

Abstract
Type I conventional dendritic cells (cDC1s) are an essential antigen-presenting population required for generating adaptive immunity against intracellular pathogens and tumors. While the transcriptional control of cDC1 development is well understood, the mechanisms by which extracellular stimuli regulate cDC1 function remain unclear. We previously demonstrated that the cytokine-responsive transcriptional regulator STAT3 inhibits polyinosinic:polycytidylic acid (poly I:C) -induced cDC1 maturation and cDC1-mediated anti-tumor immunity in murine breast cancer indicating an intrinsic, suppressive role for STAT3 in cDC1s. To probe transcriptional mechanisms regulating cDC1 function, we generated novel RNA-sequencing (RNA-seq) datasets representing poly I:C-, IL-10-, and STAT3-mediated gene expression responses in murine cDC1s. Bioinformatic analyses indicated that poly I:C stimulates multiple inflammatory pathways independent of STAT3, while IL-10-activated STAT3 uniquely inhibits the poly I:C-induced type I interferon (IFN-I) transcriptional response. We validated this mechanism using purified cDC1s deficient for STAT3 or IFN signaling. Our data reveal IL-10-activated STAT3 suppresses production of IFN-β and IFN-γ, accrual of tyrosine phosphorylated STAT1, and IFN-stimulated gene expression in cDC1s following poly I:C exposure. Moreover, we found that maturation of cDC1s in response to poly I:C is dependent on the IFN-I receptor, but not the type II IFN receptor, or IFN-λ. Taken together, we elucidate an essential role for STAT3 in restraining autocrine IFN-I signaling in cDC1s elicited by poly I:C stimulation and we provide novel RNA-seq datasets that will aid in further delineating inflammatory and anti-inflammatory mechanisms in cDC1s.
Introduction
Type I conventional dendritic cells (DCs; cDC1s) are the primary antigen-presenting cells responsible for coordinating CD8+ T cell-mediated immunity against tumors, viruses, and other intracellular pathogens (1). These critical responses are orchestrated by specific functions of cDC1s including cross-presentation of extracellular antigen on MHC-I and activation of cytotoxic CD8+ T cells (1). Moreover, cDC1s induce CD4+ T helper 1 (Th1) polarization and promote formation of tri-cell clusters in lymph nodes (LNs), comprising cDC1s, Th1, and CD8+ T cells, which are required for induction of optimal CD8+ T cell-mediated memory responses (2, 3). Recent studies indicate cDC1-induced Th1 cells “license” cDC1s via CD40 to drive cDC1-mediated CD8+ T cell activation (4). In the tumor microenvironment (TME), cDC1s promote CD8+ T cell recruitment via their production of T cell chemoattractants such as CXCL10 (5, 6), while cDC1 abundance in tumor-draining LNs (TdLNs) is critical to maintain tumor antigen-specific CD8+ T effector cells (7). Consistently, the presence of cDC1s in human tumors correlates positively with patient survival (8, 9), and is predictive of responsiveness to immunotherapy (10).
Therapeutic strategies targeting cDC1s are being investigated as new cancer treatments. For example, increasing cDC1 abundance and enhancing cDC1 maturation in the TME through delivery of the DC-stimulating cytokine FLT3-ligand (FLT3L) and the double-stranded RNA mimetic polyinosinic:polycytidylic acid (poly I:C), respectively, improves responsiveness to immunotherapy with PD-1 (11). Moreover, poly I:C administration increases the efficacy of radiation treatment in a process that requires both cDC1s and CD8+ T cells (12). Blockade of the inhibitory receptor TIM3 has recently been shown to promote IFN signaling, which is important to elicit productive cDC1-mediated anti-tumor immunity (13–15). In addition, cDC1s have been utilized as cell therapy for the treatment of cancer (16–18). Vaccination with in vitro-derived cDC1s controls primary and metastatic murine tumors, and prevents tumor growth upon rechallenge, indicating cDC1-mediated induction of systemic, long-term anti-tumor immunity (16). Nonetheless, while cDC1s are a promising new focus of cancer therapy, the intrinsic molecular regulation of cDC1 function must be better resolved to optimize these approaches.
We demonstrated recently that expression of STAT3 or the IL-10 receptor inhibits the anti-tumor efficacy of a cDC1 vaccine in murine breast cancer (17). Moreover, we found IL-10 and STAT3 restrain co-stimulatory molecule and cytokine expression in cDC1s upon poly I:C stimulation in vitro (17). Furthermore, tumor vaccination with poly I:C-stimulated STAT3-deficient cDC1s results in significant increases in Th1 cells and tumor antigen-specific CD8+ T cells in the TME and TdLNs, and promotes systemic control of murine breast tumors (17). These data collectively indicate an immunosuppressive role for IL-10-STAT3 signaling in cDC1s in the setting of cancer vaccination. STAT3 is also required in CD11c+ cells to inhibit intestinal inflammation in vivo, consistent with an immunosuppressive role in DCs (19). While IL-10 is known to employ cell type-specific anti-inflammatory responses, the immunosuppressive mechanisms mediated by STAT3 have been studied primarily in myeloid subsets such as neutrophils and macrophages (20–22). Thus, the molecular pathway(s) by which STAT3 restrains cDC1 function is unclear.
In this study, we generated novel RNA-sequencing (RNA-seq) datasets to evaluate STAT3-mediated transcriptional regulation by IL-10 and poly I:C in cDC1s. Bioinformatic analyses revealed that IL-10-activated STAT3 selectively inhibits a type I IFN (IFN-I) transcriptional response in cDC1s upon poly I:C treatment. We validated this experimentally by demonstrating STAT3 controls poly I:C-mediated autocrine IFN-I signaling in cDC1s. Our study provides new insight into the transcriptional regulation of cDC1s in response to inflammatory and anti-inflammatory stimuli, with implications for cDC1-mediated immunity. Moreover, our work provides rationale for the development of novel therapeutic strategies to inhibit STAT3 in cDC1s for the purpose of increasing IFN-I signaling and adaptive immune responses.
Materials and Methods
Mouse strains
C57BL/6J, B6.129S2-Ifnar1tm1Agt/Mmjax (Ifnar1−/−), B6.129S7-Ifngr1tm1Agt/J (Ifngr1−/−), and B6.Cg-Tg(Itgax-cre)1–1Reiz/J mice (CD11c-Cre+) mice were acquired from the Jackson Laboratory (Bar Harbor, ME, USA). CD11c-Cre+ mice were bred with Stat3fl/fl mice to generate CD11c-Cre+ Stat3fl/fl mice (Stat3Δ/Δ) and CD11c-Cre− Stat3fl/fl (Stat3fl/fl) littermate controls, as described previously (17). All mice were maintained in a specific pathogen-free animal facility at The University of Texas MD Anderson Cancer Center and treated according to Institutional Animal Care and Use Committee approved protocols.
In vitro generation of cDC1s
cDC1s were derived from cultured mouse bone marrow (BM) as described previously (17). Briefly, BM cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% heat-inactivated FBS (Atlanta Biologicals, Atlanta, GA, USA), 1 mM sodium pyruvate (Thermo Fisher Scientific), 50 μM 2-ME (Thermo Fisher Scientific), 1% penicillin-streptomycin (PS) (Thermo Fisher Scientific), 50 ng/mL human FLT3L (PeproTech, Rocky Hill, NJ, USA), and 2 ng/mL murine GM-CSF (PeproTech). BM cultures were initiated at a density of 1.5 × 106 cells/mL. On day 5, an additional 5 mL of RPMI 1640 medium (containing 10% heat-inactivated FBS, 1 mM sodium pyruvate, 50 μM 2-ME, 1% PS) was added per 10 mL of culture. On day 9, non-adherent cells were collected and transferred to fresh RPMI 1640 medium (containing 10% heat-inactivated FBS, 1 mM sodium pyruvate, 50 μM 2-ME, 1% PS, 50 ng/mL human FLT3L, and 2 ng/mL murine GM-CSF) at a density of 3 × 105 cells/mL. On day 17, non-adherent cells were collected and cDC1s (CD11c+ CD45R− CD24+ CD172α− CD103+) were purified by FACS on a FACSAria III or FACSAria Fusion (BD Biosciences, Palo Alto, CA, USA).
Cytokine, poly I:C, and antibody blocking treatments in vitro
cDC1s were treated with 10 ng/mL IL-10 (PeproTech), 10 ng/mL IFN-β (R&D Systems, Minneapolis, MN USA), 10 ng/mL IFN-γ (PeproTech), 10 ng/mL IFN-λ2 (R&D Systems), 3 μg/mL anti–IFN-λ (R&D Systems), or 20 μg/mL poly I:C (Millipore Sigma), as indicated in the Figure legends.
