

Abstract
Cell reprogramming has wide applications in tissue regeneration, disease modelling and personalized medicine. In addition to biochemical cues, mechanical forces also contribute to the modulation of the epigenetic state and a variety of cell functions through distinct mechanisms that are not fully understood. Here we show that millisecond deformation of the cell nucleus caused by confinement into microfluidic channels results in wrinkling and transient disassembly of the nuclear lamina, local detachment of lamina-associated domains in chromatin and a decrease of histone methylation (histone H3 lysine 9 trimethylation) and DNA methylation. These global changes in chromatin at the early stage of cell reprogramming boost the conversion of fibroblasts into neurons and can be partially reproduced by inhibition of histone H3 lysine 9 and DNA methylation. This mechanopriming approach also triggers macrophage reprogramming into neurons and fibroblast conversion into induced pluripotent stem cells, being thus a promising mechanically based epigenetic state modulation method for cell engineering.
Cell reprogramming technologies can be used to derive desirable cell types and have wide applications in regenerative medicine, disease modelling and drug screening1–3. Direct reprogramming enables the conversion of one cell type into another desired cell type by circumventing the pluripotent stage and time-consuming differentiation process, as exemplified in the conversion of fibroblasts into induced neuron (iN) cells4 and other cell types5–8. However, the low efficiency of these conversion processes presents a barrier for biomedical applications9.
A critical step in cell reprogramming is to overcome the epigenetic barrier of heterochromatin and turn on the endogenous genes for cell type conversion. Most previous studies have focused on the roles of transcriptional factors and biochemical factors in cell reprogramming10,11, but the effects of biophysical factors are much less understood. Cells experience mechanical stimuli at both short and long timescales, from seconds to days, which may result in mechano-chemical signalling, cytoskeleton reorganization and chromatin changes in a context-dependent manner12–22. To directly determine the effect of nuclear deformation on chromatin remodelling, we investigated whether and how mechanically squeezing suspended cells could regulate the epigenetic state and cell reprogramming, and explored this approach for mechanoceutical applications.
Microchannel-induced nuclear deformation
Microfluidic devices have been used to study cell deformation, gene transfer and cancer cells23–26; however, smaller microchannels are needed to deform the cell nucleus without compromising cell viability. Therefore, we developed microfluidic devices with various sizes of parallel constriction microchannels to deform cells, and optimized the microchannel dimension (Fig. 1a and Supplementary Fig. 1). Based on the size of mouse fibroblasts (19.7 ± 4.5 μm) and their nuclei (10.5 ± 1.2 μm; Fig. 1b), we tested microchannels with the same height (15 μm) but different widths (3, 5, 7 and 9 μm). Cells passing through wide microfluidic channels (200 μm) did not show significant nuclear deformation (Supplementary Fig. 2) and were used as a control in all studies.
Fig. 1 |. Microchannel-induced nuclear deformation.
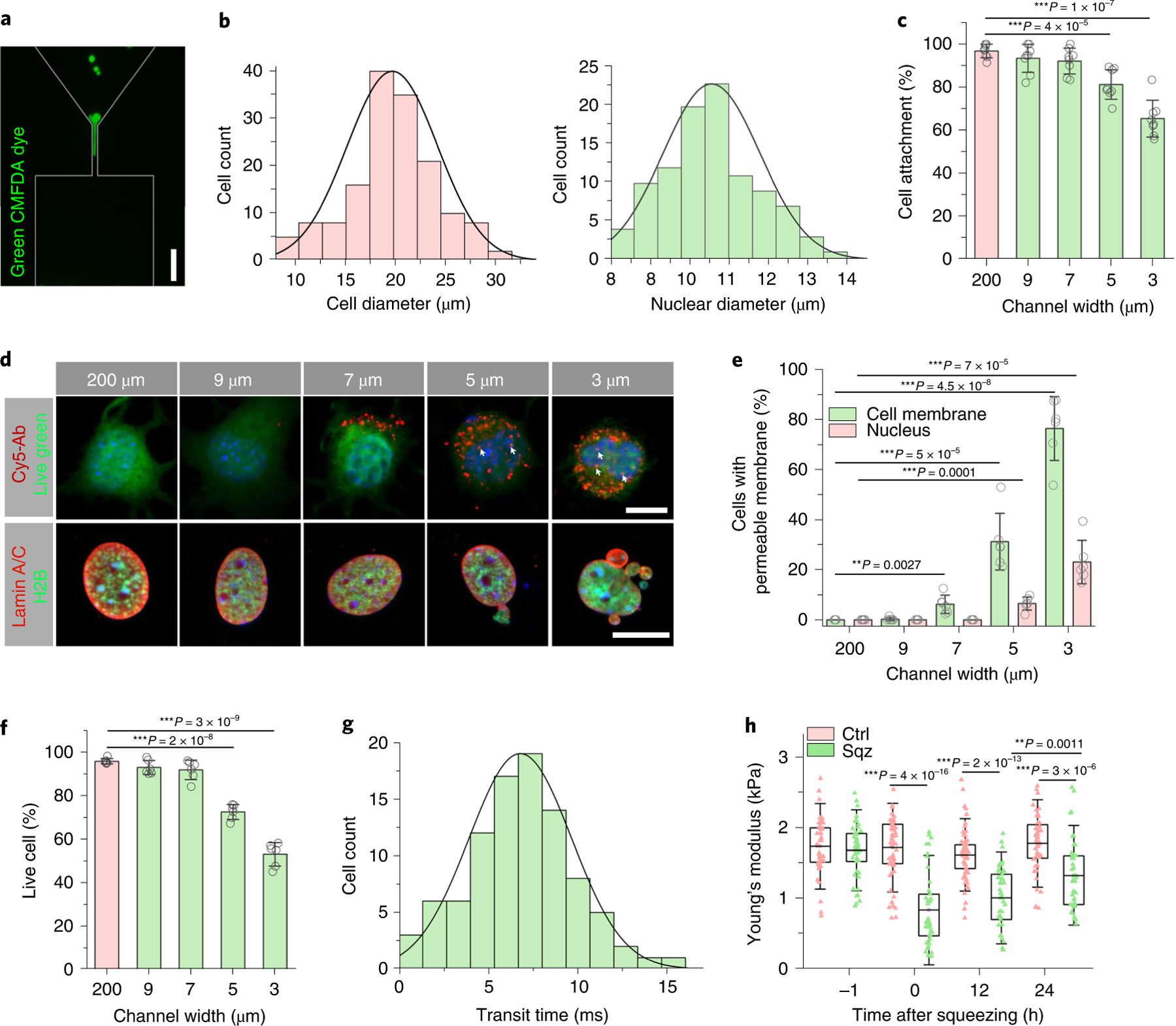
a, Immunofluorescent images of fibroblasts labelled with CellTracker Green CMFDA (5-chloromethylfluorescein diacetate) passing through the microchannels. Scale bar, 50 μm. b, Quantification of cell diameter based on TC20 Cell Counter (Bio-Rad; n = 100) and quantification of nuclear diameter based on Hoechst staining (n = 100). c, The percentage of cells attached after BAM-transduced fibroblasts were deformed using microdevices with various channel widths (n = 8). Statistical significance was determined by a one-way ANOVA and Tukey’s multiple comparison test compared to the control (200 μm channels). d, Immunofluorescent images of cells that were deformed by using microdevices with different microchannel widths. The permeability or non-specific transport of the cell membrane and nuclear envelope was examined 3 hours after cells passed through the microchannel by using Cy5-Ab (top). Live green dye was used to label live cells. White arrows indicate Cy5-Ab in the nuclei. Scale bar, 10 μm. Immunofluorescent images (bottom) of H2B-GFP fibroblasts that were deformed by using microdevices with different microchannel widths, seeded onto fibronectin-coated glass slides for 3 hours and stained with lamin A/C. Scale bar, 10 μm. e, The percentage of cells with extracellular Cy5-Ab penetrating the cell membrane (green) and nuclear envelope (red) was quantified in live cells (n = 6). Statistical significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. f, The percentage of live cells (determined by a LIVE/DEAD staining kit) after BAM-transduced fibroblasts were deformed using microdevices with various channel widths (n = 6). Statistical significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. g, Quantification of transit time in milliseconds as cells passed through the microchannels (n = 94). h, Quantification of Young’s modulus of cells at the indicated time points before and after mechanical squeezing, as measured by AFM (n = 42). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test (NS, not significant). Ctrl, control; Sqz, squeezed. In c, e and f, data represent mean ± s.d. In h, box plots show the ends at the quartiles and the mean as a horizontal line in the box, and the whiskers represent the s.d.
Cells were collected at the outlet of the microdevices and seeded onto fibronectin-coated wells. The cells passing through bigger channels (200, 9 and 7 μm wide) had negligible unattached cells after 3 hours, but 5-μm-wide and 3-μm-wide microchannels caused significant cell damage (Fig. 1c). To determine the effect of nuclear deformation on cell membrane integrity, a fluorescently tagged antibody (Cy5-Ab) was added to the culture media of fibroblasts in suspension before and during the introduction of the fibroblasts into the microfluidic device. Cy5-Ab could be detected in 31% and 76% of cells passing through 5-μm-wide and 3-μm-wide channels, respectively, suggesting a cell membrane disruption24,27 (Fig. 1d–e). This cell membrane disruption was also detected in 6% of cells passing through 7-μm-wide microchannels but not in 9-μm-wide or 200-μm-wide channels. Additionally, Cy5-Ab was detected in 6.5% and 23% of nuclei in the cells passing through 5-μm-wide and 3-μm-wide microchannels but not bigger microchannels, indicating that microchannels of less than 5 μm induced nuclear envelope rupture or nuclear transport (Fig. 1d,e). Furthermore, by using fibroblasts expressing histone 2B (H2B) tagged with green fluorescent protein (H2B-GFP), we observed that 3-μm-wide and 5-μm-wide channels, but not 7-μm-wide and 9-μm-wide channels, induced significant nuclear blebbing or segregation after squeezing (Fig. 1d and Supplementary Fig. 3). Consistently, in fibroblasts expressing a GFP with a nuclear localization signal (NLS-GFP) as a reporter, there was a diffusion of NLS-GFP into the cytoplasm in >5% of cells passing through 5 μm and 3 μm microchannels, indicating that nuclear rupture had occurred (Supplementary Fig. 4).
