

Abstract
Tumor stem cells (TSCs), capable of self‐renewal and continuous production of progeny cells, could be potential therapeutic targets. We have recently reported that chromatin remodeling regulator Brg1 is required for maintenance of murine intestinal TSCs and stemness feature of human colorectal cancer (CRC) cells by inhibiting apoptosis. However, it is still unclear how BRG1 suppression changes the underlying intracellular mechanisms of human CRC cells. We found that Brg1 suppression resulted in upregulation of the JNK signaling pathway in human CRC cells and murine intestinal TSCs. Simultaneous suppression of BRG1 and the JNK pathway, either by pharmacological inhibition or silencing of c‐JUN, resulted in even stronger inhibition of the expansion of human CRC cells compared to Brg1 suppression alone. Consistently, high c‐JUN expression correlated with worse prognosis for survival in human CRC patients with low BRG1 expression. Therefore, the JNK pathway plays a critical role for expansion and stemness of human CRC cells in the context of BRG1 suppression, and thus a combined blockade of BRG1 and the JNK pathway could be a novel therapeutic approach against human CRC.
Keywords: apoptosis, colorectal cancer, epigenetics, prognosis, tumor stem cell
It is reported that BRG1 suppression causes regression of human colorectal cancer cells. Moreover, simultaneous suppression of BRG1 and the JNK pathway resulted in even stronger inhibition of the expansion of human CRC cells compared to Brg1 suppression alone. Therefore, combined blockade of BRG1 and the JNK pathway could be a novel therapeutic approach against human CRC.
Abbreviations
- 4‐OHT
4‐hydroxytamoxifen
- BRG1
Brahma‐related gene 1
- BRM
Brahma
- CRC
colorectal cancer
- DA
Dclk1 CreERT2‐IRES‐EGFP/+ ; Apc Min/+ ; Brg1 flox/wt or Dclk1 CreERT2‐IRES‐EGFP/+ ; Apc Min/+ ; Brg1 wt/wt
- DAB
Dclk1 CreERT2‐IRES‐EGFP/+ ; Apc Min/+ ; Brg1 flox/flox
- ER
endoplasmic reticulum
- GSEA
Gene Set Enrichment Analysis
- HAB
Hnf1b CreERT2/+ ; Apc Min/+ ; Brg1 flox/flox
- hCRC
human colorectal cancer
- IHC
immunohistochemical
- qRT‐PCR
quantitative real‐time PCR
- siBRG1
siRNA targeting BRG1; siJUN, siRNA targeting JUNs
- TCGA
The Cancer Genome Atlas
- TSC
tumor stem cell
1. INTRODUCTION
Colorectal cancer is the third most common type of cancer worldwide and the second most common cause of cancer‐related death. 1 Chemotherapy is a main therapeutic option against inoperable CRCs. However, they finally acquire resistance to conventional cytotoxic chemotherapy. Thus, there is an unmet need to invent a novel therapeutic approach. In this context, drug discovery related to epigenetic modifications represents a promising field for the treatment of chemoresistant CRC. Epigenetic modifications including methylation and histone modifications and chromatin remodeling play important roles in initiation and progression of CRC. 2 , 3
Switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complexes play important roles in transcriptional regulation, DNA replication, and damage repair in an ATP‐dependent manner. 4 , 5 They contain either one of the two mutually exclusive catalytic ATPase subunits: SMARCA4 (BRG1, also known as Brahma‐related gene 1) or SMARCA2 (BRM, also known as Brahma). 6 Although human BRG1 negative cancers exist and harbor malignant behavior, 7 , 8 , 9 BRG1 has recently been shown to be overexpressed in many tumor types and is associated with tumor aggressiveness and poor prognosis, including hCRC. 10 , 11 , 12 , 13 We previously showed that Brg1 is essential for acinar cell‐derived pancreatic cancer formation by inhibiting apoptosis. 14 Previous reports, including our report, have shown that hCRCs strongly express BRG1. 12 , 15 , 16 As in other types of cancer, expression of BRG1 in hCRC is associated with recurrence, metastasis, and poor prognosis. 12 , 13 , 16 We have recently reported that Brg1 plays an essential role for maintenance of murine intestinal TSCs and for cell survival and stemness features of hCRC cells. 16 Continuous ablation of Brg1 in intestinal TSCs maintains suppression of intestinal tumors accompanied by increased apoptosis and loss of their capacity for self‐renewal in mice. However, it is still unclear how BRG1 suppression changes the underlying intracellular mechanisms of hCRC cells. In this study, we investigated the underlying molecular mechanism by which BRG1 suppression affects stemness of hCRC cells to develop a novel therapeutic approach against hCRC.
2. MATERIALS AND METHODS
2.1. Animals
The following mouse strains were used. Dclk1 CreERT2‐IRES‐EGFP mice were generated by our group, as previously described. 17 Brg1 flox mice were kindly gifted by David Reisman (University of Florida), with permission from Pierre Chambon (University of Strasbourg Institute for Advanced Study). 18 Hnf1b CreERT2 mice were also kindly gifted by Jorge Ferrer (Imperial College). 19 Apc Min mice (JAX strain 002020) were obtained from the Jackson Laboratory. Mice were housed under specific pathogen‐free conditions at the animal facilities of Kyoto University. Mice were maintained against a C57BL/6 background. Apc Min mice were maintained by breeding Apc Min male mice to C57BL/6J female mice. For induction of CreER‐mediated recombination in vivo, 200 μl of 20 mg/ml tamoxifen (Sigma‐Aldrich) in corn oil was intraperitoneally injected.
2.2. Human subjects
Specimens of surgically resected CRC were obtained from patients at Kyoto University Hospital. Clinicopathologic data were collected from medical records and pathological reports of Kyoto University Hospital. The TNM classification was decided in accordance with the UICC 8th classification.
