

Abstract
Purpose
Patients with MYC-amplified medulloblastoma (MB) have poor prognosis and frequently develop recurrence, thus new therapeutic approaches to prevent recurrence are needed.
Experimental Design
We evaluated OLIG2 expression in a panel of mouse Myc-driven MB tumors, patient MB samples, and patient-derived xenograft (PDX) tumors and analyzed radiation sensitivity in OLIG2−high and OLIG2−low tumors in PDX lines. We assessed the effect of inhibition of OLIG2 by OLIG2-CRISPR or the small molecule inhibitor CT-179 combined with radiotherapy on tumor progression in PDX models.
Results
We found that MYC-associated MB can be stratified into OLIG2−high and OLIG2−low tumors based on OLIG2 protein expression. In MYC-amplified MB PDX models, OLIG2−low tumors were sensitive to radiation and rarely relapsed, whereas OLIG2−high tumors were resistant to radiation and consistently developed recurrence. In OLIG2−high tumors, irradiation eliminated the bulk of tumor cells; however, a small number of tumor cells comprising OLIG2− tumor cells and rare OLIG2+ tumor cells remained in the cerebellar tumor bed when examined immediately post-irradiation. All animals harboring residual resistant tumor cells developed relapse. The relapsed tumors mirrored the cellular composition of the primary tumors with enriched OLIG2 expression. Further studies demonstrated that OLIG2 was essential for recurrence, as OLIG2 disruption with CRISPR-mediated deletion or with the small-molecule inhibitor CT-179 prevented recurrence from the residual radioresistant tumor cells.
Conclusion
Our studies reveal that OLIG2 is a biomarker and an effective therapeutic target in a high-risk subset of MYC-amplified MB, and OLIG2 inhibitor combined with radiotherapy represents a novel effective approach for treating this devastating disease.
INTRODUCTION
Medulloblastoma (MB) is one of the most common pediatric brain malignancies (1). Integrative genomic studies show that MB is a heterogenous disease comprising four major molecular subgroups, termed WNT, SHH, Group 3, and Group 4, which are distinct clinically, transcriptionally, and genetically (2–4). More recently, MB has been further stratified into twelve different subtypes (5). These stratifications have facilitated the diagnosis, prognosis, and optimization of treatments for specific patient subgroups. Group 3γ MB characterized by MYC amplification or overexpression is classified as a high-risk disease (2). Therefore, MYC-amplified MB patients are currently treated with intensive therapies, including surgical resection and craniospinal irradiation (CSI), followed by intense chemotherapy. Despite these intensive treatments, many patients still succumb to the disease and those who survive suffer severe treatment-related side effects (6–9). Therefore, there is an urgent need to stratify MYC-amplified MB patients to identify those who will not respond favorably to currently available treatments and require novel treatment modalities. Improved stratification will also facilitate de-escalation of aggressive therapies for patients expected to respond well to conventional therapies and thus avoid unnecessary treatment-related side effects (10).
The cellular and genomic heterogeneity of MB and the persistence of a subpopulation of cancer stem like cells (CSLCs) following conventional therapy are believed to be the main causes of treatment failure, resulting in incurable tumor recurrence (11–14). Treatment of “very high-risk” MYC-amplified MB will likely require the elimination of therapy-resistant tumor cells that are responsible for regenerating tumors. To target these cells, we must first develop strategies to identify and distinguish them based on their unique cellular and molecular characteristics. Prior studies show that recurrent MB is often genetically distinct from the initial tumors (14,15). However, analyzing tumors early in the process of recurrence has been problematic, as minimal residual disease post-therapy cannot be sampled in patients. In this study, we circumvented this problem by using MYC-amplified MB patient-derived xenograft (PDX) mouse models where we were able to isolate tumor cells that persisted after radiotherapy, study their role in tumor recurrence, and develop a strategy to block tumor relapse.
OLIG2 is a basic helix-loop-helix transcription factor that is highly expressed in glioblastoma, astrocytoma and oligodendrogliomas, and it serves as a diagnostic marker for glial tumors (16). OLIG2 also plays a critical role in gliomagenesis and tumor phenotype plasticity (17–21). Furthermore, OLIG2 expression has been shown to correlate with survival in patients with SHH-MBs 16. Here, we evaluated whether OLIG2 can serve as a biomarker to stratify risk in MYC-amplified MB. Based on our studies, we classified MYC-amplified MB PDX tumors into two subgroups: OLIG2−high tumors with high radioresistance and OLIG2−low tumors with high radiosensitivity. We also investigated how OLIG2 contributed to recurrence in MYC-amplified MB by analyzing the radioresistant tumor cells in OLIG2−high PDX tumors or OLIG2−low PDX tumors with forced overexpression of OLIG2. Finally, we found that genetic and pharmacologic inhibition of OLIG2 following radiotherapy improved outcomes in high-risk MYC-amplified MB PDX models.
METHODS AND MATERIALS
Animals
NOD-SCID-IL2Rg null mice (NSG) were purchased from Jackson Laboratory. Rag2-Il2rg double knockout mice (R2G2) were purchased from Envigo. Mice were maintained in the Animal Facility at Children’s National Health System (CNHS). All animal studies were performed in accordance with national guidelines and regulations, and with the approval of the Institutional Animal Care and Use Committee (IACUC) at CNHS.
Generation of Patient-derived Xenograft Tumor Cells Stably Expressing mCherry-Luciferase Reporters
To generate MB tumor cells stably expressing mCherry-Luciferase reporters, freshly isolated MYC-amplified MB xenograft tumor cells were plated in a 24-well plate and infected with lentivirus encoding mCherry-Luciferase reporters overnight. The tumor cells were then collected, washed with PBS twice and transplanted into the cerebella of 4–6 week old NSG or R2G2 mice as previously described (22). Both males and females were used in the studies. Tumor growth was monitored weekly by bioluminescence imaging using IVIS spectrum. Once animals developed tumors, mCherry+ tumor cells were sorted by FACS and retransplanted into the cerebella of new recipients to generate tumors composed solely of cells expressing mCherry-Luciferase. Each mouse was transplanted with 5×104 cells and were monitored weekly for tumor growth by bioluminescence imaging. PDX lines used for this study include: MB002 (23); Med-211-FH and Med-411-FH (24); RCMB20, RCMB40 and RCMB28 (25). PDX lines were generated by implanting patient cells directly into the cerebella of immune-compromised mice and propagating them from mouse to mouse without in vitro passaging. The identity and subgroup of each line was validated by gene expression and/or methylation analysis. We did not perform testing for mycoplasma.
Irradiation
To irradiate cells, freshly isolated PDX MB cells were cultured in a 24-well plate for 2 hours with stem cell media, composed of a 1:1 mixture of Neurobasal Media-Vitamin A (Gibco) and Ham’s F-12-based medium (Gibco), supplemented with Non-Essential Amino Acids (Gibco, 1:100), B27 (Gibco, 1:50), 2 mM glutamine, 5 mM HEPES (Gibco), 20 ng/mL of bFGF (Peprotech), 20 ng/mL of EGF (Peprotech), and 20 ng/mL of heparin (Sigma Aldrich). Cells were then irradiated for the indicated dose using a Faxitron CellRad X-ray irradiation cabinet. The irradiated cells were then cultured for 7 days and imaged under a Leica DMIL LED microscope.
To irradiate mice, tumor-bearing animals were irradiated with an RS 2000 Biological System Irradiator (Rad Source Technologies) when the luciferase signals from bioluminescence monitoring reached up to 1 × 106−7 radiance per second. The RS 2000 delivers photons at a constant dose rate of 2 Gy/min using x-ray irradiation as opposed to gamma irradiation. Each mouse was restrained in a lead shielding (Braintree Scientific) without anesthesia, allowing radiation exposure to only the brain and spine while shielding the rest of the body. Mice were irradiated with 2 Gy per dose at a rate of approximately 200 rads/minute. Irradiation was repeated every other day for a total cumulative dose of up to 18 Gy. Mice were imaged once per week with a bioluminescent imaging system to monitor tumor development. To assess the effect of irradiation on MYC-amplified MB, tumor bearing animals were treated with 18 Gy radiation as described above. Following a two-week recovery period, one cohort of animals was sacrificed, and tissues collected to assess tumor progression. Another cohort of animals were maintained until they showed symptoms of tumor relapse, including weight loss and ataxia, typically 7–8 weeks post-irradiation. Once animals showed symptoms, they were euthanized by inhalation of CO2 followed by cervical dislocation or decapitation, and brain tissues were collected for analysis or re-transplantation. For each cohort in the study, allowing for a tumor take rate of 75%, we used 8 mice per treatment group in order to end up with 6 treated mice. With this number of mice, we had 90% power (α =5%) to detect a 33% improvement in survival between groups.
Intrathecal (IT) Drug Delivery
To achieve sufficient concentrations in the cerebellum, CT-179 was delivered by IT injection via the cisterna magna. A 10 μL Neuros syringe attached to a 33-gauge beveled needle and with a stopper 3 mm from the tip of the needle was used for IT injection. An anesthetized mouse was loaded in the prone position on a stereotactic frame connected to an anesthesia gas mask to keep the mouse under 1.5–2% isoflurane anesthesia. The head of mouse was restrained between two ear bars, and a mask was used to align and support the front of the head. The height of the ear bars and the tooth bar were adjusted so that the line connecting the most prominent aspects of the cranium and the spine formed a 15-degree angle with the horizontal line. In this position, the cisterna magna is nearly the highest point of the mouse body. The head and neck were shaved to remove the fur. 3 μL of CT-179 (10 μg/ μL) dissolved in artificial CSF was loaded into the Neuros syringe and was placed onto the syringe holder in the micromanipulator connected to the stereotactic frame at an angle of 35°, then the space between the occiput and C1 vertebrae was palpated with either an index finger or a cotton-tipped swab. These two anatomical landmarks were used to determine the midline of the cisterna magna. An indentation was observed, which was used to define the puncture site. Mark the puncture site with a marker. The needle was then inserted into the puncture site to a depth of 2.6 – 2.8 mm at a 35° angle, and 3 μL CT-179 was injected at a rate of 3 μL/minute using an electronic infusion pump. IT injection was repeated either once or twice a week. Mice were monitored and their weight recorded daily.
