

Abstract
Background:
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a potentially lethal cardiac arrhythmia syndrome triggered by catecholamines released during exercise, stress, or sudden emotion. Variants in the calsequestrin-2 gene (CASQ2), encoding the major calcium (Ca) binding protein in the sarcoplasmic reticulum (SR), are the second most common cause of CPVT. Recently, several CASQ2 gene variants, such as CASQ2-K180R, have been linked to an autosomal dominant form of Casq2-linked CPVT (CPVT2) but the underlying mechanism is not known.
Methods:
A K180R mouse model was generated using CRIPSR/Cas9. Heterozygous (HET) and homozygous (HOM) K180R mice were studied using telemetry ECG recordings in vivo. Ventricular cardiomyocytes were isolated and studied using fluorescent Ca indicators and patch clamp. Expression levels and localization of SR Ca handling proteins were evaluated using western blotting and immunostaining. Intra-SR Ca kinetics were quantified using low-affinity Ca indicators.
Results:
K180R mice exhibit an autosomal-dominant CPVT phenotype following exercise or catecholamine stress. Upon catecholamine stress, K180R ventricular cardiomyocytes exhibit increased spontaneous SR Ca release events, triggering delayed afterdepolarizations and spontaneous beats. K180R had no effect on levels of Casq2, Casq2 polymers, or other SR Ca handling proteins. Intra-SR Ca measurements revealed that K180R impaired dynamic intra-SR Ca buffering, resulting in a more rapid rise of free Ca in the SR during diastole. Steady-state SR Ca buffering and total SR Ca content were not changed. Consistent with the reduced dynamic intra-SR buffering, K180R causes reduced SR Ca release refractoriness.
Conclusions:
CASQ2-K180R causes CPVT2 via a heretofore unknown mechanism that differs from CASQ2 variants associated with autosomal-recessive CPVT2. Unlike autosomal recessive CASQ2 variants, K180R impairs the dynamic buffering of Ca within the SR without affecting total SR Ca content or Casq2 protein levels. Our data provide insight into the molecular mechanism underlying autosomal-dominant CPVT2.
Keywords: Catecholaminergic polymorphic ventricular tachycardia, cardiac calsequestrin, CPVT2
Graphical Abstract
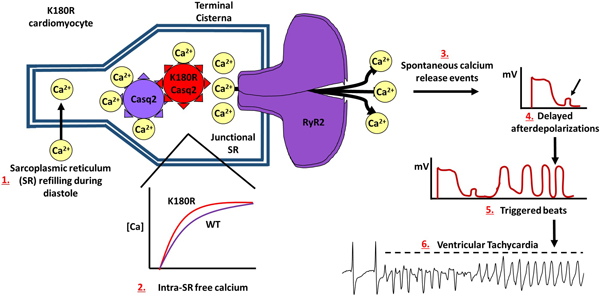
Summary
CPVT2 is a lethal, stress-induced cardiac channelopathy caused by mutations in the intra-SR Ca handling protein, Casq2. Most of the mutations that have been discovered follow an autosomal recessive inheritance pattern and disrupt Ca homeostasis by decreasing Casq2 protein levels. Recent studies have found that autosomal dominant CPVT2 exists, and patients experience similar symptoms. It had been suggested that autosomal dominant CPVT2 also decreases Casq2 levels, but no mechanism has been reported. Here we utilize an autosomal dominant Casq2 mutation, K180R, to uncover a novel mechanism for autosomal dominant CPVT2. K180R causes CPVT by altering the dynamic calcium buffering capabilities of the SR, leading to a decrease in the SR Ca release refractoriness, and increased spontaneous release of Ca. Unlike autosomal recessive CPVT2, K180R affects Ca handling without decreasing protein levels of Casq2 or reducing SR Ca content. These finding suggest a novel mechanism for autosomal dominant CPVT2.
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal, stress-induced cardiac channelopathy. First described in 1995, CPVT is characterized by polymorphic ventricular arrhythmias that are triggered by catecholamines released during exercise, stress, or sudden emotion in individuals with structurally normal hearts.1 Six different CPVT disease genes have been identified, which account for 60–75% of CPVT cases.2,3 All six CPVT genes (RYR2, CASQ2, TRDN, CALM1, CALM2, CALM3) encode proteins that are directly involved in regulating SR calcium (Ca) release during excitation-contraction (E-C) coupling. Variants in CASQ2 are the second most common cause of CPVT, termed CPVT2.4,5 Casq2 was first associated with CPVT when missense variants were discovered in a family with autosomal recessive CPVT in 2001 and nonsense variants in a similar family in 2002.6 CPVT2 was thought to be an autosomal recessive disease only,6–8 until in 2016 investigators reported a novel CASQ2 variant (K180R) in a family with autosomal dominant inheritance of CPVT.9 Since then, several other CASQ2 variants have been associated with autosomal dominant CPVT2.10
CASQ2 encodes cardiac calsequestrin (Casq2), the most abundant Ca binding protein located in the junctional SR. Casq2 is a high capacity, low affinity Ca binding protein that binds 40–50 Ca ions through its 60–70 negatively charged amino acid residues.11 Functionally, Casq2 regulates the amount of Ca released from the SR through RyR2 during E-C coupling by buffering intra-SR Ca.12,13 Casq2 is known to form homo-polymers in a Ca-dependent manner.14 The polymerization and depolymerization of Casq2 is thought to have an important role in the regulation of RyR2 during the E-C coupling cycle by affecting the termination of Ca release from the SR during the EC coupling cycle through the binding of Casq2 monomers to RyR2 Ca release channels.15 Studies involving skeletal calsequestrin (Casq1) have found that the depletion of Ca in the SR causes Casq1 to undergo major conformation changes, disable the ability of Casq1 to bind calcium, and increases the diffusional mobility of Casq1.16 While it is still not well understood how important polymerization is for the function of Casq2, Casq1 polymerization studies provide a plausible mechanism for how Casq2 is regulating Ca release and termination.
Casq2 is important for supporting excitation-contraction coupling in the heart, where electrical activation couples to mechanical force via the secondary messenger Ca.13 Membrane depolarization during the cardiac action potential opens L-type Ca channels, which bring Ca into the cell. Ca binds to and opens cardiac ryanodine receptors (RyR2) located in the terminal cisternae of the SR, which form the junctional SR, a process known as Ca-induced Ca-release (CICR).17 During systole, cytosolic Ca initiates myofilament contraction before being taken back up into the SR or pumped out into extracellular space during diastole. Supported mostly by experimental studies in mouse CPVT models,18–21 the current understanding of cellular CPVT pathophysiology is that catecholamines released during stress or exercise activate β-adrenergic receptor signaling, leading to a cellular chain reaction that culminates into pathological Ca release during diastole and Ca-triggered action potentials.
The simplest explanation as to why variants in Casq2 cause CPVT is that they impair Casq2’s ability to buffer Ca in the SR, resulting in a much faster rise of intra-SR free Ca concentration close to RyR2 release channels. Although intra-SR Ca kinetics have not yet been measured experimentally in Casq2 CPVT models, there is extensive evidence that absence of Casq2 in mice leads to hyperactive RyR2 channels, impaired Ca-release termination, shortened Ca release refractory period, and enhanced spontaneous release of Ca.18,22,23 While impaired Ca buffering is the generally accepted mechanism for Casq2 nonsense variants that result in loss of Casq2, missense variants (e.g. R33Q) may alter Casq2 interaction with RyR2 in addition to reducing Ca buffering.24
Given that all previously reported CASQ2 variants cause either a complete loss or a severe reduction of Casq2 protein, autosomal-dominant CPVT2 variants are proposed to have a dominant negative effect that would lead to a reduction in Casq2 levels and thus cause CPVT.9 Supported by a novel crystal structure of a polymer filament of Casq2,25 it was hypothesized that autosomal-dominant Casq2 variants affect the ability of Casq2 to polymerize. Using the new filament, the authors mapped two autosomal dominant variants (K180R, and S173I) to understand how they would affect Casq2 function. The analysis also utilized the filament to identify novel Ca binding sites that bridged both intra and inter-dimer interfaces of Casq2, suggesting that Ca is involved in the ability for Casq2 to form dimers and those dimers to form subsequent filaments.25 Both autosomal dominant variants were unable to form polymers in an in vitro turbidity assay and were found to be a part of the novel Ca binding sites. Yet, the exact mechanism of how autosomal dominant variants cause CPVT is unknown. To answer this question, we generated and studied a K180R knockin mouse model. We find that unlike previously reported CASQ2 variants associated with autosomal-recessive CPVT2, K180R causes CPVT2 via a heretofore unknown mechanism: K180R impairs the rate of Ca binding to the Casq2 polymer within the SR without altering total SR Ca buffering or SR Ca content.
Methods
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The use of animals was approved by the Animal Care and Use Committee of Vanderbilt University. Experimental group sizes were based on previous studies examining CPVT mouse models.18 K180R mice were generated using CRISPR/Cas9 and utilized to assay CPVT susceptibility via multiple stress tests. Isolated ventricular myocytes underwent Ca handling analysis using Ca sparks and waves protocols. Western blotting, immunostaining and electron microscopy were used to investigate jSR protein levels and localization. Single cell experiments were used to measure spontaneous Ca release events, total SR Ca content, and SR Ca release refractoriness. Intra-SR and cytosolic Ca buffering properties were assessed by simultaneously measuring intra-SR and cytosolic free Ca using low and high-affinity Ca indicators, respectively. Statistical tests used are reported in the figure legends. Results from isolated cardiomyocytes were compared using a hierarchical statistical model following the procedures by Sikkel et al 2017.26 Briefly, a linear mixed effects (hierarchical) model was used to calculate Bonferroni-adjusted p-values clustered at the animal level to account for random effects between isolations. All p-values reported in the MS are corrected for multiple comparisons. Details such as statistical tests used, normality testing, exact raw and corrected p-values, are provide in the statistical summary table published online. An expanded method section is provided in the Supplemental Material document.
