

Abstract
Endoplasmic reticulum (ER) Ca2+ homeostasis relies on an appropriate balance between efflux- and influx-channel activity responding to dynamic changes of intracellular Ca2+ levels. Dysregulation of this complex signaling network has been shown to contribute to neuronal and photoreceptor death in neuro- and retinal degenerative diseases, respectively. In mice with cone cyclic nucleotide-gated (CNG) channel deficiency, a model of achromatopsia/cone dystrophy, cones display early-onset ER stress-associated apoptosis and protein mislocalization. Cones in these mice also show reduced cytosolic Ca2+ level and subsequent elevation in the ER Ca2+-efflux-channel activity, specifically the inositol-1,4,5-trisphosphate receptor type 1 (IP3R1), and deletion of IP3R1 results in preservation of cones. This work investigated how preservation of ER Ca2+ stores leads to cone protection. We examined the effects of cone specific deletion of IP3R1 on ER stress responses/cone death, protein localization, and ER proteostasis/ER-associated degradation. We demonstrated that deletion of IP3R1 improves trafficking of cone-specific proteins M-/S-opsin and phosphodiesterase 6C to cone outer segments and reduces localization to cone inner segments. Consistent with the improved protein localization, deletion of IP3R1 results in increased ER retrotranslocation protein expression, reduced proteasome subunit expression, reduced ER stress/cone death, and reduced retinal remodeling. We also observed the enhanced ER retrotranslocation in mice that have been treated with a chemical chaperone, supporting the connection between improved ER retrotranslocation/proteostasis and alleviation of ER stress. Findings from this work demonstrate the importance of ER Ca2+ stores in ER proteostasis and protein trafficking/localization in photoreceptors, strengthen the link between dysregulation of ER Ca2+ homeostasis and ER stress/cone degeneration, and support an involvement of improved ER proteostasis in ER Ca2+ preservation-induced cone protection; thereby identifying IP3R1 as a critical mediator of ER stress and protein mislocalization and as a potential target to preserve cones in CNG channel deficiency.
Keywords: Inositol-1,4,5-trisphosphate receptor; ER Ca2+ stores; ER stress; CNG channel; cone photoreceptors; retinal degeneration
Introduction
Cone photoreceptor cyclic nucleotide-gated (CNG) channels are essential for phototransduction and cellular Ca2+ homeostasis (1). These channels are opened upon binding of cyclic guanosine monophosphate (cGMP) and maintain Ca2+ and Na+ influx into cone outer segments (OS) (2). Mutations in genes encoding the channel subunits CNGA3 and CNGB3 account for 80% of all cases of achromatopsia and are associated with progressive cone dystrophies (3, 4). These diseases are characterized by severely impaired daylight vision, lack of color discrimination, photophobia, and slow progressing degeneration of cones.
Mice lacking functional cone CNG channels, Cnga3−/− and Cngb3−/−, mimic the phenotype in human patients, displaying early-onset cone degeneration and impaired cone function (5, 6). These mice also show cone opsin mislocalization, i.e., reduced opsin localization to the OS and increased opsin localization to the inner segment (IS) and other photoreceptor regions (5, 6). Cnga3−/− and Cngb3−/− mice lacking Nrl, a rod-specific neural-retina leucine-zipper transcriptional factor conferring a cone-dominant retina (7), Cnga3−/−/Nrl−/− and Cngb3−/−/Nrl−/−, display similar phenotype as that in their respective single knockout lines (8, 9), and thus allow one to examine the cellular and biochemical mechanisms of cone degeneration (cones comprise only 2–3% of the total photoreceptor population in wild-type mouse retina). Studies with these models show that cone death in CNG channel-deficient mice involves endoplasmic reticulum (ER) stress-associated apoptosis (8, 9).
As a non-selective cation channel in the OS of photoreceptors, the CNG channel plays a pivotal role in cellular Ca2+ homeostasis. Although permeable to Na+ and Ca2+, cone CNG channels have been shown to have higher Ca2+ affinity than the channel in rods (10). Thus, cones lacking a functional CNG channel suffer from cellular calcium perturbation/cytoplasmic Ca2+ reduction. This has been demonstrated by the measurement of intracellular Ca2+ levels using calcium imaging (11). The cytosolic Ca2+ reduction in these mice has also been supported by the remarkable elevation in cellular cGMP levels (8, 12), because biosynthesis of cGMP is highly regulated by the Ca2+-guanylate cyclase activating protein-guanylate cyclase (Ca2+-GCAP-GC) axis (13, 14); cytosolic Ca2+ level negatively regulates production of cGMP.
Besides cellular Ca2+ signaling, ER Ca2+ homeostasis plays significant roles in regulating protein folding/trafficking, proteostasis/ER-associated degradation (ERAD), and unfolded protein responses (UPR)/ER stress signaling when these processes are disrupted (15–17). ER Ca2+ homeostasis/ER Ca2+ stores is primarily regulated by the two ER Ca2+ channels, the inositol-1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor (RyR), for Ca2+ efflux out of the ER into the cytosol, and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), for Ca2+ influx into the ER. ER Ca2+ channels are highly regulated by cytosolic Ca2+ levels; their activity is increased when cytosolic Ca2+ level is low. There are three isoforms of IP3R: IP3R1, IP3R2 and IP3R3. Transcript expression studies revealed that the levels of IP3R1 mRNA in the mouse retinas were approximately 6- to 10-fold higher than the expression levels of IP3R3 mRNA, whereas IP3R2 mRNA was not detectable in the retinal tissues (11). Consistent with the perturbation of cytosolic Ca2+ homeostasis, cones in mice lacking CNG channel show dysregulation of ER Ca2+ homeostasis and IP3R1. Expression/activity of IP3R1 was significantly increased in CNG channel-deficient retinas (8, 11, 18), likely resulted from decreased cytosolic Ca2+ level, as a compensatory effort to increase cytosolic Ca2+ level. The potential contribution of ER Ca2+ dysregulation/activity of IP3R1 to cone degeneration was supported by findings in which cone specific deletion of IP3R1 improved cone survival in Cnga3−/− mice (11).
The present study was designed to understand the cellular/molecular mechanisms underlying cone preservation after deletion of IP3R1. We found that protein localization to cone OS was significantly improved in CNG channel-deficient mice after deletion of IP3R1 and cone apoptosis was reduced. Correlating with these alterations, deletion of IP3R1 resulted in significant increase in the expression of ER retrotranslocation proteins, decrease in ER stress responses and downstream death signaling, and decrease in Müller glial cell activity. Similar findings were obtained in mice that have been treated with a chemical chaperone. This work demonstrates the importance of ER Ca2+ stores in ER retrotranslocation/proteostasis and protein localization, supports the link between depletion of ER Ca2+ stores and ER stress/cone degeneration, and supports a potential involvement of improved ER proteostasis in ER Ca2+ store preservation-induced cone protection.
Materials and Methods
The Cnga3−/− (5), Cnga3−/−/Nrl−/− (8), Nrl−/− (7), Cnga3−/−/Itpr1flox/flox/HrgpCre (11), and Cnga3−/−/Nrl−/−/Ryr2flox/flox/HrgpCre (19) mouse lines were generated as described previously. The Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre, Nrl−/−/Itpr1flox/flox/HrgpCre, and Itpr1flox/flox/HrgpCre lines were generated by cross-breeding. The wild-type (C57BL/6J) line was obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were housed under cyclic, 12-h light-dark conditions, with ~7-foot candles of illumination during the light cycle. Animal maintenance and experiments were approved by the local Institutional Animal Care and Use Committee (University of Oklahoma Health Sciences Center, Oklahoma City, OK) and conformed to the guidelines on the care and use of animals accepted by the Society for Neuroscience and the Association for Research in Vision and Ophthalmology (Rockville, MD). Mice of either sex were used in the experiments.
Primary antibody information is listed in Table 1. Biotinylated peanut agglutinin (PNA) was purchased from Vector Laboratories, Inc. (Burlingame, CA, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibody was obtained from Kirkegaard & Perry Laboratories Inc. (Gaithersburg, MD), fluorescent-conjugated goat anti-rabbit antibody was purchased from Invitrogen (A21428), Streptavidin-Cy3 was purchased from Sigma-Aldrich (S6402), and 4’,6-Diamidino-2-phenylindole (DAPI) was purchased from Sigma-Aldrich (D9542). Other reagents were obtained from Sigma, Bio-Rad, Invitrogen, Abcam, and Tocris Biosciences.
Table 1.
List of primary antibodies
| Antibody | Provider | Catalog No. | Dilutions used in IB or IF |
|---|---|---|---|
| M-opsin | EMD Millipore | AB5405 | 1: 500 (IB) 1: 200 (IF) |
| S-opsin | Dr. Muna Naash from University of Houston | 1: 200 (IF) | |
| PDE6C | Abgent | AP9728 | 1: 200 (IF) |
| p-eIF2α | Cell signaling | 3398 | 1: 500 (IB) |
| eIF2α | Cell signaling | 9722 | 1: 500 (IB) |
| p-Ire1α | Abcam | ab48187 | 1: 500 (IB) |
| p-creb | Cell signaling | 91985 | 1: 500 (IB) |
| Creb | Cell signaling | 9197 | 1: 500 (IB) |
| GFAP | Dako | Z0334 | 1: 500 (IB) 1: 500 (IF) |
| Derl-1 | Abcam | ab176732 | 1: 500 (IB) |
| Syvn1 | Proteintech | 13473–1-AP | 1: 500 (IB) |
| Sel1L | LSBio | LS-C747272 | 1: 500 (IB) |
| Herpud1 | Thermo Fisher Scientific | PA5–29469 | 1: 500 (IB) |
| PA28α | Cell signaling | 2408 | 1: 500 (IB) |
| PSMD11 | Cell signaling | 14303 | 1: 500 (IB) |
| CHOP | Cell signaling | 2985 | 1: 500 (IB) |
| TBP | Thermo Fisher Scientific | MA1–10883 | 1: 1000 (IB) |
| β-actin | Abcam | ab6276 | 1: 2000 (IB) |
| SQSTM1/p62 | Cell signaling | 5114 | 1: 500 (IB) |
| LC3B | Cell signaling | 2775 | 1: 500 (IB) |
Eye preparation, immunofluorescence labeling, and confocal microscopy
Retinal whole mounts or cross sections were prepared for immunofluorescence labeling, as described previously (20). For retinal whole mount preparations, eyes were enucleated, marked at the superior pole with a green dye, and fixed in 4% paraformaldehyde (PFA; Polysciences, Inc., Warrington, PA) for 30 min at room temperature, followed by removal of the cornea and lens. The eyes were then fixed in 4% paraformaldehyde in PBS for 4 – 6 h at room temperature, and retinas were isolated and the superior portion was marked for orientation with a small cut. For retinal cross-sections, mouse eyes were enucleated (the superior portion of the cornea was marked with green dye prior to enucleation) and fixed in Prefer (Anatech Ltd., Battle Creek, MI) for 25–30 min at room temperature. Paraffin sections (5-μm thickness) passing vertically through the retina (along the vertical meridian passing through the optic nerve head) were prepared using a Leica microtome (Leica Biosystems, Buffalo Grove, IL).