RNA sequencing (RNA-seq)
Total RNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany). Following purification, RNA abundance, purity, and quality were estimated via NanoDrop (OD 260/280 ≈ 2, OD 260/230 ≈ 2) (Thermo Fisher Scientific). At least 1 μg of RNA per individual biological sample was sent to a commercial sequencing vendor (Novogene, Beijing, China), where it was further assessed for quality by agarose gel electrophoresis, NanoDrop (Thermo Fisher Scientific), and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Subsequently, the RNA underwent poly (A) enrichment and was reverse transcribed to generate 250–300 base pair cDNA libraries. The cDNA libraries were sequenced using the Illumina HiSeq platform (Illumina, San Diego, CA, USA). Sequencing data in fastq file format containing 40–61 × 106 raw reads per sample was then processed and analyzed as follows. RNA-seq by Expectation Maximization (RSEM) was used for transcript quantification and the data were normalized by limma-voom (23–25). Normalized expression values were used for evaluation by Gene Set Enrichment Analysis (GSEA) (26). Differentially expressed genes (DEGs) between indicated groups were determined by Welch’s T test and the uncorrected p value < 0.05. DEGs were analyzed by Ingenuity Pathway Analysis (IPA) using the following cutoffs: fold change ≥ |2| and p < 0.05 (27). The sequencing data generated in this study have been submitted to Gene Expression Omnibus under accession number GSE188995 and can be accessed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE188995
Gene expression determination by quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA) and reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). qRT-PCR was performed using SYBR Green (Millipore Sigma, Darmstadt, Germany) or PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) and the CXF384 Touch Real-Time PCR Detection System with the following protocol: cDNA denaturation at 95°C for 10 seconds; annealing and extension at 60°C for 20 seconds. Target transcript expression was normalized to Rpl13a expression. The following primers were used: Rpl13a forward (F), 5’-GGCTGAAGCCTACCAGAAAG-3’; Rpl13a Reverse (R), 5’-TTCTCCTCCAGAGTGGCTGT-3’; Cxcl10 F, 5’-CCCACGTGTTGAGATCATTG-3’; Cxcl10 R, 5’-GAGGCTCTCTGCTGTCCATC-3’; Ifnb1 F, 5’-CTGAGGCATCAACTGACAGG-3’; Ifnb1 R 5’-GGAAAGATTGACGTGGGAGA-3’; Cxcl11 F, 5’-GGCTTCCTTATGTTCAAACAGGG-3’; Cxcl11 R, 5’-GCCGTTACTCGGGTAAATTACA-3’; Ccrl2 F, 5’-CCCCGGACGATGAATATGATG-3’; Ccrl2 R, 5’-CACCAAGATAAACACCGCCAG-3’; Ripk2 F, 5’-GCCATTGAGATTCCGCATCCT-3’; Ripk2 R, 5’-AACTTCGTGATTGAGAGAGTGAC-3’; Rsad2 F, 5’-AGCATTAGGGTGGCTAGATCC-3’; Rsad2 R, 5’-CTGAGTGCTGTTCCCATCTTC-3’; Ifit3 F, 5’-CCTACATAAAGCACCTAGATGGC-3’; Ifit3 R, 5’-ATGTGATAGTAGATCCAGGCGT-3’; Isg15 F, 5’-GGTGTCCGTGACTAACTCCAT-3’; Isg15 R, 5’-CTGTACCACTAGCATCACTGTG-3’; Ifit2 F, 5’-CTGGGGAAACTATGCTTGGGT-3’; Ifit2 R, 5’-ACTCTCTCGTTTTGGTTCTTGG-3’; Ch25h F, 5’-TGCTACAACGGTTCGGAGC-3’; Ch25h R, 5’-AGAAGCCCACGTAAGTGATGAT −3’; Irf7 F, 5’-CCACGGAAAATAGGGAAGAAG-3’; Irf7 R, 5’-ACTAGAAAGCAGAGGGCTTGG-3’.
Immunoblotting
Whole cell lysates were separated by SDS-PAGE; proteins were transferred to nitrocellulose membranes for immunoblotting assays. Membranes were incubated overnight with the following primary antibodies, as indicated in the Figure legends (all purchased from Cell Signaling Technology, Danvers, MA, USA): tyrosine phosphorylated STAT1 (Tyr701) (clone D4A7); STAT1 (polyclonal); tyrosine phosphorylated STAT3 (Tyr705) (polyclonal); STAT3 (clone D3Z2G); and GAPDH (clone 14C10). After primary antibody incubation the membranes were washed thoroughly, and subsequently incubated for 1 hour with horseradish peroxidase-conjugated secondary antibody (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Following secondary antibody incubation, membranes were washed thoroughly, followed by treatment with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). Protein expression was evaluated by X-ray film exposure or use of the Bio-Rad Chemidoc (Bio-Rad).
Fluorescent antibody staining and flow cytometry
Single-cell suspensions were incubated in PBS containing 2 mM EDTA and 2% FBS (FACS buffer). Prior to fluorescent antibody staining, single-cell suspensions were exposed to rat anti-mouse CD16/32 antibody (Tonbo Biosciences, San Diego, California, USA) for 15–30 minutes at 4°C. Single-cell suspensions were then incubated for 20 minutes at 4°C with fluorescently conjugated antibodies against the following cell surface markers: CD11c (N418; BV-421–conjugated); CD86 (GL-1), CD45R (RA3–6B2), or CD40 (3/23) (all APC-conjugated); CD172α (P84), MHC I (AF6–88.5) or CD80 (16–10A1) (all FITC-conjugated); CD24 (M1/69; Percp-Cy5.5-conjugated); CD103 (2E7; PE-conjugated); or MHC II (M5/114.15.2; PE-Cy7-conjugated). All antibodies were purchased from BioLegend (San Diego, Ca, USA), Tonbo Biosciences, Thermo Fisher Scientific, or BD Biosciences. Ghost Dye Violet 510 (Tonbo Biosciences) was used to identify dead cells and was included when staining for cell surface markers. After staining, single-cell suspensions were washed with FACS buffer and analyzed using a BD LSR Fortessa (BD Biosciences) and FlowJo v10 software (FlowJo, Ashland, OR, USA).
Cytokine detection
Chemokines and cytokines were measured in supernatants from cultured cDC1s using the mouse ProcartaPlex panel 1A kit modified to include IFN-β detection reagents, in accordance with the manufacturer’s instructions (Invitrogen) on a Luminex 200 machine (Luminex, Austin, TX, USA). Cytokines and chemokines analyzed: CXCL5, G-CSF, GM–CSF, CXCL1, IFN-α, IFN-β, IFN-γ, IL-1α, IL-1β, IL-10, IL-12p70, IL-13, IL-15/IL-15R, IL-17A, IL-18, IL-2, IL-22, IL-23, IL-27, IL-28, IL-3, IL-31, IL-4, IL-5, IL-6, IL-9, CXCL10, LIF, M-CSF, CCL2, CCL7, CCL3, CCL4, CCL5, and TNF-α.
Statistical Analyses
All statistical analyses, with the exception of those performed in RNA-seq studies, were performed using Prism 9 software (GraphPad Software, San Diego, CA, USA). Differences between 2 groups were analyzed by Welch’s T test. Differences between multiple groups were tested by one-way or two-way ANOVA with Bonferroni’s multiple comparison test. Data are shown as the mean ± SEM of the indicated biological replicates (n). Results were considered significant when p < 0.05.
Results
Genome-wide transcriptional profiling of cDC1s
To study roles for STAT3 in regulating the transcriptional state of cDC1s, we utilized an in vitro culture system to generate cDC1s from BM of CD11c-Cre+ Stat3fl/fl mice (Stat3-deficient; Stat3Δ/Δ) and Stat3-sufficient (Stat3fl/fl) controls. Studies from our lab and others have shown cDC1s derived from this in vitro system resemble cDC1s from in vivo sources, including similar expression of cell surface markers, DC lineage-defining transcriptional regulators, and antigen cross-presentation function (16, 17, 28). Moreover, in prior work, we demonstrated Stat3fl/fl and Stat3Δ/Δ cDC1s are produced in similar amounts and show comparable phenotypes following in vitro generation (17). We confirmed efficient STAT3 depletion from Stat3Δ/Δ cDC1s, as evidenced by reduction in IL-10-responsive STAT3 tyrosine phosphorylation (pSTAT3) and total STAT3 protein (Figure 1A).
Figure 1. RNA sequencing identifies STAT3-dependent and -independent states in cDC1s.

(A) Whole cell lysates from Stat3fl/fl and Stat3Δ/Δ cDC1s treated with IL-10 or PBS for 0.5 hours were subjected to immunoblotting for the indicated proteins (top). Immunoblot results were quantified by densitometry analysis as indicated (bottom). Samples derived from 3 independent experiments, n = 3. (B - D) RNA-seq was performed on Stat3fl/fl and Stat3Δ/Δ cDC1s following treatment with IL-10, poly I:C, IL-10 + poly I:C, or PBS for 6 hours from independent biological samples, n = 3 per genotype and treatment condition. (B) PCA of RNA-seq results. (C) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. DEGs were identified using the following cutoff: |FC| ≥ 2 and p < 0.05. (D) Normalized expression of the top 20 DEGs from (C) in Stat3Δ/Δ (left) or Stat3fl/fl (right) cDC1s. * p < 0.05.
We next determined STAT3-mediated transcriptional responses using RNA-seq. FACS-purified Stat3fl/fl and Stat3Δ/Δ cDC1s were stimulated with IL-10, poly I:C, IL-10 + poly I:C, or PBS for 6 hours. Total RNA was isolated from 3 independent biological samples per genotype and treatment condition and used to derive unique cDNA libraries for RNA-seq. The sequencing produced 40–59 × 106 reads per sample, with mapping rates of 91.0–93.8% to the murine reference genome GRCm38/mm10 (data not shown). Pearson correlations between the 3 independent biological samples for each group were greater than 0.9, suggesting high quality cDNA libraries yielded concordant results (data not shown). Principal component analysis (PCA) of the data revealed 4 distinct clusters among the Stat3-sufficient cDC1 treatment groups (Figure 1B). These corresponded to each treatment condition, indicating distinct transcriptional states induced by IL-10, poly I:C, and IL-10 + poly I:C in Stat3-sufficient cDC1s. By contrast, PCA showed 2 distinct clusters for Stat3-deficient cDC1s (Figure 1B). One cluster corresponded to PBS- and IL-10-treated Stat3-deficient cDC1s. This cluster was also located adjacent to the PBS-treated Stat3fl/fl cDC1 cluster (Figure 1B), suggesting that the transcriptional state induced by IL-10 in cDC1s is mediated almost entirely by STAT3. The second cluster comprised the poly I:C and IL-10 + poly I:C-treated Stat3Δ/Δ cDC1s, which was situated proximal to the poly I:C-treated Stat3fl/fl cDC1 cluster (Figure 1B). These results suggest the transcriptional state induced upon concurrent IL-10 + poly I:C exposure is also STAT3-dependent. By contrast, the transcriptional profiles of poly I:C-treated cDC1s appear to be acquired in a STAT3-independent manner (Figure 1B).
Basal and IL-10-STAT3-mediated transcriptional responses in cDC1s
To characterize STAT3-dependent transcriptional control in cDC1s, we identified DEGs from the RNA-seq data and assessed pathway responses by GSEA and IPA (26, 27). Comparison of results from PBS-treated Stat3-sufficient and -deficient cDC1s revealed less than 100 DEGs between the two groups (Figures 1C and 1D), while Stat3-deficient cDC1s displayed negative enrichment of IFN-mediated transcriptional pathways by GSEA (Figures S1A). Thus, deletion of Stat3 has a modest effect on the transcriptome of cDC1s in the absence of cytokine or TLR stimulation.
