

SUMMARY
Tubulin post-translational modifications (PTMs) alter microtubule properties by affecting the binding of microtubule-associated proteins (MAPs). Microtubule detyrosination, which occurs by proteolytic removal of the C-terminal tyrosine from ɑ-tubulin, generates the oldest known tubulin PTM but we lack comprehensive knowledge of MAPs that are regulated by this PTM. We developed a screening pipeline to identify proteins that discriminate between Y- and ΔY-microtubules and found that Echinoderm-microtubule associated protein like 2 (EML2) preferentially interacts with Y-microtubules. This activity depends on a Y-microtubule interaction motif built from WD40 repeats. We show that EML2 tracks the tips of shortening microtubules, a behavior not previously seen among human MAPs in vivo, and influences dynamics to increase microtubule stability. Our screening pipeline is readily adapted to identify proteins that specifically recognize a wide range of microtubule PTMs.
Keywords: Microtubule, tyrosination, post-translational modification, Echinoderm microtubule-associated protein, WD-repeat
Graphical Abstract
eTOC Blurb
Tubulin tyrosination/detyrosination is a reversible post-translational modification of tubulin. Hotta et al. describe a screening pipeline to identify “readers” of these post-translational modification states and identify EML2 as a tyrosinated microtubule-specific microtubule rescue factor that localizes to shrinking microtubule plus ends.
INTRODUCTION
Intracellular organization is facilitated by the microtubule cytoskeleton, a dynamic array of filamentous polymers assembled from heterodimers of ɑ,β-tubulin. Microtubules are heterogeneous in their composition, owing to the existence of many tubulin isotypes and post-translational modifications (PTMs) that modify the chemical and structural properties of the microtubule 1. One of the best characterized microtubule PTMs is detyrosination, in which the terminal tyrosine is enzymatically cleaved from the C-terminal tail (CTT) of ɑ-tubulin by the heterodimeric enzyme vasohibin (VASH) complexed with small vasohibin binding protein (SVBP) 2,3. Recently, Microtubule Associated Tyrosine Carboxy Peptidase (MATCAP) was identified as a second tubulin carboxypeptidase responsible for microtubule detyrosination. Loss of both MATCAP and VASH/SVBP in the brain of mice completely blocks the formation of ΔY-ɑ-tubulin, suggesting that we now have complete knowledge of the enzymes that generate this form of ɑ-tubulin 4. Detyrosinated tubulin can be further processed to Δ2-tubulin upon removal of the penultimate glutamate residue by cytosolic carboxypeptidases. While the generation of Δ2 ɑ-tubulin is thought to be irreversible, detyrosination can be reversed by tubulin tyrosine ligase (TTL) 5, creating a Y/ΔY cycle.
Detyrosination of a subset of microtubules is thought to be important for processes including intracellular trafficking and cell division. For example, kinetochore-microtubules are progressively detyrosinated as cells progress from prometaphase to metaphase, providing tracks for the kinetochore kinesin motor CENP-E 6. In cardiomyocytes, detyrosinated microtubules anchor to Z-disks of sarcomeres, a connection that enables microtubules to provide a restoring force to cardiomyocytes at the end of a beat cycle 7. Detyrosinated microtubules also promote the fusion of lysozomes and autophagosomes, via kinesin-1, and thereby contribute to the initiation of autophagy 8.
The Y/ΔY cycle does not affect the intrinsic properties of microtubules per se, but rather influences the cohort of microtubule-associated proteins (MAPs) and motor proteins that associate with microtubules 9. A number of proteins have been identified that can discriminate between Y- and ΔY microtubules (Y-readers and ΔY-readers, respectively). For example, cytoplasmic linker protein (CLIP)-170, CLIP-115 and p150glued (a dynactin subunit) share a glycine-rich CAP-Gly domain, which interacts with Y-αCTT using electrostatic and hydrophobic ring stacking interactions, making these proteins Y-readers10. A microtubule depolymerase, kinesin-13 (MCAK; mitotic centromere-associated kinesin) prefers Y-microtubules over ΔY-microtubules, which consequently makes ΔY-microtubules more stable and long-lived11. On the other hand, other kinesins such as kinesin-1 KIF5C and kinesin-7 CENP-E preferentially associate with ΔY-microtubules 12–14. Aside from the CAP-Gly domain, where structural information has shed light on how this module engages the Y-αCTT, mechanisms that allow proteins to discriminate between the Y/ΔY state have not been established and the full repertoire of MAPs and motors that can read the Y/ΔY code is unknown. To address these deficiencies, we established a screening pipeline to identify proteins that bind microtubules in a manner that depends on the Y versus ΔY state.
RESULTS
A screening pipeline to identify candidate proteins that discriminate between the Y/ΔY-αCTT state of microtubules
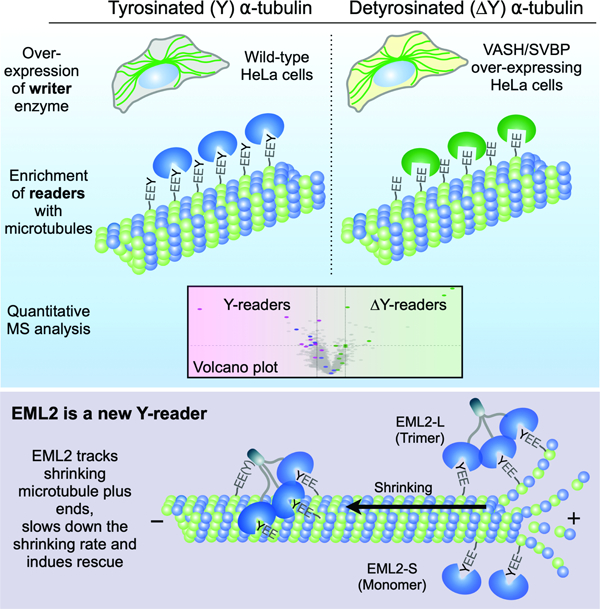
To isolate MAPs that may selectively bind to microtubules in a Y/ΔY-sensitive manner, we prepared MAPs from wild-type HeLa cells and HeLa cells engineered to overexpress VASH1-SVBP. As ɑ-tubulin is largely tyrosinated in HeLa cells 15, we utilized a stable cell line overexpressing VASH1/SVBP (VASH OE) for stoichiometric detyrosination (Figures 1B, S1A and S1B). Although Y-tubulin was detected in the lysate of VASH OE cells (Figure S1B), further detyrosination took place during microtubule assembly upon addition of Taxol and GTP to cell lysates. This caused Y-tubulin to become undetectable in microtubule pellets (Figure 1B), generating a binary screening platform to identify proteins that associate with Y- versus ΔY-microtubules (Figure 1A). Microtubules were assembled in HeLa or VASH1 OE cell extracts, sedimented by centrifugation, and the proteins in the pellet (Figure 1C) were tandem mass-tagged (TMT) 16 and subjected to mass spectrometry (MS). The microtubule interactomes in Y versus ΔY cell extracts demonstrated that CAP-Gly-containing proteins were enriched in the Y-microtubule pellet (Figure 1D, blue dots), an expected result that was also validated by immunoblotting (Figures 1E and S2). We used CAP-Gly proteins as standards to define thresholds of 1.4 and 0.2 on the x- and y-axis of the volcano plot, respectively, to select candidate Y/ΔY-readers. Additional proteins enriched in Y-microtubule pellets include CKAP2, TCP1-eta, and a splice variant of echinoderm microtubule-associated protein like 2 (EML2-S) (Figures 1D, 1F and S2). Our screening pipeline also identified proteins enriched in ΔY-microtubule pellets including SPECC1L and ɑB-crystallin (Figures 1F and S2). We observed good agreement between enrichment scores obtained by TMT analysis and quantitative immunoblotting (Figures 1E, 1F and 1G), suggesting that small fold-changes can be meaningful in isobaric labeling proteomics. Our pipeline to identify proteins differentially enriched in Y-versus ΔY-microtubules is thus sensitive and robust.
Fig. 1. A screening pipeline to identify candidate proteins that discriminate between the Y/ΔY-αCTT state of microtubules.
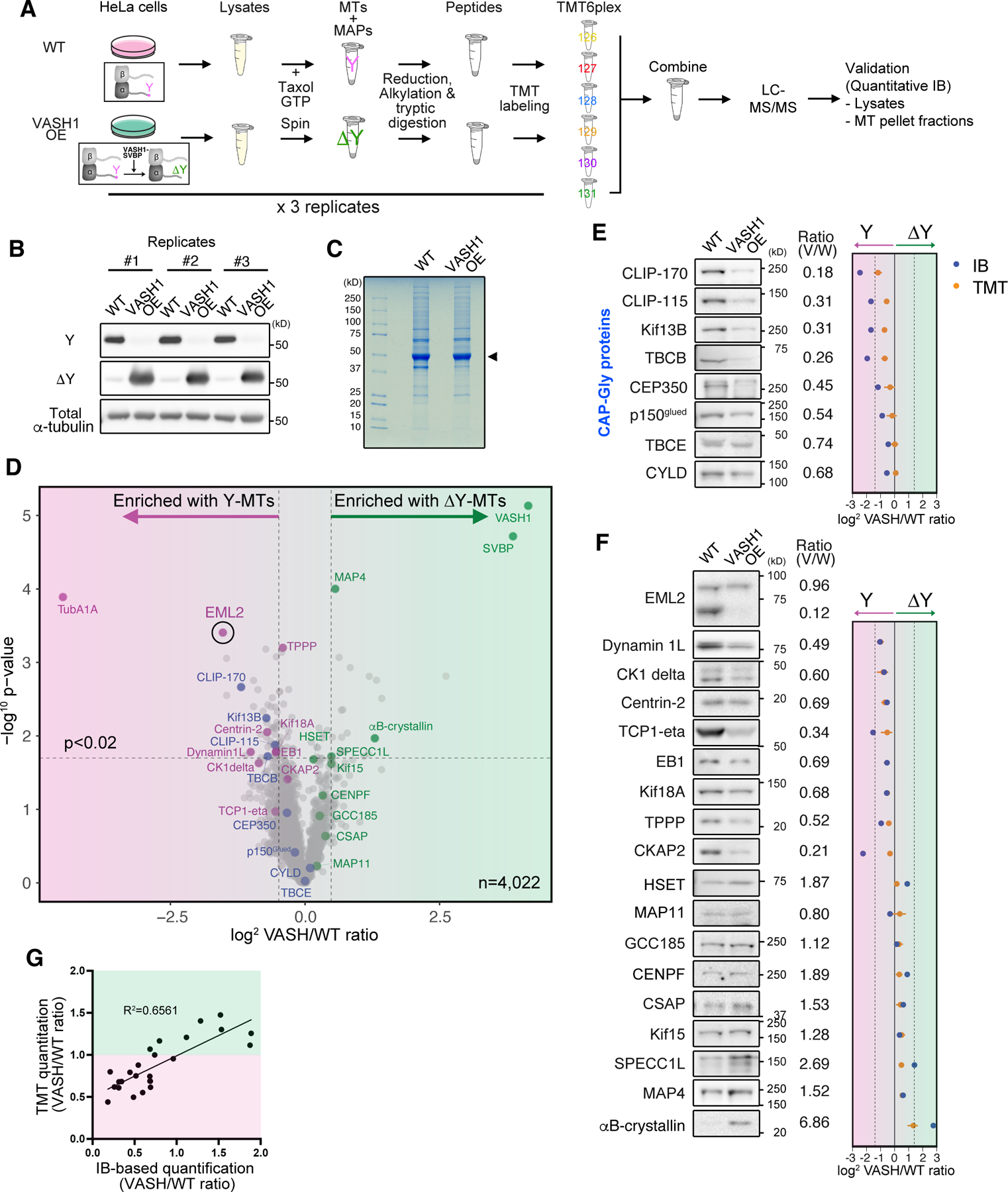
(A) Schematic of the Y/ΔY reader screen. MTs, microtubules, IB, immunoblot analysis. (B) Immunoblot analysis for tubulin Y/ΔY state in the microtubule pellet fractions prepared from wild-type and HeLa cells overexpressing VASH1-SVBP. (C) SDS-PAGE analysis of microtubule pellet fractions. Arrowhead indicates tubulin. (D) Volcano plot depicting proteins co-sedimented with Y- or ΔY-microtubules shown in magenta or green background, respectively. X axis represents log2 relative abundance of each protein in VASH1 OE over wild-type samples (VASH/WT). Y axis shows -log10 p-value. Thresholds of 1.4 (x axis) and 0.02 (y axis) are indicated by dotted lines. Blue dots highlight CAP-Gly proteins. Magenta and green dots represent proteins enriched with Y- and ΔY-readers, respectively. (E and F) Immunoblot analysis of CAP-Gly proteins (E) and Y/∆Y-reader candidates (F) in microtubule pellet fractions. Relative intensity of the bands of VASH1 OE over WT samples (normalized against corresponding total ɑ-tubulin band intensity) is shown next to each blot as well as in the graphs at the right (blue dots). In the same graphs, relative abundance of each hit calculated from the TMT analysis was indicated by orange dot with error bar (SD, n = 3). (G) Comparison of TMT- and immunoblot-based quantitation of relative protein abundance in microtubule pellet fractions. Background color indicates Y-enrichment (magenta) and ΔY-enrichment (green) based on the TMT analysis.
See also Figures S1 and S2 and Data S1.
We used a second approach to identify proteins that preferentially bind Y-versus ΔY-microtubules. Sequences corresponding to the full-length or ΔY ɑ-tubulin CTT of TubA1A were fused to GST (Figure S1C) and the resulting fusion proteins immobilized on glutathione agarose. These resins were used as affinity reagents to capture interacting proteins from CHL-1 cell lysates; CHL-1 cells express high levels of ΔY-tubulin 3, suggesting that these cells may contain readers of both Y- and ΔY-microtubules. Again, mass spectrometry revealed a CAP-Gly protein (CLIP-115) to be Y-microtubule-specific, consistent with previous findings showing that CAP-Gly domains can interact with tubulin solely through the ɑ-tubulin CTT 17. Although we were not able to identify MAPs that specifically associate with the ΔY-ɑ-tubulin CTT, we again detected EML2 as a protein that preferentially binds Y-ɑ-tubulin CTT (Figure S1G; Data S1). Since EML2 was identified in two independent approaches, and it exhibited one of the greatest enrichments in the WT microtubule fractions in the TMT analysis, we focused on EML2 to gain more insight into its ability to interact preferentially with Y-microtubules and function.
The ability of EML2-S to discriminate between Y- and ΔY-microtubules is unique among the EML proteins
Echinoderm microtubule-associated protein (EMAP) is the founding member of a large family of MAPs and associates with microtubules during interphase and mitosis in urchin embryos 18. Mammals express 6 EML proteins 19, with EML1, 2, 3, and 4 sharing similar domain organization consisting of N-terminal coiled‐coil and basic regions that promote trimerization and microtubule binding 20,21, and a C-terminal array of WD (tryptophan-aspartate) repeats (Figure 2A). Structural studies of EML1 show that the WD repeats fold into 13 individual β-sheets that form the blades of two β-propeller structures 22. The first β-propeller is assembled from 7 contiguous WD repeats, but the second β-propeller is atypical in that its 12th blade is partially formed from a hydrophobic EML protein (HELP) motif that is located upstream of sequences that form the first β-propeller. Together, the two-β-propeller architecture is referred to as a TAPE (tandem atypical propeller in EML) domain. In humans, EML1 and EML2 appear to be expressed ubiquitously, and for all EMLs, there is clear evidence of differential splicing 23.
Fig. 2. The ability of EML2-S to discriminate between Y- and ΔY-microtubules is unique among the EML proteins.
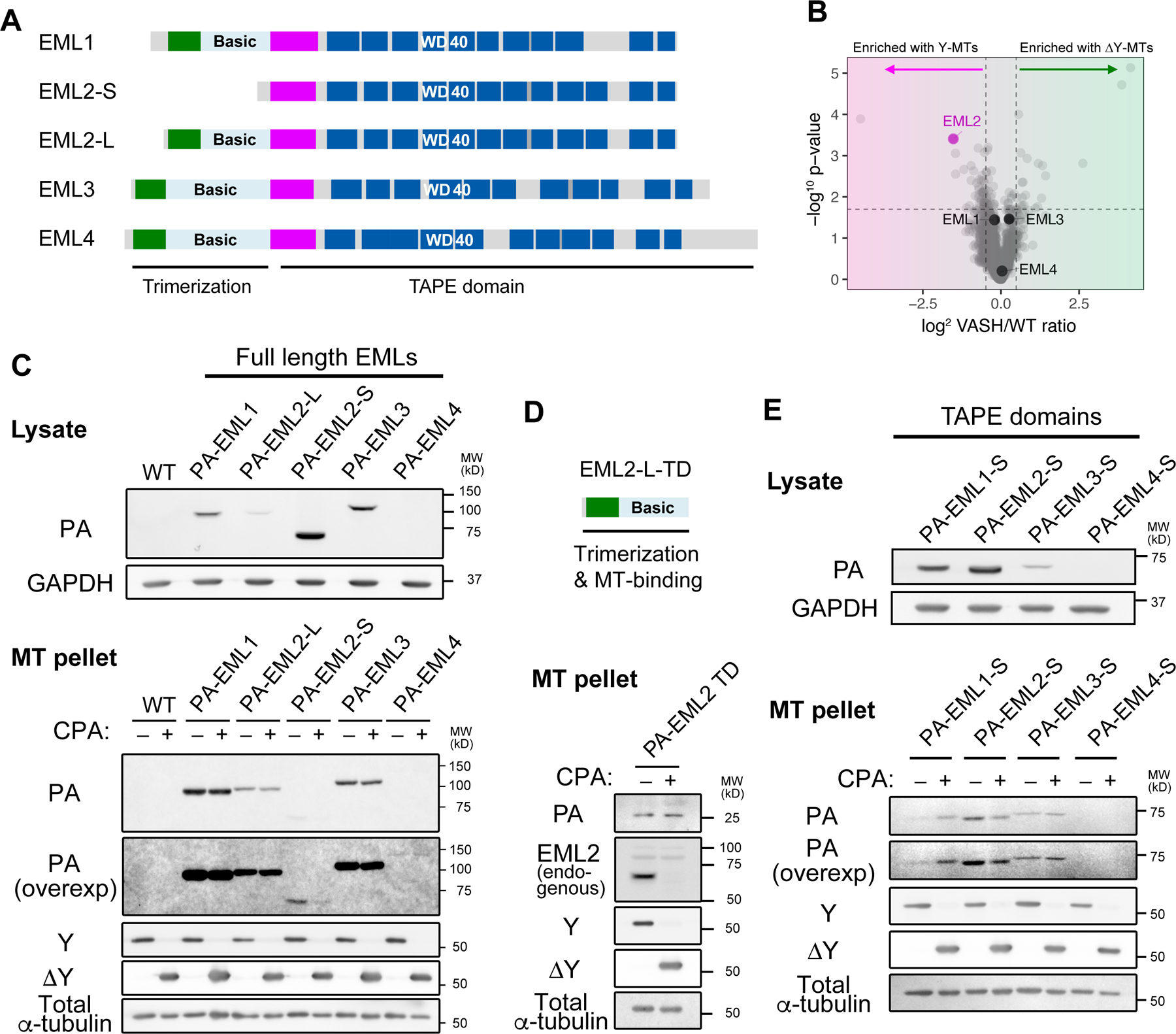
(A) Domain organization of human EML proteins. CC, coiled-coil, Basic, basic and disordered region; HELP; hydrophobic Echinoderm-MAP like protein motif. Dotted lines indicate thresholds as shown in Fig. 1D. (B) Volcano plot of TMT analysis highlighting EML1, 2, 3 and 4. (C) Immunoblot analysis for lysates of HeLa cells overexpressing PA-tagged full length EML1, 2-L, 2-S, 3 and 4 and the microtubule pellet fractions prepared from these lysates treated with (ΔY-conditions) or without (Y-conditions) CPA. (D) Schematic of the EML2-L TD construct and the microtubule co-sedimentation assay using lysate prepared from HeLa cells overexpressing PA-tagged EML2-L-TD protein. Endogenous EML2 blot was shown as a positive control. (E) The same assay shown in (C) and (D), performed with the TAPE domain constructs for EML1, 3 and 4 (PA-EML1-S, 3-S and 4-S).
