

Summary
Receptor-interacting protein kinase-3 (RIPK3) and mixed lineage kinase domain-like (MLKL) proteins are key regulators of necroptosis, a highly pro-inflammatory mode of cell death, which has been involved in various human diseases. Necroptotic-independent functions of RIPK3 and MLKL also exist, notably in the adipose tissue but remain poorly defined. Using knock-out (KO) cell models, we investigated the role of RIPK3 and MLKL in adipocyte differentiation. Mlkl-KO abolished white adipocyte differentiation via a strong expression of Wnt10b, a ligand of the Wnt/β-catenin pathway, and a downregulation of genes involved in lipid metabolism. This effect was not recapitulated by the ablation of Ripk3. Conversely, Mlkl and Ripk3 deficiencies did not block beige adipocyte differentiation. These findings indicate that RIPK3 and MLKL have distinct roles in adipogenesis. The absence of MLKL blocks the differentiation of white, but not beige, adipocytes highlighting the therapeutic potential of MLKL inhibition in obesity.
Subject areas: Biological sciences, Molecular biology, Cell biology, Stem cells research
Graphical abstract
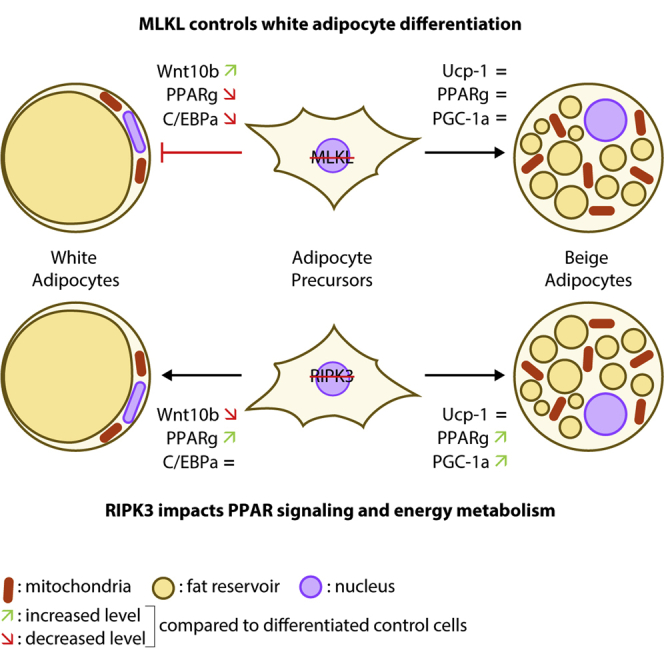
Highlights
-
•
Mlkl deficiency inhibits white, but not beige, adipocyte differentiation
-
•
MLKL deficiency suppresses the expression of master regulators of adipogenesis
-
•
Mlkl deficiency up-regulates Wnt10b expression
-
•
Ripk3 deficiency does not alter white and beige adipocyte differentiation
Biological sciences; Molecular biology; Cell biology; Stem cells research
Introduction
Obesity is the first non-infectious epidemic outbreak in history. The World Health Organization (WHO) estimates that more than one billion people worldwide are overweight, including 300 million who are obese. Obesity increases the risk of several serious chronic diseases, such as type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), cardiovascular diseases (CVD), and certain types of cancer (Field et al., 2001). Adipose tissue has been historically considered an inert tissue, which was only devoted to the storage and mobilization of lipids in response to various hormonal signals. However, over the last two decades, a considerable number of studies have demonstrated that it is an organ in its own right, with secretory functions regulating its own activity but also the whole-body energy metabolism (Kershaw and Flier, 2004).
Obesity causes major cellular alterations in the white adipose tissue (WAT), mainly adipocyte hypertrophy (i.e., an increase in the size of pre-existing adipocytes), leading to lipid-laden, dysfunctional adipocytes, which undergo cell death and contribute to adipose tissue inflammation (Sun et al., 2011). Adipocyte death is recognized as an important early event in the development of obesity complications, especially T2D (Cinti et al., 2005). Distant effects of adipose tissue inflammation such as the trigger of liver inflammation and hepatocyte injury have been documented and can contribute to the progression of diseases such as NAFLD (Gautheron et al., 2020). Therefore, a better understanding of the molecular mechanisms controlling and linking the death to inflammatory processes is necessary to identify new therapeutic targets in obesity and associated co-morbidities.
Until recently, two main forms of cell death were recognized: apoptosis, which occurs in a highly controlled manner, and necrosis which is accidentally triggered. However, during the past few years, it became clear that programmed cell death was not restricted to apoptosis, and comprised other forms of regulated cell death (Vanden Berghe et al., 2014). Necroptosis is one of them, combining the molecular machinery of the extrinsic apoptotic pathways with an execution similar to necrosis (Vanden Berghe et al., 2014). Unlike apoptosis that requires the activation of aspartate-specific proteases known as caspases (McIlwain et al., 2013), necroptosis is primarily driven by the activation of the receptor-interacting protein kinase (RIPK) 1 and 3, followed by the activation of the pseudo kinase mixed lineage kinase domain-like (MLKL) (Vanden Berghe et al., 2014). Since its discovery in 2009, necroptosis has been involved in many human diseases by mediating cell death and inflammation including ischemic reperfusion injuries (Baidya et al., 2020, 2021; Linkermann et al., 2013), neurodegeneration (Zhang et al., 2017), NAFLD (Afonso et al., 2015; Gautheron et al., 2014, 2015), progressive atherosclerotic lesions (Zhe-Wei et al., 2018), and so forth. However, reports of necroptotic-independent functions of necroptosis mediators which can regulate the interplay between cellular metabolism and death have recently emerged. Notably, we identified a paramount role of RIPK3 in maintaining WAT homeostasis in mice fed a high-fat diet (HFD) (Gautheron et al., 2016). Thus, Ripk3-ablation led to a caspase-8 mediated inflammation and apoptosis, driving insulin resistance and glucose intolerance in mice (Gautheron et al., 2016). The protective function of RIPK3 against insulin resistance seemed to be restricted to the adipose tissue, stressing the need to study its function specifically in adipocytes. Opposite results were reported in Mlkl-deficient mice, who were protected against dyslipidemia following obesogenic diets in comparison to Ripk3−/− mice (Rasheed et al., 2020; Saeed et al., 2019). Moreover, Mlkl deficiency prevented obesity-induced insulin resistance and glucose intolerance in obese mice with significantly less body weight gain and lower visceral adipose tissue compartments than in wild-type littermates (Xu et al., 2019). In line with these latter findings, mice treated with RIPA-56 (i.e., an inhibitor of RIPK1) showed downregulation of MLKL expression in liver and reduced body weight gain when fed an HFD (Majdi et al., 2020). Necroptosis inhibition by targeting MLKL has also shown beneficial effects on hepatic steatosis both in vivo and in vitro (Aoudjehane et al., 2020; Majdi et al., 2020; Wu et al., 2020b). Both MLKL and RIPK3 interact with enzymes participating in mitochondrial lipid metabolism (Yang et al., 2018; Zhang et al., 2009), and an upregulation of peroxisome-proliferator-activated receptor γ (PPARγ) results from their deletion (Afonso et al., 2021; Wu et al., 2020a). Therefore, there is evidence to suggest that RIPK3 and MLKL may have cell-death-independent effects in influencing the whole body and adipose-specific metabolism.
In the present study, to address RIPK3 and MLKL differential roles in adipose tissue homeostasis and adipogenesis, we applied functional genomic approaches in vitro using the 3T3-L1 murine cell line and primary human adipose stem cells (ASC). The impact of the loss of Ripk3 or Mlkl on adipocyte differentiation and function was evaluated by developing CRISPR/Cas9-mediated genome-editing approaches. Using the 3T3-L1 cell culture model, we demonstrated that Mlkl deficiency inhibits early white adipocyte differentiation by up-regulating the Wnt/β-catenin pathway and suppressing the expression of master gene regulators involved in adipogenesis. The absence of differentiation upon Mlkl deficiency could be recapitulated in human cells using ASC. In contrast, Ripk3 deficiency does not alter adipocyte differentiation but impacts fatty acid metabolism by upregulating the metabolic pathways that are found to be downregulated by Mlkl ablation. Finally, we show that neither Mlkl nor Ripk3 deficiency prevents beige adipocyte differentiation in 3T3-L1 cells. Altogether, our results expand our understanding of the regulation of adipogenesis and stress by necroptosis mediators and highlight MLKL inhibition as a potential therapeutic approach in obesity-related diseases.
