

Abstract
Neutrophils are critical for mediating inflammatory responses. Inhibiting neutrophil recruitment is an attractive approach for preventing inflammatory injuries, including myocardial ischemia-reperfusion (I/R) injury, which exacerbates cardiomyocyte death after primary percutaneous coronary intervention in acute myocardial infarction. Here, we found out that a neutrophil exocytosis inhibitor Nexinhib20 inhibits not only exocytosis but also neutrophil adhesion by limiting β2 integrin activation. Using a microfluidic chamber, we found that Nexinhib20 inhibited interleukin 8 (IL-8)-induced β2 integrin-dependent human neutrophil adhesion under flow. Using a dynamic flow cytometry assay, we discovered that Nexinhib20 suppresses intracellular calcium flux and β2 integrin activation after IL-8 stimulation. Western blots of Rac-1-GTP pull-down assays confirmed that Nexinhib20 inhibited Rac-1 activation in leukocytes. An in vitro competition assay showed that Nexinhib20 antagonized the binding of Rac-1 and GTP. Using a mouse model of myocardial I/R injury, Nexinhib20 administration after ischemia and before reperfusion significantly decreased neutrophil recruitment and infarct size. Our results highlight the translational potential of Nexinhib20 as a dual-functional neutrophil inhibitory drug to prevent myocardial I/R injury.
Introduction
Neutrophils are the most abundant leukocytes in humans and serve as the first responders to inflammation and infection (1). An intrinsic neutrophil defect leads to pathologies, such as leukocyte adhesion deficiency syndromes (2, 3). On the other hand, abnormal activation of neutrophils is critically involved in most inflammatory diseases, such as ischemia-reperfusion (I/R) injury (4–6) and auto-immune diseases (7–10).
β2 integrin activation is critical for neutrophil recruitment (1, 11–13). Therefore, regulating β2 integrin signaling is a potential path to reduce inflammatory injury. Several GTPases are involved in β2 integrin signaling, such as ras homolog gene family (Rho) GTPases (14–17) and Ras-related protein 1 (Rap1) GTPases (18–22). Several neutrophil exocytosis inhibitors (Nexinhibs) were identified by Förster resonance energy transfer (FRET)-based screens that targeted the interaction of the small GTPase Rab27a and its effector JFC1 (23); The small GTPase Rab27a is an essential regulator of neutrophil exocytosis (24). Molecular docking analysis showed that Nexinhib20 might interact with an epitope formed by I10, K11, R90, D91, M93, Y122, S123, I181, R184, M185, and S188 of Rab27a (23). Nexinhibs did not interact with another small GTPase, Rab11 (23). Whether Nexinhibs affect the function of other GTPases, especially those involved in the integrin activation signaling pathway, is unknown. Identifying a Nexinhib that inhibits both integrin activation and exocytosis may serve as a dual-functional drug for treating inflammatory diseases.
Myocardial I/R injury exacerbates cardiomyocyte death after primary percutaneous coronary intervention in acute myocardial infarction. Neutrophils are recruited to cardiac tissue during myocardial I/R injury (5, 6) where they worsen injury (25, 26). They mediate cardiomyocyte death by causing vascular plugging, releasing degradative enzymes, and generating reactive oxygen species (ROS) (25). Neutrophil depletion in mice (27) and dogs (28) with myocardial I/R injury showed significant benefits in reducing infarct size. Inhibiting or deleting myeloperoxidase, which is mainly expressed by neutrophils (29), improves myocardial function after I/R injury (30, 31). The neutrophil recruitment cascade includes rolling, slow-rolling, arrest, spreading, intravascular crawling, trans-endothelial migration, and migration to the site of inflammation (1, 32, 33); β2 integrins play critical roles in most steps of the neutrophil recruitment cascade (1, 32, 33). Blocking neutrophil recruitment in mouse knockouts of β2 integrin (CD18) or its ligand, intercellular adhesion molecule 1 (ICAM-1), significantly reduced infarct size after myocardial I/R injury (34). Similar results were observed in β2 integrin antibody blocking experiments in primate (35), pig (36), dog (37–39), rabbit (40, 41), and rat hearts (42, 43). Thus, targeting β2 integrin activation might be a potential path to reduce myocardial I/R injury.
Here, we tested whether Nexinhib20 could inhibit human neutrophil adhesion and β2 integrin activation by targeting Ras-related C3 botulinum toxin substrate 1 (Rac-1) GTPase, and whether Nexinhib20 could limit neutrophil recruitment and decrease infarct size after mouse myocardial I/R injury.
Materials and Methods
Reagents
Recombinant human P-selectin-Fc, ICAM-1-Fc, and IL-8 were purchased from R&D Systems. The Alexa Fluor 488 (AF488)-conjugated and unconjugated conformation-specific monoclonal antibodies mAb24 to human β2-I-like-domain (which reports the headpiece-opening), unconjugated mouse anti-human CD18 mAb (blocking, clone TS1/18), AF594-conjugated rat anti-mouse CD31 mAb, allophycocyanin (APC)-conjugated rat anti-mouse CD115 mAb, PE-conjugated rat anti-mouse Ly6G mAb, AF700-conjugated rat anti-mouse CD45 mAb, unconjugated mouse IgG1 isotype control, APC-conjugated rat anti-mouse IgG1 secondary mAb, and Zombie Yellow Fixable Viability Kit were purchased from Biolegend. The KIM127 mAb to human β2-IEGF-domain, which reports the ectodomain extension, was purified at the Lymphocyte Culture Center at the University of Virginia from hybridoma supernatant (American Type Culture Collection). KIM127 was directly labeled by DyLight 550 (DL550) using DyLight antibody labeling kits from Thermo Fisher Scientific. Nexinhib20 was purchased from Tocris. Casein blocking buffer, Fluo-4 AM, and Pierce protease inhibitor mini-tablets were purchased from Thermo Fisher Scientific. Ghost Dye Blue 516 was purchased from Tonbo Biosciences. Polymorphprep was purchased from Accurate Chemical. Roswell Park Memorial Institute (RPMI) medium 1640 without phenol red and phosphate-buffered saline (PBS) were purchased from Gibco. Human serum albumin (HSA) and fetal bovine serum (FBS) were purchased from Gemini Bio Products. Formalin and non-fat milk were purchased from Fisher Scientific. The Rac-1 Activation Assay Biochem Kit, which contains PAK-PBD protein beads, purified His-tagged Rac-1 protein, GTPγS (non-hydrolysable GTP analog), GDP, and several buffers, were purchased from Cytoskeleton, Inc. A bulk custom order of purified His-tagged Rac-1 protein was purchased from Quintarabio. N-formylmethionyl-leucyl-phenylalanine (fMLP), triphenyl tetrazolium chloride (TTC), Phorbol-12-myristate-13-acetate (PMA), polybrene, paraformaldehyde (PFA), and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich. 2× Laemmli sample buffer and Mini-PROTEAN TGX precast gels were purchased from BioRad. Mouse monoclonal anti-Rac-1 antibody was purchased from BD Biosciences. Horseradish peroxidase (HRP)-conjugated horse anti-mouse antibody was purchased from Cell Signaling Technology. Trappsol (2-Hydroxypropyl-β-cyclodextrin) was purchased from Cyclodextrins CTD, Inc. Enhanced Chemiluminescence (ECL) Ultra was purchased from Lumigen. Penicillin, streptomycin, and amphotericin B solutions were purchased from Hyclone. The total ROS Assay kit was purchased from Invitrogen.
Human neutrophil isolation
Heparinized whole blood samples were obtained from healthy human donors after informed consent, as approved by the Institutional Review Board of the La Jolla Institute for Immunology in accordance with the Declaration of Helsinki. Informed consent was obtained from all donors. Neutrophils were isolated using a Polymorphprep (a mixture of sodium metrizoate and Dextran 500) density gradient. Briefly, human blood was applied to Polymorphprep, centrifuged at 500 g for 35 minutes at 20–25 °C, resulting in neutrophils concentrated in a layer between peripheral blood mononuclear cells and erythrocytes. After washing with PBS twice, the neutrophils (>95% purity by flow cytometry, no visible activation by microscopy) were resuspended in RPMI-1640 without phenol red plus 2% HSA and used within four hours. Neutrophils were incubated with FcR blocking reagents for 10 minutes at room temperature (RT) before all the experiments.
Microfluidic device
The assembly of the microfluidic devices used in this study and the coating of coverslips with recombinant human P-selectin-Fc and ICAM-1-Fc with or without IL-8 have been described previously (44). Briefly, coverslips were coated with P-selectin-Fc (2 μg·ml−1) and ICAM-1-Fc (10 μg·ml−1) without or with IL-8 (10 μg·ml−1) for two hours and then blocked for one hour with casein (1%) at RT. After coating, coverslips were sealed with polydimethylsiloxane chips by magnetic clamps to create flow chamber channels ~29 μm high and ~300 μm across. By modulating the pressure between the inlet well and the outlet reservoir, 6 dyn·cm−2 wall shear stress was applied in all experiments.
Microfluidic perfusion assay
To study the rolling and arrest of neutrophils, isolated human neutrophils (5 × 106 cells ml−1) were perfused in the microfluidic device over a substrate of recombinant human P-selectin-Fc and recombinant human ICAM-1-Fc with or without IL-8 under a shear stress of 6 dyn cm−2. Neutrophils were incubated with Nexinhib20 (10 μM) or vehicle (DMSO) for one hour at RT before being perfused into the microfluidic devices. Time-lapse images (one frame per second) were taken by an IX71 inverted research microscope (Olympus America) with a 40× NA 0.9 air objective during the perfusion to quantify rolling velocity. The quantification was done using the “Manual tracking” plugin in FIJI-ImageJ v2.0. Cell tracks (Fig. 1A) and rolling velocity were obtained (Fig. 1B,C). After perfusion with neutrophils for 10 minutes, the microfluidic device was washed with RPMI-1640 without phenol red plus 2% HSA for 5 minutes. Then, the arrested neutrophils were counted in nine fields-of-view per group (Fig. 1D).
Figure 1.
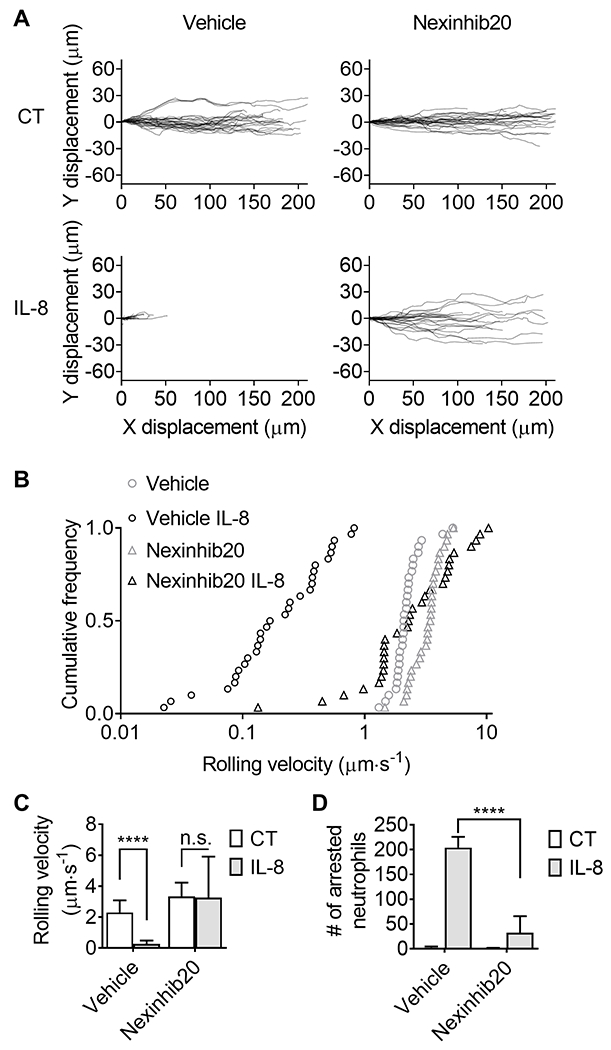
Nexinhib20 inhibits neutrophil adhesion in response to IL-8. Purified human neutrophils were rolled on the substrate of P-selectin and ICAM-1 with or without IL-8 under a shear stress of 6 dyn cm−2. A: The tracks of rolling neutrophils (n=30 cells from 3 individual experiments) treated with Nexinhib20 (10 μM, RT 1 hour) or vehicle control (CT). B: Cumulative frequency and C: neutrophil rolling velocity (Mean±SD; n=30 cells from 3 individual experiments). D: The number of arrested neutrophils (Mean±SD) in n=9 fields-of-view from 3 individual experiments. n.s. (non-significant) p>0.05, ****p<0.0001 by 2-way ANOVA followed by Tukey’s multiple comparisons test.