See also Figure S3.
We detected EML1, 2, 3 and 4 in our Y- and ΔY-interactomes. Despite their well-conserved domain organization (Figure 2A), only EML2 was more abundant in the Y-microtubule fractions (Figure 2B). Further analysis suggested that peptides that correspond to the shortest EML2 isoform are enriched with Y-microtubules (Figures S3A and S3B). To identify which isoform of EML2 can function as a Y-reader, we immunoblotted the Y/ΔY microtubule pellet fractions with an antibody specific to EML2 TAPE domain. The higher molecular weight (MW) band, which corresponds to full-length EML2 (EML2-L), was found in both Y- and ΔY-pellets whereas the lower MW band, which corresponds to isoform 1 (EML2-S, produced by alternative splicing), showed preferential binding to Y-microtubules (Figures 1F and S3C). Association of EML2-S with Y-microtubules was reproduced when using ΔY-microtubules prepared from WT HeLa lysate incubated with carboxypeptidase A (CPA, See STAR Methods; Figure S3D). EML2-S is expected to be a monomer in solution, as it only contains the TAPE domain of EML2 (Figure 2A).
We generated epitope-tagged forms of EML1, EML2-L, EML2-S, EML3 and EML4 to validate EML2-S as a Y-microtubule-specific reader, opting to use the PA-tag 24 since GFP- or SNAP-tags negatively impacted the ability of EML2-S to bind microtubules (Figure S4A). Lysates from cells expressing doxycycline-induced PA-tagged EML proteins were subjected to a microtubule co-sedimentation assay and only EML2-S showed microtubule binding that was sensitive to CPA (Figure 2C), supporting the idea that EML2-S is a Y-microtubule reader. PA-tagged EML2-L was co-pelleted with both Y- and ΔY-microtubules equally. These results agree with immunoblot analysis for the endogenous EML2 proteins (Figures 1F, S2, S3C and S3D). Despite its relatively low expression level, PA-EML2-L was pelleted with microtubules more efficiently than PA-EML2-S. We hypothesized that this is because EML2-L has a second microtubule binding domain in the N-terminal coiled-coil/basic region, which also serves as a trimerization domain (TD) 20,21. A PA-tagged EML2-TD construct co-pelleted with both Y- and ΔY-microtubules (Figure 2D), suggesting that the TD dominantly causes EML2-L to bind microtubules irrespective of their tyrosination state.
In humans, only EML2 appears to be alternatively spliced to produce a short, monomeric isoform. Because the sequences of the EML TAPE domains are highly similar (Figures 2A and S6G), we tested whether the TAPE domains of other EML proteins also preferentially bind Y-microtubules. We transiently expressed PA-tagged TAPE domains of EML1, 3 and 4 (referred to as PA-EML1-S, EML3-S and EML4-S) in HeLa cells and assessed their Y/ΔY preference with the co-sedimentation assay. While EML4-S failed to express in HeLa cells, EML1-S and EML3-S were expressed and, surprisingly, showed behaviors that were different from EML2-S: EML1-S was slightly more enriched in the ΔY-microtubule sample, and EML3-S was co-pelleted equally well with Y-versus ΔY-microtubules. When the founding member of the EML proteins, sea urchin EMAP, was tested with the same assay, we found that PA-tagged EMAP was pelleted equally well with Y- and ΔY-microtubules (Figure S3E). Taken together, these results show that 1) EML2-S preferentially associates with Y-microtubules, 2) the N-terminal microtubule binding domain of EML2-L does not have preference for Y- or ΔY-microtubules, 3) the TAPE domain of other EML proteins, including EMAP, do not have the ability to preferentially bind Y-microtubules.
EML2-S is a Y-αCTT reader
To examine the microtubule-binding properties of EML2-S in cells, we overexpressed PA-tagged EML2-L and S transiently in HeLa cells and performed immunofluorescence staining. While EML2-L and EML2-S decorated microtubules similarly during interphase (Figures 3A and S4B), EML2-S was largely excluded from the midbody in cytokinetic cells (Figures 3B and 3C). The midbody is known to contain high levels of ΔY-microtubules (Figure S4C), so EML2-S’s exclusion from this structure is consistent with its binding preference for Y-microtubules. We also found that EML2-L-TD localized on both interphase and midbody microtubules (Figures S4D and S4E), supporting the above conclusion that the N-terminal microtubule binding domain of EML2 does not have a Y/ΔY preference.
Fig. 3. EML2-S is a Y-αCTT reader.
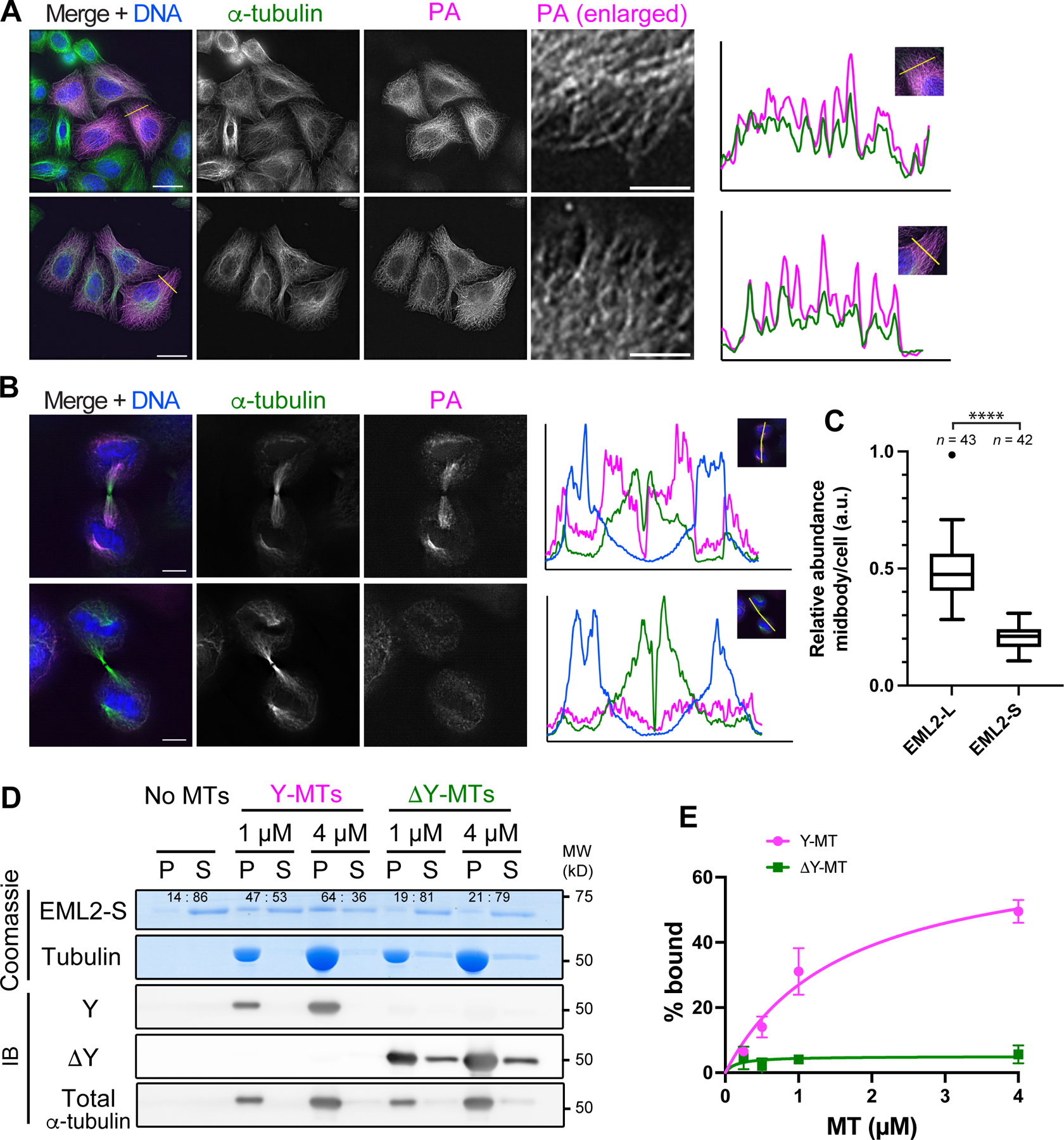
(A, B) Immunofluorescence staining of PA-EML2-L and PA-EML2-S in interphase HeLa cells (A) or cells undergoing cytokinesis (B). In (A), boxed regions in the PA-EML2 channel are enlarged, and line profiles are analyzed along the yellow lines in the merge channels. In (B), line profiles are analyzed on the white lines placed along midbody microtubules as shown in the inserted images on the line profiles. Scale bars, 20 µm [whole cell images of (A)] and 5 µm [enlarged images of (A) and in (B)]. (C) Box plots of relative abundance of PA-EML2 normalized against tubulin in the midbody microtubules over the entire cell area. The box indicates 75th, 50th and 25th percentile. Whiskers and an outlier (shown by a dot) are plotted by the Tukey method. ****, p <0.001. (D) Microtubule co-pelleting assay using purified His-EML2-S and Y- or ΔY-HeLa microtubules. P, pellet; S, supernatant. The ratios of His-EML2-S between pellet and supernatant fractions are quantified and shown on the Coomassie-gel. (E) Quantification of His-EML2-S in the pellet fractions in (D). After baseline (No MTs) subtraction, fractions (%) of His-EML2-S bound to microtubules are plotted with SD (n = 3).
See also Figure S4.
To verify that the Y-microtubule preference of EML2-S is direct, i.e., not mediated by other protein factors, we examined the ability of recombinant EML2-S to bind Y- or ΔY-microtubules in vitro. EML2-S was expressed in and purified from insect cells and assessed for its ability to co-sediment with Y-microtubules or ΔY-microtubules generated with CPA (Figures S4F and S4G). EML2-S showed increased binding to Y-microtubules as compared to ΔY-microtubules (Figures 3D and 3E), allowing us to conclude that EML2-S is a bona fide reader of Y-microtubules, similar to CAP-Gly domain-containing proteins.
Identification of the Y-αCTT recognition motif of the EML2-S TAPE domain
The CTT of ɑ-tubulin is rich in glutamate residues and its binding with MAPs is typically characterized by strong electrostatic interactions. To identify where and how the CTT could interact with EML2-S, we constructed a homology model for EML2-S (see STAR Methods) and then performed Poisson-Boltzmann calculations to look at the protein electrostatics. When the electrostatic potential is mapped on to the protein surface, we observed a large, highly basic patch in the N-terminal β-propeller domain of EML2 (blue surface in Figure 4A). The potential in this region largely comes from four basic residues – R69, R314, R316 and R341 (Figure 4B). At the end of this “R-patch” are a group of aromatic/hydrophobic residues – L209, Y254 and L256, resulting in a putative binding site reminiscent of that found in CAP-Gly domains 17,25. The R-patch would be predicted to have very strong interactions with the glutamates of the CTT, and the C-terminal tyrosine could have both hydrophobic packing and ring-stacking interactions. Molecular docking and subsequent simulation show that the αCTT binds strongly to this region. Figure 4C shows an example of the binding, but since this interaction is largely due to electrostatics, it is highly dynamic and variable and does not exhibit the lock-and-key binding commonly seen in drug interactions.
Fig. 4. Identification of the Y-αCTT recognition motif of the EML2-S TAPE domain.
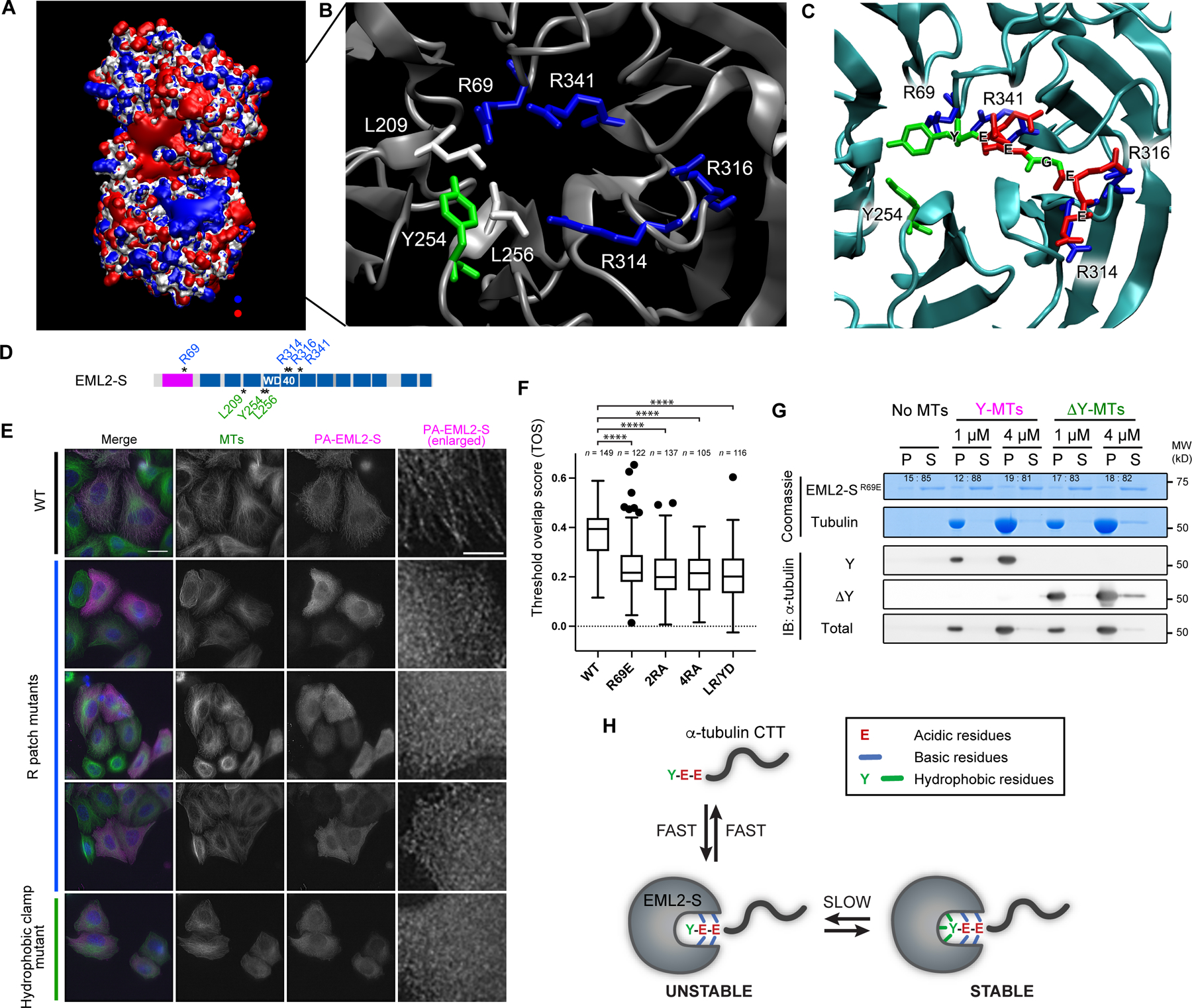
(A) The electrostatic potential of EML2-S. Potential contours are shown for +5/-5 kT/e in blue and red respectively. (B) Molecular details of the positive electrostatic patch in the N-terminal β-propeller. Blue is basic residues; green is aromatic, and white is hydrophobic. Residue numbers are for EML2-S. (C) A molecular model of αCTT (EEGEEY) interacting with EML2-S based off molecular docking and MD simulations. (D) Schematic of EML2-S residues targeted for mutagenesis of the R-patch and hydrophobic clamp. Residues in the R-patch and the hydrophobic clamp are shown in blue and green, respectively. (E) Immunofluorescent staining of HeLa cells overexpressing EML2-S mutant proteins. Scale bars, 20 µm (whole cell images) and 5 µm (enlarged images). (F) Box plots of a colocalization metrics, threshold overlap score (TOS) between EML2-S mutants and microtubules in HeLa cells. The box indicates 75th, 50th and 25th percentile. Whiskers and outliers (shown by dots) are plotted by the Tukey method. ****, p <0.001. (G) In vitro microtubule co-pelleting assay using His-EML2-SR69E mutant and Y/ΔY-HeLa microtubules. EML2-S band intensity was quantified and shown on the Coomassie gel. (H) Model of EML2-S:Y-αCTT binding. A fast but unstable electrostatic interaction between the glutamate residues in the CTT and the R-patch occurs first, and binding is then stabilized by hydrophobic interaction with ɑ-tubulin’s C-terminal tyrosine.