Immunofluorescence labeling was performed as described previously (20). Briefly, retinal whole mounts or sections were blocked with Hanks’ balanced salt solution containing 5% BSA and 0.5% Triton X-100 for 1 h at room temperature or overnight at 4°C. Prior to blocking, antigen retrieval was performed in 10 mm sodium citrate buffer (pH 6.0) in a 70°C water bath. Primary-antibody incubation was performed for 2 h at room temperature or overnight at 4°C (see Table 1 for antibody information). Slides were mounted and coverslipped after fluorescence-conjugated secondary-antibody incubation and wash steps. Immunofluorescence labeling was then imaged using an Olympus FV1000 confocal laser-scanning microscope and FluoView imaging software (Olympus, Melville, NY). For evaluations of cone OS protein cellular localization, confocal images of 10 layers were stacked with the Z-stack function in the ImageJ software (https://imagej.nih.gov/ij/) to obtain a maximal immunofluorescence density. Fluorescence density levels of the immunolabeling in the OS, IS, outer nuclear layer (ONL), and outer plexiform layer (OPL) were measured, and the density levels at each region relative to the total fluorescence density were calculated and averaged from at least three sections per eye from at least five animals per condition.
TUNEL assay
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) was performed to analyze photoreceptor apoptotic death as described previously (18), using paraffin-embedded retinal sections and an in situ cell death fluorescein detection kit (Roche Applied Science, Ref. 11684795910). Immunofluorescence labeling was imaged using an Olympus FV1000 confocal laser-scanning microscope, and TUNEL-positive cells in the ONL passing through the optic nerve were counted and averaged from at least three sections per eye from at least four animals per condition.
Retinal protein preparation, SDS-PAGE, and western blot analysis
Retinal protein preparation, SDS-PAGE, and western blot analysis were performed as described previously (18). Briefly, retinas were homogenized in homogenization buffer A [0.32 M sucrose, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, and 3 mM EDTA containing protease and phosphatase inhibitors] (Roche Applied Science, catalog no. 04693159001 and 04906837001, respectively), and homogenates were centrifuged at 1,200 g for 10 min at 4°C. The resulting supernatant was then centrifuged at 21,000 g for 35 min at 4°C to separate cytosolic (supernatant) and membrane (pellet) fractions. The resulting membrane pellet was resuspended in homogenization buffer B [0.32 M sucrose, 20 mM HEPES, pH 7.4, 3 mM EDTA, and 0.1% Triton X-100 containing protease and phosphatase inhibitors], sonicated twice for 15 s on ice at a medium speed using an XL2000 Ultrasonic Cell Disruptor (Misonix, Farmingdale, NY, USA), with a 30-s recovery between disruptions, and incubated for 1 h at 4°C with gentle agitation. After incubation, the homogenate was centrifuged at 21,000 g for 35 min at 4°C, and the resulting supernatant was used as the membrane fraction. All protein concentrations were determined by a protein-assay kit from Bio-Rad Laboratories. Retinal protein samples (20 μg protein per sample) were then subjected to SDS-PAGE and transferred to PVDF membranes, followed by blocking in 5% bovine serum albumin (BSA) for 1 h at room temperature. Immunoblots were incubated with primary antibody overnight at 4°C. After washing in Tris-buffered saline with 0.1% Tween 20, immunoblots were incubated with horseradish peroxidase-conjugated secondary antibody (1:20,000) for 1 h at room temperature. SuperSignal® West Dura Extended Duration chemiluminescent substrate (Thermo Fisher Scientific, catalog no. 34076) was used to detect binding of the primary antibodies to their cognate antigens. An Li-Cor Odyssey CLx Imager and Li-Cor software (Li-Cor Biosciences, Lincoln, NE, USA) were used for detection and densitometric analysis.
PCR array
Total RNA preparation and reverse transcription were performed as described previously (9). The Mouse Unfolded Protein Response RT2 Profiler PCR Array (Qiagen, Hiden, Germany), which profiles the expression of 84 genes involved in unfolded protein binding, protein folding, endoplasmic-reticulum-associated protein degradation, and heat shock proteins, was used under the manufacturer’s instructions.
TUDCA treatment
Treatment of Cnga3−/−/Nrl−/− mice with tauroursodeoxycholic acid (TUDCA) was performed as described previously (9, 21). Briefly, TUDCA (500 mg/kg, body weight, TCI America) or vehicle (0.15 M NaHCO3, pH 7.0) was given to Cnga3−/−/Nrl−/− mice by subcutaneous injection every 3 days for 12 days, starting at P5. Retinas were collected at the end of the treatment for western blotting analysis.
Statistical analysis
One-way analysis of variance and unpaired Student’s t test were used to evaluate significant differences between multiple groups and two groups, respectively. Statistical analyses and graph generation were performed using GraphPad Prism® software (GraphPad Software, San Diego) for Windows.
Results
Deletion of IP3R1 reduces ER stress, downstream death signaling, and cone apoptosis in CNG channel-deficient retinas.
To demonstrate the contribution of IP3R1 activity to ER stress, we evaluated the effects of cone-specific deletion of IP3R1. Mouse retinas at postnatal day 15 (P15) and P30 were analyzed for ER stress markers by immunoblotting. As shown in Figure 1A, Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre retinas showed significantly reduced phospho-eukaryotic-initiation factor 2α (p-eIF2α) levels at P15 and P30 when compared to age-matched Cnga3−/−/Nrl−/− controls, approaching the baseline level found in the age-matched control Nrl−/−/Itpr1flox/flox/HrgpCre mice (the expression level in Nrl−/−/Itpr1flox/flox/HrgpCre retina was not different from that in Nrl−/− retina, data not shown). In addition, phospho-serine/threonine-protein kinase/endoribonuclease 1α (p-IRE1α) level was significantly reduced in Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre retinas at P30 when compared to age-matched Cnga3−/−/Nrl−/− controls, and also approach baseline level (Fig. 1A). Furthermore, we analyzed the effects of IP3R1 deletion on transcriptional regulation of ER stress signaling, and found nearly complete return to baseline mRNA expression levels of each marker tested (Fig. 1B). These results suggest that reducing IP3R1 activity attenuates ER-stress responses at both transcriptional and translational levels in CNG channel-deficient retinas.
Figure 1. Deletion of IP3R1 reduced UPR/ER stress in CNG channel-deficient retinas.
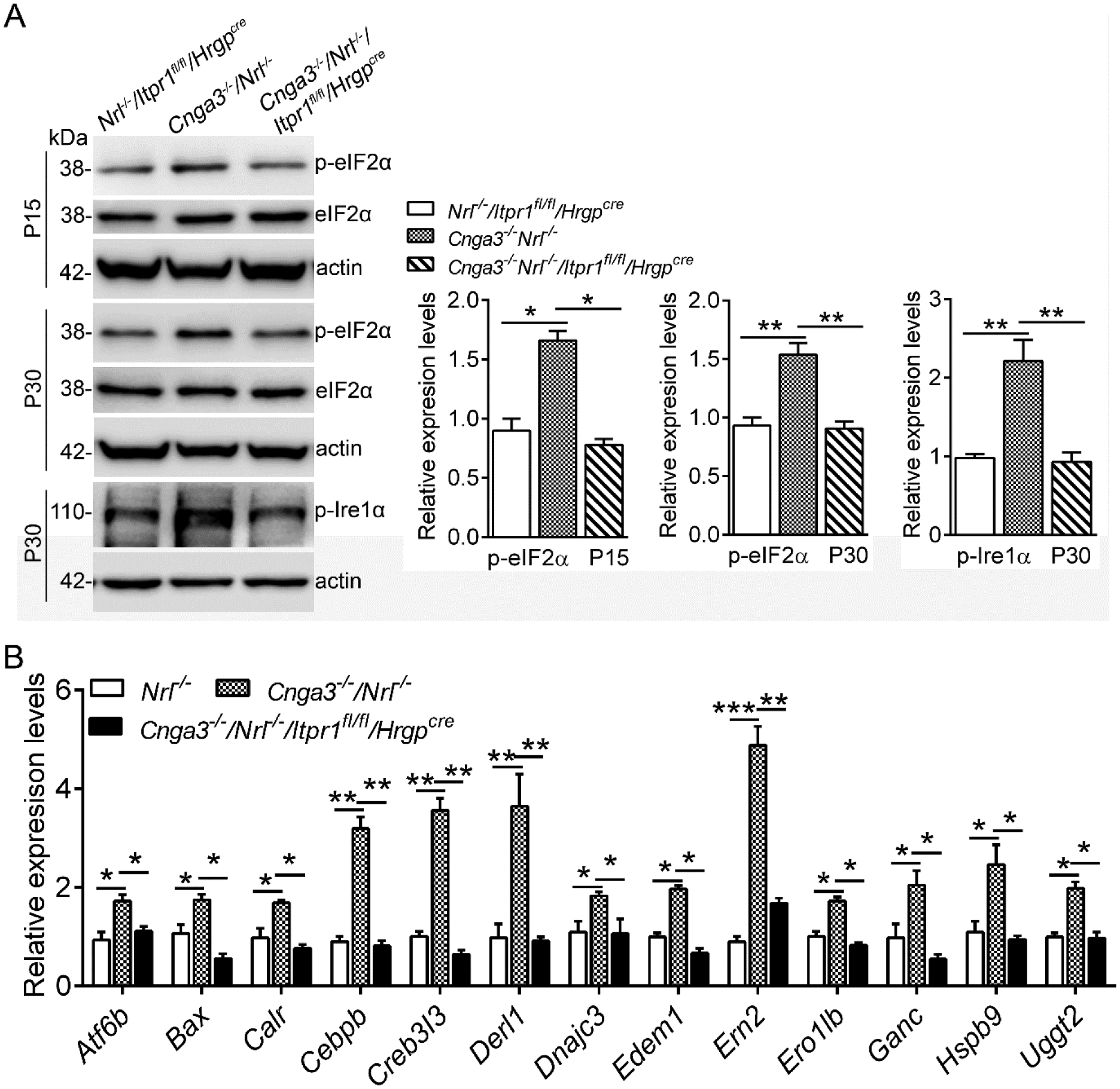
A. Expression levels of ER stress marker proteins in mouse retinas at P15 and P30 were analyzed by immunoblotting. Shown are representative immunoblotting images of these detections and corresponding quantitative analysis, following normalization to internal loading control β-actin. B. Expression of UPR genes was evaluated in mouse retinas at P15 by PCR array. Shown are PCR array results. Data are presented as mean ± SEM of 3–4 independent assays using retinas from 8–10 mice (*p < 0.05, **p < 0.01, ***p < 0.001).