Results
Differential expression of mixed lineage kinase domain-like and RIPK3 in visceral adipose tissue of obese mice
We previously reported a strong induction of RIPK3 protein expression in the visceral adipose tissue (visWAT) but not in the liver nor in the skeletal muscle of mice fed an HFD (Gautheron et al., 2016). Additionally, increased Mlkl mRNA levels were found in the liver, muscle, and visceral adipose tissue of obese mice (Xu et al., 2019). To further understand the differential regulation of RIPK3 and MLKL in adipose tissue, we evaluated the expression pattern of MLKL and RIPK3 in the visWAT of 20-week-old mice with genetic-induced obesity. We used the leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice, in which obesity is triggered by defects in the production or transduction of leptin, respectively (MacReady, 2014). As expected, RIPK3 was highly expressed in the visWAT of both genetic models compared to wild-type (WT) mice (Figures 1A and 1B). Noteworthy, RIPK3 was not expressed in the visWAT of WT C57/BL6 mice (Figures 1A and 1B). Conversely, MLKL was expressed in the visWAT of WT C57/BL6 mice and also, although to a lesser extent, increased in the visWAT of ob/ob and db/db mice (Figures 1A and 1B). Higher RIPK3 levels in visWAT were associated with an increase of the monocyte chemoattractant protein-1 (MCP-1) in the visWAT of both genetic obese murine models compared to control mice (Figure 1C), suggesting that the immunological fraction of the adipose tissue may also contribute to the increased expression of RIPK3 in these pathological conditions (Figures 1A–1C). Finally, we investigated the expression pattern of MLKL and RIPK3 in the subcutaneous adipose tissue (subWAT) of these mice. We did not observe any significant increases, but instead a slight reduction, in the subWAT of both genetic models compared to WT mice, suggesting that RIPK3 and MLKL upregulation in genetic-induced obesity occur specifically in the visWAT (Figures 1D and 1E). Taken together, these results highlight the time-dependent expression of the necroptosis mediators in adipose tissue, which may impact either its development or remodeling.
Figure 1.

RIPK3 is more expressed than MLKL in visceral adipose tissue of genetically obese mouse models
Visceral and subcutaneous adipose tissue was obtained by the dissection of 14-week-old male control mice (c57bl/6) (n = 4), leptin-deficient (ob/ob) (n = 4) and leptin receptor-deficient (db/db) mice (n = 4).
(A) Protein expression of RIPK3, MLKL, and Tubulin obtained by Western blotting from visceral adipose tissue. Numbers on the left correspond to molecular weight markers (kDa).
(B) The Western blots related to Figure 5A were quantified using FIJI software and normalized to the value of control mice. Results are expressed as means ± SEM. ∗∗p < 0.01, ∗p < 0.05.
(C) mRNA levels of F4/80, Mcp-1, and Tnfα were assessed by RT-qPCR and shown relative to control mice. Results are expressed as means ± SEM. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗p < 0.05.
(D) Protein expression of RIPK3, MLKL, and Tubulin obtained by Western blotting from subcutaneous adipose tissue. Numbers on the left correspond to molecular weight markers (kDa).
(E) The Western blots related to Figure 5D were quantified using FIJI software and normalized to the value of control mice. Results are expressed as means ± SEM.
Mlkl and Ripk3 knockout in pre-adipocytes abolishes TNF-induced cell death
To first characterize the necroptotic function of RIPK3 and MLKL in pre-adipocytes, a CRISPR/Cas9-mediated knockout (KO) approach was developed. We used the murine 3T3-L1 pre-adipocytes as a cellular model owing to their ability to differentiate into mature adipocytes after stimulation (Green and Meuth, 1974). A custom-designed single-guide RNA (sgRNA)/Cas9 expression vector targeting the first exon of Mlkl or that of Ripk3 was used. 3T3-L1 cells transfected with a Cas9/scramble gRNA plasmid were used as a control (CTL). The efficiency of RIPK3 and MLKL knockout was confirmed by Western blot analysis, which showed an almost complete loss of their expression (Figures 2A and 2B). In addition, Sanger sequencing of Mlkl or Ripk3 exon 1 in genomic DNA from KO cells revealed a high level of on-target indels with 79.3% of insertions and 13.4% of deletions for Mlkl, and 90.5% of deletions for Ripk3 (Figures S1 and S2). Treatment of CTL and both KO cells with 20 ng/mL of Tumor Necrosis Factor (TNF)-α alone for 24 h did not result in a significant loss of cell viability as measured by microscopic analysis (Figure 2C) and MTT assays (Figure 2D). However, when added after pretreatment with zVAD, a pan-caspase inhibitor, TNF-α stimulation induced approximately 40% of necroptotic death in CTL cells. CTL cells became all necrotic when a cIAP1/2 inhibitor (BV6) was added to zVAD (Figures 2C and 2D). In contrast, Ripk3 or Mlkl ablation efficiently rescued 3T3-L1 cells from necroptosis induced by zVAD + TNF-α or by zVAD + BV6+TNF-α (Figures 2C and 2D). We enhanced cell death even more by blocking overall mRNA translation, including pro-survival factors, e.g., via NF-κB, using Cycloheximide (CHX). The majority of CTL cells died when pre-treated with CHX + BV6+ZVAD combined with TNF-α stimulation, whereas a significant part (20-35%) of both KO cell lines survived (Figures 2C and 2D). Of note, a slight increase in the proliferation rate was detected in RIPK3-deficient cells (Figure 2E). Altogether, these data validate our experimental setup and provide further evidence that the use of CRISPR/Cas9 did not globally deregulate the physiological functions of 3T3-L1 cells. They also confirmed that the ablation of either Mlkl or Ripk3 can prevent necroptotic death in 3T3-L1 pre-adipocytes, and validate the use of these KO cells to study the necroptotic independent functions of MLKL and RIPK3 in adipogenesis.
Figure 2.

Mlkl and Ripk3 knockout prevent TNF-induced cell death in 3T3-L1 pre-adipocytes
Data were obtained in 3T3-L1 cells with a CRISPR/Cas-9-mediated Mlkl- or Ripk3-knockout (KO), and 3T3-L1 cells transfected with a Cas9/scramble gRNA plasmid corresponding to control (CTL) cells. p values were determined by ANOVA with the Geisser-Greenhouse correction and Tukey’s multiple comparisons test.
(A and B) Validation of RIPK3 (A) and MLKL (B) KO in 3T3-L1 pre-adipocytes. Numbers on the left correspond to molecular weight markers (kDa). Western blot images are representative of three independent experiments.
(C) Morphological features of cultured 3T3-L1 cells treated for 19 h with zVAD (20 μM), zVAD + BV6 (10 μM) and zVAD + BV6+CHX (0.5 μg/mL) all in combination with TNF-α (20 ng/mL).
(D) Cell survival analyses using an MTT assay after 24 h of the indicated dose treatment. Results are expressed as means ± SEM of three independent experiments. ∗∗∗∗p < 0.0001; ns, non-significant.
(E) Cell survival analyses using an MTT assay at the basal condition for CTL, Ripk3-KO, and Mlkl-KO cells. Results are expressed as means ± SEM of three independent experiments. ∗∗p < 0.01. See also Figures S1 and S2.
Mlkl deficiency, but not Ripk3 ablation, prevents white adipocyte differentiation
To explore the roles of MLKL and RIPK3 in white adipocyte differentiation, we investigated the kinetics of expression of adipogenesis and fatty acid storage markers during a 10-day white adipocyte differentiation protocol (Figure S3A). As shown in Figure 3, CTL cells, which expressed scrambled sgRNA or were Ripk3-KO, differentiated into adipocytes within 10 days (D10) (Figure 3A) and displayed a strong accumulation of lipid droplets in the cytoplasm as well as an increase in the triglyceride content (Figures 3B and 3C). Ripk3-KO adipocytes even showed a 20% higher triglyceride content than CTL adipocytes at D10 (Figure 3C). In contrast, Mlkl deficiency led to a strong and significant decrease in lipid droplet formation associated with a 60% reduction of triglyceride content (Figures 3A–3C). Next, we assessed the expression of adipocyte markers to evaluate adipocyte differentiation. Mlkl ablation suppressed the expression of CCAAT/enhancer-binding protein alpha (C/EBPα), fatty acid synthase (FAS), and stearoyl-CoA desaturase (SCD1), whereas their expression was globally unaffected by Ripk3 ablation (Figure 3D). The mRNA levels of these factors were significantly decreased in Mlkl-KO cells starting 3 days post-induction of differentiation and remained low at a later stage of differentiation (i.e., 10 days post-induction) (Figure 3D). Of note, neither Ripk3 nor Mlkl ablation affected the ability of cells to enter cell-cycle arrest, which is needed prior to adipocyte differentiation (Figure S4). The percentage of CTL and KO cells in G0/G1 phases increased as the cells reached confluence, while that in S and G2/M phases disappeared (Figure S4).
Figure 3.

Mlkl but not Ripk3 deficiency suppresses white adipocyte differentiation of 3T3-L1 cells
(A–D) Data were obtained in 3T3-L1 cells with a CRISPR/Cas-9-mediated Mlkl- or Ripk3-KO, and 3T3-L1 cells transfected with a Cas9/scramble gRNA plasmid corresponding to CTL cells. p values were determined by ANOVA with the Geisser-Greenhouse correction and Tukey’s multiple comparisons test.
