

Abstract
Background
Chemotherapy induces a cachectic‐like phenotype, accompanied by skeletal muscle wasting, weakness and mitochondrial dysfunction. Peroxisome proliferator‐activated receptor‐gamma coactivator‐1 alpha (PGC1α), a regulator of mitochondrial biogenesis, is often reduced in cachectic skeletal muscle. Overexpression of PGC1α has yielded mixed beneficial results in cancer cachexia, yet investigations using such approach in a chemotherapy setting are limited. Utilizing transgenic mice, we assessed whether overexpression of PGC1α could combat the skeletal muscle consequences of cisplatin.
Methods
Young (2 month) and old (18 month) wild‐type (WT) and PGC1α transgenic male and female mice (Tg) were injected with cisplatin (C; 2.5 mg/kg) for 2 weeks, while control animals received saline (n = 5–9/group). Animals were assessed for muscle mass and force, motor unit connectivity, and expression of mitochondrial proteins.
Results
Young WT + C mice displayed reduced gastrocnemius mass (male: −16%, P < 0.0001; female: −11%, P < 0.001), muscle force (−6%, P < 0.05, both sexes), and motor unit number estimation (MUNE; male: −53%, P < 0.01; female: −51%, P < 0.01). Old WT + C male and female mice exhibited gastrocnemius wasting (male: −22%, P < 0.05; female: −27%, P < 0.05), muscle weakness (male: −20%, P < 0.0001; female: −17%, P < 0.01), and loss of MUNE (male: −82%, P < 0.01; female: −62%, P < 0.05), suggesting exacerbated cachexia compared with younger animals. Overexpression of PGC1α had mild protective effects on muscle mass in young Tg + C male only (gastrocnemius: +10%, P < 0.05); however, force and MUNE were unchanged in both young Tg + C male and female, suggesting preservation of neuromuscular function. In older male, protective effects associated with PGC1α overexpression were heighted with Tg + C demonstrating preserved muscle mass (gastrocnemius: +34%, P < 0.001), muscle force (+13%, P < 0.01), and MUNE (+3‐fold, P < 0.05). Similarly, old female Tg + C did not exhibit muscle wasting or reductions in MUNE, and had preserved muscle force (+11%, P < 0.05) compared with female WT + C. Follow‐up molecular analysis demonstrated that aged WT animals were more susceptible to cisplatin‐induced loss of mitochondrial proteins, including PGC1α, OPA1, cytochrome‐C, and Cox IV.
Conclusions
In our study, the negative effects of cisplatin were heighted in aged animals, whereas overexpression of PGC1α was sufficient to combat the neuromuscular dysfunction caused by cisplatin, especially in older animals. Hence, our observations indicate that aged animals may be more susceptible to develop chemotherapy side toxicities and that mitochondria‐targeted strategies may serve as a tool to prevent chemotherapy‐induced muscle wasting and weakness.
Keywords: Skeletal muscle, Cisplatin, Chemotherapy, PGC1α, cachexia
Introduction
Despite recent progress, cancer remains a clinical concern, with nearly two million new cases and over 600 000 deaths expected in 2022 in the USA alone. 1 As many as 80% of cancer patients will develop cachexia, a multi‐organ wasting syndrome, thought to be the ultimate cause of death in nearly 30% of cases. 2 , 3 Besides skeletal muscle wasting and weakness, development of cachexia can also impair patients' functional abilities, reduce treatment tolerance, and lower overall survival. 4 , 5 Unfortunately, anticancer regimens have robust off‐target effects and are known to promote a cachexia‐like phenotype. Indeed, we and others have shown that chemotherapy, including cisplatin, directly promotes loss of body weight, muscle mass and strength, as well as motor unit connectivity. 6 , 7 , 8 , 9 , 10 , 11 Unfortunately, no approved treatment options exist to combat this debilitating co‐morbidity of cancer or to counteract anti‐cancer treatment toxicities.
We and others have suggested that cancer and chemotherapy can promote mitochondrial abnormalities, which in turn promote skeletal muscle dysfunction in cachexia. 6 , 12 , 13 , 14 The regulator of mitochondrial biogenesis, peroxisome proliferator‐activated receptor‐gamma coactivator‐1 alpha (PGC1α), has received much attention with respect to cancer cachexia, with genetic PGC1α muscle overexpression yielding mixed results in experimental C26‐induced and LLC‐induced cachexia. 15 , 16 , 17 While the effectiveness of targeting PGC1α in cancer‐induced cachexia is up for debate, less is known with respect to counteracting chemotherapy‐induced muscle wasting and weakness.
While few studies have investigated mitochondrial strategies to counteract chemotherapy‐induced muscle dysfunctions, even less have considered the potential differences that sex or age may have on cachexia. For example, evidence suggests that male cancer patients have higher prevalence of cachexia, though few experimental investigations have carried out phenotypic differences in response to cancer or chemotherapy. 18 Regarding age, and given the rarity of cancer in young individuals, it has recently been suggested that experimental cancer cachexia research should incorporate older, age‐appropriate animals, instead of young adult mice up to 14 weeks of age, as in the case of the widely used C26 or LLC mouse models. 19 , 20 Moreover, few studies have examined phenotypic cachexia differences between younger and older animals. Interestingly, recent work suggests that older animals may be more susceptible to cancer‐induced cachexia compared with younger animals, yet whether this also occurs with chemotherapy is unknown. 21
While most cachexia investigations have placed emphasis on combating muscle mass, recent work has suggested that loss of skeletal muscle function precedes muscle wasting, demonstrating a greater need for assessing the occurrence and mechanisms of muscle weakness in cachexia. 22 , 23 Along these lines, work from our group and others recently reported indices of functional denervation in models of cancer‐induced and chemotherapy‐induced cachexia. 9 , 24 In particular, we demonstrated that both tumours and anti‐cancer drugs promote reductions in the number of functionally connected motor units, and that the loss of motor unit number estimation (MUNE) is associated with skeletal muscle wasting and weakness. 9 Interestingly, mitochondrial function has known roles in the maintenance of neuromuscular junctions (NMJs), yet whether overexpression of skeletal muscle PGC1α is sufficient to preserve motor unit connectivity in the presence of chemotherapy is unknown. 25 , 26 , 27
In the present study we carried out phenotypic assessments in response to cisplatin treatment of young and old, male and female wild‐type (WT) or transgenic mice overexpressing skeletal muscle PGC1α (Tg). Utilizing a battery of tests, we assessed cisplatin‐induced alterations in body weight, lean and fat mass, in vivo and ex vivo muscle function, and electrophysiological parameters. Our present findings suggest that cisplatin induces a cachectic‐like phenotype in both male and female mice, which is exacerbated in older animals. Despite having mild effects on muscle mass in young animals, overexpression of PGC1α proved effective in combating cisplatin‐induced loss of muscle function and motor unit connectivity. Such protection was even greater in older animals, likely due to the substantial loss of muscle mitochondrial proteins caused by cisplatin in this experimental group.