The effects of IP3R1 deletion were further examined by evaluating the expression of CHOP, a member of the C/EBP (CCAAT enhancer-binding protein) transcription factor family induced in ER stress (22) and involved in ER stress-associated apoptosis (23, 24). We have previously shown increased expression/activation of CHOP in CNG channel-deficient retina (8). Nuclear protein preparations from mouse retina at P15 were analyzed for CHOP expression. The analysis showed that deletion of IP3R1 completely abolished CHOP activation in Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre retinas (Fig. 2A).
Figure 2. Deletion of IP3R1 reduced ER stress downstream signaling and apoptosis in CNG channel-deficient retinas.
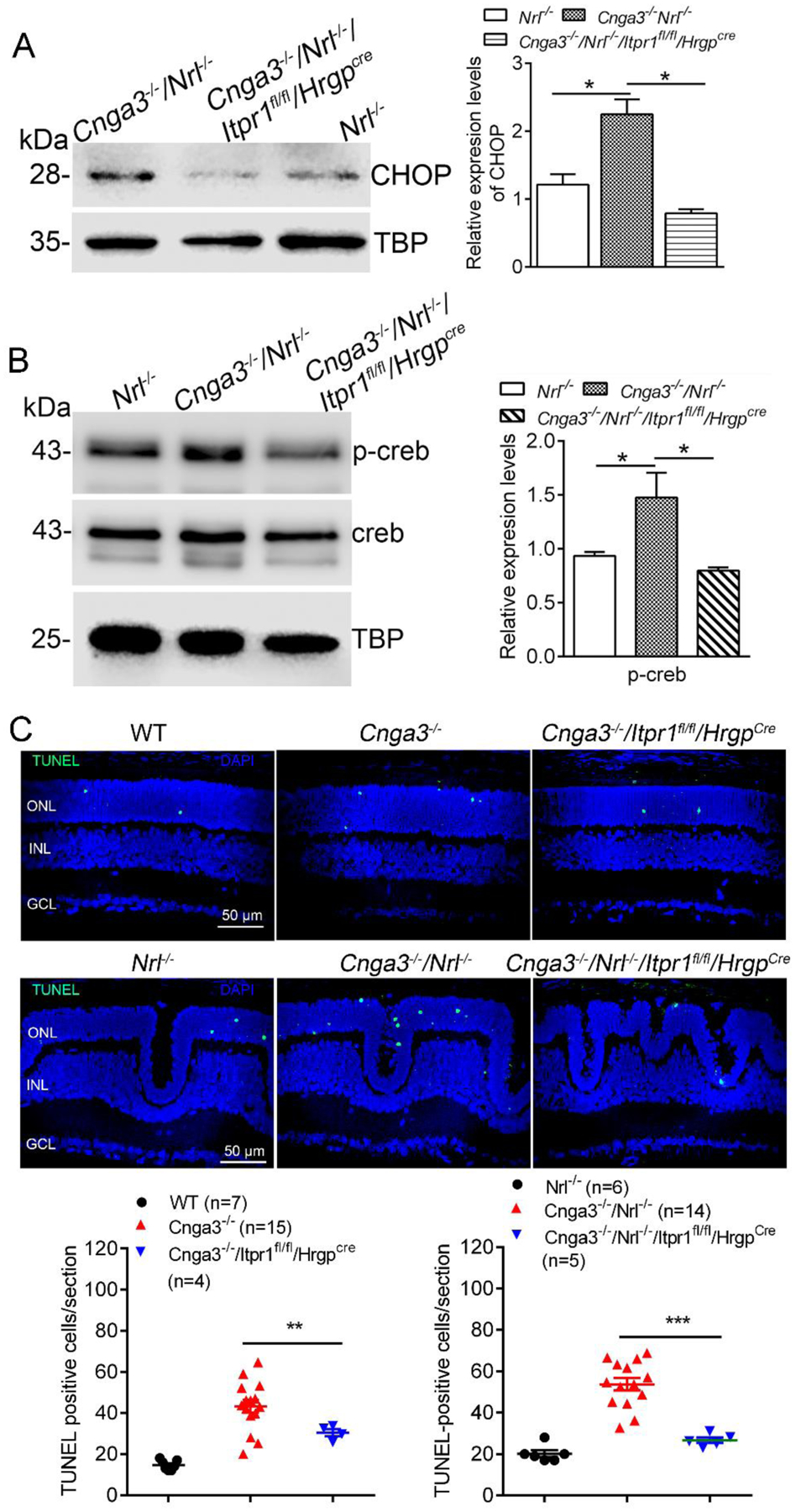
A-B. Expression of CHOP and p-Creb were evaluated in mouse retinas by immunoblotting. Shown are representative immunoblotting images for CHOP at P15 (A) and p-Creb at P30 (B) and the corresponding quantitative analysis. C. Photoreceptor apoptosis was evaluated by TUNEL labeling on retinal sections of CNG channel-deficient mice at P15. Shown are representative confocal images of TUNEL labeling and correlating quantitative analysis. ONL, outer nuclear layer; INL, inner nuclear layer. Data are presented as mean ± SEM of 3–4 independent assays using retinas/eye sections from 4–10 mice (*p < 0.05, **p < 0.01, ***p < 0.001).
The basic/leucine zipper transcription factor Creb (cyclic adenosine monophosphate response element binding protein) is activated by UPR/ER stress signaling (25). We have shown increased expression/activation of Creb signaling in CNG channel-deficient retina (26). In the present study, we evaluated whether deletion of IP3R1 affects Creb signaling. Nuclear protein preparations from mouse retina at P30 were analyzed for Creb expression/activity. The analysis showed that the phospho-Creb level was increased by about 50% in Cnga3−/−/Nrl−/− mice, compared with Nrl−/− retinas, and deletion of IP3R1 abolished this elevation (Fig. 2B).
To confirm the contribution of IP3R1 activity to cone apoptosis, we evaluated whether deletion of IP3R1 reduces apoptotic cone death by TUNEL. The analysis revealed that the retinal sections of Cnga3−/−/Itpr1flox/flox/HrgpCre mice and Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre mice at P15 display a significant reduction in the number of TUNEL-positive cells when compared with their respective age-matched controls (Fig. 2C).
Deletion of IP3R1 increases cone survival and reduces Müller glial cell activation in CNG channel-deficient retinas.
In addition, we analyzed PNA-labeled retinal sections to evaluate cone survival in these mice at 2 months and found improved survival after IP3R1 deletion (Fig. 3A). Improved cone survival was also shown by increased expression level of M-opsin in Cnga3−/−/Nrl−/−/Itpr1flox/flox/HrgpCre mice, compared with Cnga3−/−/Nrl−/− mice (Fig. 3B).
Figure 3. Deletion of IP3R1 increased cone density and expression levels of M-opsin in CNG channel-deficient retinas.
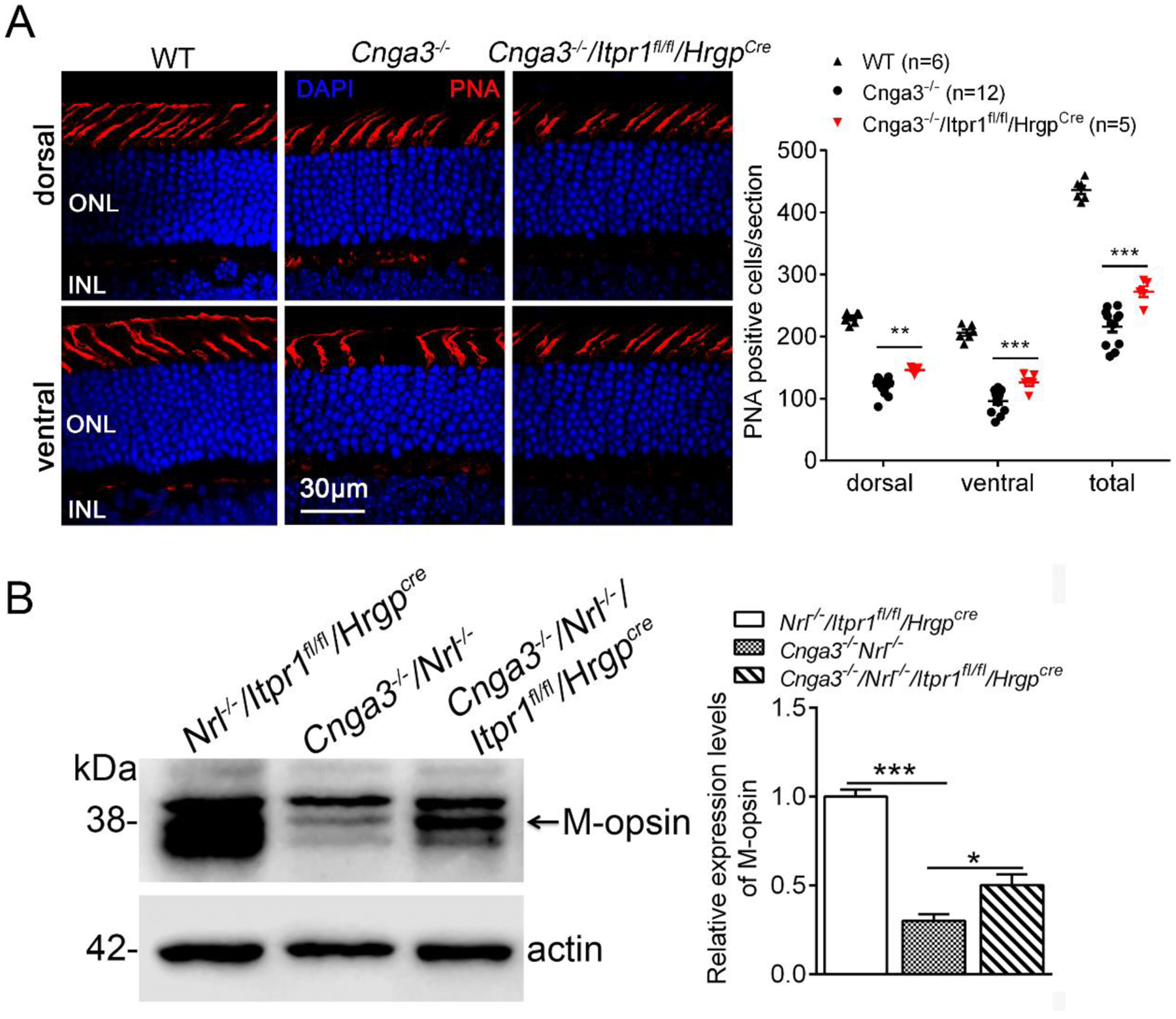
A. Cone density was evaluated by PNA labeling on retinal sections prepared from mice at 2 months. Shown are representative confocal images of PNA labeling on retinal sections and corresponding quantitative analysis. ONL, outer nuclear layer. B. Expression levels of M-opsin were analyzed by immunoblotting at P30. Shown are representative immunoblotting images and the corresponding quantitative analysis. Data are presented as mean ± SEM of 3–4 independent assays using retinas/eye sections from 5–12 mice (**p < 0.01, ***p < 0.001).