Analysis of the IL-10 response in Stat3-sufficient cDC1s revealed 295 DEGs, including Socs3 (Figures 2A and 2B), consistent with its prior identification as a STAT3 target gene (29). We also noted upregulation of genes involved in pathogen responses or migration, including Muc1 and Plet1 (Figures 2A and 2B) (30, 31). Pathway analyses revealed enrichment of the cholesterol homeostasis and IL-6-JAK-STAT3 pathways (Figures 2C and S1B). In addition, IPA upstream regulator analysis identified several cytokines including IL-1β and IFN-γ, as well as the transcriptional regulators STAT3, other STATs, and sterol regulatory element-binding protein 1 (SREBF1) as factors predicted to mediate the transcriptional changes observed in Stat3-sufficient cDC1s exposed to IL-10 (Figure S1C).
Figure 2. Bioinformatic analyses reveal IL-10- and STAT3-dependent transcriptional responses in cDC1s.

RNA-seq and DEG analysis was performed as indicated in Fig. 1. (A - C) The IL-10-driven transcriptional response in Stat3fl/fl cDC1s, assessed by comparison of IL-10 versus PBS treatments. (A) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (B) Normalized expression of the top 20 DEGs from (A) in Stat3fl/fl cDC1s treated with IL-10 (left) or PBS (right). (C) GSEA enrichment plots of gene sets enriched in IL-10 treated Stat3fl/fl cDC1s. (D - F) The IL-10-driven transcriptional response in Stat3Δ/Δ cDC1s, assessed by comparison of IL-10 versus PBS treatments. (D) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (E) Normalized expression of the top 20 DEGs from (D) in Stat3Δ/Δ cDC1s treated with IL-10 (left) or PBS (right). (F) The GSEA Apoptosis enrichment plot, enriched in IL-10-treated Stat3Δ/Δ cDC1s. (G - I) The STAT3-dependent, IL-10-driven transcriptional response, assessed by comparison of Stat3Δ/Δ versus Stat3fl/fl cDC1s treated with IL-10. (G) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (H) Normalized expression of the top 20 DEGs from (G) in Stat3fl/fl (left) or Stat3Δ/Δ cDC1s (right) treated with IL-10. (I) GSEA enrichment plots of gene sets negatively enriched in IL-10-treated Stat3Δ/Δ cDC1s. (A, D, G) Highlighted genes include significant IL-10-STAT3-regulated genes and factors in the IL-10-STAT3 signaling cascade.
By contrast, only 85 DEGs were identified in Stat3-deficient cells stimulated with IL-10 (Figure 2D). These included Socs3 and other genes activated by IL-10 in Stat3-sufficient cells (e.g., Muc1, Rap1gap) (Figures 2D and 2E), suggesting their regulation by residual STAT3 or alternative signaling pathways. Nonetheless, the GSEA Apoptosis pathway was the sole enriched pathway (Figure 2F), while IPA upstream regulator analysis indicated STAT1 and IFN-γ as potential factors mediating IL-10-dependent transcriptional responses in Stat3-deficient cDC1s (Figure S1D). Comparison of data from IL-10-treated Stat3Δ/Δ cDC1s versus IL-10-treated Stat3fl/fl cDC1s identified 173 DEGs (Figures 2G and 2H), as well as suppression of IFN gamma response and IL-6-JAK-STAT3 pathways in Stat3-deficient cDC1s (Figures 2I and S1E). Moreover, IPA predicted an IL-1β-mediated transcriptional response in Stat3-sufficient cDC1s upon IL-10 stimulation, as judged by negative enrichment of the Il1b upstream regulator pathway in Stat3-deficient versus Stat3-sufficient cDC1s (Figure S1F). Collectively, these analyses indicate that the transcriptional state induced by IL-10 in cDC1s is largely STAT3-dependent and involves protection from apoptosis. In addition, the results suggest IL-10 activates specific pathogen responses, cholesterol homeostasis pathways, and IL-1β-mediated signaling in cDC1s via STAT3.
Roles for STAT3 in poly I:C-mediated transcriptional responses in cDC1s
The transcriptional response to poly I:C in Stat3-sufficient cDC1s was characterized by upregulation of 5066 DEGs including a variety of inflammatory cytokines and chemokines, such as Il12a, Ifnb1, and Il6, as well as numerous inflammatory pathways (Figures 3A to 3C, and S1G). Consistently, inflammatory cytokines and their associated transcription factors were the top upstream regulators predicted by IPA (Figure S1H). We found increased DEGs (n=5319) as well as similar enrichment of inflammatory transcriptional pathways in poly I:C-treated Stat3-deficient cDC1s (Figures 3D to 3F, and S1I), while upstream regulator analysis of poly I:C-treated Stat3-deficient cDC1s identified primarily inflammatory cytokines and transcription factors (Figure S1J). By contrast, direct comparison of the transcriptional response in poly I:C-treated Stat3Δ/Δ cDC1s versus Stat3fl/fl cDC1s revealed only 127 DEGs as well as inhibition of the GSEA Inflammatory Response pathway in Stat3-deficient cDC1s (Figures 3G to 3I). However, IPA did not identify any potential upstream cytokines or transcriptional regulators (data not shown). Taken together, these results indicate the transcriptional profile induced by poly I:C in cDC1s is dominated by inflammatory responses, as expected. While STAT3 appears to promote the poly I:C-mediated inflammatory transcriptional state modestly, the majority of poly I:C-induced transcriptional changes are STAT3-independent.
Figure 3. Characterization of the poly I:C-induced transcriptional response in cDC1s.

RNA-seq and DEG analysis was performed as indicated in Fig. 1. (A - C) The poly I:C-driven transcriptional response in Stat3fl/fl cDC1s, assessed by comparison of poly I:C versus PBS treatments. (A) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (B) Normalized expression of the top 20 DEGs from (A) in Stat3fl/fl cDC1s treated with poly I:C (left) or PBS (right). (C) GSEA enrichment plots of gene sets enriched in poly I:C-treated Stat3fl/fl cDC1s. (D - F) The poly I:C-driven transcriptional response in Stat3Δ/Δ cDC1s, assessed by comparison of poly I:C versus PBS treatments. (D) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (E) Normalized expression of the top 20 DEGs from (D) in Stat3Δ/Δ cDC1s treated with poly I:C (left) or PBS (right). (F) GSEA enrichment plots of gene sets enriched in poly I:C-treated Stat3Δ/Δ cDC1s. (G - I) The STAT3-dependent, poly I:C-driven transcriptional response, assessed by comparison of Stat3Δ/Δ versus Stat3fl/fl cDC1s treated with poly I:C. (G) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (H) Normalized expression of the top 20 DEGs from (G) in Stat3Δ/Δ (left) or Stat3fl/fl cDC1s (right) treated with poly I:C. (I) The GSEA Inflammatory Response enrichment plot, negatively enriched in poly I:C-treated Stat3Δ/Δ cDC1s. (A, D, G) Highlighted genes include significant poly I:C-regulated genes and factors in the IL-10-STAT3 signaling cascade.
IL-10-STAT3-mediated transcriptional responses in poly I:C-treated cDC1s
To assess means by which IL-10-responsive STAT3 activation alters poly I:C-mediated transcriptional responses, we evaluated DEGs and transcriptional pathways in Stat3-sufficient and -deficient cDC1s treated with IL-10 and poly I:C concurrently (IL-10 + poly I:C) compared to cells exposed to poly I:C alone. In Stat3-sufficient cDC1s these analyses revealed 1250 DEGs including IL-10-mediated suppression of inflammatory genes such as Ifnb1 and Il12a (Figures 4A and 4B), which is consistent with our prior work (17). We also found Tlr3 was inhibited by co-treatment with IL-10 (Figure 4A). Consistent with these data, pathway analyses indicated IL-10 restrains poly I:C-mediated IFN-I transcriptional responses (Figures 4C and S1K). By contrast, IL-10 regulated only 190 DEGs in Stat3-deficient cDC1s, without affecting poly I:C-mediated inflammatory DEGs, inflammatory transcriptional pathways, or IFN transcriptional responses (Figures 4D, 4E, and S1L). These data align with our PCA results (Figure 1B), and indicate IL-10 utilizes STAT3 to modulate the poly I:C response in cDC1s.
Figure 4. Analysis of IL-10-STAT3-mediated effects on the poly I:C-induced transcriptional response in cDC1s.

RNA-seq and DEG analysis was performed as indicated in Fig. 1. (A - C) The effect of IL-10 on the poly I:C-induced transcriptional response in Stat3fl/fl cDC1s, assessed by comparison of IL-10 + poly I:C versus poly I:C treatments. (A) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (B) Normalized expression of the top 20 DEGs from (A) in Stat3fl/fl cDC1s treated with IL-10 + poly I:C (left) or poly I:C (right). (C) The GSEA IFN Alpha Response enrichment plot, negatively enriched in IL-10 + poly I:C-treated Stat3fl/fl cDC1s. (D and E) The effect of IL-10 on the poly I:C-induced transcriptional response in Stat3Δ/Δ cDC1s, assessed by comparison of IL-10 + poly I:C versus poly I:C treatments. (D) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (E) Normalized expression of the top 20 DEGs from (D) in Stat3Δ/Δ cDC1s treated with IL-10 + poly I:C (left) or poly I:C (right). (F - H) The STAT3-dependent effects of IL-10 on the poly I:C-driven transcriptional response, assessed by comparison of Stat3Δ/Δ versus Stat3fl/fl cDC1s treated with IL-10 + poly I:C. (F) Volcano plot displaying log2 fold change and significance of transcripts from DEG analysis. (G) Normalized expression of the top 20 DEGs from (F) in Stat3Δ/Δ (left) or Stat3fl/fl cDC1s (right) treated with IL-10 + poly I:C. (H) The GSEA IFN Alpha Response enrichment plot, enriched in IL-10 + poly I:C-treated Stat3Δ/Δ cDC1s. (A, D, F) Highlighted genes include significant IL-10-STAT3-regulated genes, ISGs, and factors in the IL-10-STAT3 signaling cascade.
