

Abstract
Background
Patients with multiple sclerosis (MS) experience reduced exercise tolerance that substantially reduces quality of life. The mechanisms underpinning exercise intolerance in MS are not fully clear. This study aimed to determine the contributions of the cardiopulmonary system and peripheral muscle in MS‐induced exercise intolerance before and after exercise training.
Methods
Twenty‐three patients with MS (13 women) and 20 age‐matched and sex‐matched healthy controls (13 women) performed a cardiopulmonary exercise test. Muscle fibre type composition, size, succinate dehydrogenase (SDH) activity, capillarity, and gene expression and proteins related to mitochondrial density were determined in vastus lateralis muscle biopsies. Nine MS patients (five women) were re‐examined following a 12 week exercise training programme consisting of high‐intensity cycling interval and resistance training.
Results
Patients with MS had lower maximal oxygen uptake compared with healthy controls (V̇O2peak, 25.0 ± 8.5 vs. 35.7 ± 6.4 mL/kg/min, P < 0.001). The lower gas exchange threshold (MS: 14.5 ± 5.5 vs. controls: 19.7 ± 2.9 mL/kg/min, P = 0.01) and slope of V̇O2 versus work rate (MS: 9.5 ± 1.7 vs. controls: 10.8 ± 1.1 mL/min/W, P = 0.01) suggested an intramuscular contribution to exercise intolerance in patients with MS. Muscle SDH activity was 22% lower in MS (P = 0.004), and strongly correlated with several indices of whole‐body exercise capacity in MS patients (e.g. V̇O2peak, Spearman's ρ = 0.81, P = 0.002), but not healthy controls (ρ = 0.24, P = 0.38). In addition, protein levels of mitochondrial OXPHOS complexes I (−40%, P = 0.047) and II (−45%, P = 0.026) were lower in MS patients versus controls. Muscle capillary/fibre ratio correlated with V̇O2peak in healthy controls (ρ = 0.86, P < 0.001) but not in MS (ρ = 0.35, P = 0.22), and did not differ between groups (1.41 ± 0.30 vs. 1.47 ± 0.38, P = 0.65). Expression of genes involved in mitochondrial function, such as PPARA, PPARG, and TFAM, was markedly reduced in muscle tissue samples of MS patients (all P < 0.05). No differences in muscle fibre type composition or size were observed between groups (all P > 0.05). V̇O2peak increased by 23% following exercise training in MS (P < 0.001); however, no changes in muscle capillarity, SDH activity, gene or protein expression were observed (all P > 0.05).
Conclusions
Skeletal muscle oxidative phenotype (mitochondrial complex I and II content, SDH activity) is lower in patients with MS, contributing to reduced exercise tolerance. However, skeletal muscle mitochondria appeared resistant to the beneficial effects of exercise training, suggesting that other physiological systems, at least in part, drive the improvements in exercise capacity following exercise training in MS.
Keywords: Exercise capacity, Exercise therapy, Mitochondria, Multiple sclerosis, Oxidative metabolism, Skeletal muscle
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disorder of the central nervous system and the leading cause of non‐traumatic neurological disability among young adults. 1 Following leukocyte invasion, multifocal inflammatory demyelinating lesions can be observed throughout the central nervous system, culminating in diverse symptoms including sensory, motor, and visual deficits, as well as fatigue, cognitive impairments, and depression. In most patients, MS initially manifests as a relapsing–remitting disease and advances into a secondary progressive phase in which neurodegeneration reflected by brain atrophy is the best predictor of clinical disability.
Patients with MS display limited physical capacity, which may in part be related to their habitually lower voluntary physical activity. 2 , 3 Patients with MS therefore have an increased risk for developing chronic diseases related to a sedentary lifestyle, such as metabolic and cardiovascular diseases (e.g. type 2 diabetes mellitus, obesity, and heart failure). 4 Lower peak O2 uptake (V̇O2peak) and physical inactivity in patients with MS have been hypothesized to impact MS pathology and progression. 2 , 5 Moreover, disease severity negatively correlates with markers of exercise tolerance among ambulatory MS patients. 6 , 7 Both muscle strength and aerobic capacity are lower in patients with MS. 2 , 5 , 8 Exercise training results in improvements in exercise capacity and markers of health‐related quality of life in patients with MS, 7 , 9 but the mechanisms underpinning training adaptations in this population are not well understood. 10 , 11 , 12 Understanding the limiting factors contributing to the exercise intolerance observed in MS, the mechanisms underlying their alterations with training, and the most effective intervention to maximize adherence 13 will aid increasing the confidence of health‐care providers in prescribing exercise to patients with MS. Concurrent resistance and high‐intensity interval training is well‐tolerated by patients with MS 14 , 15 and is effective in improving certain cardiovascular risk factors and exercise capacity in this population. 15 , 16 The efficacy of concurrent resistance and aerobic exercise training in this population therefore deserves further study. 16
Peripheral alterations in the skeletal muscle of patients with MS likely contribute to their exercise intolerance. Skeletal muscles from MS patients may undergo a slow‐to‐fast muscle phenotype shift and are prone to atrophy 17 , 18 ; however, this is not consistently observed. 19 Moreover, a lower muscle oxidative capacity has been inferred from slowed phosphocreatine recovery kinetics, 20 impaired muscle metabolic stability, 21 slower pulmonary O2 uptake (V̇O2) kinetics, 22 and accelerated quadriceps muscle fatigue during electrical muscle stimulation. 21 , 23 Because these alterations also result from muscle deconditioning, it remains to be determined whether similar adaptations are direct consequences of the pathophysiology of MS per se 18 , 21 or are indirectly driven by deconditioning. If deconditioning is the primary driving force for these skeletal muscle impairments then they should be alleviated by an exercise training intervention.
In order to determine limitations to exercise tolerance in MS, we simultaneously studied whole‐body exercise capacity and skeletal muscle characteristics in MS patients. Patients with MS and age‐matched and sex‐matched healthy controls underwent cardiopulmonary exercise testing to determine contributing factors to exercise tolerance. Additionally, in skeletal muscle biopsies, we assessed important structural and metabolic components of exercise capacity. To determine whether exercise training improves skeletal muscle structure and function in patients with MS, a subgroup was tested again after a 12 week combined high‐intensity cycling interval and resistance exercise training intervention. We hypothesized that patients with MS display a lower V̇O2peak compared with healthy controls, with concomitant intramuscular differences in fibre oxidative capacity, size and capillary density compared with controls. We also hypothesized that a 12 week combined high‐intensity cycling interval and resistance exercise training programme improves exercise tolerance and cellular variables of aerobic function in patients with MS.
Methods
Participants
Twenty‐three patients with MS (10 men and 13 women) and 20 healthy controls (7 men and 13 women) were recruited via local advertisement. Participants were excluded if they were <18 years, participated in another study, had contraindications to perform physical exercise, and, in patients with MS, suffered a relapse within 6 months prior to the study or had an EDSS > 6 (non‐ambulatory). All participants gave written informed consent. Ethical approval was obtained from the local Ethical Committee of the Jessa Hospital and Hasselt University. All study procedures were performed in accordance with the Declaration of Helsinki (trial registration number NCT02466165).
Body weight and height were measured and participants completed the Physical Activity Scale for Individuals with Physical Disabilities (PASIPD). Maximal voluntary muscle strength was measured as previously described (supporting information). 15
Exercise capacity
A maximal cardiopulmonary exercise test was performed on a braked cycle ergometer (eBike BasicVR, General Electric GmbH) with a metabolic cart (Jaeger OxyconVR, Erich Jaeger GmbH) to determine gas exchange and ventilatory variables breath‐by‐breath. Participants cycled at a frequency of ~70 rpm throughout and started the test at 20 (women) or 30 W (men) for 1 min. Thereafter, power output increased in steps of 10 (women) or 15 (men) W/min until task failure, defined as the point at which cadence dropped below 60 rpm. Heart rate was monitored using a 12‐lead ECG device (custo cardio 400, Custo med). A detailed analysis procedure is described in the supporting information. 24
Muscle sampling
Resting muscle biopsies from the mid‐section of the vastus lateralis muscle were obtained using the Bergström needle technique. In patients, biopsies were taken from the weakest leg based on isokinetic dynamometry measurements (System 3, Biodex, ENRAF‐NONIUS). In healthy controls, biopsies were taken from a random leg. Visible connective tissue was carefully removed, and part of the biopsy was immediately snap‐frozen in liquid nitrogen for molecular techniques. A part of the biopsy was aligned longitudinally, embedded in Tissue Tek O.C.T. compound, and frozen in liquid nitrogen‐cooled isopentane for histology. All samples were stored at −80°C until further analysis.