Results
Generation of K180R mice
The K180R variant results in a relatively conservative amino acid change from lysine to arginine. However, the lysine residue at position 180 within Casq2 is conserved across species (Fig. 1A) suggesting that K180 could be important to the structural properties of Casq2. To determine if K180R could produce a phenotype, we generated a genetic mouse model using CRISPR/Cas9. The gRNA was designed to target K180 on exon 11. C57BL/6 mouse zygotes were injected with the Cas9 enzyme, the gRNA, and a repair template to replace the lysine with an arginine. Sequencing the offspring of the mice (Fig. 1B) identified three founders carrying the K180R mutation. Heterozygous offspring from the founder lines displayed a CPVT phenotype in vivo (Fig. S1). Two founder lines (L1 and L3, Fig. S1) were backcrossed into C57BL/6 mice for 10 generations before being used in the study. Of the total mice used, 40% of the mice came from founder line 1 and 60% came from founder line 3. At least one genotype from each founder line was used in all experiments to avoid conclusions being drawn from just one line. No mice were excluded from the in vivo experiments.
Figure 1: K180R mice display a CPVT phenotype.
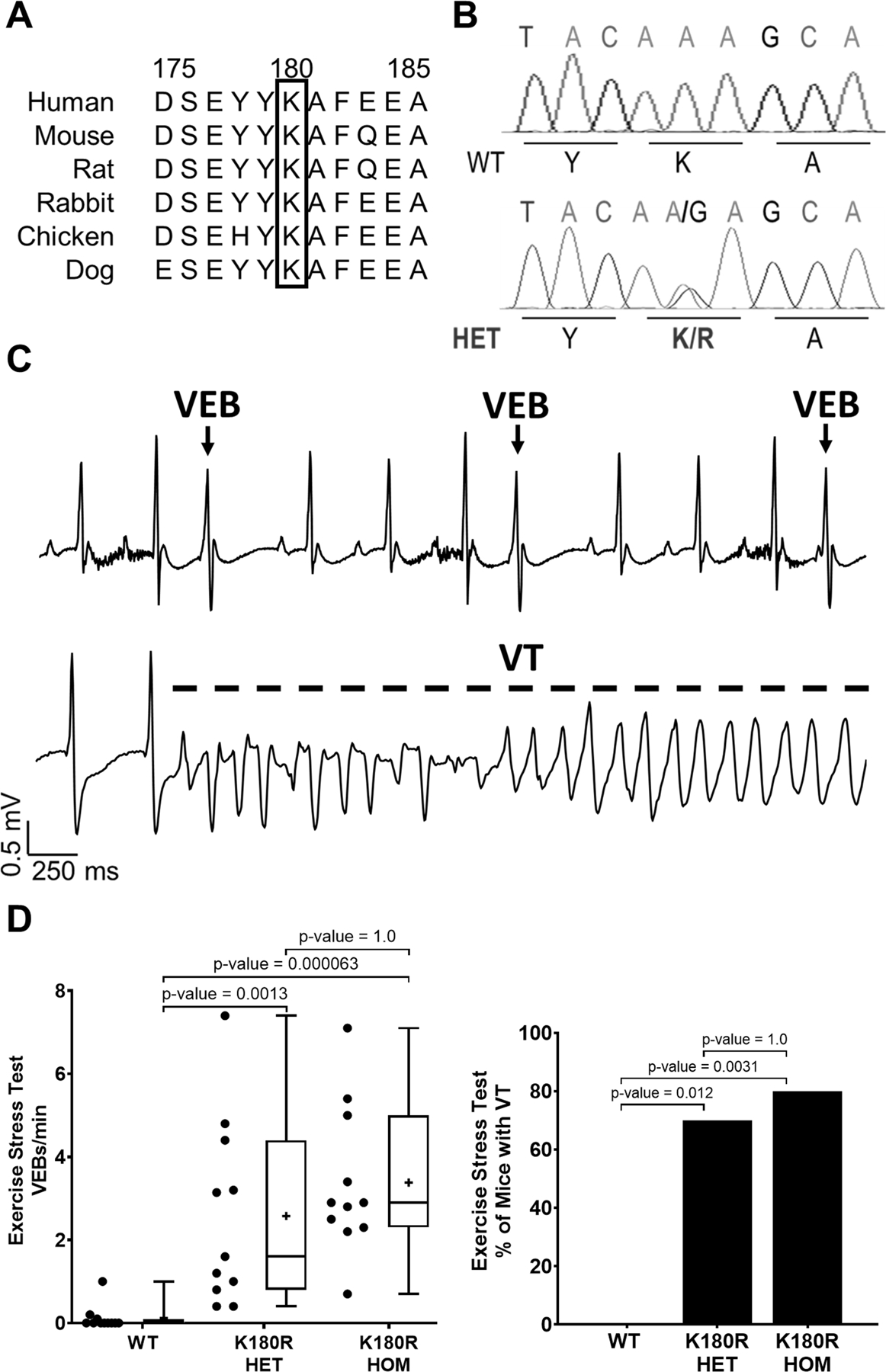
A. Amino acid 180 is conserved across species in CASQ2. B. K180R knockin mice are heterozygous for the AGG mutation. C. Example EKG traces from a K180R HET mouse, immediately after completing an exercise stress test, displaying characteristics of CPVT including ventricular ectopic beats (VEBs) and an episode of ventricular tachycardia (VT) D. Summary data of exercise-induced ventricular ectopy (VEB/min) and VT incidence. Ventricular arrhythmias were quantified during the 10 min period immediately after completion of the treadmill exercise test. n = 11 mice per group. Data displayed as a box and whisker plot. Median is displayed as a line within the box and whisker plot. Mean is displayed as a plus sign within the box and whisker plot. VEB/min data analyzed using Kruskal-Wallis test with Dunn’s multiple comparisons. VT data analyzed using Fisher’s exact test with pairwise Fisher exact test.
K180R mice display a CPVT phenotype
Arrhythmia susceptibility was investigated by two different stress tests. Mice between the ages of 12–18 weeks had ECG telemeters implanted before undergoing an exercise stress test or a catecholamine stress test with isoproterenol and caffeine. For each experimental animal, the number of VEBs or VT episodes was counted during a 10-minute recording period immediately after the stress test (either exercise or catecholamine injection). A similar CPVT phenotype was observed when K180R mice were given a catecholamine challenge (Median VEB/min: WT – 0.0+/−0.022, HET – 0.55+/−0.22, HOM – 1.35+/−1.16, n = 11 per group, p-values: WT vs HET – 0.0196, WT vs HOM – 0.0005, HET vs HOM – 0.87, Kruskal-Wallis test with Dunn’s multiple comparisons). Both heterozygous (HET) and homozygous (HOM) K180R mice displayed characteristics of CPVT, including ventricular ectopic beats (VEBs) and a high incidence of ventricular tachycardia (VT) following the exercise stress test (Fig. 1C–D). The rate of exercise-induced ventricular ectopy and VT incidence was not significantly different between K180R HET and HOM mice (Fig. 1C–D), consistent with the autosomal-dominant disease caused by the K180R variant in humans. Nor was the rate of exercise-induced ventricular ectopy significantly different between male and female K180R mice (Median VEB/min: Male – 2.2+/−0.90, n=7, Female – 2.9+/−0.49, n=15, p-value = 0.55 by Mann-Whitney test). Taken together, these results establish that K180R causes an autosomal-dominant CPVT phenotype in mice.
K180R increases the rate of spontaneous SR Ca release, delayed afterdepolarizations, and triggered beats in ventricular cardiomyocytes
It is generally accepted that genetic variants associated with CPVT render RyR2 channels hyperactive, resulting in increased likelihood of spontaneous SR Ca release,18,20 which will cause delayed afterdepolarizations that can trigger ectopic beats. Given that the K180R variant produces a CPVT phenotype in vivo (Fig. 1), we examined Ca handling and electrical activity in isolated K180R cardiomyocytes. In the first set of experiments, the rate of spontaneous RyR2 openings was quantified by measuring spontaneous Ca sparks and Ca waves after sarcolemma permeabilization with saponin. This experimental design allows complete control of cytosolic free [Ca], thereby directly examining the rate of RyR2 opening for a given cytosolic free Ca concentration. Compared to WT cardiomyocytes, HET and HOM K180R cardiomyocytes exhibited a significant increase in spark frequency (Fig. 2A–B). Spark characteristics, including spark amplitude, spark mass, and full duration half max (FDHM) were not significantly different between groups (Fig. S2). Spark-mediated SR Ca leak was significantly higher in K180R HET and HOM cardiomyocytes (Fig. S2). Consistent with an increased RyR2 activity, Ca wave frequency was also increased in K180R HET and HOM compared to WT cardiomyocytes (Fig. 2C–D), however wave amplitude remained similar between groups (Fig. S3).
Figure 2: K180R causes spontaneous SR Ca release events and delayed afterdepolarizations in single cardiomyocytes.