Increased Müller glial cell activity is associated with retinal degeneration and is commonly assessed by glial fibrillary acidic protein (GFAP) immunolabeling (27–29). Increased GFAP activity has been documented in retinas of Cnga3−/− and Cnga3−/−/Nrl−/− mice (18, 19, 30) as a consequence of progressive cone degeneration. We examined GFAP expression in CNG channel-deficient mice at P30 after deletion of IP3R1. The results in Figure 4 show reduced GFAP immunofluorescence to near baseline in Cnga3−/−/Itpr1flox/flox/HrgpCre retinal sections when compared to age-matched Cnga3−/− mice (Fig. 4A). We also measured GFAP protein levels in these mice using immunoblotting and similar trends were obtained (Fig. 4B).
Figure 4. Deletion of IP3R1 reduced activation of Müller glial cells in CNG channel-deficient retinas.
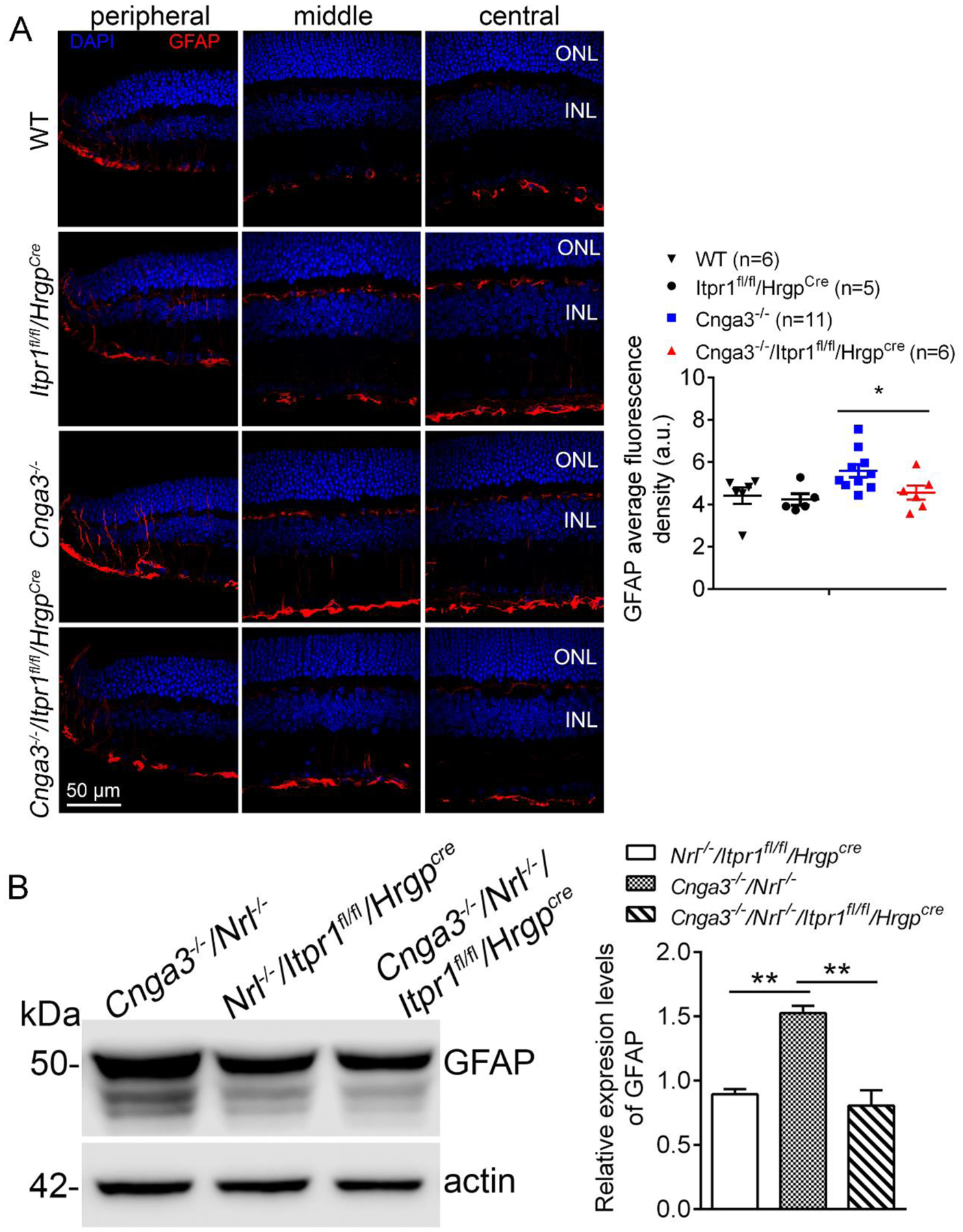
A. GFAP immunofluorescence labeling was performed on the retinal sections prepared from mouse eyes at P30. Shown are representative confocal images of immunofluorescence labeling of GFAP on the peripheral, middle, and central regions of the retinal sections and corresponding quantification of immunofluorescence intensity. ONL, outer nuclear layer; INL, inner nuclear layer; RGC, retinal ganglion cell. B. Expression levels of GFAP were evaluated in the mouse retinas by immunoblotting at P30. Shown are representative immunoblotting images of GFAP and corresponding quantitative analysis. Data are presented as mean ± SEM of 3–4 independent assays using retinas/eye sections from 5–12 mice (**p < 0.01).
Deletion of IP3R1 improves cone opsin localization to OS in CNG channel-deficient cones.
To evaluate the contributions of IP3R1 activity to cone opsin mislocalization, we examined the effects of IP3R1 deletion. As shown in Figure 5A, M-opsin localization to cone OS was significantly increased in Cnga3−/−/Itpr1flox/flox/HrgpCre mice at 4 months (from 17–31% in Cnga3−/− mice to 30–40% in Cnga3−/−/Itpr1flox/flox/HrgpCre mice). Furthermore, M-opsin levels were significantly decreased in cone IS in Cnga3−/−/Itpr1flox/flox/HrgpCre mice, providing additional evidence for improved M-opsin trafficking (Fig. 5A). Similar findings were obtained with S-opsin labeling. Deletion of IP3R1 significantly improved S-opsin localization to cone OS and reduced S-opsin mislocalization to cone IS, ONL, and OPL when compared to age-matched Cnga3−/− controls (Fig. 5B).
Figure 5. Deletion of IP3R1 increased OS localization of cone opsin in CNG channel-deficient retinas.
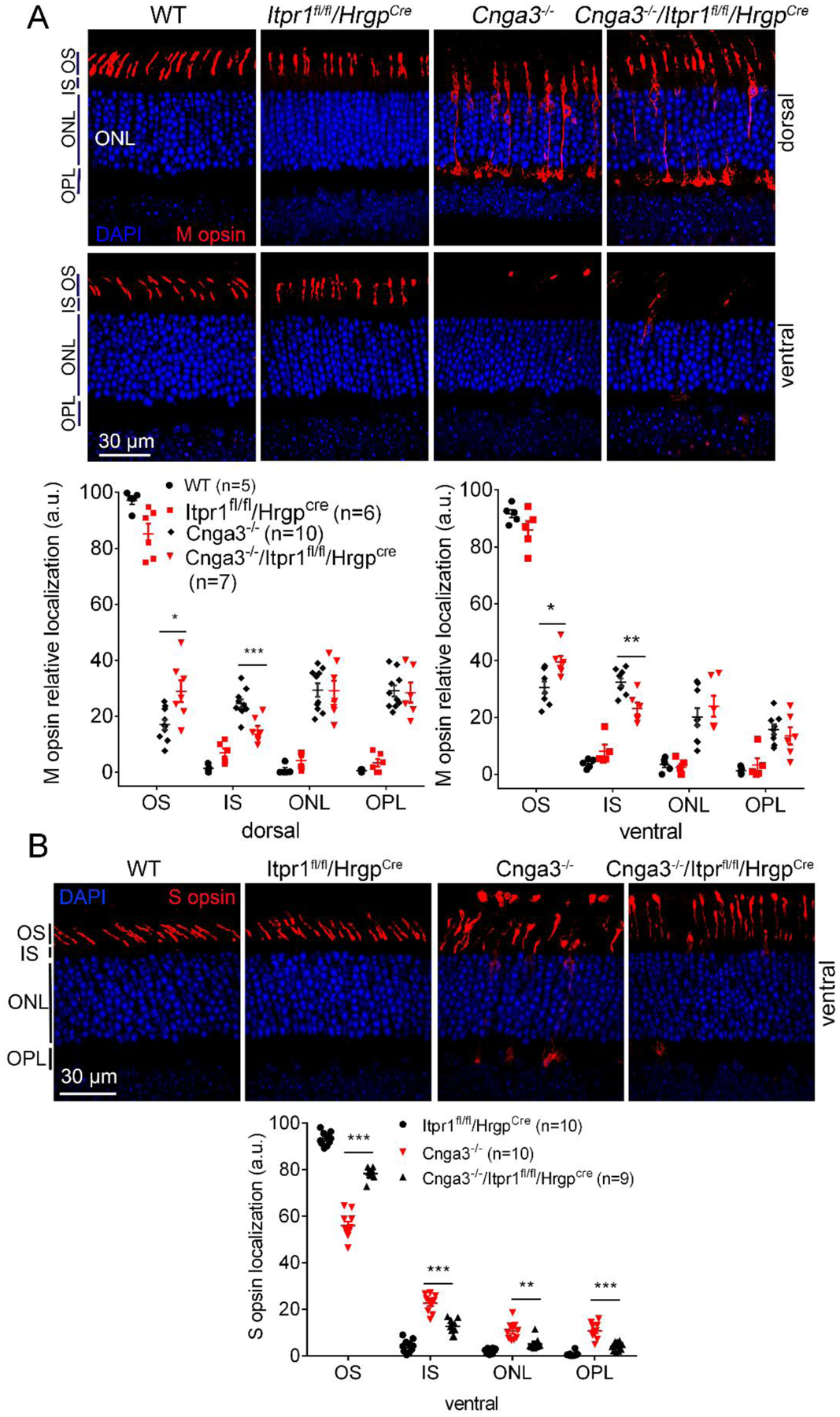
M- and S-cone opsin localization were evaluated by immunofluorescence labeling on retinal cross sections prepared from mice at P30. Shown are representative confocal images of immunofluorescence labeling of M-opsin (A) and S-opsin (B) and corresponding quantitative analysis of immunofluorescence intensity level at different regions of the retinal cross sections. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer. Data are presented as mean ± SEM of 3–4 independent assays using retinas/eye sections from 5–10 mice (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Deletion of IP3R1 improves PDE6C localization to OS in CNG channel-deficient cones.