Muscle fibre type composition and fibre size
Transverse sections (10 μm, Microm HM 550 cryostat, Thermo Scientific) were stained against myosin heavy chain isoform I, IIa, IIx, and cell membranes (with laminin). Detailed description of the immunohistochemistry protocol is presented in the supporting information. Sections were imaged at ×10 magnification using a Leica DM4000 B LED microscope and a Leica EL6000 external light source (Leica Microsystems). Muscle fibre type composition, fibre cross‐sectional area (FCSA, μm2), and fibre minimal Feret's diameter (μm) were quantified manually using ImageJ software (National Institutes of Health, NIH). No pure type IIx fibres were observed, therefore type I, IIa, and IIa/x fibres were quantified. On average, 326 ± 161 muscle fibres were analysed per biopsy.
Muscle succinate dehydrogenase activity
Under conditions of unlimited oxygen supply, succinate dehydrogenase (SDH) activity is linearly related to the maximum rate of O2 uptake by the muscle fibre. 25 Maximal SDH activity was determined in muscle sections by histochemistry as published previously 26 and detailed in the supporting information. SDH activity was analysed using ImageJ (NIH), pooled per fibre type, via serial sections stained for fibre type, and also weighted for muscle fibre type composition. SDH activity was expressed in ΔA660 per μm tissue thickness per second of staining time (ΔA660/μm/s). A minimum of 10 fibres per fibre type was included per participant (on average: 41 ± 24 type I, 38 ± 26 type IIa, and 23 ± 13 type IIa/x fibres).
Muscle capillarity
To determine muscle capillarity, muscle sections were stained against laminin (cell membranes) and CD31 (endothelial marker). Details of the immunohistochemistry protocol can be found in the supporting information. Capillary/fibre ratio and capillary density (expressed per mm2) were calculated from a random area (1.02 ± 0.45 mm2) containing at least 100 fibres (ImageJ, NIH).
Protein concentrations
Detailed description of the western immuno‐blot protocols to determine protein levels of PGC‐1α and five subunits of mitochondrial complexes in snap‐frozen muscle tissue are presented in the supporting information.
Gene expression analysis
Detailed description of the RNA isolation, cDNA synthesis and quantitative polymerase chain reaction (qPCR) protocols are presented in the supporting information. Gene expression of mitochondria‐related gene transcripts (Table S1) was calculated with the 2−ΔΔCt method, normalized to the geometric mean of RPL13A and GAPDH expression, and expressed as fold changes.
Exercise training intervention
Following baseline muscle biopsy sampling and exercise testing, a subgroup of MS patients (n = 9; four men and five women) were enrolled in a 12 week exercise training programme consisting of high‐intensity cycling interval and resistance training. Patients attended five sessions every 2 weeks with one‐on‐one supervision from a physical therapist. Cycle interval training progressed from five 1 min bouts to five 2 min bouts at the highest power output attained during the incremental exercise test, as described previously. 15 Machine‐based upper and lower body resistance exercises progressed from 1 × 10 to 2 × 20 repetitions at an individual maximal attainable load for each subject. 15 Lower limb exercises were performed separately with each leg.
Statistical analysis
Data are presented as mean ± SD. Normality of the data was tested by Shapiro–Wilk tests (for intervention effects, the difference scores were used). Baseline characteristics of both groups were compared with independent t‐tests and χ 2 tests, where appropriate. Muscle fibre type, size, and fibre type‐specific SDH activity were analysed by a repeated measures fibre type × group (3 × 2) mixed model with Bonferroni‐adjusted post hoc tests. Differences in fibre type composition was assessed by independent t‐tests. For all other outcomes, MS patients and healthy subjects were compared by means of independent t‐tests or Mann–Whitney U‐tests when data were not normally distributed. Correlational analyses were run with Spearman correlation coefficients. Multiple linear regression analysis was used to compute the contribution of different muscle parameters to exercise capacity in each group. Multicollinearity was assessed by the variance inflation factor (VIF) diagnostic. The effects of the training intervention were assessed by paired t‐tests or Wilcoxon matched pairs signed‐rank tests, where appropriate. Statistics were performed with IBM SPSS Statistics v25 (NY, USA) and the significance level was set at α = 0.05.
Results
Maximal exercise capacity and aerobic function are impaired in MS
Participant characteristics are given in Table 1. Patients with MS had a significantly lower peak oxygen uptake (V̇O2peak) compared with healthy controls (−27%; P = 0.009, Figure 1A, Tables S2 and S3), which correlated negatively with the Expanded Disability Status Scale (EDSS, Spearman's ρ = −0.639, P = 0.003, Figure 1B). Peak minute ventilation (V̇Epeak) as a proportion of predicted maximal voluntary ventilation (V̇E%MVV) was lower in patients with MS, indicating no respiratory limitation at peak exercise. This is confirmed by the lack of difference in other variables related to respiratory function, such as V̇Epeak, tidal volume (VTpeak), breathing frequency (BFpeak) and the slope of V̇E versus CO2 output (V̇CO2; all P > 0.05, Table S2). Peak heart rate (HRpeak; P = 0.014) and peak O2 pulse (i.e. V̇O2peak/HRpeak; P = 0.043) were both significantly lower in MS compared with controls (Figure 1C,D, Table S2), indicative of impaired maximal cardiac function in patients with MS. Because peak O2 pulse conflates both maximal peripheral O2 extraction and cardiac stroke volume, both lower cardiac output and muscle O2 extraction at maximal exercise potentially contributed to the lower V̇O2peak observed in patients with MS.
Table 1.
Baseline subject characteristics
| Controls (n = 20) | MS (n = 23) | MS intervention study (n = 9) | |
|---|---|---|---|
| Age (year) | 49 ± 12 | 53 ± 8 | 52 ± 8 |
| Gender (m/f) | 7/13 | 10/13 | 4/5 |
| Weight (kg) | 68 ± 12 | 72 ± 12 | 68 ± 15 |
| BMI (kg/m2) | 23.9 ± 2.2 | 24.5 ± 3.5 | 23.0 ± 3.4 |
| Smoking (yes/no) | 2/18 | 5/18 | 0/9 |
| PASIPD | 21.6 ± 11.7 | 16.3 ± 11.9 | 20.9 ± 13.4 |
| EDSS | / | 3.1 ± 1.6 | 2.6 ± 1.5 |
| Type MS (RR/SP/PP/unknown) | / | 17/4/1/1 | 6/3/0/0 |
Baseline subject characteristics of healthy controls and patients with MS (whole group and those who participated in the training intervention). BMI, body mass index; EDSS, Expanded Disability Status Scale; MS, multiple sclerosis; PASIPD, Physical Activity Scale for Individuals with Physical Disabilities; PP, primary progressive; RR, relapsing remitting; SP, secondary progressive. No significant differences were found for any variable (MS vs. controls, all P > 0.05).
Figure 1.