2.3. Histological analysis
Mice tissues were fixed with 4% buffered paraformaldehyde solution overnight at 4°C. They were then paraffin‐embedded and sectioned (5 μm thickness). Sections were deparaffinized and rehydrated. Antigen retrieval for all primary Abs was achieved by boiling in 10 mM citrate buffer at pH 6.0 for 15 min. For immunohistochemistry, sections were incubated with primary Abs overnight at 4°C, followed by incubation with biotinylated secondary Ab for 1 h at room temperature. Immunoperoxidase labeling was undertaken with Vectastain ABC kit (Vector Laboratories; catalog no. PK‐6102), and sections were then colored with diaminobenzidine substrate (Dako; catalog no. K3468) and counterstained with hematoxylin. The primary Abs used in this study were obtained from the indicated suppliers as follows: rabbit anti‐p‐c‐Jun (1:200; 3270, Cell Signaling Technology) and rabbit anti‐Brg1 (1:200; ab110641, Abcam).
2.4. RNA extraction and qRT‐PCR
Total RNA was extracted from tissues or cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized using the ReverTra Ace qPCR RT Kit (Toyobo). Quantitative RT‐PCR was carried out using FastStart SYBR Green Master (Roche Applied Science) and Light Cycler 96 (Roche Applied Science). The Cq values were measured in triplicate. The expression levels were standardized by comparing to the levels of GAPDH. Primers were designed using Primerbank and are listed in Table S1.
2.5. Spheroid establishment and culture
The extracted mice intestines were washed several times with PBS and the intestinal tumors were dissected. Tumor cells were dissociated using 2.5 mg collagenase from Clostridium histolyticum (Sigma‐Aldrich) with 2.5 ml advanced DMEM/F‐12 (Thermo Fisher Scientific). After dissociation, tumor cells were collected by centrifugation and embedded in Matrigel (Corning). For spheroid culture of tumor cells, advanced DMEM/F‐12 and 10% FBS (Sigma‐Aldrich) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (FUJIFILM Wako Pure Chemical Corporation), and GlutaMAX (Thermo Fisher Scientific) was added to each well. To establish the spheroids (P0), 10 mmol/L Y‐27632 (Tocris Bioscience) was added to the culture medium. For spheroid culture of intestinal tumors of Dclk1 CreERT2‐IRES‐EGFP/+ ; Apc Min/+ ; Brg1 flox/flox spheroid, 20 ng/ml murine interleukin‐13 (PeproTech) and 1 mM valproic acid (Fujifilm Wako Pure Chemical Corporation) was added to the culture medium.
For induction of Cre‐mediated recombination in vitro, 1 μmol/L 4‐OHT (Sigma‐Aldrich) was added to the culture medium.
2.6. Cell lines
DLD‐1 (CCL‐221) and HCT 116 (CCL‐247) cells were obtained from ATCC. Culture medium was made from DMEM/F‐12 (Fujifilm Wako Pure Chemical Corporation) and supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were incubated at 37°C with 5% CO2. Cells that had undergone 5–10 passages were used for experiments. Mycoplasma was confirmed negative using an e‐Myco Mycoplasma PCR Detection Kit (iNtRON Biotechnology). SP600125 was purchased from Fujifilm Wako Pure Chemical Corporation.
2.7. Small interfering RNA transfection
Human CRC cell lines were transfected with 10 nmol/L siRNA targeting BRG1 and JUN (SMARTpool: ON‐TARGETplus SMARCA4 siRNA; Dharmacon), and siRNA nontargeting control (ON‐TARGETplus nontargeting pool; Dharmacon) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) and Opti‐MEM (Thermo Fisher Scientific). We added this mixture into dishes or wells overnight and changed to fresh medium the next day. Due to unexpected toxicity, penicillin and streptomycin were excluded from the culture medium with siRNA.
2.8. Crystal violet staining
Twenty thousand cells were seeded in 35‐mm plates overnight prior to intervention. After removal of the culture medium of cell lines, cells were gently washed with PBS. They were fixed and stained with a mixture of 6.0% v/v glutaraldehyde (Sigma‐Aldrich) and 0.4% w/v crystal violet (Merck) for 30 min. Plates were carefully washed with water so as not to tear off the fixed cells on the plates. Images were taken using an optical microscope.
2.9. Western blot analysis
Cell lines were homogenized in Cell Lysis Buffer (Cell Signaling Technology). We measured the protein concentration using a Bio‐Rad protein assay (Bio‐Rad) and added sample buffer solution with reducing reagent (6×) for SDS‐PAGE (Nacalai Tesque) to each sample after matching the concentration. Proteins were separated by SDS‐PAGE and transferred to nitrocellulose membranes. After blocking with blocking buffer (Nacalai Tesque), membranes were incubated with primary Abs overnight at 4°C. The next day, membranes were incubated with HRP‐conjugated secondary Abs (1:5000 anti‐rabbit, 7074; 1:5000 anti‐mouse, 7076; both Cell Signaling Technology). The immobilized peroxidase activity was detected using SuperSignal West Pico PLUS (Thermo Fisher Scientific). The primary Abs used in this study were obtained from the indicated suppliers as follows: rabbit anti‐p‐c‐Jun (1:500, #3270; Cell Signaling Technology), rabbit anti‐c‐Jun (1:1000, #9165; Cell Signaling Technology), and mouse anti‐β‐actin (1:10,000, A1978; Sigma‐Aldrich).
2.10. Cell proliferation assay
Cells were seeded in 96‐well plates (1000 cells/well) overnight prior to intervention and were then maintained in the presence of 100 μl culture medium. We added 20 μl CellTiter 96 Aqueous One Solution (Promega) per well. Plates were incubated for 1 h before absorbance was measured at 490 nm with a Sunrise microplate reader (Tecan).