Methods
Animals
Use of animals for all experimental studies was approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine and was in compliance with the National Institutes of Health Guidelines for Use and care of Laboratory Animals and with the 1964 Declaration of Helsinki and its later amendments. Young (2 months) and old (18 months) male and female WT C57BL/6J or C57BL/6‐Tg (Ckm‐Ppargc1a)31Brsp/J (mPGC1α: referred to as Tg throughout the manuscript; The Jackson Laboratory, Bar Harbour, ME, USA) were used in separate experiments and divided into four experimental groups (n = 5–9/group): WT administered intraperitoneal injections of sterile saline; WT administered cisplatin [(WT + C) 2.5 mg/kg; nine total injections] in sterile saline; Tg administered intraperitoneal injections of sterile saline; Tg administered intraperitoneal injections of cisplatin [(Tg + C) 2.5 mg/kg; nine total injections] in sterile saline (Supporting Information, Figure S1). 7 , 9 At time of euthanasia, skeletal muscles were harvested, weighed, and snap frozen in liquid nitrogen and stored at −80°C for further studies.
Body composition
All experimental animals were assessed for lean and fat mass (i.e. body composition) at baseline, 1 week post treatment, and the day before sacrifice in un‐anaesthetized physically restrained manner via Echo medical systems' EchoMRI‐100 (EchoMRI, Houston, USA), as carried out previously. 6
Muscle contractility
In order to assess changes in strength upon cisplatin treatment, animals underwent in vivo plantarflexion torque assessment (Aurora Scientific Inc, Canada) at baseline and 2 days prior to sacrifice as carried out previously. 9 Peak twitch torque was established to determine maximal stimulus intensity (< 1.5 mA) and torque was assessed at 100 Hz (0.2 ms). At time of sacrifice, extensor digitorum longus (EDL) muscles were dissected, and tendons were sutured to stainless‐steel hooks to assess whole‐muscle contractility. 9 Force frequency relationships of the EDL were then assessed via a supramaximal frequency stimulation protocol (10, 25, 40, 60, 80, 100, 125, and 150 Hz for 350 ms). Following completion of the force frequency protocol, muscles rested for 5 min and then underwent a 60‐contraction fatiguing protocol at 60 Hz (every 3 s). All in vivo and ex vivo force data was collected and subsequently analysed using Dynamic Muscle Control/Data Acquisition and Dynamic Muscle Control Data Analysis programs (Aurora Scientific).
In vivo electrophysiology
Electrophysiological functional assessment was performed on the triceps surae muscles in anaesthetized mice 1 day before sacrifice using the Sierra Summit 3–12 Channel EMG (Cadwell Laboratories Incorporated, Kennewick, WA, USA), as carried out previously. 9 Supramaximal stimulations (continuous current: <10 mA; pulse duration: 0.1 ms) were used to obtain peak‐to‐peak and baseline‐to‐peak compound muscle action potentials (CMAP). An incremental stimulation technique 9 was used to obtain peak‐to‐peak single motor unit (SMUP) potentials. CMAP amplitudes (baseline‐to‐peak) were used for comparison between experimental groups, and MUNE was determined by the following equation: MUNE = CMAP amplitude (peak‐to‐peak)/average SMUP (peak‐to‐peak).
Western blotting
Quadriceps muscles (~50 mg) were homogenized on ice, protein was extracted, and samples were prepared for western blotting as performed previously. 9 Extracted proteins (30 μg) were electrophoresed, transferred onto nitrocellulose membranes, and prepared for antibody incubations as carried out previously. 9 Antibodies used were PGC1α (#AB3242) from MilliporeSigma (Burlington, MA, USA); OPA1 (#80471), Mitofusin‐2 (#9482), VDAC (#4866), cytochrome‐C (#11940) and Cox IV (#4844) from Cell Signalling Technologies (Danvers, MA, USA); and α‐Tubulin (#12G10) from Developmental Studies Hybridoma Bank (Iowa City, IA, USA). Membranes were then incubated with either anti‐rabbit IgG (H + L) DyLight 800 or anti‐mouse IgG (H + L) DyLight 680 secondary antibodies (Cell Signalling Technologies, Danvers, MA, USA), and analysed with Odyssey's Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA). Total proteins were normalized to the tubulin loading control.
Statistics
Two‐way analysis of variance (ANOVA) tests, with genotype and treatment factors, were performed to determine differences between age and sex‐matched experimental groups. Separate two‐way ANOVA tests were performed within WT groups to determine age‐dependent effects of cisplatin treatment. Post‐hoc comparisons were accomplished via a Tukey's test, with statistical significance set a priori at P ≤ 0.05. If normality and heteroscedasticity tests failed, main effect statistics were reported and Student's t‐test pairwise comparisons were used to determine differences within genotypes. This was the case for nearly all western blotting data as Tg animals were consistently displaying robust increases in mitochondrial proteins. All statistics were performed using GraphPad Prism 8.4.1 and data are presented as means ± SD.
Results
Cisplatin exacerbates weight loss in aged mice, which is combatted by skeletal muscle PGC1α overexpression
We and others have reported that cisplatin promotes significant in vivo weight loss, skeletal muscle wasting and mitochondrial alterations, including reduced PGC1α. 7 , 9 , 11 , 28 , 29 , 30 Whether this effect is worsened in aged animals or whether overexpression of skeletal muscle PGC1α is sufficient to combat cisplatin‐induced cachexia remains unknown. Thus, we treated young (2 months) and old (18 months) male and female WT and Tg mice with cisplatin for 2 weeks. In young WT animals, cisplatin promoted losses in body weight [male: −13% (Figure 1A,B); female: −12% (Figure 1F,G)], carcass weight [male: −17% (Figure 1C); female: −13% (Figure 1H)], fat content [male: −17% (Figure 1D); female: −18% (Figure 1I)], gonadal fat [male: −39% (Figure S2A); female: −51%; (Figure S2F)], and lean mass [male: −14% (Figure 1E); female: −13% (Figure 1J)] compared with untreated WT littermates, altogether indicative of cachexia. Interestingly, 18‐month‐old WT mice appeared more susceptible to cisplatin‐induced cachexia than young animals. Aged WT + C male saw reductions in body weight (−34%), carcass weight (−32%), as well as fat mass (−54%) and lean mass (−19%) compared with aged WT male, suggestive of exacerbated cachexia when compared with young WT mice (Figure 1K–O; Table S1). Similarly, aged WT + C female displayed heightened cisplatin‐induced cachexia, as reflected by weight loss (−27%), reductions in carcass weight (−26%), and losses in fat mass (−57%) and lean mass (−14%) compared with 18‐month‐old untreated female (Figure 1P–T; Table S2). Overexpression of skeletal muscle PGC1α had negligible effects in young animals. Male and female Tg + C yielded no significant changes compared with WT + C animals, though mean changes in body weight, fat mass, and lean mass were generally smaller compared with genotype controls (Figure 1A–J). The most striking observations were noticed in aged Tg + C male, exhibiting significant improvements in body weight, carcass weight and lean mass compared with WT + C mice (Figure 1K–O). Meanwhile, overexpression of PGC1α was mildly effective in aged female, with carcass weight remaining unchanged in Tg + C versus Tg (Figure 1R). However, similar to young animals, aged female Tg + C saw significant losses in body weight, fat mass and lean mass, though these reductions were generally smaller than that of 18‐month‐old WT + C mice (Figure 1Q–T). Together these results suggest that aged animals are more susceptible to cisplatin‐induced weight loss, and that muscle‐restricted overexpression of PGC1α efficacy increases with age, particularly in male mice.