Mice
C57BL/6J wild-type mice (000664; JAX) were originally obtained from the Jackson Laboratory. LysM-EGFP or Lyz2-EGFP mice (45) were originally obtained from Albert Einstein College of Medicine through a material transfer agreement. Mice were fed a standard rodent chow diet and were housed in microisolator cages in a pathogen-free facility in the Center for Comparative Medicine at UConn Health. All experiments followed the UConn Health Institutional Animal Care and Use Committee (IACUC) guidelines, and approval for the use of rodents was obtained from the UConn Health IACUC according to criteria outlined in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. Both male and female mice aged from 12 to 16 weeks were used in the experiments.
Ischemia-reperfusion injury
Mice were subjected to 35 minutes of myocardial ischemia and 1 (for multi-photon microscopy and flow cytometry) or 22-26 (for TTC-phthalo-blue staining) hours of reperfusion. The reason we use two time points to harvest is because neutrophil recruitment happens 1 hour after the reperfusion (5), and the infarct size can be significantly quantified by TTC-phthalo-blue staining after about 24-hour reperfusion (46). Briefly, anesthesia was induced with an intraperitoneal injection of ketamine hydrochloride (125 mg kg−1) and xylazine (12.5 mg kg−1). Mice were intubated with 24G×3/4” Surflo i.v. catheter and ventilated using MiniVent 845 (Harvard Apparatus).
Surgeries were performed under an SMZ168 Stereo Zoom microscope (Motic). Ischemia was achieved by ligating the left anterior descending coronary artery (LAD) using a 6-0 silk suture with a section of PE-10 tubing placed over the LAD, 1 mm from the tip of the normally positioned left atrium. One critical problem in drug administration is water solubility, which greatly affects drug absorption and bioavailability (47). In our study, we used Trappsol (2-hydroxypropyl-β-cyclodextrin) as a cosolvent for in vivo administration to increase Nexinhib20 solubility. In clinics, the primary percutaneous coronary intervention aims to be performed less than 90 min (within 60 min is preferable) after the patient arrives. To mimic a prevention treatment of reperfusion injury before the primary percutaneous coronary intervention, which is feasible in the clinics, Nexinhib20 (100 mM, 10 μL in DMSO mixed with 190 μL 10% Trappsol per mouse) or vehicle control were administered i.p. 30 minutes before the reperfusion. After occlusion for 35 minutes, reperfusion was initiated by releasing the ligature and removing the PE-10 tubing. The chest wall was closed, the animal extubated, and body temperature was maintained by use of a 37°C warm pad. Hearts were harvested 1 or 22-26 hours later. The loosened suture was left in place and then retied for the purpose of evaluating the ischemic area. Sham control and no drug administered control were performed as well.
Flow cytometry
Isolated human neutrophils (2 × 106 cells mL−1 in RPMI-1640 without phenol red plus 2% HSA) were incubated with Nexinhib20 (10 μM) or vehicle (DMSO) for one hour at RT before being assayed. To monitor the dynamics of β2 integrin activation, 400 μL of 2.5 × 105 cells·mL−1 neutrophils were assessed by an LSRII analyzer (BD Biosciences, San Jose, CA) for 10 s. After adding 0.5 μg mL−1 AF488-conjugated mAb24 and DL550-conjugated KIM127 (final concentration), cells were put back into the analyzer for another 5 minutes. Then, after adding 1 μg·mL−1 IL-8, cells were put back into the analyzer for another 10 minutes. The curves showing the dynamics of integrin activation (Fig. 2A,B) were generated by FlowJo software (version 10.6). The antibody specificities were validated in our previous study using β2 integrin knockout cells and β2-integrin-activation-deficient talin-1 knockout cells (48). Compensations were performed before all experiments.
Figure 2.
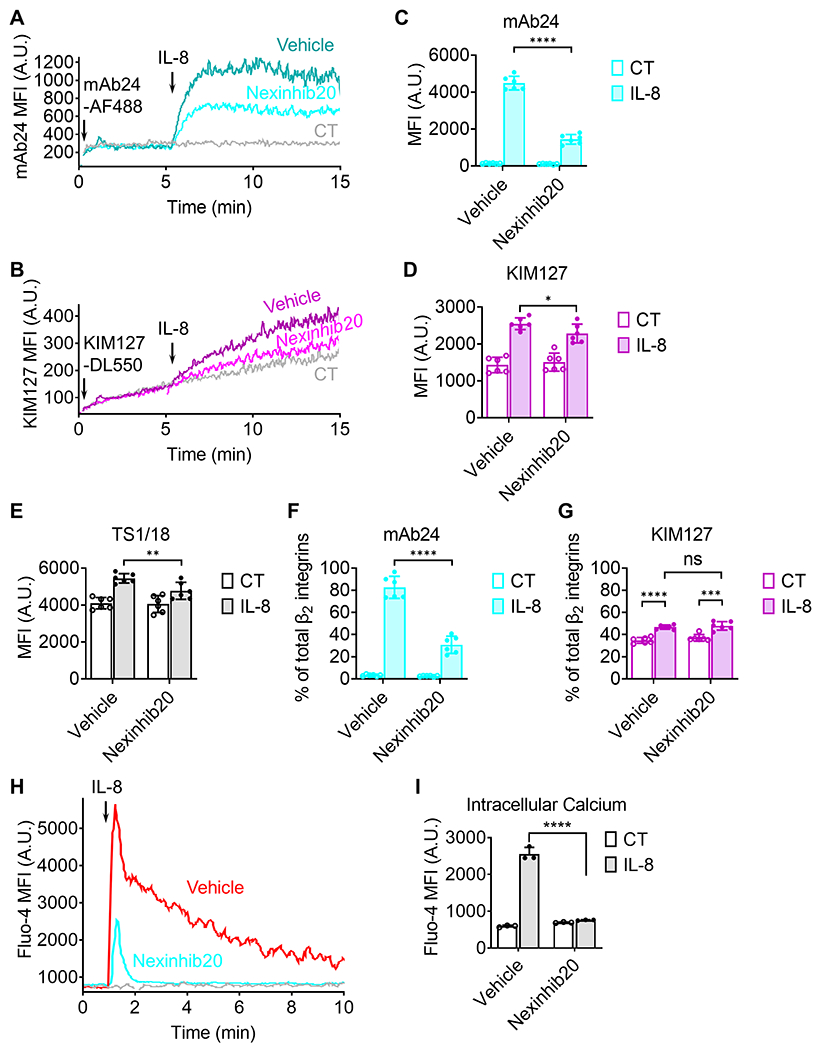
Nexinhib20 inhibits β2 integrin activation and intracellular Ca2+ signal after stimulation by IL-8. A-B: Homogenous binding assay: typical graphs showing the dynamic expression (the moving average of median fluorescence intensity, MFI) of mAb24 (A, high-affinity β2 integrins) and KIM127 (B, extended β2 integrins) epitopes on purified human neutrophils pretreated with Nexinhib20 (10 μM, RT 1 hour, the cyan curve in A or magenta curve in B) or vehicle control (DMSO, the blue curve in A or purple curve in B). Fluorescent-labeled antibody (mAb24-AF488 in A or KIM127-DL550 in B) was added 10 seconds after initiation to stain neutrophils. IL-8 was added 5 minutes after initiation to induce integrin activation (high-affinity and extension). The background, in which neutrophils were not stimulated with IL-8, was shown as control (CT) in gray curves. C-E: Bar graphs showing the MFI of mAb24 (C), KIM127 (D), or TS1/18 (E, total β2 integrins) on neutrophils treated with vehicle (DMSO) or Nexinhib20 10 min after IL-8 stimulation (IL-8) or vehicle (PBS, CT). MFI of isotype control staining was subtracted as the background. Mean±SD, n = 6 replicates from 3 individual experiments. F-G: Percentage of high-affinity (F, mAb24) and extended (G, KIM127) β2 integrins on neutrophils treated with vehicle (DMSO) or Nexinhib20 10 min after IL-8 stimulation (IL-8) or vehicle (PBS, CT). Since mAb24, KIM127, and TS1/18 are all IgG1 isotypes, and we used the same secondary antibody, the percentage of high-affinity and extended β2 integrins can be calculated by dividing the MFI of mAb24 and KIM127 by the MFI of TS1/18. Mean±SD, n = 6 replicates 3 individual experiments. H: A typical graph showing the dynamics (the moving average of Fluo-4 MFI) of intracellular Ca2+ in neutrophils treated with Nexinhib20 (10 μM, RT 1 hour, the cyan curve) or vehicle control (DMSO, the red curve) stimulated by IL-8 (added at minute 1) or not (the gray curve). I: Intracellular calcium of neutrophils by Fluo-4 MFI (Mean±SD; n=3 individual experiments) without (CT) or with IL-8 simulation and without or with Nexinhib20. ns (non-significant) p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by unpaired student’s t-test.
To quantify the percentage of mAb24 and KIM127 epitopes and assess inhibition of β2 integrin exocytosis, pan-CD18 mAb24 TS1/18, which has the same isotype (mouse IgG1) as mAb24 and KIM127, was used. Isolated human neutrophils (5 × 105 cells·mL−1 in RPMI-1640 without phenol red plus 2% HSA) were incubated with Nexinhib20 (10 μM) or vehicle (DMSO) for one hour at RT before being assayed. Neutrophils were mixed with unconjugated mAb24 (1 μg mL−1), KIM127 (1 μg mL−1), TS1/8 (1 μg mL−1), or mouse IgG1 isotype control (1 μg mL−1), and incubated with 1 μg mL−1 IL-8 at RT for 10 min. After incubation, neutrophils were fixed by 1% PFA at 4°C for 10 min. After two washes with PBS, cells were incubated with APC-conjugated rat anti-mouse IgG1 secondary mAb (1 μg mL−1) at RT for 10 min. After two washes with PBS, cell fluorescence was assessed with an LSRII (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (version 10.6). The quantifications of mAb24, KIM127, TS1/18, and isotype mean fluorescence intensities (MFI) (Fig. 2C–E) were analyzed by FlowJo software (version 10.6) and obtained from six replicates. MFI of isotype controls was subtracted as background signal. Since mAb24, KIM127, and TS1/18 are all IgG1 isotypes and we used the same secondary antibody, the percentage of high-affinity and extended β2 integrins (Fig. 2F and G) can be calculated by dividing the MFI of mAb24 and KIM127 by the MFI of TS1/18.
To monitor the dynamics of intracellular calcium (Ca2+) flux, neutrophils (2 × 106 cells mL−1 in RPMI-1640 without phenol red plus 2% HSA) were incubated with Fluo-4 (4 μg mL−1) for one hour at RT. After washes, neutrophils were resuspended in RPMI-1640 without phenol red plus 2% HSA and assessed by an LSRII analyzer (BD Biosciences, San Jose, CA). One minute after analyzing, 1 μg mL−1 IL-8 was added to the cells. Cells were put back into the analyzer for another 9 minutes. The curves showing the dynamics of intracellular Ca2+ flux (Fig. 2H) were generated by FlowJo software (version 10.6). The quantification of Fluo-4 MFI (Fig. 2I) was analyzed by FlowJo software (version 10.6) and obtained from three individual experiments.
To assess the viability of neutrophils (Supplemental Fig. 1B), neutrophils (2 × 106 cells mL−1 in RPMI-1640 without phenol red plus 2% HSA) were incubated with different concentrations (0, 10, 20, 50, and 100 μM) of Nexinhib20 at RT for one hour. After washes, neutrophils were incubated with Ghost Dye Blue 516 at RT for 15 minutes. After washes, cell fluorescence was assessed with an LSRII (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (version 10.6).