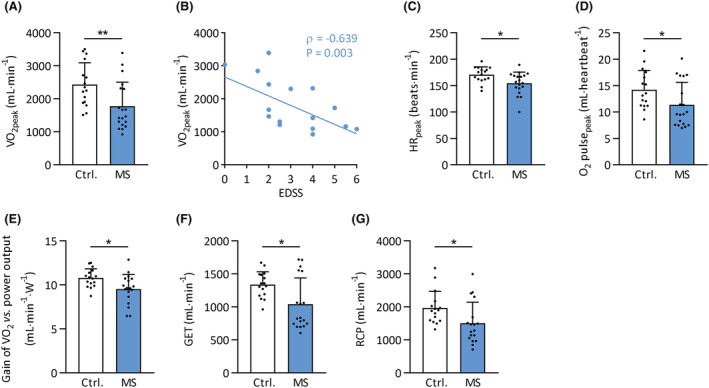
Lower exercise capacity in MS. Variables determined during cardiopulmonary exercise testing in MS patients and healthy controls. Black circles represent individual participant values. (A) Peak oxygen uptake (V̇O2peak) in MS patients versus controls. (B) Relationship between V̇O2peak and score on the expanded disability status scale (EDSS) within the MS patients. (C) Peak heart rate (HRpeak) in MS patients versus controls. (D) Peak O2 pulse (O2 pulsepeak), determined as V̇O2peak/HRpeak in MS patients versus controls. (E) Gain of the relationship between V̇O2 and change in external power output in MS patients versus controls. (F) Gas exchange threshold (GET) in MS versus controls. (G) Respiratory compensation point (RCP) in MS versus controls. *P < 0.05; **P < 0.01. Mean ± SD.
Submaximal aerobic function is impaired in MS despite normal cardiac and pulmonary function
The gain of the V̇O2/power output relationship (in mL/min/W) was lower in patients with MS compared with healthy controls (P = 0.013, Figure 1E, Table S2), indicating a blunted oxidative metabolic response for a given external workload. The gas exchange threshold and respiratory compensation point (i.e. submaximal indices of whole‐body blood acid–base homeostasis during submaximal exercise) were both lower in patients with MS (P = 0.011 and P = 0.027, respectively; Figure 1F,G, Table S2). The cardiovascular and respiratory responses to submaximal exercise, indicated by the V̇O2‐HR and V̇E‐V̇CO2 slopes respectively, were not different between MS and controls (P > 0.05, Table S2). As submaximal exercise was associated with impaired oxidative metabolism without altered cardiovascular or respiratory function in patients with MS, an impaired skeletal muscle metabolic response must be implicated.
Skeletal muscle fibre type composition and size
Vastus lateralis fibre type composition and size were compared between groups using immunohistochemistry (Figure 2A). No differences in the relative composition of type I, IIa, or IIa/x fibre types between groups were observed (all P > 0.05, Figure 2B). Moreover, FCSA (Figure 2C) or minimal Feret's diameter (Figure S1) did not differ between groups for any fibre type (P > 0.05). Hence, exercise intolerance in MS could not be explained by differences in muscle fibre type composition or size.
Figure 2.
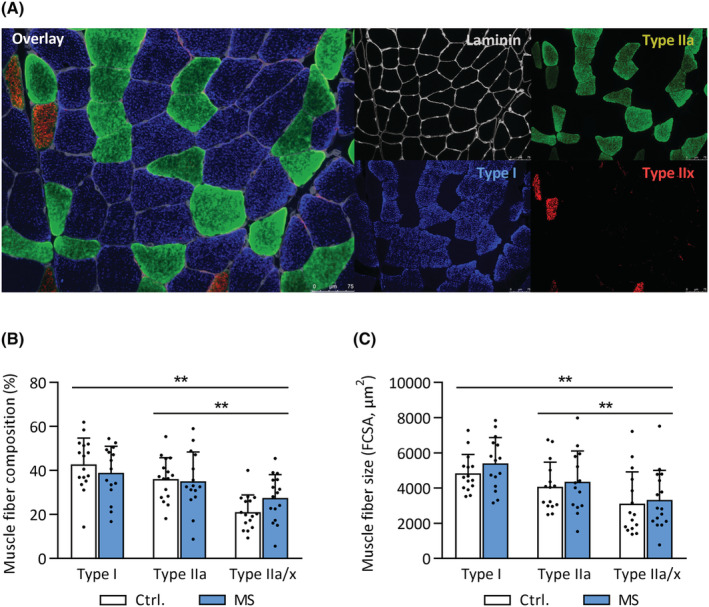
Normal muscle fibre size and composition in MS. Vastus lateralis biopsies were obtained from MS patients and healthy controls. (A) Representative immunohistochemistry images showing the presence of myosin heavy chain type I, IIa, and IIx, delineated by laminin‐stained cell membranes. Scale bar: 75 μm. (B) Muscle fibre type composition of type I, IIa, and IIa/x fibres in MS and controls. (C) Fibre cross‐sectional area (FCSA) of type I, IIa, and IIa/x fibres in MS and controls. **P < 0.01 for main effect of fibre type (from mixed model analysis). Mean ± SD.
Impaired exercise capacity in MS is related to reduced muscle fibre oxidative capacity
In patients with MS, SDH activity (Figure 3A) was lower in type I (−23%; P = 0.012) and type IIa fibres (−25%; P = 0.008), but not in type IIa/x fibres (−13%; P = 0.21, Figure 3B). The overall SDH activity, weighted for muscle fibre type composition, was lower in patients with MS compared with healthy controls (−22%; P = 0.004, Figure 3C), indicating a lower overall oxidative capacity in skeletal muscle tissue of patients with MS. Weighted fibre SDH activity correlated strongly with V̇O2peak (Spearman's ρ = 0.81, Figure 3D), the gain of the V̇O2 versus power output relationship (Spearman's ρ = 0.85, Figure 3E), and with O2 pulsepeak (Spearman's ρ = 0.65, Figure 3F) in patients with MS. The correlations between SDH activity and V̇O2peak were also observed separately in each fibre type in MS (Spearman's ρ = 0.78–0.90, Figure 3G–I). No significant correlations between SDH activity and any exercise‐related variable were observed in healthy controls, suggesting that a lower muscle oxidative capacity constrains exercise tolerance in patients with MS, but not in healthy controls. Moreover, protein content of subunits of mitochondrial OXPHOS complexes I (−40%; P = 0.047) and II (−45%; P = 0.026) were lower in MS patients compared with controls. Protein content of subunits for complexes III, IV and V (Figure 3J,K) and total average mitochondrial OXPHOS protein content was not different between groups (Figure 3L), albeit with large variability for some complexes. These data suggest specific impairments in mitochondrial complex I and complex II content and activity in skeletal muscle in MS patients, although a lower total mitochondrial protein content cannot be completely ruled out.
Figure 3.
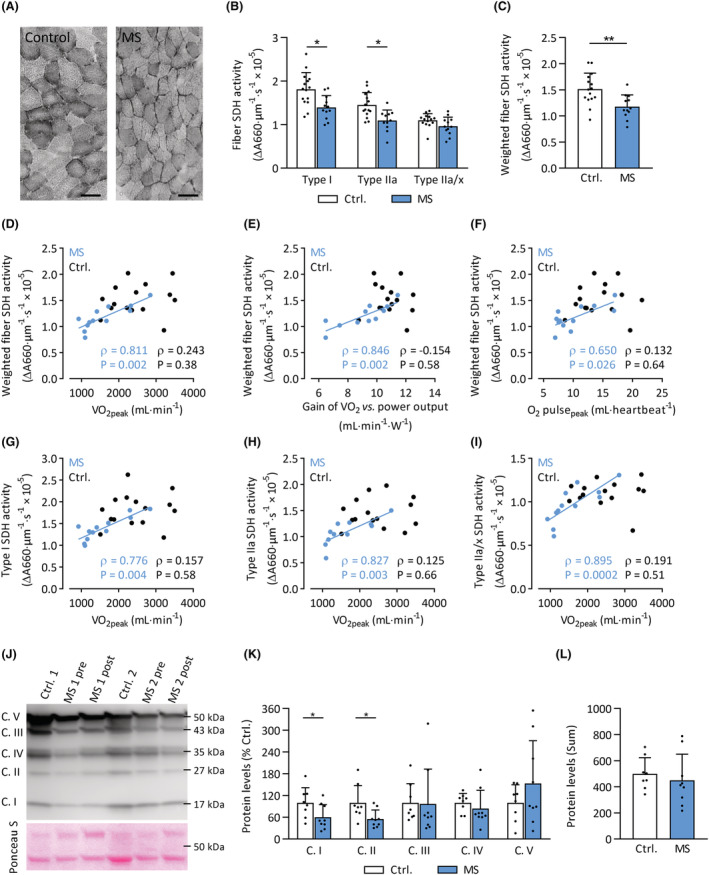
Lower muscle fibre SDH activity and mitochondrial complex I and II protein levels in MS. (A) Grey‐scale images stained for SDH activity from representative MS and control subjects. Scale bar: 100 μm. (B) Fibre SDH activity in type I, type IIa, and type IIa/x muscle fibres of MS patients (blue bars) and controls (clear bars). (C) SDH activity weighted for muscle fibre composition in MS patients (blue bars) compared with controls (clear bars). Relationships between weighted SDH activity and (D) peak oxygen uptake (V̇O2peak); (E) gain of the V̇O2 versus power output relationship; (F) peak O2 pulse (O2 pulsepeak). Relationship between V̇O2peak and SDH activity in type I, IIa, and IIa/x (G–I) in MS patients (blue circles) and controls (black circles). (J) Representative Western immunoblot with subunits for complex (C.) I, II, III, IV, and V protein bands at different molecular weights, and Ponceau S loading control. (K) Quantification of subunits of mitochondrial complex protein levels, relative to controls. (L) Total mitochondrial OXPHOS protein levels in MS and controls, calculated as the sum of individual complexes. *P < 0.05, **P < 0.01. Mean ± SD.