See also Figure S5.
To experimentally test the importance of amino acid residues in the R-patch and hydrophobic “clamp”, we performed mutagenesis (Figure 4D) and analyzed the ability of mutants to bind interphase microtubules in HeLa cells. For the R-patch, the EML2-S charge-reversal mutant R69E failed to interact with microtubules in cells, as did 2RA (R69A/R341A) and 4RA (R69A/R314A/R316A/R341A) mutants. For the hydrophobic clamp, a double mutant in which residues L209 and Y254 were changed to R and D, respectively, also failed to bind microtubules in cells (Figures 4E, 4F and S5). These data support a model in which the N-terminal β-propeller of EML2-S binds the CTT of ɑ-tubulin using a combination of electrostatic and hydrophobic interactions. To further investigate this, we expressed and purified recombinant EML2-SR69E and examined its ability to bind Y- or ΔY-microtubules in vitro using a co-sedimentation assay. Similar to its behavior in cells, EML2-SR69E showed only weak interaction with either Y- or ΔY-microtubules (Figure 4G). Collectively, these data suggest that the interaction of EML2-S with microtubules is initiated by electrostatic interactions and is stabilized by interaction of the C-terminal Y with L209 and Y254 (Figure 4H). With just the electrostatic interaction, we speculate that the detyrosinated CTT is unable to stay in place (Figures 3D and 3E). A similar mechanism underlies the interaction of the 2nd CAP-Gly domain of CLIP170 with the ΔY-ɑCTT 25.
EML2-S is a MAP that stabilizes microtubules
To gain insight into the function of EML2, we reconstituted microtubule dynamics in vitro by growing microtubules from stable GMPCPP microtubule “seeds” and visualized their dynamics in the absence or presence of purified EML2-S protein. In this assay, we used brain tubulin, in which more than the half of the ɑ-tubulin is tyrosinated (Figure S6A), consistent with a previous work26. In the absence of EML2-S, microtubules exhibited typical in vitro dynamics with fast shrinkage rates, and low rescue frequencies (Figures 5A, 5B and S6B) 27. Addition of purified EML2-S resulted in a reduction in microtubule shrinkage rate and catastrophe frequency as well as a dramatic increase in rescue frequency (Figure 5B). Microtubule growth rates were not affected by EML2-S and templated nucleation rates increased only minimally (Figure S6C). Together, these changes in microtubule dynamics lead to increased average microtubule length over time (Figure S6D).
Fig. 5. EML2-S is a MAP that stabilizes microtubules.
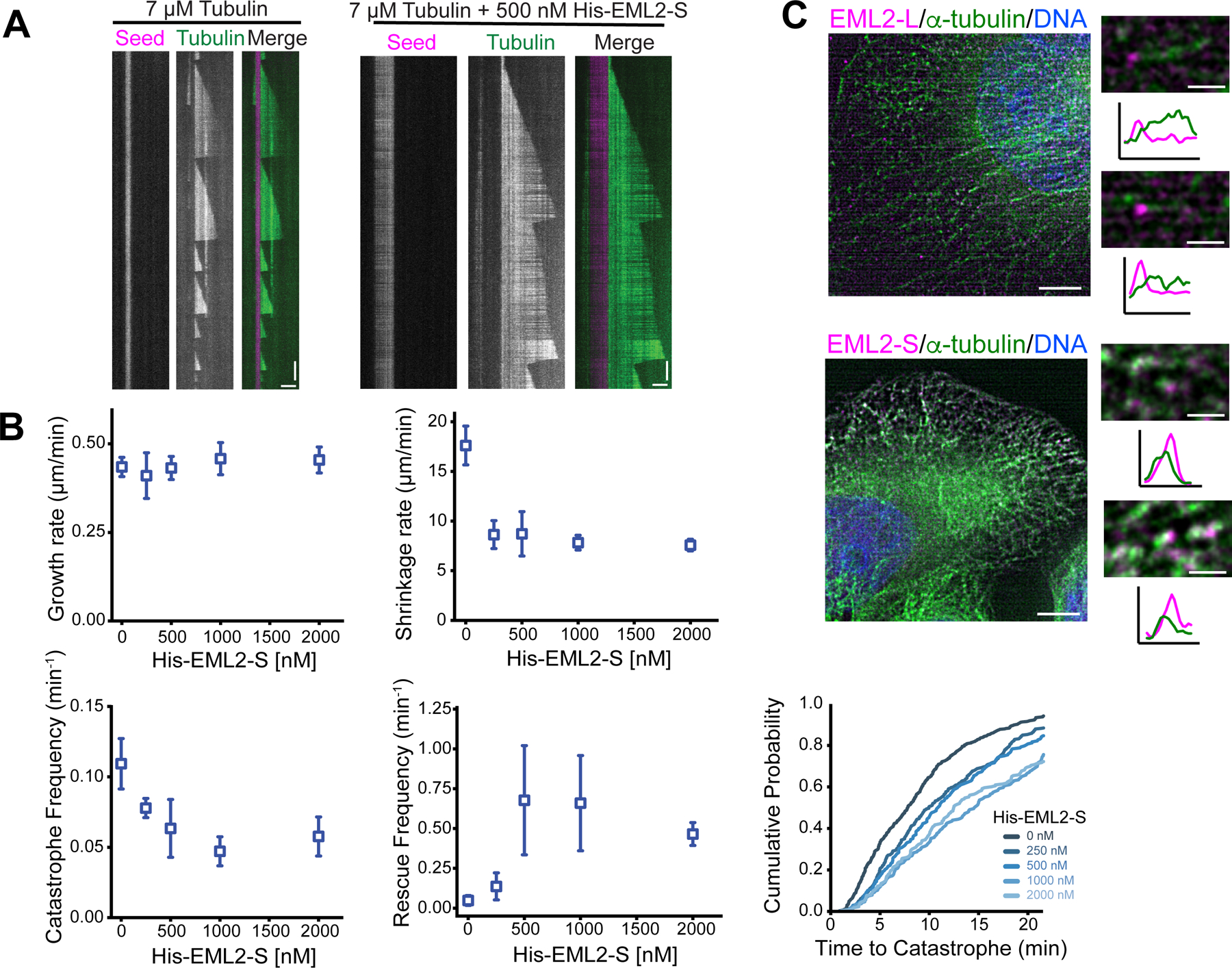
(A) In vitro microtubule dynamics assay using a constant concentration of brain tubulin (7 µM) with or without purified His-EML2-S. Seeds are shown in magenta and dynamic microtubules in green. Vertical bars, 3 min; horizontal bars, 3 µm. (B) Measurements of dynamics parameters. Mean ± SD (from 3–5 independent experiments). (C) Immunofluorescent staining of PA-EML2-L and S in HeLa cells briefly exposed to nocodazole. Boxed regions were enlarged and shown with line profiles analyzed along each microtubule. Arrowheads indicate EML2 enriched at microtubule ends. Magenta, PA-tagged EML2 (L/S); green, ɑ-tubulin; blue, DNA. Bars, 5 µm (whole cell images) and 1 µm (enlarged images).
See also Figure S6.
EML2-S affected microtubule dynamics parameters that typically reflect events at the microtubule plus end, and we therefore speculated that EML2-S may be located at the plus end. However, since fusion of a large tag to either end of EML2-S disrupts its ability to bind microtubules (Figure S4A), we were unable to visualize where EML2-S localizes on microtubules to influence polymerization/depolymerization dynamics in the in vitro assay. PA-tagged EML2-L and EML2-S localized along microtubules as shown by indirect immunofluorescence (Figure 3A), but whether they are enriched at the microtubule ends was unclear. Since EML2-S slowed microtubule shortening rates and increased the frequency of rescue, we hypothesized that EML2-S may enrich on shortening microtubule plus ends. We thus examined EML2-S localization in fixed cells that had been exposed to nocodazole, a drug that induces microtubule disassembly. Brief exposure (16 seconds) of cells to nocodazole caused thinning and fragmentation of interphase microtubule arrays (Figure 5C). Both EML2-L and EML2-S, detected with a PA-tag antibody, localized at microtubule tips suggesting that both isoforms localize to the plus ends of shortening microtubules.
mNeonGreen-tagged EML2-L tracks shrinking microtubule plus ends and slows the microtubule shrinkage rate
To further study the localization and function of EML2 at microtubule plus ends, we expressed mNeonGreen (mNG)-tagged EML2-L in HeLa cells and carried out live imaging. mNG-EML2-L localized to the microtubule lattice but also at ends of microtubules that appeared to be shortening (Figure 6A; Video S1). To investigate this, we examined the localizations of mNG-EML2-L and EB3-mCherry, a protein that tracks growing microtubule ends, in HeLa cells. Strikingly, mNG-EML2-L localized to microtubule ends that were not decorated with EB3-mCherry, indicating that mNG-EML2-L concentrates at the tips of shortening microtubules (Figure 6B; Videos S2 and 3). The decoration of mNG-EML2-L at the shortening microtubule ends was also observed in cells expressing mCherry-TubA1A and mNG-EML2-L (Figure S6E; Video S4), suggesting that the mutual exclusion of EML2 and EB3 is not likely due to a steric interference between two end-binding proteins.
Fig. 6. mNeonGreen-tagged EML2-L tracks shrinking microtubule plus ends and slows the microtubule shrinkage rate.
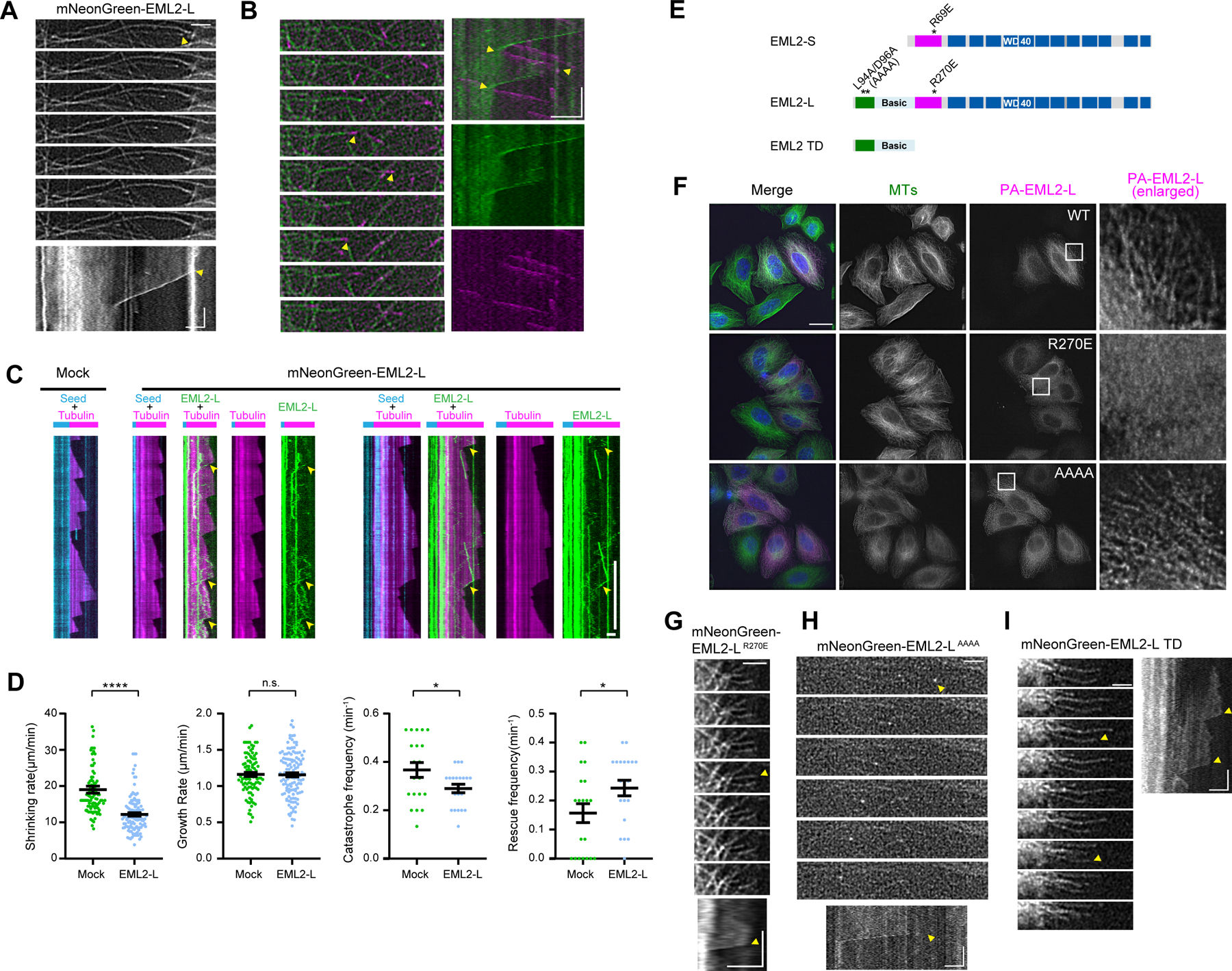
(A) Time lapse images and kymograph of mNG-EML2-L enriched at shortening microtubule ends in HeLa cells. Arrowheads mark the position of shortening microtubule ends. Yellow arrowheads indicate the beginning of a microtubule shortening event. Vertical bar, 30 s; horizontal bars, 2 µm. (B) Time lapse images and kymograph of mNG-EML2-L and EB3-mCherry co-expressed in HeLa cells. Arrowheads mark the position of microtubule ends. Rescue and catastrophe are indicated by yellow arrowheads. Vertical bar, 30 s; horizontal bars, 2 µm. (C) In vitro microtubule dynamics assay with lysates prepared from COS7 cells expressing mNG-EML2-L. Cyan, microtubule seeds; magenta, dynamic microtubules; green, mNG-EML2-L. Vertical bars, 5 min; horizontal bars, 2 µm. Arrowheads indicate enrichment of EML2-L at a shrinking microtubule end. (D) Quantification of the microtubule dynamics. n=~100 events for shrinking rate and growth rate analysis. n=~20 events for catastrophe frequency and rescue frequency analysis from 2–3 independent experiments. Mean ± SEM. *, p<0.05; ****, p <0.001 and n.s., not significant (two-tailed t test) (E) Schematic of EML2-L residues targeted for mutagenesis and truncation. One of the EML2-S mutants analyzed in Fig. 4 (R69E) is shown as a reference. (F) Immunofluorescence of HeLa cells overexpressing PA-tagged mutant EML2-L proteins. Scale bars, 20 µm (whole cell images) and 5 µm (enlarged images). (G-I) Time lapse images and kymograph of mNG-EML2-L R270 (G), mNG-EML2-L AAAA (H) and mNG-EML2-L TD proteins (I). Arrowheads mark the position of shrinking microtubule ends. Yellow arrowheads indicate the beginning of microtubule shortening events. Vertical bar, 30 s; horizontal bars, 2 µm.
As a first step to understand the mechanism by which EML2-L tracks shortening microtubule ends, we performed an in vitro assay where we imaged dynamic microtubules in the absence or presence of cell lysates containing mNG-EML2-L 28. mNG-EML2-L underwent one-dimensional diffusion along the surface of the microtubule and became enriched at shrinking microtubule ends (Figure 6C, yellow arrowheads). In the presence of mNG-EML2-L, the shrinkage rate decreased significantly (mock control, 19.02 ± 0.96µm/min [n = 93]; with mNG-EML2-L, 12.20 ± 0.46 µm/min [n = 117] [mean ± SE]) while the growth rate remained unaffected (mock control, 1.16 ± 0.03 µm/min [n = 99]; with mNG-EML2-L, 1.16 ± 0.03 µm/min [n = 126] [mean ± SE]; Figure 6D), implicating EML2-L as a regulator of microtubule dynamics. In addition, mNG-EML2-L reduced the frequency of microtubule catastrophes (mock control, 0.37 ± 0.03 min−1 [n = 20]; with mNG-EML2-L, 0.29 ± 0.02 min−1 [n = 20] [mean ± SE]) and increased the rescue frequency (mock control, 0.16 ± 0.03 min−1 [n = 20]; with mNG-EML2-L, 0.24 ± 0.03 min−1 [n = 20] [mean ± SE]; Figure 6D). Importantly, these data show that EML2-L and EML2-S appear to similarly affect microtubule dynamics, indicating that the TD does not contribute significantly to the ability of EML2-L to regulate microtubule dynamics.
To understand which microtubule-binding domain is responsible for the shortening end-binding behavior of EML2-L, we generated mutant versions of EML2-L (Figure 6E). First, we produced an R270E mutant in the TAPE domain (corresponding to EML2-S R69E), which has a charge-reversal mutation in the R-patch. Second, we generated an L94A/D96A double mutant within the conserved “ALAD” sequence of TD to weaken the microtubule binding ability of this region (“AAAA” mutant 21). Third, we made a construct that contains the TD alone. Immunofluorescence of PA-tagged versions of mutant EML2-L proteins expressed in HeLa cells showed that microtubule binding of the EML2-LR270E mutant was drastically reduced (Figure 6F). As shown previously 21, the EML2-LAAAA mutant localized to microtubules, albeit with reduced efficiency (Figure 6F). These results suggest that the β-propeller microtubule binding domain of EML2-L is critical for microtubule binding, even in the presence of the N-terminal microtubule binding domain (Figure S4D).