Figure 1.
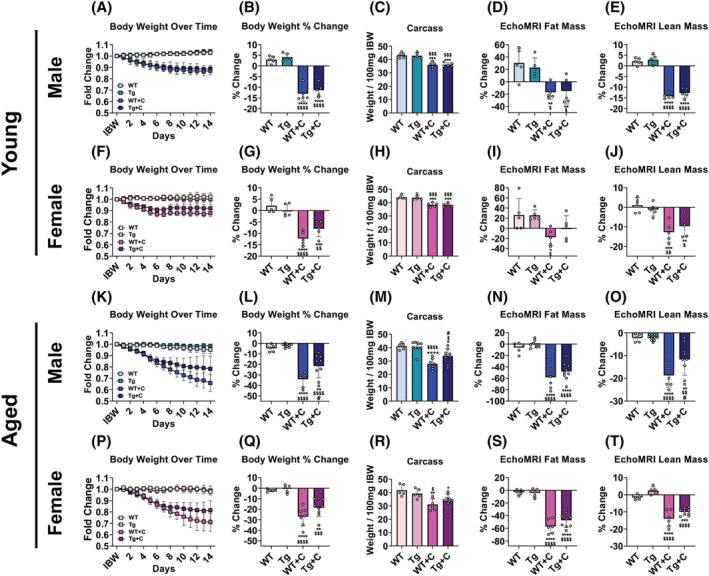
Cisplatin exacerbates weight loss in aged mice which is combatted by overexpression of skeletal muscle PGC1α. Body weight (BW) curves, BW percent (%) change at time of sacrifice (vs. Day 1), carcass weights normalized to initial body weight (IBW), fat content % change (vs. Day 1), and lean content % change (vs. Day 1) of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A–E): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); row 2 (F–J): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); row 3 (K–O): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); row 4 (P–T): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WT; $ P < 0.05, $$ P < 0.01, $$$ P < 0.001, $$$$ P < 0.0001 versus Tg; # P < 0.05 versus WT + C.
PGC1α overexpression provides greater protection against cisplatin‐induced muscle wasting in aged mice
Similar to body composition, aged WT animals experienced heighted cisplatin‐induced muscle wasting compared with young WT animals. Cisplatin promoted muscle wasting in young WT male (gastrocnemius: −16%; tibialis anterior: −12%; quadriceps: −17%) and WT female (gastrocnemius: −11%; tibialis anterior: −11%) compared with age‐matched WT controls (Figure 2A–F). Meanwhile, muscle mass reductions witnessed in aged WT + C male (gastrocnemius: −22%; tibialis anterior: −20%; quadriceps: −28%) and WT + C female (gastrocnemius: −27%; tibialis anterior: −36%; quadriceps: −25%) consistently reached greater significance than that of young WT + C (Figure 2G–L; Tables S1 and S2). Similar to body weight, overexpression of PGC1α had mild to negligible effects on muscle mass in young mice. Young male Tg + C animals displayed increases in gastrocnemius (+10%) and tibialis anterior (+15%) compared with WT + C, although there was no protection in quadriceps weight, and gastrocnemius muscles in Tg + C were significantly wasted (−12%) compared with Tg (Figure 2A–C). Young female Tg + C displayed significant losses in gastrocnemius (−8%) and tibialis anterior (−9%) weights compared with Tg, and did not display protection compared with WT + C in any of the assessed muscles (Figure 2D–F). In contrast, PGC1α overexpression proved protective in maintaining skeletal muscle in aged mice. Skeletal muscles (gastrocnemius: +34%; tibialis anterior: +33%; quadriceps: +39%) were significantly protected in aged male Tg + C compared with aged male WT + C (Figure 2G–I). Though aged female Tg + C did not significantly differ from WT + C, muscles in Tg + C were not reduced compared with Tg or WT (Figure 2J–L). Taken together these results demonstrate that cisplatin causes heightened muscle wasting in aged animals, which is counteracted by overexpression of skeletal muscle PGC1α.
Figure 2.
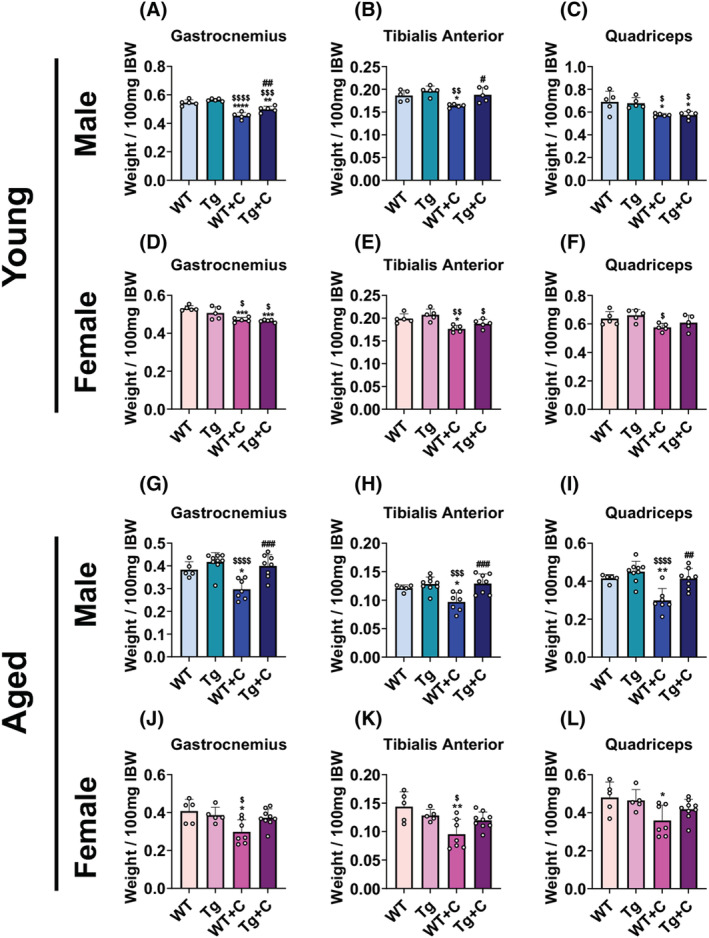
Cisplatin exacerbates muscle wasting in aged mice which is combatted by elevated skeletal muscle PGC1α. Gastrocnemius, tibialis anterior, and quadriceps weights normalized to initial body weight (IBW) of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A–C): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); row 2 (D–F): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); row 3 (G‐I): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); row 4 (J–L): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WT; $ P < 0.05, $$ P < 0.01, $$$ P < 0.001, $$$$ P < 0.0001 versus Tg; # P < 0.05, ## P < 0.01, ### P < 0.001 versus WT + C.