Additionally, capillary density was significantly lower in MS muscle tissue (P = 0.026, Figure 4A,B); however, there were no differences in the capillary/fibre ratio (P = 0.65, Figure 4C). In contrast to SDH activity, muscle capillarity did not correlate with any whole‐body exercise variable in patients with MS (Figure S2), suggesting that impaired peripheral O2 supply is not a primary contributor to exercise intolerance in MS. In healthy controls, however, capillary/fibre ratio showed a strong positive association with V̇O2peak (Figure 4D). Furthermore, while capillary density scaled linearly with fibre SDH activity in healthy controls, such a relationship was absent in patients with MS (Figure 4E). This indicates that the tight coupling between muscle O2 supply and oxidative capacity in healthy controls is absent in patients with MS.
Figure 4.
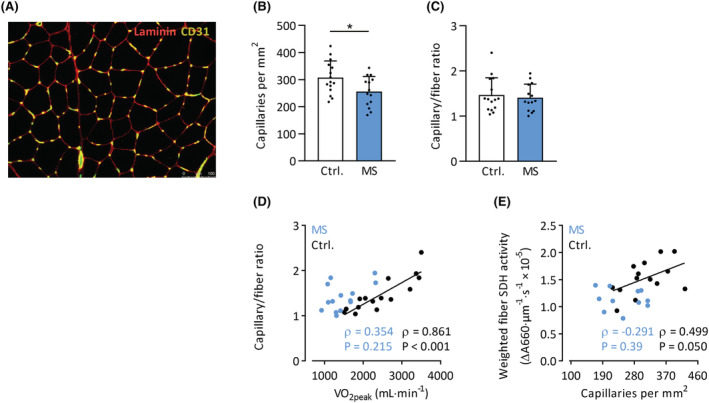
Muscle capillarity in MS patients and controls. (A) Representative immunohistochemical section stained for capillaries (CD31+) and membranes (laminin). Scale bar: 100 μm. Capillary density (B) and capillary/fibre ratio (C) in MS patients and controls. Relationships between V̇O2peak and muscle capillary/fibre ratio (D) and muscle capillary density and SDH activity (E). *P < 0.05. Mean ± SD.
A multiple linear regression analysis was conducted to investigate which skeletal muscle variables were the major contributors to exercise capacity (V̇O2peak) in both groups. The combination of weighted SDH activity, percentage type I fibre composition, type I fibre size, capillary density, and capillary/fibre ratio explained 64% of the variability in V̇O2peak in healthy controls (P = 0.017) and 71% in patients with MS (P = 0.036). In healthy controls, only the capillary/fibre ratio contributed significantly to the model (P = 0.045), whereas in MS, SDH activity (P = 0.020) was the only significant contributor.
Collectively, our data therefore indicate that intramuscular impairments in skeletal muscle oxidative capacity, rather than capillarity, are the primary constraint to exercise tolerance and aerobic function in MS; even when muscle fibre type composition and fibre size are preserved.
Subtle changes in mitochondrial signalling in muscle tissue of MS patients
No overall differences between patients with MS and healthy controls were observed in protein and gene expression levels of PGC‐1α (PPARGC1A), a transcriptional co‐activator involved in skeletal muscle mitochondrial biogenesis and capillarity/angiogenesis (Figure 5A–D). Gene expression of the transcription factors PPARA (−64%; P < 0.001), PPARG (−45%; P = 0.024) and TFAM (−46%; P < 0.001) was lower in skeletal muscle of MS patients (Figure 5D). PPARA gene expression positively correlated with SDH activity of type I and IIa fibres and overall weighted SDH activity (Spearman's ρ = 0.63–0.65, P < 0.05). Such associations were absent for the other tested genes or PGC‐1α protein levels. Gene expression of COX4I1, CS, and SDHB, genes involved in mitochondrial metabolism, were not different between groups (Figure 5E). Thus, lower exercise tolerance and muscle SDH activity in patients with MS cannot be explained by alterations in PGC‐1α protein or gene expression but possibly by lower PPARA, PPARG, and TFAM signalling.
Figure 5.
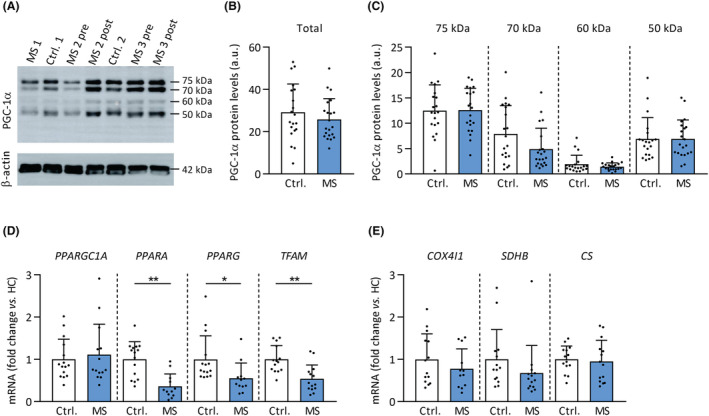
Protein and gene expression levels of mitochondrial‐related genes in MS. PGC‐1α protein content and mRNA expression of mitochondrial transcription factors and genes by qPCR were determined in vastus lateralis biopsies of MS patients and healthy controls. (A) Representative Western immunoblot with PGC‐1α protein bands at different molecular weights. Total (B) and individual bands (C) of PGC‐1α protein levels in MS and controls. (D) Quantitative mRNA expression analyses of mitochondria‐related transcription factors and genes (E). *P < 0.05, **P < 0.01. Mean ± SD.
Exercise training improves exercise capacity and strength in patients with MS
In the subset of MS patients that completed the exercise training programme (n = 9, EDSS ≤ 5, Table 1), V̇O2peak improved following exercise training (P = 0.001, Figure 6A, Table S4). Improvements in markers of submaximal aerobic function, that is, the gas exchange threshold (P = 0.048, Figure 6B, Table S4) and respiratory compensation point (P = 0.008, Figure 6C, Table S4), were also observed. Moreover, O2 pulsepeak (P = 0.004, Figure 6D, Table S4), V̇Epeak (P = 0.003, Figure 6E, Table S4), BFpeak (P = 0.022, Figure 6F, Table S4), and the V̇E‐V̇CO2 slope (P = 0.005, Figure 6G, Table S4) were all improved after exercise training. The gain of the V̇O2 versus power output relationship, however, was unchanged following training (P = 0.079, Figure 6H, Table S4). Hence, improvements in both cardiac output and respiratory function contributed to the enhanced exercise capacity and aerobic function following exercise training. In addition, MS patients were able to improve muscle strength (+19%; P = 0.0028, Table S3).
Figure 6.