Next, we expressed mNG-tagged versions of the mutant EML2-L proteins in HeLa cells and subjected these cells to live imaging. Again, mNG-EML2-LR270E did not localize on microtubules efficiently (Figure S6F). Occasionally, in cells that expressed the mutant protein at high levels, we detected microtubule-localization of mNG-EML2-LR270E, but this protein did not enrich at microtubule ends (Figure 6G; Video S5). mNG-EML2-LAAAA did not express well, but it decorated microtubules and enriched at shortening microtubule ends (Figures 6H and S6F; Video 5). Finally, mNG-EML2-L-TD decorated the microtubule lattice but we did not detect clear enrichment at microtubule ends (Figures 6I and S6F; Video S5). Taken together, we conclude that the shortening end-binding feature of EML2-L requires the Y-ɑCTT binding module in the TAPE domain, and that the TD may play a minor role in this behavior.
Although EML1–4 are highly conserved and residues we identified as being important for EML2-S to interact with Y-microtubules are preserved in EML1, 3 and 4 (Figure S6G), the TAPE domains of these EMLs differ in their ability to discriminate between Y- and ΔY-microtubules (Figure 2E), suggesting that EML proteins differ in their functional properties. To test this idea, we fused EML1, 3 and 4 to mNG, and imaged them by live cell microscopy. All proteins localized to microtubules, but only mNG-EML1 showed a slight enrichment on microtubule ends (Video S6). Interestingly, mNG-EML3 uniquely caused microtubule curling and fragmentation. Therefore, EML1–4 all engage microtubules, but each isoform has unique activities when bound to microtubules. A more rigorous analysis of EML isoforms will be the subject of future work.
DISCUSSION
We describe a systematic screening pipeline to identify MAPs that can associate with microtubules of different PTM states. Such proteins, termed “readers”, are hypothesized to translate tubulin PTMs into a physiological output. Here, we applied our screening platform to microtubules that differ in their Y/ΔY state. Despite being the first discovered tubulin PTM, we lack a comprehensive list of readers for Y- or ΔY-microtubules. Slow progress in this area may reflect the fact that the enzymes that detyrosinate ɑ-tubulin (“writers”) have only recently been discovered 3,4. By combining writer (VASH1/SVBP) overexpression with quantitative mass spectrometry (TMT analysis), we succeeded in creating a highly-sensitive binary screen in which microtubules were either fully tyrosinated or detyrosinated, and this allowed us to identify EML2 as Y-microtubule-specific microtubule stabilizer. Notably, however, we observed that differences in the affinity of MAPs for Y-versus ΔY -microtubules were not dramatic, even under our conditions. For example, the well-known Y-microtubule reader, CLIP-170, displayed only 2-fold change (VASH/WT = 0.45) in our TMT analysis. Polyvalency of MAPs for their microtubule substrate, where MAPs engage the ɑCTT and the microtubule lattice, may provide one explanation for this observation. The success of future screens targeted at other tubulin PTMs is likely to depend on the degree to which PTMs are written.
The pipeline we describe here requires consideration of several factors during data interpretation. First, overexpression of VASH1/SVBP may affect the state of other PTMs that occur within CTTs, e.g., poly-glutamylation. This concern is not likely to be significant for HeLa cells, which do not have a large amount of poly-glutamylated tubulin29, but should be kept in mind when this screening approach is applied to cell types that do (e.g., neurons). Second, the detyrosination promoted by VASH1/SVBP during microtubule assembly generated a small but detectable level of Δ2-tubulin (Figure S1B), making it possible that some of the proteins enriched in the VASH OE samples prefer the Δ2-state of α-tubulin rather than ΔY-microtubules. Third, VASH/SVBP overexpression may change the expression level of MAPs in cells, although we confirmed that this does not seem to be the case for most of proteins that we tested (Figure S2). Lastly, it is critical to recognize that proteins identified through this screening approach may not directly discriminate between microtubules that are differentially modified, but rather may hitchhike with true reader proteins. In vitro reconstitution is essential to validate candidate readers, as we demonstrated with EML2-S in this study (Figures 3D and 3E).
Our work reveals that the WD-repeats of EML2 form a Y-ɑCTT-binding module, which is noteworthy as only the CAP-Gly domain was previously known to serve as a Y-reader domain. Humans encode 921 WD repeat proteins that carry out a broad range of biological functions 30. A subset of these are MAPs and motor proteins, e.g., KIF21B 31, WDR47 32 and LRRK2 33, but the structural mechanism(s) that enables microtubule association is not known in any of these cases. We show here that the CAP-Gly and TAPE domains share general features that allow them to bind the Y-ɑCTT. Like EML2-S, some CAP-Gly proteins, e.g., CLIPs, use a combination of electrostatic and hydrophobic interactions to engage the Y-ɑCTT. In the case of CAP-Gly domains, synergy between electrostatic and hydrophobic interactions dramatically increase the strength of Y-ɑCTT interactions, and similar to EML2-S charge-neutralization mutants of residues that coordinate ionic interactions with glutamates in the Y-ɑCTT significantly reduce CAP-Gly:Y-ɑCTT interaction strength 25. Intriguingly, CAP-Gly domains use their ability to bind the Y-ɑCTT to track growing microtubule plus ends, but EML2, instead uses its Y-ɑCTT recognition motif to track shrinking microtubule ends. This difference may stem from the interaction of CAP-Gly proteins with other proteins in the +TIP network, which concentrate on growing microtubule plus ends34. Notably, the N-terminus of CLIP-170, a CAP-Gly protein, has been shown to promote rescue in vitro35, but how this activity functions in the context of the full-length protein and how it compares to EML2 remains to be tested.
In addition to showing that the TAPE domain of EML2 is a Y-ɑCTT recognition motif, our work clarifies some confusion regarding the biochemical properties of EML proteins. Previous work showed that the TAPE domain of EML1 is able to associate with tubulin dimers, but not microtubules 21,22. Moreover, recent work suggests that EML2 does not bind microtubules or influence microtubule dynamics in vitro 36. Both studies used EML constructs that fuse fluorescent tags to the TAPE domain. Here, we demonstrate that the addition of large tags to either the N- or C-termini of EML2-S abolish microtubule binding (Figure S4A), and that small tags (e.g., PA or 6xHis) are necessary to study its microtubule binding properties. Unfortunately, this prevented us from visualizing EML2-S on microtubules in real-time both in cells and in vitro. Additional work is needed to identify tagging strategies that overcome this obstacle.
Earlier work suggested that EML2-S could be a microtubule destabilizer 37. In contrast, our in vitro dynamics assay showed that EML2-S stabilizes microtubules by slowing microtubule shortening and increasing rescues. It is difficult to pinpoint the reason(s) for the discrepancies between the two studies because of the many differences in experimental conditions and setups. Eichenmuller et al. used Chlamydomonas axonemes as nucleation sources and video-enhanced DIC to record MT dynamics, whereas we used GMPCPP seeds and imaged fluorescently-labeled MTs with total internal reflection fluorescence microscopy. Moreover, Eichenmuller et al. used 13–16 µM brain tubulin and 500 to 800 nM of ELP70 (EML2-S). In contrast, we tested the activity of EML2-S on 7 µM brain tubulin, with concentrations of EML2-S ranging from 250 nM to 2 µM. In our hands, the effects of EML2-S on microtubule dynamics saturates at <500 nM (Fig 5B). Unfortunately, raw data or conditions including the buffer used, pH, and ionic strength were not provided in the earlier study, so it is not possible to compare these important parameters. An additional consideration is that although we both used brain tubulin, it is possible that the PTM state of tubulin used in the assays may contribute to observed differences. For example, the level of ΔY-ɑ-tubulin is likely to impact the ability of EML2-S to alter microtubule dynamics. Lastly, we showed that EML2-S does not destabilize microtubules in cells (Figure 3A) which is more consistent with the idea that EML2-S is a microtubule stabilizer rather than a destabilizer.
The ability of EML2-L to track shortening microtubule ends is relatively unique among MAPs. In vitro, the yeast Dam1 kinetochore ring complex, human Ska1 complex and microtubule severing protein spastin all track shortening microtubule plus ends 38–40. Similarly, a multivalent peptide construct featuring four lysine-alanine heptarepeats designed to imitate MAPs can diffuse on lattice and track disassembling MT ends 41. In plant cells, a plant specific MAP, SPIRAL2, and NIMA-related kinase 6 (NEK6) are known to track shortening microtubule plus ends in vivo 42,43. Thus, to our knowledge, EML2-L is the first human MAP to track shortening microtubule plus ends in cells.
The mechanism by which EML2-L and -S engage microtubules is complex. Although the TAPE domain is selective for Y-microtubules, our data show that the TD binds Y- and ΔY-microtubules equally well. How does EML2-L utilize these two microtubule binding domains? Mutation of two conserved residues in the TD of EML2-L does not negate binding of this construct to microtubules, as shown for EML1 21. In contrast, a TAPE domain mutation (EML2-L R270E) abolishes microtubule binding of EML2-L (Figures 6F and 6G). These results suggest that the β-propeller’s microtubule binding domain (the Y-αCTT recognition site) is the dominant microtubule binding domain in EML2-L. We speculate that EML2-L first engages the microtubule lattice via the Y-αCTT recognition domain, and that microtubule binding is then stabilized by the TD. Interestingly, although EML1–4 are similar in domain structure, their affinity for Y-versus ΔY-microtubules (Figure 2E) and their localizations in cells differ (Video S6). Moreover, our data show that sea urchin EMAP is not selective for Y-microtubules (Figure S3E). Sequence comparisons of the EMLs have not revealed an obvious reason for why the TAPE domain of EML2 is selective for Y-microtubules, so further structural studies are needed to determine the underlying mechanism.
The screening pipeline described here can be modified to discover readers of other tubulin PTMs, e.g., poly-glutamylation, acetylation, or poly-glycylation, provided that the modifying enzyme is known. Application of this approach to the Y/ΔY cycle enabled us to reveal the complexity of proteins that bind microtubules in a manner that is sensitive to the tyrosination state of ɑ-tubulin. Although we show that cultured cells express a battery of potential Y/ΔY readers, we expect that additional readers will be uncovered in cell types that are more dependent on tubulin PTMs to carry out their physiological functions, such as neurons or cardiomyocytes.
STAR Methods
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ryoma Ohi (oryoma@umich.edu).
MATERIALS AVAILABILITY
Plasmids and cell lines generated in this study are available upon request.
DATA AND CODE AVAILABILITY
Raw LC/MS data and result tables are available through ProteomeXchange with identifier PXD030448. All other data are available in the main text or the supplementary materials.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E.coli strain XL1-Blue (Agilent, Cat# 200236) and DH5α (Invitrogen, 18258–012) were used throughout the molecular cloning. For the production of recombinant GST-CTT proteins, E. coli strain Rosetta 2 (DE3) pLysS (Millipore, Cat# 71403–3) was used. E.coli cells were cultured in standard LB medium supplemented with appropriate antibiotics at 37°C. Cell lines (HeLa, CHL-1, COS7) were cultured in DMEM medium (Thermo Fisher Scientific, Cat# 11965118) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Cat# S11150) and penicillin-streptomycin (Thermo Fisher Scientific, Cat# 15140122). Knock-in HeLa lines for EMLs 1, 2-L, 2-S, 3 and 4, mCherry-TubA1A and VASH1-SVBP 15 were cultured in DMEM medium with 10% tetracycline-negative FBS (R&D Systems, Cat# S10350), penicillin-streptomycin and 1 µg/ml puromycin (Sigma-Aldrich, Cat# P8833). Cells were maintained in the presence of 5% CO2 at 37°C. Expression of transgene(s) in knock-in cells was initiated by the addition of 2 μg/ml doxycycline (Thermo Fisher Scientific, Cat# BP26531). To evaluate the expression level and the size of the expressed proteins by immunoblot analysis, induced or transfected (for conditions, see METHOD DETAILS) cells were lysed with lysis buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 2 mM EDTA, 150 mM NaCl, 1% NP40 and protease inhibitors) with a brief sonication followed by clarification. Sf9 cells were cultured in Grace’s insect medium (Thermo Fisher, Cat# 11605–094) supplemented with 10% FBS, 0.1% Pluronic F-68 (Thermo Fisher, Cat# 24040–032) and antibiotic-antimycotic (Thermo Fisher, Cat# 15240062). High Five cells were maintained in Insect-XPRESS medium (Lonza, Cat# 12–730Q) with antibiotic-antimycotic at 27°C. Cells were not authenticated and are negative for mycoplasma contamination.
METHOD DETAILS
Plasmid construction
Oligonucleotides and plasmids used in this study are summarized in the KEY RESOURCES TABLE in STAR Methods. All PCR and mutagenesis were performed with Prime STAR Max DNA polymerase (Takara bio. Cat# R045B). Unless otherwise noted, Gibson assembly (NEB) was used to insert DNA fragments into vectors. cDNAs were obtained from Horizon Discovery: EML1 (Clone ID 5533599), EML2 (Clone ID 5177401; partial sequence), EML3 (Clone ID 3915493) and EML4 (Clone ID 9021713). A 2,061-nucleotide cDNA of sea urchin EMAP (uniport: Q26613) was synthesized by gBlocks (Integrated DNA Technologies) with codon-optimization. A 36-nucleotide sequence corresponding to the PA tag (5’-GGCGTTGCCATGCCAGGTGCCGAAGATGATGTGGTG-3’) was inserted right after the start codon of pEGFP-C1 and pEGFP-N1 resulting pPA-EGFP-C1 and pPA-EGFP-N1. EGFP of these vectors were modified to the monomeric EGFP with A207K mutation using an oligonucleotide TH460.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-tyrosinated α-tubulin, rat monoclonal, clone YL1//2 | Accurate Chemical and Scientific | Cat# YSRTMCA77G |
| Anti-detyrosinated α-tubulin, rabbit monoclonal, clone RM444 | RevMAb Biosciences | Cat# 31–1335-00 |
| Anti-Δ2 α-tubulin, rabbit monoclonal, clone RM447 | RevMAb Biosciences | Cat# 31–1339-00 |
| Anti-α-tubulin, mouse monoclonal, clone DM1α | Millipore Sigma | Cat# 05–829 |
| Anti-α-tubulin conjugated with FITC, mouse monoclonal, clone DM1α | Sigma-Aldrich | Cat# F2168 |
| Anti-CLIP-170, rabbit polyclonal | Novus Biologicals | Cat# NBP1–85568 |
| Anti-CLIP-115, rabbit | Boster | Cat# A07287 |
| Anti-Kif13B, rabbit polyclonal | Sigma-Aldrich | Cat# HPA025023 |
| Anti-TBCB, mouse monoclonal, clone E-1 | Santa Crutz | Cat# sc-390783 |
| Anti-CEP350, rabbit polyclonal | Novus Biologicals | Cat# NB100–59810 |
| Anti-p150glued, mouse monoclonal | BD Transduction Laboratories | Cat# 610474 |
| Anti-TBCE, rabbit polyclonal | Sigma-Aldrich | Cat# SAB1408635 |
| Anti-CYLD, mouse monoclonal, clone 733 | Thermo Fisher | Cat# 43–7700 |
| Anti-EML2, rabbit polyclonal | Sigma-Aldrich | Cat# HPA012757 |
| Anti-EML2, rabbit polyclonal | Proteintech | Cat# 13529–1-AP |
| Anti-Dynamin 1L, rabbit polyclonal | Thermo Fisher | Cat# PA1–16987 |
| Anti-CK1 delta, goat polyclonal | abcam | Cat# ab48031 |
| Anti-Centrin, rabbit | Gayek and Ohi44 | N/A |
| Anti-TCP1 eta, rabbit polyclonal | Novus Biologicals | Cat# NBP1–88144 |
