

Abstract
In both mice and humans, complement and Th17 cells have been implicated in periodontitis, an oral microbiota-driven inflammatory disease associated with systemic disorders. A recent clinical trial showed that a complement C3 inhibitor (AMY-101) causes sustainable resolution of periodontal inflammation, the main effector of tissue destruction in this oral disease. Although both complement and Th17 are required for periodontitis, it is uncertain how these immune components cooperate in disease development. Here we dissected the complement-Th17 relationship in the setting of ligature-induced periodontitis (LIP), a model that previously established that microbial dysbiosis drives Th17 cell expansion and periodontal bone loss. Complement was readily activated in the periodontal tissue of LIP-subjected mice but not when the mice were placed on broad-spectrum antibiotics. Microbiota-induced complement activation generated critical cytokines, IL-6 and IL-23, which are required for Th17 cell expansion. These cytokines as well as Th17 accumulation and IL-17 expression were significantly suppressed in LIP-subjected C3-deficient mice relative to wild-type controls. As IL-23 has been extensively studied in periodontitis, we focused on IL-6 and showed that LIP-induced IL-17 and bone loss required intact IL-6 receptor signaling in the periodontium. LIP-induced IL-6 was predominantly produced by gingival epithelial cells that upregulated C3a receptor upon LIP challenge. Experiments in human gingival epithelial cells showed that C3a upregulated IL-6 production in cooperation with microbial stimuli that upregulated C3a receptor expression in ERK1/2- and JNK-dependent manner. In conclusion, complement links the periodontal microbiota challenge to Th17 cell accumulation and thus integrates complement- and Th17-driven immunopathology in periodontitis.
Introduction
Periodontitis, a prevalent inflammatory disease of the tooth-supporting tissues (‘periodontium’), is a major cause of tooth loss in adults and is associated with increased risk of systemic comorbidities (1–3). Whereas dysbiosis of the periodontal microbiota is necessary for disease development, it is the host inflammatory response to this microbial challenge that is responsible for periodontal tissue destruction and likely for disease progression and chronification (4). Although periodontitis remains a significant public health and economic burden (5–7), current therapies aiming to control the dysbiotic microbiota by mechanical debridement and frequently adjunctive antibiotics, are often ineffective particularly in highly susceptible individuals (5, 8–10). On the other hand, the concept of host modulation therapy as an adjunctive treatment in periodontitis has gained momentum in an effort to enhance clinical outcomes beyond those achieved by conventional approaches (9, 11, 12). In this regard, a recent placebo-controlled, double-blind phase 2a clinical trial has shown that treatment of patients with a complement C3-targeted inhibitor (AMY-101) resulted in significantly reduced gingival inflammation and markers of periodontal tissue destruction (13).
The activation of complement, a network of fluid-phase or cell surface-associated proteins (proenzymes, convertase enzymes, effectors, receptors, and regulators), triggers and regulates immune and inflammatory responses for surveillance and homeostasis; however, when dysregulated or overactivated, complement can drive destructive inflammation (14, 15). The initiation pathways for complement activation (classical, lectin or alternative) all converge at the third component (C3). Therefore, regardless of initiation mechanism, C3 blockade can inhibit inflammation by blocking the enzymatic generation of downstream effector molecules, including the proinflammatory anaphylatoxins C3a and C5a (16). Consistently, mice deficient in C3 (C3–/–) are protected against experimental periodontal inflammation and bone loss relative to wild-type (WT) littermate controls (17), whereas local treatment with Cp40 (clinically developed as AMY-101), an improved third generation analog of the compstatin family of C3 inhibitors (16), protects non-human primates from both experimental (17) and naturally occurring (18, 19) periodontitis. As alluded to above, these preclinical studies were corroborated by the AMY-101 trial which showed pronounced and sustainable resolution (lasting for at least 90 days after treatment initiation) of gingival inflammation in human subjects (13).
Besides complement, CD4+ Th17 cells have also been implicated in periodontal disease pathogenesis in mice and humans (20). Th17 cells are a major cellular source of IL-17, a proinflammatory cytokine that drives inflammatory bone loss (20, 21). The expansion of Th17 cells in the mouse gingiva during active periodontitis was linked to quantitative and qualitative alterations (dysbiosis) in the local periodontal microbiota, whereas intensive preventive treatment with broad-spectrum antibiotics diminished Th17 cell expansion and bone loss (20). Consistently, individuals with genetically defective Th17 cell development display reduced periodontal inflammation and bone loss relative to age- and gender-matched controls (having normal numbers of Th17 cells) or periodontitis patients (20).
Given that both complement and Th17 are required for periodontitis, in the present study we aimed to dissect the relationship of these immune components in an experimental periodontal disease setting. Since complement is a major initiator of inflammation (15), we hypothesized that Th17 cells act downstream of complement activation, in other words, complement mediates between the dysbiotic microbiota and Th17 cells. Indeed, our findings showed that microbiota-induced complement activation generates critical Th17-inducing cytokines, IL-6 and IL-23, thereby linking the periodontal microbiota to Th17 cell expansion and inflammatory bone loss.
Materials and Methods
Mice
All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. C57BL/6 wild-type (WT) mice were purchased from The Jackson Laboratory. C57BL/6 C3−/− and C3+/+ WT littermate controls were bred under specific pathogen-free conditions. The C3−/− mice were originally provided by Dr. Rick Wetsel (University of Texas) (22). Mice were maintained in individually ventilated cages, provided sterile food and water ad libitum, and were used for experiments at the age of 8–9 weeks.
Ligature-induced periodontitis
Ligature placement generates a subgingival biofilm-retentive milieu leading to inflammation and bone loss in conventional (but not germ-free) mice or other animals (20, 23–29). Ligature-induced periodontitis (LIP) in mice was induced as previously described (23). Briefly, a 5–0 silk ligature was tied around the maxillary left second molar, whereas the contralateral molar tooth was left unligated to serve as baseline control. The mice were euthanized 3 or 5 days later. Defleshed maxillae were used to measure bone heights (i.e., the distances from the cementoenamel junction (CEJ) to the alveolar bone crest (ABC)) at six predetermined points on the ligated site as previously specified (23). Measurements were made using a dissecting microscope fitted with a video image marker measurement system (Nikon Instruments). To calculate bone loss, the six-site total CEJ–ABC distance for the ligated site of each mouse was subtracted from the six-site total CEJ–ABC distance of the contralateral unligated site. The results were presented in millimeters, and negative values indicate bone loss relative to the baseline (unligated control). In intervention experiments, blocking Ab or isotype control was microinjected, one day prior to LIP, into the palatal gingiva of the ligated second maxillary molar, as previously described (30). In some experiments, mice were administered an antibiotic cocktail in their drinking water (Vancomycin, 0.5 g/L; Doripenem, 0.25 g/L, Neomycin sulfate, 1 g/L; VDN) for 2 weeks prior to and during LIP. The periodontal bacterial load was determined by quantitative real-time PCR using 16S rRNA gene primers (20, 31).
Antibodies
MAb to C3a receptor (C3aR) (cat# sc-133172, clone D-12, IgG2a, κ, 1:50) was purchased from Santa Cruz Biotechnology, Dallas, TX. MAbs against mouse CD45 (cat# 103128, clone 30-F11, rat IgG2b, κ, 1:100), C5a receptor-1 (C5aR1, CD88) (cat# 135808, clone 20/70, rat IgG2b, κ, 1:100), IL-17A (cat# 506908, clone TC11–18H10.1, rat IgG1, κ, 1:100), CD4 (cat# 100412, clone GK1.5, rat IgG2b, κ, 1:100), TCRβ (cat# 109224, clone H57–597, armenian hamster IgG, 1:100), IL-6Rα chain (CD126) (cat# 115815, clone D7715A7, rat IgG2b, κ, 5 μg/mouse), Ly6G (cat# 127616, clone 1A8, rat IgG2a, κ, 1:100), EpCAM (cat# 118208, clone G8.8, rat IgG2a, κ, 1:100), rat IgG2b, κ isotype control (cat# 400644, clone RTK4530, 1:100 or 5 μg/mouse), PE mouse IgG2a, κ isotype control (cat# 400213, clone MOPC-173, 1:50), APC rat IgG2b, κ isotype control (cat# 400611, clone RTK4530, 1:100), Alexa Fluor 700 rat IgG2b, κ isotype control (cat# 400628, clone RTK4530, 1:100), FITC rat IgG2a, κ isotype control (cat# 400505, clone RTK2758, 1:100), were from Biolegend, San Diego, CA. MAbs to EpCAM (cat# 563478, clone G8.8, rat IgG2a, κ, 1:100), IL-6 (cat# 554401, clone MP5–20F3, rat IgG1, 1:100), APC rat IgG2a κ isotype control (cat# 554690, clone R35–95, 1:100) and PE rat IgG1, κ isotype control (cat# 554685, clone R3–34, 1:100) were obtained from BD Bioscience, Franklin Lakes, NJ.