The rupture of the cell membrane and nuclear envelope may induce DNA damage and cell death28,29. Live/dead cell staining and PrestoBlue assays showed that 7-μm-wide and 9-μm-wide channels did not induce noticeable cell death (Fig. 1f and Supplementary Fig. 5). By contrast, 3-μm-wide and 5-μm-wide channels decreased cell viability and induced significant (20–50%) DNA damage (Supplementary Fig. 6). Based on these findings, we used 7 μm microchannels for the rest of the studies. We also optimized the flow rates in 7 μm microchannels and showed that a flow rate of up to 20 μl min−1 did not affect the cell viability (Supplementary Fig. 7) or cause cell aggregation. Therefore, we used 20 μl min−1 as an optimal flow rate, which resulted in an average of 6.8 milliseconds (ms) for a cell to pass through a 7-μm-wide microchannel (Fig. 1g), resulting in a transient nuclear deformation.
As shown by atomic force microscopy (AFM) measurement (Fig. 1h), this transient cell squeezing decreased the stiffness of the cells, measured by indenting the plasma membrane of the cells above the nucleus. Additionally, the cell nucleus was more elongated after squeezing (Supplementary Fig. 8). After 24 hours, we observed a gradual recovery of elastic modulus and nucleus shape.
Nuclear deformation on iN reprogramming
To determine whether microchannel-induced nuclear deformation had any effect on the direct conversion of fibroblasts into iN cells, adult mouse fibroblasts were transduced with doxycycline (Dox)-inducible lentiviral constructs containing the three reprogramming factors BRN2, ASCL1 and MYT1L (BAM) as depicted in the timeline for the reprogramming experimental procedure (Fig. 2a). Two days later, Dox was added (designated as day 0) to induce the expression of BAM transgenes and proteins within a few hours. To determine the timing of mechanical squeezing, cells were introduced into microfluidic devices with 7-μm-wide channels before or after administering Dox. Then the cells were collected and seeded onto fibronectin-coated glass coverslips and cultured in serum-free N2B27 medium (Fig. 2a). Seven days after induced mechanical deformation, cultures were fixed and stained for β-III tubulin (Tubb3) to determine the reprogramming efficiency. The reprogramming efficiency of cells going through 200-μm-wide channels (as the control group) was not significantly different from the cells under static culture conditions (Supplementary Fig. 9). Administering Dox at least 6 hours before subjecting the cells to nuclear deformation produced the highest reprogramming efficiency (approximately eightfold) compared to the control group (Fig. 2b). These results suggested that the presence of BAM proteins within the first few hours after squeezing was critical and that a transient nuclear deformation could enhance the reprogramming efficiency. Consistent with the effects of microchannel size on cell viability (Fig. 1f and Supplementary Fig. 5), microchannels smaller than 7 μm (that is, 5 μm and 3 μm) decreased cell viability and thus compromised reprogramming efficiency, while 9 μm microchannels might not induce sufficient nuclear deformation and resulted in a lower reprogramming efficiency than 7 μm microchannels (Fig. 2c).
Fig. 2 |. Microchannel-induced nuclear deformation boosted neuronal marker expression and enhanced iN reprogramming efficiency.
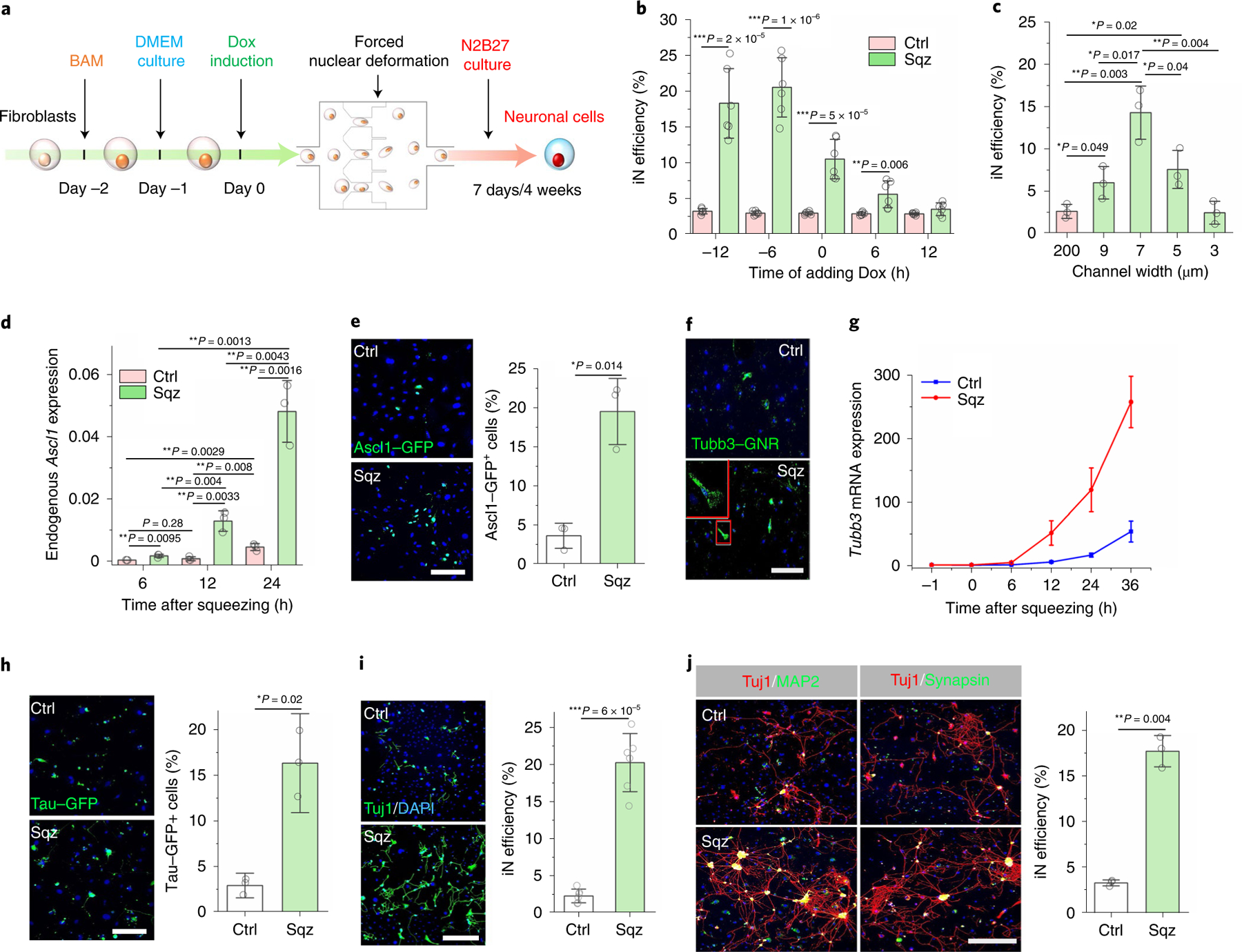
a, Experimental timeline for nuclear-deformation-induced iN reprogramming. b, Timing effect of Dox administration on microchannel-induced iN reprogramming. Dox was added to BAM-transduced fibroblasts at the indicated time points before or after mechanical squeezing, and the reprogramming efficiency was determined at day 7 (n = 6). c, Effect of microchannel width on iN reprogramming efficiency at day 7 based on Tuj1 staining (n = 3). d, Endogenous Ascl1 mRNA expression level in cells at the indicated time points after nuclear deformation by 7 μm microchannels or passing 200 μm channels (control, Ctrl) where Ascl1 expression was normalized to 18S rRNA (n = 3). e, Immunofluorescent images of fibroblasts transduced with BAM and an Ascl1 promoter–GFP construct that were either mechanically deformed or kept as a control at day 1. Scale bar, 200 μm. Quantification of Ascl1 promoter–GFP+ cells at day 1 (n = 3). Significance was determined by a two-tailed, unpaired t-test. f, Immunofluorescent images show Tubb3 mRNA in mechanically deformed and control cells at 12 hours after mechanical squeezing, as detected by using a Tubb3 mRNA GNR biosensor. Scale bar, 100 μm. g, Relative Tubb3 mRNA expression in mechanically deformed and control cells at the indicated time points after nuclear deformation (n = 3). h, Immunofluorescent images show Tau–GFP+ cells at day 7 in mechanically deformed and control cells, as quantified in the bar graph (n = 3). Scale bar, 200 μm. Significance was determined by a two-tailed, unpaired t-test. i, Reprogramming efficiency of BAM-transduced fibroblasts that were either deformed or kept as a control at day 7 (n = 6). Scale bar, 200 μm. Significance was determined by a two-tailed, unpaired t-test. j, Representative images of Tuj1+ cells expressing MAP2 and synapsin at four weeks after nuclear deformation. Scale bar, 200 μm. Reprogramming efficiency of BAM-transduced fibroblasts at four weeks after nuclear deformation as determined by synapsin+ staining (n = 3). Significance was determined by a two-tailed, unpaired t-test. In b–d, statistical significance was determined by a one-way ANOVA and Tukey’s multiple comparison test compared to control. Data in bar graphs represent mean ± s.d.