| Anti-EB1, mouse monoclonal | BD Transduction Laboratories | Cat# 610535 |
| Anti-Kif18A, rabbit | Du et al.45 | N/A |
| Anti-TPPP, goat polyclonal | Thermo Fisher | Cat# PA5–19243 |
| Anti-CKAP2, mouse monoclonal, clone B-12 | Santa Crutz | Cat# sc-398286 |
| Anti-KifC1 (HSET), mouse monoclonal, clone 2B9 | Thermo Fisher | Cat# H00003833-M01 |
| Anti-MAP11, rabbit polyclonal | Sigma-Aldrich | Cat# HPA019359 |
| Anti-GCC185, rabbit polyclonal | Sigma-Aldrich | Cat# HPA035849 |
| Anti-CENPF, rabbit | Novus Biologicals | Cat# NBP2–56124 |
| Anti-CSAP | Backer et al.46 | N/A |
| Anti-Kif15 | Sturgill et al. 47 | N/A |
| Anti-SPECC1L, rabbit polyclonal | Proteintech | Cat# 25390–1-AP |
| Anti-MAP4, rabbit polyclonal | abcam | Cat# ab89650 |
| Anti-αB-crystallin, rabbit polyclonal | Thermo Fisher | Cat# PA1–16950 |
| Anti-PA-tag, rat monoclonal, clone NZ-1 | FUJIFILM Wako Pure Chemical | Cat# 016–25861 |
| Anti-GAPDH, mouse monoclonal, clone G-9 | Santa Crutz | Cat# sc-365062 |
| Anti-rat Alexa 594, goat | Thermo Fisher | Cat# A-11007 |
| Anti-rabbit Alexa 647, goat | Thermo Fisher | Cat# A-21245 |
| Anti-rat Alexa 680, goat | Thermo Fisher | Cat# A-21096 |
| Anti-rabbit Alexa 680, goat | Jackson ImmunoResearch Laboratorie | Cat# 711–625-152 |
| Anti-mouse Alexa 700, goat | Thermo Fisher | Cat# A-21036 |
| Anti-rabbit Alexa 700, goat | Thermo Fisher | Cat# A-21038 |
| Anti-goat Alexa 790, donkey | Thermo Fisher | Cat# A-11370 |
| Anti-mouse DyLight 800, goat | Thermo Fisher | Cat# SA5–10176 |
| Anti-rabbit IRDye 800CW, goat | LI-COR | Cat# 926–32211 |
| Bacterial and Virus Strains | ||
| XL1-Blue | Agilent | Cat# 200236 |
| DH5α | Invitrogen | Cat# 18258–012 |
| Rosetta2(DE3)pLysS | Millipore | Cat# 71403–3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM, high glucose | Thermo Fisher | Cat# 11965118 |
| Opti-MEM I Reduced Serum Medium | Thermo Fisher | Cat# 31985070 |
| Leibovitz's L-15 Medium, no phenol red | Thermo Fisher | Cat# 21083027 |
| Grace's Insect Medium | Thermo Fisher | Cat# 11605094 |
| Insect-XPRESS Protein-free Insect Cell Medium with L-glutamine | Lonza | Cat# 12–730Q |
| Fetal bovine serum (FBS) | Atlanta Biologicals | Cat# S11150 |
| Fetal Bovine Serum (FBS), TET Tested | R&D Systems | Cat# S10350 |
| Penicillin and streptomycin | Thermo Fisher | Cat# 15140122 |
| Antibiotic-Antimycotic | Thermo Fisher | Cat# 15240062 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Doxycycline hydrochloride | Thermo Fisher | Cat# BP26531 |
| Pluronic F-68 Non-ionic Surfactant | Thermo Fisher | Cat# 24040032 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668030 |
| PrimeSTAR max DNA polymerase | Takara Bio. | Cat# R045B |
| T4 Polynucleotide Kinase (PNK) | New England Biolabs | Cat# M0201S |
| T4 DNA ligase | New England Biolabs | Cat# M0202S |
| Carboxypeptidase A (CPA) from bovine pancreas | Sigma-Aldrich | Cat# C9268 |
| Paclitaxel (taxol) | Sigma-Aldrich | Cat# T7191 |
| Dithiothreitol (DTT) | Sigma-Aldrich | Cat# D9779 |
| GTP | Sigma-Aldrich | Cat# G8877 |
| Urea | GE Healthcare | Cat# 17–1319-01 |
| Nocodazole | Sigma-Aldrich | Cat# M1404 |
| SNAP-Cell, Oregon Green | New England BioLabs | Cat# S9104S |
| IPTG | Sigma-Aldrich | Cat# I6758 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 04693159001 |
| PMSF | Sigma-Aldrich | Cat# 78830 |
| Benzonase nuclease | Sigma-Aldrich | Cat# E1014 |
| Lysozyme | Sigma-Aldrich | Cat# L6876 |
| Glutathione Sepharose 4B | GE Healthcare | Cat# 17075601 |
| Reduced glutathione | Sigma-Aldrich | Cat# G4251 |
| SIGMAFAST Protease Inhibitor Cocktail Tablets, EDTA-Free | Sigma-Aldrich | Cat# S8830 |
| Ni-NTA agarose | QIAGEN | Cat# 30230 |
| His-tagged TEV protease | This study | N/A |
| Novex WedgeWell 4 to 20%, Tris-Glycine, Mini protein gels | Invitrogen | Cat# XP04200BOX |
| Fractogel EMD SO3- (M) resin | Millipore-Sigma | Cat# 1168820010 |
| CF640R NHS-Ester | Biotium | Cat# 92108 |
| TAMRA; 5-(and-6)-Carboxytetramethylrhodamine, Succinimidyl Ester, mixed isomers | Thermo Fisher | Cat# C1171 |
| Pluronic-F127 | Sigma-Aldrich | Cat# P2443 |
| ATP | Sigma-Aldrich | Cat# A2383 |
| GMPCPP | Jena Bioscience | Cat# NU-405S |
| NeutrAvidin | Thermo Fisher | Cat# 31000 |
| BSA-biotin | Sigma-Aldrich | Cat# A8549 |
| His-EML2-S protein | This study | N/A |
| HeLa tubulin | PTL paper | N/A |
| HeLa tubulin with CPA-treatment | This paper | N/A |
| Bovine brain tubulin | PTL paper | N/A |
| CF640R-labeled bovine brain tubulin | This study | N/A |
| TAMRA-labeled bovine brain tubulin | This study | N/A |
| X-rhodamine-labeled porcine brain tubulin | Cytoskeleton Inc. | Cat# TL620M |
| Hilyte647-labeled porcine brain tubulin | Cytoskeleton Inc. | Cat# TL670M |
| GST protein | This study | N/A |
| GST-CTT Y protein | This study | N/A |
| GST-CTT ∆Y protein | This study | N/A |
| Critical Commercial Assays | ||
| TMTsixplex Isobaric Label Reagent Set, 1 x 0.8 mg | Thermo Fisher | Cat# 90061 |
| Bio-Rad Protein Assay (Bradford protein assay) | Bio-Rad | Cat# 5000006 |
| Deposited Data | ||
| Raw LC/MS data and search result tables | ProteomeXchange repository | PXD030448 |
| Experimental Models: Cell Lines | ||
| HeLa Kyoto | Shuh Narumiya | RRID: CVCL_1922 |
| Knock-in HeLa Kyoto cell line expressing mCherry-TubA1A | This study | N/A |
| Knock-in HeLa Kyoto cell line co-expressing PA-VASH1-GFP and SVBP-FLAG | Hotta et al.15 | N/A |
| Knock-in HeLa Kyoto cell line expressing PA-EML1 | This study | N/A |
| Knock-in HeLa Kyoto cell line expressing PA-EML2-L |
This study | N/A |
| Knock-in HeLa Kyoto cell line expressing PA-EML2-S | This study | N/A |
| Knock-in HeLa Kyoto cell line expressing PA-EML3 |
This study | N/A |
| Knock-in HeLa Kyoto cell line expressing PA-EML4 | This study | N/A |
| CHL-1 | Thijn Brummelkamp | RRID: CVCL_1122 |
| COS7 | This study | RRID:CVCL_0224 |
| Sf9 | This study | RRID:CVCL_0549 |
| High Five | This study | N/A |
| Oligonucleotides | ||
| TH460, AGCACCCAGTCCAAGCTGAGCAAAGAC, mutagenesis to generate monomeric EGFP with A207K mutation | This study | N/A |
| TH511, ACCCTCTCCTTCAACCGATGAATTAAGCTTGAGCTC, Insertion of TubA1A CTT to pGEX-KGT | This study | N/A |
| TH512, gaggaagaaggagaggaatactgactgactgacgatctgcct, Insertion of TubA1A CTT(Y) to pGEX-KGT | This study | N/A |
| TH513, gaggaagaaggagaggaatgactgactgacgatctgcct, Insertion of TubA1A CTT(∆Y) to pGEX-KGT | This study | N/A |
| TH581, taccgcgggcccgggatccaccggtcgccaccATGGTGAGCAAGGGCGAGGAGG, Inserting mCherry to pEM791-TubA1A | This study | N/A |
| TH582, cgagatctgagtccggacttgtacaGCTCGTCCATG, Inserting mCherry to pEM791-TubA1A | This study | N/A |
| TH607, gatctcgagctcaagcttcgATGCTGGAACGAAGGGCGTTGCTATG, Cloning the partial cDNA of EML2 into pPA-EGFP-C1 or the full length EML2-L into pmNeonGreen-C1 | This study | N/A |
| TH608, cgcggtaccgtcgactgcagaattcTTGCGGCCGCCCTGGTCCAC, Cloning the partial cDNA of EML2 into pPA-EGFP-C1 | This study | N/A |
| TH609, GAGGATGGCTCCGTGAAAATGTTCCTG, Correction of L222 to V | This study | N/A |
| TH610, ggtggaccagggcggccgcAAgGTCAGCCGCCTGGGC, Adding EML2 C-terminal 155 aa | This study | N/A |
| TH611, cgcggtaccgtcgactgcagTCAGACCACCCGCCACTG, Adding EML2 C-terminal 155 aa | This study | N/A |
| TH640, gatctcgagctcaagcttcgATGAGTAGCTTTGGAGCTGGCAAAACCAAAGAAGTTATC, Cloning EML2-S into pPA-EGFP-C1 | This study | N/A |
| TH641, taccgcgggcccgggatccaATGGGCGTTGCCATGCCA, Cloning PA-EMLs into pEM791 | This study | N/A |
| TH642, ctgcacctgagtgtttacttTCAGACCACCCGCCACTG, Cloning PA-EML2-L/S into pEM791 | This study | N/A |
| TH647, gtgccgaagatgatgtggtgCTGGAACGAAGGGCGTTG, Cloning EML2-L into pPA-EGFP-N1 | This study | N/A |
| TH648, ccgcctgaaccgcctccaccGACCACCCGCCACTGTAG, Cloning EML2-L/S into pPA-EGFP-N1 | This study | N/A |
| TH649, gtgccgaagatgatgtggtgAGTAGCTTTGGAGCTGGC, Cloning EML2-S into pPA-EGFP-N1 | This study | N/A |
| TH655, acgtaggcctttgaattccgTCAGACCACCCGCCACTG, Cloning EML2-S into pFastBac-HTA | This study | N/A |
| TH656, tgtattttcagggcgccatgagtagctttggagctggcaaaac, Cloning EML2-S into pFastBac-HTA | This study | N/A |
| TH657, tgggctgggtcctgcaggcgtgggcgttgccatgccag, Cloning EML2-L/S into pSNAPf | This study | N/A |
| TH658, taacctcgagtttaaacgcgttagaccacccgccactgtag, Cloning EML2-L/S into pSNAPf | This study | N/A |
| TH661, CAGCACCACATCATCTTCGGCAC, Removal of EGFP from pPA-EGFP-C1-EML2-L | This study | N/A |
| TH662, GAACGAAGGGCGTTGCTATGGCA, Removal of EGFP from pPA-EGFP-C1-EML2-L | This study | N/A |
| TH664, CACCACATCATCTTCGGCACCTG, Removal of EGFP from pPA-EGFP-C1-EML2-S | This study | N/A |
| TH665, agtagctttggagctGGCAAAACC, Removal of EGFP from pPA-EGFP-C1-EML2-S | This study | N/A |
| TH670, caccacatcatcttcggcacc, Amplifying pPA vector backbone | This study | N/A |
| TH671, tccggactcagatctcgagctc, Amplifying pPA vector backbone | This study | N/A |
| TH672, ggtgccgaagatgatgtggtggaggacggcttctccagctac, Cloning EML1 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH674, ggtgccgaagatgatgtggtggacggggccgcggggcccggt, Cloning EML3 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH675, agctcgagatctgagtccggatcaaacgtcgagggaggaggcgggggacaggg, Cloning EML3 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH676, ggtgccgaagatgatgtggtggacggtttcgccggcagtctc, Cloning EML4 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH677, agctcgagatctgagtccggattaggacgagggcgaagggtc, Cloning EML4 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH702, agctcgagatctgagtccggaCtaaatgacgcgccactgcatg, Cloning EML1 cDNA into pPA-EGFP-C1 | This study | N/A |
| TH703, ctgcacctgagtgtttacttCTAAATGACGCGCCACTGC, Cloning PA-EML1 into pEM791 | This study | N/A |
| TH704, ctgcacctgagtgtttacttTCAAACGTCGAGGGAGGAGG, Cloning PA-EML3 into pEM791 | This study | N/A |
| TH705, ctgcacctgagtgtttacttTTAGGACGAGGGCGAAGG, Cloning PA-EML4 into pEM791 | This study | N/A |
| TH745, CAAATAAAGGTTGGCCGCGCAGTCTCGG, 2 R to A mutagenesis | This study | N/A |
| TH746, TTTGGCCCTGTGgcCACCGTGGCAGA, 2 R to A mutagenesis | This study | N/A |
| TH747, TGGAGGGGGCgcTGATgcGCGGGTGGTCCTCT, Additional 2 R to A mutagenesis | This study | N/A |
| TH748, CAAATAAAGGTTGGCCTCGCAGTCTCGG, R69E (EML2-S)/R270E (EML2-L) mutagenesis | This study | N/A |
| TH750, GGTGGCCACCCTTACAGCCTC, LR/YD mutagenesis | This study | N/A |
| TH753, GAAACCGAAGgATGTGCTGTGTGTG, LR/YD mutagenesis | This study | N/A |
| TH780, gatctcgagctcaagcttcgATGGAGGACGGCTTCTCC, Cloning EML1 into pmNeonGreen-C1 | This study | N/A |
| TH781, cgcggtaccgtcgactgcagCTAAATGACGCGCCACTG, Cloning EML1 into pmNeonGreen-C1 | This study | N/A |
| TH782, gatctcgagctcaagcttcgATGGACGGGGCCGCGGGG, Cloning EML3 into pmNeonGreen-C1 | This study | N/A |
| TH783, cgcggtaccgtcgactgcagTCAAACGTCGAGGGAGGAGGCGG, Cloning EML3 into pmNeonGreen-C1 | This study | N/A |
| TH784, gatctcgagctcaagcttcgATGGACGGTTTCGCCGGC, Cloning EML4 into pmNeonGreen-C1 | This study | N/A |
| TH785, cgcggtaccgtcgactgcagTTAGGACGAGGGCGAAGGG, Cloning EML4 into pmNeonGreen-C1 | This study | N/A |
| TH796, ggtgccgaagatgatgtggtgGGCAAAAAGAACAGTGAAAGCA, Cloning EML1-S into pPA-EGFP-C1 | This study | N/A |
| TH797, ggtgccgaagatgatgtggtgTTCCTTCGAGGGCGCCCCATTAC, Cloning EML3-S into pPA-EGFP-C1 | This study | N/A |
| TH798, ggtgccgaagatgatgtggtgAAAGTTACCAAAACTGCAGAC, Cloning EML4-S into pPA-EGFP-C1 | This study | N/A |
| TH870, GACGCAGAGCAGCCGCCGCCGCCGCCTTTAG, L94A/D96A (AAAA) mutagenesis | This study | N/A |
| TH902, gtgccgaagatgatgtggtgatgagcaacagaccgtcaac, Cloning EMAP cDNA into pPA-EGFP-C1 | This study | N/A |
| TH903, gctcgagatctgagtccggattacaccacgctccattg, Cloning EMAP cDNA into pPA-EGFP-C1 | This study | N/A |
| oMG89, catggacgagctgtacaagTCCGGACTCAGATCTCGAGTAatgcgtgagtgcatctcc, Cloning TubA1A cDNA into pEM791 | This study | N/A |
| oMG90, ggcagagggaaaaagatctcttagtattcctctccttcttc, Cloning TubA1A cDNA into pEM791 | This study | N/A |
| Recombinant DNA | ||
| EML1 cDNA | Horizon Discovery | Clone ID 5533599 |
| EML2 cDNA | Horizon Discovery | Clone ID 5177401 |
| EML3 cDNA | Horizon Discovery | Clone ID 3915493 |
| EML4 cDNA | Horizon Discovery | Clone ID 9021713 |
| TubA1A cDNA | Horizon Discovery | Clone ID 3528520 |
| pEGFP-C1 | Clontech | N/A |
| pmEGFP-C1 | This study | N/A |
| pPA-EGFP-C1-EML2-L | This study | N/A |
| pPA-EGFP-C1-EML2-S | This study | N/A |
| pPA-EGFP-N1-EML2-L | This study | N/A |
| pPA-EGFP-N1-EML2-S | This study | N/A |
| pSNAPf | New England BioLabs | Cat# N9183S |
| pSNAPf-EML2-L | This study | N/A |
| pSNAPf-EML2-S | This study | N/A |
| pPA-EML2-L | This study | N/A |
| pPA-EML2-S | This study | N/A |
| pPA-EML2-L TD | This study | N/A |
| pPA-EML2-S R69E | This study | N/A |
| pPA-EML2-S R69A/R341A (2RA) | This study | N/A |
| pPA-EML2-S R69A/R314A/R316A/R341A (4RA) | This study | N/A |
| pPA-EML2-S L209R/Y254D (LR/YD) | This study | N/A |
| pPA-EML2-L R270E | This study | N/A |
| pPA-EML2-L AAAA | This study | N/A |
| pPA-EML1 | This study | N/A |
| pPA-EML3 | This study | N/A |
| pPA-EML4 | This study | N/A |
| pPA-EML1-S | This study | N/A |
| pPA-EML3-S | This study | N/A |
| pPA-EML4-S |
This study | N/A |
| pPA-EMAP | This study | N/A |
| pEM791 | Eugene Makeyev; Khandelia et al.48 | N/A |
| pEM791-PA-EML1 | This study | N/A |
| pEM791-PA-EML2-L | This study | N/A |
| pEM791-PA-EML2-S | This study | N/A |
| pEM791-PA-EML3 | This study | N/A |
| pEM791-PA-EML4 | This study | N/A |
| pEM791-mCherry-TubA1A | This study | N/A |
| pmNeonGreen-C1 | Allele Biotechnology | N/A |
| pmNeonGreen-C1-EML2-L | This study | N/A |
| pmNeonGreen-C1-EML2-L R270E | This study | N/A |
| pmNeonGreen-C1-EML2-L AAAA | This study | N/A |
| pmNeonGreen-C1-EML2-L TD | This study | N/A |
| pmNeonGreen-C1-EML1 | This study | N/A |
| pmNeonGreen-C1-EML3 | This study | N/A |
| pmNeonGreen-C1-EML4 | This study | N/A |
| pFastBac-HT A | Thermo Fisher | Cat# 10584027 |
| pFastBac-EML2-S | This study | N/A |
| pFastBac-EML2-S R69E | This study | N/A |
| pGEX-KGT | This study | N/A |
| pGEX-KGT-αCTT(Y) | This study | N/A |
| pGEX-KGT-αCTT(∆Y) | This study | N/A |
| Software and Algorithms | ||
| Fiji/imageJ, version | NIH image | https://imagej.nih.gov/ij/; RRID: SCR_003070 |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com |
| OriginPro2020 | OriginLab | https://www.originlab.com/2020 |
| Python 3 | Python | https://www.python.org |
| Proteome Discoverer v2.1, v2.3 | Thermo Fisher | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com |
| FragPipe version 17.1 | Alexey Nesvizhskii lab | https://msfragger.nesvilab.org/ |
| MSFragger version 3.4 | Alexey Nesvizhskii lab; Kong et al.49 | https://philosopher.nesvilab.org/ |
| Philosopher version 4.1.1 | Alexey Nesvizhskii lab; da Veiga Leprevost et al.50 | https://fragpipe.nesvilab.org/ |
| TMT-Integrator version 3.2.1 | Alexey Nesvizhskii lab | https://tmt-integrator.nesvilab.org/ |
To clone EML2-L, EML2 cDNA was PCR-amplified with primers TH607 and TH608 and inserted into pPA-EGFP-C1 that had been linearized with EcoRI (pPA-EGFP-C1-EML2-partial). Since the inserted EML2 sequence lacks C-terminal 155-amino acid (aa) of EML2-L (isoform 3 [850 aa] on uniport database), a corresponding 465-nucleotide fragment was synthesized by gBlocks (Integrated DNA Technologies). gBlocks fragment was PCR amplified with primers TH610 and TH611 and inserted into EcoRI-treated pPA-EGFP-C1-EML2-partial. To match the database sequence of EML2 isoform 3, a single amino acid substitution of EML2L cDNA was corrected (L222V) using an oligonucleotide TH609. This vector, pPA-EGFP-C1-EML2-L, was further used as template to amplify EML2-S (uniport isoform 1 [649 aa]) using TH640 and TH611, and the amplified fragment was cloned into EcoRI-treated pPA-EGFP-C1 to assemble pPA-EGFP-C1-EML2-S. These constructs were used as templates to make C-terminally-GFP-tagged EML2 constructs (pPA-EGFP-N1) with primers TH647 and TH648 (EML2-L) or TH649 and TH648 (EML2-S), or N-terminally-SNAP-tagged constructs (BamHI site of pSNAPf; NEB, Cat# N9183S) using primers TH657 and TH658 (for both EML2-L and S).