2.11. Microarray analysis
We collected three samples with Control and three samples with siBRG1 per cell line. We used two cell lines (DLD‐1 and HCT 116). The quality of RNA extracted from cell lines was examined using a Nanodrop (Thermo Fisher Scientific). The RNA samples were hybridized using a Clariom S Assay (Thermo Fisher Scientific). Normalization was undertaken using Affymetrix Power Tools Software (Thermo Fisher Scientific). The gene expression data of the cell line was analyzed using GSEA software and the Molecular Signature Database, which was provided by the Broad Institute of MIT and Harvard. A gene set was enrolled at the Broad Institute. All original microarray data were deposited in the Gene Expression Omnibus at NCBI (accession number: GSE 169627).
2.12. Kaplan–Meier curve
For Figure 5A,B, the RNA sequencing dataset of colon adenocarcinomas combining data (n = 380) in the TCGA dataset was downloaded from cBioportal. For Figure 5A, the highest 25% and the lowest 25% of c‐JUN expression data with colon adenocarcinomas were extracted from the TCGA dataset (n = 95). For Figure 5B, the high 50% and the low 50% of c‐JUN expression data from colon adenocarcinomas with lower BRG1 expression (n = 95) was extracted from the TCGA dataset (n = 47, respectively). The analysis including the log–rank test was carried out using EZR version 1.51.
FIGURE 5.
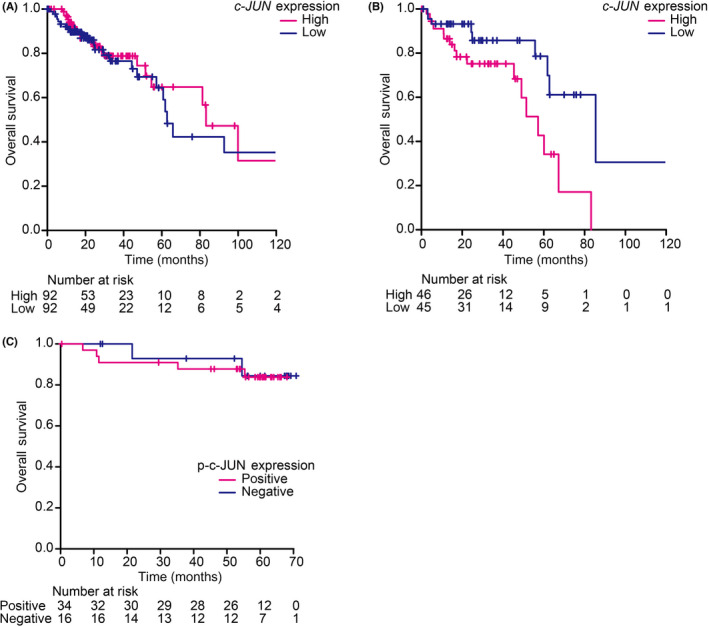
JNK pathway is crucial for expansion and stemness of human colorectal cancer (CRC) cells in the context of BRG1 suppression. (A) Kaplan–Meier analysis of CRC patients between the c‐JUN high and low expression groups (p = 0.653). (B) Kaplan–Meier analysis of CRC patients with low BRG1 expression between the c‐JUN high and low expression groups (p = 0.0131). (C) Kaplan–Meier analysis of CRC patients between p‐c‐JUN positive and p‐c‐JUN negative expression groups (p = 0.825). Analyzed by log–rank test. *p < 0.05, **p < 0.01, ***p < 0.001
FIGURE 1.
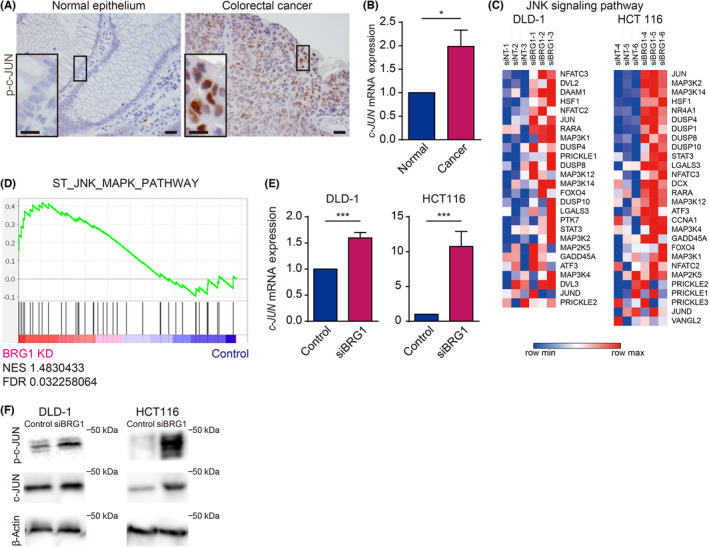
Activation of the JNK pathway in murine intestinal tumors and human colorectal cancer (hCRC) was stimulated by BRG1 suppression. (A) Immunostaining for BRG1 in human colon epithelium and CRC. Scale bars = 50 μm and 20 μm. (B) Quantitative real‐time (qRT)‐PCR analysis of BRG1 in hCRC and surrounding normal epithelium (p = 0.0255, n = 8). Means ± SEM are shown. (C) Heatmap of microarray analysis of JNK signaling pathway in DLD‐1 (left) and HCT 116 (right) cells silenced with si nontargeting RNA or siBRG1 through Morpheus (https://software.broadinstitute.org/morpheus) (n = 3 independent experiments). (D) Gene Set Enrichment Analysis of JNK pathway in hCRC cells silenced with control and siBRG1. FDR, false discovery rate; NES, normalized enrichment score. (E) qRT‐PCR analysis of c‐JUN expression in DLD‐1 and HCT 116 cells silenced with control or siBRG1 (DLD‐1, p = 0.0045; HCT 116, p = 0.0041; n = 3–4). (F) Western blot analysis for p‐c‐JUN and c‐JUN expression in DLD‐1 and HCT 116 cells silenced with control or siBRG1. Analyzed by Student' s t‐test. *p < 0.05, **p < 0.01, ***p < 0.001
2.13. Statistics
All values are presented as mean ± SEM, unless otherwise described. A two‐tailed unpaired Student's t‐test after F‐test was used for statistical analysis of continuous data. The χ2‐test was used for statistical analysis of categorical data. All statistical analyses were undertaken using GraphPad Prism version 6.07 for Windows (GraphPad Software). Those p values <0.05, <0.01, and <0.001 were considered statistically significant.