Flow cytometry and cell sorting
Gingival tissue was dissected around the area of ligature placement and digested for 1h at 37°C with RPMI 1640 medium (cat# 11875093, Gibco, Grand Island, NY) supplemented with collagenase IV (3.2 mg/ml, cat# LS004188,Worthington, Lakewood, NJ) and DNase (0.15 μg/ml, cat# LS002006, Worthington) (32). Single-cell suspensions were obtained by mashing the tissue against a strainer using plungers and filtered for staining and flow cytometry. Before staining, cells were incubated with purified anti-mouse CD16/32 (cat# 101302, clone 93, Biolegend, 1:100) to block Fcγ III/II receptors for 10 minutes at 4°C. For intracellular IL-17 staining, cells were stimulated with PMA (50 ng/ml; cat# P8139–5MG, Sigma-Aldrich, Rockville, MD) and ionomycin (500 ng/ml; cat# I9657–5MG, Sigma-Aldrich) in the presence of GolgiPlug Protein Transport Inhibitor (cat# 555029, BD Bioscience, 1:1000) at 37°C and 5% CO2. After 4 h, cells were incubated with Live/Dead fixable dye (cat#L34955, Invitrogen, Waltham, MA, 1:1000) to exclude dead cells and stained with Abs to surface markers (CD45, CD4, and TCRβ). The cells were then washed, fixed and permeabilized with intracellular fixation and permeabilization buffer set (cat# 00-5523-00, eBioscience, Santa Clara, CA) and stained intracellularly with FITC-conjugated Ab to IL-17 for 1 h. For intracellular IL-6 staining, cells were stimulated with LPS (1 μg/ml) in the presence of GolgiPlug Protein Transport Inhibitor for 4 h at 37°C and 5% CO2. Cells were incubated with Live/Dead fixable dye to exclude dead cells and stained with Abs against CD45 and EpCAM, then fixed and permeabilized with intracellular fixation and permeabilization buffer set. The cells were stained with PE-conjugated Ab to IL-6 for 1 h. For cell sorting, cells were stained with PE-EpCAM (cat# 118206, clone G8.8, Biolegend, 1:100) for 15 minutes and separated into EpCAM+ and EpCAM– cells by using EasySep Mouse PE positive selection kit (cat# 17666, Stemcell, Cambridge, MA). To identify complement receptor-expressing cells, the cells were stained with Abs against CD45, EpCAM, C3aR and C5aR1. To quantify neutrophils in the gingival tissue, single-cell suspensions were stained with Live/Dead fixable dye to gate out dead cells and then stained with anti-CD45 and anti-Ly6G Abs (33). Cell acquisition was performed on a NovoCyte flow cytometer (ACEA Biosciences, Santa Clara, CA). Data were analyzed with NovoExpress software (ACEA Biosciences).
Immunofluorescence histochemistry
Mouse maxillae with intact surrounding tissue were fixed in 4% paraformaldehyde for 1 day at 4°C, decalcified in formic acid for 2 weeks, followed by immersing in 30% sucrose in PBS and then embedded in optimal cutting temperature (OCT) compound. Coronal sections (8-μm thick) were blocked with 5% goat normal serum in Tris-buffered saline and 0.05% Tween 20 (TBST) before incubation with goat anti-mouse C3d Ab (cat# AF2655-SP, R&D Systems, Minneapolis, MN, IgG) followed by AlexaFluor 647-conjugated donkey anti-goat IgG (cat# A32849, Invitrogen, 1:1000). The tissues were counterstained with DAPI. Images were captured using a Nikon Eclipse NiE automated fluorescent microscope.
Quantitative real-time PCR for cytokine gene expression
Total RNA was extracted from gingival tissues using the GeneJET RNA Purification Kit (cat# K0731, Thermo-Fisher Scientific, Waltham, MA) and quantified by NanoDrop spectrometry at 260 and 280 nm. The RNA was reverse-transcribed using the High-Capacity cDNA Archive kit (cat# 4368814, Applied Biosystems, Waltham, MA) and real-time PCR with cDNA was performed using the ABI 7500 Fast System, according to the manufacturer’s protocol (Applied Biosystems). Data were analyzed using the comparative (ΔΔCt) method. TaqMan probes for detection and quantification of cytokine genes investigated in this paper (Supplemental Table 1) were purchased from Thermo-Fisher Scientific. In LIP experiments assessing gingival cytokine mRNA expression in ligated sites, the data were normalized to that of Gapdh and are shown as fold change relative to the contralateral unligated control sites (baseline), set as 1.
Microbial stimulation of human gingival epithelial cells
P. gingivalis (strain 33277) was purchased from ATCC and cultured in GAM broth (Nissui, Tokyo, Japan) under anaerobic conditions at 37°C. Human immortalized gingival keratinocytes (HIGK; donated by Richard J. Lamont, University of Louisville, Louisville, KY), generated by transfection of primary gingival epithelial cells with the E6/E7 gene from human papilloma virus (34), were maintained at 37°C and 5% CO2 in Keratinocyte Growth Medium-2 (cat# CC-3107, Lonza, Basel, Switzerland). The cells were seeded into 96-wells plates at a density of 2 × 105 cells per well for 18 h and then challenged with heat-killed (65°C, 1 h) P. gingivalis at a multiplicity of infection (MOI) of 10:1, in the presence or absence of different concentrations of recombinant human C3a or C5a (cat# 3677-C3–025 or 2037-C5–025/CF, R&D Systems) or the synthetic microbial lipopeptide Pam3Cys (cat# tlrl-pms, InvivoGen, San Diego, CA). In experiments designed to block TLR2, the cells were treated with anti-human TLR2 Ab (maba2-htlr2) or isotype control (maba2-ctrl), both from InvivoGen. In experiments designed to block signaling molecules, the following inhibitors were used: PD98059 (cat# tlrl-pd98, MEK/ERK inhibitor), SP600125 (cat# tlrl-sp60, JNK inhibitor), SB202190 (cat# tlrl-sb90, p38 MAPK inhibitor), purchased from InvivoGen, and SN50 (cat# sc-3060, NFκB inhibitor) obtained from Santa Cruz Biotechnology. The IL-6 concentration in stimulated HIGK culture supernatants was measured using an ELISA kit according to the manufacturer’s protocol (cat# D6050, R&D Systems).
Immunoblotting analysis
HIGK were seeded into 6-wells plates at 2×105/ml, cultured overnight, and treated with Pam3Cys (1 μg/ml) for different time lengths indicated in figure 5F. Cell lysates were prepared using RIPA lysis buffer (cat# sc-24948A, Santa Cruz Biotechnology) and protein concentrations were determined using the Bradford dye assay (cat# B6916, Sigma-Aldrich). Proteins were separated by standard SDS-PAGE on 12% acrylamide gels (Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride membranes (Bio-Rad) by electroblotting. The membranes were incubated in blocking buffer (5% nonfat dry milk, 10 mM Tris [pH 7.5], 100 mM NaCl, and 0.05% Tween 20) followed by probing with Abs, and visualization with horseradish peroxidase-conjugated secondary Ab and chemiluminescence using the Millipore ECL system. Images were captured using a FluorChem M imaging system (ProteinSimple, Minneapolis, MN) or iBright 1500 (Invitrogen). Abs used included polyclonal Abs against total ERK1/2 (cat# 9102S, 1:1000), JNK (cat# 9252S, 1:1000), and p38 MAPK (cat# 9212S, 1:1000) as well as monoclonal Abs against phospho-ERK1/2 (Thr202/Tyr204, cat# 4370S, clone D13.14.4E, 1:1000), phospho-JNK (Thr183/Tyr185, cat# 4668S, clone 81E11, 1:500), phospho-p38 (Thr180/Tyr182, cat# 9215S, clone 3D7, 1:1000) and total GAPDH (cat# 5174S, clone D16H11, 1:1000). All Abs were purchased from Cell Signaling, Danvers, MA.
Figure 5. C3a-induced IL-6 release in human gingival epithelial cells.