Elevated PGC1α protects against cisplatin‐induced muscle weakness in young and aged mice
As we previously reported that cisplatin promotes loss of muscle strength in young animals, we wanted to assess if cisplatin exacerbates muscle weakness in older mice, and whether muscle PGC1α overexpression is sufficient to combat such event. 9 In vivo plantarflexion assessment demonstrated a 6% baseline‐to‐final drop in torque in young WT + C male and female, while ex vivo contractility of the EDL only revealed a decline in male WT + C specific force (−18%) (Figures 3A–D and S3). In line with heightened muscle wasting, plantarflexion torque assessment demonstrated greater weakness in aged WT + C male (−20%) and female (−17%) than young WT + C. Meanwhile, EDL contractility revealed that only aged female WT + C (−14%) had greater losses in specific force, with old WT + C male reduced (−20%) to a similar extent as young WT + C when compared with untreated controls (Figures 3G,H,J,K and S3, Tables S1 and S2). Interestingly, despite having mild and inconsistent protection over muscle mass in young animals, overexpression of PGC1α was sufficient to protect against declines in in vivo muscle strength and reductions in ex vivo specific force in both young male and female Tg + C (Figures 3A–D and S3). In aged animals, Tg + C male (+13%) and female (+11%) were protected compared with WT + C, while EDL specific force was no different between Tg + C and Tg in either sex (Figures 3G,H,J,K and S3). Interestingly, fatigue assessment of the EDL did not reveal higher fatigability in WT + C compared with WT, although Tg and Tg + C generally displayed lower fatigability across sex and age (Figures 3K,F,I,L and S3).
Figure 3.
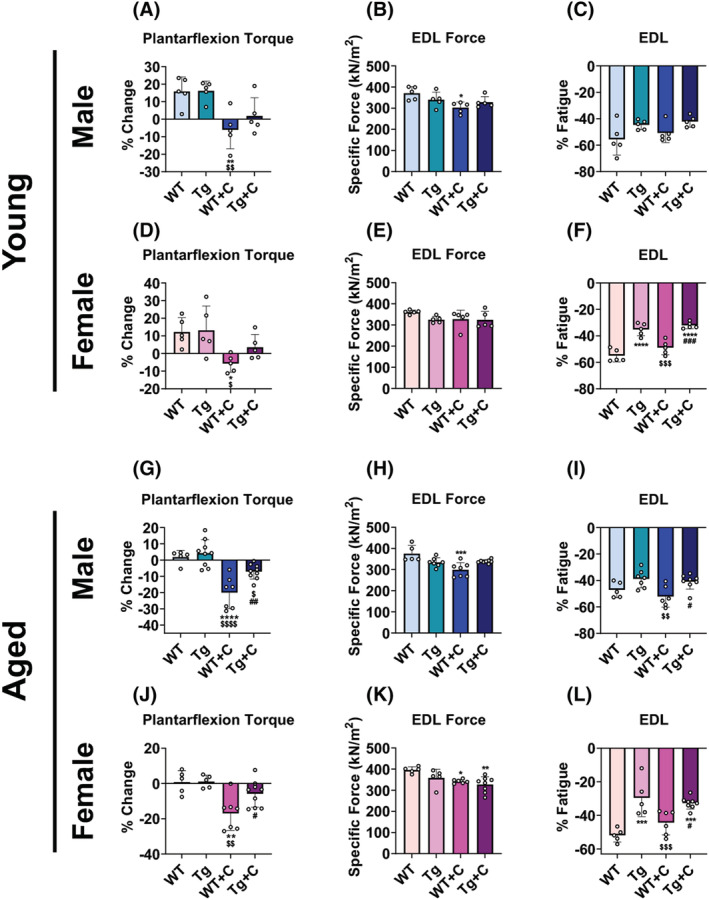
High PGC1α protects against cisplatin‐induced muscle weakness in young and aged mice. Plantarflexion torque % change (vs. Day 1), extensor digitorum longus (EDL) ex vivo specific force (expressed as kN/m2) at 125 Hz, and EDL % fatigue of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A–C): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); row 2 (D–F): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); row 3 (G–I): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); row 4 (J–L): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WT; $ P < 0.05, $$ P < 0.01, $$$ P < 0.001, $$$$ P < 0.0001 versus Tg; # P < 0.05, ## P < 0.01, ### P < 0.001 versus WT + C.
PGC1α overexpression preserves MUNE in young and aged mice treated with cisplatin
We recently reported that cancer and chemotherapy, including cisplatin, promote functional loss of connected motor units. 9 Hence, we assessed whether this was also heighted in aged animals treated with chemotherapy, and whether overexpression of PGC1α would preserve motor unit connectivity. Similar to our previous findings, CMAP levels were unchanged in young animals treated with cisplatin, nor were they changed in aged animals. SMUP was increased in young WT + C (male: +145%; female: +119%), while MUNE was reduced (male: −53%; female: −51%) compared with young untreated WT mice (Figure 4A–F). Interestingly, in line with the muscle phenotype, indices of motor unit connectivity were also exacerbated, consistently reaching greater levels of significance in aged WT animals treated with cisplatin. Aged WT + C male displayed larger increases in SMUP (+7‐fold) and marked reductions in MUNE (−82%) compared with WT untreated male (Figure 4G–I; Table S1). Similarly, aged WT female treated with cisplatin exhibited larger increases in SMUP (+240%) and reductions in MUNE (−62%) than did younger female when compared with untreated WT animals (Figure 4J–L; Table S2). In the case of PGC1α overexpression, Tg + C mice demonstrated consistent protection in SMUP and MUNE parameters. SMUP levels were reduced in young male Tg + C (−46%), young female Tg + C (−56%), aged male Tg + C (−79%), and aged female Tg + C (−56%) compared with WT + C animals (Figure 4B,E,H,K). In the case of MUNE, young male Tg + C were no different from any other group, while young female Tg + C were protected (+76%) compared with WT + C (Figure 4C,F). For aged animals, male Tg + C presented increased MUNE (3‐fold) compared with WT + C, yet were also lower that untreated aged male Tg (−50%), while aged female Tg + C were no different from WT + C, yet also displayed no significant reductions in MUNE compared with untreated WT or Tg animals (Figure 4I,L). Together these results suggest that disrupted motor unit connectivity with cisplatin is heighted with age but can be protected against by overexpression of skeletal muscle PGC1α.
Figure 4.
