

Abstract
We describe the Mass Spectrometry Adduct Calculator (MSAC), an automated Python tool to calculate the adduct ion masses of a parent molecule. Here, adduct refers to a version of a parent molecule [M] that is charged due to addition or loss of atoms and electrons resulting in a charged ion, for example, [M + H]+. MSAC includes a database of 147 potential adducts and adduct/neutral loss combinations and their mass-to-charge ratios (m/z) as extracted from the NIST/EPA/NIH Mass Spectral Library (NIST17), Global Natural Products Social Molecular Networking Public Spectral Libraries (GNPS), and MassBank of North America (MoNA). The calculator relies on user-selected subsets of the combined database to calculate expected m/z for adducts of molecules supplied as formulas. This tool is intended to help researchers create identification libraries to collect evidence for the presence of molecules in mass spectrometry data. While the included adduct database focuses on adducts typically detected during liquid chromatography–mass spectrometry analyses, users may supply their own lists of adducts and charge states for calculating expected m/z. We also analyzed statistics on adducts from spectra contained in the three selected mass spectral libraries. MSAC is freely available at https://github.com/pnnl/MSAC.
Graphical Abstract

INTRODUCTION
Mass spectrometry is a frequently used tool for measuring and putatively identifying molecules, from proteins to metabolites. An important part of the molecular identification process is matching a mass-to-charge ratio (m/z) feature detected from mass spectrometry analysis of a mixture to an associated library entry of a compound. Here, we define “feature” as the unprocessed spectroscopic signature of an analyte ion described by one or more separation coordinates and/or spectra (e.g., retention time/index and m/z, for LC- or GC-MS, or with drift time for ion mobility (IMS) coupled methods). A review of small molecule identification by De Vijlder et al.1 separates analytes into four classes: known and expected, known and unexpected, unknown and expected, and unknown and unexpected. Calculation of adduct masses for a given set of molecules assists with identification of the first two classes of analytes.
Previous targeted studies using adducts for identification focused on only a few commonly formed adducts, for example, [M + H]+, [M − H]−, [M + Na]+, and [M + K]+. Here, adduct refers to a version of a parent molecule [M] that is charged due to addition or loss of atoms and electrons resulting in a charged ion, for example, [M + H]+.2 A study by Huang et al. gives an often-cited list of 44 adducts commonly observed in electrospray ionization mass spectra.3 This list is included in the previously most comprehensive adduct tool available, an Excel-based mass spectrometry adduct calculator offered by the Fiehn lab.4 However, there is a need for an adduct mass calculation method that is compatible with Python-based analysis and library creation pipelines,5,6 rather than manual entry into an Excel spreadsheet. Other software and tools, such as LipidBlast,7 CAMERA,8 and MZmine,9 have short preset adduct options without a mechanism for extension. Thus, there is also need for easily customizable adduct lists.
Here, we present the Mass Spectrometry Adduct Calculator (MSAC), a Python package that flexibly and rapidly calculates a variety of adduct masses based on user-defined criteria and user-supplied parent molecule lists. While existing adduct mass calculators rely on online interfaces or downloadable spreadsheets, MSAC uses a command-line interface that can be easily integrated into any mass spectral data analysis pipeline. MSAC provides a simple avenue for researchers to customize output for adducts of interest. A default adduct list provides 147 potential adducts previously observed in small molecule spectra found in NIST/EPA/NIH Mass Spectral Library (NIST17),10 Global Natural Products Social Molecular Networking Public Spectral Libraries (GNPS),11 and MassBank of North America (MoNA),12 all repositories of LC-MS spectra. We also report frequency data for these 147 adducts. When neutral losses are considered, there are 2336 unique loss/adduct pairs.
IMPLEMENTATION
MSAC is a fully automated Python tool that allows input of multiple molecules at once. Users input a comma-separated values (.csv) file including a column of exact masses for all molecules and can optionally provide the number of adducts to be considered; MSAC outputs a .csv file containing masses for potential adducts for each molecule. The original input data are preserved. MSAC is freely available in the Supporting Information and at https://github.com/pnnl/MSAC.
METHODS
MSAC is implemented in Python (version 3+) and makes use of NumPy13 (version 1.18.1+) for vectorized math and Pandas14 (version 1.0.0+) for data manipulation. The adduct ion portion of the mass calculation is done using the Python program molmass.15 Adduct ions not included in the original list can be specified in the input .csv, and their masses will automatically be determined. MSAC is efficient for most data sets, but if the input file is greater than several gigabytes, it is advised to split files into smaller pieces.