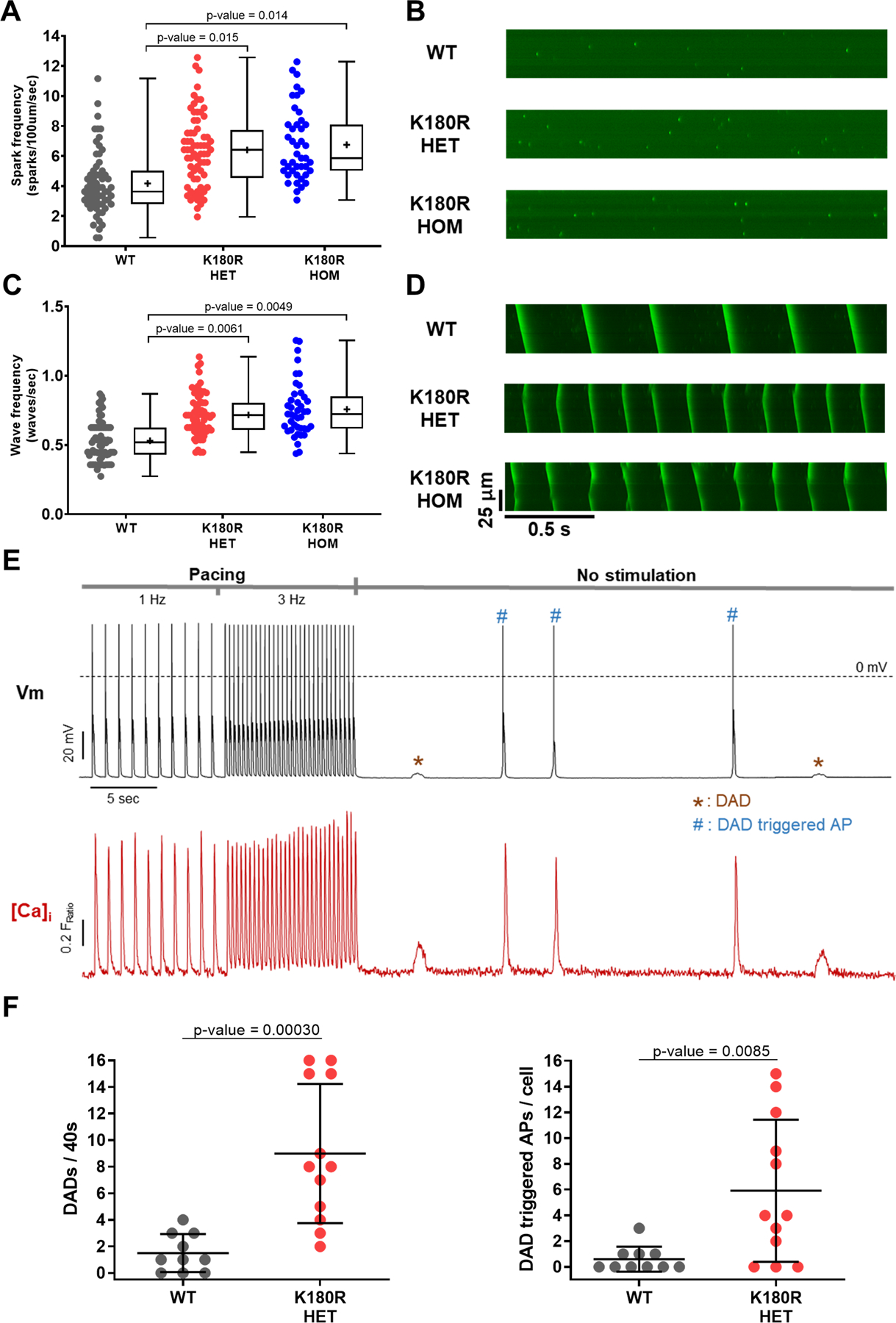
Intracellular Ca handling was examined in permeabilized (panels A-D) and intact (panels E-F) cardiomyocytes.
A-D: Saponin-permeabilized cardiomyocytes were incubated in an internal solution containing the Ca indicator (Fluo-4) and free [Ca] of 70 nm for spark measurements (panels A&B) and 190 nm for wave measurements (panels C&D). Cells were imaged using an inverted confocal microscope in line-scan mode. No isoproterenol was present for these experiments. A. Ca spark frequency in WT, K180R HET, and K180R HOM cardiomyocytes. WT; n = 63 cells from 3 hearts. K180R HET; n = 68 cells from 3 hearts. K180R HOM; n = 43 cells from 2 hearts. B. Example line-scan images of Ca sparks in K180R cardiomyocytes. C. Ca wave frequency in WT, K180R HET, and K180R HOM cardiomyocytes. WT n = 71 cells from 3 hearts; K180R HET n = 67 cells from 3 hearts; K180R HOM n = 42 cells from 2 hearts. D. Example line-scan images of Ca waves. Data displayed as a box and whisker plot. Median is displayed as a line within the box and whisker plot. Mean is displayed as a plus sign within the box and whisker plot.
E-F: Intact cardiomyocytes were loaded with the Ca indicator Fura-2AM and incubated in normal Tyrode solution containing 2 mM Ca and 1 µM isoproterenol. Membrane potential (Vm) was measured using patch clamp in current clamp mode. Intracellular Ca was measured using a dual-beam excitation fluorescence photometry. E. Examples of simultaneous recordings of [Ca]i and Vm from a K180R HET cardiomyocyte stimulated at 1 and 3 Hz (for 10 s each) followed by 40 s pause. Dotted line indicates 0 mV. For each cardiomyocyte, spontaneous Ca release events and the resulting delayed afterdepolarizations (DAD)s were quantified during the 40s pause. DADs are indicated by * and #, which sometimes generated a DAD-triggered action potential (AP, #). F. Frequency of DADs and DAD-triggered APs. WT n = 10 cells from 5 hearts; K180R HET n = 12 cells from 6 hearts. Data are presented as mean ± SD and were analyzed using a hierarchical statistical model with Bonferroni correction.
In the second set of experiments, we examined Ca handling and electrical activity in intact cardiomyocytes stimulated with the beta-adrenergic agonist isoproterenol to simulate catecholamine stress. Compared to WT, both K180R HET and HOM exhibited significantly higher rates of spontaneous Ca release events (Fig. S4), with no differences between the K180R HET and HOM groups. Interestingly, the time to the first spontaneous Ca release event (i.e., spontaneous Ca release latency) was significantly shorter in K180R cardiomyocytes compared to WT (Median: WT – 27.1 s, HET – 8.6 s, and HOM – 11.7 s; p-value = 0.014 and 0.015 vs WT, analyzed using a hierarchical statistical model with Bonferroni correction). We next examined Ca handling and SR Ca content in intact cardiomyocytes paced at 1.0 Hz. In contrast to autosomal-recessive CPVT due to loss of Casq2 protein, Ca handling parameters and SR Ca content of K180R-HET cardiomyocytes were not significantly different from WT (Fig. S5 and Table S1). To examine the effect of spontaneous Ca release on electrical activity, we next measured membrane potential and intracellular Ca simultaneously in current-clamped ventricular cardiomyocytes (Fig. 2E–F). The frequency of delayed afterdepolarizations and triggered action potentials was significantly increased in K180R HET cardiomyocytes (Fig. 2F), whereas action potential parameters were not altered (Table S2). Taken together, these results establish that the K180R variant in Casq2 causes a dominant gain of function in the RyR2 Ca release unit which generates pro-arrhythmic DADs and DAD-triggered beats in response to catecholaminergic stimulation.
K180R does not affect expression levels of Casq2 and other junctional SR proteins
All previously reported Casq2 variants cause a decrease in the level of Casq2 protein,29 and it had been suggested that K180R decreases Casq2 levels through a dominant negative mechanism.9 Furthermore, any reduction in Casq2 protein reduces other junctional SR (jSR) proteins including triadin and junctin, which may independently contribute to RyR2 hyperactivity in CPVT.18 To investigate jSR protein levels, protein lysate was isolated from mouse cardiomyocytes and analyzed via western blotting (Fig. 3A). Cardiomyocytes from two autosomal-recessive CPVT models (Casq2 and triadin knockout (KO) mice) were used as positive controls. The absence of one of these jSR proteins can decrease the levels of other jSR proteins.18,30 GAPDH was used as a loading control to compare protein levels between groups. Consistent with previous reports, Casq2 KO cardiomyocytes had a significant decrease in Casq2, triadin, and junctin protein levels. Knocking out triadin also significantly reduced Casq2, triadin, and junctin levels. In contrast, the K180R mutation had no effect on Casq2 protein levels (Fig. 3B). Recent in vitro studies suggested that the K180R variant may affect the ability of Casq2 to form polymers.10,31 Based on the Casq2 polymers visible on western blot, polymer levels were not significantly decreased in K180R HET and HOM mouse lysates (Fig. 3B). Other CRU protein levels (RyR2, triadin, and junctin) were also not significantly different in the K180R HET and HOM mouse cardiomyocytes (Fig. 3C). Taken together, these results indicate that the K180R mutations causes CPVT without altering jSR proteins or Casq2 polymers and therefore via a novel mechanism.
Figure 3: Levels of cardiac junctional SR proteins are not altered in K180R mice.
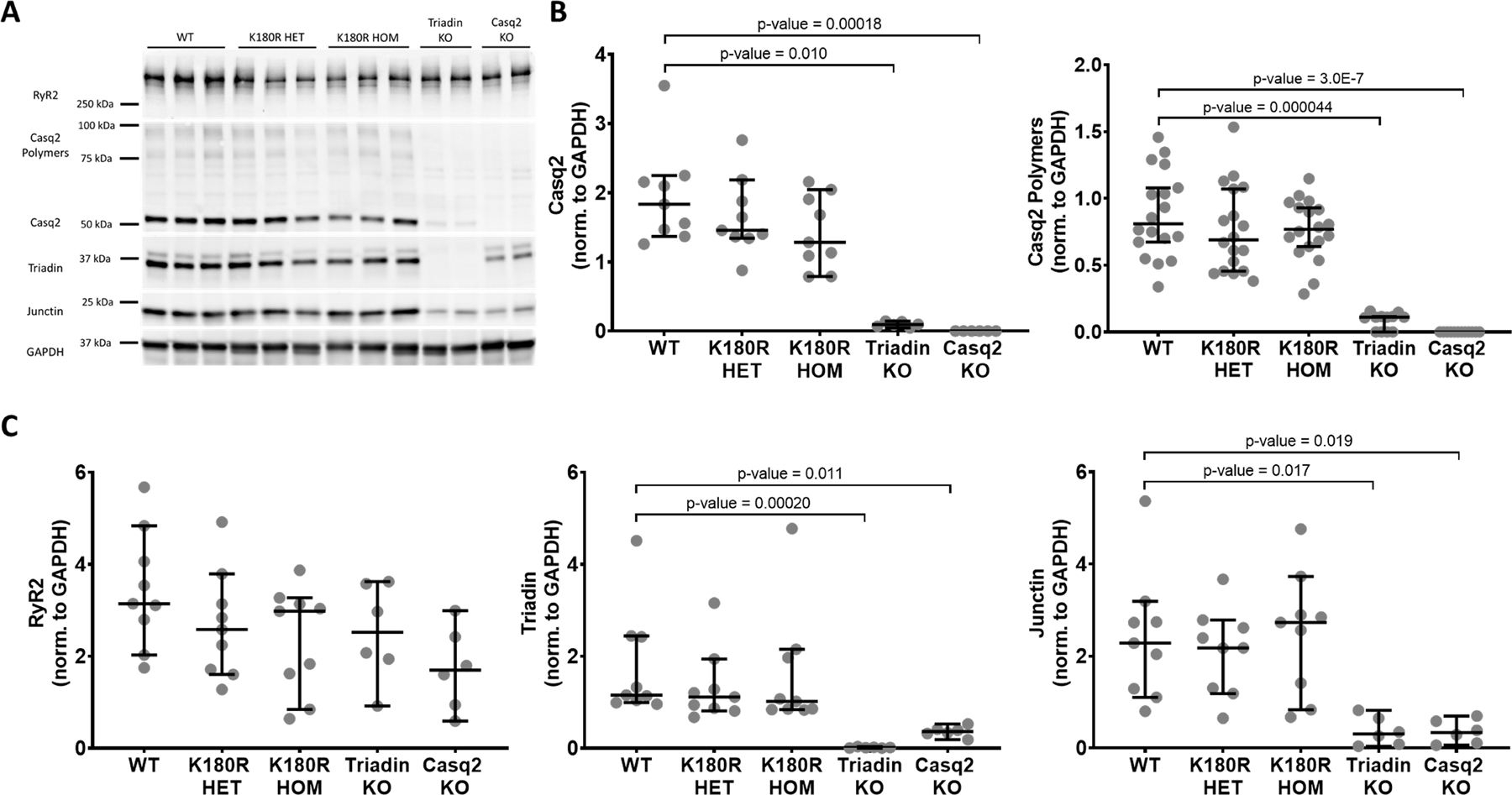
Ventricular cellular lysate was collected from isolated mouse hearts and analyzed using western blotting. A. Representative raw western blot image. Triadin knockout and Casq2 knockout mice were used as controls. B. Quantification of Casq2 and Casq2 polymers band density. There was no significant difference between Casq2 and Casq2 polymer protein levels in K180R mice and WT mice. C. Quantification of junctional SR proteins. There was no significant difference in junctional SR protein levels in K180R mice compared to WT. All values were normalized to GAPDH. n = 9 lysates for WT, K180R HET, and K180R HOM. N = 6 lysates for Triadin KO and Casq2 KO. Each lysate was collected from an independent animal. Data displayed as mean ± SD. Data were analyzed using Kruskal-Wallis test with Dunn’s multiple comparison test.