To further evaluate the impact of IL-10-STAT3 signaling, we compared transcriptional responses in IL-10 + poly I:C-treated Stat3-deficient and -sufficient cDC1s directly. These analyses revealed DEGs encoding inflammatory cytokines and chemokines, as well as Tlr3, were upregulated in Stat3-deficient cDC1s exposed to IL-10 + poly I:C versus Stat3-sufficient cells (Figure 4F and 4G). Moreover, GSEA indicated the IFN Alpha Response pathway was specifically enriched in Stat3-deficient cDC1s (Figure 4H). Consistently, IPA upstream regulator analysis indicated IFNs and IFN regulatory factors (IRFs) as the top factors predicted to mediate the transcriptional state in Stat3-deficient cDC1s upon concomitant exposure to IL-10 and poly I:C (Figure S1M). These results suggest IL-10-STAT3 signaling can suppress Tlr3 expression in poly I:C-treated cDC1s, yet indicate the major function for the IL-10-STAT3 pathway is selective inhibition of poly I:C-induced IFN signaling.
Evaluation of IFN and IFN-stimulated gene (ISG) expression in cDC1s
Since our RNA-seq studies were performed on cDC1s stimulated in vitro, the results suggest a cDC1-intrinsic mechanism leads to induction of IFN signaling upon poly I:C treatment. To evaluate this further, we examined activation (tyrosine phosphorylation) of STAT1 (pSTAT1), the major IFN-responsive signal transducer, in cDC1s by immunoblotting. These assays were conducted 6 hours following treatment of Stat3-sufficient and -deficient cDC1s with IL-10, poly I:C, IL-10 + poly I:C, or PBS, to correspond to the time point analyzed in our RNA-seq studies. We found poly I:C treatment led to pSTAT1 accrual in cDC1s (Figures 5A and S2A), suggesting activation of IFN signaling. Importantly, pSTAT1 was induced to similar amounts in poly I:C-treated Stat3fl/fl and Stat3Δ/Δ cDC1s relative to total STAT1 protein or our internal loading control GAPDH, consistent with a STAT3-independent poly I:C response (Figures 5A and S2A). pSTAT1 was undetectable in PBS- or IL-10-stimulated cells, as expected (Figures 5A and S2A). By contrast, poly I:C-induced pSTAT1 was inhibited by concomitant treatment with IL-10 in Stat3-sufficient cDC1s but was unaffected in Stat3-deficient cDC1s (Figures 5A and S2A). These results show IL-10-STAT3 signaling suppresses poly I:C-mediated STAT1 activation, consistent with our RNA-seq and bioinformatic studies.
Figure 5. IL-10-STAT3-mediated control of poly I:C-induced IFN signaling in cDC1s.
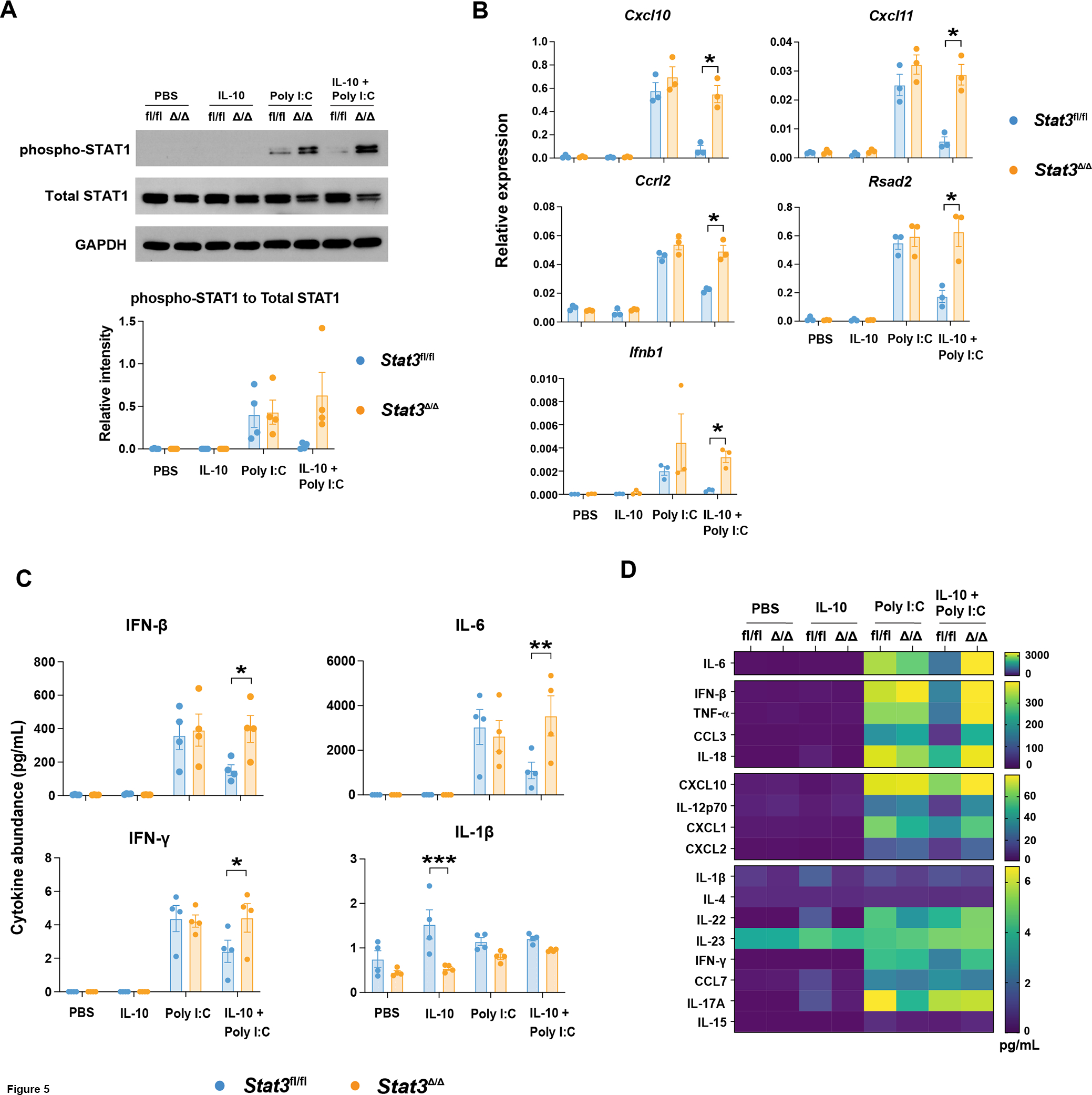
Stat3fl/fl and Stat3Δ/Δ cDC1s were treated with IL-10, poly I:C, IL-10 + poly I:C, or PBS for 6 hours. (A) Whole cell lysates from Stat3fl/fl and Stat3Δ/Δ cDC1s were subjected to immunoblotting for pSTAT1, total STAT1, or GAPDH. Representative immunoblot results (top) for the indicated proteins in Stat3fl/fl (fl/fl) and Stat3Δ/Δ (Δ/Δ) cDC1s, or cumulative data quantified by densitometry (bottom) from 3 independent experiments, n = 4 per genotype and condition. (B) Relative transcript expression of the indicated genes, as assessed by qRT-PCR, combined from 3 independent experiments, n = 3 per genotype and treatment condition. (C) Representative cytokines from multiplex analysis of cDC1 cell culture supernatants. Data from 4 independent experiments, n = 4 per genotype and treatment condition. (D) Mean concentration of all detectable factors analyzed as indicated in (C). * p < 0.05, ** p < 0.01, *** p < 0.001.
To confirm STAT3-mediated regulation of IFNs and ISGs in cDC1s, we analyzed gene expression responses in Stat3fl/fl and Stat3Δ/Δ cDC1s by qRT-PCR. As expected, we observed poly I:C-responsive induction of representative ISGs including Ccrl2, Ch25h, Cxcl10, Cxcl11, Irf7, Ifit2, Ifit3, Isg15, Ripk2, and Rsad2 (Figures 5B and S2B). These ISGs were induced by poly I:C to a similar degree in Stat3fl/fl and Stat3Δ/Δ cDC1s, in line with STAT3-independent poly I:C transcriptional responses (Figures 5B and S2B). Concurrent treatment with IL-10, however, suppressed poly I:C-responsive expression of Cxcl10, Cxcl11, Ccrl2, and Rsad2 in a STAT3-dependent manner (Figure 5B), consistent with IL-10-STAT3-mediated inhibition of poly I:C-responsive pSTAT1 (Figures 5A and S2A). In addition, Ch25h, Irf7, Ifit2, Ifit3, and Ripk2 displayed a similar trend towards IL-10-STAT3-mediated inhibition (Figure S2B). By contrast, Isg15 was not repressed by IL-10-STAT3 signaling (Figure S2B). We also detected Ifnb1 induction by poly I:C as well as inhibition of this response by IL-10-STAT3 (Figure 5B). Taken together, our results suggest autocrine IFN signaling and IFN-mediated transcriptional responses are induced in cDC1s upon poly I:C exposure, while concurrent IL-10-STAT3 signaling selectively blocks this IFN response.
IFN production in poly I:C-stimulated cDC1s
To identify potential IFNs involved in autocrine signaling, we assessed IFN production from cDC1s stimulated with poly I:C, IL-10, or IL-10 + poly I:C for 6 hours in vitro using multiplex cytokine analyses of culture supernatants. These assays showed production of IFN-β as well as modest amounts of IFN-γ from poly I:C-stimulated Stat3-sufficient cDC1s (Figures 5C and 5D). We confirmed our cDC1 culture was homogeneous and lacked detectable amounts of major IFN-γ-producing subsets such as CD3+ T and NK1.1+ NK cells (data not shown), supporting direct IFN-γ production by cDC1s. In addition, IFN-β and IFN-γ were produced at similar amounts in Stat3-sufficient and -deficient cDC1s upon poly I:C treatment (Figures 5C and 5D). By contrast, concomitant IL-10 exposure inhibited IFN-β and IFN-γ production in Stat3-sufficient cDC1s but did not affect production from Stat3-deficient cDC1s (Figures 5C and 5D). Moreover, although our RNA-seq data suggested poly I:C stimulates expression of Ifnl2, IFN-λ was undetectable across all treatment conditions (data not shown). IFN-α was also undetectable (data not shown). Together, these results indicate poly I:C stimulates rapid secretion of IFN-β as well as modest amounts of IFN-γ from cDC1s, and production of these cytokines is suppressed by concurrent IL-10 exposure via a STAT3-dependent mechanism.