MSAC assumes that the user-provided masses are monoisotopic. Mass calculations performed by MSAC account for the mass of the electron in charged molecules.16 Both positive and negative ionization modes are supported and are specified by the charge on the input adduct.
In default form, MSAC is called from the command line with the required input of a .csv file containing a column of monoisotopic mass. The default name of the mass column is “mass”, but users can specify different column names via the –m, or –mass_col, flag. If the user does not have exact mass calculated but does have chemical formula information, the monoisotopic mass can be calculated when the name of the formula column is provided with the –f, or –formula_col, flag. If users choose to supply their own list of adducts, they need two pieces of information to create their own adduct list: the adduct type (e.g., [M − H]−) and charge (e.g., −1). Users can then make a .csv file with an “adduct” column and a “charge” column and specify this file with the –a flag on the command line. Other optional inputs include specifying an output name with –o, or –outname.
The default adduct list example_data/adduct_only_list.csv, used when the user does not specify a list, consists of the most common adducts for small molecules found in NIST17, GNPS, and MoNA. If interested in neutral losses, use the –n flag, –neutral_losses_included, to use example_data/adduct_list_full.csv. When using the provided lists, users may also specify a percentile above which adducts will be calculated, i.e., to calculate the X%ile most common adducts. For instance, entering a value of 0.5 with the –c, or –coverage_cutoff, flag will output the top 50th percentile of the most common adducts based on number of appearances in NIST17, GNPS, and MoNA. The default value of 1 gives the entire list.
However, the supplied list of adducts includes mass losses that may be impossible for a given compound that does not contain the requisite atoms. When a parent structure is specified via formula using the –r, or –restrict, flag, MSAC checks all mass losses in adducts against the parent structure. If the parent structure does not contain the atoms necessary to produce the specified loss, MSAC labels the adduct as being impossible to form by outputting a NaN value instead of a mass, represented in an Excel sheet by a blank cell.
RESULTS AND DISCUSSION
We conducted an analysis of adducts across the range of masses represented in the NIST17, GNPS, and MoNA databases, focusing on adducts of parent molecules under 1000 Da (but collecting statistics on those up to 2000 Da). A total of 665,070 spectra were extracted from NIST17’s tandem MS/MS libraries. The GNPS Public Spectral Library of 74,179 spectra was obtained from University of California San Diego’s GNPS website. A total of 131,877 spectra were downloaded from MoNA’s LC-MS/MS spectra database.
From this analysis, we identified 147 different adducts that appeared coupled to a small molecule in more than one spectrum. The databases also included 923 unique neutral losses. Altogether, there are 2336 unique adduct/neutral loss pairs. Our results were similar to those from a previous paper that characterized adduct frequency for the NIST17 database alone.17 The most commonly found adduct for each ion mode was the hydrogen adduct, as shown in Table 1. Single protonation accounted for 74.0% of adducts seen in positive mode spectra, while in negative mode spectra 80.7% of spectra were single deprotonations.
Table 1.
Percent Occurrence of Top Five Most Common Adducts among Collected Spectra of 78 Unique Adducts Found in Positive Mode and 69 Found in Negative Mode
| Mode | Adduct | Percent coverage (%) |
|---|---|---|
| Positive | ||
| M + H | 74.0 | |
| M + Na | 6.7 | |
| M + 2H | 5.6 | |
| 2M + H | 2.5 | |
| M + 3H | 2.2 | |
| Negative | ||
| M − H | 80.7 | |
| 2M − H | 5.8 | |
| M − 2H | 5.7 | |
| M + Cl | 2.5 | |
| M + HCO2 | 1.2 |
Spectra were collected from each database’s text file if they contained information on the adduct’s parent mass, name, and the International Union of Pure and Applied Chemistry hashed International Chemical Identifier (InChIKey). Some entries had additional information such as SMILES, name synonyms, and/or formula. Charge was obtained by parsing the adduct information (e.g., [M + H]+ is parsed as a charge of +1) or manually annotating adducts in cases where charge information is missing (e.g., when adduct type was input as M + H without the charge state superscript).
Solvents are shown to change ionization of molecules processed with mass spectrometry, and buffers can suppress formation of certain ions.18,19 Due to the community-sourced nature of GNPS and MoNA, solvent and buffer data are not readily available and so beyond the scope of our analysis. Our analysis makes no attempt to distinguish adducts resulting from molecules of interest from adducts resulting from buffer/solvent effects.
Adduct Charge Prevalence.
Table 2 shows how many spectra in the examined databases are from adducts of a particular charge. Spectra from adducts with charges of +1 or −1 are by far the most common in the databases, representing 89% of all spectra. Less than 0.001% of unique molecules across all three databases have absolute charge states greater than 2.