To determine whether nuclear deformation by microchannels facilitated the activation of endogenous neuronal genes, we monitored the effect of nuclear deformation on neuronal marker expression by quantitative polymerase chain reaction (qPCR) and live cell imaging. Among three transgenes, Ascl1 is a pioneer factor30,31 and was studied as an example. While endogenous Ascl1 expression in the control group showed a negligible basal level at 6 hours and a twofold increase at 12 hours, squeezing cells triggered 4.6-fold to 15.4-fold induction of endogenous Ascl1 expression at 6 and 12 hours, respectively, when compared with the control group (Fig. 2d). Furthermore, to directly monitor the temporal activation of endogenous Ascl1 protein, fibroblasts were transduced with a GFP driven by Ascl1 promoter (Ascl1-GFP) and subjected to the reprogramming process. Consistently, we found a significant increase in the number of Ascl1-GFP+ cells at day 1 in the squeezed cells compared to the control (Fig. 2e). On the other hand, the transgene expression of Ascl1 showed a slight increase (less than twofold) within 24 hours after squeezing (Supplementary Fig. 10a), and the overall gene expression of BRN2 and MYT1L had less than 2.5-fold increase (Supplementary Fig. 10b–c).
We also monitored the expression of other neuronal markers such as Tubb3. We first used gold nanorod (GNR) biosensors32 with a complementary sequence to detect mRNA expression of Tubb3 in living cells. Tubb3 mRNA expression was detectable as early as 12 hours in squeezed cells but rarely in the control group (Fig. 2f and Supplementary Fig. 11). Consistently, qPCR analysis showed significantly higher Tubb3 expression in the squeezed cells than the control group after 12 hours following nuclear deformation (Fig. 2g). In addition, we performed reprogramming experiments by using fibroblasts isolated from transgenic mice expressing an enhanced GFP (EGFP) reporter driven by the promoter of neuronal gene Tau. The number of Tau–GFP+ cells in the squeezed group at day 4 was approximately six times greater than that in the control group (Fig. 2h). We then used Tuj1 (for neuron-specific class III beta-tubulin, Tubb3) staining and neuron morphology analysis to determine reprogramming efficiency at various time points. At one week and two weeks after nuclear deformation, the iN reprogramming efficiency was significantly higher in the mechanically squeezed group compared with the control (Fig. 2i and Supplementary Fig. 12). Further characterization of the derived cells revealed that iN cells expressed the mature neuronal markers microtubule associated protein 2 (MAP2) and synapsin at four weeks after nuclear deformation (Fig. 2j). The terminally differentiated neurons from the control and squeezed groups had similar morphology (Supplementary Fig. 13). After six weeks, iN cells resulting from mechanical squeezing treatment showed calcium fluctuations, indicating a mature neuronal phenotype (Supplementary Video 1)33.
Nuclear deformation on chromatin changes
While nuclear deformation resulted in a modest transgene expression, which could also be achieved by increasing the titre of viral constructs, it could not fully account for the eightfold induction of reprogramming efficiency. Therefore, we investigated the potential involvement of epigenetic changes that controls the on/off state of phenotypic genes. To investigate whether nuclear-deformation-enhanced reprogramming efficiency was due to the changes in chromatin and epigenetic state, we first utilized a fluorescence resonance energy transfer (FRET) biosensor targeted at the nucleosome to monitor the levels of a heterochromatin mark histone H3 lysine 9 trimethylation (H3K9me3). We found that the H3K9me3 FRET signal significantly decreased in fibroblasts passing through 7 μm microchannels (Fig. 3a). To observe the temporal change in the same cells, we slowed the squeezing process by lowering the pressure and flow rate in the microdevice. Cells passing through 200 μm channels did not show significant change in H3K9me3 FRET signal (Supplementary Fig. 14), whereas 7 μm microchannels decreased H3K9me3 FRET signal within 1 minute (Supplementary Fig. 15).
Fig. 3 |. Nuclear deformation induced the demethylation of histone and DNA.
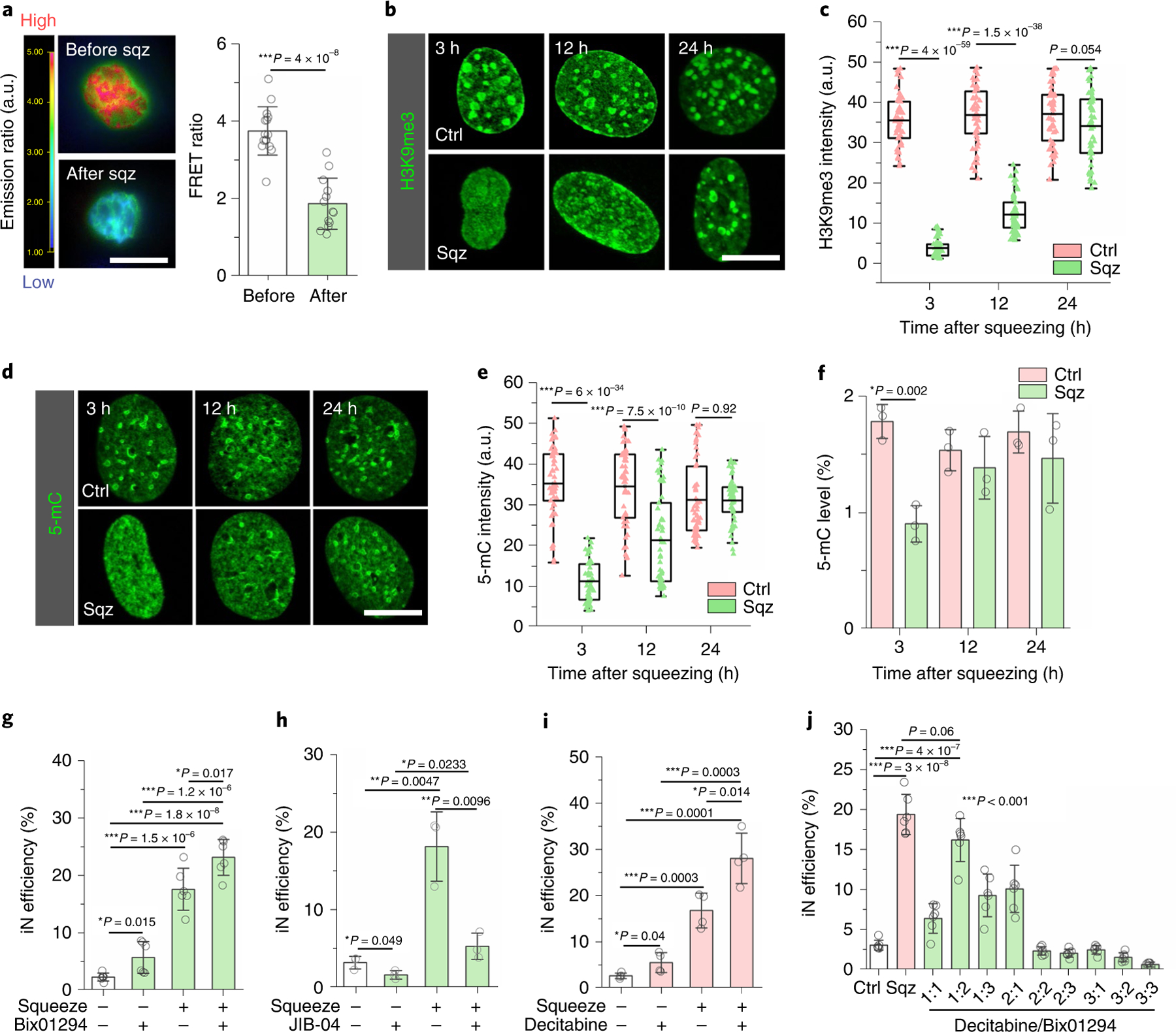
a, Images of fibroblasts transduced with an H3K9me3 FRET sensor before and after passing through 7 μm microchannel. Red colour indicates higher level of H3K9me3, and green indicates a lower FRET signal. Scale bar, 10 μm. Quantification of FRET ratio (n = 13). Significance was determined by a two-tailed, unpaired t-test. b, Immunostaining of H3K9me3 in the nucleus at the indicated time points after mechanical squeezing. Scale bar, 10 μm. c, Quantification of H3K9me3 intensity in control and squeezed cells at the indicated time points (based on experiments in b; n = 53). Significance was determined by a two-tailed, unpaired t-test for each time point. d, Representative images of 5-mC staining in control and squeezed cells at various time points. Scale bar, 10 μm. e, Quantification of 5-mC intensity in control and squeezed cells at the indicated time points (based on experiments in d; n = 53). Significance was determined by a two-tailed, unpaired t-test for each time point. f, Quantification of 5-mC level by MethylFlash Global DNA Methylation (5-mC) ELISA Easy Kit in control and squeezed cells at the indicated time points (n = 3). Significance was determined by a two-tailed, unpaired t-test for each time point. g, Reprogramming efficiency of BAM-transduced fibroblasts that were pretreated with 1 μM Bix01294 (or solvent control) for 24 hours followed by mechanical deformation (n = 6). h, Reprogramming efficiency of BAM-transduced fibroblasts that were pretreated with 100 nM JIB-04 (or solvent control) for 24 hours followed by mechanical deformation (n = 3). i, Reprogramming efficiency of BAM-transduced fibroblasts that were pretreated with 0.5 μM decitabine (or solvent control) followed by mechanical deformation (n = 4). j, Reprogramming efficiency of BAM-transduced fibroblasts that were squeezed by 7 μm microchannels, flowed through 200 μm channels (Ctrl) or treated with different ratios of decitabine to Bix01294:1:0.2 μM, 2:0.4 μM and 3:0.6 μM (n = 6). In a and f–j, data represent mean ± s.d. In c and e, box plots show the ends at the quartiles and the mean as a horizontal line in the box, and the whiskers represent the s.d. In g–j, significance was determined by a one-way ANOVA and Tukey’s multiple comparison test.