2.14. Study approval
All mouse experiments were approved by the animal research committee of Kyoto University (180260) and carried out in accordance with Japanese government regulations. Analyses for human subjects were approved by the ethical committee of Kyoto University Hospital (#G1200‐1, R2904) and carried out in accordance with the Declaration of Helsinki.
3. RESULTS
3.1. Activation of the JNK pathway augmented by BRG1 suppression in hCRC cells
We investigated the expression pattern of p‐c‐JUN by IHC analysis in hCRC. p‐c‐JUN is the activated form of c‐JUN by N‐terminal phosphorylation, and the most popular marker of the JNK pathway activation. 20 The IHC analysis of hCRC showed that p‐c‐JUN expression was not observed in the normal colon epithelium (Figure 1A, left), whereas hCRC specimen showed positive expression of p‐c‐JUN (Figure 1A, right). Quantitative RT‐PCR analysis showed that the expression level of c‐JUN mRNA was significantly higher in hCRCs than in the surrounding normal colorectal epithelium (Figure 1B).
Our recent work indicated that the expression of BRG1 is suppressed by siRNA technique at nearly 10% in hCRC cells (DLD‐1 and HCT 116) and that BRG1 silencing results in impaired cell growth, loss of stemness feature, and increased apoptosis in hCRC cells. 16 In the report, we undertook a microarray analysis of control and BRG1‐silenced hCRC cells (GSE 169627). The heatmap showed that expression of the JNK pathway genes was upregulated in BRG1‐silenced DLD‐1 and HCT 116 cells (Figure 1C). Our Kyoto Encyclopedia of Genes and Genomes pathway analysis also showed that Brg1 suppression affected gene expression patterns of the ER and MAPK signaling pathways. 16 Of the 50 most upregulated genes in BRG1‐silenced hCRC cells, we observed JUN (c‐JUN) and MAP3K14, which were known to be JNK pathway‐related genes, and DDIT3 (CHOP) and HMOX1, which were known to be ER stress‐related genes. 16 Gene Set Enrichment Analysis revealed that there was a positive enrichment of gene sets related to the JNK pathway (“ST_JNK_MAPK_PATHWAY”) in BRG1‐silenced cells (Figure 1D). These findings indicate that the JNK pathway was upregulated in the context of BRG1 suppression in hCRC cells. The qRT‐PCR analysis verified the upregulated expression of c‐JUN in BRG1‐silenced hCRC cells (Figure 1E). We found that the activated level of p‐c‐JUN was significantly increased in BRG1‐silenced DLD‐1 and HCT 116 cells (Figure 1F). Protein level of c‐JUN was elevated in BRG1‐silenced DLD‐1 and HCT 116 cells (Figure 1F).
Therefore, these findings indicate that the JNK pathway was activated to some extent at the basal level in hCRCs and the activation of the JNK pathway was further reinforced by BRG1 suppression.
3.2. Activation of the JNK pathway reinforced by Brg1 ablation in murine intestinal tumors
We next investigated whether activation of the JNK pathway was augmented by Brg1 ablation in murine intestinal tumors. Hnf1b is expressed in intestinal tumor cells and spheroids generated from intestinal tumors in Apc Min mice. 16 Therefore, we first generated HAB mice and established spheroids from intestinal tumors of HAB mice (Figure 2A,B). Through this model, we could ablate Brg1 in tumor cells of spheroids by adding 4‐OHT in the medium. Regarding Brg1 KO efficacy in HAB spheroids, we had verified the low expression of Brg1, which was shown in our previous report. 16 We determined the expression level of c‐Jun mRNA in spheroids from intestinal tumors from HAB mice compared to controls. Expression of c‐Jun was significantly upregulated in Brg1 KO spheroids from HAB mice compared to controls (Figure 2C). Immunohistochemistry for p‐c‐JUN also revealed that the JNK pathway was upregulated in Brg1 KO spheroids from HAB mice (Figure 2D).
FIGURE 2.