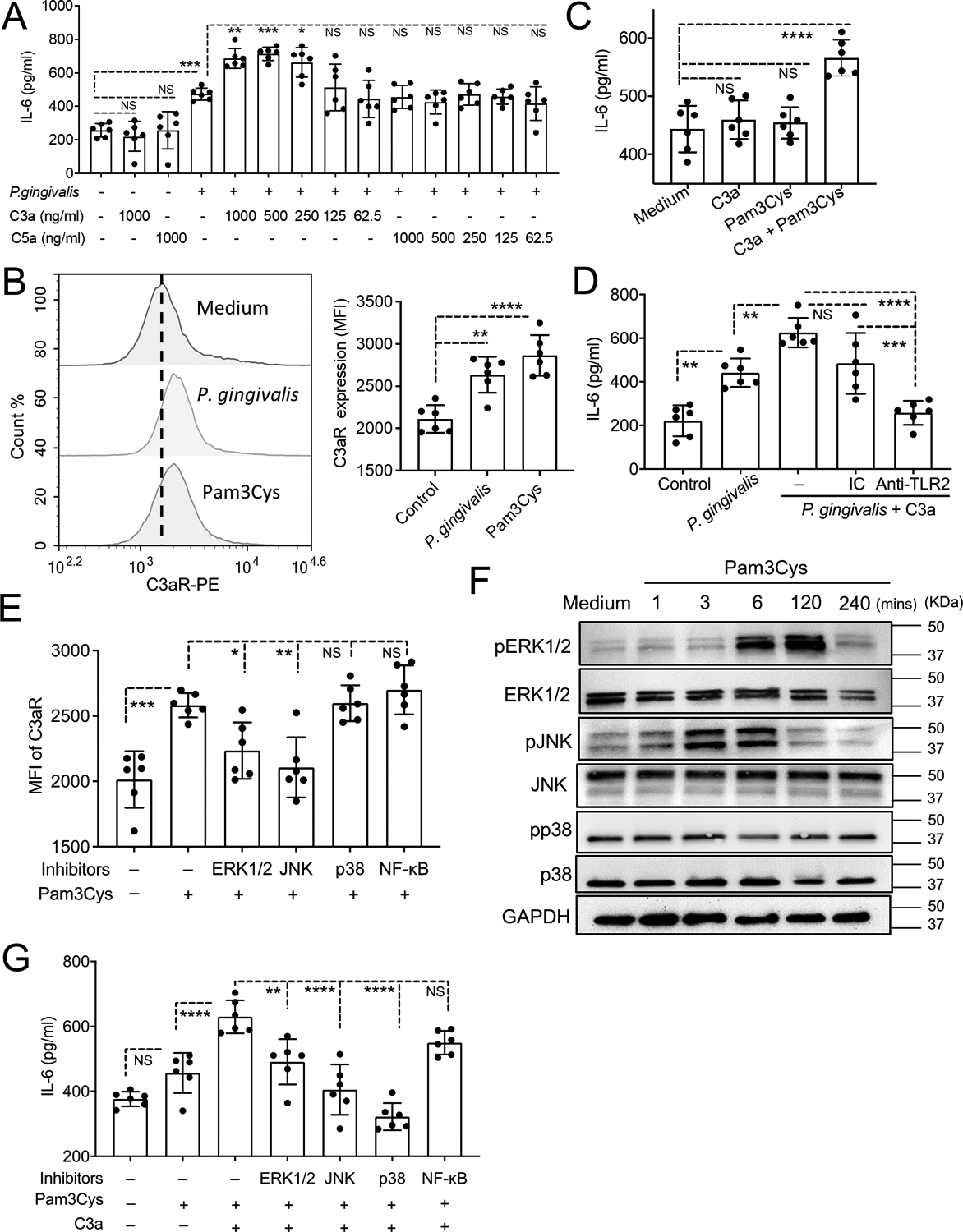
(A) Human immortalized gingival keratinocytes (HIGK) were stimulated, or not, with heat-killed P. gingivalis (MOI=10:1), C3a, or C5a and combinations thereof (at the indicated concentrations). Culture media were collected after 24-h incubation and assayed for IL-6 by ELISA. (B) HIGK were stimulated for 24 h with heat-killed P. gingivalis (MOI=10:1) or Pam3Cys lipopeptide (1 μg/ml) and C3aR expression was measured by FACS. Representative histogram (left) and bar graphs for MFI of C3aR expression (right). (C) HIGK were stimulated, or not, with C3a (500 ng/ml), Pam3Cys (1 μg/ml), or both, and IL-6 was measured in collected culture supernatants after 24-h incubation. (D) HIGK were stimulated for 24 h, or not, with P. gingivalis (MOI=10:1) alone or with C3a (500 ng/ml), in the presence or absence of 10 μg/ml anti-TLR2 neutralizing Ab or isotype control (IC), which were added 2 h prior to stimulation. IL-6 release was assayed by ELISA. (E) HIGK were pretreated with PD98059 (10 μM; MEK/ERK inhibitor), SP600125 (50 μM; JNK inhibitor), SB202190 (20 μM; p38 MAPK inhibitor), or SN50 (50 μM, NF-κB inhibitor). After 1 h, Pam3Cys (1 μg/ml) was added in the cultures and C3aR expression (MFI) was determined by FACS after 24 h-incubation. (F) HIGK were stimulated with Pam3Cys (1 μg/ml) for the indicated time lengths. Total protein was extracted and immunoblot analysis was performed with specific Abs against phosphorylated and total ERK1/2, JNK, and p38 MAPK as well as against GAPDH (loading control). (G) HIGK were pretreated with Pam3Cys (1 μg/ml) for 4 h, and then exposed to PD98059 (10 μM; MEK/ERK inhibitor), SP600125 (50 μM; JNK inhibitor), SB202190 (20 μM; p38 MAPK inhibitor), or SN50 (50 μM, NFκB inhibitor). After 1 h, C3a (500 ng/ml) was added in the cultures. Culture media were collected after 24 h and assayed for IL-6 by ELISA. Data are means ± SD (n = 6 cultures per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One-way ANOVA and Tukey’s (A,D,E,G) or Dunnett’s (B,C) multiple comparisons tests. NS, not significant.
Statistical analysis
After confirming normality, data were evaluated by 2-tailed, unpaired Student’s t test (comparisons of two groups only) or by one-way ANOVA (comparison of more than two groups) followed by Tukey’s or Dunnett’s multiple-comparison test, as appropriate. All statistical analyses were performed using GraphPad Prism software. P values less than 0.05 were considered to be statistically significant. Experiments were performed at least twice for verification and data were pooled.
Results
Bacterial accumulation is required for complement activation, inflammation, and bone loss in experimental periodontitis
To address our hypothesis that complement links the dysbiotic microbiota to Th17 cell expansion in periodontitis, we used the ligature-induced periodontitis (LIP) model. The use of the LIP model has separately implicated complement and Th17 in destructive periodontal inflammation in mice and these findings were confirmed in non-human primates and human patients (13, 17, 18, 20, 26). We and collaborators also showed that LIP-induced dysbiosis of the periodontal microbiome and bone loss are prevented by broad-spectrum systemic antibiotic combination (vancomycin-doripenem-neomycin; VDN), further confirming that the LIP model mimics the microbe-driven nature of this human inflammatory disease (20). To assess the contribution of the microbiota to complement activation, mice were administered VDN in drinking water for 2 weeks and subsequently subjected to LIP in the continuous presence of VDN treatment. As expected from our earlier work (20), VDN suppressed bacterial accumulation by ~3 log10 units (Figure 1A) and inhibited bone loss (Figure 1B), relative to mice given plain water. Moreover, in the present study VDN inhibited periodontal inflammation as evidenced by reduced neutrophil infiltration of the gingival tissue and decreased expression of gingival pro-inflammatory cytokines, including IL-6, IL-23, and IL-17 (Figure 1C). Staining of periodontal tissue sections for the complement activation fragment C3d revealed C3d deposition at and adjacent to the junctional epithelium (i.e., at the periodontal microbe-host interface) in ligated sites of mice given plain water (Figure 1D, middle) but not in ligated sites of mice given VDN-containing water (Figure 1D, bottom), which resembled unligated sites (Figure 1D, top). Therefore, LIP-induced bacterial accumulation appears to be required for complement activation.
Figure 1: Antibiotic suppression of bacterial accumulation inhibits LIP-induced complement activation and periodontal inflammation.
