

SUMMARY
Bacteria encode reverse transcriptases (RTs) of unknown function that are closely related to group II intron-encoded RTs. We found that a Pseudomonas aeruginosa group II intron-like RT (G2L4 RT) with YIDD instead of YADD at its active site functions in DNA repair in its native host and when transferred into Escherichia coli. G2L4 RT has biochemical activities strikingly similar to those of human DNA repair polymerase θ and uses them for translesion DNA synthesis and double-strand break repair (DSBR) via microhomology-mediated end-joining (MMEJ). We also found that a group II intron RT can function similarly in DNA repair, with reciprocal active-site substitutions showing isoleucine favors MMEJ and alanine favors primer extension in both enzymes. These DNA repair functions utilize conserved structural features of non-LTR-retroelement RTs, including human LINE-1 and other eukaryotic non-LTR-retrotransposon RTs, suggesting such enzymes may have inherent ability to function in DSBR in a wide range of organisms.
Keywords: Alt-EJ, alternative end joining, DNA repair polymerase, insect R2 element, high-throughput sequencing, non-retroviral reverse transcriptase, targetron, thermostable group II intron reverse transcriptase
Graphical Abstract
In-Brief:
DNA-repair roles for evolutionarily conserved reverse transcriptase families strengthen the link between genome stability and transposable element biology
INTRODUCTION
Reverse transcriptases (RTs) are best known for their crucial roles in the replication of human pathogens, such as retroviruses and hepatitis B virus, and as tools for biotechnological applications, such as high-throughput RNA sequencing (RNA-seq) and RT-qPCR (Martín-Alonso et al., 2021). However, RTs are found in all domains of life and are common in bacteria, where they are thought to have evolved from an RNA-dependent RNA polymerase (Lambowitz and Belfort, 2015). The most prevalent bacterial RTs are those encoded by mobile group II introns, retrotransposons that are evolutionary ancestors of spliceosomal introns and the spliceosome, as well as retrovirus and other retroelements in eukaryotes (Lambowitz and Zimmerly, 2011; Lambowitz and Belfort, 2015). Extant bacteria also harbor a variety of other RTs, all of which are closely related to group II intron RTs and some of which have been found to perform cellular functions. The latter include diversity-generating retroelement RTs, CRISPR-associated RTs, abortive phage infection RTs, and retron RTs, which were shown recently to function in phage defense systems (Liu et al., 2002; Wang et al., 2011; Silas et al., 2016; Gao et al., 2020; Millman et al., 2020). In addition to these characterized enzymes, bacteria contain families of unexplored group II intron-like RTs that are encoded by free-standing conserved ORFs in bacterial genomes and whose biochemical activities and biological functions remain unknown (Kojima and Kanehisa, 2008; Zimmerly and Wu, 2015).
Group II intron and other bacterial RTs belong to a larger family of non-LTR-retroelement RTs, which includes human LINE-1 and other eukaryotic non-LTR-retrotransposons RTs (Xiong and Eickbush, 1990). These non-LTR-retroelement RTs are homologous to retroviral RTs but have distinctive conserved structural features that impact RT activity, including an N-terminal extension (NTE) with an RT0 loop, two insertions, RT2a and RT3a, between universally conserved RT sequence blocks (RT1–7), and a larger thumb domain with three instead of two α-helices (Xiong and Eickbush, 1990; Blocker et al., 2005; Stamos et al., 2017; Figure 1A). A crystal structure of a full-length group II intron RT (Geobacillus stearothermophilus GsI-IIC RT) in complex with template-primer and incoming dNTP showed that group II intron RTs are similar to retroviral RTs in folding into a hand-like structure with fingers, palm, and thumb forming a cleft that binds the template-primer at the RT active site, but with the NTE/RT0 loop and RT2a insertions contributing to tighter binding pockets for the template/primer and incoming dNTP that could increase the fidelity and processivity of these enzymes (Mohr et al., 2013; Stamos et al., 2017; Figure 1B). The NTE/RT0 loop also plays a key role in a proficient group II intron RT template-switching activity that is dependent upon a short base-pairing interaction between the 3’ ends of the donor and acceptor nucleic acids (Mohr et al., 2013; Stamos et al., 2017; Lentzsch et al., 2019, 2021).
Figure 1. Characteristics of P. aeruginosa G2L4 RT.
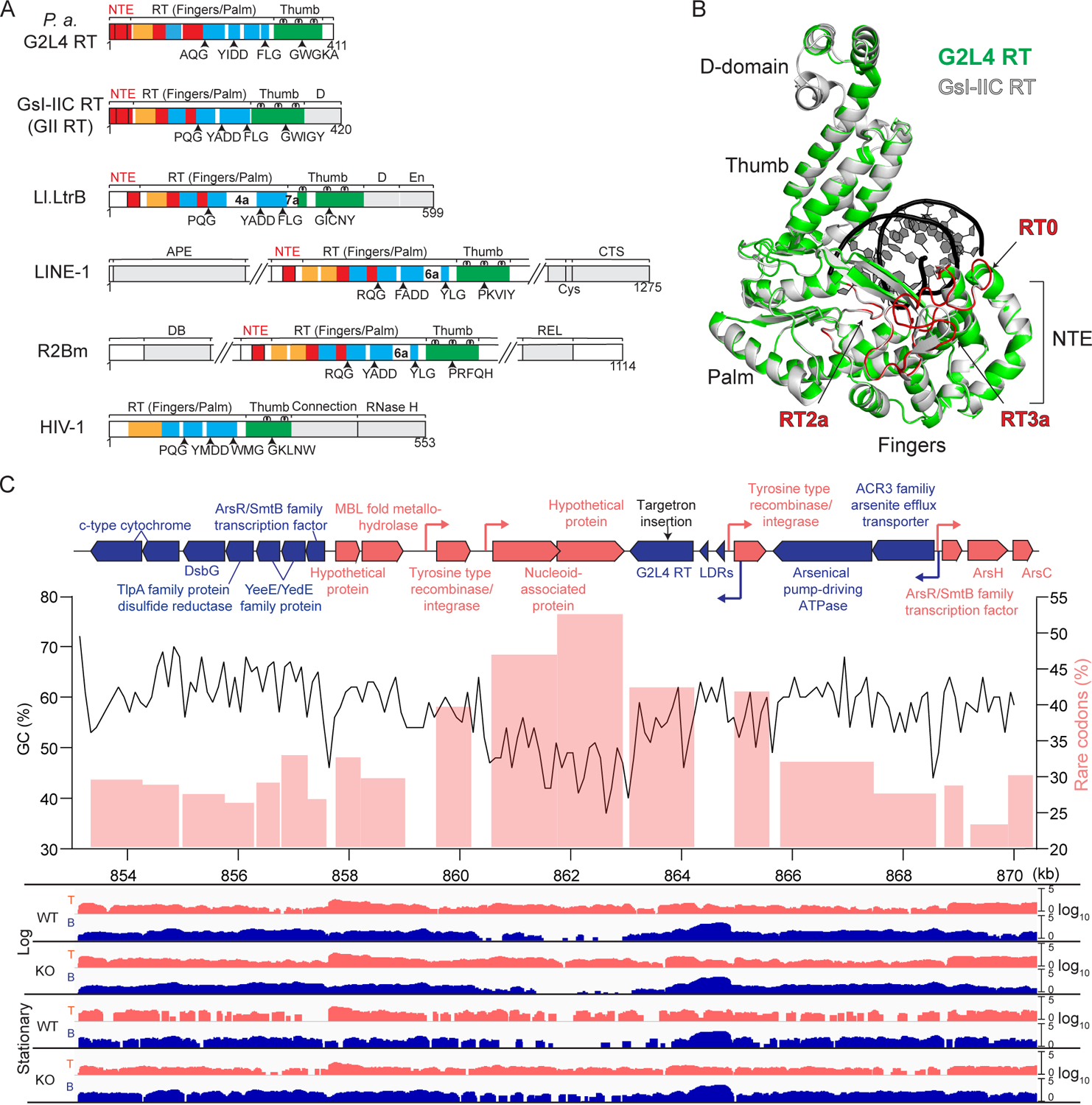
(A) Schematics comparing P. aeruginosa G2L4 RT to RTs encoded by group II introns G. stearothermophilus GsI-IIC (GII) and Lactococcus lactis Ll.LtrB (LtrA protein), human LINE-1 and Bombyx mori R2Bm non-LTR retrotransposons, and retrovirus HIV-1. Protein regions: RT1–7, conserved sequences blocks found in all RTs; NTE/RT0, RT2a and RT3a insertions relative to retroviral RTs (red); fingers (orange); palm (blue); thumb (green); other domains (gray).
(B) Three-dimensional model of G2L4 RT (green) constructed by I-TASSER (Yang and Zhang, 2015) superimposed on the crystal structure of GII RT (gray; PDB: 6AR1). Primer-template (black).
(C) Genomic region encompassing the G2L4 RT ORF in P. aeruginosa AZPAE12409. Top, map of a 17.2-kb region containing the G2L4 RT gene. Genes with protein-coding sequences on the top and bottom strand and their predicted promoters (bent arrows) are shown in red and blue, respectively. Targetron insertion site (black arrow). Middle, GC content calculated over a 500-bp sliding window (black line) and rare codon usage (pink bars) in regions around the G2L4 RT ORF. Bottom, TGIRT-seq coverage plots of cellular RNAs in the WT and G2L4 RT KO strains in log and stationary phases
All RTs have an active site containing a conserved F/YxDD motif, whose aspartates bind catalytic Mg2+ ions (Argos, 1988; Xiong and Eickbush, 1990). In group II intron RTs, this motif is typically F/YADD, with the conserved alanine part of a network of structural features that could impact fidelity and processivity (Stamos et al., 2017). However, in some families of bacterial RTs, this conserved alanine is replaced by a different conserved amino acid (I, V, M, H, S, or R; Zimmerly and Wu, 2015).
Here, we found via gene disruption and complementation that a Pseudomonas aeruginosa RT belonging to a family denoted Group II-Like 4 (G2L4; Zimmerly and Wu, 2015) with a conserved YIDD at its active site functions in DNA repair in its native host. Further analyses showed: (i) G2L4 RT can function in both translesion DNA synthesis and DSBR via MMEJ; (ii) a group II intron RT (GsI-IIC RT, denoted GII RT) with YADD at its active site can function similarly in DNA repair; (iii) an isoleucine at the active site favors MMEJ at the expense of primer extension; and (iv) the MMEJ activity of both enzymes is dependent upon the RT0 loop, a conserved structural feature of non-LTR-retroelement RTs. Our findings demonstrate that RTs have the previously unsuspected ability to function in DSBR and suggest that non-LTR-retroelement RTs may have an inherent ability do so in a wide range of organisms.
RESULTS
Characteristics of G2L4 RTs
A BLASTP search of GenBank identified 503 G2L4 RTs with 238 unique sequences in gram negative α, β, γ, and a few δ proteobacteria. Among the γ proteobacteria, many G2L4 RTs were found in Pseudomonas spp., and we focused on a member of this group (WP_034031052) found in P. aeruginosa strain AZPAE12409 (Kos et al., 2015). The genomic region encoding this G2L4 RT has the characteristics of a horizontally transferred genetic element, including lower GC content and genes whose codon usage differs from that of neighboring host genes (Figure 1C). Most G2L4 RT ORFs (376 of 503) in different bacteria were preceded by two palindromic ~140-bp long direct-repeats (LDRs) separated by an ~240-bp spacer, whose sequences were conserved in different strains (Figures 1C and S1A). Further, the G2L4 ORF and its upstream LDRs were inserted in different genomic regions in different Pseudomonas spp. strains, frequently in proximity to ORFs encoding putative tyrosine recombinases or other DNA integrases (Figure 1C), suggesting that G2L4 RT might be associated with an independently mobile genetic element.
Like group II intron and other non-LTR-retroelement RTs, G2L4 RT contains an NTE with a RT0 loop and RT2a and RT3a regions, which are absent in retroviral RTs (Figures 1A). The predicted secondary and tertiary structures of the G2L4 RT closely matched the known structure of GII RT, with the major differences being a longer RT3a insertion and small insertions downstream of RT6 and in the thumb domain (Figures 1B and S1B). The RT0 loop, which plays a crucial role in the template-switching activity of non-LTR-retroelement RTs (see above), is structurally similar to that of group II intron RTs but differs in having conserved serine residues (Figure S1C). G2L4 RTs also differ from group II intron RTs in lacking C-terminal DNA-binding (D) and DNA endonuclease (En) domains, which bind and cleave DNA target sites during group II intron retrohoming (Figure 1A; San Filippo and Lambowitz, 2002).
Analysis of G2L4 RT knock out strains
To investigate the function of the G2L4 RT in its native host, we used targetron mutagenesis (Yao and Lambowitz, 2007) to disrupt the G2L4 RT ORF in P. aeruginosa AZPAE12409. We obtained two G2L4 RT disruptants in which the targetron had inserted at the same site in the antisense orientation relative to the G2L4 ORF (Figures 1C, S2A, and S2B). Whole genome sequencing showed that one disruptant (KO2) had no other changes, while the other (KO1) had a single missense mutation in an ORF encoding cell division protein ZapA (Figure S2C; Supplemental File). The wild-type (WT) and both KO strains had similar growth rates through log and stationary phases in complete medium, indicating that G2L4 RT is not an essential gene in its native host (Figure S2D). We used the KO2 strain lacking the secondary mutation for further analysis.
To assess the effect of the G2L4 RT disruption on gene expression, we analyzed the transcriptomes of the G2L4 WT and KO strains in log (15 h) and stationary (30 h) phases by using TGIRT-seq, an RNA-seq method that enables simultaneous profiling of all RNA biotypes without size selection (Nottingham et al., 2016). In the resulting TGIRT-seq datasets, 70–80% of the reads mapped to protein-coding genes, with the remainder mapping to small non-coding RNAs (Table S1, Supplemental file). The most abundant sncRNAs were tRNAs, RNase P RNA, and tmRNA, which releases mRNAs from stalled ribosomes (Müller et al., 2021). In both the WT and KO strains, the proportion of tRNA reads decreased in stationary phase, while the proportion of tmRNA reads increased, consistent with its regulation by RpoS, a bacterial stress response sigma factor that is up regulated in stationary phase (Supplemental file; Himeno et al., 2014). Coverage plots showed relatively uniform shallow read depth on both strands over the G2L4 RT coding region, but with the bottom strand showing 20-fold higher read depth over the 140-bp LDRs preceding the RT (Figure 1C).
Volcano plots comparing the relative abundance of different RNAs in the WT and KO strains showed differences in both log and stationary phases, but with more differentially expressed genes and larger fold changes in stationary phase (Figure S2E, Supplemental File). A notable difference in the KO strain in log phase was the higher expression level of tRNAs that recognize rare codons, including those used in the G2L4 RT ORF (Figure S2E). This finding suggests that G2L4 RT disruption may activate a pathway that up regulates tRNAs recognizing rare codons, possibly part of a global stress response.
Among the differentially expressed protein-coding genes between log and stationary phase were three encoding transcriptional regulators. The gene encoding sigma factor AlgU, which induces osmotic, oxidative, and temperature stress responses, was expressed at higher levels in the KO than the WT strain in both log and stationary phases; the gene encoding sigma factor RpoS, which is induced in stationary phase for tolerance of high osmolarity, DNA damage, and oxidative stress, was up regulated in stationary phase in both the WT and KO strains; and the gene encoding the LexA repressor, a DNA damage sensor whose cleavage after interaction with RecA at double-strand breaks (DSBs) triggers an SOS response, was down regulated in stationary phase in both the WT and the KO strain (Figure 2A; Cirz et al., 2006; Schurr and Deretic, 1997, Jaishankar and Srivastava, 2017; Kreuzer, 2013). The altered expression levels of these three transcriptional regulators were correlated with the up regulation of pathways and genes related to DNA repair, oxidative stress and SOS responses in the KO compared to the WT strain in log and/or stationary phases, but most pronounced in the KO strain in stationary phase (Figures 2A, 2B, Table S2, Supplemental file). The exacerbated DNA damage responses overseen by the three transcriptional regulators (AlgU, RpoS, and LexA) in log and/or stationary phase in the KO strain suggested that G2L4 might function in DNA repair.
Figure 2. G2L4 and GII RT function in DNA repair in vivo.
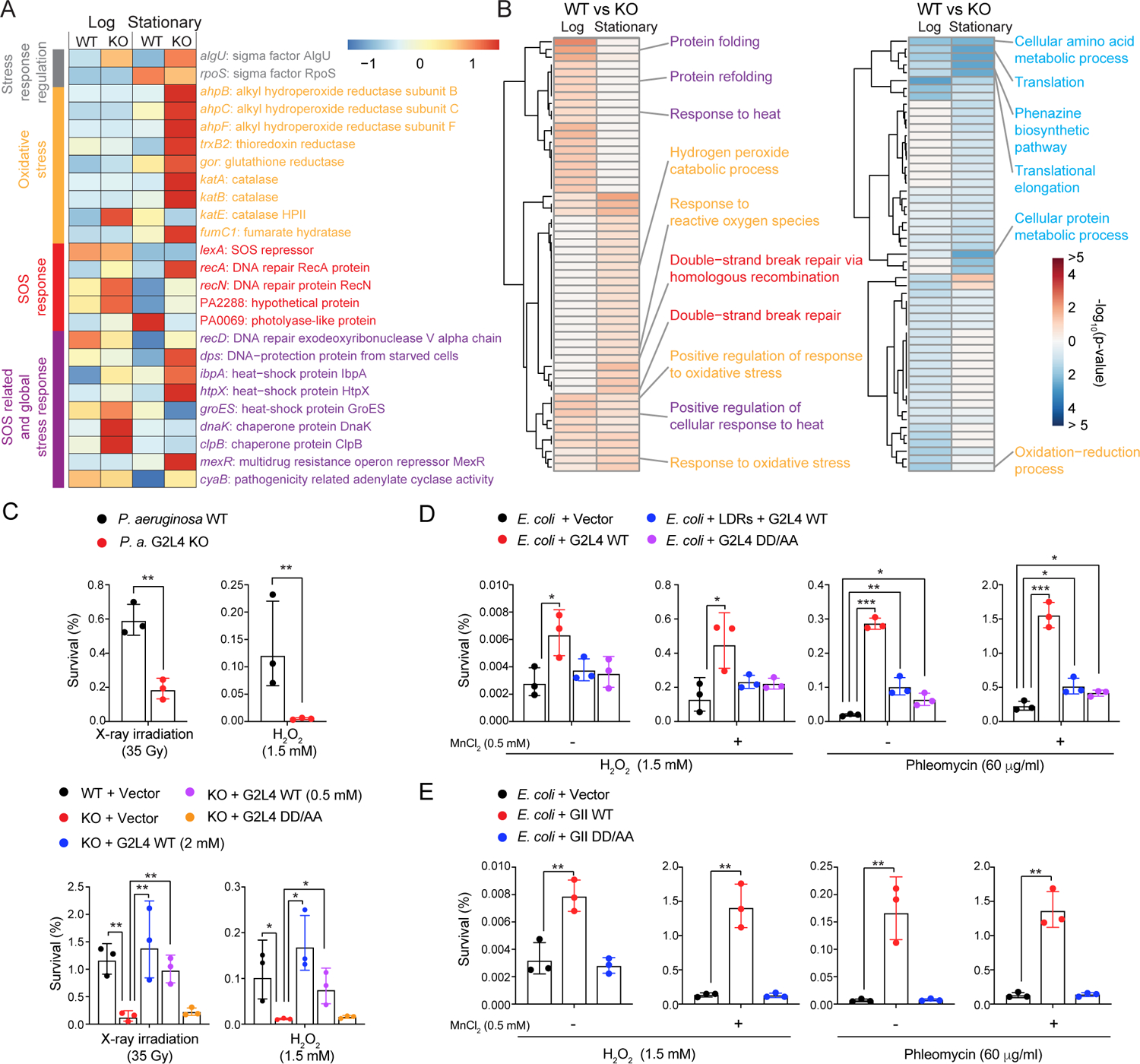
(A) Heatmap of significantly up or down regulated cellular stress and DNA damage response genes in G2L4 RT KO versus WT strains of P. aeruginosa AZPAE12409 in log and stationary phase. The color scale is based on the Z score for the mean of the sample-medians of the four replicates divided by the standard deviation of the sample-medians. Normalized read counts are listed in the Supplemental File.
(B) Heatmap of significantly enriched GO terms among up regulated (left, red) and down regulated (right, blue) genes in G2L4 RT KO versus WT strains in log and stationary phase. The color scale shows the −log10(p-value) of enriched GO terms for genes that were significantly up- or down regulated (log2FC>0 or FC<0, respectively, adjusted p-value ≤0.05). p-values for enriched GO terms are listed in the Supplemental File.
(C) Survival of P. aeruginosa AZPAE12409 WT and KO strains after X-ray irradiation or H2O2 treatment without (top) or with (bottom) expression of WT or RT-deficient mutant DD/AA G2L4 RTs. WT G2L4 RT was expressed at two different concentrations of m-toluic acid (0.5 and 2 mM) to vary the protein expression level.
(D) Survival of E. coli HMS174 (DE3) expressing WT and DD/AA mutant G2L4 RTs after treatment with H2O2 or phleomycin compared to a vector control.
(E) Survival of E. coli HMS174 (DE3) expressing WT and DD/AA mutant GII RTs after treatment with H2O2 or phleomycin compared to a vector control.
Analyses in panels A and B were based on four TGIRT-seq datasets for each strain and condition. Survival assays were repeated three times, with the error bars indicating the standard deviation. p-values <0.05, *; <0.01, **; <0.001, ***.
G2L4 RT functions in DNA repair in vivo
To investigate if G2L4 RT functions in DNA repair in its native host, we compared survival of the P. aeruginosa WT and G2L4 RT KO strains after inducing DNA damage and found that the KO strain was 3-fold more sensitive to X-ray irradiation, which causes DSBs, and 10-fold more sensitive to H2O2, which causes oxidative damage, including 8-oxoguanine and apurinic/apyrimidinic (AP) site lesions (Driessens et al., 2009; Poetsch, 2020; Figure 2C top). In both cases, survival was restored to at or near WT levels by expressing WT G2L4 RT from a plasmid using an m-toluic acid inducible promoter, but not by a vector control nor by an RT-deficient G2L4 RT in which the conserved aspartates at the RT active site were replaced with alanines (denoted DD/AA; Figure 2C bottom). By varying the concentration of the m-toluic acid inducer, we confirmed rescue after X-ray irradiation by WT G2L4 RT over a range of protein expression levels, including the lower expression level of the G2L4 RT DD/AA mutant protein (Figures 2C, S2F, S2G).