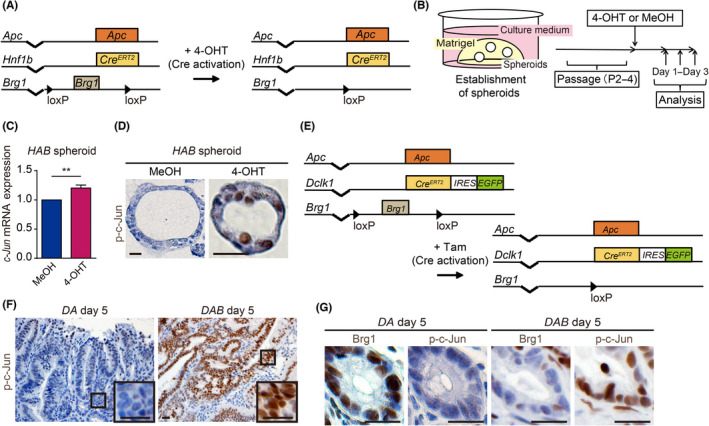
Increased activation of JNK pathway in murine intestinal tumors by Brg1 suppression. (A) Genetic strategy for Brg1 ablation in intestinal tumor cells following tamoxifen (4‐OHT) induction in Hnf1b CreERT2/+; Apc Min/+; Brg1 flox/flox (HAB) mice. (B) Experimental schedule for establishment of HAB spheroids and analysis. (C) Quantitative real‐time PCR analysis of c‐Jun expression in HAB spheroids treated with MeOH or 4‐OHT (p = 0.0080; n = 4). (D) Immunohistochemistry for p‐c‐JUN in HAB spheroids treated with MeOH or 4‐OHT. Scale bars = 20 μm. (E) Genetic strategy for Dclk1+ intestinal tumor cells specific Brg1 deletion following tamoxifen (Tam) induction in Dclk1 CreERT2‐IRES‐EGFP/+; Apc Min/+; Brg1 flox/flox (DAB) mice. (F) Immunohistochemistry for p‐c‐JUN in intestinal tumors of Dclk1 CreERT2‐IRES‐EGFP/+; Apc Min/+; Brg1 flox/wt or Dclk1 CreERT2‐IRES‐EGFP/+; Apc Min/+; Brg1 wt/wt (DA) and DAB mice 5 days after the last Tam injection. Scale bars = 100 μm and 20 μm. (G) Immunohistochemistry for Brg1 and p‐c‐Jun in serial sections of intestinal tumors in DA and DAB mice at day 5 after the last Tam injection. Scale bars = 20 μm. Analyzed by Student' s t‐test. *p < 0.05, **p < 0.01, ***p < 0.001
To further determine whether activation of the JNK pathway was reinforced by Brg1 ablation in murine intestinal tumors in mice, we next generated DAB mice along with Dclk1 CreERT2‐IRES‐EGFP/+ ; Apc Min/+ ; Brg1 wt/wt or Dclk1 CreERT2‐IRES‐EGFP/+; Apc Min/+; Brg1 wt/wt (DA) mice (Figure 2E). Because we previously reported that Dclk1 is an intestinal TSC marker, 17 we could ablate Brg1 specifically in intestinal TSCs by injecting tamoxifen intraperitoneally. 16 We previously reported that most intestinal tumor cells were Brg1 negative in DAB mice on day 5 after tamoxifen injection, resulting in the collapse of intestinal tumors. 16 Immunostaining for p‐c‐Jun revealed that p‐c‐Jun was highly expressed in intestinal tumor cells in DAB mice on day 5 after tamoxifen injection, whereas p‐c‐Jun expression was rarely detected in intestinal tumor cells in DA mice (Figure 2F). Brg1 negative intestinal tumor clusters were positive for p‐c‐Jun expression in DAB mice (Figure 2G).
These findings suggests that the JNK pathway is activated by Brg1 ablation also in murine intestinal tumors.
3.3. JNK pathway crucial for expansion and stemness of BRG1‐silenced hCRC cells
Given that the JNK pathway was upregulated in the context of BRG1 suppression in hCRC cells, we sought to clarify its role in BRG1‐suppressed hCRC cells in the context of BRG1 suppression. To this end, we first examined whether treatment with SP600125, a selective JNK pathway inhibitor, 21 affects the expansion of siRNA‐mediated BRG1‐silenced hCRC cells. We validated the efficiency of siBRG1 by qRT‐PCR (Figure S1). Western blot analysis showed that c‐JUN activation was sufficiently inhibited by SP600125 treatment (Figure 3A). We carried out an MTS assay and found that SP600125 does not influence cell viability or proliferation, or expression of stemness‐related genes in hCRC cells without treatment of siBRG1 (Figure 3B,C). We next incubated hCRC cells with siBRG1 and SP600125 and compared them with siBRG1 and vehicle control. Notably, cell expansion was significantly suppressed in the siBRG1 and SP600125 group compared to the siBRG1 and vehicle control group (Figure 3D,E). Expression of stem cell markers was significantly downregulated in the siBRG1 and SP600125 group compared to the siBRG1 and vehicle control group (Figure 3F). These results indicate that the JNK pathway is crucial for the expansion of BRG1‐silenced hCRC cells.
FIGURE 3.