(E–H) Data were obtained in human adipose stem cells (ASC) with a CRISPR-Cas9-mediated MLKL-KO, and ASC transduced with a Cas9/scramble gRNA plasmid corresponding to CTL cells. p values were determined using Student’s t test. (A) The 3T3-L1 pre-adipocytes were studied during adipocyte differentiation for 10 days. Representative images of fluorescence microscopy after staining of intracellular lipids (Oil Red O) and nuclei (DAPI, blue). Images are representative of three independent experiments. D0 - D10: Day 0 to Day 10. (B) Quantification of Oil Red O fluorescence normalized to DNA content (DAPI). Results are expressed as means ± SEM of three independent experiments. ∗∗p < 0.01; ∗p < 0.05. (C) Intracellular triglyceride content measured at D10 in CTL, Ripk3-KO, and Mlkl-KO cells. Measurements are representative of seven independent experiments. Results are expressed as means ± SEM. ∗∗p < 0.01. (D) 3T3-L1 cells were induced to differentiate from pre-adipocytes into adipocytes as described. Time course assessment of mRNA expression levels of the adipocyte markers C/ebpα, Fasn, and Scd1 for CTL, Ripk3-KO and Mlkl-KO cells from D0 to D10. # indicates that mRNA levels of Mlkl-KO cells were significantly decreased compared with CTL and Ripk3-KO cells. (E) Validation of MLKL KO in ASC. Numbers on the left correspond to molecular weight markers (kDa). Western blot images are representative of three independent experiments. (F) Representative images of fluorescence microscopy of human ASC after staining of Oil Red O and nuclei with DAPI. Images are representative of three independent experiments of differentiated CTL and MLKL-KO ASC at day 20 after differentiation onset (D20). (G) Quantification of Oil Red O fluorescence normalized to DNA content (DAPI). Results are expressed as means ± SEM of three independent experiments. ∗∗∗p < 0.001. (H) Protein expression of adipocyte markers obtained by Western blotting during in vitro adipocyte differentiation of ASC cells. Numbers on the left correspond to molecular weight markers (kDa). Western blot images are representative of three independent experiments. PPARγ: peroxisome proliferator-activated receptor-gamma; C/EBPα: CCAAT/enhancer-binding protein-alpha; FAS: fatty acid synthase. See also Figures S3–S6.
Importantly, the effects of Mlkl ablation evidenced in 3T3-L1 cells were recapitulated in human adipose stem cells (ASC), which can be differentiated into adipocytes using an appropriate cocktail (Figure S5). The efficiency of MLKL knockout was confirmed by Western blot analysis, which led to a complete loss of its expression compared to control (CTL) ASC (Figure 3E). In addition, Sanger sequencing of MLKL exon 4 in genomic DNA from KO cells revealed a high level of on-target indels with 96% of insertions and deletions (Figure S6). We found that MLKL-KO ASC displayed a lower accumulation of lipid droplet in their cytoplasm than CTL ASC (Figures 3F and 3G). The effect of MLKL ablation on adipocyte was further evaluated by an expression study of adipogenic and mature adipocyte markers during the differentiation process. As compared to CTL ASC, MLKL-KO ASC displayed a sharp decrease in the expression of adipogenic markers, including the transcription factors PPARγ and C/EBPα at D20 (Figure 3H). The expression of mature adipocyte markers, such as fatty acid synthase (FAS), perilipin, and adiponectin was also markedly decreased in MLKL-KO cells (Figure 3H). Altogether, these findings suggest divergent roles of MLKL and RIPK3 in controlling adipocyte differentiation and fatty acid metabolism.
Mlkl deficiency does not prevent beige adipocyte differentiation
Because MLKL loss-of-function improves the metabolic profile and reduces body weight gain when mice are fed a high-fat diet (Majdi et al., 2020; Rasheed et al., 2020; Saeed et al., 2019; Xu et al., 2019), we next studied the impact of Mlkl ablation on beige adipocyte differentiation. The 3T3-L1 cells were differentiated into beige adipocytes by adding to the white adipocyte differentiation medium rosiglitazone and triiodothyronine (T3) (Asano et al., 2014; Wang et al., 2014) (Figure S3B). As shown in Figure 4, CTL cells expressing scrambled sgRNA differentiated into beige adipocytes under these conditions within 10 days. They displayed a massive accumulation of lipid droplets in the cytoplasm as well as increased triglyceride content (Figures 4A and 4B). As shown by immunofluorescence, the presence of lipid accumulation correlated with an increase of perilipin-1, a protein-coating lipid storage droplet in adipocytes (Figures 4A and 4C). Moreover, immunofluorescence studies confirmed the increased abundance of UCP-1 protein and mRNA levels, further indicating the presence of beige adipocytes (Figures 4D–4F). Mlkl deficiency did not alter neither lipid droplet formation nor accumulation of triglycerides when cells were grown in the beige adipocyte culture medium (Figures 4A and 4B). In agreement, Ucp-1 and Pparγ mRNA levels were also increased in Mlkl-KO beige adipocytes compared with white adipocytes, together with increased expression of Pcg-1 (Figures 4F and 4G), which plays a critical role to establish the thermogenic pathway in beige adipocytes (Uldry et al., 2006). Of note, Ripk3 deficiency did not alter beige adipocyte differentiation (Figures S7A and S7B). Moreover, the addition of rosiglitazone to the beige adipocyte differentiation medium was not sufficient to explain the discrepancies in-between white and beige differentiation capacities of Mlkl-KO cells, as a short-term rosiglitazone boost (i.e., less than 3 days) did not overcome MLKL requirement for white adipocyte differentiation (Figure S7C). Altogether, these data suggest that MLKL is a limiting factor for white but not beige adipocyte differentiation, highlighting a specific metabolic role of MLKL in white adipose tissue expansion.
Figure 4.

Mlkl deficiency does not alter differentiation into beige adipocytes
Data were obtained in 3T3-L1 cells with a CRISPR/Cas-9-mediated Mlkl-KO and 3T3-L1 cells transfected with a Cas9/scramble gRNA plasmid corresponding to CTL cells. p values were determined by ANOVA with the Geisser-Greenhouse correction and Tukey’s multiple comparisons test. Results are expressed as means ± SEM.
(A) Representative images of fluorescence microscopy after staining of Oil Red O (Red) or Perilipin-1 (Green) and nuclei (Blue). Images are representative of three independent experiments of differentiated CTL and Mlkl-KO cells at D10.
(B) Triglyceride content measured in CTL and Mlkl-KO cells at 10 days post-differentiation (White vs. beige adipocytes). # indicates that triglyceride levels in white adipocyte-oriented Mlkl-KO cells are significantly decreased compared to all other groups.
(C) Immunofluorescence quantification of Perilipin-1. # indicates that immunofluorescence level of Perilipin-1 is significantly decreased compared to all other groups.
(D) Representative images of fluorescence microscopy after staining Ucp-1 (Green) and nuclei (Blue). Images are representative of three independent experiments of differentiated CTL and Mlkl-KO cells at D10.
(E) Immunofluorescence quantification of UCP-1. # indicates that immunofluorescence level of UCP-1 is significantly decreased compared to the groups of beige adipocytes.
(F) Ucp-1 mRNA expression levels in white and beige adipocytes at D10. ∗∗p < 0.001, ∗p < 0.05.
(G) Pparγ, Pgc-1α, and Pgc-1β mRNA expression levels in white and beige adipocytes at D3. ∗p < 0.05. See also Figures S3 and S7.