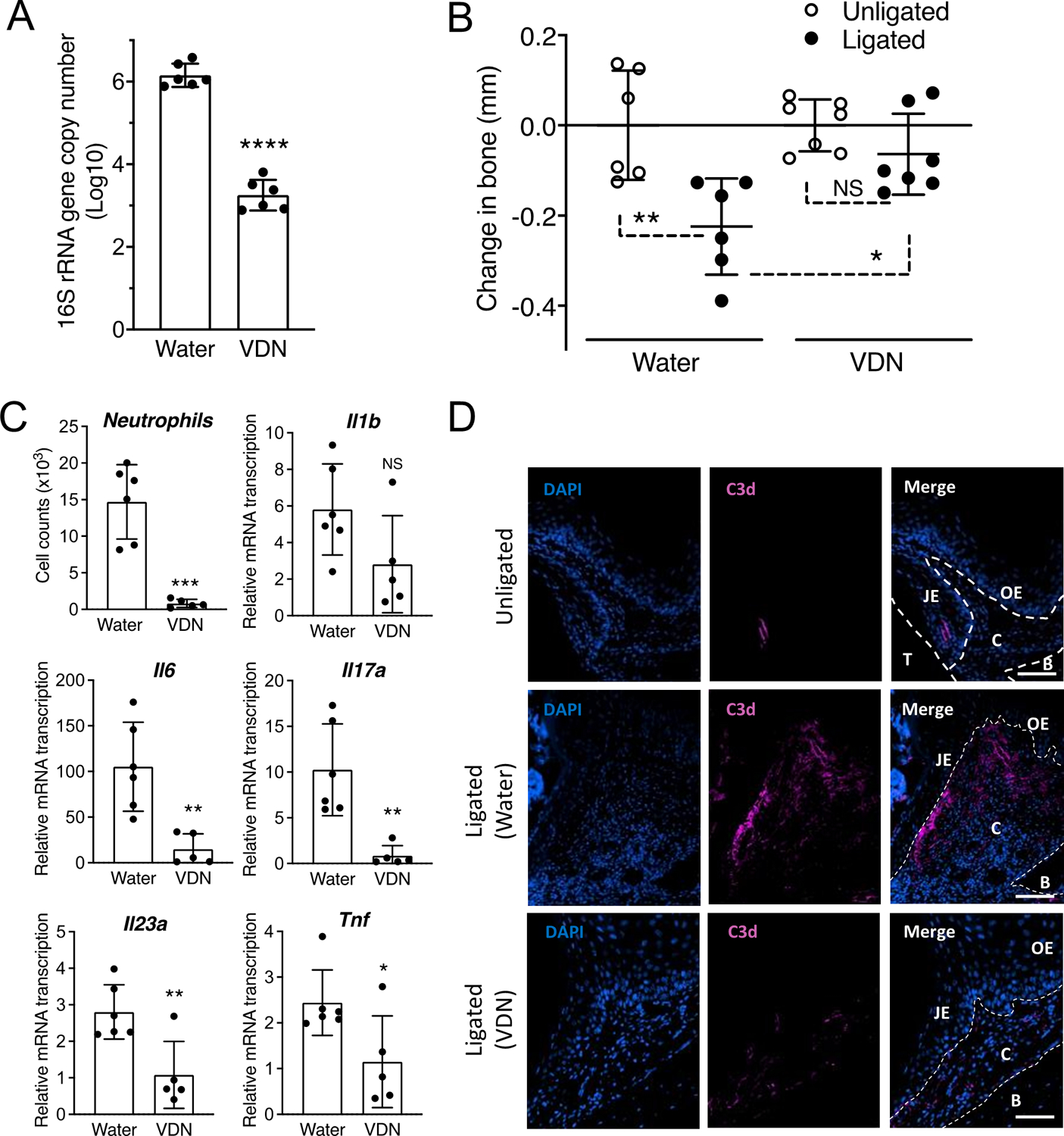
Mice were given drinking water with or without antibiotics (Vancomycin, 0.5 g/L; Doripenem, 0.25 g/L, Neomycin sulfate, 1 g/L; VDN) for 2 weeks and then subjected to ligature-induced periodontitis (LIP) for 3 days by ligating a maxillary second molar and leaving the contralateral tooth unligated to serve as baseline control. (A) Ligature-associated periodontal microbial load determined by quantitative real-time PCR (qPCR) of the 16S rRNA gene. (B) Bone loss determination relative to the unligated baseline. (C) Gingival tissues were processed and analyzed for determining the numbers of neutrophils (CD45+Ly6G+) by FACS and the relative gingival mRNA expression of indicated cytokines determined by qPCR. (D) Periodontal tissue sections from ligated and unligated control sites were stained with DAPI and anti-C3d mAb (scale bars, 100 μm). Data are means ± SD (n = 5–7 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 between indicated groups (A,C: Unpaired Student’s t-test; B: One-way ANOVA with Tukey’s multiple comparison test). C, Connective tissue; B, Bone; JE, Junction epithelium; OE, Oral epithelium; T, Tooth. NS, not significant.
C3 deficiency blocks induction of cytokines required for Th17 expansion
We previously showed that C3–/– mice, which are unable to activate the complement cascade regardless of the initiation pathway involved (35), are protected from LIP-induced bone loss, although we did not analyse the host inflammatory response in that model (17). In the present study, we showed that C3–/– mice not only developed significantly less bone loss than their WT (C3+/+) littermate controls (Figure 2A), but also displayed reduced gingival mRNA expression of several pro-inflammatory and pro-osteoclastogenic cytokines, including IL-6 and IL-23, but not IL-1β (Figure 2B). In general, all of these three cytokines have been implicated in the development and expansion of Th17 cells (36). However, we have shown that the expansion of gingival Th17 cells in response to the oral dysbiotic microbiota strictly requires both IL-6 and IL-23, albeit not IL-1 (20). Therefore, both VDN treatment (Figure 1C) and C3 deficiency (Figure 2B) block the induction of critical cytokines (IL-6 and IL-23) that are indispensable for Th17 expansion in the LIP model (20). Consistent with this, Th17 cell frequencies and absolute numbers were significantly decreased in the gingival tissue of LIP-subjected C3–/– mice as compared to LIP-subjected C3+/+ controls (Figure 2C).
Figure 2: C3 deficiency inhibits LIP-induced periodontal inflammation and Th17 cell accumulation.
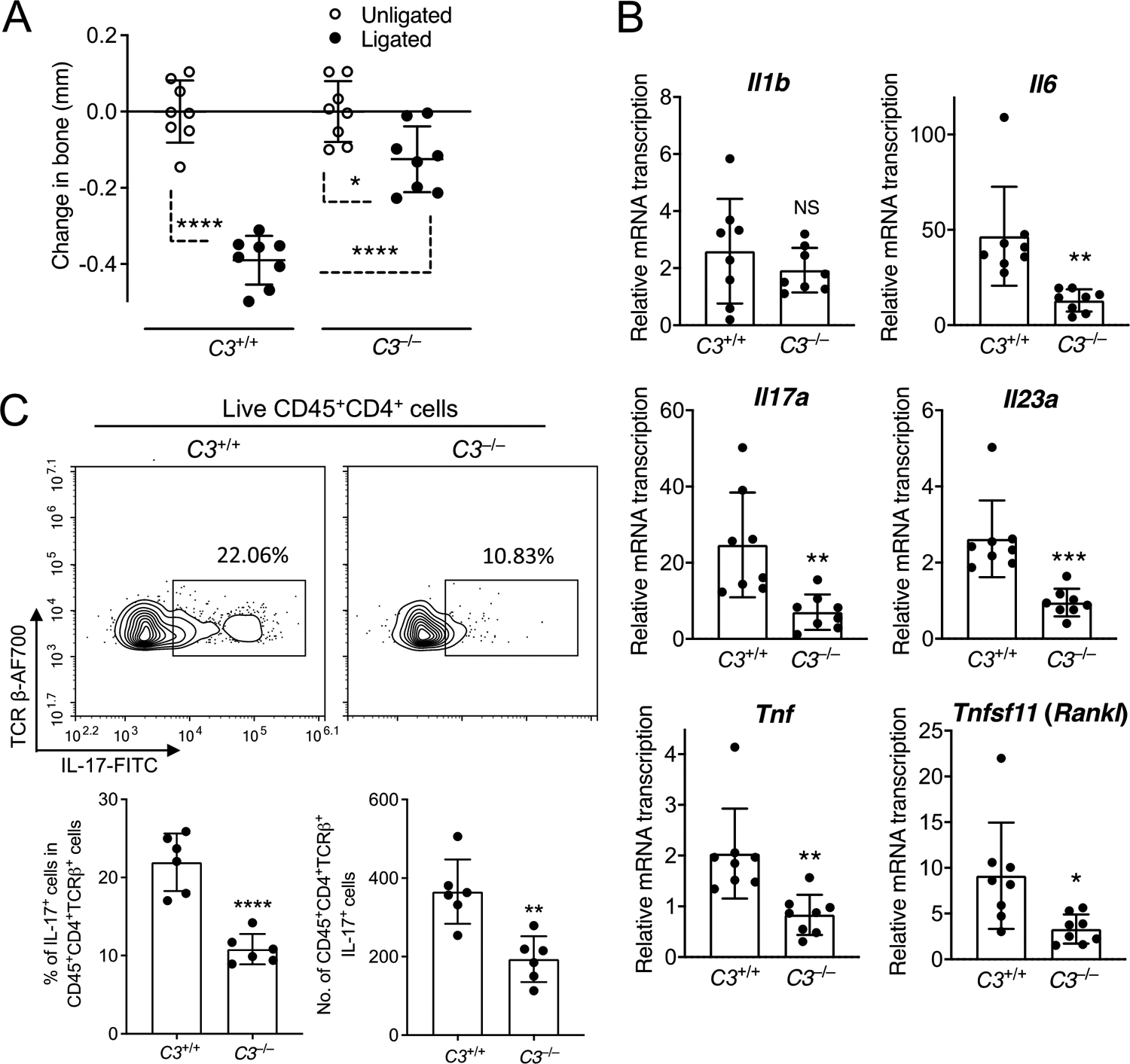
C3–/– mice and C3+/+ littermate controls were subjected to ligature-induced periodontitis (LIP) for 5 days by ligating a maxillary second molar and leaving the contralateral tooth unligated to serve as baseline control. (A) Bone loss determination relative to the unligated baseline. (B) Relative gingival mRNA expression of indicated cytokines determined by quantitative real-time PCR. (C) Representative FACS plots of Th17 cells in gingival tissue (top) and bar graphs showing percentage of Th17 cells in CD4+ T cells (bottom left) and Th17 absolute numbers (bottom right) in the gingival tissue of C3–/– mice and C3+/+ controls on day 5 after LIP. Data are means ± SD (A,B: n = 8 mice per group; C: n = 6 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 between indicated groups (A: One-way ANOVA with Tukey’s multiple comparison test; B,C: Unpaired Student’s t-test). NS, not significant.