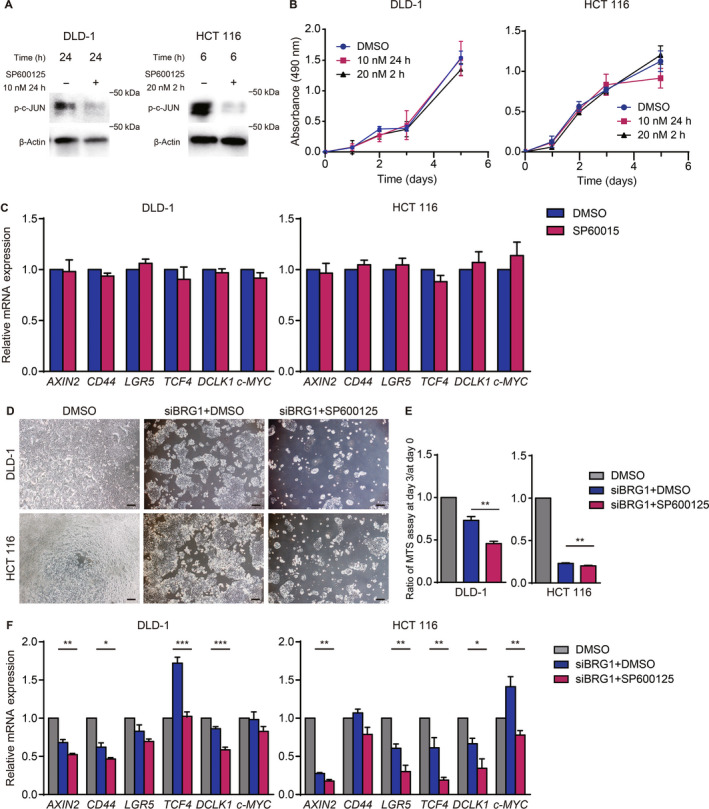
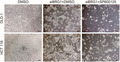
JNK pathway is crucial for expansion and stemness of human colorectal cancer cells in the context of BRG1 suppression. (A) Western blot analysis for p‐c‐JUN expression in DLD‐1 and HCT 116 cells treated with DMSO or SP600125 at 10 nM for 24 h and at 20 nM for 2 h. (B) MTS assay of DLD‐1 and HCT 116 cells treated with DMSO or SP600125 at 10 nM for 24 h and at 20 nM for 2 h (n = 3, 4). Means ± SEM are shown. (C) Quantitative real‐time (qRT)‐PCR analysis of DLD‐1 and HCT 116 cells 2 days after treatment with DMSO or SP600125 at 10 nM for 24 h (n = 4). Means ± SEM are shown. (D) Images of DLD‐1 and HCT 116 cells treated with siBRG1 + DMSO or siBRG1 + SP600125 on day 3 after siRNA treatment. Scale bars = 100 μm. (E) Ratio of MTS assay on day 3 / day 0 of DLD‐1 and HCT 116 cells silenced with DMSO, siBRG1 + DMSO, and siBRG1 + SP600125 at 10 nM for 24 h (DLD‐1, p = 0.007; HCT 116, p = 0.002; n = 5). (F) qRT‐PCR analysis of DLD‐1 and HCT 116 cells silenced with DMSO, siBRG1 + DMSO, and siBRG1 + SP600125 at 10 nM for 24 h on day 2 after siRNA treatment (n = 5). Analyzed by Student's t‐test. *p < 0.05, **p < 0.01, ***p < 0.001
Next, we undertook dual silencing of BRG1 and c‐JUN in hCRC cells by siRNA. The qRT‐PCR analysis validated that c‐JUN expression was silenced to approximately 20% in DLD‐1 and HCT 116 cells compared with controls (Figure 4A). Silencing of c‐JUN alone did not affect cell expansion or expression of stemness‐related genes in hCRC cells (Figure 4B,C). Dual silencing of BRG1 and c‐JUN inhibited the expansion of DLD‐1 and HCT 116 cells compared with silencing of BRG1 alone (Figure 4D,E). Moreover, the expression of stem cell markers was significantly downregulated in BRG1 and c‐JUN dual‐silenced hCRC cells compared to BRG1‐silenced CRC cells (Figure 4F). These results further indicated that the JNK pathway is crucial for the expansion and stemness of hCRC cells in the absence, but not in the presence, of BRG1.
FIGURE 4.
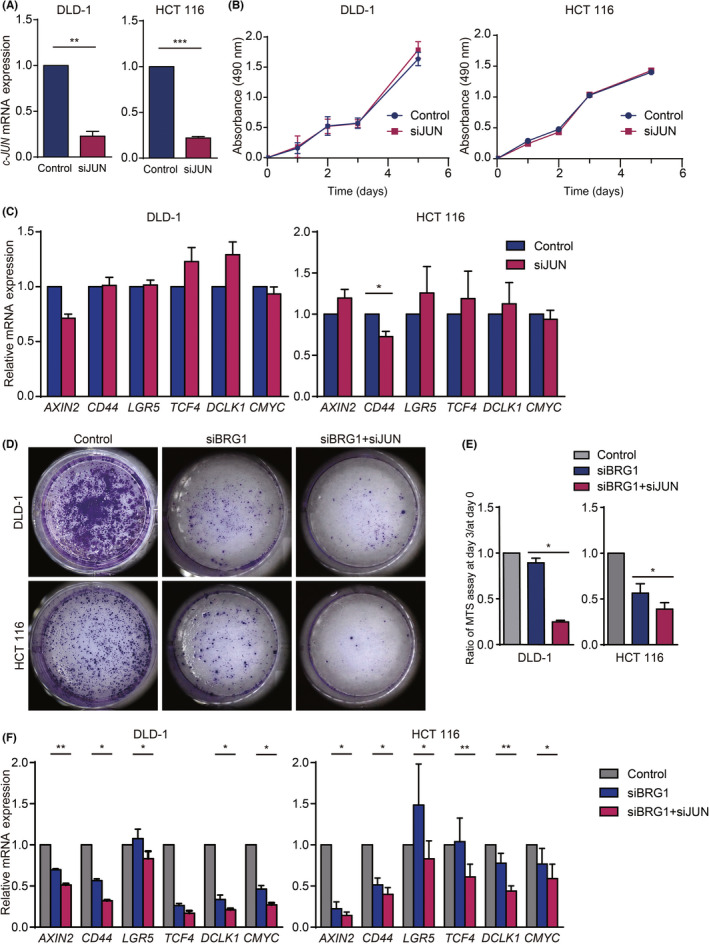
JNK pathway was crucial for expansion and stemness of human colorectal cancer cells in the context of BRG1 suppression. (A) Quantitative real‐time (qRT)‐PCR analysis of c‐JUN expression in DLD‐1 and HCT 116 cells silenced with control (si nontargeting RNA) or siJUN (DLD‐1, p = 0.0046; HCT 116, p < 0.0001; n = 3, 4). Means ± SEM are shown. (B) MTS assay of DLD‐1 and HCT 116 cells treated with control or siJUN (n = 4, 3). Means ± SEM are shown (HCT116 control: day 1, 0.288 ± 0.002, day 2, 0.477 ± 0.009, day 3, 1.027 ± 0.003, day 5, 1.403 ± 0.001; siJUN: day 1, 0.244 ± 0.005, day 2, 0.430 ± 0.005, day 3, 1.037 ± 0.004, day 5, 1.429 ± 0.011). (C) qRT‐PCR analysis of HCT 116 cells silenced with control and siJUN on day 2 after siRNA treatment (n = 5). Means ± SEM are shown. (D) Crystal violet staining of DLD‐1 and HCT 116 cells silenced with control alone, siBRG1 alone, and siBRG1 + siJUN on day 3 after siRNA treatment. (E) Ratio of MTS assay on day 3 / day 0 of DLD‐1 and HCT 116 cells silenced with control alone, siBRG1 alone, and siBRG1 + siJUN (DLD‐1, p = 0.0104; HCT 116, p = 0.0117; n = 3–4). (F) qRT‐PCR analysis of DLD‐1 and HCT 116 cells silenced with control alone, siBRG1 alone, and siBRG1 + siJUN on day 2 after siRNA treatment (n = 3). Analyzed by Student' s t‐test. *p < 0.05, **p < 0.01, ***p < 0.001
3.4. High C‐JUN expression correlated with worse prognosis for survival in CRC patients with low BRG1 expression
We next investigated the correlative relationship between c‐JUN expression and prognosis for survival in CRC patients with low BRG1 expression using the TCGA dataset. Consistent with our data of hCRC cells, there was no significant difference in prognosis for survival in total CRC patients between the group with high c‐JUN expression and the group with low c‐JUN expression (Figure 5A). Next, patients were classified according to the expression of BRG1, and only patient cohort with low BRG1 expression was further analyzed. Interestingly, among the patients with low BRG1 expression, the high c‐JUN expression group had a significantly worse prognosis for survival compared to the low c‐JUN expression group (Figure 5B). These findings further support our notion that the JNK pathway is crucial for the expansion of hCRC cells when BRG1 expression is low.