We carried out two different treatment strategies in our studies. 1) 2–3 weeks after transplantation, tumor-bearing mice were randomized into 4 groups and treated with Control; CSI; CT-179; and CSI + CT-179. For CSI treatment, animals were irradiated with 2 Gy every other day for a total cumulative dose of up to 18 Gy. In CSI + CT-179 group, mice were treated with CT-179 by IT injection once a week for 3 weeks after the completion of radiotherapy. Animals were sacrificed, and tumor tissues were collected when they showed symptoms of a brain tumor or the mice in CSI + CT-179 group were sacrificed together with the mice in CSI alone group to compare the tumor size in these two groups. 2) We further determined whether increasing the frequency and duration of CT-179 by IT injection help improve efficacy. 2–3 weeks after transplantation, tumor-bearing mice were randomized into the 4 groups and treated with Control; CSI; CT-179; and CSI + CT-179. The mice in the group of CSI + CT-179 were treated with 2 Gy CSI every other day, along with intrathecal injection of CT-179 twice per week (10 μg/ μL in 3 μL) until they displayed signs of morbidity or toxicity (>20% weight loss). For each cohort in the study, allowing for a tumor take rate of 75%, we used 8 mice per treatment group in order to end up with 6 treated mice. With this number of mice, we had 90% power (α =5%) to detect a 33% improvement in survival between groups.
Flank Tumors Treated with OLIG2 Inhibitor
To implant flank tumors, 2X105 MYC-amplified MB xenograft cells were suspended in growth-factor-reduced Matrigel (BD BioSciences) at a ratio 1:1 and injected subcutaneously into the flanks of 6–8 weeks old shaved NSG mice. Tumors were measured with calipers every two days, and tumor volumes were calculated as length × width2× 0.52 with all measurements in millimeters. When tumor volume reached ∼100mm3 (usually ~15 days after inoculation), mice were randomly divided into 2 groups and were treated with water or CT-179 (105 mg/kg) for 12 days consecutively by oral gavage. 24 hours after the last dose, animals were euthanized and tumors were harvested and weighed. CT-179 can be dissolved in distilled water at a concentration of 30 mg/mL and produces a clear solution.
Intracranial Tumors Treated with OLIG2 Inhibitor Delivered via Oral Gavage
To assess the effect of OLIG2 inhibitor on the growth of intracranial tumors, 2–3 weeks after transplantation, tumor-bearing mice were randomized into the following treatment groups: Control (Ctrl); cranial-spinal irradiation (CSI); CT-179; and CSI + CT-179. For CSI treatment, animals were irradiated with 2 Gy every other day for a total cumulative dose of up to 18 Gy. In CSI + CT-179 group, after the completion of 18 Gy CSI treatment, the animals were treated with CT-179. CT-179 was administered by oral gavage daily (105 mg/kg body weight) until they displayed signs of morbidity or toxicity (>20% weight loss), whereupon they were euthanized. For each cohort in the study, allowing for a tumor take rate of 75%, we used 8 mice per treatment group in order to end up with 6 treated mice. With this number of mice, we had 90% power (α =5%) to detect a 33% improvement in survival between groups.
Statistics
Statistical analysis was performed using Excel or GraphPad Prism software. All data are presented as means ± SD or means ± SEM as indicated. Comparisons between different groups were made using Student’s t test or ANOVA as appropriate. The statistical significance of Kaplan-Meier survival curves was assessed using the log-rank (Mantel-Cox) test. p Values of 0.05 or lower were considered statistically significant for all experiments. Power calculations were performed using an online tool (https://clincalc.com/stats/samplesize.aspx) and the stated parameters.
Data Availability
The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible with GEO series accession numbers GSE188789.
RESULTS
MYC-amplified MB tumors can be classified into two subgroups based on OLIG2 protein expression levels
In our previous studies, we showed that Myc-driven MB, generated from Sox2+ astrocyte progenitor cells of the early postnatal cerebellum, had a high level of OLIG2 expression (26). To determine whether OLIG2 represents a molecular marker in Myc-driven MB, we examined a panel of mouse Myc-driven MB tumors derived from Sox2+ cells (26). We observed two subsets of tumors with variable OLIG2 expression: one subset had 50–80% OLIG2+ cells, and another subset had less than 1% OLIG2+ cells (Figures 1A and 1B). Expression of MYC varied among these tumors but did not correlate with OLIG2 expression (Figure 1C). To correlate the mouse model to human disease, we examined OLIG2 expression in six MYC-amplified MB PDX tumors that expressed similar levels of MYC protein. Three of these PDX tumor lines showed high OLIG2 expression, whereas OLIG2 was rarely detectable in the other three PDX tumors (Figures 1D–F). We then evaluated OLIG2 protein expression in samples resected from patients, including four MYC-amplified MB and five non-MYC-amplified MB tumors. Among the four MYC-amplified tumors, three had widespread OLIG2 expression. One MYC-amplified tumor and all non-MYC-amplified tumors contained rare or no OLIG2+ cells (Figure 1G and Figure S1A). These findings indicate that MYC-amplified MB can be divided into two subsets: OLIG2−high and OLIG2−low. We further examined the correlation of OLIG2 expression to patient survival with the dataset by Cavalli et al (5) and found that OLIG2 expression correlated significantly with poor outcomes among patients with Group 3γ MB and SHH MB, but not Group 4 MB (Figure 1H–J) or Group 3α or Group 3β MB (Figure S1B and S1C). Thus OLIG2−high tumors may represent a high-risk subgroup of MB.
Figure 1. OLIG2 protein expression divides MYC-driven MB into two subgroups.
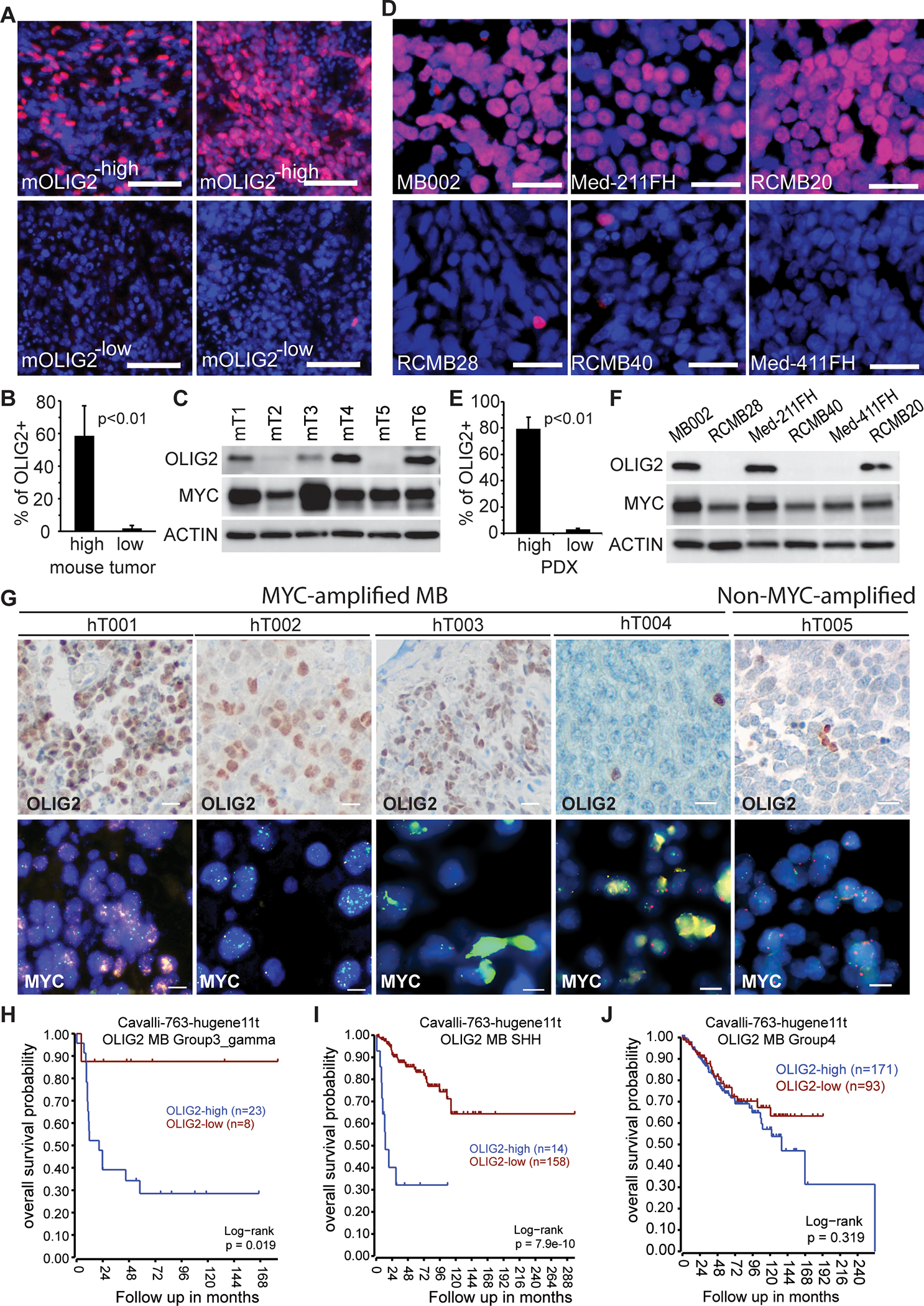
(A) Immunofluorescence histochemistry staining (IHC) of OLIG2 (red) in four representative mouse Myc-driven MB tumors. Nuclei counterstained with DAPI. Scale bars = 50 μm.
(B) Quantification of OLIG2+ cells in OLIG2high (n=5) and OLIG2low (n=5) mouse Myc-driven MB tumors. P values determined by Student T test.
(C) Western blot analysis of OLIG2 and MYC expression in six mouse MYC-driven MB tumors.
(D) IHC staining of OLIG2 (red) in MYC-amplified MB PDXs. Nuclei counterstained with DAPI. Scale bars = 25 μm.