To gain additional evidence for the contribution of IP3R1 activity to cone protein mislocalization, we analyzed the effects of IP3R1 deletion on the trafficking of another cone-specific protein, the phosphodiesterase 6C (PDE6C). The data in Figure 6 show that Cnga3−/−/Itpr1flox/flox/HrgpCre retinas have improved PDE6C localization to cone OS when compared to age-matched Cnga3−/− controls (Fig. 6). Together with the opsin localization data, these results indicate IP3R1 activity contributes to impairment of global protein trafficking found in CNG channel deficiency and deletion of IP3R1 improves protein trafficking.
Figure 6. Deletion of IP3R1 increased OS localization of PDE6C in CNG channel-deficient retinas.
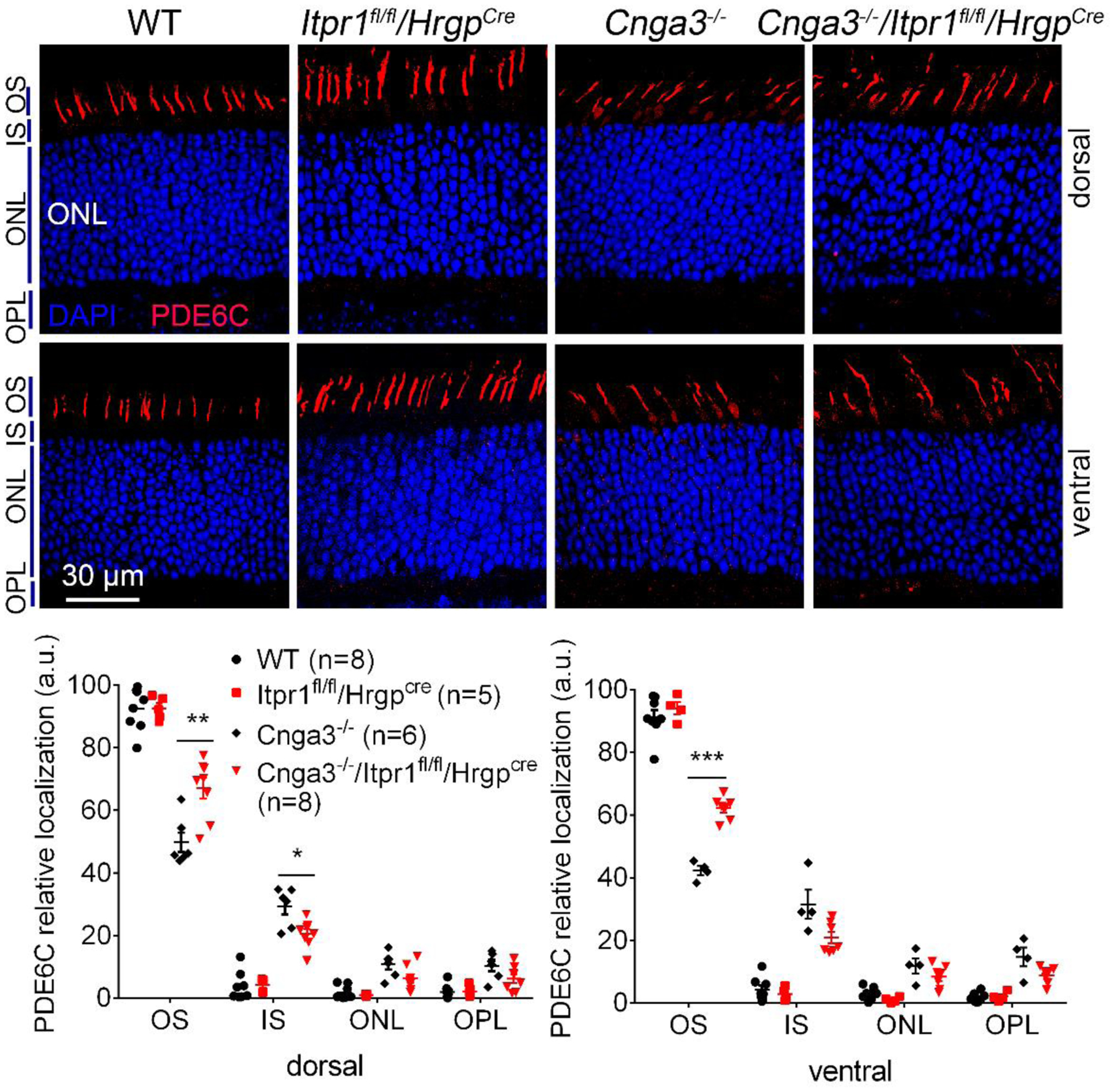
PDE6C localization was evaluated by immunofluorescence labeling on retinal cross sections prepared from mice at P30. Shown are representative confocal images of immunofluorescence labeling of PDE6C (A) and corresponding quantitative analysis of immunofluorescence intensity level at different regions of the retinal cross sections (B). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer. Data are presented as mean ± SEM of 3–4 independent assays using retinas/eye sections from 5–8 mice (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Deletion of IP3R1 increases expression of ER retrotranslocation proteins in CNG channel-deficient retinas.
ER protein retrotranslocation and the subsequent ubiquitination-mediated degradation via proteasomes is a critical mechanism to maintain ER proteostasis/ER function and regulate UPR/ER stress. ER retrotranslocation is a process involving multiple machinery proteins, including Syvn1 (E3 ubiquitin-protein ligase synoviolin 1, directing ubiquitination and targeting to proteasomes for degradation) (31, 32), Sel1L (ERAD E3 ligase adaptor subunit) (33, 34), Herpud1 (homocysteine inducible ER protein with ubiquitin like domain 1) (35, 36), and Derlin-1 (degradation in ER protein 1) (37, 38). We examined expression of these proteins in CNG channel-deficient retina at P15 and P30, and the effects of IP3R1 deletion. We found that the expression levels of Syvn1, Sel1L, and Herpud1 were unchanged in Cnga3−/−/Nrl−/− mice, compared with Nrl−/− controls. However, deletion of IP3R1 significantly increased expression of these proteins (Fig. 7A). Interestingly, expression of Derlin-1 was increased in CNG channel-deficient retina and deletion of IP3R1 completely abolished this upregulation (Fig. 7B). To assess whether these observations are associated with ER Ca2+ stores but not the IP3R1 channel itself, we included a second mouse line with deletion of RyR2 (Cnga3−/−/Nrl−/−/Ryr2flox/flox/HrgpCre) and obtained similar findings (Fig. 7A). In a separate experiment, we examined whether a chemical chaperone that reduces ER stress also increases expression of the ER retrotranslocation proteins. We treated Cnga3−/−/Nrl−/− mice with TUDCA, a well-known chemical chaperone that has been proven to reduce ER stress and photoreceptor cell death (21, 39, 40), including ER stress/cone death in Cnga3−/−/Nrl−/− retinas (9, 11). Immunoblotting analysis showed that expression levels of Syvn1, Sel1L, and Herpud1 were significantly increased after TUDCA treatment, compared with vehicle-treated controls. However, TUDCA treatment did not alter the expression of Derlin-1 (Fig. 7C).
Figure 7. Deletion of IP3R1 or treatment with TUDCA increased expression of ER retrotranslocation proteins in CNG channel-deficient retinas.