To assess the neutrophil recruitment in myocardial I/R injury, LysM-EGFP mice underwent 35 minutes of ischemia and 1 hour of reperfusion. To mimic a prevention treatment of reperfusion injury before the primary percutaneous coronary intervention, which is feasible in the clinics, Nexinhib20 (100 mM, 10 μL per mouse) or vehicle control was administered i.p. 30 minutes prior to the reperfusion. After the 1-hour reperfusion, the mouse heart was harvested and perfused with ice-cold PBS to remove residual blood and unbound leukocytes, transferred into an ice-cold gentleMACS C tube, cut into ~1 mm3 pieces, suspended with 5 mL PBS plus 2% FBS, 2 mM EDTA, and 0.08 μg mL−1 APC-conjugated anti-CD115 mAb, and homogenized five times by the ‘m_Heart_01’ program of the gentleMACS Dissociator (Miltenyi). The cell suspension was filtered by 70 μm nylon mesh strainer (Fisher), centrifuged at 500x g, 4°C for 5 minutes, resuspended in 200 μL 1:300 diluted Zombie Yellow fixable viability dye, and incubated on ice for 15 minutes. After centrifuging at 500x g, 4°C for 5 minutes, cells were resuspended in 200 μL ice-cold PBS containing 1.25 μg mL−1 AF700-conjugated anti-CD45 mAb and 1 μg mL−1 PE-conjugated anti-Ly6G mAb and incubated on ice for 10 minutes. After being fixed with 1% PFA and washes with ice-cold PBS, cell fluorescence was assessed with an LSRII (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (version 10.6).
Peripheral blood of the above mice was also collected. 100 μL was mixed with 200 μL 1:300 diluted Zombie Yellow fixable viability dye and incubated on ice for 15 minutes. After centrifuging at 500x g, 4°C for 5 minutes, cells were resuspended in 200 μL ice-cold PBS containing 1.25 μg mL−1 AF700-conjugated anti-CD45 mAb, 1 μg mL−1 PE-conjugated anti-Ly6G mAb, and 2 μg mL−1 APC-conjugated anti-CD115 mAb, and incubated on ice for 10 minutes. After being fixed with 1% PFA, red blood cells were lysed with deionized water for 30 seconds (stopped by adding 10× PBS). Leukocyte fluorescence was assessed with an LSRII (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (version 10.6).
Reactive oxygen species (ROS) production
ROS production of isolated human neutrophils was quantified by using the Total ROS Assay kit from Invitrogen. A black, clear bottom, non-treated 96-well plate was used in this assay. Before the assay, the 96-well plate was coated with 10 μg mL−1 human ICAM-1-Fc at RT for 2 hours and washed twice with PBS. Isolated human neutrophils (2 × 106 cells mL−1) were incubated with Nexinhib20 (10 μM) or vehicle (DMSO) for one hour at RT before being assayed. After centrifuging at 300x g, RT for 2 min, cells were resuspended at 106 cells mL−1 in the ROS Assay Stain Solution from the kit and incubated with 2 μg mL−1 mouse anti-human CD18 blocking mAb (TS1/18 to block neutrophil adhesion) (49) or isotype control at RT for 10 min. The 100 μL well−1 neutrophils (3 replicates per group) were seeded into the ICAM-1-coated 96-well plate. The background ROS before stimulation was measured by Cytation 1 Cell Imaging Multi-Mode Reader (Filter set: Green, Ex: 485/20nm, Em: 528/20 nm, BioTek, Santa Clara, CA). Then 100 nM PMA was added to each well, and ROS production was measured by Cytation 1 Cell Imaging Multi-Mode Reader every 5 minutes.
Cell culture
The HL60 cells and CXCR2-expressing HL60 cells (HL60-CXCR2)(50) were gifts from Dr. Orion D. Weiner at the University of California San Francisco and Dr. Ann Richmond at the Vanderbilt University School of Medicine, respectively. HL60-CXCR2 cells were selected with G418 (0.5 μg mL−1) to maintain CXCR2 expression. Cells were maintained in culture medium (RPMI-1640, 10% FBS, 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin, and 250 ng mL−1 amphotericin B) at 37°C and 5% CO2. In most experiments, cells were differentiated with 1.3% DMSO for 7 days before assays. Cells were checked monthly for mycoplasma infection using the e-Myco plus Mycoplasma PCR Detection Kit.
Rac-1-GTP pull-down and western blots
Differentiated HL60 or HL60-CXCR2 cells or isolated human neutrophils (2 × 106 cells mL−1 in RPMI-1640 without phenol red plus 2% HSA) were incubated with Nexinhib20 (10 μM) or vehicle (DMSO) for one hour at RT. After washes, cells were resuspended in RPMI-1640 without phenol red (107 cells mL−1) and incubated with or without stimulators (100 nM fMLP for HL60, 1 μg mL−1 IL-8 for HL60-CXCR2) at RT for 1 minute. Cells were lysed by 1:1 addition of 2× Triton X-100 lysis buffer (final concentration: 1% Triton X-100, 50 mM HEPES pH 7.0, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, plus protease inhibitor mixture — one Pierce protease inhibitor mini-tablet per 5 mL 2× buffer) on ice for 5 minutes. After centrifuging at 16,000x g, 4°C for 8 minutes, supernatants were saved as protein samples.
For the in vitro Rac-1/GTP binding competition assays, purified His-tagged Rac-1 (0.08 μg mL−1 in 1× Triton X-100 lysis buffer) were mixed with loading buffer (from the Rac-1 activation assay biochem kit, 1:10) and Nexinhib20 (1, 3, 10, 30, 100, 300, and 1000 μM) or vehicle (DMSO). Then samples were incubated with GTPγS (0.4 μM) or GDP (0.8 mM) at RT for 15 minutes. The reaction was stopped by transferring samples to 4°C and adding the stop buffer (from the Rac-1 activation assay biochem kit, 1:10).
The Rac-1-GTP pull-down was performed using Rac-1 activation assay biochem kit following manufacturer’s instructions. Briefly, protein samples were immediately incubated with p21 activated kinase 1-p21 binding domain (PAK-PBD) beads (10 μL per 1 mL sample) for one hour at 4°C. Then beads were pelleted by centrifugation at 5000x g, 4°C for 8 minutes. After removal of most of the supernatant, beads were washed twice with 500 μL washing buffer from the kit. Beads were resuspended with 2× Laemmli sample buffer and boiled for two minutes.
Protein samples (before and after the Rac-1-GTP pull-down) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. Membranes were blocked for ~30 min in tris-buffered saline with 0.1% tween 20 (TBST) plus 5% non-fat milk. After blocking, membranes were incubated overnight with mouse monoclonal anti-Rac-1 antibody diluted 1:2000 in TBST at 4°C, and HRP-conjugated horse anti-mouse antibody diluted 1:5000 in TBST plus 5% non-fat milk at RT for one hour. ImageQuant LAS 4000 (GE) was used to image membranes after adding ECL Ultra (Fig. 3).
Figure 3.
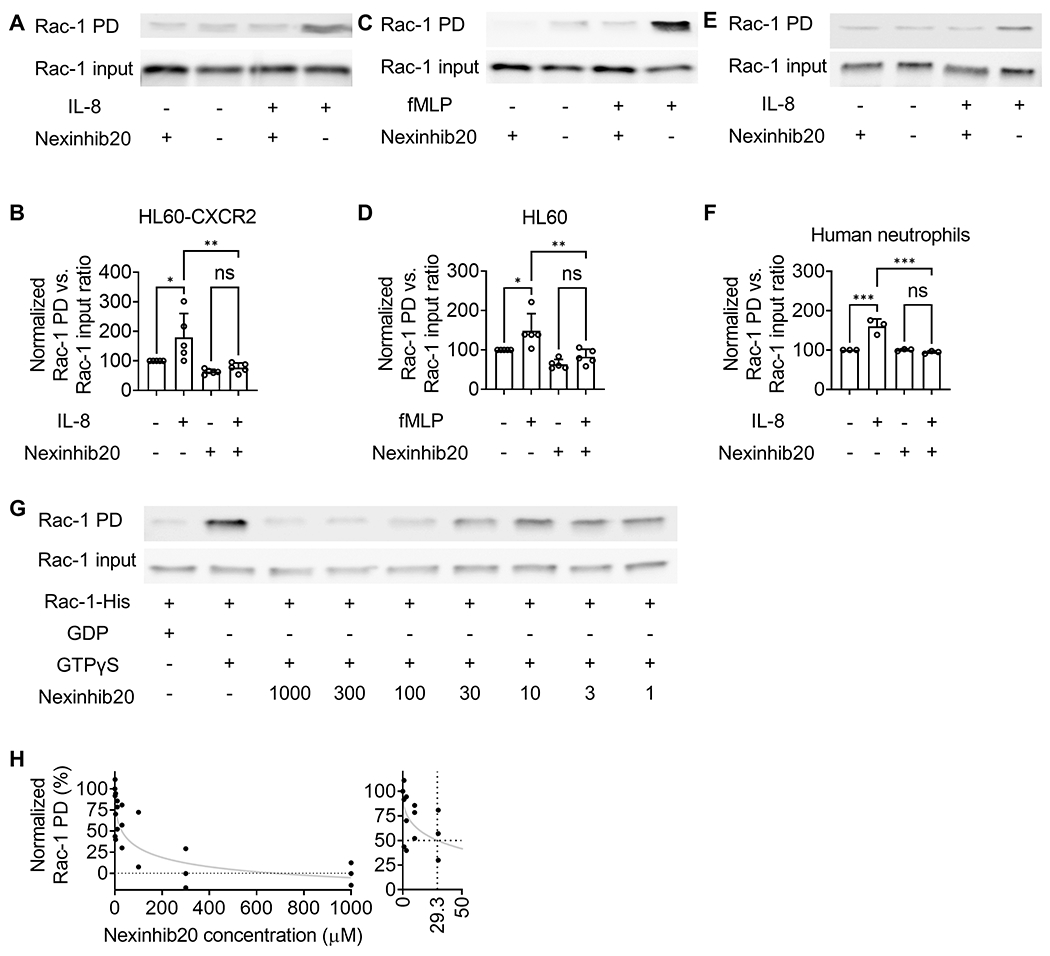
Nexinhib20 inhibits Rac-1 activation by antagonizing GTP binding. A-B: A representative western blot image (A) and quantifications (B) of active PAK-PBD pulled-down Rac-1-GTP (Rac-1 PD) and total Rac-1 (Rac-1 input) in Nexinhib20 incubated (+) (10 μM, RT 1 hour) or control (−) HL60-CXCR2 cells stimulated with (+) or without (−) IL-8 (1 μg mL−1, 1 minute, RT). C-D: A representative western blot image (C) and quantifications (D) of active PAK-PBD pulled-down Rac-1-GTP (Rac-1 PD) and total Rac-1 (Rac-1 input) in Nexinhib20-incubated (+) (10 μM, RT 1 hour) or control (−) HL60 cells stimulated with (+) or without (−) fMLP (100 nM, 1 minute, RT). E-F: A representative western blot image (E) and quantifications (F) of active PAK-PBD pulled-down Rac-1-GTP (Rac-1 PD) and total Rac-1 (Rac-1 input) in Nexinhib20-incubated (+) (10 μM, RT 1 hour) or control (−) human neutrophils stimulated with (+) or without (−) IL-8 (1 μg mL−1, 1 minute, RT). Mean±SD from n=5 independent experiments in B and D, and n=3 independent experiments in F. ns (non-significant) p>0.05, *p<0.05, **p<0.01 by one-way ANOVA followed by Tukey’s multiple comparisons test. G: A representative western blot image showing the amount of PAK-PBD pulled-down Rac-1-GTP (Rac-1 PD) when purified His-tag Rac-1 was incubated with GTPγS (non-hydrolysable GTP analog) in vitro in the presence of different concentrations of Nexinhib20 (shown in μM) or not (the same amount of DMSO vehicle added). His-tag Rac-1 incubated with GDP was used as a negative control. H: The fitting curve (Absolute IC50, X is concentration in Prism) showing the inhibition efficiency of Nexinhib20 on the Rac-GTP interaction. Individual values from n=3 independent experiments are shown. The values were normalized by setting GDP-added samples to 0 and GTPγS-added vehicle samples to 100. Zoomed-in graph (right) showing that the IC50 was around 29.3 μM.