K180R does not affect subcellular localization of Casq2
Casq2 is found in the junctional SR where it co-localizes with RyR2, triadin, and junctin. Together, these four proteins form the SR component of the Ca release unit.32,33 CPVT caused by loss of triadin is likely caused not only by a decrease in Casq2 protein levels, but also impaired localization of Casq2 to the jSR 30. With Casq2 protein levels being normal in K180R mice, one potential mechanism for K180R causing altered Ca handling could be abnormal subcellular localization of Casq2 or other CRU proteins. To test this hypothesis, K180R mouse cardiomyocytes were isolated, plated, fixed, and stained for Casq2, RyR2, and triadin (Fig. 4A). There were no significant differences for Casq2-RyR2 or triadin-RyR2 co-localization in K180R HET cardiomyocytes compared to WT cells (Fig. 4B). On the other hand, Triadin KO cardiomyocytes, consistent with previous studies, exhibited decreased Casq2-RyR2 co-localization. To corroborate the results of our immunostaining experiments, we next examined electron micrographs of ventricular sections from WT and K180R HET hearts. K180R HET and HOM hearts had normal SR structure, and Casq2 polymers were visible in all jSR cross-sections examined (Fig. S6). Taken together, the normal localization of Casq2 in K180R HET myocytes indicates that K180R localizes correctly in the jSR and likely can interact with other jSR proteins.
Figure 4: K180R does not change intra-SR localization of Casq2.
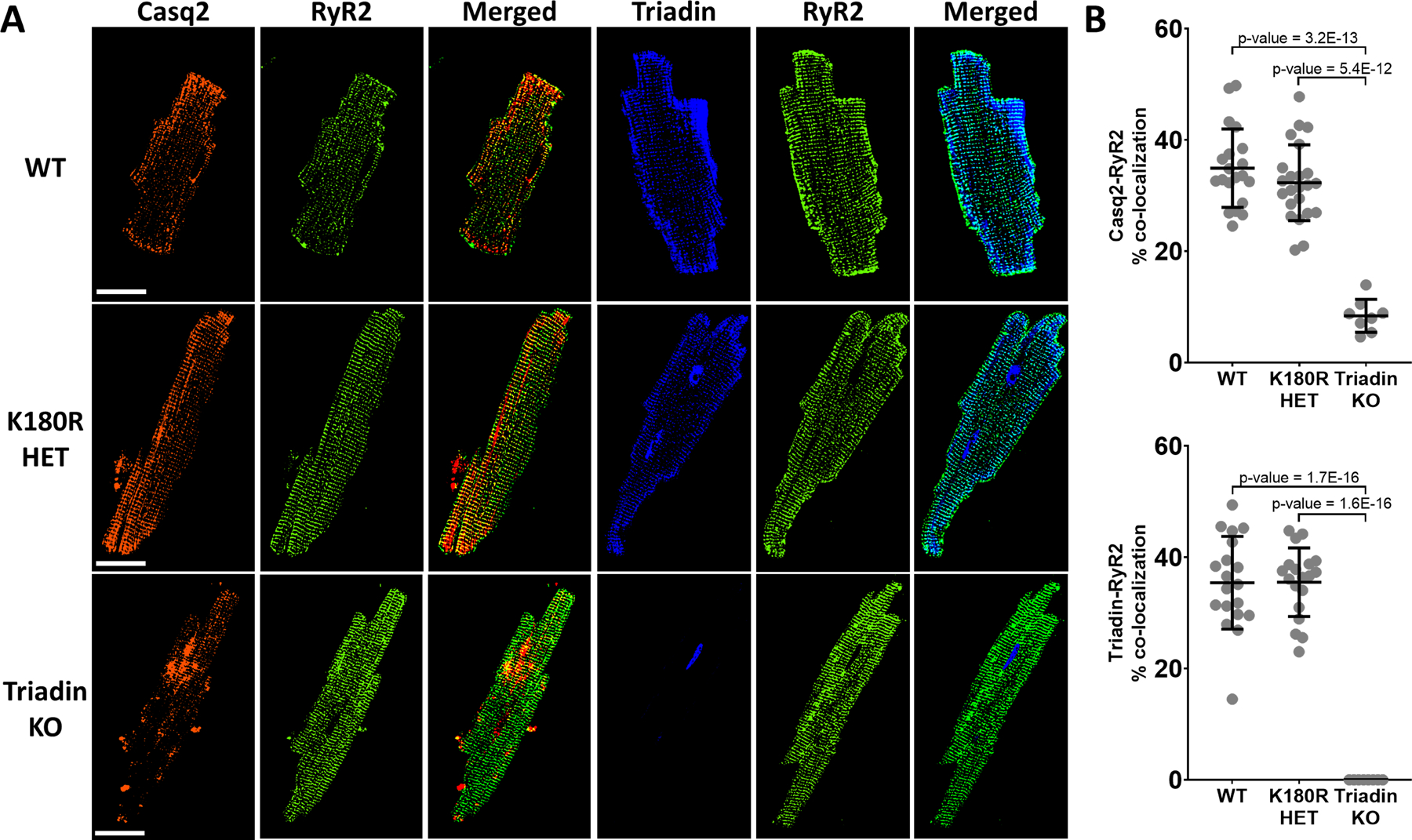
Cardiomyocytes isolated from K180R mice were stained for Casq2 and RyR2 or triadin and RyR2 to analyze co-localization between junctional SR proteins. A. Immunostaining displaying representative images for each genotype. B. Co-localization analysis was performed and displayed as a percent of overlapping pixels between images. WT n = 20 cells from 2 hearts, K180R HET n = 23 cells from 2 hearts, Triadin KO n = 8 cells from 2 hearts (top panel). WT n = 19 cells from 2 hearts, K180R HET n = 19 cells from 2 hearts, Triadin KO n = 8 cells from 2 hearts (bottom panel). Data displayed as mean ± SD. Data analyzed using a hierarchical statistical model with Bonferroni correction. Scale bar = 50 µM.
K180R does not alter total SR Ca levels under steady-state conditions
Given its role as the major Ca buffer in the SR, a decrease in Casq2 protein causes a reduction in total SR Ca content.18 Our experiments in intact cardiomyocytes suggested that K180R does not alter total SR Ca content (Fig. S5;). To quantify total SR Ca content more accurately, we measured NCX currents in voltage-clamped K180R cardiomyocytes generated during the rapid application of caffeine, which releases the total amount of Ca stored in the SR (Fig. 5A). The integral of NCX currents generated by caffeine-induced SR Ca release is a well-established quantitative measure of total SR Ca content.34 In contrast to Casq2 KO cardiomyocytes, which exhibited a significant decrease in the NCX integral, the NCX integral of K180R cardiomyocytes was not significantly different from WT cardiomyocytes (Fig. 5B). Taken together, our data indicate that unlike autosomal-recessive CPVT2 cardiomyocytes, K180R cardiomyocytes have normal total SR Ca content.
Figure 5: K180R does not change total SR Ca content.