Similarly in E. coli HMS174 (DE3), WT G2L4 RT expressed from a plasmid increased resistance to both H2O2 and phleomycin, which induces DSBs (3- to 10-fold compared to the vector control; Figure 2D; Merrikh et al., 2009). As Mn2+ was known to enhance the activities of DNA repair polymerases (Hutfilz et al., 2019), we tested whether adding MnCl2 to the growth medium increased survival from DNA damage in E. coli expressing G2L4 RT and found this was the case (note different y-axis scales; Figure 2D). In E. coli, the ability of G2L4 RT to increase cell survival from H2O2 or phleomycin was strongly decreased but not completely abolished for the RT-deficient DD/AA mutant (Figure 2D). The latter finding indicates that the RT activity of G2L4 RT plays a major role in DNA damage survival, but leaves open the possibility that other activities of this protein may also contribute, as discussed further below. Co-expression of G2L4 RT with the upstream LDRs modulated its ability to increase cell survival from H2O2- and phleomycin-treatment in E. coli without decreasing the protein expression level (Figures 2D and S2H). Collectively, these results indicate that G2L4 RT functions in DNA repair in both its native host and E. coli.
G2L4 and GII RTs have robust DNA polymerase activity
To investigate if G2L4 RT has the enzymatic activities required for a DNA repair polymerase and how these activities compare to those of a group II intron RT, we carried out parallel biochemical assays with purified G2L4 and GII RT, a thermostable group II intron RT (TGIRT) that retains high activity at lower temperatures (Mohr et al., 2013). In addition to the WT enzymes, we tested both proteins with reciprocal I/A substitutions and DD/AA mutations at the RT active site. All of the purified proteins ran as a single major band in a Coomassie blue-stained gel (Figure S2I). Based on pilot experiments showing that G2L4 RT prefers low salt concentrations (Figures S3A–D) and the finding above that supplemental Mn2+ increased DNA damage survival in E. coli expressing G2L4 RT, the assays were done in reaction medium containing 20 mM NaCl and 10 mM MgCl2 at 37°C in the absence or presence of 1 mM MnCl2. These assay conditions are similar to those used for human DNA polymerase θ, which repairs double-strand breaks via MMEJ (Kent et al., 2015; Chandramouly et al., 2021).
First, we assayed the DNA polymerase and RT activities of G2L4 and GII RTs by primer extension using 3’-blocked 50-nt DNA or RNA oligonucleotide templates of identical sequence with different length DNA primers annealed at their 3’ ends. We found that WT G2L4 had high primer extension activity on both the DNA and RNA template with primers up to 5-nt long, but differed from GII RT in being unable to efficiently use primers ≥10 nt (Figures 3A–D). Time courses with the 5-nt DNA primer showed that WT G2L4 RT prefers DNA over RNA templates, with the rate of primer extension on both increased ~6-fold in the presence of Mn2+ (Figures S3E and S3F). Parallel assays showed that WT GII RT could efficiently use both short and long primers (Figures 3C and 3D), but time courses revealed some preference for shorter primers, particularly on the DNA template, and little effect of added Mn2+ with either template (Figures S3G and S3H). As expected, the primer extension activity of both enzymes was abolished by DD/AA mutations at the RT active site (Figure 3A–D).
Figure 3. Biochemical activities of WT and mutant G2L4 and GII RTs.
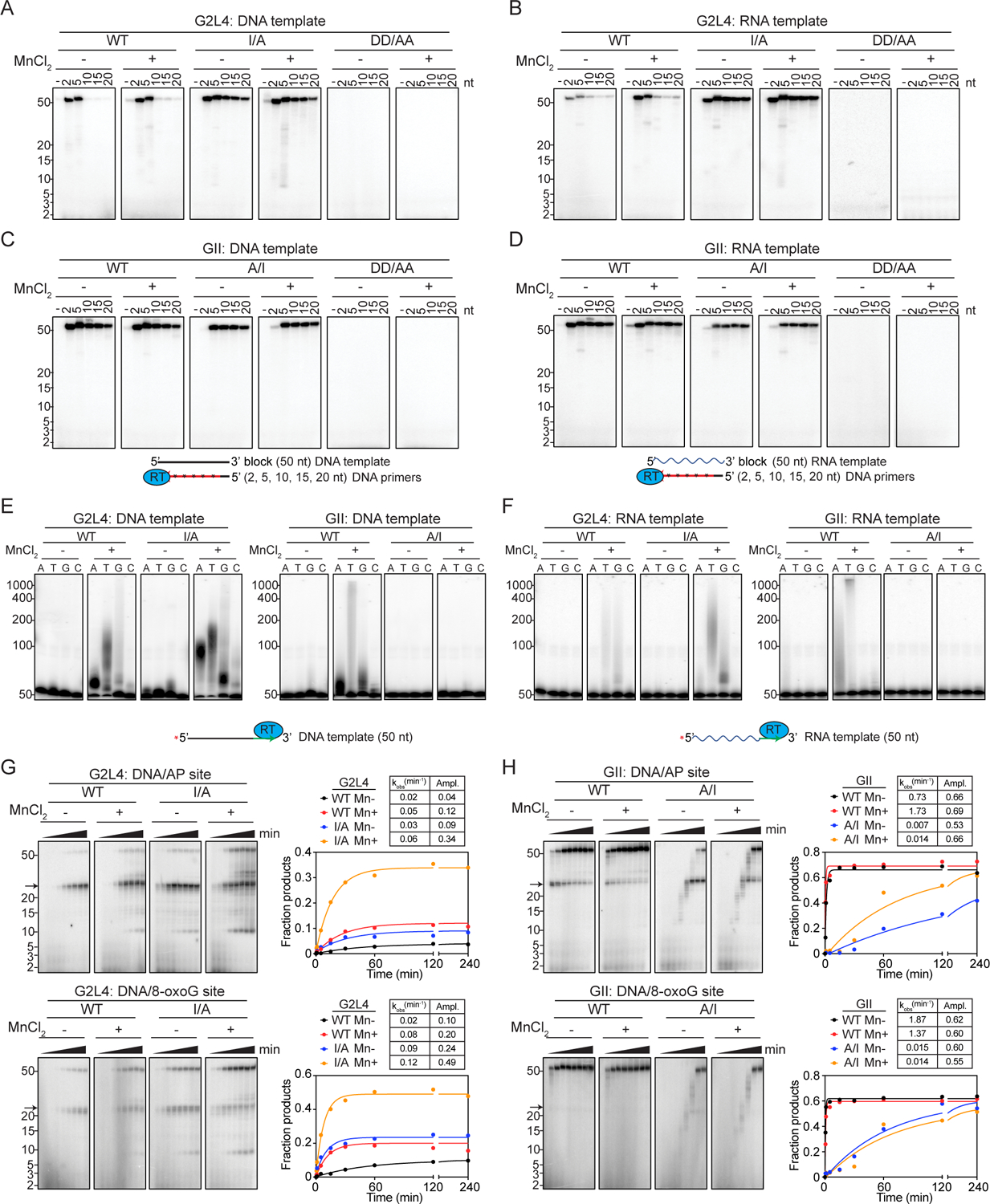
(A-D) Primer extension assays with 50-nt 3’-blocked DNA or RNA templates annealed to primers of various lengths. Reactions were initiated by adding 1 mM labeled dNTPs (1 mM each of dATP, dCTP, dGTP, and dTTP plus trace [α–32P]-dTTP) and incubated for 240 min at 37°C.
(E and F) Terminal transferase assays with 5’-labeled 50-nt DNA or RNA templates (see above) without a 3’-blocking group, and 1 mM of the indicated dNTP incubated for 20 min at 37°C.
(G and H) Translesion DNA synthesis time courses with 50-nt 3’-blocked DNA templates containing an AP site or 8-oxoguanine 23 nt from the 3’ end. The tables show rate constants (kobs) and amplitudes (Ampl.) for production of the labeled 50-nt DNA product obtained by fitting the data to a first-order rate equation.
The numbers to the left of the gels indicate the positions of size markers in a parallel lane.
Notably, I/A substitution at the G2L4 RT active site increased the rate of primer extension and alleviated the strict requirement of G2L4 RT for short primers, enabling more efficient use of primers up to 20 nt (Figures 3A, 3B; time courses Figures S3I, S3J). The mutant enzyme still preferred DNA over RNA templates, but added Mn2+ had less effect. By contrast, the reciprocal A/I substitution in GII RT decreased the rate of primer extension (Figures 3C, 3D; time courses Figures S3K, S3L). These findings indicate that the larger I residue at the active site of G2L4 RT dictates its strong preference for short DNA primers, while substitution of the smaller A residue enables use of longer primers and a higher rate of primer extension on both DNA and RNA templates. The finding that both G2L4 and GII RTs have robust DNA polymerase activity was expected for G2L4 RT functioning as a DNA repair polymerase and raised the possibility that the GII RT might also be capable of functioning as a DNA repair polymerase. Prompted by these biochemical assays, we confirmed that expression of WT GII RT in E. coli increased cell survival after treatment with H2O2 or phleomycin (3- to 20-fold compared to the vector control), with the ability to do so inhibited by DD/AA mutations at the RT active site (Figures 2E, S2J).
G2L4 and GII RT have Mn2+-stimulated terminal transferase activity
Human DNA repair polymerase θ, which functions in DSBR via MMEJ, has a Mn2+-dependent terminal transferase activity that extends single-stranded 3’-DNA overhangs at 5’-resected DSB sites until they can base pair with short complementary regions (microhomologies) in the 3’ overhang on the opposite side of the break (Kent et al., 2016). We found that WT G2L4 and GII RTs also have a Mn2+-stimulated terminal transferase activity with nucleotide preferences A=T>G>>C for G2L4 RT and with both enzymes preferring single-stranded (ss) DNA over RNA substrates (Figures 3E, 3F; time courses Figure S4). The G2L4 I/A mutation had little effect on terminal transferase activity, while the reciprocal A/I substitution in GII RT strongly inhibited this activity (Figures 3E, 3F).
G2L4 and GII RT read through DNA lesions
Human DNA polymerase θ has a translesion DNA synthesis activity that enables it to bypass DNA lesions in damaged DNA (Seki et al., 2004). To investigate if G2L4 and GII RTs have a similar activity, we did primer extension assays using the 50-nt DNA template containing lesions known to be induced by oxidative damage (8-oxoguanine or AP sites; Driessens et al., 2009; Poetsch, 2020), positioned 23 nt from its 3’ end. The reactions were done with a short 5-nt DNA primer, which can be used efficiently by both enzymes (see above).
We found that WT G2L4 RT was impeded by these lesions (pause site at ~23 nt; arrow), but could read through both types to produce full-length 50-nt DNA products with this ability strongest for the I/A mutant in the presence Mn2+, likely reflecting its higher primer extension activity (Figure 3G). WT GII RT, which has higher primer extension activity than G2L4 RT, was more efficient in reading though the lesion sites, as judged by the ratio of 50- to 23-nt bands, while the ability to give full-length product was much lower for the GII A/I mutant due to its low primer extension activity (Figure 3H). These findings indicate that both the G2L4 and GII RTs can read through damaged DNA templates with the ability of both enzymes to do so favored by an A residue at the active site, which enables higher primer extension activity.
G2L4 and GII RT promote snap-back DNA synthesis
Human DNA polymerase θ functions in DSBR by an error prone process (Alternative End Joining; Alt-EJ), which involves annealing microhomologies between single-stranded 3’ overhangs resulting from 5’-DNA strand resection on both sides of a double-strand break and then using the 3’ ends of the annealed strands as primers to fill in the single-stranded gaps (Black et al., 2016; Ramsden et al., 2022). The ability of DNA polymerase θ to anneal short microhomologies enables a distinctive biochemical activity termed snap-back DNA replication in which the enzyme uses the unblocked 3’ end of a DNA template as a primer to initiate DNA synthesis at short stretches of complementary nucleotides located farther upstream (Kent et al., 2016; Black et al., 2019).
To assay the snap-back replication activity of G2L4 and GII RTs, we used 5’-labeled 50-nt DNA and RNA oligonucleotides identical to those used above for primer extension assays but with unblocked 3’ ends and no added primer (Figures 4A, 4B). The products were analyzed in a non-denaturing 12% polyacrylamide gel, which makes it possible to distinguish double-stranded snap-back products from the single-stranded template and longer MMEJ products in MMEJ assays below (Kent et al., 2015).
Figure 4. Snap-back DNA synthesis by WT and mutant G2L4 and GII RTs.
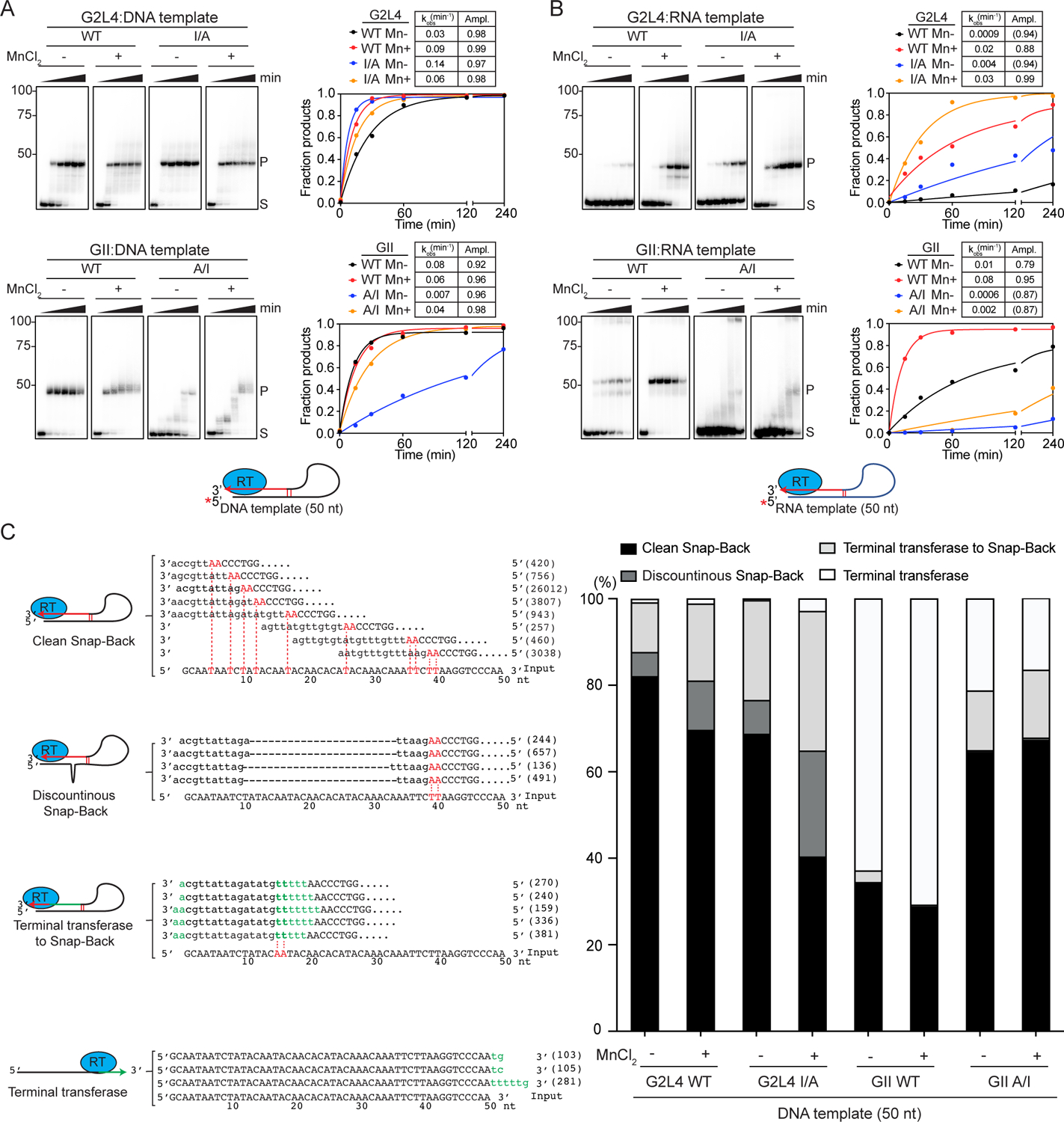
(A and B) Snap-back DNA synthesis assays using 5’-labeled 50-nt DNA or RNA templates with unblocked 3’ ends. The numbers to the left of gels indicate the positions of size markers in a parallel lane. The plots show the fraction of substrate (S) that was converted to products extending up to the major product band (P), and the tables show rate constants (kobs) and amplitudes (Ampl.) obtained by fitting the data to a first-order rate equation.
(C) High-throughput sequencing of snap-back DNA synthesis products. Schematics of different products along with sequences and read counts in parentheses for WT G2L4 RT without MnCl2 are shown to the left. Template and product nucleotides are in upper- and lower-case letters, respectively. Nucleotides involved in short base-pairing interactions between the 3’ AA and internal regions of the DNA template are in red, and nucleotides added by terminal transferase activity to the 3’ end of the template are in green. The stacked bar graphs show the proportions of different products in different samples.
The WT and I/A mutant G2L4 and GII RTs all gave products with electrophoretic mobility expected for snap-back DNA products, with a preference for the DNA over the RNA template (Figures 4A and 4B). WT G2L4 and GII RTs and G2L4 I/A mutant RT had robust snap-back DNA synthesis activities on the DNA template, while the GII A/I mutant RT had lower activity and gave intermediate size products at short time points, likely reflecting its decreased rate of primer extension. Mn2+ stimulated snap-back DNA synthesis on DNA and RNA templates in most cases, exceptions being WT GII and G2L4 I/A RTs, the two proteins with A at the active site, whose activity on DNA templates was not strongly affected by adding Mn2+ (Figures 4A and 4B).
We sequenced the products of snap-back replication on DNA substrates by using a TGIRT-based template switching method to obtain full-length DNA copies of the product flanked by Illumina adapter sequences. Based on the sequences, we classified the products into four categories, called Clean Snap Back, Discontinuous Snap Back, Terminal Transferase to Snap Back, and Terminal Transferase (Figure 4C). Clean Snap Back products were primed via base pairing between the 3’ A or AA residues of the template and upstream sites and continued uninterrupted to the 5’ end of the template. Remarkably, even a single A-T base pair was sufficient for priming. Discontinuous Snap Back and Terminal Transferase to Snap Back products were initiated similarly by the 3’ end of the template priming at short upstream microhomologies, but with the former having deletions due to the enzyme skipping over part of the template after the initial priming event and the latter containing non-templated nucleotides added by terminal transferase activity to the 3’ end of the template prior to annealing to upstream AA residues (Figure 4C). The remaining sequences contained only non-coded nucleotides added by terminal transferase activity to the 3’ end of the template without continuing to snap back DNA synthesis.
Comparison of the sequencing data for different proteins showed that the WT and I/A mutant G2L4 RTs produced the highest proportion of snap-back products (>95% of sequences), with WT G2L4 RT in the absence of Mn2+ giving the highest proportion of clean snap-back products and the I/A mutation or presence of Mn2+ decreasing the proportion of clean snap-back products (Figure 4C). WT GII RT produced fewer total and clean snap-back products on the DNA substrate, but the A/I substitution increased the proportions of both to levels close to those of WT G2L4 RT (Figure 4C). These findings show that both G2L4 and GII RTs have snap-back DNA synthesis activity, reflecting the ability to anneal and extend short microhomologies between the 3’ end and upstream regions of DNA templates and that this activity is favored by an I at the active site in both enzymes.
G2L4 and GII RT function in MMEJ in vitro
Next, we tested whether the G2L4 and GII RTs could perform MMEJ in a classical DSBR assay using 5’-labeled partially double-stranded DNA substrates with 15-nt single-stranded 3’ overhangs (Figure 5). This assay requires the annealing of short microhomologies at the 3’ ends of the single-stranded 3’ overhangs and then using the annealed 3’ ends as primers for DNA synthesis to fill in the resulting single-stranded (ss) DNA gaps (schematic Figure 5A bottom).
Figure. 5. MMEJ by WT and mutant G2L4 and GII RTs.