Finally, we undertook IHC analysis for p‐c‐JUN in resected hCRC specimens in our institution. Sixty‐eight percent (34/50) of hCRC specimens were positive for p‐c‐JUN (Figure 1B). There were no significant differences in clinicopathologic features between p‐c‐JUN positive hCRC and p‐c‐JUN negative hCRC (Table S2). Overall survival did not significantly differ between p‐c‐JUN positive hCRC and p‐c‐JUN negative hCRC (p = 0.825; Figure 5C). These results also suggested that activation of the JNK pathway alone is not a prognostic factor for hCRC.
These data further strengthen the human relevance of our data, providing evidence that the JNK pathway plays a crucial role for CRC in the context of BRG1 suppression. Therefore, a combined blockade of BRG1 and the JNK pathway could be a novel therapeutic approach against hCRC.
4. DISCUSSION
In this study, we found that the JNK pathway was upregulated in both hCRC cells and murine intestinal tumors by BRG1 suppression. Furthermore, we showed for the first time that the JNK pathway plays a critical role in the expansion and stemness of hCRC cells in the absence, but not in the presence, of BRG1. It is notable that suppression of BRG1 combined with the inhibition of the JNK pathway led to even stronger inhibition of expansion of hCRC cells than did Brg1 suppression alone. Therefore, our findings suggest that a combined blockade of BRG1 and the JNK pathway could be a novel therapeutic approach for hCRC.
Our data showed that JNK pathway blockade even strengthened the phenotype of BRG1‐suppressed hCRC cells, which includes loss of cell expansion and stemness features. The JNK pathway has been shown to regulate stemness in normal murine intestinal epithelial cells and human pancreatic cancer cells. 22 , 23 In this study, we showed that simultaneous suppression of BRG1 and the JNK pathway resulted in downregulated expression of stemness‐related genes in hCRC cells compared to Brg1 suppression alone, whereas JNK inhibition alone did not affect the expansion of hCRC cells or the expression of stemness‐related genes. This suggests that increased expression of JNK pathway genes is a compensatory mechanism to survive the fatal phenotypes (i.e., loss of expansion and stemness) induced by BRG1 suppression.
In this study, our findings provide important clinical relevance for the development of therapeutic strategies for hCRC. We showed that suppression of BRG1, combined with the inhibition of the JNK pathway, led to even stronger inhibition of the expansion of hCRC cells compared to Brg1 suppression alone. Consistently, it is notable that high c‐JUN expression was correlated with worse prognosis for survival in hCRC patients with low BRG1 expression. Therefore, enhancing the antitumor effect of BRG1 inhibition through the use of combination therapy involving both BRG1 suppression and JNK inhibitors could be a new therapeutic strategy for hCRC. In particular, inhibition of the JNK pathway could be effective for hCRC patients with low BRG1 expression.
In conclusion, activation of the JNK signaling was augmented by BRG1 suppression in hCRC cells and murine intestinal tumors. The JNK pathway is critical for the expansion and stemness of hCRC cells in the context of BRG1 suppression. Consistently, high c‐JUN expression correlated with worse prognosis for survival in hCRC patients with low BRG1 expression. Therefore, a combined blockade of BRG1 and the JNK pathway could be a novel therapeutic strategy against hCRC.
AUTHOR CONTRIBUTIONS
T.Y. and A.F. conceived and designed the study. M.O., M.N., M.S., Y.F., T.M., O.A., M.N., S.O., K.M., N.G., Y.H., Y.M., M.T., T. M., and Y.N. conducted the experiments and analyzed the data. K.K. and S.T. contributed reagents, materials, and analytic tools. T.Y. wrote the manuscript, and A.F. and H.S. revised the manuscript.