To determine whether the epigenetic changes persisted after squeezing and whether squeezing induced changes in other epigenetic marks, we performed immunostaining analysis of heterochromatin and euchromatin marks at multiple time points within the first 24 hours after cells passed through the microchannels. Consistently, we observed a significant decrease in H3K9me3 at 3 hours and 12 hours after nuclear deformation, which returned to the same level as the cells in the control group after 24 hours (Fig. 3b,c). Western blotting analysis also confirmed the global decrease of H3K9me3 induced by nuclear deformation (Supplementary Fig. 16). These results indicated that nuclear deformation resulted in a transient reduction of heterochromatin. On the other hand, the levels of acetylated histone marks including AcH3 and H3K9ac, and histone methylation marks including H3K4me1, H4K20me3 and H3K27me3, did not show significant global changes in response to forced nuclear deformation (Supplementary Fig. 17). Western blotting analysis further confirmed that heterochromatin mark H3K27me3 did not show significant changes after squeezing, although H3K27me3 was inhibited by its inhibitor EED226 (Supplementary Fig. 18).
In addition to histone modifications, DNA methylation influences chromatin organization, which is critical for cell reprogramming34. To investigate the effect of nuclear deformation on DNA methylation, we analysed DNA condensation and the level of 5-methylcytosine (5-mC), a DNA methylation marker, in fibroblasts squeezed by microchannels. As shown in Fig. 3d–f, both immunostaining analysis and enzyme-linked immunoassay (ELISA; Supplementary Fig. 19) showed that nuclear deformation significantly decreased DNA methylation for at least 12 hours. These results indicated that nuclear deformation caused chromatin modifications in both H3K9me3 and 5-mC.
To test whether the decrease in H3K9me3 played a role in iN conversion, BAM-transduced fibroblasts were treated with a H3K9-specific histone methyltransferase (HMT) inhibitor (Bix01294) for 24 hours. The inhibitor specifically suppressed H3K9me3 in a dose-dependent manner, and we selected a concentration of Bix01294 (1 μM) that did not affect cell viability (Supplementary Figs. 20–22). As shown in Fig. 3g, Bix01294 partially mimicked the squeezing effect on the reprogramming efficiency. Additionally, pretreatment with Bix01294, together with mechanical deformation, slightly increased the reprogramming efficiency compared to the squeezed-only group (Fig. 3g), suggesting that a 24-hour HMT inhibition by Bix01294 led to only a marginal enhancement. Furthermore, to determine whether the decrease of H3K9me3 was required for nuclear-deformation-induced iN reprogramming, BAM-transduced fibroblasts were pretreated with JIB-04 (100 nM), an H3K9-specific histone demethylase (HDMT) inhibitor, for 24 hours before being introduced into the microdevice. JIB-04, which significantly increased H3K9me3 (Supplementary Fig. 20), not only reduced the reprogramming efficiency compared to the control, but also strikingly suppressed nuclear-deformation-induced iN reprogramming (Fig. 3h). Additionally, in fibroblasts pretreated with JIB-04 for 24 hours and then introduced into the 7 μm channels, the decrease of H3K9me3 by squeezing was much less than the group without JIB-04 treatment (Supplementary Figs. 23 and 24). These results suggest that the decrease in H3K9me3 levels was required for the mechanical squeezing effect on iN reprogramming, which might be mediated by HDMTs. On the other hand, pretreatment with the DNA methyltransferase (DNMT) inhibitor decitabine (0.5 μM; Supplementary Figs. 20 and 25) slightly enhanced iN reprogramming efficiency, and the pretreatment with decitabine before squeezing further enhanced squeezing-induced iN reprogramming (Fig. 3i), demonstrating that DNA demethylation also played a role in iN reprogramming.
To investigate whether the combined effects of suppressing H3K9me3 and DNA methylation could match the reprogramming efficiency induced by mechanical squeezing, BAM-transduced fibroblasts were either subjected to transient nuclear deformation or treated with different combinations and concentrations of decitabine and Bix01294. As shown in Supplementary Figs. 26 and 27, Bix01294 or decitabine alone, at its optimal concentration, increased iN conversion only by about twofold. Interestingly, 0.2 μM decitabine combined with 0.4 μM Bix01294 (1:2) significantly increased the iN reprogramming efficiency in comparison to other combinations tested and showed similar efficiency when compared with the mechanically squeezed group (Fig. 3j), suggesting that the suppression of H3K9me3 and DNA methylation may be the major mediators of microchannel-induced iN reprogramming efficiency.
We further investigated whether ion channels were involved in squeezing-induced mechanotransduction leading to improved reprogramming efficiency. As shown in Supplementary Figs. 28–33, the inhibition of Na+, K+ and Ca2+ ion channels and the manipulation of extracellular pH during the squeezing process did not significantly affect the iN reprogramming efficiency.
Lamin-mediated epigenetic changes
The observations that mechanical squeezing forced nuclear deformation and a decrease in the elastic modulus of the cells suggested that the structural changes of the nucleus could mediate mechanotransduction through the nuclear lamina. Indeed, lamin A/C staining showed that nuclear deformation by 7 μm microchannels induced a transient increase in nuclear wrinkling that lasted for at least 12 hours (Fig. 4a,b), more than that in cells passing through 200 μm channels (Supplementary Fig. 34). In addition, mechanical squeezing by 7 μm microchannels caused a transient decrease in lamin A/C assembly at the nuclear periphery (Fig. 4c) and a slight shift of lamin A/C to nucleoplasm within 3 hours (Supplementary Fig. 35). This increase in nucleoplasmic lamin A/C after mechanical squeezing might be attributed, at least in part, to changes in lamin A/C phosphorylation35 as we observed a slight increase in phosphorylated lamin A/C at 30 minutes after mechanical squeezing (Supplementary Fig. 36). On the other hand, mechanical squeezing had no significant effect on the total protein levels of lamin A/C and lamin B1 (Supplementary Fig. 36). Interestingly, lamin B1 did not show a complete colocalization with lamin A/C following squeezing, especially within blebbing regions induced by 5 μm microchannels (Supplementary Fig. 37), and lamin B1 assembly at the nuclear periphery was not notably affected by mechanical squeezing using 7 μm microchannels as compared with lamin A/C (Supplementary Figs. 38 and 39). This finding is consistent with the reports that lamin A/C and lamin B1 form different structural layers36 and that lamin A/C but not lamin B1 regulates nuclear mechanics37.
Fig. 4 |. Lamin A/C-mediated microchannel-induced epigenetic changes and iN reprogramming.
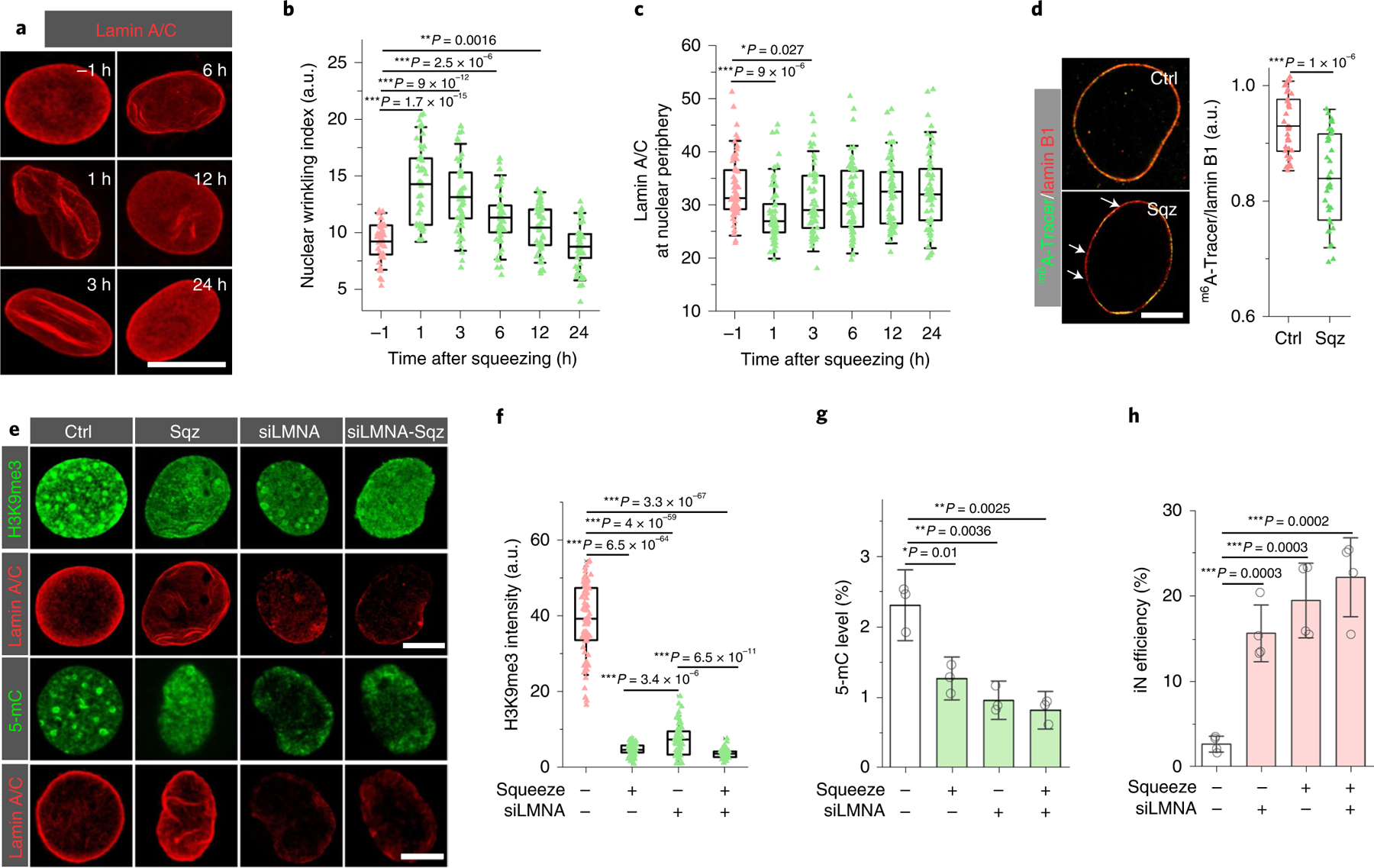
a, Representative images of lamin A/C staining in squeezed cells at the indicated time points after being squeezed by 7 μm microchannels. Scale bar, 10 μm. b, Quantification of nuclear wrinkling at the indicated time points before and after mechanical squeezing (n = 50). Nuclear wrinkling was defined as the immunofluorescence intensity of lamin A/C wrinkles within the nuclear region. Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. c, Quantification of lamin A/C at the nuclear periphery at the indicated time points before and after mechanical squeezing with 7 μm microchannels (n = 61). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. d, Immunofluorescent images of lamin B1 and m6A-Tracer in control and squeezed fibroblasts at 3 hours after mechanical deformation. Scale bar, 5 μm. Bar graph shows the ratio of the signal intensity of m6A-Tracer and lamin B1 in the control and squeezed cells at 3 hours (n = 34). Significance was determined by a two-tailed, unpaired t-test. e, Immunostaining of H3K9me3 and 5-mC in cells treated with random siRNA and siRNA for lamin A (siLMNA) at 3 hours after mechanical squeezing (siLMNA-Sqz). Scale bars, 5 μm. f, Quantification of H3K9me3 intensity in control and squeezed cells after lamin A knockdown (based on experiments in e; n = 69). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. g, Quantification of 5-mC level in control and lamin A-silenced fibroblasts at 3 hours after mechanical squeezing (n = 3). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. h, Reprogramming efficiency of control and lamin A-silenced BAM-transduced fibroblasts at day 7 after mechanical deformation (n = 4). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. In g and h, data represent mean ± s.d. In b, c, d and f, box plots show the ends at the quartiles and the mean as a horizontal line in the box, and the whiskers represent the s.d.
