/s0090-6980(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 54. Halade GV, Kain V, De La Rosa X, Lindsey ML. Metabolic transformation of fat in obesity determines the inflammation resolving capacity of splenocardiac and cardiorenal networks in heart failure. Am J Physiol Heart Circ Physiol 322: H953–H970, 2022. doi: 10.1152/ajpheart.00684.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, Hansson GK, Back M. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation 123: 1316–1325, 2011. doi: 10.1161/CIRCULATIONAHA.110.966846. [DOI] [PubMed] [Google Scholar]
- 56. Tsai MY, Cao J, Steffen BT, Weir NL, Rich SS, Liang S, Guan W. 5-Lipoxygenase gene variants are not associated with atherosclerosis or incident coronary heart disease in the multi-ethnic study of atherosclerosis cohort. J Am Heart Assoc 5: e002814, 2016. doi: 10.1161/JAHA.115.002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stratton R, Shiwen X. Role of prostaglandins in fibroblast activation and fibrosis. J Cell Commun Signal 4: 75–77, 2010. doi: 10.1007/s12079-010-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Claesson H-E, Odlander B, Jakobsson P-J. Leukotriene B4 in the immune system. Int J Immunopharmacol 14: 441–449, 1992. doi: 10.1016/0192-0561(92)90174-j. [DOI] [PubMed] [Google Scholar]
- 59. Rimm EB, Appel LJ, Chiuve SE, Djousse L, Engler MB, Kris-Etherton PM, Mozaffarian D, Siscovick DS, Lichtenstein AH, American Heart Association Nutrition Committee of the Council on L, Cardiometabolic H, Council on E, Prevention, Council on Cardiovascular Disease in the Y, Council on C, Stroke N, and Council on Clinical Cardiology. Seafood long-chain n − 3 polyunsaturated fatty acids and cardiovascular disease: a science advisory from the American Heart Association . Circulation 138: e35–e47, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuppe C, Ramirez Flores RO, Li Z, Hayat S, Levinson RT, Liao X, et al. Spatial multi-omic map of human myocardial infarction. Nature 608: 766–777, 2022. doi: 10.1038/s41586-022-05060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S5 and Figs. S1–S6: https://doi.org/10.6084/m9.figshare.19293287.v3.