

Abstract
The persistence of the global COVID-19 pandemic caused by the SARS-CoV-2 virus has continued to emphasize the need for point-of-care (POC) diagnostic tests for viral diagnosis. The most widely used tests, lateral flow assays used in rapid antigen tests, and reverse-transcriptase real-time polymerase chain reaction (RT-PCR), have been instrumental in mitigating the impact of new waves of the pandemic, but fail to provide both sensitive and rapid readout to patients. Here, we present a portable lens-free imaging system coupled with a particle agglutination assay as a novel biosensor for SARS-CoV-2. This sensor images and quantifies individual microbeads undergoing agglutination through a combination of computational imaging and deep learning as a way to detect levels of SARS-CoV-2 in a complex sample. SARS-CoV-2 pseudovirus in solution is incubated with acetyl cholinesterase 2 (ACE2)-functionalized microbeads then loaded into an inexpensive imaging chip. The sample is imaged in a portable in-line lens-free holographic microscope and an image is reconstructed from a pixel superresolved hologram. Images are analyzed by a deep-learning algorithm that distinguishes microbead agglutination from cell debris and viral particle aggregates, and agglutination is quantified based on the network output. We propose an assay procedure using two images which results in the accurate determination of viral concentrations greater than the limit of detection (LOD) of 1.27·103 copies·mL−1, with a tested dynamic range of 3 orders of magnitude, without yet reaching the upper limit. This biosensor can be used for fast SARS-CoV-2 diagnosis in low-resource POC settings and has the potential to mitigate the spread of future waves of the pandemic.
Graphical Abstract
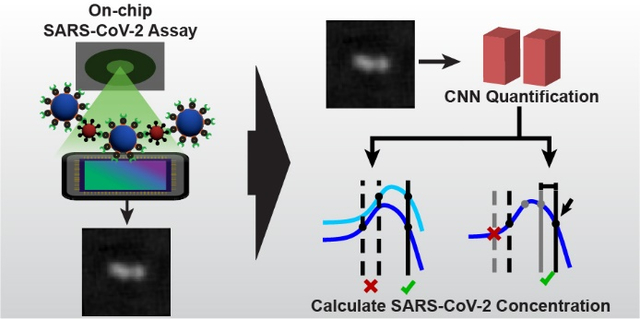
1. Introduction
As the pandemic caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) persists and the virus continues to mutate to evade the human immune response, there continues to be a need for powerful point-of-care (POC) tests to diagnose infection and limit the impact of new viral mutations.1 Currently, the main biosensors used in clinical settings are lateral flow assays (LFAs), used in rapid antigen tests, and the gold standard technique of reverse-transcriptase real-time polymerase chain reaction (RT-PCR) amplification, used to detect viral genetic material.2–6 LFAs for SARS-CoV-2 can be implemented in POC settings or as take-home tests and give a readout of results within minutes. However, their drawback is a relatively high limit of detection (LOD), as there needs to be a lot of viral antigen present to receive a positive test. Widely used LFAs for SARS-CoV-2 have a LOD of 3·106 copies·mL−1, which contributes to a large proportion of tests returning false negatives and can contribute to the spread of SARS-CoV-2 as infected individuals assume they are not contagious and fail to limit exposing others accordingly.5–7 RT-PCR by contrast has a very low LOD, from 560 copies·mL−1 to 1,065 copies·mL−1 depending on the individual test.8 This enables it to diagnose SARS-CoV-2 infection even before patients become symptomatic. However, this sensitivity comes at a cost of a slow turnaround time. RT-PCR tests typically take days to come back with results, and it requires specialized equipment, training, and personnel to perform.9,10 A turnaround time of days can result in patients spreading the virus before receiving results.11,12
To address these issues, several groups have been experimenting with alternative POC biosensors. One promising approach is an agglutination assay.13–18 It is performed by coating latex or polystyrene microspheres with a functional capture molecule, typically an antibody, and mixing these microbeads with the test sample. In the presence of the target biomolecule or pathogen, beads will bind together, aggregating and resulting in bead precipitation from suspension. In conventional agglutination assays, agglutination is seen qualitatively, not quantitatively, and typically requires the target to be cultured or amplified in some way to get enough agglutination to be visible. Agglutination assays are commonly used in food safety applications, as well as in the diagnosis of infectious diseases, and, barring the need for complex analyte amplification techniques, can be used in POC settings.19,20
Recently, lens-free holographic microscopy (LFHM) has been combined with agglutination assays that make these tests more sensitive, quantitative, and easier to perform in POC settings than conventional qualitative agglutination assays.21–23 In these in-line LFHM systems, a coherent light source is used to generate an interference pattern from a sample placed between the source and the sensor and an image of the sample is computationally reconstructed based on the interference pattern.24,25 This enables LFHM to maintain a wide field of view (FOV), essentially the size of the image sensor itself, while achieving a high resolution.26,27 To achieve the sub-micron resolution necessary to resolve microbeads for agglutination assays, pixel superresolution LFHM designs and corresponding algorithms have been utilized.28–30 This resolution is necessary to detect subtle changes in agglutination of microbeads, and has been used to achieve nanogram per milliliter LODs from these devices.21
Here, we show a portable LFHM-agglutniation assay sensor based on our previous benchtop quantitative large-area binding (QLAB) assay.21 Furthermore, our new sensor has been optimized for SARS-CoV-2 pseudovirus sensing and is coupled with a deep-learning algorithm that can distinguish beads in the sample from cell debris and viral particle aggregates in order to aid in computational speed and accuracy of agglutination quantification. This sensor can resolve and track 2 μm diameter latex microspheres undergoing Brownian motion in solution to detect subtle agglutination changes in a sample of over 10,000 beads. Compared to similar biosensors,23 the one we have developed here utilizes a unique quantification method for individually resolved beads in clusters, is robust in handling samples polluted with unpredictable debris, exhibits a large dynamic range, and is accurate in quantifying analyte concentration. The biosensor is able to provide a POC readout of SARS-CoV-2 pseudovirus concentration within 3 hrs of sample collection and has a LOD within an order of magnitude of RT-PCR tests.
2. Materials and Methods
2.1. SARS-CoV-2 Pseudovirus
The pseudovirus used for these experiments is a pseudotype HIV-1-derived lentiviral particle bearing SARS-CoV-2 spike protein. The particle has a lentiviral backbone and expresses luciferase as a reporter. Viral particles were produced in HEK293T cells engineered to express ACE2, the SARS-CoV-2 receptor, as previously described.31–33 Cells were lysed using the Bright-Glo Luciferase Assay System (Cat: E2610, Promega, Madison, WI, USA). Lysate was transfered to 96-well Costar flat-bottom luminometer plates where relative luciferase units (RLUs) were detected using Cytation 5 Cell Imaging Multi-Mode Reader (BioTek, Winooski, VT, USA). Luciferase luminescence scales linearly with the concentration of pseudovirus copies in a given sample, and enabled calculation of pseudoviral copies·mL−1.32 For these experiments, the initial pseudoviral concentration was determined to be 3·106 copies·mL−1.
Vesicular stomatitis virus G (VSV-G), a lentivirus similar to the SARS-CoV-2 pseudovirus, but which does not bear the ACE2 binding spike, was used as a control to confirm assay specificity. VSV-G concentrations were not separately quantified using luminescence, but the virus was produced similarly to the SARS-CoV-2 pseudovirus, and a wide range of dilution concentrations were tested to confirm a lack of agglutination.