Multi-photon microscopy
Mice underwent 35 minutes of ischemia and 1 hour of reperfusion. To mimic a prevention treatment of reperfusion injury before the primary percutaneous coronary intervention, which is feasible in the clinics, Nexinhib20 (100 mM, 10 μL per mouse) or vehicle control were administered i.p. 30 minutes prior to the reperfusion. After the reperfusion, the mouse heart was harvested and perfused with PBS to remove residual blood and unbound leukocytes and was incubated with anti-CD31-AF594 mAb (10 μg/mL, 250 μL per heart) to label the coronary artery sequentially. The explanted heart was immersed in PBS and imaged by a multi-photon microscope immediately. The Bruker’s upright multi-photon microscope (#4269) was equipped with a Mai Tai High-Performance Ti:sapphire femtosecond pulsed laser (tuning range 690-1020 nm, set to 780 nm excitation in this assay) and a 20× NA 0.95 water immersion objective. The bandpass filters in front of the corresponding four different photomultiplier tube detectors are 660/40, 595/50, 525/50, and 460/50 nm. The 595/50 nm channel and 525/50 nm channel were used for EGFP and AF594 imaging, respectively. Three-dimensional z-stack series (5 μm interval, 10-20 stacks) images of the coronary artery were acquired (Fig. 4A). The mean fluorescence intensity of EGFP within the coronary artery was quantified by FIJI-ImageJ v2.0.
Figure 4.
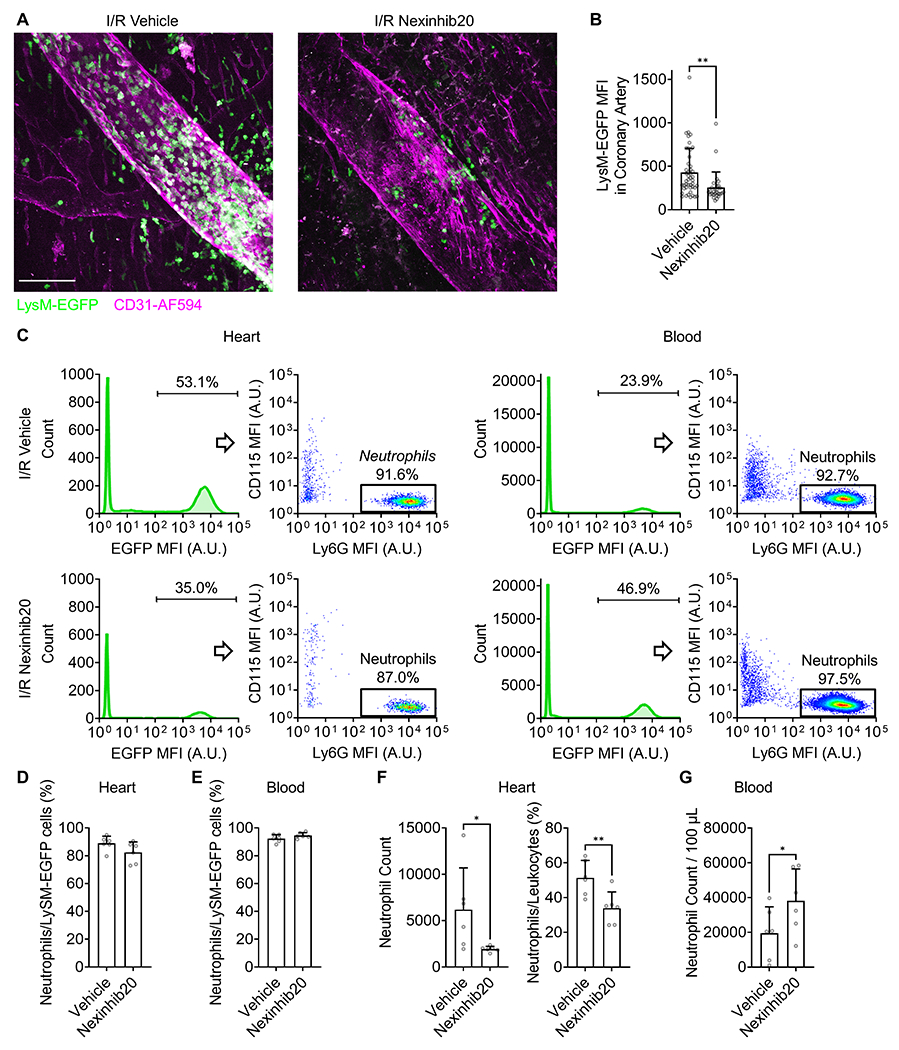
Nexinhib20 limits neutrophil recruitment in the heart during mouse myocardial I/R injury. A: Representative multi-photon microscopy images showing the recruitment of EGFP-labeled leukocytes (most of them are neutrophils) at the coronary artery (CD31-AF594 labeled) of LysM-EGFP myocardial I/R (35/60 minutes) mice without (left) or with (right) Nexinhib20 (1 μmol per mouse) administration. In these images, peripheral blood in the heart was washed out by infusing PBS through the aorta; Thus, LysM-EGFP+ cells visualized in the images were adhered to the vessel wall or infiltrated into the tissue. B: Mean±SD of EGFP MFI in coronary arteries of mice administered with vehicle control and Nexinhib20. n=46 and 28 fields-of-view from 9 vehicle control and 7 Nexinhib20 treated mice, respectively, obtained in 7 individual experiments. C: Representative flow cytometry plots showing percentages of Ly6G+ neutrophils in LysM-EGFP+ leukocytes in I/R heart (left) and blood (right) of mice without (top) or with (bottom) Nexinhib20 (1 μmol per mouse) administration. D-E: Mean±SD of neutrophil percentages in LysM-EGFP+ leukocytes in I/R heart (D) and blood (E) of mice treated with Nexinhib20 (1 μmol per mouse) or vehicle. n=6 mice obtained in 3 individual experiments. F: Mean±SD of neutrophil counts (left) and percentages in CD45+ live leukocytes (right) in I/R heart of mice treated with Nexinhib20 (1 μmol per mouse) or vehicle. n=6 mice obtained in 3 individual experiments. G: Mean±SD of neutrophil counts in blood of myocardial I/R mice treated with Nexinhib20 (1 μmol per mouse) or vehicle. n=6 mice obtained in 3 individual experiments. *p<0.05, **p<0.01 by unpaired Student’s t-test.
TTC-phthalo-blue staining
To assess the ischemic area at risk after 22-26 hours of reperfusion, hearts were excised, infused with PBS and freshly prepared 10% phthalo-blue (PBS with 0.75% tween 20) through the aorta and coronary arteries in a retrograde fashion, frozen at −20°C for 10 minutes, and sliced into five to six 1 mm cross-sections with the aid of a pre-freeze acrylic matrix (ZIVIC Labs). The heart sections were incubated with freshly prepared 1% TTC solution (Sigma-Aldrich) at 37°C for 10 minutes and fixed with formalin. Viable myocardium stained red, and infarcted tissue appeared white. Images (Fig. 5A) were acquired by an MU130 color-complementary metal-oxide-semiconductor (CMOS) camera (AmScope) equipped on an SMZ168 Stereo Zoom microscope (Motic). The infarct area (white), the area at risk (red and white), and the total left ventricle area from each section were measured using ZEN v3.1 (Zeiss). Ratios of infarct area/area at risk (Fig. 5B) and of area at risk/left ventricle (Fig. 5C) were calculated and expressed as percentages.
Figure 5.
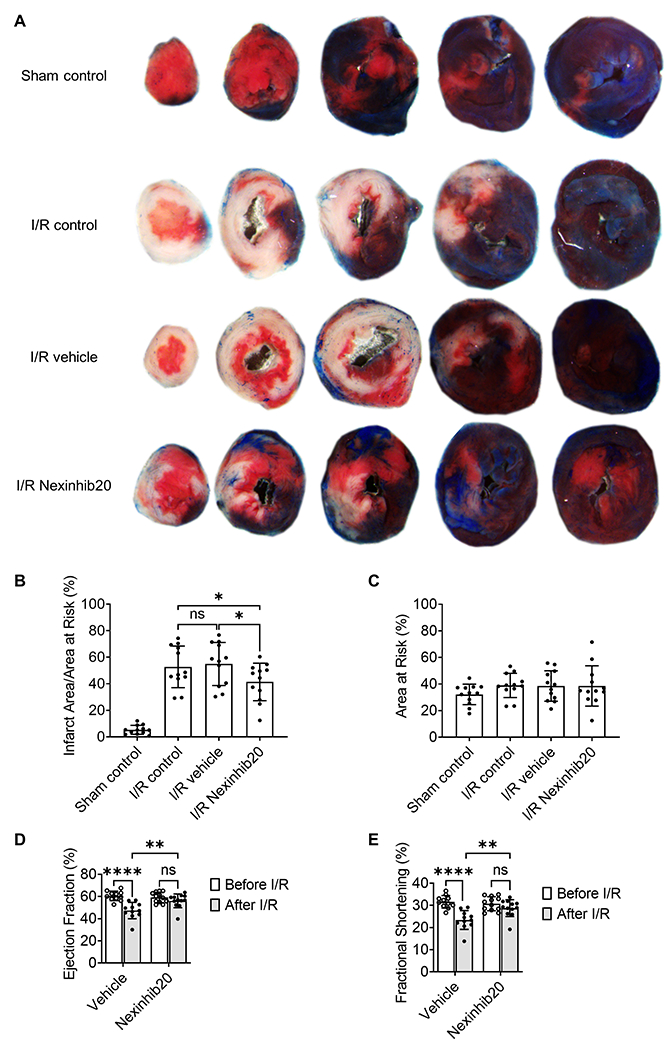
Nexinhib20 decreases the infarct area in mouse myocardial I/R injury. A: Representative images showing TTC-phthalo-blue-stained heart serial sections from myocardial I/R (35 min/24 hours) mice administered with Nexinhib20 (I/R Nexinhib20, the bottom row) or vehicle control (I/R vehicle, the third row). Sham control (the top row) and myocardial ischemia-reperfusion mice without any administration (I/R control, the second row) are also shown. B: Mean±SD of the infarct area percentage in the area of risk from n=12 mice per group obtained in 24 individual experiments. C: Mean±SD of the area of risk percentage from n=12 mice per group obtained in 24 individual experiments. ns (non-significant) p>0.05, *p<0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test. D-E: Analysis of left ventricle echocardiogram before and after myocardial I/R (35 min/24 hours). Mean±SD of ejection fraction (D) and fraction shortening (E) from n=11 vehicle-treated mice and n=12 Nexinhib20-treated mice obtained in 5 individual experiments. ns p>0.05, **p<0.01, **** p<0.0001 by two-way ANOVA followed by Tukey’s multiple comparisons test.
Left ventricle echocardiogram
To quantify left ventricle function, we performed echocardiograms on mice before and seven days after myocardial I/R injury. Mice were anesthetized with 2% isoflurane i.n. and placed on a heating pad. Chest hair was removed using an electric shaver and animals were fixated on their backs. Echocardiography loops were recorded in B and two-dimensional-targeted M modes in longitudinal and short-axis views on a Vevo 3100 High-Resolution Imaging System equipped with an MX550D transducer (VisualSonics, Toronto, ON, Canada). Mice were fixed on a heated table and heart rate was monitored during the procedure. Systole and diastole were defined based on concomitant electrocardiography (ECG) recordings. The end-systolic time point for left ventricle diameter measurement was defined as the maximum ventricle contraction just before the complete closure of the aortic valve. End-diastole was defined as the maximum left ventricle dilation and filling just before mitral valve closing (when visible) and aortic valve opening. Left ventricular ejection fraction was determined by left ventricle tracing relating the end-systolic left ventricle area as the minimal left ventricle cross-sectional area to the end-diastolic left ventricle area as the maximum left ventricle cross-sectional area in long-axis views. Fractional shortening was assessed by using VevoLab software (VisualSonics).