We further examined the changes of H3K9me3 and 5-mC in relation to lamin A/C following squeezing and showed that the decrease of lamin A/C at the nuclear periphery was accompanied by the decrease of H3K9me3 and 5-mC (Supplementary Fig. 40 and Figs. 3a–f and 4c). In addition, we examined the colocalization of the nuclear lamina and lamina-associated domains (LADs), which are enriched in heterochromatin (for example, H3K9me3) and anchor chromatins to the nuclear lamina38,39. We used an m6A-Tracer GFP reporter to label LADs40 and used lamin B1 to label nuclear lamina, as its distribution of lamin B1 at nuclear periphery did not change significantly following squeezing (Supplementary Figs. 38 and 39). As shown in Fig. 4d and Supplementary Fig. 41, after cells passed through 7 μm microchannels, part of the m6A-Tracer GFP signal was dislodged from the nuclear periphery, suggesting that a partial detachment between LADs and the nuclear lamina had occurred.
To investigate whether lamin disruption mediated nuclear-deformation-induced epigenetic changes during iN reprogramming, we silenced lamin A/C by using a small interfering RNA (siRNA) in BAM-transduced fibroblasts 24 hours prior to mechanical squeezing (Supplementary Figs. 42 and 43). As shown in Fig. 4e, lamin A/C knockdown mimicked the effects of mechanical squeezing, which caused a lamin disassembly at the nuclear periphery and a decrease in H3K9me3 and 5-mC (Fig. 4f,g). Moreover, lamin A/C knockdown enhanced the iN reprogramming efficiency of non-deformed cells to an extent similar to the cells that were mechanically squeezed, but lamin A/C was not silenced (Fig. 4h). The iN cells derived after lamin A/C knockdown expressed MAP2 and synapsin at four weeks after mechanical deformation (Supplementary Fig. 44). Taken together, these results suggested that lamin A/C played an important role in regulating the demethylation of histone and DNA induced by forced nuclear deformation.
Scaling up the mechanopriming process
To determine whether microfluidic-device-induced nuclear deformation could regulate the reprogramming of different cell types, we performed similar experiments by using macrophages transduced with BAM and fibroblasts transduced with OCT4, SOX2, KLF4 and c-MYC (OSKM). Interestingly, we found that both iN reprogramming from macrophages and iPSCs (induced pluripotent stem cells) reprogramming from fibroblasts were significantly enhanced after nuclear deformation (Fig. 5a,b), suggesting that mechanical reprogramming can be utilized as a general approach to prime the epigenetic state of cells to promote cell reprogramming.
Fig. 5 |. Development of mechanical reprogramming technologies by nuclear deformation.
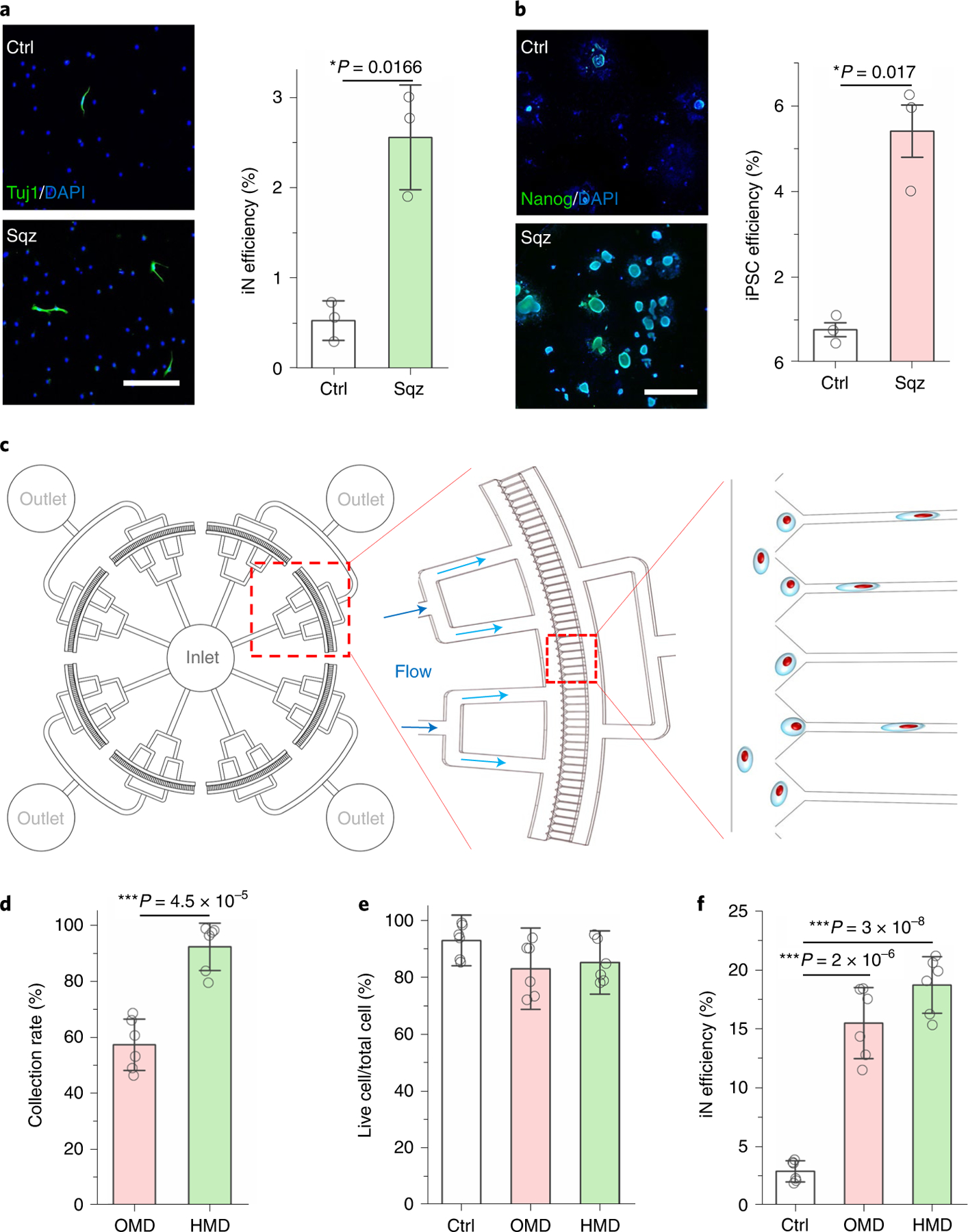
a, Immunofluorescent images show Tuj1+ iN cells generated from BAM-transduced mouse macrophages that were squeezed by 5 μm microchannels or flowed through 200 μm channels (Ctrl). Scale bar, 200 μm. Reprogramming efficiency of BAM-transduced macrophages after mechanical squeezing (n = 3). Statistical significance was determined by a two-tailed, unpaired t-test. b, Immunofluorescent images show Nanog+ colonies generated from OSKM-transduced mouse fibroblasts that were squeezed by 7 μm microchannels or flowed through 200 μm channels (Ctrl). Scale bar, 1 mm. Reprogramming efficiency of OSKM-transduced mouse fibroblasts into iPSCs after mechanical squeezing as determined by normalizing the number of Nanog+ colonies to the number of cells initially seeded (n = 3). Statistical significance was determined by a two-tailed, unpaired t-test. c, Schematic illustrating the design of the high-throughput device. d, Quantification of the percentage of cells that were collected after passing through OMD or HMD (n = 6). Statistical significance was determined by a two-tailed, unpaired t-test. e, The percentage of live cells after BAM-transduced fibroblasts were deformed using OMD or HMD, or flowed through 200 μm channels (Ctrl; n = 6). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. f, Reprogramming efficiency of BAM-transduced fibroblasts that were either kept as a control or deformed (7 μm microchannels) using OMD or HMD at day 7 (n = 6). Significance was determined by a one-way ANOVA and Tukey’s multiple comparison test. Data in bar graphs represent mean ± s.d.