Table 2.
Counts of Spectra and Unique Molecules Represented by These Spectra in NIST17, GNPS, and MONA by Charge
| Charge | Spectra | Unique molecules |
|---|---|---|
| −6 | 18 | 1 |
| −5 | 11 | 1 |
| −4 | 41 | 2 |
| −3 | 973 | 5 |
| −2 | 9163 | 204 |
| −1 | 142,811 | 12,150 |
| 1 | 489,827 | 23,463 |
| 2 | 36,721 | 1340 |
| 3 | 12,667 | 13 |
| 4 | 7763 | 1 |
| 5 | 4671 | 1 |
| 6 | 2941 | 1 |
| 7 | 1513 | 1 |
| 8 | 509 | 1 |
| 9 | 154 | 1 |
Charge and Mass.
Most spectra in the combined databases are from molecules under 750 Da (Figure 1a). Thus, available data in these databases are skewed toward small molecules. The mass distribution of multiply charged adducts in parent molecules under 2000 Da is more widely distributed than that of singly charged adducts. Multiply charged adducts make up less than 15% of all adducts represented in NIST17, GNPS, and MoNA. Of adducts with a charge greater than 2 and mass over 1000, over 1264 are peptides, determined from a syntax search of common peptide naming conventions.
Figure 1.
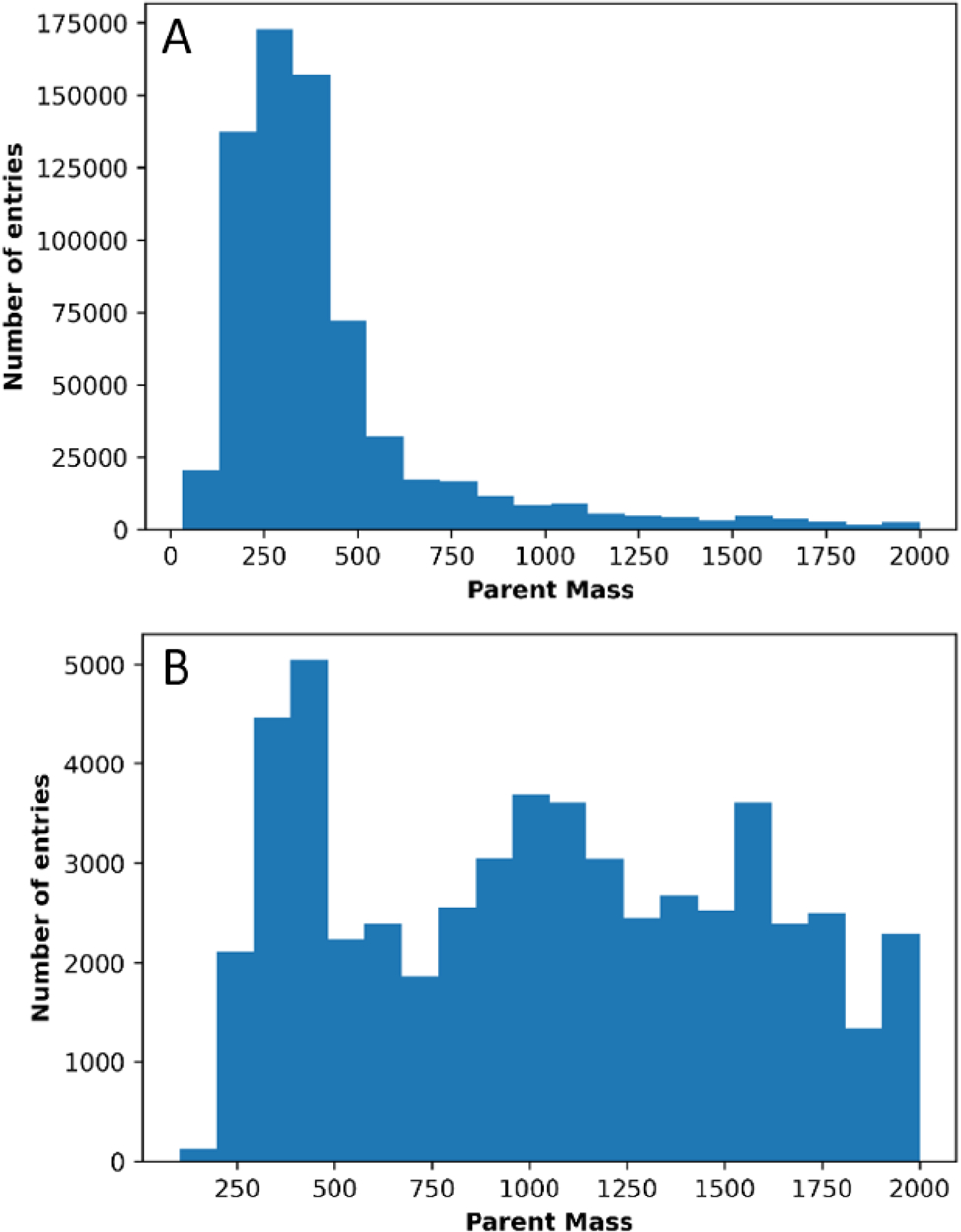
Mass distribution of (a) all and (b) multiply charged adducts in NIST17, GNPS, and MoNA.