To generate pPA-EML2-L and S (N-terminal PA-tag), EGFP was removed from pPA-EGFP-C1-EML2-L and S. pPA-EGFP-C1-EML2-L and S were PCR-amplified with primers TH661 and TH662 (EML2-L) or TH664 and TH665 (EML2-S) that had been phosphorylated using T4 polynucleotide kinase (NEB, Cat# M0201S), and then the PCR products were ligated using T4 DNA ligase (NEB, Cat# M0202S). Site-directed mutagenesis was performed against pPA-EML2-S for R69E, R69A/R341A (2RA), R69A/R314A/R316A/R341A (4RA) and L209R/Y254D (LR/YD) using primers TH748 (R69E), TH745 and TH746 (2RA), TH747 (additional 2 sites for 4RA) and TH750 and TH753 (LR/YD), respectively. pPA-EML2-L R270E and pPA-EML2-L L94A/D96A (AAAA mutant) were generated with primers TH748 and TH870, respectively. pPA-EML2-L TD construct was generated by mutating Lys209 of EML2-L to a stop codon in pPA-EML2-L by site-directed mutagenesis using primer TH731.
cDNAs of EML1, 3 and 4 and EMAP were amplified using TH672 and TH702 (EML1), TH674 and TH675 (EML3), TH676 and TH677 (EML4) and TH902 and TH903 (EMAP) and inserted into pPA-EGFP-C1 that had been PCR-amplified with TH670 and TH671 (EGFP was excluded) to generate pPA-EML1, 3 and 4 and EMAP (N-terminal PA-tag). To make inducible HeLa cell lines for PA-tagged EMLs, corresponding pPA-EML constructs were used as templates to generate PA-EML fragments with the following primers: TH641 and TH703 (PA-EML1), TH641 and TH642 (PA-EML2-L and S), TH641 and TH704 (PA-EML3) and TH641 and TH705 (PA-EML4). PA-EML fragments were then inserted into the pEM791 vector that had been digested with AgeI and BsrGI. The resulting pEM791-PA-EML vectors were used to establish knock-in HeLa cell lines expressing PA-EMLs in a doxycycline inducible manner using recombination mediated cassette exchange 48. To generate the TAPE domain constructs, pPA-EML1-S, 3-S and 4-S, cDNA fragments lacking N-terminal 166 (EML1), 206 (EML3) and 281 (EML4) amino acids were cloned in the PA-tag vector. Primers used are TH796 and TH702 (EML1-S), TH797 and TH765 (EML3-S), and TH798 and TH677 (EML4-S).
To assemble pmNeonGreen-EML2-L, EML1, EML3 and EML4 (N-terminal mNeonGreen-tag), inserts were amplified with TH607 and TH611 (EML2-L), TH780 and TH781 (EML1), TH782 and TH783 (EML3) and TH784 and TH785 (EML4) and assembled into EcoRI-treated pmNeonGreen-C1. Mutant versions of pmNeonGreen-EML2-L were generated as described for the pPA-EML2-L above. pmCherry-N1-EB3 was described previously 51. To assemble pEM791-mCherry-TubA1A, first TubA1A cDNA was amplified with primers oMG89 and oMG90, and cloned into pEM791 vector between BsrGI and BglII sites. Then, to swap GFP with mCherry, an mCherry fragment amplified with TH581 and TH582 was inserted to the resulting vector between AgeI and BsrGI sites.
pFastBac-EML2-S was assembled from an EML2-S fragment amplified with TH655 and TH656 and BamHI-digested pFastBac-HT A (Thermo Fisher Scientific). To introduce the R69E mutation, TH748 was used.
To generate GST-CTT(Y), GST-CTT(ΔY) constructs, a modified version of pGEX-KG vector where the thrombin site had been replaced with TEV protease recognition site, was used (pGEX-KGT). The vector was PCR-amplified with phosphorylated primers TH511 and TH512 (Y) and TH511 and TH513 (ΔY), and ligated.
Cell cycle analysis
Cellular DNA content was analyzed with flow cytometry. HeLa cells overexpressing VASH1-SVBP in the presence of doxycycline for 4 days and WT HeLa cells were fixed in cold ethanol, treated with 100 µg/ml RNase Type I-A and 50 µg/ml propidium iodide, and analyzed by BD LSR Fortessa Cell Analyzer (BD Biosciences). Cell cycle was analyzed with FlowJo v10.8.1.
Microtubule co-sedimentation assay using lysates
Microtubule co-sedimentation was performed based on a published method 52. For the Y-conditions, WT HeLa cells were used. For the ΔY conditions, VASH1-SVBP knock-in HeLa cells were cultured in the presence of 2 μg/ml doxycycline for 4 days. Cells were harvested with trypsin, rinsed in DPBS (Thermo Fisher Scientific, Cat# 14190144) twice and suspended in ice-cold BRB80 (80 mM PIPES, 1 mM EGTA, 1 mM MgCl2 pH 6.8) supplemented with protease inhibitor cocktail (Complete mini EDTA free; Roche, Cat# 04693159001) and 1 mM DTT (Sigma-Aldrich, Cat# D9779). After sonication for 10 sec x 4 times, lysates were cleared by centrifugation at 100,000 x g for 1 hour at 4°C. To induce microtubule assembly, 10 µM Taxol (Sigma-Aldrich, Cat# T7191) and 1 mM GTP (Sigma-Aldrich, Cat# G8877) were added and incubated at 37°C for 25 min. The assembly mix was layered on a pre-warmed sucrose cushion (5% sucrose in BRB80 with 10µM Taxol and 1 mM GTP) and centrifuged at 80,000 x g for 30 min at 37°C. Microtubule pellets were rinsed with BRB80-Taxol/GTP twice, resuspended in BRB80 supplemented with 8 M urea and incubated on ice for 10 min. Protein concentration was measured by Bradford Protein Assay (Bio-Rad, Cat# 5000006). The assay was repeated three times on different days and a total of 6 samples (each 25 µg of tubulin + MAPs) were subjected to a TMT analysis. For CPA-mediated tubulin detyrosination in the WT HeLa lysate, 1/400 the lysate volume of CPA (Sigma-Aldrich, Cat# C9268) was added to the assembly mix and incubated on ice for 3 min prior to the microtubule assembly incubation.
GST-CTT pulldown
pGEX-KGT-ɑCTT constructs were transformed into BL21 (DE3). IPTG-induced gene expression occurred for 4 h at 25°C. Bacteria were pelleted, rinsed in PBS once and resuspended in lysis buffer (PBS, 1 mM MgCl2, 0.5% Triton-X100, 1 mM ATP, 1 mM DTT, 1 mM PMSF, 0.1 mg/ml lysozyme (Sigma-Aldrich, Cat# L6876) and Benzonase nuclease (Sigma-Aldrich, Cat# E1014). Bacterial lysates were sonicated and centrifuged (100,000 x g, 30 min, 4°C). Cleared lysates were loaded onto Glutathione sepharose 4B columns (GE Healthcare, Cat# 17075601) that had been pre-equilibrated with the lysis buffer. Columns were washed with 10 column volumes each of wash buffer 1 (PBS, 1 mM MgCl2, 1 mM ATP, 1 mM DTT, and 0.2 mM PMSF) and wash buffer 2 (PBS, 1 mM DTT, and 0.2 mM PMSF), and GST-CTT proteins were eluted with elution buffer (PBS with 10 mM reduced glutathione). Glutathione was removed by repeated ultrafiltration using Amicon Ultra-4 10K (Merck Millipore, Cat# UFC801024D) and protein concentration in the final fractions were measured with Bradford protein assay.
The pulldown assay was performed as follows. Glutathione Sepharose 4B resin (24 µl for each reaction) was rinsed in PBS 5 times prior to the addition of 370 µg of GST, GST-CTT (Y and ΔY). GST-resin conjugates were incubated on ice for 1 h and excess proteins were removed by 3 times of wash with the pulldown buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl). CHL-1 cell lysates were prepared in lysis buffer (pulldown buffer supplemented with protease inhibitor cocktail [Complete mini EDTA free], 0.2 mM PMSF and 1 mM DTT). After sonication, lysates were cleared by centrifugation (56,000 x g, 20 min, 4°C). Lysates (corresponding to cells grown on two 15-cm dishes) were added to each GST-resin conjugate and gently mixed for 30 min at 4°C. For the negative control, lysates were replaced with lysis buffer. Resins were washed with the lysis buffer twice followed by the pulldown buffer three times. Bound proteins were eluted with GST proteins with 60 mM reduced glutathione in the pulldown buffer and analyzed on a precast gel.
For the identification of bound proteins with MS analysis, the above elution step was modified to avoid introducing overwhelming amounts of GST and GST-CTT proteins to the MS. After rinsing the resins with the pulldown buffer, resins were resuspended in pulldown buffer supplemented with 1 mM EDTA and 1 mM DTT. His-tagged TEV protease was added and incubated overnight at 4°C. After quick spin, the supernatants were transferred to new tubes and mixed with Ni-NTA resins that had been equilibrated with the pulldown buffer. After 2 h of incubation at 4°C, supernatants were recovered and subjected to the MS analysis.
Mass spectrometry
For the 6 samples generated in the microtubule co-sedimentation assay, each 25 µg protein sample was digested and labeled with TMT 6-plex isobaric tags following the manufacturer’s protocol (Thermo Fisher). Samples were first diluted to ~1 M urea, reduced with 5 mM DTT for 30 min at 45°C, then alkylated with 15 mM 2-chloracetamide for 30 min at room temperature in the dark. Sequencing grade modified trypsin (Promega) was added at a 1:25 enzyme:protein for overnight (~16 hr) digestion at 37°C with constant mixing. Digestion was stopped by acidification, and peptides desalted using SepPak C18 cartridges according to the manufacturer’s protocol (Waters, Cat# WAT023501). Samples were completely dried using a vacufuge and reconstituted in 100 ul of 0.1M TEAB. TMT 6-plex reagents were dissolved in 41 µl anhydrous acetonitrile, to which each digest was added and incubated at room temperature for 1 h. Reactions were quenched by adding 8 µl of 5% hydroxylamine for 15 min. Labeled samples were mixed together and dried via vacufuge. Offline high pH reversed-phase fractionation of the combined sample into 8 fractions was performed according to the manufacturer’s protocol (Thermo Fisher, Cat# 84868). Fractions were dried and reconstituted in 9 µl of 0.1% formic acid/2% acetonitrile in preparation for LC-MS/MS analysis. For each fraction, a 2 µl aliquot was analyzed by LC-MS/MS using a RSLC Ultimate 3000 nano-UPLC (Dionex) with a 50 cm, 75 µm i.d. C18 column (Thermo Fisher Cat # ES903) and an Orbitrap Fusion (Thermo Fisher). A 3-hour gradient at 300 nl/min using 0.1% formic acid/acetonitrile (2–22% acetonitrile in 150 min; 22–32% acetonitrile in 40 min; 20 min wash at 90% followed by 50 min re-equilibration) was used, and eluent was introduced in the mass spectrometer via an EasySpray source (Thermo Fisher). The mass spectrometer was set to collect MS1 scans (Orbitrap; 120K resolution; AGC target 2x105; max IT 100 ms) followed by Top Speed MS2 scans (0.5 m/z isolation width, collision induced dissociation; ion trap; NCE 35; AGC 5x103; max IT 100 ms). For multinotch-MS3, the top 10 precursors from each MS2 were fragmented by HCD followed by Orbitrap analysis (NCE 55; 60K resolution; AGC 5x104; max IT 120 ms, 100–500 m/z scan range). Proteome Discoverer (v2.3; Thermo Fisher) was used for data analysis. Spectral files were searched against the SwissProt human protein database (20353 protein sequences downloaded 06/20/2019) using the following search parameters: MS1 and MS2 tolerance were set to 10 ppm and 0.6 Da, respectively; carbamidomethylation of cysteines and TMT labeling of lysine and N-termini of peptides were considered static modifications; oxidation of methionine and deamidation of asparagine and glutamine were considered variable. Identified proteins and peptides were filtered to retain only those passing a 1% FDR threshold. Quantification was performed using high-quality MS3 spectra (average signal-to-noise ratio >6 and <30% isolation interference). For the analysis of EML2 isoforms, a separate search was conducted using the same SwissProt human database with EML2 isoforms 1, 2, and 3 added (UniProt identifiers O95834–1, O95834–2, and O95834–3). Abundances of peptides unique to isoforms 2 and 3 were compared between VASH and WT samples.
For the supernatants from the GST-CTT pulldown, cysteines were reduced then alkylated with 10 mM DTT at 45° C for 30 min and 65 mM 2-chloroacetamide for 30 min at room temperature in the dark, respectively. Sequencing grade modified trypsin (Promega) was added at a 1:50 enzyme:protein for overnight digestion at 37°C with constant mixing. Digestion was stopped by acidification, and peptides desalted using SepPak C18 cartridges according to the manufacturer’s protocol (Waters, Cat# WAT023501). Eluate was dried using vacufuge, then peptides were reconstituted in 8 µl of 0.1% formic acid/2% acetonitrile solution in preparation for LC-MS/MS analysis. For each sample, 2 µl of the peptide solution were resolved on a 50 cm, 75 µm i.d. C18 column (Thermo Fisher, Cat# ES903) using a 0.1% formic acid/2% acetonitrile (Buffer A) and 0.1% formic acid/95% acetonitrile (Buffer B) gradient at 300 nl/min over a period of 180 min (2–22% buffer B in 110 min, 22–40% in 25 min, 40–90% in 5 min followed by holding at 90% buffer B for 5 min and equilibration with Buffer A for 25 min). Eluent was directly introduced into Orbitrap Fusion tribrid mass spectrometer (Thermo Fisher) using an EasySpray source. MS1 scans were acquired at 120K resolution (AGC target 2x105; max IT 100 ms). HCD MS/MS spectra were acquired using the Top speed method following each MS1 scan (NCE ~32%; AGC target 5x104; max IT 50 ms, 15K resolution) with 0.8 m/z isolation width. Spectra were searched against a SwissProt human database (28476 reviewed entries; downloaded on 08/29/2018) appended with GST-CTT Y protein using Proteome Discoverer (v2.1, Thermo Fisher). Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.2 Da; two missed cleavages were allowed; carbamidomethylation of cysteine was set as a fixed modification, and oxidation of methionine, deamidation of asparagine and glutamine were set as variable modifications. False discovery rates (FDRs) were estimated with Percolator, and peptides and proteins were filtered to 1% FDR.
Immunofluorescence staining
HeLa cells were seeded on coverslips 24 h prior to transfection using lipofectamine 2000 (Invitrogen, Cat# 11668030). Each 1–3 µg of plasmid was used per well on a 6-well plate. After 3 h of incubation with plasmid-transfection reagent in Opti-MEM (Thermo Fisher Scientific, Cat# 31985–070), cells were rinsed and cultured in DMEM containing FBS and antibiotics overnight and subjected to staining. For nocodazole treatment, coverslips were soaked in culture media containing 664 nM nocodazole (Sigma-Aldrich, Cat# M1404) for 16 sec prior to the fixation. Staining was performed as described 15. Antibodies were applied in the following order: Anti-PA tag antibody (clone NZ-1, Fujifilm Wako Pure Chemical, Cat# 016–25861, diluted at 1:500, 45 min), anti-rat Alexa Fluor 594 (Thermo Fisher, Cat# A11007, diluted at 1:2,000, 35 min) and anti-ɑ-tubulin DM1ɑ conjugated with FITC (Sigma-Aldrich, Cat# F2168, diluted at 1:500, 30 min). When ∆Y-tubulin was co-stained, anti-detyrosinated α-tubulin antibody (Clone RM444; RevMAb Biosciences, Cat# 31–1335-00, final concentration 1 µg/ml, 45 min) and then anti-rabbit Alexa Fluor 647 (Thermo Fisher, Cat# A21245, diluted at 1:2,000, 35 min) were applied between Rat Alexa Fluor 594 and DM1α-FITC staining. Images were obtained with a DeltaVision microscope equipped with an Olympus Plan Apo N 60x/1.42 oil immersion lens and deconvolved. Images of single optical sections were presented. Line profile was analyzed with Fiji/ImageJ (line width, 5 pixels across interphase microtubules in Figure 3A; 15 pixels along midbody microtubules in Figure 3B). To quantitatively evaluate the accumulation of PA-EML2-L or PA-EML2-S in the midbody microtubules, first, the cell boundary was manually traced, and midbody regions were determined by the 0.5 percentile thresholding of the blurred microtubule images with Gaussian Filter (Sigma = 6 pixels). Then the PA intensities were measured and normalized against ɑ-tubulin intensities in both regions. The relative PA intensity in the midbody divided by the relative PA intensity in the cell was calculated as an indicator of the accumulation of PA-EML2 in the midbody. For colocalization analysis between microtubules and PA-EML2-S, first, the cell boundary was manually defined, then fluorescent signals inside the cells were measured and averaged to confirm that the expression level of EML2-S and microtubule density do not differ among mutant constructs tested. The colocalization indicator, threshold overlap score (TOS), with the top 25 percentile was analyzed using the ImageJ plugin EzColocalization 53. For statistical analyses, see QUANTIFICATION AND STATISTICAL ANALYSIS in STAR Methods.