To scale up the mechano-preconditioning of cells for reprogramming, we developed a higher-throughput microfluidic device (HMD) containing ten times more microchannels (400 microchannels) than the original microfluidic device (OMD) with 36 microchannels (Fig. 5c). The design was validated by fluid flow simulation, showing the velocity profile in different sizes of channels in the devices (Supplementary Fig. 45). HMD allowed the mechanical processing and collection of 10,000 cells within a minute, which was five times faster than OMD. Compared with OMD, this HMD significantly increased the yield of cell collection (Fig. 5d); HMD also maintained high cell viability and significantly improved iN efficiency (Fig. 5e,f).
Outlook
Our findings suggest that nuclear deformation has a mechanopriming effect to help overcome the epigenetic barrier during the reprogramming process9. Although the combination of chemical inhibitors for H3K9 and DNA methylation may reach the same iN efficiency as mechanical squeezing, the chemical inhibitors require hours of treatment and induce significant DNA damage, while mechanical squeezing causes little DNA damage and increases the cell proliferation that is critical for cell reprogramming (Supplementary Figs. 46 and 47), highlighting the advantage of cell squeezing as a mechanoceutical treatment. In addition, squeezing did not significantly affect the activities of HMTs and DNMTs as compared with the chemicals (Supplementary Fig. 48), suggesting that this mechanical squeezing modulates H3K9me3 and 5-mC through different mechanisms. Furthermore, although mechanical squeezing did not induce a global change in histone mark acetylated histone 3 (AcH3; Supplementary Fig. 17), the specific inhibition of histone deacetylase by valproic acid slightly increased iN reprogramming efficiency, which was additive to mechanical squeezing (Supplementary Figs. 49–51), suggesting that the enhancement of an open-chromatin state may further facilitate iN reprogramming. Additionally, we found that squeezing did not promote chemical-induced iN reprogramming (Supplementary Fig. 52) via small molecule compounds (forskolin, ISX-9, CHIR99021 and IBET-151)41, which may be explained by the fact that the chemical-induced reprogramming process requires a complex signalling network to activate neuronal genes and different kinetics and time period for epigenetic modulations.
Heterochromatin is often anchored to nuclear lamina through LADs that are abundant for heterochromatin marks such as H3K9me3 and are repressive for gene expression38,42,43. Mechanical force-induced nuclear deformation may partially induce nuclear lamina reorganization, wrinkling, lamin A/C disassembly at the nuclear periphery (Fig. 4c and Supplementary Fig. 35) and a partial detachment of heterochromatin from the nuclear lamina (Fig. 4d and Supplementary Fig. 41), which relocates heterochromatin towards the interior of the nucleus, accompanied by the downregulation of heterochromatin marks. Our data also suggest that transient biophysical modulation of the epigenetic state is universal and independent of cell type and reprogramming factors. This nuclear deformation in suspended cells is distinctly different from cell culture models because this approach decouples nuclear deformation from various cellular structural changes in adherent cells and the mechanical loading is transient and active. It also appears to be more effective than local magnetic twisting on the surface of adherent cells, which is insufficient to overcome the heterochromatin barrier44.
The simplicity of this mechanical approach by squeezing suspended cells provides direct evidence on the effect of nuclear deformation on chromatin remodelling. We also examined the roles of other major mediators of mechanotransduction45,46 and showed that yes1 Associated Transcriptional Regulator (YAP) and Piezo-type mechanosensitive ion channel component 1(Piezo-1) did not play a major role in mediating microchannel-induced epigenetic changes and iN reprogramming. YAP translocation into the nucleus upon cell adhesion was not affected by the squeezing process (Supplementary Fig. 53). In addition, the knockdown of Piezo-1 by siRNA had a negligible effect on various histone marks including H3K9me3, H3K27me3, AcH3, H3K27ac and 5-mC (Supplementary Figs. 54–56), but slightly decreased the microchannel-induced iN reprogramming efficiency (Supplementary Fig. 57), suggesting that Piezo-1 may contribute to iN reprogramming independent of epigenetic modulation.
Another highlight of this work is the translation of mechanobiology findings into cell engineering applications. Microfabricated devices provide a well-controlled microenvironment and real-time process control with a minimal benchtop space requirement47. Scalable microfluidic devices can be used to engineer a variety of cells such as fibroblasts, stem cells and immune cells, and to facilitate the conversion of cell types from one to another. In addition, this system can be used to investigate how mechanical deformation of cells in a three-dimensional matrix and microcirculation regulate epigenetic changes in tissue remodelling and diseases.
Methods
All experiments were performed in accordance with relevant guidelines and ethical regulations approved by the University of California, Los Angeles (UCLA), Institutional Biosafety Committee (BUA-2016-222).
Cell isolation, culture and reprogramming.
Mice utilized in these studies were housed under specific pathogen-free conditions and 12-hour-light/12-hour-dark cycles with a control of temperature (20–26 °C) and humidity (30–70%). All experiments, including breeding, maintenance and euthanasia of animals, were performed in accordance with relevant guidelines and ethical regulations approved by the UCLA Institutional Animal Care and Use Committee (protocol nos ARC-2016-036 and ARC-2016-101).
Fibroblasts were isolated from ear tissues of adult (1 month old, male and female) C57BL/6 mice (Jackson Laboratory, 002052), Tau-EGFP reporter mice (Jackson Laboratory, 004779) and R26-M2rtTA;Col1a1-tetO-H2B-GFP compound mutant mice (Jackson Laboratory, 016836), and expanded in fibroblast medium: DMEM medium (Gibco, 11965), 10% foetal bovine serum (FBS; Gibco, 26140079) and 1% penicillin/streptomycin (Gibco, 15140122). For all experiments, passage-2 cells were used and synchronized upon reaching 80% confluency using DMEM with 1% FBS for 24 hours before the transduction with viruses containing BAM constructs. The following day (day 0), the medium was changed to mouse embryonic fibroblast (MEF) medium containing Dox (2 ng ml−1, Sigma) to initiate the expression of the transgenes and thus, reprogramming. After 6 hours, transduced fibroblasts were passaged and subjected to microfluidic deformation using microchannels of various widths. Cells were then seeded onto glass slides coated with 0.1 mg ml−1 fibronectin (Thermo Fisher, 33016015) overnight at a density of 3,000 cells cm−2. Twenty-four hours later (day 1), cells were cultured in N2B27 medium: DMEM/F12 (Gibco, 11320033), N-2 supplement (Gibco, 17502048), B-27 supplement (Gibco, 17504044), 1% penicillin/streptomycin and Dox (2 ng ml−1, Sigma) and half medium changes were performed every two days. On day 7 after microfluidic deformation, cells were fixed and stained for Tuj1 to determine the reprogramming efficiency. The iN cells were identified based on positive Tuj1 staining and a neuronal morphology. The reprogramming efficiency was determined as the percentage of iN cells on day 7 relative to the number of the cells initially seeded. For long-term studies where maturation and functionality of the iN cells were examined, cells were kept in culture for five weeks. Reprogramming of iPSC from wild-type fibroblasts was performed as described previously18.
Macrophages for reprogramming experiments were derived from differentiated monocytes. Monocytes were isolated from the bone marrow of adult C57BL/6 mice and expanded in monocyte medium: RPMI 1640 (Gibco, 11875093), 10% FBS (Gibco, 26140079) and 1% penicillin/streptomycin (Gibco, 15140122). The next day, macrophage-colony stimulating factor (M-CSF; 50 ng ml−1, Thermo Fisher, PMC2044) was added to the medium and cells were cultured for an additional two days. Cells were then washed three times with phosphate buffered saline (PBS) before transduction with viruses containing BAM constructs.
Microfabrication of the microfluidic device.
The moulds of designed microfluidic devices for cell squeezing were fabricated via photolithography. A 15-μm-thick layer of SU-8 2015 (Microchem Corporation, 3,300 r.p.m.) was spun-coated onto a four-inch silicon wafer, followed by a standard photolithography process according to the manufacturer’s instruction. A base and curing agent of polydimethylsiloxane (PDMS; Sylgard 184, Dow Corning) was mixed in a 10:1 weight ratio and degassed in a vacuum chamber for one hour to remove air bubbles before being poured onto the mould. After curing at 65 °C for 4 hours, the PDMS mould was punched to make inlets and outlets for tubing connections. The PDMS mould and precleaned glass were bonded after treatment with oxygen plasma for 30 seconds. The bonded chips were baked at 65 °C for 10 minutes to enhance the bonding.
Lentiviral preparation and transduction.