(E) Quantification of OLIG2+ cells in OLIG2−high (MB002, Med-211FH and RCMB20) and OLIG2−low (RCMB28, RCMB40 and Med-411FH) human MYC-amplified MB PDX tumors. P values determined by Student T test.
(F) Western blot analysis of OLIG2 and MYC expression in MYC-amplified MB PDX tumors.
(G) OLIG2 or MYC expression in MYC-amplified MB tumors resected from patients, as assessed by IHC (scale bars = 25 μm) or FISH (scale bars = 10 μm).
(H-J) Correlation of OLIG2 expression with patient survival in Group 3γ MB, SHH-MB and Group 4 MB.
(See also Figure S1).
OLIG2−high and OLIG2−low MYC-amplified MBs show distinct responses to irradiation
In glioblastoma and SHH-MB, OLIG2+ cells have been shown to resist radiotherapy (19,27). To examine the role of OLIG2 in MYC-amplified MB tumors, we isolated fresh tumor cells from xenografts of one OLIG2−high PDX cell line (MB002) and one OLIG2−low PDX line (RCMB28), irradiated them with 0, 2 or 4 Gy and cultured them using a medium favorable to stem cell growth. As shown in Figure S1D, unirradiated control cells from both PDX lines formed spheres within 7 days of culturing. Many of the irradiated OLIG2−high tumor cells died; however, a fraction of the cells survived (Figure S1D, top panels) and were able to form secondary spheres (Figure S1E, top panel). In contrast, the OLIG2−low tumor cells were more sensitive to radiation as all died by 7 days post-irradiation at 4Gy and could not form secondary spheres (Figure S1D and Figure S1E bottom panels). To assess the response to irradiation in vivo, we transplanted tumor cells from five different MYC-amplified MB PDX lines, including three OLIG2−high (MB002, Med-211FH, and RCMB20) and two OLIG2−low (RCMB28 and RCMB40), into the cerebella of 4 to 6-week-old R2G2 immunodeficient mice (28). Before transplantation, tumor cells were stably infected with a lentivirus encoding both mCherry and luciferase reporters. mCherry facilitates tumor cell detection in tissue sections and by flow cytometry, while the luciferase reporter enables tumor growth monitoring in vivo using bioluminescence imaging. Two to three weeks after transplantation when the luciferase signals reached 1 × 106–7 radiance/second, animals received 9 fractions of 2 Gy CSI every other day for a cumulative dose of 18 Gy, which is relevant to the treatment of patients with brain tumors (29). All three OLIG2−high tumors initially responded to irradiation with significantly lower luciferase signal and longer survival compared to untreated controls, but then consistently relapsed (Figures 2A–I). In contrast, the two OLIG2−low PDX lines showed persistent radio-sensitivity with marked tumor regression that was rarely followed by recurrence over a 3-month monitoring period (Figures 2J–O).
Figure 2. OLIG2−high and OLIG2−low MYC-amplified MB tumors show distinct responses to craniospinal irradiation.
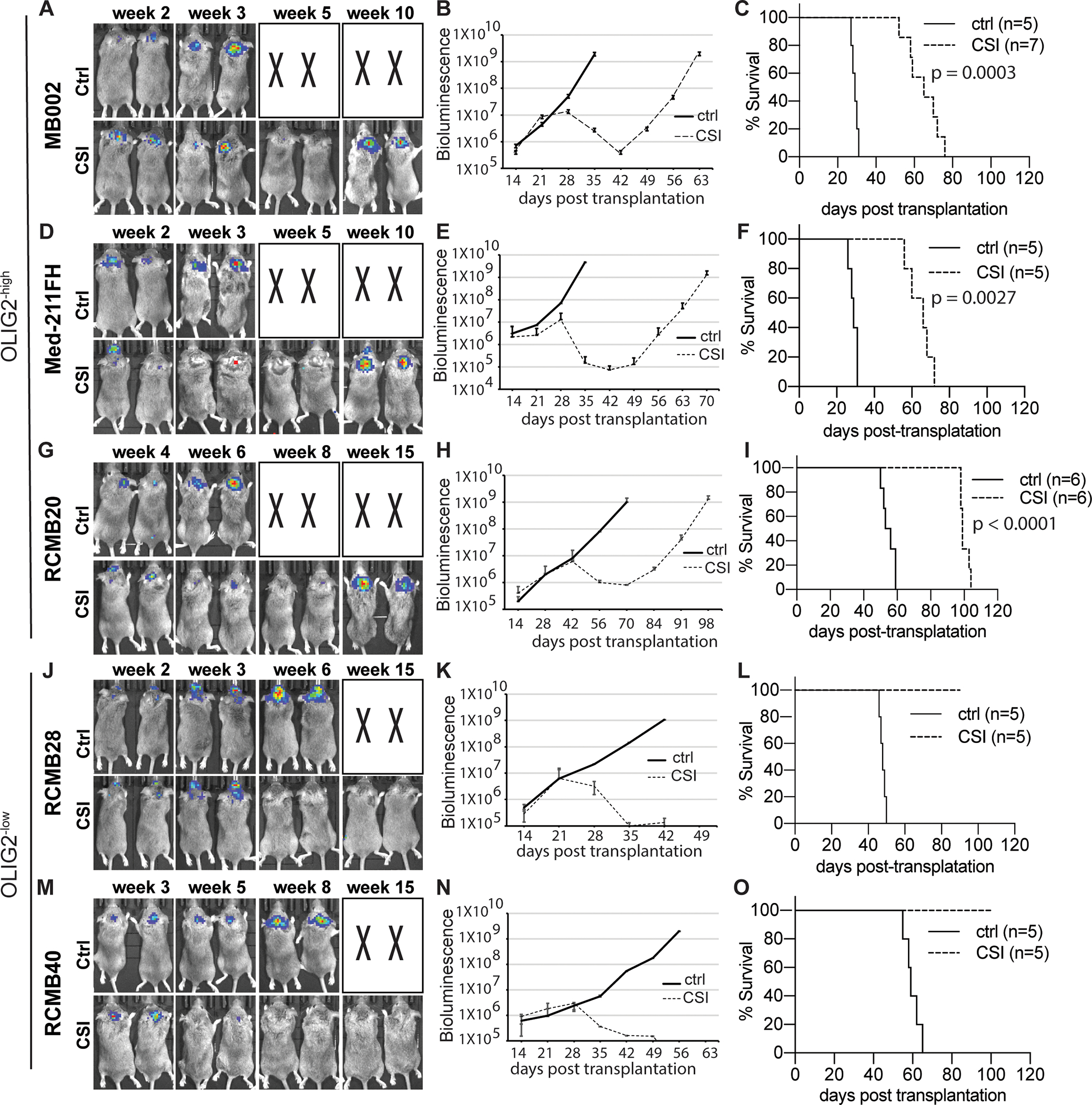
(A, D, G, J, M) IVIS images of representative tumor bearing mice with or without irradiation. Craniospinal irradiation, CSI; Control, ctrl. X denotes animals euthanized before they could be imaged. (B, E, H, K N) Bioluminescence signaling in radiance (p/sec/cm2/sr) over time after transplantation for the corresponding tumors.
(C, F, I, L, O) Corresponding survival analysis (n=5–7 each group) by Kaplan Meier.
Microscopic analyses of the brains of untreated and irradiated MB002 PDX mice also showed results consistent with bioluminescent imaging studies. We observed large tumors in the cerebellum of untreated symptomatic animals around 30 days post-transplantation (Figures 3A–a and b, and 3B–a and b). Irradiated tumors collected immediately after the completion of radiotherapy (around 32 days post-transplantation) comprised of only a small number of mCherry+ cells in the cerebellar tumor bed (Figures 3A–c and d, and 3B–c and d). We stained the tumor tissues with antibodies specific for markers of cell proliferation (Ki67), apoptosis (cleaved-caspase 3, CC3), and stem cells/glia (OLIG2). 12% of primary tumor cells were Ki67+ whereas radioresistant tumor cells in the cerebellar tumor bed were completely Ki67− (Figures 3C, D), indicating that the radioresistant tumor cells were relatively quiescent. CC3 expression was observed in the untreated tumors but not in the residual radioresistant tumor cells (Figure 3C, E). Although the bulk of tumor cells in untreated tumors were OLIG2+, only rare OLIG2+ cells were seen in the residual tumors post-irradiation (Figure 3C, F). A similar profile of radioresistant tumor cells in the cerebellar bed was observed in another OLIG2−high line (Med-211FH) (Figure S2). Collectively, these data show that radiation altered the cellular composition of OLIG2−high PDX tumors, with a reduced OLIG2+ population and persisting quiescent residual tumor cells.
Figure 3. Characterization of radioresistant tumor cells.