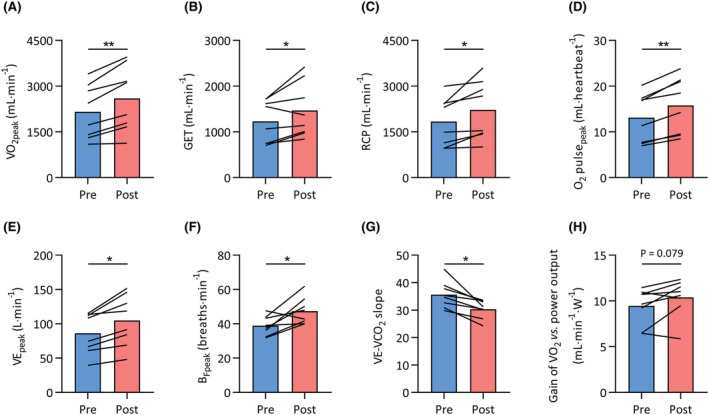
Improved exercise capacity following exercise training in MS. Whole‐body exercise capacity was assessed before (pre, blue bars) and after (post, red) a 12 week exercise training intervention in a subset of patients with MS. (A) Peak oxygen uptake (V̇O2peak), (B) gas exchange threshold (GET), (C) respiratory compensation point (RCP), (D) peak oxygen pulse (O2 pulsepeak), (E) peak ventilation (V̇Epeak), (F) peak breathing frequency (BFpeak), (G) slope of the relationship between ventilation and carbon dioxide output (V̇E‐V̇CO2 slope), and (H) gain of the relationship between V̇O2 and external power output. *P < 0.05, **P < 0.01.
Exercise training does not improve skeletal muscle characteristics in MS
This subgroup of MS patients had significantly lower SDH activity at baseline (Table S5). Surprisingly, SDH activity (P = 0.753; Figure 7A) and protein levels of mitochondrial OXPHOS complexes (all P ≥ 0.100; Figures 3J and 7B) did not increase in response to the training intervention. Similarly, no changes in muscle fibre type, size, and capillary density were observed (Figure 7C–E). In addition, there were no differences in gene and protein expression following exercise training, as PGC‐1α protein levels (Figure 7F) and gene expression of various mitochondria‐related transcription factors and enzymes (Figure 7G) did not change following exercise training. Moreover, no significant correlation existed between changes in V̇O2peak and molecular outcomes (e.g. SDH activity and RNA levels). Thus, despite an improved whole‐body exercise capacity, skeletal muscle of patients with MS did not respond to the applied exercise training regimen.
Figure 7.
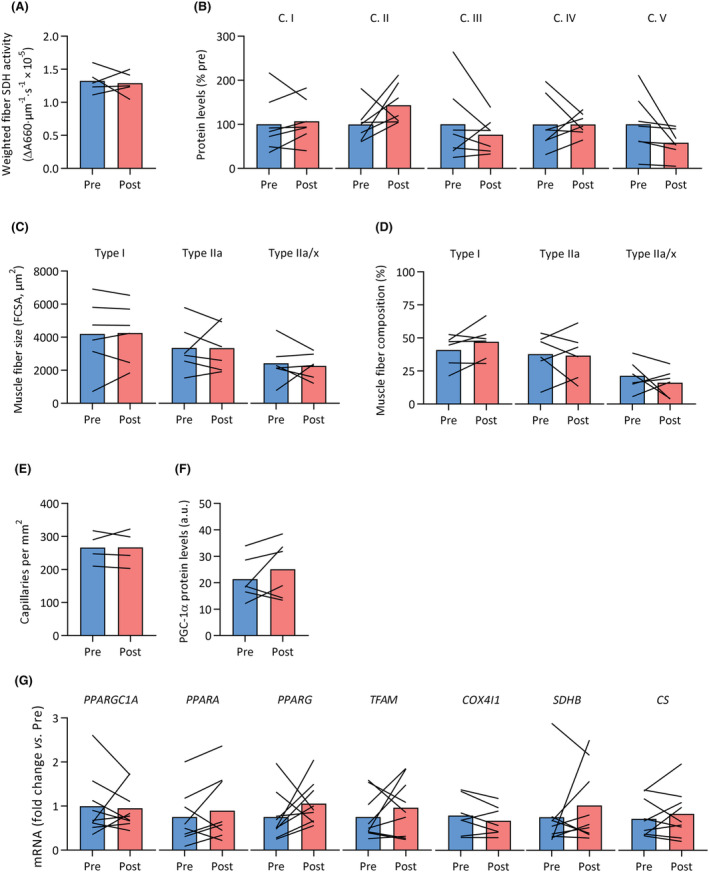
Skeletal muscle fibre composition, fibre size, oxidative capacity, mitochondrial OXPHOS protein levels, capillarity and mitochondrial signalling did not respond to exercise training in MS. Vastus lateralis biopsies were obtained from patients with MS before (pre, blue) and after (post, red) a 12 week exercise training intervention. No significant differences were observed in (A) SDH activity weighted for muscle fibre type, (B) mitochondrial OXPHOS protein levels (complex [C.] I to V), (C) fibre cross‐sectional area (FCSA) for type I, IIa and IIa/x fibres, (D) fibre type composition, (E) capillary density, (F) PGC‐1α protein content, and (G) mRNA expression of various mitochondrial transcription factors and genes.
Discussion
This study demonstrates that patients with MS have a lower exercise capacity when compared with their healthy counterparts with similar levels of subjectively measured physical activity. This is associated with reductions in key variables of aerobic function and a blunted aerobic response to exercise training. MS patients had lower mitochondrial oxidative capacity and capillary density compared with healthy controls, and mitochondrial oxidative capacity explained a large proportion of the variation in V̇O2peak in patients with MS. Twelve weeks of exercise training substantially improved whole‐body exercise capacity, but surprisingly, no improvements in mitochondrial oxidative capacity or capillary density were observed, suggesting that skeletal muscles from patients with MS appear somewhat resistant to the beneficial effects of exercise training. By extension, these findings suggest that skeletal muscle dysfunction is an intrinsic aspect of MS pathophysiology, rather than primarily related to deconditioning. Instead, improvements in the cardiovascular and ventilatory systems underlie the improvement in whole‐body exercise capacity following exercise training.
Exercise capacity and intramuscular impairments in MS patients
Consistent with previous studies, our data demonstrate that V̇O2peak and peak power output are substantially impaired in MS. 6 , 7 We found no evidence for impaired pulmonary function at maximal exercise, but a lower central O2 transport capacity at maximal exercise. However, because most daily life activities are conducted at submaximal intensities, submaximal parameters of aerobic function likely possess more relevance to daily activities of patients with MS. 22 , 27 We observed that the gain of the V̇O2 response (i.e. ΔV̇O2/ΔWatts) was constrained in patients with MS, consistent with slower pulmonary V̇O2 kinetics. 22 The lower gas exchange threshold, respiratory compensation point, and higher peak RER in MS patients support the notion that these patients become more reliant on non‐oxidative metabolism at lower metabolic rates, likely leading to greater depletion of intramuscular phosphocreatine stores and lactate accumulation. 20 , 21 , 27
Submaximal cardiac and pulmonary function did not differ between groups. As the derangements in submaximal aerobic function in MS could not be attributed to cardiovascular or ventilatory deficiencies, we reasoned that an impaired muscle metabolic response must be contributory. Indeed, SDH activity (reflective of muscle oxidative capacity 21 , 25 , 26 ) was lower in type I and IIa fibres of MS patients, and weighted SDH activity was strongly correlated with V̇O2peak and the gain of the V̇O2 versus power output relationship in MS patients. The strong correlation between capillarity and V̇O2peak in the present study in healthy controls is consistent with the general consensus that V̇O2peak is constrained by convective and diffusive O2 delivery in healthy humans. 28 The absence of such a relationship in MS and the presence of a strong correlation between SDH activity and V̇O2peak in these patients, however, indicate the importance of lower skeletal muscle oxidative capacity in defining exercise limitation in MS. Moreover, SDH activity was correlated with O2 pulsepeak, suggesting a role for impaired muscle O2 extraction consequent to reduced muscle oxidative capacity, as opposed to a reduction in stroke volume, in determining the reduction in this parameter.