2.2. Portable Lens-free Holographic Microscope
The LFHM system is based on an in-line imaging method that has been previously described and termed the Quantitative Large-Area Binding (QLAB) sensor,21 but now fully contained in a light-weight portable housing weighing less than 800 g (Fig. 1). Briefly, the light source of the system consists of a 15 green light-emitting diode (LED) array positioned 15 cm above the sample. LEDs illuminate one at a time for 120 ms each, with a delay of 15 ms between each LED, resulting in 15 sub-pixel shifted images captured over 2.025 seconds. LFHM spatial coherence is provided by 180 μm diameter hole punches placed just below each LED, and temporal coherence is provided by a bandpass filter with central wavelength 532 nm and bandwidth 3 nm. At the base of the biosensor is a complementary metal-oxide-semiconductor (CMOS) monochromatic image sensor (ON Semiconductor AR1335) with a pixel width of 1.1 μm.
Fig. 1.
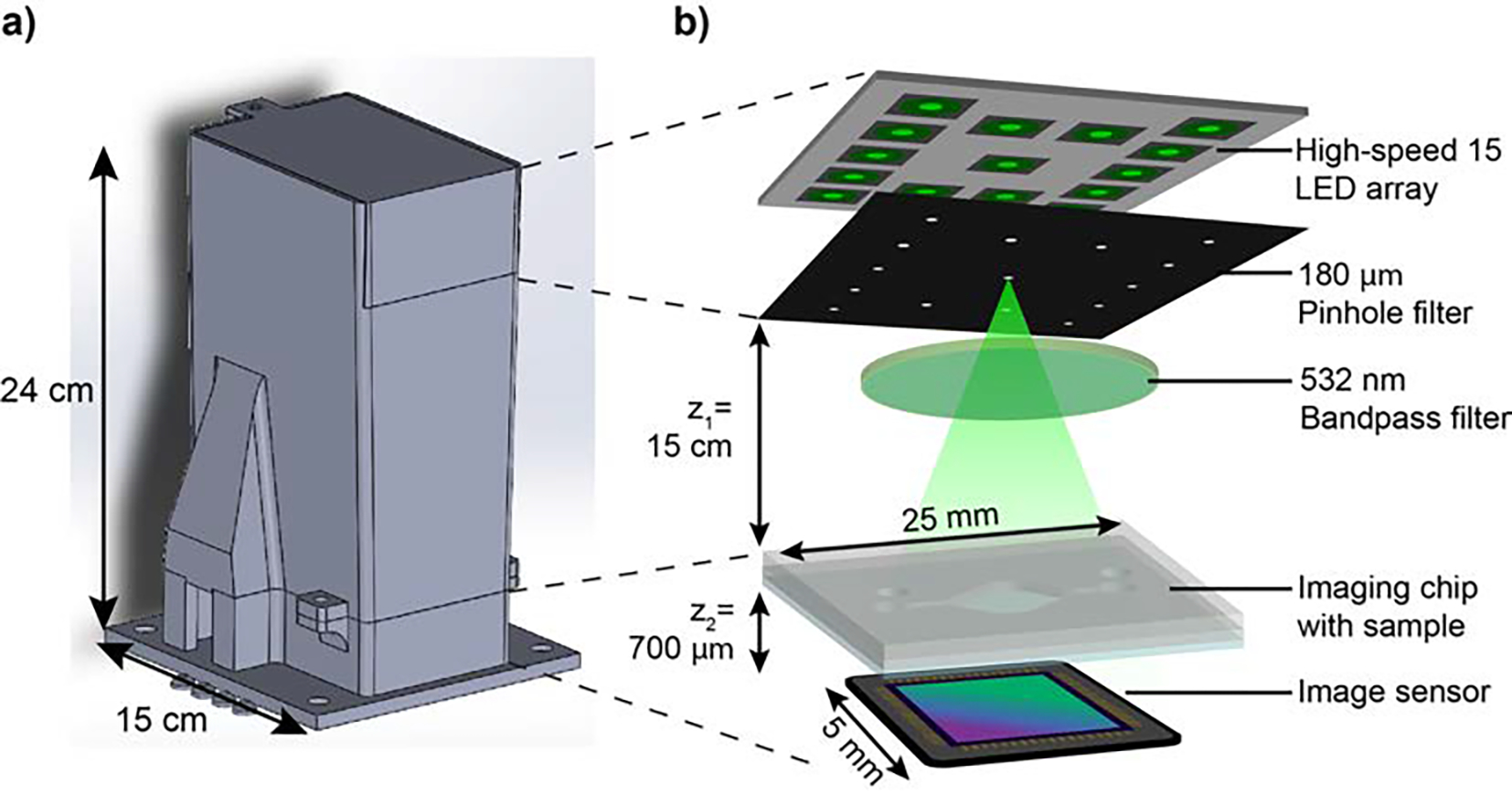
Portable QLAB Sensor. a) CAD design of all components of the portable sensor housing. The housing was 3D printed from black PLA. b) Functional components of the LFHM inside the sensor.
The liquid sample is loaded into a large-area (65 mm2) microfludic chamber, or imaging chip, constructed out of 2 layers of clear, laser-cut polycarbonate and a single No.1 glass coverslip constructed in advance of performing the assay (Figure S1†). The center layer forms the boundary and thickness of the open chamber and is cut from a 125 μm polycarbonate sheet. Inlet and outlet ports are cut from a 250 μm thick upper polycarbonate sheet. The coverslip serves as the bottom of the chip and is placed closest to the image sensor. To ensure optical clarity of the coverslip and to remove dust and other particles, coverslips were treated using a piranha solution. For this procedure, 30% H2O2 was mixed with sulfuric acid (H2SO4) in a 1:3 ratio, then coverslips were placed into this piranha solution for 1 hr. Treated coverslips were washed with Milli-Q ultrapure water before being dried and assembled into the finished imaging chip. All layers were adhered to each other using UV-curable adhesive (Norland Products 7230B).
For POC use, a custom housing was designed and then printed in a FlashForge Creator 3 3D printer with black polylactic acid (PLA) (Fig. 1a). The housing was designed to optimize deployment in a portable setting by blocking all ambient light from the image sensor for maximum optical signal-to-noise ratio. The entire top portion of the housing is hinged, allowing the device to be opened for easy placement of the microfluidic chip over the image sensor, and then closed again for imaging. The footprint of the device is only 15 × 15 cm, and images were captured using this setup paired with a laptop computer outside of the environment in which the LFHM was initially tested and constructed. Figure S2† shows images of the fully assembled LFHM components inside the housing. The total cost of this prototype device is $1,382, with the majority of the cost allocated to a development board attached to the image sensor. Future iterations of this device would not include this board, reducing the cost to $517. The cost could further be reduced to $267 by using a different image sensor, such as a Sony IMX519. The imaging chips can be fabricated for as little as $0.11 each.
2.3. Particle preparation and agglutination assay
The protocol for performing the agglutination assay is depicted in Figure 2. Polystyrene microspheres 2 μm in diameter and conjugated with streptavidin (Nanocs PS2u-SV-1) were diluted to a concentration of 0.005% or 0.01% weight/volume (w/v) in 1× phosphate-buffered saline (PBS). Solid biotinylated acetylcholinesterase 2 (ACE2) (Sino Biological Inc. 10108-H08H-B) was added to the microbead suspension to a final concentration of 5.0 or 10.0 μg·mL−1 of ACE2. This concentration corresponds to approximately 50,000 molecules of ACE2 per microsphere. Microspheres and biotinylated ACE2 were incubated for 2 hrs at 25°C on a shaker at 1200 rpm. Functionalized beads were stored at 4°C before use.
Fig. 2.