Further analysis of the cytokine multiplex data indicated that other inflammatory chemokines and cytokines, such as CXCL10 and IL-6, were produced by cDC1s in response to poly I:C, as expected (Figures 5C, 5D, and S2C). Many of these factors were inhibited by concurrent treatment with IL-10 in a STAT3-dependent manner, consistent with our RNA-seq data and prior studies (Figures 4B, 4G, 5C, 5D, and S2C) (17). We also detected a modest STAT3-dependent increase in production of IL-1β from cDC1s upon IL-10 treatment, in agreement with our bioinformatic analysis and previous findings (Figures 5C, 5D, S1C, and S1F) (17), thus suggesting cDC1s respond to IL-10 by secreting IL-1β and stimulating IL-1β-mediated transcriptional responses. In addition, our data suggest IFN-β and IFN-γ are potential factors mediating a poly I:C-stimulated, autocrine IFN signaling loop that results in STAT1 phosphorylation and ISG induction in cDC1s.
Type I and type II, but not type III IFNs, induce ISG expression in cDC1s
Our RNA-seq results suggested cDC1s express type I, type II, and to a lesser extent type III IFN receptor mRNAs (data not shown), yet it was unclear whether cDC1s respond to these IFNs. To test this, along with potential for autocrine signaling, we performed direct stimulation of cDC1s with IFN-β, IFN-γ, or IFN-λ in vitro. Immunoblotting experiments demonstrated that IFN-β and IFN-γ induced pSTAT1 in cDC1s (Figures 6A and S3A). Although IFN-λ also stimulated pSTAT1, this response appeared weaker than that elicited by either IFN-β or IFN-γ, as judged by differential exposure times for the respective immunoblots (Figures 6A, 6B and S3B). Total STAT1 was unaltered by IFN treatment (Figures 6A and 6B). Thus, these results indicate cDC1s are capable of responding to type I, II, or III IFNs by eliciting STAT1 activation.
Figure 6. IFN-mediated signaling responses in cDC1s.
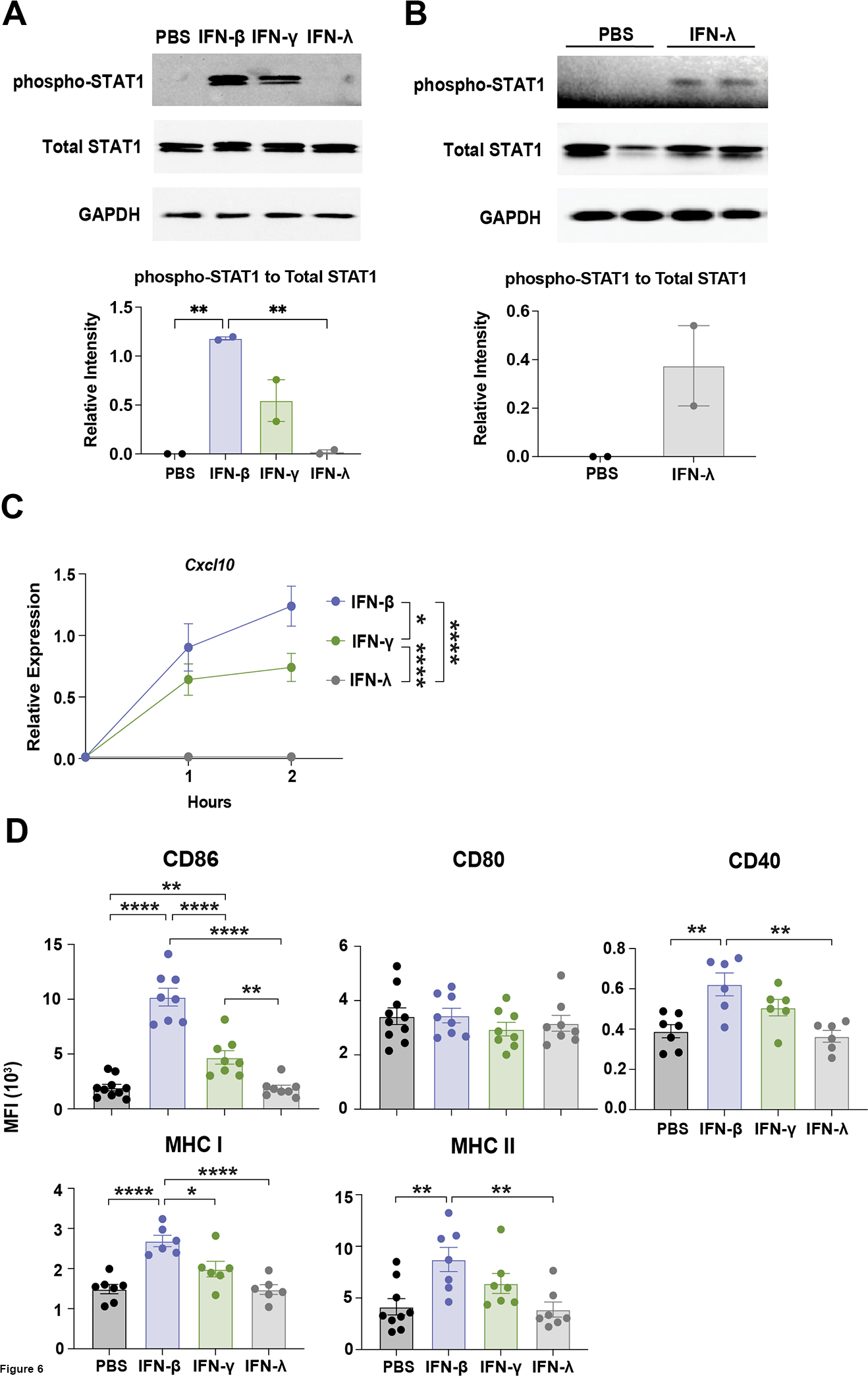
cDC1s were treated with IFN-β, IFN-γ, IFN-λ, or PBS as indicated. (A and B) Whole cell lysates from cDC1s treated with the indicated IFNs or PBS for 0.5 hours were subjected to immunoblotting for pSTAT1, total STAT1, or GAPDH, as shown. (A) Representative immunoblot results (top) for the indicated proteins, or cumulative data quantified by densitometry (bottom) from 2 independent experiments, n = 2 per condition. pSTAT1 immunoblot (top) exposed for 20 seconds. (B) Immunoblot (top) analysis and quantification by densitometry (bottom) of samples from 2 independent experiments, n = 2 per condition. pSTAT1 immunoblot (top) exposed for 20 minutes. (C) Cxcl10 expression over time in cDC1s following the indicated treatments, determined by qRT-PCR. Data combined from 3 independent experiments, n = 7 (0 hour), n = 8 (1 and 2 hour). (D) Surface expression of the indicated markers, assessed by flow cytometry, after IFN treatment for 16 hours. CD86 and CD80 data combined from 5 independent experiments, n = 10 (PBS), n = 8 (IFN-β, IFN-γ, and IFN-λ). MHC II data combined from 5 independent experiments, n = 9 (PBS), n = 7 (IFN-β, IFN-γ, and IFN-λ). CD40 and MHC I data combined from 4 independent experiments, n = 7 (PBS), n = 6 (IFN-β, IFN-γ, and IFN-λ). * p < 0.05, ** p < 0.01, **** p < 0.0001.
To further assess IFN responses in cDC1s, we asked whether stimulation with type I, II, or III IFNs induced expression of the representative ISG Cxcl10, as well as co-stimulatory and antigen presentation molecules in cDC1s. These assays showed IFN-β, and to a lesser extent IFN-γ, stimulated Cxcl10 expression, while IFN-λ treatment had no effect on Cxcl10 (Figure 6C). Similarly, cell surface expression of CD86 was induced by IFN-β treatment, and to a lesser degree by IFN-γ, but was refractory to IFN-λ stimulation (Figure 6D). By contrast, cell surface expression of CD80 was unresponsive to IFN stimulation (Figure 6D). Moreover, CD40, MHC I, and MHC II were induced by IFN-β, and modestly but not significantly by IFN-γ stimulation, while these factors were unaffected by IFN-λ (Figure 6D). Collectively, these results indicate that cDC1s are responsive to exogenous IFN-β, and to a lesser extent IFN-γ, while exogenous IFN-λ induces a weak pSTAT1 signal but is unable to promote ISG expression or canonical cDC1 maturation responses. Thus, the data suggest IFN-β and IFN-γ as potential factors mediating autocrine IFN signaling in poly I:C-stimulated cDC1s.