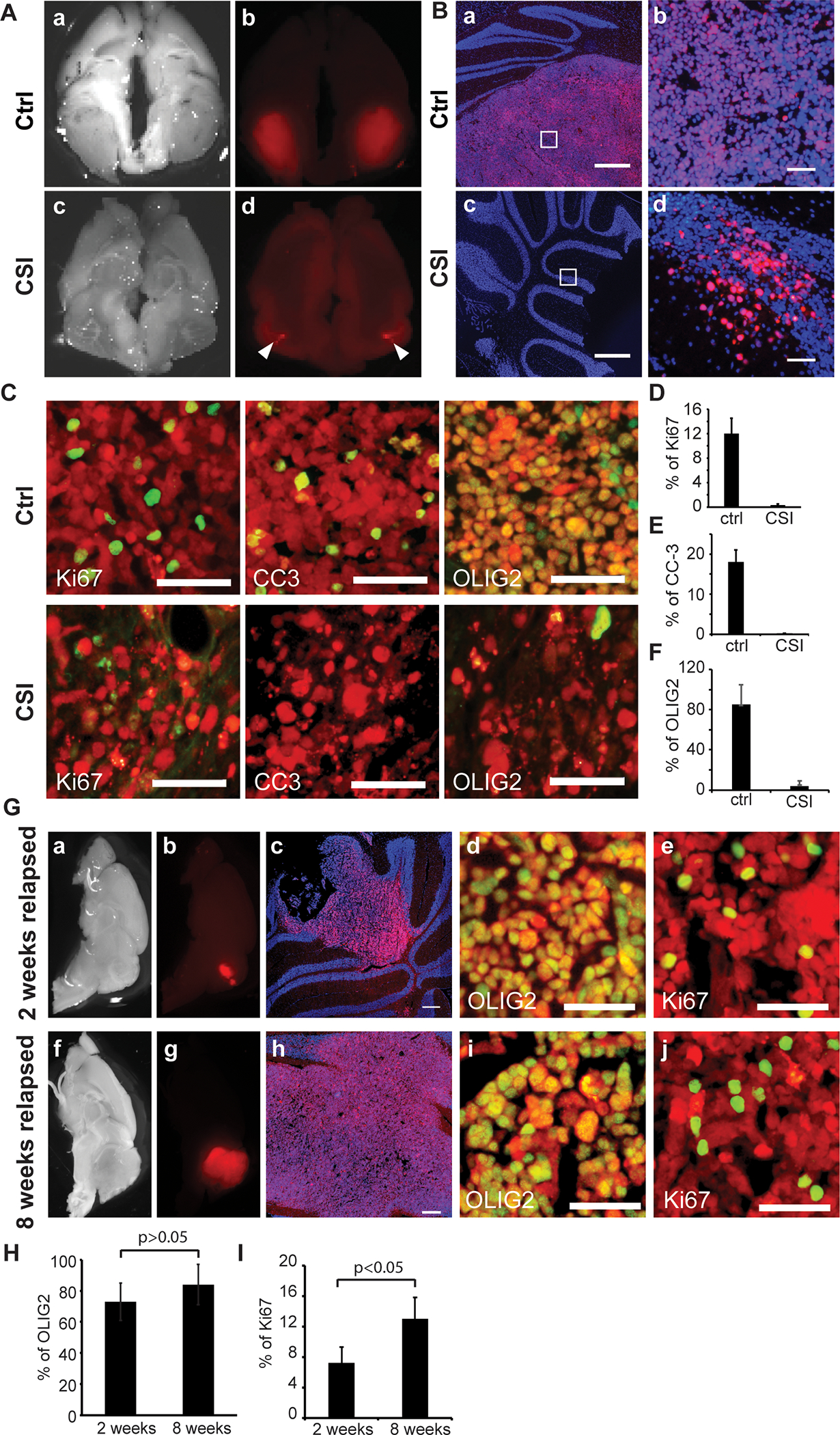
(A) Images of brain tissues from MB002 tumor-bearing animals unirradiated control (a, b) and irradiated (c, d). Tumor cells are indicated in red (mCherry). The images in A (a and c) are in bright field.
(B) Sagittal sections from unirradiated control (a, b) and irradiated (c, d) tumors stained with DAPI (blue). Scale bars = 500 μm in a and c. Images b and d are magnified images of the inset boxes in a and c, respectively. Scale bars = 50 μm in b and d. Tumor cells are indicated in red (mCherry).
(C) IHC staining with antibodies (green) against Ki67, CC3 or OLIG2 in the untreated control tumors (top panels) or residual radioresistant tumors (bottom panels). Tumor cells are indicated in red (mCherry). Scale bars = 50 μm.
(D-F) Quantification of %ki67+ (D), %CC-3+ (E) or %OLIG2+ (F) cells out of total mCherry+ tumor cells in irradiated (CSI; n=5) and nonirradiated (ctrl; n=5) tumors.
(G) Post-irradiation progressive tumors collected at early (2 weeks post-irradiation) or fully recurrent stages (8 weeks post-irradiation). Images of brain tissues in bright field (a, f) or mCherry (b, g). Tumor cells are indicated in red (mCherry). Sagittal sections were stained with DAPI (blue, c, h), OLIG2 (green, d, i) or ki67 (green, e, j). Scale bars = 250 μm in c and h; Scale bars = 50 μm in d, e, i, and j.
(H) Quantification of %OLIG2+ cells out of total mCherry+ tumor cells in relapsed tumors at 2 weeks (n=3) and 8 weeks (n=3). P values determined by Student T test.
(I) Quantification of %ki67+ cells out of total mCherry+ tumor cells in relapsed tumors at 2 weeks (n=3) and 8 weeks (n=3). P values determined by Student T test.
(See also Figure S2).
In mice with OLIG2−high MB002 tumors maintained for 2 weeks after completion of radiotherapy, we observed tumor progression in the cerebellum (Figure 3G–a–c). Many tumor cells in the early tumor lesion were OLIG2+ and Ki67+ (Figure 3G–d and e). When the PDX mice were maintained longer post-irradiation, all developed large tumors (Figure 3G–f–h). The tumors had elevated OLIG2 expression and the number of proliferating cells in recurrent tumors was similar to untreated controls (Figure 3G–i and j, 3H and 3I).
To examine if recurrent tumors (tumors recurring in symptomatic animals following the completion of irradiation treatment, typically 7–8 weeks after the last dose) arising from radioresistant cells (the surviving tumor cells immediately after the completion of irradiation) have comparable radiosensitivity to cells from untreated tumors, we transplanted an equal number of cells from recurrent and untreated tumors into the cerebella of R2G2 mice. Two weeks after transplantation, the mice were irradiated with 2 Gy every other day for a cumulative dose of 18 Gy. We found that recurrent tumors were less sensitive to irradiation than the untreated tumors (Figures S3A and S3B), indicating that the radioresistant phenotype is retained by the recurrent tumor cells. These data demonstrate that following initial radiation treatment, recurring tumors resist radiotherapy. Therefore, to improve patient outcome after radiation therapy, new effective therapeutic approaches that can completely eradicate the residual radioresistant tumor cells are needed (30–32).
Tumorigenic potential of transplanted radioresistant tumor cells in the cerebellar bed
Our studies demonstrated that most residual radioresistant tumor cells in the cerebellar bed were OLIG2− and only a few were OLIG2+. However, the post-irradiation progressive tumors consisted mostly of OLIG2+ tumor cells. These results indicate that the relapsed tumors either developed from the rare OLIG2+ radioresistant tumor cells remaining in the cerebellar bed, or the OLIG2− radioresistant tumor cells gave rise to OLIG2+ tumor cells during tumor recurrence. To test the tumorigenic potential of radioresistant tumor cells in vivo, mCherry+ residual radioresistant tumor cells or untreated tumor cells were sorted by flow cytometry and cultured for 2 weeks in the presence of neurosphere-promoting growth factors bFGF and EGF. We then dissociated the neurospheres and transplanted them into the cerebella of NSG mice. All mice transplanted with 2,000 residual radioresistant tumor cells or untreated tumor cells developed tumors in 7–9 weeks. However, animals transplanted with 500 untreated tumor cells survived significantly longer than those transplanted with an equal number of radioresistant tumor cells (Figures 4A). Only 2 out of 5 mice transplanted with 500 untreated tumor cells developed tumors over 20 weeks of monitoring (Figure 4A), indicating that neurospheres from the radioresistant tumor cells were thus relatively more tumorigenic than the untreated tumor cells when transplanted. We also examined whether the uncultured residual radioresistant tumor cells could develop tumors after transplantation. We injected 2,000 freshly FAC-sorted radioresistant tumor cells into the cerebella of a cohort of mice and none of them developed tumors. Conversely, all animals that received 2,000 freshly FAC-sorted cells from untreated tumors developed tumors within 9–10 weeks (Figure S4). These findings suggest that the quiescent post-irradiation residual tumor cells rely on molecular signals that derive either from their initial tumor microenvironment or exogenous stimulation in culture to develop into tumors post-transplantation.
Figure 4. Characterization of the tumors derived from the radioresistant tumor cells in the cerebellar bed.
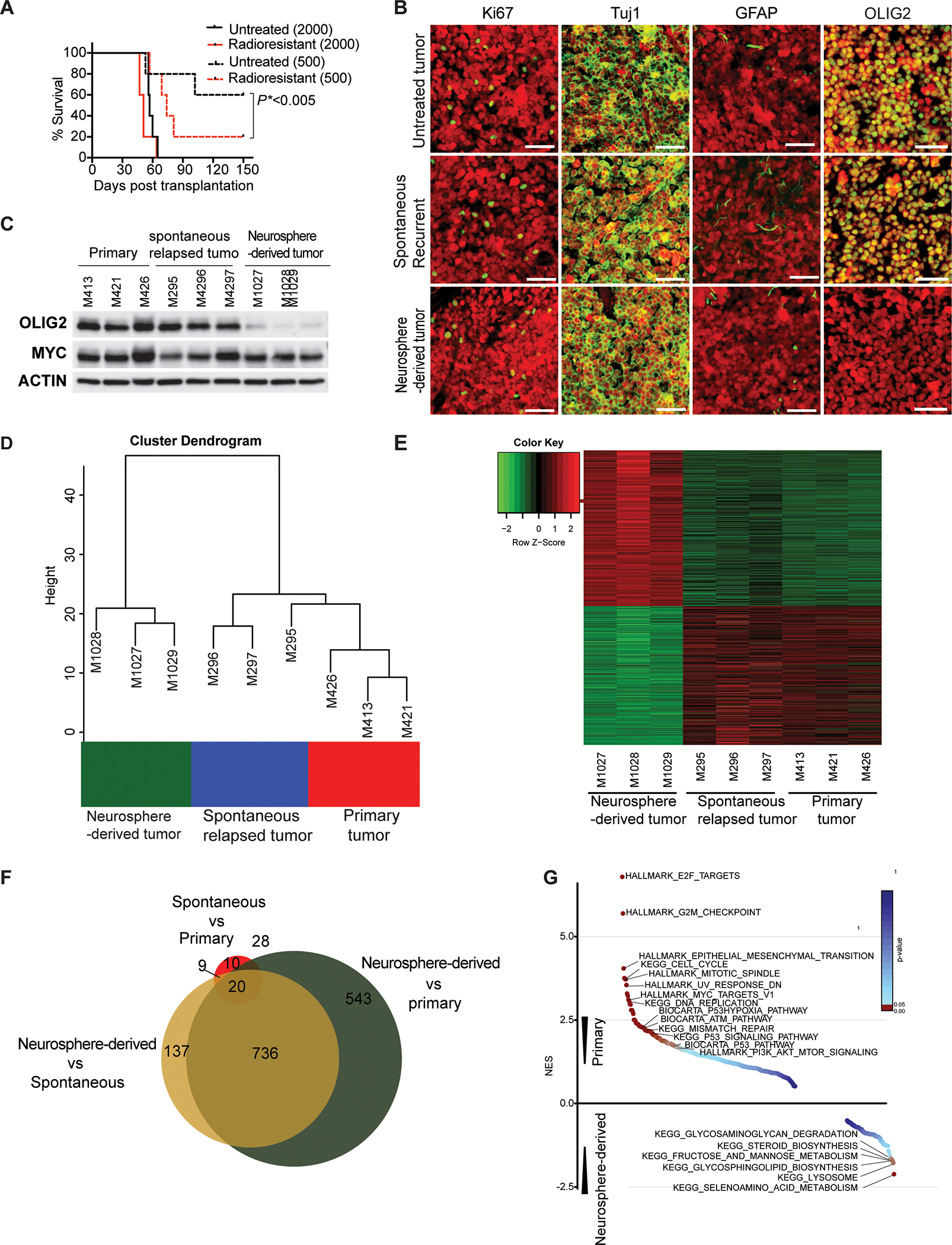
(A) Survival curves for animals transplanted with untreated cells or residual radioresistant neurospheres. Survival analysis was performed using Kaplan Meier. The p value was calculated for the radioresistant tumors compared to untreated tumors transplanted with 500 cells.