Mixed lineage kinase domain-like and RIPK3 modulate regulatory networks involved in adipogenesis
To further investigate the differential impact of Mlkl and Ripk3 ablations in white adipocyte differentiation, gene expression profiles were determined by RNA sequencing of the 3T3-L1 pre-adipocytes deleted for either Mlkl or Ripk3 and induced to adipocyte differentiation for 7 days (Figure S4A). Three biological replicates with 25-45 million reads per replicate were generated for undifferentiated (D0) or differentiated (D7) adipocytes per genotype. The good sequencing quality score (Figure S8A) and a high consistency in gene expression distribution among samples confirmed the low technical variability across samples (Figure S8B). Additionally, principal component analysis demonstrated expected grouping among replicates in-between samples (Figure S8C). A total of 5,167 and 3,055 differentially expressed genes (DEG), with a false discovery rate (FDR) below 0.05, were identified between undifferentiated (D0) Ripk3-KO or Mlkl-KO vs. CTL cells, respectively. Differentiation from pre-adipocytes to adipocytes resulted in drastic changes in DEG, with more than 12,000 DEG observed in CTL and Ripk3-KO cells D7 vs. D0 (Figure 5A). However, the differentiation protocol seemed to have a minimal effect on Mlkl-KO cells with only 1,891 DEG at D7 compared to D0 (Figure 5A). This was further confirmed in the Fragments Per Kilobase Million (FPKM) hierarchical clustering heatmap, indicating that Mlkl-KO cells at D7 post-differentiation are clustering with undifferentiated rather than differentiated Ripk3-KO or CTL cells (Figure 5B). In addition, by comparing transcriptomes of differentiated cells (at D7) a total of 5,696 and 7,288 DEG were identified in Ripk3-KO or Mlkl-KO vs. CTL, respectively (Figure 5C). To identify signatures specific to each genotype, we then performed gene ontology and functional enrichment analysis using STRING, Reactome, KEGG, and Wikipathways databases. At D0 and compared to CTL cells, the transcriptome of Mlkl-KO cells is specifically enriched for genes involved in regulatory networks controlling extracellular matrix organization and ossification, suggesting that Mlkl-KO pre-adipocytes might be oriented more toward trans-differentiation to bone cells formation rather than adipogenesis (Figures S9A and S9B) (Zhang et al., 2012). Interestingly, Ripk3-KO pre-adipocytes present a completely different signature for which pathways are enriched for “DNA replication” and “G1 to S cell cycle control” (Figures S9C and S9D). We further analyzed differences in gene regulatory network at D7 of differentiation. Wikipathways enrichment analysis revealed that most of the significantly enriched pathways contained down-regulated genes in Mlkl-KO cells vs. CTL cells (Figure 5D). The signature of Mlkl-KO cells indicates a clear reduction in the expression of genes involved in the early or late stages of adipogenesis (C/ebpα, Pparγ, Adipoq, Plin1, Lpl, Fabp4, Lipe) (Figure 5E), PPAR signaling pathways, fatty acid and cholesterol biosynthesis, fatty acid-beta oxidation and TCA cycle (Figure S10). Interestingly, C/ebpα and Pparγ down-regulated expressions in our dataset correlated with an increase in expression of Wnt10b a known inhibitor of adipogenesis (Figure 5E) (Cawthorn et al., 2012). We could further confirm by RT-qPCR that the expression of genes involved in fatty acid metabolism and PPAR signaling pathway was, indeed, reduced in Mlkl-KO cells at 7 days post-differentiation (Figure 5F). On the contrary, Ripk3-KO differentiated cells present an enrichment in pathways involved in PPAR signaling, fatty acid beta-oxidation, electron transport chain, and TCA cycle compared to CTL cells, suggesting an increase in the expression of genes implicated in mitochondria bioenergetics and function (Figures 5D and 5G, and S11). In agreement, we observed a significant increase in the mitochondrial metabolic rate of Ripk3-KO cells using the MTT assay (Figure 2E). Taken together, these data suggest that Mlkl but not Ripk3 deficiency impaired adipogenesis of 3T3-L1 cells by reducing the expression of pro-adipogenic factors, genes involved in fatty acid metabolism, and possibly through the inhibitory effect of Wnt10b expression.
Figure 5.

Gene expression profiles of Mlkl and Ripk3 knocked-out cells after the induction of white adipocyte differentiation
Data were obtained in 3T3-L1 cells with a CRISPR/Cas-9-mediated Mlkl- or Ripk3-KO, and 3T3-L1 cells transfected with a Cas9/scramble gRNA plasmid corresponding to CTL cells. The analysis was performed by comparing 3T3-L1 pre-adipocytes with cells 7 days post-differentiation.
(A) Differentially expressed genes (DEG) were analyzed between differentiated or undifferentiated cells for each genotype. Gray bars represent the total of significantly DEGs (up- or down-regulated). Red bars represent significantly up-regulated DEGs and blue bars represent significantly down-regulated genes.
(B) Hierarchical clustering heatmap of fragments per kilobase million (FPKM) values of differentially expressed genes across conditions. Red represents high FPKM values and blue represents low FPKM values. Color descending from red to blue indicates log 10 (FPKM +2) from high to low expression.
(C) Volcano plots of DEGs comparing Mlkl- or Ripk3-KO to CTL at 7 days post-differentiation. Red and green dots represent up- and down-regulated DEGs, respectively, with FDR <0.05.
(D) Significantly enriched Wikipathways in Mlkl- or Ripk3-KO compared to CTL datasets at 7 days post-differentiation.
(E) Protein-protein interaction network generated using STRING-db for Wikipathways WP447: Adipogenesis genes in Mlkl-KO vs CTL cells at D7.
(F) Fasn, Pparγ, Fabp4, Scd1, and Wnt10β mRNA levels were assessed by RT-qPCR. Results are expressed as means ± SEM. ∗∗∗∗p < 0.0001; ∗∗p < 0.01; ∗p < 0.05.
(G) Protein-protein interaction network generated using STRING-db for Wikipathways WP1269: Fatty acid beta-oxidation in Ripk3-KO vs CTL cells at D7. See also Figures S4 and S8–S11.
Discussion
Despite a progressive awareness of the population toward the global obesity epidemic, obesity is the main risk factor for several diseases, including diabetes mellitus, NAFLD, cardiovascular diseases, and certain forms of cancer. The present study aimed to demonstrate that the necroptosis regulators MLKL and RIPK3 could have necroptotic-independent functions in modulating adipogenesis, a key event to control adipose tissue expansion and remodeling. Our main findings demonstrate that MLKL, but not RIPK3, plays a determinant role in adipogenesis in pre-adipocytes. Notably, we show that Mlkl ablation markedly inhibits white adipocyte differentiation while surprisingly preserving the orientation of pre-adipocytes toward the beige phenotype. Therefore, this study highlights a novel function of MLKL in regulating adipogenesis.
To uncover the role of RIPK3 and MLKL during adipogenesis we used the murine 3T3-L1 pre-adipocytes in vitro model. 3T3-L1 cells are often used to study adipocyte differentiation and are known to recapitulate the pro-adipogenic transcriptional programs observed in vivo during WAT development (Birsoy et al., 2011; Sun et al., 2020). To study the function of MLKL and RIPK3 during adipogenesis, we deleted both genes using the CRISPR/Cas9 technology. To decrease potential off-target mutations resulting from Cas9 integration into the genome, we favored a transient transfection with a plasmid containing both the Cas9 and gRNA targeting Ripk3 or Mlkl genes. Moreover, to avoid the selection of clones with off-target mutations, we analyzed a heterogeneous population of cells issued from FACS sorting. As expected, we observed that the gene expression pattern of Glu1, Glud1, Pgam5, or Camk2, all known targets of RIPK3 (Wang et al., 2012; Yang et al., 2018; Zhang et al., 2016), were upregulated in the Ripk3-KO cells while being downregulated in Mlkl-KO cells compared to CTL cells (Figure S12).
To decipher how MLKL exerts its cell death-independent function in adipocyte differentiation, we attempted to study its phosphorylation profile in 3T3-L1 cells. We tested several commercial antibodies against known phosphorylated forms of MLKL, but failed to detect any phosphorylation using these antibodies. How can the lack of phosphorylation be explained? Most MLKL phosphorylation sites have been identified in the context of necroptosis. For instance, MLKL is phosphorylated on Ser-345 to translocate, accumulate in the plasma membrane, and cause necroptosis (Rodriguez et al., 2016). The apparent lack of cell death during adipocyte differentiation can likely explain the absence of phosphorylation on the known phospho-sites. Thus, we can hypothesize that the phospho-sites of MLKL, if needed for adipocyte differentiation, might be different from those typically targeted during necroptosis. Overall, this highlights the need for a large-scale proteomic analysis to identify MLKL phospho-sites during adipocyte differentiation.
Our study suggests a necroptotic-independent function of MLKL in regulating gene expression/mRNA stabilization, among them Wnt10b. Indeed, preceding necroptosis induction, MLKL is also located in the nucleus without being associated with the nuclear membrane (Yoon et al., 2016). Noticeably, preventing MLKL translocation at the plasma membrane using Necrosulfonamide (NSA) has no impact on its nuclear translocation, suggesting that the transit of MLKL to either the plasma membrane or the nucleus is independent (Yoon et al., 2016). Recently, MLKL was found to act as part of an RNA binding protein complex in association with RNA-binding motif protein 6 (RBM6) to stabilize a variety of transcripts including several adhesion molecules and demonstrating that MLKL is, indeed, involved in post-transcriptional regulation (Dai et al., 2020). We sought to establish the localization of MLKL in adipocytes (Figure S13). Immunofluorescence analysis revealed that MLKL resides in both cytoplasm and nucleus in adipocytes (Figure S13), suggesting that MLKL may regulate the stability/degradation of Wnt10b mRNA in order to drive adipocyte differentiation. However, further studies are needed to confirm this assumption and to decipher potential mechanisms.
Mlkl−/− mice were previously reported to resist HFD-induced obesity compared to WT littermates (Xu et al., 2019). These data were also in line with our previous report showing that the inhibition of RIPK1 in mice reduced MLKL expression, which correlated with a lower weight gain compared with wild-type littermates when fed an HFD (Majdi et al., 2020). Similarly, it has been reported in 2020 in a PISA (Pathobiology for investigators, students, & academicians) meeting (https://pisa20.asip.org; abstract A073) that an 8-week exposure to either RIPK1 or MLKL antisense oligonucleotides (ASO) therapy in obese mice markedly reduced weight and fat mass compared to controls. Therefore, these in vivo data corroborate a potential role of MLKL in promoting fat mass accumulation by regulating adipogenesis; however, conditional murine models, which are still lacking, are mandatory to confirm these observations and to analyze adipose tissue-specific functions of MLKL. In this line of work, our study provides further mechanistical explanation over the role of MLKL in controlling white adipocyte differentiation. Our data suggest that MLKL might differentially regulate white and beige adipose tissue development, stressing the need to study white vs. beige adipose tissue repartition, energy expenditure, and thermoregulation in adipose-specific Mlkl-deficient mice.