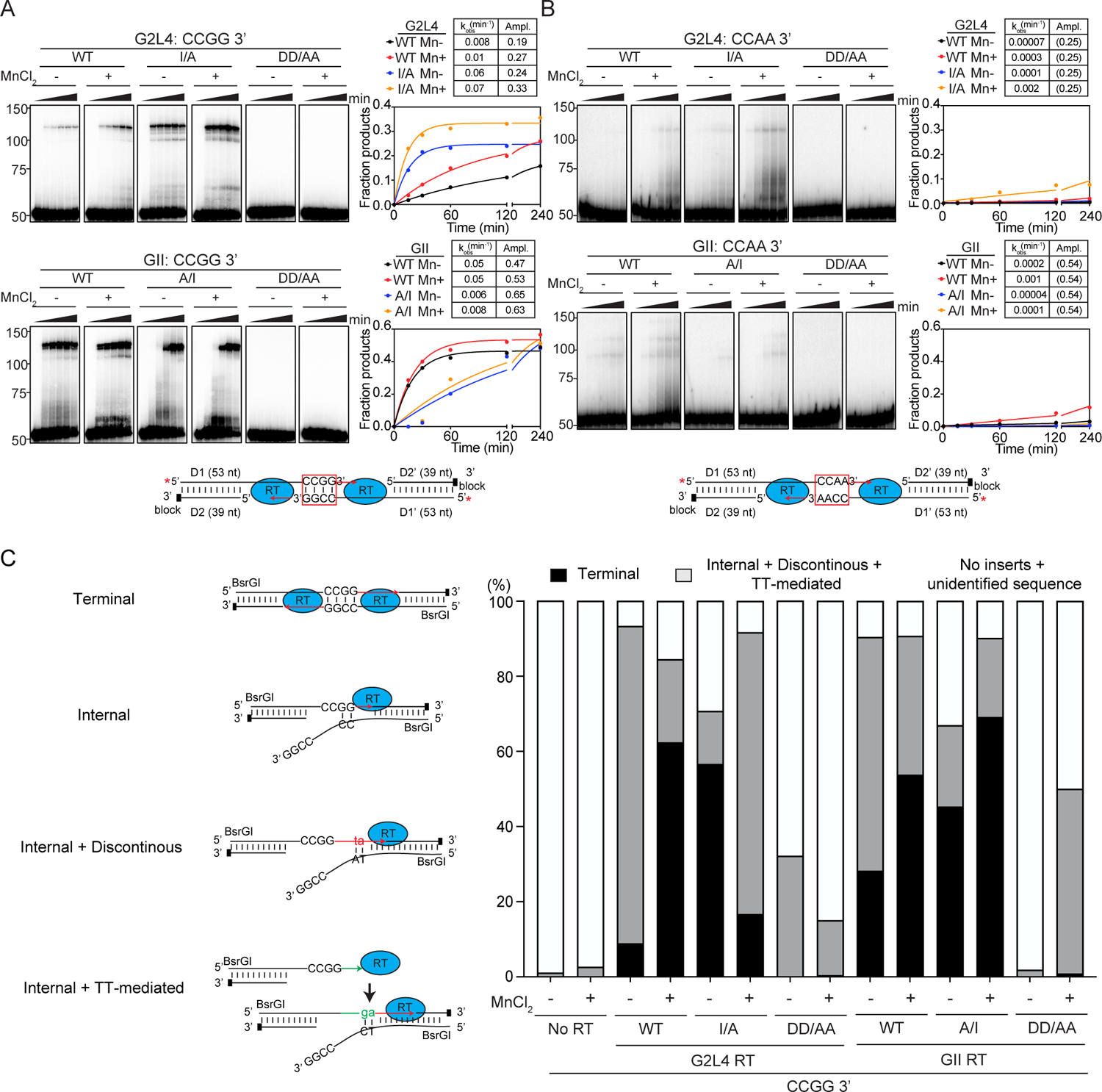
(A and B) Biochemical assays. Reactions were done with pre-annealed double-stranded DNAs having 3’ overhangs ending with complementary CCGG-3’ (panel A) or non-complementary CCAA-3’ (panel B) sequences. The pre-annealed oligonucleotides are denoted D1/D2 (left side) and D1’/D2’ (right side), with D1/D1’ 5’-labeled (red star) and D2/D2’ 3’-blocked. The numbers to the left of the gels indicate the positions of size markers in a parallel lane. The plots show the fraction of substrate converted to products running between the 100- and 150-nt size markers. Tables show rate constants (kobs) and amplitudes (Ampl.) obtained by fitting the data to a first-order rate equation. Values in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the experiment.
(C) High-throughput sequencing of MMEJ products with DNA substrates having CCGG-3’ microhomologies. Schematics of different products are shown to the left. Red and green indicate nucleotides at the 3’ end of nascent DNA or added by terminal transferase (TT) to the 3’ end of the nascent DNA, respectively. The stacked bar graphs show the proportions of different products in different samples.
In assays with DNA substrates having 3’ overhangs with complementary CCGG-3’ sequences, both the WT G2L4 and I/A mutant RTs gave products of the size expected for MMEJ (~102 bp, but running higher in the non-denaturing gel), with the rate of product formation higher for the mutant than the WT enzyme, and Mn2+ increasing activity of both enzymes (Figure 5A, top). The GII WT and A/I mutant RTs also gave MMEJ products with this substrate, but with the rate of product formation lower for the mutant than the WT enzyme (Figure 5A, bottom). The slower rates of product formation for G2L4 and GII A/I mutant RTs, the two enzymes with I at the active site, likely reflect that primer extension activity is rate-limiting after annealing of the microhomologies. Similar results were obtained with DNA substrates having 3’ overhangs with complementary TTAA-3’ sequences, but with lower rates and amplitudes and different effects of added Mn2+ (Figure S5A). By contrast, little or no product was seen for any of the proteins with DNA substrates having 3’ overhangs with non-complementary CCAA-3’ sequences, confirming the dependence of the activity on base pairing of complementary 3’ overhangs (Figure 5B). Human DNA polymerase θ prefers MMEJ substrates with a 5’ phosphate on the resected strand (Kent et al., 2015), but this was not the case for the G2L4 or GII RTs (Figure S5B). For both G2L4 and GII RTs, DD/AA mutations abolished detectable MMEJ product formation, indicating that primer extension is required to stabilize the products after annealing of the microhomologies (Figures 5A, 5B, S5A, S5B).
For high-throughput sequencing, we did MMEJ reactions with DNA substrates having 3’ overhangs with complementary CCGG-3’ sequences and cloned the resulting in vitro products in E. coli using BsrGI sites at the oligonucleotide termini to enable completion of partially single-stranded products. As observed in similar assays with DNA polymerase θ (Kent et al., 2016), the MMEJ products could be divided into two major categories: terminal (bidirectional) products (82–86 nt), resulting from annealing of the 3’ microhomologies and filling in the single-stranded gaps in both directions, and internal (unidirectional) products (<82 nt or >86 nt), resulting from annealing of the 3’ microhomologies and filling in the single-stranded gaps on one side only. The internal products included those with discontinuities in DNA synthesis, including recopying parts of the templates, or in which 3’ nucleotides added by terminal transferase were used to initiate internally within the 3’ overhang. WT G2L4 and GII A/I mutant RTs (the two enzymes with I at the active site) in the presence of Mn2+ were the combinations that gave the highest proportions of terminal MMEJ products (62–69%), followed closely by G2L4 I/A mutant RT in the absence of Mn2+ and WT GII RT in the presence of Mn2+ (Figure 5C). Notably, the DD/AA mutants of both enzymes gave no terminal MMEJ products, but did give internal MMEJ products above the levels of the No RT control, likely reflecting that the strand annealing activity of these enzymes can contribute to the formation of MMEJ products even in the absence of RT activity (Figure 5C).
Additional MMEJ assays showed that G2L4 and GII RT function similarly to DNA polymerase θ in binding to single-stranded regions preceding microhomologies (Kent et al., 2016; Black et al., 2019), with WT GII and the G2L4 I/A mutant RTs, the two enzymes with an A at the active site, better able to bind directly to and initiate DNA synthesis from longer annealed microhomologies (Figure S5C–E). The differing effects of I/A substitutions at the active sites of G2L4 and GII RTs may reflect a balance between their opposing effects on the strand annealing and primer extension activities required for MMEJ, with I favoring strand annealing and A compensating for less favorable strand annealing by enabling higher primer extension activity.
RT0 loop-dependent strand annealing contributes to MMEJ
The finding that G2L4 and GII RTs function in DSBR by annealing short microhomologies recalled that group II intron and other non-LTR-retroelement RTs have a proficient end-to-end template-switching activity that requires the annealing of short base-pairing interactions between the donor and acceptor nucleic acids (Bibillo and Eickbush, 2004; Mohr et al., 2013). Previous findings showed that this activity is dependent upon the RT0 loop, a distinctive conserved structural feature of non-LTR-retroelement RTs (see above), with deletions in the RT0 loop inhibiting the template-switching activity but not the primer extension activity of both GII and insect R2 element RTs (Jamburuthugoda and Eickbush, 2014; Stamos et al., 2017; Lentzsch et al., 2019). An X-ray crystal structure of a template-switching complex of GII RT revealed the structural basis for this activity by showing that the annealing of short base-pairing interactions between the donor and acceptor nucleic acids occurs in a binding pocket that is formed by the RT0 and fingertips loops and is absent in retroviral RTs (Lentzsch et al., 2021).
To investigate if RT0 loop-dependent strand annealing contributes to MMEJ, we constructed a G2L4 mutant (G2L4 ΔRT0) in which the RT0 loop was replaced with a glycine and compared its biochemical activities to those of the previously described GII RT ΔRT0 mutant. We found that the ΔRT0 mutants of both enzymes retained high primer extension activity on both DNA and RNA templates, with the ΔRT0 loop mutation surprisingly enabling G2L4 RT to use the longer 20-nt DNA primer almost as efficiently as the 5-nt DNA primer (Figures 6A, 6B, S6A, S6B; time courses Figures S6C, S6D). Notably, the RT0 loop deletion in both RTs strongly inhibited Mn2+-dependent terminal transferase activity, suggesting that this mutation affects the ability to bind the 3’ end of a ssDNA in a position to function as a primer at the active site (Figure 6C; time course gels Figures S6E, S6F).
Figure 6. Effect of deleting the RT0 loop on biochemical activities of G2L4 and GII RTs.
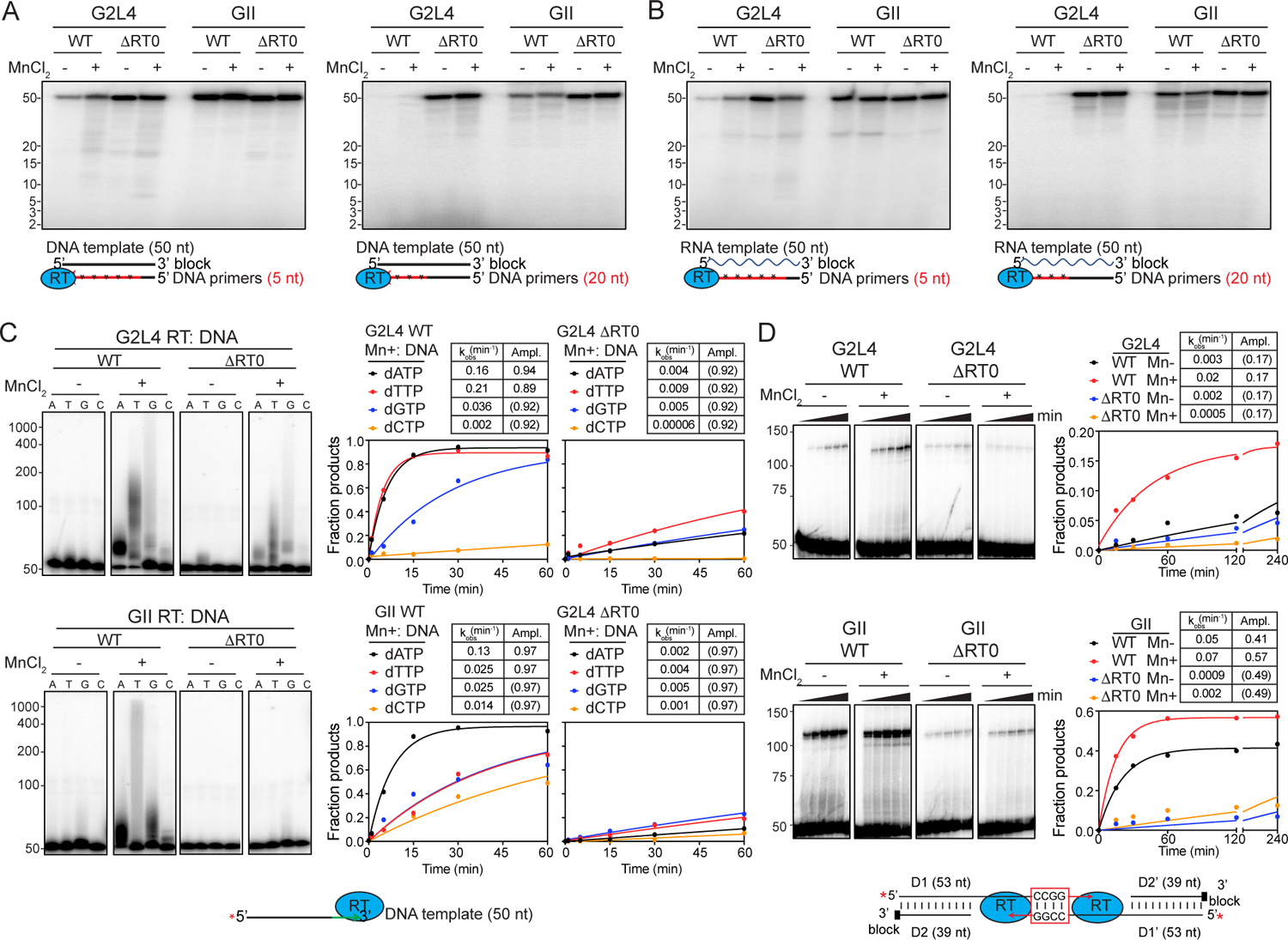
(A and B) Primer extension assays with 3’-blocked 50-nt DNA or RNA templates incubated for 20 min at 37°C.
(C) Terminal transferase assays. The gels (left) show a 20-min time point, and the plots (right) show time courses for gels shown in Figures S6E, S6F.
(D) MMEJ assays using DNA substrates with CCGG-3’ microhomologies.
The Tables shows kinetic paraments calculated as described in the legends of Figures 3 to 5. The numbers to the left of the gels indicate the positions of size markers in a parallel lane.
Because the ΔRT0 mutants retain high primer extension activity, MMEJ assays provide a means of assessing the contribution of the RT0 loop to the strand annealing activity used in MMEJ. For both enzymes, the ΔRT0 loop mutation strongly inhibited MMEJ (Figures 6D and S6G for CCGG-3’ and TTAA-3’ microhomologies, respectively), indicating that the presence of the RT0 loop is crucial for the strand annealing activity of both G2L4 and GII RTs.
G2L4 and GII RTs repair double-strand breaks in chromosomal DNA
Finally, to investigate how G2L4 and GII RTs repair DSBs in bacterial chromosomes, we used CRISPR/Cas9 (Chen et al., 2018) to introduce a targeted DSB in the E. coli thyA gene, which encodes thymidylate synthase and enables both positive and negative selections for thyA mutants (Figures 7A, S7A, S7B). As chromosomal DSBs are lethal in E. coli, we first tested whether the expression of WT or mutant G2L4 and GII RTs increases cell survival after co-expression of Cas9 and a single-stranded guide RNA (sgRNA) directed to introduce a DSB at a site within the thyA gene of E. coli HMS174 (DE3). The surviving bacteria were plated on medium containing thymine to enable growth of cells with thyA mutations.
Figure 7. Repair of CRISPR/Cas9-induced double-strand breaks in the E. coli thyA gene by G2L4 and GII RTs.
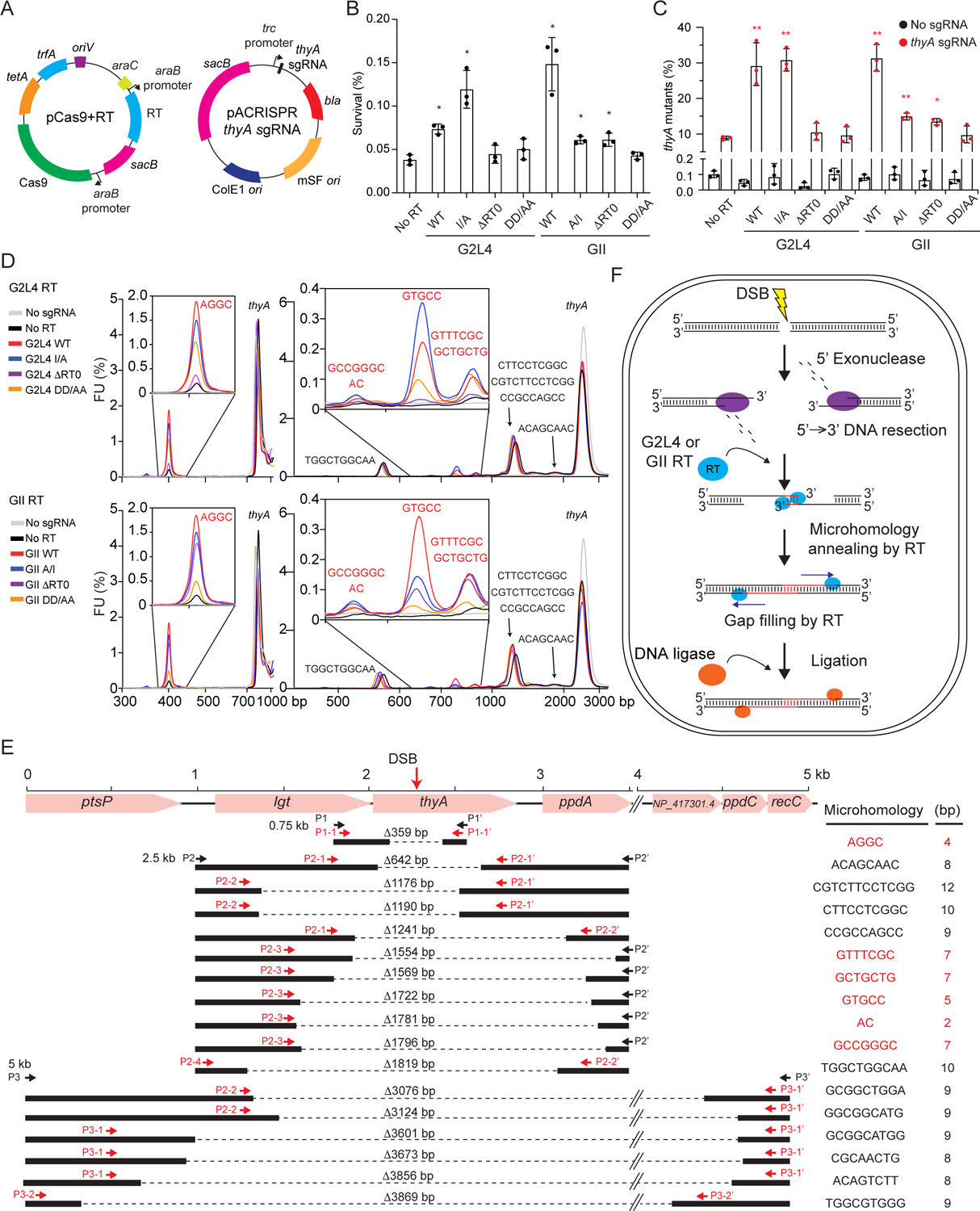
(A) Plasmids used in the experiment. pCas9+RT is a derivative of pCasPA that co-expresses Cas9 and G2L4 or GII RTs using arabinose-inducible araB promoters, and pACRISPR thyA sgRNA is a derivative of pACRISPR that expresses the thyA sgRNA from a constitutive trc promoter (Chen et al., 2018).
(B) Cell survival after CRISPR/Cas9-induced DSBs in the E. coli thyA gene with or without expression of WT of mutant G2L4 or GII RTs. Percent survival was measured in plating assays on medium containing thymine. p-values were calculated relative to the No RT vector control.
(C) Percentage of thyA mutants after repair of CRISPR/Cas9 induced DSBs. The mutation rate was measured as percent survival in plating assays on medium containing thymine + trimethoprim. p-values were calculated relative to the No RT vector control.
The bar graphs in panels B and C show average values for three repeats with the error bars indicating the standard deviation. p-values <0.05, *; <0.01, **.
(D) Bioanalyzer traces of PCR products obtained from genomic DNA using primers that amplify 750 bp (P1 and P1’, left) and 2.5-kb (P2 and P2’, right) regions around the DSB site in the thyA gene in cells expressing WT or mutant G2L4 (top) or GII (bottom) RTs compared to No RT and No guide RNA vector controls. Bioanalyzer traces were aligned via the peak corresponding to the full-length thyA gene.
(E) Sequences of MMEJ products resulting from DSBR in cells expressing WT G2L4 or GII RT and thyA sgRNA. Initial and nested PCR products obtained with the indicated primers (black and red arrows, respectively) were gel purified, cloned in E. coli HMS174 (DE3), and analyzed by Sanger sequencing (>10 clones for each sequence; Figure S7F and Table S3). Sequences of MMEJ junctions whose use was increased by WT G2L4 or GII RT expression are in red.
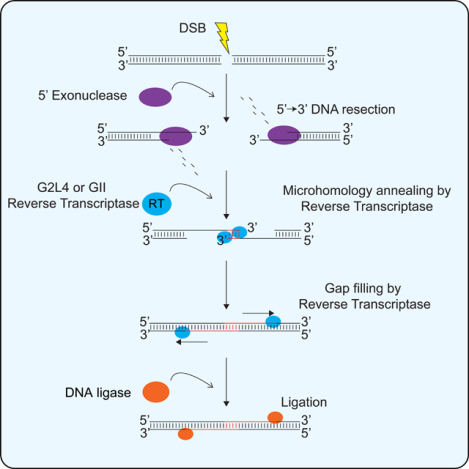
(F) Model for G2L4 and GII RT-mediated DSBR. After DNA cleavage resulting in a DSB, a 5’ to 3’ exonuclease (e.g., RecBCD, RecJ, or ExoVII; Lovett, 2011) resects the 5’ strand on opposite sides of the break, resulting in single-stranded 3’-DNA overhangs. G2L4 or GII RTs promote MMEJ by annealing microhomologies on opposite sides of the DSB and using the 3’ ends of the annealed strands as primers to fill in the single-stranded gaps. The annealed microhomologies are depicted as being at the 3’ end of the 3’ overhangs as in the MMEJ assays reported here, but could also be at internal positions within the 3’ overhangs.
The results paralleled those of previous genetic assays, with the frequency of surviving bacteria relative to the No RT control increased 2- to 3-fold by WT G2L4, WT GII, and G2L4 I/A RTs, marginally by GII A/I and ΔRT0 RTs, and not significantly above background by G2L4 ΔRT0 or G2L4 and GII DD/AA mutant RTs (Figure 7B). Reciprocally, plating the treated cells on medium containing thymine with added trimethoprim, which selects for thyA mutants, showed ~3-fold increased frequencies of thyA mutations relative to the No RT control in cells expressing WT G2L4, WT GII, and G2L4 I/A RTs, smaller increases for GII A/I and ΔRT0 RTs, and not significantly above background for the other mutant RTs (Figure 7C). All proteins were expressed at high levels, except G2L4 ΔRT0 and DD/AA mutants, possibly limiting the ability to detect low residual activity of these enzymes (Figure S7C). These findings show that expression of G2L4 and GII RTs increases cell survival after induction of a DSB and that the surviving cells have increased frequencies of thyA mutations, as expected for DNA repair by MMEJ.