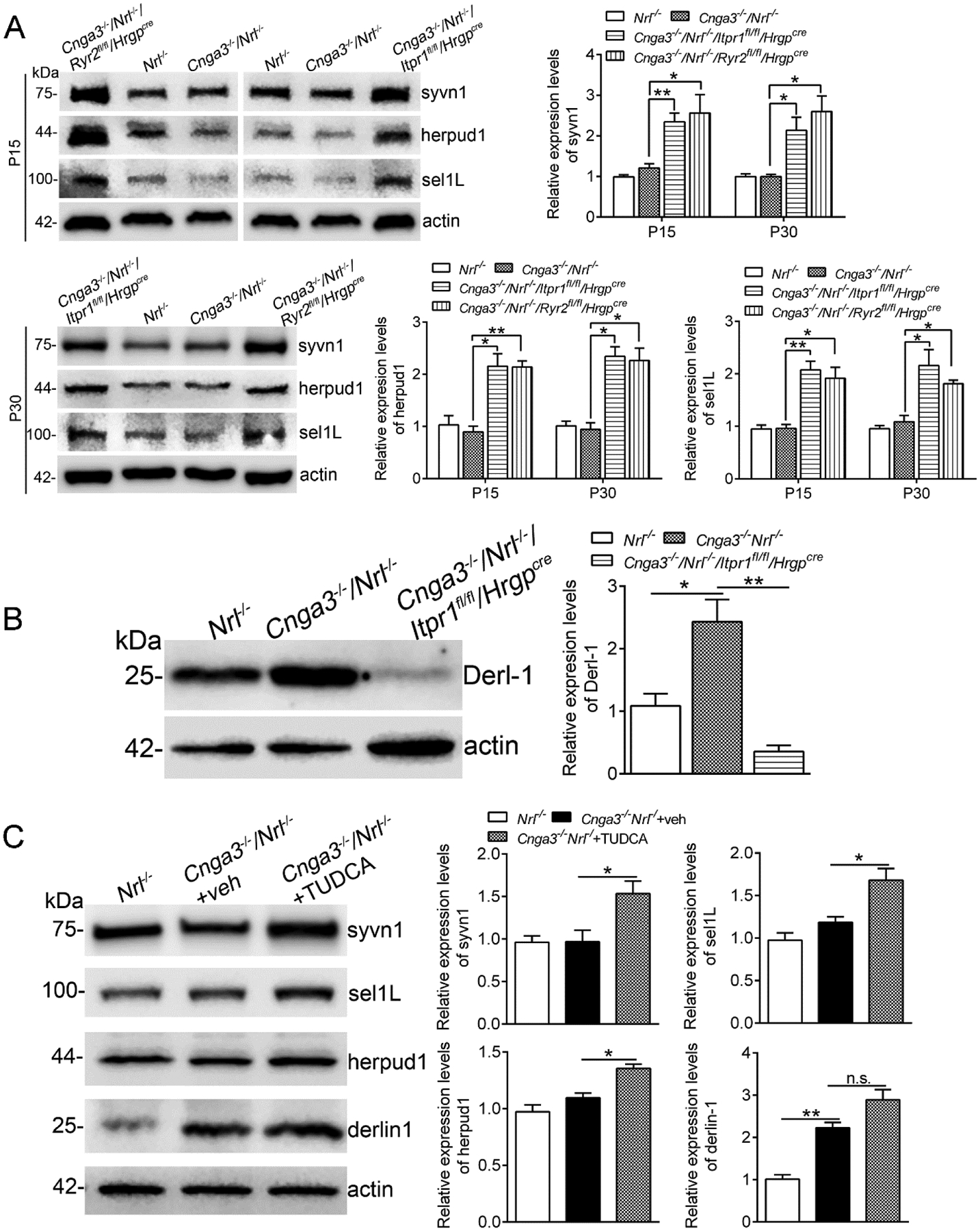
Expression levels of ER retrotranslocation proteins in mouse retinas at P15 and P30 (A-B), and after TUDCA treatment (C) were analyzed by immunoblotting. Shown are representative immunoblotting images of these detections and corresponding quantitative analysis, following normalization to internal loading control β-actin. Data are presented as mean ± SEM of 3–4 independent assays using retinas from 8–10 mice (*p < 0.05, **p < 0.01).
Deletion of IP3R1 decreases expression of the proteasome subunit in CNG channel-deficient retinas.
We extended our evaluations to expression of the proteasome machinery. The proteasome subunits proteasome activator 28α (PA28α) and PA28β form a heteroheptameric complex and function by binding to the 20S proteasome complex (41), whereas PSMD11 (proteasome 26S subunit, non-ATPase 1) is a component of the 19S regulator of the proteasomes. We examined expression of PA28α and PSMD11 in CNG channel-deficient retinas after deletion of IP3R1. We found that the expression levels of PA28α was not different between Cnga3−/−/Nrl−/− and age-matched Nrl−/− retinas. However, deletion of IP3R1 or RyR2 significantly reduced expression levels of PA28α in CNG channel-deficient retinas, and this reduction was observed at P15 but not P30 (Fig. 8A). In contrast to changes in PA28α, expression levels of PSMD11 remained unchanged among the different genotypes (Fig. 8A). Similar to findings in mice with IP3R1 deletion, PA28α expression level was significantly reduced after TUDCA treatment whereas PSMD11 remained unchanged (Fig. 8B).
Figure 8. Deletion of IP3R1 or treatment with TUDCA decreased expression of the proteasome subunit in CNG channel-deficient retinas.
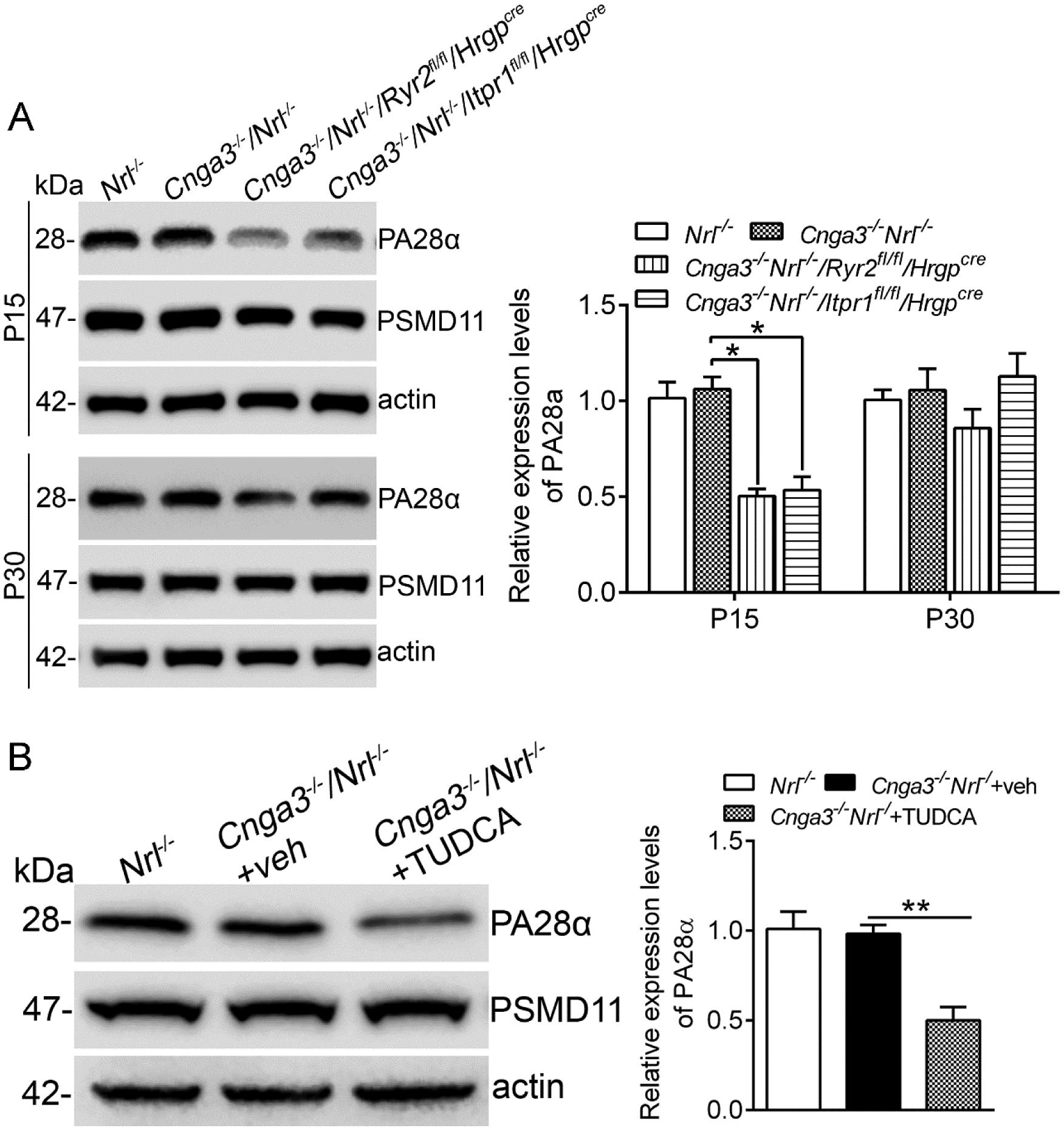
Expression levels of the proteasome subunits in mouse retinas at P15 and P30 (A) and after TUDCA treatment (B) were analyzed by immunoblotting. Shown are representative immunoblotting images of these detections and corresponding quantitative analysis, following normalization to internal loading control β-actin. Data are presented as mean ± SEM of 3–4 independent assays using retinas from 8–10 mice (*p < 0.05, **p < 0.01).
Deletion of IP3R1 does not affect autophagy activity in CNG channel-deficient retinas.
It has been known that UPR/ER stress induces/increases autophagy (42, 43). We examined markers for autophagy in CNG channel-deficient retinas and the effects of IP3R1 deletion. As shown in Figure 9, the expression levels of SQSTM1/p62 and LC3B II were significantly elevated in Cnga3−/−/Nrl−/− retinas. However, deletion of IP3R1 or RyR2 did not induce further alterations (Fig. 9A). Similar findings were observed in TUDCA-treated Cnga3−/−/Nrl−/− retinas. Expression levels of SQSTM1/p62 and LC3B II remained the same in mice with and without TUDCA treatment (Fig. 9B).
Figure 9. Deletion of IP3R1 did not affect autophagy activity in CNG channel-deficient retinas.