Dox-inducible lentiviral vectors for Tet-O-FUW-Brn2, Tet-O-FUW-Ascl1, Tet-O-FUW-Myt1l and FUW-rtTA plasmids were used to transduce fibroblasts for ectopic expression of BRN2, ASCL1, MYT1L, GFP and reverse tetracycline transactivator (rtTA). The STEMCCA lentiviral vector was used for the ectopic expression of OSKM18. The Ascl1-eGFP lentiviral vector (Genecopoeia, MPRM39894-LvPF02) was used to monitor the activation of the Ascl1 promoter. Lentivirus was produced by using established calcium phosphate transfection methods, and Lenti-X Concentrator (Clontech, 631232) was utilized to concentrate viral particles according to the manufacturer’s protocol. Stable virus was aliquoted and stored at −80 °C. Fibroblasts were plated and synchronized for 24 hours before viral transduction in the presence of polybrene (8 μg ml−1, Sigma, H9268). Cells were incubated with the virus for 24 hours before performing microfluidic deformation experiments.
Cell viability assays.
After cells passed through the microdevice, 10 × 103 fibroblasts were plated and allowed to attach for 3 hours in a 96-well plate. Live and dead assays were performed using the LIVE/DEAD Cell Imaging Kit (Invitrogen, R37601) according to the manufacturer’s protocol. Cells were incubated with an equal volume of ×2 working solution for 15 minutes at room temperature. Epifluorescence images were collected using a Zeiss Axio Observer Z1 inverted fluorescence microscope and analysed using ImageJ.
Cell viability was assayed using the PrestoBlue Cell Viability Reagent (Invitrogen, A13261) according to the manufacturer’s protocol. Cells were incubated with the PrestoBlue Reagent for 2 hours. Absorbance was measured by a plate reader (Infinite 200PRO) at excitation/emission = 560 nm/590 nm. Results were normalized to control (that is, cell passing through >200 μm channels) samples.
DNA damage assays.
After cells passed through the microdevice, 5 × 103 fibroblasts were plated and allowed to attach for 3 hours in a 96-well plate. DNA damage assays were performed using the HCS DNA Damage Kit (Invitrogen, H10292) according to the manufacturer’s protocol. Cells were fixed with 4% paraformaldehyde solution for 15 minutes at room temperature and permeabilized by 0.25% Triton X-100 in PBS for another 15 minutes at room temperature. Cells were washed three times with PBS and incubated in 1% bovine serum albumin (BSA) solution for 1 hour, followed by phospho-H2AX (pH2AX) antibody (1:1,000) for 1 hour at room temperature and then Alexa Fluor 555 goat anti-mouse IgG (H+L) secondary (1:5,000) with Hoechst 33342 (1:6,000) for another 1 hour at room temperature after removing the antibody. Epifluorescence images were collected using a Zeiss Axio Observer Z1 inverted fluorescence microscope and analysed using ImageJ. Results were normalized to control samples (that is, cell passing through channels of >200 μm), and cells treated with 200 nM lipopolysaccharide served as a positive control.
Immunofluorescence staining and microscopy.
Samples collected for immunofluorescence staining at the indicated time points were washed once with PBS and fixed in 4% paraformaldehyde for 15 minutes. Samples were washed three times with PBS for 5 minutes each and permeabilized using 0.5% Triton X-100 for 10 minutes. After three subsequent PBS washes, samples were blocked with 5% normal donkey serum (Jackson Immunoresearch, 017000121) in PBS for 1 hour. Samples were incubated with primary antibodies (Supplementary Table 1) in antibody dilution buffer (1% normal donkey serum + 0.1% Triton X-100 in PBS) for either 1 hour or overnight at 4 °C followed by three PBS washes and a 1 hour incubation with donkey anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher, A-21202); donkey anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 546 (Thermo Fisher, A-10036); donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher, A-21206); and/or donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 546 (Thermo Fisher, A-10040). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher, D3571) in PBS for 10 minutes. Epifluorescence images were collected using a Zeiss Axio Observer Z1 inverted fluorescence microscope and analysed using ImageJ. Confocal images were collected using a Leica SP8-STED/FLIM/FCS Confocal and analysed using ImageJ.
For DNA methylation staining, samples were fixed with ice-cold 70% ethanol for 5 minutes followed by three PBS washes. Samples were then treated with 1.5 M HCl for 30 minutes and washed thrice with PBS. The immunostaining procedure proceeded from the donkey serum blocking step as aforementioned.
Average lamin and histone marker intensities per nuclei were quantified using an ImageJ macro. Gaussian blur, thresholding, watershed and analyse particle functions were applied to the DAPI channel to create individual selections for each nucleus. This mask was applied to the corresponding stain image to measure the average fluorescence intensity within each nucleus.
Chemical treatment of cells.
To determine the role of H3K9 methylation in microfluidic-device-induced iN reprogramming, BAM-transduced fibroblasts were treated with the H3K9 methyltransferase inhibitor Bix01294 (Cayman Chemical, 13124) or demethylase inhibitor JIB-04 (Cayman Chemical, 15338) at the indicated concentrations for 24 hours prior to introduction into the microdevice. Parallel conditions with dimethyl sulfoxide (DMSO) serving as a control. The iN reprogramming efficiency was determined via Tuj1 staining seven days after squeezing.
To determine the involvement of ion channels in microfluidic-device-induced iN reprogramming, calcium channel blocker amlodipine (Cayman Chemical, 14838), potassium channel blocker quinine (Cayman Chemical, 23958) and sodium channel blocker procainamide (Cayman Chemical, 24359) were used to inhibit calcium, potassium and sodium ion channels, respectively. BAM-transduced fibroblasts were treated with small molecule blockers at the indicated concentrations for 12 hours prior to being introduced into the microfluidic device. Parallel conditions with DMSO served as a control. The iN reprogramming efficiency was determined via Tuj1 staining seven days after squeezing.
To determine the effect of pH on iN reprogramming induced by forced nuclear deformation, BAM-transduced fibroblasts were treated with DMEM medium at different pH levels (pH = 6.5, 7.5 and 8.5) for 1 hour prior to being introduced into the microdevice. The iN reprogramming efficiency was determined via Tuj1 staining at day 7 after squeezing.
DNA methylation assay.
After cells passed through the device, cells were collected, and 10 × 105 cells were plated in 60 mm dishes. At different time points, cells were trypsinized and DNA was extracted by Invitrogen PureLink Genomic DNA mini kit (Invitrogen, K1820-01). The 5-mC level was analysed by the MethylFlash Global DNA Methylation (5-mC) ELISA Easy Kit (Epigentek, P-1030) according to the manufacturer’s instructions. Briefly, 100 ng of sample DNA was bonded into the assay wells and incubated with a 5-mC detection complex solution for 60 minutes. Then colour developer solution was added into assay wells, and the absorbance at 450 nm was measured by using a plate reader (Infinite 200Pro, 30050303).
Reverse transcription and qPCR (RT-qPCR).
After cells passed through the device, cells were collected, and 10 × 105 cells were plated in 60 mm dishes. At different time points, TRIzol Reagent (Invitrogen, 15596026) was used to lyse cells, and RNA was isolated as described previously48. After RNA extraction, a ThermoScientific Maxima First Strand cDNA Synthesis Kit (Thermo Fisher, K1641) was used for first-strand cDNA synthesis. Then RT-qPCR was performed using a CFX96 Real-Time PCR Detection System to detect the gene expression levels of Ascl1 endogenous (forward primer, CAACCGGGTCAAGTTGGTCA; reverse primer, CTCATCTTCTTGTTGGCCGC), Ascl1 exogenous (forward primer, CTCGCC AACGACGTCAATG; reverse primer, CCCTGTAGGTTGGCTGTCTG), Brn2 (forward primer, AGGGCGCAAACGGAAAA; reverse primer, GGCTTAGGGCATTTGAGGAAA), Myt1l (forward primer, CTACAAGATGGACGTGGACTCTGA; reverse primer, GGAACTCGAACCCCTTTGG) and Tubb3 (forward primer, GCGCCTTTGGACACCTATTC; reverse primer, CACCACTCTGACCAAAGATAAAGTTGT), where 18S (forward primer, GCCGCTAGAGGTGAAATTCTTG; reverse primer, CATTCTTGGCAAATGCTTTCG) level was used for normalization.
Lamin A siRNA knockdown and m6A-Tracer transfection.
For lamin A siRNA knockdown, 1 × 106 cells were plated in 60 mm dishes for 24 hours. RNA interference was performed using ON-TARGETplus siLMNA (Dharmacon, L-040758-00-0005), and transfections were carried out using Lipofectamine Stem Transfection Reagent (Thermo Fisher, STEM00008) according to the manufacturer’s protocol. Briefly, 250 μl Opti-MEM medium (Thermo Fisher, 31985062) was mixed with 7.5 μl Lipofectamine stem reagent and incubated at 37 °C for 15 minutes. At the same time, 5 μg siRNA was diluted in 250 μl Opti-MEM medium and incubated at 37 °C for 15 minutes. These two solutions were mixed and the DNA–lipid complexes to cells were added to 1.5 ml DMEM culture medium and incubated at 37 °C for 48 hours. The media were then replaced with DMEM medium with 10% FBS and 1% penicillin/streptomycin. One day after transfection, RNA was isolated and RT-qPCR was performed to detect LMNA (forward primer, GTCTCGAATCCGCATTGACA; reverse primer, TGGCTGCCAACTGCTTTTG) mRNA expression to determine whether lamin A had been silenced.