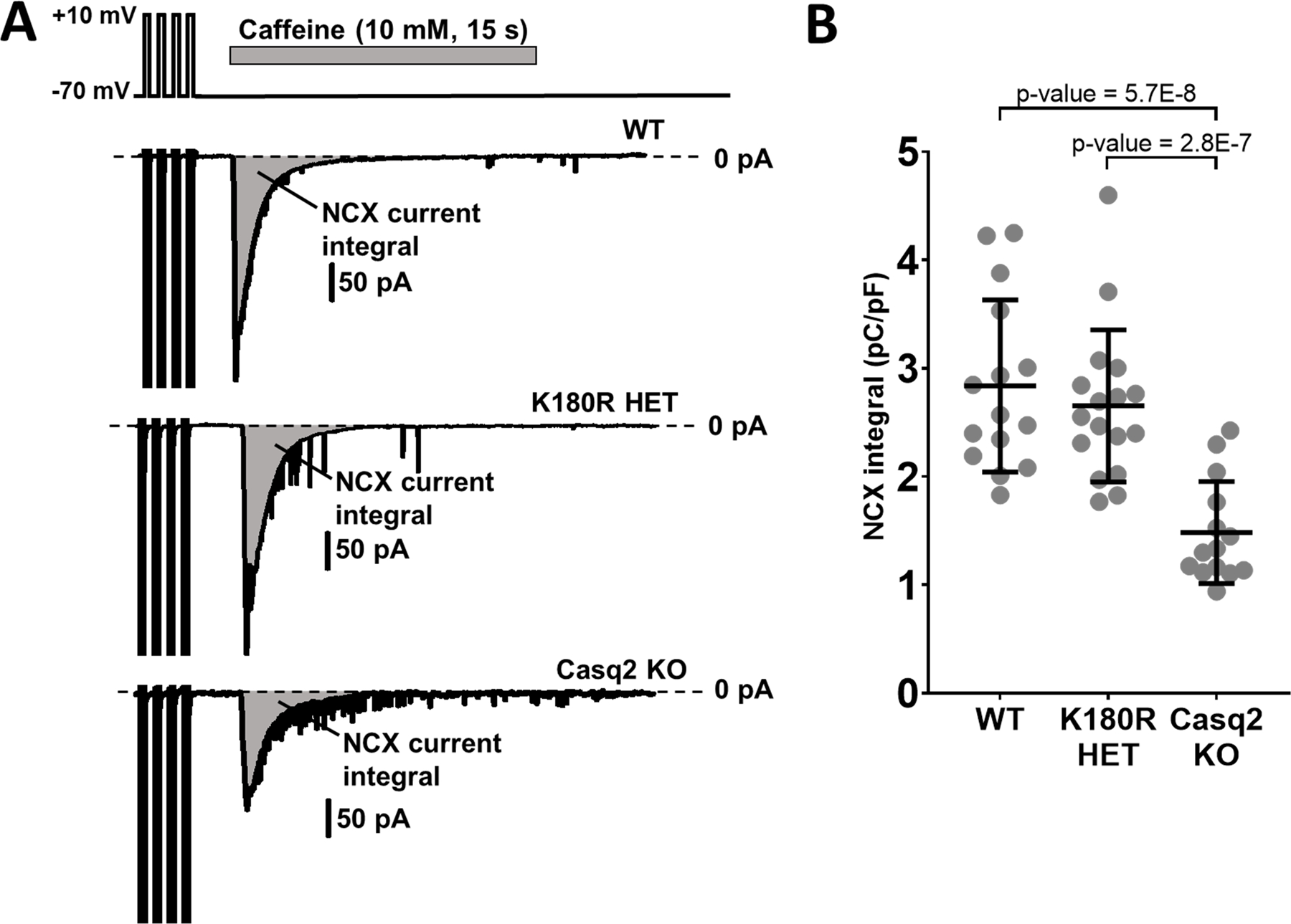
Total SR Ca content was quantified by integration of NCX current elicited in response to rapid caffeine application. Following four brief membrane depolarizations from −70 mV to +10 mV at 2 Hz to ensure consistent SR Ca loading, caffeine (10 mM) was applied using a rapid solution exchanger for 15 s at a holding potential of −70 mV. Isoproterenol (1 µM) was added to the external solution to activate beta-adrenergic receptors. NCX current integrals were normalized by cell capacitance, a measure of cell size and expressed as pC/pF. A. Representative traces of the NCX currents from 3 groups. B. NCX integral summary data. WT n = 15 cells from 5 hearts; K180R HET n = 17 cells from 4 hearts; Casq2 KO n = 14 cells from 5 hearts. Data displayed as mean ± SD. Data analyzed using a hierarchical statistical model with Bonferroni correction.
K180R decreases SR Ca release refractoriness
Casq2 is known to regulate the refractory period during each E-C coupling cycle. By buffering free Ca in the SR, Casq2 prevents premature release of Ca during diastole. Absence of Casq2 decreases the refractory period of Ca release which leads to premature Ca release and arrythmias.35 Our experiments showed that the K180R variant produces CPVT in vivo and premature spontaneous Ca release in vitro without affecting Casq2 levels or total SR Ca content. To test if K180R alters SR Ca release refractoriness, isolated cardiomyocytes were loaded with the Ca fluorescent indicator fluo-4, patched in voltage clamp mode, and underwent an established stimulation protocol.22 To avoid confounding effects from reduced Ca currents at short coupling intervals, we used ICa tail currents to trigger Ca release, as detailed in our previous report.22 Tail currents allowed us to maintain a maximal and constant Ca current trigger and hence isolated RyR2 release from potential changes in the Ca trigger current. Ca transients from WT and K180R HET mice were recorded and normalized to first peak amplitude (Fig. 6A). The amplitude from the premature beat (S2) was measured and compared to the amplitude of the first peak (S1) to calculate a fractional recovery of the Ca transient (ratio of S2/S1, Fig. 6B). The ratio was then plotted as a function of the coupling interval between S1 and S2 to assess the amount of Ca release occurring during different periods of diastole. K180R HET myocytes had a significantly higher fractional recovery of Ca transients at shorter coupling intervals compared to WT myocytes (Fig. 6C). At the same time, the S1 Ca transient and SR Ca content were not significantly different between the two groups (Fig. S7). Hence, the higher amount of Ca being released at shorter coupling intervals indicates that the K180R variant impairs SR Ca release refractoriness, which would render the cell prone to release Ca prematurely.
Figure 6: SR Ca release refractoriness is impaired in K180R cardiomyocytes.
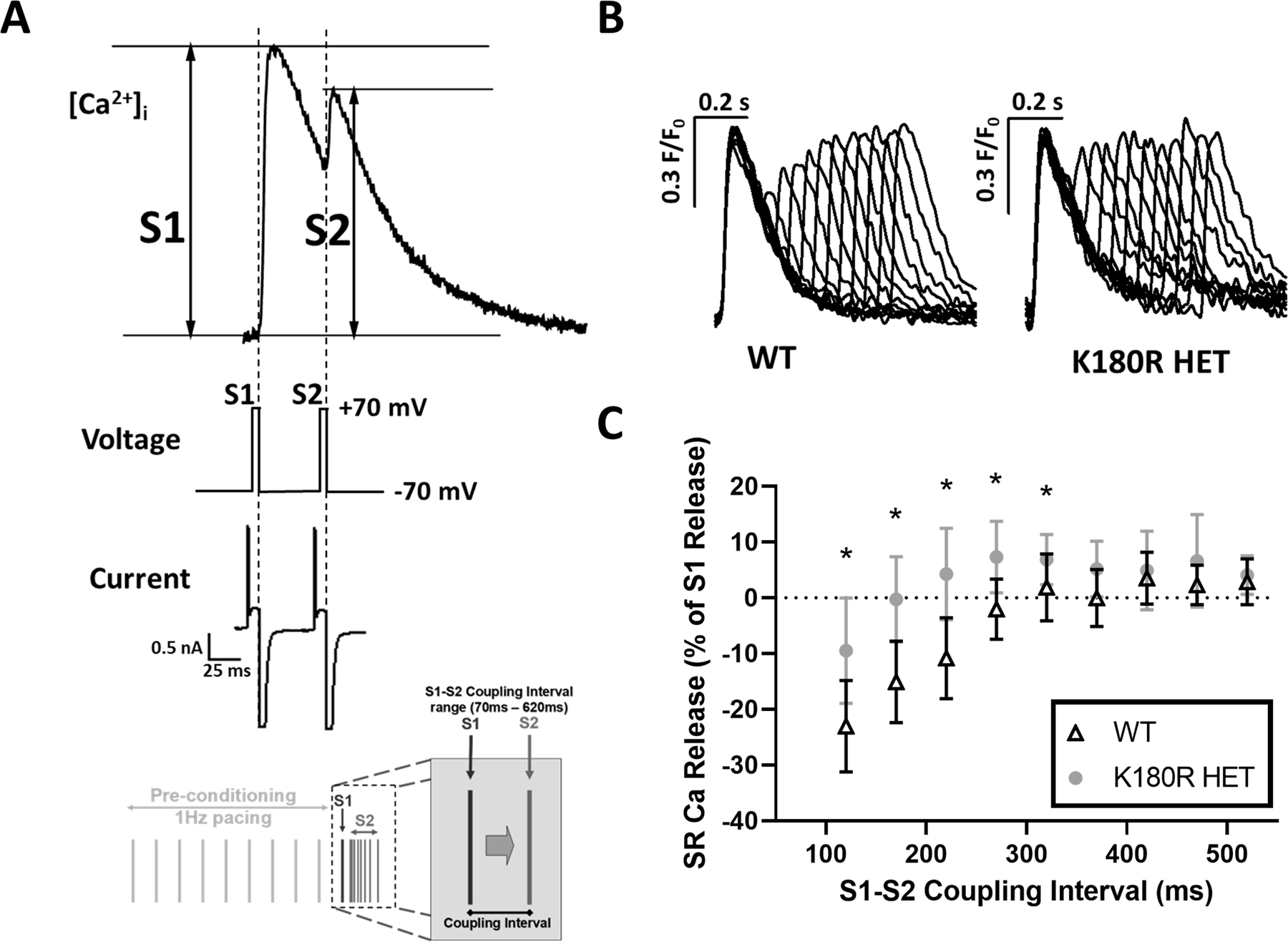
To measure the refractoriness of SR Ca release, isolated cardiomyocytes were loaded with the Ca indicator fluo-4, patched in voltage clamp mode, and a voltage stimulation protocol used as shown in bottom of panel A. Isoproterenol was not used in this experiment. SR Ca release was triggered by L-type Ca tail currents elicited by stepping membrane potential from +70 to −70mV. A. Example [Ca]i fluorescent trace, voltage protocol, and membrane currents elicited during the S1 and S2 voltage step, illustrating how S1 and S2 transient amplitudes were calculated following the restitution protocol. B. Representative Ca transients for WT and K180R HET cells. Traces were normalized to the amplitude of the first peak (S1) and superimposed. C. Average S2 SR Ca release fraction plotted as a function of varying the S1–S2 coupling interval. WT n = 8 cells from 3 hearts; K180R HET n = 10 cells (9 at 120 ms) from 3 hearts. Data displayed as mean ± SD. Data was analyzed using a hierarchical statistical model with Bonferroni correction. *P-values = 0.0039, 0.00034, 0.00041, 0.0026, and 0.045, respectively.