(B) IHC staining with antibodies (green) against Ki67, Tuj1, GFAP or OLIG2 in a post-irradiation neurospheres-derived tumor, spontaneously relapsed tumor, and primary tumor. Scale bars = 50 μm. Tumor cells are indicated in red (mCherry).
(C) Western blot analysis of OLIG2 and MYC expression in primary tumors, spontaneously relapsed tumors, and post-irradiation neurosphere-derived tumors.
(D, E) Unsupervised hierarchical clustering and heat map of batch effect adjusted primary tumors, spontaneously relapsed tumors, or post-irradiation neurosphere-derived tumors.
(F) Comparison of differential gene expression among primary tumors, spontaneously relapsed tumors, or post-irradiation neurosphere-derived tumors.
(G) Gene set enrichment analysis of primary tumors, spontaneously relapsed tumors, or post-irradiation neurosphere-derived tumors.
(See also Figures S3 and S4 and Table S1).
To determine whether the tumors derived from post-irradiation neurospheres resemble primary tumors and spontaneously relapsed tumors, we compared their cellular phenotypes examined by immunohistochemistry staining. All three types of tumors demonstrated similar levels of proliferation (Ki67) and neural and glia differentiation (Tuj1 and GFAP, respectively) (Figure 4B). However, the tumors derived from implanted post-irradiation neurospheres did not express OLIG2, while many tumor cells in the spontaneously relapsed tumors were OLIG2+, mirroring the primary tumors (Figure 4B). Western blot analysis confirmed low OLIG2 expression in the post-irradiation neurospheres-derived tumors and high OLIG2 expression in both the primary tumors and spontaneously relapsed tumors, with no significant difference in MYC expression between these three types of tumors (Figure 4C).
To further characterize tumor recurrence, we used bulk RNA-seq to compare average gene expression patterns of tumors derived from implanted post-irradiation neurospheres to those of primary tumors and spontaneously relapsed tumors post-radiotherapy in vivo. Spontaneously relapsed tumors closely resembled the primary tumors and differed significantly from tumors derived from implanted post-irradiation neurospheres (Figures 4D and 4E). Filtering by 2-fold and higher changes in gene expression and p-value with FDR correction <0.0001, we identified 44 upregulated and 23 downregulated genes in the spontaneously relapsed tumors compared to primary tumors, and 817 upregulated and 509 downregulated genes in tumors derived from implanted post-irradiation neurospheres compared to primary tumors (Table S1). Among these genes, 736 (57%) genes were also differentially expressed between tumors derived from implanted post-irradiation neurospheres and spontaneously relapsed tumors (Figure 4F). Gene set enrichment analysis (GSEA)(33) showed that pathways associated with p53, cell cycle, DNA replication, and DNA damage were relatively downregulated in tumors derived from implanted post-irradiation neurospheres and metabolic pathways were upregulated (Figure 4G). These results indicate that tumors derived from implanted post-irradiation neurospheres did not phenocopy the histopathological and molecular features of the primary and spontaneously relapsed tumors. Transplantation of OLIG2− radioresistant tumor cells passaged through sphere culture was thus sufficient to generate tumors, but did not recapitulate the process of in vivo recurrence, warranting further analysis of the recurrence process.
Overexpression of OLIG2 makes OLIG2−low MYC-amplified MB cells resistant to radiation
To determine the role of OLIG2 in tumor progression and radioresistance, we evaluated whether overexpression of OLIG2 promotes growth of OLIG2−low MYC-amplified MB and drives radiation resistance in the tumor cells. We transduced RCMB28 (OLIG2−low tumor) PDX cells with lentivirus encoding OLIG2−GFP and transplanted the cells into the cerebella of R2G2 mice. Another cohort of mice were transplanted with RCMB28 cells transduced with control virus encoding only a GFP reporter. 2–3 weeks post-transplantation, we treated the mice with 2 Gy CSI every other day for a cumulative dose of 18 Gy. None of the animals transplanted with control virus-infected tumor cells developed relapse post-irradiation over a 4-month monitoring period, consistent with our observation in Figure 2J–L. In contrast, 4 out of 6 (67%) mice transplanted with OLIG2−GFP virus-infected OLIG2−low tumor cells relapsed 15–20 weeks post-irradiation, indicating that OLIG2 induces radioresistance in OLIG2−low MYC-amplified MB. Histologic analyses of the brains of irradiated animals identified large, relapsed tumors in the cerebella of animals transplanted with OLIG2−GFP virus-infected tumor cells, and no tumors in animals transplanted with control tumor cells (Figures 5A and 5B). GFP expression was observed only in a fraction of relapsed tumor cells (Figure 5B), which is likely due to the low efficiency of virus infection (Figure 5C). Apoptosis was observed in the GFP− tumor region (arrow in Figure 5B), but not in the GFP+ tumor region. Histologic analysis revealed that most tumor cells in the apoptotic area were GFP− (OLIG2−) and CC3+ (Figures 5D and 5E), suggesting that the tumor cells without OLIG2 expression were sensitive to irradiation and underwent apoptosis. OLIG2+ tumor cells were highly proliferative (Figure 5F, Square 1 from Figure 5B). We also observed a significant amount of OLIG2− tumor cells post-irradiation that were Ki67+ (Figure 5F, Square 2 from Figure 5B), suggesting that OLIG2+ tumor cells may generate a tumor microenvironment that allows adjacent OLIG2− tumor cells to resist irradiation. We further examined the impact of OLIG2 expression levels on radioresistance. We purified OLIG2−GFP+ tumor cells from the mice that received OLIG2−GFP virus-infected tumor cells and then re-transplanted the purified OLIG2−GFP+ tumor cells into the cerebella of new hosts. Again, none of the animals transplanted with control tumor cells relapsed post-irradiation (n=5), but 100% mice (n=6) transplanted with purified OLIG2+ tumor cells developed relapse post-treatment. The mice transplanted with purified OLIG2-expressing tumor cells had median survival of 112 days (Figures 5G and 5H), whereas the mice transplanted with OLIG2-expressing tumor cells without purification had median survival of 157 days (Figure S5), indicating that OLIG2 levels positively correlate to radioresistance. Flow cytometry analysis shows that the tumors were composed of approximately 100% and 97% GFP+ cells on average in animals transplanted with purified OLIG2-lentivirus infected tumor cells treated with or without irradiation, respectively (Figures 5I–5M). We next assessed the impact of radiation on tumor size or OLIG2, Ki67 or CC3 expression between OLIG2-lentivirus infected tumors without radiation and relapsed tumors post-irradiation. No significant difference in these features was observed (Figures 5N–5Q). Taken together, our data show that OLIG2 expression induces radioresistance in OLIG2−low MB.
Figure 5. Overexpression of OLIG2 induces radioresistance in OLIG2−low MYC-amplified MB.