We hypothesized that the lower skeletal muscle oxidative capacity in patients with MS was driven by lower mitochondrial biogenesis, with a crucial role for the transcriptional co‐activator PGC‐1α. 29 However, we did not observe any basal differences in PGC‐1α protein content, mitochondrial mRNA transcripts, or total mitochondrial protein content between groups. Nonetheless, the consistently lower PPARA, PPARG, and TFAM gene expression (−45% to 65%) suggests that subtle changes in cellular signalling may contribute to the observed impairment in oxidative capacity. The lower protein levels of subunits of mitochondrial complexes I and II in MS patients indicate that the lower skeletal muscle oxidative capacity was due to specific defects in individual complexes of the electron transport system, rather than an overt reduction in overall mitochondrial content.
Deconditioning versus MS pathology
Skeletal muscle weakness in MS patients is often suggested to be caused by physical inactivity‐induced deconditioning, as opposed to primary effects of the condition per se. 18 , 21 We tested the hypothesis that skeletal muscle dysfunction in MS is primarily related to muscle deconditioning (as opposed to MS pathophysiology) in the present study by (i) comparing MS patients with a group of healthy controls with similar levels of physical activity and by (ii) subjecting MS patients to an exercise training regimen similar to that which has previously been shown to result in substantial skeletal muscle mitochondrial adaptations and improvements in muscle size in healthy individuals. 30 , 31 Hence, if the skeletal muscle alterations that we observed at baseline were due to physical inactivity and/or deconditioning, we would have expected an improvement in these variables following the exercise training intervention. That this was not the case strongly suggests that MS pathophysiology per se, and not deconditioning, is the primary mediator of the skeletal muscle myopathy phenotype observed in MS. 18 , 21 , 22 , 32 , 33 Moreover, the present data demonstrate that muscle mitochondrial alterations precede the changes in muscle FCSA and fibre type composition in MS pathology observed by other studies. 8 , 17
Despite the lack of skeletal muscle plasticity to exercise training in patients with MS, however, improvements in the performance of other systems (i.e. nervous, cardiovascular, and pulmonary systems) were able to compensate for the lower oxidative capacity, thereby improving whole‐body aerobic function. For instance, O2 pulsepeak, V̇Epeak, BFpeak, and the V̇E‐V̇CO2 slope all improved following exercise training, indicating improved cardiovascular and pulmonary performance. Another possibility is that motor unit recruitment was augmented at greater exercise intensities during the incremental test. In support of this latter suggestion, muscle strength was improved by our training intervention whereas muscle FCSA was unchanged, suggesting that the improvements in muscle strength were the results of neuromuscular adaptations. Indeed, a greater capacity for motor unit recruitment at peak exercise would enable a greater V̇O2peak and exercise capacity and retain a consistency with the notion that mitochondrial oxidative capacity constrained exercise tolerance at baseline.
Lack of plasticity of skeletal muscle in MS
We found that oxidative capacity constrained exercise tolerance in MS at baseline, whereas the cardiovascular and pulmonary systems were able compensate for the inflexibility of skeletal muscle oxidative capacity following training, enabling an improved aerobic capacity. The causes of the inability of skeletal muscle oxidative capacity to adapt appropriately to exercise training, however, are presently unclear. Adenosine monophosphate‐activated protein kinase (AMPK) activation during repeated contractions increases mitochondrial biogenesis via activation of PGC‐1α. 34 As PPARGC1A mRNA increases following acute and chronic exercise results in higher PGC‐1α protein levels in healthy individuals, 35 we were surprised that gene expression and protein levels of PGC‐1α were not different after the exercise training intervention in patients with MS. That patients with MS have no changes in AMPK phosphorylation following endurance exercise 36 suggests that a defective AMPK signalling cascade could, in part, explain the lack of adaptation to exercise training in the present study. Alternatively, the lack of adaptations in MS muscle observed following training herein might be related to greater levels of systemic inflammation. Training adaptations are often blunted in patients with chronic diseases and the elderly, and this blunting of the adaptive response has been linked with higher levels of inflammatory markers and/or states of hyperglycaemia (e.g. previous studies 37 , 38 , 39 , 40 ). Although dependent on disease stage and activity, patients with MS may exhibit higher levels of low‐grade systemic inflammation. How low‐grade inflammation creates an inhibitory environment that blunts adaptations to exercise training in patients with MS deserves further research. Because some of the patients in the present study also took various drugs known to affect skeletal muscle (such as statins and metformin), we cannot exclude the possibility that certain medication classes (or other unmeasured lifestyle factors in these patients, such as nutrition) alter the local metabolic environment, mitochondrial function and blunt skeletal muscle training adaptations.
Limitations
A limitation of the present study is that the sample size was lower for certain comparisons following the training intervention due to limited tissue availability, and lower inclusion of patients. These patients had relatively low EDSS scores. Despite this, we did not see a training‐induced improvement in skeletal muscle mitochondrial function or FCSA following training, similar to the blunted adaptive responses to exercise training in patients with chronic diseases and the elderly. 37 , 38 , 39 , 40 Post hoc power analysis revealed that 276 patients would have to be included to observe a significant difference in SDH activity following training (α = 0.05, 1‐β = 0.80, one‐tailed; G*Power 3.1.9.2). Such an unfeasibly high number, therefore, likely indicates that the true effect of exercise training on muscle SDH activity in this population is either absent or physiologically insignificant.
A second limitation of the present study is that the chosen concurrent resistance and high‐intensity interval aerobic training intervention may have been suboptimal for eliciting clear improvements in aerobic and mitochondrial function. Concurrent aerobic and resistance exercise in healthy people results in substantial upregulation of molecular pathways related to intracellular metabolism, mitochondrial function and angiogenesis. 30 , 31 Five weeks aerobic and resistance exercise training increases endurance capacity, citrate synthase activity, and PGC1‐α/VEGF mRNA content in healthy individuals. 31 Hence, our 12 week training intervention should have provided ample stimulus to increase skeletal muscle oxidative capacity; that this did not change highlights the reduced plasticity of skeletal muscle in patients with MS. Although we did not include functional measures such as a 6 minute walk test, performance on such functional tests are strongly correlated with V̇O2peak and muscle strength, and thus an improvement in functional outcomes in the present study may be directly inferred from training‐induced improvements in V̇O2peak and muscle strength (see supporting information).
The lack of a healthy control group undergoing the same intervention is a limitation of the present study. Concurrent resistance and high‐intensity interval exercise training interventions have previously been shown to result in clear improvements in functional outcomes such as V̇O2peak in untrained healthy elderly individuals (i.e. ~20% improvement, 41 ), as well as robust molecular changes predicted to impact mitochondrial biogenesis and angiogenesis (see supporting information for additional literature references). These molecular changes in skeletal muscle in healthy individuals following concurrent rehabilitation training were not observed in our patients with MS, suggesting that the functional improvements must have been brought about via a different mechanism.
The subjective assessment of physical activity using the PASIPD may be considered a limitation (see reference 3 and supporting information). Objectively measuring physical activity using accelerometry would have provided greater confidence that both groups were similarly active. However, the PASIPD indicated that at the group level, MS and healthy controls did not differ for physical activity, and groups were matched for age and sex.
Due to the heterogeneous sample population, and current best clinical practice, it was impossible to individually assess the contribution of the different MS phenotypes (e.g. RRMS and SPMS), disease progression (newly diagnosed vs. chronic), or medications. This could also have affected our multiple regression analysis. Our observation that V̇O2peak correlates with EDSS supports the hypothesis that exercise capacity variables and muscular alterations may differ among patients and/or fluctuate over time. Controlled animal experiments have confirmed that skeletal muscle characteristics can change during different disease phases. 42 Studying the individual contributors of the confounding factors in a such a human patient requires careful future study designs.
Finally, the lack of functional measurements of skeletal muscle mitochondria, such as high‐resolution respirometry or muscle [phosphocreatine] recovery kinetics using 31P magnetic resonance spectroscopy, constitutes a limitation of the present study. Such measurements will provide fruitful avenues for future research in this area.