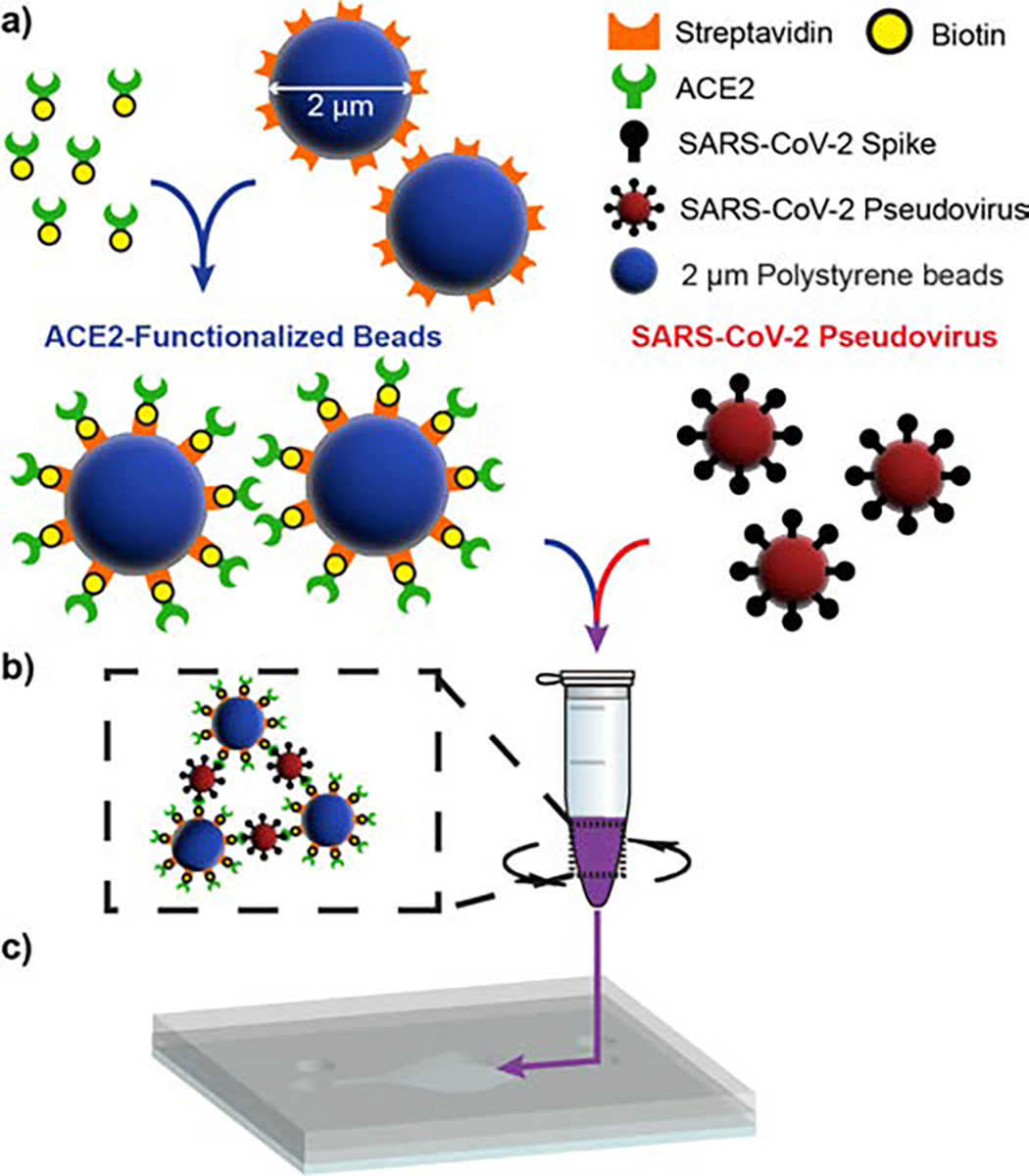
Agglutination assay procedure. a) 2 μm polystyrene beads conjugated with streptavidin are incubated with biotinylated ACE2, yielding ACE2-functionalized microbeads. b) Functionalized beads are incubated with SARS-CoV-2 pseudovirus within a 1.5 mL test tube on a shaker at 1200 rpm, resulting in microbead agglutination. c) The completed assay is loaded into the imaging chip via micropipette direct injection.
Extracted pseudovirus or VSV-G was filtered using a syringe filter with a 0.22 μm pore size to remove larger cell debris from the sample. The filtered virus was diluted in Dulbecco’s Modified Eagle Medium (DMEM) in half-log dilutions ranging from 3·106 copies·mL−1 to 3·102 copies·mL−1, with an extra negative control of pure DMEM. Functionalized microbeads were mixed 1:1 with each pseudovirus dilution for triplicate samples per dilution, creating 30 samples with a microbead concentration of 0.0025% or 0.005% and pseudovirus concentration ranging from 1.5·106 copies·mL−1 to 1.5·102 copies·mL−1, including the 3 negative controls. In this procedure, only 40 μL of viral sample is required per test. A single microbead sample was reserved in pure PBS for comparison. Samples were incubated for 2 hrs at 25°C on a shaker at 1200 rpm and then 25 μL were micropipetted into the imaging chip. The chamber was sealed using UV-curable adhesive, allowed to sediment for 15 minutes to ensure particles were at the bottom plane of the chip, and placed inside our portable LFHM for on-chip imaging of the completed agglutination reaction. Sealing the chip is only necessary for preventing evaporation when storing the chip to make repeated measurements at later times.
2.4. Image Processing and Analysis
To process the low-resolution (LR), sub-pixel shifted holograms captured of the SARS-CoV-2 agglutination assay, the following workflow was employed. LR holograms are first divided into 5 × 7 partially overlapping patches. A PSR technique that has been optimized for small targets was used to synthesize a high resolution (HR) hologram from the LR holograms for each patch in parallel.28 HR hologram patches are then back-propagated to the sample plane. Cardinal-neighbor regularization (weight = 200) and twin-image noise suppression were used to improve the signal-to-noise ratio of the back-propagated HR reconstructions. Then, the reconstructed HR patches are stitched back together to create a single image of the full FOV that is used for assay analysis. This image processing is performed using the University of Arizona’s high-performance computing clusters, which are accessed remotely on a portable takes on average 20 minutes per image. Performing the data processing on the laptop alone without access to a cluster takes approximately 42 minutes. Similar holographic reconstruction tasks using parallel processing on a graphical processing unit (GPU) have demonstrated approximately and order of magnitude improvement in processing time, and so processing time could potentially be reduced to just a few minutes.34–36 Figure 3 depicts LR holograms and their fully processed HR reconstructions.
Fig. 3.

Image reconstruction process. The top image is a single LR hologram captured with the portable LFHM. Green boxes represent the FOV of a conventional microscope using different objective lenses. Scale bar = 1 mm. The second row is a small region of interest showing a comparison of a LR hologram (left) to the reconstructed HR image after PSR and back propagation (right). Scale bar = 100 μm. The bottom row is a further zoomed-in region of this image, depicting the LR hologram (left) and HR reconstruction of fully resolved beads (right). Scale bar = 10 μm.