Induction of inflammatory bone loss requires IL-6 receptor signaling
IL-23 has been extensively studied in periodontitis and Ab-mediated neutralization of this cytokine inhibits periodontal bone loss in animal models, as well as gingival inflammation in humans with an aggressive form of periodontal disease (33, 37–40). We have thus focused on the other complement-dependent cytokine that is required for Th17 expansion in the periodontal tissue, namely IL-6 (20). To determine whether IL-6 is indeed an important complement-dependent cytokine required for induction of periodontitis, LIP-subjected mice were locally microinjected, or not, with a blocking mAb to the α subunit of the IL-6 receptor (anti-IL-6Rα) or an isotype control. Anti-IL-6Rα mAb treatment significantly inhibited bone loss relative to the isotype control (Figure 3A). The anti-IL-6Rα-mediated inhibition of bone loss was associated with reduced expression of mRNA for IL-17 and receptor-activated NF-κB ligand (RANKL) (Figure 3B), suggesting that these pro-osteoclastogenic factors are induced downstream of IL-6 receptor signaling.
Figure 3. Induction of maximal inflammatory bone loss requires IL-6 receptor signaling.
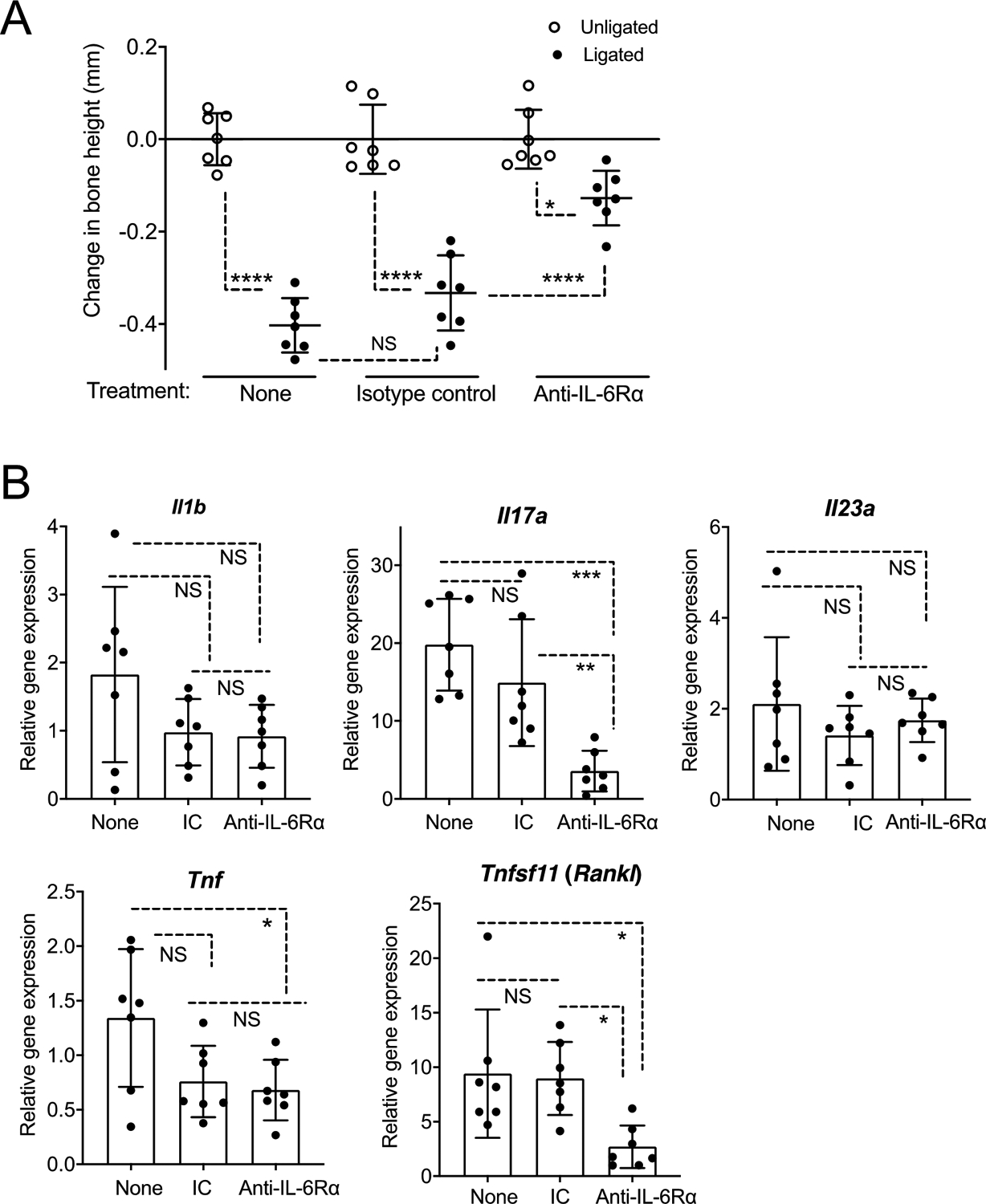
Groups of mice were subjected to ligature-induced periodontitis (LIP) for 5 days by ligating a maxillary second molar and leaving the contralateral tooth unligated to serve as baseline control. One day prior to LIP (day −1), the mice were microinjected, or not, with anti-IL-6Rα mAb or isotype control. (A) Bone loss determination relative to corresponding unligated baseline. (B) Relative gingival mRNA expression of indicated cytokines determined by quantitative real-time PCR. Data are means ± SD (n = 7 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 between indicated groups (One-way ANOVA with Tukey’s multiple comparison test). IC, isotype control, NS, not significant.
LIP-induced IL-6 is predominantly produced by epithelial cells that upregulate C3aR expression
Since IL-6 is complement-dependent and complement is activated at or adjacent to gingival epithelial cells (Figure 1D, middle), we hypothesized that gingival epithelial cells might be a target of complement for IL-6 induction. Indeed, flow cytometric analysis of gingival tissue cell suspensions from LIP-subjected mice (gating strategy in Supplemental figure 1) showed that IL-6+ cells were predominantly epithelial (EpCAM+) cells, rather than CD45+ immune cells (Figure 4A). Moreover, EpCAM+ cells (isolated from gingival tissue cells through positive selection) expressed significantly higher IL-6 mRNA levels than EpCAM– cells (Figure 4B). To identify complement receptor(s) on gingival epithelial cells that might mediate IL-6 induction, we performed a LIP experiment and analyzed C3aR and C5aR1 expression on CD45−EpCAM+ cells by FACS. The counts and frequency of C3aR+ epithelial (CD45−EpCAM+) cells were significantly upregulated upon LIP (P < 0.0001 in ligated vs. unligated sites), whereas no significant differences were observed with regard to the counts and frequency of C5aR1+ epithelial (CD45−EpCAM+) cells in ligated and unligated sites (Figure 4 C,D). These data suggested that gingival epithelial cells selectively upregulate C3aR in response to the LIP challenge.
Figure 4. LIP-induced IL-6 is predominantly produced by epithelial cells that also express C3aR.