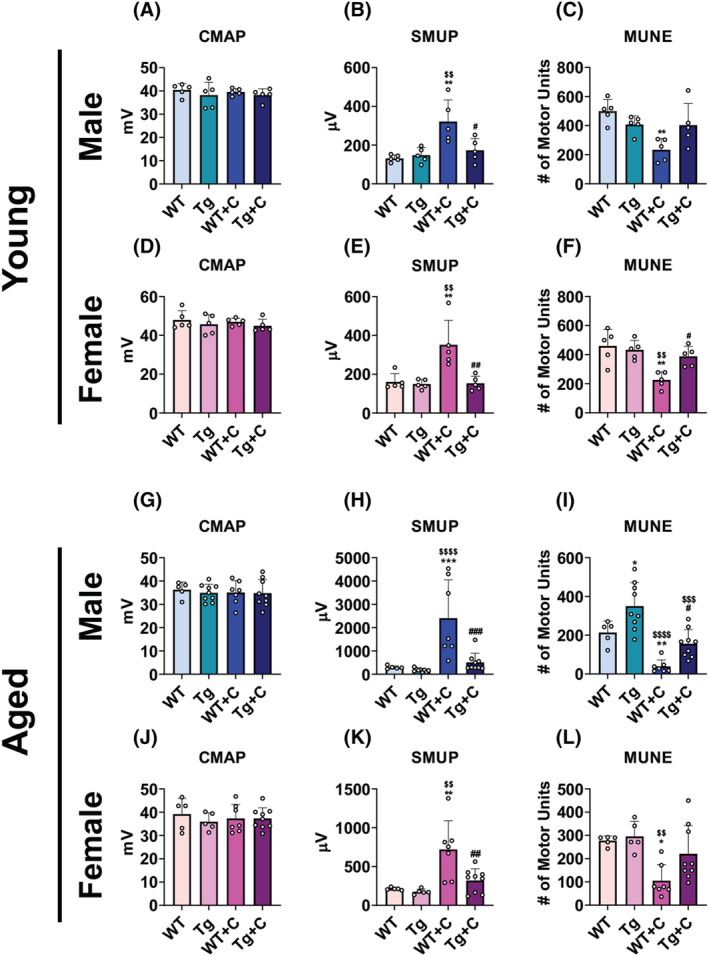
Overexpression of muscle PGC1α preserves MUNE in young and aged mice treated with cisplatin. Compound muscle action potential [CMAP: Millivolts (mV)], single motor unit potential [SMUP; microvolts (μV)], and motor unit number estimation (MUNE) of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A–C): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); row 2 (D–F): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); row 3 (G–I): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); row 4 (J–L): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *P < 0.05, **P < 0.01, ***P < 0.001 versus WT; $$ P < 0.01, $$$ P < 0.001, $$$$ P < 0.0001 versus Tg; # P < 0.05, ## P < 0.01, ### P < 0.001 versus WT + C.
Mitochondrial disruptions by cisplatin are exacerbated with age
As we identified heightened wasting and weakness in aged animals treated with cisplatin, we also assessed whether mitochondrial proteins were reduced to a greater extent. Generally, main effects were seen in PGC1α transgenic animals regardless of age, sex, or treatment for all measured mitochondrial proteins including PGC1α, OPA1, Mitofusin‐2, VDAC, cytochrome‐C, and Cox IV (Figure 5A–X). The one exception in which there was no main effect of transgenic animals was for Mitofusin‐2 in aged male mice. Otherwise, mitochondrial proteins were increased in Tg compared with WT animals (Figure 5A–X). Interestingly, despite previous evidence that cisplatin leads to reductions in PGC1α, 28 , 29 young male WT + C did not display reductions in PGC1α compared with untreated WT animals (Figure 5A). Further, young male WT + C did not show reductions in any mitochondrial proteins assessed, including OPA1, VDAC, cytochrome‐C, and Cox IV, while Mitofusin‐2 levels were increased compared with untreated WT animals (Figure 5B–F). Similar to young male, young female WT + C, did not display reductions in the expression of Mitofusin‐2, VDAC, cytochrome‐C, or Cox IV (Figure 5I–L), whereas OPA1 was significantly reduced (−21%) and PGC1α presented with a tendency for reductions versus untreated WT counterparts (Figure 5G,H). Unlike younger WT animals treated with cisplatin, aged WT mice displayed substantial and consistent reductions in nearly all assessed mitochondrial proteins. Aged male WT + C exhibited reductions in PGC1α (−42%), OPA1 (−45%), Mitofusin‐2 (−54%), cytochrome‐C (−56%) and Cox IV (−55%) compared with WT, while VDAC was non‐significantly reduced (Figure 5M–R; Table S1). Meanwhile, aged female WT + C had reduced PGC1α (61%), OPA1 (−67%), VDAC (−56%), cytochrome‐C (−55%) and Cox IV (−75%) compared with WT, whereas Mitofusin‐2 was elevated (+3‐fold) (Figure 5S–X; Table S2). These results demonstrate that aged animals, which appear to be more susceptible to cisplatin‐induced cachexia, consistently also suffer marked reductions in mitochondrial proteins.
Figure 5.
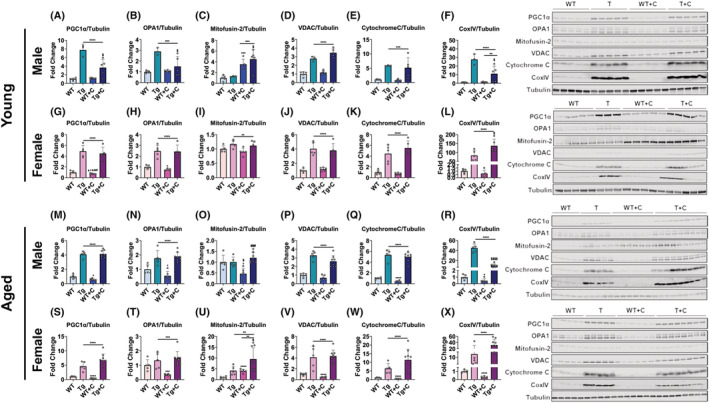
Overexpression of PGC1α preserves muscle mitochondrial proteins in mice treated with cisplatin. Representative western blotting and quantification [expressed as fold change versus wild‐type (WT)] for PGC1α, OPA1, Mitofusin‐2, VDAC, cytochrome‐C, and Cox IV of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A–F): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 3–5); row 2 (G–L): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); row 3 (M–R): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–8); row 4 (S–X): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Tubulin was used as loading control. Data are expressed as mean ± SD. Significance of the differences: *P < 0.05, ***P < 0.001, ****P < 0.0001, versus WT; $ P < 0.05, $$$$ P < 0.0001 versus Tg; ### P < 0.001 versus WT + C; ― spanning Tg‐Tg + C indicates main effect of genotype: **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WT mice; ― spanning WT + C‐Tg + C indicates main effect of treatment: **P < 0.01, ***P < 0.001 versus untreated mice.