Two methods of feature analysis were used to quantify the agglutination assay from these HR reconstructions. In one method, image features (monomers and clusters) were isolated by applying a binary threshold to the reconstructed HR image, and then finding connected features. Feature area and eccentricity were calculated and a boundary in this parameter space was automatically determined to separate monomer features from clusters as previously described.21 Cluster size was then extrapolated based on the given feature’s area. Intensity (brightness) and size (area) thresholds were selected to include as many true beads as possible, while excluding non-bead features. To further optimize thresholding, a range of intensity and size thresholds were scanned through and the combination of values that resulted in calculated monomer and dimer areas most similar to expected areas were selected. Bound ratio (BR) was calculated with the following equation based on the results of the thresholding analysis.
| (1) |
The LOD for the overall assay was determined by calculating the mean and standard deviation of the BR for the negative control sample and using Equation 2, which combines the mean and standard of deviation of the negative control data points, to determine the BRLOD cutoff. The range of SARS-CoV-2 concentrations with a BR above this cutoff determine the dynamic range of this assay, while the LOD is given by the lowest concentration where BR ≥ BRLOD. Similarly, any BR that falls below the lower limit of detection cutoff, BRLLOD (Eq. 3), is also within the dynamic range of this assay.
| (2) |
| (3) |
2.5. Residual Convolutional Neural Network
The second method of image analysis was implementation of a deep convolutional neural network (CNN) with residual connections to classify image features (Fig. 4). This network was designed to account for the complex imaging conditions present in the SARS-CoV-2 agglutination assay that contains cell debris, viral particles, and other contaminants. To accomplish this, a 4-block deep CNN with residual connections was designed with the MATLAB Deep Learning Toolbox. This network updates convolutional filters, weights, and biases according to the built-in stochastic gradient descent with momentum optimizer, and employs L2 regularization to prevent overfitting. Classification loss is calculated using the following equation and used to update values in the network:
| (4) |
where N is the number of samples, K is the number of classes, wi is the weight for class i, tni is the indicator that the nth sample belongs to the ith class, and yni is probability that the network associates the nth input with class i.
Fig. 4.
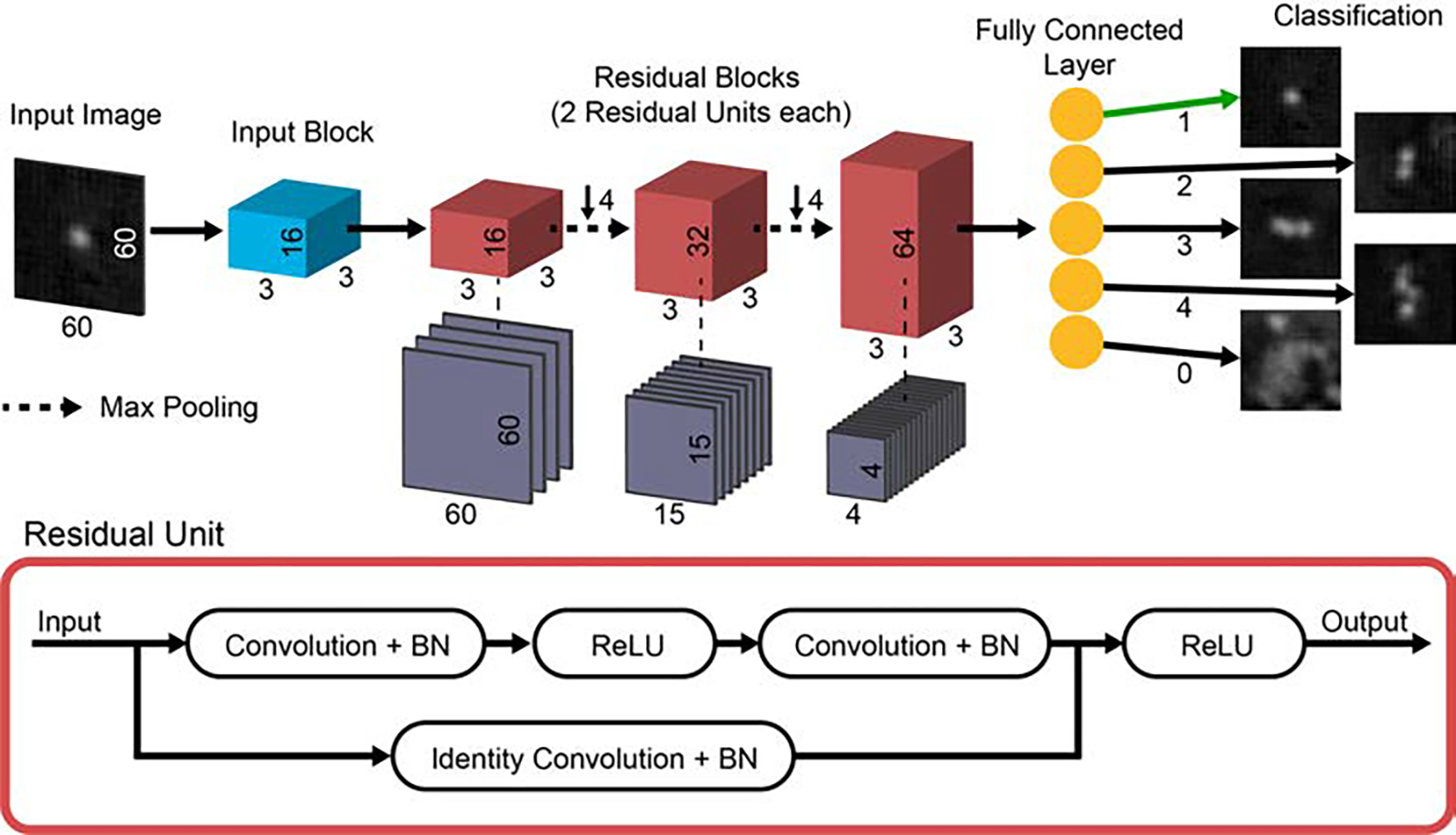
Residual CNN architecture. Input images of size 60 × 60 pixels are fed individually into the network. Each image passes through the input block, consisting of a convolutional layer with 16 3 × 3 filters, a batch normalization (BN) layer, and ReLU activation layer. Then the data, now 16 channels wide, is passed to the first of 3 residual blocks. Each residual block consists of 2 residual units, and connections between the blocks contain pooling layers that reduce the data size by a factor of 42. Each residual block increases the width of the network by a factor of 2. Finally, data is passed into a fully connected layer which outputs a classification for the input image. Examples of the classification result for 5 input images are shown in the last column of the diagram.
Training of this network was accomplished by using a single intensity threshold to identify features of interest in several agglutination assay images and cropping a subset (< 1% of the total features in any given image) of these features into small images 60 × 60 pixels in size. These 1,410 unique images were hand-classified into 5 categories: features consisting of 1, 2, 3, or 4 microspheres, as well as a fifth category for cell debris or unknown features that should not contribute to the calculation of BR. We disregard cluster sizes greater than 4 because their size cannot be measured as accurately and we are optimizing our sensor for very low concentrations of SARS-CoV-2, where very large cluster sizes are rare. To augment the training dataset, the cropped and hand-classified feature images were rotated and mirrored, to yield a total of 11,280 images. Of these images, 75% were used for training and 25% were reserved for validation. Training data was fed into the network in a random order each epoch to ensure generalizability of the training. Validation was performed every 2 epochs. A 2-core Intel® Xeon® Gold 5218 2.29 GHz processor was used to train the network. To prevent overtraining, training was halted after the classification accuracy of the validation image set stopped improving.
3. Results and Discussion
3.1. Network training outcomes
CNN training took 1,980 iterations or 30 epochs (Fig. 5a–b). Each iteration consisted of a batch of 128 training images. After 30 epochs, validation accuracy ceased improving so the training was halted to prevent overtraining (training accuracy and loss diverging from validation accuracy and loss). The final validation accuracy of the trained CNN was 82.06%. Due to the complexity of appearance of the debris in the training images, varied bead configurations for any given cluster size (e.g. linearly arranged, beads touching all other beads in the cluster, etc.), and slight variations in focus of the individual features, the image data was highly heterogeneous. Because of this heterogeneity, CNN validation accuracy could not be further improved without sacrificing generalizability to the broader intended dataset. To account for this, the CNN was designed to place features with low maximum softmax probabilities or activations, or ones that could be classified incorrectly, into smaller feature size categories, rather than larger ones (Fig. 5c). The net effect of this “rounding down” network behavior is the undervaluation of BR, as cluster size tends to be undercounted rather than overcounted. Thus, even though validation accuracy never reached 100%, we are confident that the network is not artificially inflating the BR for any samples and that our calculation of LOD for our assay is therefore conservative. Furthermore, since this behavior is consistent among all samples tested, we do not suspect that the validation error had a significant impact on our assay. Precision and recall measurements for each classification category are shown in Figure 5, with averages across all categories of 83.21% and 82.04% respectively.