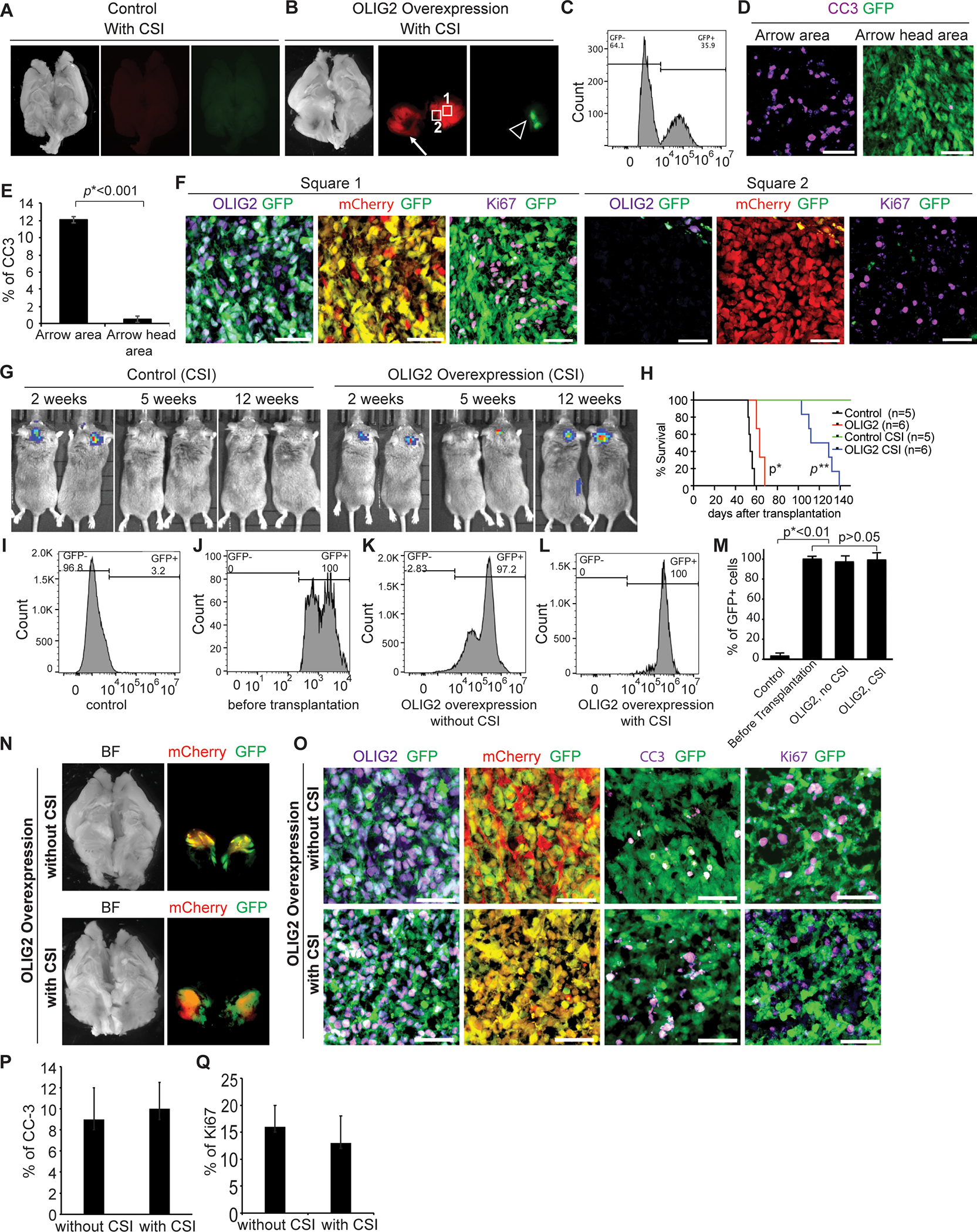
(A) No tumor was detected in mice transplanted with control RCMB28 tumor cells post-irradiation. Sagittal images of whole brain (bright field). Tumor cells indicated in red (mCherry), OLIG2−GFP-lentivirus infected cells indicated in green (GFP).
(B) Large tumors were observed in mice receiving OLIG2−GFP-lentivirus infected tumor cells post-irradiation. Sagittal images of whole brain (bright field). The arrow points to an area of apoptosis. Tumor cells indicated in red (mCherry), OLIG2−GFP-lentivirus infected cells indicated in green (GFP).
(C) Analysis of the efficiency (GFP) of viral transduction of pWPI-OLIG2-GFP in mCherry+ tumor cells by flow cytometry before transplantation.
(D, E) Apoptosis in the GFP+ (arrow) and GFP− (arrowhead) tumor regions in B. In D, tissue sections were immunostained for CC-3 (violet). OLIG2−GFP-lentivirus infected cells indicated in green (GFP). E, quantification of %CC-3+ cells in apoptotic and OLIG2−GFP-lentivirus infected regions of pWPI-OLIG2−GFP transduced irradiated RCMB28 tumors (n=3). P values determined by Student T test.
(F) Ki67 expression (violet) was detected in both GFP+ (square 1) and GFP− (square 2) tumor regions in mice receiving OLIG2−GFP-lentivirus infected tumor cells post-irradiation. OLIG2 expression (violet) was co-expressed with GFP.
(G) IVIS images of representative mice transplanted with control tumor cells or OLIG2−GFP-lentivirus infected tumor cells with CSI.
(H) Survival of animals transplanted with control tumor cells or purified OLIG2−GFP-lentivirus-infected tumor cells with or without CSI. Log-rank test p*=0.0007, OLIG2 group vs. control group; p**=0.0014, OLIG2 CSI group vs. control CSI group.
(I-M). Flow cytometry assay of OLIG2-GFP+ cells: isotype control (I), purification before transplantation (J), tumors without irradiation (K), and relapsed tumors post-irradiation (L). Quantification of GFP expression (M). P values determined by Student T test.
(N) Comparison of tumors from mice transplanted with OLIG2−GFP-virus infected tumor cells without CSI (top) and relapsed tumors post-irradiation (bottom). Sagittal images of whole brain (bright field). Tumor cells indicated in red (mCherry), OLIG2−GFP-lentivirus infected cells indicated in green (GFP).
(O) IHC of GFP (green), mCherry (red), OLIG2 (violet), Ki67 (violet), and CC3 (violet) in the tumors from mice transplanted with OLIG2-expressing RCMB28 tumor cells with (bottom panels) or without irradiation (top panels). Scale bars= 50 μm.
(P, Q). Quantification of the percentage of CC-3+ (P) or Ki67+ (Q) cells out of total mCherry+ tumor cells in nonirradiated (n=3) and irradiated (n=5) tumors.
(See also Figures S5).
Targeting OLIG2 effectively blocks OLIG2−high MYC-amplified MB recurrence
Since our data show that OLIG2 induces radioresistance and promotes tumor recurrence, we reasoned that OLIG2 may be a therapeutic target in MYC-amplified MB to prevent post-irradiation relapse. We attempted to knockdown OLIG2 by infecting MYC-amplified MB with 3 distinct CRISPR/Cas9 gRNAs against OLIG2 and found OLIG2-CRISPR1 exhibited the most effective knockdown as examined by Western blot (Figure 6A). Knockdown of OLIG2 with OLIG2-CRISPR1 in combination with irradiation significantly delayed the progression of OLIG2−high tumor (MB002) (Figures 6B), even though a small fraction of persisting OLIG2+ tumor cells were detected in the relapsed tumors (Figures 6C), supporting the clinical potential of disrupting OLIG2 expression.
Figure 6. Targeting OLIG2 prevents recurrence of OLIG2−high MYC-amplified MB in vivo.
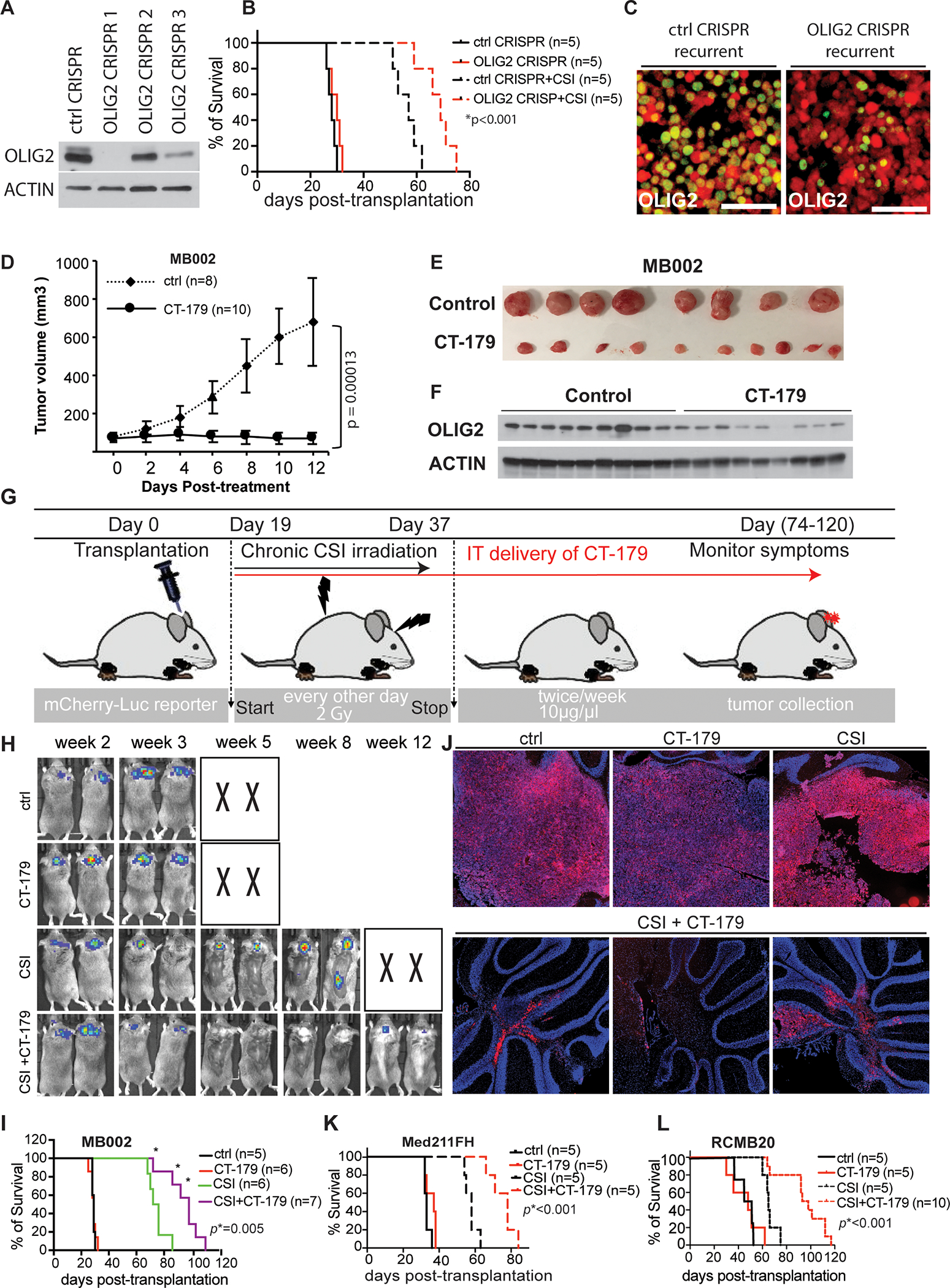
(A) Analysis of OLIG2-CRISPR mediated knock down of OLIG2 protein in MB002 tumor by western blot. Control, ctrl.
(B) Mice survival curves for animals bearing MB002 tumors with overexpression of OLIG2-CRISPR1. Survival analysis was performed using Kaplan Meier. The p value is for the OLIG2-CRISPR+CSI compared to control-CRISPR+CSI.