Beige adipocytes rely heavily on mitochondrial respiration to maintain their normal physiological function of thermogenesis (Cedikova et al., 2016). Previous work demonstrated that PPARγ is a key regulator of both white and beige adipocytes. However, ectopic expression of PPARγ alone gives rise to white adipocytes (Uldry et al., 2006). PGC-1, a co-activator of PPARγ, is also involved in mitochondrial biogenesis, and known to induce UCP-1 expression, a key regulator of thermogenesis and beige adipocyte function (Uldry et al., 2006). We demonstrated previously that Mlkl-deficient hepatocytes displayed higher PGC-1 levels, which were associated with increased mitochondrial respiration and biogenesis (Majdi et al., 2020). In the present study, we showed that Mlkl ablation did not impair Pgc-1 mRNA level induction in the beige adipocyte differentiation medium. Together with PPARγ, PGC-1 can drive beige adipocyte differentiation in Mlkl-deficient cells. Similarly, Ripk3-deficient tumor-associated macrophages or Ripk3−/− mice had increased mitochondrial biogenesis and respiration (Afonso et al., 2021; Majdi et al., 2020; Wu et al., 2020a). Altogether, these data highlight that neither Ripk3 nor Mlkl ablation impacts negatively on mitochondrial biogenesis and activity, which intrinsically accompanies beige adipocyte differentiation.
We herein report that Ripk3 ablation slightly increased triglyceride content in white adipocytes. Moreover, Ripk3-KO adipocytes had an enrichment in pathways involved in PPAR signaling and fatty acid beta-oxidation compared to CTL adipocytes. These data are in line with our recent study showing that the deletion of Ripk3 impacted on hepatic lipidome by upregulating PPARγ in a murine model of NAFLD (Afonso et al., 2021). In this study, PPARγ was also negatively correlated with hepatic RIPK3 in patients with NAFLD (Afonso et al., 2021). Moreover, the inhibition of RIPK3 using a Glaxo Smith Kline inhibitor increased the expression of PPARγ as well as triglyceride accumulation in ASC (Yang et al., 2022). Thus, RIPK3 appears as a lipid metabolism regulator, which activity might be targeted (e.g., by knocking down its expression or by the pharmacological inhibition of its kinase activity) to enhance lipid metabolism. However, deletion or inhibition of RIPK3 often triggers apoptosis, which may be deleterious in a pathological context (Orozco and Oberst, 2017). This phenomenon has been shown both in the liver and adipose tissue of obese mice (Gautheron et al., 2016; Roychowdhury et al., 2016). In that specific context, MLKL seems to be a more privileged target to counteract obesity. However, specific RIPK3 inhibitors deprived of pro-apoptotic effects could represent a relevant strategy.
The transcription factors that control the cascade of events leading to mature adipocytes are well characterized. For instance, it is established that the master regulators PPARγ and C/EPBα coordinate the adipocyte program. Nevertheless, secreted or circulating extra-cellular factors may regulate preadipocyte differentiation as well (Rosen and MacDougald, 2006). One of the signaling pathways that involve secreted factors is the Wnt/β-catenin pathway (Nusse and Clevers, 2017). Wnt ligands are an evolutionarily conserved family of secreted, cysteine-rich glycoproteins, whose role in cell proliferation, differentiation, and polarity during embryogenesis is well established (Nusse and Clevers, 2017). More recently, Wnt signaling has been shown to modulate other physiological and developmental processes, including adipocyte differentiation (Prestwich and Macdougald, 2007). In pre-adipocytes, Wnt/β-catenin signaling pathway maintains the cells in an undifferentiated state primarily by suppressing key adipogenic transcription factors C/EBPα and PPARγ (Prestwich and Macdougald, 2007). Among Wnt ligands, Wnt10b is expressed mostly in pre-adipocytes and its levels decline rapidly after the induction of differentiation (Ross et al., 2000). On the contrary, its overexpression in 3T3-L1 cells stabilizes β-catenin and blocks adipogenesis (Christodoulides et al., 2009). The association between Wnt10b and obesity has emerged with the identification of mutations resulting in the loss of Wnt10b function (Christodoulides et al., 2006). These mutations abrogated the ability of Wnt10b to activate canonical WNT signaling in vitro and blocked the differentiation of 3T3-L1 cells (Christodoulides et al., 2006). In the present study, we found that Mlkl deficiency in 3T3-L1 cells triggers a high expression of Wnt10b in 3T3-L1 pre-adipocytes and 7 days after the addition of the white adipocyte differentiation medium. However, Wnt10b overexpression could be a mere consequence of the downregulation of the pro-adipogenic factors seen in these cells. Indeed, PPARγ agonists induce β-catenin inhibition, while the inhibition of the canonical Wnt/β-catenin pathway activates PPARγ (Lecarpentier et al., 2017). A similar cross-talk can be found in-between C/EBPα and the Wnt/β-catenin pathway (Chung et al., 2012). Interestingly, in our dataset Wnt10b is already up-regulated in Mlkl-KO pre-adipocytes, while Pparγ is unchanged compared to CTL cells. This strongly suggests that the impact of Mlkl deficiency on the Wnt/β-catenin pathway may occur before the engagement of cells toward adipogenesis. Overall, we propose that MLKL can act as a molecular switch between these pathways in order to control adipogenesis. However, additional investigations are needed to decipher if the inhibition of MLKL regulates directly or indirectly the Wnt/β-catenin pathway.
In summary, we have shown that the ablation of Mlkl, but not Ripk3, specifically blocks white adipocyte differentiation. This study uncovers a novel metabolic function of MLKL independent of its major involvement in the necroptosis pathway that could be targeted in obesity and related diseases for therapeutic purposes.
Limitations of the study
This study essentially relies on human and mouse pre-adipocytes cellular models. Therefore, in vivo mouse models, such as adipose-cell-specific Mlkl deficiency, are needed to corroborate these data and to study white vs. beige adipose tissue distribution, energy expenditure, and thermoregulation. Specifically, compensatory mechanisms owing to the absence of MLKL in mice could happen and contradict our results. To overcome such discrepancies, the development of an inducible knockout mouse should be preferred. Further mechanistic studies are needed to corroborate and elucidate MLKL and Wnt10b interactions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-β-actin | Protein Tech | Cat# 66009-1-lg |
| Anti-Adiponectin | Thermo Fisher Scientific | Cat# MA1-054 |
| Anti-C/EBPα | Protein Tech | Cat# 18311-1-AP |
| Anti-FAS | Cell Signaling Technology | Cat# 3180 |
| Anti-GAPDH | Cell Signaling Technology | Cat# 97166 |
| Anti-MLKL | Abcam | Cat# ab172868 |
| Anti-Perilipin-1 | Abcam | Cat# ab3526 |
| Anti-PPARγ | Protein Tech | Cat# 16643-1-AP |
| Anti-RIPK3 | Biorbyt | Cat# orb74415 |
| Anti-Tubulin | Protein Tech | Cat# 66031-1-lg |
| Anti-UCP1 | Cell Signaling Technology | Cat# 54262 |
| Anti-rabbit-HRP | GE Healthcare | Cat# NA934V |
| Anti-mouse-HRP | GE Healthcare | Cat# NA931V |
| Biological samples | ||
| Adipose Stem Cells (ASC) | Pr. Fève Lab at CRSA, Paris | N/A |
| Fetal calf serum | Sigma-Aldrich | #F7524 |
| Newborn calf serum | Biosera | #CA-1151500 |
| Chemicals, peptides, and recombinant proteins | ||
| 3-isobutyl-1-methyl xanthine (IBMX) | Sigma-Aldrich | #I7018 |
| cIAP1/2 inhibitor, BV-6 | MedChemExpress | #HY-16701 |
| Cycloheximide | Sigma-Aldrich | #C4859 |
| DAPI | Sigma-Aldrich | #D1306 |
| Dexamethasone | Sigma-Aldrich | #D4902 |
| Insulin | Sigma-Aldrich | #I0516 |
| MTT | Sigma-Aldrich | #M2003-1G |
| Oil Red-O | Sigma-Aldrich | #O0625 |
| Pan-caspase inhibitor, Z-VAD(OMe)-FMK | MedChemExpress | #HY-16658 |
| Propidium iodine (PI) | Sigma-Aldrich | #P4170 |
| Rosiglitazone | Sigma-Aldrich | #D2408 |
| TNF-α | PeproTech | #315-01A |
| Triiodothyronine (T3) | Sigma-Aldrich | #T6397 |
| Critical commercial assays | ||
| MycoAlertTM PLUS Mycoplasma Detection Kit | Lonza | #LT07-701 |
| Nucleospin RNAkit | Macherey-Nagel | #740955 |
| Trigyceride Infinity Kit | Thermo Fisher Scientific | #TR22421 |
| TurboFect | Thermo Fisher Scientific | #R0532 |
| Deposited data | ||
| RNA sequencing | Novogene Europe | GSE201450 |
| Experimental models: Cell lines | ||
| 3T3-L1 | ATCC | CL-173 |
| Experimental models: Organisms/strains | ||
| Leptin-deficient obese model | Janvier labs | B6.V-Lepob/ob/JRj |
| Long-form leptin receptor-deficient obese model | Janvier labs | BKS-Lepr/db/db/JOrlRj |
| Wild type | Janvier labs | C57BL/6JRj |
| Oligonucleotides | ||
| See Table S2 | ||
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | plasmid #48138 |
| Software and algorithms | ||
| Prism | Graphpad Software | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jérémie Gautheron at jeremie.gautheron@inserm.fr.