Pharmacokinetics
Pharmacokinetics was performed through a service provided by the Shanghai Institute of Materia Medica. Nexinhib20 (160 mM, 5 μL in DMSO mixed with 95 μL 10% Trappsol per mouse) was administered i.p. to three 18-19 g male mice. Blood samples (20 μL) were collected at 3 min, 15 min, 45 min, 2 h, 4 h, 8 h, and 24 h through femoral vein phlebotomy. 200 μL of methanol:acetonitrile (1:1, v/v) with internal standard was added to 20 μL of plasma and vortexed thoroughly. After centrifuging at 11000 rpm, RT for 5 min, 20 μL of the supernatant was collected and mixed with 20 μL of water for analysis. Samples were analyzed by a TQ-S triple quadrupole mass spectrometer (Waters, Milford, MA, USA). An Acquity Uplc Beh C18 Column (1.7 μm, 2.0 mm × 50 mm, Waters) was used for the analysis. Gradient elution was applied consisting of 5 mM aluminum ammonium sulfate dodecahydrate containing 0.1% formic acid and methanol:acetonitrile (1/9, v/v) containing 0.1% formic acid.
Statistics
Statistical analysis was performed using PRISM software (version 8.30, GraphPad Software). Data analysis was performed using student’s t-test, one-way ANOVA followed by Tukey’s multiple comparisons test, or 2-way ANOVA followed by Šídák’s or Tukey’s multiple comparisons test, which are indicated in Figure Legends. P values less than 0.05 were considered significant.
Results
Nexinhib20 inhibits IL-8-induced neutrophil adhesion
Nexinhib20 inhibits exocytosis without inducing apoptosis or cell death (23). Here, to further analyze whether Nexinhib20 could be toxic to neutrophils, we tested the viability of neutrophils after Nexinhib20 treatment using flow cytometry. We showed that neutrophil viability remained close to 100% even when incubated with 100 μM Nexinhib20 for one hour at room temperature (RT, Supplemental Fig. 1A). This is consistent with the previous study that Nexinhib20 did not induce a significant increase in cell death after 1 and 4 hours of incubation compared to DMSO vehicle controls (23). We incubated neutrophils with 10 μM Nexinhib20 for one hour at room temperature in most of our cellular experiments unless stated otherwise.
To assess the impact of Nexinhib20 on neutrophil adhesion, we performed microfluidic assays as described previously (44, 51). As expected, neutrophils rolled on the substrate of human P-selectin and ICAM-1 under a shear stress of 6 dyn cm−2 (Fig. 1A, upper left), which is a typical shear stress in postcapillary venules that commonly show neutrophil recruitment during inflammation (52). Upon addition of IL-8 to the substrate, neutrophils stopped rolling (arrest), and reduced the 100-second rolling distance from ~200 μm to ~40 μm (Fig. 1A, bottom left). When neutrophils were incubated with Nexinhib20 before the perfusion, they failed to arrest (Fig. 1A, bottom right). After quantifying the rolling velocity of these neutrophils (Fig. 1B,C), we found that IL-8 stimulation did not slow down the rolling velocity of Nexinhib20-treated neutrophils. After 10 minutes of rolling and 5 minutes of washing, we quantified the arrested neutrophils, and found that Nexinhib20 significantly decreased the number of arrested neutrophils from ~200 cells per field-of-view to ~20 cells per field-of-view (~90%, Fig. 1D). Thus, Nexinhib20 inhibited adhesion of human neutrophils to P-selectin and ICAM-1 in a microfluidic model of physiological flow conditions.
Nexinhib20 limits β2 integrin exocytosis and activation
Since β2 integrins are critical for neutrophil adhesion (33, 53, 54), Nexinhib20 was tested for its potential to inhibit β2 integrin expression and activation on neutrophils. Nexinhib20 was developed as a neutrophil exocytosis inhibitor that decreases exocytosis of the integrin αMβ2 (Mac-1, CD11b/CD18) α subunit CD11b (23). It has been shown that after 30 mins of stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) and fMLP, CD11b on human neutrophils was upregulated to ~2-fold compared to unstimulated cells. Pretreatment with Nexinhib20 diminished this CD11b upregulation. Here, we assessed the effect of Nexinhib20 on total β2 subunit (CD18) surface expression (Fig. 2E). We found that total CD18 expression was upregulated by ~40% after 10 min with 1 μg mL−1 IL-8 stimulation. As expected, this β2 integrin exocytosis was inhibited significantly by Nexinhib20 treatment (Fig. 2E). Secondly, we tested β2 integrin activation, which has two major conformational changes of the β2 integrin extracellular domain - headpiece-opening to acquire high-affinity (H+) binding to ligands and extension (E+) that allows binding ligands in trans (13, 33). H+ and E+ β2 integrins can be monitored by using the conformation-specific antibodies mAb24 (55–57) and KIM127 (58, 59), respectively. Please note that both lymphocyte function-associated antigen 1 (LFA-1, CD11a/CD18, αLβ2) and Mac-1 were detected. Time-resolved flow cytometry showed that IL-8 induced dramatic increases of both mAb24 (Fig. 2A) and KIM127 (Fig. 2B) binding, and that Nexinhib20 treatment inhibited these effects. Since the time-resolved flow cytometry cannot remove free mAb in the cell suspension which generates background noise, we also use standard flow cytometry with fixation and washing to remove free mAb and get more accurate quantification (Fig. 2C–G). Isotype control (mouse IgG1) was used to determine background noise in the standard flow cytometry assay. After 10 min of 1 μg mL−1 IL-8 stimulation, we found that mAb24 staining increased to ~10-fold, and KIM127 staining increased to ~2-fold. Nexinhib20 inhibited the IL-8-induced elevation of mAb24 (Fig. 2C) by ~75% and KIM127 (Fig. 2D) by ~20%. Since TS1/18 (Fig. 2E) is the same isotype (mouse IgG1) as mAb24 and KIM127 and the same secondary antibody was used, the percentage of high-affinity and extended β2 integrins can be calculated by dividing the MFI of mAb24 and KIM127, respectively, by the MFI of TS1/18. We found that Nexinhib20 reduced the percentage of high-affinity (mAb24, Fig. 2F) but not extended β2 integrins (KIM127, Fig. 2G), suggesting that Nexinihib20 inhibits β2 integrin high-affinity activation but not extension activation. These results demonstrated that Nexinhib20 significantly limited both the exocytosis and activation of β2 integrins on human neutrophils, which are critical events for neutrophil adhesion.
Nexinhib20 inhibits chemokine-induced calcium (Ca2+) flux
Intracellular Ca2+ transients are involved in the chemokine-triggered integrin inside-out activation signaling pathway (60). Ca2+ and diacylglycerol (DAG) activate Rap1 GTPases, which are critical for integrin inside-out activation (18, 22), through calcium and DAG-regulated guanine nucleotide exchange factors (CalDAG-GEFs) (22). The short inside-out Ca2+ signal can be triggered by IL-8 through its receptor CXCR2 (61, 62). The disassociated Gβγ activates Ras-related C3 botulinum toxin substrate 1 (Rac-1) and phospholipase C β (PLCβ) sequentially and induces intracellular Ca2+ flux (16). Using the intracellular Ca2+ dye Fluo-4 and time-resolved flow cytometry, we evaluated transient elevation of Fluo-4 fluorescence in neutrophils upon IL-8 stimulation (Fig. 2H, the red trace). Nexinhib20 treatment potently blocked the IL-8-induced Ca2+ signal (Fig. 2H, the blue trace; Figure 2I).
Nexinhib20 suppresses Rac-1 activation in cells
In the integrin inside-out activation signaling pathway, Rac-1 is an upstream signaling molecule of intracellular Ca2+ flux. In this pathway, the activation of G-protein-coupled receptors dissociates G protein to Gα and Gβγ subunits (63). Gβγ activates Rac-1 through P-Rex1 and Vav1 (64), then activates phospholipase C β2 (PLCβ2) and PLCβ3, induces intracellular Ca2+ flux and downstream signaling molecules mentioned above to activate β2 integrins (16). Rac-1 knockout neutrophils showed defects in inside-out signaling-triggered adhesion (16). Thus, we tested if Nexinhib20 can inhibit Rac-1. The neutrophil-like cell line HL60 and HL60 cells stably expressing CXCR2 (HL60-CXCR2) were used in the Rac-1 activity assays. Using the p21-activated kinase 1-p21 binding domain (PAK-PBD) bead pull-down assay, which enriches for the active GTP form of Rac-1 (Rac-1-GTP), followed by anti-Rac-1 western blots, we found that Nexinhib20 significantly inhibited the IL-8 (Fig. 3A–B) or fMLP (Fig. 3C–D) induced Rac-1 activation in HL60-CXCR2 or HL60 cells, respectively. Quantification showed that IL-8 (Fig. 3B) and fMLP (Fig. 3D) stimulation increased the amount of Rac-1-GTP by ~80% and ~50%, respectively, and Nexinhib20 treatment eliminated these increases (Fig. 3B,D). To further confirm our findings in human neutrophils, we performed the Rac-1 pull-down assay using Nexinhib20 or vehicle-treated human neutrophils (Fig. 3E,F). Similar to HL60 data, we showed that IL-8 stimulation increases the amount of Rac-1-GTP by ~60%. Nexinhib20 treatment eliminated these increases. Thus, Nexinhib20 inhibited Rac-1 activity in cells, which was consistent with the inhibition of intracellular Ca2+ flux and integrin activation in neutrophils.
Nexinhib20 antagonizes the Rac-1-GTP interaction
Nexinhib20 was discovered by a screen for inhibitors of Ras-related protein Rab27a-synaptotagmin-like 1 (SYTL1 or JFC1) interaction and was expected to directly bind Rab27a by molecular docking analysis (23). Rac-1 was reported to interact with JFC1 as well (65). Thus, we hypothesized that Nexinhib20 directly binds Rac-1 or competes for Rac-1-GTP binding. To test this, we performed in vitro binding assays using purified His-tagged Rac-1 protein (Fig. 3G,H). Incubating with the non-hydrolysable GTP analog GTPγS produced active Rac-1-GTP that was enriched by PAK-PBD beads (Fig. 3G, the second column, vehicle control). His-tagged Rac-1 protein incubated with GDP was used as a negative control (Fig. 3G, the first column). In the presence of Nexinhib20, the binding of His-tagged Rac-1 and GTPγS was significantly inhibited in a dose-dependent manner (Fig. 3G). After calculating the fitting curve of the inhibition percentage (Fig. 3G), we found that the IC50 of Nexinhib20 to Rac-1-GTP binding was ~29.3 μM. These data suggested that Nexinhib20 could antagonize the Rac-1-GTP interaction and may directly bind to Rac-1. This direct inhibition indicated that the upstream P-Rex1 and Vav1 for Rac-1 activation might not be relevant in the Nexinhib20 inhibition of neutrophil integrin activation.
Nexinhib20 limits adhesion-independent human neutrophil ROS production
Nexinhib20 was shown to inhibit neutrophil extracellular superoxide anion production by ~50% (23). Here we tested the effect of Nexinhib20 on neutrophil total ROS production and its adhesion-dependency (Supplemental Fig. 1B–C). After 50 min of 100 nM PMA stimulation, Nexinhib20-treated neutrophils showed significantly reduced total ROS production compared to vehicle controls (Supplemental Fig. 1B). Interestingly, this inhibition is not adhesion-dependent because CD18 blockade, which was shown to reduce adhesion (51) and spreading (49) of neutrophils, did not inhibit ROS production in both vehicle control neutrophils (Supplemental Fig. 1C) and Nexinhib20-treated neutrophils (data not shown).
Nexinhib20 reduces neutrophil recruitment to the coronary artery during reperfusion
Neutrophils are critically involved in myocardial I/R injury (25, 26). Intravital imaging has shown that neutrophils are recruited abundantly to the coronary artery 60 minutes after reperfusion (5). To test whether Nexinhib20 inhibits neutrophil recruitment in myocardial I/R injury in vivo, we performed multi-photon imaging on explanted hearts after 35 minutes of ischemia and 60 minutes of reperfusion (Fig. 4). To test this possibility, LysM-EGFP mice were used in our study, and were also used to monitor neutrophil recruitment in hearts (5, 66, 67). We observed profound accumulation of LysM-GFP+ cells in the coronary artery in vehicle controls (Fig. 4A, left panel). To mimic a prevention treatment of reperfusion injury before the primary percutaneous coronary intervention, which is feasible in clinics, Nexinhib20 was administered 30 minutes before the reperfusion. Nexinhib20 treatment significantly reduced the number of LysM-GFP+ leukocytes (Fig. 4A, right panel). Quantification of EGFP fluorescence in coronary arteries confirmed that Nexinhib20 significantly limited LysM-GFP+ leukocyte recruitment to the coronary artery during reperfusion (Fig. 4B).