Discussion
Chemotherapy remains a preferred treatment option for most cancers, despite having robust off target effects. There is now agreement that chemotherapy can independently cause a cachectic‐like phenotype, characterized by skeletal muscle wasting and weakness, 6 , 7 , 8 , 12 , 31 which, in turn, reduce chemotherapy tolerance, often leading to cessation of treatment and resulting in poorer outcomes in cancer patients. 4 , 5 As there are no currently approved therapies to combat cachexia and improve treatment tolerance, quality of life, and survival in the nearly two million individuals that are diagnosed with cancer every year in the USA, identification of potential strategies to preserve muscle mass and function is of the utmost importance.
Cachexia research has placed emphasis on preserving skeletal muscle mass, especially because preservation of muscle mass improves survival in experimental models of cachexia. 11 , 32 However, recent works have demonstrated that muscle dysfunction may precede wasting, highlighting the importance of functional assessment in cachexia. 22 , 23 Meanwhile, cancer patients may have persistent weakness and fatigability for years following cancer remission, 33 stressing the importance of understanding the mechanisms that impose muscle deficits during cachexia progression. Interestingly, much like muscle weakness, mitochondrial dysfunction may also precede loss of muscle mass in experimental cachexia. 14 , 22 Several routinely used chemotherapeutics are known to induce skeletal muscle weakness and loss of muscle mitochondrial proteins, including PGC1α. 6 , 8 , 9 , 28 , 29 In the present studies we assessed whether overexpression of skeletal muscle PGC1α could preserve muscle mass and function in mice treated with cisplatin. Our findings highlight that cisplatin‐induced cachexia occurs in both male and female mice and is exacerbated in older animals compared with younger mice, with the former benefitting more from the protective effects associated with elevated muscle PGC1α.
With several recent reviews highlighting molecular, physiological, and phenotypic differences between male and female, 18 , 34 , 35 biological sex differences have become an area of interest in cachexia research. Phenotypically, male patients seem to have higher prevalence of cachexia, with a study from Baracos et al. 36 demonstrating muscle wasting in 61% of male and only 31% of female with non‐small cell lung cancer (see review 18 for further examples). Similarly, in the APCMin/+ mouse model of colorectal cancer, male exhibit greater body weight loss than female. 37 However, to our knowledge few studies have observed phenotypic sex differences in response to chemotherapy alone. In the present study both male and female WT mice were susceptible to cisplatin‐induced cachexia. In young WT mice, male (−13%) and female (−12%) had comparable reductions in body weight. Similarly, with respect to lean mass assessments by EchoMRI, male and female displayed analogous losses of 14% and 13%, respectively. Moreover, young male and female WT mice also experienced equivalent declines in plantarflexion torque, suggesting that cisplatin has similar cachexia‐like consequences regardless of sex. As we decipher between sex differences in cachexia research, chemotherapy regimens are certainly another variable to consider. It is important to note, that in the present study animals were not inoculated with tumours, thus representing a limitation of our approach. As our laboratory has previously shown that cachexia can be exacerbated by the combination of cancer and chemotherapy in male mice, 31 further investigations should also begin to investigate if this holds true in female mice.
In similar fashion to identifying potential sex response differences during cachexia, age of the experimental animals used is another important consideration. Recent reports have contended that age‐appropriate (i.e. older) animals should be included into experimental cancer research, especially because cancer in young individuals is far rarer. 19 , 20 This is also true of cachexia research, where a majority of tumour‐induced cachexia models (e.g. C26 or LLC) utilize young adult mice up to 14 weeks of age, approximately equivalent to a 25 year old human. 19 Few studies have examined the differences in cancer cachexia development between young and old mice, and to our knowledge none have examined the age‐associated phenotypic effects of chemotherapy‐induced cachexia. Here, we show that cisplatin‐induced losses in body weight, fat mass, lean mass, skeletal muscle size and skeletal muscle function all tend to be exacerbated in older (18 month) male and female WT animals compared with traditionally used younger (2 month) mice. In line with this, recent work from Geppert et al. demonstrated that older C57BL/6N (16 month vs. 2 month) and C57BL/6J (20 month vs. 3 month) LLC‐tumour bearing mice were more susceptible to develop body weight loss. 21 However, much like our present study did not include tumour implantations, the work from Geppert et al. did not include chemotherapeutic regimens. Whether the heightened cachexia observed with cancer or chemotherapy alone in aged mice would be compounded by a combination of cancer and chemotherapy is unknown. Future cachexia investigations should consider incorporating combinations of cancer and chemotherapy in aged animals to help clarify this point. Moreover, the present study was only limited to one anti‐cancer treatment. Given that we and others have shown cachexia‐like phenotypes in young animals treated with a range of chemotherapeutics, future studies should consider examining if the negative consequences of these anti‐cancer treatments are consistently heighted with age. 6 , 8 , 10 , 12 , 38
Identification of therapeutic targets to combat skeletal muscle wasting and weakness caused by cancer and chemotherapy continues to progress, yet skeletal muscle mitochondria have remained a focus point. Work from our group and others have demonstrated that cachectic muscle is associated with the loss of mitochondrial proteins. Regarding experimental chemotherapy‐induced cachexia, loss of skeletal muscle PGC1α has been consistent across several chemotherapeutic regimens, including folfiri, folfox, sorafenib, doxorubicin and cisplatin. 6 , 8 , 28 , 29 , 38 Interestingly, in contrast to previous work, young WT male treated with cisplatin in our study did not display reductions in skeletal muscle PGC1α. We presently measured protein in quadriceps muscles, while prior work assessed PGC1α levels in gastrocnemius, soleus, and tibialis anterior muscles, which may account for discrepancy of results. 28 , 29 Further, these two studies were conducted in rats, while our present work was conducted in mice. Meanwhile, young treated female WT animals did present reductions in mitochondrial proteins. With recent reviews highlighting that female have greater mitochondrial content and activity, it is plausible that female may be more susceptible to chemotherapy‐induced mitochondrial dysfunction. 18 , 35 Strikingly, and regardless of sex, older WT animals displayed dramatic loss of muscle mitochondrial proteins. As we also observed heighted muscle wasting and weakness in older mice treated with cisplatin, which was largely protected against in PGC1α, our data suggests that mitochondrial targeted strategies may be more advantageous in older animals. Interestingly, a majority of cachexia studies using PGC1α transgenic mice, mitochondrial targeting agents (e.g. SS‐31, MitoQ, and trimetazidine), or exercise resulting in either mild benefits or negligible results have been conducted in young animals. 13 , 15 , 16 , 17 , 39 , 40 Interestingly, tumours have shown to grow larger in young PGC1α transgenic compared with WT mice, yet whether this occurs in older animals, where tumours tend to grow slower than in younger mice, has not been investigated. 17 , 21 As we presently demonstrate improved anti‐cachectic benefits of muscle PGC1α overexpression in older animals, future studies should consider examining whether other mitochondria‐targeted strategies have improved efficacy when performed in older animals, particularly in the presence of cancer and chemotherapy.