Poly I:C-induced cDC1 maturation selectively requires IFN-I signaling
To delineate the role of type I, II, and III IFNs in the context of poly I:C treatment, we utilized cDC1s lacking required receptor subunits for type I IFNs (Ifnar1−/−) or type II IFN (Ifngr1−/−) (32, 33). We also employed an IFN-λ blocking antibody to suppress potential IFN-λ autocrine signaling. Importantly, Ifnar1- or Ifngr1-deficiency did not alter the production of cDC1s in vitro (Figures S3C and S3D). Upon poly I:C treatment, we found that Ifnar1−/− cDC1s failed to induce CD86, CD80, CD40, MHC I, or MHC II (Figure 7A). By contrast, neither Ifngr1-deficiency nor concomitant treatment with anti–IFN-λ altered poly I:C-induced costimulatory or MHC molecule expression (Figures 7B and 7C). Similarly, Ifnar1−/− cDC1s did not respond to poly I:C by inducing pSTAT1 or expression of specific ISGs, including Cxcl10, Irf7, Ccrl2, or Ifit3, while Ifngr1-deficiency had no effect on these responses (Figures 8A, 8B, 8C, S3E, and S3F). Total STAT1 abundance was not changed by Ifnar1- or Ifngr1-deficiency (Figures 8A, 8B, and S3E). Interestingly, although Isg15 was induced by poly I:C in an Ifnar1-dependent manner (Figure S3F), this gene was the sole ISG assayed that was not sensitive to IL-10-STAT3-mediated inhibition (Figure S2B), indicating some IFN-I-dependent ISGs may not be targets of IL-10-STAT3. Taken together, our results indicate that poly I:C-induced IFN signaling in cDC1s is mediated by an autocrine IFN-I mechanism. Our data suggest poly I:C-elicited IFN-β plays a key role in this autocrine pathway, while IL-10-STAT3 signaling effectively dampens IFN-β production and IFN-I transcriptional responses in cDC1s.
Figure 7. Ifnar1-mediated expression of MHC and co-stimulatory molecules in poly I:C-treated cDC1s.
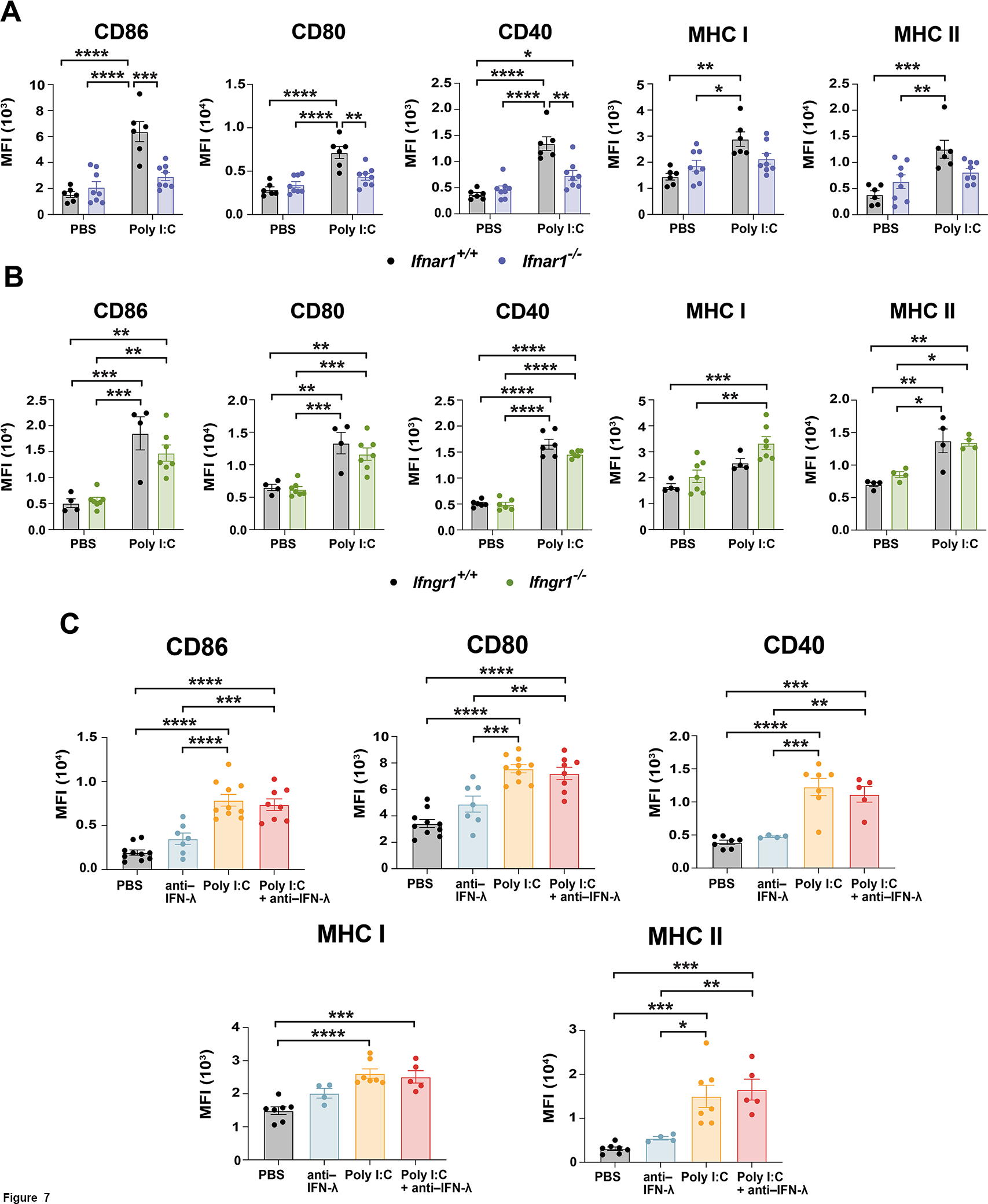
(A – C) Cell surface expression profiles were determined by flow cytometry. (A and B) Ifnar1+/+, Ifnar1−/−, Ifngr1+/+ and Ifngr1−/− cDC1s were exposed to poly I:C or PBS for 16 hours, as indicated. (A) Data combined from 3 independent experiments, n = 6 (Ifnar1+/+), n = 8 (Ifnar1−/−). (B) CD86, CD80, and MHC I data pooled from 3 independent experiments, n = 4 (Ifngr1+/+), n = 7 (Ifngr1−/−). CD40 and MHC II data combined from 2 independent experiments, n = 6 (CD40), n = 4 (MHC II). (C) cDC1s were treated with poly I:C or PBS for 16 hours in the absence or presence of IFN-λ antibody (anti–IFN-λ), as indicated. CD86 and CD80 data pooled from 4 independent experiments, n = 10 (PBS, poly I:C), n = 8 (poly I:C + anti–IFN-λ), n = 7 (anti–IFN-λ). CD40, MHC I, and MHC II data combined from 3 independent experiments, n = 7 (PBS, poly I:C), n = 5 (poly I:C + anti–IFN-λ), n = 4 (anti–IFN-λ). * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Figure 8. Ifnar1-dependent signaling and ISG induction in poly I:C-treated cDC1s.
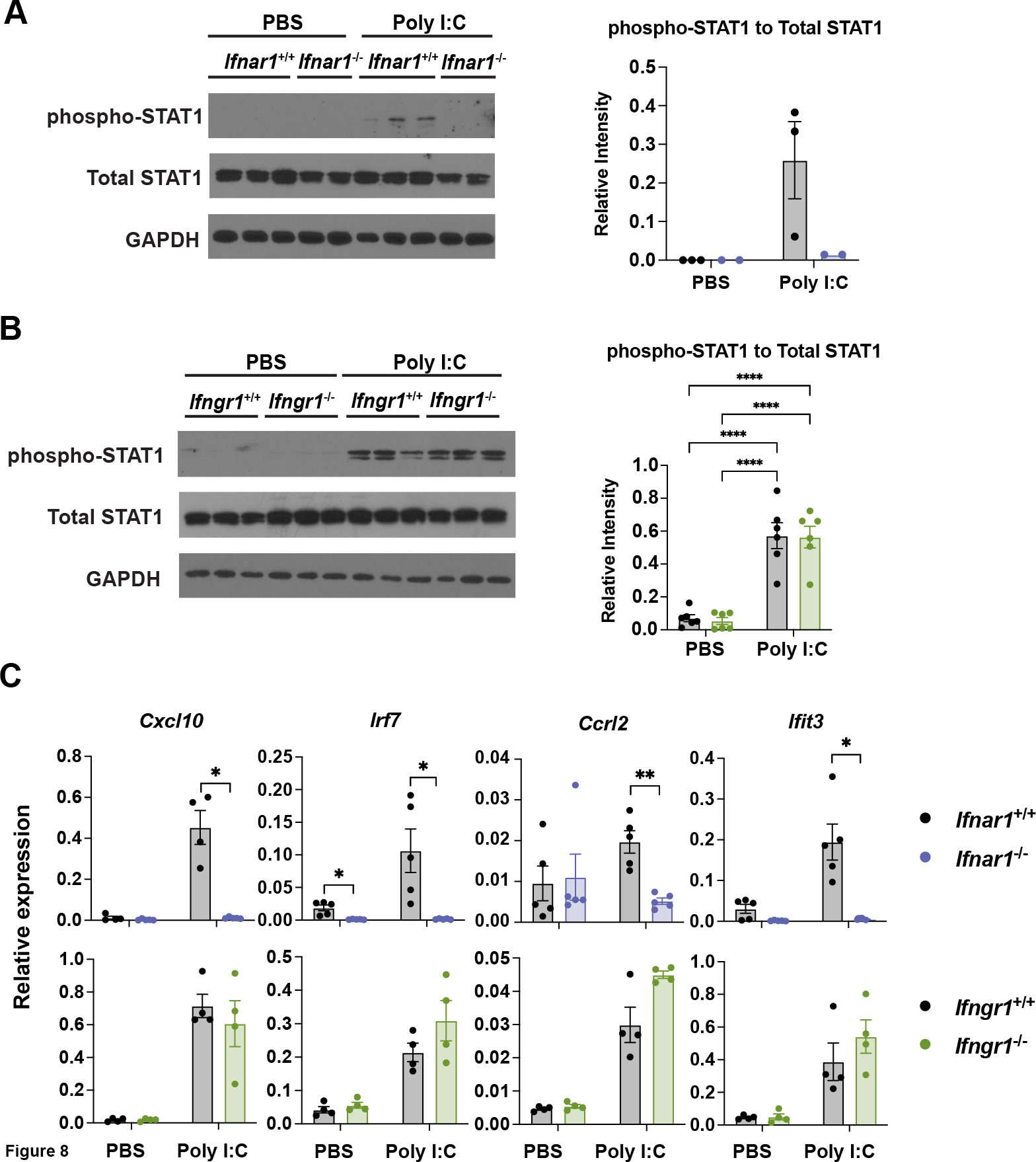
Ifnar1+/+, Ifnar1−/−, Ifngr1+/+ and Ifngr1−/− cDC1s were treated with poly I:C or PBS for 6 hours. (A and B) Whole cell lysates were subjected to immunoblotting for pSTAT1, total STAT1, or GAPDH. (A) Representative immunoblot results (left) for the indicated proteins in Ifnar1+/+ and Ifnar1−/− cDC1s, and quantification by densitometry (right) from 1 of 2 independent experiments, n = 3 (Ifnar1+/+), n = 2 (Ifnar1−/−). (B) Representative immunoblot (left) for the indicated proteins in Ifngr1+/+ and Ifngr1−/− cDC1s, or cumulative data quantified by densitometry (right) from 2 independent experiments, n = 6 per genotype and condition. (C) Relative transcript expression of the indicated genes, as determined by qRT-PCR. Data combined from 2 independent experiments, n = 5 (Ifnar1+/+, Ifnar1−/−), n = 4 (Ifngr1+/+, Ifngr1−/−). * p < 0.05, ** p < 0.01, **** p < 0.0001.