(C) IHC staining for OLIG2 (green) in the relapsed tumors with overexpression of control (ctrl CRISPR) and OLIG2-CRISPR1. Tumor cells are indicated in red (mCherry)
(D-F) CT-179, an OLIG2-small molecule inhibitor, restricts MB002 subcutaneous tumor growth. Animals were euthanized when their body weight decreased by ~20% from initial weight. (D) Tumor volume was measured using calipers every 2 days during the course of the treatment. Data are shown as mean ± SD for tumor volume. p values were determined using one-way ANOVA followed by Tukey post-hoc test. Tumor volumes on day 12 of treatment were used in statistical analysis. (E) Tumor images at the time of sacrifice. (F) Western blot analysis showing that OLIG2 expression in flank tumors was reduced by CT-179 treatment.
(G) Schematic showing CT-179 treatment twice weekly for intracranial tumors. IT, intrathecal injection
(H-J) Animals bearing MB002 tumors treated with vehicle, CT-179, CSI alone or CSI + CT-179 by intrathecal injection. (H) Bioluminescence images of representative mice (X denotes animals euthanized before they could be imaged). (I) Kaplan Meier survival curves. p values were determined by Log-rank (Mantel-Cox) test. (J) Images of brains from representative animals. Red is mCherry, indicating tumor cells. Tissues were counterstained with DAPI. Animals were euthanized when their body weight decreased by ~20% from initial weight. The p value is for the CST+ CT-179 compared to CSI.
(K-L) Kaplan Meier survival curves for animals bearing Med-211FH (K) or RCMB20 (L) tumors treated with vehicle, CT-179, CSI alone or CSI + CT-179 by intrathecal injection. The p value is for the CST+ CT-179 compared to CSI.
(See also Figures S6 and S7).
Hence, we tested the in vitro inhibitory effects of two small molecule inhibitors of OLIG2, CT-179 and CT-767, in an OLIG2−high (MB002) and an OLIG2−low (RCMB28) human MB PDX line. Cells were cultured and treated with CT-179, CT-767 or vehicle (DMSO) for 72 hours and cell viability was measured using the Cell-Titer-Glo Assay. MB002 OLIG2−high cell viability was significantly reduced by both CT-179 and CT-767 with IC50s 0.9 μM and 0.08 μM, respectively (Figures S6A and S6B). In contrast, OLIG2−low RCMB28 cell growth was moderately inhibited by CT-179 with IC50 6 μM (Figure S6C) and the cells did not respond to CT-767 (Figure S6D). Since MB002 OLIG2−high cells were sensitive to CT-179 and CT-767, we measured the OLIG2 protein level to see whether the inhibitory effect was achieved by reducing OLIG2 expression. As shown in Figures S6E and S6F, CT-179 treatment resulted in significant dose-dependent reductions in OLIG2 protein level, whereas CT-767 treatment moderately reduced OLIG2 protein level and the effect was not dose dependent.
We then tested CT-179 in vivo using two OLIG2−high and one OLIG2−low PDX lines in flank tumor models. After implanting the cells and allowing the tumor volume to reach ~100 mm3 (usually ~14–20 days), mice were randomized into two groups and treated with vehicle (water) or CT-179 by oral gavage. Mice with flank tumors were treated with 105 mg/kg CT-179 for 14 consecutive days. Twenty-four hours after the last dose, animals were euthanized, and tumors were harvested and evaluated. CT-179 significantly reduced the growth of OLIG2−high (Figures 6D and 6E; Figures S6G–I) but did not affect OLIG2−low subgroup tumors (Figures S6K–M). Compared with control, CT-179 treatment resulted in a significant reduction in the level of OLIG2 protein (Figure 6F; Figure S6J).
Previous studies have reported that CT-179 readily crosses the blood-brain barrier (BBB), achieves high concentrations in the brain, and significantly prolongs survival of GBM mice (34). Therefore, we first studied whether systemic administration of CT-179, given via oral gavage, can inhibit the growth of intracranial MYC-amplified MB. We compared survival of mice bearing MB002 PDX tumors treated with vehicle (water), irradiation with 2 Gy every other day for a cumulative dose of 18 Gy, CT-179 105 mg/kg, and irradiation + CT-179. However, systemic CT-179 (oral gavage) neither blocked the growth of intracranial OLIG2−high MYC-amplified MB in vivo (Figure S7A) nor decreased the OLIG2 protein level in tumors (Figure S7B), suggesting that tumor exposure to CT-179 via oral administration may be insufficient. Therefore, we next opted for intrathecal (IT) delivery directly to the cisterna magna. We first injected an artificial cerebrospinal fluid (CSF) tracer (fluorescein isothiocyanate (FITC)-dextran-500) to validate our technique, as described in the literature (data not shown) (35–37). We then randomized tumor-bearing mice into three groups: vehicle (artificial CSF), craniospinal irradiation (CSI), and CSI + CT-179. We then injected 3 μl of CT-179 (10 μg/ μl) or vehicle by IT into tumor bearing mice and repeated the injection weekly for three weeks. This dose was empirically determined in pilot studies as optimal for high effectiveness and low toxicity. After the last dose of CT-179 treatment, all animals were sacrificed when they lost more than 20% of their initial weight and brains were collected to evaluate tumor development. Animals treated with CT-179 following CSI survived significantly longer with smaller tumors compared to those exposed to CSI alone or control (Figures S7C and S7D).
We next examined if increasing the frequency and duration of CT-179 by IT injection, from once a week for three weeks to twice a week and continuing until the mice succumbed to the tumor, help improve efficacy further (Figure 6G). Again, animals treated with an increased dose of CT-179 alone did not show prolonged survival compared to control animals. However, CSI radiotherapy plus increased CT-179 administration significantly prolonged animal survival compared to CSI alone and yielded a significant additional survival advantage compared to the low-dose treatment (Figures 6H and 6I). Mice in the groups treated with vehicle (artificial cerebrospinal fluid), CT-179 alone or irradiation alone were sacrificed once they presented with symptoms including weight loss and ataxia. To compare the sizes of progressive tumors in the mice post-treatment with irradiation alone to irradiation plus CT-179, a cohort of mice in these two groups were sacrificed at the same time points. Large tumors were observed in the mice treated with control, CT-179 alone, or CSI alone at the end point of tumor development, whereas only small tumor lesions were seen in the combination treatment group, CSI + CT-179, when the tumors were harvested at the same time points as the CSI alone group (Figure 6J). Histological analysis showed that after treatment with an increased dose of CT-179 following CSI, the treated tumors were composed mostly of OLIG2− tumor cells and rare OLIG2+ tumor cells. Ki67+ cells were also rare in the remaining tumor cells (Figures S7E and S7F). Since animals lost body weight due to either the frequent anesthesia with isoflurane or the toxicity of long-term treatment (over 60 days) with CT-179, we had to stop CT-179 treatment or reduce the dose of CT-179 when animals presented with loss of body weight. These animals then eventually developed tumor recurrence. The inhibitory effect of the combination of CT-179 and radiotherapy was further validated in another two OLIG2−high PDX tumors (Med-211FH and RCMB20) (Figures 6K and 6L). These results suggest that targeting OLIG2 pharmacologically by CT-179 in combination with radiotherapy effectively prevents the progression of OLIG2−high MYC-amplified MB tumors.
DISCUSSION
Medulloblastoma heterogeneity, both between patients and within individual tumors, influences prognosis, response to therapy, and probability of recurrence (38–42). The genetic makeup and the cellular/molecular heterogeneity in MYC-amplified MB are poorly defined, which limits our ability to develop effective therapies. To overcome this limitation, we leveraged mouse and human PDX models to demonstrate that MYC-amplified MB is heterogenous and OLIG2 is a determinant of high-risk MYC-amplified MB. Our study indicates that OLIG2 is a potential biomarker to select high-risk patients for therapy intensification, while stratifying the low-risk patients for therapy de-escalation. We also provide evidence that OLIG2 protein is a druggable target that is amenable to inhibition by the small molecule inhibitor CT-179, which prevents post-irradiation tumor recurrence effectively.
Mouse model Myc-driven MB tumors show variable OLIG2 protein expression despite deriving from the same cell population in the postnatal cerebellum and sharing the same oncogenic driver (26). Like the mouse model, human MYC-amplified MB also varies in OLIG2 expression. This variation is clinically relevant, as OLIG2−high and OLIG2−low subsets of MYC-amplified MB respond differently to irradiation. Residual tumor cells persisted in all animals implanted with OLIG2−high MYC-amplified MB at the end of radiotherapy, and these animals subsequently developed recurrence consistently if maintained for adequate time. The residual tumor cells that persisted immediately following the completion of radiotherapy were mostly OLIG2− with rare OLIG2+ cells. In contrast, progressive tumors that developed in the weeks following the completion of radiotherapy were predominantly OLIG2+.
The residual radioresistant tumor cells displayed certain CSLC features, including unlimited self-renewal activity, quiescence, and high tumorigenicity post-transplantation, consistent with previous studies (43–46). Our studies further reveal that the residual tumor cells have the ability to change state, from quiescent to proliferative, in response to environmental conditions. However, the tumors generated from transplantation of post-radiotherapy cultured neurospheres did not contain OLIG2+ cells, in contrast to both primary tumors and spontaneously relapsed tumors post-irradiation in vivo. Thus, our studies show that OLIG2− CSLCs that emerged through neurosphere culture could not fully recapitulate OLIG2+ cells during tumor development, suggesting that they may not confer tumor recurrence directly. However, it is unclear whether OLIG2− CSLCs have a crosstalk with the rare radioresistant OLIG2+ cells to drive tumor relapse corporately. It will be of great interest in future studies to determine the role of OLIG2− CSLCs in tumor recurrence.