Although macrophages and monocytes may also be highlighted by EGFP in the LysM-EGFP mice, it has been shown that ~90% of EGFP positive cells in I/R heart are Ly6G+ neutrophils (66). To further explore the components of LysM-GFP+ leukocytes in our experimental setting, we used flow cytometry to quantify the percentage of Ly6G+ neutrophils in the I/R heart and blood circulation in mice treated with Nexinhib20 or not (Fig. 4C–E). Consistent with the multi-photon imaging data (Fig. 4A–B), there were fewer LysM-EGFP+ leukocytes recruited to the heart and more LysM-EGFP+ leukocytes retained in the blood circulation of Nexinhib20-treated mice compared to vehicle controls (Fig. 4C). In heart LysM-EGFP+ leukocytes, ~73-95% of them were Ly6G+ neutrophils, regardless of Nexinhib20 treatment (Fig. 4D). In blood LysM-EGFP+ leukocytes, ~88-98% of them were Ly6G+ neutrophils, regardless of Nexinhib20 treatment (Fig. 4E). These results are consistent with a previous report (66).
If we define CD45+Ly6G+ cells as neutrophils, we find that ~6000 neutrophils were recruited to the heart after 35-minute ischemia and 60-minute reperfusion in mice administered vehicle control (Fig. 4F). Nexinhib20 administration significantly reduced heart neutrophil counts to ~2000 (Fig. 4F). The percentage of neutrophils in heart leukocytes was also reduced from ~50% to ~30% by Nexinhib20 administration (Fig. 4F). Since Nexinhib20 limited neutrophil recruitment, neutrophils retained in the blood circulation were doubled in Nexinhib20-treated myocardial I/R mice compared to vehicle controls (Fig. 4G). These data suggested that Nexinhib20 inhibited neutrophil recruitment in vivo in this mouse preclinical model of myocardial I/R injury.
Nexinhib20 prevents myocardial I/R injury
Neutrophils mediate cardiomyocyte death by causing vascular plugging, releasing degradative enzymes, and generating ROS (25, 26). Since we showed that Nexinhib20 limits neutrophil recruitment to the coronary artery and Nexinhib20 was discovered as a neutrophil exocytosis inhibitor that inhibits degradative enzyme release (23) and ROS production (Supplemental Fig. 1B), we reasoned that it might be useful as a dual-functioning drug to treat myocardial I/R injury. As expected, we found that Nexinhib20 administration significantly decreased infarct size (white area in the TTC-phthalo-blue-staining) after myocardial I/R injury compared to no-drug and vehicle controls (Fig. 5A). Sham controls with little to no infarction were shown as well. Quantification of infarct area/area at risk ratios showed that Nexinhib20 significantly reduced the infarct area percentage from ~50%, which were shown in mice administered with vehicle, to ~40% (Fig. 5B). Quantifications of the area of risk percentage confirmed the stability and reproducibility of our surgical procedure (Fig. 5C). These data suggested that Nexinhib20 has potential to treat myocardial I/R injury.
Then we quantified heart function using a left ventricle echocardiogram. We performed the echocardiogram before and 7 days after myocardial I/R injury. Ejection fraction and fractional shortening were measured to quantify left ventricle function (Fig. 5D and E). Ejection fraction is a measurement, expressed as a percentage, of how much blood the left ventricle pumps out with each contraction. Fractional shortening shows the percentage of size differences of the left ventricle as a parameter of how well the left ventricle is contracting, i.e., reducing its size during systole. We found that in the vehicle-treated mice, the ejection fraction (Fig. 5D) and fractional shortening (Fig. 5E) were reduced by ~25% and ~30%, respectively, after myocardial I/R injury, indicating a loss of left ventricle function. In Nexinhib20-treated mice, there is no significant reduction of either ejection fraction (Fig. 5D) or fractional shortening (Fig. 5E) after myocardial I/R injury. Compared to vehicle-treated mice, Nexinhib20-treated mice have significant improvement in left ventricle function 7 days after myocardial I/R injury (Fig. 5D and E).
Discussion
Nexinhib20 was discovered as a neutrophil exocytosis inhibitor, and we confirmed this by testing β2 integrin exocytosis after IL-8 stimulation (Supplemental Fig. 1B). Importantly, we determined that Nexinhib20 also inhibited neutrophil adhesion (Fig. 1) and β2 integrin activation (Fig. 2) without any effect on cell viability (Supplemental Fig. 1A). Thus, Nexinhib20 was confirmed as a dual-functional neutrophil inhibitor. We then found that Rac-1 is a target of Nexinhib20 (Fig. 3). Nexinhib20 inhibited Rac-1 activation in cells by antagonizing the Rac-1-GTP interaction with an IC50 of 29.3 μM. Since Nexinhib20 was also reported to specifically inhibit the interaction between the small GTPase Rab27a and its effector JFC1 and neutrophil exocytosis with an IC50 of 0.33 μM, it is likely that Nexinhib20 exerts a sequential and concentration-dependent inhibition. Rab27a is critical for neutrophil exocytosis (23, 68–70), adhesion molecule presentation (23, 71), migration (72), and ROS production (70). Rac-1 is important for neutrophil integrin activation (16, 73), adhesion (16, 73), migration (16, 65, 73–76), and phagocytosis (77, 78). Nexinhib20 has the potential to work as an anti-inflammatory drug by blocking neutrophil function. Whether Rab27a or Rac-1 is more important and whether they crosstalk during Nexinhib20 inhibition of neutrophil function remains to be further investigated.
Since it has been shown that Nexinhib20 inhibits both Rab27a and Rac-1, this raises concern about whether Nexinhib20 has poor specificity and may inhibit many other GTPases. All small Rab GTPases share a common mechanism of GTP-dependent binding to their respective effectors. However, each pair is characterized by highly specific binding properties, and therefore, it is unlikely that Nexinhib20 would have a high affinity for other GTPases. In fact, the binding affinity of Nexinhib20 to Rac-1 is much lower than Rab27a as we showed a ~90-fold IC50 for Rac-1 compared to Rab27a. It has also been shown that another GTPase, Rab11, was not inhibited by Nexinhib20 (23). Nevertheless, we cannot exclude the possibility that Nexinhib20 may also inhibit other GTPases, especially other Rho family GTPases, such as Rac-2, Cdc42, and RhoA.
We have shown that Nexinhb20 inhibited the interaction of recombinant Rac-1 protein and GTPγS in a dose-dependent manner. This suggests that Nexinhib20 may directly bind Rac-1 and interact with key amino acids of the GTP-binding site. The crystal structure of Rac-1 complexed with a GTP analogue, guanosine-5-(βγ-imino)triphosphate (GMPPNP), has been determined (79). The GTP binding site includes the phosphate-binding loop residues 10-17 and residues 57~61; the guanine base recognition motif residues 116-119 and 158-160; and the effector loop, residues 28-38, which interacts with the ribose and the magnesium ion. Whether Nexinhib20 directly interacts with these residues remains to be further investigated.
Nexinhib20 has been reported to inhibit recruitment of neutrophils to the liver and kidney in a lipopolysaccharide (LPS)-induced systemic inflammation model (23) and to lung lumen and parenchyma in an acute LPS-induced lung injury mouse model (80). Our study was the first to show that Nexinhib20 reduced neutrophil recruitment to the coronary artery during myocardial I/R injury (Fig. 4), which was accomplished with multi-photon microscopy. This method could directly visualize neutrophil recruitment in a very accurate manner. The multi-photon microscopy assay also provided information in a sample with ~100 μm thickness, which was more expansive than quantification using ~8–10 μm histology sections. By combining optical clearing and light-sheet microscopy, neutrophil recruitment in the entire area at risk can be visualized and quantified (6). The attempt to use this method will be limited by instruments and experience with whole tissue optical clearing and staining. Another method to quantify neutrophil recruitment is flow cytometry of heart single-cell suspensions (81).
Nexinhib20 showed both anti-exocytosis and anti-adhesion activities, suggesting that it might be a dual-functional drug for myocardial I/R injury. We showed that Nexinhib20 improves myocardial I/R injury in mice by reducing infarct size by ~20% (Fig. 5A,B) and almost completely restoring the left ventricle function (Fig. 5D,E). Although antibodies against β2 integrins showed benefits in myocardial I/R injury in multiple species (35–43), the clinical trial using a β2 integrin antibody to treat myocardial I/R injury failed (82). This might be due to the long half-life of antibodies in patient circulation that also inhibits the resolution of inflammation after myocardial I/R injury: It has been shown that accurate clearance of dead cells is a prerequisite for favorable MI healing, whereas failed resolution promotes unfavorable cardiac remodeling, which may ultimately result in heart failure (83). The clearance of dead cardiomyocytes and inflammatory neutrophils is orchestrated by macrophages, which are thought to derive from recruited Ly6Chi monocytes (84–86), and β2 integrin antibody can block the recruitment of monocytes (87). Meanwhile, during the clearance of dead cells, macrophages or monocytes must migrate to dead cells and perform phagocytosis. β2 integrins are critical for both cell migration (88, 89) and phagocytosis (33, 90). Thus, a small molecule drug like Nexinhib20 that inhibits β2 integrin function for a shorter period (several hours) compared to antibodies (several weeks to months (91)) might be an advantage in treating acute inflammatory diseases like myocardial I/R injury. This is because administering a small molecule drug can alleviate the pro-inflammatory responses during acute inflammation and then degrade after several hours, so it will not block the later resolution of inflammation (92). We have studied the pharmacokinetics of Nexinhib20 after i.p. injection on mice (Supplemental Fig. 2), and have shown that Nexinhib20 was degraded quickly within 2 hours, which is during the acute inflammation phase. Four hours after administration, the Nexinhib20 concentration is lower than ~28.8 μg mL−1 (~96 nM), which may not block the recruitment of monocytes/macrophages and their mediation of inflammation resolution and healing. This needs to be validated in future investigations. Furthermore, Nexinhib20-mediated inhibition of neutrophil exocytosis (23) and ROS production (Supplemental Fig. 1B) would also contribute to the attenuation of the I/R injury. Since we demonstrated that Nexinhib20 could prevent myocardial I/R injury, our work highlights its possible use for other acute inflammatory diseases involving neutrophils, such as noninfectious acute lung injury (93), I/R injury after transplantation (94, 95), ischemic stroke (96), systemic inflammatory response to severe injury (97), and multiple organ dysfunction syndrome (97).
We have identified Nexinhib20 as an antagonist of the Rac-1-GTP interaction. Since Rac-1 is critically involved in the functions of many cells, Nexinhib20 may be used for treatments targeting other cells. Since our study focused on myocardial I/R injury, it is important to discuss the role of Rac-1 in cardiomyocytes. Rac-1 is not only important for leukocyte activation and ROS production but it is also essential for ROS production by cardiomyocytes (98). Cardiomyocyte-specific overexpression of an active Rac mutation aggravated myocardial I/R injury (99), and myocardial I/R-induced ventricular arrhythmia was significantly decreased in cardiac-specific Rac-1 knockdown mice (100). Another Rac-1 inhibitor NSC23766 decreased I/R-induced ventricular arrhythmia (100). Active Rac-1 was upregulated in failing myocardium of patients with ischemic cardiomyopathy and dilated cardiomyopathy (101). Statin treatment decreased myocardial Rac1-GTPase activity (101). Rac-1 activation was involved in the hypertrophic response of cardiomyocytes (102–104), hyperglycemia-induced apoptosis of cardiomyocytes in diabetes (105), and doxorubicin-induced cardiotoxicity (106). Besides cardiomyocytes, shear stress-induced Rac-1 activation in endothelial cells is responsible for ICAM-1 expression, which is critical for the recruitment of neutrophils and inflammatory responses (107). Inhibition of Rac1 GTPase decreases vascular oxidative stress, improves endothelial function, and attenuates atherosclerosis development in mice (108). Overall, most studies supported that inhibition of Rac-1 was beneficial to most cardiomyopathies, therefore, Rac-1-specific inhibitors, such as NSC23766 or statin may help as well. Chemical modulation may also help to increase the affinity of Nexinhib20 to Rac-1 that increases inhibition efficiency.