To see if DSBR occurred by MMEJ, we PCR-amplified 0.75-, 2.5-, and 5-kb regions around the thyA gene DSB site from cells expressing WT or mutant G2L4 or GII RTs or vector controls with or without the thyA-directed sgRNA. Bioanalyzer traces showed that expression of the sgRNA resulted in a series of shorter PCR products expected for DSBR after DNA resection resulting in deletions of chromosomal DNA in and around the thyA gene (Figures 7D, 7E, S7D, S7E). The size distribution of the DSBR products was similar in all cases. However, the relative abundance of a subset of products was increased substantially by the expression of WT and I/A mutant G2L4 and WT GII RTs and to lesser extents but still above the No RT control for GII ΔRT0 and DD/AA mutant RTs, possibly reflecting residual strand-annealing activity of these proteins (see insets Figures 7D, S7E). The bioanalyzer profiles of the PCR products for each strain and condition and the RT-dependence of specific peaks were confirmed in a full repeat of the experiments starting with induction of the DSB.
To characterize the deletion junctions, we Sanger sequenced size-selected, gel-purified products from nested PCRs for WT G2L4 and GII RTs using primer pairs that enabled resolution and assignment of each peak (Fig S7F, Table S3). The results showed that the repaired DSBs had deletions of 359 to 3,869 bp encompassing the region targeted by the sgRNA and that all the deletions had junction sequences that mapped to short (2 to 12 nt) sequence duplications in the genome (Figures 7E, S7F). Notably, the subset of products enhanced by the G2L4 and GII RTs resulted from the annealing of short (2 to 7 nt) microhomologies on either side of the break (sequences in red), while those not enhanced by these enzymes resulted from the annealing of longer microhomologies (8–12 nt, sequences in black; Figures 7D, 7E, S7D–S7F, Table S3), the latter presumably resulting from DSBR by endogenous cellular enzymes. Collectively, these findings show that G2L4 and GII RTs function in repairing chromosomal DSBs by the MMEJ mechanism elucidated in the biochemical assays (Figure 7F).
Discussion
Here we found that G2L4 RT, a genomically encoded group II intron-like RT with a YIDD instead of YADD at its active site, functions in DNA repair in its native host and that a group II intron-encoded RT (GII RT), has innate ability to function in DNA repair. The DNA repair activities of these enzymes are remarkably similar to those of human DNA Pol θ, which has both DNA polymerase and limited RT activity, the ability to read through DNA lesions, a Mn2+-dependent terminal transferase activity that enables extension of 3’ ends in search of microhomologies, and the ability to anneal short (≤6 nt) microhomologies between 3’-DNA overhangs and use the annealed 3’ ends as primers to fill in the resulting single-stranded gaps (Seki et al., 2004; Black et al., 2016). The similarities to Pol θ extend to the ability to switch between templated and non-templated nucleotide addition during DNA synthesis, the ability to bind the 3’ terminus of ssDNA, and a requirement for a ssDNA region upstream of the annealed microhomology (Kent et al., 2016; Black et al., 2019). GII RT was somewhat better than G2L4 RT in being able to bind directly to and initiate DNA synthesis from longer annealed microhomologies, a difference governed largely by the I/A residues at the RT active site (Figures 3, S3, S5). When expressed in E. coli, both G2L4 and GII RT enhanced DSBR by MMEJ at chromosomal DNA sites with short microhomologies that were used inefficiently by endogenous cellular enzymes.
Group II intron RTs were shown previously to copy DNA templates, but typically prefer RNA templates in primer extension assays, likely reflecting a steric preference against initiating from B-form DNA template/DNA primer duplexes that fit poorly into the RT active site (Stamos et al., 2017). Similar steric preferences likely contribute to the findings that both G2L4 and GII RT prefer shorter more malleable DNA primers with that preference being particularly stringent for G2L4 RT, which was unable to efficiently initiate DNA synthesis from primers ≥10 nt (Figure 3). This more stringent preference for shorter primers was governed largely by the non-canonical I residue at the active site, with I/A substitution in G2L4 RT enabling it to use longer primers (Figure 3). Reciprocal substitutions in both enzymes showed that A at the active site enables a higher rate of primer extension while I at the active site decreases the rate but not the processivity or amplitude of primer extension (Figures 3, S3).
The ability of both G2L4 and GII RT to read through DNA lesions, such as abasic sites and 8-oxoguanine, was unsurprising in light of previous studies, which showed that group II intron RTs differ from retroviral RTs in their ability to read through and distinctively mis-incorporate at RNA post-transcriptional modifications that affect base pairing with the incoming dNTP, enabling GII RT (sold commercially as TGIRT-III) to be used for mapping base modifications in naturally occurring RNAs (Katibah et al., 2014) and DMS-induced modifications in RNA structure mapping (Zubradt et al., 2017). The findings that group II intron and group II intron-like RTs have robust DNA polymerase activity and perform a DNA-based function like DSBR suggest that additional biological functions and biotechnological applications of group II intron-related RTs may be on the horizon.
The RT0 loop was shown previously to play a key role in annealing short base-pairing interactions between the donor and acceptor nucleic acids in end-to-end template switching by group II intron and non-LTR-retrotransposon RTs (Jamburuthugoda and Eickbush, 2014; Stamos et al., 2017; Lentzsch et al., 2019). Our findings that deletions in the RT0 loops of G2L4 and GII RT inhibit MMEJ without inhibiting primer extension activity (Figures 6, 7) indicate that it also plays a role in strand annealing of microhomologies during MMEJ. Mechanistically, MMEJ and template switching are analogous in requiring the annealing of short microhomologies between two nucleic acid substrates and using the 3’ end of one of the annealed strands to prime DNA synthesis on the other. A difference, however, is that end-to-end template switching by group II intron RTs is optimal for annealing of a single base pair, while longer base-pairing interactions are inhibitory (Lentzsch et al., 2019), likely reflecting that the 3’ ends of the donor and acceptor nucleic acid bind after RT core closure in a tightly constrained binding pocket formed by the RT0 and fingertips loops (Lentzsch et al., 2021). By contrast, the annealing of longer microhomologies, such as those typically used for MMEJ, is more akin to the mechanism used for binding and annealing primers for primer extension, as evidenced by the findings that WT G2L4 and GII A/I RT with I at the active site favor the use of shorter primers and microhomologies, while GII and G2L4 I/A RTs with A at the active site can more efficiently use longer primers or microhomologies (Figures 3, S3, S5). Although the RT0 loop deletions that inhibited MMEJ by G2L4 and GII RTs did not inhibit primer extension activity, they did inhibit the terminal transferase activity of these enzymes (Figures 6, S6), suggesting that the RT0 loop may be required for binding the 3’ end of the priming strand at the RT active site, potentially a critical first step for strand annealing by these enzymes (Kent et al., 2015).
Although the DNA repair activities of GII RT appear to be as good or better than those of G2L4 RT, mobile group II introns RTs bind group II intron RNAs co-transcriptionally in order to promote RNA splicing and remain tightly bound to the excised the intron RNA to promote reverse splicing into DNA target sites during retrohoming (Saldanha et al., 1999; Gu et al., 2010). Some group II intron RTs can mobilize other group II intron RNAs in trans, indicating that they remain functionally active as free proteins and might contribute to DNA repair in their host cells (Lambowitz and Belfort, 2015). Other group II intron RTs, however, may be sequestered to at least some degree by binding to the intron RNA co-transcriptionally and thus impeded from functioning in DNA repair in their host cells.
A likely evolutionary scenario is that free-standing bacterial RTs that perform host functions evolved from the RT of a mobile group II intron that integrated into a bacterial genome and became immobilized by mutations in the intron RNA. Because group II intron mobility is deleterious to the host cell, mutations that immobilize the intron RNA are favored by purifying selection resulting in numerous examples of mobility-compromised group II introns that remain integrated in bacterial genomes (Robart and Zimmerly, 2005; Mohr et al., 2010; Leclercq and Cordaux, 2012). After acquiring a host function that contributes to cell survival, the RT would be subject to positive selection for additional mutations that enhance that function, with integration into a horizontally transferred genetic element, as found here for G2L4 RT (Figure 1C), facilitating dissemination of such beneficial enzymes to other bacteria.
In the case of G2L4 RT, the potential to function in DSBR may have pre-existed in the ancestral group II intron RT, with subsequent dissociation from the intron RNA enabling the protein to evolve to better perform that function with less constraint on biochemical activities required for intron mobility. Our results indicate that the substitution of I for A at the RT active site was a key adaptation that enabled G2L4 RT to better perform its host function in DSBR by favoring the strand annealing activity required for MMEJ. However, the finding that the reciprocal A/I substitution in GII RT inhibits primer extension activity indicates that additional changes in the protein were needed to accommodate the bulkier I at the active site with less effect on primer extension activity. Other group II intron-like bacterial RTs that evolved to perform host functions also have conserved substitutions in the F/YxDD motif at the RT active site, which may likewise enable them to better perform their host function, including M in diversity generating retroelement RTs associated with a higher frequency of nucleotide substitutions (Wu et al., 2018).
Finally, the close structural similarity between group II intron and other non-LTR-retroelement RTs and our finding that the MMEJ activity of G2L4 and GII RTs is dependent upon the RT0 loop, a distinctive conserved structural feature of non-LTR-retroelement RTs, suggest that non-LTR-retroelement RTs may have an inherent ability to function in DSBR in a wide range of organisms. That LINE-1 RT has the strand annealing activity required for MMEJ is indicated by previous findings of short microhomologies at the junctions of inversions and deletions that occurred during LINE-1 retrotransposition in cultured cells (Ostertag and Kazazian, 2001; Gilbert et al., 2005). A driving force for the evolution of DSBR activity in non-LTR-retroelement RTs is suggested by the finding that the ability of both a bacterial group II intron and human LINE-1 element to proliferate to higher copy numbers in bacterial cells correlates with the ability of the bacterial host strain to repair DSBs, which are a side product of the retromobility of these elements (Lee et al., 2018). Further, numerous previous findings have shown that human LINE-1 elements have a close personal relationship with DSBs, including inducing them during retrotransposition and contributing to RNA-mediated DSB repair by using both LINE-1 endonuclease-induced and spontaneous DSB sites for templated insertions of processed pseudogenes and other cDNAs (Esnault et al., 2000; Morrish et al., 2002, 2007; Onozawa et al., 2014). Our findings extend the previously known connections between LINE-1 elements and DSBs by suggesting that non-LTR-retrotransposon RTs may function not only in producing cDNAs that are integrated at DSBs, but may also play an active role in repairing DSBs by mechanisms similar to those elucidated here for G2L4 and GII RTs. In this way, human LINE-1 and other non-LTR retroelement RTs may not only mitigate damage caused by their retrotransposition, but may also provide a benefit to their host organisms in exchange for proliferating within their genomes.
Limitations of study
Our findings and others and from the literature suggest that MMEJ may be an inherent activity of LINE-1 and other non-LTR-retroelement RTs, but further studies are needed to demonstrate this directly. Our findings indicate that substitution of isoleucine for alanine in the YADD motif at the active site played a major role in adapting G2L4 RT to function in MMEJ by favoring strand annealing activity. However, additional as yet unidentified changes were likely needed to accommodate the larger isoleucine at the active site and adapt the enzyme to function more efficiently in DSBR. Finally, the contributions of host enzymes to G2L4 and GII RT-mediated DNA repair pathways and vice versa remain to be elucidated (e.g., by epistasis analysis).
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Alan M. Lambowitz (lambowitz@austin.utexas.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Datasets for P. aeruginosa whole-genome sequencing, TGIRT-seq, and sequencing of Snap-Back DNA synthesis and MMEJ products in biochemical experiments have been deposited in the Sequence Read Archive (SRA) under accession number PRJNA814398. A gene counts table, dataset metadata file, and scripts used for data processing and plotting have been deposited in GitHub: https://github.com/reykeryao/Seung. Unprocessed gel images, bioanalyzer traces, Sanger sequencing traces, and repeats of biochemical experiments have been deposited in Mendeley data, V3, doi: 10.17632/7dbyk67546.3.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
Pseudomonas aeruginosa AZPAE12409, which is naturally resistant to chloramphenicol (CapR), was obtained from Entasis Therapeutics (Kos et al., 2015). E. coli HMS174 (DE3) (F− recA1 hsdR(rK12− mK12+) Rif R (DE3)) was purchased from Novagen. E. coli S17.1 (recA pro hsdR RP42-Tc∷Mu-Km∷Tn7 integrated into the chromosome StrR; SpcR; TmpR) was purchased form ATCC. E. coli Rosetta 2 (F− ompT hsdSB(rB− mB−) gal dcm pRARE2 (CapR)) and Rosetta 2 (DE3) (F− ompT hsdSB(rB− mB−) gal dcm (DE3) pRARE2 (CapR)) were purchased from Novagen.
METHOD DETAILS
DNA and RNA oligonucleotides
The DNA and RNA oligonucleotides used in this study are listed in Table S4. All were purchased in RNase-free, HPLC or PAGE-purified form from Integrated DNA Technologies (IDT) or Dharmacon. Oligonucleotides were 5’-labeled with [γ–32P]-ATP (6,000 Ci/mmol; Perkin Elmer) by using T4 polynucleotide kinase (New England Biolabs) and cleaned up by using an Oligo Clean & Concentrator or RNA Clean & Concentrator Kit (Zymo Research), all according to the manufacturer’s protocols. Quantifications for labeled DNA and RNA oligonucleotides were performed by Qubit ssDNA and RNA Assay kit as manufacturer’s protocols respectively.
Recombinant plasmids
Recombinant plasmids used in this study are listed in Table S5. The targetron expression plasmid pBL1 is a derivative of the broad host range expression vector pJB866 (Blatny et al., 1997), which expresses the Ll.LtrB-ΔORF targetron using an m-toluic acid-inducible promoter and carries a TetR marker (Yao and Lambowitz, 2007).
pBL1-MCS is a derivative of pBL1 used as an intermediate in the construction of plasmids that express G2L4 and GII RTs in P. aeruginosa and E. coli. It was derived from pBL1 by replacing the 3-kb XhoI + KpnI fragment containing the targetron cassette with a 42-nt DNA segment containing a multi-cloning site (KpnI/SpeI/BamHI/HindIII/BsrGI/XhoI) oligonucleotide.
pBL1-MBP-8XHis or pBL1-MBP-RT-8XHis plasmids used for expressing WT and mutant G2L4 and GII RTs in P. aeruginosa and E. coli were constructed by PCR amplifying the RT ORFs from pMal-RT plasmids (see below) with primers that introduce flanking KpnI and SpeI sites and then cloning the resulting ~2.4-kb PCR products between the KpnI and SpeI sites of pBL1-MCS. The long direct repeat (LDR) region upstream of the G2L4 RT ORF in P. aeruginosa AZPAE12409 was inserted into pBL1-MBP-G2L4 RT-8XHis by PCR amplifying a 658-bp region of genomic DNA containing the LDRs with Gibson forward and reverse primers that append flanking KpnI sites and inserting the KpnI-digested PCR product into the KpnI site of pBL1-MBP-G2L4 RT-8XHis by using NEBuilder HiFi DNA Assembly (New England Biolabs) according to the manufacturer’s protocol. The proteins expressed from these plasmids have an N-terminal maltose-binding protein (MBP) tag, which stabilizes and increases the solubility of expressed group II intron RTs (Mohr et al., 2013), and a C-terminal 8XHis tag used for detection by immunoblotting.
pMal-RT plasmids used to express G2L4 in E. coli for protein purification were derivatives of pMal-c5X (New England Biolabs), which carries an AmpR marker and uses an IPTG-inducible tac promoter to expresses recombinant proteins with a factor Xa cleavable maltose-binding protein (MBP) tag. pMal-GII RT WT and GII RT 23–31/4G (denoted GII ΔRT0 RT) were described previously (Mohr et al., 2013; Stamos et al., 2017). pMal-G2L4 RT was constructed by cloning a G-Block (IDT) containing a codon-optimized G2L4 RT ORF flanked by HpaI and BamHI sites between the XmnI and BamHI sites of pMal-c5X. Other G2L4 and GII RT mutant plasmids were derived from pMal-G2L4 RT or pMal-GII RT by using a Q5 mutagenesis kit (New England Biolabs). In the G2L4 ΔRT0 RT mutant, amino acids 24 to 32 (corresponding to positions 23 to 31 in GII RT), were replaced by a glycine.
pKS-SacB used for cloning in vitro MMEJ products (Figure 5) is a derivative of pBluescriptII KS(+) (Agilent), which was constructed by PCR amplifying the sacB gene of pACRISPR (Addgene plasmid #113348; Chen et al., 2018) with SacB forward and reverse primers that introduce flanking EcoRV sites, and then cloning the resulting PCR Product into the EcoRV site of pBluescriptII KS(+).
pCas9+RT ORF plasmids used for CRISPR/Cas9 in vivo DSBR assays in E. coli were constructed via an intermediate plasmid pCas9SX derived by replacing the lambda red ORF of pCasPA (Addgene #113347; Chen et al., 2018) with a 47-bp DNA region with flanking SpeI and XbaI sites by using a Q5 mutagenesis kit (New England Biolabs). MBP-RT-8XHis ORFs were inserted into pCas9SX by PCR amplifying the pBL1-MBP-RT-8XHis ORFs with primers that introduce flanking SpeI and XbaI sites and cloning the PCR product between the corresponding sites of pCas9SX.
pACRISPR thyA sgRNA expressing plasmids were constructed as described (Chen et al., 2018). A sgRNA for the thyA gene was designed with on-line tools (http://chopchop.cbu.uib.no) and the corresponding DNA sequence was inserted into pACRISPR.
All insertions and PCR amplified regions of plasmids used in this study were confirmed by Sanger sequencing.
Bioinformatic analysis of G2L4 RT ORF in AZPAE12409 P. aeruginosa
At the outset of this study, we used the protein sequence of a G2L4 RT (ABB74237) from Nitrospira multiformis ATCC 25196 to search Genbank using BLASTP (Altschul et al., 1997) and identified >100 G2L4 RT ORFs in gram negative α, β, γ and a few δ proteobacteriales from which a G2L4 protein (WP_034031052) from P. aeruginosa strain AZPAE12409 was selected for further analysis. More recent Genbank searches revealed 238 unique G2L4 RT proteins and a total of 503 G2L4 RTs including identical proteins in different strains. Analysis of the genomic neighborhood of G2L4 RTs with MUMmer3 (Kurtz et al., 2004) revealed two ~140 bp direct repeats within 1-kb upstream of the G2L4 RT ORF in 75% (376/503) of the sequences (Supplemental File).
The GC content across the region of the P. aeruginosa AZPAE12409 genome containing the G2L4 RT ORF (Figure 1C) was calculated across a 500-bp sliding window by using a Python script. The number of rare codons in the G2L4 RT and neighboring ORFs was determined from the P. aeruginosa PAO1 codon table (https://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=208964) and defined as codons that constitute <1% of the codons. Promoters were predicted by using BacPP (bacterial promoter prediction, http://www.bacpp.bioinfoucs.com; de Avila E Silva et al., 2011). The secondary structure of G2L4 RT was predicted by using HHpred (https://toolkit.tuebingen.mpg.de/tools/hhpred).
Targetron gene knock-out of G2L4 RT in P. aeruginosa
Targetron disruption of the G2L4 RT ORF in P. aeruginosa AZPAE12409 was done by using the broad-host range targetron expression vector pBL1 with targetrons designed and constructed as described (https://sites.cns.utexas.edu/lambowitz/targetron-design; Yao and Lambowitz, 2007). pBL1-G2L4 targetron plasmids were transformed into E. coli S17.1 and introduced into P. aeruginosa AZPAE12409 via conjugation. For this purpose, the P. aeruginosa recipient and E. coli donor carrying the pBL1 TetR targetron construct were grown separately in 50-mL conical tubes (Sarstedt) containing 5-mL Luria Bertani (LB) medium with tetracycline (25 μg/mL) added for the E. coli culture and shaken (250 rpm) at 37°C until O.D.600 = 0.3–0.4. The P. aeruginosa and E. coli cultures were then mixed at a 1:10 ratio, and cells were collected by filtration on a 25-mm diameter membrane filter (0.45-μm pore size; Millipore). For conjugation, the membrane was placed on a LB agar plate for 3 h at room temperature and then transferred to 5-mL of LB medium in a 50-mL conical tube (Sarstedt) and vortexed vigorously to separate the conjugating cells. Aliquots were plated on a LB agar plate containing tetracycline (100 μg/mL) and chloramphenicol (25 μg/mL) to which the P. aeruginosa strain is naturally resistant and incubated at 37°C for 14–16 h to select P. aeruginosa colonies carrying the TetR targetron plasmid. A single P. aeruginosa colony containing the targetron plasmid was picked and grown in LB medium containing tetracycline (100 μg/mL) overnight at 37°C. The culture was then diluted 1:100 into 5-mL LB medium plus tetracycline (100 μg/mL) in a 50-mL conical tube (Sarstedt) and incubated at 37°C with shaking (250 rpm) until O.D.600 = 0.3–0.4, at which point 2 mM m-toluic acid was added to induce targetron expression. After incubating at 30°C without shaking overnight, cells were plated on LB agar containing tetracycline (100 μg/mL), and disruptants were identified by colony PCR using primers flanking the predicted targetron insertion site in the G2L4 RT ORF. Twelve colonies were picked of which two (KO1 and KO2) contained the targetron insertion. After curing the targetron expression plasmid by growing cells in LB medium without tetracycline, single targetron insertion at the desired site in the disruptants was confirmed by Southern hybridization and whole genome sequencing (see below).
Southern hybridization
Genomic DNA was isolated from WT and G2L4 knock-out P. aeruginosa AZPAE12409 by using a Quick-DNA Fungal/Bacterial Miniprep Kit (Zymo Research) according to the manufacturer’s protocol. The DNA was digested with PstI and EcoRI and run in a 1% agarose gel alongside a 1-kb Plus DNA Ladder (Invitrogen) that was 5’-labeled with [γ–32P]-ATP (6000 Ci/mmol; Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs). After electrophoresis, the gel was blotted onto an Amersham Hybond-XL (Cytiva) membrane by overnight capillary transfer. The membrane was washed 3 times with 25 mL 6X SSC, dried, and UV irradiated to cross-link the DNA to the membrane (120 mJ; Stratalinker UV Crosslinker 2400). Hybridization was done with a 5’-labeled targetron probe (200 bp PCR product obtained using G2L4 RT targetron probe primers; Table S4) in a hybridization tube with Amersham Rapid-hybridization Buffer (Cytiva) for 2.5 h at 60°C. After washing twice with 2X SSC plus 0.1 % SDS, the membrane was dried and scanned with a phosphorimager (Typhoon FLA 9500; GE Healthcare).