The widespread expression of OLIG2 in both untreated and post-irradiation progressive tumors suggests that OLIG2+ cells are required for robust tumor formation and that OLIG2 may be an effective therapeutic target. Our lentiviral OLIG2 transduction studies substantiate the findings that OLIG2 promotes recurrence. Forced expression of OLIG2 potentiated tumor growth and induced radioresistance in OLIG2−low MYC-amplified MB PDX, indicating that OLIG2 underlies therapy-resistance and recurrence. These data are consistent with prior reports that OLIG2 confers radiation and chemo-resistance in glioma cells (17,19). Findings from the use of OLIG2 CRISPR and antagonist CT-179 also support the crucial role of OLIG2 in recurrence in MYC-amplified MB. While blocking OLIG2 with CT-179 alone was not sufficient to inhibit tumor growth, consistently treating with CT-179 after radiotherapy significantly stalled tumor recurrence even in the presence of OLIG2− CSLCs. However, tumors treated with CSI + CT-179 still generated OLIG2+ post-irradiation progressive tumors if CT-179 treatment was stopped or the dose was reduced, indicating that continuous treatment with CT-179 is necessary to prevent tumor relapse. Although prior studies show the important role of OLIG2 in SHH-MB, where OLIG2+ progenitors are tumor-initiating cells and genetic OLIG2 deletion delays SHH-MB growth in mouse tumors(47), our data show for the first time that OLIG2 is a biomarker in high risk MYC-amplified MB and can be targeted by the small molecule CT-179 in human OLIG2+ MYC-amplified MB, unveiling a new approach to prevent or delay recurrence after radiotherapy. Moreover, we demonstrated that CT-179 is not bioavailable when orally administered. Hence, CT-179 is an effective druggable candidate only when intrathecally administered along with radiotherapy. OLIG2 is also differentially expressed in other types of pediatric brain tumors including pilocytic astrocytoma and diffuse intrinsic pontine glioma (DIPG)(47–49). Thus, our studies may provide the framework for elucidating the mechanisms underlying therapeutic resistance and developing a new approach to treating these cancers.
Supplementary Material
Translational Relevance.
Our studies demonstrated that OLIG2-expressing tumor cells were highly enriched in therapy-resistant and recurrent MYC-amplified MB and high levels of OLIG2 correlated with poor prognosis. Genetic or pharmacological inhibition of OLIG2 in combination with radiotherapy significantly suppressed the progression of OLIG2−high tumors in patient-derived xenograft mouse models, indicating that OLIG2 represents a novel therapeutic target in high-risk MB. CT-179 is a highly potent and selective small molecule OLIG2 inhibitor. The Food and Drug Administration (FDA) has granted Rare Pediatric Disease (RPD) designation to CT-179 for MB treatment. Our studies provide strong evidence for the potential of CT-179 to act as an adjunctive therapy for patients with high-risk MB. OLIG2 is also highly expressed in other types of pediatric brain tumors including pilocytic astrocytoma and diffuse intrinsic pontine glioma. Thus, patients with these cancers may also benefit from CT-179 treatment.
Acknowledgements
The authors would like to acknowledge Dr. Didier Trono for pWPI-IRES-GFP and Dr. Kevin Eggan for pMXs-OLIG2 plasmids. We thank Dr. Greg Stein at Curtana Pharmaceuticals to provide the small molecule inhibitors of CT-179 and CT-767. We also thank Drs. Yoon-Jae Cho, Robert Wechsler-Reya, Xiao-Nan Li, and James Olson for the PDX lines. This work was supported by funds from the Children’s National Health Systems, NIH RO1 CA241192 (Y.P.), St. Baldrick’s Foundation (Y.P.), and Matthew Larson Foundation for pediatric brain tumors (Y.P.).
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest.
References
- 1.Packer RJ, Rood BR, MacDonald TJ. Medulloblastoma: present concepts of stratification into risk groups. Pediatr Neurosurg 2003;39(2):60–7 doi 10.1159/000071316. [DOI] [PubMed] [Google Scholar]
- 2.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123(4):465–72 doi 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012;488(7409):49–56 doi 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012;123(4):473–84 doi 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017;31(6):737–54 e6 doi 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer 2012;12(12):818–34 doi 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers 2019;5(1):11 doi 10.1038/s41572-019-0063-6. [DOI] [PubMed] [Google Scholar]
- 8.Gopalakrishnan V, Tao RH, Dobson T, Brugmann W, Khatua S. Medulloblastoma development: tumor biology informs treatment decisions. CNS Oncol 2015;4(2):79–89 doi 10.2217/cns.14.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 2011;29(11):1424–30 doi 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalski JM, Janss AJ, Vezina LG, Smith KS, Billups CA, Burger PC, et al. Children’s Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol 2021;39(24):2685–97 doi 10.1200/JCO.20.02730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manoranjan B, Venugopal C, McFarlane N, Doble BW, Dunn SE, Scheinemann K, et al. Medulloblastoma stem cells: where development and cancer cross pathways. Pediatr Res 2012;71(4 Pt 2):516–22 doi 10.1038/pr.2011.62. [DOI] [PubMed] [Google Scholar]
- 12.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 13.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432(7015):396–401 doi 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 14.Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016;529(7586):351–7 doi 10.1038/nature16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrissy AS, Cavalli FMG, Remke M, Ramaswamy V, Shih DJH, Holgado BL, et al. Spatial heterogeneity in medulloblastoma. Nat Genet 2017;49(5):780–8 doi 10.1038/ng.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, et al. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 2004;63(5):499–509 doi 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
- 17.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007;53(4):503–17 doi 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SK, Fiorelli R, Kupp R, Rajan S, Szeto E, Lo Cascio C, et al. Post-translational Modifications of OLIG2 Regulate Glioma Invasion through the TGF-beta Pathway. Cell Rep 2016;16(4):950–66 doi 10.1016/j.celrep.2016.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 2011;19(3):359–71 doi 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kupp R, Shtayer L, Tien AC, Szeto E, Sanai N, Rowitch DH, et al. Lineage-Restricted OLIG2-RTK Signaling Governs the Molecular Subtype of Glioma Stem-like Cells. Cell Rep 2016;16(11):2838–45 doi 10.1016/j.celrep.2016.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, et al. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A 2014;111(14):5248–53 doi 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016;29(3):311–23 doi 10.1016/j.ccell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res 2014;20(4):912–25 doi 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morfouace M, Shelat A, Jacus M, Freeman BB, 3rd, Turner D, Robinson S, et al. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 2014;25(4):516–29 doi 10.1016/j.ccr.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brun SN, Markant SL, Esparza LA, Garcia G, Terry D, Huang JM, et al. Survivin as a therapeutic target in Sonic hedgehog-driven medulloblastoma. Oncogene 2015;34(29):3770–9 doi 10.1038/onc.2014.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao R, Murad N, Xu Z, Zhang P, Okonechnikov K, Kool M, et al. MYC Drives Group 3 Medulloblastoma through Transformation of Sox2(+) Astrocyte Progenitor Cells. Cancer Res 2019;79(8):1967–80 doi 10.1158/0008-5472.CAN-18-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malawsky DS, Weir SJ, Ocasio JK, Babcock B, Dismuke T, Cleveland AH, et al. Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile. Commun Biol 2021;4(1):616 doi 10.1038/s42003-021-02099-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Envigo. Rag2/IL2RG Double Knockout (R2G2). Available from: http://wwwenvigocom/resources/data-sheets/11029-envigo-r705-rms-rag2-model-sheet-us-4pppdf [cited 2017 September 14, 2017].
- 29.Stripay JL, Merchant TE, Roussel MF, Tinkle CL. Preclinical Models of Craniospinal Irradiation for Medulloblastoma. Cancers (Basel) 2020;12(1) doi 10.3390/cancers12010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R, Smith KS, Deng M, Terhune C, Robinson GW, Orr BA, et al. Clinical Outcomes and Patient-Matched Molecular Composition of Relapsed Medulloblastoma. J Clin Oncol 2021;39(7):807–21 doi 10.1200/JCO.20.01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu APY, Smith KS, Kumar R, Paul L, Bihannic L, Lin T, et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021. doi 10.1016/j.ccell.2021.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg TJ, Marques C, Pantazopoulou V, Johansson E, von Stedingk K, Lindgren D, et al. The Irradiated Brain Microenvironment Supports Glioma Stemness and Survival via Astrocyte-Derived Transglutaminase 2. Cancer Res 2021;81(8):2101–15 doi 10.1158/0008-5472.CAN-20-1785. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alton GB GR, Knowles S, Stein G, Kesari S. CT179 degradesthe olig2 transcription factor in glioblastoma stem-like cells and prolongs surviva. Experimental and Molecular Therapeutics (American Association for Cancer Research 2017:1174. [Google Scholar]
- 35.Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 2013;33(46):18190–9 doi 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 2012;4(147):147ra11 doi 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reijneveld JC, Taphoorn MJ, Voest EE. A simple mouse model for leptomeningeal metastases and repeated intrathecal therapy. J Neurooncol 1999;42(2):137–42 doi 10.1023/a:1006237917632. [DOI] [PubMed] [Google Scholar]
- 38.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res 2015;21(6):1258–66 doi 10.1158/1078-0432.CCR-14-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013;501(7467):328–37 doi 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15(2):81–94 doi 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 41.Bernstock JD, Mooney JH, Ilyas A, Chagoya G, Estevez-Ordonez D, Ibrahim A, et al. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: clinical and translational implications. J Neurosurg 2019:1–9 doi 10.3171/2019.5.JNS19364. [DOI] [PubMed] [Google Scholar]
- 42.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 doi 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexander J, LaPlant QC, Pattwell SS, Szulzewsky F, Cimino PJ, Caruso FP, et al. Multimodal single-cell analysis reveals distinct radioresistant stem-like and progenitor cell populations in murine glioma. Glia 2020;68(12):2486–502 doi 10.1002/glia.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell 2014;14(3):275–91 doi 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Arnold CR, Mangesius J, Skvortsova II, Ganswindt U. The Role of Cancer Stem Cells in Radiation Resistance. Front Oncol 2020;10:164 doi 10.3389/fonc.2020.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer 2008;8(7):545–54 doi 10.1038/nrc2419. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, He X, Liu X, Zhang F, Huang LF, Potter AS, et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019;36(3):302–18 e7 doi 10.1016/j.ccell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson JL, Muraleedharan R, Oatman N, Klotter A, Sengupta S, Waclaw RR, et al. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro Oncol 2017;19(8):1068–78 doi 10.1093/neuonc/now299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otero JJ, Rowitch D, Vandenberg S. OLIG2 is differentially expressed in pediatric astrocytic and in ependymal neoplasms. J Neurooncol 2011;104(2):423–38 doi 10.1007/s11060-010-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible with GEO series accession numbers GSE188789.