We and others have recently reported that cancer and chemotherapy promote loss of NMJ proteins and reduced presynaptic staining, suggestive of disrupted skeletal muscle innervation in cachexia. 9 , 24 Moreover, we demonstrated cancer and chemotherapy, including cisplatin, promote a loss of functionally connected motor units (i.e. MUNE), which associates with muscle weakness. 9 Interestingly, mitochondria have been implicated in regulating NMJ stability, fragmentation, and formation in neuromuscular disease. 25 , 26 Our current results demonstrate that cisplatin, which causes greater loss of mitochondrial proteins in aged animals, also induces exacerbated loss of MUNE in aged WT animals. Meanwhile, overexpression of PGC1α largely preserves MUNE in combination with cisplatin treatment, suggesting that maintenance of mitochondria is likely sufficient to preserve neuromuscular function.
In summary, our present study demonstrates that cisplatin treatment promotes weight loss, muscle wasting, muscle weakness and loss of motor unit connectivity in both male and female mice. We also demonstrate that the negative effects of cisplatin on body weight, muscle mass, muscle force, and motor unit connectivity are exacerbated in aged animals. Our approach including overexpression of skeletal muscle PGC1α proved effective in reducing muscle weakness and loss of MUNE caused by cisplatin. Taken together, our results suggest that mitochondrial therapeutic strategies may be able to counteract skeletal muscle dysfunction caused by chemotherapy, with potentially greater efficacy in older animals. Future cachexia studies should investigate whether translational mitochondria‐targeted approaches are sufficient to preserve skeletal muscle mass and function in older animals bearing cancer, alone or in combination with chemotherapy.
Conflict of interest
The authors have declared that no conflict of interest exists.
Supporting information
Table S1. Statistical comparisons of young and aged male wild‐type animals treated with cisplatin.
Table S2. Statistical comparisons of young and aged female wild‐type animals treated with cisplatin.
Figure S1. Schematic representation of the experimental model. (A) Young (2‐month) and old (18‐month), male and female, wild‐type (WT) C57BL/6J or C57BL/6‐Tg(Ckm‐Ppargc1a)31Brsp/J (mPGC1α: referred to as Tg) were injected intraperitoneally with cisplatin (C; 2.5 mg/kg). The control, untreated mice received equal volumes of sterile saline. The red arrows indicate the day of cisplatin treatment. (B) Representative western blotting of PGC1α for WT and Tg animals. (C) Summary of group sample sizes.
Figure S2. Cisplatin exacerbates fat wasting in aged mice. Fat, liver, heart, kidney, and spleen weights normalized to initial body weight (IBW) of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A‐E): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); Row 2 (F‐J): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); Row 3 (K‐O): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); Row 4 (P–T): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. WT; $ p < 0.05, $$p < 0.01, $$$p < 0.001, $ $ $ $p < 0.0001 vs. Tg; #p < 0.05 vs. WT + C.
Figure S3. Protection against cisplatin‐induced muscle weakness by overexpression of PGC1α. Force frequency relationship of extensor digitorum longus (EDL) and EDL % fatigue over 60 contractions at 60 Hz of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A‐B): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); Row 2 (C‐D): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); Row 3 (E‐F): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); Row 4 (G‐H): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *p < 0.05, **p < 0.01, ***p < 0.001 WT + C vs. WT.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. This study was supported by the Department of Surgery and the Department of Otolaryngology – Head & Neck Surgery at Indiana University, by grants from the V Foundation for Cancer Research (V2017‐021), the American Cancer Society (Research Scholar Grant 132013‐RSG‐18‐010‐01‐CCG), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR079379) to AB. JRH was supported by a T32 Institutional Training Grant from NIH (AR065971). The #12G10 anti‐Tubulin monoclonal antibody (developed by Frankel J and Nelsen EM at University of Iowa) was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA, USA.
Huot J. R., Pin F., Chatterjee R., and Bonetto A. (2022) PGC1α overexpression preserves muscle mass and function in cisplatin‐induced cachexia, Journal of Cachexia, Sarcopenia and Muscle, 13, 2480–2491, 10.1002/jcsm.13035
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 3. Melstrom LG, Melstrom KA Jr, Ding XZ, Adrian TE. Mechanisms of skeletal muscle degradation and its therapy in cancer cachexia. Histol Histopathol 2007;22:805–814. [DOI] [PubMed] [Google Scholar]
- 4. von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers‐update 2014. J Cachexia Sarcopenia Muscle 2014;5:261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruggeman AR, Kamal AH, LeBlanc TW, Ma JD, Baracos VE, Roeland EJ. Cancer Cachexia: Beyond Weight Loss. J Oncol Pract 2016;12:1163–1171. [DOI] [PubMed] [Google Scholar]
- 6. Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A. Chemotherapy‐related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016;7:43442–43460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Essex AL, Pin F, Huot JR, Bonewald LF, Plotkin LI, Bonetto A. Bisphosphonate Treatment Ameliorates Chemotherapy‐Induced Bone and Muscle Abnormalities in Young Mice. Front Endocrinol (Lausanne) 2019;10:809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huot JR, Essex AL, Gutierrez M, Barreto R, Wang M, Waning DL, et al. Chronic Treatment with Multi‐Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers (Basel) 2019;11. 10.3390/cancers11040571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huot JR, Pin F, Bonetto A. Muscle weakness caused by cancer and chemotherapy is associated with loss of motor unit connectivity. Am J Cancer Res 2021;11:2990–3001. [PMC free article] [PubMed] [Google Scholar]
- 10. Hain BA, Xu H, Wilcox JR, Mutua D, Waning DL. Chemotherapy‐induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun 2019;2:1–12. [PMC free article] [PubMed] [Google Scholar]
- 11. Chen JA, Splenser A, Guillory B, Luo J, Mendiratta M, Belinova B, et al. Ghrelin prevents tumour‐ and cisplatin‐induced muscle wasting: characterization of multiple mechanisms involved. J Cachexia Sarcopenia Muscle 2015;6:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front Physiol 2016;7:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ballaro R, Lopalco P, Audrito V, Beltra M, Pin F, Angelini R, et al. Targeting Mitochondria by SS‐31 Ameliorates the Whole Body Energy Status in Cancer‐ and Chemotherapy‐Induced Cachexia. Cancers (Basel) 2021;13. 10.3390/cancers13040850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown JL, Rosa‐Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2017;8:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ballaro R, Beltra M, De Lucia S, Pin F, Ranjbar K, Hulmi JJ, et al. Moderate exercise in mice improves cancer plus chemotherapy‐induced muscle wasting and mitochondrial alterations. FASEB J 2019;33:5482–5494. [DOI] [PubMed] [Google Scholar]