For m6A-Tracer transfection, 1 × 106 cells were plated in 60 mm dishes for 24 hours followed by transfection with m6A-Tracer GFP (AddGene, 139403) and DAM-lamin B1 (AddGene, 119764) using Lipofectamine 3000 reagent (Thermo Fisher, L3000015) according to the manufacturer’s protocol. Briefly, 250 μl Opti-MEM medium (Thermo Fisher, 31985062) was mixed with 7.5 μl Lipofectamine 3000 reagent and incubated at 37 °C for 5 minutes. At the same time, 5 μg DNA was diluted in 250 μl Opti-MEM medium and incubated at 37 °C for 15 minutes. These two solutions were mixed and the DNA–lipid complexes to cells were added to 1.5 ml DMEM medium without FBS and penicillin/streptomycin and incubated at 37 °C for 24 hours. The media were then replaced with DMEM medium with 10% FBS and 1% penicillin/streptomycin. Three days after transfection, m6A-Tracer GFP-labelled fibroblasts were subjected to microfluidic deformation experiments.
FRET biosensor.
Lentiviruses of H3K9me3 were produced from Lenti-X 293T cells (Clontech Laboratories, 632180) cotransfected with a pSin containing biosensor and the viral packaging plasmids pCMV-△8.9 and pCMV-VSVG using the ProFection Mammalian Transfection System (Promega, catalog no. E1200). Viral supernatant was collected 48 hours after transfection, filtered with 0.45 μm filter (Sigma-Millipore). Primary fibroblasts were transduced with the virus, and the cells expressing the biosensor were sorted using flow cytometry (Sony, SH800). Images of the FRET experiment were taken with a Nikon Eclipse Ti inverted microscope equipped with a cooled charge-coupled device (CCD) camera, a 420DF20 excitation filter, a 450DRLP dichroic mirror and two emission filters controlled by a filter changer (480DF30 for ECFP and 535DF35 for FRET) as described previously49,50. The images were acquired, and the enhanced cyan fluorescent protein (ECFP)/FRET ratio was calculated and visualized by MetaFluor 7.8 (Molecular Devices).
GNR locked nucleic acid (LNA) probe for mRNA.
To detect Tubb3 mRNA expression in living cells after cells were passed through the device, cells were collected and plated in a 24-well plate at 2,000 cells per well, and GNR–LNA complexes specific to Tubb3 was added to culture media as described previously32. Briefly, GNR–LNA complexes were made by mixing 1.5 μl LNA probe (10 μM), 2.5 μl GNR and 46 μl Tris-EDTA buffer, and incubating at 37 °C for 15 minutes. The GNR–LNA complex solution (50 μl) and fresh culture medium (450 μl) were then mixed and added to the cells. After a four-hour incubation, cells were washed with PBS, and fresh culture medium was added. Cells were incubated at 37 °C in the dark for an additional 60 minutes prior to performing live cell imaging. Epifluorescence images were collected using a Zeiss Axio Observer Z1 inverted fluorescence microscope.
AFM measurement of cell mechanical property.
To determine the elastic modulus of cells after passing through the device, mechanical measurements of single cells were performed using AFM (JPK Nanowizard 4a) with tipless cantilevers (NPO-10, Bruker), a highly sensitive cantilever with nominal spring constants k = 0.06 N m−1 and a sample Poisson’s ratio of 0.499, found at the UCLA Nano & Pico Characterization facility. During the measurement, cells were cultured on a glass-bottom dish with prewarmed PBS and set on a temperature-controlled stage at 37 °C. The measurement was performed by indenting the plasma membrane of the cells above the nucleus. The force–distance curves were recorded and the elastic modulus of cells was calculated by NanoScope Analysis using the Hertz model.
Western blotting.
Equal amounts of total protein (50 μg) from each sample were separated in a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride membrane at 120 V for 2 hours at room temperature. The blot was blocked with 5% nonfat dry milk suspended in ×1 TBS (25 mM Tris, 137 mM NaCl and 2.7 mM KCl) for 1 hour. Membranes were incubated sequentially with primary antibodies (Supplementary Table 1) and secondary antibodies (anti-mouse IgG horseradish peroxidase (HRP)-linked antibody (Cell Signaling, 7076S) and anti-mouse IgG HRP-linked antibody (Cell Signaling, 7074S)). Bands were scanned using a densitometer (Bio-Rad) and quantified using the Quantity One v.4.6.3 software (Bio-Rad).
Statistics and reproducibility.
All data are presented as mean ± 1 s.d., where sample size (n) is ≥3. Comparisons among values for groups greater than two were performed by using a one-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test. For two group analysis, a two-tailed, unpaired Student’s t-test was used to analyse differences. For all cases, P values less than 0.05 were considered statistically significant. Origin 2018 software was used for all statistical evaluations. At least three independent experiments were performed to confirm findings.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank M. Wernig at Stanford University for providing the constructs of BAM for reprogramming experiments and C.M. Ho for his suggestions on the design of microdevices. We were supported in part by a UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Innovation Award and research grants from the National Institutes of Health (HL121450 and GM143485 to S.L. and GM140106 to S.K.K.) and the National Science Foundation (BMMB-1906165 to A.C.R.). We acknowledge the use of instruments at the Nano & Pico Characterization Lab and Advanced Light Microscopy and Spectroscopy Lab at the California NanoSystems Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41563-022-01312-3.
Code availability
Codes utilized for image analysis are available on the lab website (https://li-lab.seas.ucla.edu/requestform/).
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41563-022-01312-3.
Peer review information Nature Materials thanks Quasar Padiath and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper, source data and Supplementary Information files. Additional images or videos are available from the corresponding author upon request. Source data are provided with this paper.
References
- 1.Aydin B & Mazzoni EO Cell reprogramming: the many roads to success. Annu. Rev. Cell Dev. Biol 35, 433–452 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Karagiannis P et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev 99, 79–114 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Yang Y, Liu J & Qian L Direct cell reprogramming: approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol 22, 410–424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang ZP et al. Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ieda M et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagliuca FW et al. Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabo E et al. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468, 521–526 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Huang P et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 14, 370–384 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Vierbuchen T et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu W et al. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K & Yamanaka S A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol 17, 183–193 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Soto J, Chen B, Yang L & Li S Cell engineering: biophysical regulation of the nucleus. Biomaterials 234, 119743 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Uhler C & Shivashankar GV Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol 18, 717–727 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Li Y et al. Biophysical regulation of histone acetylation in mesenchymal stem cells. Biophys. J 100, 1902–1909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Y, Soto J & Li S Mechanical regulation of histone modifications and cell plasticity. Curr. Opin. Solid State Mater. Sci 24, 100872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Discher DE et al. Matrix mechanosensing: from scaling concepts in ’omics data to mechanisms in the nucleus, regeneration, and cancer. Annu. Rev. Biophys 46, 295–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miroshnikova YA, Nava MM & Wickström SA Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci 130, 2243–2250 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Downing TL et al. Biophysical regulation of epigenetic state and cell reprogramming. Nat. Mater 12, 1154–1162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang P et al. WDR5 modulates cell motility and morphology and controls nuclear changes induced by a 3D environment. Proc. Natl Acad. Sci. USA 115, 8581–8586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Y et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun 5, 4619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nava MM et al. Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damodaran K et al. Compressive force induces reversible chromatin condensation and cell geometry–dependent transcriptional response. Mol. Biol. Cell 29, 3039–3051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding X et al. High-throughput nuclear delivery and rapid expression of DNA via mechanical and electrical cell-membrane disruption. Nat. Biomed. Eng 1, 0039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denais CM et al. Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul CD, Mistriotis P & Konstantopoulos K Cancer cell motility: lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gossett DR et al. Hydrodynamic stretching of single cells for large population mechanical phenotyping. Proc. Natl Acad. Sci. USA 109, 7630–7635 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naim B et al. Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J. Biol. Chem 282, 3881–3888 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Raab M et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Irianto J et al. DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr. Biol 27, 210–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wapinski OL et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wapinski OL et al. Rapid chromatin switch in the direct reprogramming of fibroblasts to neurons. Cell Rep 20, 3236–3247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S, Riahi R, Li N, Zhang DD & Wong PK Single cell nanobiosensors for dynamic gene expression profiling in native tissue microenvironments. Adv. Mater 27, 6034–6038 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Song Y et al. Asymmetric cell division of fibroblasts is an early deterministic step to generate elite cells during cell reprogramming. Adv. Sci 8, 2003516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Carvalho DD, You JS & Jones PA DNA methylation and cellular reprogramming. Trends Cell Biol 20, 609–617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho S et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell 49, 920–935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nmezi B et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl Acad. Sci. USA 116, 4307–4315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lammerding J et al. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem 281, 25768–25780 (2006). [DOI] [PubMed] [Google Scholar]
- 38.van Steensel B & Belmont AS Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collas P, Ali TML, Brunet A & Germier T Finding friends in the crowd: three-dimensional cliques of topological genomic domains. Front. Genet 10, 602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kind J et al. Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Li X et al. Small-molecule-driven direct reprogramming of mouse fibroblasts into functional neurons. Cell Stem Cell 17, 195–203 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Guelen L et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Becker JS, Nicetto D & Zaret KS H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet 32, 29–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Chen J, Mohagheghian E & Wang N Force-induced gene up-regulation does not follow the weak power law but depends on H3K9 demethylation. Sci. Adv 6, eaay9095 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elosegui-Artola A et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Monroe TO et al. YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev. Cell 48, 765–779 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gill NK et al. A scalable filtration method for high throughput screening based on cell deformability. Lab Chip 19, 343–357 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Sia J, Yu P, Srivastava D & Li S Effect of biophysical cues on reprogramming to cardiomyocytes. Biomaterials 103, 1–11 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Peng Q et al. Coordinated histone modifications and chromatin reorganization in a single cell revealed by FRET biosensors. Proc. Natl Acad. Sci. USA 115, E11681–E11690 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L et al. Integration of FRET and sequencing to engineer kinase biosensors from mammalian cell libraries. Nat. Commun 12, 5031 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper, source data and Supplementary Information files. Additional images or videos are available from the corresponding author upon request. Source data are provided with this paper.