K180R alters dynamic intra-SR Ca buffering
Given that K180R reduced the time to the first spontaneous Ca release event (Fig. 2) and the refractoriness of SR Ca release (Fig. 6), we reasoned that K180R impairs the ability of the SR to buffer Ca appropriately following Ca release. To quantify intra-SR Ca buffering, we next measured cytosolic and intra-SR free Ca simultaneously. A low-affinity esterified Ca indicator, Calbryte 520L-AM, was loaded into the SR of the cardiomyocytes. A high-affinity Ca indicator, Calbryte 590 potassium salt, was used to measure free Ca in the cytosol (Fig. 7A) and was loaded via permeabilization of the cell membrane with saponin. Ca waves in the cytosol and corresponding Ca depletions in the SR were recorded concurrently via confocal line scans (Fig. 7A, B). The amplitude of the cytosolic Ca transient and the corresponding Ca depletion in the SR was not different between the groups (Fig. 7B–D). Notably, following Ca release, the free Ca concentration in the SR rose more quickly in K180R compared to WT cardiomyocytes, as evidenced by a shorter time to 90% recovery of the SR Ca depletion transient (Fig. 7B–D). On the other hand, the rate of Ca uptake into the SR, measured by the time to 90% decay of the cytosolic Ca transient (Fig. 7D), was identical in both groups. In other words, for the same amount of Ca entering the SR, the free SR Ca concentration increased significantly faster in K180R compared to WT cardiomyocytes. These results directly demonstrate that K180R impairs the dynamic buffering of Ca by Casq2. Given that the amplitude of the SR Ca depletion signal (i.e., the maximal change in intra-SR free Ca concentration during the SR Ca release event) was not different between the groups, this result indicates that steady-state SR Ca buffering is not altered by K180R, consistent with our finding that total SR Ca content is unchanged in K180R cardiomyocytes (Figs. 5 and S5). To exclude the possibility that the higher wave frequency could affect intra-SR Ca kinetics, we next measured intra-SR Ca handling in intact cardiomyocytes at a fixed pacing rate of 1 Hz (Fig. 8). Corroborating our results from the permeabilized cardiomyocytes, the K180R variant caused a more rapid rise of intra-SR Ca after each paced beat without altering the amplitude of the intra SR Ca depletion signal (Fig. 8). Taken together, our results indicate that the K180R variant causes premature Ca release by impairing the rate of Ca binding to Casq2 (i.e., dynamic Ca buffering) without changing steady-state Ca buffering by Casq2.
Figure 7: K180R decreases dynamic SR Ca buffering without affecting steady-state SR Ca buffering.
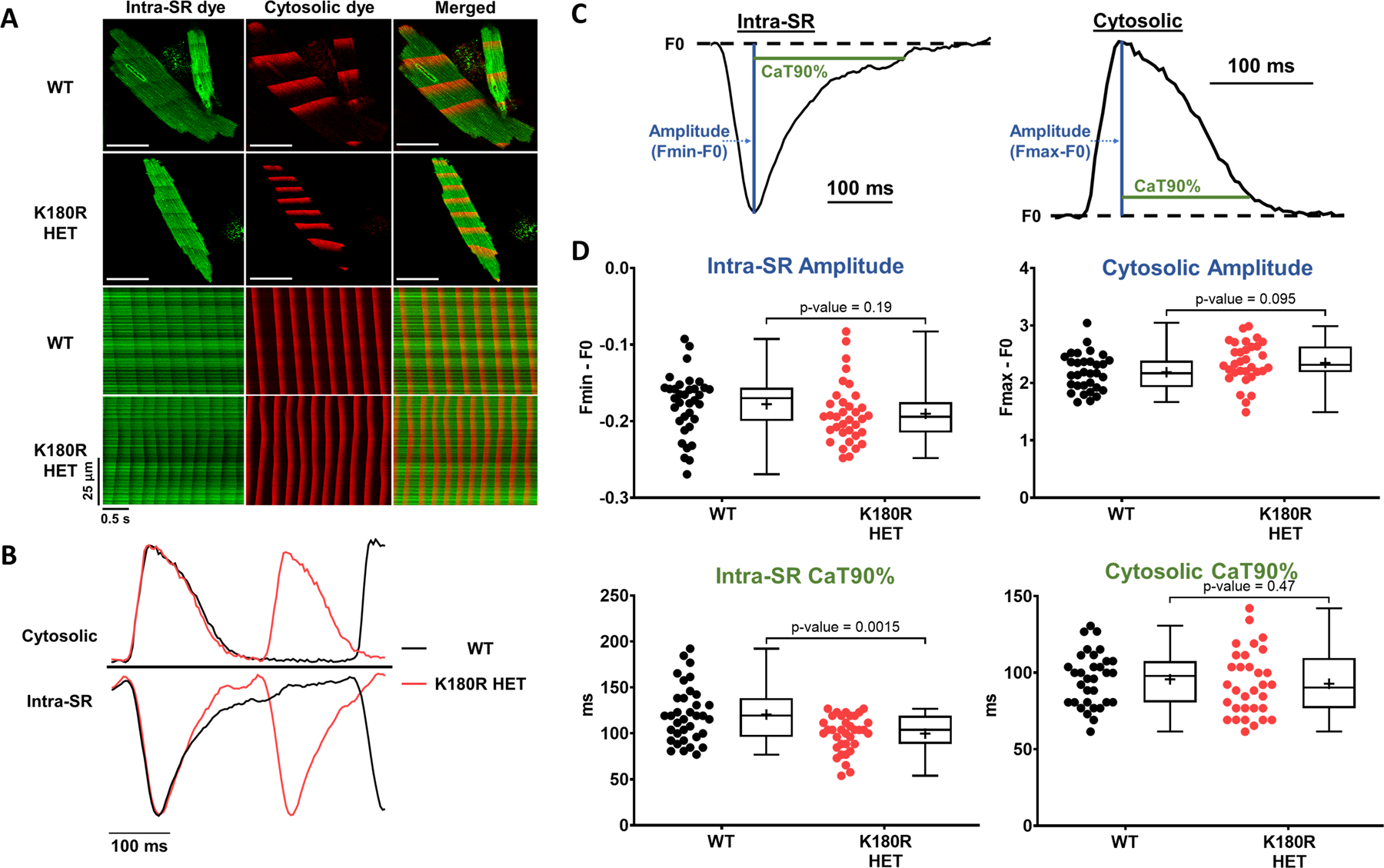
Cytosolic and intra-SR free Ca was measured simultaneously using a dual-indicator approach. Briefly, cardiomyocytes were first loaded with an esterified low-affinity Ca indicator that accesses the SR (Calbryte-520L AM). Cells were then permeabilized and loaded with the high-affinity Ca indicator Calbryte-590, potassium salt, to measure cytosolic Ca levels. Cells were imaged using an inverted confocal microscope in line-scan mode. A. Top: Representative images of indicator loading and wave propagation in permeabilized cardiomyocytes. Bottom: Example line scan images for SR and cytosolic fluorescent Ca indicators during Ca waves. Scale bar = 50 µm. B. Representative example trace of fluorescent signals generated from the line scans, illustrating the faster rise of intra-SR Ca in K180R HET cardiomyocytes. C. To compare Ca dynamics between groups, cytosolic Ca transients and intra-SR Ca depletion transients were parameterized by the transient amplitude and time to 90% recovery (CaT90%). D. Summary data of Ca dynamics. Intra-SR data: WT n = 34 cells from 3 hearts; K180R HET n = 35 cells from 3 hearts. Cytosolic data: WT n = 31 cells from 3 hearts for; K180R HET n = 32 cells from 3 hearts. Data reported as mean ± SD. Data analyzed using a hierarchical statistical model with Bonferroni correction.
Figure 8: Intra-SR Ca dynamics in intact K180R cardiomyocytes paced at 1 Hz.
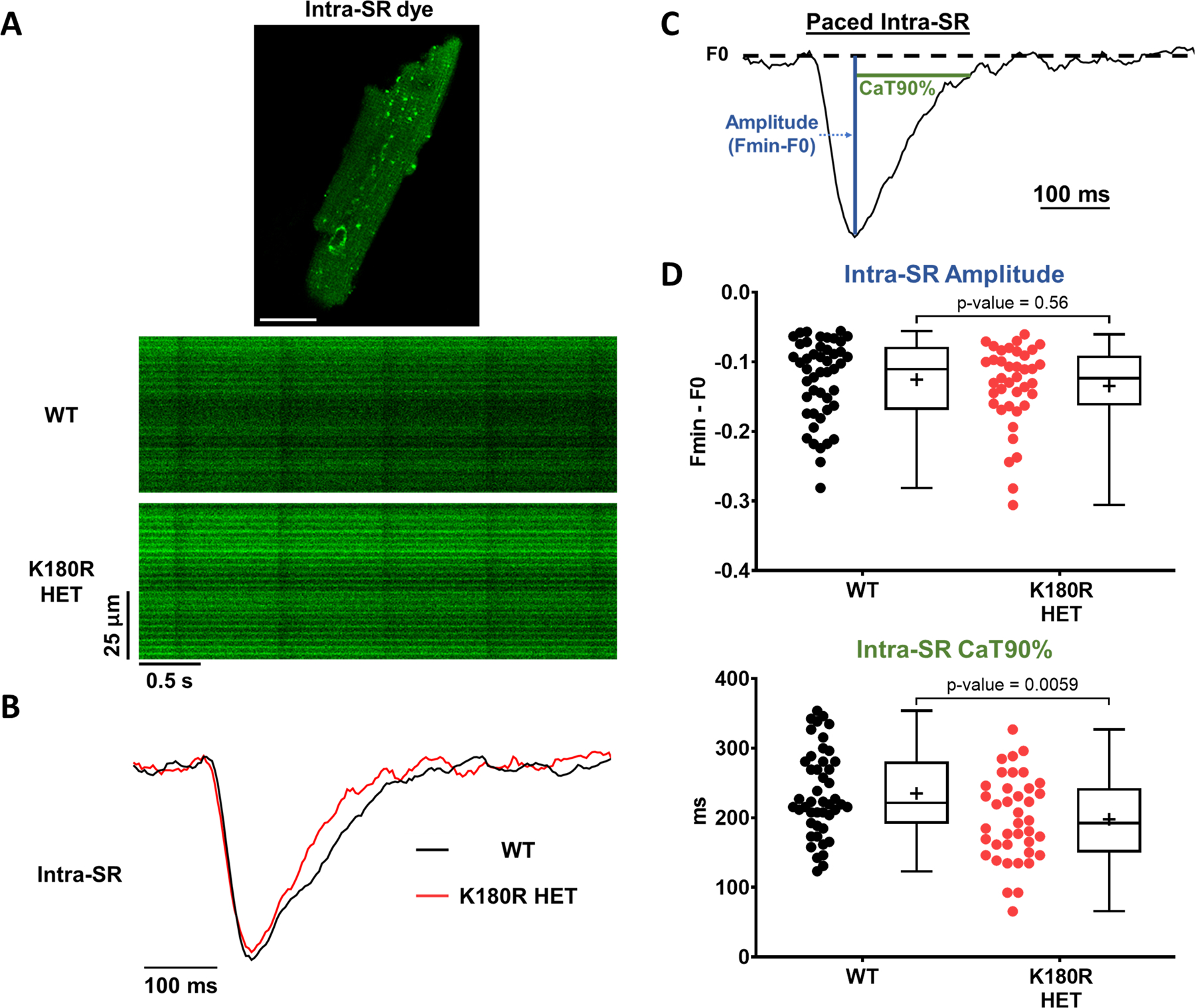
Intra-SR Ca was measured in intact cardiomyocytes using the low-affinity Ca indicator Mag-Fluo-4 AM. Cells were incubated in 2 mM Ca normal Tyrode solution without isoproterenol and paced at 1 Hz. Cells were imaged using an inverted confocal microscope in line-scan mode and analyzed using ImageJ. A. Example image of an isolated cardiomyocyte loaded with Mag-Fluo-4 AM (top), with representative line scans paced at 1 Hz (bottom). B. Representative example trace of fluorescent signals generated from the line scans, illustrating the faster rise of intra-SR Ca in K180R HET cardiomyocytes. C. To compare Ca dynamics between groups, cytosolic Ca transients and intra-SR Ca depletion transients were parameterized by the transient amplitude and time to 90% recovery (CaT90%). D. Summary data of intra-SR Ca dynamics. WT n = 47 (amplitude) or 46 (CaT90%) cells from 3 hearts; K180R HET n = 39 cells from 3 hearts. Data reported as mean ± SD. Data analyzed using a hierarchical statistical model with Bonferroni correction.