Materials availability
Unique materials generated in this study is available upon complete materials transfer agreement.
Experimental model and subject details
Animal model
12-week-old-male homozygous ob/ob mice (B6.V-Lepob/ob/JRj; leptin-deficient obese model), db/db mice (BKS-Lepr/db/db/JOrlRj; long-form leptin receptor-deficient obese model), and C57BL/6JRj wildtype mice were obtained from Janvier Labs (Le Genest-Saint-Isle, France). All experiments were conducted in the CRSA animal facility (Agreement No. C-75-12-01), according to the European Communities Council Directive (2010/63/UE) for the care and use of animals for experimental procedures. All were approved (No. B751201) by Ethics Committee of Animal Experiments Charles Darwin.
Cell culture
3T3-L1 pre-adipocytes purchased from ATCC with undetected mycoplasma contamination (MycoAlertTM PLUS Mycoplasma Detection Kit; #LT07-701; Lonza, Bale, Switzerland) were maintained in an undifferentiated state in high-glucose (25 mmol/L) Dulbecco’s Modified Eagle’s Medium (DMEM; #11960085; Thermo Fisher Scientific) supplemented with 10% newborn calf serum (#CA-1151500; Biosera, MI, USA) and 1% Penicillin/Streptomycin (P/S). White adipocyte differentiation was induced by treating 2-day post-confluent cultures with high-glucose (4.5 g/L) DMEM supplemented with 10% fetal calf serum (FCS) (#F7524; Sigma-Aldrich, MI, USA), 1% P/S, 1 μmol/L dexamethasone (#D4902; Sigma-Aldrich), 500 μmol/L 3-isobutyl-1-methyl xanthine (IBMX) (#I7018; Sigma-Aldrich) and 0.17 μmol/L insulin (#I0516; Sigma-Aldrich) for three days. The medium was then replaced with high-glucose DMEM supplemented with 10% FCS, 1% P/S and 0.17 μmol/L insulin, and changed to fresh medium every day until the 6th day. Beige adipocyte differentiation was performed as white adipocyte differentiation with the supplementation of 1 nmol/L rosiglitazone (#D2408; Sigma-Aldrich) and 2 nmol/L triiodothyronine (T3) (#T6397; Sigma-Aldrich) (Asano et al., 2014; Ohno et al., 2012).
Human adipose stem cells (ASC) were isolated from surgical samples of sub-cutaneous abdominal adipose tissue from a control woman with normal BMI. Adipose tissue samples were enzymatically digested with collagenase B (0.2%). After centrifugation, stromal vascular fraction was filtered, rinsed, plated and cultured in α-MEM with 10% FCS, 2 mmol/L glutamine, 1% P/S (10,000 UI/mL), 1% Hepes and Fibroblast Growth Factor-2 (145 nmol/L). After 24 h, only ASC adhered to plastic surfaces, while other cells were removed after culture medium replacement. ASC were maintained in an undifferentiated state in high-glucose (4.5 g/L) DMEM supplemented with 10% newborn calf serum and 1% P/S. All culture conditions were kept constant throughout the experiments. ASC differentiation was induced as described before (Gautheron et al., 2021, 2022). Briefly, 2-day post-confluent cultures were treated with high-glucose (25 mmol/L) DMEM supplemented with 10% FCS, 1% P/S, 1 μmol/L dexamethasone, 1 μmol/L rosiglitazone, 250 μmol/L IBMX and 0.17 μmol/L insulin for ten days. The medium was then replaced with high-glucose DMEM supplemented with 10% FCS, 1% P/S, 1 μmol/L rosiglitazone and 0.17 μmol/L insulin, and changed to fresh medium every 2 days until the 20th day.
Method details
CRISPR/Cas9-mediated deletion of Ripk3 or Mlkl in 3T3-L1 pre-adipocytes
pSpCas9(BB)-2A-GFP (PX458) was a gift from Zhang lab (Addgene, MA, USA; plasmid #48138) and was used to transfect 3T3-L1 cells with Cas9 along with the targeting guide RNAs (gRNAs). gRNAs were designed and checked for efficiency (http://cistrome.org/SSC) and specificity (http://crispr.mit.edu). We used the web-based tool, CRISPOR (http://crispor.tefor.net/) to avoid off-target sequences (Table S1). Subsequently, gRNAs were cloned in the plasmid and transfected into cells using TurboFect (#R0532; Thermo Fisher Scientific) according to the manufacturer’s instructions. Forty-eight hours post-transfection, cells were sorted by flow cytometry (Cell Sorting Core Facility, CRSA) and cells with the highest GFP positivity were transferred into a 24-well plate and propagated. The percentage of on-target recombination including insertions and deletions (indels) in the genomic DNA from these cell populations was evaluated by Sanger sequencing followed by analysis using the Tide web-based tool (https://tide.nki.nl) (Figures S1 and S2). The gRNA sequences used in this study were the following:
| gRNA | Sense primer | Antisense primer |
|---|---|---|
| Ripk3 | 5′-TGGTGCGTCAGCGGTTCCTC-3′ | 5′-GAGGAACCGCTGACGCACCA-3′ |
| Mlkl | 5′-CGTCTAGGAAACCGTGTGCA-3′ | 5′-TGCACACGGTTTCCTAGACG-3′ |
CRISPR/Cas9-mediated deletion of MLKL in human ASCs
The lentiviral plasmid plentiCRISPRv2 was a gift from Zhang lab (Addgene, MA, USA; plasmid #52961) and contains the puromycin resistance, hSpCas9 and the chimeric gRNAs. The gRNA targeting MLKL was designed and checked for efficiency (http://cistrome.org/SSC) and specificity (http://crispr.mit.edu). The web-based tool, CRISPOR (http://crispor.tefor.net) was used to evaluate potential off-target sequences (Table S1). Subsequently, the gRNA was cloned into plentiCRISPRv2 and lentivirus were produced by the VVTG platform (SFR Necker, France). ASC were infected with viral particles at a minimal titer of 108 transducing units per mL. Forty-eight hours post-infection, the cells were selected with 5 μg/mL puromycin dihydrochloride (#P9620; Sigma-Aldrich). Surviving cells were propagated and the heterogeneous cell pool was used for experiments. The percentage of on-target recombination including insertions and deletions (indels) in the genomic DNA from the KO cells was evaluated by Sanger sequencing followed by analysis using the Synthego web-based tool (https://ice.synthego.com) (Figure S6). The gRNA sequences used in this study were the following:
| gRNA | Sense primer | Antisense primer |
|---|---|---|
| MLKL | 5′-AGTGAGGCAGACTTTCAATA-3′ | 5′-TATTGAAAGTCTGCCTCACT-3′ |
RNA sequencing
Total RNA was extracted from 3 replicates of undifferentiated (D0) and differentiated (D7) 3T3-L1 cells using Qiagen RNeasy mini kit per manufacturer’s instructions (Qiagen, Hilden, Germany). RNA integrity (RIN) was assessed using Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA with a RIN above 9 were used to establish the sequencing libraries. PolyA mRNA selection, cDNA libraries preparation and sequencing were conducted by Novogene Co., LTD (Beijing, China) using the Illumina NOVAseq 6000 plateform and 150 bp paired-end reads sequencing. Sequencing depth ranged from 26’310’118 to 46’400’698 reads per sample. Reads’ quality check was examined using FastQC v0.11.9. Clean reads were obtained using Fastp v0.20.1 to remove low-quality reads and read adapters. Clean reads were mapped on the USCS Mus musculus mm10 reference genome using HISAT2 v2.0.5 (Kim et al., 2019). Transcripts’ abundance was quantified using HTSeq v0.6.1 (Anders et al., 2015). The differential expression analysis was performed using the DEseq2 package (Dillies et al., 2013). Genes with an adjusted p-value <0.05 were further used for analysis. R software and ggplot2 v3.3.4 were used to generate volcano plots and perform the principal component analysis in-between Ripk3-KO, Mlkl-KO and CTL replicates. Gene ontology, pathways enrichment analysis and protein-protein interaction networks were generated using STRING Reactome (www.reactome.org) (Szklarczyk et al., 2021), KEGG (Kanehisa and Goto, 2000) and Wikipathways databases (https://wikipathways.org). RNAseq data are deposited on the NCBI Gene Expression Omnibus (GEO) repository under the accession number GSE201450.