In conclusion, patients with MS display a lower maximal and submaximal aerobic function, when compared with age‐matched and sex‐matched healthy controls. At maximal exercise, there was evidence of a central cardiovascular impairment, however, during submaximal exercise impaired central O2 delivery could not account for the hampered aerobic function. Skeletal muscle mitochondrial impairments (mitochondrial complex I and II content, SDH activity) appeared to be primarily responsible for the exercise limitation in these patients. Skeletal muscles of patients with MS possessed an inherently lower plasticity with respect to the beneficial effects of exercise training, but other physiological systems compensated to offset the inflexibility of muscle oxidative capacity following training. As training interventions similar to the one employed herein have been shown to cause robust adaptations in skeletal muscle in healthy individuals, we conclude that the lower oxidative capacity in skeletal muscle in MS is not related to deconditioning, but is an inherent feature of MS pathophysiology or of the effects of common drugs prescribed to treat MS patients. A better understanding of the causes of the inflexibility of skeletal muscle in MS will provide avenues for future therapies aimed at optimizing exercise therapy in patients with MS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Table S1. Primer sequences.
Table S2. Cardiopulmonary exercise test variables in MS patients and healthy controls.
Table S3. Variables derived from cardiopulmonary exercise testing, separated by sex.
Table S4. Cardiopulmonary exercise test variables before and after exercise training in MS patients.
Table S5. Baseline muscle fibre SDH activity in healthy controls, all MS patients, and the MS training subgroup.
Figure S1, related to Figure 2. Muscle fibre minimal Feret's diameter in biopsies from MS patients and healthy controls. Vastus lateralis biopsies were obtained from MS patients and healthy controls. Muscle fibre minimal Feret's diameter of type I, IIa and IIa/x fibres in MS and controls.
** P < 0.01 for main effect of fibre type (from mixed model analysis).
Figure S2, related to Figure 4. Muscle capillarity and cardiopulmonary exercise test variables. Vastus lateralis biopsies were obtained from MS patients and healthy controls to determine muscle capillarity by immunohistochemistry. Correlations are displayed between muscle capillary density and (a) peak oxygen uptake (V̇O2peak), (b) the gain of the relationship between V̇O2 vs. external power output, and (c) peak O2 pulse (O2 pulsepeak). Figure panels (d‐e) display the correlations between capillary/fibre ratio and (d) the gain of the relationship between V̇O2 vs. external power output, and (e) peak O2 pulse (O2 pulsepeak). Note that the relation between capillary/fibre ratio and V̇O2peak is shown in Figure 4d.
Figure S3. Changes in muscle strength following exercise training in MS patients. Maximal isometric voluntary knee extensor strength before and after the exercise intervention in MS patients. ** P < 0.01.
Acknowledgements
We are grateful to MS‐Liga Vlaanderen (Belgium) for financial support and to Wendy Noort (Vrije Universiteit Amsterdam, The Netherlands) for her advice and assistance for the histochemistry analyses. Figures were created using GraphPad Prism v7.04. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.
Spaas J., Goulding R. P., Keytsman C., Fonteyn L., van Horssen J., Jaspers R. T., Eijnde B. O., and Wüst R. C. I. (2022) Altered muscle oxidative phenotype impairs exercise tolerance but does not improve after exercise training in multiple sclerosis, Journal of Cachexia, Sarcopenia and Muscle, 13, 2537–2550, 10.1002/jcsm.13050
Jan Spaas and Richie P. Goulding shared first authors.
Bert O. Eijnde and Rob C. I. Wüst shared last authors.
Contributor Information
Bert O. Eijnde, Email: bert.opteijnde@uhasselt.be.
Rob C.I. Wüst, Email: r.wust@vu.nl.
References
- 1. Ontaneda D, Thompson AJ, Fox RJ, Cohen JA. Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function. Lancet 2017;389:1357–1366. [DOI] [PubMed] [Google Scholar]
- 2. Dalgas U, Stenager E. Exercise and disease progression in multiple sclerosis: can exercise slow down the progression of multiple sclerosis? Ther Adv Neurol Disord 2012;5:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casey B, Coote S, Galvin R, Donnelly A. Objective physical activity levels in people with multiple sclerosis: meta‐analysis. Scand J Med Sci Sports 2018;28:1960–1969. [DOI] [PubMed] [Google Scholar]
- 4. Marrie RA. Comorbidity in multiple sclerosis: Past, present and future. Clin Invest Med 2019;42:E5–E12. [DOI] [PubMed] [Google Scholar]
- 5. Motl RW, Dlugonski D, Pilutti L, Sandroff B, McAuley E. Premorbid physical activity predicts disability progression in relapsing–remitting multiple sclerosis. J Neurol Sci 2012;323:123–127. [DOI] [PubMed] [Google Scholar]
- 6. Heine M, Wens I, Langeskov‐Christensen M, Verschuren O, Eijnde BO, Kwakkel G, et al. Cardiopulmonary fitness is related to disease severity in multiple sclerosis. Mult Scler J 2016;22:231–238. [DOI] [PubMed] [Google Scholar]
- 7. Langeskov‐Christensen M, Heine M, Kwakkel G, Dalgas U. Aerobic capacity in persons with multiple sclerosis: a systematic review and meta‐analysis. Sports Med 2015;45:905–923. [DOI] [PubMed] [Google Scholar]
- 8. Kent‐Braun J, Ng AV, Castro M, Weiner MW, Gelinas D, Dudley GA, et al. Strength, skeletal muscle composition, and enzyme activity in multiple sclerosis. J Appl Physiol 1997;83:1998–2004. [DOI] [PubMed] [Google Scholar]
- 9. Platta ME, Ensari I, Motl RW, Pilutti LA. Effect of Exercise Training on Fitness in Multiple Sclerosis: A Meta‐Analysis. Arch Phys Med Rehabil 2016;97:1564–1572. [DOI] [PubMed] [Google Scholar]
- 10. Motl RW, Sandroff BM, Kwakkel G, Dalgas U, Feinstein A, Heesen C, et al. Exercise in patients with multiple sclerosis. Lancet Neurol 2017;16:848–856. [DOI] [PubMed] [Google Scholar]
- 11. Kalb R, Brown TR, Coote S, Costello K, Dalgas U, Garmon E, et al. Exercise and lifestyle physical activity recommendations for people with multiple sclerosis throughout the disease course. Mult Scler J 2020;26:1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dalgas U, Hvid LG, Kwakkel G, Motl RW, de Groot V, Feys P, et al. Moving exercise research in multiple sclerosis forward (the MoXFo initiative): developing consensus statements for research. Mult Scler J 2020;26:1303–1308. [DOI] [PubMed] [Google Scholar]
- 13. Dennett R, Madsen LT, Connolly L, Hosking J, Dalgas U, Freeman J. Adherence and drop‐out in randomized controlled trials of exercise interventions in people with multiple sclerosis: a systematic review and meta‐analyses. Mult Scler Relat Disord 2020;43:102169. [DOI] [PubMed] [Google Scholar]
- 14. Surakka J, Romberg A, Ruutiainen J, Aunola S, Virtanen A, Karppi SL, et al. Effects of aerobic and strength exercise on motor fatigue in men and women with multiple sclerosis: a randomized controlled trial. Clin Rehabil 2004;18:737–746. [DOI] [PubMed] [Google Scholar]
- 15. Keytsman C, Hansen D, Wens I, O. Eijnde B. Impact of high‐intensity concurrent training on cardiovascular risk factors in persons with multiple sclerosis–pilot study. Disabil Rehabil 2017;41:1–6. [DOI] [PubMed] [Google Scholar]
- 16. Dalgas U, Stenager E, Ingemann‐Hansen T. Multiple sclerosis and physical exercise: recommendations for the application of resistance‐, endurance‐and combined training. Mult Scler J 2008;14:35–53. [DOI] [PubMed] [Google Scholar]