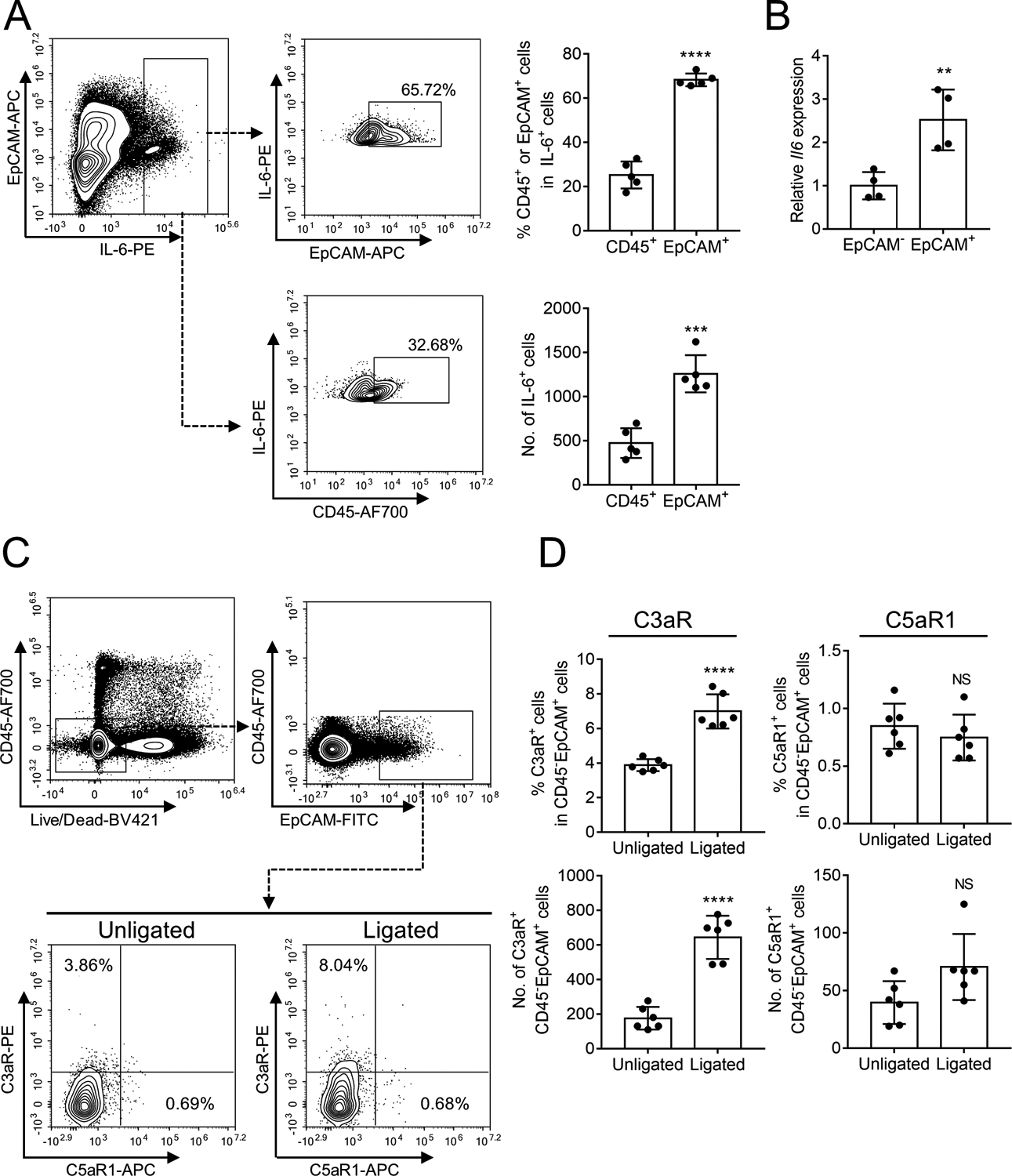
(A-B) Gingival tissues were collected from ligated sites of ligature-induced periodontitis (LIP)-subjected mice and processed for FACS analysis to detect IL-6-expressing leukocytes (CD45+) or epithelial cells (EpCAM+). (A) Representative FACS plots (left) and bar graphs showing percentage of IL-6-expressing cells in CD45+ cells and EpCAM+ cells (top right) and absolute numbers of IL-6+CD45+ cells and IL-6+EpCAM+ cells (bottom right) in the gingival tissue on day 5 after LIP. (B) Gingival cells were sorted into EpCAM+ and EpCAM– cells and analyzed for IL-6 mRNA expression by quantitative real-time PCR. (C,D) Gingival tissues were collected from LIP-subjected mice and processed for FACS analysis to detect expression of C3aR and C5aR1 in CD45−EpCAM+ cells. (C) Representative FACS plots and (D) bar graphs showing percentage of C3aR-expressing or C5aR1-expressing cells in CD45−EpCAM+ cells (top) and absolute numbers of C3aR+CD45−EpCAM+ cells or C5aR1+CD45−EpCAM+ cells (bottom) in unligated and ligated sites on day 5 after LIP. Data are means ± SD (n = 4–6 mice per group). **P < 0.01, ***P < 0.001, ****P < 0.0001 between indicated groups (Unpaired Student’s t-test). NS, not significant.
The ability of C3a to induce IL-6 release in human gingival epithelial cells requires priming with bacteria or agonists thereof
Our in vivo experiments suggested that C3aR activation might be associated with IL-6 production in gingival epithelial cells (Figure 4). To directly investigate this possibility and understand the mechanism(s) involved, we used human immortalized gingival keratinocytes (HIGK), a widely used model for human gingival epithelial cells (34, 41). Initial experiments showed that C3a by itself (even at 1000 ng/ml) failed to induce IL-6 release from HIGK. Since complement activation in the periodontium occurs in the setting of concomitant microbial challenge, we tested different concentrations of C3a in the presence of a model periodontal pathogen, Porphyromonas gingivalis. In the range of 250–1000 ng/ml, C3a significantly increased P. gingivalis-induced IL-6, whereas C5a was without effect in this regard (Figure 5A). Upon LIP, gingival epithelial cells expressed C3aR in ligated sites (active periodontitis) but not in unligated sites (healthy periodontium) (Figure 4C). Therefore, we reasoned that the ability of C3a to enhance IL-6 release in P. gingivalis-challenged, but not in unchallenged, HIGK might be due to differential expression of C3aR; that is, C3aR expression being relatively low in unchallenged cells. Indeed, we showed that the synthetic microbial lipopeptide Pam3Cys (a TLR2 agonist), as well as P. gingivalis, significantly upregulated C3aR expression in HIGK (Figure 5B). We next demonstrated that C3a could induce IL-6 release in HIGK when combined with Pam3Cys, but not when C3a was tested alone (Figure 5C). Because Pam3Cys also failed to induce IL-6 release by itself, we concluded that the IL-6 release induced by combined treatment with C3a and Pam3Cys may represent a synergistic interaction, with Pam3Cys upregulating C3aR expression and C3a activating C3aR. In other words, combined TLR2 and C3aR signaling mediates IL-6 release in HIGK. Consistently, the enhanced IL-6 release by C3a and P. gingivalis was abrogated by a neutralizing Ab to TLR2 (Figure 5D). Taken together, these data indicate that C3a can induce IL-6 release in HIGK in a manner dependent on bacterial stimulation (whole bacteria or agonists thereof), which can upregulate the expression of C3aR and thus prime HIGK to detect and respond to C3a.
It is uncertain how TLR2 might regulate C3aR expression in gingival epithelial cells. In an exploratory experiment, we tested inhibitors of several signaling molecules that have been shown to mediate TLR2-induced signaling in gingival epithelial cells (42–44). Specifically, we tested the abilities of PD98059 (inhibitor of the MEK/ERK pathway), SP600125 (JNK inhibitor), SB202190 (p38 MAPK inhibitor) and SN50 (NF-κB inhibitor) to inhibit Pam3Cys-induced C3aR expression in the HIGK model. PD98059 and SP600125, but not the other two inhibitors tested, suppressed C3aR upregulation by Pam3Cys (Figure 5E). Consistently, immunoblot analysis showed that Pam3Cys induced phosphorylation of ERK1/2 and JNK, but not of p38 MAPK (Figure 5F). Therefore, ERK1/2 and JNK appeared to mediate Pam3Cys-induced upregulation of C3aR. In line with this notion, inhibition of ERK1/2 (by PD98059) and JNK (by SP600125) also suppressed IL-6 release in HIGK stimulated by both Pam3Cys and C3a (Figure 5G). In the same experiment, IL-6 release was not affected by NF-κB inhibition (SN50) but was diminished by p38 MAPK inhibition (SB202190) (Figure 5G). Although p38 MAPK was not involved in C3aR upregulation, it has been shown to be activated downstream of C3aR activation in different cell types (45, 46). Thus, p38 MAPK appears to participate in the C3aR signaling pathway leading to IL-6 induction in HIGK.
Discussion
Previous work has suggested that the periodontal dysbiotic microbiota and complement engage in reciprocally reinforced interactions, where bacteria activate complement that in turn induces inflammation, which can fuel further microbial growth (10, 17, 31, 47, 48). This is because inflammation generates tissue breakdown products utilized as nutrients by the bacteria, thus creating a nutritionally supportive environment for the persistence of dysbiosis (4, 48). We now showed that the microbiota-complement interaction additionally generates an inflammatory environment that leads to the expansion of pathogenic Th17 cells in the periodontium. Indeed, LIP-subjected C3-deficient mice exhibited significantly reduced gingival expression of IL-6 and IL-23 accompanied by decreased frequency and absolute numbers of Th17 cells relative to WT littermate controls. By acting upstream of these inflammatory effector cells and mediators, complement is an ideal target for host-modulation in periodontitis, a concept that has been confirmed in a recent interventional trial with the C3-targeted inhibitor, AMY-101 (13).