FUNDING INFORMATION
This work was supported in part by Grants‐in‐Aid KAKENHI (JP19H03639, 21H02902), a research program that is part of the Project for Development of Innovative Research on Cancer Therapeutics (P‐Direct) from the Ministry of Education, Culture, Sports, Science, and Technology and the Japan Society for the Promotion of Science. It was also supported by the Japan Agency for Medical Research and Development, the Project for Cancer Research and Therapeutic Evolution (P‐CREATE; 19cm0106142h0002, 20cm0106177h0001, 21cm0106283h0001) and AMED‐PRIME (20gm6010022h0003). This study was also supported by the Princess Takamatsu Cancer Research Fund (13–‐24,514, 17–‐24,924, 21–‐25,332), the Mochida Foundation (2017bvAg), the Mitsubishi Foundation (201,910,037, 281,119), the Uehara Foundation (201720143), the Naito Foundation (32924–‐1) the Takeda Foundation (201749741), and Sumitomo Dainippon Pharma.
DISCLOSURE
Y.F. is an employee of Sumitomo Dainippon Pharma. S.T. is partially supported by Sumitomo Dainippon Pharma. The other authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: Analyses for human subjects were approved by the ethical committee of Kyoto University Hospital (#G1200‐1, R2904) and conducted in accordance with the Declaration of Helsinki.
INFORMED CONSENT
N/A.
REGISTRY AND THE REGISTRATION OF THE STUDY
N/A.
ANIMAL STUDIES
All mouse experiments were approved by the animal research committee of Kyoto University (180260) and performed in accordance with Japanese government regulations.
Supporting information
Figure S1
Table S1
Table S2
ACKNOWLEDGMENTS
We thank Shoko Yokoyama for mouse bleeding and technical support, and all members of the A.F. laboratory for technical assistance and helpful discussions. We also thank D. Reisman of the University of Florida, with permission from P. Chambon, for sharing Brg1 flox mice and J. Ferrer for sharing Hnf1b CreERT2 mice.
Yoshikawa T, Fukuda A, Omatsu M, et al. JNK pathway plays a critical role for expansion of human colorectal cancer in the context of BRG1 suppression. Cancer Sci. 2022;113:3417‐3427. doi: 10.1111/cas.15520
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Jung G, Hernandez‐Illan E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17:111‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vaiopoulos AG, Athanasoula K, Papavassiliou AG. Epigenetic modifications in colorectal cancer: molecular insights and therapeutic challenges. Biochim Biophys Acta. 2014;1842:971‐980. [DOI] [PubMed] [Google Scholar]
- 4. Euskirchen G, Auerbach RK, Snyder M. SWI/SNF chromatin‐remodeling factors: multiscale analyses and diverse functions. J Biol Chem. 2012;287:30897‐30905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5:752‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481‐492. [DOI] [PubMed] [Google Scholar]
- 7. Le Loarer F, Watson S, Pierron G, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF‐deficient sarcomas. Nat Genet. 2015;47:1200‐1205. [DOI] [PubMed] [Google Scholar]
- 8. Matsubara D, Kishaba Y, Ishikawa S, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci. 2013;104:266‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tischkowitz M, Huang S, Banerjee S, et al. Small‐cell carcinoma of the ovary, hypercalcemic type‐genetics, new treatment targets, and current management guidelines. Clin Cancer Res. 2020;26:3908‐3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerrero‐Martinez JA, Reyes JC. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci Rep. 2018;8:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun A, Tawfik O, Gayed B, et al. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate. 2007;67:203‐213. [DOI] [PubMed] [Google Scholar]
- 12. Lin S, Jiang T, Ye L, et al. The chromatin‐remodeling enzyme BRG1 promotes colon cancer progression via positive regulation of WNT3A. Oncotarget. 2016;7:86051‐86063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pyo JS, Son BK, Oh D, Kim EK. BRG1 is correlated with poor prognosis in colorectal cancer. Hum Pathol. 2018;73:66‐73. [DOI] [PubMed] [Google Scholar]
- 14. Tsuda M, Fukuda A, Roy N, et al. The BRG1/SOX9 axis is critical for acinar cell‐derived pancreatic tumorigenesis. J Clin Invest. 2018;128:3475‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe T, Semba S, Yokozaki H. Regulation of PTEN expression by the SWI/SNF chromatin‐remodelling protein BRG1 in human colorectal carcinoma cells. Br J Cancer. 2011;104:146‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoshikawa T, Fukuda A, Omatsu M, et al. Brg1 is required to maintain colorectal cancer stem cells. J Pathol. 2021;255:257‐269. [DOI] [PubMed] [Google Scholar]
- 17. Nakanishi Y, Seno H, Fukuoka A, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2013;45:98‐103. [DOI] [PubMed] [Google Scholar]
- 18. Sumi‐Ichinose C, Ichinose H, Metzger D, Chambon P. SNF2beta‐BRG1 is essential for the viability of F9 murine embryonal carcinoma cells. Mol Cell Biol. 1997;17:5976‐5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Solar M, Cardalda C, Houbracken I, et al. Pancreatic exocrine duct cells give rise to insulin‐producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17:849‐860. [DOI] [PubMed] [Google Scholar]
- 20. Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c‐Jun mediated by MAP kinases. Nature. 1991;353:670‐674. [DOI] [PubMed] [Google Scholar]
- 21. Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N‐terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681‐13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sancho R, Nateri AS, de Vinuesa AG, et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843‐1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okada M, Shibuya K, Sato A, et al. Targeting the K‐Ras‐‐JNK axis eliminates cancer stem‐like cells and prevents pancreatic tumor formation. Oncotarget. 2014;5:5100‐5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2