Discussion
cDC1s are essential for the coordination of T cell-mediated immunity against intracellular pathogens and tumors. Although transcriptional mechanisms regulating cDC1 development have been elucidated (34), molecular and transcriptional responses that control cDC1 function remain understudied. Recently, we found that the transcriptional regulator STAT3 inhibits poly I:C-mediated cDC1 maturation as well as the activity of cDC1s used as a cell therapy in murine breast cancer (17). To understand molecular control of cDC1s by STAT3, we utilized RNA-seq to probe global transcriptional responses regulated by poly I:C, IL-10, and STAT3. Our bioinformatic analyses revealed that poly I:C stimulates many inflammatory pathways in cDC1s, as expected, and these are activated primarily via STAT3-independent mechanisms. By contrast, we found IL-10-STAT3 signaling selectively inhibits the IFN-I transcriptional response induced by poly I:C. Using cDC1s deficient for STAT3 or IFN signaling, we demonstrated STAT3 mediates suppression of poly I:C-induced IFN-β secretion, autocrine IFN-I signaling, and expression of multiple ISGs, such as the T cell-recruiting factor CXCL10. Taken together, we provide important datasets for understanding transcriptional control of cDC1 function by the inflammatory factor poly I:C and the anti-inflammatory cytokine IL-10. In addition, we elucidate a unique immunosuppressive role for STAT3 in cDC1s mediated by inhibition of poly I:C-activated IFN-I signaling.
IL-10 has well-established anti-inflammatory activity in monocytes, monocyte-derived DCs (moDCs), and BM-derived macrophages (BMDMs), as indicated by potent suppression of TLR-induced inflammatory responses (21, 22, 35–37). Interestingly, a recent study employing RNA-seq analyses of distinct myeloid and DC populations indicated IL-10 elicits unique anti-inflammatory transcriptional responses upon stimulation with the TLR4 agonist LPS (20). For instance, IL-10 selectively inhibits LPS-activated NF-κB transcriptional pathways in macrophages, while IL-10 suppresses IFN and IRF transcriptional responses in splenic cDCs, neutrophils, and mast cells (20). These data suggest discrete cell-intrinsic mechanisms dictate the IL-10-mediated anti-inflammatory response in different myeloid and DC subsets exposed to LPS. Distinct mechanisms of IL-10-mediated inhibition may be executed, in part, via cell-specific differences in TLR-induced signaling cascades, which have been reported recently (38). Nonetheless, these studies have not resolved inflammatory or anti-inflammatory transcriptional pathways in specific cDC populations including cDC1 and cDC2. Using the agonist poly I:C, which targets TLR3, a TLR primarily expressed by cDC1, we find IL-10 selectively blocks IFN-mediated transcriptional responses in cDC1s. While poly I:C can also activate intracellular nucleic acid sensors such as MDA-5 and RIG-I (39), it remains unclear whether IL-10-STAT3 mediated inhibition in cDCs uniquely targets the TLR3 pathway or is capable of more broadly suppressing inflammatory responses elicited by a variety of pattern-recognition receptors including other TLRs or intracellular pattern recognition molecules. These points will be important to resolve in future studies, including additional RNA-seq profiling studies and analysis of cDC1-intrinsic IL-10 signaling in vivo.
As a major IL-10 signal transducer, STAT3 elicits potent anti-inflammatory functions in myeloid cells and DCs (21, 22, 37), yet specific signaling mechanisms controlling inflammatory responses in well-defined immune subsets have been elusive. We demonstrated previously that STAT3 inhibits LPS-responsive inflammatory signaling in BMDMs by mediating an autocrine IL-6 pathway that results in STAT3-dependent transcriptional repression of Ube2n/Ubc13 (22). Ubc13 is a critical E2 ubiquitin-conjugating enzyme required for TRAF6 activation and TLR-responsive induction of the NF-κB and MAPK signaling cascades. Accordingly, autocrine IL-6 signaling and STAT3-mediated inhibition of Ube2n effectively dampens TLR signaling in BMDMs (22). Although poly I:C-stimulated cDC1s also produce abundant quantities of IL-6, intrinsic transcriptional responses elicited by poly I:C in the absence of exogenous cytokine appear to be largely independent of STAT3, as judged by activation of similar pathways in poly I:C-treated Stat3-sufficient and -deficient cDC1s. Moreover, we did not find evidence for STAT3-mediated inhibition of Ube2n expression in cDC1s (data not shown). These results suggest poly I:C-activated IL-6 production from cDC1s does not induce an autocrine IL-6-STAT3-dependent anti-inflammatory response in cDC1s, distinct from roles for this pathway in BMDMs (22). This concept is concordant with our data indicating specific STAT3-mediated inhibition of poly I:C-elicited IFN-I transcriptional responses, which are regulated primarily by TRIF and IRF3 (40, 41). Therefore, our results collectively support a model in which TLR agonist-induced autocrine cytokine signals regulate distinct outcomes in cDC1s and BMDMs, while cytokine-activated STAT3 signaling exerts anti-inflammatory states in both subsets via different mechanisms.
Prior work has shown that STAT3 is phosphorylated upon IFN-I stimulation and acts as a negative regulator of IFN-I signaling in monocytes and BMDMs (42, 43). We did not detect significant differences in ISG expression or IFN-responsive maturation in Stat3-sufficient versus Stat3-deficient cDC1s treated with poly I:C, indicating STAT3 does not inhibit poly I:C-induced IFN-signaling in cDC1s in the absence of exogenous cytokine. These results suggest an exogenous factor such as IL-10 must be present in the microenvironment to induce STAT3 anti-inflammatory signaling in cDC1s. These findings are similar to results from others, which have shown IL-10 treatment of monocytes inhibits accrual of pSTAT1 and ISG expression induced upon IFN-β or IFN-γ exposure (44). Future studies will be needed to determine whether IL-10 exposure results in the inhibition of IFN-I signaling in cDC1s in vivo.
In addition to controlling poly I:C-elicited type I IFN signaling, our data reveal an unexpected role for IL-10 and STAT3 in promoting Il1b expression and IL-1β secretion from cDC1s. We found IL-1β production was induced in cDC1s by IL-10 stimulation in the absence of poly I:C treatment, suggesting direct regulation of Il1b/IL-1β by the IL-10-STAT3 signaling cascade. These data agree with our RNA-seq results, which implicate IL-10 and STAT3 in induction of transcriptional responses mediated by SREBF1, an upstream IL-1β regulator (45). Interestingly, recent reports link IL-1β and cholesterol metabolism (46, 47). These findings are consistent with IL-10-STAT3-mediated control of cholesterol synthesis pathways, which we also uncovered in our bioinformatic analyses. For example, IL-1β expression induced upon intracellular bacterial infection was found to promote cholesterol accumulation in macrophages (47). Furthermore, cholesterol accumulation in DCs has been shown to enhance inflammasome activation and IL-1β secretion (46). Potential consequences of IL-1β-mediated responses following IL-10-induced STAT3 signaling in cDC1s is of great interest and requires further evaluation.
Interestingly, our results suggest distinct IFN-I-mediated mechanisms of cDC1 maturation, a key step in cDC1-induced priming and activation of T lymphocytes. While IFN-β exposure was sufficient to stimulate CD86, CD40, MHC I, and MHC II expression on cDC1s, IFN-β did not induce CD80 expression appreciably. Moreover, we found that poly I:C-elicited CD80 expression was dependent upon Ifnar1. Taken together, our results suggest that CD80 is unique among the maturation markers we examined by requiring both IFN-I- and TLR-elicited signaling to induce its upregulation. While prior reports have demonstrated distinct roles for CD80 and CD86 in mediating immune and inflammatory responses (48, 49), determining the consequence(s) of their differential regulation on cDC1-mediated immune functions will require further study. More broadly, it is also imperative to understand cDC1-intrinsic STAT3 function in regulating immune responses in vivo. Our data suggest STAT3 plays an important role in modulating cDC1 function in microenvironments abundant in both IL-10 and TLR agonists, such as the TME and colon. However, determining the role of cDC1-intrinsic STAT3 in these contexts will require the generation of novel research tools such as cDC1-specific deletion of Stat3 in mice, an important area for future studies.
Collectively, our results provide new insight into the transcriptional mechanisms by which the immunosuppressive and tumor-associated cytokine IL-10, and its major signal transducer STAT3, control cDC1 function. Our findings also suggest cancer immunotherapies may benefit from combination treatment with STAT3 inhibitors to enhance IFN signaling, cDC1 maturation, and cDC1-mediated anti-tumor immunity. Lastly, our study provides novel transcriptomic datasets that will aid in further delineation of inflammatory and anti-inflammatory molecular mechanisms regulating cDC1 function.
Supplementary Material
Key Points.
RNA-seq profiling delineates cDC1 responses to extracellular stimuli.
Poly I:C activates autocrine IFN-I signaling in cDC1s.
IL-10 inhibits autocrine IFN-I responses in cDC1s via STAT3.
Acknowledgments
We would like to thank the MD Anderson South Campus Flow Cytometry Core Facility for their experimental assistance, Bailey Harmon for contribution to the RNA-seq analyses, and Dr. Matthew M. Gubin for critical review of this manuscript.