Discussion
In the heart, Casq2 regulates SR free Ca levels and Ca release during the excitation-contraction (E-C) coupling. Variants in CASQ2 can cause a lethal arrhythmia syndrome, catecholaminergic polymorphic ventricular tachycardia type 2 (CPVT2). In autosomal recessive CPVT2, CASQ2 variants cause CPVT by drastically reducing or completely abolishing Casq2 protein. The absence of Casq2 reduces the ability of the SR to buffer Ca appropriately, resulting in a propensity to release Ca prematurely and cause delayed afterdepolarizations, triggered beats and ultimately CPVT.29 The first autosomal dominant variant in Casq2, K180R, was reported in 2016.9 Analogous to autosomal recessive variants, it was expected that K180R would decrease Casq2 protein levels and cause CPVT via a dominant negative mechanism whereby mutant K180R Casq2 would bind to WT Casq2, ultimately resulting in the degradation of the Casq2 multimer. However, this proposed dominant negative hypothesis had not been examined and the pathophysiology of the K180R variant was unknown. Here, we developed a K180R mouse model to study the pathophysiology of the K180R-linked autosomal dominant CPVT. We find that K180R produces a CPVT phenotype in both mice and isolated cardiomyocytes. Analogous to autosomal recessive Casq2 variants, K180R also decreases Ca release refractoriness. However, the underlying mechanism is fundamentally different, because K180R does NOT alter Casq2 protein levels, Casq2 polymers, or steady-state Ca buffering in the SR. Rather, K180R appears to reduce the rate of Ca binding to Casq2, resulting in a more rapid rise of free Ca within the SR after a heartbeat. In other words, K180R alters the dynamic ability of Casq2 to regulate buffering of Ca ions as they move in and out of the SR during the E-C coupling cycle. Static buffering (e.g., steady-state Ca levels in the SR) appears to be not altered by the K180R variant, which is fundamentally different from autosomal-recessive Casq2 variants that reduce Casq2 protein levels and hence total SR Ca buffering. By affecting the dynamic buffering, K180R is decreasing the SR Ca release refractoriness of the cell. As Ca is re-entering the SR during diastole, the SR is now prone to having a faster rise in free Ca levels, which can lead to premature Ca release. We acknowledge that several in vitro data sets were based on a relatively small number of mice, which may limit the robustness of individual datasets. Nevertheless, the main experimental findings (i.e., K180R myocytes exhibit increased spontaneous SR Ca release, increased rate of rise in intra-SR free [Ca], no change in total SR Ca content) was confirmed in multiple independent experimental approaches, demonstrating the robustness of the overall conclusions.
How does the K180R cause an impairment of dynamic SR Ca buffering? While the reported findings give insight into the pathophysiology of the SR Ca handling caused by K180R Casq2, the variant itself is quite small and is a conserved amino acid change within the structure of the protein. Recent studies have suggested that the K180R variant may affect the polymerization of Casq2.10,31 Casq2 has been found to form monomers, dimers, and polymers through a Ca dependent process.14 The polymerization of Casq2 plays an important role in the ability of Casq2 to bind Ca and to provide a structural component for the SR.36 A recent breakthrough allowed for the crystallization of the Casq2 polymer and was studied to gain insight into how Casq2 can cause CPVT. The study investigated potential autosomal dominant variants in CASQ2 that have been linked to families with CPVT.31 Based on the results of in vitro turbidity assays, the authors concluded that K180R and the other autosomal dominant CASQ2 variants decreased the ability of Casq2 to polymerize. Further investigation of the crystallized polymer mapped the K180 amino acid onto a potential inter-dimerization site of Casq2. Based on the finding of impaired Casq2 polymerization, the authors hypothesized that the K180R variant will cause the depletion of part of the wild type and variant protein populations due to their inability to polymerize in the SR appropriately. The removal of WT or variant protein would affect the protein levels of Casq2 in K180R cells. However, we found that this hypothesis is not correct. Based on our western blot analysis, Casq2 protein levels are not decreased in K180R HET or HOM mice (Fig. 3). Furthermore, K180R-Casq2 is also capable of forming polymers in vivo, as evidenced by our western blot and electron microscopy data (Fig. 3, Fig. S5). Polymerization is critical for trafficking and retention of Casq2 to the SR, since alterations in Casq2 polymerization, due to the addition of tags or fluorescent markers, lead to an inability of the protein to localize to the SR appropriately.37–39 If K180R was disrupting the polymerization of Casq2, it would be expected that Casq2 would not be retained in the SR. Based on our results from K180R HET and HOM mice, this is not the case. Rather, K180R Casq2 appears to be trafficked correctly and able to polymerize. Our data indicate that in vivo, the K180R mutations cause a more subtle defect in the Casq2 polymer, whereby the rate of Ca binding to Casq2 is reduced. As previously discussed, Casq2 is a high-capacity, low-affinity Ca-binding protein that binds 40–50 Ca ions mainly through the 60–70 negatively charged amino acid residues within the monomer.11 The initial polymerization studies suggested that the ability for Casq2 to bind Ca may also be dependent on polymerization state with an increase in Ca affinity as the polymer forms.36 The recent Casq2 polymer crystal structure also revealed novel potential Ca binding sites within the polymer and found a large cavity within the center of the polymer where Ca ions could bind.31 Currently, there are no studies that have measured the Ca binding affinity (Kd) of the different states of Casq2. The location of K180 within the Casq2 polymer is proposed to be an inter-dimer region. The same region was also found to have two potential Ca binding sites located near K180.31 It is possible that K180R could affect the ability of the Casq2 polymer to bind Ca given its involvement in the inter-dimer region. This mechanism would help explain how K180R is affecting the dynamic buffering capabilities of the SR. As the polymerization of Casq2 is a Ca dependent process, when Ca is re-entering the SR during diastole, K180R could be increasing the Ca Kd of the polymer. A decreased Ca affinity is consistent with the more rapid SR Ca refilling kinetics that we observed in K180R HET cardiomyocytes (Figs. 7 and 8). The faster refilling would then lead to a decrease in SR Ca release refractoriness (Fig. 6), an increase in the potential for spontaneous Ca release events (Fig. 2), and ultimately generates CPVT (Fig. 1).
The results of our experiments have broader implications regarding the pathophysiology of CASQ2 variants. Recently, a large multi-center study was conducted to look for prevalence of autosomal-dominant CASQ2 variants within CPVT families.10 Of 36 probands, 12 had heterozygous CASQ2 variants. The penetrance of heterozygous variants was lower than that of homozygous variants, 33.3% vs. 97.1%, but heterozygous patients still experienced similar symptoms and severity of arrhythmias as homozygous patients (e.g., aborted cardiac arrest). The study identified several other autosomal-dominant CASQ2 variants that could be the target of future studies to confirm if other autosomal-dominant variants follow a similar mechanism as K180R.
Here we have shown that K180R is an autosomal-dominant CASQ2 variant that is able to produce CPVT in mice. The K180R variant disrupts cellular Ca handling by altering the dynamic intra-SR buffering of Ca without reducing the levels of Casq2 or other junctional SR proteins. K180R had no effect on the static Ca buffering of the SR. The impaired dynamic buffering increases the rate of Ca rise in the SR and thereby decreases SR Ca release refractoriness. The reduced SR Ca release refractory period can lead to spontaneous Ca release, delayed after depolarizations, triggered beats, and CPVT. Hence, our experimental work has uncovered a novel mechanism responsible for autosomal-dominant CPVT2 and thereby provides a template to investigate if other heterozygous variants in CPVT2 follow a similar pathophysiology.
Supplementary Material
Novelty and Significance.
What is known?
Catecholaminergic polymorphic ventricular tachycardia type 2 (CPVT2) is a lethal, stress-induced cardiac channelopathy that is caused by mutations in the gene encoding calsequestrin (Casq2)
Autosomal recessive mutations are the more common cause of CPVT2, but autosomal dominant mutations are increasingly recognized and cause a similar phenotype.
Autosomal recessive mutations in Casq2 decrease Casq2 protein levels leading to hyperactive cardiac ryanodine receptor (RyR2) channels, impaired calcium (Ca) release termination, shortened Ca release refractory period, and enhanced spontaneous release of Ca.
What new information does this article contribute?