Fig. 5.
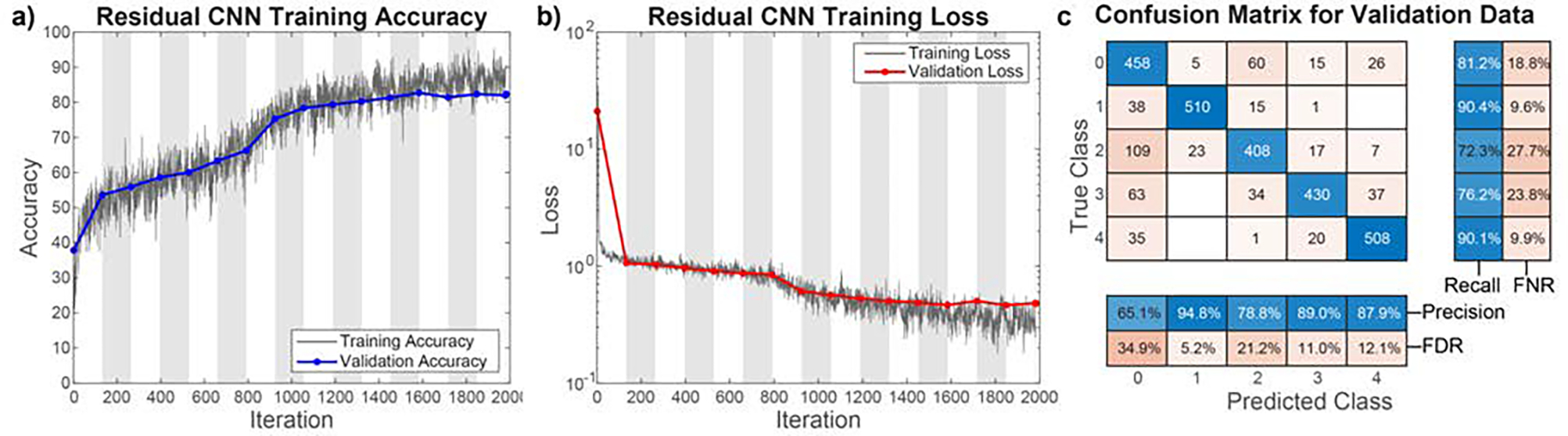
Results of CNN training. a) Accuracy of the training and validation data over the course of training. Validation accuracy does not diverge from the training accuracy over the course of training, indicating the network is not overtraining. b) Loss calculation for the training and validation data over the same training timespan. Again, validation loss does not diverge from training loss, indicating the network has not been overtrained. The training duration of 1,980 iterations corresponds to 30 epochs. White and grey chart backgrounds denote 2 epochs in width each. c) Confusion matrix for validation data classification. The blocks at the right and bottom show the total correct and incorrect classifications in each row and column of the confusion matrix. 63.83% of all incorrectly classified features are classified as smaller than they actually are, resulting in a slight undervaluation of bound ratio. Precision and recall for each classification category are shown on the bottom and right respectively, matched with false discovery rate (FDR) and false negative rate (FNR) respectively. Average precision was 83.21% and average recall was 82.04%.
To ensure the network was not overfit to augmented data, a test data set of 1,200 images without augmentation was analyzed using the CNN (Table 1). Unlike the training and validations sets, the test data set did not have an equal number of images in each category, and instead the category distribution was representative of the distribution seen in real images. The accuracy for this real-world application of the network was 88.58%. Since the images of this test set consist predominately of single beads (874 out of the 1,200), this high accuracy is consistent with the accuracy values shown in Fig. 5c. To better compare this result to the training and validation accuracy, images in test data set were removed and features of size 3 and 4 taken from other sample images were added such that the categories were balanced and large enough to provide an adequate comparison (150 images total). The resulting accuracy of 81.33% is within a percent of the validation accuracy and definitively confirms that this network is not overfit and performs well for real images.
Table 1.
Network output results for test data sets. Training and validation images are drawn from an augmented data set. Test data are unaugmented. Balanced test data include equal numbers of images with 0, 1, 2, 3, and 4 beads, while representative test data include a distribution of bead cluster sizes that is representative of real samples.
| Image Data Set | Network Output Accuracy |
|---|---|
|
| |
| Training | 89.28% |
| Validation | 82.06% |
| Test (Balanced) | 81.33% |
| Test (Representative) | 88.58% |
The network also exhibited higher maximum softmax probability values for correctly identified features than for incorrectly identified features (Table 2). In this case, maximum softmax probability values represent the relative activation of the network output layer neurons and give an indication of the “certainty” the network has for a given prediction, with high values indicative of a high degree of certainty that the classification is indeed accurate.37 This network behavior was unexpected, but unsurprising as the network training is designed to minimize training loss (a more complex measure of network error that includes all softmax probabilities), rather than maximizing accuracy. Low maximum softmax probability for incorrectly classified images represents lower loss than high maximum softmax probability for incorrect classifications. While softmax probabilities provide some indication of certainty, and can be used to derive useful statistics, they should not be used as direct measures of statistical confidence.37
Table 2.
Network maximum softmax probability values for image classification of training and validation data
| Mean maximum softmax probability (correctly classified) | Mean maximum softmax probability (incorrectly classified) | |
|---|---|---|
|
| ||
| Training | 90.31% | 63.11% |
| Validation | 90.27% | 68.02% |
The training time for this network was 175 s. Feature classification using the trained CNN takes 14.11 s for an average full field of view sample, which is 2.74× faster than the previous thresholding-based classification, which takes 38.65 s for an average full image.
3.2. Pseudovirus sensing
The SARS-CoV-2 agglutination assay dilutions were imaged and analyzed following the procedures described in Section 2. The optimized thresholding analysis and CNN-based results are shown in Figure 6a and b, respectively. The BRLOD cutoff for thresholding analysis was calculated to be 34.13%. According to this calculation, the lowest sample concentration in Figure 6a whose mean minus one standard error of the mean (SEM) falls above the LOD cutoff corresponds to 1.5·102 copies·mL−1. However, for an imaging chamber with 25 μL of sample, one would expect < 4 viral particles in the chamber. Therefore, this point is very unlikely to be a true LOD and only falls above the LOD cutoff due to the high variability in the thresholding analysis process. Additionally, the VSV-G specificity control shows this same high variability as its concentration changes, even though the BR for this control should remain constant at or near the BR of the negative SARS-CoV-2 pseudovirus control at 31.70%. Overall, thresholding analysis had a very high average standard of deviation for all non-control points of 10.66%. This makes determination of a true LOD from these data impossible, as there appears to be no clear trend as either virus concentration increases. For this dataset, thresholding analysis fails as it is not robust enough to account for the heterogeneous nature of a sample with cell debris and higher levels of non-specific binding.
Fig. 6.