Live cell imaging
HeLa cells were plated on glass bottom dishes coated with poly-L-lysine (MatTek, Cat# P35GC-1.5-14-C) 24 h prior to transfection. Each 0.1–1 µg of plasmid per dish was used for transfection with Lipofectamine 2000. Cells were subject to imaging the next day. SNAP-tag was labeled with a SNAP-tag substrate (SNAP-Cell Oregon Green, NEB, Cat# S9104S) according to the manufacturer's instructions. Prior to imaging, culture medium was switched to L-15 medium (Thermo Fisher Scientific, Cat# 21083027) containing 10% FBS, penicillin-streptomycin. Images were captured at 37°C using a DeltaVision Elite microscope equipped with an 60X objective, a 1.59X magnifier, a Photometrics Prime 95B sCMOS camera, and an environmental chamber (GE Healthcare). Time lapse images for NeonGreen-tagged EMLs and EB3-mCherry were acquired at each 50 msec exposure every 1 sec for a total of 5–15 minutes. Deconvolved single plane images were used for the kymograph analysis using Multi Kymograph plug-in of Fiji/ImageJ and generation of movies.
Immunoblot analysis
Proteins were separated on 10% acrylamide gels except for gels in Figures 1C and S1E being done with precast gradient gels (Thermo Fisher, Cat# XP04205BOX). Proteins were transferred onto nitrocellulose membranes and briefly stained with Ponceau S (Sigma-Aldrich, Cat# P7170). Blocking was performed with 5% skim milk solution in PBS supplemented with 0.05 % tween 20 (PBST) for 1 hour at room temperature. Primary antibody reaction was performed at 4°C overnight. After washing with PBST for 10 min x 3 times, secondary antibodies were applied and incubated for 1 hour at room temperature. After another three times of wash, membranes were imaged with Azure c600 imager (Azure Biosystems). Antibodies used are listed in the KEY RESOURCES TABLE in STAR Methods. The final concentrations of antibodies are as follows: anti-tyrosinated α-tubulin YL1/2, 1:3,000; anti-detyrosinated α-tubulin RM444, 0.1 µg/ml; anti-∆2 α-tubulin RM447, 0.5 µg/ml; anti-α-tubulin DM1α, 1:3,000; anti-CLIP-170, 1:250; anti-CLIP-115, 1:500; anti-Kif13B, 1:250; anti-TBCB, 1:500; anti-CEP350, 1:500; anti-p150glued, 1:400; anti-TBCE, 1:500; anti-CYLD, 1:500; anti-EML2, 1:200 (Sigma-Aldrich) and 1:1,000 (Proteintech); anti-Dynamin 1L, 1:500; anti-CK1 delta, 1:500; anti-Centrin, 1:1,000; anti-TCP1 eta, 1:250; anti-EB1, 1:1,000; anti-Kif18A, 1:1,000; anti-TPPP, 1:1,000; anti-CKAP2, 1:500; anti-KifC1 (HSET), 1:500; anti-MAP11, 1:500; anti-GCC185, 1:500; anti-CENPF, 1:1,000, anti-CSAP, 1:200; anti-Kif15, 1:2,000; anti-SPECC1L, 1:500; anti-MAP4, 1:800; anti-αB-crystallin, 1:1,000; anti-PA-tag NZ-1, 1:5,000; anti-GAPDH, 1:2,000. Quantification was performed with the gel analyzer function of Fiji/ImageJ. First, band intensities of each reader in the microtubule pellet fractions (WT and VASH1 OE) were measured and normalized against corresponding total ɑ-tubulin (DM1ɑ), and then relative abundance was calculated (VASH1 OE over WT). To compare the IB- and TMT-based quantification methods (Fig 1E, F and G), a TMT score averaged from three independent preparations was compared with a relative abundance calculated for a representative blot for each protein shown in Fig. S2 (except for ɑB-crystallin and SPECC1L being excluded in the analysis shown in Fig. 1G).
Purification of tubulin from HeLa cells
Tubulin was purified from HeLa Kyoto cells using TOG affinity chromatography as described previously 15,54,55. To obtain detyrosinated HeLa tubulin, cleared HeLa lysate was supplemented with CPA (1/100 the cell pellet volume) and incubated on ice for 20 min before being loaded on a TOG column. Concentration of tubulin was calculated by measuring A280 on NanoDrop (Thermo Fisher) and using an extinction coefficient of tubulin (115,000 M−1cm−1). HeLa tubulin was used in microtubule co-pelleting assays with purified His-EML2-S, and brain tubulin was used in microtubule dynamics assays (see below).
Purification of EML2-S protein from insect cells
Production of recombinant His-EML2-S protein was performed based on the Bac-to-bac Baculovirus expression system (Thermo Fisher). Baculoviral amplification was carried out in Sf9 cells, and P3 virus was infected to High Five cells. Three days after the addition of P3 virus, cells were harvested and stored at −80°C until use.
Protein was first purified with Ni-NTA affinity chromatography and optionally further subjected to the polishing step with size exclusion chromatography. Insect cells were thawed, suspended in lysis buffer (PNI buffer [50 mM Sodium phosphate pH 7.4, 500 mM NaCl, 20 mM imidazole] supplemented with 0.2 mM PMSF [Sigma-Aldrich, Cat# 78830], and protease inhibitor cocktail [SIGMAFAST, Sigma-Aldrich, Cat# S8820]) and sonicated. After centrifugation at 18,5000 x g for 20 min at 4°C), lysate was mixed with Ni-NTA agarose resin that had been pre-equilibrated with lysis buffer and incubated for 1 hour at 4°C with gentle agitation. Agarose resin was washed in wash buffer 1 (PNI supplemented with 1 mM MgCl2, 1 mM ATP and protease inhibitor) and wash buffer 2 (PNI with protease inhibitor) three times for each. Protein was eluted by PNI buffer whose imidazole concentration was elevated to 200 mM. For His-EML2-S proteins intended to the use in the in vitro microtubule co-sedimentation assay, imidazole was removed by PD-10 desalting columns (GE Healthcare, Cat# 17085101) and protein was concentrated with Amicon Ultra-4 10K (Merck Millipore, Cat# UFC801024D). For further polishing of the protein for the in vitro dynamics assay, Ni-NTA eluate was loaded onto HiLoad Superdex 200 size exclusion column equilibrated with 10 mM Hepes pH 7.5, 300 mM KCl and 1 mM DTT. A peak fraction corresponding to the expected molecular weight of His-EML2-S (73.8 kD) was pooled and concentrated. Protein concentration was measured by Bradford protein assay using BSA as a standard. Purified protein was aliquoted, snap frozen and stored at −80°C until use.
Microtubule co-pelleting assay using purified proteins
HeLa tubulin (Y and ΔY) and His-EML2-S were thawed and centrifuged at 100,000 x g for 10 min at 4°C to remove any protein aggregates. Microtubule assembly was initiated by adding 1 mM GTP to cleared tubulin solution (5–10 mg/ml) and incubated at 37°C for 15 min. Taxol was added to the reaction at final concentrations of 0.1, 1.0 and 10 µM successively with 10 min intervals. After 15 min of final incubation, microtubules were pelleted by centrifugation at 25,000 x g at 25°C for 15 min and resuspended in BRB80 supplemented with 10 µM Taxol (BRB80-Taxol). To measure microtubule concentration, a small volume of microtubule solution was diluted into ice-cold BRB80 supplemented with 50 mM KCl and 1M CaCl2, and incubated on ice to completely disassemble microtubules. Tubulin concentration was calculated from the A280 measurements and the extinction coefficient of tubulin (115,000 M−1cm−1). Binding of EML2-S to microtubules was carried out by incubating 500 nM His-EML2-S protein and 0.25, 0.5. 1,0 and 4 µM of Y- or ΔY-microtubules in BRB80-Taxol for 20 min at room temperature. After centrifugation at 25,000 x g at 25°C for 20 min, supernatants and pellets were separated, and SDS-PAGE sample buffer was added to each fraction. For each reaction, a total of 814 ng or 136 ng of His-EML2-S protein (pellet + supernatant) were separated on SDS-PAGE gels for Coomassie staining or immunoblot analysis, respectively. Gels were stained with colloidal Coomassie, destained with water and scanned with a flatbed scanner (CanoScan 8800F, Canon). Blots were images with Azure c600 (Azure Biosystems). Densitometric analysis was performed using the gel analyzer function of Fiji/ImageJ. Data analysis and non-linear fitting were performed on GraphPad Prism 9.
Homology modeling and protein electrostatics
EML2-S and EML1 have 69% sequence identity and 91% sequence homology, with no insertions or deletions in the β-propeller domains. This high degree of homology makes structural modeling relatively simple and using the x-ray structure of EML1 (pdb 4ic8) 22 we created a homology using basic threading of the EML2-S sequence into the EML1 structure. Missing atoms and hydrogens were added using VMD 56. The electrostatic potential was calculated using APBS 57. Calculations were performed at 50 mM NaCl using the linearized Poisson-Boltzmann equation, single Debye-Hückel boundary conditions, a protein dielectric of 2.0, and CHARMM partial charges 58. The potential was saved with ~0.5 Å grid spacing and visualized using VMD. A 6-residue peptide corresponding to the αCTT (EEGEEY) was docked to EML2-S using Autodock VINA 59. We performed unbiased docking using a large docking grid that covered most of the protein, but all docking poses interacted at our site of interest. The best docking pose (protein + peptide) was then put through molecular dynamics simulations to look at stability and dynamics. Using NAMD, we carried out NpT simulations using the CHARMM36 forcefield 60,61. The system was solvated with a box of TIP3P water molecules with 10-Å padding around the protein. Na+ and Cl− were added to both neutralize the systems and set the ionic concentration to 50 mM. Following minimization, heating and equilibration, the system was simulated at 300 K and 1 atm of pressure. To allow for 2-fs time steps, bonded hydrogens were fixed. For long-range electrostatics, Particle Mesh Ewald was employed with a 10-Å cutoff and 8.5-Å switch distance for van der Waals interactions.
In vitro microtubule dynamics assay using purified His-EML2-S protein
Tubulin was purified from bovine brains as previously described 62 with the modification of using Fractogel EMD SO3- (M) resin (Millipore-Sigma) instead of phosphocellulose. Tubulin was labeled using CF640R NHS-Ester (Biotium) and tetramethylrhodamine (TAMRA, Invitrogen) as described 63. An additional cycle of polymerization/depolymerization was performed before use. Protein concentrations were determined using a DS-11 FX spectrophotometer (DeNovix, Inc.).
To visualize dynamic microtubules, we reconstituted microtubule growth from GMPCPP double stabilized microtubule ‘seeds’ 28. Cover glass was cleaned in acetone, sonicated in 50% methanol, sonicated in 0.5 M KOH, exposed to air plasma (Plasma Etch) for 3 min, then silanized by soaking in 0.2% Dichlorodimethylsilane (DDS) in n-Heptane for 2 hours. Five microliter flow channels were constructed using two pieces of silanized cover glasses (22 X 22 mm and 18 X 18 mm) held together with double-sided tape and mounted into custom-machined cover slip holders. GMPCPP seeds were prepared by polymerizing a 1:4 molar ratio of TAMRA labeled:unlabeled tubulin in the presence of guanosine-5’-[(α, β)-methyleno]triphosphate (GMPCPP, Jena Biosciences) in two cycles, as described 28. Channels were first incubated with anti-TAMRA antibodies (Invitrogen) and then blocked with 5% Pluronic F-127. Flow channels were washed 3x with BRB80 before incubating with GMPCPP seeds. On each day of experiments tubes of unlabeled and CF640R labelled tubulin was thawed and mixed at a 1:17 molar ratio and then sub-aliquoted and refrozen in liquid nitrogen. For consistency in microtubule growth dynamics, one sub-aliquot of tubulin was used for each experiment. Microtubule growth from GMPCPP seeds was achieved by incubating flow channels with tubulin in imaging buffer: BRB80, 1 mM GTP, 0.1 mg/mL BSA, 0.01% Methylcellulose, 10 mM dithiothreitol, 250 nM glucose oxidase, 64 nM catalase, and 40 mM D-glucose, with the addition of purified His-EML2-S.
Data was acquired with a customized Zeiss Axio Observer 7 equipped with a Laser TIRF III, 405/488/561/638 nm lasers, and an Alpha Plan-Apo 100x/1.46 Oil DIC M27 Objective with objective heater 25.5/33 S1 set to 35° C. Images were recorded on a Prime 95B CMOS camera (Photometrics) with a pixel size of 110 nm. Image acquisition was controlled using ZEN 2.3 (Zeiss). Images were acquired at 3 sec intervals.
All images were processed and analyzed using Fiji/ImageJ. If needed, prior to analysis images were corrected for stage drift using a drift correct script (Hadim). Microtubule dynamics were analyzed using kymographs. Growth and shrinkage rates were measured by manually drawing lines on kymographs and measuring the slope of growth or shrinkage. Catastrophe frequency was calculated by counting the total number of catastrophe events over the total time of all microtubule growth within a channel. Rescue frequency was calculated by counting the number of rescue events per total time or distance of microtubule disassembly. For statistical analyses, see QUANTIFICATION AND STATISTICAL ANALYSIS in STAR Methods.
In vitro microtubule dynamics assay using cell lysates
To prepare extracts for microtubule dynamics assays, COS-7 cells 16 h post-transfection were harvested and centrifuged at low-speed at 4°C. The cell pellet was washed with PBS and resuspended in ice-cold BRB80 buffer freshly supplemented with 1 mM ATP, 1 mM PMSF and protease inhibitors cocktail. The cells were lysed by sonication (Fisher Scientific, Sonic Dismembrator Model 500) using 10% power, 4x10 sec on ice. After centrifugation for 10 min at 20,000 x g at 4°C, aliquots of the supernatant were snap frozen in liquid nitrogen and stored at −80 °C until further use.
The concentration of mNG-EML2-L in the COS-7 lysates was measured by a dot-blot, in which the same volumes of COS-7 lysates and a series of diluted known concentration of His-EML2-S protein were spotted onto a nitrocellulose membrane that was air-dried for 1 h and immunoblotted with a primary antibody to EML2 (Proteintech, Cat# 13529–1-AP) overnight at 4°C and secondary antibody 680nm-anti rabbit (Jackson ImmunoResearch Laboratories Inc.) at room temperature for 1h. The fluorescence intensity of the spots on the nitrocellulose membrane was detected by Azure c600 and quantified based on the standard curve of known concentration of EML2 protein using Fiji/ImageJ (NIH).
For the microtubule dynamics assay, a flow cell (~10 μl volume) was assembled by attaching a clean #1.5 coverslip (Fisher Scientific) to a glass slide (Fisher Scientific) with two strips of double-sided tape. Microtubule seeds containing 10% biotin-labeled tubulin and 10% X-rhodamine-labeled (Cytoskeleton Inc.) were generated by polymerization in the presence of GMPCPP (Jena Bioscience, Cat# NU-405S) and then immobilized on the coverslip by incubating the flow chamber sequentially with the following solutions: (1) 1 mg/ml BSA-biotin (Sigma, Cat# A8549), (2) blocking buffer (1 mg/ml BSA in BRB80), (3) 0.5 mg/ml NeutrAvidin (ThermoFisher, Cat# 31000), (4) blocking buffer, (5) GMPCPP-stabilized microtubule seeds, (6) blocking buffer. Microtubule growth was then initiated by flowing in 10 µM brain tubulin containing 12.5% Hilyte647-labeled tubulin (Cytoskeleton Inc.) together with 10 μl untransfected COS-7 cell lysate (Mock) or 10 μl COS-7 cell lysate expressing mNG-EML2-L protein (~16 nM) in reaction buffer [BRB80 supplemented with 1 mM GTP, 3 mM MgCl2, 1 mM DTT, 1 mg/ml casein, 0.1% methylcellulose (Sigma) and oxygen-scavenging system (12.5 mM glucose, 0.05 mg/ml catalase, 0.25 mg/ml glucose oxidase)]. The flow cells were sealed with molten paraffin wax and imaged by TIRF microscopy. The temperature was set at 35°C in a temperature-controlled chamber (Tokai Hit). Time-lapse images were acquired in 488 nm, 561 nm and 640 nm channels at a rate of every 2 s for 15 min. To determine the shrinking rate of microtubule plus ends, maximum intensity projections were generated, and kymographs (width= 3 pixels) were prepared using Fiji/ImageJ2 and displayed with time on the y axis and distance on the x axis. Only microtubule shrinking event with a slope over a three-pixel length were analyzed. For statistical analyses, see QUANTIFICATION AND STATISTICAL ANALYSIS in the STAR Methods.
All images were processed and analyzed using Fiji/ImageJ. For figure assembly, images were linearly adjusted in brightness and contrast using Photoshop (Adobe). All final figures were assembled using Illustrator (Adobe).
QUANTIFICATION AND STATISTICAL ANALYSIS
For the TMT analysis shown in Figure 1, quantification and statistical analyses are performed as described in the legend or the Method details. For the comparison of relative abundance of EML2 peptides (Figure S3A), abundance ratios (shown in Figure S3B) are averaged, standard deviation was calculated, and t test (Two tailed; statistical significance, P < 0.05) was performed with Microsoft Excel.