Grant Support:
This work was supported by the NIH NIAID (R01AI109294 and R01AI133822 to S.S.W.), the Cancer Prevention and Research Institute of Texas (CPRIT) Research Training award (RP170067 to T.T.C., N.L.D., and A.B; and RP210028 to L.M.K.), and the NIH NCI MD Anderson Cancer Center Core grant (P30CA016672; supporting the MD Anderson South Campus Flow Cytometry Core Facility).
Abbreviations:
- BM
bone marrow
- BMDM
bone marrow-derived macrophage
- cDC1s
type I conventional dendritic cell
- DC
dendritic cell
- DEG
differentially expressed gene
- FLT3L
FLT3 ligand
- GSEA
Gene Set Enrichment Analysis
- IFN-I
type I IFN
- IPA
Ingenuity Pathway Analysis
- IRF
Interferon regulatory factor
- ISG
IFN-stimulated gene
- LN
lymph node
- moDC
monocyte-derived DC
- PCA
Principle component analysis
- poly I:C
polyinosinic:polycytidylic acid
- PS
penicillin-streptomycin
- pSTAT1
tyrosine phosphorylated STAT1
- pSTAT3
tyrosine phosphorylated STAT3
- qRT-PCR
quantitative RT-PCR
- RNA-seq
RNA sequencing
- RSEM
RNA-seq by expectation maximization
- TdLN
tumor-draining lymph node
- Th1
T helper 1
- TME
tumor microenvironment
References
- 1.Chrisikos TT, Zhou Y, Slone N, Babcock R, Watowich SS, and Li HS. 2019. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol. Immunol. 110: 24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, and Mueller SN. 2015. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 43: 554–565. [DOI] [PubMed] [Google Scholar]
- 3.Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, Garbi N, Kaisho T, Germain RN, and Kastenmüller W. 2015. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell 162: 1322–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson JT, Bagadia P, Liu T, Briseño CG, Li L, Gillanders WE, Wu GF, Yokoyama WM, Murphy TL, Schreiber RD, and Murphy KM. 2020. cDC1 prime and are licensed by CD4+ T cells to induce anti-tumour immunity. Nature 584: 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulido Á. de M., Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B. 2018. TIM-3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 33: 60–74.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spranger S, Dai D, Horton B, and Gajewski TF. 2017. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31: 711–723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan S-L, Gibbons G, Smith OC, Kim JY, Westcott P, Hwang WL, Freed-Pastor WA, Eng G, Cuoco MS, Rogers P, Park JK, Burger ML, Rozenblatt-Rosen O, Cong L, Pauken KE, Regev A, and Jacks T. 2021. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 54: 2338–2353.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, van’t Veer LJ, Sperling AI, Wolf DM, and Krummel MF. 2014. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hubert M, Gobbini E, Couillault C, Manh T-PV, Doffin A-C, Berthet J, Rodriguez C, Ollion V, Kielbassa J, Sajous C, Treilleux I, Tredan O, Dubois B, Dalod M, Bendriss-Vermare N, Caux C, and Valladeau-Guilemond J. 2020. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 5: eaav3942. [DOI] [PubMed] [Google Scholar]
- 10.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, Alvarado MD, Wolf DM, Bogunovic D, Bhardwaj N, Daud AI, Ha PK, Ryan WR, Pollack JL, Samad B, Asthana S, Chan V, and Krummel MF. 2018. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 24: 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, Ginhoux F, and Merad M. 2016. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44: 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blair TC, Bambina S, Alice AF, Kramer GF, Medler TR, Baird JR, Broz ML, Tormoen GW, Troesch V, Crittenden MR, and Gough MJ. 2020. Dendritic cell maturation defines immunological responsiveness of tumors to radiation therapy. J. Immunol. 204: 3416–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Mingo Pulido Á, Hänggi K, Celias DP, Gardner A, Li J, Batista-Bittencourt B, Mohamed E, Trillo-Tinoco J, Osunmakinde O, Peña R, Onimus A, Kaisho T, Kaufmann J, McEachern K, Soliman H, Luca VC, Rodriguez PC, Yu X, and Ruffell B. 2021. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54: 1154–1167.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, and Schreiber RD. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vatner RE, and Janssen EM. 2019. STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol. 110: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Slone N, Chrisikos TT, Kyrysyuk O, Babcock RL, Medik YB, Li HS, Kleinerman ES, and Watowich SS. 2020. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103+ conventional dendritic cells. J. Immunother. Cancer 8: e000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chrisikos TT, Zhou Y, Li HS, Babcock RL, Wan X, Patel B, Newton K, Mancuso JJ, and Watowich SS. 2020. STAT3 inhibits CD103+ cDC1 vaccine efficacy in murine breast cancer. Cancers (Basel) 12: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wculek SK, Amores-Iniesta J, Conde-Garrosa R, Khouili SC, Melero I, and Sancho D. 2019. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 7: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, and Schindler C. 2010. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol. 184: 2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchins AP, Takahashi Y, and Miranda-Saavedra D. 2015. Genomic analysis of LPS-stimulated myeloid cells identifies a common pro-inflammatory response but divergent IL-10 anti-inflammatory responses. Sci. Rep. 5: 9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray PJ 2005. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. USA 102: 8686–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Hu H, Greeley N, Jin J, Matthews AJ, Ohashi E, Caetano MS, Li HS, Wu X, Mandal PK, McMurray JS, Moghaddam SJ, Sun S-C, and Watowich SS. 2014. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nat. Commun. 5: 5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B, and Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinfor. 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Law CW, Chen Y, Shi W, and Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat M-L, Smyth GK, and Ritchie ME. 2015. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucl. Acids Res. 43: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krämer A, Green J, Pollard J Jr, and Tugendreich S. 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stüve P, Puttur F, Merad M, and Sparwasser T. 2014. Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood 124: 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillmer EJ, Zhang H, Li HS, and Watowich SS. 2016. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 31: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams MA, Bauer S, Lu W, Guo J, Walter S, Bushnell TP, Lillehoj EP, and Georas SN. 2010. Deletion of the mucin-like molecule Muc1 enhances dendritic cell activation in response to Toll-like receptor ligands. J. Innate Immun. 2: 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karrich JJ, Romera-Hernández M, Papazian N, Veenbergen S, Cornelissen F, Aparicio-Domingo P, Stenhouse FH, Peddie CD, Hoogenboezem RM, den Hollander CWJ, Gaskell T, Medley T, Boon L, Blackburn CC, Withers DR, Samsom JN, and Cupedo T. 2019. Expression of Plet1 controls interstitial migration of murine small intestinal dendritic cells. Eur. J. Immunol. 49: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264: 1918–1921. [DOI] [PubMed] [Google Scholar]
- 33.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilček J, Zinkernagel RM, and Aguet M. 1993. Immune response in mice that lack the interferon-γ receptor. Science 259: 1742–1745. [DOI] [PubMed] [Google Scholar]
- 34.Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, Kretzer NM, Durai V, and Murphy KM. 2016. Transcriptional control of dendritic cell development. Ann. Rev. Immunol. 34: 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knödler A, Schmidt SM, Bringmann A, Weck MM, Brauer KM, Holderried TAW, Heine A-K, Grünebach F, and Brossart P. 2009. Post-transcriptional regulation of adapter molecules by IL-10 inhibits TLR-mediated activation of antigen-presenting cells. Leukemia 23: 535–544. [DOI] [PubMed] [Google Scholar]
- 36.Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, and Locati M. 2013. Negative regulation of Toll-like receptor 4 signaling by IL-10–dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 110: 11499–11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasmi K. C. el, Smith AM, Williams L, Neale G, Panopolous A, Watowich SS, Häcker H, Foxwell BMJ, and Murray PJ. 2007. Cutting Edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J. Immunol. 179: 7215–7219. [DOI] [PubMed] [Google Scholar]
- 38.Sin WX, Yeong JPS, Lim TJF, Su IH, Connolly JE, and Chin KC. 2020. IRF-7 mediates type I IFN responses in endotoxin-challenged mice. Front. Immunol. 11: 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlee M, and Hartmann G. 2016. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 16: 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, and Akira S. 2003. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171: 4304–4310. [DOI] [PubMed] [Google Scholar]
- 41.Doyle SE, Vaidya SA, O’Connell R, Dadgostar H, Dempsey PW, Wu TT, Rao G, Sun R, Haberland ME, Modlin RL, and Cheng G. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17: 251–263. [DOI] [PubMed] [Google Scholar]
- 42.Ho HH, and Ivashkiv LB. 2006. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 281: 14111–14118. [DOI] [PubMed] [Google Scholar]
- 43.Wang W-B, Levy DE, and Lee C-K. 2011. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 187: 2578–2585. [DOI] [PubMed] [Google Scholar]
- 44.Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, Larner AC, and Finbloom DS. 1999. Interleukin-10 inhibits expression of both interferon α– and interferon γ– induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 93: 1456–1463. [PubMed] [Google Scholar]
- 45.Im S-S, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, and Osborne TF. 2011. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 13: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, Wang N, Randolph GJ, D’Agati VD, Yvan-Charvet L, and Tall AR. 2017. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 25: 1294–1304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tumurkhuu G, Dagvadorj J, Porritt RA, Crother TR, Shimada K, Tarling EJ, Erbay E, Arditi M, and Chen S. 2018. Chlamydia pneumoniae hijacks a host autoregulatory IL-1β loop to drive foam cell formation and accelerate atherosclerosis. Cell Metab. 28: 432–448.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nolan A, Kobayashi H, Naveed B, Kelly A, Hoshino Y, Hoshino S, Karulf MR, Rom WN, Weiden MD, and Gold JA. 2009. Differential role for CD80 and CD86 in the regulation of the innate immune response in murine polymicrobial sepsis. PLoS One 4: e6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nuriya S, Yagita H, Okumura K, and Azuma M. 1996. The differential role of CD86 and CD80 co-stimulatory molecules in the induction and the effector phases of contact hypersensitivity. Int. Immunol. 8: 917–926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.