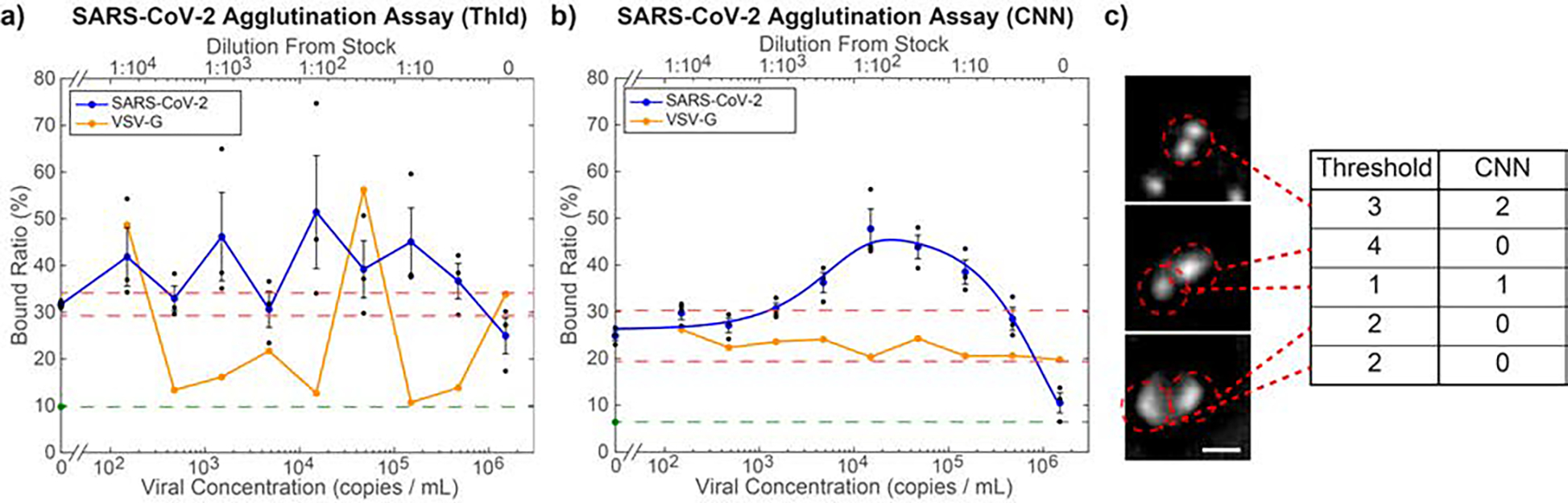
Quantification of microbead agglutination in the presence of SARS-CoV-2 pseudovirus. a) Optimized threshold-based quantification of the agglutination assay, which fails to find a clear trend in BR as a function of concentration. Over all non-control virus concentrations, the BR shows an average standard deviation of 10.66%, indicating inconsistency in the threshold-based quantification. Orange points indicate measurement results for VSV-G, which is used as a negative control. As the viral concentration of the VSV-G was not independently measured, the dilutions of the sample relative to the stock solution (top axis) were matched to the same dilutions of the SARS-CoV-2 pseudovirus, whose stock concentration was independently measured, yielding the bottom axis values. b) CNN-based agglutination quantification. Average standard of deviation for the BR of all non-control virus concentrations is 3.89%. The blue curve is a best fit to Eq. 5 (R2 = 0.974). The LOD based on this curve is 1,270 viral copies·mL−1. Orange points are negative control measurements using VSV-G samples diluted as specified in panel (a). The lack of a significant response shows that this assay is specific to SARS-CoV-2. For both graphs, the red dashed lines are the upper and lower LOD cutoffs, while the green point and dashed line indicate the BR in pure PBS. Black points indicate triplicate samples for each concentration and error bars are standard error of the mean. c) Example images classified by thresholding vs CNN. The CNN correctly identified each one of these features, while thresholding did not. Scale bar = 5 μm.
For CNN-based analysis (Fig. 6b), the BRLOD cutoff was calculated to be 30.33%. The mean BR corresponding to 1.5·103 copies·mL−1 is the first to fall above the LOD cutoff. An empirical curve of best fit, plotted in Figure 6b, was determined according to the following equation:
| (5) |
The functional form of this empirical curve is useful for inferring the analyte concentration of an unknown sample from its BR, as described below, but is not intended as a physical model of the binding process, which is a more complex relationship.21 The coefficients and 95% confidence intervals are: A = 46.86±4.11, b = 5.982±0.125, C = 20.60±4.86, d = 3.753±0.328 (R2 = 0.974). Since measurements of BR are compared to this curve, the LOD can be determined by where the curve first exceeds the BRLOD, which occurs at a concentration of 1.27·103 copies·mL−1.
Unlike the thresholding analysis (Fig. 6a), the CNN-based analysis (Fig. 6b) exhibits a clear peak in BR, where higher viral concentration ultimately leads to bead saturation and therefore reduced binding, which was observed previously in agglutination assays for other proteins.21 As a result, it is not possible to determine the exact concentration of virus in a sample from a single BR measurement when the BR > 19.40%. Nonetheless, a BR > BRLOD = 30.33% would be an unambiguous positive result, which is most relevant for rapid COVID diagnosis. The CNN resolves the variability seen in the VSV-G specificity control and those points are seen to lie within the LOD cutoffs, correctly interpreted as a negative result.
Interestingly, the BR for the highest concentrations of SARS-CoV-2 fell well below the BR for the negative control samples that still contained DMEM (black points at zero concentration in Fig. 6b). This can be explained by DMEM causing non-specific binding.13 For comparison, negative control samples of PBS without DMEM (green point at zero concentration in Fig. 6b), exhibit a significantly lower BR than the negative control samples with DMEM. At very high viral concentrations, the beads in the sample become saturated with viral particles before the beads can collide with one another. In this way, the viral particles effectively act as blockers for both specific and non-specific bead-to-bead binding. Hence, for high viral concentrations, the BR trends toward the BR found in PBS in the absence of DMEM. This behavior indicates that the use of fully saturated beads as a control for non-specific binding23 would be inappropriate for this type of agglutination assay because it fails to account for non-specific binding that occurs as a result of bead-to-bead interactions in different media. Additionally, this result means that our assay can distinguish between low levels and very high levels of virus by defining a lower LOD cutoff as specified in Eq. 3. For the CNN-based assay with 0.0025% bead concentration, BRLLOD = 19.40%, which corresponds to viral concentrations of 8.45·105 copies·mL−1 and greater on the best-fit curve.
Overall, compared to traditional image processing based on thresholding, CNN-based analysis enables successful and robust quantification of BR from complex pseudovirus samples, and extends the assay’s dynamic range by enabling sensing of higher pseudovirus concentrations whose BR falls below BRLLOD. Figure 6c shows a selection of features that were incorrectly classified by thresholding, but correctly classified by the CNN. Unfortunately, there is still a blind spot between 1.5·105 copies·mL−1 and 1.5·106 copies·mL−1, where a false negative result would occur since BRLLOD < BR < BRLOD for these concentrations.
One method to reduce this blind spot and infer specific viral concentrations from BR measurements throughout the dynamic range is to perform a second BR measurement on the same original sample, but with a higher bead concentration. Our previous work with agglutination assay-based sensing has shown that increasing bead concentration shifts the binding curve from left to right.21 Here, we performed this second measurement on an unfiltered pseudovirus sample with a 0.005% bead concentration instead of 0.0025% (Fig. 7a). The higher negative control BR can be explained by a higher number of bead-bead interactions in the higher bead concentration, resulting in more non-specific binding. Since the CNN analyzes small images of individual features, the network performance was not impacted by a higher bead concentration or a lack of pseudovirus filtration because those only increased the number of features classified without changing their appearance. The only effect an unfiltered sample had was a slight increase in average standard deviation of non-control BR measurements: 4.45% compared to 3.89% for filtered sample at a lower bead concentration (representative images shown in Fig.S3†). A curve was fitted using Eq. 5 with the following best-fit coefficients: A = 59.79±4.00, b = 6.429±0.132, C = 20.88±4.60, d = 4.077±0.301 (R2 = 0.962). This leads to a LOD of 4.81·103 copies·mL−1, were this measurement to be used in isolation.