Neutrophil-mediated tissue damage after I/R injury is a multifactorial process that depends on β2 integrin-dependent neutrophil adhesion, recruitment, and secretion. Here, we show that Nexinhib20, in addition to exocytosis, also inhibits human neutrophil adhesion and β2 integrin activation by targeting Rac1 GTPase. Importantly Nexinhib20 decreased neutrophil recruitment in vivo and decreased infarct size after mouse myocardial I/R injury further validating that inhibition of neutrophil recruitment and activation increases the likelihood of a favorable outcome during myocardial tissue damage.
Supplementary Material
Key Points:
Nexinhib20 inhibits neutrophil adhesion and β2 integrin activation;
Nexinhib20 antagonizes the binding of Rac-1 and GTP;
Nexinhib20 prevents mice from myocardial I/R injury.
Acknowledgments
The clonal HL60 cell line was generated by Alba Diz-Munoz (EMBL Heidelberg) and a generous gift from the laboratory of Orion Weiner (the University of California San Francisco). The CXCR2-expressing HL60 cells (HL60-CXCR2) was a gift from Dr. Ann Richmond at the Vanderbilt University School of Medicine. We acknowledge Dr. Klaus Ley from the La Jolla Institute for Immunology for supporting reagents and instruments to perform neutrophil rolling and integrin activation assays. We acknowledge Dr. Wolfgang Peti from the Department of Molecular Biology & Biophysics at UConn Health for providing the necessary instruments. We acknowledge Dr. Penghua Wang and Dr. Tingting Geng from the Department of Immunology at UConn Health for providing the Cytation 1 Cell Imaging Multi-Mode Reader for the ROS production assay. We acknowledge Dr. Lixia Yue and Pengyu Zhong from the Pat and Jim Calhoun Cardiology Center at UConn Health for reviewing and providing suggestions for this manuscript. We acknowledge Dr. Feifei Lin from the Shanghai Institute of Materia Medica for the service of Nexinhib20 pharmacokinetics. We acknowledge Dr. Christopher “Kit” Bonin and Dr. Geneva Hargis from UConn School of Medicine for their help with scientific writing and editing of this manuscript.
Grants
This research was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute, USA (R01HL145454, R41HL156322, R44HL152710, and R00HL153678), a Career Development Award from American Heart Association (18CDA34110426), an AAI Fellowship for Career Reentry from the American Association of Immunologists, and a startup fund from UConn Health.
Footnotes
Disclosures
The authors declare no competing financial interest.
References:
- 1.Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, and Catz SD 2018. Neutrophils: New insights and open questions. Sci Immunol 3. [DOI] [PubMed] [Google Scholar]
- 2.Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, Wang H-V, Sperandio M, and Fassler R 2009. Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nature medicine 15: 300–305. [DOI] [PubMed] [Google Scholar]
- 3.Fagerholm SC, Guenther C, Llort Asens M, Savinko T, and Uotila LM 2019. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front Immunol 10: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yago T, Petrich BG, Zhang N, Liu Z, Shao B, Ginsberg MH, and McEver RP 2015. Blocking neutrophil integrin activation prevents ischemia-reperfusion injury. J Exp Med 212: 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ, and Kreisel D 2016. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merz SF, Korste S, Bornemann L, Michel L, Stock P, Squire A, Soun C, Engel DR, Detzer J, Lorchner H, Hermann DM, Kamler M, Klode J, Hendgen-Cotta UB, Rassaf T, Gunzer M, and Totzeck M 2019. Contemporaneous 3D characterization of acute and chronic myocardial I/R injury and response. Nat Commun 10: 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang CC, Cheng WJ, Korinek M, Lin CY, and Hwang TL 2019. Neutrophils in Psoriasis. Front Immunol 10: 2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemeth T, and Mocsai A 2012. The role of neutrophils in autoimmune diseases. Immunol Lett 143: 9–19. [DOI] [PubMed] [Google Scholar]
- 9.Gupta S, and Kaplan MJ 2016. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12: 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, and Monteiro MC 2017. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med 15: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margraf A, Ley K, and Zarbock A 2019. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol 40: 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolaczkowska E, and Kubes P 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175. [DOI] [PubMed] [Google Scholar]
- 13.Sun H, Hu L, and Fan Z 2021. beta2 integrin activation and signal transduction in leukocyte recruitment. Am J Physiol Cell Physiol 321: C308–C316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soede RD, Zeelenberg IS, Wijnands YM, Kamp M, and Roos E 2001. Stromal cell-derived factor-1-induced LFA-1 activation during in vivo migration of T cell hybridoma cells requires Gq/11, RhoA, and myosin, as well as Gi and Cdc42. J Immunol 166: 4293–4301. [DOI] [PubMed] [Google Scholar]
- 15.Giagulli C, Scarpini E, Ottoboni L, Narumiya S, Butcher EC, Constantin G, and Laudanna C 2004. RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity 20: 25–35. [DOI] [PubMed] [Google Scholar]
- 16.Block H, Stadtmann A, Riad D, Rossaint J, Sohlbach C, Germena G, Wu D, Simon SI, Ley K, and Zarbock A 2016. Gnb isoforms control a signaling pathway comprising Rac1, Plcbeta2, and Plcbeta3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood 127: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arana E, Vehlow A, Harwood NE, Vigorito E, Henderson R, Turner M, Tybulewicz VL, and Batista FD 2008. Activation of the small GTPase Rac2 via the B cell receptor regulates B cell adhesion and immunological-synapse formation. Immunity 28: 88–99. [DOI] [PubMed] [Google Scholar]
- 18.Katagiri K, Shimonaka M, and Kinashi T 2004. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-gamma1. J Biol Chem 279: 11875–11881. [DOI] [PubMed] [Google Scholar]
- 19.Quinn MT, Parkos CA, Walker L, Orkin SH, Dinauer MC, and Jesaitis AJ 1989. Association of a Ras-related protein with cytochrome b of human neutrophils. Nature 342: 198–200. [DOI] [PubMed] [Google Scholar]
- 20.Ebisuno Y, Katagiri K, Katakai T, Ueda Y, Nemoto T, Inada H, Nabekura J, Okada T, Kannagi R, Tanaka T, Miyasaka M, Hogg N, and Kinashi T 2010. Rap1 controls lymphocyte adhesion cascade and interstitial migration within lymph nodes in RAPL-dependent and -independent manners. Blood 115: 804–814. [DOI] [PubMed] [Google Scholar]
- 21.Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, Critchley DR, Ginsberg MH, Fassler R, and Ley K 2012. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 119: 4275–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghandour H, Cullere X, Alvarez A, Luscinskas FW, and Mayadas TN 2007. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood 110: 3682–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, Biris N, Gavathiotis E, Rosen H, and Catz SD 2016. Identification of Neutrophil Exocytosis Inhibitors (Nexinhibs), Small Molecule Inhibitors of Neutrophil Exocytosis and Inflammation: DRUGGABILITY OF THE SMALL GTPase Rab27a. J Biol Chem 291: 25965–25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catz SD 2014. The role of Rab27a in the regulation of neutrophil function. Cell Microbiol 16: 1301–1310. [DOI] [PubMed] [Google Scholar]
- 25.Yellon DM, and Hausenloy DJ 2007. Myocardial reperfusion injury. N Engl J Med 357: 1121–1135. [DOI] [PubMed] [Google Scholar]
- 26.Puhl SL, and Steffens S 2019. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front Cardiovasc Med 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, and Steffens S 2017. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38: 187–197. [DOI] [PubMed] [Google Scholar]
- 28.Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL, and Lucchesi BR 1986. Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J 112: 682–690. [DOI] [PubMed] [Google Scholar]
- 29.Aratani Y 2018. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys 640: 47–52. [DOI] [PubMed] [Google Scholar]
- 30.Vasilyev N, Williams T, Brennan ML, Unzek S, Zhou X, Heinecke JW, Spitz DR, Topol EJ, Hazen SL, and Penn MS 2005. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 112: 2812–2820. [DOI] [PubMed] [Google Scholar]
- 31.Ali M, Pulli B, Courties G, Tricot B, Sebas M, Iwamoto Y, Hilgendorf I, Schob S, Dong A, Zheng W, Skoura A, Kalgukar A, Cortes C, Ruggeri R, Swirski FK, Nahrendorf M, Buckbinder L, and Chen JW 2016. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling After Experimental Myocardial Infarction. JACC Basic Transl Sci 1: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liew PX, and Kubes P 2019. The Neutrophil’s Role During Health and Disease. Physiological reviews 99: 1223–1248. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Zhi K, Hu L, and Fan Z 2021. The Activation and Regulation of beta2 Integrins in Phagocytes and Phagocytosis. Front Immunol 12: 633639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palazzo AJ, Jones SP, Girod WG, Anderson DC, Granger DN, and Lefer DJ 1998. Myocardial ischemia-reperfusion injury in CD18- and ICAM-1-deficient mice. Am J Physiol 275: H2300–2307. [DOI] [PubMed] [Google Scholar]
- 35.Aversano T, Zhou W, Nedelman M, Nakada M, and Weisman H 1995. A chimeric IgG4 monoclonal antibody directed against CD18 reduces infarct size in a primate model of myocardial ischemia and reperfusion. J Am Coll Cardiol 25: 781–788. [DOI] [PubMed] [Google Scholar]
- 36.Kupatt C, Wichels R, Deiss M, Molnar A, Lebherz C, Raake P, von Degenfeld G, Hahnel D, and Boekstegers P 2002. Retroinfusion of NFkappaB decoy oligonucleotide extends cardioprotection achieved by CD18 inhibition in a preclinical study of myocardial ischemia and retroinfusion in pigs. Gene Ther 9: 518–526. [DOI] [PubMed] [Google Scholar]
- 37.Horwitz LD, Kaufman D, and Kong Y 1997. An antibody to leukocyte integrins attenuates coronary vascular injury due to ischemia and reperfusion in dogs. Am J Physiol 272: H618–624. [DOI] [PubMed] [Google Scholar]
- 38.Arai M, Lefer DJ, So T, DiPaula A, Aversano T, and Becker LC 1996. An anti-CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48-hour reperfusion. J Am Coll Cardiol 27: 1278–1285. [DOI] [PubMed] [Google Scholar]
- 39.Perez RG, Arai M, Richardson C, DiPaula A, Siu C, Matsumoto N, Hildreth JE, Mariscalco MM, Smith CW, and Becker LC 1996. Factors modifying protective effect of anti-CD18 antibodies on myocardial reperfusion injury in dogs. Am J Physiol 270: H53–64. [DOI] [PubMed] [Google Scholar]
- 40.Williams FM, Kus M, Tanda K, and Williams TJ 1994. Effect of duration of ischaemia on reduction of myocardial infarct size by inhibition of neutrophil accumulation using an anti-CD18 monoclonal antibody. Br J Pharmacol 111: 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka M, Brooks SE, Richard VJ, FitzHarris GP, Stoler RC, Jennings RB, Arfors KE, and Reimer KA 1993. Effect of anti-CD18 antibody on myocardial neutrophil accumulation and infarct size after ischemia and reperfusion in dogs. Circulation 87: 526–535. [DOI] [PubMed] [Google Scholar]
- 42.Lefer DJ, Shandelya SM, Serrano CV Jr., Becker LC, Kuppusamy P, and Zweier JL 1993. Cardioprotective actions of a monoclonal antibody against CD-18 in myocardial ischemia-reperfusion injury. Circulation 88: 1779–1787. [DOI] [PubMed] [Google Scholar]
- 43.McDonagh PF, Wilson DS, Iwamura H, Smith CW, Williams SK, and Copeland JG 1996. CD18 antibody treatment limits early myocardial reperfusion injury after initial leukocyte deposition. J Surg Res 64: 139–149. [DOI] [PubMed] [Google Scholar]
- 44.Fan Z, Kiosses WB, Sun H, Orecchioni M, Ghosheh Y, Zajonc DM, Arnaout MA, Gutierrez E, Groisman A, Ginsberg MH, and Ley K 2019. High-Affinity Bent beta2-Integrin Molecules in Arresting Neutrophils Face Each Other through Binding to ICAMs In cis. Cell Rep 26: 119–130 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faust N, Varas F, Kelly LM, Heck S, and Graf T 2000. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96: 719–726. [PubMed] [Google Scholar]