Genomic DNA sequence analysis of P. aeruginosa AZPAE12409 WT and G2L4 KO strains
Glycerol stocks of P. aeruginosa WT and G2L4 RT knock-out strains were inoculated into 5-mL LB medium in a 50-mL conical tube (Sarstedt) and incubated at 37°C for 16–18 h with shaking (200 rpm). The culture was then centrifuged at 4000 × g for 5 min, and genomic DNA was extracted by using a Monarch Genomic DNA Purification Kit (New England Biolabs) according to the manufacturer’s protocol. 1 μg of each genomic DNA was submitted to the Genome Sequencing and Analysis Facility (GSAF) at the University of Texas at Austin and sequencing libraries were prepared and sequenced on an Illumina MiSeq v2 instrument to obtain ~1 million 2 × 250 nt paired end reads per sample. Reads were mapped to a customized P. aeruginosa AZPAE12409 reference genome, which contains the targetron inserted at the designated location and the pBL1 vector used to express the targetron, using BWA with the default settings (Li and Durbin, 2010). The genomic DNA coverage was calculated as mean coverage of 500-bp bins along the genomic sequence and plotted using R. Variants were called using freeBayes on bam files from genomic alignment of the WT or KO dataset, with the following settings: --ploidy 1 --min-mapping-quality 30 --min-alternate-count 10 (Garrison and Marth, 2012). The statistical test of KO-specific variants (point mutations) against the WT was analyzed by VarScan (Koboldt et al., 2009).
P. aeruginosa growth curves
Glycerol stocks of P. aeruginosa WT and G2L4 RT knock-out strains were streaked on LB agar and incubated at 37°C overnight. The next day, a single colony was inoculated into 5-mL LB medium in a 50-mL conical tube (Sarstedt) and incubated at 37°C overnight with shaking (200 rpm). A 1-mL aliquot of the overnight culture was then added to 100-mL LB in a 250-mL Erlenmeyer flask and incubated at 37°C with shaking (200 rpm). 0.5-mL samples of P. aeruginosa WT and G2L4 RT KO cultures were collected every 6 h for up to 72 h, serially diluted, and plated on LB agar. The plates were incubated overnight at 37°C, and colonies were counted to calculate colony forming units (CFU) per mL.
TGIRT-seq of P. aeruginosa WT and G2L4 RT disruptant whole-cell RNAs
P. aeruginosa WT and G2L4 RT knock-out strains were grown as described above, and 500 μL samples were collected at 15 and 30 h corresponding to log and mid-stationary phase, respectively. Total cellular RNA was extracted by using a Monarch Total RNA Miniprep kit (New England Biolabs), and rRNA-depleted by using riboPOOL (siTOOLs biotech), both according to the manufacturer’s protocols. After clean-up using an RNA Clean & Concentrator Kit (Zymo Research), the RNA was fragmented at 95°C for 5 min by using a Next Magnesium RNA Fragmentation Module (New England Biolabs), and cleaned up by using a MinElute PCR Purification Kit (Qiagen). TGIRT-seq libraries were prepared as described (Xu et al., 2019, 2021), and a 1-μL aliquot was analyzed on an Agilent 2100 Bioanalyzer using a High Sensitivity DNA kit to assess quality and concentration. The TGIRT-seq libraries were sequenced via Illumina NextSeq500 to obtain ~20 million 2 × 75 nt paired end reads per sample at the University of Texas MD Anderson Cancer Center, Science Park. Datasets were obtained for four independent replicates for each strain and condition (Table S1).
Reads were mapped to both a P. aeruginosa AZPAE12409 reference genome, which was incomplete and computationally curated with only limited information about predicted genes, and to the model P. aeruginosa strain PAO1 reference genome, which was complete and had detailed gene annotation (Pseudomonas Genome Database; Winsor et al., 2011). For read mapping, Illumina TruSeq adapters and PCR primer sequences were trimmed from the reads with Cutadapt v3.2 (sequencing quality score cut-off at 20; p-value <0.01), and reads <15-nt after trimming were discarded (Martin, 2011). The processed reads were mapped separately to the reference genomes for each of the P. aeruginosa strains by using Bowtie 2 v2.2.5 with local alignment (settings: --local -N 1 -D 20 -L 20 -X 1000 --no-mixed --no-discordant) and intersected with P. aeruginosa PAO1 and AZPAE12409 gene annotations by BEDTools v2.29.2 (Langmead and Salzberg, 2012; Quinlan, 2014). Finally, gene counts from the two P. aeruginosa strains were combined by using a customized R script. If a read pair mapped only to a PAO1 or AZPAE12409 gene, the gene annotation of the mapped strain was used, but if a read pair mapped to both the PAO1 and AZPAE12409 strains, the more complete gene annotation of the PAO1 strain was used.
Differential gene expression was analyzed by using DESeq2 with p values calculated by the Wald test and adjusted by the Benjamini-Hochberg procedure (Love et al., 2014). Volcano plots were plotted using R. GO term enrichment analysis was done by using the goseq package in R, with p-values calculated by a hyper-geometric test. Heatmaps were plotted by using the pheatmap package in R (https://cran.r-project.org/package=pheatmap; Young et al., 2010). Coverage plots and read alignments were created by using Integrative Genomics Viewer v2.6.2 (IGV). Genes with >100 mapped reads were down-sampled to 100 mapped reads for visualization in IGV (Robinson et al., 2011). Authors acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing high performance computing resources that have contributed to the research results reported in this paper (URL: http://www.tacc.utexas.edu).
P. aeruginosa and E. coli cell survival assays
P. aeruginosa AZPAE12409 WT and KO strains, which had been electroporated with pBL1-MBP-RT-8XHis plasmids or vector controls, were plated on LB medium containing tetracycline (100 μg/mL) and incubated overnight at 37°C. A single colony was picked and grown in LB containing tetracycline (100 μg/mL) overnight with shaking (200 rpm) at 37°C. The culture was then diluted 1:100 into 5-mL LB with tetracycline (100 μg/mL) in a 50-mL conical tube (Sarstedt) and incubated at 37°C with shaking (200 rpm) until O.D.600 = 1.0, at which point G2L4 RT expression was induced with m-toluic acid (2 mM final) for 2 h with shaking (200 rpm) at 37°C. P. aeruginosa WT and G2L4 RT KO strains lacking pBL1 were grown in the same medium as pBL1-containing strains without tetracycline until O.D.600 = 0.5–0.6. The P. aeruginosa WT and G2L4 RT KO strains or strains expressing WT or mutant G2L4 RTs from plasmids (see above) were diluted at 1:100 ratio into M63 minimal medium (22 mM KH2PO4, 40 mM K2HPO4, 15 mM (NH4)2SO4) supplemented with 2 mM MgSO4, 0.2% glucose and 0.5% casamino acids for cell survival assays described below.
E. coli HMS174 (DE3) cells, which had been transformed with pBL1-MBP-RT-8XHis plasmids or vector controls, were processed similarly for cell survival assays except that tetracycline concentration was 25 μg/mL, protein expression was induced with m-toluic acid (2 mM final) at 18°C for 19–21 h with shaking (100 rpm), and after induction cells were diluted 1:100 in modified M9 minimal medium (33.7 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 9.35 mM NH4Cl) supplemented with 0.4 % glucose, 2 mM MgSO4, 2 mM MgCl2, 0.1 mM CaCl2, 1 μg/mL thiamine, 2 mM m-toluic acid with or without 0.5 mM MnCl2.
For X-ray irradiation assays, 0.5 mL of the P. aeruginosa cells that had been diluted into M63 minimal medium were pipetted into single wells in a 24-well plate (Falcon) and exposed to 35 Gy X-rays using a 43855D RX-650 X-Ray Generator (Faxitron) according to the manufacturer’s protocol, while a second control plate containing P. aeruginosa cells was not irradiated. The X-ray irradiated and non-irradiated control cells were serially diluted in M63 medium and plated on LB agar plates for P. aeruginosa WT and G2L4 RT KO strains or LB agar plates containing tetracycline (100 μg/mL) for P. aeruginosa strains containing pBL1-MBP-RT-8XHis plasmids expressing WT or mutant G2L4 RTs.
For chemical cell survival assays, 1-mL of P. aeruginosa or E. coli cells diluted as above into M63 and M9 minimal medium, respectively, were incubated with or without 1.5 mM hydrogen peroxide (Sigma-Aldrich) or 60 μg/mL phleomycin (InvivoGen) in 15-mL tubes (Sarstedt) for 1.5 h at 37°C with shaking (250 rpm). The cells were then serially diluted into minimal medium (M63 for P. aeruginosa and M9 for E. coli), and plated on LB agar plates or LB agar plates containing tetracycline (100 μg/mL for P. aeruginosa or 25 μg/mL for E. coli). After overnight incubation at 37°C, colonies were counted, and survival determined as the proportion of colonies surviving after DNA damage compared to untreated controls. p-values were calculated by student’s unpaired t-test in Prism 9.0 (GraphPad Software).
Immunoblotting
Immunoblot analysis was done with parallel cultures to those used for cell survival assays. Instead of diluting into minimal medium for the cell survival assays, 5 mL of P. aeruginosa or E. coli HMS174 (DE3) cells expressing wild-type and mutant G2L4 and GII RTs in LB medium were centrifuged at 4°C, 4,000 × g for 10 min, and the pellets were lysed by resuspending in 300-μL of lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.1% Triton X-100 and 20% glycerol). The lysed cells were transferred to a 1.5-mL microcentrifuge tube and sonicated three times for 5 sec at 30% amplitude using a Branson Sonifier 250 (Branson Ultrasonics) followed by centrifugation at 4°C, 15,500 × g for 15 min. Protein concentrations in the lysates were measured with a Quick Start Bradford Protein Assay Kit (Bio-Rad) and a SmartSpec Plus Spectrophotometer (Bio-Rad) according to manufacturer’s protocols, and 75 μL of the supernatant was transferred to a 1.5-mL tube, mixed with 25 μL 4X sample buffer (200 mM Tris-HCl pH 6.8, 400 mM DTT, 8 % SDS, 6 mM bromophenol blue, 40% glycerol), and incubated at 95°C for 5 min. Protein samples (50 μg) and Color Prestained Protein Standard, Broad Range ladder (10–250 kDa; New England Biolabs) were loaded on a NuPAGE 4–12 % Bis-Tris gel, and electrophoresis was done in 1X MES running buffer (Thermo Fisher Scientific) at 150 V for 1 h by using an XCell Surelock Electrophoresis Cell according to the manufacturer’s protocol. For membrane transfer, an Immuno-Blot PVDF Membrane (Bio-Rad) was pre-soaked for 30 sec in 100% methanol, and membrane transfer was performed in 1X NuPAGE Transfer Buffer (Thermo Fisher Scientific) by using a Xcell II Blot Module according to the manufacturer’s protocol. The membrane was blocked by incubating in 15 mL of blocking solution (5% Blotting Grade Blocker Non-Fat Dry Milk; Bio-Rad) in 1X TBS-T (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20) in a rectangular tray at 4°C on an orbital shaker (55 rpm) for 1 h. For primary antibody treatment, 15 mL 6x His-Tag Monoclonal antibody (MA1–21315; Invitrogen) diluted 1:1500 in blocking solution supplemented with 0.1% NaN3 was added to the membrane and incubated at 4°C on an orbital shaker at 55 rpm for 14–16 h. The membrane was then washed three times with 15 mL 1X TBS-T with shaking (55 rpm) for 10 min at room temperature. For secondary antibody treatment, the membrane was incubated with Donkey anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP (SA1–100; Invitrogen) diluted 1:5000 in 1X TBS-T with shaking (55 rpm) for 1 h at room temperature, followed by three washes with 1X TBS-T. The antibody-treated membrane was then incubated with 15 mL Clarity Western ECL Substrate (Bio-Rad) at room temperature with shaking (55 rpm) for 5 min and exposed to CL-Xposure Film (Thermo Fisher Scientific), which was then developed with an X-ray film processor (Konica Minolta SRX-101A).
Protein purification
Recombinant proteins used for biochemical assays were expressed from pMal-RT plasmids (see above and Table S5). For each protein preparation, E. coli Rossetta2 CapR cells (Novagen) containing freshly transformed expression constructs were plated on LB agar containing ampicillin (100 μg/mL) and chloramphenicol (25 μg/mL) and incubated at 37°C for 14–16 h. A single colony was inoculated into 20 mL of LB containing the same antibiotics and incubated in a 50-mL conical tube (Sarstedt) at 37 °C with shaking (250 rpm) for 14–16 h, then diluted 1:50 into 1 L of LB with the same antibiotics in a 4 L Erlenmeyer flask and incubated at 37°C with shaking (220 rpm) until the O.D.600 reached 0.8–1.0. Protein expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) (100 μM for G2L4 RT constructs and 1 mM for GII RT constructs), followed by incubation at 18°C with shaking (100 rpm) for 19–21 hr. Bacteria were harvested at 4,000 × g for 25 min in a JLA-8.1000 rotor in an Avanti J-E centrifuge (Beckman), and the pellet was transferred to a 50-mL conical tube (Sarstedt) and resuspended in 45 mL of lysis buffer containing 20 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.1% Triton X-100 and 20% glycerol. The resuspended cells were sonicated on ice at 80% amplitude for three 1 min intervals with 1 min pause between bursts using a Branson Sonifier 250 (Branson Ultrasonics) followed by centrifugation at 15,500 × g for 25 min in a JA 25.50 rotor in an Avanti J-E centrifuge (Beckman). The supernatant was transferred to a 50-mL conical tube, and polyethyleneimine (final concentration of 0.04%) was added, mixed by inverting 2–3 times, and incubated on ice for 10 min to precipitate nucleic acids. Precipitates were removed by centrifugation as above, and the supernatant was filtered through a 0.2-μm poresize nylon membrane (Fisher). The filtrate was loaded onto a 5-mL MBPTrap HP column (Cytiva) at a flow rate of 5 mL/min using an ÅKTA START FPLC (Cytiva). The column was washed with 5 column volumes of buffer A (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.1% β-mercaptoethanol and 10% glycerol) followed by 5 column volumes of buffer B (20 mM Tris-HCl pH 7.5, 1.5 M NaCl, 0.1% β-mercaptoethanol, 10% glycerol), and then again with 5 column volumes of buffer A. The protein was eluted from the column with 10 column volumes of elution buffer (buffer A plus 10 mM maltose). 2 mL fractions were collected. 15-μL of each column fraction was mixed with 5-μL 4X sample buffer and loaded on a NuPAGE 4–12% Bis-Tris gel and gel electrophoresis was performed as described above for Immunoblotting. The gel was stained with 0.25% Coomassie brilliant blue R (Sigma-Aldrich) to identify recombinant proteins. Peak fractions were pooled and loaded onto a 5-mL HiTrap Heparin HP column (Cytiva) at a flow rate of 5 mL/min using an ÅKTA START FPLC (Cytiva). The column was washed with 5 column volumes of buffer A. Bound proteins were eluted using 10 column volumes of a 0.1 to 1.5 M NaCl gradient collecting 2 mL fractions. Column fractions containing the protein were identified by SDS-PAGE and Coomassie staining (see above). Fractions containing protein were pooled and concentrated to 10 μM into storage buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 50% glycerol for G2L4 RTs and 20 mM Tris-HCl pH 7.5, 500 mM KCl, 50% glycerol for GII RTs) with an Amicon Ultra-15 (30k) concentrator (Millipore) according to the manufacturer’s protocol.
Primer extension assays
The templates for primer extension assays were 50-nt DNA or RNA oligonucleotides (Table S4) with 3’-ends blocked by an inverted dT residue (IDT). Templates were pre-annealed to DNA primers of different lengths (Table S4) by mixing 1 μM template with 400 μM of 2 nt, 200 μM of 5 nt DNA primers or 2 μM of 10, 15 or 20 nt DNA primers in 100 μL of TE (10 mM Tris-HCl pH 7.5, 1 mM EDTA) and heating to 95°C for 3 min followed by cooling to 25°C at 0.1°C/min in a T100 thermal cycler (Bio-Rad). The assays were performed either as time courses (up to 240 min) in 80 μL of reaction medium or as single time points (20 min) in 20 μL of reaction medium containing 500 nM WT or mutant G2L4 or GII RT, 250 nM template-primer complex (unless indicated otherwise), 20 mM NaCl, 10 mM MgCl2, and 20 mM Tris-HCl pH 7.5, with or without 1 mM MnCl2. After pre-incubating the RT with the annealed template-primer substrate for 30 min at room temperature, the reactions were initiated by adding 1 mM dNTPs (1 mM each of dATP, dCTP, dGTP and dTTP) plus 1 μCi [α–32P]-dTTP (3,000 Ci/mmol; Perkin Elmer) and incubated at 37°C for times indicated in Figure Legends for individual experiments. For time courses, aliquots (10 μL) were removed at each time point and quenched by adding 2 μL of 6X stop solution (25 mM EDTA (Sigma-Aldrich), 0.5 U/μL proteinase K (New England Biolabs)) and incubating for 15 min at 37°C. The samples were then mixed with an equal volume of 2X loading dye (95% formamide, 0.25% SDS, 25 mM EDTA and 0.1% xylene cyanole and bromophenol blue), and analyzed by electrophoresis in a denaturing 20% polyacrylamide gel, with 5’-labeled DNA primer and template oligonucleotides as size markers. The gel was scanned with a phosphorimager (Typhoon FLA 9500; GE Healthcare), and the scanned image was processed with ImageJ. The amount of labeled dTTP incorporated into the product was determined by quantitating bands using ImageQuant TL 8.1. In order to account for the size difference of the primer extension products generated by primers of different length from the same template, the amount of label was multiplied by the dTTP concentration and divided by the number of T residues per extension product to determine the concentration of extended product, which was then plotted as a fraction relative to the template concentration. Time course data were fit to a first-order rate equation using Prism 9.0. For reactions that were slow and progressed approximately linearly during the observation time, such that the reaction end point could not be determined from the data, the fitting to a first-order rate equation was done with the end point (reaction amplitude) forced to the same value as for corresponding reactions that had defined end points. Amplitude values that were forced during fitting are indicated by parentheses in the tables next to the corresponding plots.
Terminal transferase assays
The substrates for terminal transferase assays were the same 50-nt DNA or RNA oligonucleotides used as templates in primer extension assays (Table S4) but without the 3’-blocking group. Terminal transferase assays were done either as time courses (up to 60 min) in 80 μL of reaction medium or as single time points (20 min) in 20-μL of reaction medium. The RT (500 nM) was preincubated with 5’-labeled oligonucleotide substrate (10 nM) in reaction medium containing 20 mM NaCl, 10 mM MgCl2, and 20 mM Tris-HCl pH 7.5 with or without 1 mM MnCl2 for 30 min at room temperature, and the reaction was initiated by adding 1 mM of a single dNTP (final concentration). The reactions were incubated at 37°C for times indicated for individual experiments, and quenched as described above for primer extension assay. The products were analyzed in a denaturing 6% polyacrylamide gel with a 5’-labeled RiboRuler Low Range RNA Ladder (Thermo Scientific) as size markers, and the gel was dried and scanned with a phosphorimager (Typhoon FLA 9500; GE Healthcare), and the scanned image was processed by ImageJ. The products were quantified with ImageQuant TL 8.1, and the data were analyzed as described above for primer extension assays.
Snap-back replication assays
The substrates for snap-back replication assays were 50-nt DNA or RNA oligonucleotides (see Table S4) without 3’ blockers. Snap-back replication assays were done as time courses by preincubating 10 nM 5’-labeled 50-nt DNA or RNA oligonucleotide with 500 nM enzyme in 80 μl of the same reaction medium used for primer extension and terminal transferase assays (see above) for 30 min at room temperature and then initiating the reactions by adding 1 mM dNTPs (an equimolar mix of 1 mM each of dATP, dCTP, dGTP and dTTP). A 10-μL aliquot was taken at each time point (up to 240 min). The reactions were quenched by adding 2 μL of 6X stop solution (see above for primer extension assays) and analyzed by electrophoresis in a non-denaturing 12% polyacrylamide gel with a 5’-labeled Low Molecular Weight DNA Ladder (New England Biolabs) run in a parallel lane. After electrophoresis, the gel was dried and scanned with a phosphorimager (Typhoon FLA 9500; GE Healthcare), and the scanned image was processed by ImageJ. The products were quantified by ImageQuant TL 8.1, and data were analyzed as described above for primer extension assays.
For high-throughput sequencing of snap-back replication products, the reactions were scaled up to use 1 μM of the 50-nt DNA oligonucleotide substrate and 1 μM RT together with 1 mM dNTPs in 100 μl of reaction medium and incubated at 37°C for 3 h. The reaction was terminated by adding 20 μL of 6X stop solution (see above), and the products were cleaned-up with an Oligo Clean & Concentrator Kit (Zymo Research). Nucleic acid concentrations were determined with a Nanodrop One (Thermo Scientific), and 50 ng of product was used to prepare TGIRT-seq libraries for high-throughput sequencing.
High-throughput sequencing libraries were constructed by using a variation of the TGIRT-seq method (Xu et al., 2019, 2021). First strand DNA synthesis was initiated at the 3’ end of the snapback DNA product by template switching from an RNA/DNA heteroduplex consisting of a 34-nt RNA containing an Illumina R2 adapter sequence annealed to a complementary 35-nt DNA leaving a single-nucleotide 3’ overhang (an equimolar mix of A, C, G, and T) that can base pair to the 3’ end of the DNA product, resulting in a full-length DNA copy of the product with an the reverse complement of the Illumina R2 adapter (denoted R2R) seamlessly linked to its 3’ end. After clean up using a MinElute PCR Purification Kit (Qiagen), the second-strand DNA synthesis was done by annealing 200 nM of a snap-back-specific R1R DNA oligonucleotide whose 3’ end was complementary to 3’ end of the DNA product, followed by a single-cycle of PCR using Phusion High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs) (98°C for 10 sec pre-denaturation followed by 98°C for 5 sec, 60°C for 10 sec, and 72°C for 15 sec). After two rounds of clean up with 1.4X AMPure XP beads (Beckman Coulter) with elution in 25 μL of double-distilled H2O, the products were amplified by PCR using Phusion High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs) with 200 nM of Illumina multiplex and index barcode primers (98°C for 10 sec pre-denaturation followed by 12 cycles of 98°C for 5 sec, 60°C 10 sec, 72°C 15 sec). The resulting TGIRT-seq libraries were cleaned up by using 1.4X AMPure XP beads (Beckman Coulter) and eluted in 25 μL double-distilled H2O, with 1 μL analyzed on an Agilent 2100 Bioanalyzer using a High Sensitivity DNA chip (Agilent) to assess product profiles and concentrations. The remainder of the library was sequenced on an Illumina MiSeq v2 instrument to obtain ~1 million 2 × 75 nt paired end reads per sample at the Genome Sequencing and Analysis Facility (GSAF) at the University of Texas at Austin.