The Casq2-K180R genetic variant causes an autosomal dominant CPVT phenotype in mice.
K180R increases spontaneous sarcoplasmic reticulum (SR) Ca release events, delayed afterdepolarizations and impairs SR Ca release refractoriness by altering the dynamic Ca buffering in the SR.
Unlike autosomal recessive CPVT2, K180R causes CPVT without affecting static SR Ca buffering or decreasing Casq2 protein levels.
Acknowledgements
We would like to thank Dr. Eric Delpire, Department of Anesthesiology, Vanderbilt University Medical Center, and the Vanderbilt Genome Editing Resource (VGER) for their help in generating the Casq2-K180R mouse model.”
Sources of funding
This research was supported by the US National Institutes of Health grants NHLBI R35 HL144980 (to BCK) and F30-HL145917 (to M.W.). This research was also supported by an American Heart Association grant 19SFRN34830019 (to BCK). Imaging was supported by Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY008126)
Non-standard Abbreviations and Acronyms
- Ca
calcium
- Casq2
cardiac calsequestrin
- CICR
calcium induced calcium release
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CRU
calcium release unit
- DAD
delayed afterdepolarizations
- E-C
excitation-contraction
- EKG/ECG
electrocardiogram
- FDHM
full duration at half maximum
- FWHM
full width at half maximum
- HET
heterozygous
- HOM
homozygous
- HRP
horseradish peroxidase
- jSR
junctional SR
- kDa
kilodalton
- NCX
sodium-calcium exchanger
- NT
normal Tyrode
- PVC
premature ventricular contraction
- RyR2
cardiac ryanodine receptor
- SCR
spontaneous calcium release events
- SR
sarcoplasmic reticulum
- VEB
ventricular ectopic beat
- VT
ventricular tachycardia
- WT
wild-type
Footnotes
This manuscript was sent to Francesco Violi, Senior Guest Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures
The authors have declared that no conflict of interest exists.
References
- 1.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995;91:1512–1519. [DOI] [PubMed] [Google Scholar]
- 2.Pérez-Riera AR, Barbosa-Barros R, de Rezende Barbosa MPC, Daminello-Raimundo R, de Lucca AA, de Abreu LC. Catecholaminergic polymorphic ventricular tachycardia, an update. Ann Noninvasive Electrocardiol 2018;23:e12512. doi: 10.1111/anec.12512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roston TM, Yuchi Z, Kannankeril PJ, Hathaway J, Vinocur JM, Etheridge SP, Potts JE, Maginot KR, Salerno JC, Cohen MI, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace 2018;20:541–547. doi: 10.1093/europace/euw389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 2001;69:1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation 2001;103:2822–2827. doi: 10.1161/01.cir.103.23.2822 [DOI] [PubMed] [Google Scholar]
- 6.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2002;91:e21–26. [DOI] [PubMed] [Google Scholar]
- 7.de la Fuente S, Van Langen IM, Postma AV, Bikker H, Meijer A. A case of catecholaminergic polymorphic ventricular tachycardia caused by two calsequestrin 2 mutations. Pacing Clin Electrophysiol 2008;31:916–919. [DOI] [PubMed] [Google Scholar]
- 8.Kirchhefer U, Wehrmeister D, Postma AV, Pohlentz G, Mormann M, Kucerova D, Muller FU, Schmitz W, Schulze-Bahr E, Wilde AA, et al. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J Mol Cell Cardiol 2010;49:95–105. [DOI] [PubMed] [Google Scholar]
- 9.Gray B, Bagnall RD, Lam L, Ingles J, Turner C, Haan E, Davis A, Yang PC, Clancy CE, Sy RW, et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016;13:1652–1660. doi: 10.1016/j.hrthm.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng K, Titus EW, Lieve KV, Roston TM, Mazzanti A, Deiter FH, Denjoy I, Ingles J, Till J, Robyns T, et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of. Circulation 2020;142:932–947. doi: 10.1161/CIRCULATIONAHA.120.045723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem 1994;135:61–70. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 1997;272:23389–23397. [DOI] [PubMed] [Google Scholar]
- 13.Bers DM. Cardiac excitation-contraction coupling. Nature 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 14.Park H, Wu S, Dunker AK, Kang C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem 2003;278:16176–16182. [DOI] [PubMed] [Google Scholar]
- 15.Györke S, Stevens SC, Terentyev D. Cardiac calsequestrin: quest inside the SR. J Physiol 2009;587:3091–3094. doi: 10.1113/jphysiol.2009.172049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manno C, Figueroa LC, Gillespie D, Fitts R, Kang C, Franzini-Armstrong C, Rios E. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc Natl Acad Sci U S A 2017;114:E638–E647. doi: 10.1073/pnas.1620265114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol 1985;85:247–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. The Journal of clinical investigation 2006;116:2510–2520. doi: 10.1172/JCI29128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res 2005;96:e77–82. [DOI] [PubMed] [Google Scholar]
- 21.Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res 2008;103:298–306. doi: [DOI] [PubMed] [Google Scholar]
- 22.Kryshtal DO, Gryshchenko O, Gomez-Hurtado N, Knollmann BC. Impaired calcium-calmodulin-dependent inactivation of Cav1.2 contributes to loss of sarcoplasmic reticulum calcium release refractoriness in mice lacking calsequestrin 2. Journal of molecular and cellular cardiology 2015;82:75–83. doi: 10.1016/j.yjmcc.2015.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, et al. Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circulation research 2007;101:617–626. doi: 10.1161/CIRCRESAHA.107.157552 [DOI] [PubMed] [Google Scholar]
- 24.Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 2006;98:1151–1158. doi: 10.1161/01.RES.0000220647.93982.08 [DOI] [PubMed] [Google Scholar]
- 25.Titus EW, Deiter FH, Shi C, Wojciak J, Scheinman M, Jura N, Deo RC. The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. bioRxiv 2019:672303. doi: 10.1101/672303 [DOI] [PMC free article] [PubMed]
- 26.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE, MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res 2017;113:1743–1752. doi: 10.1093/cvr/cvx151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol 1997;272:H657–668. doi: 10.1152/ajpheart.1997.272.2.H657 [DOI] [PubMed] [Google Scholar]
- 28.Bers DM. Excitation-contraction coupling and cardiac contractile force Dordrecht; Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 29.Wleklinski MJ, Kannankeril PJ, Knollmann BC. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. The Journal of physiology 2020;598:2817–2834. doi: 10.1113/JP276757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proceedings of the National Academy of Sciences of the United States of America 2009;106:7636–7641. doi: 10.1073/pnas.0902919106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Titus EW, Deiter FH, Shi C, Wojciak J, Scheinman M, Jura N, Deo RC. The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. Nat Struct Mol Biol 2020;27:1142–1151. doi: 10.1038/s41594-020-0510-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem 1995;270:30787–30796. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi YM, Jones LR. Identification of triadin 1 as the predominant triadin isoform expressed in mammalian myocardium. J Biol Chem 1999;274:28660–28668. [DOI] [PubMed] [Google Scholar]
- 34.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca²⁺ release and reduces rate-dependent Ca²⁺-alternans in intact mouse hearts. J Mol Cell Cardiol 2012;52:21–31. doi: 10.1016/j.yjmcc.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem 2004;279:18026–18033. [DOI] [PubMed] [Google Scholar]
- 37.Milstein ML, Houle TD, Cala SE. Calsequestrin isoforms localize to different ER subcompartments: evidence for polymer and heteropolymer-dependent localization. Exp Cell Res 2009;315:523–534. [DOI] [PubMed] [Google Scholar]
- 38.McFarland TP, Milstein ML, Cala SE. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J Mol Cell Cardiol 2010;49:556–564. doi: S0022–2828(10)00231–2 [pii] 10.1016/j.yjmcc.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sleiman NH, McFarland TP, Jones LR, Cala SE. Transitions of protein traffic from cardiac ER to junctional SR. J Mol Cell Cardiol 2015;81:34–45. doi: 10.1016/j.yjmcc.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, et al. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem 2005;280:37183–37194. [DOI] [PubMed] [Google Scholar]
- 41.Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D. Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am J Physiol 1997;272:H1053–1061. doi: 10.1152/ajpheart.1997.272.2.H1053 [DOI] [PubMed] [Google Scholar]
- 42.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol 2010;99:1–26. doi: 10.1016/B978-0-12-374841-6.00001-3 [DOI] [PubMed] [Google Scholar]
- 43.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 2007;293:C1073–1081. doi: 00586.2006 [pii] 10.1152/ajpcell.00586.2006 [DOI] [PubMed] [Google Scholar]
- 44.Galimberti ES, Knollmann BC. Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin. J Mol Cell Cardiol 2011;51:760–768. doi: 10.1016/j.yjmcc.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shankar TA-O, Ramadurai DKA, Steinhorst K, Sommakia S, Badolia R, Thodou Krokidi A, Calder D, Navankasattusas S, Sander PA-O, Kwon OS, et al. Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis [DOI] [PMC free article] [PubMed]
- 46.Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011;4:128–135. doi: 10.1161/CIRCEP.110.959916 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The use of animals was approved by the Animal Care and Use Committee of Vanderbilt University. Experimental group sizes were based on previous studies examining CPVT mouse models.18 K180R mice were generated using CRISPR/Cas9 and utilized to assay CPVT susceptibility via multiple stress tests. Isolated ventricular myocytes underwent Ca handling analysis using Ca sparks and waves protocols. Western blotting, immunostaining and electron microscopy were used to investigate jSR protein levels and localization. Single cell experiments were used to measure spontaneous Ca release events, total SR Ca content, and SR Ca release refractoriness. Intra-SR and cytosolic Ca buffering properties were assessed by simultaneously measuring intra-SR and cytosolic free Ca using low and high-affinity Ca indicators, respectively. Statistical tests used are reported in the figure legends. Results from isolated cardiomyocytes were compared using a hierarchical statistical model following the procedures by Sikkel et al 2017.26 Briefly, a linear mixed effects (hierarchical) model was used to calculate Bonferroni-adjusted p-values clustered at the animal level to account for random effects between isolations. All p-values reported in the MS are corrected for multiple comparisons. Details such as statistical tests used, normality testing, exact raw and corrected p-values, are provide in the statistical summary table published online. An expanded method section is provided in the Supplemental Material document.