- 17. Wens I, Dalgas U, Vandenabeele F, Krekels M, Grevendonk L, Eijnde BO. Multiple sclerosis affects skeletal muscle characteristics. PLoS ONE 2014;9:e108158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kent‐Braun JA, Ng AV, Castro M, Weiner MW, Gelinas D, Dudley GA, et al. Strength, skeletal muscle composition, and enzyme activity in multiple sclerosis. J Appl Physiol (1985) 1997;83:1998–2004. [DOI] [PubMed] [Google Scholar]
- 19. Carroll CC, Gallagher PM, Seidle ME, Trappe SW. Skeletal muscle characteristics of people with multiple sclerosis. Arch Phys Med Rehabil 2005;86:224–229. [DOI] [PubMed] [Google Scholar]
- 20. Kentbraun JA, Kent‐Braun JA, Sharma KR, Miller RG, Weiner MW. Postexercise phosphocreatine resynthesis is slowed in multiple‐sclerosis. Muscle Nerve 1994;17:835–841. [DOI] [PubMed] [Google Scholar]
- 21. Sharma KR, Kent‐Braun J, Mynhier MA, Weiner MW, Miller RG. Evidence of an abnormal intramuscular component of fatigue in multiple sclerosis. Muscle Nerve 1995;18:1403–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansen D, Wens I, Kosten L, Verboven K, Eijnde BO. Slowed exercise‐onset Vo2 kinetics during submaximal endurance exercise in subjects with multiple sclerosis. Neurorehabil Neural Repair 2013;27:87–95. [DOI] [PubMed] [Google Scholar]
- 23. de Haan A, de Ruiter CJ, van der Woude LHV, Jongen PJH. Contractile properties and fatigue of quadriceps muscles in multiple sclerosis. Muscle Nerve 2000;23:1534–1541. [DOI] [PubMed] [Google Scholar]
- 24. Goulding RP, Roche DM, Marwood S. Prior exercise speeds pulmonary oxygen uptake kinetics and increases critical power during supine but not upright cycling. Exp Physiol 2017;102:1158–1176. [DOI] [PubMed] [Google Scholar]
- 25. Van der Laarse W, Diegenbach P, Elzinga G. Maximum rate of oxygen consumption and quantitative histochemistry of succinate dehydrogenase in single muscle fibres ofXenopus laevis. J Muscle Res Cell Motil 1989;10:221–228. [DOI] [PubMed] [Google Scholar]
- 26. van der Zwaard S, van der Zwaard S, Brocherie F, Kom BLG, Millet GP, Deldicque L, et al. Adaptations in muscle oxidative capacity, fiber size, and oxygen supply capacity after repeated‐sprint training in hypoxia combined with chronic hypoxic exposure. J Appl Physiol 2018;124:1403–1412. [DOI] [PubMed] [Google Scholar]
- 27. Goulding RP, Rossiter HB, Marwood S, Ferguson C. Bioenergetic Mechanisms Linking V̇O2 Kinetics and Exercise Tolerance. Exerc Sport Sci Rev 2021;49:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagner PD. Determinants of maximal oxygen transport and utilization. Annu Rev Physiol 1996;58:21–50. [DOI] [PubMed] [Google Scholar]
- 29. Handschin C, Spiegelman BM. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008;454:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fernandez‐Gonzalo R, Lundberg TR, Tesch PA. Acute molecular responses in untrained and trained muscle subjected to aerobic and resistance exercise training versus resistance training alone. Acta Physiologica 2013;209:283–294. [DOI] [PubMed] [Google Scholar]
- 31. Lundberg TR, Fernandez‐Gonzalo R, Tesch PA. Exercise‐induced AMPK activation does not interfere with muscle hypertrophy in response to resistance training in men. J Appl Physiol 2014;116:611–620. [DOI] [PubMed] [Google Scholar]
- 32. Garner DJ, Widrick JJ. Cross‐bridge mechanisms of muscle weakness in multiple sclerosis. Muscle Nerve 2003;27:456–464. [DOI] [PubMed] [Google Scholar]
- 33. De Ruiter CJ, Jongen PJH, van der Woude LHV, de Haan A. Contractile speed and fatigue of adductor pollicis muscle in multiple sclerosis. Muscle Nerve 2001;24:1173–1180. [DOI] [PubMed] [Google Scholar]
- 34. Sanchez AM, Candau RB, Csibi A, Pagano AF, Raibon A, Bernardi H. The role of AMP‐activated protein kinase in the coordination of skeletal muscle turnover and energy homeostasis. Am J Physiol‐Cell Physiol 2012;303:C475–C485. [DOI] [PubMed] [Google Scholar]
- 35. Granata C, Jamnick NA, Bishop DJ. Training‐induced changes in mitochondrial content and respiratory function in human skeletal muscle. Sports Med 2018;48:1809–1828. [DOI] [PubMed] [Google Scholar]
- 36. Hansen D, Wens I, Vandenabeele F, Verboven K, Eijnde BO. Altered signaling for mitochondrial and myofibrillar biogenesis in skeletal muscles of patients with multiple sclerosis. Transl Res 2015;166:70–79. [DOI] [PubMed] [Google Scholar]
- 37. Constantin D, Menon MK, Houchen‐Wolloff L, Morgan MD, Singh SJ, Greenhaff P, et al. Skeletal muscle molecular responses to resistance training and dietary supplementation in COPD. Thorax 2013;68:625–633. [DOI] [PubMed] [Google Scholar]
- 38. Perry BD, Caldow MK, Brennan‐Speranza TC, Sbaraglia M, Jerums G, Garnham A, et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: roles of inflammatory pathways, physical activity and exercise. Exerc Immunol Rev 2016;22:94. [PMC free article] [PubMed] [Google Scholar]
- 39. Durham WJ, Casperson SL, Dillon EL, Keske MA, Paddon‐Jones D, Sanford AP, et al. Age‐related anabolic resistance after endurance‐type exercise in healthy humans. FASEB J 2010;24:4117–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jankowski CM, Wilson MP, MaWhinney S, Reusch J, Knaub L, Hull S, et al. Blunted muscle mitochondrial responses to exercise training in older adults with HIV. J Infect Dis 2021;224:679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferrari R, Kruel LFM, Cadore EL, Alberton CL, Izquierdo M, Conceição M, et al. Efficiency of twice weekly concurrent training in trained elderly men. Exp Gerontol 2013;48:1236–1242. [DOI] [PubMed] [Google Scholar]
- 42. Spaas J, van Noten P, Keytsman C, Nieste I, Blancquaert L, Derave W, et al. Carnosine and skeletal muscle dysfunction in a rodent multiple sclerosis model. Amino Acids 2021;53:1749–1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences.
Table S2. Cardiopulmonary exercise test variables in MS patients and healthy controls.
Table S3. Variables derived from cardiopulmonary exercise testing, separated by sex.
Table S4. Cardiopulmonary exercise test variables before and after exercise training in MS patients.
Table S5. Baseline muscle fibre SDH activity in healthy controls, all MS patients, and the MS training subgroup.
Figure S1, related to Figure 2. Muscle fibre minimal Feret's diameter in biopsies from MS patients and healthy controls. Vastus lateralis biopsies were obtained from MS patients and healthy controls. Muscle fibre minimal Feret's diameter of type I, IIa and IIa/x fibres in MS and controls.
** P < 0.01 for main effect of fibre type (from mixed model analysis).
Figure S2, related to Figure 4. Muscle capillarity and cardiopulmonary exercise test variables. Vastus lateralis biopsies were obtained from MS patients and healthy controls to determine muscle capillarity by immunohistochemistry. Correlations are displayed between muscle capillary density and (a) peak oxygen uptake (V̇O2peak), (b) the gain of the relationship between V̇O2 vs. external power output, and (c) peak O2 pulse (O2 pulsepeak). Figure panels (d‐e) display the correlations between capillary/fibre ratio and (d) the gain of the relationship between V̇O2 vs. external power output, and (e) peak O2 pulse (O2 pulsepeak). Note that the relation between capillary/fibre ratio and V̇O2peak is shown in Figure 4d.
Figure S3. Changes in muscle strength following exercise training in MS patients. Maximal isometric voluntary knee extensor strength before and after the exercise intervention in MS patients. ** P < 0.01.