To analyze the product sequences, Illumina TruSeq adapters and PCR primer sequences were trimmed from the reads with Cutadapt v3.2 (sequencing quality score cut-off at 20; p-value <0.01), and reads <15-nt after trimming were discarded (Martin, 2011). After merging the trimmed pair-ended reads by using BBMerge (Bushnell et al., 2017), the template sequence (5’-GCAATAATCTATACAATACAACACATACAAACAAATTCTTAAGGTCCCAA-3’) was trimmed from the 5’ ends of the merged reads by using Cutadapt, and downstream sequences were sorted to collect unique sequences. These unique sequences were then aligned to the reverse complement of the template sequence (5’-TTGGGACCTTAAGAATTTGTTTGTATGTGTTGTATTGTATAGATTATTGC-3’) using ClustalW (https://www.genome.jp/tools-bin/clustalw) with default settings and manually adjusted to correct minor misalignments.
Microhomology-mediated end-joining assays
Initial MMEJ assays were by done using a double-stranded DNA with a 15-nt single-stranded 3’ overhang with or without a 3’ terminal self-complementary microhomology sequence mimicking 5’-strand resected double-stranded DNAs on either side of a double-strand break. A 5’-labeled 53-nt oligonucleotide (D1) ending with a 4-nt microhomology or a control lacking the microhomology was annealed to an unlabeled complementary 39-nt DNA oligonucleotide with a 3’ blocker (D2) at a ratio of 1:2 in 100 μL of TE by heating to 95°C for 3 min followed by cooling to 25°C at 0.1°C/min in a T100 thermal cycler (Bio-Rad). In Figures, the left- and right-hand substrates though identical are denoted D1/D2, and D1’/D2’, respectively.
Biochemical assays of MMEJ activity were done with 10 nM of the annealed substrate and 500 nM enzyme, which had been preincubated in reaction medium for 30 min at room temperature. The reaction was initiated by adding 1 mM dNTPs, incubated at 37°C for times indicated for individual experiments (up to 240 min), and quenched as described above for the primer extension assays. The products were analyzed by electrophoresis in a non-denaturing 12% polyacrylamide gel against a 5’-labeled Low Molecular Weight DNA Ladder (New England Biolabs). The gel was scanned by a phosphorimager (Typhoon FLA 9500; GE Healthcare), and the scanned image was processed by ImageJ.. Some experiments used substrates that varied the length and sequence of the 3’ microhomology (4-bp CCGG-3’ or TTAA-3’; 10-bp CCCCCGGGGG-3’; non-complementary CCAA-3’) or changed the length of the single-stranded gap flanking the microhomology by varying the length of the D2 oligonucleotide (27, 33 and 39 nt). The products were quantified by ImageQuant TL 8.1, and data were analyzed as described above for primer extension assays.
For high-throughput sequencing of MMEJ products, the reactions were scaled up to use 1 μM of unlabeled annealed oligonucleotides and 1 μM of enzyme in 50 μL of reaction medium. The samples were incubated at 37°C for 4 h, and terminated by adding 10 μL of 6X stop solution and incubating for 15 min at 37°C. The products were cleaned up using 1.8X AMPure XP Beads and eluted with 25 μL of double-distilled H2O, digested with BsrGI (New England Biolabs), and ligated into the BsrGI site of BsrGI-linearized pKS-SacB. After transforming the ligated plasmids into E. coli HMS174 (DE3), cells were incubated overnight in 5 mL of LB containing 50 μg/mL carbenicillin and 6% sucrose, to select cells containing plasmids in which the sacB gene was inactivated by an insertion into the BsrGI site. The plasmids were isolated by using a Monarch Plasmid Miniprep kit (New England Biolabs) according to the manufacturer’s protocol and PCR amplified using primers MMEJ R1 and MMEJ R2R, which are complementary to sequences that flank the sacB BsrGI cleavage site and add Illumina R1 and R2R sequences to either end of the PCR product. The PCR was done with 1–2 ng of plasmid and 200 nM of each primer in Phusion High-Fidelity PCR Master Mix (New England Biolabs) with pre-denaturation at 98°C for 5 sec followed by 12 cycles of 98°C for 5 sec, 65°C for 10 sec, and 72°C for 15 sec. After PCR amplification, the products were cleaned up with 0.4X AMPure XP Beads to remove the plasmid, followed by 1.4X AMPure XP Beads clean-up to remove the primers. The PCR products were eluted in 25 μL double-distilled H2O, and 1 μL was analyzed on an Agilent 2100 Bioanalyzer with a High Sensitivity DNA chip to confirm the product and determine product concentration. For sequencing, Illumina multiplex and bar code primers were added by PCR (1 μL of MMEJ product and 200 nM primers in Phusion High-Fidelity PCR Master Mix (New England Biolabs) with 98°C, 5 sec pre-denaturation followed by 12 cycles of 98°C for 5 sec, 60°C for 10 sec, and 72°C for 15 sec). After 1.4X AMPure XP Beads clean-up to remove primer dimers, 1 μL of the product was analyzed on an Agilent 2100 Bioanalyzer with High Sensitivity DNA chip to assess the product profile and concentration, and the libraries were sequenced on an Illumina MiSeq v2 instrument to obtain ~1 million 2 ×150 nt paired end reads of each sample at the University of Texas MD Anderson Cancer Center, Science Park.
For the analysis of product sequences, Illumina TruSeq adapter and PCR primer sequences were trimmed from the reads with Cutadapt v3.2 (sequencing quality score cut-off at 20; p-value <0.01), and reads <15 nt after trimming were discarded (Martin, 2011). Trimmed pair-ended reads were then merged by using BBMerge (Bushnell et al., 2017). Sequences between two BsrGI sites that were longer than 45 nt were analyzed by using a custom R script to categorize the type of the MMEJ products.
CRISPR/Cas9-induced thyA DSBR assay
The ability of WT and mutant G2L4 and GII RTs to repair double-strand breaks in E. coli HMS174 (DE3) was assessed by using CRISPR/Cas9 to introduce a DSB in the E. coli thyA gene. CRISPR/Cas9 components and G2L4 and GII RTs were expressed in E. coli HMS174 (DE3) using a two-plasmid system based on that described by Chen et al. (2018). First, a pCas9+RT TetR-based plasmid (Figure 7A), which expresses Streptococcus pyogenes Cas9 and WT or mutant G2L4 and GII RTs using independent L-arabinose-inducible promoters, was transformed into E. coli HMS174 (DE3) chemically competent cells via heat shock, and the transformed cells were incubated in 5 mL of LB medium containing tetracycline (25 μg/mL) in a 50-mL conical tube (Sarstedt) at 37°C with shaking (250 rpm) for 14–16 h. Then, 1 mL of the culture was transferred into 100 mL LB containing tetracycline (25 μg/mL) and incubated at 37°C until O.D.600 was 1.0. Expression of the RT and Cas9 proteins was induced by adding L-arabinose (2 mg/mL) and incubating at 18°C with shaking (100 rpm) for 19–21 h. To introduce the second plasmid pACRISPR thyA sgRNA expressing thyA guide RNAs, 25 mL of the overnight culture was centrifuged at 4000 × g for 15 min at 4°C, and the cell pellet was gently resuspended in 20 mL of ice-cold 10% glycerol, then centrifuged and resuspended in 10% glycerol twice more, with the final pellet resuspended in 500 μL of ice-cold 10% glycerol. 50-μL portions of the cells were then electroporated with 1 μg pACRISPR vector or pACRISPR-thyA sgRNA plasmids in a 2-mm cuvette at 3.0 kv, 200 Ω, 25 μF using a Gene Pulser Xcell Electroporation System (Bio-Rad). The bacteria were recovered in 1-mL fresh SOC (Super Optimal broth with Catabolite repression) medium supplemented with 2 mg/mL L-arabinose, 0.5 mM MnCl2, 25 μg/mL tetracycline, 100 μg/mL thymine and incubated at 37°C with shaking (250 rpm) for 1 h. To determine survival and mutation frequencies, the cells were serially diluted in SOC medium and plated on 2X yeast tryptone (YT) plates supplemented with 2 mg/mL L-arabinose, 10 mM MgCl2, 0.5 mM MnCl2, 25 μg/mL tetracycline, 50 μg/mL carbenicillin and 100 μg/mL thymine with or without trimethoprim (200 μg/mL), the latter to select against cells containing a functional thyA gene. Colonies were counted after 16–48 h at 37°C. p-values were calculated by student’s unpaired t-test in Prism 9.0.
To characterize E. coli chromosomal thyA genes with repaired DSBs, a 100-μL portion of the culture after electroporation of the thyA sgRNA plasmid was inoculated into 5 mL 2X YT medium containing 2 mg/mL L-arabinose, 10 mM MgCl2, 0.5 mM MnCl2, 25 μg/mL tetracycline, 50 μg/mL carbenicillin and 100 μg/mL thymine plus 200 μg/mL trimethoprim, which selects for thyA mutants, and incubated in a 50-mL conical tube (Sarstedt) at 250 rpm and 37°C for 60–72 h. The cells were then centrifuged at 4000 × g for 10 min, and genomic DNA was extracted by using a Monarch Genomic DNA Purification Kit (New England Biolabs). The thyA gene region was amplified from the genomic DNA (5 ng) with Phusion High-Fidelity PCR Master Mix (New England Biolabs) using 200 nM of forward and reverse primers that give amplicons of 0.75-kb (primers, P1-P1’), 2.5-kb (primers, P2-P2’), or 5-kb (primers, P3-P3’) encompassing the thyA gene DSB site (Figure 7E, S7D, and Table S3). PCR conditions were 98°C for 10 sec pre-denaturation followed by 25 cycles of 98°C for 10 sec, 60°C for 30 sec, and 72°C for 30 sec for the 0.75-kb amplicon, 72 °C for 1.5 min for the 2.5-kb amplicon, or 72 °C for 2.5 min for the 5-kb amplicons. The PCR products were analyzed by electrophoresis at 120 V in agarose gels containing Tris-acetate-EDTA buffer (2% agarose for the 750-bp amplicon and 1% agarose for the 2.5-kb and 5-kb amplicons). Size-selected PCR products were extracted by using a Monarch DNA gel extraction kit (New England Biolabs), and 1 ng of the extracted PCR products was analyzed on an Agilent 2100 Bioanalyzer using an Agilent High Sensitivity DNA kit. Nested PCRs of gel-purified products from the initial PCRs used primer sets that flanked deletion junctions inferred from the location of peaks in Bioanalyzer traces from the initial PCR and gave amplicons of 161–708 bp (Figures 7E, S7F and Table S3). An additional PCR was done directly from genomic DNA with primers closer to the deletion junctions to confirm the assignment and RT-dependence of closely spaced peaks (Figure S7E).
For sequencing, size-selected PCR products from nested PCRs using primers indicated in Table S3 were extracted from gel slices by using a Monarch DNA Gel Extraction kit (New England Biolabs) according to the manufacturer’s protocol (a single band for the 0.75-kb amplicon, two groups of larger and smaller bands for the 2.5- and 5-kb amplicons), cloned in E. coli HMS174 (DE3) by using a PCR Cloning Kit (New England Biolabs), and analyzed by Sanger sequencing with >10 clones obtained for each sequence.
QUANTIFICATION AND STATISTICAL ANALYSIS
ImageQuant TL ver. 8.1 (General Electric) was used for quantitation of in vitro primer extension, terminal transferase, snap-back replication, and MMEJ assays. Excel ver. 16 (Microsoft) was used to determine mean, median, and standard deviation values. Prism 9.0 (Graphpad Software) was used for curve fitting of primer extension, terminal transferase, snap-back replication and MMEJ assays in order to determine kobs and Amplitude values and to calculate p values using a student’s unpaired t-test. Bedtools v2.29.2 was used to determine gene counts. R (v4.0.3) package DESeq2 was used to normalize gene counts, identify differentially expressed genes, and calculate Benjamini-Hochberg procedure adjusted p values. The R package goseq was used to identify enriched Gene Ontology (GO) terms in differentially expressed genes between KO and WT and to calculate their p values based on hypergeometric distribution.
Supplementary Material
Table S1, related to Figure 1. TGIRT-seq datasets and read mapping statistics
Table S2, related to Figure 2. Proteins encoded by differentially expressed transcription factor and stress response genes in WT and G2L4 KO strains in log and stationary phase
Table S3, related to Figure 7. Sanger sequencing of PCR products containing deletion junctions
Table S4, related to STAR Methods. Oligonucleotides
Table S5, related to STAR Methods. Recombinant plasmids
Figure S1, related to Figure 1. Conservation of the long direct repeat and spacer sequences preceding the G2L4 RT ORF in different P. aeruginosa strains and characteristics of the G2L4 RT ORF (A) ClustalW alignment of the region upstream of the G2L4 RT ORF containing the ~140-bp direct repeats and ~240-bp spacer from five P. aeruginosa strains. The repeat and spacer sequences are delineated above the alignment. Identical nucleotides in ≥80% of the aligned sequences are shown as white letters on a black background. A consensus sequence is shown at the bottom. The sources for the aligned sequences were: P. aeruginosa AZPAE12409 whole-genome sequencing data GCA_000797005.1; P. aeruginosa PABL068, whole-genome sequencing data GCA_003411275.2; P. aeruginosa GAR02, GenBank accession NZ_JABUGS010000001; P. aeruginosa AUS183, GenBank accession NZ_NSZP01000001.1.; P. aeruginosa AR_0352, GenBank accession QMGJ01000001.1.
(B) Sequence alignment and predicted secondary structure of the GII and G2L4 RTs. Secondary structure was predicted by using HHPred (Zimmermann et al., 2018). Conserved sequence motifs found in all RTs (RT1 to 7) and the thumb domain are delineated above. The RT0 loop and the RT2a and RT3a insertions are in blue boxes. The YxDD motif at the RT active site motif is boxed in green. Red boxes indicate insertions in G2L4 RT relative to GII RT. α-helices, H/h (red); β-sheets, E/e (blue); Coiled coil, C/c (black). Upper- and lower-case letters indicate higher and lower confidence predictions, respectively (Gabler et al., 2020).
(C) Weblogos of G2L4 RT sequences. Two hundred thirty-eight unique G2L4 RT sequences were aligned with ClustalW, and the alignment was manually refined. Sequences in boxed regions of G2L4 RT in panel (B) were used to generate sequence logos with default parameters (Crooks et al., 2004).
Figure S2, related to Figure 2 and biochemical assays. Disruption of the G2L4 RT ORF by targetron insertion, immunoblots showing expression levels of WT and mutant G2L4 and GII RTs in in vivo DNA damage survival assays, and Coomassie blue-stained gel of purified proteins used in biochemical experiments. (A) Identification of targetron disruptants of the G2L4 RT ORF in P. aeruginosa AZPAE12409 by colony PCR, using primers flanking the targetron-insertion site (Yao and Lambowitz, 2007). Colony PCR products were run in a 1% agarose gel against Quick-Load 1-kb Plus DNA Ladder (New England Biolabs). Colonies 6 and 10 (red) gave larger PCR products reflecting the targetron insertion.
(B) Southern hybridization showing a single targetron insertion in the G2L4 RT ORF. After curing the pBL1 plasmid by growth in Luria-Bertani (LB) medium without tetracycline, genomic DNA of P. aeruginosa AZPAE12409 WT and G2L4 RT KO strains was digested with PstI and EcoRI, which are predicted to generate a 1.5-kb band containing the integrated targetron. The pBL1 vector digested with the same enzymes produced a 3.6-kb DNA fragment. The digested DNAs were run in a 1% agarose gel against a 5’-labeled 1-Kb Plus DNA Ladder (Invitrogen), and Southern hybridization using a 5’-labeled targetron probe was done as described in Methods.
(C) Comparison of whole-genome sequencing (WGS) coverage of the KO1 and KO2 knockout strains. Fold change (FC) was calculated as the ratio of coverage between the WT and KO strains in 500-bp bins arranged by contig numbers. The lines highlighted in gray correspond to contig 110, which contains the targetron insertion site (I.S.) in the KO strains. (D) Growth curve of P. aeruginosa AZPAE12409 WT and G2L4 RT KO strains. Single colonies of each strain were inoculated into LB medium and incubated at 37°C, 200 rpm, overnight. The next day, cultures were diluted 1:100 into fresh LB medium and incubated further at 37°C, 200 rpm. Cells were harvested every 6 h up to 72 h, serially diluted, and plated on LB agar plates to determine the number of colony forming units (CFUs)/mL. The error bars indicate the standard deviation for three repeats using separate cultures.
(E) Volcano plots showing differences in gene expression in the G2L4 WT and KO strains in log and stationary phases. Read counts were DESeq2 normalized. RNAs showing significant differential expression between the G2L4 RT KO and WT strains (adjusted p value ≤0.05 calculated by Wald test and adjusted by the Benjamini-Hochberg procedure using DESeq2) are color coded.
(F) Immunoblot of WT and mutant G2L4 RTs expressed from pBL1-MBP-G2L4–8XHis versus a vector control expressing MBP with a C-terminal 8XHis tag in cultures of P. aeruginosa AZPAE12409 WT and KO strains grown in parallel to those used for cell survival assays. Concentrations of m-toluic acid used to induce protein expression are shown above each lane.
(G) X-ray irradiation cell survival assays and correlated immunoblots at different expression levels of G2L4 RT induced by different concentrations of m-toluic acid. The assays were repeated three times, with the error bars indicating the standard deviation. p-values for significant differences relative to WT G2L4 induced by 2 mM m-toluic acid were calculated by standard t-test. **, p-value <0.01.
(H) Immunoblot of WT and mutant G2L4 RTs expressed from pBL1-MBP-RT-8XHis and a vector control after induction with 2 mM m-toluic acid in E. coli HMS174 (DE3) cultures grown in parallel to those used for cell survival assays.
(I) Coomassie blue-stained gel of purified WT and mutant G2L4 or GII RT proteins used in biochemical assays. Proteins were expressed with a N-terminal MBP tag and C-terminal 8XHis tag and purified as described in Methods.
(J) Immunoblot of WT and mutant GII RTs expressed from pBL1-MBP-RT-8XHis and a vector control after induction with 2 mM m-toluic acid in E. coli HMS174 (DE3) cultures grown in parallel to those used for cell survival assays.
In panels F-J, proteins were run in a NuPAGE 4–12 % polyacrylamide gel (Invitrogen) against Prestained Protein Standard size markers (M, New England Biolabs) in a parallel lane. Immunoblots were probed with an α–6XHis-tag antibody (Invitrogen) that recognizes the 8XHis tag.
Figure S3, related to Figure 3A–D. NaCl concentration dependence of WT G2L4 RT primer extension activity and effects of primer length and MnCl2 on the primer extension activity of WT and mutant G2L4 and GII RTs (A-D) NaCl concentration dependence of primer extension activity of WT G2L4 RT assayed with 50-nt DNA or RNA templates with 3’ ends blocked with an inverted dT and 5-nt or 20-nt DNA primers in reaction medium containing 10 mM MgCl2 and 20 mM Tris-HCl pH 7.5 at 37°C.
(E-L) Primer extension activity of WT and mutant G2L4 RT or GII RTs with 50-nt DNA or RNA templates with 3’ ends blocked with an inverted dT and 5-nt or 20-nt DNA primers in reaction medium containing 20 mM NaCl, 10 mM MgCl2 and 20 Tris-HCl pH 7.5 at 37°C in the absence or presence of 1 mM MnCl2.
Primer extension reactions were done as described in Methods, and the products were analyzed in a denaturing 20% polyacrylamide gel with 5’-labeled DNA template and primer oligonucleotides as size markers in a parallel lane. The tables above the plots show rate constants (kobs) and amplitudes (Ampl.) for the production of the labeled 50-nt DNA product in panels A-J and for products larger than 5 or 20 nt for the slower GII A/I RT in panels K and L, obtained by fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S4, related to Figure 3E and F. Terminal transferase time courses for WT G2L4 and GII RTs (A and B) Terminal transferase assays with WT G2L4 or GII RT using a DNA substrate.
(C and D) Terminal transferase assays with WT G2L4 and GII RTs using an RNA substrate. Terminal transferase time courses were done as described in Methods using 5’-labeled 50-nt DNA or RNA substrates (the 50-nt DNA or RNA templates used in primer extension reactions without a 3’-blocking group) in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled RiboRuler Low Range RNA Ladder (Thermo Scientific) run in a parallel lane. Tables above the plots show the rate constants (kobs) and amplitudes (Ampl.) for production of all labeled products >50 nt obtained by fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S5, related to Figure 5. MMEJ time courses for WT G2L4 and GII RTs with TTAA-3’ microhomologies, effect of adding a 5’ phosphate to the D2/D2’ oligonucleotide corresponding to the resected 5’ end at a double-strand break, and effects of different length microhomologies and 3’ single-stranded gaps (A) MMEJ reactions using DNA substrates with 3’ overhangs having complementary TTAA-3’ sequences.
(B) MMEJ reactions using DNA substrates with 3’ overhangs having complementary CCGG-3’ sequences as in Figure 5A, but with a 5’ phosphate (red circled P in the schematic) at the 5’ end of the D2/D2’ oligonucleotide corresponding to resected 5’ ends at a double-strand break.
(C) MMEJ assay with CCGG-3’ (4 bp) microhomologies and 6-nt single-stranded gaps.
(D) MMEJ assay with CCGG-3’ (4 bp) microhomologies and 17-nt single-stranded gaps.