- 46.Bohl S, Medway DJ, Schulz-Menger J, Schneider JE, Neubauer S, and Lygate CA 2009. Refined approach for quantification of in vivo ischemia-reperfusion injury in the mouse heart. Am J Physiol Heart Circ Physiol 297: H2054–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Savjani KT, Gajjar AK, and Savjani JK 2012. Drug solubility: importance and enhancement techniques. ISRN Pharm 2012: 195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun H, Fan Z, Gingras AR, Lopez-Ramirez MA, Ginsberg MH, and Ley K 2020. Frontline Science: A flexible kink in the transmembrane domain impairs beta2 integrin extension and cell arrest from rolling. J Leukoc Biol 107: 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu W, Hsu AY, Wang Y, Lin T, Sun H, Pachter JS, Groisman A, Imperioli M, Yungher FW, Hu L, Wang P, Deng Q, and Fan Z 2021. Mitofusin-2 regulates leukocyte adhesion and beta2 integrin activation. J Leukoc Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sai J, Walker G, Wikswo J, and Richmond A 2006. The IL sequence in the LLKIL motif in CXCR2 is required for full ligand-induced activation of Erk, Akt, and chemotaxis in HL60 cells. J Biol Chem 281: 35931–35941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, Deutsch U, Ginsberg M, Groisman A, and Ley K 2016. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat Commun 7: 12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ley K, and Gaehtgens P 1991. Endothelial, not hemodynamic, differences are responsible for preferential leukocyte rolling in rat mesenteric venules. Circ Res 69: 1034–1041. [DOI] [PubMed] [Google Scholar]
- 53.Lawrence MB, and Springer TA 1991. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell 65: 859–873. [DOI] [PubMed] [Google Scholar]
- 54.von Andrian UH, Chambers JD, McEvoy LM, Bargatze RF, Arfors KE, and Butcher EC 1991. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Natl Acad Sci U S A 88: 7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamata T, Tieu KK, Tarui T, Puzon-McLaughlin W, Hogg N, and Takada Y 2002. The role of the CPNKEKEC sequence in the beta(2) subunit I domain in regulation of integrin alpha(L)beta(2) (LFA-1). J Immunol 168: 2296–2301. [DOI] [PubMed] [Google Scholar]
- 56.Lu C, Shimaoka M, Zang Q, Takagi J, and Springer TA 2001. Locking in alternate conformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci U S A 98: 2393–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dransfield I, and Hogg N 1989. Regulated expression of Mg2+ binding epitope on leukocyte integrin alpha subunits. EMBO J 8: 3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MK, Andrew D, Rosen H, Brown D, Ortlepp S, Stephens P, and Butcher EC 1992. Antibody against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J Immunol 148: 1080–1085. [PubMed] [Google Scholar]
- 59.Lu C, Ferzly M, Takagi J, and Springer TA 2001. Epitope mapping of antibodies to the C-terminal region of the integrin beta 2 subunit reveals regions that become exposed upon receptor activation. J Immunol 166: 5629–5637. [DOI] [PubMed] [Google Scholar]
- 60.Fan Z, and Ley K 2015. Leukocyte arrest: Biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology 52: 353–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schorr W, Swandulla D, and Zeilhofer HU 1999. Mechanisms of IL-8-induced Ca2+ signaling in human neutrophil granulocytes. Eur J Immunol 29: 897–904. [DOI] [PubMed] [Google Scholar]
- 62.Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, and Simon SI 2008. Calcium flux in neutrophils synchronizes beta2 integrin adhesive and signaling events that guide inflammatory recruitment. Ann Biomed Eng 36: 632–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neves SR, Ram PT, and Iyengar R 2002. G protein pathways. Science 296: 1636–1639. [DOI] [PubMed] [Google Scholar]
- 64.Lawson CD, Donald S, Anderson KE, Patton DT, and Welch HC 2011. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol 186: 1467–1476. [DOI] [PubMed] [Google Scholar]
- 65.Ramadass M, Johnson JL, Marki A, Zhang J, Wolf D, Kiosses WB, Pestonjamasp K, Ley K, and Catz SD 2019. The trafficking protein JFC1 regulates Rac1-GTP localization at the uropod controlling neutrophil chemotaxis and in vivo migration. J Leukoc Biol 105: 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W, Nava RG, Bribriesco AC, Zinselmeyer BH, Spahn JH, Gelman AE, Krupnick AS, Miller MJ, and Kreisel D 2012. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest 122: 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, Liu X, Hassan A, Tanaka S, Cicka M, Hsiao HM, Ruiz-Perez D, Bredemeyer A, Gross RW, Mann DL, Tyurina YY, Gelman AE, Kagan VE, Linkermann A, Lavine KJ, and Kreisel D 2019. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest 129: 2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munafo DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B, and Catz SD 2007. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J 402: 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brzezinska AA, Johnson JL, Munafo DB, Crozat K, Beutler B, Kiosses WB, Ellis BA, and Catz SD 2008. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic 9: 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson JL, Brzezinska AA, Tolmachova T, Munafo DB, Ellis BA, Seabra MC, Hong H, and Catz SD 2010. Rab27a and Rab27b regulate neutrophil azurophilic granule exocytosis and NADPH oxidase activity by independent mechanisms. Traffic 11: 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson JL, Hong H, Monfregola J, and Catz SD 2011. Increased survival and reduced neutrophil infiltration of the liver in Rab27a- but not Munc13-4-deficient mice in lipopolysaccharide-induced systemic inflammation. Infect Immun 79: 3607–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Strom M, Hume AN, Tarafder AK, Barkagianni E, and Seabra MC 2002. A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. J Biol Chem 277: 25423–25430. [DOI] [PubMed] [Google Scholar]
- 73.Lawson CD, and Burridge K 2014. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5: e27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Filippi MD, Szczur K, Harris CE, and Berclaz PY 2007. Rho GTPase Rac1 is critical for neutrophil migration into the lung. Blood 109: 1257–1264. [DOI] [PubMed] [Google Scholar]
- 75.Mazaki Y, Onodera Y, Higashi T, Horinouchi T, Oikawa T, and Sabe H 2017. ARF1 recruits RAC1 to leading edge in neutrophil chemotaxis. Cell Commun Signal 15: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, and Kwiatkowski DJ 2003. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol 170: 5652–5657. [DOI] [PubMed] [Google Scholar]
- 77.Nakaya M, Kitano M, Matsuda M, and Nagata S 2008. Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc Natl Acad Sci U S A 105: 9198–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cox D, Chang P, Zhang Q, Reddy PG, Bokoch GM, and Greenberg S 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J Exp Med 186: 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirshberg M, Stockley RW, Dodson G, and Webb MR 1997. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat Struct Biol 4: 147–152. [DOI] [PubMed] [Google Scholar]
- 80.Mejias JC, Forrest OA, Margaroli C, Frey Rubio DA, Viera L, Li J, Xu X, Gaggar A, Tirouvanziam R, and Roy K 2019. Neutrophil-targeted, protease-activated pulmonary drug delivery blocks airway and systemic inflammation. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aronoff L, Epelman S, and Clemente-Casares X 2018. Isolation and Identification of Extravascular Immune Cells of the Heart. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F, and Investigators H-M 2002. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol 40: 1199–1204. [DOI] [PubMed] [Google Scholar]
- 83.Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, and Thorp EB 2013. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 113: 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nahrendorf M, and Swirski FK 2013. Monocyte and macrophage heterogeneity in the heart. Circ Res 112: 1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dutta P, and Nahrendorf M 2015. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol 35: 1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rurik JG, Aghajanian H, and Epstein JA 2021. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ Res 128: 1766–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schenkel AR, Mamdouh Z, and Muller WA 2004. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol 5: 393–400. [DOI] [PubMed] [Google Scholar]
- 88.Kovacs M, Nemeth T, Jakus Z, Sitaru C, Simon E, Futosi K, Botz B, Helyes Z, Lowell CA, and Mocsai A 2014. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med 211: 1993–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meerschaert J, and Furie MB 1994. Monocytes use either CD11/CD18 or VLA-4 to migrate across human endothelium in vitro. J Immunol 152: 1915–1926. [PubMed] [Google Scholar]
- 90.Jaumouille V, Cartagena-Rivera AX, and Waterman CM 2019. Coupling of beta2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat Cell Biol 21: 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Isho B, Abe KT, Zuo M, Jamal AJ, Rathod B, Wang JH, Li Z, Chao G, Rojas OL, Bang YM, Pu A, Christie-Holmes N, Gervais C, Ceccarelli D, Samavarchi-Tehrani P, Guvenc F, Budylowski P, Li A, Paterson A, Yue FY, Marin LM, Caldwell L, Wrana JL, Colwill K, Sicheri F, Mubareka S, Gray-Owen SD, Drews SJ, Siqueira WL, Barrios-Rodiles M, Ostrowski M, Rini JM, Durocher Y, McGeer AJ, Gommerman JL, and Gingras AC 2020. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sansbury BE, and Spite M 2016. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circ Res 119: 113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grommes J, and Soehnlein O 2011. Contribution of neutrophils to acute lung injury. Mol Med 17: 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamura K, Kageyama S, and Kupiec-Weglinski JW 2019. The Evolving Role of Neutrophils in Liver Transplant Ischemia-Reperfusion Injury. Curr Transplant Rep 6: 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaturvedi S, Yuen DA, Bajwa A, Huang YW, Sokollik C, Huang L, Lam GY, Tole S, Liu GY, Pan J, Chan L, Sokolskyy Y, Puthia M, Godaly G, John R, Wang C, Lee WL, Brumell JH, Okusa MD, and Robinson LA 2013. Slit2 prevents neutrophil recruitment and renal ischemia-reperfusion injury. J Am Soc Nephrol 24: 1274–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, and Sharp FR 2015. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 35: 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mortaz E, Alipoor SD, Adcock IM, Mumby S, and Koenderman L 2018. Update on Neutrophil Function in Severe Inflammation. Front Immunol 9: 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gregg D, Rauscher FM, and Goldschmidt-Clermont PJ 2003. Rac regulates cardiovascular superoxide through diverse molecular interactions: more than a binary GTP switch. Am J Physiol Cell Physiol 285: C723–734. [DOI] [PubMed] [Google Scholar]
- 99.Talukder MA, Elnakish MT, Yang F, Nishijima Y, Alhaj MA, Velayutham M, Hassanain HH, and Zweier JL 2013. Cardiomyocyte-specific overexpression of an active form of Rac predisposes the heart to increased myocardial stunning and ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 304: H294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang L, Lu X, Gui L, Wu Y, Sims SM, Wang G, and Feng Q 2016. Inhibition of Rac1 reduces store overload-induced calcium release and protects against ventricular arrhythmia. J Cell Mol Med 20: 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, and Laufs U 2003. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 108: 1567–1574. [DOI] [PubMed] [Google Scholar]
- 102.Clerk A, Pham FH, Fuller SJ, Sahai E, Aktories K, Marais R, Marshall C, and Sugden PH 2001. Regulation of mitogen-activated protein kinases in cardiac myocytes through the small G protein Rac1. Mol Cell Biol 21: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, and Liao JK 2006. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A 103: 7432–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, Hikoso S, Kashiwase K, Takeda T, Watanabe T, Mano T, Matsumura Y, Ueno H, and Hori M 2003. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-kappa B. J Biol Chem 278: 20770–20777. [DOI] [PubMed] [Google Scholar]
- 105.Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, Arnold JM, and Peng T 2009. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 58: 2386–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma J, Wang Y, Zheng D, Wei M, Xu H, and Peng T 2013. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res 97: 77–87. [DOI] [PubMed] [Google Scholar]
- 107.Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, and Schwartz MA 2002. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J 21: 6791–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zimmer S, Goody PR, Oelze M, Ghanem A, Mueller CF, Laufs U, Daiber A, Jansen F, Nickenig G, and Wassmann S 2021. Inhibition of Rac1 GTPase Decreases Vascular Oxidative Stress, Improves Endothelial Function, and Attenuates Atherosclerosis Development in Mice. Front Cardiovasc Med 8: 680775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.