The gingival epithelial cells of the subgingival crevice constitute a physical barrier to the periodontal microbiota but also form an interactive interface that signals the microbial challenge to the underlying cells and defense mechanisms of the immune system (49–53). Little is known about the interaction of complement effectors with gingival epithelial cells in the context of periodontitis. However, complement has been shown to regulate inflammatory responses of epithelial cells in other tissues (54–57). Here we showed that LIP induced C3aR expression and complement-dependent IL-6 production in gingival epithelial cells in vivo. Using an in vitro model of human gingival epithelial cells, we demonstrated that microbial stimulation (P. gingivalis or a synthetic microbial agonist of TLR2) upregulated the expression of C3aR, which in turn mediated IL-6 release in response to recombinant human C3a. Combined with the in vivo data, this finding suggests that microbial stimulation primes gingival epithelial cells to sense and respond to local C3a by releasing IL-6. In the LIP model, the gingival epithelial cells were a major source of IL-6 production, which was significantly reduced by either broad-spectrum antibiotics or by C3 deficiency. Thus, microbiota-driven complement activation generates C3a which acts on C3aR-expressing gingival epithelial cells to generate IL-6 that is crucial for Th17 expansion, as shown earlier by us and collaborators (20). In the absence of experimental periodontitis (unligated sites), C3aR expression was essentially absent from gingival epithelial cells. Therefore, complement appears to initiate periodontal disease by acting at the microbiome-gingival epithelial interface and causing the release of IL-6 required for Th17 expansion.
The ability of inflammation to exacerbate microbial dysbiosis (4) conceivably stimulates further complement activation, thereby potentially generating a feed-forward loop involving the dysbiotic microbiota, complement and other inflammatory pathways. However, the primary cause of complement activation in the setting of periodontitis should be the microbial challenge, consistent with the fact that complement is activated promptly upon infection and is a key inducer of inflammation (58). At least in principle, the periodontal microbial challenge could initiate either the lectin pathway by mannose-containing carbohydrates (present on bacteria and other microorganisms) or the alternative pathway that can be initiated on microbial pathogen surfaces (35).
In the mouse LIP model, only Th17 cells, but not γδ T cells or other types of IL-17-secreting cells, were shown to be expanded in response to the challenge by the local dysbiotic microbiota (20). When LIP-subjected mice were placed under broad-spectrum antibiotics, Th17 cells failed to expand beyond steady-state levels, correlating with significantly decreased inflammation and bone loss (20). Interestingly, the expansion of Th17 cells required induction of IL-6 and IL-23, which are C3-dependent cytokines according to the present study, but not of IL-1, which was not affected by C3 deficiency here. Th17-derived IL-17 can mediate bone immunopathology by inducing matrix metalloproteinases (MMP) and RANKL, thereby contributing to degradation of both connective tissue and the underlying alveolar bone, in both human periodontitis and experimental periodontitis in animal models (20, 26, 59, 60). The C3-targeted compstatin-based inhibitor AMY-101 was shown to significantly reduce the gingival crevicular fluid (inflammatory periodontal tissue exudate) levels of IL-17 and RANKL in non-human primates (18) and of MMP-8 and MMP-9 in humans (13). Based on our current findings, the ability of AMY-101 to restrain the production of these inflammatory and tissue-destructive molecules might be mediated in part by inhibition of Th17 accumulation.
The importance of complement in enhancing Th17 development was shown also in different experimental systems. In a mouse model of systemic inflammation, i.p. injection of C5a and LPS synergistically induced IL-6 release in serum that in turn promoted Th17 cell development (61). In another study, activation of serum complement, via all three pathways, resulted in C5aR1-dependent initiation of Th17 cell differentiation and expansion and development of autoimmune arthritis in genetically autoimmune-susceptible SKG mice (62).
We have previously shown that C5aR1 and TLR2 agonists (C5a and Pam3Cys, respectively) synergize for the induction of proinflammatory cytokines in the mouse periodontal tissue (30). Our present findings show that C3aR and TLR2 agonists also cooperate, since both C3a and Pam3Cys were required for IL-6 release by gingival epithelial cells. The production of IL-6 in the in vitro HIGK system was dependent particularly on p38 MAPK, but also on ERK1/2 and JNK that appeared to mediate Pam3Cys-induced C3aR upregulation. C3aR-dependent upregulation of IL-6 may not only be important for Th17 expansion but also for disease development, since anti-IL-6Rα treatment inhibited periodontal bone loss. Consistent with this important role of C3aR in periodontal disease pathogenesis, C3aR-deficient mice were protected (relative to WT controls) from periodontal bone loss, although a different model was used in our earlier study (P. gingivalis-induced periodontitis) (31).
In summary, complement links the periodontal microbiota challenge to Th17 expansion and bone loss, a function that is mediated, at least in part, by the ability of C3a to promote the release of IL-6 from gingival epithelial cells. A therapeutic implication of our findings is that the success of the AMY-101 trial in blocking periodontal inflammation (13) might in part be attributed to restraining the abundance of Th17 cells. By integrating complement- and Th17-driven immunopathology in periodontitis, our study provides a deeper understanding of the mechanisms that drive this oral inflammatory disease.
Supplementary Material
Key points.
Ligature-induced periodontitis activates complement in microbiota-dependent manner
Complement C3 deficiency inhibits LIP-induced IL-6 and IL-23 and Th17 expansion
Complement links the periodontal microbiota to Th17 expansion and bone loss
Financial Support
This work was supported by grants from the US National Institutes of Health (AI068730 to JDL and DE015254 and DE021685 to GH).
Abbreviations used in this article
- ABC
alveolar bone crest
- CEJ
cementoenamel junction
- C3aR
C3a receptor
- C5aR1
C5a receptor-1
- HIGK
human immortalized gingival keratinocytes
- IC
isotype control
- LIP
ligature-induced periodontitis
- MMP
matrix metalloproteinase
- RANKL
receptor-activated NF-κB ligand
- VDN
vancomycin-doripenem-neomycin
- WT
wild-type
Footnotes
Conflict of interest
J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs such as AMY-101). J.D.L. is inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. J.D.L. and G.H. have a joint patent that describes the use of complement inhibitors for therapeutic purposes in periodontitis. J.D.L. is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives such as APL-2/pegcetacoplan/Empaveli/Aspaveli). The other authors declare no competing interest.
References
- 1.Kinane DF, Stathopoulou PG, and Papapanou PN. 2017. Periodontal diseases. Nat Rev Dis Primers 3: article No.17038. [DOI] [PubMed] [Google Scholar]
- 2.Hajishengallis G, and Chavakis T. 2021. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol 21: 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Wang H, Yu X, Saha G, Kalafati L, Ioannidis C, Mitroulis I, Netea MG, Chavakis T, and Hajishengallis G. 2022. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell 185: 1709–1727.e1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamont RJ, Koo H, and Hajishengallis G. 2018. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol 16: 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peres MA, Macpherson LMD, Weyant RJ, Daly B, Venturelli R, Mathur MR, Listl S, Celeste RK, Guarnizo-Herreño CC, Kearns C, Benzian H, Allison P, and Watt RG. 2019. Oral diseases: a global public health challenge. The Lancet 394: 249–260. [DOI] [PubMed] [Google Scholar]
- 6.Collaborators, G. D. a. I. I. a. P. 2018. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392: 1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botelho J, Machado V, Leira Y, Proença L, Chambrone L, and Mendes JJ. 2021. Economic burden of periodontitis in the United States of America and Europe – an updated estimation. J Periodontol 93: 373–379. [DOI] [PubMed] [Google Scholar]
- 8.Tonetti MS, Chapple IL, and P. Working Group 3 of Seventh European Workshop on. 2011. Biological approaches to the development of novel periodontal therapies--consensus of the Seventh European Workshop on Periodontology. J Clin Periodontol 38 Suppl 11: 114–118. [DOI] [PubMed] [Google Scholar]
- 9.Tonetti MS, Jepsen S, Jin L, and Otomo-Corgel J. 2017. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J Clin Periodontol 44: 456–462. [DOI] [PubMed] [Google Scholar]