Figure 2 shows the percent abundance of absolute charge values in each mass range. The final column includes all molecules with parent masses above 1000 Da, and multiple charges above 2 are overwhelmingly represented in this category. Molecules under 200 Da have few double charges in comparison to the number of singly charged adducts.
Figure 2.

Percentage of adducts with a particular absolute charge for each mass bin from 0 to 1100 Da, in mass increments of 100 Da. Percentages are computed relative to all adducts in a particular mass bin. The largest bin includes all masses above 1000.
By default, MSAC makes no assumptions about what adducts may or may not form with a given input, except when the formula is specified with –r. Chemically infeasible adducts are unlikely to be found in downstream analysis. A recent study by Liigand et al. used computed molecular descriptors to predict whether a parent molecule would ionize under specific operation modes.20 A future update could include calculations of adduct likelihood by incorporating quantum chemical calculations, machine learning, and/or cheminformatic predictions based on molecular structure or chemical class.
CONCLUSIONS
The Mass Spectrometry Adduct Calculator is presented here as a lightweight tool for calculating potential adduct masses based on a known exact molecular mass. We selected default adducts based on frequency in publicly available mass spectral databases. This software is available at https://github.com/pnnl/MSAC.
Adduct mass calculation is an important step in downstream mass spectrometry analysis. In targeted analysis, researchers can use MSAC-calculated masses to create identification libraries to match to features observed by mass spectrometry analysis. This process can be automated with instrument vendor software or open-source tools such as Data Extraction for Integrated Multidimensional Spectrometry (DEIMoS) and the Multi-Attribute Matching Engine (MAME),6 instrument-agnostic Python packages for mass spectrometry data analysis. For example, MSAC has been coupled to a mass spectrometry data analysis tool, DEIMoS, to generate target masses for detection of potential adducts in LC-IMS-MS data. We have used this method to automate the process of building identification libraries for standards-free identification. In this case, annotated features were validated by comparing retention time across adducts for a single compound.
Supplementary Material
ACKNOWLEDGMENTS
Support for this work was provided by the National Institutes of Health, National Institute of Environmental Health Sciences Grant U2CES030170 and by the PNNL Laboratory Directed Research and Development program, the m/q Initiative. Pacific Northwest National Laboratory (PNNL) is a multiprogram national laboratory operated by Battelle Memorial Institute for the U.S. Department of Energy under Contract DE-AC05-76RL01830.
Footnotes
The authors declare no competing financial interest.
DATA AND SOFTWARE AVAILABILITY
This software is available at https://github.com/pnnl/MSAC.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.1c00579.
MSAC software (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.1c00579
Contributor Information
Madison R. Blumer, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Christine H. Chang, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Evangelina Brayfindley, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Jamie R. Nunez, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Sean M. Colby, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Ryan S. Renslow, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Thomas O. Metz, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
REFERENCES
- (1).De Vijlder T; Valkenborg D; Lemiere F; Romijn EP; Laukens K; Cuyckens F A Tutorial in Small Molecule Identification Via Electrospray Ionization-Mass Spectrometry: The Practical Art of Structural Elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).HolČapek M; Jirásko R; Lísa M Basic Rules for the Interpretation of Atmospheric Pressure Ionization Mass Spectra of Small Molecules. Journal of Chromatography A 2010, 1217, 3908–921. [DOI] [PubMed] [Google Scholar]
- (3).Huang N; Siegel M; Kruppa G; Laukien F Automation of a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer for Acquisition, Analysis, and E-Mailing of High-Resolution Exact-Mass Electrospray Ionization Mass Spectral Data. J. Am. Soc. Mass Spectrom. 1999, 10, 1166–1173. [Google Scholar]
- (4).Fiehn O Mass Spectrometry Adduct Calculator. https://fiehnlab.ucdavis.edu/staff/kind/metabolomics/ms-adduct-calculator (accessed October 2021).