- 16. Pin F, Busquets S, Toledo M, Camperi A, Lopez‐Soriano FJ, Costelli P, et al. Combination of exercise training and erythropoietin prevents cancer‐induced muscle alterations. Oncotarget 2015;6:43202–43215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Pickrell AM, Zimmers TA, Moraes CT. Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer‐induced cachexia. PLoS One 2012;7:e33426. 10.1371/journal.pone.0033426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhong X, Zimmers TA. Sex Differences in Cancer Cachexia. Curr Osteoporos Rep 2020;18:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S, Lai X, Deng Y, Song Y. Correlation between mouse age and human age in anti‐tumor research: Significance and method establishment. Life Sci 2020;242:117242. 10.1016/j.lfs.2019.117242 [DOI] [PubMed] [Google Scholar]
- 20. Beheshti A, Benzekry S, McDonald JT, Ma L, Peluso M, Hahnfeldt P, et al. Host age is a systemic regulator of gene expression impacting cancer progression. Cancer Res 2015;75:1134–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geppert J, Walth AA, Terron Exposito R, Kaltenecker D, Morigny P, Machado J, et al. Aging Aggravates Cachexia in Tumor‐Bearing Mice. Cancers (Basel) 2021;14. 10.3390/cancers14010090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. VanderVeen BN, Fix DK, Montalvo RN, Counts BR, Smuder AJ, Murphy EA, et al. The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin‐6. Exp Physiol 2019;104:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. VanderVeen BN, Hardee JP, Fix DK, Carson JA. Skeletal muscle function during the progression of cancer cachexia in the male Apc (Min/+) mouse. J Appl Physiol (1985) 2018;124:684–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sartori R, Hagg A, Zampieri S, Armani A, Winbanks CE, Viana LR, et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci Transl Med 2021;13. 10.1126/scitranslmed.aay9592 [DOI] [PubMed] [Google Scholar]
- 25. De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, et al. Familial amyotrophic lateral sclerosis‐linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 2007;16:2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magrane J, Sahawneh MA, Przedborski S, Estevez AG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci 2012;32:229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu H, Ranjit R, Richardson A, Van Remmen H. Muscle mitochondrial catalase expression prevents neuromuscular junction disruption, atrophy, and weakness in a mouse model of accelerated sarcopenia. J Cachexia Sarcopenia Muscle 2021;12:1582–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sirago G, Conte E, Fracasso F, Cormio A, Fehrentz JA, Martinez J, et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin‐induced cachexia. Sci Rep 2017;7:13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bae JH, Seo DY, Lee SH, Shin C, Jamrasi P, Han J, et al. Effects of exercise on AKT/PGC1‐alpha/FOXO3a pathway and muscle atrophy in cisplatin‐administered rat skeletal muscle. Korean J Physiol Pharmacol 2021;25:585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia JM, Cata JP, Dougherty PM, Smith RG. Ghrelin prevents cisplatin‐induced mechanical hyperalgesia and cachexia. Endocrinology 2008;149:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pin F, Barreto R, Couch ME, Bonetto A, O'Connell TM. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle 2019;10:140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach‐Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti‐cancer treatments. Skelet Muscle 2016;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meeske K, Smith AW, Alfano CM, McGregor BA, McTiernan A, Baumgartner KB, et al. Fatigue in breast cancer survivors two to five years post diagnosis: a HEAL Study report. Qual Life Res 2007;16:947–960. [DOI] [PubMed] [Google Scholar]
- 34. Montalvo RN, Counts BR, Carson JA. Understanding sex differences in the regulation of cancer‐induced muscle wasting. Curr Opin Support Palliat Care 2018;12:394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosa‐Caldwell ME, Greene NP. Muscle metabolism and atrophy: let's talk about sex. Biol Sex Differ 2019;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baracos VE, Reiman T, Mourtzakis M, Gioulbasanis I, Antoun S. Body composition in patients with non‐small cell lung cancer: a contemporary view of cancer cachexia with the use of computed tomography image analysis. Am J Clin Nutr 2010;91:1133S–1137S. [DOI] [PubMed] [Google Scholar]
- 37. Hetzler KL, Hardee JP, Puppa MJ, Narsale AA, Sato S, Davis JM, et al. Sex differences in the relationship of IL‐6 signaling to cancer cachexia progression. Biochim Biophys Acta 2015;1852:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hulmi JJ, Nissinen TA, Rasanen M, Degerman J, Lautaoja JH, Hemanthakumar KA, et al. Prevention of chemotherapy‐induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Molinari F, Pin F, Gorini S, Chiandotto S, Pontecorvo L, Penna F, et al. The mitochondrial metabolic reprogramming agent trimetazidine as an 'exercise mimetic' in cachectic C26‐bearing mice. J Cachexia Sarcopenia Muscle 2017;8:954–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pin F, Huot JR, Bonetto A. The Mitochondria‐Targeting Agent MitoQ Improves Muscle Atrophy, Weakness and Oxidative Metabolism in C26 Tumor‐Bearing Mice. Front Cell Dev Biol 2022;10:861622. 10.3389/fcell.2022.861622 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Statistical comparisons of young and aged male wild‐type animals treated with cisplatin.
Table S2. Statistical comparisons of young and aged female wild‐type animals treated with cisplatin.
Figure S1. Schematic representation of the experimental model. (A) Young (2‐month) and old (18‐month), male and female, wild‐type (WT) C57BL/6J or C57BL/6‐Tg(Ckm‐Ppargc1a)31Brsp/J (mPGC1α: referred to as Tg) were injected intraperitoneally with cisplatin (C; 2.5 mg/kg). The control, untreated mice received equal volumes of sterile saline. The red arrows indicate the day of cisplatin treatment. (B) Representative western blotting of PGC1α for WT and Tg animals. (C) Summary of group sample sizes.
Figure S2. Cisplatin exacerbates fat wasting in aged mice. Fat, liver, heart, kidney, and spleen weights normalized to initial body weight (IBW) of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A‐E): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); Row 2 (F‐J): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); Row 3 (K‐O): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); Row 4 (P–T): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. WT; $ p < 0.05, $$p < 0.01, $$$p < 0.001, $ $ $ $p < 0.0001 vs. Tg; #p < 0.05 vs. WT + C.
Figure S3. Protection against cisplatin‐induced muscle weakness by overexpression of PGC1α. Force frequency relationship of extensor digitorum longus (EDL) and EDL % fatigue over 60 contractions at 60 Hz of wild‐type (WT) and PGC1α transgenic (Tg) mice treated with cisplatin (C: 2.5 mg/kg) or vehicle for 14 days. Row 1 (A‐B): 2‐month‐old male WT, Tg, WT + C, Tg + C (n = 5); Row 2 (C‐D): 2‐month‐old female WT, Tg, WT + C, Tg + C (n = 5); Row 3 (E‐F): 18‐month‐old male WT, Tg, WT + C, Tg + C (n = 5–9); Row 4 (G‐H): 18‐month‐old female WT, Tg, WT + C, Tg + C (n = 5–9). Data are expressed as mean ± SD. Significance of the differences: *p < 0.05, **p < 0.01, ***p < 0.001 WT + C vs. WT.