Fig. 7.
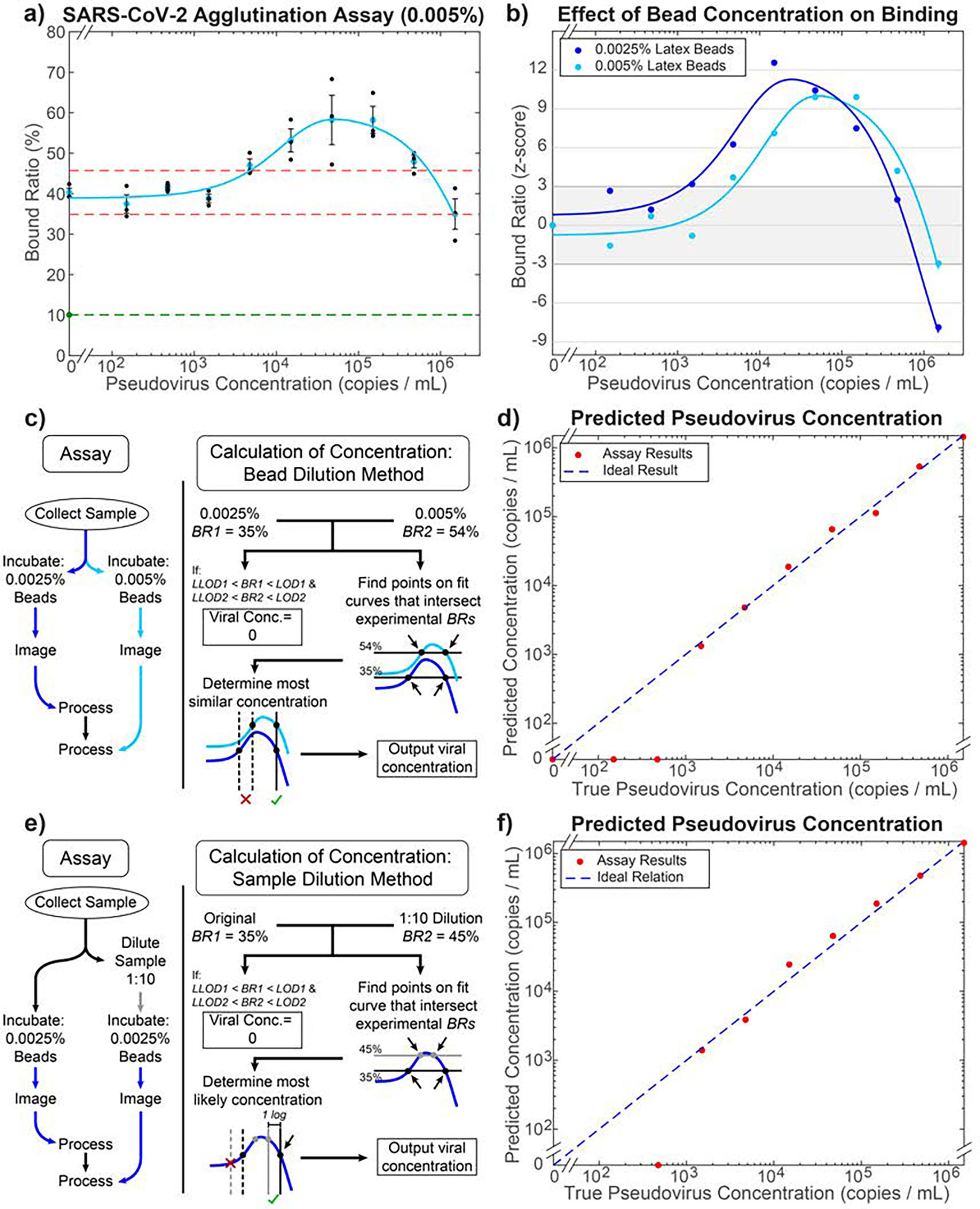
Inferring a unique analyte concentration using two independent measurements. a) CNN-based agglutination quantification on unfiltered pseudovirus lysates with 0.005% bead concentration. The LOD cutoffs (red dashed lines) are calculated as 45.72% and 34.86%. The light blue curve is fitted using Eq. 5 (R2 = 0.962). The green point and dashed line indicate the BR in pure PBS. b) Comparison of binding curves for 0.0025% (blue) and 0.005% (light blue) bead assays normalized by their z-score: how many standard deviations a measurement is away from the negative control value. A z-score of ±3 corresponds to the BRLOD and BRLLOD for each assay. c) Flowchart of combined assay to achieve accurate quantification of viral concentration using the bead dilution method. d) Results of quantification based on the method in (c) performed on the two mean BR values for each viral concentration. The R2 is 0.993, calculated based on the log of the y-values above the LOD compared to ideal result. e) Flowchart of assay using the sample dilution method. f) Results of quantification based on the method in (e) performed on the mean BR values from the 0.0025% bead curve for each viral concentration. The R2 is 0.989, calculated based on the log of the y-values above the LOD compared to ideal result.
By combining the binding results from the two bead concentrations, the exact viral concentration of almost any given sample (above the LOD) can be inferred due to the relative shift in binding curves for the two concentrations (Fig. 7b). To incorporate this into the assay workflow, the procedure depicted in Fig. 7c was performed. For example, a BR of 35% using a single 0.0025% bead concentration assay would yield 2 possible viral concentrations of 3.20·103 copies·mL−1 or 2.80·105 copies·mL−1. However, when combined with a BR of 54% from the 0.005% bead concentration assay, the true viral concentration of 2.80·105 copies·mL−1 would be selected. The outcome of this process for all mean points is illustrated in Fig. 7d (R2 = 0.993 for values within the assay’s dynamic range). The effect of this extra step on assay time would only be an additional 20 min for image processing, as the incubation and imaging could be done for both samples concurrently while the additional final calculation step occurs in a matter of 1–2 seconds, resulting in a total assay time of less than 3 hrs. Although this procedure is successful in inferring concentration for most of the experiments, a small blind spot remains, corresponding to where both best-fit curves fall between the BRLOD and BRLLOD cutoffs at concentrations greater than the LOD: 7.26·105 copies·mL−1 to 8.45·105 copies·mL−1. We expect that this approach could completely remove the blind spot if an even higher bead concentration were used for the second measurement.
A second alternate method of performing this assay, which completely resolves any blind spot using only one binding curve, is described in Figure 7e–f. By diluting the original sample by 1:10 and performing the agglutination assay on this dilution as well as the initial sample, both using the same bead concentration, we receive 2 points along the binding curve that can be used to determine which side of the peak the points correspond to. While this gives a slightly less accurate measurement of the true concentration (R2 = 0.989 for values within the assay’s dynamic range) and adds an extra dilution step over the two-bead concentration method, it nevertheless shows that the blind spot observed in this assay can be fully resolved through at most one additional measurement.
4. Conclusions
Here, we have shown a portable, LFHM biosensor capable of detecting SARS-CoV-2 pseudovirus concentrations at least as low as 1,270 copies·mL−1 within 3 hours, using only 80 μL of viral sample per test. This LOD is within an order of magnitude of widely used RT-PCR tests for SARS-CoV-2 and greatly improves upon the LOD of 3·106 copies·mL−1 for SARS-CoV-2 LFAs. Additionally, we have developed a deep-learning based categorization method that can accommodate heterogeneous solutions by distinguishing cell debris and other non-bead particles from microbead clusters, improving on traditional algorithms in speed, accuracy, and versatility. We also show that the choice of negative control beads (fully saturated vs. unsaturated) for high-sensitivity agglutination assays is important by showing that fully saturated beads fail to take into account non-specific binding that occurs as a result of exposure to a different liquid medium, potentially leading to a miscalculation of LOD. Finally, we showed two methods by which two measurements of BR can be used to compensate for the blind spots of a single individual assay and accurately determine the exact viral load of the sample across a dynamic range of 3 orders of magnitude in concentration. Future work will include reducing sample incubation time to enable more effective POC deployment, developing additional machine learning algorithms to assist with computational analysis, and further testing of the proposed assay to ensure these results are consistent when this assay is applied with patient samples.