- (5).Colby SM; Thomas DG; Nunez JR; Baxter DJ; Glaesemann KR; Brown JM; Pirrung MA; Govind N; Teeguarden JG; Metz TO; Renslow RS Isicle: A Quantum Chemistry Pipeline for Establishing in Silico Collision Cross Section Libraries. Anal. Chem. 2019, 91, 4346–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nuñez JR; Colby SM; Thomas DG; Tfaily MM; Tolic N; Ulrich EM; Sobus JR; Metz TO; Teeguarden JG; Renslow RS Evaluation of In Silico Multifeature Libraries for Providing Evidence for the Presence of Small Molecules in Synthetic Blinded Samples. J. Chem. Inf. Model. 2019, 59, 4052–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kind T; Liu KH; Lee DY; DeFelice B; Meissen JK; Fiehn O Lipidblast in Silico Tandem Mass Spectrometry Database for Lipid Identification. Nat. Methods 2013, 10, 755–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kuhl C; Tautenhahn R; Bottcher C; Larson TR; Neumann S Camera: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pluskal T; Castillo S; Villar-Briones A; Oresic M Mzmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinf. 2010, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).NIST/NIH/EPA Mass Spectral Library: Standard Reference Database 1, NIST 17. Standard Reference Data Program; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar]
- (11).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu W-T; Crusemann M; Boudreau PD; Esquenazi E; Sandoval-Calderon M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu C-C; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw C-C; Yang Y-L; Humpf H-U; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; Boya P,CA; Torres-Mendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining OB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodriguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard P-M; Phapale P; Nothias L-F; Alexandrov T; Litaudon M; Wolfender J-L; Kyle JE; Metz TO; Peryea T; Nguyen D-T; VanLeer D; Shinn P; Jadhav A; Muller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BØ; Pogliano K; Linington RG; Gutierrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Horai H; Arita M; Kanaya S; Nihei Y; Ikeda T; Suwa K; Ojima Y; Tanaka K; Tanaka S; Aoshima K; Oda Y; Kakazu Y; Kusano M; Tohge T; Matsuda F; Sawada Y; Hirai MY; Nakanishi H; Ikeda K; Akimoto N; Maoka T; Takahashi H; Ara T; Sakurai N; Suzuki H; Shibata D; Neumann S; Iida T; Tanaka K; Funatsu K; Matsuura F; Soga T; Taguchi R; Saito K; Nishioka T Massbank: A Public Repository for Sharing Mass Spectral Data for Life Sciences. J. Mass Spectrom. 2010, 45, 703–14. [DOI] [PubMed] [Google Scholar]
- (13).Harris CR; Millman KJ; van der Walt SJ; Gommers R; Virtanen P; Cournapeau D; Wieser E; Taylor J; Berg S; Smith NJ; Kern R; Picus M; Hoyer S; van Kerkwijk MH; Brett M; Haldane A; Del Rio JF; Wiebe M; Peterson P; Gerard-Marchant P; Sheppard K; Reddy T; Weckesser W; Abbasi H; Gohlke C; Oliphant TE Array Programming with Numpy. Nature 2020, 585, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McKinney W Python for Data Analysis: Data Wrangling with Pandas, Numpy, and Ipython; O’Reilly Media, 2013.
- (15).Gohlke C Molmass, version 2020.6.10; PyPi, 2020.
- (16).Ferrer I; Thurman EM Importance of the Electron Mass in the Calculations of Exact Mass by Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2538–9. [DOI] [PubMed] [Google Scholar]
- (17).Vinaixa M; Schymanski EL; Neumann S; Navarro M; Salek RM; Yanes O Mass Spectral Databases for Lc/Ms- and Gc/Ms-Based Metabolomics: State of the Field and Future Prospects. TrAC, Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar]
- (18).Zhou S; Hamburger M Effects of Solvent Composition on Molecular Ion Response in Electrospray Mass Spectrometry: Investigation of the Ionization Processes. Rapid Commun. Mass Spectrom. 1995, 9, 1516–1521. [Google Scholar]
- (19).Matsuura K; Takashina H Effects of Functional Groups of Acrylic Acid Derivatives as Derivatization Reagents for Thiol Compounds on Molecular Ion Responses in Electrospray Ionization-Mass Spectrometry. J. Mass Spectrom. 1998, 33, 1199–1208. [Google Scholar]
- (20).Liigand J; Wang T; Kellogg J; Smedsgaard J; Cech N; Kruve A Quantification for Non-Targeted Lc/Ms Screening without Standard Substances. Sci. Rep. 2020, 10, 5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.