For the quantification of PA-EML2-L/S localization on midbody microtubules (Figure 3) and the colocalization analysis between PA-EML2-S mutants and microtubules (Figure 4), statistical analyses were performed with GraphPad Prism 9 (GraphPad) using Wilcoxon rank sum exact test (Two tailed; statistical significance, P < 0.05). Data were obtained for numbers indicated in each graph from at least three independent experiments. Detailed information is seen in the legends.
For the in vitro microtubule dynamics assay using purified His-EML2-S (Figures 5A and 5B), all functions were fitted and graphed with OriginPro2020 (OriginLab) or Python 3 using a JupyterLab Notebook. Mean and standard deviation were calculated using Excel. Detailed information is available in the legend and Method details section.
For the in vitro microtubule dynamics assay using cell lysate (Figures 6C and 6D), statistical analyses were performed, and graphs were generated using GraphPad Prism software (GraphPad). Comparisons of microtubule dynamic parameters between in the absence (mock control) and in the presence of mNG-EML2-L conditions were carried out using a two-tailed t test. Detailed information is seen in Results and the legend.
Supplementary Material
Video S1. Live imaging of mNeonGreen-EML2-L transiently expressed in HeLa cells, related to Figure 6. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Videos S2 and S3. Live imaging of mNeonGreen-EML2-L and EB3-mCherry transiently expressed in HeLa cells, related to Figure 6. Whole cell image (S2) and enlarged image (S3). mNeonGreen-EML2-L (green) and EB3-mCherry (magenta) were co-expressed in HeLa cells. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Video S4 Live imaging of mNeonGreen-EML2-L and mCherry-TubA1A in HeLa cells, related to Figure 6. mNeonGreen-EML2-L (green) was transiently expressed in HeLa cells expressing mCherry-TubA1A (magenta). Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Video S5. Live imaging of mNeonGreen-EML2-L mutants transiently expressed in HeLa cells, related to Figure 6. R270E (upper), AAAA (middle) and TD mutants (lower). Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bars, 5 µm.
Video S6. Live imaging of mNeonGreen-EML1, 2-L, 3 and 4 transiently expressed in HeLa cells, related to Figure 6. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bars, 5 µm.
Data S1. MS analysis for proteins pulled down with GST-CTT proteins, related to Figure 1.
Proteins detected in GST-CTT (Y/ΔY) pulldown but absent in the control GST pulldown are highlighted as follows: GST-CTTY-specific binding proteins (Y-CTT readers), magenta; GST-CTTΔY-specific binding proteins (ΔY-CTT-readers), green; and general CTT binding proteins, yellow.
Highlights.
A screening method for readers of tyrosinated/detyrosinated tubulin is established
EML2-S preferentially associates with tyrosinated microtubules
EML2’s β-propeller contains a recognition motif for the tyrosinated α-tubulin C-terminal tail
EML2 is a microtubule stabilizing factor that tracks shrinking microtubule plus ends
ACKNOWLEDGMENTS
The authors thank Dr. Venkatesha Basrur and Kevin Conlon of the Proteomics Resource Facility at the University of Michigan for providing technical expertise in mass spectrometry, and the Flow Cytometry Core at the University of Michigan Biomedical Research Core Facilities for technical assistance in cell cycle analysis. The S.B. laboratory is funded by CIHR PJT-156193 and NSERC RGPIN-2017–04649. Work in the laboratories of K.V. and A.I.N. are supported by NIH grants R35 GM131744 and R01 GM094231, respectively. Work in the laboratory of R.O. was supported by NIH grant R01 GM086610, start-up funds at the University of Michigan, a Research Stimulus grant awarded to R.O. by the University of Michigan, and a MICHR Accelerating Synergy grant awarded to R.O. and K.V.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Authors declare no competing interests.
References
- 1.Roll-Mecak A (2020). The Tubulin Code in Microtubule Dynamics and Information Encoding. Dev Cell 54, 7–20. 10.1016/j.devcel.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aillaud C, Bosc C, Peris L, Bosson A, Heemeryck P, Van Dijk J, Le Friec J, Boulan B, Vossier F, Sanman LE, et al. (2017). Vasohibins/SVBP are tubulin carboxypeptidases (TCPs) that regulate neuron differentiation. Science 358, 1448–1453. 10.1126/science.aao4165. [DOI] [PubMed] [Google Scholar]
- 3.Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, Mazouzi A, Stickel E, Celie P, Altelaar M, Knipscheer P, Perrakis A, Blomen VA, and Brummelkamp TR (2017). Vasohibins encode tubulin detyrosinating activity. Science 358, 1453–1456. 10.1126/science.aao5676. [DOI] [PubMed] [Google Scholar]
- 4.Landskron L, Bak J, Adamopoulos A, Kaplani K, Moraiti M, van den Hengel LG, Song JY, Bleijerveld OB, Nieuwenhuis J, Heidebrecht T, et al. (2022). Posttranslational modification of microtubules by the MATCAP detyrosinase. Science 376, eabn6020. 10.1126/science.abn6020. [DOI] [PubMed] [Google Scholar]
- 5.Schroder HC, Wehland J, and Weber K (1985). Purification of brain tubulin-tyrosine ligase by biochemical and immunological methods. J Cell Biol 100, 276–281. 10.1083/jcb.100.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barisic M, Silva e Sousa R, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, Janke C, Grishchuk EL, and Maiato H (2015). Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 348, 799–803. 10.1126/science.aaa5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, and Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. 10.1126/science.aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohan N, Sorokina EM, Verdeny IV, Alvarez AS, and Lakadamyali M (2019). Detyrosinated microtubules spatially constrain lysosomes facilitating lysosome-autophagosome fusion. J Cell Biol 218, 632–643. 10.1083/jcb.201807124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Kholina E, Szyk A, Fedorov VA, Kovalenko I, Gudimchuk N, and Roll-Mecak A (2021). alpha-tubulin tail modifications regulate microtubule stability through selective effector recruitment, not changes in intrinsic polymer dynamics. Dev Cell 56, 2016–2028 e2014. 10.1016/j.devcel.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinmetz MO, and Akhmanova A (2008). Capturing protein tails by CAP-Gly domains. Trends Biochem Sci 33, 535–545. 10.1016/j.tibs.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Peris L, Wagenbach M, Lafanechere L, Brocard J, Moore AT, Kozielski F, Job D, Wordeman L, and Andrieux A (2009). Motor-dependent microtubule disassembly driven by tubulin tyrosination. J Cell Biol 185, 1159–1166. 10.1083/jcb.200902142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn S, Morrison EE, Liverpool TB, Molina-Paris C, Cross RA, Alonso MC, and Peckham M (2008). Differential trafficking of Kif5c on tyrosinated and detyrosinated microtubules in live cells. J Cell Sci 121, 1085–1095. 10.1242/jcs.026492. [DOI] [PubMed] [Google Scholar]
- 13.Cai D, McEwen DP, Martens JR, Meyhofer E, and Verhey KJ (2009). Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol 7, e1000216. 10.1371/journal.pbio.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konishi Y, and Setou M (2009). Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci 12, 559–567. 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- 15.Hotta T, Haynes SE, Blasius TL, Gebbie M, Eberhardt EL, Sept D, Cianfrocco M, Verhey KJ, Nesvizhskii AI, and Ohi R (2021). Parthenolide Destabilizes Microtubules by Covalently Modifying Tubulin. Curr Biol 31, 900–907 e906. 10.1016/j.cub.2020.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, and Hamon C (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 17.Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, Akhmanova A, and Steinmetz MO (2007). Structure-function relationship of CAP-Gly domains. Nat Struct Mol Biol 14, 959–967. 10.1038/nsmb1291. [DOI] [PubMed] [Google Scholar]
- 18.Suprenant KA, Dean K, McKee J, and Hake S (1993). EMAP, an echinoderm microtubule-associated protein found in microtubule-ribosome complexes. J Cell Sci 104, 445–450. [DOI] [PubMed] [Google Scholar]
- 19.Fry AM, O'Regan L, Montgomery J, Adib R, and Bayliss R (2016). EML proteins in microtubule regulation and human disease. Biochem Soc Trans 44, 1281–1288. 10.1042/BST20160125. [DOI] [PubMed] [Google Scholar]
- 20.Adib R, Montgomery JM, Atherton J, O'Regan L, Richards MW, Straatman KR, Roth D, Straube A, Bayliss R, Moores CA, and Fry AM (2019). Mitotic phosphorylation by NEK6 and NEK7 reduces the microtubule affinity of EML4 to promote chromosome congression. Sci Signal 12. 10.1126/scisignal.aaw2939. [DOI] [PubMed] [Google Scholar]
- 21.Richards MW, O'Regan L, Roth D, Montgomery JM, Straube A, Fry AM, and Bayliss R (2015). Microtubule association of EML proteins and the EML4-ALK variant 3 oncoprotein require an N-terminal trimerization domain. Biochem J 467, 529–536. 10.1042/BJ20150039. [DOI] [PubMed] [Google Scholar]
- 22.Richards MW, Law EW, Rennalls LP, Busacca S, O'Regan L, Fry AM, Fennell DA, and Bayliss R (2014). Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc Natl Acad Sci U S A 111, 5195–5200. 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lepley DM, Palange JM, and Suprenant KA (1999). Sequence and expression patterns of a human EMAP-related protein-2 (HuEMAP-2). Gene 237, 343–349. 10.1016/s0378-1119(99)00335-2. [DOI] [PubMed] [Google Scholar]
- 24.Fujii Y, Kaneko M, Neyazaki M, Nogi T, Kato Y, and Takagi J (2014). PA tag: a versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein Expr Purif 95, 240–247. 10.1016/j.pep.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Mishima M, Maesaki R, Kasa M, Watanabe T, Fukata M, Kaibuchi K, and Hakoshima T (2007). Structural basis for tubulin recognition by cytoplasmic linker protein 170 and its autoinhibition. Proc Natl Acad Sci U S A 104, 10346–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreitzer G, Liao G, and Gundersen GG (1999). Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin-dependent mechanism. Mol Biol Cell 10, 1105–1118. 10.1091/mbc.10.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker RA, O'Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, and Salmon ED (1988). Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J Cell Biol 107, 1437–1448. 10.1083/jcb.107.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gell C, Bormuth V, Brouhard GJ, Cohen DN, Diez S, Friel CT, Helenius J, Nitzsche B, Petzold H, Ribbe J, et al. (2010). Microtubule dynamics reconstituted in vitro and imaged by single-molecule fluorescence microscopy. Methods Cell Biol 95, 221–245. 10.1016/S0091-679X(10)95013-9. [DOI] [PubMed] [Google Scholar]
- 29.Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW, and Janke C (2010). Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biol 189, 945–954. 10.1083/jcb.201001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y, and Kim SH (2020). WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System. Endocrinol Metab (Seoul) 35, 494–506. 10.3803/EnM.2020.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Riel WE, Rai A, Bianchi S, Katrukha EA, Liu Q, Heck AJ, Hoogenraad CC, Steinmetz MO, Kapitein LC, and Akhmanova A (2017). Kinesin-4 KIF21B is a potent microtubule pausing factor. eLife 6. 10.7554/eLife.24746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, Roos M, McGillewie L, Bar S, Minocha S, et al. (2017). WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A 114, E9308–E9317. 10.1073/pnas.1713625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, Ellisman M, and Dauer WT (2012). LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet 21, 890–899. 10.1093/hmg/ddr526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhmanova A, and Steinmetz MO (2008). Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9, 309–322. 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 35.Arnal I, Heichette C, Diamantopoulos GS, and Chretien D (2004). CLIP-170/tubulin-curved oligomers coassemble at microtubule ends and promote rescues. Curr Biol 14, 2086–2095. S0960982204009212 [pii] 10.1016/j.cub.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 36.Jijumon AS, Bodakuntla S, Genova M, Bangera M, Sackett V, Besse L, Maksut F, Henriot V, Magiera MM, Sirajuddin M, and Janke C (2022). Lysate-based pipeline to characterize microtubule-associated proteins uncovers unique microtubule behaviours. Nat Cell Biol 24, 253–267. 10.1038/s41556-021-00825-4. [DOI] [PubMed] [Google Scholar]
- 37.Eichenmuller B, Everley P, Palange J, Lepley D, and Suprenant KA (2002). The human EMAP-like protein-70 (ELP70) is a microtubule destabilizer that localizes to the mitotic apparatus. J Biol Chem 277, 1301–1309. 10.1074/jbc.M106628200. [DOI] [PubMed] [Google Scholar]
- 38.Kuo YW, Trottier O, Mahamdeh M, and Howard J (2019). Spastin is a dual-function enzyme that severs microtubules and promotes their regrowth to increase the number and mass of microtubules. Proc Natl Acad Sci U S A 116, 5533–5541. 10.1073/pnas.1818824116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, et al. (2012). The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell 23, 968–980. 10.1016/j.devcel.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westermann S, Wang HW, Avila-Sakar A, Drubin DG, Nogales E, and Barnes G (2006). The Dam1 kinetochore ring complex moves processively on depolymerizing microtubule ends. Nature 440, 565–569. nature04409 [pii] 10.1038/nature04409. [DOI] [PubMed] [Google Scholar]
- 41.Drechsler H, Xu Y, Geyer VF, Zhang Y, and Diez S (2019). Multivalent electrostatic microtubule interactions of synthetic peptides are sufficient to mimic advanced MAP-like behavior. Mol Biol Cell 30, 2953–2968. 10.1091/mbc.E19-05-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takatani S, Ozawa S, Yagi N, Hotta T, Hashimoto T, Takahashi Y, Takahashi T, and Motose H (2017). Directional cell expansion requires NIMA-related kinase 6 (NEK6)-mediated cortical microtubule destabilization. Sci Rep 7, 7826. 10.1038/s41598-017-08453-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao M, Wakamatsu Y, Itoh TJ, Shoji T, and Hashimoto T (2008). Arabidopsis SPIRAL2 promotes uninterrupted microtubule growth by suppressing the pause state of microtubule dynamics. J Cell Sci 121, 2372–2381. 10.1242/jcs.030221. [DOI] [PubMed] [Google Scholar]
- 44.Gayek AS, and Ohi R (2014). Kinetochore-microtubule stability governs the metaphase requirement for Eg5. Mol Biol Cell 25, 2051–2060. 10.1091/mbc.E14-03-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Y, English CA, and Ohi R (2010). The kinesin-8 Kif18A dampens microtubule plus-end dynamics. Curr Biol 20, 374–380. S0960–9822(09)02212-X [pii] 10.1016/j.cub.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 46.Backer CB, Gutzman JH, Pearson CG, and Cheeseman IM (2012). CSAP localizes to polyglutamylated microtubules and promotes proper cilia function and zebrafish development. Mol Biol Cell 23, 2122–2130. 10.1091/mbc.E11-11-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sturgill EG, and Ohi R (2013). Kinesin-12 differentially affects spindle assembly depending on its microtubule substrate. Curr Biol 23, 1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khandelia P, Yap K, and Makeyev EV (2011). Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci U S A 108, 12799–12804. 10.1073/pnas.1103532108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang HY, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat Methods 17, 869–870. 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller PM, Folkmann AW, Maia AR, Efimova N, Efimov A, and Kaverina I (2009). Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol 11, 1069–1080. 10.1038/ncb1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller LM, Xiao H, Burd B, Horwitz SB, Angeletti RH, and Verdier-Pinard P (2010). Methods in tubulin proteomics. Methods Cell Biol 95, 105–126. 10.1016/S0091-679X(10)95007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stauffer W, Sheng H, and Lim HN (2018). EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep 8, 15764. 10.1038/s41598-018-33592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hotta T, Fujita S, Uchimura S, Noguchi M, Demura T, Muto E, and Hashimoto T (2016). Affinity Purification and Characterization of Functional Tubulin from Cell Suspension Cultures of Arabidopsis and Tobacco. Plant Physiol 170, 1189–1205. 10.1104/pp.15.01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Widlund PO, Podolski M, Reber S, Alper J, Storch M, Hyman AA, Howard J, and Drechsel DN (2012). One-step purification of assembly-competent tubulin from diverse eukaryotic sources. Mol Biol Cell 23, 4393–4401. 10.1091/mbc.E12-06-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, and Mackerell AD Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 59.Trott O, and Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Henin J, Jiang W, et al. (2020). Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 153, 044130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang J, and MacKerell AD Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weingarten MD, Lockwood AH, Hwo SY, and Kirschner MW (1975). A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72, 1858–1862. 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, and Mitchison T (1991). Preparation of modified tubulins. Methods Enzymol 196, 478–485. 10.1016/0076-6879(91)96041-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Live imaging of mNeonGreen-EML2-L transiently expressed in HeLa cells, related to Figure 6. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Videos S2 and S3. Live imaging of mNeonGreen-EML2-L and EB3-mCherry transiently expressed in HeLa cells, related to Figure 6. Whole cell image (S2) and enlarged image (S3). mNeonGreen-EML2-L (green) and EB3-mCherry (magenta) were co-expressed in HeLa cells. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Video S4 Live imaging of mNeonGreen-EML2-L and mCherry-TubA1A in HeLa cells, related to Figure 6. mNeonGreen-EML2-L (green) was transiently expressed in HeLa cells expressing mCherry-TubA1A (magenta). Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bar, 5 µm.
Video S5. Live imaging of mNeonGreen-EML2-L mutants transiently expressed in HeLa cells, related to Figure 6. R270E (upper), AAAA (middle) and TD mutants (lower). Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bars, 5 µm.
Video S6. Live imaging of mNeonGreen-EML1, 2-L, 3 and 4 transiently expressed in HeLa cells, related to Figure 6. Time lapse images were acquired every 1 sec. Movie frame rate, 30 frame per sec. Scale bars, 5 µm.
Data S1. MS analysis for proteins pulled down with GST-CTT proteins, related to Figure 1.
Proteins detected in GST-CTT (Y/ΔY) pulldown but absent in the control GST pulldown are highlighted as follows: GST-CTTY-specific binding proteins (Y-CTT readers), magenta; GST-CTTΔY-specific binding proteins (ΔY-CTT-readers), green; and general CTT binding proteins, yellow.
Data Availability Statement
Raw LC/MS data and result tables are available through ProteomeXchange with identifier PXD030448. All other data are available in the main text or the supplementary materials.