(E) MMEJ assay with CCCCCGGGGG-3’ (10 bp) microhomologies with 17-nt single-strand gaps. MMEJ reaction time courses were done using partially double-stranded 5’-labeled (red star) DNA substrates with 3’ overhangs having complementary sequences at their 3’ ends in the presence or absence of 1 mM MnCl2.
The numbers to the left of the gels indicate the positions of 5’-labeled size markers (Low Molecular Weight DNA Ladder; New England Biolabs) run in a parallel lane. The plots show the fraction of substrate that was converted to products running between the 100- and 150-nt size markers. Tables above the plots show rate constants (kobs) and amplitudes (Ampl.) obtained from fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Among the substrates tested, the MMEJ activity of WT G2L4 was highest on the CCGG-3’ substrate with an 11-nt single-stranded gap (Figure 5A). G2L4 MMEJ activity was strongly decreased on substrates with a longer microhomology (10 bp) or shorter single-stranded gap (6 nt), but substitution of A for I at the active site enabled higher activity with such substrates (Figure S5C and S5E). By contrast, MMEJ activity of WT GII RT was better than that of WT G2L4 RT on DNA substrates with a 10-bp microhomology and 6-nt single-stranded gap with the major effect of the A/I mutation being to decrease the rate and/or increase the lag phase for initiation of primer extension (Figure S5C and 5E).
Figure S6, related to Figure 6. Effect of deleting the RT0 loop on the biochemical activities of G2L4 and GII RTs (A-D) Primer extension assays for WT and ΔRT0 mutant G2L4 and GII RTs with DNA and RNA templates with no primer (−) or different length DNA primers with or without 1 mM MnCl2. Primer extension reactions were done as described in Figure S3 and Methods with 3’-blocked 50-nt DNA or RNA templates pre-annealed with different length DNA primers (100 μM 2 nt; 50 μM 5 nt; 500 nM 10, 15, 20-nt primers, respectively). Reactions were done at 37°C for 20 min (panels A and B) or as 240-min time courses (panels C and D). The numbers to the left of the gels indicate the positions of 5’-labeled DNA template and primer oligonucleotides run as size markers in a parallel lane.
(E and F) Terminal transferase reactions time courses for WT (top) or ΔRT0 mutant (bottom) G2L4 or GII RT using a 5’-labeled 50-nt DNA substrate. Plots of the reaction time courses for production of labeled products >50 nt are shown in Figure 6C. Terminal transferase time courses were done as described in Methods using a 5’-labeled 50-nt DNA substrate corresponding to the DNA template used in primer extension reactions without a 3’-blocking group in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled RiboRuler Low Range RNA Ladder (Thermo Scientific) run in a parallel lane.
(G) MMEJ assay by WT or ΔRT0 G2L4 and GII RTs using 5’-labeled DNA substrates having a 3’ overhang with a TTAA-3’ microhomology. MMEJ reaction time courses were done as described in Methods using partially double-stranded 5’-labeled (red star) DNA substrates with 3’ overhangs having complementary TTAA-3’ sequences at their 3’ ends in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled Low Molecular Weight DNA Ladder (New England Biolabs) run in a parallel lane. The plots show the fraction of substrate that was converted to products running between the 100- and 150-nt size markers.
Tables above the plots show the rate constants (kobs) and amplitudes (Ampl.) obtained from fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S7, related to Figure 7. G2L4 and GII RT-mediated repair of CRISPR/Cas9-induced DSBs in the E. coli thyA gene (A) Sequence of a segment of the E. coli HMS174 (DE3) thyA gene showing the CRISPR/Cas9 cleavage site (arrow) and thyA guide RNA (sgRNA; underlined) with the PAM sequence (boxed).
(B) E. coli thymidylate synthase pathway showing the basis for trimethoprim selection for thyA mutations. ThyA catalyzes the reductive methylation of 2’-deoxyuridine-5’-monophosphate (dUMP) to 2’-deoxythymidine-5’-monophosphate (dTMP) by using 5,10-methylenetetrahydrofolate (mTHF) as the methyl donor and reductant and yielding dihydrofolate (DHF) as a by-product. Trimethoprim (TMP) blocks the conversion of dihydrofolate to tetrahydrofolate (THF), which is needed for other cellular processes, resulting in cell growth arrest (Sangurdekar et al., 2011).
(C) Immunoblot showing expression levels of WT and mutant G2L4 or GII RTs and MBP-8XHis from the No RT vector control after induction of pCas9+G2L4 and GII RT plasmids in E. coli HMS174 (DE3).
(D) Bioanalyzer traces of PCR products obtained from genomic DNA using primers P3 and P3’ (Fig. 7 and Table S4) that amplify a 5-kb region around the DSB site in the thyA gene in cells expressing WT or mutant G2L4 (top) or GII (bottom) RTs compared to No RT and No guide RNA vector controls. Bioanalyzer traces were aligned via the peak corresponding to the full-length thyA gene.
(E) Bioanalyzer traces of PCR products obtained from genomic DNA using primers (P2–3 and P2’; Table S4) that better resolve a set of closely spaced peaks in Figure 7D.
(F) Sequencing of deletion junctions resulting from DSBR by MMEJ by WT G2L4 RT. The left-hand panels show bioanalyzer traces of nested PCR products obtained using the primer pairs indicated at the top left of each trace. Gel-purified, size-selected nested PCR products corresponding to the region of the bioanalyzer trace highlighted in yellow were cloned in E. coli HMS174 (DE3), and analyzed by Sanger sequencing (>10 clones for each sequence). The right-hand panels show Sanger sequencing traces across the deletion junction. The schematics below the bioanalyzer traces show annealed microhomologies between direct repeat sequences on either side of the break. The numbers in parentheses indicate E. coli K12 genomic coordinates. The same sequences were obtained for the same peaks in the WT GII RT bioanalyzer traces (Mendeley Data, V3, doi: 10.17632/7dbyk67546.3). *, unidentified peak. Potential microhomologies outside the sequenced regions are in light gray.
Supplemental File, related to Figures 1, 2, 4, and 5. High-throughput sequencing data mapping statistics and gene counts.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 6x His-Tag Monoclonal antibody | Invitrogen | MA1–21315 |
| Donkey anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP | Invitrogen | SA1–100 |
| Bacterial and virus strains | ||
| Pseudomonas aeruginosa AZPAE12409 | Kos et al., 2015 | Entasis Therapeutics |
| Pseudomonas aeruginosa AZPAE12409 G2L4 RT KO | This study | N/A |
| E. coli HMS174 (DE3) | Novagen | 69453 |
| E. coli S17.1 | ATCC | 47055 |
| E. coli Rosetta 2 | Novagen | 71402 |
| E. coli Rosetta 2 (DE3) | Novagen | 71400 |
| Chemicals, peptides, and recombinant proteins | ||
| [γ-32P]-ATP (6,000 Ci/mmol) | Perkin Elmer | NEG035C005MC |
| [α-32P]-dTTP (3,000 Ci/mmol) | Perkin Elmer | BLU005H250UC |
| T4 polynucleotide kinase | NEB | M0201 |
| T4 polynucleotide kinase | Epicenter | P0503K |
| Proteinase K Molecular biology grade | NEB | P8107S |
| Phusion PCR master mix | NEB | M0531L |
| TGIRT-III Enzyme | InGex | TGIRT10 |
| Tetracycline | Millipore Sigma | 87128 |
| Chloramphenicol | Millipore Sigma | C0378 |
| Carbenicillin | Research Products International | C46000 |
| m-toluic acid | Acros Organics | AC139050010 |
| Phleomycin | InvivoGen | ant-ph-1 |
| Hydrogen peroxide solution | Millipore Sigma | 88597 |
| Blotting Grade Blocker Non-Fat Dry Milk | Bio-Rad | 1706404XTU |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | GoldBio | I2481C |
| Trimethoprim | Millpore Sigma | 92131 |
| Magnesium chloride solution | Millpore Sigma | 68475 |
| Manganese (II) chloride tetrahydrate | Acros Organics | AC223610500 |
| RiboRuler Low Range RNA Ladder | Thermo Scientific | SM1831 |
| Quick-Load 1kb Plus DNA Ladder | NEB | N0469S |
| 1 Kb Plus DNA Ladder | Invitrogen | 10787018 |
| Critical commercial assays | ||
| Oligo Clean and Concentrator kit | Zymo | D4060 |
| RNA Clean and Concentration kit | Zymo | R1015 |
| Quick-DNA Fungal/Bacterial Miniprep Kit | Zymo | D6005 |
| Amersham Hybond-XL | Cytiva | RPN203S |
| Amersham Rapid-hybridization Buffer | Cytiva | RPN1635 |
| Monarch Plasmid Miniprep kit | NEB | T1010L |
| Monarch Genomic DNA Purification Kit | NEB | T3010L |
| Monarch Total RNA Miniprep Kit | NEB | T2010S |
| riboPOOL | siTOOLs biotech | N/A |
| Next Magnesium RNA Fragmentation Module | NEB | E6150S |
| MinElute kit | QIAgen | 28004 |
| Ampure XP beads | Beckman-Coulter | A63881 |
| 2100 Bioanalyzer High Sensitivity DNA kit | Agilent | 5067–4626 |
| Quick Start Bradford Protein Assay Kit | Bio-Rad | 5000201 |
| NuPAGE 4–12 % Bis-Tris gel | Invitrogen | NP0322PK2 |
| Color Prestained Protein Standard, Broad Range ladder (10–250 kDa) | NEB | P7719S |
| Immuno-Blot PVDF Membrane | Bio-Rad | 1620177 |
| NuPAGE Transfer Buffer (20X) | Thermo Fisher Scientific | NP00061 |
| Clarity Western ECL Substrate | Bio-Rad | 1705060 |
| CL-Xposure Film | Thermo Fisher Scientific | 34090 |
| MBPTrap HP column | Cytiva | 28918780 |
| HiTrap Heparin HP column | Cytiva | 17040703 |
| Amicon Ultra-15 (30k) concentrator | Millipore | UFC903024 |
| Qubit ssDNA Assay kit | Life Technologies | Q10212 |
| Qubit RNA HS Assay kit | Life Technologies | Q32852 |
| NEB PCR Cloning Kit | NEB | E1202S |
| NEBuilder HiFi DNA Assembly | NEB | E2621S |
| Q5 mutagenesis kit | NEB | E0554S |
| Monarch DNA Gel Extraction Kit | NEB | T1020L |
| Deposited data | ||
| Figure 2, raw TGIRT-seq data | This paper | SRA: PRJNA814398 |
| Figure 4, high-throughput snap-back replication data | This paper | SRA: PRJNA814398 |
| Figure 5, high-throughput MMEJ data | This paper | SRA: PRJNA814398 |
| Figure 7, S7, bioanalyzer traces and sanger sequencing data | This paper | DOI: 10.17632/7dbyk67546.3 |
| Unprocessed imaging data | This paper | DOI: 10.17632/7dbyk67546.3 |
| Repeats of biochemical experiments | This paper | DOI: 10.17632/7dbyk67546.3 |
| Oligonucleotides | ||
| See Table S4 for Oligonucleotides | ||
| Recombinant DNA | ||
| See Table S5 for Recombinant plasmids | ||
| Software and algorithms | ||
| Phython 3.10 | Python Software Foundation | https://www.python.org/downloads/release/python-3100/ |
| Biopython | Cock et al., 2009 | https://biopython.org |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| ImageQuant TL 8.1 | Cytiva | N/A |
| BBMerge | Bushnell et al., 2017 | https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0185056 |
| BWA | Li and Durbin, 2010 | https://doi.org/10.1093/bioinformatics/btp698 |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MUMmer3 | Kurtz et al., 2004 | https://genomebiology.biomedcentral.com/articles/10.1186/gb-2004-5-2-r12 |
| CLUSTALW | Thompson et al., 1994 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC308517/ |
| Cutadapt | Martin 2011 | https://doi.org/10.14806/ej.17.1.200 |
| Bedtools | Quinlan, 2014 | https://doi.org/10.1093/bioinformatics/btq033 |
| IGV | Robinson et al., 2011 | https://www.nature.com/articles/nbt.1754 |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| R | R Core Team, 2021 | https://www.R-project.org/ |
| R package: DESeq2 | Love et al., 2014 | https://doi.org/10.1186/s13059-014-0550-8 |
| R package: goseq | Young et al. 2010 | https://genomebiology.biomedcentral.com/articles/10.1186/gb-2010-11-2-r14 |
| R package: pheatmap | Raivo Kolde, 2019 | https://CRAN.R-project.org/package=pheatmap |
| freeBayes | Garrison and Marth, 2012 | https://arxiv.org/abs/1207.3907 |
| VarSacn | Koboldt et al., 2009 | https://doi.org/10.1093/bioinformatics/btp373 |
| MacVector 18.0 | MacVector | www.macvector.com |
| Prism v9.0 | GraphPad Software | www.graphpad.com |
| Other | ||
| Code for analysis of the TGIRT-seq dataset | This paper | https://github.com/reykeryao/Seung |
| Pseudomonas aeruginosa AZPAE12409 genome reference | The Pseudomonas Genome Database | https://www.pseudomonas.com/ |
| Pseudomonas aeruginosa PAO1 genome reference | The Pseudomonas Genome Database | https://www.pseudomonas.com/ |
| BacPP (bacterial promoter prediction) | de Avila Silva et al., 2011 | http://www.bacpp.bioinfoucs.com |
| HHpred | Zimmermann et al., 2018 | https://toolkit.tuebingen.mpg.de/tools/hhpred |
Highlights:
A Pseudomonas group II intron-like reverse transcriptase (RT) functions in DNA repair
A group II intron-encoded RT can function similarly in DNA repair
Both RTs repair double-strand breaks via microhomology-mediated end-joining
DNA repair uses conserved structural features of non-LTR-retroelement RTs
ACKNOWLEGMENTS
This work was supported by Welch Foundation grant F-1607 and NIH grant R35 GM136216 to AML. RR is supported by NIH grant R35 GM131777. We thank Kyle Miller for use of his X-ray generator, Jennifer Stamos for comments on the manuscript, and Douglas Wu for help with whole-genome sequence analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A.M.L., G.M., and the University of Texas are minority equity holders in and receive royalties from InGex, a company that sells TGIRT enzymes. The other authors declare no competing interests.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos P (1988). A sequence motif in many polymerases. Nucleic Acids Res. 16, 9909–9916. doi: 10.1093/nar/16.21.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Avila E Silva S, Echeverrigaray S, and Gerhardt GJ (2011) BacPP: bacterial promoter prediction—a tool for accurate sigma-factor specific assignment in enterobacteria. J. Theor. Biol 287, 92–99. doi: 10.1016/j.jtbi.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Bibillo A, and Eickbush TH (2004). End-to-end template jumping by the reverse transcriptase encoded by the R2 retrotransposon. J. Biol. Chem 279, 14945–14953. doi: 10.1074/jbc.M310450200. [DOI] [PubMed] [Google Scholar]
- Black SJ, Ozdemir AY, Kashkina E, Kent T, Rusanov T, Ristic D, Shin Y, Suma A, Hoang T, Chandramouly G, et al. (2019). Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun 10, 4423. doi: 10.1038/s41467-019-12272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black S, Kashkina E, Kent T, and Pomerantz R (2016). DNA Polymerase θ: A unique multifunctional end-joining machine. Genes 7, 67. doi: 10.3390/genes7090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatny JM, Brautaset T, Winther-Larsen HC, Karunakaran P, and Valla S (1997). Improved broad-host-range RK2 vectors useful for high and low regulated gene expression levels in gram-negative bacteria. Plasmid 38, 35–51. doi: 10.1006/plas.1997. [DOI] [PubMed] [Google Scholar]
- Blocker FJ, Mohr G, Conlan LH, Qi L, Belfort M, and Lambowitz AM (2005). Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA 11, 14–28. doi: 10.1261/rna.7181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B, Rood J, and Singer E (2017). BBMerge - Accurate paired shotgun read merging via overlap. PLoS One. 12, e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, Zhao J, McDevitt S, Rusanov T, Hoang T, Borisonnik N, Treddinick T, Lopezcolorado FW, Kent T, Siddique LA, et al. (2021). Polθ reverse transcribes RNA and promotes RNA-templated DNA repair. Science Advances 7, eabf1771. doi: 10.1126/sci-adv.abf1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang Y, Zhang Y, Pi Y, Gu T, Song L, Wang Y, and Ji Q (2018). CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and Cytidine deaminase-mediated base editing in Pseudomonas Species. iScience 6, 222–231. doi: 10.1016/j.isci.2018.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz RT, O’Neill BM, Hammond JA, Head SR, and Romesberg FE (2006). Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic Ciprofloxacin. J. Bacteriol 188, 7101–7110. doi: 10.1128/JB.00807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJ (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 25, 1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De, Deken X, Van, Sande J, Dumont JE, Miot F, and Corvilain B (2009). Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr. Relat. Cancer 16, 845–856. doi: 10.1677/ERC-09-0020. [DOI] [PubMed] [Google Scholar]
- Esnault C, Maestre J, and Heidmann T (2000). Human LINE retrotransposons generate processed pseudogenes. Nat. Genet 24, 363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Söding J, Lupas AN, and Alva V (2020). Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics 72, e108. doi: 10.1002/cpbi.108. [DOI] [PubMed] [Google Scholar]
- Gao L, Altae-Tran H, Böhning F, Makarova KS, Segel M, Schmid-Burgk JL, Koob J, Wolf YI, Koonin EV, and Zhang F (2020). Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084. doi: 10.1126/science.aba0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, and Marth G (2012). Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907. doi: 10.48550/arXiv.1207.3907 [DOI] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, and Moran J. v. (2005). Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol. Cell. Biol 25, 7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SQ, Cui X, Mou S, Mohr. S, Yao J, and Lambowitz AM (2010). Genetic identification of potential RNA-binding regions in a group II intron-encoded reverse transcriptase. RNA 16, 732–747. doi: 10.1261/rna.2007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Kurita D, and Muto A (2014). tmRNA-mediated trans-translation as the major ribosome rescue system in a bacterial cell. Front. Genet 5, 66. doi: 10.3389/fgene.2014.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutfilz CR, Wang NE, Hoff CA, Lee JA, Hackert BJ, Courcelle J, and Courcelle CT (2019). Manganese is required for the rapid recovery of DNA synthesis following oxidative challenge in Escherichia coli. J. Bacteriol 201, e00426–19. doi: 10.1128/JB.00426-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaishankar J, and Srivastava P (2017). Molecular basis of stationary phase survival and applications. Front. Microbiol 8, 2000. doi: 10.3389/fmicb.2017.02000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamburuthugoda VK, and Eickbush TH (2014) Identification of RNA binding motifs in the R2 retrotransposon-encoded reverse transcriptase. Nucleic Acids Res. 42, 8405–8415. doi: 10.1093/nar/gku514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katibah GE, Qin Y, Sidote DJ, Yao J, Lambowitz AM, and Collins K (2014). Broad and adaptable RNA structure recognition by the human interferon-induced tetratricopeptide repeat protein IFIT5. Proc. Natl. Acad. Sci. U.S.A 111, 12025–12030. doi: 10.1073/pnas.1412842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, and Pomerantz RT (2015). Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol 22, 230–237. doi: 10.1038/nsmb.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent T, Mateos-Gomez P, Sfeir A, and Pomerantz RT (2016) Polymerase θ is a robust terminal transferase that oscillates between three different mechanisms during end-joining. eLife 5, e13740. doi: 10.7554/eLife.13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, and Larson DE (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima KK, and Kanehisa M (2008). Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol 25, 1395–1404. doi: 10.1093/molbev/msn081. [DOI] [PubMed] [Google Scholar]
- Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, Corbeil J, and Gardner H (2015). The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother 59, 427–436. doi: 10.1128/AAC.03954-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN (2013). DNA Damage Responses in Prokaryotes: Regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb. Perspect. Biol 5, a012674. doi: 10.1101/cshperspect.a012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, and Salzberg SL (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, and Belfort M (2015). Mobile bacterial group II introns at the crux of eukaryotic evolution. Microbiol. Spectr 3, MDNA3-0050-2014. doi: 10.1128/microbiolspec.MDNA3-0050-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, and Zimmerly S (2011). Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol 3, a003616. doi: 10.1101/cshperspect.a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, and Cordaux R (2012). Selection-driven extinction dynamics for group II introns in Enterobacteriales. PLoS One 7, e52268. doi: 10.1371/journal.pone.0052268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Sherer NA, Kim NH, Rajic E, Kaur D, Urriola N, Martini KM, Xue C, Goldenfeld N, and Kuhlman TE (2018). Testing the retroelement invasion hypothesis for the emergence of the ancestral eukaryotic cell. Proc. Natl. Acad. Sci. U.S.A 115, 12465–12470. doi: 10.1073/pnas.1807709115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentzsch AM, Yao J, Russell R, and Lambowitz AM (2019). Template-switching mechanism of a group II intron-encoded reverse transcriptase and its implications for biological function and RNA-Seq. J. Biol. Chem 294, 19764–19784. doi: 10.1074/jbc.RA119.011337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentzsch AM, Stamos JL, Yao J, Russell R, and Lambowitz AM (2021). Structural basis for template switching by a group II intron-encoded non-LTR-retroelement reverse transcriptase. J. Biol. Chem 297, 100971. doi: 10.1016/j.jbc.2021.100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Deora R, Doulatov SR, Gingery M, Eiserling FA, Preston A, Maskell DJ, Simons RW, Cotter PA, Parkhill J, et al. (2002). Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 295, 2091–2094. doi: 10.1126/science.1067467. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett ST (2011). The DNA exonucleases of Escherichia coli. EcoSal Plus 4. doi: 10.1128/ecosalplus.4.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.1089/cmb.2017.0096. [DOI] [Google Scholar]
- Martín-Alonso S, Frutos-Beltrán E, and Menéndez-Arias L (2021). Reverse transcriptase: From transcriptomics to genome editing. Trends Biotechnol. 39,194–210. doi: 10.1016/j.tibtech.2020.06.008. [DOI] [PubMed] [Google Scholar]
- Merrikh H, Ferrazzoli AE, Bougdour A, Olivier-Mason A, and Lovett ST (2009). A DNA damage response in Escherichia coli involving the alternative sigma factor, RpoS. Proc. Natl. Acad. Sci. U.S.A 106, 611–616. doi: 10.1073/pnas.0803665106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppen-heimer-Shaanan Y, and Sorek R (2020). Bacterial retrons function in anti-phage defense. Cell 183, 1551–1561. doi: 10.1016/j.cell.2020.09.065. [DOI] [PubMed] [Google Scholar]