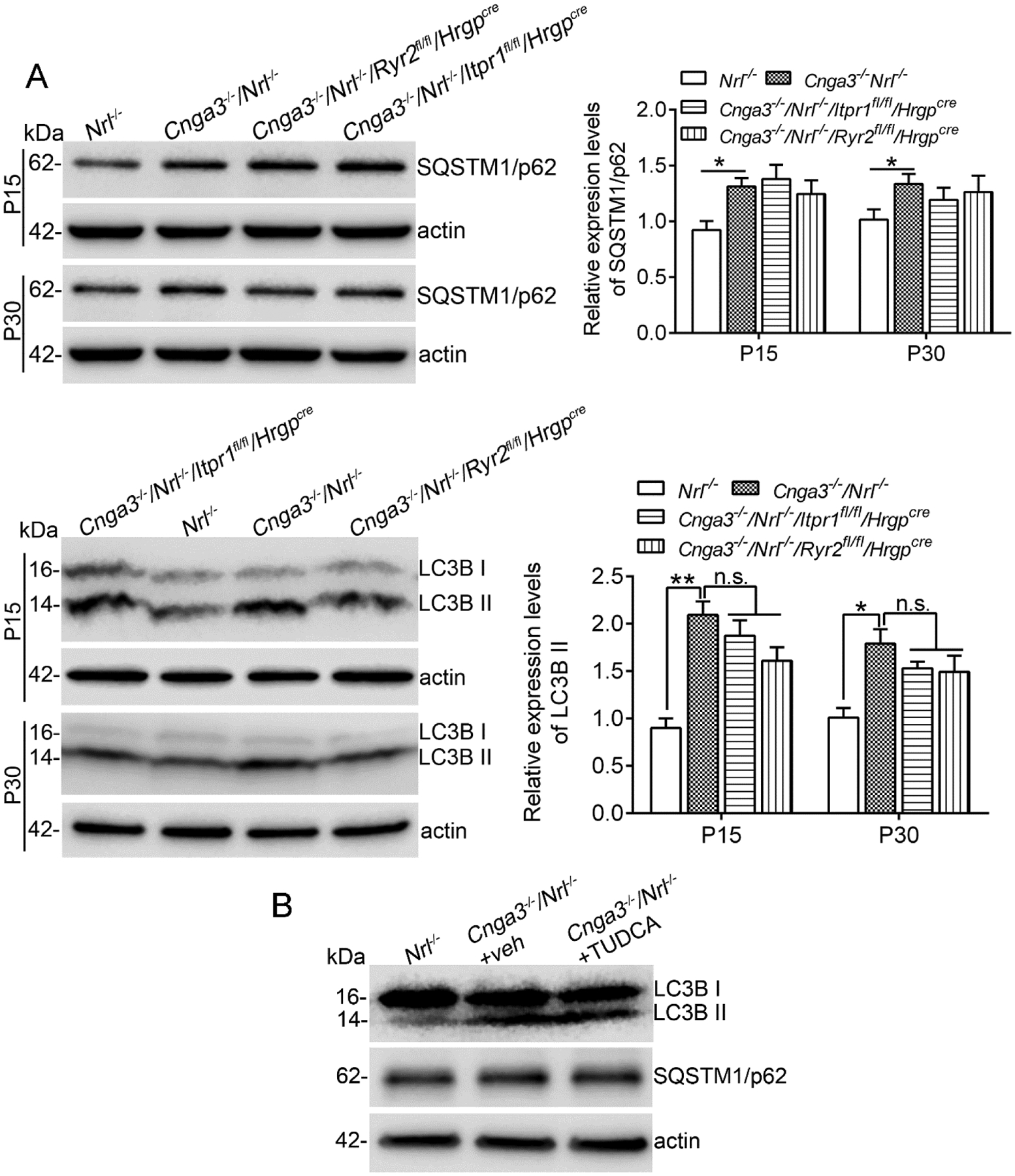
Expression levels of autophagy marker proteins in mouse retinas (A) and after TUDCA treatment (B) were analyzed by immunoblotting. Shown are representative immunoblotting images of these detections and corresponding quantitative analysis, following normalization to internal loading control β-actin. Data are presented as mean ± SEM of 3–4 independent assays using retinas from 8–10 mice (*p < 0.05, **p < 0.01).
Discussion
In our previous work, we have documented ER stress-associated apoptotic cone death and protein mislocalization in CNG channel-deficient mice. Retinas of these mice show elevation of all three arms of the ER stress pathways, i.e., elevated levels of phospho-eIF2α and phospho-IRE1α and increased cleavage of ATF6 (8, 11, 18), as well as increased nuclear localization of CHOP (8, 18). CNG channel-deficient cones also show reduced cytosolic Ca2+ levels at early ages and a subsequent increase in the expression/activity of IP3R1 (11). Furthermore, pharmacological inhibition of IP3R using chemical inhibitors significantly reduces ER stress/cone death and improves protein OS localization, and deletion of IP3R1 improved cone survival (11). The present study expanded on the previous work and explored the mechanism(s) underlying how deletion of IP3R1 leads to cone protection. Based on the known connections between ER Ca2+ dysregulation and ER stress (44) and our previous findings, we hypothesized that IP3R1 deletion would preserve ER Ca2+ stores/improve ER Ca2+ homeostasis, resulting in improved protein trafficking/proteostasis and subsequent reduction in ER stress. Consistent with our predictions, the present work demonstrated improved localization of M-/S-opsin and PDE6C to cone OS and reduced localization to cone IS after deletion of IP3R1, reduced ER stress responses/cone apoptosis, reduced Müller glial cell activation, and early-onset cone protection. Moreover, deletion of IP3R1 increased expression of the ER retrotranslocation proteins and reduced expression of the proteasome proteins, suggesting that the protective effects of IP3R1 deletion may be related to a potentially improved ER proteostasis. Thus, targeting IP3R1 is sufficient to rebalance ER Ca2+ homeostasis in CNG channel deficiency, improve ER proteostasis, and reduce ER stress/cone apoptosis, representing a novel strategy for cone protection.
The potential mechanism underlying improved protein trafficking/OS localization and reduced ER stress after IP3R1 deletion may involve an increased ER retrotranslocation/improved proteostasis. The expression levels of the retrotranslocation complex proteins Syvn 1, Sel1L and Herpud1 were significantly increased after IP3R1 deletion. Similar findings were observed in mice that have been treated with TUDCA. Increased ER retrotranslocation would relieve ER protein accumulation and facilitate the degradation of some death signaling molecules. Syvn1 is known to reduce ER stress-associated apoptosis through increased clearance of IRE1α and CHOP (45, 46). Consistent with this finding, the present work show reduced levels of phospho-IRE1α and CHOP after IP3R1 deletion. We presume that deletion of IP3R1 improves ER protein retrotranslocation/ER proteostasis, which in turn reduces ER stress/cone death and improves protein trafficking/cone survival. Furthermore, the reduction in ER stress activity including improved protein folding/trafficking globally may subsequently reduce proteasome activity because of a decreased demand for protein clearance. This prediction was supported by the reduction of the proteasome component PA28α. Previous studies have shown upregulation of the proteasome components in retinal degeneration resulting from misfolding mutations in rhodopsin (P23H mutation) (47), and proteasome overload has been implicated as a mechanism of photoreceptor death (48). Nevertheless, our findings suggest that preservation of ER Ca2+ stores is sufficient to improve ER proteostasis and reduce ER stress and the subsequent proteasome burden. Although the intermediate signaling connecting ER Ca2+ stores with ER retrotranslocation machinery and proteasome capacity requires further exploration, enhancing ER retrotranslocation may represent a potential approach to improve ER proteostasis/relieve ER stress in retinal degeneration. Indeed, over expression of Syvn1 has been shown to reduce retinal degeneration in diabetic retinopathy (49).
ER stress and ER stress-associated apoptotic cone death is an early event in CNG channel deficiency (8, 18). The present study showed that the reduction in phospho-IRE1α and phospho-eIF2α was observed as early as P15 after IP3R1 deletion, supporting a decrease in ER stress at early age. Furthermore, cone preservation/increased cone density was already apparent at 2 months. When combined with previous results showing improved cone density at older ages (4 and 8 months) (11), our findings suggest that early intervention to reduce ER stress in CNG-channel deficiency can lead to early-onset, sustained cone preservation.
Although ER stress responses were nearly completely reversed in CNG channel-deficient mice after deletion of IP3R1 and cone apoptosis was greatly reduced, protein localization and cone density were only partially improved/resumed. These observations suggest that the stabilization of ER Ca2+ stores was likely incomplete. The contributions of other ER Ca2+ channels, like RyR2, cannot be overlooked due to their up-regulation in CNG channel deficiency and the contribution to protein mislocalization and ER stress, as shown previously (11, 19). It can be predicted that deletion of both IP3R1 and RyR2 would enhance the improvement of protein trafficking and cone survival. The IS membrane Ca2+ channels, such as stromal interaction molecule (STIM) proteins and transient receptor potential channels (TRPCs) (50, 51), may contribute to disruption of ER Ca2+ homeostasis in the channel-deficient mice. In addition, cone OS malformation is a hallmark of CNG channel deficiency (5, 52) and presents a persistent mechanical barrier to protein transport.
Autophagy activity was elevated in CNG channel-deficient retina. However, deletion of IP3R1 did not suppress activation of autophagy. This finding suggests that preservation of ER Ca2+ store may not be sufficient enough to correct autophagy in CNG channel deficiency. It may also suggest that autophagy in CNG channel deficiency does not primarily involve an ER stress-related mechanism although ER stress has been shown to induce autophagy (53). The sustained elevation of autophagy activity after IP3R1 deletion may contribute in part to the partial rescue of cone survival/density.
In summary, the present study demonstrates that IP3R1 deletion improves cone protein trafficking to OS and reduces ER stress/apoptosis, leading to cone preservation. The improved ER retrotranslocation/proteostasis after deletion of IP3R1 may underlie the observed cone protection. Findings from the present work identify IP3R1 activity as a key mediator of dysfunctional ER Ca2+ homeostasis and subsequent ER stress, protein mislocalization, and cone death in CNG channel deficiency, revealing a novel mechanism of photoreceptor degeneration. Importantly, these results support early intervention to stabilize cone Ca2+ dynamics and that doing so leads to improvement in photoreceptor viability. Altered Ca2+ signaling, ER stress, and dysfunctional proteostasis are common phenotypes of many different types of retinal degenerative diseases. Understanding the molecular components contributing to these disease-promoting processes will help develop novel strategies to stabilize cytosolic-ER Ca2+ dynamics and slow photoreceptor death in retinal degeneration.
Acknowledgments
We thank Drs. Martin Biel, Anand Swaroop, Katsuhiko Mikoshiba, and Yun-Zheng Le for providing Cnga3−/−, Nrl−/−, Itpr1flox/flox, and Hrgpcre mouse lines. We thank the Histology, Microscopy, and Image Analysis Core Facility at OUHSC for technical assistance.
This work was supported by grants from the National Eye Institute (R01EY027754 and P30EY021725) and the Oklahoma Center for the Advancement of Science and Technology.
Abbreviations used:
- cGMP
cyclic guanosine monophosphate
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CNG
cyclic nucleotide-gated
- Creb
cyclic adenosine monophosphate response element binding protein
- Derlin-1
degradation in ER protein 1
- ER
endoplasmic reticulum
- eIF2α
eukaryotic translation initiation factor 2 alpha
- ERAD
ER-associated degradation
- GFAP
glial fibrillary acidic protein
- Herpud1
homocysteine inducible ER protein with ubiquitin like domain 1
- HRGP
human red/green pigment
- IP3R 1
inositol-1,4,5-trisphosphate receptor type 1
- Ire1α
serine/threonine-protein kinase/endoribonuclease 1α
- LC3B
microtubule-associated proteins 1A/1B light chain 3B
- PA28α
proteasome activator complex subunit 1
- PDE
phosphodiesterase
- PNA
peanut agglutinin lectin
- PSMD11
26S proteasome non-ATPase regulatory subunit 11
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- RyR2
ryanodine receptor type 2
- Sel1L
ERAD E3 ligase adaptor subunit
- SQSTM1/P62
sequestosome-1
- Syvn1
E3 ubiquitin-protein ligase synoviolin 1
- TBP
TATA binding protein
- TUDCA
tauroursodeoxycholic acid
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end-labeling
- UPR
unfolded protein response
Footnotes
All authors declare that they have no conflict of interest.