- 10.Hajishengallis G, Hasturk H, Lambris JD, Apatzidou DA, Belibasakis GN, Bostanci N, Corby PM, Cutler CW, D’Aiuto F, Hajishengallis E, Huber-Lang M, Ioannidou E, Kajikawa T, Kantarci A, Korostoff JM, Kotsakis GA, Maekawa T, Mastellos DC, Moutsopoulos NM, Myneni S, Nagelberg R, Nilsson B, Papapanou PN, Papathanasiou E, Potempa J, Risitano A, Sahingur SE, Saito A, Sculean A, Stavropoulos A, Teles FR, Tonetti M, and Yancopoulou D. 2021. C3-targeted therapy in periodontal disease: moving closer to the clinic. Trends Immunol 42: 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balta MG, Papathanasiou E, Blix IJ, and Van Dyke TE. 2021. Host Modulation and Treatment of Periodontal Disease. J Dent Res 100 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajishengallis G, Chavakis T, and Lambris JD. 2020. Current understanding of periodontal disease pathogenesis and targets for host-modulation therapy. Periodontol 2000 84: 14–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasturk H, Hajishengallis G, C. Forsyth Institute Center for, s. Translational Research, Lambris JD, Mastellos DC, and Yancopoulou D. 2021. Phase IIa clinical trial of complement C3 inhibitor AMY-101 in adults with periodontal inflammation. J Clin Invest 131: e152973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricklin D, Reis ES, and Lambris JD. 2016. Complement in disease: a defence system turning offensive. Nat Rev Nephrol 12: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, and Lambris JD. 2017. Novel mechanisms and functions of complement. Nat Immunol 18: 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mastellos DC, Ricklin D, and Lambris JD. 2019. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov 18: 707–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, Lambris JD, and Hajishengallis G. 2014. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J. Immunol 192 6020–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maekawa T, Briones RA, Resuello RR, Tuplano JV, Hajishengallis E, Kajikawa T, Koutsogiannaki S, Garcia CA, Ricklin D, Lambris JD, and Hajishengallis G. 2016. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol 43: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kajikawa T, Briones RA, Resuello RRG, Tuplano JV, Reis ES, Hajishengallis E, Garcia CAG, Yancopoulou D, Lambris JD, and Hajishengallis G. 2017. Safety and Efficacy of the Complement Inhibitor AMY-101 in a Natural Model of Periodontitis in Non-human Primates. Mol Ther Methods Clin Dev 6: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, Morell RJ, Freeman AF, Lazarevic V, Trinchieri G, Diaz PI, Holland SM, Belkaid Y, Hajishengallis G, and Moutsopoulos NM. 2018. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med 10: eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, Ciero PA, Krauss JL, Li F, Rauner M, Hofbauer LC, Choi EY, Chung KJ, Hashim A, Curtis MA, Chavakis T, and Hajishengallis G. 2012. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol 13: 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, and Colten HR. 1999. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology 42: 135–149. [DOI] [PubMed] [Google Scholar]
- 23.Abe T, and Hajishengallis G. 2013. Optimization of the ligature-induced periodontitis model in mice. Journal of immunological methods 394: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, Chen GY, Nunez G, Giannobile WV, Raes J, and Inohara N. 2013. Induction of bone loss by pathobiont-mediated nod1 signaling in the oral cavity. Cell Host Microbe 13: 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rovin S, Costich ER, and Gordon HA. 1966. The influence of bacteria and irritation in the initiation of periodontal disease in germfree and conventional rats. J Periodontal Res 1: 193–204. [DOI] [PubMed] [Google Scholar]
- 26.Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, Iwakura Y, Nakashima T, Okamoto K, and Takayanagi H. 2018. Host defense against oral microbiota by bone-damaging T cells. Nat Commun 9: 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, Mitroulis I, Chavakis T, and Hajishengallis G. 2015. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med 7: 307ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, and Little SR. 2013. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A 110: 18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, Grzybek M, von Renesse J, Czogalla A, Troullinaki M, Ferreira A, Doreth C, Ruppova K, Chen LS, Hosur K, Lim JH, Chung KJ, Grossklaus S, Tausche AK, Joosten LAB, Moutsopoulos NM, Wielockx B, Castrillo A, Korostoff JM, Coskun U, Hajishengallis G, and Chavakis T. 2019. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol 20: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, and Hajishengallis G. 2012. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 189: 5442–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, and Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutzan N, Abusleme L, Konkel JE, and Moutsopoulos NM. 2016. Isolation, Characterization and Functional Examination of the Gingival Immune Cell Network. J Vis Exp: 53736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kajikawa T, Wang B, Li X, Wang H, Chavakis T, Moutsopoulos NM, and Hajishengallis G. 2020. Frontline Science: Activation of metabolic nuclear receptors restores periodontal tissue homeostasis in mice with leukocyte adhesion deficiency-1. J Leukoc Biol 108: 1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda D, Bigler L, Lee P, and Blanton R. 1996. HPV immortalization of human oral epithelial cells: a model for carcinogenesis. Exp Cell Res 226: 164–169. [DOI] [PubMed] [Google Scholar]
- 35.Ricklin D, Hajishengallis G, Yang K, and Lambris JD. 2010. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korn T, Bettelli E, Oukka M, and Kuchroo VK. 2009. IL-17 and Th17 Cells. Annu Rev Immunol 27: 485–517. [DOI] [PubMed] [Google Scholar]
- 37.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, Uzel G, Chen W, Chavakis T, Holland SM, and Hajishengallis G. 2014. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med 6: 229ra240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allam JP, Duan Y, Heinemann F, Winter J, Gotz W, Deschner J, Wenghoefer M, Bieber T, Jepsen S, and Novak N. 2011. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol 38: 879–886. [DOI] [PubMed] [Google Scholar]
- 39.Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, Uzel G, Axelrod KC, Lisco A, Notarangelo LD, Hajishengallis G, Notarangelo LD, and Holland SM. 2017. Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1. N Engl J Med 376: 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bunte K, and Beikler T. 2019. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tribble GD, Mao S, James CE, and Lamont RJ. 2006. A Porphyromonas gingivalis haloacid dehalogenase family phosphatase interacts with human phosphoproteins and is important for invasion. Proc Natl Acad Sci U S A 103: 11027–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asai Y, Ohyama Y, Gen K, and Ogawa T. 2001. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect. Immun 69: 7387–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji S, Shin JE, Kim YS, Oh J-E, Min B-M, and Choi Y. 2009. Toll-Like Receptor 2 and NALP2 Mediate Induction of Human Beta-Defensins by Fusobacterium nucleatum in Gingival Epithelial Cells. Infect Immun 77: 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imamura K, Kokubu E, Kita D, Ota K, Yoshikawa K, Ishihara K, and Saito A. 2016. Role of mitogen-activated protein kinase pathways in migration of gingival epithelial cells in response to stimulation by cigarette smoke condensate and infection by Porphyromonas gingivalis. J Periodontal Res 51: 613–621. [DOI] [PubMed] [Google Scholar]
- 45.Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, Liu L, and Zhou H. 2016. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflamm 13: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Napier BA, Brubaker SW, Sweeney TE, Monette P, Rothmeier GH, Gertsvolf NA, Puschnik A, Carette JE, Khatri P, and Monack DM. 2016. Complement pathway amplifies caspase-11–dependent cell death and endotoxin-induced sepsis severity. J Exp Med 213: 2365–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, Hashim A, Hoch S, Curtis MA, Nussbaum G, Lambris JD, and Hajishengallis G. 2014. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 15: 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrero ER, Fernandes S, Verspecht T, Ugarte-Berzal E, Boon N, Proost P, Bernaerts K, Quirynen M, and Teughels W. 2018. Dysbiotic Biofilms Deregulate the Periodontal Inflammatory Response. J Dent Res: 22034517752675. [DOI] [PubMed] [Google Scholar]
- 49.Maekawa T, Kulwattanaporn P, Hosur K, Domon H, Oda M, Terao Y, Maeda T, and Hajishengallis G. 2017. Differential Expression and Roles of Secreted Frizzled-Related Protein 5 and the Wingless Homolog Wnt5a in Periodontitis. J Dent Res 96: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eskan MA, Hajishengallis G, and Kinane DF. 2007. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun 75: 892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimada E, Kataoka H, Miyazawa Y, Yamamoto M, and Igarashi T. 2012. Lipoproteins of Actinomyces viscosus induce inflammatory responses through TLR2 in human gingival epithelial cells and macrophages. Microbes Infect 14: 916–921. [DOI] [PubMed] [Google Scholar]
- 52.Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, and Ojcius DM. 2010. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol 12: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Attar A, Alimova Y, Kirakodu S, Kozal A, Novak MJ, Stromberg AJ, Orraca L, Gonzalez-Martinez J, Martinez M, Ebersole JL, and Gonzalez OA. 2018. Activation of Notch-1 in oral epithelial cells by P. gingivalis triggers the expression of the antimicrobial protein PLA2-IIA. Mucosal Immunol 11: 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandez-Godino R, and Pierce EA. 2018. C3a triggers formation of sub-retinal pigment epithelium deposits via the ubiquitin proteasome pathway. Sci Rep 8: 9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lueck K, Busch M, Moss SE, Greenwood J, Kasper M, Lommatzsch A, Pauleikhoff D, and Wasmuth S. 2015. Complement Stimulates Retinal Pigment Epithelial Cells to Undergo Pro-Inflammatory Changes. Ophthalmic Res 54: 195–203. [DOI] [PubMed] [Google Scholar]
- 56.Peng Q, Li K, Smyth LA, Xing G, Wang N, Meader L, Lu B, Sacks SH, and Zhou W. 2012. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, Nemenoff RA, Quigg RJ, and Holers VM. 2007. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J Immunol 178: 1819–1828. [DOI] [PubMed] [Google Scholar]
- 58.Hajishengallis G, and Lambris JD. 2010. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol 31: 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miossec P, and Kolls JK. 2012. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11: 763–776. [DOI] [PubMed] [Google Scholar]
- 60.Zenobia C, and Hajishengallis G. 2015. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000 69: 142–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang C, Zhang X, Miwa T, and Song WC. 2009. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood 114: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hashimoto M, Hirota K, Yoshitomi H, Maeda S, Teradaira S, Akizuki S, Prieto-Martin P, Nomura T, Sakaguchi N, Kohl J, Heyman B, Takahashi M, Fujita T, Mimori T, and Sakaguchi S. 2010. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med 207: 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.