- Mohr G, Ghanem E, and Lambowitz AM (2010). Mechanisms used for genomic proliferation by thermophilic group II introns. PLoS Biol. 8, e1000391. doi: 10.1371/journal.pbio.1000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Ghanem E, Smith W, Sheeter D, Qin Y, King O, Polioudakis D, Iyer VR, Hunicke-Smith S, Swamy S, et al. (2013). Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA 19, 958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish TA, García Pérez JL, Stamato TD, Taccioli GE, Sekiguchi J, and Moran JV (2007). Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature 446, 208–212. doi: 10.1038/nature05560. [DOI] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, and Moran JV (2002). DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet 31, 159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- Müller C, Crowe-McAuliffe C, and Wilson DN (2021). Ribosome rescue pathways in bacteria. Front. Microbiol 12, 652980. doi: 10.3389/fmicb.2021.652980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottingham RM, Wu DC, Qin Y, Yao J, Hunicke-Smith S, and Lambowitz AM (2016). RNA-seq of human reference RNA samples using a thermostable group II intron reverse transcriptase. RNA 22, 597–613. doi: 10.1261/rna.055558.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onozawa M, Zhang Z, Kim YJ, Goldberg L, Varga T, Bergsagel PL, Kuehl WM, and Aplan PD (2014). Repair of DNA double-strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc. Natl. Acad. Sci. U.S.A 111, 7729–7734. doi: 10.1073/pnas.1321889111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, and Kazazian HH Jr (2001). Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 11, 2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch AR (2020). The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J 18, 207–219. doi: 10.1016/j.csbj.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden DA, Carvajal-Garcia J, and Gupta GP (2022). Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat. Rev. Mol. Cell Biol 23, 125–140. doi: 10.1038/s41580-021-00405-2. [DOI] [PubMed] [Google Scholar]
- Kolde Raivo (2019). pheatmap: Pretty Heatmaps. R package Version 1.0.12. Available online at: https://CRAN.R-project.org/package=pheatmap
- R Core Team. (2021). R: A Language and Environment for Statistical Computing. Vienna, Austria. Available online at: https://www.R-project.org/ [Google Scholar]
- Robart AR, and Zimmerly S (2005). Group II intron retroelements: function and diversity. Cytogenet. Genome Res 110, 589–597. doi: 10.1159/000084992. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha R, Chen B, Wank H, Matsuura M, Edwards J, and Lambowitz AM (1999). RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry 38, 9069–9083. doi: 10.1021/bi982799l. [DOI] [PubMed] [Google Scholar]
- San Filippo J, and Lambowitz AM (2002). Characterization of the C-terminal DNA-binding/DNA endonuclease region of a group II intron-encoded protein. J. Mol. Biol 324, 933–951. doi: 10.1016/s0022-2836(02)01147-6. [DOI] [PubMed] [Google Scholar]
- Sangurdekar DP, Zhang Z, and Khodursky AB (2011). The association of DNA damage response and nucleotide level modulation with the antibacterial mechanism of the anti-folate drug Trimethoprim. BMC Genomics 12, 583. doi: 10.1186/1471-2164-12-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr MJ, and Deretic V (1997). Microbial pathogenesis in cystic fibrosis: co-ordinate regulation of heat-shock response and conversion to mucoidy in Pseudomonas aeruginosa. Mol. Microbiol 24, 411–420. doi: 10.1046/j.1365-2958.1997.3411711.x. [DOI] [PubMed] [Google Scholar]
- Seki M, Masutani C, Yang LW, Schuffert A, Iwai S, Bahar I, and Wood RD (2004). High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 23, 4484–4494. doi: 10.1038/sj.emboj.7600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silas S, Mohr G, Sidote DJ, Markham LM, Sanchez-Amat A, Bhaya D, Lambowitz AM, and Fire AZ (2016). Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science 351, aad4234. doi: 10.1126/science.aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Lentzsch AM, and Lambowitz AM (2017). Structure of a thermostable group II intron reverse transcriptase with template-primer and its functional and evolutionary implications. Mol. Cell 68, 926–939. doi: 10.1016/j.molcel.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Villion M, Semper C, Coros C, Moineau S, and Zimmerly S (2011). A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids Res. 39, 7620–7629. doi: 10.1093/nar/gkr397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor GL, Lam D, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock REW, Brinkman FSL (2011). Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Gingery M, Abebe M, Arambula D, Czornyj E, Handa S, Khan H, Liu M, Pohlschroder M, Shaw KL, Du A, Guo H, Ghosh P, Miller JF, and Zimmerly S (2018). Diversity-generating retroelements: natural variation, classification and evolution inferred from a large-scale genomic survey. Nucleic Acids Res. 46, 11–24. doi: 10.1093/nar/gkx1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, and Eickbush TH (1990). Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9, 3353–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Nottingham RM, and Lambowitz AM (2021). TGIRT-seq protocol for the comprehensive profiling of coding and non-coding RNA biotypes in cellular, extracellular vesicle, and plasma RNAs. Bio-Protocol 11, e4239. doi: 10.21769/BioProtoc.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yao J, Wu DC, and Lambowitz AM (2019). Improved TGIRT-seq methods for comprehensive transcriptome profiling with decreased adapter dimer formation and bias correction. Sci. Rep 9, 621–627. doi: 10.1038/s41598-019-44457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, and Zhang Y (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, and Lambowitz AM (2007). Gene targeting in gram-negative bacteria by use of a mobile group II intron (“Targetron”) expressed from a broad-host-range vector. Appl. Environ. Microbiol 73, 2735–2743. doi: 10.1128/AEM.02829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerly S, and Wu L (2015). An unexplored diversity of reverse transcriptases in bacteria. Microbiol. Spectr 3, MDNA3-0058-2014. doi: 10.1128/microbiolspec.MDNA3-0058-2014. [DOI] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam S, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, and Alva V (2018). A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J. Mol. Biol 430, 2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 7582. doi: 10.1038/nmeth.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. TGIRT-seq datasets and read mapping statistics
Table S2, related to Figure 2. Proteins encoded by differentially expressed transcription factor and stress response genes in WT and G2L4 KO strains in log and stationary phase
Table S3, related to Figure 7. Sanger sequencing of PCR products containing deletion junctions
Table S4, related to STAR Methods. Oligonucleotides
Table S5, related to STAR Methods. Recombinant plasmids
Figure S1, related to Figure 1. Conservation of the long direct repeat and spacer sequences preceding the G2L4 RT ORF in different P. aeruginosa strains and characteristics of the G2L4 RT ORF (A) ClustalW alignment of the region upstream of the G2L4 RT ORF containing the ~140-bp direct repeats and ~240-bp spacer from five P. aeruginosa strains. The repeat and spacer sequences are delineated above the alignment. Identical nucleotides in ≥80% of the aligned sequences are shown as white letters on a black background. A consensus sequence is shown at the bottom. The sources for the aligned sequences were: P. aeruginosa AZPAE12409 whole-genome sequencing data GCA_000797005.1; P. aeruginosa PABL068, whole-genome sequencing data GCA_003411275.2; P. aeruginosa GAR02, GenBank accession NZ_JABUGS010000001; P. aeruginosa AUS183, GenBank accession NZ_NSZP01000001.1.; P. aeruginosa AR_0352, GenBank accession QMGJ01000001.1.
(B) Sequence alignment and predicted secondary structure of the GII and G2L4 RTs. Secondary structure was predicted by using HHPred (Zimmermann et al., 2018). Conserved sequence motifs found in all RTs (RT1 to 7) and the thumb domain are delineated above. The RT0 loop and the RT2a and RT3a insertions are in blue boxes. The YxDD motif at the RT active site motif is boxed in green. Red boxes indicate insertions in G2L4 RT relative to GII RT. α-helices, H/h (red); β-sheets, E/e (blue); Coiled coil, C/c (black). Upper- and lower-case letters indicate higher and lower confidence predictions, respectively (Gabler et al., 2020).
(C) Weblogos of G2L4 RT sequences. Two hundred thirty-eight unique G2L4 RT sequences were aligned with ClustalW, and the alignment was manually refined. Sequences in boxed regions of G2L4 RT in panel (B) were used to generate sequence logos with default parameters (Crooks et al., 2004).
Figure S2, related to Figure 2 and biochemical assays. Disruption of the G2L4 RT ORF by targetron insertion, immunoblots showing expression levels of WT and mutant G2L4 and GII RTs in in vivo DNA damage survival assays, and Coomassie blue-stained gel of purified proteins used in biochemical experiments. (A) Identification of targetron disruptants of the G2L4 RT ORF in P. aeruginosa AZPAE12409 by colony PCR, using primers flanking the targetron-insertion site (Yao and Lambowitz, 2007). Colony PCR products were run in a 1% agarose gel against Quick-Load 1-kb Plus DNA Ladder (New England Biolabs). Colonies 6 and 10 (red) gave larger PCR products reflecting the targetron insertion.
(B) Southern hybridization showing a single targetron insertion in the G2L4 RT ORF. After curing the pBL1 plasmid by growth in Luria-Bertani (LB) medium without tetracycline, genomic DNA of P. aeruginosa AZPAE12409 WT and G2L4 RT KO strains was digested with PstI and EcoRI, which are predicted to generate a 1.5-kb band containing the integrated targetron. The pBL1 vector digested with the same enzymes produced a 3.6-kb DNA fragment. The digested DNAs were run in a 1% agarose gel against a 5’-labeled 1-Kb Plus DNA Ladder (Invitrogen), and Southern hybridization using a 5’-labeled targetron probe was done as described in Methods.
(C) Comparison of whole-genome sequencing (WGS) coverage of the KO1 and KO2 knockout strains. Fold change (FC) was calculated as the ratio of coverage between the WT and KO strains in 500-bp bins arranged by contig numbers. The lines highlighted in gray correspond to contig 110, which contains the targetron insertion site (I.S.) in the KO strains. (D) Growth curve of P. aeruginosa AZPAE12409 WT and G2L4 RT KO strains. Single colonies of each strain were inoculated into LB medium and incubated at 37°C, 200 rpm, overnight. The next day, cultures were diluted 1:100 into fresh LB medium and incubated further at 37°C, 200 rpm. Cells were harvested every 6 h up to 72 h, serially diluted, and plated on LB agar plates to determine the number of colony forming units (CFUs)/mL. The error bars indicate the standard deviation for three repeats using separate cultures.
(E) Volcano plots showing differences in gene expression in the G2L4 WT and KO strains in log and stationary phases. Read counts were DESeq2 normalized. RNAs showing significant differential expression between the G2L4 RT KO and WT strains (adjusted p value ≤0.05 calculated by Wald test and adjusted by the Benjamini-Hochberg procedure using DESeq2) are color coded.
(F) Immunoblot of WT and mutant G2L4 RTs expressed from pBL1-MBP-G2L4–8XHis versus a vector control expressing MBP with a C-terminal 8XHis tag in cultures of P. aeruginosa AZPAE12409 WT and KO strains grown in parallel to those used for cell survival assays. Concentrations of m-toluic acid used to induce protein expression are shown above each lane.
(G) X-ray irradiation cell survival assays and correlated immunoblots at different expression levels of G2L4 RT induced by different concentrations of m-toluic acid. The assays were repeated three times, with the error bars indicating the standard deviation. p-values for significant differences relative to WT G2L4 induced by 2 mM m-toluic acid were calculated by standard t-test. **, p-value <0.01.
(H) Immunoblot of WT and mutant G2L4 RTs expressed from pBL1-MBP-RT-8XHis and a vector control after induction with 2 mM m-toluic acid in E. coli HMS174 (DE3) cultures grown in parallel to those used for cell survival assays.
(I) Coomassie blue-stained gel of purified WT and mutant G2L4 or GII RT proteins used in biochemical assays. Proteins were expressed with a N-terminal MBP tag and C-terminal 8XHis tag and purified as described in Methods.
(J) Immunoblot of WT and mutant GII RTs expressed from pBL1-MBP-RT-8XHis and a vector control after induction with 2 mM m-toluic acid in E. coli HMS174 (DE3) cultures grown in parallel to those used for cell survival assays.
In panels F-J, proteins were run in a NuPAGE 4–12 % polyacrylamide gel (Invitrogen) against Prestained Protein Standard size markers (M, New England Biolabs) in a parallel lane. Immunoblots were probed with an α–6XHis-tag antibody (Invitrogen) that recognizes the 8XHis tag.
Figure S3, related to Figure 3A–D. NaCl concentration dependence of WT G2L4 RT primer extension activity and effects of primer length and MnCl2 on the primer extension activity of WT and mutant G2L4 and GII RTs (A-D) NaCl concentration dependence of primer extension activity of WT G2L4 RT assayed with 50-nt DNA or RNA templates with 3’ ends blocked with an inverted dT and 5-nt or 20-nt DNA primers in reaction medium containing 10 mM MgCl2 and 20 mM Tris-HCl pH 7.5 at 37°C.
(E-L) Primer extension activity of WT and mutant G2L4 RT or GII RTs with 50-nt DNA or RNA templates with 3’ ends blocked with an inverted dT and 5-nt or 20-nt DNA primers in reaction medium containing 20 mM NaCl, 10 mM MgCl2 and 20 Tris-HCl pH 7.5 at 37°C in the absence or presence of 1 mM MnCl2.
Primer extension reactions were done as described in Methods, and the products were analyzed in a denaturing 20% polyacrylamide gel with 5’-labeled DNA template and primer oligonucleotides as size markers in a parallel lane. The tables above the plots show rate constants (kobs) and amplitudes (Ampl.) for the production of the labeled 50-nt DNA product in panels A-J and for products larger than 5 or 20 nt for the slower GII A/I RT in panels K and L, obtained by fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S4, related to Figure 3E and F. Terminal transferase time courses for WT G2L4 and GII RTs (A and B) Terminal transferase assays with WT G2L4 or GII RT using a DNA substrate.
(C and D) Terminal transferase assays with WT G2L4 and GII RTs using an RNA substrate. Terminal transferase time courses were done as described in Methods using 5’-labeled 50-nt DNA or RNA substrates (the 50-nt DNA or RNA templates used in primer extension reactions without a 3’-blocking group) in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled RiboRuler Low Range RNA Ladder (Thermo Scientific) run in a parallel lane. Tables above the plots show the rate constants (kobs) and amplitudes (Ampl.) for production of all labeled products >50 nt obtained by fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S5, related to Figure 5. MMEJ time courses for WT G2L4 and GII RTs with TTAA-3’ microhomologies, effect of adding a 5’ phosphate to the D2/D2’ oligonucleotide corresponding to the resected 5’ end at a double-strand break, and effects of different length microhomologies and 3’ single-stranded gaps (A) MMEJ reactions using DNA substrates with 3’ overhangs having complementary TTAA-3’ sequences.
(B) MMEJ reactions using DNA substrates with 3’ overhangs having complementary CCGG-3’ sequences as in Figure 5A, but with a 5’ phosphate (red circled P in the schematic) at the 5’ end of the D2/D2’ oligonucleotide corresponding to resected 5’ ends at a double-strand break.
(C) MMEJ assay with CCGG-3’ (4 bp) microhomologies and 6-nt single-stranded gaps.
(D) MMEJ assay with CCGG-3’ (4 bp) microhomologies and 17-nt single-stranded gaps.
(E) MMEJ assay with CCCCCGGGGG-3’ (10 bp) microhomologies with 17-nt single-strand gaps. MMEJ reaction time courses were done using partially double-stranded 5’-labeled (red star) DNA substrates with 3’ overhangs having complementary sequences at their 3’ ends in the presence or absence of 1 mM MnCl2.
The numbers to the left of the gels indicate the positions of 5’-labeled size markers (Low Molecular Weight DNA Ladder; New England Biolabs) run in a parallel lane. The plots show the fraction of substrate that was converted to products running between the 100- and 150-nt size markers. Tables above the plots show rate constants (kobs) and amplitudes (Ampl.) obtained from fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Among the substrates tested, the MMEJ activity of WT G2L4 was highest on the CCGG-3’ substrate with an 11-nt single-stranded gap (Figure 5A). G2L4 MMEJ activity was strongly decreased on substrates with a longer microhomology (10 bp) or shorter single-stranded gap (6 nt), but substitution of A for I at the active site enabled higher activity with such substrates (Figure S5C and S5E). By contrast, MMEJ activity of WT GII RT was better than that of WT G2L4 RT on DNA substrates with a 10-bp microhomology and 6-nt single-stranded gap with the major effect of the A/I mutation being to decrease the rate and/or increase the lag phase for initiation of primer extension (Figure S5C and 5E).
Figure S6, related to Figure 6. Effect of deleting the RT0 loop on the biochemical activities of G2L4 and GII RTs (A-D) Primer extension assays for WT and ΔRT0 mutant G2L4 and GII RTs with DNA and RNA templates with no primer (−) or different length DNA primers with or without 1 mM MnCl2. Primer extension reactions were done as described in Figure S3 and Methods with 3’-blocked 50-nt DNA or RNA templates pre-annealed with different length DNA primers (100 μM 2 nt; 50 μM 5 nt; 500 nM 10, 15, 20-nt primers, respectively). Reactions were done at 37°C for 20 min (panels A and B) or as 240-min time courses (panels C and D). The numbers to the left of the gels indicate the positions of 5’-labeled DNA template and primer oligonucleotides run as size markers in a parallel lane.
(E and F) Terminal transferase reactions time courses for WT (top) or ΔRT0 mutant (bottom) G2L4 or GII RT using a 5’-labeled 50-nt DNA substrate. Plots of the reaction time courses for production of labeled products >50 nt are shown in Figure 6C. Terminal transferase time courses were done as described in Methods using a 5’-labeled 50-nt DNA substrate corresponding to the DNA template used in primer extension reactions without a 3’-blocking group in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled RiboRuler Low Range RNA Ladder (Thermo Scientific) run in a parallel lane.
(G) MMEJ assay by WT or ΔRT0 G2L4 and GII RTs using 5’-labeled DNA substrates having a 3’ overhang with a TTAA-3’ microhomology. MMEJ reaction time courses were done as described in Methods using partially double-stranded 5’-labeled (red star) DNA substrates with 3’ overhangs having complementary TTAA-3’ sequences at their 3’ ends in the presence or absence of 1 mM MnCl2. The numbers to the left of the gels indicate the positions of a 5’-labeled Low Molecular Weight DNA Ladder (New England Biolabs) run in a parallel lane. The plots show the fraction of substrate that was converted to products running between the 100- and 150-nt size markers.
Tables above the plots show the rate constants (kobs) and amplitudes (Ampl.) obtained from fitting the data to a first-order rate equation. Values of Ampl. in parentheses indicate that the amplitude was fixed at the given value because the reaction did not reach an end point during the measurement time.
Figure S7, related to Figure 7. G2L4 and GII RT-mediated repair of CRISPR/Cas9-induced DSBs in the E. coli thyA gene (A) Sequence of a segment of the E. coli HMS174 (DE3) thyA gene showing the CRISPR/Cas9 cleavage site (arrow) and thyA guide RNA (sgRNA; underlined) with the PAM sequence (boxed).
(B) E. coli thymidylate synthase pathway showing the basis for trimethoprim selection for thyA mutations. ThyA catalyzes the reductive methylation of 2’-deoxyuridine-5’-monophosphate (dUMP) to 2’-deoxythymidine-5’-monophosphate (dTMP) by using 5,10-methylenetetrahydrofolate (mTHF) as the methyl donor and reductant and yielding dihydrofolate (DHF) as a by-product. Trimethoprim (TMP) blocks the conversion of dihydrofolate to tetrahydrofolate (THF), which is needed for other cellular processes, resulting in cell growth arrest (Sangurdekar et al., 2011).
(C) Immunoblot showing expression levels of WT and mutant G2L4 or GII RTs and MBP-8XHis from the No RT vector control after induction of pCas9+G2L4 and GII RT plasmids in E. coli HMS174 (DE3).
(D) Bioanalyzer traces of PCR products obtained from genomic DNA using primers P3 and P3’ (Fig. 7 and Table S4) that amplify a 5-kb region around the DSB site in the thyA gene in cells expressing WT or mutant G2L4 (top) or GII (bottom) RTs compared to No RT and No guide RNA vector controls. Bioanalyzer traces were aligned via the peak corresponding to the full-length thyA gene.
(E) Bioanalyzer traces of PCR products obtained from genomic DNA using primers (P2–3 and P2’; Table S4) that better resolve a set of closely spaced peaks in Figure 7D.
(F) Sequencing of deletion junctions resulting from DSBR by MMEJ by WT G2L4 RT. The left-hand panels show bioanalyzer traces of nested PCR products obtained using the primer pairs indicated at the top left of each trace. Gel-purified, size-selected nested PCR products corresponding to the region of the bioanalyzer trace highlighted in yellow were cloned in E. coli HMS174 (DE3), and analyzed by Sanger sequencing (>10 clones for each sequence). The right-hand panels show Sanger sequencing traces across the deletion junction. The schematics below the bioanalyzer traces show annealed microhomologies between direct repeat sequences on either side of the break. The numbers in parentheses indicate E. coli K12 genomic coordinates. The same sequences were obtained for the same peaks in the WT GII RT bioanalyzer traces (Mendeley Data, V3, doi: 10.17632/7dbyk67546.3). *, unidentified peak. Potential microhomologies outside the sequenced regions are in light gray.
Supplemental File, related to Figures 1, 2, 4, and 5. High-throughput sequencing data mapping statistics and gene counts.
Data Availability Statement
Datasets for P. aeruginosa whole-genome sequencing, TGIRT-seq, and sequencing of Snap-Back DNA synthesis and MMEJ products in biochemical experiments have been deposited in the Sequence Read Archive (SRA) under accession number PRJNA814398. A gene counts table, dataset metadata file, and scripts used for data processing and plotting have been deposited in GitHub: https://github.com/reykeryao/Seung. Unprocessed gel images, bioanalyzer traces, Sanger sequencing traces, and repeats of biochemical experiments have been deposited in Mendeley data, V3, doi: 10.17632/7dbyk67546.3.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.