Supplementary Material
Acknowledgements
This work utilized High Performance Computing (HPC) resources supported by the University of Arizona TRIF, UITS, and Research, Innovation, and Impact (RII) and maintained by the UArizona Research Technologies department. Research reported in this publication was supported by the National Institute of Aging of the National Institutes of Health under award number T32AG061897. Partial support was also provided by National Science Foundation grant number ECCS-2114275.
Footnotes
Conflicts of interest
Z.X. and E.M. are inventors on intellectual property related to lensfree holographic microscope sensors.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 00.0000/00000000.
Notes and references
- 1.Desai S, Rashmi S, Rane A, Dharavath B, Sawant A and Dutt A, Briefings in Bioinformatics, 2021, 22, 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vandenberg O, Martiny D, Rochas O, van Belkum A and Kozlakidis Z, Nat Rev Microbiol, 2021, 19, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Böger B, Fachi MM, Vilhena RO, Cobre AF, Tonin FS and Pontarolo R, American Journal of Infection Control, 2021, 49, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sethuraman N, Jeremiah SS and Ryo A, JAMA, 2020, 323, 2249. [DOI] [PubMed] [Google Scholar]
- 5.Peto T and O. behalf of the UK COVID-19 Lateral Flow Oversight Team, EClinicalMedicine, 2021, 36, 100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant BD, Anderson CE, Williford JR, Alonzo LF, Glukhova VA, Boyle DS, Weigl BH and Nichols KP, Anal. Chem, 2020, 92, 11305–11309. [DOI] [PubMed] [Google Scholar]
- 7.Pickering S, Batra R, Merrick B, Snell LB, Nebbia G, Douthwaite S, Reid F, Patel A, Kia Ik MT, Patel B, Charalampous T, Alcolea-Medina A, Lista MJ, Cliff PR, Cunningham E, Mullen J, Doores KJ, Edgeworth JD, Malim MH, Neil SJD and Galão RP, The Lancet Microbe, 2021, 2, e461–e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan JF-W, Yip CC-Y, To KK-W, Tang TH-C, Wong SC-Y, Leung K-H, Fung AY-F, Ng AC-K, Zou Z, Tsoi H-W, Choi GK-Y, Tam AR, Cheng VC-C, Chan K-H, Tsang OT-Y and Yuen K-Y, J Clin Microbiol, 2020, 58, e00310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heid CA, Stevens J, Livak KJ and Williams PM, Genome Res, 1996, 6, 986–994. [DOI] [PubMed] [Google Scholar]
- 10.Eftekhari A, Alipour M, Chodari L, Maleki Dizaj S, Ardalan M, Samiei M, Sharifi S, Zununi Vahed S, Huseynova I, Khalilov R, Ahmadian E and Cucchiarini M, Microorganisms, 2021, 9, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochańczyk M, Grabowski F and Lipniacki T, Dynamics of COVID-19 pandemic at constant and time-dependent contact rates, Epidemiology preprint, 2020.
- 12.Chen L, Wang G, Long X, Hou H, Wei J, Cao Y, Tan J, Liu W, Huang L, Meng F, Huang L, Wang N, Zhao J, Huang G, Sun Z, Wang W and Zhou J, The Journal of Molecular Diagnostics, 2021, 23, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molina-Bolívar JA and Galisteo-González F, Journal of Macromolecular Science, Part C: Polymer Reviews, 2005, 45, 59–98. [Google Scholar]
- 14.Marra CM, Maxwell CL, Dunaway SB, Sahi SK and Tantalo LC, J Clin Microbiol, 2017, 55, 1865–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominic RS, Prashanth HV, Shenoy S and Baliga S, J Lab Physicians, 2009, 1, 067–068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De la fuente L, Anda P, Rodriguez I, Hechemy KE, Raoult D and Casal J, Journal of Medical Microbiology, 1989, 28, 69–72. [DOI] [PubMed] [Google Scholar]
- 17.Friedman CA, Wender DF and Rawson JE, Pediatrics, 1984, 73, 27–30. [PubMed] [Google Scholar]
- 18.Xu X, Jin M, Yu Z, Li H, Qiu D, Tan Y and Chen H, J Clin Microbiol, 2005, 43, 1953–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You DJ, Geshell KJ and Yoon J-Y, Biosensors and Bioelectronics, 2011, 28, 399–406. [DOI] [PubMed] [Google Scholar]
- 20.Fronczek CF, You DJ and Yoon J-Y, Biosensors and Bioelectronics, 2013, 40, 342–349. [DOI] [PubMed] [Google Scholar]
- 21.Xiong Z, Potter CJ and McLeod E, ACS Sens, 2021, 6, 1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Ray A, Wei Q, Feizi A, Tong X, Chen E, Luo Y and Ozcan A, ACS Photonics, 2019, 6, 294–301. [Google Scholar]
- 23.Luo Y, Joung H-A, Esparza S, Rao J, Garner O and Ozcan A, Lab Chip, 2021, 21, 3550–3558. [DOI] [PubMed] [Google Scholar]
- 24.McLeod E and Ozcan A, Rep. Prog. Phys, 2016, 79, 076001. [DOI] [PubMed] [Google Scholar]
- 25.McLeod E, Dincer TU, Veli M, Ertas YN, Nguyen C, Luo W, Greenbaum A, Feizi A and Ozcan A, ACS Nano, 2015, 9, 3265–3273. [DOI] [PubMed] [Google Scholar]
- 26.Ozcan A and McLeod E, Annu. Rev. Biomed. Eng, 2016, 18, 77–102. [DOI] [PubMed] [Google Scholar]
- 27.Baker M, Liu W and McLeod E, Opt. Express, 2021, 29, 22761. [DOI] [PubMed] [Google Scholar]
- 28.Xiong Z, Melzer JE, Garan J and McLeod E, Opt. Express, 2018, 26, 25676. [DOI] [PubMed] [Google Scholar]
- 29.Bishara W, Su T-W, Coskun AF and Ozcan A, Opt. Express, 2010, 18, 11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishara W, Sikora U, Mudanyali O, Su T-W, Yaglidere O, Luckhart S and Ozcan A, Lab Chip, 2011, 11, 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samavati L and Uhal BD, Front. Cell. Infect. Microbiol, 2020, 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, Chu HY, Tortorici MA, Veesler D, Murphy M, Pettie D, King NP, Balazs AB and Bloom JD, Viruses, 2020, 12, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Meng X, Zhang F, Xiang Y and Wang J, Emerging Microbes & Infections, 2021, 10, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isikman SO, Bishara W, Sikora U, Yaglidere O, Yeah J and Ozcan A, Lab Chip, 2011, 11, 2222–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isikman SO, Bishara W, Mavandadi S, Yu FW, Feng S, Lau R and Ozcan A, Proc. Natl. Acad. Sci. U.S.A, 2011, 108, 7296–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenbaum A, Luo W, Su T-W, Göröcs Z, Xue L, Isikman SO, Coskun AF, Mudanyali O and Ozcan A, Nat Methods, 2012, 9, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendrycks D and Gimpel K, International Conference on Learning Representations (ICLR), 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.