References
- 1.Kaupp UB, and Seifert R (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82, 769–824 [DOI] [PubMed] [Google Scholar]
- 2.Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, Spegal R, Anastasi M, Zrenner E, Sharpe LT, and Wissinger B (2000) Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 9, 2107–2116 [DOI] [PubMed] [Google Scholar]
- 3.Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla E, Zrenner E, Sharpe LT, and Wissinger B (1998) Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet 19, 257–259 [DOI] [PubMed] [Google Scholar]
- 4.Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, and Dryja TP (2005) Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum Mutat 25, 248–258 [DOI] [PubMed] [Google Scholar]
- 5.Biel M, Seeliger M, Pfeifer A, Kohler K, Gerstner A, Ludwig A, Jaissle G, Fauser S, Zrenner E, and Hofmann F (1999) Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc Natl Acad Sci U S A 96, 7553–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Morris L, Fliesler SJ, Sherry DM, and Ding XQ (2011) Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest Ophthalmol Vis Sci 52, 3557–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, and Swaroop A (2001) Nrl is required for rod photoreceptor development. Nat Genet 29, 447–452 [DOI] [PubMed] [Google Scholar]
- 8.Thapa A, Morris L, Xu J, Ma H, Michalakis S, Biel M, and Ding XQ (2012) Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J Biol Chem 287, 18018–18029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma H, Thapa A, Morris LM, Michalakis S, Biel M, Frank MB, Bebak M, and Ding XQ (2013) Loss of cone cyclic nucleotide-gated channel leads to alterations in light response modulating system and cellular stress response pathways: a gene expression profiling study. Hum Mol Genet 22, 3906–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackos DH, and Korenbrot JI (1999) Divalent cation selectivity is a function of gating in native and recombinant cyclic nucleotide-gated ion channels from retinal photoreceptors. J Gen Physiol 113, 799–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler MR, Ma H, Yang F, Belcher J, Le YZ, Mikoshiba K, Biel M, Michalakis S, Iuso A, Krizaj D, and Ding XQ (2017) Endoplasmic reticulum (ER) Ca(2+)-channel activity contributes to ER stress and cone death in cyclic nucleotide-gated channel deficiency. J Biol Chem 292, 11189–11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Morris L, Thapa A, Ma H, Michalakis S, Biel M, Baehr W, Peshenko IV, Dizhoor AM, and Ding XQ (2013) cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J Neurosci 33, 14939–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olshevskaya EV, Ermilov AN, and Dizhoor AM (2002) Factors that affect regulation of cGMP synthesis in vertebrate photoreceptors and their genetic link to human retinal degeneration. Mol Cell Biochem 230, 139–147 [PubMed] [Google Scholar]
- 14.Baehr W, and Palczewski K (2009) Focus on Molecules: Guanylate cyclase-activating proteins (GCAPs). Experimental Eye Research 89, 2–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorlach A, Klappa P, and Kietzmann T (2006) The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants & redox signaling 8, 1391–1418 [DOI] [PubMed] [Google Scholar]
- 16.Mekahli D, Bultynck G, Parys JB, De Smedt H, and Missiaen L (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sammels E, Parys JB, Missiaen L, De Smedt H, and Bultynck G (2010) Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium 47, 297–314 [DOI] [PubMed] [Google Scholar]
- 18.Ma H, Butler MR, Thapa A, Belcher J, Yang F, Baehr W, Biel M, Michalakis S, and Ding XQ (2015) cGMP/Protein Kinase G Signaling Suppresses Inositol 1,4,5-Trisphosphate Receptor Phosphorylation and Promotes Endoplasmic Reticulum Stress in Photoreceptors of Cyclic Nucleotide-gated Channel-deficient Mice. J Biol Chem 290, 20880–20892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma H, Yang F, Butler MR, Rapp J, Le YZ, and Ding XQ (2019) Ryanodine Receptor 2 Contributes to Impaired Protein Localization in Cyclic Nucleotide-Gated Channel Deficiency. eNeuro 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma H, Thapa A, Morris L, Redmond TM, Baehr W, and Ding XQ (2014) Suppressing thyroid hormone signaling preserves cone photoreceptors in mouse models of retinal degeneration. Proc Natl Acad Sci U S A 111, 3602–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang T, Baehr W, and Fu Y (2012) Chemical chaperone TUDCA preserves cone photoreceptors in a mouse model of Leber congenital amaurosis. Invest Ophthalmol Vis Sci 53, 3349–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Lee B, and Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281, 7260–7270 [DOI] [PubMed] [Google Scholar]
- 23.Szegezdi E, Logue SE, Gorman AM, and Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishitoh H (2012) CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 151, 217–219 [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi D, Tanimoto K, and Nakayama K (2016) CREB is activated by ER stress and modulates the unfolded protein response by regulating the expression of IRE1alpha and PERK. Biochem Biophys Res Commun 469, 243–250 [DOI] [PubMed] [Google Scholar]
- 26.Yang F, Ma H, Butler MR, and Ding XQ (2020) Potential contribution of ryanodine receptor 2 upregulation to cGMP/PKG signaling-induced cone degeneration in cyclic nucleotide-gated channel deficiency. FASEB J 34, 6335–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Raad S, Szczesny PJ, Munz K, and Reme CE (1996) Light damage in the rat retina: glial fibrillary acidic protein accumulates in Muller cells in correlation with photoreceptor damage. Ophthalmic Res 28, 99–107 [DOI] [PubMed] [Google Scholar]
- 28.Bringmann A, and Reichenbach A (2001) Role of Muller cells in retinal degenerations. Front Biosci 6, E72–92 [DOI] [PubMed] [Google Scholar]
- 29.Zhao TT, Tian CY, and Yin ZQ (2010) Activation of Muller cells occurs during retinal degeneration in RCS rats. Adv Exp Med Biol 664, 575–583 [DOI] [PubMed] [Google Scholar]
- 30.Michalakis S, Geiger H, Haverkamp S, Hofmann F, Gerstner A, and Biel M (2005) Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest Ophthalmol Vis Sci 46, 1516–1524 [DOI] [PubMed] [Google Scholar]
- 31.Carvalho P, Stanley AM, and Rapoport TA (2010) Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaneko M, Ishiguro M, Niinuma Y, Uesugi M, and Nomura Y (2002) Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett 532, 147–152 [DOI] [PubMed] [Google Scholar]
- 33.Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR 3rd, Long Q, and Qi L (2014) Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A 111, E582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneko M, Yasui S, Niinuma Y, Arai K, Omura T, Okuma Y, and Nomura Y (2007) A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett 581, 5355–5360 [DOI] [PubMed] [Google Scholar]
- 35.Huang CH, Chu YR, Ye Y, and Chen X (2014) Role of HERP and a HERP-related protein in HRD1-dependent protein degradation at the endoplasmic reticulum. J Biol Chem 289, 4444–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paredes F, Parra V, Torrealba N, Navarro-Marquez M, Gatica D, Bravo-Sagua R, Troncoso R, Pennanen C, Quiroga C, Chiong M, Caesar C, Taylor WR, Molgo J, San Martin A, Jaimovich E, and Lavandero S (2016) HERPUD1 protects against oxidative stress-induced apoptosis through downregulation of the inositol 1,4,5-trisphosphate receptor. Free Radic Biol Med 90, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadowaki H, Satrimafitrah P, Takami Y, and Nishitoh H (2018) Molecular mechanism of ER stress-induced pre-emptive quality control involving association of the translocon, Derlin-1, and HRD1. Sci Rep 8, 7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Y, Shibata Y, Yun C, Ron D, and Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429, 841–847 [DOI] [PubMed] [Google Scholar]
- 39.Mantopoulos D, Murakami Y, Comander J, Thanos A, Roh M, Miller JW, and Vavvas DG (2011) Tauroursodeoxycholic acid (TUDCA) protects photoreceptors from cell death after experimental retinal detachment. PLoS One 6, e24245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Sanchez L, Lax P, Pinilla I, Martin-Nieto J, and Cuenca N (2011) Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Invest Ophthalmol Vis Sci 52, 4998–5008 [DOI] [PubMed] [Google Scholar]
- 41.Song X, von Kampen J, Slaughter CA, and DeMartino GN (1997) Relative functions of the alpha and beta subunits of the proteasome activator, PA28. J Biol Chem 272, 27994–28000 [DOI] [PubMed] [Google Scholar]
- 42.Smith M, and Wilkinson S (2017) ER homeostasis and autophagy. Essays Biochem 61, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi Z, and Chen L (2019) Endoplasmic Reticulum Stress and Autophagy. Adv Exp Med Biol 1206, 167–177 [DOI] [PubMed] [Google Scholar]
- 44.Krebs J, Agellon LB, and Michalak M (2015) Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem Biophys Res Commun 460, 114–121 [DOI] [PubMed] [Google Scholar]
- 45.Gao B, Lee SM, Chen A, Zhang J, Zhang DD, Kannan K, Ortmann RA, and Fang D (2008) Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO reports 9, 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yagishita N, Ohneda K, Amano T, Yamasaki S, Sugiura A, Tsuchimochi K, Shin H, Kawahara K, Ohneda O, Ohta T, Tanaka S, Yamamoto M, Maruyama I, Nishioka K, Fukamizu A, and Nakajima T (2005) Essential role of synoviolin in embryogenesis. J Biol Chem 280, 7909–7916 [DOI] [PubMed] [Google Scholar]
- 47.Lobanova ES, Finkelstein S, Li J, Travis AM, Hao Y, Klingeborn M, Skiba NP, Deshaies RJ, and Arshavsky VY (2018) Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat Commun 9, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lobanova ES, Finkelstein S, Skiba NP, and Arshavsky VY (2013) Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci U S A 110, 9986–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S, He H, Ma QS, Zhang Y, Zhu Y, Wan X, Wang FW, Wang SS, Liu L, and Li B (2015) Experimental study of the protective effects of SYVN1 against diabetic retinopathy. Sci Rep 5, 14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldridge WH, Kurennyi DE, and Barnes S (1998) Calcium-sensitive calcium influx in photoreceptor inner segments. J Neurophysiol 79, 3012–3018 [DOI] [PubMed] [Google Scholar]
- 51.Szikra T, Cusato K, Thoreson WB, Barabas P, Bartoletti TM, and Krizaj D (2008) Depletion of calcium stores regulates calcium influx and signal transmission in rod photoreceptors. J Physiol 586, 4859–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding XQ, Harry CS, Umino Y, Matveev AV, Fliesler SJ, and Barlow RB (2009) Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum Mol Genet 18, 4770–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yorimitsu T, Nair U, Yang Z, and Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281, 30299–30304 [DOI] [PMC free article] [PubMed] [Google Scholar]