Real-time quantitative PCR (RT-qPCR)
Total RNA was purified from cells using Nucleospin RNAkit (#740955, Macherey-Nagel, Düren, Germany). The quantity and quality of RNA were determined spectroscopically using a nanodrop (Thermo Fisher Scientific). Total RNA (2 μg) was used to synthesize cDNA using the M-MLV reverse transcriptase kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The cDNA samples (2 μL) were used for RT-qPCR in a total volume of 10 μL using SYBR Green Reagent (Roche Diagnostics, Meylan, France) and specific primers, on a LightCycler 96 Roche Instrument. All RT-qPCRs were performed in duplicate. Data were generated and analyzed using the LightCycler 96 software 1.1.0. All values were normalized for the level of hydroxymethylbilane synthase (Hmbs) or hypoxanthine guanine phosphoribosyltransferase (Hprt) mRNAs. The list of primer sequences is available in Table S2.
Western blot
Cells were homogenized in NP-40 lysis buffer to obtain protein lysates. Thirty micrograms of protein extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride membrane and analyzed by immunoblotting. Adipose tissues were dissociated and homogenized with ceramic beads and NP-40 lysis buffer using a Bead Ruptor (#19-042E, OMNI International, GA, USA).
Cell viability assay
Cells were incubated with different combination of a pan-caspase inhibitor, Z-VAD(OMe)-FMK (20 μmol/L, #HY-16658, MedChemExpress, NJ, USA), a cIAP1/2 inhibitor, BV-6 (10 μmol/L, #HY-16701, MedChemExpress) and an inhibitor of protein synthesis, cycloheximide (CHX) (0.5 μg/mL, #C4859, Sigma-Aldrich) 1 h before TNF-α stimulation (20 ng/mL, #315-01A, PeproTech, Neuilly-sur-Seine, Paris). Cell viability was determined by using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium(MTT) colorimetric assay (Thermo Fisher Scientific), which measures cell metabolic activity. It is based on the ability of nicotinamide adenine dinucleotide phosphate (NADPH) to reduce the MTT to its insoluble formazan end product, which has a purple color. Cells were incubated with 0.5 mg/mL of MTT reagent (Thermo Fisher Scientific) for 2 h. Once MTT crystals were developed and controlled under light microscopy, they were dissolved in DMSO and quantified by measuring absorbance at 540 nm.
Cell cycle arrest assay
The 3T3-L1 cells were harvested, washed with PBS, and fixed with precooled 70% ethanol in the dark at −20°C for 1 h. Then, the fixed cells were washed with PBS and treated with RNase-A (#R4875, Sigma-Aldrich) at 37°C for 30 min. Finally, the cells were stained with propidium iodine (PI, #P4170, Sigma-Aldrich) at 4°C for additional 30 min and then measured through Gallios (Beckman-Coulter) FACS caliber.
Quantification of intracellular triglyceride content
Intracellular lipids were extracted from cells using hexane/isopropyl alcohol (3:2). Cells were washed and incubated with 500 μL hexane/isopropyl alcohol (3:2, vol/vol, per well in 12-well culture plates), in a shaker (80 rpm/minute) at room temperature for 60 minutes. The contents of all wells were then transferred into a glass tube for nitrogen evaporation of the organic solvent. After evaporation, lipids were resuspended in isopropyl alcohol and transferred into duplicate 96-well plates for analysis after drying. Triglycerides were measured using InfinityTM Triglyceride kit (Thermo Fisher Scientific) according to manufacturer’s instructions. The absorbance of each well was measured using a Tecan microplate reader (TECAN) and converted to concentration based on a standard curve. Results were normalized to the cell protein content.
Oil Red-O staining, image processing, and quantification
Intracellular lipids were stained by Oil Red-O (#O0625; Sigma-Aldrich). Cells were washed with PBS and fixed with 4% PFA in PBS, for 10 min. Fixed cells were incubated with Oil Red-O solution for 1 h at room temperature and then with DAPI (#D1306, Thermo Fischer Scientific) for 5 min. Fluorescence images were generated with IX83 Olympus microscope, acquired with Cell-Sens V1.6 and analyzed with FIJI software. Images of 8–10 different areas per condition were visualized by fluorescence microscopy using mCherry and DAPI filters, followed by computer image analysis using FIJI software. Briefly, analysis was performed by threshold converting the 8-bit Red-Green-Blue image into a binary image, which consists only of pixels representing lipid droplets (i.e., red). Importantly, after separation, the binary image was manually compared with the original image for consistency and correct binary conversion. The area occupied by lipid droplets in the image was displayed by FIJI software as surface area in μm2 and normalized to cell number by semi-automated counting of DAPI-stained nuclei.
Immunofluorescence
Cells were plated on coverslips in 6-well plates. After differentiation, cells were washed and fixed with 4% PFA for 10 min. Cells were then quenched (NH4Cl) and permeabilized (PBS-0.5% triton, 10 min). After blocking with a solution of PBS-0.5% Tween-5% BSA, the cells were incubated overnight with primary antibody (see above key resources table). All cells were counterstained with DAPI for 5 min. Fluorescence images were generated with IX83 Olympus microscope (using EGFP and DAPI filters), acquired with Cell-Sens V1.6 and analyzed with FIJI software.
Quantification and statistical analysis
Statistical analysis
Data are presented as means ± SEM (standard error of the mean). GraphPad Prism software (GraphPad Software) was used to calculate statistical significance. Gaussian distribution was tested with the D’Agostino-Pearson normality test. p values were determined by analysis of variance (ANOVA) with the Geisser-Greenhouse correction and Tukey’s multiple comparisons test. Differences between 2 groups were determined using Student’s t test. p < 0.05 was considered statistically significant.
Acknowledgments
Jérémie Gautheron is funded by the Fondation pour la Recherche Médicale (FRM – ARF20170938613 & EQU202003010517), the Société Francophone du Diabète (SFD – R19114DD), the Mairie de Paris (Emergences – R18139DD) and the Agence Nationale de la Recherche (ANR-21-CE18-0002-01). Cecília M. P. Rodrigues is funded by the national funds through Fundação para a Ciência e a Tecnologia to CMPR (grants SAICTPAC/0019/2015 - LISBOA-01-0145-FEDER-016405 and PTDC/MED-FAR/29097/2017 - LISBOA-01-0145-FEDER-029097) and the EU H2020 Marie Sklodowska-Curie 722619 grant.
Author contributions
J.M. performed, analyzed, and interpreted most of the experiments; C.B. analyzed and interpreted the RNA sequencing experiments; M.A. and J-L. D. provided technical and supervisory assistance; MB. A., P-A. S., G.C., T.A-S., C.H., I.J., B.F., V.R., and CMP. R. provided important intellectual input; C.B. and J.G. wrote the article; J.G. conceived and designed the study, and obtained funding for the study. C.H., I.J., B.F., and V.R. critically revised the article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105166.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Gene expression data are available through the Gene Expression Omnibus (GEO) repository under the accession number GSE201450. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Afonso M.B., Rodrigues P.M., Carvalho T., Caridade M., Borralho P., Cortez-Pinto H., Castro R.E., Rodrigues C.M.P. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015;129:721–739. doi: 10.1042/CS20140732. [DOI] [PubMed] [Google Scholar]
- Afonso M.B., Rodrigues P.M., Mateus-Pinheiro M., Simão A.L., Gaspar M.M., Majdi A., Arretxe E., Alonso C., Santos-Laso A., Jimenez-Agüero R., et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021;70:2359–2372. doi: 10.1136/gutjnl-2020-321767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoudjehane L., Gautheron J., Le Goff W., Goumard C., Gilaizeau J., Nget C.S., Savier E., Atif M., Lesnik P., Morichon R., et al. Novel defatting strategies reduce lipid accumulation in primary human culture models of liver steatosis. Dis. Model. Mech. 2020;13:dmm042663. doi: 10.1242/dmm.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano H., Kanamori Y., Higurashi S., Nara T., Kato K., Matsui T., Funaba M. Induction of beige-like adipocytes in 3T3-L1 cells. J. Vet. Med. Sci. 2014;76:57–64. doi: 10.1292/jvms.13-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidya R., Crawford D.H.G., Gautheron J., Wang H., Bridle K.R. Necroptosis in hepatosteatotic ischaemia-reperfusion injury. Int. J. Mol. Sci. 2020;21:E5931. doi: 10.3390/ijms21165931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidya R., Gautheron J., Crawford D.H.G., Wang H., Bridle K.R. Inhibition of MLKL attenuates necroptotic cell death in a murine cell model of ischaemia injury. J. Clin. Med. 2021;10:E212. doi: 10.3390/jcm10020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K., Berry R., Wang T., Ceyhan O., Tavazoie S., Friedman J.M., Rodeheffer M.S. Analysis of gene networks in white adipose tissue development reveals a role for ETS2 in adipogenesis. Development. 2011;138:4709–4719. doi: 10.1242/dev.067710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn W.P., Bree A.J., Yao Y., Du B., Hemati N., Martinez-Santibañez G., MacDougald O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone. 2012;50:477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedikova M., Kripnerová M., Dvorakova J., Pitule P., Grundmanova M., Babuska V., Mullerova D., Kuncova J. Mitochondria in white, brown, and beige adipocytes. Stem Cells Int. 2016;2016:6067349. doi: 10.1155/2016/6067349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulides C., Lagathu C., Sethi J.K., Vidal-Puig A. Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 2009;20:16–24. doi: 10.1016/j.tem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulides C., Scarda A., Granzotto M., Milan G., Dalla Nora E., Keogh J., De Pergola G., Stirling H., Pannacciulli N., Sethi J.K., et al. WNT10B mutations in human obesity. Diabetologia. 2006;49:678–684. doi: 10.1007/s00125-006-0144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S.S., Lee J.S., Kim M., Ahn B.Y., Jung H.S., Lee H.M., Kim J.W., Park K.S. Regulation of Wnt/beta-catenin signaling by CCAAT/enhancer binding protein beta during adipogenesis. Obesity. 2012;20:482–487. doi: 10.1038/oby.2011.212. [DOI] [PubMed] [Google Scholar]
- Cinti S., Mitchell G., Barbatelli G., Murano I., Ceresi E., Faloia E., Wang S., Fortier M., Greenberg A.S., Obin M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- Dai J., Zhang C., Guo L., He H., Jiang K., Huang Y., Zhang X., Zhang H., Wei W., Zhang Y., et al. A necroptotic-independent function of MLKL in regulating endothelial cell adhesion molecule expression. Cell Death Dis. 2020;11:282. doi: 10.1038/s41419-020-2483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillies M.A., Rau A., Aubert J., Hennequet-Antier C., Jeanmougin M., Servant N., Keime C., Marot G., Castel D., Estelle J., et al. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2013;14:671–683. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- Field A.E., Coakley E.H., Must A., Spadano J.L., Laird N., Dietz W.H., Rimm E., Colditz G.A. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch. Intern. Med. 2001;161:1581–1586. doi: 10.1001/archinte.161.13.1581. [DOI] [PubMed] [Google Scholar]
- Gautheron J., Gores G.J., Rodrigues C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020;73:394–408. doi: 10.1016/j.jhep.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J., Lima L., Akinci B., Zammouri J., Auclair M., Ucar S.K., Ozen S., Altay C., Bax B.E., Nemazanyy I., et al. Loss of thymidine phosphorylase activity disrupts adipocyte differentiation and induces insulin-resistant lipoatrophic diabetes. BMC Med. 2022;20:95. doi: 10.1186/s12916-022-02296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J., Morisseau C., Chung W.K., Zammouri J., Auclair M., Baujat G., Capel E., Moulin C., Wang Y., Yang J., et al. EPHX1 mutations cause a lipoatrophic diabetes syndrome due to impaired epoxide hydrolysis and increased cellular senescence. Elife. 2021;10:e68445. doi: 10.7554/eLife.68445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J., Vucur M., Luedde T. Necroptosis in nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2015;1:264–265. doi: 10.1016/j.jcmgh.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J., Vucur M., Reisinger F., Cardenas D.V., Roderburg C., Koppe C., Kreggenwinkel K., Schneider A.T., Bartneck M., Neumann U.P., et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014;6:1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheron J., Vucur M., Schneider A.T., Severi I., Roderburg C., Roy S., Bartneck M., Schrammen P., Diaz M.B., Ehling J., et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat. Commun. 2016;7:11869. doi: 10.1038/ncomms11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H., Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw E.E., Flier J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecarpentier Y., Claes V., Vallée A., Hébert J.L. Interactions between PPAR gamma and the canonical Wnt/beta-catenin pathway in type 2 diabetes and colon cancer. PPAR Res. 2017;2017:5879090. doi: 10.1155/2017/5879090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A., Hackl M.J., Kunzendorf U., Walczak H., Krautwald S., Jevnikar A.M. Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant. 2013;13:2797–2804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- MacReady N. Leptin: 20 years later. Lancet Diabetes Endocrinol. 2014;2:864. doi: 10.1016/S2213-8587(14)70224-4. [DOI] [PubMed] [Google Scholar]
- Majdi A., Aoudjehane L., Ratziu V., Islam T., Afonso M.B., Conti F., Mestiri T., Lagouge M., Foufelle F., Ballenghien F., et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 2020;72:627–635. doi: 10.1016/j.jhep.2019.11.008. [DOI] [PubMed] [Google Scholar]
- McIlwain D.R., Berger T., Mak T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R., Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Ohno H., Shinoda K., Spiegelman B.M., Kajimura S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S., Oberst A. RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol. Rev. 2017;277:102–112. doi: 10.1111/imr.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich T.C., Macdougald O.A. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr. Opin. Cell Biol. 2007;19:612–617. doi: 10.1016/j.ceb.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed A., Robichaud S., Nguyen M.A., Geoffrion M., Wyatt H., Cottee M.L., Dennison T., Pietrangelo A., Lee R., Lagace T.A., et al. Loss of MLKL (mixed lineage kinase domain-like protein) decreases necrotic Core but increases macrophage lipid accumulation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020;40:1155–1167. doi: 10.1161/ATVBAHA.119.313640. [DOI] [PubMed] [Google Scholar]
- Rodriguez D.A., Weinlich R., Brown S., Guy C., Fitzgerald P., Dillon C.P., Oberst A., Quarato G., Low J., Cripps J.G., et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23:76–88. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen E.D., MacDougald O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- Ross S.E., Hemati N., Longo K.A., Bennett C.N., Lucas P.C., Erickson R.L., MacDougald O.A. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S., McCullough R.L., Sanz-Garcia C., Saikia P., Alkhouri N., Matloob A., Pollard K.A., McMullen M.R., Croniger C.M., Nagy L.E. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology. 2016;64:1518–1533. doi: 10.1002/hep.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed W.K., Jun D.W., Jang K., Oh J.H., Chae Y.J., Lee J.S., Koh D.H., Kang H.T. Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J. Gastroenterol. Hepatol. 2019;34:2206–2218. doi: 10.1111/jgh.14740. [DOI] [PubMed] [Google Scholar]
- Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. J. Clin. Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Yu Z., Yang S., Jiang C., Kou Y., Xiao L., Tang S., Zhu T. A transcriptomic analysis reveals novel patterns of gene expression during 3T3-L1 adipocyte differentiation. Front. Mol. Biosci. 2020;7:564339. doi: 10.3389/fmolb.2020.564339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P., et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Wang C.Z., Wei D., Guan M.P., Xue Y.M. Triiodothyronine regulates distribution of thyroid hormone receptors by activating AMP-activated protein kinase in 3T3-L1 adipocytes and induces uncoupling protein-1 expression. Mol. Cell. Biochem. 2014;393:247–254. doi: 10.1007/s11010-014-2067-6. [DOI] [PubMed] [Google Scholar]
- Wang Z., Jiang H., Chen S., Du F., Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Wu L., Zhang X., Zheng L., Zhao H., Yan G., Zhang Q., Zhou Y., Lei J., Zhang J., Wang J., et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol. Res. 2020;8:710–721. doi: 10.1158/2326-6066.CIR-19-0261. [DOI] [PubMed] [Google Scholar]
- Wu X., Poulsen K.L., Sanz-Garcia C., Huang E., McMullen M.R., Roychowdhury S., Dasarathy S., Nagy L.E. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 2020;73:616–627. doi: 10.1016/j.jhep.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Du X., Liu G., Huang S., Du W., Zou S., Tang D., Fan C., Xie Y., Wei Y., et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019;23:14–23. doi: 10.1016/j.molmet.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Qi Z., Yang X., Gao Q., Hu Y., Yuan X. Inhibition of RIP3 increased ADSC viability under OGD and modified the competency of adipogenesis, angiogenesis, and inflammation regulation. Biosci. Rep. 2022;42 doi: 10.1042/BSR20212808. BSR20212808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wang Y., Zhang Y., He X., Zhong C.Q., Ni H., Chen X., Liang Y., Wu J., Zhao S., et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018;20:186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- Yoon S., Bogdanov K., Kovalenko A., Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–260. doi: 10.1038/cdd.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang S., Tang M.B., Luo H.Y., Shi C.H., Xu Y.M. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 2017;8:e2905. doi: 10.1038/cddis.2017.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F., Liu Y., Zheng W., Shang H., Zhang J., et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Y., Li X., Qian S.W., Guo L., Huang H.Y., He Q., Liu Y., Ma C.G., Tang Q.Q. Down-regulation of type I Runx2 mediated by dexamethasone is required for 3T3-L1 adipogenesis. Mol. Endocrinol. 2012;26:798–808. doi: 10.1210/me.2011-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhe-Wei S., Li-Sha G., Yue-Chun L. The role of necroptosis in cardiovascular disease. Front. Pharmacol. 2018;9:721. doi: 10.3389/fphar.2018.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Gene expression data are available through the Gene Expression Omnibus (GEO) repository under the accession number GSE201450. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.